

Abstract
Inhibitors of Apoptosis Proteins (IAPs) are conserved E3‐ligases that ubiquitylate substrates to prevent apoptosis and activate the NF‐kB survival pathway, often deregulated in cancer. IAPs‐mediated regulation of NF‐kB signaling is based on the formation of protein complexes by their type‐I BIR domains. The XIAP‐BIR1 domain dimerizes to bind two TAB1 monomers, leading to downstream NF‐kB activation. Thus, impairment of XIAP‐BIR1 dimerization could represent a novel strategy to hamper cell survival in cancer. To this aim, we previously reported NF023 as a potential inhibitor of XIAP‐BIR1 dimerization. Here we present a thorough analysis of NF023 binding to XIAP‐BIR1 through biochemical, biophysical and structural data. The results obtained indicate that XIAP‐BIR1 dimerization interface is involved in NF023 binding, and that NF023 overall symmetry and the chemical features of its central moiety are essential for an efficient interaction with the protein. Such strategy provides original hints for the development of novel BIR1‐specific compounds as pro‐apoptotic agents.
Keywords: apoptosis, drug discovery, structure-activity relationship, in silico docking, protein structures
1. Introduction
Inhibitor of apoptosis proteins (IAPs) regulate apoptosis and cell survival.1 IAPs are E3 ligases that ubiquitylate different substrates for the regulation of the NF‐κB survival pathway;2 furthermore, they are able to prevent both caspase‐dependent and ‐independent apoptosis.[3][4] In this context, X‐chromosome linked IAP (XIAP) is the only member of the IAP family known to directly inhibit caspases.5 In humans, IAPs are characterized by the presence of one or more Baculoviral IAP Repeat (BIR) domains, essential for protein‐protein interactions and dimerization.3 BIRs are Zinc‐finger domains composed of ∼70 residues and can be classified into type I and type II, with the latter group displaying a unique peptide binding cleft for the accommodation of N‐terminal tetrapeptides of substrate proteins.[2][3] Type II BIR domains have been extensively studied for the development of Smac‐mimetics (SM), specific IAP inhibitors currently in advanced clinical trial as anti‐cancer agents.6, 7, 8 Although SM have been shown to be very efficient in sensitizing tumor cells to apoptosis, some cancer cell lines present SM‐resistance due to IAP‐dependent re‐activation of NF‐κB.9 We thus decided to explore alternative mechanisms to interfere with IAP‐involving signaling in NF‐κB regulation. IAPs role in this survival pathway is based on the formation of different protein‐protein complexes, regulating ubiquitin‐dependent signal transduction cascades.2 Type I BIR domains from different IAPs have been recognized as a pivotal platform for the assembly of such complexes. In this context, we focused our attention on the interaction of XIAP‐BIR1 homodimer with TAK‐Binding protein 1 (TAB1) that leads to NF‐κB activation through the recruitment of the kinase TAK1.10 Inhibition of XIAP‐BIR1 dimerization and the consequent assembly of the XIAP‐BIR1/TAB1 complex could in fact represent a novel strategy for the development of anti‐cancer drugs. In a previous work we identified NF023 as a promising XIAP‐BIR1 inhibitor.11 Here, the analysis of the interaction between NF023 and four XIAP‐BIR1 mutants (i. e. R62S, D71A, R82S and V86E) demonstrates that NF023 can bind to the domain in an additional site beyond the one observed in the crystal structure. The new binding site explains the perturbative effect of NF023 on the equilibrium between monomeric and dimeric forms of the protein in solution.11
Furthermore we here report an exhaustive structure−activity relationship analysis (SAR) of the interaction of XIAP‐BIR1 with 7 different NF023 analogs: compounds 6, 7, 10 12 and 3, 5 a, 5 b, and 9, here reported for the first time. The three tetrasodium ureidobis(naphthalenedisulphonate)s (3, 5 a, 5 b) and the disodium naphthalenedisulphonate (9) were synthesized from the corresponding arylamines (and triphosgene for 3, 5 a, and 5 b only). The length and symmetry of ureidosulphonates (i. e., NF023, 5 a, and 5 b) are of importance for their binding to XIAP‐BIR1 and provide details for the design of a new class of compounds as pro‐apoptotic agents.
2. Results and Discussion
2.1. Synthesis and Spectral Characteristics of Sodium Organosulphonates 3, 5 a, 5 b and 9
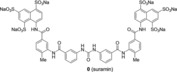
NF023 is an analog of suramin, which showed trypanocidal activity and became the drug of choice for treatment of African trypanosomiasis and onchocerciasis.13 As shown in Figure 1, sodium organosulphonate NF023 and sodium suramin share a common feature of a symmetric framework that contains an ureido (NH−CO−NH) functionality in the center as the “head” (Figure 1).14 Both compounds possess six sulphonate groups as “fingers” and two naphthalene rings as the “palms”. Nevertheless, suramin is built by two benzamido linkers on both flanks as the “shoulders” and two tolylamido linkers as the “arms.” Organosulphonate NF023 has only the two benzamido linkers, lacking the two tolylamido linkers.
Figure 1.
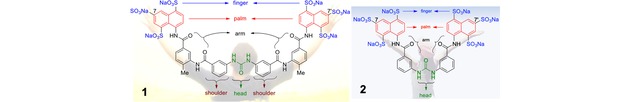
The longer framework of sodium suramin (left panel) in comparison with its shorter analog sodium (ureido)naphthylsulphonate (NF023, right panel).
Crystals of triphosgene (1, (Cl3CO)2CO) have been chosen as a safer substitute for phosgene,15 to couple three molecular arylamines for production of their respective symmetric ureido salts.16,17 First, an aniline salt 2, 4 a or 4 b in 1.3 equivalents was added to water with pH value 3.0. It was allowed to react with 2.0 equivalents of triphosgene (1) in toluene through a condensation process at 25 °C (Scheme 1). The desired symmetric ureido products 3, 5 a and 5 b were produced in 89 %, 84 % and 86 % yields, respectively. The structures of all these sodium ureido salts 3, 5 a and 5 b were identified on the basis of their spectroscopic characteristics (see Supporting Information).
Scheme 1.
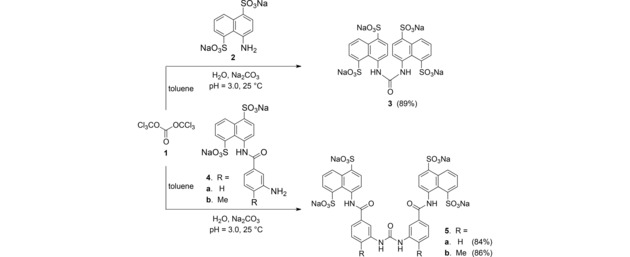
Synthesis of tetrasodium ureidobis(naphthalenedisulphonate)s 3, 5 a and 5 b.
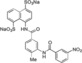
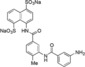
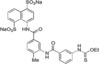
For the study of structure–activity relationship, an asymmetrical disodium salt 9 became our synthetic target. Coupling of aniline disodium salt 7, obtained from the corresponding nitro compound 6,12 with potassium ethyl xanthate (8) in the presence of potassium triiodide in a water solution with pH 11.0 at 25 °C for 30 min, led to the formation of ethyl thiocarbamate 9 in 67 % yield (Scheme 2). Spectroscopic analysis of 9 are shown in Supporting Information.
Scheme 2.
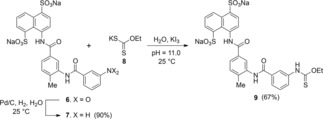
Synthesis of a disodium salt of thiocarbamate 9
2.2. Mutants Selection from In Silico Docking Data
The published crystallographic data of XIAP‐BIR1 in complex with NF023 (PDB: 4MTZ11) show a binding mode that is not congruent with the interference on the protein dimerization observed in solution.11 With the aim to select a new potential NF023 binding site in solution, we identified, through in silico docking, a favored conformation of NF023 along XIAP‐BIR1 dimerization surface (Figure 2A; docking energy of −9.13 kcal mol−1).11 Such conformation allowed us to select a set of BIR1 mutants likely able to impair NF023 binding: R62S, D71A, R82S and V86E (known to prevent XIAP‐BIR1 dimerization10) (Figure 2B).
Figure 2.
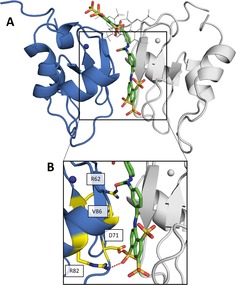
In silico prediction of NF023 binding to XIAP‐BIR1. (A) Comparison of NF023 binding modes as seen in docked conformation (shown as sticks, with carbon, oxygen, nitrogen and sulfur atoms in green, red, blue and orange, respectively) or in the crystal structure (shown as grey lines; PDB: 4MTZ11). The two monomers of the XIAP‐BIR1 dimer are shown as blue and grey cartoons. Zn ions are shown as spheres with colors of the corresponding protein chain. (B) Zoomed panel with mutated residues shown as sticks, with yellow carbon atoms. Red dashed lines represent predicted interactions with NF023.
The 4 protein mutants were thus expressed and purified as described (see Materials and Methods). DLS measurements confirmed that all mutants, with the expected exception of V86E, were able to undergo homodimerization as wild type XIAP‐BIR1.
2.3. Binding Assays of XIAP‐BIR1 Mutants to NF023
The binding of NF023 to XIAP‐BIR1 (wild type and mutant variants), was assessed by MicroScale Thermophoresis (MST). Increasing concentrations of NF023 were used to titrate wild type XIAP‐BIR1 (100 nM) yielding an estimated dissociation constant (K d) of 25±5 μM. This value is consistent with that previously reported,11 indicating that protein labelling does not interfere with NF023 binding. Analogous experiments were therefore performed on all the mutant variants of XIAP‐BIR1 (Table 1).
Table 1.
Affinities of XIAP‐BIR1 forms for NF023 determined by MST measurements.
| XIAP‐BIR1 form | Kd vs NF023 (μM) |
|---|---|
| Wild type | 25±5 |
| R62S | 1,145±202 |
| D71A | >10,000 |
| R82S | 1,790±425 |
| V86E | 135±30 |
Overall, the tested mutations, which are located along XIAP‐BIR1 dimerization interface, have a negative effect on NF023 binding, indicating that the compound actually binds to this protein surface, as predicted by in silico docking (Figure 2). With respect to the other three tested mutations, which are in positions predicted to interact with NF023, the V86E one presents a milder effect on XIAP‐BIR1 affinity vs NF023. In fact, even if V86 faces one of the two NF023 naphthalene moieties (Figure 2B), it was not predicted to interact with the compound. Probably, the glutamate steric hindrance and polarity in this position should not compromise NF023 binding. Such hypothesis was confirmed by the crystal structure of V86E mutant, where E86 side chain orientation is compatible with the predicted NF023 binding to the protein (PDB: 6QCI, Table 2, Figure S1).
Table 2.
X‐ray data collection and refinement statistics for the XIAP‐BIR1 V86E and XIAP‐BIR1/5 a structures.
| Structure | XIAP‐BIR1V86E | XIAP‐BIR1/5 a |
|---|---|---|
| Space group | P21 | P21 |
| Unit‐cell parameters [Å;°] | a=36.5, b=72.7, c=70.2; β=96.2 | a=36.5, b=75.8, c=70.0; β=90.0 |
| Solvent content (%) | 36 | 39 |
| Number of molecules per asymmetric unit | 4 | 4 |
| Resolution (Å) | 36.35–2.30 | 33.33–1.90 |
| Number of unique reflections | 16,267 (1,191) a | 30,098 (2,231) |
| Completeness (%) | 99.6 (99.9) | 99.9 (100) |
| Multiplicity | 3.7 (3.8) | 7.1 (7.2) |
| Rmeas (%) | 23.6 (87.0) | 7.9 (61.1) |
| Average I/σ (I) | 4.4 (1.58) | 15.8 (2.49) |
| Rfactor (%) | 26.3 | 24.0 |
| Rfree (%) | 30.4 | 28.9 |
| r.m.s. bond lengths (Å) | 0.009 | 0.015 |
| r.m.s. bond angles (°) | 1.64 | 1.85 |
| Average B factor (Å2) | 47.8 | 30.0 |
| Residues in most favored regions (%) | 93.5 | 97 |
| Residues in additionally allowed regions (%) | 6.5 | 3 |
| PDB‐ID | 6QCI | 6GJW |
aValues in parentheses are for the highest resolution shell: 2.36–2.30 and 1.95–1.90, for XIAP‐BIR1V86E and XIAP‐BIR1/5 a, respectively.
2.4. XIAP‐BIR1 Binding and SEC Assays Using Compounds 5 a and 5 b
Since NF023 appears to bind to XIAP‐BIR1 dimerization surface, we hypothesized that compounds with higher affinity for the protein might induce the disruption of the dimeric assembly, which was not observed at high concentration of BIR1 in presence of NF023.11 Two known NF023‐analog compounds (5 a and 5 b)13 were analyzed following the quenching of protein tryptophan fluorescence (Trp73) upon binding. The K d of 5 a and 5 b for the XIAP‐BIR1 domain was of 9±2 and 11±2 μM, respectively, showing a two‐fold improvement with respect to NF023 (24±4 μM11).
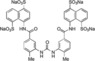
In these two compounds, the sulphonate group in position 3 of each naphthalene is missing. Such sulfonate is not involved in the interaction with the protein as observed in the in silico docking model. Moreover, in compound 5 b two methyl groups are present in the para position.
The capability of these two compounds to disrupt the dimer was checked through SEC experiments on XIAP‐BIR1 (sequence‐based molecular weight (Mw) 12.3 kDa) at initial protein concentrations of 40 and 1000 μM, in the absence/presence of 2.50 mM of 5 a or 5 b, using a Superdex75 10/300 column. In the absence of the compounds, the apparent Mw of XIAP‐BIR1 is 11.4 kDa and 15.7 kDa, respectively. Such evidence suggests the presence of a monomer/dimer equilibrium that shifts towards the dimer, as a function of protein concentration. Addition of 5 a/5 b affected the XIAP‐BIR1 monomer/dimer equilibrium to a certain extent, hindering the dimerization process (i. e. the highest XIAP‐BIR1 apparent Mw measured in the presence of 5 b is 12.8 kDa; data not shown). Such results may suggest that 5 a/5 b bind to XIAP‐BIR1 dimerization interface, thus interfering with the protein dimerization process.
2.5. Structural Characterization of XIAP‐BIR1/5 a Complex
Co‐crystallization experiments of the purified BIR1 domain in the presence of saturating amounts of compounds 5 a and 5 b were set up. Crystals of the BIR1/5 a complex diffracted to a maximum resolution of 1.9 Å (Table 2).
The crystals displayed a slightly larger unit cell with respect to the apoprotein (PDB: 4OXC11). After molecular replacement (MOLREP18) and a few refinement cycles, additional electron density in the Fo−Fc map allowed to model the 5 a molecule between the facing β2–β3 loops of each of the two XIAP‐BIR1 dimers in the crystal asymmetric unit (Figure 3).
Figure 3.
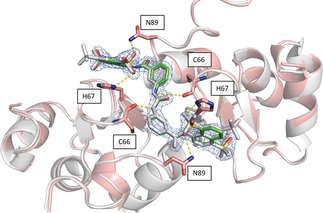
5 a interacts with XIAP‐BIR1 dimer with the same binding mode as NF023. Superposition of the XIAP‐BIR1/5 a structure, in pink cartoons and green sticks, with XIAP‐BIR1/NF023 complex (PDB: 4MTZ11), represented in grey: 5 a displays the same positioning and interactions (yellow dashed lines) as NF023; interacting residues are shown as sticks with pink carbon atoms; Zn ions are represented as spheres; the blue cage represents 5 a electron density.
The binding mode of 5 a with the BIR1 domain is almost identical to that observed for NF02311 (Figure 3) with 5 a sitting above the association interface of the dimer and establishing nearly symmetrical interactions with both subunits.
In particular, 5 a central N atom establishes a hydrogen‐bond with the main chain carbonyl O atom of Cys66 (average distance 3.4 Å), whereas the sulphonates of the two opposite naphthalene‐disulphonate palms are H‐bonded to the side chains of His67 and Asn89 (average distances 3.2 Å).
In conclusion, the binding mode observed in crystal structures is different from the in silico docking prediction on the BIR1 monomer along the dimerization interface, and is in contrast with protein‐mutations data and SEC experiments. We speculated that this discrepancy might be due to XIAP‐BIR1 homodimers that are already present at the high protein concentrations used for crystallography.11 Meanwhile, NF023 or 5 a may not be able to cause the disruption of a pre‐formed dimer and to bind in the correct position.
2.6. SAR Analysis
In order to improve binding, we decided to identify the smallest NF023 like fragment with a comparable binding capability. This approach was also used with the idea that the frameworks of Suramin and NF023 (Figure 1) can be modified to allow their analogs to bind protein more efficiently. To this aim, thermal shift (Table S1) and thermophoresis assays were performed on wild type XIAP‐BIR1 using NF023, Suramin, tetrasodium organosulphonates (i. e., 3, 5 a and 5 b), their smaller analogs (i. e., 6, 7, 9 and 10) and NAF2 (Table 3).
Table 3.
XIAP‐BIR1 affinity for NF023 like molecules moieties.
| Sample | Compounds Chemical Structure | K d of Protein‐Ligand Complex (μM) |
|---|---|---|
| XIAP‐BIR1/NF023 |

|
25±5 |
| XIAP‐BIR1/Suramin |
|
612±109 |
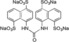
| XIAP‐BIR1/3 |
|
>10000 |
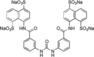
| XIAP‐BIR1/5 a |
|
11±2 |
| XIAP‐BIR1/5 b |
|
9±2 |
| XIAP‐BIR1/6 |
|
525±50 |
| XIAP‐BIR1/7 |
|
553±140 |
| XIAP‐BIR1/9 |
|
6810±2000 |
| XIAP‐BIR1/10 |
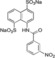
|
8190±1800 |
| XIAP‐BIR1/NAF2 |

|
no binding |
The measured K d values confirmed an affinity in the micromolar range for NF023 and its analogs. By contrast, the smallest compounds, containing only NF023 polar disulphonate naphthalene palms (i. e. 9, 10 and NAF2 and tetrasulphonate naphthalene without arms 3) have little or no affinity for the protein. This result might suggest an involvement of NF023 diphenyl‐urea central moiety in the interaction with the protein. Indeed, the comparable amidic group steric hindrance of 6 and 7 central moieties with respect to NF023, leads to a significant affinity, nevertheless in the high micromolar range. The addition of a tail in 9, with respect to 6 and 7, significantly changes the chemical nature resulting in a drastic decrease of the affinity for XIAP‐BIR1. In contrast, the longer central moiety of Suramin also leads to an affinity in the high micromolar range. Taken together, these results suggest that the chemical features of the ureido “head” of NF023 (in terms of length and symmetry) are essential for binding to the protein. Negative results emerged from compounds that lacked an ureido head and thus possessed only half of the full framework (compounds 6, 7, 9, 10 and NAF2). Moreover, either the tolylamido “arms” or the benzamido “shoulders” should be kept, since the lack of both components (compound 3) leads to very poor affinity.
To further corroborate such hypothesis, we evaluated the affinity of 6, which represents half of NF023 symmetric molecule with a modified central moiety, for the D71A mutant variant of XIAP‐BIR1. Such protein point‐mutation showed to have the most dramatic effect on the protein interaction with NF023. In contrast, the mutation did not alter the affinity for 6, resulting in a K d of 537±60 μM, which is comparable with that observed using wild type XIAP‐BIR1 (Table 3). Given the position of D71 in the lower part of XIAP‐BIR1 dimerization interface (Figure 2), these results suggest that contacts established between compound 6 and the upper part of this protein surface are sufficient to have an appreciable binding (with a K d in the high micromolar range). These data corroborate the hypothesis that symmetric divalent molecules, presenting a diphenyl urea central moiety and hosting two naphthylsulphonate heads, are needed to engage a full network of interactions with the whole protein dimerization surface, leading to a higher affinity.
The information gained so far set the bases for the design of a new class of compounds directed at the NF‐κB pathway modulation and for the development of new therapeutic strategies hampering cancer cells survival.
Experimental Section
Chemicals and Reagents
Hexasodium (ureido)naphthylsulphonate NF023, hexasodium Suramin and 1,5‐naphthalenedisulphonate NAF2 were purchased from Sigma‐Aldrich, dissolved in H2O at a stock concentration of 100 mM and stored at −20 °C. All other reagents were of the highest commercially available grade.
Chemical Synthesis of NF023 Analogues and Derivatives
Synthesis of sulphonates 6, 7 and 10 was carried out as previously described.12
Chemical Synthesis of Sulphonates 3,5 a, 5 b and 9
General Procedure. All reactions were carried out in oven‐dried glassware (120 °C) under an atmosphere of nitrogen unless indicated otherwise. Acetone, acetic acid, diethylether, methanol and toluene were purchased from Mallinckrodt. 4‐Amino‐1,5‐naphthalenedisulphonic acid disodium salt and triphosgene were purchased from TCI Chemical Co. Potassium triiodide, phosphorus pentoxide and sodium carbonate were purchased from Sigma‐Aldrich.
Analytical thin layer chromatography (TLC) was performed on precoated plates (silica gel 60 F‐254) purchased from Merck Inc. High performance liquid chromatography (HPLC) was performed on two Waters 515 HPLC pumps equipped with a Waters 2489 UV/Visible Detector and a Thermo 5 μm Hypersil ODS (250×4.6 mm I.D.). Purity of all compounds was >98.4 %, as checked by HPLC.
Proton NMR spectra were obtained on a Varian Mercury‐400 (400 MHz) spectrometer or a Bruker AV‐400 (400 MHz) by use of dimethylsulphoxide‐d 6 as solvent. Carbon‐13 NMR spectra were obtained on a Varian Mercury‐400 (100 MHz) spectrometer or a Bruker AV‐400 (100 MHz). The residual solvent peaks, d H 2.50 ppm and d C 39.5 ppm for DMSO‐d 6 was used as reference. High‐resolution mass spectra were obtained by means of a VARIAN‐901 mass spectrometer.
Standard Procedure for the Preparation of Tetrasodium ureidobis(naphthalenedisulphonate)s 3, 5 a and 5 b. Aniline salts 2, 4 a and 4 b were dissolved in water with pH=3.0 by addition of Na2CO3 solution (2.0 M). To this solution triphosgene (1) in toluene was slowly added maintaining pH=3.0. Afterward the reaction mixture was stirred at 25 °C for 6.0 h and the toluene layer was discarded. The water was removed over P2O5 under reduced pressure at 45 °C to give crude solids. The solids were purified by dissolution in methanol and the precipitated inorganic salts were filtered off. The methanol was removed under reduced pressure and the residue was dried under reduced pressure over P2O5 to give the desired product. 1H NMR data (DMSO‐d 6, 400 MHz) are reported in Supporting Information.
Tatrasodium 4,4′‐Carbonylbis(imino)bis‐1,5‐naphthalenedisulphonate (3). The standard procedure was followed by use of 4‐amino‐1,5‐naphthalenedisulphonic acid disodium salt (2, 60.1 mg, 185 μmole, 2.0 equiv), water (2.0 mL), triphosgene (1, 36.6 mg, 0.120 mmol, 1.30 equiv) and toluene (2.0 mL). The desired product 3 19 (47.9 mg, 66.5 μmol) was obtained in 89 % yield as white solids.
Tatrasodium 4,4′‐[Carbonylbis(imino‐3,1‐phenylenecarbonylimino)]bis‐1,5‐naphthalenedisulphonate (5 a). The standard procedure was followed by use of aniline derivative 4 a 12 (100.2 mg, 215.2 μmol, 2.0 equiv), water (2.0 mL), triphosgene (1, 41.5 mg, 140 μmol, 1.30 equiv) and toluene (2.0 mL). The desired product 5 a 19 (86.6 mg, 90.4 μmol) was obtained in 84 % yield as brown solids.
Tatrasodium 4,4′‐(Carbonylbis[imino‐3,1‐(4‐methylphenylene)carbonylimino])bis‐1,5‐naphthalenedisulphonate (5 b). The standard procedure was followed by use of aniline derivative 4 b 12 (100.3 mg, 208.5 μmol, 2.0 equiv), water (2.0 mL), triphosgene (1, 40.2 mg, 136 μmol, 1.3 equiv) and toluene (2.0 mL). The desired product 5 b 16,17 (88.4 mg, 89.7 μmol) was obtained in 86 % yield as brown solids.
Disodium 4‐(3‐[3‐(Ethoxylthioxoimino)benzamido]‐4‐tolylamido)naphthalene‐1,5‐disulphonate (9). Aniline salt 7 12 (59.9 mg, 99.9 μmol, 1.0 equiv) and potassium ethyl xanthate (8, 16.1 mg, 0.100 mmol, 1.0 equiv) were dissolved in water (1.0 mL) and the pH was adjusted to 11.0 by addition of aqueous NaOH solution (10 N). To this stirring solution was slowly added potassium triiodide solution (0.27 mL, 0.38 M solution, 0.10 mmol, 1.0 equiv) over 20‐min period. Once the addition was completed, the mixture was stirred for another 30 min to complete the reaction. The aqueous layer was washed with diethyl ether (3×1.0 mL). Then the water in the aqueous solution was removed under vacuum over P2O5 to give crude solids, which were then dissolved in methanol followed by filtration. After methanol was evaporated under reduced pressure, the residue was dried over P2O5(s) under vacuum to give thiocarbamate 9 19 (46.1 mg, 67.0 μmol) in 67 % yield as pale yellow powders.
In Silico Docking Data Analysis
All the NF023 poses found in the grid covering the whole XIAP‐BIR1 domain (as previously reported11), were analyzed with Python Molecule Viewer 1.4.5. Based on the predicted locations of NF023 on the XIAP‐BIR1 surface, the R62S, D71A, R82S and V86E mutations were selected to impair NF023 binding to the protein.
Cloning, Expression and Purification of Wild Type and Mutant Forms of XIAP‐BIR1
The human XIAP‐BIR1 domain was cloned in pET28b, as already described.11 The R62S, D71A, R82S and V86E point mutations were obtained using the Q5® Site‐directed mutagenesis kit (New England Biolabs), according to the manufacturer's instructions. The purification of wild type and mutant forms of XIAP‐BIR1 was performed as already described,11 to obtain highly pure protein samples. DLS analysis confirmed the high quality of the samples obtained, revealing a sample size polydispersity lower than 20 % for all protein constructs.
XIAP‐BIR1 Binding Assays
Tryptophan assays. In vitro binding assays were performed at 24 °C in 50 mM Tris HCl, pH 8.0 containing 200 mM NaCl, 10 % glycerol, 10 mM DTT, using a Varian Cary Eclipse Fluorescence Spectrophotometer and recording Trp fluorescence data between 300 and 400 nm (excitation wavelength=280 nm). The initial protein concentration was set at 5 μM, in order to obtain a significant fluorescence peak at 336 nm. Two‐fold dilution series of NF023 or its tetrasodium bis(naphthalenedisulphonate) analogs, 5 a and 5 b (not fluorescent under our experimental conditions), were prepared, in order to have a final concentration range from 0.2 to 106 μM. 8 μl of each compound dilution were mixed with 180 μl of the 5 μM protein solution and fluorescence was measured in a 200 μl quartz cuvette. The values of fluorescence vs. inhibitor concentration were fitted with GraFit5 (Erithacus Software Limited, 2010) using the three parameters (M, m, Kd) equation:
where F is the fluorescence intensity, [PT]/[IT] are the total protein/inhibitor concentrations, M/m is the max/min of fluorescence and [PI] is the concentration of the protein bound to the inhibitor.
Thermophoresis assays. In MicroScale Thermophoresis (MST) an infrared laser is used for local heating, then molecule mobility in the temperature gradient is analyzed via fluorescence of the target molecule appropriately labeled. In our case, we labeled lysine residues of all XIAP‐BIR1 forms, using the covalent dye and protocol provided by the manufacturer (NanoTemper). Two‐fold dilution series were prepared to have a final concentration range from 30 nM to 1.0 mM for compounds with high affinity and from 300 nM to 10 mM for those with lower affinity. The protein concentration was kept constant at 100 nM in all experiments, for all tested XIAP‐BIR1 forms. Assays were performed at 24 °C in 50 mM TrisHCl, pH 8 containing 200 mM NaCl, 10 % glycerol, 10 mM DTT and 0.05 % Tween20, using premium‐treated glass capillaries and the instrument Monolith NT.115 (NanoTemper) with 20 % LED intensity and medium MST power. The K d of protein‐ligand complexes engaged by XIAP‐BIR1 forms with the tested compounds was t calculated with the following equation:
where F indicates the measured fluorescence, while U and B represent the response values of the unbound and bound states, respectively.
Size Exclusion Chromatography (SEC) Assays
To monitor XIAP‐BIR1 monomer/dimer equilibrium in the absence/presence of 2.5 mM 5 a or 5 b, analytical SEC experiments were performed at different protein concentrations. During the SEC experiments sample volumes of 50 μl, with XIAP‐BIR1 concentrated at 0.04 mM or 1 mM, were injected on a Superdex 75 Increase 10/300 column (GE Healthcare, volume=24 ml) attached to an ÄKTA Pure system in 20 mM Tris‐HCl pH 7.5, 200 mM NaCl, and 10 mM DTT. A 1 : 30 dilution ratio (injected sample volume/eluted peak volume) was estimated for all tested samples. Low molecular weight standards, from GE Healthcare, were used to calibrate the column.
Crystallization and Crystallographic Data Collection
Crystallization of XIAP‐BIR1 V86E mutant (6 mg/ml) and co‐crystallizations of XIAP‐BIR1 (9 mg/ml) with a final concentration of 2.5 mM of compounds 5 a or 5 b were performed at 20 °C with sitting drop set up, using an Oryx‐8 crystallization robot (Douglas Instruments, East Garston, UK), from a 1 : 1 mixture of V86E and precipitant solution, and from a 2 : 1 mixture of XIAP‐BIR1/compound and precipitant solution, to final drop volumes of 0.3 μl. The V86E mutant crystallized in 0.2 M Lithium sulphate, 0.1 M Tris pH 8.5, 30 % PEG4000. Small prismatic crystals of XIAP‐BIR1/5 a were found in 8 % PEG8000, 100 mM Tris pH 8.5. The crystals were analyzed at the ESRF synchrotron (Grenoble, FR) on beam‐line ID23‐2 and BM14, respectively, after being immersed in a cryoprotectant solution containing 20 % glycerol and respective mother liquor (added with 2.5 mM 5 a for the wild type protein) and flash‐frozen in liquid nitrogen. The diffraction experiment allowed to collect datasets at a resolution of 2.3 and 1.9 Å for XIAP‐BIR1V86E and XIAP‐BIR1/5 a, respectively.
Structure Determination and Refinement
Diffraction analysis revealed that the XIAP‐BIR1 V86E and the XIAP‐BIR1/5 a crystals belonged to the monoclinic space group P21, with 4 molecules in the asymmetric unit.
The crystal structures were solved by molecular replacement (MOLREP18), using chain A from the XIAP‐BIR1 structure (PDB: 4OXC11) as search model. The four independent molecules in the crystal asymmetric unit were subjected to rigid‐body refinement (R/Rfree=44.5/44.7 % and 43.8/44.5 %, for XIAP‐BIR1 V86E and XIAP‐BIR1/5 a), and subsequently refined using REFMAC5.20 A random set comprising 5 % of the data was omitted from refinement for R‐free calculation. Manual rebuilding with Coot21 and additional refinement with REFMAC522 were subsequently performed. In the case of XIAP‐BIR1/5 a crystal, inspection of difference Fourier maps at this stage showed strong residual density, located on the top of each XIAP‐BIR1 dimer (chain A/D and B/C), near residues Arg62, Cys66, His67 and Asn89 compatible with one 5 a molecule, that was accordingly modeled.
The refined XIAP‐BIR1 V86E structure shows between 71 and 77 residues out of the 111 of our XIAP‐BIR1 construct, the V86E replacement is clearly visible; the refined XIAP‐BIR1/5 a complex model displays 78 residues out of the 111; the first 32 N‐terminal residues (21 belonging to the expression vector and 11 to the N‐terminal sequence of XIAP‐BIR1) and the last C‐terminal one are missing in the electron density. Data collection and refinement statistics are summarized in Table 2. The stereochemical quality of the models was checked using the program Procheck.23 Atomic coordinates and structure factors have been deposited in the Protein Data Bank24 with accession codes 6QCI and 6GJW, for XIAP‐BIR1 V86E and the XIAP‐BIR1/5 a, respectively. PyMol Graphic System (Schrödinger, LLC) was used for figures preparation.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. Delia Tarantino (University of Milan) for help in MST measurements and the ESRF facility for diffraction experiments. We also thank the Italian Association for Cancer Research (AIRC‐MFAG Grant Nr 17083, awarded to EM), the Ministry of Education (Grant Nrs 106 N501CE1 and 107QR001I5), the Ministry of Science and Technology (MOST, Grant Nrs 106‐2113‐M‐007‐011‐MY2 and 107‐3017‐F‐007‐002) of R.O.C. for funding.
L. Sorrentino, F. Cossu, M. Milani, B. Malkoc, W.-C. Huang, S.-C. Tsay, J. Ru Hwu, E. Mastrangelo, ChemistryOpen 2019, 8, 476.
Contributor Information
Prof. Jih Ru Hwu, Email: jrhwu@mx.nthu.edu.tw, Phone: (+886) 35725813.
Dr. Eloise Mastrangelo, Email: eloise.mastrangelo@unimi.it, Phone: (+39) 0250314898.
References
- 1. Deveraux Q. L., Reed J. C., Genes Dev. 1999, 13, 239–252. [DOI] [PubMed] [Google Scholar]
- 2. Gyrd-Hansen M., Darding M., Miasari M., Santoro M. M., Zender L., Xue W., Tenev T., da Fonseca P. C. A., Zvelebil M., Bujnicki J. M., Nat. Cell Biol. 2008, 10, 1309–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Salvesen G. S., Duckett C. S., Nat. Rev. Mol. Cell Biol. 2002, DOI 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 4. Wilkinson J. C., Wilkinson A. S., Galbán S., Csomos R. A., Duckett C. S., Mol. Cell. Biol. 2008, 28, 237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deveraux Q. L., Takahashi R., Salvesen G. S., Reed J. C., Nature 1997, 388, 300–304. [DOI] [PubMed] [Google Scholar]
- 6. Cossu F., Milani M., Mastrangelo E., Vachette P., Servida F., Lecis D., Canevari G., Delia D., Drago C., Rizzo V., J. Mol. Biol. 2009, DOI 10.1016/j.jmb.2009.04.033. [DOI] [PubMed] [Google Scholar]
- 7. Cossu F., Mastrangelo E., Milani M., Sorrentino G., Lecis D., Delia D., Manzoni L., Seneci P., Scolastico C., Bolognesi M., Biochem. Biophys. Res. Commun. 2009, DOI 10.1016/j.bbrc.2008.10.139. [DOI] [PubMed] [Google Scholar]
- 8. Lecis D., Mastrangelo E., Belvisi L., Bolognesi M., Civera M., Cossu F., Cesare M. De, Delia D., Drago C., Manenti G., Manzoni L., Milani M., Moroni E., Perego P., Potenza D., Rizzo V., Scavullo C., Scolastico C., Servida F., Vasile F., Seneci P., Bioorg Med Chem. 2012, 20, 6709–6723. [DOI] [PubMed] [Google Scholar]
- 9. Petersen S. L., Peyton M., Minna J. D., Wang X., Proc. Mont. Acad. Sci. 2010, 107, 11936–11941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu M., Lin S. C., Huang Y., Kang Y. J., Rich R., Lo Y. C., Myszka D., Han J., Wu H., Mol. Cell 2007, 26, 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cossu F., Milani M., Grassi S., Malvezzi F., Corti A., Bolognesi M., Mastrangelo E., Proteins Struct. Funct. Bioinf. 2015, DOI 10.1002/prot.24766. [DOI] [PubMed] [Google Scholar]
- 12. Croci R., Pezzullo M., Tarantino D., Milani M., Tsay S. C., Sureshbabu R., Tsai Y. J., Mastrangelo E., Rohayem J., Bolognesi M., PLoS One 2014, 9, e91765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hawking F., Adv. Pharmacol. 1978, 15, 289–322. [DOI] [PubMed] [Google Scholar]
- 14. Tsay S.-C., Gupta N. K., Bachurin S. O., Hwu J. R., Org. Med. Chem. Int. J. 2017, 2, 555593–555597. [Google Scholar]
- 15. Borak J., Diller W. F., J. Occup. Environ. Med. 2001, 43, 110–119. [DOI] [PubMed] [Google Scholar]
- 16. Kassack M. U., Braun K., Ganso M., Ullmann H., Nickel P., Böing B., Müller G., Lambrecht G., Eur. J. Med. Chem. 2004, 39, 345–357. [DOI] [PubMed] [Google Scholar]
- 17. Trapp J., Meier R., Hongwiset D., Kassack M. U., Sippl W., Jung M., ChemMedChem 2007, 2, 1419–1431. [DOI] [PubMed] [Google Scholar]
- 18. Vagin A., Teplyakov A., J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar]
- 19. Albulescu I. C., Van Hoolwerff M., Wolters L. A., Bottaro E., Nastruzzi C., Yang S. C., Tsay S. C., Hwu J. R., Snijder E. J., Van Hemert M. J., Antiviral Res. 2015, 121, 39–46. [DOI] [PubMed] [Google Scholar]
- 20. Winn M. D., Isupov M. N., Murshudov G. N., Acta Crystallogr. Sect. D 2001, 57, 122–133. [DOI] [PubMed] [Google Scholar]
- 21. Emsley P., Cowtan K., Acta Crystallogr. Sect. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 22. Murshudov G. N., Vagin A. A., Dodson E. J., Acta Crystallogr. Sect. D 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- 23. Laskowski R., Rullmann J. A., MacArthur M., Kaptein R., Thornton J., J. Biomol. NMR 1996, 8, DOI 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 24. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E., Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary