

Abstract
Succinate dehydrogenase (SDH) is a mitochondrial enzyme that plays an important role in both the Krebs cycle and the electron transport chain. SDH inactivation is associated with tumorigenesis in certain types of tumor. SDH consists of subunits A, B, C and D (SDHA, SDHB, SDHC, and SDHD, respectively). Immunohistochemistry for SDHB is a reliable method for detecting the inactivation of SDH by mutations in SDHA, SDHB, SDHC, SDHD and SDH complex assembly factor 2 (SDHAF2) genes with high sensitivity and specificity. SDHB immunohistochemistry has been used to examine the inactivation of SDH in various types of tumors. However, data on central nervous system (CNS) tumors are very limited. In the present study, we investigated the loss of SDHB immunoexpression in 90 cases of CNS tumors. Among the 90 cases of CNS tumors, only three cases of hemangioblastoma showed loss of SDHB immunoexpression. We further investigated SDHB immunoexpression in 35 cases of hemangioblastoma and found that 28 (80%) showed either negative or weak-diffuse pattern of SDHB immunoexpression, which suggests the inactivation of SDH. Our results suggest that SDH inactivation may represent an alternative pathway in the tumorigenesis of hemangioblastoma.
Introduction
Succinate dehydrogenase (SDH) is an important mitochondrial enzyme that participates in the Krebs cycle and the electron transport chain1,2. It consists of four subunits: SDHA, SDHB, SDHC, and SDHD. Each subunit is encoded by the corresponding SDHA, SDHB, SDHC, and SDHD gene in the nucleus and is incorporated at the inner mitochondrial membrane. A functional unit, SDH complex assembly factor 2 (SDHAF2), which is encoded by the SDHAF2 gene, is also required for its enzymatic activity2–4.
Interestingly, in addition to its pivotal role in normal aerobic respiration, SDH has tumor-suppressive effects5–7. SDH inactivation results in the accumulation of succinate and induces the stabilization of hypoxia-inducible factor (HIF) via competitive inhibition of HIF prolyl-hydroxylases. Stabilized HIF activates pseudo-hypoxic signaling and leads to angiogenesis, the dysregulation of cellular proliferation, and adhesion5,8–11. The accumulation of succinate may also be associated with alteration of epigenomic landscapes favoring oncogenesis through the inhibition of histone demethylation12.
The inactivation of SDH can be caused by any mutation of SDHA, SDHB, SDHC, SDHD, or SDHAF2 (SDHx genes)2–4. Germline mutations in SDHx genes were first believed to be limited to familial paraganglioma/pheochromocytoma7. However, it has since been reported in other solid tumors, such as gastrointestinal stromal tumors (GISTs)13,14, renal cell carcinomas (RCCs)15–17, pituitary adenomas (PAs)18–20, and pancreatic neuroendocrine tumors (NETs)19.
Immunohistochemistry for SDHB is a reliable method for detecting SDHx mutations with high sensitivity and specificity21–24. Various types of tumors have been evaluated to determine the status of SDHx mutations using SDHB immunohistochemistry15,19,21–27. However, data on SDHx mutations of central nervous system (CNS) tumors are very limited19,28,29. Furthermore, to the best of our knowledge, the loss of SDHB immunoexpression has not been explored in various types of CNS tumors.
In the present study, we performed SDHB immunohistochemistry in various types of CNS tumors and found a significant proportion of hemangioblastomas with loss of SDHB immunoexpression.
Results
SDHB immunohistochemistry in CNS tumors
To screen for the inactivation of SDHB across CNS tumors, we performed SDHB immunohistochemistry using TMA blocks including 17 cases of glioblastoma, 7 of astrocytoma, 9 of oligodendroglioma, 9 of ependymoma, 10 of meningioma, 6 of hemangiopericytoma, 7 of central neurocytoma, 12 of PA, 5 of craniopharyngioma, 3 of schwannoma, and 3 of hemangioblastoma. In all, 81 cases (90%) of CNS tumors showed positive staining for SDHB in the whole tumor, and 6 cases (6.7%) revealed strong granular SDHB immunoreactivity in part of the tumor area. Among the 90 cases of CNS tumors, only 3 (3.3%) showed no immunoexpression of SDHB protein (Table 1). Interestingly, all three cases were hemangioblastoma (Fig. 1).
Table 1.
SDHB immunonegativity in 90 cases of central nervous system tumors.
| Tumor type | SDHB immunonegativity |
|---|---|
| Glioblastoma | 0/17 |
| Astrocytoma | 0/7 |
| Oligodendroglioma | 0/9 |
| Ependymoma | 0/9 |
| Central neurocytoma | 0/7 |
| Schwannoma | 0/3 |
| Meningioma | 0/10 |
| Hemangiopericytoma | 0/6 |
| Hemangioblastoma | 3/3 |
| Craniopharyngioma | 0/5 |
| Pituitary adenoma | 0/12 |
Figure 1.

SDHB immunohistochemistry showed strong granular cytoplasmic positivity in central nervous system tumors except for hemangioblastoma and control pheochromocytoma with an SDHB mutation. (a) Glioblastoma, (b) Astrocytoma, (c) Oligodendroglioma, (d) Ependymoma, (e) Central neurocytoma, (f) Meningioma, (g) Hemangiopericytoma, (h) Craniopharyngioma, (i) Pituitary adenoma, (j) Schwannoma, (k) Hemangioblastoma, (l) Control: Pheochromocytoma with an SDHB mutation. Bar indicates 100 µm.
SDHB immunohistochemistry in hemangioblastomas
To examine SDHB immunoexpression in hemangioblastoma, we performed SDHB immunohistochemistry in 35 hemangioblastoma cases with two different primary antibodies against SDHB. The clinical characteristics of the 35 patients with hemangioblastoma are summarized in Table 2. First, we performed SDHB immunohistochemistry with a primary rabbit polyclonal antibody (HPA002868). Among the 35 cases, 9 (25.7%) showed negative staining for SDHB, whereas 7 (20%) showed strong granular staining in the cytoplasm. Among the seven cases of strong granular positivity, two showed partial loss of SDHB. The remaining 19 cases (54.3%) revealed a week-diffuse pattern of SDHB immunostaining (Fig. 2) (Table 3). Next, we performed an additional SDHB immunohistochemistry with the different primary mouse monoclonal antibody (ab14714) and compared the results of both SDHB immunostainings. SDHB expression patterns in 9 of negative and 7 of strong granular staining were almost identical in both SDHB immunostainings. However, among 19 cases of a week-diffuse pattern of SDHB immunostaining with a primary rabbit polyclonal antibody, only 10 (28.6%) revealed a week-diffuse pattern of SDHB immunostaining, whereas remaining 9 (25.7%) showed negative staining (Fig. 3) (Table 3).
Table 2.
Clinical characteristics of 35 patients with hemangioblastoma.
| Male:Female | 25:10 |
| Mean age (years ± SD) | 41 ± 8.5 |
| Tumor location | |
| Cerebellum (%) | 29 (82.9) |
| Spinal Cord (%) | 6 (17.1) |
| Association with VHL | |
| Sporadic (%) | 33 (94.3) |
| VHL (%) | 2 (5.7) |
Figure 2.
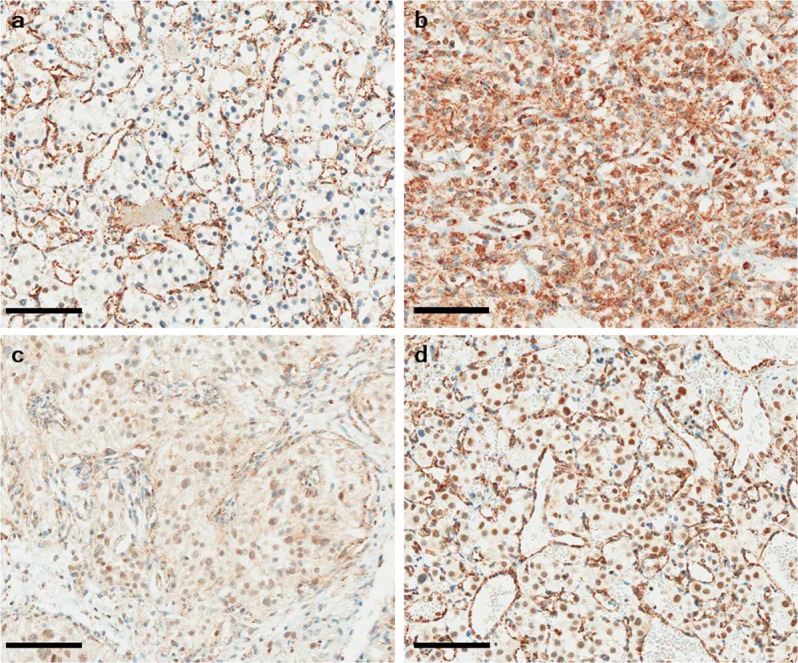
SDHB immunohistochemistry of hemangioblastoma. (a) Hemangioblastoma with SDHB immunonegativity shows no immunoexpression in the cytoplasm in the presence of strong granular staining of capillary endothelial cells (internal control). (b) Hemangioblastoma with strong granular cytoplasmic positivity. Hemangioblastoma with a weak-diffuse pattern of SDHB immunoexpression showing mild cytoplasmic (c) and/or nuclear blush staining (d). Bar indicates 100 µm.
Table 3.
Results of SDHB immunohistochemical staining in 35 cases of hemangioblastoma.
| Case no | Sex | Age | Location | SDHB (HPA002868) | SDHB (ab14714) |
|---|---|---|---|---|---|
| 1 | M | 58 | Cerebellum | Strong | Strong |
| 2 | F | 47 | Cerebellum | Negative | Negative |
| 3 | M | 25 | Cerebellum | Weak-diffuse | Weak-diffuse |
| 4 | M | 27 | Cerebellum | Weak-diffuse | Weak-diffuse |
| 5 | M | 42 | Cerebellum | Negative | Negative |
| 6 | M | 41 | Cerebellum | Weak-diffuse | Negative |
| 7 | M | 23 | Spinal cord | Weak-diffuse | Negative |
| 8 | M | 55 | Cerebellum | Negative | Negative |
| 9 | M | 41 | Spinal cord | Negative | Negative |
| 10 | M† | 30 | Cerebellum | Weak-diffuse | Weak-diffuse |
| 11 | F | 29 | Cerebellum | Strong | Strong |
| 12 | M | 35 | Cerebellum | Strong | Strong |
| 13 | M | 26 | Cerebellum | Weak-diffuse | Negative |
| 14 | M | 48 | Cerebellum | Weak-diffuse | Negative |
| 15 | F | 36 | Spinal cord | Weak-diffuse | Negative |
| 16 | M† | 30 | Cerebellum | Weak-diffuse | Weak-diffuse |
| 17 | M | 46 | Spinal cord | Negative | Negative |
| 18 | M | 15 | Spinal cord | Weak-diffuse | Weak-diffuse |
| 19 | M | 39 | Cerebellum | Strong | Strong |
| 20 | F | 75 | Cerebellum | Weak-diffuse | Weak-diffuse |
| 21 | F | 53 | Cerebellum | Weak-diffuse | Weak-diffuse |
| 22 | F | 15 | Cerebellum | Weak-diffuse | Negative |
| 23 | M | 42 | Cerebellum | Negative | Negative |
| 24 | F | 32 | Cerebellum | Negative | Negative |
| 25 | M | 51 | Cerebellum | Weak-diffuse | Weak-diffuse |
| 26 | M | 54 | Cerebellum | Strong‡ | Strong‡ |
| 27 | F | 69 | Cerebellum | Weak-diffuse | Negative |
| 28 | M | 56 | Cerebellum | Weak-diffuse | Weak-diffuse |
| 29 | M | 44 | Cerebellum | Negative | Negative |
| 30 | M | 44 | Cerebellum | Negative | Negative |
| 31 | M | 22 | Spinal cord | Strong | Strong |
| 32 | F | 32 | Cerebellum | Weak-diffuse | Weak-diffuse |
| 33 | M | 37 | Cerebellum | Weak-diffuse | Negative |
| 34 | F | 51 | Cerebellum | Weak-diffuse | Negative |
| 35 | M | 71 | Cerebellum | Strong‡ | Strong‡ |
†VHL-associated case. ‡Cases showing strong granular SDHB immunostaining with partial negative staining.
Figure 3.

SDHB immunohistochemistry of hemangioblastoma with two different primary antibodies. (a) Hemangioblastoma with SDHB immunonegativity, (b) strong granular cytoplasmic positivity, and (c) a weak-diffuse pattern and/or nuclear blush staining using a primary rabbit polyclonal antibody (HPA002868). (d–f) reveal results of SDHB immunostaining with a different primary mouse monoclonal antibody (ab14714). SDHB expression patterns of (d,e) are consistent with those of corresponding (a,b) areas, respectively. (f) show negative SDHB staining. Bar indicates 100 µm.
Mutation analyses in hemangioblastomas
We performed mutational analyses by direct sequencing in 10 cases. Among 10 cases, 4 cases were negative for SDHB immunostaining and remaining 6 were cases with a weak-diffuse pattern of SDHB immunostaining. We did not detect any pathogenic SDHB mutations except for three cases of a mutated exon 4 and one case of a mutated exon 1, which failed to amplify. Interestingly, we found an SDHB c.18C > A single nucleotide variant in all nine cases of hemangioblastoma, which was present in exon 1. We did not observe any pathogenic mutations in previously reported missense mutation sites of SDHA (Chr5:254599, Chr5:256509, or Chr5:223646 on Assembly GRCh37)30.
Discussion
SDH was the first mitochondrial enzyme identified as a tumor suppressor5,7. Among the SDH complex, SDHA and SDHB are hydrophilic catalytic subunits, whereas SDHC and SDHD are hydrophobic and anchor the catalytic subunits to the inner mitochondrial membrane2–4. If any component of the SDH complex is lost, SDHB protein is released into the cytoplasm and rapidly degraded3,4,24. Remarkably, SDHB immunohistochemistry shows negative immunoexpression in the presence of bi-allelic inactivation of any of SDHx mutation and has been suggested to be a surrogate marker for SDHx mutation21–24. Because of its wide expression and fundamental role in cell biology, its inactivation may be associated with other neoplasms beyond paraganglioma/pheochromocytoma3,5,8,31. Various types of tumors were reported in SDHx mutation carriers14,15,17,20,25,31 or have been identified in a series of tumors that have not been genetically characterized19,27,31. SDHB immunonegativity has been reported in pheochromocytoma/paraganglioma, GISTs, RCCs, PAs, pancreatic NETs, prostate cancer, stomach cancer, and testicular seminoma15,20,22–24,27,31. However, data on CNS tumors are very limited. A retrospective cohort study on SDHx mutation carriers19 reported a case of meningioma in a patient with SDHA germline mutation and a case of oligodendroglioma in a patient with SDHD germline mutation. However, these tumors showed positive SDHB immunoexpression, which suggests the absence of SDH inactivation. A case of atypical meningioma was reported in a patient with a germline mutation in the SDHB gene and molecular analyses with tumor tissue confirmed an SDHB mutation in the meningioma. However, SDHB immunohistochemistry was not performed29. A recent study suggested that oligodendrogliomas with a 1p19q deletion are associated with the downregulation of SDHB expression, but SDHB immunohistochemistry was not performed28. In the present study, we performed SDHB immunohistochemistry on various types of CNS tumors and observed that all cases of oligodendroglioma (9 cases) and meningioma (10 cases) showed strong granular immunopositivity. Previous studies have reported that PA may harbor mutations in SDHx genes and exhibit SDHB immunonegativity18,20. However, SDH inactivation in PA is very rare (only 0.3%)4,32. In the present study, we did not detect any loss of SDHB immunoexpression in PAs. Unexpectedly, we found SDHB immunonegativity in hemangioblastoma.
Hemangioblastomas arising in the CNS are benign tumors composed of large and vacuolated stromal cells and numerous thin-walled blood vessels. CNS hemangioblastomas most often occur in the cerebellum, followed by the brainstem and spinal cord33,34. Approximately 25% of hemangioblastomas are associated with von Hippel-Lindau (VHL) disease, whereas the remaining 75% of cases are sporadic33. In VHL-related hemangioblastomas, bi-allelic inactivation of the VHL gene can induce HIF stabilization. As a result, HIF induces the activation of genes related to the tumorigenesis of VHL disease33,35,36. Recent studies have suggested that the inactivation of VHL plays a dominant role not only in the pathogenesis of familial hemangioblastomas but also in the sporadic form30,37. However, a significant proportion of sporadic hemangioblastomas still exist without VHL inactivation, which suggests that alternative pathways may be involved in the tumorigenesis of sporadic hemangioblastomas37. In the present study, 80% of hemangioblastomas showed either negative or weak-diffuse pattern of SDHB immunoexpression, which suggests the inactivation of SDH. Clinical manifestations of the SDHx mutation are very similar to those of VHL disease. Paraganglioma/pheochromocytoma, RCC, and pancreatic NET can be caused by both disease entities3,4,19,31,35,36. Furthermore, the inactivation of SDH and VHL can share a common pathway via HIF stabilization5,10,11,38. Therefore, our results suggest that SDH inactivation may represent one alternative pathway involved in the tumorigenesis of sporadic hemangioblastoma.
SDH inactivation-related tumors can be caused irrespective of the type of SDHx mutation. However, there are some correlations between the tumor type and mutation frequency. SDHB and SDHD mutations are common in pheochromocytoma/paraganglioma19,24,38, and SDHA mutations and SDHC promoter hypermethylation are relatively common in GISTs4. In RCC, SDHB mutations are more common3,4,31. PA is frequently associated with SDHA mutations4. Although several recent studies have performed comprehensive molecular analyses on a series of hemangioblastomas30,39,40, data related to SDHx mutations in hemangioblastoma are rare. Shankar et al. performed molecular analyses of hemangioblastomas using deep-coverage DNA sequencing. They reported inactivation of the VHL gene in 78% of sporadic hemangioblastomas, but no other gene was significantly mutated30. In the supplementary data of that study, we found seven cases of hemangioblastoma with missense mutations in SDHA and SDHB genes. However, missense mutations in the SDHA gene were benign or uncertain significance and only one mutation in the SDHB gene was associated with conflicting interpretations of pathogenicity. In the present study, we performed direct sequencing on whole exons of the SDHB gene and on previously reported missense mutation sites in the SDHA gene30. However, we did not find any pathogenic mutation. We further performed SDHA immunohistochemistry, but did not observe any loss of SDHA immunoexpression (Supplementary Fig. S1). Only one type of SDHB polymorphism was found (c.18C > A single nucleotide). This polymorphism is one of well-known polymorphisms and found with a frequency of 2.7% in Danish patients with neuroendocrine cancer41. In the present study, because of our limited analytical methods, we could not demonstrate an association between SDHB immunonegativity and causal mutations in SDHx genes. Therefore, to elucidate how the inactivation of SDHB is related to mutations in SDHx genes, further comprehensive genomic studies, including epigenomic analyses of SDHx genes, is needed.
SDHB immunohistochemical results should be interpreted with caution, because false negative immunostaining may be associated with tissue quality, poor fixation, and/or immunohistochemical technique. Therefore, it is important to use an internal positive control in non-neoplastic cells, such as endothelial, stromal, or inflammatory cells, throughout the tumor before interpreting SDHB immunohistochemistry. If there is no internal positive control, SDHB staining should not be interpreted irrespective of the status of SDHB immunoexpression in the tumor4,23,24. In the present study, all 35 cases of hemangioblastoma were compared to the internal positive control. SDHB immunostaining must be considered positive (normal) when strong granular cytoplasmic positivity (mitochondrial pattern) is present. However, the interpretation of a weak-diffuse staining pattern can be challenging4,22,23. This pattern has been reported 3.7–11.5% of paragangliomas/pheochromocytomas and was frequently identified in cases with SDHD and SDHB mutations4,22–24. Therefore, a week-diffuse pattern of SDHB immunostaining, particularly when this contrasts markedly with true mitochondrial (strong granular cytoplasmic) staining in internal positive controls, should correctly be considered negative and indicative of SDH deficiency4,22–24. In the present study, we initially observed a weak-diffuse pattern of SDHB in 19 cases (54.3%) of hemangioblastoma. However, an additional SDHB immunohistochemistry with a different antibody showed only 10 (28.6%) of them were a weak-diffuse pattern. Our results suggest that an additional SDHB immunohistochemistry with a different antibody could be beneficial when SDHB immunostaining showed a weak-diffuse pattern and support the idea that a week-diffuse pattern of SDHB immunostaining, in internal positive controls, should correctly be considered negative and indicative of SDH deficiency4,22–24. Since the inactivation of SDH involves the HIF-1α pathway5,10,11,38, we additionally performed HIF-1α immunostaining with 9 cases of a weak-diffuse pattern of SDHB and found that all of the cases showed revealed an increased expression of HIF-1α suggesting the inactivation of SDH (Supplementary Fig. S2).
Decreased SDHB expression also could be observed in tumors associated with VHL disease (germline VHL mutation)4,21. In our study, we included two cases of hemangioblastoma associated with VHL disease and observed a weak-diffuse pattern of SDHB immunoexpression. Overall, 5~15% of tumors without SDHx gene mutations are interpreted as being SDHB immunonegative21–24. However, SDHB immunonegativity in tumors without SDHx gene mutations may be associated with limitations in the molecular methods or epigenetic changes3,4,22. Therefore, to elucidate the mechanism involved in the loss of SDHB immunoexpression in hemangioblastoma, further comprehensive molecular genetic analyses of SDHx mutations, including promoter methylation and/or VHL testing, should be performed.
Conclusion
To the best of our knowledge, this is first study to evaluate SDHB immunohistochemistry in various types of CNS tumors. Among the CNS tumors, we found that hemangioblastoma was associated with SDHB immunonegativity, which suggests the inactivation of SDH. However, to elucidate the association between SDHB inactivation and hemangioblastoma, further comprehensive molecular analyses, including epigenetic analyses, should be conducted.
Materials and Methods
Patients and tumor tissues
This study was approved by the Institutional Review Board Committee of the Ajou University Medical Center (Approval No. AJIRB-BMR-OBS-16-187) and all experiments were performed in accordance with our institutional guidelines and regulations. Anonymized tissue microarray (TMA) tissue from various types of CNS tumors was used for SDHB immunohistochemistry. The surgical pathology records of all patients with hemangioblastoma between June 1994 and December 2016 were reviewed. Patients whose pathology specimens and ancillary tests were available for review were included. All slides of each case were reviewed and a representative block was selected for ancillary testing.
Immunohistochemistry
Immunohistochemistry was performed on representative sections (4 µm thick) of formalin-fixed, paraffin-embedded (FFPE) tissues using a BenchMark XT automated immunohistochemistry stainer (Ventana Medical Systems, Tucson, AZ, USA). The Ventana staining procedure included pretreatment with a cell conditioner (pH 8) for 92 min, followed by incubation with the diluted SDHB primary rabbit polyclonal antibody (CAT# HPA002868, LOT# B105404, Sigma-Aldrich Corp; St Louis, MO, USA; 1:400) at 37 °C for 48 min. To confirm results of SDHB immunohistochemistry, immunohistochemistry using a different primary mouse monoclonal antibody against SDHB (CAT# ab14714, LOT# GR3256027-1, Abcam Inc; Cambridge, MA, USA; 1:500) was also performed. The primary antibodies were detected using an OptiView DAB IHC Detection kit (Ventana Medical Systems) following incubation with hematoxylin and a bluing reagent (4 min each). Subsequently, slides were removed from the immunostainer, washed in water containing a drop of dishwashing detergent, and mounted. The evaluation of SDHB immunoexpression was conducted by a single experienced pathologist (JH Kim) without prior knowledge of the clinicopathological data. SDHB was scored as positive if the cytoplasm showed a strong granular staining. SDHB was scored negative only if the cytoplasm was negative in parallel with positive staining for the internal control (capillary endothelial cells). Cases in which tumor cells revealed a weak cytoplasmic or nuclear blush staining without the presence of definite granular mitochondrial staining were classified as weak-diffuse pattern22,23.
Mutation analyses
Genomic DNA was extracted from FFPE tumor tissues using a QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. DNA fragments of the SDHB gene corresponding to each exon and its flanking intron were amplified by PCR using the following gene-specific primers:
SDHB_1_F 5′-ATGCGCCGCTACTGCTACTGCGCTATT-3′
SDHB_1_R 5′-TGAGGCCTTGCCCTATGCTTCCT-3′
SDHB_2_F 5′-AATCCAGCGTTACATCTGTTGTGCCA-3′
SDHB_2_R 5′-AAGCATGTCCCTAAATCAAA-3
SDHB_3_F 5′-GAACTTTACATAAATACCACTGGA-3′
SDHB_3_R 5′-CTATCAGCTTTGGCCAGC-3′
SDHB_4_F 5′-ACCTCTGTCAGAGGAATGTTGCAT-3′
SDHB_4_R 5′-CTACTGACTAGAAGAGGAGCCTTA-3′
SDHB_5_F 5′-TGATGATGGAATCTGATCCT-3′
SDHB_5_R 5′-CAGATTGAAACAATAAATAGGGA-3′
SDHB_6_F 5′-CCTCTCTTTTCTCCCCATAC-3′
SDHB_6_R 5′-CAGCAATCTATTGTCCTCTTG-3′
SDHB_7_F 5′-AGCTAATCATCCCTGGTTTT-3′
SDHB_7_R 5′-TTGTGAGCACATGCTACTTC-3′
SDHB_8_F 5′-GTGGGTTTTCCCTTTCAGTT-3′
SDHB_8_R 5′-CGGCAAGTAAAGGAACAGGT-3′.
We also performed PCR on previously reported missense mutation sites of SDHA (Chr5:254599, Chr5:256509, and Chr5:223646 on Assembly GRCh37) in hemangioblastoma30 using the following primers:
SDHA_1F 5′-AACAGTTTGCAAGGGGAAATTACT -3′
SDHA_1R 5′-TAGATCCTTACCCCCTAAGCCA -3′
SDHA_14F 5′-GATGGTGTTTCTGGCCTCAG -3′
SDHA_14R 5′-TGTCGGAGTGCCTTTTTCAG -3′
SDHA_15F 5′-GAGAATCTTAAAGTTCACATGCC -3′
SDHA_15R 5′-GAGTGCAGAAGCGTATGAAGAC -3′
The amplified PCR products were purified and sequenced using a 3500xL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Sequence data were compared to a reference sequence (GenBank: NG_012340.1).
Ethical approval
This study was approved by the Institutional Review Board Committee of the Ajou University Medical Center (Approval No. AJIRB-BMR-OBS-16-187) and was performed according to our institutional guidelines and regulations (For this type of study formal consent is not required in our regulation).
Supplementary information
Acknowledgements
This work was supported by the faculty research fund (Ajou translational research fund 2017) of Ajou University School of Medicine to Se-Hyuk Kim and Jang-Hee Kim, and by National Research Foundation of Korea to Jang-Hee Kim (NRF-2016R1D1A1B02010452).
Author Contributions
Design and contributed to analysis and interpretation of data; J.-H.K., S.-H.K., T.H.R., H.Y. and K.B.L.; Molecular experiments and analysis; S.Y.J. and J.R.; Provided patients samples and clinical data analysis; T.H.R. and S.-H.K.; Immunohistochemistry and analysis; S.H.P. and, J.-H.K. Writing manuscript; T.H.R., H.Y., S.-H.K. and J.-H.K. All authors reviewed the manuscript.
Data Availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Tae Hoon Roh and Hyunee Yim contributed equally.
Contributor Information
Se-Hyuk Kim, Email: nsksh@ajou.ac.kr.
Jang-Hee Kim, Email: drjhk@ajou.ac.kr.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-42338-z.
References
- 1.Lancaster CR. Succinate:quinone oxidoreductases: an overview. Biochimica et biophysica acta. 2002;1553:1–6. doi: 10.1016/S0005-2728(01)00240-7. [DOI] [PubMed] [Google Scholar]
- 2.Aldera AP, Govender D. Gene of the month: SDH. Journal of clinical pathology. 2018;71:95–97. doi: 10.1136/jclinpath-2017-204677. [DOI] [PubMed] [Google Scholar]
- 3.Mannelli M, et al. DIAGNOSIS of ENDOCRINE DISEASE: SDHx mutations: beyond pheochromocytomas and paragangliomas. European journal of endocrinology. 2018;178:R11–R17. doi: 10.1530/EJE-17-0523. [DOI] [PubMed] [Google Scholar]
- 4.Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology. 2018;72:106–116. doi: 10.1111/his.13277. [DOI] [PubMed] [Google Scholar]
- 5.Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nature reviews. Cancer. 2005;5:857–866. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- 6.Gimm O, Armanios M, Dziema H, Neumann HP, Eng C. Somatic and occult germ-line mutations in SDHD, a mitochondrial complex II gene, in nonfamilial pheochromocytoma. Cancer research. 2000;60:6822–6825. [PubMed] [Google Scholar]
- 7.Baysal BE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 8.Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochimica et biophysica acta. 2011;1807:1432–1443. doi: 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 9.King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25:4675–4682. doi: 10.1038/sj.onc.1209594. [DOI] [PubMed] [Google Scholar]
- 10.Dahia PL. & Familial Pheochromocytoma, C. Transcription association of VHL and SDH mutations link hypoxia and oxidoreductase signals in pheochromocytomas. Annals of the New York Academy of Sciences. 2006;1073:208–220. doi: 10.1196/annals.1353.023. [DOI] [PubMed] [Google Scholar]
- 11.Dahia PL, et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS genetics. 2005;1:72–80. doi: 10.1371/journal.pgen.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Letouze E, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 13.Janeway KA, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McWhinney SR, Pasini B, Stratakis CA. International Carney, T. & Carney-Stratakis Syndrome, C. Familial gastrointestinal stromal tumors and germ-line mutations. The New England journal of medicine. 2007;357:1054–1056. doi: 10.1056/NEJMc071191. [DOI] [PubMed] [Google Scholar]
- 15.Gill AJ, et al. Renal tumors and hereditary pheochromocytoma-paraganglioma syndrome type 4. The New England journal of medicine. 2011;364:885–886. doi: 10.1056/NEJMc1012357. [DOI] [PubMed] [Google Scholar]
- 16.Ricketts CJ, et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat. 2010;31:41–51. doi: 10.1002/humu.21136. [DOI] [PubMed] [Google Scholar]
- 17.Solis DC, et al. Penetrance and clinical consequences of a gross SDHB deletion in a large family. Clinical genetics. 2009;75:354–363. doi: 10.1111/j.1399-0004.2009.01157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xekouki P, et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in humans and mice. The Journal of clinical endocrinology and metabolism. 2015;100:E710–719. doi: 10.1210/jc.2014-4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niemeijer ND, et al. Succinate Dehydrogenase (SDH)-Deficient Pancreatic Neuroendocrine Tumor Expands the SDH-Related Tumor Spectrum. The Journal of clinical endocrinology and metabolism. 2015;100:E1386–1393. doi: 10.1210/jc.2015-2689. [DOI] [PubMed] [Google Scholar]
- 20.Xekouki P, et al. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: a new association for SDH? The Journal of clinical endocrinology and metabolism. 2012;97:E357–366. doi: 10.1210/jc.2011-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papathomas TG, et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: a multicenter interobserver variation analysis using virtual microscopy: a Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T) Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2015;28:807–821. doi: 10.1038/modpathol.2015.41. [DOI] [PubMed] [Google Scholar]
- 22.Castelblanco E, et al. Usefulness of negative and weak-diffuse pattern of SDHB immunostaining in assessment of SDH mutations in paragangliomas and pheochromocytomas. Endocrine pathology. 2013;24:199–205. doi: 10.1007/s12022-013-9269-4. [DOI] [PubMed] [Google Scholar]
- 23.Gill AJ, et al. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Human pathology. 2010;41:805–814. doi: 10.1016/j.humpath.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 24.van Nederveen FH, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. The Lancet. Oncology. 2009;10:764–771. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ni Y, et al. Germline and somatic SDHx alterations in apparently sporadic differentiated thyroid cancer. Endocrine-related cancer. 2015;22:121–130. doi: 10.1530/ERC-14-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Renella R, et al. Exploring the association of succinate dehydrogenase complex mutations with lymphoid malignancies. Familial cancer. 2014;13:507–511. doi: 10.1007/s10689-014-9725-4. [DOI] [PubMed] [Google Scholar]
- 27.Miettinen M, et al. Mapping of succinate dehydrogenase losses in 2258 epithelial neoplasms. Applied immunohistochemistry & molecular morphology: AIMM. 2014;22:31–36. doi: 10.1097/PAI.0b013e31828bfdd3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gladitz J, Klink B, Seifert M. Network-based analysis of oligodendrogliomas predicts novel cancer gene candidates within the region of the 1p/19q co-deletion. Acta neuropathologica communications. 2018;6:49. doi: 10.1186/s40478-018-0544-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shiwa T, et al. A Patient with an Extra-adrenal Pheochromocytoma and Germ-line SDHB Mutation Accompanied by an Atypical Meningioma. Internal medicine. 2015;54:2355–2360. doi: 10.2169/internalmedicine.54.4663. [DOI] [PubMed] [Google Scholar]
- 30.Shankar GM, et al. Sporadic hemangioblastomas are characterized by cryptic VHL inactivation. Acta neuropathologica communications. 2014;2:167. doi: 10.1186/s40478-014-0167-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papathomas TG, et al. Non-pheochromocytoma (PCC)/paraganglioma (PGL) tumors in patients with succinate dehydrogenase-related PCC-PGL syndromes: a clinicopathological and molecular analysis. European journal of endocrinology. 2014;170:1–12. doi: 10.1530/EJE-13-0623. [DOI] [PubMed] [Google Scholar]
- 32.Gill AJ, et al. Succinate dehydrogenase deficiency is rare in pituitary adenomas. The American journal of surgical pathology. 2014;38:560–566. doi: 10.1097/PAS.0000000000000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Louis, D. N. et al. WHO classification of tumours of the central nervous system. Revised 4th edition. edn, (International Agency For Research On Cancer, 2016).
- 34.Shin Y, Kim S, Lee HW, Bang H, Suh YL. Supratentorial hemangioblastoma with unusual features. Korean J Pathol. 2014;48:462–465. doi: 10.4132/KoreanJPathol.2014.48.6.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaelin WG. Von Hippel-Lindau disease. Annu Rev Pathol. 2007;2:145–173. doi: 10.1146/annurev.pathol.2.010506.092049. [DOI] [PubMed] [Google Scholar]
- 36.Woodward ER, Maher ER. Von Hippel-Lindau disease and endocrine tumour susceptibility. Endocrine-related cancer. 2006;13:415–425. doi: 10.1677/erc.1.00683. [DOI] [PubMed] [Google Scholar]
- 37.Takayanagi S, et al. Differences in genetic and epigenetic alterations between von Hippel-Lindau disease-related and sporadic hemangioblastomas of the central nervous system. Neuro-oncology. 2017;19:1228–1236. doi: 10.1093/neuonc/nox034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fishbein L, et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer cell. 2017;31:181–193. doi: 10.1016/j.ccell.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma D, et al. Whole exome sequencing identified genetic variations in Chinese hemangioblastoma patients. American journal of medical genetics. Part A. 2017;173:2605–2613. doi: 10.1002/ajmg.a.38350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mehrian-Shai R, et al. Identification of genomic aberrations in hemangioblastoma by droplet digital PCR and SNP microarray highlights novel candidate genes and pathways for pathogenesis. BMC genomics. 2016;17:56. doi: 10.1186/s12864-016-2370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bennedbaek M, et al. Identification of eight novel SDHB, SDHC, SDHD germline variants in Danish pheochromocytoma/paraganglioma patients. Hered Cancer Clin Pract. 2016;14:13. doi: 10.1186/s13053-016-0053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).