

Abstract
To produce a new category of anti-cancer compounds, a facile and environmentally sustainable method for preparing diversified bis-quinazolinones was demonstrated using recyclable deep eutectic solvent (DES) under ultrasonic irradiation. The reactions were performed smoothly with a wide scope of substrates affording the desired derivatives in good-to-excellent yields under an atom-economical pathway. Particularly, halogen substituents that are amenable for further synthetic elaborations are well tolerated. Furthermore, the ‘greenness’ of the protocol was assessed within the scope of several green metrics and found to display an excellent score in the specified parameters. Cytotoxic activity of all novel bis-quinazolinones was investigated utilizing two cancer cell lines: breast (MCF-7) and lung (A549) cell lines and their IC50 values were determined. Most of the prepared derivatives displayed fascinating inhibitory activity with IC50 values in a low micromolar range. Remarkably, the derivative 7e [3,3'-(sulfonylbis(4,1-phenylene))bis(2-methyl-6-nitroquinazolin-4(3H)-one)] showed superior potency against MCF-7 and A549 cancer cell lines, with IC50 values of 1.26 µM and 2.75 µM, respectively. Moreover, this derivative was found to have low toxicity to the normal breast cell line (MCF-10A) and could serve as a promising lead candidate for further development.
Keywords: sono-synthesis, bis-quinazolin-4-ones, anti-cancer agents, deep eutectic solvent
1. Introduction
As an important category of nitrogen-containing heterocycles, quinazolines are prevalent in many naturally occurring alkaloids as well as marketed drugs [1–4]. Some quinazolines which are well known as drugs, such as mecloqualone (Casfen), mebroqualone and methaqualone (Quaalude) possessed anxiolytic, calmative and hypnotic properties and are used for treating insomnia [5,6]. Presently, the quinazoline moiety is recognized to have a broad scope of beneficial biological activities, for instance, anti-cancer, anti-inflammatory, anti-tumour, protein kinase inhibitor, anti-microbial cholinesterase inhibitor, antifolate, antiviral and are the essential moiety of HIV reverse transcriptase inhibitors [7–11]. Proportional with the importance of this heterocyclic motif, considerable synthetic protocols have been reported to develop access to quinazolines with diversified substituents [12–15]. Nevertheless, most of them are restricted with a multistep procedure and low atom-economy to prepare quinazoline compounds. Also, in several cases, the utilization of metal–catalyst may enhance the reaction efficiency; metal contaminations are still an issue, essentially when the outcomes are manufactured for the consumption of humans [16–19]. Latterly, many endeavours have been reported to demonstrate easy-to-handle, cost-effective and eco-friendly protocols for the preparation of quinazolinones [20–22]. Notwithstanding, to the best of our knowledge there have been restricted articles to date on the assembly of bis-quinazolinones [23,24]. Subsequently, the development of an effective and greener methodology for the construction of bis-quinazolinones is extremely in demand. Recently, more attempts have been made for designing eco-friendly solvents that could be reusable and/or smoothly biodegradable. For example, green solvents such as supercritical liquids [25], water [26], polyethylene glycol [27] and ionic liquids [28–30] have emerged to supersede numerous organic solvents. Nevertheless, in some cases, the utilization of these solvents is limited because of their poor stability and solubility of organic substances. Lately, deep eutectic solvents (DESs), the environmentally benign solvent systems, have attracted considerable attention [31,32]. DESs possess several noteworthy merits when compared with conventional solvents, comprising a broad range of liquid temperatures, renewability, water tolerance, low toxicity, biodegradability, low volatility and high solvation capacity for an extensive number of organic substances [33–35]. DESs are easily prepared from mixing an organic hydrogen bond donor (e.g. tartaric acid) and a hydrogen bond acceptor (e.g. choline chloride) [31,32]. The obtained mixture possesses high stability and remains in a high-entropy liquid state even at low temperatures; this may be attributed to the newly formed hydrogen-bond network between its components [31,32,36,37]. Numerous reports have demonstrated their utilization in several organic conversions [38–40]. Because of these special advantages, DESs opened novel perspectives to prepare new substances. The use of ultrasonic irradiation as the green energy source is of a significant utility in the field of pharmaceutical and green chemistry. The impacts of sonication on organic synthesis are imputed to cavitations, which generate extremely high local pressure and temperature inside the formed bubbles. When these bubbles collapse, a sufficient amount of energy was generated for performing the chemical reaction and allowing the process to occur easily with a high yield in a very short time [41,42]. In spite of that, to the best of our knowledge there have been no reports to date on the preparation of bis-quinazolinones using deep eutectic solvents under ultrasonic irradiation. In continuation to our ongoing efforts to develop greener synthetic pathways for organic conversions [43–46], herein I wish to report a DES-mediated protocol for greener and atom-economical preparation of novel series of bis-quinazolinones by using commercially available and inexpensive materials under appropriate catalyst-free and ultrasonic conditions.
2. Results and discussion
At the outset, the present study was commenced with optimization of the reaction conditions using 2-methyl-4H-benzo[d][1,3]oxazin-4-one (1a) and trans-cyclohexane-1,4-diamine (2a) as model substrates (table 1). The initial endeavour utilizing conventional refluxing conditions in CH3CN and in the presence of K2CO3 as the basic catalyst [23,24] afforded the hitherto unreported 3,3′-((1R,4R)-cyclohexane-1,4-diyl)bis(2-methylquinazolin-4(3H)-one) (3a) but in a poor yield; instead, a considerable amount of starting materials remain unchanged (table 1, entry 1). Next, on performing the above reaction under ultrasonic irradiation (60 W) at 50°C for 20 min, furnished 40% of the product (table 1, entry 2). Anticipating further improvement in the yield, a series of base and acid catalysts, such as Cs2CO3, AcOH/NaOAc and p-toluenesulfonic acid (table 1, entries 3–5), were screened under ultrasonic irradiation (60 W); of these, p-TSA was found to be the most effective catalyst for this conversion (table 1, entry 5). Furthermore, a moderate yield was obtained under catalyst-free and ultrasound conditions, which suggest an apparent over-activity by the used catalyst (table 1, entry 6). Such an observation is also confirmed by literature studies, which proposed that ultrasonic irradiation may replace a catalyst under definite conditions [47,48]. In order to acquire more acceptable results, other reaction variables were assessed. First, the role of additional solvents was investigated and found that the yield of 3a was improved to be 70% when performing water as a solvent at 50°C (table 1, entry 7), and slightly increased to be 77% in a longer reaction time at 95°C (with table 1, entry 8). In addition, ethanol furnished a moderate yield at 50°C (table 1, entry 9), while other solvents such as i-PrOH and toluene were substantially less efficient and THF was ineffectual (table 1, entries 10–12). To evaluate the current protocol from the greener edge, deep eutectic solvents (DESs) were used as the reaction medium for this conversion. To acquire derivative 3a (table 1), we turned our venture by using certain types of DESs with the model substrates 1a (2.0 equiv.) and 2a (1.0 equiv). As displayed in table 1, only a small amount of the required product (3a) was obtained when the reaction proceeded in sucrose–choline chloride mixture (1 : 1) at its melting temperature (table 1, entry 13). Considering that the model reaction works well under acidic conditions, three acidic DESs were examined with a view to acquiring more satisfactory results. Interestingly, a considerable improvement was obtained when DESs such as lactic acid–choline chloride (1 : 2), oxalic acid–choline chloride (1 : 1) and l-(+)-tartaric acid–choline chloride (1 : 2) were used as the reaction solvent (table 1, entries 14–16). This screening disclosed that the utilization of l-(+)-tartaric acid–choline chloride (1 : 2) melt afforded the superior result (table 1, entry 16). In a quest to further improve the reaction yield, the model substrates were sonicated at two operating temperatures, i.e. 85 and 90°C and the required product (3a) was obtained in 89% and 99% yields, respectively (table 1, entries 16 and 17). Thus, the optimal temperature was established to be 90°C, which achieved the best yield of the desirable product. Decreasing the sonication time to 10 min diminished the isolated yield to 91% (table 1, entry 18).
Table 1.
Optimization of the reaction conditions for the preparation of compound 3a. 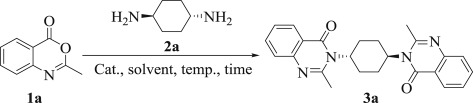
| entry | catalyst | solvent | temp. (°C)/method | time (min) | yield (%)a |
|---|---|---|---|---|---|
| 1 | K2CO3 | CH3CN | Refluxb | 300 | 25 |
| 2 | K2CO3 | CH3CN | 50/USc | 20 | 40 |
| 3 | Cs2CO3 | CH3CN | 50/US | 20 | 43 |
| 4 | NaOAc | AcOH | 50/US | 20 | 45 |
| 5 | p-TSA | CH3CN | 50/US | 20 | 57 |
| 6 | — | CH3CN | 50/US | 20 | 65 |
| 7 | — | H2O | 50/US | 20 | 70 |
| 8 | — | H2O | 95/US | 35 | 77 |
| 9 | — | EtOH | 50/US | 20 | 68 |
| 10 | — | i-PrOH | 50/US | 20 | 57 |
| 11 | — | toluene | 50/US | 20 | 38 |
| 12 | — | THF | 50/US | 20 | 15 |
| 13 | — | sucrose–choline chloride (1 : 1) | 80/US | 20 | 17 |
| 14 | — | lactic acid–choline chloride (1 : 2) | 80/US | 20 | 73 |
| 15 | — | oxalic acid–choline chloride (1 : 1) | 80/US | 20 | 75 |
| 16 | — | l-(+)-tartaric acid–choline chloride (1 : 2) | 85/US | 20 | 89 |
| 17 | — | l-(+)-tartaric acid–choline chloride (1 : 2) | 90/US | 15 | 99 |
| 18 | — | l-(+)-tartaric acid–choline chloride (1 : 2) | 90/US | 10 | 91 |
aIsolated yield.
bThe reaction was carried out under reflux conditions.
cThe reactions were carried out under ultrasonic irradiation (US) conditions.
After optimization of the current protocol, the impact of several ultrasonic irradiation powers on the yield and rate of model reaction were also studied (table 2). The results display that the power of sonication has a considerable impact on the reaction system. Thus, the sonication of the model substrates in l-(+)-tartaric acid–choline chloride (1 : 2) at 30 W afforded the desired product (3a) in a moderate yield throughout 60 min (table 2, entry 1). While, both reaction rate and yield were improved by increasing the sonication power to 60 W (table 2, entries 2–4). Moreover, on sonication of model reactants at 70 W, neither the reaction rate nor the reaction yield was improved (table 2, entry 5). Noteworthy, on carrying out the model reaction under reflux conditions, the reaction effectiveness is obviously diminished (table 2, entry 6). The utilization of ultrasound technique reduces the reaction duration and enhances the reaction yield possibly through increasing the energy and collision speed of the reactants. Consequently, sonication (60 W) of 1a (2 mmol) and 2a (1 mmol) in l-(+)-tartaric acid–choline chloride (1 : 2) at 90°C for 15 min was established as the optimum conditions, affording the desired product (3a) in 99% yield (table 1, entry 17).
Table 2.
The impact of ultrasonic irradiation power on the synthesis of 3a.
| entry | power (W) | time (min) | yield (%)a |
|---|---|---|---|
| 1 | 30 | 60 | 76 |
| 2 | 40 | 50 | 80 |
| 3 | 50 | 30 | 82 |
| 4 | 60 | 15 | 99 |
| 5 | 70 | 15 | 99 |
| 6 | silent (reflux)b | 320 | 78 |
aIsolated yield.
bThe reaction was carried out under reflux conditions.
After achieving an optimized strategy for this new reaction, the substrates' versatility and scope with regard to diamines and benzoxazin-4-ones were then investigated for the assembly of various bis-quinazolinones (schemes 1 and 2). First, the electronic impacts of R groups on 2-methyl-4H-benzo[d][1,3]oxazin-4-ones (1a–e) were studied. Generally, the aryl motif linked to an electron donating substituent (e.g. –CH3) afforded better yields than that linked to electron withdrawing substituents (e.g. –Cl, –F and –NO2).
Scheme 1.

Reaction of derivatives 2a–c under the standard conditions.
Scheme 2.

Reaction of derivatives 2d and 2e under the standard conditions.
Furthermore, diverse diamines, comprising aromatic and aliphatic diamines, were well tolerated. Interestingly, all the reactions proceeded smoothly to provide the corresponding bis-quinazolinones depending on the reaction time. trans-Cyclohexane-1,4-diamine (2a) afforded the required products (3a–e) in short reaction time with high yields. Also, p-xylylenediamine (2b) and ethylene diamine (2c) could not reduce the reaction rate remarkably. Furthermore, p-phenylene diamine (2d) and dapsone (2e) produced the corresponding derivatives in moderate-to-high yields but in longer reaction time (scheme 2). Owing to the fact that heterocycles bearing halogens are significant building blocks in the assembly of plentiful pharmaceuticals and natural products [49,50], an assortment of halo-substituted bis-quinazolinones, e.g. chloro 3c (96%), 4c (95%), 5c (95%), 6c (95%), 7c (97%) and fluoro 3d (96%), 4d (92%), 5d (94%), 6d (97%), 7d (93%), were successfully prepared.
The steric impact of the utilized diamines had an obvious effect on the effectiveness of these reactions. For instance, when the present method was put in an application for o-phenylene diamine (2f), however, bis-quinazolinone derivative (9, 67%) was isolated along with the mono-quinazolinone derivative [23,24] (8, 33%). Whereas, by increasing the reaction time to 25 min and molar ratio of 1a to 2.5 mmol, the reaction yield was improved to 88% with the complete consumption of the mono-quinazolinone derivative 8 (monitored by TLC). The comparable result was acquired when m-phenylene diamine (2 g) was utilized as the substrate; the reaction afforded a complex mixture of compounds 10 [51] and 11 under optimum conditions. By increasing both time (30 min) and molar ratio of 1a (2.5 mmol), compound 10 disappeared and the reaction afforded derivative 11 in 92% yield (scheme 3). The structures of all the novel bis-quinazolinones were interpreted on the basis of spectral analyses (IR, NMR spectroscopy and mass spectrometry).
Scheme 3.

Reaction of derivatives 2f and 2g under the standard conditions.
In order to further display the practicality of the above-mentioned investigations, the current protocol was extended for large-scale preparation of bis-quinazolin-4(3H)-one (3a) (scheme 4). As outlined in scheme 4, the reaction of trans-cyclohexane-1,4-diamine (2a, 1.14 g, 10.0 mmol) with benzoxazine (1a, 3.22 g, 20.0 mmol), using l-(+)-tartaric acid–choline chloride (1 : 2) as the solvent, was scaled up. Under the optimal conditions, the required product 3a was obtained in 98% yield by crystallization from dioxane. Lastly, to assess the current protocol on the ‘greenness’ scale, green metrics [52,53] such as E-factor (EF), atom economy (AE), reaction mass efficiency (RME), process mass intensity (PMI), yield economy (YE) and carbon efficiency (CE) were studied. As summarized in scheme 4, the current protocol achieved a good combination of AE (91.74%), CE (100%), EF (0.02), RME (98.67%), PMI (1.11) and YE (6.53%), which makes it an ideal sustainable and green process. The results might be beneficial from environmental and industrial perspectives.
Scheme 4.

Calculated green metrics for the scaled-up preparation of 3,3'-((1R,4R)-cyclohexane-1,4-diyl)bis(2-methylquinazolin-4(3H)-one) 3a.
According to the aforementioned experiments and the reported literature [54], a proposed mechanism for the reaction between diamines (2a–g) and benzoxazines (1a–e) was established as presented in scheme 5. The first step could be the ring opening via nucleophilic attack of amine (NH2) to carbonyl carbon under the presence of l-(+)-tartaric acid–choline chloride. Next, the formed intermediate underwent intramolecular nucleophilic attack of the nitrogen (–NH–) of the amide group to carbonyl carbon. Finally, the required product was formed through dehydration [54]. From the proposed mechanism, it is obviously concluded that, l-(+)-tartaric acid–choline chloride performs a dual function; as solvent and as catalyst.
Scheme 5.

A plausible mechanism for the preparation of bis-quinazolin-4-ones 3–11.
Recyclability of the solvent is a needful merit for industrial processes. The recyclability of l-(+)-tartaric acid–choline chloride in this investigation was proceeded in the model substrates; 2-methyl-4H-benzo[d][1,3]oxazin-4-one (1a) and trans-cyclohexane-1,4-diamine (2a) under optimal conditions (figure 1). After consumption of reactants, the reaction contents were cooled to ambient temperature and diluted with ethanol. The desired product was filtered and washed with ethanol. After removal of ethanol under vacuum, DES was recovered and re-used for the next cycles. The recyclability proceeded five times without considerably lowering the reaction efficiency (figure 1).
Figure 1.
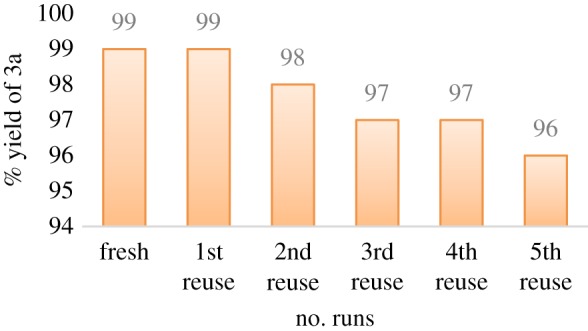
Recyclability of DES for the synthesis of 3a.
3. Assessment of in vitro human cancer cell lines growth inhibition and the structure and activity relationships (SARs)
The unprecedented derivatives (3–7 and 11) were tested for their in vitro cytotoxicity against human MCF-7 (breast cancer), A549 (lung cancer) cancer cell lines and one type of normal cell line MCF10A (normal breast cell line) utilizing the standard MTT (3,4-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay according to Mosmann's methodology [55] (figure 2), whereas sorafenib was performed as a standard compound. Two concentrations (10 and 30 µM in triplicate) of the derivatives were utilized and after 48 h of drug treatment the results were analyzed. The obtained results were tabulated as mean ± s.e.m. With standard errors below 10%, the reproducibility between replicate wells is within the acceptable values. Concentrations less than 10 µM have negligible impacts on the cytotoxicity. The investigation of the SARs displayed that the substituent R groups in the quinazolinone moieties have a significant role in their biological properties. The results summed up in electronic supplementary material, S59, table S3 indicate that all of the tested derivatives have good-to-excellent cytotoxic activities, with the IC50 values at the micromolar range (figure 2). Regarding the MCF-7 breast cancer cell line, compounds 7a–e elicited potent anti-breast cancer activity (IC50 = 1.26–5.78 µM), compared with sorafenib (IC50 = 4.03 µM). This reactivity could be related to the high potency of both dapsone and quinazolinone moieties, when compared with the other derivatives. Noteworthy, compounds 4a–e comprising p-xylylene moiety resulted in good-to-excellent activities with IC50 of 2.75–6.73 µM. Bis-quinazolin-4-one derivatives 5a–e (IC50 = 5.21–13.55 µM) were less active than the cyclohexane derivatives 3a–e (IC50 = 4.55–12.70 µM). Whereas, 1,4-phenylene derivatives 6a–e (2.73–7.48 µM) were slightly more active than cyclohexane derivatives 3a–e. Similarly, derivative 11 possessed good activity with an IC50 value of 5.42 µM. Moreover, substitution with electron withdrawing groups (Cl, F and NO2) displayed good activity, while electron donating group (CH3) reduced the activity. A comparison of the substituents on the quinazolinone ring indicated that nitro groups significantly enhanced the cytotoxic activity and determined the order of potency. For instance, derivative 7e with NO2-substitution at the quinazolinone moiety was found to be the most potent in the series with IC50 value of 1.26 µM against MCF-7, being approximately thrice as potent as sorafenib. Likewise, derivatives 7c and 7d possessed a more important activity than unsubstituted quinazolinone moiety (7a) and methyl substituted derivative (7b).
Figure 2.

In vitro studies of the synthesized compounds (3–7 and 11) against human cancer cell lines (MCF-7 and A549).
Concerning the A549 lung cancer cell line (electronic supplementary material, table S3), the alteration of substituents on 6-position of the quinazolinone ring could also influence the activities of these compounds. As summarized in figure 2, the potency of substituents was ordered as NO2 > F > Cl > H > CH3, indicating that those with the electron withdrawing group are more active than those with an electron donating substitute. Ten derivatives, 7a–e, 6c–e, 5e and 4e, possessed promising cytotoxic activities as indicated from their IC50 values (2.78–5.77 µM) compared with sorafenib (IC50 = 5.20 µM). Among them, derivative 7e, 3,3′-(sulfonylbis(4,1-phenylene))bis(2-methyl-6-nitroquinazolin-4(3H)-one), showed the best activity with IC50 value of 2.75 µM against A549, being approximately twice as potent as sorafenib. The series of derivatives 6 and compound 11, with aryl motif, also afforded potent activities and displayed similar tendencies in the relationship between the structure and activity. Nevertheless, the series of compounds 3 and 5 afforded slightly weaker activities (figure 2). Figure 3 clarifies the structural activity relationship of the designed derivatives.
Figure 3.
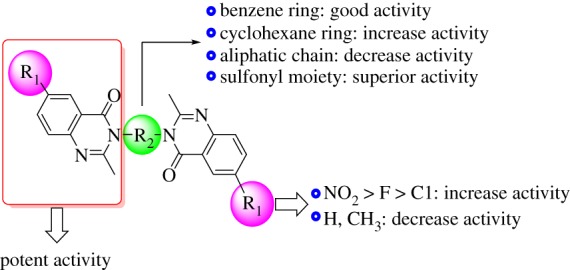
Illustration of the structural activity relationship of the designed derivatives.
In order to decide whether the cytotoxic properties of the investigated derivatives were selective for cancerous cells in contrast to non-cancerous cells, the most active derivatives, viz. 4e, 6e, 7d and 7e, were subjected to in vitro assessment against the normal breast cell line (MCF-10A). The results of the non-malignant cell (MCF-10A) demonstrated that derivatives 7e (IC50 = 7.17 ± 0.76 µM), 7d (IC50 = 5.98 ± 1.86 µM), 6e (IC50 = 5.65 ± 2.34 µM) and 4e (IC50 = 5.43 ± 2.46 µM) exhibited promising results in comparison with sorafenib (IC50 = 5.98 ± 0.49 µM) when tested under the same conditions. From the obtained results, we can conclude that these derivatives might be further used as promising anti-cancer agents.
In the present investigation, incorporation of two symmetrical pharmacophores into a single molecule resulted in designed multiple compounds possessing fascinating inhibition activities against clinically relevant objectives that are not obtainable by their monomeric analogues.
4. Conclusion
A straightforward, green and effective protocol for synthesizing new series of bis-quinazolin-4-ones from easily obtainable precursors under minutes of ultrasonic irradiation was demonstrated on a gram-scale. Noteworthy, the l-(+)-tartaric acid–choline chloride deep eutectic solvent displayed to be appropriate for these transformations as indicated by its recyclability, eco-friendly and inexpensive preparation, and simplicity of operation. Also, the sonochemical methodology performs well for both activating and deactivating starting substrates. The reaction yields are excellent and the protocol is devoid of column purification and any tedious work-up. An assortment of halo-substituted products was successfully synthesized via the current strategy that provides possibility for further cross-coupling reactions. As well, the protocol scored well in several green metrics and consequently may be of sustainable and practical benefit in the future. All the newly prepared derivatives possessed potential cytotoxic activities, with the strength of the impact depending on the substituent on the quinazolinone moiety. Among them, derivatives 7e, 3,3′-(sulfonylbis(4,1-phenylene))bis(2-methyl-6-nitroquinazolin-4(3H)-one), exhibited the most potent cytotoxic activities and possessed the ability to kill malignant cells more effectively than non-malignant cells.
5. Experiment
5.1. General information
1H, 19F and 13C NMR spectra were recorded on Bruker Ultra Shield spectrometer at 400, 376 and 100 MHz, respectively. Mass spectra were determined on the GC-MS (QP/000 EX) Shimadzu spectrometer at an ionizing voltage of 70 eV. IR spectra were recorded on the Perkin-Elmer Spectrum One spectrometer using KBr pellets. Analytical thin layer chromatography (TLC) was employed on a silica gel plate (Merck® 60F254). Sonication was performed in a SY5200DH-T ultrasound cleaner. Melting points were measured on Electrothermal IA9100 melting point apparatus (UK) using open capillary tubes and are uncorrected. All commercial reagents were used as received without any purification. Cytotoxic activities were performed by the Microanalysis Center, Faculty of Science, Cairo University. Derivatives 1a–e were synthesized by the sonication of anthranilic acid derivatives and acetic anhydride at 40°C for 10 min.
5.2. Synthesis of DESs
The DESs were synthesized by mixing choline chloride with several hydrogen bond donors such as sucrose, lactic acid, oxalic acid and tartaric acid, according to the molar ratio mentioned in table 1. The two components of each mixture were heated at 85°C under sonication until a clear liquid was obtained, which was directly used for bis-quinazolin-4(3H)-ones synthesis.
5.3. Typical procedure for the preparation of bis-quinazolin-4(3H)-ones 3–11
2-Methyl-4H-benzo[d][1,3]oxazin-4-ones (1a–e) (2 mmol), diamines (2a–g) (1 mmol) and l-(+)-tartaric acid–choline chloride (1 : 2) based DES (4 ml) were added to a 25 ml round bottom flask and the reaction mixture was sonicated (60 W) for 15 min (20–30 min in the case of using 2d–g) at 90°C. The progress of the reaction was monitored by TLC. After completion of the reaction, the mixture was cooled to room temperature and diluted with ethanol. The desired product was obtained by filtration and recrystallized from dioxane containing a few drops of DMF. The filtrate which mainly contained DES was dried under vacuum to get DES. DES so recovered was utilized for the next experiments. The recyclability study was performed five times and displayed no significant drop in the reaction yields.
5.3.1. 3,3′-((1R,4R)-Cyclohexane-1,4-diyl)bis(2-methylquinazolin-4(3H)-one) 3a
White crystals, yield 99%, m.p. 273–275°C; IR (KBr): ν/cm−1 1672 (C=O), 1620, 1595 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 8.37–8.35 (d, J = 5.5 Hz, 2H, Ar-H), 7.87–7.85 (d, J = 5.6 Hz, 2H, Ar-H), 7.44–7.31 (m, 4H, Ar-H), 3.74–3.61 (m, 2H, Cyclohexane-H), 2.30 (s, 6H, CH3), 1.65 (br, 4H, Cyclohexane-H), 1.44–1.31 (m, 4H, Cyclohexane-H); 13C NMR (100 MHz, DMSO-d6): δ 170.1 (C=O), 147.6 (C=N), 146.0, 134.5, 128.1, 127.5, 127.1, 126.7 (Ar-C), 52.5 (Cyclohexane-C), 31.1 (Cyclohexane-C), 20.3 ppm (CH3); MS (EI): m/z (%) 401 (M++1, 4.6), 400 (M+, 100); Anal. Calcd for C24H24N4O2: C, 71.98; H, 6.04; N, 13.99%; Found: C, 71.93; H, 5.99; N, 13.90%.
5.3.2. 3,3′-((1R,4R)-Cyclohexane-1,4-diyl)bis(2,6-dimethylquinazolin-4(3H)-one) 3b
White crystals, yield 99%, m.p. 280–281°C; IR (KBr): ν /cm−1 1678 (C=O), 1613, 1590 (C=N, C=C); 1H NMR (400 MHz, CDCl3): δ 8.25 (s, 2H, Ar-H), 7.90–7.87 (d, J = 7.5 Hz, 2H, Ar-H), 7.59–7.55 (d, J = 7.8 Hz, 2H, Ar-H), 3.69 (br, 2H, Cyclohexane-H), 2.75 (s, 6H, CH3), 2.30 (s, 6H, CH3), 1.72 (br, 4H, Cyclohexane-H), 1.20 (m, 4H, Cyclohexane-H); 13C NMR (100 MHz, CDCl3): δ 170.8 (C=O), 147.2 (C=N), 145.6, 138.7, 133.5, 128.0, 126.5, 122.1 (Ar-C), 52.4 (Cyclohexane-C), 31.0 (Cyclohexane-C), 20.8 ppm (CH3), 20.2 ppm (CH3); MS (EI): m/z (%) 429 (M++1, 7.3), 428 (M+, 100); Anal. Calcd for C26H28N4O2: C, 72.87; H, 6.59; N, 13.07%; Found: C, 72.86; H, 6.62; N, 13.01%.
5.3.3. 3,3′-((1R,4R)-Cyclohexane-1,4-diyl)bis(6-chloro-2-methylquinazolin-4(3H)-one) 3c
White crystals, yield 96%, m.p. 311–313°C; IR (KBr): ν/cm−1 1670 (C=O), 1611, 1587 (C=N, C=C); 1H NMR (400 MHz, CDCl3): δ 8.11 (s, 2H, Ar-H), 7.92–7.90 (d, J = 7.7 Hz, 2H, Ar-H), 7.65–7.64 (d, J = 5.8 Hz, 2H, Ar-H), 3.29–3.27 (m, 2H, Cyclohexane-H), 2.31 (s, 6H, CH3), 1.97–1.95 (m, 4H, Cyclohexane-H), 1.74–1.69 (m, 4H, Cyclohexane-H); 13C NMR (100 MHz, CDCl3): δ 171.0 (C=O), 144.5 (C=N), 145.0, 133.8, 131.6, 128.3, 128.2, 122.6 (Ar-C), 51.7 (Cyclohexane-C), 31.3 (Cyclohexane-C), 20.4 ppm (CH3); MS (EI): m/z (%) 472 (M++4, 10.7), 470 (M++2, 66.3), 468 (M+, 100); Anal. Calcd for C24H22Cl2N4O2: C, 61.42; H, 4.72; N, 11.94%; Found: C, 61.39; H, 4.79; N, 11.88%.
5.3.4. 3,3'-((1R,4R)-Cyclohexane-1,4-diyl)bis(6-fluoro-2-methylquinazolin-4(3H)-one) 3d
White crystals, yield 96%, m.p. 262–264°C; IR (KBr): ν/cm−1 1682 (C=O), 1616, 1597 (C=N, C=C); 1H NMR (400 MHz, CDCl3): δ 8.12 (s, 2H, Ar-H), 7.81–7.80 (d, J = 5.5 Hz, 2H, Ar-H), 7.709–7.702 (m, 2H, Ar-H), 3.79 (br, 2H, Cyclohexane-H), 2.41 (s, 6H, CH3), 1.71–1.64 (m, 4H, Cyclohexane-H), 1.33–1.26 (m, 4H, Cyclohexane-H); 13C NMR (100 MHz, CDCl3): δ 171.1 (C=O), 144.5 (C=N), 159.8, 142.0, 123.8, 121.8, 118.8, 115.2 (Ar-C), 51.8 (Cyclohexane-C), 31.6 (Cyclohexane-C), 20.7 ppm (CH3); 19F NMR (376 MHz, CDCl3): δ -125.90; MS (EI): m/z (%) 437 (M++1, 13.8), 436 (M+, 100); Anal. Calcd for C24H22F2N4O2: C, 66.05; H, 5.08; N, 12.84%; Found: C, 66.11; H, 5.01; N, 12.80%.
5.3.5. 3,3′-((1R,4R)-Cyclohexane-1,4-diyl)bis(2-methyl-6-nitroquinazolin-4(3H)-one) 3e
Light yellow crystals, yield 96%, m.p. 301–304°C; IR (KBr): ν/cm−1 1675 (C=O), 1612, 1585 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 8.80 (s, 2H, Ar-H), 8.13–8.11 (d, J = 7.5 Hz, 2H, Ar-H), 7.79–7.77 (d, J = 7.8 Hz, 2H, Ar-H), 3.71 (br, 2H, Cyclohexane-H), 2.63 (s, 6H, CH3), 1.97–1.95 (m, 4H, Cyclohexane-H), 1.74–1.69 (m, 4H, Cyclohexane-H); 13C NMR (100 MHz, DMSO-d6): δ 170.4 (C=O), 145.8 (C=N), 151.8, 143.3, 128.4, 124.8, 119.6, 117.4 (Ar-C), 52.3 (Cyclohexane-C), 31.9 (Cyclohexane-C), 20.4 ppm (CH3); MS (EI): m/z (%) 491 (M++1, 7.3), 490 (M+, 100); Anal. Calcd for C24H22N6O6: C, 58.77; H, 4.52; N, 17.13%; Found: C, 58.79; H, 4.48; N, 17.09%.
5.3.6. 3,3′-(1,4-Phenylenebis(methylene))bis(2-methylquinazolin-4(3H)-one) 4a
White crystals, yield 98%, m.p. 277–279°C; IR (KBr): ν/cm−1 1688 (C=O), 1616, 1605 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 8.32–8.29 (d, J = 7.7 Hz, 2H, Ar-H), 7.91–7.90 (d, J = 5.6 Hz, 2H, Ar-H), 7.767–7.761 (m, 2H, Ar-H), 7.659–7.650 (d, J = 5.5 Hz, 2H, Ar-H), 7.30 (s, 4H, Ar-H), 4.78 (s, 4H, CH2), 2.29 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 170.7 (C=O), 144.6 (C = N), 146.7, 134.8, 134.2, 128.2, 127.0, 126.9, 126.2, 122.5 (Ar-C), 62.5 (CH2), 20.9 ppm (CH3); MS (EI): m/z (%) 423 (M++1, 8.9), 422 (M+, 100); Anal. Calcd for C26H22N4O2: C, 73.92; H, 5.25; N, 13.26%; Found: C, 73.89; H, 5.29; N, 13.21%.
5.3.7. 3,3′-(1,4-Phenylenebis(methylene))bis(2,6-dimethylquinazolin-4(3H)-one) 4b
White crystals, yield 99%, m.p. 285–287°C; IR (KBr): ν/cm−1 1682 (C=O), 1623, 1611 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 8.39 (s, 2H, Ar-H), 7.98–7.96 (d, J = 7.7 Hz, 2H, Ar-H), 7.41–7.29 (m, 6H, Ar-H), 4.81 (s, 4H, CH2), 2.67 (s, 6H, CH3), 2.29 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 171.2 (C=O), 145.1 (C=N), 142.9, 136.7, 134.4, 132.6, 128.0, 127.5, 126.5, 121.2 (Ar-C), 62.7 (CH2), 21.8 (CH3), 20.6 ppm (CH3); MS (EI): m/z (%) 451 (M++1, 11.5), 450 (M+, 100); Anal. Calcd for C28H26N4O2: C, 74.65; H, 5.82; N, 12.44%; Found: C, 74.61; H, 5.87; N, 12.39%.
5.3.8. 3,3′-(1,4-Phenylenebis(methylene))bis(6-chloro-2-methylquinazolin-4(3H)-one) 4c
Light yellow crystals, yield 95%, m.p. 327–330°C; IR (KBr): ν/cm−1 1675 (C = O), 1615, 1602 (C = N, C = C); 1H NMR (400 MHz, DMSO-d6): δ 8.37 (s, 2H, Ar-H), 7.82–7.81 (d, J = 7.5 Hz, 2H, Ar-H), 7.29 (s, 4H, Ar-H), 7.13–7.06 (m, 2H, Ar-H), 4.81 (s, 4H, CH2), 2.25 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 171.0 (C=O), 144.8 (C=N), 144.9, 133.6, 132.2, 128.2, 127.8, 127.4, 127.1, 121.2 (Ar-C), 62.8 (CH2), 20.3 ppm (CH3); MS (EI): m/z (%) 494 (M++4, 11.1), 492 (M++2, 65.3), 490 (M+, 100); Anal. Calcd for C26H20Cl2N4O2: C, 63.55; H, 4.10; N, 11.40%; Found: C, 63.59; H, 4.05; N, 11.36%.
5.3.9. 3,3′-(1,4-Phenylenebis(methylene))bis(6-fluoro-2-methylquinazolin-4(3H)-one) 4d
White crystals, yield 92%, m.p. 217–219°C; IR (KBr): ν/cm−1 1680 (C = O), 1611, 1593 (C = N, C = C); 1H NMR (400 MHz, DMSO-d6): δ 8.01–8.00 (d, J = 5.6 Hz, 2H, Ar-H), 7.81–7.79 (m, 2H, Ar-H), 7.54–7.49 (m, 2H, Ar-H), 7.15 (s, 4H, Ar-H), 4.78 (s, 4H, CH2), 2.29 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 171.0 (C = O), 145.4 (C = N), 160.8, 141.8, 134.6, 128.1, 127.9, 124.3, 120.2, 117.8 (Ar-C), 62.8 (CH2), 20.3 ppm (CH3); 19F NMR (376 MHz, DMSO-d6): δ -125.91; MS (EI): m/z (%) 459 (M++1, 11.8), 458 (M+, 100); Anal. Calcd for C26H20F2N4O2: C, 68.11; H, 4.40; N, 12.22%; Found: C, 68.07; H, 4.48; N, 12.17%.
5.3.10. 3,3′-(1,4-Phenylenebis(methylene))bis(2-methyl-6-nitroquinazolin-4(3H)-one) 4e
Light yellow crystals, yield 93%, m.p. 350–353°C; IR (KBr): ν/cm−1 1675 (C = O), 1601, 1593 (C = N, C = C); 1H NMR (400 MHz, DMSO-d6): δ 8.69 (s, 2H, Ar-H), 8.36–8.35 (d, J = 5.6 Hz, 2H, Ar-H), 7.87–7.85 (d, J = 7.6 Hz, 2H, Ar-H), 7.29 (s, 4H, Ar-H), 4.78 (s, 4H, CH2), 2.29 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 170.7 (C = O), 145.6 (C = N), 153.1, 142.8, 134.4, 128.4, 128.2, 123.1, 118.0, 117.4 (Ar-C), 62.5 (CH2), 20.4 ppm (CH3); MS (EI): m/z (%) 513 (M++1, 2.6), 512 (M+, 100); Anal. Calcd for C26H20N6O6: C, 60.94; H, 3.93; N, 16.40%; Found: C, 61.00; H, 3.91; N, 16.38%.
5.3.11. 3,3′-(Ethane-1,2-diyl)bis(2-methylquinazolin-4(3H)-one) 5a
White crystals, yield 98%, m.p. 190–192°C; IR (KBr): ν/cm−1 1679 (C = O), 1612, 1590 (C = N, C = C); 1H NMR (400 MHz, DMSO-d6): δ 8.29–8.26 (d, J = 7.1 Hz, 2H, Ar-H), 7.78 (s, 2H, Ar-H), 7.59–7.48 (t, J = 7.4 Hz, 2H, Ar-H), 7.25–7.21 (d, J = 7.1 Hz, 2H, Ar-H), 3.78 (s, 4H, CH2), 2.30 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 171.0 (C = O), 144.9 (C = N), 143.9, 132.7, 127.4, 126.4, 126.0, 121.3 (Ar-C), 42.5 (CH2), 20.4 ppm (CH3); MS (EI): m/z (%) 347 (M++1, 1.8), 346 (M+, 100) [23,24].
5.3.12. 3,3′-(Ethane-1,2-diyl)bis(2,6-dimethylquinazolin-4(3H)-one) 5b
White crystals, yield 97%, m.p. 211–212°C; IR (KBr): ν/cm−1 1675 (C = O), 1608, 1590 (C = N, C = C); 1H NMR (400 MHz, CDCl3): δ 8.00 (s, 2H, Ar-H), 7.80–7.79 (d, J = 7.4 Hz, 2H, Ar-H), 7.50–7.49 (d, J = 7.4 Hz, 2H, Ar-H), 3.80 (s, 4H, CH2), 2.80 (s, 6H, CH3), 2.30 ppm (s, 6H, CH3); 13C NMR (100 MHz, CDCl3): δ 171.2 (C = O), 144.6 (C = N), 143.3, 137.2, 134.6, 127.9, 126.2, 122.6 (Ar-C), 42.7 (CH2), 21.9 (CH3), 20.3 ppm (CH3); MS (EI): m/z (%) 375 (M++1, 3.2), 374 (M+, 100); Anal. Calcd for C22H22N4O2: C, 70.57; H, 5.92; N, 14.96%; Found: C, 70.60; H, 5.89; N, 14.91%.
5.3.13. 3,3′-(Ethane-1,2-diyl)bis(6-chloro-2-methylquinazolin-4(3H)-one) 5c
White crystals, yield 95%, m.p. 244–246°C; IR (KBr): ν/cm−1 1680 (C=O), 1618, 1603 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 7.94 (s, 2H, Ar-H), 7.60–7.59 (d, J = 7.5 Hz, 2H, Ar-H), 7.20–7.19 (d, J = 7.4 Hz, 2H, Ar-H), 3.70 (s, 4H, CH2), 2.29 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 170.5 (C=O), 144.9 (C=N), 133.0, 132.5, 127.6, 127.2, 122.0 (Ar-C), 42.9 (CH2), 20.5 ppm (CH3); MS (EI): m/z (%) 418 (M++4, 11.2), 416 (M++2, 64.6), 414 (M+, 100); Anal. Calcd for C20H16Cl2N4O2: C, 57.85; H, 3.88; N, 13.49%; Found: C, 57.81; H, 3.91; N, 13.42%.
5.3.14. 3,3′-(Ethane-1,2-diyl)bis(6-fluoro-2-methylquinazolin-4(3H)-one) 5d
White crystals, yield 94%, m.p. 219–221°C; IR (KBr): ν/cm−1 1676 (C=O), 1609, 1601 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 7.60–7.57 (d, J = 7.7 Hz, 2H, Ar-H), 7.53–7.51 (d, J = 7.6 Hz, 2H, Ar-H), 7.39–7.35 (d, J = 7.7 Hz, 2H, Ar-H), 3.73 (s, 4H, CH2), 2.34 ppm (s, 6H, CH3); 19F NMR (376 MHz, DMSO-d6): -125.96; 13C NMR (100 MHz, DMSO-d6): δ 170.8 (C=O), 145.5 (C=N), 160.8, 143.1, 124.5, 122.1, 121.1, 113.9 (Ar-C), 42.8 (CH2), 20.5 ppm (CH3); MS (EI): m/z (%) 383 (M++1, 4.9), 382 (M+, 100); Anal. Calcd for C20H16F2N4O2: C, 62.82; H, 4.22; N, 14.65%; Found: C, 62.87; H, 4.18; N, 14.60%.
5.3.15. 3,3′-(Ethane-1,2-diyl)bis(2-methyl-6-nitroquinazolin-4(3H)-one) 5e
Yellow crystals, yield 94%, m.p. 278–279°C; IR (KBr): ν/cm−1 1683 (C=O), 1613, 1603 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 8.61 (s, 2H, Ar-H), 8.41–8.40 (d, J = 7.7 Hz, 2H, Ar-H), 7.809–7.802 (d, J = 7.8 Hz, 2H, Ar-H), 3.60 (s, 4H, CH2), 2.29 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 171.3 (C=O), 144.8 (C=N), 152.6, 143.0, 128.3, 125.3, 120.0, 115.9 (Ar-C), 42.8 (CH2), 20.5 ppm (CH3); MS (EI): m/z (%) 437 (M++1, 6.2), 436 (M+, 100); Anal. Calcd for C20H16N6O6: C, 55.05; H, 3.70; N, 19.26%; Found: C, 54.98; H, 3.74; N, 19.22%.
5.3.16. 3,3′-(1,4-Phenylene)bis(2-methylquinazolin-4(3H)-one) 6a
Yellow crystals, yield 98%, m.p. 178–180°C; IR (KBr): ν/cm−1 1687 (C=O), 1604, 1598 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 8.30–8.28 (d, J = 7.5 Hz, 2H, Ar-H), 8.09–8.01 (m, 2H, Ar-H), 7.78–7.74 (d, J = 7.6 Hz, 2H, Ar-H), 7.61–7.54 (m, 2H, Ar-H), 7.28 (s, 4H, Ar-H), 2.29 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 170.1 (C=O), 145.2 (C=N), 136.8, 134.3, 130.8, 129.2, 127.3, 126.9, 122.3, 117.2 (Ar-C), 21.2 ppm (CH3); MS (EI): m/z (%) 395 (M++1, 11.2), 394 (M+, 100) [23,24].
5.3.17. 3,3′-(1,4-Phenylene)bis(2,6-dimethylquinazolin-4(3H)-one) 6b
Yellow crystals, yield 97%, m.p. 263–265°C; IR (KBr): ν/cm−1 1676 (C=O), 1610, 1591 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 7.99 (s, 2H, Ar-H), 7.80–7.79 (d, J = 7.6 Hz, 2H, Ar-H), 7.42–7.39 (m, 2H, Ar-H), 7.23 (s, 4H, Ar-H), 2.63 (s, 6H, CH3), 2.26 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 170.9 (C=O), 144.5 (C=N), 143.1, 138.3, 133.9, 133.0, 128.1, 126.3, 121.9 (Ar-C), 23.6 (CH3), 21.3 ppm (CH3); MS (EI): m/z (%) 423 (M++1, 6.2), 422 (M+, 100); Anal. Calcd for C26H22N4O2: C, 73.92; H, 5.25; N, 13.26%; Found: C, 73.96; H, 5.22; N, 13.21%.
5.3.18. 3,3′-(1,4-Phenylene)bis(6-chloro-2-methylquinazolin-4(3H)-one) 6c
Yellow crystals, yield 95%, m.p. 287–290°C; IR (KBr): ν/cm−1 1660 (C = O), 1615, 1597 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 8.09 (s, 2H, Ar-H), 7.55–7.51 (d, J = 7.6 Hz, 2H, Ar-H), 7.46 (s, 4H, Ar-H), 7.12–7.10 (m, 2H, Ar-H), 2.30 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 170.0 (C=O), 144.7 (C=N), 144.5, 134.2, 133.7, 127.7, 127.2, 122.9, 121.7 (Ar-C), 21.5 ppm (CH3); MS (EI): m/z (%) 464 (M++2, 62.8), 462 (M+, 100); Anal. Calcd for C24H16Cl2N4O2: C, 62.22; H, 3.48; N, 12.09%; Found: C, 62.18; H, 3.51; N, 12.03%.
5.3.19. 3,3′-(1,4-Phenylene)bis(6-fluoro-2-methylquinazolin-4(3H)-one) 6d
Yellow crystals, yield 97%, m.p. 269–272°C; IR (KBr): ν/cm−1 1674 (C=O), 1622, 1599 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 7.98–7.96 (d, J = 7.7 Hz, 2H, Ar-H), 7.66–7.61 (m, 2H, Ar-H), 7.32 (s, 4H, Ar-H), 7.22–7.19 (m, 2H, Ar-H), 2.20 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 170.2 (C=O), 144.0 (C = N), 162.2, 143.3, 134.0, 125.7, 122.7, 121.2, 120.9, 112.9 (Ar-C), 21.8 ppm (CH3); MS (EI): m/z (%) 431 (M++1, 6.4), 430 (M+, 100); Anal. Calcd for C24H16F2N4O2: C, 66.97; H, 3.75; N, 13.02%; Found: C, 66.95; H, 3.78; N, 12.98%.
5.3.20. 3,3′-(1,4-Phenylene)bis(2-methyl-6-nitroquinazolin-4(3H)-one) 6e
Yellow crystals, yield 93%, m.p. 307–310°C; IR (KBr): ν/cm−1 1683 (C=O), 1614, 1593 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 8.68 (s, 2H, Ar-H), 8.22–8.20 (d, J = 5.5 Hz, 2H, Ar-H), 7.81–7.80 (d, J = 5.5 Hz, 2H, Ar-H), 7.33 (s, 4H, Ar-H), 2.26 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 170.9 (C=O), 144.2 (C = N), 152.2, 144.7, 134.6, 128.5, 124.2, 121.8, 121.5, 119.5 (Ar-C), 21.6 ppm (CH3); MS (EI): m/z (%) 485 (M++1, 3.1), 484 (M+, 100); Anal. Calcd for C24H16N6O6: C, 59.51; H, 3.33; N, 17.35%; Found: C, 59.48; H, 3.39; N, 17.31%.
5.3.21. 3,3′-(Sulfonylbis(4,1-phenylene))bis(2-methylquinazolin-4(3H)-one) 7a
Yellow crystals, yield 98%, m.p. 294–297°C; IR (KBr): ν/cm−1 1689 (C=O), 1615, 1597 (C=N, C=C), 1321 (S=O); 1H NMR (400 MHz, DMSO-d6): δ 8.10–8.07 (m, 4H, Ar-H), 7.88–7.83 (m, 4H, Ar-H), 7.58–7.54 (m, 4H, Ar-H), 7.44–7.40 (m, 4H, Ar-H), 2.22 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 170.2 (C=O), 144.8 (C=N), 145.7, 144.6, 138.5, 134.0, 128.5, 128.1, 126.1, 123.5, 121.3 (Ar-C), 21.9 ppm (CH3); MS (EI): m/z (%) 535 (M++1, 1.3), 534 (M+, 100); Anal. Calcd for C30H22N4O4S: C, 67.40; H, 4.15; N, 10.48%; Found: C, 67.44; H, 4.12; N, 10.43%.
5.3.22. 3,3′-(Sulfonylbis(4,1-phenylene))bis(2,6-dimethylquinazolin-4(3H)-one) 7b
Light yellow crystals, yield 98%, m.p. 310–313°C; IR (KBr): ν/cm−1 1680 (C=O), 1622, 1611 (C=N, C=C), 1308 (S=O); 1H NMR (400 MHz, DMSO-d6): δ 7.92–7.90 (m, 2H, Ar-H), 7.72 (s, 2H, Ar-H), 7.60–7.52 (m, 2H, Ar-H), 7.42–7.39 (d, J = 7.5 Hz, 4H, Ar-H), 7.306–7.301 (m, 4H, Ar-H), 2.40 (s, 6H, CH3), 2.21 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 170.5 (C=O), 144.2 (C=N), 144.0, 143.7, 137.5, 133.7, 128.2, 128.0, 127.8, 126.2, 123.1, 121.8 (Ar-C), 23.3 (CH3), 21.6 ppm (CH3); MS (EI): m/z (%) 563 (M++1, 1.9), 562 (M+, 100); Anal. Calcd for C32H26N4O4S: C, 68.31; H, 4.66; N, 9.96%; Found: C, 68.27; H, 4.69; N, 9.91%.
5.3.23. 3,3′-(Sulfonylbis(4,1-phenylene))bis(6-chloro-2-methylquinazolin-4(3H)-one) 7c
Yellow crystals, yield 97%, m.p. 332–335°C; IR (KBr): ν/cm−1 1689 (C=O), 1629, 1607 (C=N, C=C), 1327 (S=O); 1H NMR (400 MHz, DMSO-d6): δ 8.36 (s, 2H, Ar-H), 7.92–7.90 (m, 2H, Ar-H), 7.71–7.62 (m, 2H, Ar-H), 7.47–7.41 (d, J = 7.8 Hz, 4H, Ar-H), 7.39–7.32 (m, 4H, Ar-H), 2.29 (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 171.1 (C=O), 144.0 (C=N), 144.2, 143.9, 138.1, 133.9, 132.8, 128.5, 127.8, 122.8, 121.6 (Ar-C), 21.9 ppm (CH3); MS (EI): m/z (%) 604 (M++2, 62.9), 602 (M+, 100); Anal. Calcd for C30H20Cl2N4O4S: C, 59.71; H, 3.34; N, 9.28%; Found: C, 59.74; H, 3.32; N, 9.25%.
5.3.24. 3,3′-(Sulfonylbis(4,1-phenylene))bis(6-fluoro-2-methylquinazolin-4(3H)-one) 7d
Yellow crystals, yield 93%, m.p. 301–303°C; IR (KBr): ν/cm−1 1692 (C=O), 1621, 1602 (C=N, C=C), 1312 (S=O); 1H NMR (400 MHz, DMSO-d6): δ 7.82–7.80 (m, 2H, Ar-H), 7.69–7.59 (m, 2H, Ar-H), 7.36–7.30 (m, 4H, Ar-H), 7.16–7.15 (m, 2H, Ar-H), 7.14–7.08 (m, 4H, Ar-H), 2.25 (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 171.1 (C=O), 144.0 (C=N), 161.9, 143.6, 142.7, 138.0, 128.9, 125.6, 122.7, 120.8, 112.9 (Ar-C), 21.8 ppm (CH3); MS (EI): m/z (%) 571 (M++1, 2.5), 570 (M+, 100); Anal. Calcd for C30H20F2N4O4S: C, 63.15; H, 3.53; N, 9.82%; Found: C, 63.11; H, 3.57; N, 9.78%.
5.3.25. 3,3′-(Sulfonylbis(4,1-phenylene))bis(2-methyl-6-nitroquinazolin-4(3H)-one) 7e
Yellow crystals, yield 95%, m.p. 316–319°C; IR (KBr): ν/cm−1 1688 (C=O), 1601, 1589 (C=N, C=C), 1318 (S=O); 1H NMR (400 MHz, DMSO-d6): δ 8.66 (s, 2H, Ar-H), 8.31–8.29 (m, 2H, Ar-H), 8.00–7.98 (m, 4H, Ar-H), 7.66–7.65 (d, J = 5.5 Hz, 4H, Ar-H), 7.45–7.40 (m, 2H, Ar-H), 2.21 (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 171.1 (C=O), 144.2 (C=N), 152.9, 143.2, 137.3, 128.6, 125.0, 122.4, 122.2, 120.6, 118.5 (Ar-C), 21.8 ppm (CH3); MS (EI): m/z (%) 625 (M++1, 0.9), 624 (M+, 100); Anal. Calcd for C30H20N6O8S: C, 57.69; H, 3.23; N, 13.46%; Found: C, 57.72; H, 3.20; N, 13.41%.
5.3.26. 3-(2-Aminophenyl)-2-methylquinazolin-4(3H)-one 8
White crystals, yield 33%, m.p. 109–111°C; IR (KBr): ν/cm−1 3324–2984 (NH2), 1694 (C=O), 1611, 1594 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 8.208–8.205 (d, J = 5.7 Hz, 1H, Ar-H), 7.93–7.91 (d, J = 7.8 Hz, 1H, Ar-H), 7.73–7.70 (d, J = 5.7 Hz, 1H, Ar-H), 7.66–7.63 (m, 2H, Ar-H), 7.48–7.44 (m, 2H, Ar-H), 7.28–7.24 (m, 1H, Ar-H), 6.50 (br, 2H, NH2), 2.29 ppm (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 171.1 (C=O), 154.3 (C=N), 141.7, 137.8, 135.0, 130.9, 130.1, 128.9, 127.7, 127.4, 126.7, 121.9, 117.4, 115.0 (Ar-C), 21.0 ppm (CH3); MS (EI): m/z (%) 252 (M++1, 100), 251 (M+, 2.6) [23,24].
5.3.27. 3,3′-(1,2-Phenylene)bis(2-methylquinazolin-4(3H)-one) 9
Yellow crystals, yield 88%, m.p. 187–190°C; IR (KBr): ν/cm−1 1685 (C=O), 1621, 1588 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 8.11–8.08 (d, J = 5.7 Hz, 2H, Ar-H), 7.90–7.88 (d, J = 7.7 Hz, 2H, Ar-H), 7.72–7.68 (m, 4H, Ar-H), 7.33–7.30 (d, J = 9.8 Hz, 4H, Ar-H), 2.24 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 171.7 (C=O), 148.9 (C=N), 142.7, 133.9, 133.7, 127.0, 126.9, 126.6, 123.4, 121.2, 115.7 (Ar-C), 24.1 ppm CH3); MS (EI): m/z (%) 395 (M++1, 7.9), 394 (M+, 100) [23,24].
5.3.28. 3-(3-Aminophenyl)-2-methylquinazolin-4(3H)-one 10
White crystals, yield 33%, m.p. 121–123°C; IR (KBr): ν/cm−1 3365–2990 (NH2), 1687 (C=O), 1609, 1587 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 8.18–8.15 (d, J = 7.5 Hz, 1H, Ar-H), 7.86–7.81 (t, J = 7.8 Hz, 1H, Ar-H), 7.76–7.70 (m, 2H, Ar-H), 7.36–7.31 (m, 1H, Ar-H), 6.98–6.94 (m, 2H, Ar-H), 6.78–6.74 (m, 1H, Ar-H), 2.26 ppm (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 170.4 (C=O), 154.0 (C=N), 145.7, 144.8, 133.2, 129.9, 128.0, 127.5, 126.6, 122.4, 120.0, 118.9, 115.4, 111.6 (Ar-C), 21.4 ppm (CH3); MS (EI): m/z (%) 252 (M++1, 17.0), 251 (M+, 100) [51].
5.3.29. 3,3′-(1,3-Phenylene)bis(2-methylquinazolin-4(3H)-one) 11
Light yellow crystals, yield 92%, m.p. 236–238°C; IR (KBr): ν/cm−1 1679 (C=O), 1616, 1607 (C=N, C=C); 1H NMR (400 MHz, DMSO-d6): δ 7.92–7.90 (m, 2H, Ar-H), 7.72 (s, 1H, Ar-H), 7.34–7.28 (m, 4H, Ar-H), 7.10–7.08 (d, J = 7.5 Hz, 2H, Ar-H), 7.03–6.99 (m, 3H, Ar-H), 2.30 ppm (s, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 171.2 (C=O), 154.3 (C=N), 144.6, 134.4, 133.4, 129.6, 127.8, 127.4, 126.1, 122.4, 121.7, 114.1 (Ar-C), 21.7 ppm (CH3); MS (EI): m/z (%) 395 (M++1, 22.5), 394 (M+, 100); Anal. Calcd for C24H18N4O2: C, 73.08; H, 4.60; N, 14.20%; Found: C, 73.11; H, 4.58; N, 14.19%.
5.4. In vitro anti-cancer screening
The cytotoxic activities of all newly prepared derivatives were assessed in monolayer cultures by utilizing MTT assay. Two human cancer cell lines (MCF-7 and A549) and one normal cell line (MCF-10A), were maintained in the minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 2 ml glutamine and 100 units ml−1 penicillin in a CO2 incubator in a humidified atmosphere of 5% CO2 and 95% air. The tested derivatives were prepared prior to the experiment by dissolving in 0.1% DMSO and diluted with medium. The cells were then exposed to two concentrations of drugs (10 and 30 µM) in the volume of 100 µM/well. Cells in the control wells received the same volume of medium comprising 0.1% DMSO. After 24 h, the medium was removed and cell cultures were incubated with 100 ml MTT reagent for 5 h at 37°C. The known number of cells was incubated in a 5% CO2 incubator at 37°C in the presence of different concentrations of test compounds. After 48 h of drug incubation, the MTT solution was added in each well and the absorbance was measured by using a microplate reader at 490 nm. The experiment was performed in triplicate. Cell survival was calculated as the percentage of MTT inhibition as % growth inhibition = 100 − (mean OD of individual test group/mean OD of each control group) × 100. The IC50 values of the synthesized compounds for two cell lines are summarized in electronic supplementary material, S59, table S3).
Supplementary Material
Data accessibility
The datasets supporting this article have been uploaded as part of the electronic supplementary material.
Competing interests
I declare I have no competing interests.
Funding
This study was financially supported by the Jouf University, project no. 634/39.
References
- 1.Kshirsagar UA. 2015. Recent developments in the chemistry of quinazolinone alkaloids. Org. Biomol. Chem. 13, 9336 ( 10.1039/C5OB01379H) [DOI] [PubMed] [Google Scholar]
- 2.Michael JP. 2007. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 24, 223 ( 10.1039/b509528j) [DOI] [PubMed] [Google Scholar]
- 3.Lüth A, Löwe W. 2008. Syntheses of 4-(indole-3-yl)quinazolines—a new class of epidermal growth factor receptor tyrosine kinase inhibitors. Eur. J. Med. Chem. 43, 1478 ( 10.1016/j.ejmech.2007.09.018) [DOI] [PubMed] [Google Scholar]
- 4.Shakhidoyatov KM, Elmuradov BZ. 2014. Tricyclic quinazoline alkaloids: isolation, synthesis, chemical modification, and biological activity. Chem. Nat. Compd. 50, 781 ( 10.1007/s10600-014-1086-6) [DOI] [Google Scholar]
- 5.Mhaske SB, Argade NP. 2006. The chemistry of recently isolated naturally occurring quinazolinone alkaloids. Tetrahedron 62, 9787 ( 10.1016/j.tet.2006.07.098) [DOI] [Google Scholar]
- 6.Connolly DJ, Cusack D, O'Sullivan TP, Guiry PJ. 2005. Synthesis of quinazolinones and quinazolines. Tetrahedron 61, 10153 ( 10.1016/j.tet.2005.07.010) [DOI] [Google Scholar]
- 7.Alagarsamy V, Chitra K, Saravanan G, Raja Solomon V, Sulthana MT, Narendhar B. 2018. An overview of quinazolines: Pharmacological significance and recent developments. Eur. J. Med. Chem. 151, 628 ( 10.1016/j.ejmech.2018.03.076) [DOI] [PubMed] [Google Scholar]
- 8.Lu J, Zhan X, Chen L, Zhang L, Mao SJ. 2014. Solubility of two polymorphs of erlotinib hydrochloride in isopropanol and acetone from (273.15 to 303.15) K. J. Chem. Eng. Data 59, 2665 ( 10.1021/je500482k) [DOI] [Google Scholar]
- 9.Zhang Y, Tortorella MD, Liao J, Qin X, Chen T, Luo J, Guan J, Talley JJ, Tu Z. 2015. Synthesis and evaluation of novel erlotinib–NSAID conjugates as more comprehensive anticancer agents. ACS Med. Chem. Lett. 6, 1086 ( 10.1021/acsmedchemlett.5b00286) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou G-H, Wang L, Ma Y-D, Wang L-P, Zhang Y-Y, Jiang W. 2011. Synthesis of a quinazoline derivative: a new α1-adrenoceptor ligand for conjugation to quantum dots to study α1-adrenoceptors in living cells. Bioorg. Med. Chem. Lett. 21, 5905 ( 10.1016/j.bmcl.2011.07.122) [DOI] [PubMed] [Google Scholar]
- 11.Cao SL, Feng YP, Jiang YY, Liu SY, Ding GY, Li RT. 2005. Synthesis and in vitro antitumor activity of 4(3H)-quinazolinone derivatives with dithiocarbamate side chains. Bioorg. Med. Chem. Lett. 15, 1915 ( 10.1016/j.bmcl.2005.01.083) [DOI] [PubMed] [Google Scholar]
- 12.Zou S, Wang S, Xi C. 2018. ROTf-induced annulation of heteroatom reagents and unsaturated substrates leading to cyclic compounds. R. Soc. open sci. 5, 181389 ( 10.1098/rsos.181389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oschatz S, Brunzel T, Wu XF, Langer P. 2015. Catalyst-free synthesis of 2-aryl-1,2-dihydro-quinazolin-4(1H)-thiones from 2-aminobenzothio-amides and aldehydes in water. Org. Biomol. Chem. 13, 1150 ( 10.1039/C4OB02207F) [DOI] [PubMed] [Google Scholar]
- 14.Stevens MY, Wieckowski K, Wu P, Sawant RT, Odell LR. 2015. A microwave-assisted multicomponent synthesis of substituted 3,4-dihydroquinazolinones. Org. Biomol. Chem. 13, 2044 ( 10.1039/C4OB02417F) [DOI] [PubMed] [Google Scholar]
- 15.Bogert MT, Gortner RA, Amend CG. 1911. Researches on quinazolines (twenty-seventh paper). the synthesis of 3-aminoaryl-4-quinazolones from acylanthranils and aromatic diamines. J. Am. Chem. Soc. 33, 949 ( 10.1021/ja02219a016) [DOI] [Google Scholar]
- 16.Gholap AVA, Maity S, Schulzke C, Maiti D, Kapdi AR. 2017. Synthesis of Cu-catalysed quinazolinones using a Csp3–H functionalisation/cyclisation strategy. Org. Biomol. Chem. 15, 7140 ( 10.1039/C7OB01723E) [DOI] [PubMed] [Google Scholar]
- 17.Qiao R, Ye L, Hu K, Yu S, Yang W, Liu M, Chen J, Dinga J, Wu H. 2017. Copper-catalyzed C–O bond cleavage and cyclization: synthesis of indazolo[3,2-b]quinazolinones. Org. Biomol. Chem. 15, 2168 ( 10.1039/C6OB02352E) [DOI] [PubMed] [Google Scholar]
- 18.Maiden TMM, Harrity JPA. 2016. Recent developments in transition metal catalysis for quinazolinone synthesis. Org. Biomol. Chem. 14, 8014 ( 10.1039/C6OB01402J) [DOI] [PubMed] [Google Scholar]
- 19.Zheng Y, Song WB, Zhang SW, Xuan LJ. 2015. Ruthenium-catalyzed oxidative coupling of 2-aryl-4-quinazolinones with olefins: synthesis of pyrrolo[2,1-b]quinazolin-9(1H)-one motifs. Org. Biomol. Chem. 13, 6474 ( 10.1039/C5OB00601E) [DOI] [PubMed] [Google Scholar]
- 20.Xu G, Tong C, Cui S, Dai L. 2018. A silver catalyzed domino reaction of N-cyanamide alkenes and 1,3-dicarbonyls for the synthesis of quinazolinones. Org. Biomol. Chem. 16, 5899 ( 10.1039/C8OB01252K) [DOI] [PubMed] [Google Scholar]
- 21.Zhou J, Ji M, Yao H, Cao R, Zhao H, Wang X, Chen X, Xu B. 2018. Discovery of quinazoline-2,4(1H,3H)-dione derivatives as novel PARP-1/2 inhibitors: design, synthesis and their antitumor activity. Org. Biomol. Chem. 16, 3189 ( 10.1039/C8OB00286J) [DOI] [PubMed] [Google Scholar]
- 22.Guo S, Zhai J, Fan X. 2017. An I2-mediated cascade reaction of 2′-bromoacetophenones with benzohydrazides/benzamides leading to quinazolino[3,2-b]cinnoline or tryptanthrin derivatives. Org. Biomol. Chem. 15, 1521 ( 10.1039/C6OB02699K) [DOI] [PubMed] [Google Scholar]
- 23.Reddy CN, Mohan HR, Rao DM. 2011. Synthesis of novel quinazolino-quinolones as potential antibacterial agents. Der Pharma Chem. 3, 440. [Google Scholar]
- 24.Ajani OO, Audu OY, Germann MW, Bello BL. 2017. Expeditious synthesis and spectroscopic characterization of 2-methyl-3-substituted-quinazolin-4(3H)-one derivatives. Orient. J. Chem. 33, 562 ( 10.13005/ojc/330203) [DOI] [Google Scholar]
- 25.Ramsey E, Sun Q, Zhang Z, Zhang C, Gou W. 2009. Mini-review: green sustainable processes using supercritical fluid carbon dioxide. J. Environ. Sci. 21, 720 ( 10.1016/S1001-0742(08)62330-X) [DOI] [PubMed] [Google Scholar]
- 26.Li CJ, Chen L. 2006. Organic chemistry in water. Chem. Soc. Rev. 35, 68 ( 10.1039/B507207G) [DOI] [PubMed] [Google Scholar]
- 27.Arafa WAA, Fareed MF, Rabeh SA, Shaker RM. 2016. Ultrasound mediated green synthesis of rhodanine derivatives: synthesis, chemical behavior, and antibacterial activity. Phosphorus Sulfur Silicon Relat. Elem. 191, 1129 ( 10.1080/10426507.2016.1146276) [DOI] [Google Scholar]
- 28.Imperato G, König B, Chiappe C. 2007. Ionic green solvents from renewable resources. Eur. J. Org. Chem. 7, 1049 ( 10.1002/ejoc.200600435) [DOI] [Google Scholar]
- 29.Chen J, Xie F, Li X, Chen L. 2018. Ionic liquids for the preparation of biopolymer materials for drug/gene delivery: a review. Green Chem. 20, 4169 ( 10.1039/C8GC01120F) [DOI] [Google Scholar]
- 30.Kudłak B, Owczarek K, Namieśnik J. 2015. Selected issues related to the toxicity of ionic liquids and deep eutectic solvents—a review. Environ. Sci. Pollut. Res. Int. 22, 11975 ( 10.1007/s11356-015-4794-y) [DOI] [PubMed] [Google Scholar]
- 31.Vanda H, Dai Y, Wilson EG, Verpoorte R, Choi YH. 2018. Green solvents from ionic liquids and deep eutectic solvents to natural deep eutectic solvents. C. R. Chimie 21, 628 ( 10.1016/j.crci.2018.04.002) [DOI] [Google Scholar]
- 32.Tomé LIN, Baião V, da Silva W, Bret CMA. 2018. Deep eutectic solvents for the production and application of new materials. Appl. Mater. Today 10, 30 ( 10.1016/j.apmt.2017.11.005) [DOI] [Google Scholar]
- 33.Kazi TG, Afridi HI, Bhatti M, Akhtar A. 2019. A rapid ultrasonic energy assisted preconcentration method for simultaneous extraction of lead and cadmium in various cosmetic brands using deep eutectic solvent: A multivariate study. Ultrason. Sonochem. 51, 40 ( 10.1016/j.ultsonch.2018.10.016) [DOI] [PubMed] [Google Scholar]
- 34.Ballantyne AD, Hallett JP, Riley DJ, Shah N, Payne DJ. 2018. Lead acid battery recycling for the twenty-first century. R. Soc. open sci. 5, 171368 ( 10.1098/rsos.171368) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alcalde R, Atilhanb M, Aparicio S. 2018. On the properties of (choline chloride + lactic acid) deep eutectic solvent with methanol mixtures. J. Mol. Liq. 272, 815 ( 10.1016/j.molliq.2018.10.052) [DOI] [Google Scholar]
- 36.Zhao BY, Xu P, Yang FX, Wu H, Zong MH, Lou WY. 2015. Biocompatible deep eutectic solvents based on choline chloride: characterization and application to the extraction of rutin from Sophora japonica. ACS Sustain. Chem. Eng. 3, 2746 ( 10.1021/acssuschemeng.5b00619) [DOI] [Google Scholar]
- 37.Araujo CF, Coutinho JAP, Nolasco MM, Parker SF, Ribeiro-Claro PJA, Rudic S, Soares BIG, Vaz PD. 2017. Inelastic neutron scattering study of reline: shedding light on the hydrogen bonding network of deep eutectic solvents. Phys. Chem. Chem. Phys. 19, 17998 ( 10.1039/C7CP01286A) [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Liu Y, Zhou Z, Fu Y, Chang J. 2017. Epoxidation of soybean oil catalyzed by deep eutectic solvents based on the choline chloride–carboxylic acid bifunctional catalytic system. Ind. Eng. Chem. Res. 56, 8224 ( 10.1021/acs.iecr.7b01677) [DOI] [Google Scholar]
- 39.Khandelwal S, Tailor YK, Kumar M. 2016. Deep eutectic solvents (DESs) as eco-friendly and sustainable solvent/catalyst systems in organic transformations. J. Mol. Liq. 215, 345 ( 10.1016/j.molliq.2015.12.015) [DOI] [Google Scholar]
- 40.Li P, Sirviö JA, Haapala A, Liimatainen H. 2017. Cellulose nanofibrils from nonderivatizing urea-based deep eutectic solvent pretreatments. ACS Appl. Mater. Interfaces 9, 2846 ( 10.1021/acsami.6b13625) [DOI] [PubMed] [Google Scholar]
- 41.Banerjee B. 2017. Recent developments on ultrasound-assisted one-pot multicomponent synthesis of biologically relevant heterocycles. Ultrason. Sonochem. 35, 15 ( 10.1016/j.ultsonch.2016.10.010) [DOI] [PubMed] [Google Scholar]
- 42.Shaabani A, Hooshmand SE. 2018. Diversity-oriented catalyst-free synthesis of pseudopeptides containing rhodanine scaffolds via a one-pot sequential isocyanide-based six-component reactions in water using ultrasound irradiation. Ultrason. Sonochem. 40, 84 ( 10.1016/j.ultsonch.2017.06.030) [DOI] [PubMed] [Google Scholar]
- 43.Ibrahim HM, Arafa WAA, Behbehani H. 2018. l-Proline catalyzed one-pot synthesis of polysubstituted pyridine system incorporating benzothiazole moiety via sustainable sonochemical approach. RSC Adv. 8, 37606 ( 10.1039/C8RA07013J) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arafa WAA, Ibrahim HM. 2018. A sustainable strategy for the synthesis of bis-2-iminothiazolidin-4-ones utilizing novel series of asymmetrically substituted bis-thioureas as viable precursors. RSC Adv. 8, 10516 ( 10.1039/C8RA01253A) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arafa WAA. 2018. An eco-compatible pathway to the synthesis of mono and bis-multisubstituted imidazoles over novel reusable ionic liquids: an efficient and green sonochemical process. RSC Adv. 8, 16392 ( 10.1039/C8RA02755B) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arafa WAA, Faty RAM, Mourad AK. 2018. A new sustainable strategy for synthesis of novel series of bis-imidazole and bis-1,3-thiazine derivatives. J. Heterocycl. Chem. 55, 1886 ( 10.1002/jhet.3221) [DOI] [Google Scholar]
- 47.Ramazani A, Rouhani M, Jo SW. 2016. Catalyst-free sonosynthesis of highly substituted propanamide derivatives in water. Ultrason. Sonochem. 28, 393 ( 10.1016/j.ultsonch.2015.08.019) [DOI] [PubMed] [Google Scholar]
- 48.Puri S, Kaur B, Parmar A, Kuma H. 2013. Applications of ultrasound in organic synthesis: a green approach. Curr. Org. Chem. 17, 1790 ( 10.2174/13852728113179990018) [DOI] [Google Scholar]
- 49.Bryan MC, et al. 2018. Key green chemistry research areas from a pharmaceutical manufacturers' perspective revisited. green chem. 20, 5082 ( 10.1039/C8GC01276H) [DOI] [Google Scholar]
- 50.Henderson SH, West RA, Ward SE, Honey MA. 2018. Metal-free selective mono-halodecarboxylation of heteroarenes under mild conditions. R. Soc. open sci. 5, 180333 ( 10.1098/rsos.180333) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Staskun B, Wolfe JF. 1992. New approach to the indolo[2,1-b]quinazoline ring system by cyclization of 3-(o-chlorophenyl)-2-methyl-4(3H)-quinazolinone and its m-isomer: synthesis of the antibiotic tryptanthrin1. S. Afr. J. Chem. 45, 5. [Google Scholar]
- 52.Curzons D, Constable DJC, Mortimer DN, Cunningham VL. 2001. So you think your process is green, how do you know?—using principles of sustainability to determine what is green – a corporate perspective. Green Chem. 3, 1 ( 10.1039/b007871i) [DOI] [Google Scholar]
- 53.Jimenez-Gonzalez C, Constable DJC, Ponder CS. 2012. Evaluating the ‘greenness’ of chemical processes and products in the pharmaceutical industry—a green metrics primer. Chem. Soc. Rev. 41, 1485 ( 10.1039/C1CS15215G) [DOI] [PubMed] [Google Scholar]
- 54.Di Gioia ML, Nardi M, Costanzo P, De Nino A, Maiuolo L, Oliverio M, Procopio A. 2018. Biorenewable deep eutectic solvent for selective and scalable conversion of furfural into cyclopentenone derivatives. Molecules 23, 1891 ( 10.3390/molecules23081891) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55 ( 10.1016/0022-1759(83)90303-4) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting this article have been uploaded as part of the electronic supplementary material.