

Abstract
Low-valent early transition metals are often intrinsically highly reactive as a result of their strong propensity toward oxidation to more stable high-valent states. Harnessing these highly reducing complexes for productive reactivity is potentially powerful for C-C bond construction, organic reductions, small-molecule activation and many other reactions that offer orthogonal chemoselectivity and/or regioselectivity patterns to processes promoted by late transition metals. Recent years have seen many exciting new applications of low-valent metals through building new catalytic and/or multicomponent reaction manifolds out of classical reactivity patterns. In this Review, we survey new methods that employ early transition metals and invoke low-valent precursors or intermediates in order to identify common themes and strategies in synthesis and catalysis.
Early transition metals are excellent candidates for the design and implementation of new catalytic reactions: they are earth abundant, often highly reactive and frequently exhibit different structures and orthogonal reactivity compared with late transition metals1,2. However, owing to their electropositive, oxophilic3 nature, the organometallic chemistry of early transition metals has historically been dominated by complexes in high-valent states (FIG. 1). By contrast, there are considerably fewer examples of the application of low-valent early transition metals: such complexes are often highly reducing and require strong stabilizing ligands, such as strong π-acceptors or bulky cyclopentadienyls.
Fig. 1 |. Relative abundance of the valence states of early transition metal organometallic complexes.
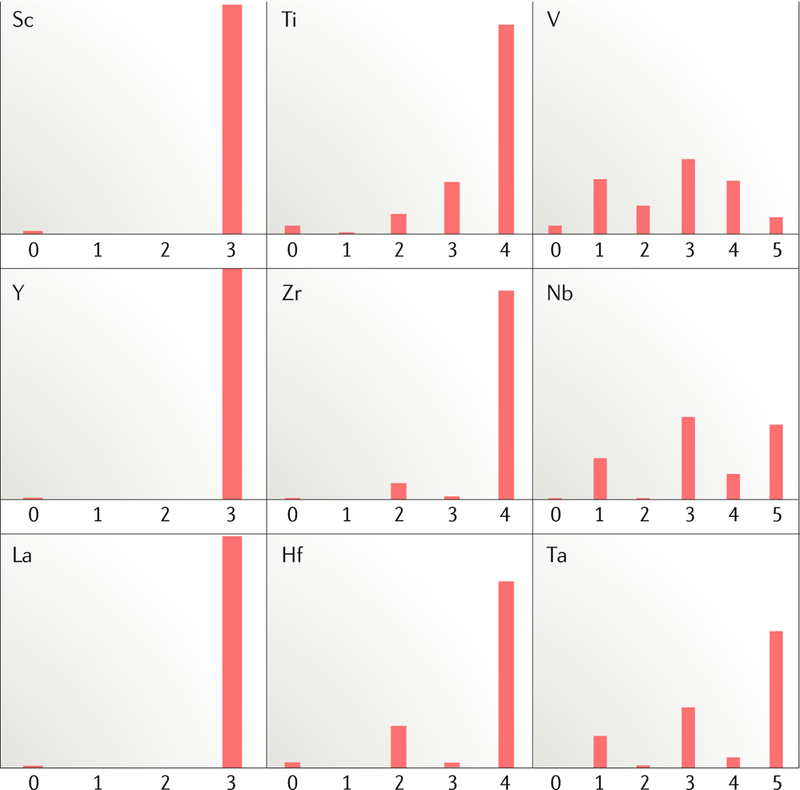
Reduced early transition metal complexes become increasingly rare as one proceeds from group 5 to earlier groups and also from period 3 to period 5. Data adapted from REF26.
Nonetheless, there is great interest in harnessing the highly reactive nature of low-valent early transition metals for practical synthesis and catalysis. There are myriad elegant examples of stoichiometric N2 activation4–17, reductive coupling18–22 and radical reactions23–25 mediated by low-valent early transition metals. The majority of this reactivity is dependent on the fact that highly reduced early metals can activate otherwise unreactive molecules, wherein metal oxidation and the formation of strong metal-ligand bonds provide a large thermodynamic driving force.
Although many seminal contributions to the field were carried out decades ago, there has been a renaissance in the field over the past 15 years enabled by new ligand designs, reagents and approaches. Early transition metals are well positioned to have considerable impact in a wide array of fields, from small-molecule activation and energy storage to photoredox catalysis and other new organic methods. This Review covers new applications of low-valent early transition metals26 (classified here as groups 3, 4 and 5, excluding V because it does not predominantly exist in its highest oxidation state) in synthesis and catalysis. In particular, this Review highlights how the unique properties of low-valent metals can access new mechanisms and enable challenging chemical transformations inaccessible by their late transition metal counterparts.
Generation of low-valent complexes
The most common routes for generating low-valent early transition metal complexes involve the reduction of metal halides with strong alkali metals27–32; reduction with other (often weaker) reducing metals, such as Mg (REFS33,34), Mn (REF.35), Al (REFS36,37) or Zn (REF.38) (providing more functional group tolerance); or p-H abstraction39–43 from a metal alkyl (Fig. 2a). Many of these reductions can be performed in situ to directly generate reactive low-valent intermediates or in the presence of π-accepting ligands to generate isolable or transiently stable ‘masked’ species that can liberate a reactive low-valent metal fragment upon loss of the re-accepting ligand. For example, the Negishi reagent, Cp2Zr(η2-butene) (1), is synthesized by the treatment of Cp2ZrCl2 with nBuLi or nBuMgBr, wherein the Cp2ZrnBu2 intermediate undergoes β-H abstraction to liberate one equivalent of butane along with the product44–46. Cp2Ti(TMS2C2) (2) (TMS2C2 = bistrimethylsilylacetylene) and related Ti and Zr analogues can be synthesized by reduction of the corresponding high-valent chlorides with Mg in the presence of the π-accepting alkyne TMS2C2 (REF47), while substituted hafnocene analogues have been synthesized using Li as a reductant48,49.
Fig. 2 |. Examples of synthetic routes to low-valent early transition metal complexes.

a | Classical reduction routes involving strong reductants. b | Representative examples of modern low-valent metal synthesis. c | New organosilane reductants for low-valent metal synthesis and catalysis. These routes demonstrate the diverse array of reduction methods available and the common ability of π-accepting ligands to stabilize low-valent early transition metal complexes. Red-coloured metals are in their highest oxidation state, orange-coloured metals are one-electron reduced, and teal-coloured metals are two-electron reduced. hv, irradiation; Dipp, 2,6-diisopropylphenyl; E1/2, half-wave potential; Fc, ferrocene; THF, tetrahydrofuran; TMSCl, trimethylsilyl chloride.
Stable low-valent coordination complexes of the early transition metals that lack strongly stabilizing ligands are uncommon. However, examples continue to emerge that challenge the limits of stability (FIG. 2b). For example, a suite of divalent group 3 amides has recently been reported50–53, including the first crystallographically characterized Sc11 compound52, extending the chemistry of low-valent Sc and Y beyond metallocene derivatives54,55. The ScII amide complex 3 exhibits remarkably different reactivity from other reduced group 3 and lanthanide complexes, for the first time revealing end-on, reversible binding of N2 to form the bridged bimetallic 4 (REF53). Bulky aromatic heterocycles derived from phosphaalkynes, which are much poorer donors than Cp-based ligands, can also stabilize ScII complexes56. An improved synthesis and full electronic characterization of py4TiCl2 (5)57, a rare example of a TiII coordination complex, have recently been reported, building upon earlier studies by several groups58,59. The chemistry of low-valent early transition metal isocyanide60–62 and arene complexes63,64 has also continued to expand, including the recent synthesis of the formally Ta0 complex 6 (REF60).
The high reactivity of strong alkali metal reductants places an inherent restriction on the solvent and reagent compatibility of reactions that employ them as reductants. Considerable progress has been made in designing new reactions and reductants that overcome this challenge. One creative workaround that was recently introduced is to reduce compounds by passing them through columns of KC8, limiting (or at least fine tuning) the contact time of the reagent and solvent with the strong reductant65. Another strategy is to use inner-sphere reductants that are less prone to over-reduction. In this context, Tsurugi and Mashima have promoted the use of organosilicon reductants, such as 3,6-bis(trimethylsilyl)-1,4-cyclohexadiene66–68 ( 7) and 1,4-bis(trimethylsilyl)-1,4-dihydropyrazine69–71 derivatives (for example, 8), to reduce group 4 and group 5 metal halides (Fig. 2c) for homogeneous catalytic applications. In the case of 8, the steric bulk of the tetramethylpyrazine by-product prevents its coordination to the reduced metal catalyst, which is critical for productive chemistry. These organosilicon reductants derive their reduction potential (ranging from +0.83 V to −0.40 V versus Cp2Fe) from the aromatization of the 1,4-diene core. These reductants have been used by the broader community for wide-ranging applications, from the in situ reduction of Ti precursors in chemical vapour deposition72 to the in situ generation of TiIII for reductive umpolung reactions73. Similarly, commercially available tris(trimethylsilyl)silane has been used to generate NbIII cyclotrimerization catalysts in situ74. A major advantage of these silicon-based reductants is that the by-products are simple to remove: Me3SiCl is less likely than alkali metal halides to coordinate to electrophilic metals, and the aromatic by-products can be removed in vacuo. Metal salt by-products are often difficult and/or tedious to remove and may interfere with productive chemistry of the reduced complex; thus, these procedures offer an important advancement for applications of reduced early metal chemistry.
Alternative reduction methods of early transition metals include single-electron transfers from weak reductants such as citrate75 or benzylamine76. Hydrogenolysis of metal alkyls has also been used to synthesize low-valent Nb (REF77) and Zr (REF78) arene complexes. Redox non-innocent ligands can also engender redox reactivity with early transition metals without formally changing the metal oxidation state67,68,79–82.
Reductive coupling reactions
Reductive coupling of unsaturated organics by d2 early transition metals has been exploited for a vast array of synthetically useful C-C bond forming reactions. Typically, these reactions involve activation of an alkene or an alkyne to form either a metallacyclopropane or metallacyclopropene, respectively. These species can be considered as 1,2-dianion equivalents and can be intercepted sequentially by two different electrophiles. Often, the resulting metallacycles can be further functionalized, forming the basis for a diverse set of coupling reactions, such as the Negishi21,22,45, Fagan-Nugent19,83–85, Kulinkovich43,86–88, Pauson-Khand89–94 and several other stoichiometric coupling reactions of alkynes, alkenes, alkyl halides and carbonyl compounds18,20. Although most d2 early metals exhibit this type of reactivity, low-valent Ti and Zr complexes — in particular, Cp2Mn (M = Ti, Zr) and (O’’Pr)2TII fragments — have seen the most development in this field, and as such the use of such reagents in the total synthesis of natural products has flourished23,95–97.
Ti(OiPr)4/RM-mediated reductive coupling reactions.
The discovery of the main group metal alkyl reduction of early metals45 inspired many reductive coupling reactions using titanium. In 1989, Kulinkovich reported the Ti(O’Pr)4-catalysed cyclopropanation of esters with Grignard reagents to form cyclopropanols86 (FIG. 3a). Shortly after, the Kulinkovich reaction was translated to amides (de Meijere modification), which results in the formation of cyclopropylamines by a similar mechanism98. Since these initial reports, several improvements have been made to this chemistry. Intermolecular alkene couplings can be achieved by using cyclohexyl or cyclopentylmagnesium chloride to activate Ti(OiPr)4, as the resulting Ti-cyclopentene (or cyclohexene) adduct 9 can be displaced by monosubstituted or tethered alkenes to make a new alkene adduct 10 (REFS99, 100) (FIG. 3a, bottom). This discovery has helped dramatically expand the scope of Kulinkovich reactions, as alkene starting materials are more widely available than Grignard reagents. The Kulinkovich cyclopropanation has an intrinsic diastereoselectivity for the cis-1,2-dialkylated cyclopropanol when using Grignard reagents larger than ethyl, and the use of chiral TADDOL (a,a,a’,a’-tetraaryl-2,2-disubstituted-1,3-dioxolane-4,5-dimethanol) ligands has allowed for good enantioselectivity88,101,102. This diastereoselectivity can be reversed when cyclopropanating homoallylic alcohols as a result of coordinating directing group effects99 or remote stereocentre influences103.
Fig. 3 |. Recent advances in two-electron reductive coupling reactions proceeding through titanacyclopropane and titanacyclopropene intermediates from Ti(OiPr)4/RM and related species.

a | Catalytic KuLinkovich cydopropanation reactions, in which ester C = O bond insertion into the Ti bond precedes pericycLic metaLLacycLe collapse. b | Stoichiometric arrested KuLinkovich-de Meijere-Like cyclopropanations, in which metaLLacycLe collapse is prevented through steric (top) or electronic (bottom) blocking. c | Alkoxide-directed regiospecific and stereospecific reductive couplings that bias regiospecific metaLLacycLe formation and subsequent insertion steps through covalent Ti-O bonding. DCE, dichLoroethane; DME, dimethoxyethane; equiv., equivalents; THF, tetrahydrofuran; TMS, trimethyLsiLyL.
A recent modification of the Kulinkovich-de Meijere reaction allows for the synthesis of carbocyclic amino ketones mediated by Ti(OiPr)4/RMgBr (REF104) (FIG. 3b, top). By using allyl-substituted lactams such as 11, the cyclopropanation step of the de Meijere mechanism can be arrested, as the bridgehead nitrogen atom of the alkene-inserted intermediate 12 cannot π-bond with the neighbouring carbon (Bredt’s rule)105. This ultimately leads to cyclic aminoketone 13 upon protic workup. Increasing the ring size of the lactam relieves ring strain in the intermediate 12; thus, larger ring systems are no longer affected by Bredt’s rule and typical de Meijere cyclopropylamine generation proceeds. Similarly, interrupted Kulinkovich-de Meijere reactions yield hydroxyl-substituted lactams (16) with homoallylic imide (14) substrates106–108, which do not undergo cyclopropanation owing to resonance stabilization of the metallacyclic intermediate 15 (FIG. 3b, bottom).
Micalizio has shown that regioselectivity and stereoselectivity in the reductive coupling of various unsaturated compounds can be induced through the use of alkoxide directing groups (FIG. 3c) — a strategy similar to that employed by Sharpless for asymmetric alkene epoxidation109. This is an advancement from the limited selectivity obtained through Fagan-Nugent chemistry: substrate-directed metallacycle-mediated cross-couplings, although requiring prefunctionalization with potentially undesirable functional groups, allow for the regiospecific assembly of target molecules. By contrast, undirected cross-couplings of the same type typically give statistical mixtures in unbiased systems110,111.
Regioselective and stereoselective coupling of alkenes with imines is possible when using Ti(OiPr)4/nBuLi as opposed to Ti(OiPr)4/RMgBr (REFS112,113). Using BuLi as the metal alkyl reductant rather than a Grignard reagent grants dramatically enhanced stability to the titanacyclopropane intermediate as a result of the interaction of LIOiPr with the metal centre, forming a tentative ‘ate’ complex113. Homoallylic alcohols cross-couple with aromatic imines to give 1,5-aminoalcohols using Ti(OiPr)4/RMgBr (REF109), but the employment of Ti(OiPr)4/n BuLi allows for both aromatic and aliphatic imines to be used successfully in the reaction114. Imines (17) couple with allylic alcohols (18) to form stereodefined homoallylic amines (19)115 (FIG. 3c, top). Alkene stereoselectivity is achieved in these reactions through coordination of the deprotonated alcohol to the preformed Ti-imine complex, which undergoes syn-carbometallation (20) and syn-elimination through boat-like transition states (21 and 22) (FIG. 3c, top right). Primary homoallylic amines are accessible through similar coupling reactions with aldehydes in the presence of LiHMDS (lithium hexamethyldisilazide), which forms the TMS-imine complex in situ. These primary homoallylic amines have been applied as starting materials for the synthesis of complex bicycles116 and pyridines117.
Substrate-directed selective alkene-alkyne-alkyne formal [2+2+2] reactions are also possible. For example, alkoxide-directed annulations can give hydroindane derivatives (26 and 27) and angularly substituted decalins118–122 (FIG. 3c, bottom). The reactions proceed through alkoxide-directed insertion of an alkyne into the titanacyclopropene 23 to make the bicyclic metallacyclopentadiene 24. The chemoselectivity of this insertion is driven by the electronic bias of the a-SiMe3 group: the Ti-C(TMS) bond is more difficult to insert into than the Ti-C(Ph) bond of 23. Next, [4+2] cycloaddition of the pendent alkene forms a bridged bicyclic metallacyclopentene 25, which can undergo allylic isomerization and be quenched through protonation with allylic transposition (26) or oxidation (27). The choice of alcohol for the quench affects the selectivity for either cis-hydroindanes or trans-hydroindanes, although the origin of this effect remains unclear119.
Other early-transition-metal-mediated reductive coupling reactions.
Metallacycle 28, formed by reducing Cp*TiCl3 (Cp* = C5Me5−) in the presence of dimethylbutadiene, can react with two equivalents of isocyanide to give an asymmetric metal-bound bis(imine) 29 (REF123) (FIG. 4a). Furthermore, upon heating 29 at 55˚C in pyridine, cyclopentenimine 30 is extruded, and Cp*TiCl(py) (N′Bu) is formed. The result is a unique formal [4+1] coupling reaction alongside cleavage of an isocyanide triple bond, such that the C atom is inserted into both C-H bonds at the 2 position to give an exo-methylene. By contrast, the reaction with the hafnium analogue of 28 results in 31, which was initially misidentified as a cyclic amidine complex124 but instead is the result of imine C-C coupling from the Hf analogue of 29. The Cp*2Zr analogue can selectively insert one equivalent of isocyanide (or CO) into the metal-butadiene bond, allowing for sequential reactions to be carried out125.
Fig. 4 |. Examples of new reductive coupling reactions.

a | Isocyanide insertion into metaiiacydopentenes can result in imido extrusion and formal [4+1] coupling with Ti, while intermediate products can be isolated with the heavier Hf congener. b | Nitrile reductive couplings lead to diazatitanacyclopentadienes that can undergo further reaction to make new π-conjugated materials. c | A general mechanistic scheme for early-transition-metal-catalysed cyclotrimerization catalysis (left) and new advances in selective heterocyclization catalysed by simple reduced Nb complexes. DCE, 1,2- dichloroethane; DME, 1,2-dimethoxyethane; equiv., equivalents; THF, tetrahydrofuran; TM, transition metal; TMS, trimethylsilyl
CpR2M(TMS2C2) (M = Ti, Zr; R = H, alkyl) can act as a masked source of CpR2MII, easily liberating TMS2C2 upon treatment with a variety of reagents47. Recently, Rosenthal has discovered that Cp*2II fragments can couple nitriles to form metalla-2,5-diazacyclopentadienes, which can undergo further reaction and exchange one nitrile partner for another unsaturated substrate (for example, a different nitrile, CO2 or H2)126–128. Nitrile-nitrile couplings are quite uncommon, although they have been observed with TiIII complexes much earlier129. Taking advantage of this reaction manifold, Tilley has recently used Cp2Ti(TMS2C2) for the coupling of tethered dinitriles, such as 32, to diazametallacyclopentadienes, such as 33 (REF100). Once coupled, 33 can be reacted further with electron-deficient alkynes, H3O+ or ECl2 (E = SO, POPh, S, Se, Te) to generate pyrazines (34), diones (35) or diazoles (36) (FIG. 4b). Although this work focused primarily on installing these moieties in polycyclic aromatic hydrocarbons, [2+2+2] pyrazine synthesis at an early transition metal had not been previously observed, providing new insight on routes to incorporate nitriles more effectively into reductive coupling reactions mediated by early transition metals. In a similar vein, stoichiometric Cp2Zr(TMS2C2) has been used as a general platform for C-C coupling and can be used to couple tethered diynes to make polycyclic aromatic hydrocarbons130–133. Cp2MII fragments can also be used to construct metallacyclocumulenes and other highly strained metallacycles47,49,134–142. Here, the steric bulk of the Cp* ligands imbue stability, which enables easy isolation of the metal complexes that feature exotic bonding motifs. Structural characterization of the products of reductive metallacyclization of 1,3-butadiynes reveals that the rich chemistry is strongly dependent on the terminal diyne substituents. Similarly, Beckhaus has coupled unsaturated organics with group 4 metal pentafulvene complexes143–148, presenting an interesting approach to the synthesis of exotic appended Cp-type ligands. Likewise, fulvenes can be homocoupled to give ansa-metallocene complexes from group 4 metal chlorides and nBuLi (REFS149,150).
There are fewer examples of group 5 metals facilitating reductive coupling reactions. Alkyne-stabilized masked TaIII hydrides can stoichiometrically insert isonitriles and alkynes into the Ta-H bond, followed by C-C reductive coupling of the bound alkyne to give butadienyl and azadiene fragments151. Dimethyltantalum corrole complexes can react with CO to give coupled diacetyl complexes152. This process is presumed to occur via insertion of two CO molecules into the two Ta-Me bonds, followed by reductive elimination of the two acyl ligands to give diacetyl, which then binds to the resulting low-valent tantalum as the ene-diolate form to formally oxidize TaIII to TaV.
Catalytic alkyne cyclotrimerization is also common with low-valent group 4–5 transition metals34,110,153. There are two limiting mechanisms for cyclotrimerization: triple insertion followed by reductive elimination through a metallacycloheptatriene (37) or [4+2] cycloaddition of the 3rd unsaturated partner to make a metallanorbornadiene (38) (FIG. 4c, left). Recent mechanistic work on a ditantalum system indicates that the [4+2] pathway is more likely154. Remarkably, alkyne cyclotrimerization reactions can be intercepted with other unsaturated substrates; for example, Obora has shown that thermally stable low-valent Nb catalysts can chemoselectively insert alkenes155–157 and nitriles158 to give cross-coupled products159 (FIG. 4c, right).
Functionalization and defunctionalization
Hydrogenation.
Catalytic hydrogenation of unsaturated substrates is challenging with d2 early transition metals because of their propensity to promote reductive coupling. Additionally, there are very few examples of direct H2 activation by d2 group 5 metals160–166. Nonetheless, there are several elegant examples of arene and alkene hydrogenation and alkyne semi-hydrogenation catalysed by d2 group 5 metals162,167,178.
β-Diketiminate NbIII imido complexes promote remarkably selective semi-hydrogenation of alkynes176 (FIG. 5a, top). Treatment of (BDI)Nb(N′Bu)(CO)2 (39) (BDI = 2,6-diisopropylbenzene-p-diketiminate) with 1-phenyl-1-propyne yields the metallacyclopropene complex 33, which does not undergo reductive coupling owing to steric encumbrance around the metal centre179. Consequently, it was found that 40 can catalyse the semi-hydrogenation of phenylpropyne to Z-p-methylstyrene under H2/CO. Two mechanistic proposals were made (FIG. 5a, right): through alkyne-CO ligand exchange, followed by σ-bond metathesis of the metallacyclopropene of 40 with H2 to yield a NbV vinyl hydride (41), from which C-H reductive elimination then liberates Z-β-methylstyrene and regenerates an arene-bound Nb111 (42) (path A), or through 1,2-H2 addition across the imido of 40 to make a Nb amido hydride (43), which can then undergo reductive H migration to vinyl amide 44 followed by a-elimination to 42 (path B). The rate and selectivity for semi-hydrogenation are highly CO-dependent, which may provide a tuneable handle in future systems. Higher CO concentration maintained selectivity but slowed the reaction, indicating that CO-alkyne exchange is kinetically relevant. However, lower loadings led to the formation of more fully hydrogenated propylbenzene, indicating that CO also plays a role in efficient product displacement.
Fig. 5 |. Catalytic (de)hydrofunctionalizations.

a | Well-defined Nb-catalysed (Z)-selective alkyne semi-hydrogenation catalysed by Nb imido complexes (top) and Ti-catalysed transfer hydrogenation (bottom). b | Selective, catalytic alkane dehydrogenation using phosphorus ylides as terminal oxidants. c | Catalytic examples of hydrodefluorination that take advantage of the formation of strong Al–F or Si–F bonds as a driving force for defluorination. Δ, heat; BDI, β-diimine; DME, dimethoxyethane; equiv, equivalents; THF, tetrahydrofuran; TON, turnover number.
Fortier recently described the synthesis of a new masked TiII synthon, 45 (FIG. 5a, bottom). This complex undergoes a relatively rare example of early transition metal d2 C-H oxidative addition160,180,194 followed by β-H abstraction to extrude H2 and make a new cyclometallated complex 46 (REF195). Remarkably, this reaction is reversible, opening up the possibility that these complexes might be useful as hydrogenation catalysts. In fact, 45 also performs a unique example of early-transition-metal-promoted transfer hydrogenation196 (FIG. 5a, bottom left), hydrogenating cyclohexene with concomitant production of 46, with the ligand serving as the terminal hydrogen donor. This interesting system can also catalytically hydrogenate cyclic alkenes and cyclic dienes, although the mechanism by which this occurs — transfer hydrogenation or a more classical dihydride mechanism — remains unknown.
Dehydrogenation.
There are several examples of C-H oxidative addition to low-valent early transition metals160,180–194,197,198, including several that lead to stoichiometric dehydrogenation188–190. The majority of dehydrogenation reactions promoted by early transition metals are, however, predicated on high-valent, electrophilic metal reactivity, such as 1,2-C-H addition across metal-ligand multiple bonds199–201. The first catalytic, selective dehydrogenation of alkanes was recently demonstrated with Ti catalyst 47 [REF.202] [FIG. 5b]. This system takes advantage of 1,2-C-H addition of the alkane C-H bond to a Ti alkylidiyne 50, followed by β-H abstraction of the resultant Ti-alkyl-alkylidene (51) to formally reduce Ti, making a new Ti11 alkyl-alkene adduct 52 (REFS40–41–203–204). Remarkably, this system is rendered catalytic by addition of a methylene transfer agent, phenyldibenzophosphole methylene ylide, H2CP(C12H8)Ph (48), which serves as an oxidant to reoxidize the TiII fragment (53) back to a TiIV alkylidene (54)202. The structure of this reagent is critical, as deleterious phenyl migration from a TiII-ylide adduct will occur with other simple arylphosphorus ylides204. Although turnover numbers in this system are poor and phosphorus ylides are impractical terminal oxidants, this system is remarkable in its selectivity, yielding only terminal alkene products. This selectivity results from the unique mechanism, which — unlike late transition metal alkane dehydrogenation catalysts — does not involve metal hydride species, which could isomerize the alkene.
There are many low-valent Ti and Zr complexes capable of the catalytic dehydrogenation of amine boranes, particularly using metallocene derivatives205–210. In a recent elegant application, a Cp*2TiCl2/nBuLi catalytic system has been shown to promote an unusual step-growth dehydropolymerization of MeNH2(BH3), enabling the first synthesis of high-molar-mass poly(N-methylaminoborane) by an early transition metal211. This system was also successful in the catalytic dehydropolymerization of several other primary amine borane derivatives. Despite computational evidence for a MII/MIV redox mechanism for amine borane dehydrogenation212, there is conflicting experimental evidence that indicates alternate mechanisms may also be involved. For example, several MIV precatalysts such as M(NMe2)4 (M = Ti, Zr)208, [iPr2Si(NDIPP)2] Zr(NMe2)2-LiCl(THF)3 (DIPP = 2,6-diisopropylphenyl)213 and K5-(Me3SiNCH2CH2)2N(CH2CH2SiMe2CH2) Zr (REF196) may promote dehydrogenation via c-bond metathesis routes, while in other cases, MIII metallocene species have been observed in situ214.
Hydrodefluorination.
C-F bond dissociation energies (BDEs) range from 110 to 130 kcal mol−1 fluoro-organics are not easily biodegraded but instead are often incinerated217,218. Thus, there is considerable impetus to design catalytic methods for C-F bond degradation219,220. Although C-F bond activation has been observed across the transition metal series221–229, early transition metals are exceptionally fluorophilic, making them excellent candidates for hydrodefluorination reactions. There are numerous examples in which low-valent early transition metals can mediate C-F bond activation to generate M-F species, including examples with Ti (REFS230–233), Zr (REFS233–239), Hf (REF240), Nb (REFS241–243) and Sc (REF244). The challenge in these systems is rendering them catalytic, as the resultant M-F species is often a considerable thermodynamic sink245.
In the first demonstration of catalytic C-F bond activation by an early metal, Cp2TiF2 was shown to defluorinate perfluorinated cycloalkanes to produce perfluorinated arenes in the presence of excess Al0 with HgCl2 (REF246) (FIG. 5c, top). In the same study, it was found that Cp2ZrF2 similarly catalyses this reaction in the presence of Mg0 and HgCl2, although in a rather exothermic fashion. This result was followed by mechanistic work using Cp*2ZrH2. Under an atmosphere of H2, it was found that Cp*2ZrH2 reacted with primary, secondary and tertiary C-F bonds to yield the corresponding alkanes and Cp*2ZrHF (REF247). Intriguingly, mechanistic evidence supported a radical mechanism in which in situ generated Cp*2ZrH abstracted F% despite the fact that the relative rate of C-F reactivity was 1° > 2° > 3° — the reverse of the pattern seen in typical radical reactions. In support of the radical mechanism, added reductant increased the rate of 1-fluorohexene hydrodefluorination threefold, while radical inhibitors such as 9,10-dihydroanthracene inhibit the rate of defluorination. Radical clock experiments of Cp*2ZrH2 with cyclopropylcarbinyl fluoride provided Cp*2ZrHF and ring-opened Cp*2Zr(“Bu)H as opposed to methylcyclopropane. It was suggested that the steric bulk of Cp*2ZrH2 may be the origin of the observed rate trends237–247–248.
There are also several notable examples of Nb-catalysedhydrodefluorination. Mixtures of NbCl5 and LiAlH4 catalyse the hydrodefluorination of aryl fluorides and benzylic fluorides249–250 (FIG. 5c, middle). In these reactions, trifluoromethyl groups were reduced fully to methyl groups under mild conditions. In the case of aryl fluoride reductions, it was proposed that the reaction proceeds through a (DME)NbCl3(arene) (DME = 1,2-dimethoxyethane) intermediate that undergoes nucleophilic substitution by hydride, but the mechanism of these reductions has not been fully established. Arnold and Bergman recently examined a more well-defined system for aryl fluoride hydrodefluorination using a d2 (BDI)NbIII(N′Bu) fragment251 (FIG. 5c, bottom). Mechanistic studies in this system indicate that (BDI)Nbm(N′Bu)(arene) (57) can oxidatively add Ar-F to generate (BDI)NbV(N′Bu)F(Ar) (55), which can be transmetallated with silanes, making a strong Si-F bond and a (BDI)NbV(NfBu)H(Ar) species capable of C-H reductive elimination. In both the NbCl5 and (BDI)Nb systems, catalytic turnover is achieved by using a terminal reductant (Si, Al) that is more fluorophilic than Nb.
Catalytic single-electron transfer
Single-electron transfers employing low-valent early transition metal reductants are frequently used in organic synthesis. The Nugent-RajanBabu reagent24–25, Cp2TiCl, is used in radical ring-opening-cyclization cascades and is the most common early transition metal reagent for single-electron transfer252. These types of cyclization cascades have been used in numerous total syntheses of natural products23–253–254. Cp2TiCl is easily generated via in situ reduction of commercially available Cp2TiCl2 by Zn or Mn dust and has recently been touted as a ‘greeN′ reagent for chemical synthesis255–257. This reagent typically exists as the chloride-bridged dimer, [Cp2TiCl]2, and forms the active monomeric species through a solvent-assisted equilibrium258.
Gansäuer expanded the use ofthe Nugent-RajanBabu reagent in the context of radical ring opening of epoxides by developing a catalytic procedure that uses a weak acid (2,4,6-collidine hydrochloride) to protonate the generated titanium alkoxide (59) and a reductant (Zn or Mn) to regenerate Cp2TiCl (REF35) (FIG. 6a, left). 2,4,6-Collidine hydrochloride is an ideal acid for epoxide reactions; it is strong enough to protonate the Ti-alkoxide bonds but weak enough to not open the epoxide ring independently. Furthermore, the liberated 2,4,6-collidene conjugate base is sterically congested, disfavouring association with titanium that would hinder catalysis. In their initial report, Mn was the preferred reductant in this reaction, as the MnCl2 by-product is insufficiently Lewis acidic to promote epoxide ring opening, although later catalytic applications have successfully used Zn. With appropriate solvent and counteranion choice, cationic [Cp2Ti] fragments can also be used as catalysts259. This catalytic single-electron reduction of epoxides has been used to functionalize arenes, alkenes and alkynes259–263 (FIG. 6a, right). Remarkably, these radical reactions are able to proceed enantioselectively when using large, terpene-derived cyclopentadienyl ligands (60) on titanium263.
Fig. 6 |. Single-electron processes catalysed by TiIII complexes.
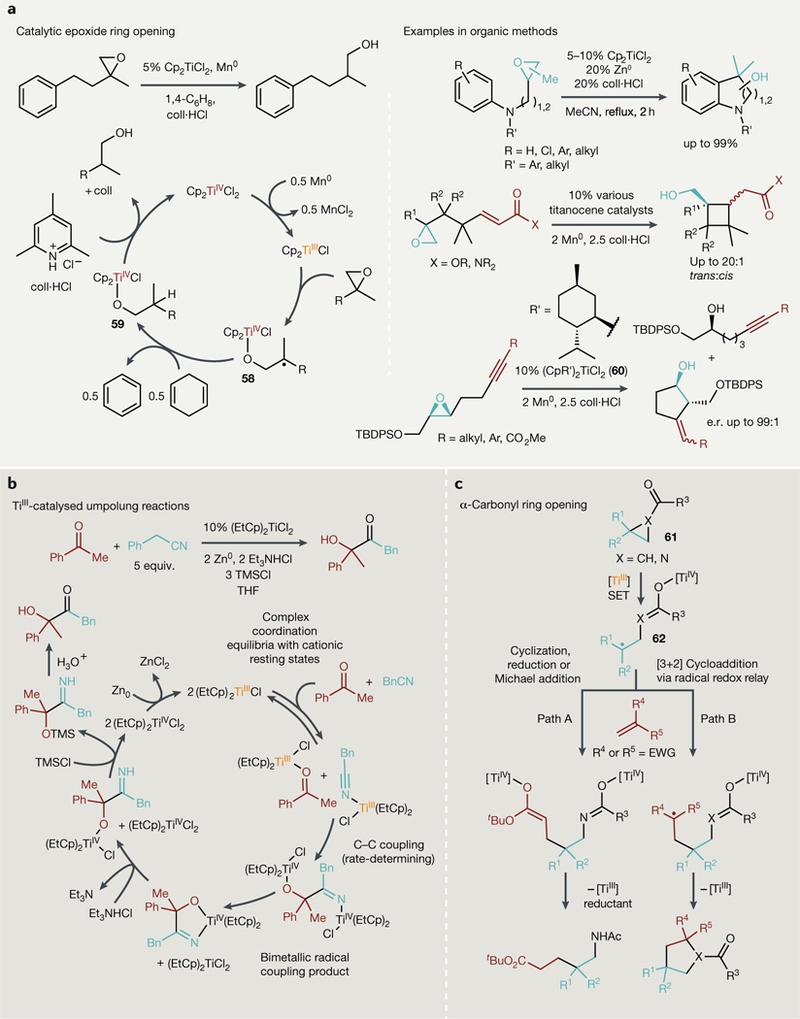
a | Catalytic epoxide ring opening using weak reductants and acids to drive turnover (Left) and examples in selective organic synthesis. b | Single-electron umpolung reactions can yield new C-C bonded products through a complex bimetallic radical coupling mechanism. c | SingLe-eLectron transfer (SET) from TiIII into carbonyls adjacent to strained rings gives access to several new aLkene coupling reactions including cyclization, Michael addition and [3+2] cycLoadditions. CoLL-HCL, 2,4,6-coLLidine hydrochloride; equiv, equivalents; e.r., enantiomeric ratio; EWG, eLectron-withdrawing group; TBDPS, tert-butyLdiphenylsiLyL; THF, tetrahydrofuran; TMS, trimethyLsiLyL.
Cp2TiCl can also be used to catalyse redox umpolung reactions for the coupling of nitriles or activated alkenes with carbonyls or imines73,264–274 (FIG. 6b). These reactions share similar mechanistic features to classical epoxide and azirine ring-opening reactions; owing to the oxophilicity of Ti, treatment of many types of carbonyl derivatives with TiIII species results in the formation of TiIV carbonyl radicals. These Ti carbonyl radicals can further react with myriad electrophiles273,275. In a detailed mechanistic study of a TiIII-catalysed acyloin-type cross-coupling of ketones and nitriles, it was found that a bimolecular and bimetallic radical recombination mechanism that circumvented free radical intermediates was operational274 (FIG. 6b, bottom). Umpolung-like conjugate additions have also been achieved with hemiaminals, yielding quaternary carbons-a to amines, and coupling or reduction of α,β-unsaturated carbonyls is also possible276–278.
Combining the above approaches, both Gansauer and Lin have each demonstrated that three-membered ring rupture can also be promoted by a-carbonyl reduction in N-acyl aziridines279,280 and cyclopropylketones281 (FIG. 6c). In the case of N-acyl aziridines (61), treatment with TiIII sources results in ring opening that, in contrast to typical aziridine ring openings, produces the more substituted carbon radical 62. These reactions take advantage of the biradical character282–288 of the resulting TiIV azaenolate or enolate 62; radical reductive couplings of the ring-opened azaenolate or enolate with alkene electrophiles can be performed catalytically in the presence of reductant (Mn0 or Zn0) and weak organic acid (collidine HBr or NEt3HCl) to form amines (path A), while redox-neutral [3+2] cycloadditions with electrophilic alkenes can also be achieved in a catalytic fashion (path B).
Cp2TiIII pinacolates, generated through Cp2TiClpromoted pinacol coupling of benzaldehyde followed by further reduction, undergo facile deoxygenation to form alkenes. This reaction challenges the classical McMurry coupling mechanism, in which TiII or Ti0 complexes were thought to promote carbonyl coupling and/or olefination289.
Single-electron strategies in small-molecule activation and photoredox catalysis.
Reduced early transition metal complexes have historically been excellent platforms for small-molecule activation because of their strong reduction potentials and propensity to form thermodynamically stable metal-ligand multiple bonds. However, these same features become a challenge when attempting to render these reactions catalytic. There are myriad examples of stoichiometric N2 activation with low-valent early transition metals. Given that this field has been thoroughly reviewed4–17, here, we focus on only several examples that highlight emerging new strategies toward catalytic applications of small-molecule activation — in particular, proton-coupled electron transfers (PCETs) in early transition metals and their applications toward N2 reduction and water oxidation.
Recent work using group 4 metallocene amide derivatives has found PCET to be a promising route to ammonia formation via M-N hydrogenolysis290,291 (FIG. 7a). This route has the potential to overcome the incredible free energy inefficiency of nitrogen reduction involving separated sources of protons and electrons (for example, [HOEt2] + [BArF24] + KC8). Experimental and computational studies have demonstrated that coordination of NH3 to (η5-C5Me4SiMe3)2M (M = Ti, Zr) fragments decreases the N-H bond dissociation free energy (BDFE) by >40 kcal mol−1 compared with free NH3. In general, the degree of N-H BDFE lowering is a function of the early transition metal redox couple, in which higher energy redox couples (for example, ZrIV/ZrIII in 64 or TiIII/TiII in 65) lead to greater activation than lower energy couples (for example, TiIV/TiIII in 63). This allows for thermodynamically favourable PCET to occur from (η5-C5Me5)(py-Ph)RhH (68) — which has a weak Rh-H bond (52.3 kcal mol−1) but is nonetheless able to split molecular H2 — to (η5Me4SiMe3)2TiCl(NR2) derivatives (R = H, Me, NH2, NMe2) (BDFEs ^ 60 kcal mol−1). Using this strategy, the catalytic hydrogenolysis of TiIV amide complex 66 to the TiIII chloride complex 67 has been realized, opening the possibility of incorporating PCET into a catalytic N2 reduction cycle with H2 as the terminal reductant.
Fig. 7 |. Single-electron strategies in small-molecule activation chemistry.

a | Hydrogenoiysis of metal amides to ammonia via proton-coupled electron transfer (PCET), enabled by coordination-induced weakening of the N-H bond. b | Photochemical model of H2O splitting with TiIII catalysts, again enabled by coordination-induced weakening of O-H bonds. c | Photoredox catalysis via ligand-to-metal charge transfer (LMCT) with (MePDP)2Zr complexes, in which charge transfer occurs in a direction opposite of state-of-the-art late transition metal photoredox catalysts, which undergo metal-to-ligand charge transfer (MLCT). hv, irradiation; BDFE, bond dissociation free energy; Cp*, C5Me5−; Epc, cathodic peak potential; equiv, equivalents; Ered, reduction potential; Fc, ferrocene; LED, light-emitting diode; MePDP, 2,6-bis (5-methyl-3-phenyl-1H-pyrrol-2-yl)-pyridine; TEMPO, (2,2,6,6-tetramethy[piperidin-1-y[)oxy[; THF, tetrahydrofuran; TMS, trimethylsilyL; TON, turnover number.
Similar to the effects seen with NH3, coordination of H2O to Cp2TiIIIX results in dramatically reduced H-OH BDEs (Abde ≈ 50 kcal mol−1), enabling water to serve as a H-atom donor292–294. This important effect can be used in the photochemical splitting of water. For example, ansa-titanocene dihydroxido complexes undergo Ti-O bond homolysis under visible light to form ansa-titanocene monohydroxido complexes and OH radicals295–297. This reaction had been observed with non-tethered titanocenes before298, but the Cp-Ti BDFE is typically smaller than the Ti-O BDFE, leading primarily to loss of Cp in those systems. A catalytic model system for the photochemical splitting of water has been demonstrated using an ansa-titanocene aquo-hydroxo complex 70 (REF 297) (FIG. 7b). Currently, this process requires TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl) oxyl) to reoxidize the bis(aquo) complex 69 to 70, which can liberate an OH radical upon irradiation. While this reaction may be catalytic, a turnover number could not be confirmed. In contrast to the above reaction, reaction of Cp*2TiOTf with water results in rapid oxidation to Cp*2Ti(OH)(OTf) and hydrogen gas without TEMPO additive299.
Building upon this work showing that Cp2MX2 can undergo ligand-to-metal charge-transfer (LMCT) processes to liberate radicals, it has also been shown that early transition metals can be used as sensitizers for single-electron photoredox catalysis. For example, metal photosensitizers have been designed from π-donor ligands on electron-poor Zr centres (FIG. 7c). As ZrIV is a d0 metal, these complexes use LMCT processes as opposed to the typical metal-to-ligand charge-transfer (MLCT) processes seen in late transition metal photosensitizers such as [Ru(bpy)3]2+ (REF 300). Zr is considerably less expensive and more earth abundant than the common late metals used in photosensitizers, making it an attractive alternative for catalysis. Zr(MePDP)2 (MePDP = 2,6-bis(5-methyl-3-phenyl-1H-pyrrol-2-yl)-pyridine) (73) has a visible excitation at 528 nm and a photoemission at 594 nm. Cyclic voltammetry (CV) experiments show a reversible reduction at −2.16 V, while Rehm-Weller analysis estimates an excited-state reduction potential of −0.07 V. This excited-state reduction potential is supported by excited-state quenching using weak organic reductants such as 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-7-methylbenzo-[d]imidazole (MeBIH). Complex 73 is able to catalyse the reductions of ethyl 2-bromo-2,2-difluoroacetate and diethyl maleate to ethyl 2,2-difluoroacetate and diethyl succinate, respectively (FIG. 7c, right). An in-depth study of the homocoupling of benzyl bromide helped support the photoredox mechanism301.
Group-transfer catalysis
As is the case with small-molecule activation reactions, engendering catalytic reactivity in group-transfer reactions is particularly challenging owing to the strength of M-E multiple bonds. Classical examples such as methylene transfer in the Tebbe olefination result in the formation of strong Ti-O double bonds, precluding practical turnover. Nonetheless, there have been several notable advances toward catalytic oxidative group-transfer reactions, in particular nitrene transfer. Most of the catalytic early transition metal nitrene transfer reactions involve oxidative imination of unsaturated organic functional groups, which stands in contrast to late-transition-metal-catalysed nitrene transfers, which often perform C-H insertions302–305 or aziridinations306–308.
Heyduk has developed several Zr and Ta complexes of aminoquinone-derived redox non-innocent ligands80,309. In these complexes, multi-electron redox changes needed for group transfer take place on the ligand framework instead of at the metal centre, allowing facile, low-temperature access to imido complexes that can undergo further reactivity such as catalytic nitrene coupling310,311, imide reduction312 and oxidative isocyanide imination (FIG. 8a, left). For example, in the (NNN) ZrCl (74)-catalysed isocyanide imination reaction, the trianionic (tris)amide (NNN)3−ligand undergoes two-electron oxidation by RN3 to yield the monoanionic (NNN)1−imido complex 75, which can insert isocyanide to form an η2-carbodiimide 76. Upon carbodiimide ligand loss, the NNN ligand framework is again reduced to the (NNN)3− form in 74 to close the catalytic cycle313. A related Ti system based on a diamide-dimine (dadi) framework has also been reported82. These (dadi) Ti complexes can access a large array of ligand oxidation states, thereby realizing a two-electron pathway for catalytic azide carbonylation by 77 (FIG. 8a, right). Productive catalysis hinges on weak CO binding to Ti resulting from the redox non-innocent ligand; one would expect TiII to exhibit strong CO binding due to backdonation into the C-O π* orbital, but because the redox events take place on the ligand, the electron density at Ti remains low throughout the catalytic cycle and thus Ti is incapable of strong backdonation.
Fig. 8 |. Group-transfer catalysis mediated by early transition metal complexes.

a | Redox non-innocent ligand frameworks enable nitrene transfer reactions by undergoing redox changes at the ligand instead of the metal. b | TiII/TiIV redox catalytic nitrene transfer in oxidative amination catalysis — a new mechanism for pyrrole formation (left) — and examples in selective synthesis (right). c | Nitrene transfer promoted by a π-loaded bis(imido)Nb catalyst, which renders the imido fragment more labile than other group 5 imido complexes. d | A closed synthetic cycle for ligand-based CO2 reduction to CO that avoids the formation of strong M-O multiple bonds. BDI, 2,6 diisopropylbenzene-β diketiminate; dadi, diamide-dimine; equiv, equivalents;THF, tetrahydrofuran; TMS, trimethylsilyl; TON, turnover number.
In contrast to the above examples of overt ligand redox non-innocence in nitrene transfer, there are several recent examples of formally TiII/TiIV-catalysed oxidative nitrene transfer reactions. These reactions yield multi-substituted pyrroles from the coupling of alkynes with azobenzene314,315 or α,β-unsaturated imines and α-iminocyclopropanes from the coupling of alkynes and alkenes with azobenzene316 (FIG. 8b). The crux of this system is that cooperative metal-ligand effects can also stabilize low-valent metals through backbonding (classical redox non-innocence) into the reagents (alkynes and diazenes) or products (pyrroles and imines), allowing for otherwise challenging reductive events, such as the reductive elimination from 82 to 83, to occur with relative ease. Although it was initially speculated that azobenzene was a critical reagent for catalytic reoxidation of 83 to 80 through the hydrazido adduct 84, recent advances have extended this chemistry into other nitrene sources such as azides317.
Computational analysis has revealed several additional surprising facets of the reductive elimination events that are critical for catalytic turnover in these TiII/TiIV reactions318,319. In general, it appears that direct reductive coupling of M-C and M-X σ-bonds from intermediates 82 or 88 is unfavourable relative to other reductive pathways. For example, in the pyrrole synthesis, manifold intrinsic bond orbital (IBO) analysis indicates that reductive elimination from the six-membered metallacycle 82 is a π-type electrocyclic ring closure (FIG. 8b, left). By contrast, in the alkene-alkyne coupling reactions, the six-membered metallacycle 88 is semi-saturated (FIG. 8b, right) and thus lacks the requisite π-orbital for electrocyclization. This opens up alternative reductive elimination pathways: p-H elimination to ultimately form α,β-unsaturated imines (86) or a pericyclic Kulinkovich-like cyclopropanation to form a-iminocyclopropanes (87). It is proposed that the α,β-unsaturated imines are formed through N-H reductive elimination, which has not previously been observed on Ti. Computational studies have proposed a more complex insertion-type pathway for the formation of this product, although experimental evidence for either pathway is currently lacking320.
In contrast to group 4 metals, group 5 metal imidos tend to be considerably less reactive. To counteract this, efforts have been made to synthesize group 5 systems that electronically saturate the metal d orbitals (π-loading), thereby increasing the polarizability of the imido bonds321. The re-loaded β-diketiminate (BDI) Nb bis(imido) complex 89 can thus perform a formally NIII/NbV-catalysed nitrene transfer reaction to generate asymmetric dialkylcarbodiimides from azides and isocyanides322 (FIG. 8c).
Outside of catalytic nitrene transfer, there are also many examples of stoichiometric atom-transfer reactions (As4 (REF.323), S8 (REFS324,325) and P4 (REF.326)). For example, reaction of either P4 or As4 with activated zirconocene derivatives results in pnictogen-silicon analogues of benzene327 and carbon-phosphorous cage compounds328. The Cummins group has also extensively studied P4 transformations329 and synthesized phosphorous-rich organic clusters mediated by a NbIII/NbV couple330.
O-atom transfer is an incredibly challenging target to achieve with oxophilic early transition metals because the strong metal-oxo bond is a thermodynamic sink. In order to circumvent this problem, Nb nitride 90 has been used to perform outer-sphere O-atom transfer from CO2 (FIG. 8d). CO2 binds to the terminal anionic nitride in 90, and the resulting oxide ion in 91 can then be transferred to a RC(O)Z (Z = halide, OAc) acceptor to form 92, an N-bound cyanate. Subsequent oxidation and reduction steps liberate CO from the bound cyanate in 93, regenerating 90. While this system is not catalytic, it remains a rare example of a closed synthetic cycle to achieve CO2 reduction331.
Summary and outlook
The field of low-valent early transition metal synthesis, reactivity and catalysis has undergone a resurgence of activity in recent years. Many of these new reactions and applications can be split into three categories: reactions providing a modern twist on classic low-valent organometallic transformations, such as catalytic ring-opening reactions and substrate-directed reductive couplings; reactions that push low-valent early transition metals into new, emerging fields, such as PCET, photoredox catalysis and catalyst-controlled radical reactions; and new modes of low-valent inorganic and organometallic reactivity, such as oxidative nitrene transfer catalysis and reversible end-on ScII N2 binding.
Each of these categories illustrates that early transition metals play a rich and evolving role in a broad array of chemical applications, and that there likely remains substantially more to discover as the field of low-valent early metal chemistry is revisited through the lens of modern organometallic chemistry. For example, accessing low-valent intermediates under mild conditions (using, for example, organosilane reductants or photoredox chemistry) is only beginning to have an impact in the field and should help to overcome many of the chemical compatibility issues associated with the strong reductants previously used to access low-valent early transition metals.
While many of the reactions discussed here are catalytic in nature, it would be unwise to neglect the importance of fundamental and stoichiometric reactivity with the early transition metals — not only can these later provide the basis for catalytic reactions (as seen in many of the examples discussed above), but the early metals are incredibly earth abundant, environmentally friendly and inexpensive and often have practical utility even in stoichiometric quantities.
Inner-sphere reductants
Reagents that act as reductants after first forming a bond to the complex of interest.
Umpolung reactions
Reactions that proceed through polarity inversion of the given functional group.
Redox non-innocent ligands
Ligands bound to a metal complex in which the oxidation state is ambiguous, wherein oxidation or reduction of the ligands may occur in tandem with or instead of metal oxidation or reduction.
Acknowledgements
Financial support was provided by the National Institutes of Health (1R35GM119457) and the Alfred P. Sloan Foundation (I.A.T. is a 2017 Sloan Fellow).
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hunt AJ, Farmer TJ & Clark JH in Element Recovery and Sustainability (ed. Hunt AJ) 1–28 (Royal Society of Chemistry, 2013). [Google Scholar]
- 2.Greenwood NN & Earnshaw A Chemistry of the Elements (Butterworth-Heinemann, Oxford, 1997). [Google Scholar]
- 3.Kepp KP A quantitative scale of oxophilicity and thiophilicity. Inorg. Chem 55, 9461–9470 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Gambarotta S & Scott J Multimetallic cooperative activation of N2. Angew. Chem. Int. Ed 43, 5298–5308 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Gambarotta S Dinitrogen fixation and activation after 30 years: a puzzle still unsolved. J. Organomet. Chem 500, 117–126 (1995). [Google Scholar]
- 6.Hidai M & Mizobe Y Recent advances in the chemistry of dinitrogen complexes. Chem. Rev 95, 1115–1133 (1995). [Google Scholar]
- 7.MacKay BA & Fryzuk MD Dinitrogen coordination chemistry: on the biomimetic borderlands. Chem. Rev 104, 385–401 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Gardiner MG & Stringer DN Dinitrogen and related chemistry of the lanthanides: a review of the reductive capture of dinitrogen, as well as mono-and di-aza containing ligand chemistry of relevance to known and postulated metal mediated dinitrogen derivatives. Materials 3, 841–862 (2010). [Google Scholar]
- 9.Studt F & Tuczek F Theoretical, spectroscopic, and mechanistic studies on transition-metal dinitrogen complexes: implications to reactivity and relevance to the nitrogenase problem. J. Comput. Chem 27, 1278–1291 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Bazhenova TA & Shilov AE Nitrogen fixation in solution. Coord. Chem. Rev 144, 69–145 (1995). [Google Scholar]
- 11.Mori M Activation of nitrogen for organic synthesis. J. Organomet. Chem 689, 4210–4227 (2004). [Google Scholar]
- 12.Fryzuk MD & Johnson SA The continuing story of dinitrogen activation. Coord. Chem. Rev 200–202, 379–409 (2000). [Google Scholar]
- 13.Burford RJ, Yeo A & Fryzuk MD Dinitrogen activation by group 4 and group 5 metal complexes supported by phosphine-amido containing ligand manifolds. Coord. Chem. Rev 334, 84–99 (2017). [Google Scholar]
- 14.Fryzuk MD Side-on end-on bound dinitrogen: an activated bonding mode that facilitates functionalizing molecular nitrogen. Acc. Chem. Res 42, 127–133 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Chirik PJ Dinitrogen functionalization with bis(cyclopentadienyl) complexes of zirconium and hafnium. Dalton Trans 0, 16–25 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Burford RJ & Fryzuk MD Examining the relationship between coordination mode and reactivity of dinitrogen. Nat. Rev. Chem 1,0026 29(2017). [Google Scholar]
- 17.Jia H-P & Quadrelli EA Mechanistic aspects of dinitrogen cleavage and hydrogenation to produce ammonia in catalysis and organometallic chemistry: relevance of metal hydride bonds and dihydrogen. Chem. Soc. Rev 43, 547–564 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Reichard HA & Micalizio GC Metallacycle-mediated cross-coupling with substituted and electronically unactivated alkenes. Chem. Sci 2,573–589 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan X & Xi C Conversion of zirconacyclopentadienes into metalloles: Fagan-Nugent reaction and beyond. Acc. Chem. Res 48, 935–946 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Micalizio GC in Comprehensive Organic Synthesis II 2nd edn Vol. 5 (ed. Knochel P) 1660–1737 (Elsevier, 2014). [Google Scholar]
- 21.Xu S & Negishi E Zirconium-catalyzed asymmetric carboalumination of unactivated terminal alkenes. Acc. Chem. Res 49, 2158–2168 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Negishi E Controlled carbometalation as a new tool for carbon-carbon bond formation and its application to cyclization. Acc. Chem. Res 20, 65–72 (1987). [Google Scholar]
- 23.Zweig JE, Kim DE & Newhouse TR Methods utilizing first-row transition metals in natural product total synthesis. Chem. Rev 117, 11680–11752. [DOI] [PubMed] [Google Scholar]
- 24.Nugent WA & RajanBabu TV Transition-metalcentered radicals in organic synthesis. Titanium(III)-induced cyclization of epoxy olefins. J. Am. Chem. Soc 110, 8561–8562 (1988). [Google Scholar]
- 25.Rosales A et al. The Nugent-RajanBabu reagent: a formidable tool in contemporary radical and organometallic chemistry. Eur. J. Org. Chem 2015, 4567–4591 (2015). [Google Scholar]
- 26.Parkin G in Comprehensive Organometallic Chemistry III Vol. 1 (eds Mingos MP & Crabtree RH)1–57 (Elsevier, 2007). [Google Scholar]
- 27.Grant LN, Miehlich ME, Meyer K & Mindiola DJ Arrested disproportionation in trivalent, mononuclear, and non-metallocene complexes of Zr(III) and Hf(III). Chem. Commun 54, 2052–2055 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Kurogi T, Miehlich ME, Halter D & Mindiola DJ, 1, 2-CH bond activation of pyridine across a transient titanium alkylidene radical and re-formation of the Ti=CHtBu moiety. Organometallics 37, 165–167 (2018). [Google Scholar]
- 29.Perera TH, Lord RL, Heeg MJ, Schlegel HB& Winter C Metallapyrimidines and metallapyrimidiniums from oxidative addition of pyrazolate N-N bonds to niobium(III), niobium(IV), and tantalum(IV) metal centers and assessment of their aromatic character. Organometallics 31,5971–5974 (2012). [Google Scholar]
- 30.Pun D, Leopold SM, Bradley CA, Lobkovsky E& Chirik PJ Bis(indenyl)hafnium chemistry: ligand-induced haptotropic rearrangement and fundamental reactivity studies at a reduced hafnium center. Organometallics 28, 2471–2484 (2009). [Google Scholar]
- 31.Chomitz WA, Sutton AD, Krinsky JL & Arnold J Synthesis and reactivity of titanium and zirconium complexes supported by a multidentate monoanionic [N2P2] ligand. Organometallics 28, 3338–3349 (2009). [Google Scholar]
- 32.Figueroa JS, Piro NA, Clough CR& Cummins CC A nitridoniobium(V) reagent that effects acid chloride to organic nitrile conversion: synthesis via heterodinuclear (Nb/Mo) dinitrogen cleavage, mechanistic insights, and recycling. J. Am. Chem. Soc 128, 940–950 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Buschel S et al. Adduct formation of [(η7-C7H7)Hf(n5-C5H5)] with isocyanides, phosphines and N-heterocyclic carbenes: an experimental and theoretical study. J. Organomet. Chem 694, 1244–1250 (2009). [Google Scholar]
- 34.Okamoto S Synthetic reactions using low-valent titanium reagents derived from Ti(OR)4 or CpTiX3 (X=O-i-Pr or Cl) in the presence of Me3SiCl and Mg. Chem. Rec 16, 857–872 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Gansauer A, Bluhm H & Pierobon M Emergence of a novel catalytic radical reaction: titanocene-catalyzed reductive opening of epoxides. J. Am. Chem. Soc 120, 12849–12859 (1998). [Google Scholar]
- 36.Galindo A et al. Alkyl alkyne mono((trimethylsilyl) cyclopentadienyl) niobium complexes. Synthesis and chemical behavior in insertion processes. X-ray crystal structures of [NbCp’(CH2SiMe3)2(Me3SiCCSiMe3)] and [NbCp’(NAr){η4-CH(SiMe3)C(SiMe3)C(CH2SiMe3) = CH(SiMe3)}], (Cp’=n5-C5H4SiMe3, Ar = 2,6-Me2C6H3). DFT studies of the model complexes [Nb(n5-C5H5) R2(HCCH)] (R = Cl, Me). Organometallics 21,293–304 (2002). [Google Scholar]
- 37.Calderazzo F, Pampaloni G, Rocchi L, Strahle J & Wurst K The reduction of the NbX5/AlX3 system with aluminum in the presence of aromatic hydrocarbons:an approach to niobium(II), niobium(I), and niobium(0) organometallics. Angew. Chem. Int. Ed 30, 102–103 (1991). [Google Scholar]
- 38.Oshiki T, Yamada A, Kawai K, Arimitsu H& Takai K Alkyne exchange reactions of silylalkyne complexes of tantalum: mechanistic investigation and its application in the preparation of new tantalum complexes having functional alkynes (PhC=CR (R = COOMe. CONMe2). Organometallics 26, 173–182 (2007). [Google Scholar]
- 39.Kamitani M, Searles K, Chen C-H, Carroll PJ& Mindiola DJ p-hydrogen abstraction of an ethyl group provides entry to titanium and zirconium ethylene complexes. Organometallics 34,2558–2566 (2015). [Google Scholar]
- 40.Crestani MG et al. Room temperature dehydrogenation of ethane, propane, linear alkanes C4–C8, and some cyclic alkanes by titaniumcarbon multiple bonds. J. Am. Chem. Soc 135, 14754–14767 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Cavaliere VN et al. Room temperature dehydrogenation of ethane to ethylene. J. Am. Chem. Soc 133, 10700–10703 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Fernandez FJ et al. β-hydrogen-containing zirconium alkyls with the doubly-bridged bis(dimethylsilanediyl) dicyclopentadienyl ligand. X-ray molecular structures of [Zr{(SiMe2)2(n5-C5H3)2}ClEt] and [Zr{(SiMe2)2(n5-C5 H3) 2}Et]2 (h-Ch 2=CH 2). Organometallics 16, 1553–1561 (1997). [Google Scholar]
- 43.Epstein OL, Savchenko AI & Kulinkovich OG Titanium(IV) isopropoxide-catalysed reaction of alkylmagnesium halides with ethyl acetate in the presence of styrene. Non-hydride mechanism of ligand exchange in the titanacyclopropanes. Tetrahedron Lett 40, 5935–5938 (1999). [Google Scholar]
- 44.Negishi E-I & Takahashi T Alkene and alkyne complexes of zirconocene. Their preparation, structure, and novel transformations. Bull. Chem. Soc. Jpn 71, 755–769 (1998). [Google Scholar]
- 45.Negishi E-I, Cederbaum FE & Takahashi T Reaction of zirconocene dichloride with alkyllithiums or alkyl grignard reagents as a convenient method for generating a “zirconocene” equivalant and its use in zirconium-promoted cyclization of alkenes, alkynes, dienes, enynes, and diynes. Tetrahedron Lett 27, 2829–2832 (1986). [Google Scholar]
- 46.Negishi E-I & Takahashi T Patterns of stoichiometric and catalytic reactions of organozirconium and related complexes of synthetic interest. Acc. Chem. Res 27, 124–130 (1994). [Google Scholar]
- 47.Rosenthal U, Burlakov VV, Arndt P, Baumann W & Spannenberg A The titanocene complex of bis(trimethylsilyl)acetylene: synthesis, structure, and chemistry. Organometallics 22, 884–900 (2003). [Google Scholar]
- 48.Beweries T et al. Complexation of bis(trimethylsilyl) acetylene by decamethylhafnocene to give the hafnacyclopropene Cp*2Hf(rη2-Me3SiC2SiMe3): an unusually strong metal-alkyne interaction. Organometallics 26, 247–249 (2007). [Google Scholar]
- 49.Beweries T et al. Synthesis of hafnacyclopentanes from hafnocene alkyne complexes: influence of styrene substituents on the C-C coupling regioselectivity. Eur. J. Inorg. Chem 2009, 1456–1459 (2009). [Google Scholar]
- 50.Fang M et al. Synthesis of the (N2)3- radical from Y2+ and its protonolysis reactivity to form (N2H2)2- via the Y[N(SiMe3)2]3/KC8 reduction system. J. Am. Chem. Soc 133, 3784–3787 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Fang M et al. Isolation of (CO)1- and (CO2)1- radical complexes of rare earths via Ln(NR2)3/K reduction and [K2(18-crown-6)2|2+ oligomerization. J. Am. Chem. Soc 6064–6067 (2012). [DOI] [PubMed]
- 52.Woen DH et al. Solution synthesis, structure, and CO2 reduction reactivity of a scandium(II) complex, {Sc[N(SiMe3)2]3}−. Angew. Chem. Int. Ed 56, 2050–2053 (2017).This study presents the first crystallographically characterized ScII compound with amide ligands that reversibly binds N2 in an end-on fashion. This example highlights the fact that with the right ligand design, even group 3 low-valent early transition metal complexes can be stabilized.
- 53.Woen DH et al. End-on bridging dinitrogen complex of scandium. J. Am. Chem. Soc 139, 14861–14864 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Schmiege BM, Ziller JW & Evans WJ Reduction of dinitrogen with an yttrium metallocene hydride precursor, [(C5Me5)2YH]2. Inorg. Chem 49, 10506–10511 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Demir S et al. Synthesis, structure, and density functional theory analysis of a scandium dinitrogen complex, [(C5Me4H)2Sc]2(µ-η|2:η|2-N2). J. Am. Chem. Soc 132, 11151–11158 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Clentsmith GKB et al. Stabilization of low-oxidation-state early transition-metal complexes bearing 1,2,4-triphosphacyclopentadienyl ligands: structure of [{Sc(P3C2tBu2)2}2]; ScII or mixed oxidation state? Angew. Chem. Int. Ed 42, 1038–1041 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Wijeratne GB et al. Electronic structure and reactivity of a well-defined mononuclear complex of Ti(II). Inorg. Chem 54, 10380–10397 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Edema JJH, Duchateau R, Gambarotta S, Hynes R & Gabe E Novel titanium(II) amine complexes L4TiCl2[L = 1/2 N,N,N′,N′-tetramethylethylenediamine (TMEDA), 1/2 N,N,N′- trimethylethylenediamine, pyridine, 1/22,2’-bipyridine]: synthesis and crystal structure of monomeric trans-(TMEDA)2TiCl2. Inorg. Chem 30, 154–156 (1991). [Google Scholar]
- 59.Araya MA, Cotton FA, Matonic JH& Murillo CA An efficient reduction process leading to titanium(II) and niobium(II): preparation and structural characterization of trans-MCl2(py)4 compounds, M = Ti, Nb, and Mn. Inorg. Chem 34, 5424–5428 (1995). [Google Scholar]
- 60.Chakarawet K et al. Ta(CNDipp)6: an isocyanide analogue of hexacarbonyltantalum(0). Angew. Chem. Int. Ed 56, 10577–10581 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Allen JM & Ellis JE Synthesis and characterization of titanium tetraisocyanide complexes, [CpTi(CNXyl)4E], E = I, SnPh3, and SnMe3. J. Organomet. Chem 693, 1536–1542 (2008). [Google Scholar]
- 62.Barybin MV et al. Homoleptic isocyanidemetalates of 4d- and 5d-transition metals: [Nb(CNXyl)6]-, [Ta(CNXyl)6]-, and derivatives thereof. J. Am. Chem. Soc 129, 1141–1150 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Jilek RE, Jang M, Smolensky ED, Britton JD& Ellis JE Structurally distinct homoleptic anthracene complexes, [M(C14H10)3]2-, M = titanium, zirconium, hafnium: tris(arene) complexes for a triad of transition metals. Angew. Chem. Int. Ed 47, 8692–8695 (2008). [DOI] [PubMed] [Google Scholar]
- 64.Sussman VJ & Ellis JE From storable sources of atomic Nb- and Ta- ions to isolable anionic tris (1,3-butadiene)metal complexes: [M(η4-C4H5)3]-, M = Nb, Ta. Angew. Chem. Int. Ed 47, 484–489 (2008). [DOI] [PubMed] [Google Scholar]
- 65.MacDonald MR et al. Expanding rare-earth oxidation state chemistry to molecular complexes of holmium(II) and erbium(II). J. Am. Chem. Soc 134, 8420–8423 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Arteaga-Muller R et al. New tantalum ligand-free catalyst system for highly selective trimerization of ethylene affording 1-hexene: new evidence of a metallacycle mechanism. J. Am. Chem. Soc 131, 5370–5371 (2009). [DOI] [PubMed] [Google Scholar]
- 67.Nishiyama H et al. Structural and electronic noninnocence of a-diimine ligands on niobium for reductive C-Cl bond activation and catalytic radical addition reactions. J. Am. Chem. Soc 139, 6494–6505 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Tsurugi H, Saito T, Tanahashi H, Arnold J & Mashima K Carbon radical generation by d0 tantalum complexes with a-diimine ligands through ligand- centered redox processes. J. Am. Chem. Soc 133, 18673–18683 (2011). [DOI] [PubMed] [Google Scholar]
- 69.Saito T et al. Reduction of (tBuN=)NbCl3(py)2 in a salt-free manner for generating Nb(IV) dinuclear complexes and their reactivity toward benzo[c] cinnoline. Inorg. Chem 54, 6004–6009 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Saito T et al. 1,4-bis(trimethylsilyl)-1,4-diaza-2,5- cyclohexadienes as strong salt-free reductants for generating low-valent early transition metals with electron-donating ligands. J. Am. Chem. Soc 136, 5161–5170 (2014).This study shows that inner-sphere organosilicon reductants offer many advantages over traditional alkyl metal halide reductants to access low-valent early transition metals: they are less prone to over-reduction, and the organic by-products are easily removed.
- 71.Gomez M, Hernandez-Prieto C, Martin A, Mena M & Santamaria C An effective route to dinuclear niobium and tantalum imido complexes. Inorg. Chem 56, 11681–11687 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Klesko JP, Thrush CM & Winter CH Thermal atomic layer deposition of titanium films using titanium tetrachloride and 2-methyl-1,4-bis(trimethylsilyl)-2,5- cyclohexadiene or 1,4-bis(trimethylsilyl)-1,4- dihydropyrazine. Chem. Mater 27, 4918–4921 (2015). [Google Scholar]
- 73.Frey G, Hausmann JN & Streuff J Titanium-catalyzed reductive umpolung reactions with a metal-free terminal reducing agent. Chem. Eur. J 21,5693–5696 (2015). [DOI] [PubMed] [Google Scholar]
- 74.Satoh Y & Obora Y Active low-valent niobium catalysts from NbCl5 and hydrosilanes for selective intermolecular cycloadditions. J. Org. Chem 76, 8569–8573 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Paradies J et al. Photogeneration of titanium(III) from titanium(IV) citrate in aqueous solution. J. Inorg. Biochem 100, 1260–1264 (2006). [DOI] [PubMed] [Google Scholar]
- 76.Chong E, Xue W, Storr T, Kennepohl P& Schafer LL Pyridonate-supported titanium(III). Benzylamine as an easy-to-use reductant. Organometallics 34, 4941–4945 (2015). [Google Scholar]
- 77.Gianetti TL et al. Diniobium inverted sandwich complexes with |j-r|6:r|6-arene ligands: synthesis, kinetics of formation, and electronic structure. J. Am. Chem. Soc 135, 3224–3236 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Plundrich GT, Wadepohl H, Clot E & Gade LH η6-arene-zirconium-PNP-pincer complexes: mechanism of their hydrogenolytic formation and their reactivity as zirconium(II) synthons. Chem. Eur. J 22, 9283–9292 (2016). [DOI] [PubMed] [Google Scholar]
- 79.Hananouchi S, Krull BT, Ziller JW, Furche F& Heyduk AF Metal effects on ligand non-innocence in group 5 complexes of the redox-active [ONO] pincer ligand. Dalton Trans 43, 17991–18000 (2014). [DOI] [PubMed] [Google Scholar]
- 80.Munha RF, Zarkesh RA & Heyduk AF Group transfer reactions of d0 transition metal complexes: redox-active ligands provide a mechanism for expanded reactivity. Dalton Trans 42, 3751–3766. (2013)This perspective shows how redox non-innocent ligands can access multiple redox states and facilitate catalytic group-transfer reactions with early transition metals.
- 81.Duan L et al. Synthesis, characterization, and reversible multielectron redox properties of a biradical yttrium complex containing bis(2-isopropylaminophenyl) amide. Eur. J. Inorg. Chem 2017, 2231–2235 (2017). [Google Scholar]
- 82.Heins SP, Wolczanski PT, Cundari TR & MacMillan SN Redox non-innocence permits catalytic nitrene carbonylation by (dadi)Ti=NAd (Ad=adamantyl). Chem. Sci 8, 3410–3418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fagan PJ, Nugent WA & Calabrese JC Metallacycle transfer from zirconium to main group elements: a versatile synthesis of heterocycles. J. Am. Chem. Soc 116, 1880–1889 (1994). [Google Scholar]
- 84.Fagan PJ, Burns EG & Calabrese JC Synthesis of boroles and their use in low-temperature Diels-Alder reactions with unactivated alkenes. J. Am. Chem. Soc 110, 2979–2981 (1988). [Google Scholar]
- 85.Fagan PJ & Nugent WA Synthesis of main group heterocycles by metallacycle transfer from zirconium. J. Am. Chem. Soc 110, 2310–2312 (1988). [Google Scholar]
- 86.Kulinkovich OG, Sviridov SV, Vasilevskii DA& Pritytskaya TS Reaction of ethylmagnesium bromide with esters of carboxylic-acid in the presence of tetraisopropoxytitanium. Zh. Org. Khim 25, 2244 (1989). [Google Scholar]
- 87.Epstein OL, Savchenko AI & Kulinkovich OG On the mechanism of titanium-catalyzed cyclopropanation of esters with aliphatic organomagnesium compounds. Deuterium distribution in the reaction products of (CD3)2CHMgBr with ethyl 3-chloropropionate in the presence of titanium tetraisopropoxide. Russ. Chem. Bull 49, 378–380 (2000). [Google Scholar]
- 88.Corey EJ, Rao SA & Noe MC Catalytic diastereoselective synthesis of cis-1,2-disubstituted cyclopropanols from esters using a vicinal dicarbanion equivalent. J. Am. Chem. Soc 116, 9345–9346 (1994). [Google Scholar]
- 89.Geis O & Schmalz H-G New developments in the Pauson-Khand reaction. Angew. Chem. Int. Ed 37, 911–914 (1998). [DOI] [PubMed] [Google Scholar]
- 90.Hicks FA & Buchwald SL Highly enantioselective catalytic Pauson-Khand type formation of bicyclic cyclopentenones. J. Am. Chem. Soc 118, 11688–11689 (1996). [Google Scholar]
- 91.Hicks FA & Buchwald SL An intramolecular titanium-catalyzed asymmetric Pauson-Khand type reaction. J. Am. Chem. Soc 121, 7026–7033 (1999). [Google Scholar]
- 92.Hicks FA, Kablaoui NM & Buchwald SL Scope of the intramolecular titanocene-catalyzed Pauson-Khand type reaction. J. Am. Chem. Soc 121 , 5881–5898 (1999). [Google Scholar]
- 93.Sturla SJ & Buchwald SL Catalytic asymmetric cyclocarbonylation of nitrogen-containing enynes. J. Org. Chem 64, 5547–5550 (1999). [DOI] [PubMed] [Google Scholar]
- 94.Kablaoui NM, Hicks FA & Buchwald SL Titanocene-catalyzed cyclocarbonylation of o-allyl aryl ketones to y-butyrolactones. J. Am. Chem. Soc 119, 4424–4431 (1997). [Google Scholar]
- 95.Campbell AD, Taylor RJK & Raynham TM The total synthesis of (−)-α-kainic acid using titanium-mediated diene metallabicyclisation methodology. Chem. Commun 0, 245–246 (1999). [Google Scholar]
- 96.Cheng X & Micalizio GC Synthesis of neurotrophic seco-prezizaane sesquiterpenes (1R,10S)-2-oxo-3, 4-dehydroneomajucin, (2S)-hydroxy-3, 4-dehydroneomajucin, and (−)-jiadifenin. J. Am.Chem. Soc 138, 1150–1153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Meng Z et al. Total synthesis and antiviral activity of indolosesquiterpenoids from the xiamycin and oridamycin families. Nat. Commun 6, 6096 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chaplinski V & de Meijere A A versatile new preparation of cyclopropylamines from acid dialkylamides. Angew. Chem. Int. Ed 35, 413–414 (1996). [Google Scholar]
- 99.Quan LG, Kim S-H, Lee JC & Cha JK Diastereoselective synthesis of trans-1,2- dialkylcyclopropanols by the Kulinkovich hydroxycyclopropanation of homoallylic alcohols. Angew. Chem. Int. Ed 41,2160–2162 (2002). [PubMed] [Google Scholar]
- 100.Kiel GR, Samkian AE, Nicolay A, Witzke RJ & Tilley TD Titanocene-mediated dinitrile coupling: a divergent route to nitrogen-containing polycyclic aromatic hydrocarbons. J. Am. Chem. Soc 140, 2450–2454 (2018).This is an example of the emerging field of nitrile couplings, demonstrating their applications in the field of polycyclic aromatics and conducting materials.
- 101.Konik YA, Kananovich DG & Kulinkovich OG Enantioselective cyclopropanation of carboxylic esters with alkyl magnesium bromides in the presence of titanium(IV) (4 R,5 R)-TADDOLates. Tetrahedron 69, 6673–6678 (2013). [Google Scholar]
- 102.Kulinkovich OG, Kananovich DG, Lopp M& Snieckus V Insight into the mechanism and stereochemistry of the transformations of alkyltitanium ate-complexes. An enhanced enantioselectivity in the cyclopropanation of the carboxylic esters with titanacyclopropane reagents. Adv. Synth. Catal 356, 3615–3626 (2014). [Google Scholar]
- 103.Barysevich MV et al. Stereoselective synthesis of a-methyl and a-alkyl ketones from esters and alkenes via cyclopropanol intermediates. Chem. Commun 54, 2800–2803 (2018). [DOI] [PubMed] [Google Scholar]
- 104.Finn PB, Derstine BP & Sieburth SM Carbocyclic amino ketones by Bredt’s rule-arrested Kulinkovich-de Meijere reaction. Angew. Chem. Int. Ed 55, 2536–2539 (2016). [DOI] [PubMed] [Google Scholar]
- 105.Fawcett FS Bredt’s rule of double bonds in atomic-bridged-ring structures. Chem. Rev 47, 219–274 (1950). [DOI] [PubMed] [Google Scholar]
- 106.Kim S-H, Park Y, Choo H & Cha JK Regio- and stereochemistry of inter- and intramolecular titanium- mediated coupling of imides and mono-substituted olefins. Tetrahedron Lett 43, 6657–6660 (2002). [Google Scholar]
- 107.Santra S, Masalov N, Epstein OL & Cha JK Diastereoselective, titanium-mediated cyclization of w-vinyl tethered imides. Org. Lett 7, 5901–5904 (2005) [DOI] [PubMed] [Google Scholar]
- 108.Lee J, Ha JD & Cha JK New synthetic method for functionalized pyrrolizidine, indolizidine, and mitomycin alkaloids. J. Am. Chem. Soc 119, 8127–8128 (1997). [Google Scholar]
- 109.Takahashi M & Micalizio GC Regio- and stereoselective cross-coupling of substituted olefins and imines. A convergent stereoselective synthesis of saturated 1,5-aminoalcohols and substituted piperidines. J. Am. Chem. Soc 129, 7514–7516 (2007).This study shows that highly regioselective and stereoselective transformations can be accomplished using alkoxide directing groups in combination with stoichiometric amounts of Ti complexes.
- 110.Hill JE, Balaich G, Fanwick PE & Rothwell IP The chemistry of titanacyclopentadiene rings supported by 2,6-diphenylphenoxide ligation: stoichiometric and catalytic reactivity. Organometallics 12, 2911–2924 (1993). [Google Scholar]
- 111.See XY et al. Generation of TiII alkyne trimerization catalysts in the absence of strong metal reductants. Organometallics 36, 1383–1390 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eisch JJ, Gitua JN, Otieno PO & Shi X Carbon-carbon bond formation via oxidative-addition processes of titanium(II) reagents with n-bonded organic substrates. Reactivity modifications by Lewis acids and Lewis bases: part 22. Organic chemistry of subvalent transition metal complexes. J. Organomet. Chem 624, 229–238 (2001). [Google Scholar]
- 113.Rassadin VA & Six Y A study of the reaction of n-BuLi with Ti(Oi-Pr)4 as a method to generate titanacyclopropane and titanacyclopropene species. Tetrahedron 70, 787–794 (2014). [Google Scholar]
- 114.Tarselli MA & Micalizio GC Aliphatic imines in titanium-mediated reductive cross-coupling: unique reactivity of Ti(O-i-Pr)4/n-BuLi. Org. Lett 11, 4596–4599 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen MZ et al. Preparation of stereodefined homoallylic amines from the reductive cross-coupling of allylic alcohols with imines. J. Org. Chem 75, 8048–8059 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yang D & Micalizio GC Convergent and stereodivergent synthesis of complex 1-aza-7- oxabicyclo[2.2.1]heptanes. J. Am. Chem. Soc 133, 9216–9219 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen MZ & Micalizio GC Three-component coupling sequence for the regiospecific synthesis of substituted pyridines. J. Am. Chem. Soc 134, 1352–1356 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Greszler SN, Reichard HA & Micalizio GC Asymmetric synthesis of dihydroindanes by convergent alkoxide-directed metallacycle-mediated bond formation. J. Am. Chem. Soc 134, 2766–2774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jeso V et al. Synthesis of angularly substituted trans-fused hydroindanes by convergent coupling of acyclic precursors. J. Am. Chem. Soc 136, 8209–8212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cheng X & Micalizio GC An annulation reaction for the synthesis of cross-conjugated triene-containing hydroindanes from acyclic precursors. Org. Lett 16, 5144–5147 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mizoguchi H & Micalizio GC Synthesis of angularly substituted trans-fused decalins through a metallacycle-mediated annulative cross-coupling cascade. Angew. Chem. Int. Ed 55, 13099–13103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mizoguchi H & Micalizio GC Synthesis of highly functionalized decalins via metallacycle-mediated cross-coupling. J. Am. Chem. Soc 137, 6624–6628 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Valadez TN, Norton JR & Neary MC Reaction of Cp*(Cl)M(diene) (M = Ti, Hf) with isonitriles. J. Am. Chem. Soc 137, 10152–10155 (2015). [DOI] [PubMed] [Google Scholar]
- 124.Hessen B, Blenkers J, Teuben JH, Helgesson G& Jagner S Carbon-carbon couplings in the reactions of unsaturated Group 4 metal s-cis-butadiene complexes with 2,6-xylyl isocyanide. Organometallics 8, 830–835 (1989). [Google Scholar]
- 125.Valadez TN, Norton JR, Neary MC & Quinlivan PJ Reaction of Cp*2Zr(2,3- dimethylbutadiene) with isonitriles and CO. Organometallics 35, 3163–3169 (2016). [Google Scholar]
- 126.Becker L, Haehnel M, Spannenberg A, Arndt P & Rosenthal U Reactions of group 4 metallocenes with monosubstituted acetonitriles: keteniminate formation versus C-C coupling. Chem. Eur. J 21, 3242–3248 (2015). [DOI] [PubMed] [Google Scholar]
- 127.Becker L, Arndt P, Jiao H, Spannenberg A& Rosenthal U Nitrile-nitrile C-C coupling at group 4 metallocenes to form 1-metalla-2,5-diaza-cyclopenta- 2,4-dienes: synthesis and reactivity. Angew. Chem. Int. Ed 52, 11396–11400 (2013). [DOI] [PubMed] [Google Scholar]
- 128.Rosenthal U Reactions of group 4 metallocene bis(trimethylsilyl)acetylene complexes with nitriles and isonitriles. Angew. Chem. Int. Ed 10.1002/anie.201805157 (2018). [DOI] [PubMed]
- 129.Rehbaum F, Thiele K-H & Trojanov SI Darstellung und molekulstruktur des (µ-alkylidenamido) titanocenkomplexes [(C5H5) 2TiCl]2[µ-{ N = C(CH2C5H5) C(CH2C6H5) = N. J. Organomet. Chem 410, 327–333 (1991). [Google Scholar]
- 130.Kiel GR, Patel SC, Smith PW, Levine DS & Tilley TD Expanded helicenes: a general synthetic strategy and remarkable supramolecular and solid-state behavior. J. Am. Chem. Soc 139, 18456–18459 (2017). [DOI] [PubMed] [Google Scholar]
- 131.Kiel GR, Ziegler MS & Tilley TD Zirconacyclopentadiene-annulated polycyclic aromatic hydrocarbons. Angew. Chem. Int. Ed 56, 4839–4844 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nitschke JR & Tilley TD Novel templating effect in the macrocyclization of functionalized diynes by zirconocene coupling. Angew. Chem. Int. Ed 40, 2142–2145 (2001). [DOI] [PubMed] [Google Scholar]
- 133.Nitschke JR, Zurcher S & Tilley TD New zirconocene-coupling route to large, functionalized macrocycles. J. Am. Chem. Soc 122, 10345–10352 (2000). [Google Scholar]
- 134.Altenburger K et al. Multiple and highly selective alkyne-isonitrile C-C and C-N couplings at group 4 metallocenes. Chem. Eur. J 22, 9169–9180 (2016). [DOI] [PubMed] [Google Scholar]
- 135.Rosenthal U, Burlakov VV, Bach MA & Beweries T Five-membered metallacycles of titanium and zirconium? Attractive compounds for organometallic chemistry and catalysis. Chem. Soc. Rev 36, 719 (2007). [DOI] [PubMed] [Google Scholar]
- 136.Beweries T et al. Reactions of decamethylhafnocene with 1,3-butadiynes: formation of hafnacyclocumulenes and C-H activation at pentamethylcyclopentadienyl ligands. Organometallics 26, 6827–6831 (2007). [Google Scholar]
- 137.Rosenthal U, Pellny P-M, Kirchbauer FG & Burlakov VV What do titano- and zirconocenes do with diynes and polyynes? Acc. Chem. Res 33, 119–129 (2000). [DOI] [PubMed] [Google Scholar]
- 138.Burlakov VV et al. The first titanacyclic five-membered cumulene. Synthesis, structure, and reactivity. Chem. Ber 128, 967–971 (1995). [Google Scholar]
- 139.Burlakov VV et al. Novel acetylene complexes of titanocene and permethyltitanocene without additional ligands. Synthesis spectral characteristics and X-ray diffraction study. J. Organomet. Chem 476, 197–206 (1994). [Google Scholar]
- 140.Rosenthal U, Burlakov VV, Arndt P, Baumann W & Spannenberg A Five-membered titana- and zirconacyclocumulenes: stable 1-metallacyclopenta- 2,3,4-trienes. Organometallics 24, 456–471.(2005) [Google Scholar]
- 141.Kaleta K et al. Reactions of group 4 metallocene alkyne complexes with carbodiimides: experimental and theoretical studies of the structure and bonding of five-membered hetero-metallacycloallenes. J. Am. Chem. Soc 133, 5463–5473 (2011). [DOI] [PubMed] [Google Scholar]
- 142.Tomaschun G et al. Group 4 metallocene complexes and cyanopyridines: coordination or coupling to metallacycles. Eur. J. Inorg. Chem 2016, 272–280 (2016) [Google Scholar]
- 143.Diekmann M et al. Chiral bis(η5:η’-pentafulvene) titanium complexes. Organometallics 25, 339–348 (2006) [Google Scholar]
- 144.Theilmann O, Saak W, Haase D & Beckhaus R Reactions of low-valent titanocene(II) fragments with trans-4,4’-azobispyridine (RN=NR, R=C5H4N): formation of tetranuclear molecular squares by trans-cis isomerization. Organometallics 28, 2799–2807 (2009). [Google Scholar]
- 145.Loose F, Schmidtmann M, Saak W & Beckhaus R Imines in the titanium coordination sphere: highly reactive titanaaziridines and larger titanacycles formed by subsequent C-C coupling reactions. Eur. J. Inorg. Chem 2015, 5171–5187 (2015). [Google Scholar]
- 146.Jaroschik F et al. Synthesis, characterization and reactivity of formal 20 electron zirconocene-pentafulvene complexes. Organometallics 36, 2004–2013 (2017). [Google Scholar]
- 147.Manβen M, Lauterbach N, Woriescheck T, Schmidtmann M & Beckhaus R Reactions of secondary amines with bis(η|5:η|’-pentafulvene)titanium complexes: formation of titanium amides and titanaaziridines. Organometallics 36, 867–876 (2017). [Google Scholar]
- 148.Fischer M, Schaper R, Jaugstetter M, Schmidtmann M & Beckhaus R Electrophilic d0 cations of group 4 metals (M=Ti, Zr, Hf) derived from monopentafulvene complexes: direct formation of tridentate Cp, O, P-ligands. Organometallics 37, 1192–1205 (2018). [Google Scholar]
- 149.Eisch JJ, Owuor FA & Shi X Novel synthesis of unbridged, sterically substituted zirconocene dichlorides from fulvenes and dialkylzirconium dichlorides via zirconium(IV) hydride transfer. Organometallics 18, 1583–1585 (1999). [Google Scholar]
- 150.Eisch JJ, Shi X & Owuor FA Novel synthesis of ansa-metallocenes via the reductive dimerization of fulvenes with group 4 metal divalent halides. Organometallics 17, 5219–5221 (1998). [Google Scholar]
- 151.Parker KDJ & Fryzuk MD Carbon-carbon bond forming reactions with tantalum diamidophosphine complexes that incorporate alkyne ligands. Organometallics 33, 6122–6131 (2014). [Google Scholar]
- 152.Ziegler JA, Buckley HL & Arnold J Synthesis and reactivity of tantalum corrole complexes. Dalton Trans 46, 780–785 (2017). [DOI] [PubMed] [Google Scholar]
- 153.Yamamoto K, Tsurugi H & Mashima K Direct evidence for a [4 + 2] cycloaddition mechanism of alkynes to tantallacyclopentadiene on dinuclear tantalum complexes as a model of alkyne cyclotrimerization. Chem. Eur. J 21, 11369–11377 (2015). [DOI] [PubMed] [Google Scholar]
- 154.Yamamoto K, Nagae H, Tsurugi H & Mashima K Mechanistic understanding of alkyne cyclotrimerization on mononuclear and dinuclear scaffolds: [4 + 2] cycloaddition of the third alkyne onto metallacyclopentadienes and dimetallacyclopentadienes. Dalton Trans 45, 17072–17081 (2016). [DOI] [PubMed] [Google Scholar]
- 155.Obora Y, Takeshita K & Ishii Y NbCl3-catalyzed [2 + 2 + 2] intermolecular cycloaddition of alkynes and alkenes to 1,3-cyclohexadiene derivatives. Org.Biomol. Chem 7, 428–431 (2009). [DOI] [PubMed] [Google Scholar]
- 156.Obora Y, Satoh Y & Ishii Y NbC^-catalyzed intermolecular [2 + 2 + 2] cycloaddition of alkynes and a, w-dienes: highly chemo- and regioselective formation of 5-w-alkenyl-1,4-substituted-1,3- cyclohexadiene derivatives. J. Org. Chem 75, 6046–6049 (2010). [DOI] [PubMed] [Google Scholar]
- 157.Satoh Y & Obora Y NbCl3-catalyzed three-component [2 + 2 + 2] cycloaddition reaction of terminal alkynes, internal alkynes, and alkenes to 3,4,5-tetrasubstituted 1,3-cyclohexadienes. Org. Lett 13, 2568–2571 (2011). [DOI] [PubMed] [Google Scholar]
- 158.Satoh Y & Obora Y Low-valent niobium-catalyzed intermolecular [2 + 2 + 2] cycloaddition of tert-butylacetylene and arylnitriles to form 2,3,6-trisubstituted pyridine derivatives. J. Org. Chem 78, 7771–7776 (2013). [DOI] [PubMed] [Google Scholar]
- 159.Satoh Y & Obora Y Niobium complexes in organic transformations: from stoichiometric reactions to catalytic [2 + 2 + 2] cycloaddition reactions. Eur. J. Org. Chem 2015, 5041–5054 (2015). [Google Scholar]
- 160.LaPointe RE, Wolczanski PT & Mitchell JF Carbon monoxide cleavage by (silox)3 Ta (silox=t-Bu3SiO-). J. Am. Chem. Soc 108, 6382–6384 (1986). [Google Scholar]
- 161.LaPointe RE & Wolczanski PT Silox hydrides(silox=t-Bu3SiO-) of group 5: do [(silox)2MH2]2 (M=Nb, Ta) complexes contain unbridged M-M bonds? J. Am. Chem. Soc 108, 3535–3537 (1986). [Google Scholar]
- 162.Steffey BD et al. Intramolecular arene hydrogenation by niobium aryloxide compounds: stereochemistry of cyclohexadiene formation. J. Am. Chem. Soc 111,378–380 (1989). [Google Scholar]
- 163.Chesnut RW, Steffey BD & Rothwell IP The metallation and hydrogenation of aryl rings in early transition metal aryloxide systems. Polyhedron 8, 1607–1610 (1989). [Google Scholar]
- 164.Bell RA, Cohen SA, Doherty NM, Threlkel RS & Bercaw JE Borohydride, hydride, halide, and carbonyl derivatives of bis(pentamethylcyclopentadienyl)niobium. Organometallics 5, 972–975 (1986). [Google Scholar]
- 165.Parkin G et al. Alpha- and beta-migratory insertion and elimination processes for alkyl complexes of permethylscandocene and permethyltantalocene. J. Mol. Catal 41, 21–39 (1987). [Google Scholar]
- 166.Millar SP, Zubris DL, Bercaw JE & Eisenberg R On the mechanism of dihydrogen addition to tantalocene complexes. J. Am. Chem. Soc 120, 5329–5330 (1998). [Google Scholar]
- 167.Tebbe FN & Parshall GW Hydride derivatives of niobocene and tantalocene. J. Am. Chem. Soc 93, 3793–3795 (1971). [Google Scholar]
- 168.Ankianiec BC, Fanwick PE & Rothwell IP Isolation of a new series of seven-coordinate hydride compounds of tantalum(V) and their involvement in the catalytic hydrogenation of arene rings. J. Am.Chem. Soc 113, 4710–4712 (1991). [Google Scholar]
- 169.Parkin BC et al. Synthesis and characterization of a series of mononuclear tantalum(V) hydride compounds containing aryloxide ligation. Organometallics 14, 3002–3013 (1995). [Google Scholar]
- 170.Yu JS, Ankianiec BC, Nguyen MT & Rothwell IP All-cis catalytic hydrogenation of polynuclear aromatic hydrocarbons by group 5 metal aryloxide compounds. J. Am. Chem. Soc 114, 1927–1929 (1992). [Google Scholar]
- 171.Visciglio VM et al. Coordination and hydrogenation of 1,3-cyclohexadiene by niobium and tantalum aryl oxide compounds: relevance to catalytic arene hydrogenation. J. Am. Chem. Soc 119, 3490–3499 (1997). [Google Scholar]
- 172.Parker KG, Noll B, Pierpont CG & Dubois MR Syntheses of tantalum(V) complexes containing tetramethylpyrrolyl, pyrrolyl, and indolyl ligands. Inorg. Chem 35, 3228–3234 (1996). [DOI] [PubMed] [Google Scholar]
- 173.Fryzuk MD, Kozak CM, Bowdridge MR & Patrick BO Cyclohexadienyl niobium complexes and arene hydrogenation catalysis. Organometallics 21, 5047–5054 (2002). [Google Scholar]
- 174.Clark JR, Fanwick PE & Rothwell IP Aryloxide ligand dependent reactivity of tantalum dihydride compounds with alkenes. J. Chem. Soc. Chem. Commun 553–554 (1995).
- 175.Mulford DR, Clark JR, Schweiger SW,Fanwick PE & Rothwell IP Reactions of alkynes and olefins with tantalum hydrides containing aryloxide ancillary ligation: relevance to catalytic hydrogenation. Organometallics 18, 4448–4458 (1999). [Google Scholar]
- 176.Gianetti TL, Tomson NC, Arnold J & Bergman RG Z-selective, catalytic internal alkyne semihydrogenation under H2/CO mixtures by a niobium(III) imido complex. J. Am. Chem. Soc 133, 14904–14907 (2011). [DOI] [PubMed] [Google Scholar]
- 177.Gianetti TL, Bergman RG & Arnold J Stoichiometric carbon-carbon bond formation mediated by well defined Nb(III) complexes. Polyhedron 84, 19–23 (2014). [Google Scholar]
- 178.Gianetti TL, La Pierre HS & Arnold J Group 5 imides and bis(imide)s as selective hydrogenation catalysts. Eur. J. Inorg. Chem 2013, 3771–3783 (2013). [Google Scholar]
- 179.Tomson NC, Arnold J & Bergman RG Synthesis, characterization, and reactions of isolable (beta-diketiminato)niobium(III) imido complexes. Organometallics 29, 5010–5025 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Brintzinger HH & Bercaw JE The nature of so-called titanocene, (C10H10Ti)2. J. Am. Chem. Soc 92, 6182–6185 (1970). [Google Scholar]
- 181.Figueroa JS, Piro NA, Mindiola DJ, Fickes MG & Cummins CC Niobaziridine hydrides. Organometallics 29, 5215–5229 (2010). [Google Scholar]
- 182.Rankin MA & Cummins CC Carbon dioxide reduction by terminal tantalum hydrides: formation and isolation of bridging methylene diolate complexes. J. Am. Chem. Soc 132, 10021–10023 (2010). [DOI] [PubMed] [Google Scholar]
- 183.Berno P & Gambarotta S Formation of a metallaaziridine ring and dinitrogen fixation promoted by a niobium amide complex. Organometallics 14, 2159–2161 (1995). [Google Scholar]
- 184.Curtis MD, Bell LG & Butler WM C-H activation. Synthesis of silyl derivatives of niobocene and tantalocene hydrides, their H/D exchange reactions with C6D6, and the structure of Cp2Ta(H)2SiMe2Ph. Organometallics 4, 701–707 (1985). [Google Scholar]
- 185.Kriegel BM, Bergman RG & Arnold J Generation of low-valent tantalum species by reversible C-H activation in a cyclometallated tantalum hydride complex. Dalton Trans 43, 10046–10056 (2014). [DOI] [PubMed] [Google Scholar]
- 186.Ballard KR, Gardiner IM & Wigley DE Evidence for intramolecular C-H bond activation and the formation of a “tucked-in” complex of hexamethylbenzene at a tantalum(III) center. J. Am. Chem. Soc 111, 2159–2162 (1989). [Google Scholar]
- 187.Bercaw JE, Marvich RH, Bell LG &Brintzinger HH Titanocene as an intermediate in reactions involving molecular hydrogen and nitrogen. J. Am. Chem. Soc 94, 1219–1238 (1972). [Google Scholar]
- 188.Yu JS, Fanwick PE & Rothwell IP Intramolecular alkane dehydrogenation and functionalization at niobium metal centers. J. Am. Chem. Soc 11, 8171–8172 (1990). [Google Scholar]
- 189.Riley PN, Clark JR, Fanwick PE & Rothwell IP Synthesis and structure of niobium and tantalum derivatives of bis(dicyclohexylphosphino)methane (dcpm). Inorg. Chim. Acta 288, 35–39 (1999). [Google Scholar]
- 190.Yu JS et al. Intramolecular dehydrogenation of alkyl groups at niobium aryloxide centers: bonding and reactivity of the ensuing niobacyclopropane ring. Organometallics 15, 4443–4449 (1996). [Google Scholar]
- 191.Steffey BD et al. Intramolecular activation of aliphatic and aromatic carbon-hydrogen bonds by tantalum(III) metal centers: synthesis and structure of the bis-metalated compounds Ta(OC6H3ButCMe2CH2)2Cl and Ta(OC6H3PhC6H4)2 (OAr-2,6-Ph2) (OAr-2,6-Ph2=2,6-diphenylphenoxide). Organometallics 8, 1419–1423 (1989). [Google Scholar]
- 192.Mindiola DJ & Cummins CC Probing the niobium metallaaziridine functionality. Organometallics 20, 3626–3628 (2001). [Google Scholar]
- 193.Figueroa JS & Cummins CC The niobaziridine-hydride functional group: synthesis and divergent reactivity. J. Am. Chem. Soc 125, 4020–4021 (2003). [DOI] [PubMed] [Google Scholar]
- 194.Figueroa JS & Cummins CC A niobaziridine hydride system for white phosphorus or dinitrogen activation and N- or P-atom transfer. Dalton Trans 35, 2161–2168 (2006). [DOI] [PubMed] [Google Scholar]
- 195.Aguilar-Calderon JR, Metta-Magana AJ, Noll B & Fortier S C(sp3)-H oxidative addition and transfer hydrogenation chemistry of a titanium(II) synthon: mimicry of late-metal type reactivity. Angew. Chem. Int. Ed 55, 14101–14105 (2016). [DOI] [PubMed] [Google Scholar]
- 196.Erickson KA, Stelmach JPW, Mucha NT & Waterman R Zirconium-catalyzed amine borane dehydrocoupling and transfer hydrogenation. Organometallics 34, 4693–4699 (2015). [Google Scholar]
- 197.Tayebani M, Feghali K, Gambarotta S & Yap G Molecular rearrangements of a low-valent niobium amide: ligand C-H bond oxidative addition and reductive elimination. Organometallics 17, 4282–4290 (1998). [Google Scholar]
- 198.Shaver MP & Fryzuk MD Phosphine- induced ancillary ligand orthometalation at a tantalum-tantalum double bond. Organometallics 24, 2606–2601 (2005). [Google Scholar]
- 199.Wolczanski PT Activation of carbon-hydrogen bonds via 1,2-RH-addition/-elimination to early transition metal imides. Organometallics 37, 505–516 (2018). [Google Scholar]
- 200.Bailey BC, Fan H, Huffman JC, Baik M-H & Mindiola DJ Intermolecular C-H bond activation reactions promoted by transient titanium alkylidynes. Synthesis, reactivity, kinetic, and theoretical studies of the Ti=C linkage. J. Am. Chem. Soc 129, 8781–8793. [DOI] [PubMed] [Google Scholar]
- 201.Bailey BC et al. Intermolecular C-H bond activation promoted by a titanium alkylidyne. J. Am. Chem. Soc 127, 16016–16017 (2005). [DOI] [PubMed] [Google Scholar]
- 202.Solowey DP et al. A new and selective cycle for dehydrogenation of linear and cyclic alkanes under mild conditions using a base metal. Nat. Chem 9, 1126–1132 (2017).In this study, the mechanism for dehydrogenation is proposed to avoid metal hydride intermediates, resulting in high terminal selectivity, as the terminal alkenes cannot isomerize.
- 203.Hickey AK et al. Dehydrogenation of hydrocarbons with metal-carbon multiple bonds and trapping of a titanium(II) intermediate. Dalton Trans 43, 9834–9837 (2014). [DOI] [PubMed] [Google Scholar]
- 204.Kamitani M et al. Phosphinoalkylidene and -alkylidyne complexes of titanium: intermolecular C-H bond activation and dehydrogenation reactions. J. Am. Chem. Soc 137, 11872–11875 (2015). [DOI] [PubMed] [Google Scholar]
- 205.Clark TJ, Russell CA & Manners I Homogeneous, titanocene-catalyzed dehydrocoupling of amine-borane adducts. J. Am. Chem. Soc 128, 9582–9583 (2006). [DOI] [PubMed] [Google Scholar]
- 206.Pun D, Lobkovsky E & Chirik PJ Amineborane dehydrogenation promoted by isolable zirconium sandwich, titanium sandwich and N2 complexes. Chem. Commun 43, 3297–3299 (2007). [DOI] [PubMed] [Google Scholar]
- 207.Sloan ME et al. Homogeneous catalytic dehydrocoupling/dehydrogenation of amine-borane adducts by early transition metal, group 4 metallocene complexes. J. Am. Chem. Soc 132, 3831–3841 (2010). [DOI] [PubMed] [Google Scholar]
- 208.Beweries T, Hansen S, Kessler M, Klahn M& Rosenthal U Catalytic dehydrogenation of dimethylamine borane by group 4 metallocene alkyne complexes and homoleptic amido compounds. Dalton Trans 40, 7689–7692 (2011). [DOI] [PubMed] [Google Scholar]
- 209.Beweries T et al. Catalytic and kinetic studies of the dehydrogenation of dimethylamine borane with an iPr substituted titanocene catalyst. ChemCatChem 3, 1865–1868 (2011). [Google Scholar]
- 210.Thomas J, Klahn M, Spannenberg A & Beweries T Group 4 metallocene catalysed full dehydrogenation of hydrazine borane. Dalton Trans 42, 14668–14672 (2013). [DOI] [PubMed] [Google Scholar]
- 211.Jurca T et al. Step-growth titanium-catalysed dehydropolymerisation of amine-boranes. Chem. Sci 9, 3360–3366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Luo Y & Ohno K Computational study of titanocene-catalyzed dehydrocoupling of the adduct Me2NH-BH3: an intramolecular, stepwise mechanism. Organometallics 26, 3597–3600 (2007). [Google Scholar]
- 213.Lummis PA, McDonald R, Ferguson MJ &Rivard E Synthesis, characterisation, and dehydrocoupling ability of zirconium complexes bearing hindered bis(amido)silyl ligands. Dalton Trans 44, 7009–7020 (2015). [DOI] [PubMed] [Google Scholar]
- 214.Helten H et al. Paramagnetic titanium(III) and zirconium(III) metallocene complexes as precatalysts for the dehydrocoupling/dehydrogenation of amine-boranes. Angew. Chem. Int. Ed 52, 437–440 (2013). [DOI] [PubMed] [Google Scholar]
- 215.Richmond TG in Topics in Organometallic Chemistry Vol. 3 (eds Murai S et al.) 243–269 (1999). [Google Scholar]
- 216.Blanksby SJ & Ellison GB Bond dissociation energies of organic molecules. Acc. Chem. Res 36, 255–263 (2003). [DOI] [PubMed] [Google Scholar]
- 217.Giesy JP & Kannan K Global distribution of perfluorooctane sulfonate in wildlife. Environ. Sci. Techno} 35, 1339–1342 (2001). [DOI] [PubMed] [Google Scholar]
- 218.de Boer J, Wester PG, Klamer HJC, Lewis WE & Boon JP Do flame retardants threaten ocean life? Nature 394, 28–29 (1998). [DOI] [PubMed] [Google Scholar]
- 219.Schine KP & Sturges WT CO2 is not the only gas. Science 315, 1804–1805 (2007). [DOI] [PubMed] [Google Scholar]
- 220.Perutz RN A catalytic foothold for fluorocarbon reactions. Science 321, 1168–1169 (2008). [DOI] [PubMed] [Google Scholar]
- 221.Kiplinger JL, Richmond TG & Osterberg CE Activation of carbon-fluorine bonds by metal complexes. Chem. Rev 94, 373–431 (1994). [Google Scholar]
- 222.Richmond TG Organometallic transformations demonstrate that fluorocarbons are reactive molecules. Angew. Chem. Int. Ed 39, 3241–3244 (2000). [PubMed] [Google Scholar]
- 223.Klahn M & Rosenthal U An update on recent stoichiometric and catalytic C-F bond cleavage reactions by lanthanide and group 4 transition-metal complexes. Organometallics 31, 1235–1244 (2012). [Google Scholar]
- 224.Driver TG Niobium-catalyzed activation of aryl trifluoromethyl groups and functionalization of C-H bonds: an efficient and convergent approach to the synthesis of N-heterocycles. Angew. Chem. Int. Ed 48, 7974–7976 (2009). [DOI] [PubMed] [Google Scholar]
- 225.Amii H & Uneyama KC-F Bond activation in organic synthesis. Chem. Rev 109, 2119–2183 (2009). [DOI] [PubMed] [Google Scholar]
- 226.Clot E et al. C-F and C-H bond activation of fluorobenzenes and fluoropyridines at transition metal centers: how fluorine tips the scales. Acc. Chem. Res 44, 333–348 (2011). [DOI] [PubMed] [Google Scholar]
- 227.Kuehnel MF, Lentz D & Braun T Synthesis of fluorinated building blocks by transition-metal- mediated hydrodefluorination reactions. Angew. Chem. Int. Ed 52, 3328–3348 (2013). [DOI] [PubMed] [Google Scholar]
- 228.Ahrens T, Kohlmann J, Ahrens M & Braun T Functionalization of fluorinated molecules by transition-metal-mediated C-F bond activation to access fluorinated building blocks. Chem. Rev 115, 931–972 (2015). [DOI] [PubMed] [Google Scholar]
- 229.Eisenstein O, Milani J & Perutz RN Selectivity of C-H activation and competition between C-H and C-F bond activation at fluorocarbons. Chem. Rev 117, 8710–8753 (2017). [DOI] [PubMed] [Google Scholar]
- 230.Bouwkamp MW et al. Structure of the decamethyl titanocene cation, a metallocene with two agostic C-H bonds, and its interaction with fluorocarbons. J. Am. Chem. Soc 124, 12956–12957 (2002). [DOI] [PubMed] [Google Scholar]
- 231.Piglosiewicz IM, Kraft S, Beckhaus R, Haase D & Saak W Selective C-H and C-F bond activation reactions of pyridine and fluoropyridines — formation of binuclear µ-X titanocene complexes (X = H, F) with a-functionalized N-heterocycles. Eur. J. Inorg. Chem 2005, 938–945 (2005). [Google Scholar]
- 232.Burk MJ, Staley DL & Tumas W Oxidatively induced reductive elimination. A novel titanium complex resulting from C-F bond activation. J. Chem. Soc. Chem. Commun 809–810 (1990).
- 233.Rosenthal U et al. Bis(trimethylsilyl)acetylene complexes of titanocenes and zirconocenes: their recent chemistry and reactions with Lewis acids. Eur. J. Inorg. Chem 2004, 4739–4749 (2004). [Google Scholar]
- 234.Kiplinger JL & Richmond TG Selective room temperature hydrogenolysis of aromatic fluorocarbons mediated by a low-valent zirconium complex. Chem. Commun 1115–1116 (1996).
- 235.Fujiwara M, Ichikawa J, Okauchi T & Minami T Vinylic C-F bond activation with low-valent zirconocene: the generation and cross-coupling reactions of 1-fluorovinylzirconocene. Tetrahedron Lett 40, 7261–7265 (1999). [Google Scholar]
- 236.O’Connor PE, Berg DJ & Barclay T Photolytic zirconium benzyl bond cleavage and subsequent aryl C-F activation in zirconium complexes of fluorinated aryl diamides. Organometallics 21,3947–3954 (2002). [Google Scholar]
- 237.Jones WD Activation of C-F bonds using Cp*2ZrH2: a diversity of mechanisms. Dalton Trans 2003, 3991–3995 (2003). [Google Scholar]
- 238.Jager-Fiedler U et al. Reactions of zirconocene bis(trimethylsilyl)acetylene complexes with fluorinated pyridines: C-H versus C-F bond activation. Eur. J. Inorg. Chem 2005, 2842–2849 (2005). [Google Scholar]
- 239.Edelbach BL, Kraft BM & Jones WD Generation of perfluoropolyphenylene oligomers via carbon- fluorine bond activation by Cp2Zr(C6F5)2: a dual mechanism involving a radical chain and release of tetrafluorobenzyne. J. Am. Chem. Soc 121, 10327–10331 (1999). [Google Scholar]
- 240.Rieth RD, Brennessel WW & Jones WD Activation of aromatic, aliphatic, and olefinic carbon-fluorine bonds using Cp*2HfH2. Eur. J. Inorg. Chem 2007, 2839–2847 (2007). [Google Scholar]
- 241.Fuchibe K & Akiyama T Low-valentniobium-mediated double activation of C-F/C-H bonds: fluorene synthesis from o-arylated a, a, a-trifluorotoluene derivatives. J. Am. Chem. Soc 128, 1434–1435 (2006). [DOI] [PubMed] [Google Scholar]
- 242.Gianetti TL, Bergman RG & Arnold J Disassembly of a benzylic CF3 group mediated by a niobium(III) imido complex. J. Am. Chem. Soc 135, 8145–8148 (2013). [DOI] [PubMed] [Google Scholar]
- 243.Nechayev M, Gianetti TL, Bergman RG& Arnold J C-F sp2 bond functionalization mediated by niobium complexes. Dalton Trans 44, 19494–19500 (2015). [DOI] [PubMed] [Google Scholar]
- 244.Huang W & Diaconescu PL Aromatic C-F bond activation by rare-earth-metal complexes. Organometallics 36, 89–96 (2017). [Google Scholar]
- 245.Simoes JAM & Beauchamp JL Transition metal- hydrogen and metal-carbon bond strengths: the keys to catalysis. Chem. Rev 90, 629–688 (1990). [Google Scholar]
- 246.Kiplinger JL & Richmond TG Group IV metallocene-mediated synthesis of fluoroaromatics via selective defluorination of saturated perfluorocarbons. J. Am. Chem. Soc 118, 1805–1806 (1996). [Google Scholar]
- 247.Kraft BM, Lachicotte RJ & Jones WD Aliphatic carbon-fluorine bond activation using (C5Me5)2ZrH2. J. Am. Chem. Soc 122, 8559–8560 (2000). [DOI] [PubMed] [Google Scholar]
- 248.Kraft BM, Lachicotte RJ & Jones WD Aliphatic and aromatic carbon-fluorine bond activation with Cp*2ZrH2: mechanisms of hydrodefluorination. J. Am. Chem. Soc 123, 10973–10979 (2001). [DOI] [PubMed] [Google Scholar]
- 249.Fuchibe K, Ohshima Y, Mitomi K & Akiyama T Low-valent niobium-catalyzed reduction of a, a, a-trifluorotoluenes. Org. Lett 9, 1497–1499 (2007). [DOI] [PubMed] [Google Scholar]
- 250.Fuchibe K, Mitomi K, Suzuki R & Akiyama T C-C coupling reactions of superstrong CF3 groups with C(sp2)-H bonds: reactivity and synthetic utility of zero-valent niobium catalyst. Chem. Asian J 3, 261–271 (2008). [DOI] [PubMed] [Google Scholar]
- 251.Gianetti TL, Bergman RG & Arnold J Carbon-fluorine bond cleavage in fluoroarenes via a niobium(III) imido complex: from stoichiometric to catalytic hydrodefluorination. Chem. Sci 5, 2517–2524 (2014).This study shows that the well-characterized (BDI)Nb platform allows for a more detailed understanding of the mechanism of Nb-catalysed hydrodefluorination.
- 252.Barrero AF et al. Couplings of benzylic halides mediated by titanocene chloride: synthesis of bibenzyl derivatives. J. Org. Chem 72, 2251–2254 (2007). [DOI] [PubMed] [Google Scholar]
- 253.Cha JY, Yeoman JTS & Reisman SE A concise total synthesis of (−)-maoecrystal Z. J. Am. Chem. Soc 133, 14964–14967 (2011). [DOI] [PubMed] [Google Scholar]
- 254.Plesniak MP, Huang H-M & Procter DJ Radical cascade reactions triggered by single electron transfer. Nat. Rev. Chem 1, 0077 (2017). [Google Scholar]
- 255.Castro Rodriguez M et al. Cp2TiCl: an ideal reagent for green chemistry? Org. Process Res. Dev 21, 911–923 (2017). [Google Scholar]
- 256.Richrath RB et al. Cp2TiX complexes for sustainable catalysis in single-electron steps. Chem. Eur. J 24, 6371–6379 (2018). [DOI] [PubMed] [Google Scholar]
- 257.Morcillo SP et al. Recent applications of Cp2TiCl in natural product synthesis. Org. Chem. Front 1 ,15–33 (2014). [Google Scholar]
- 258.Enem^rke RJ, Larsen J, Skrydstrup T & Daasbjerg K Revelation of the nature of the reducing species in titanocene halide-promoted reductions. J. Am. Chem. Soc 126, 7853–7864 (2004). [DOI] [PubMed] [Google Scholar]
- 259.Gansauer A et al. Cationic titanocene(III) complexes for catalysis in single-electron steps. Angew. Chem. Int. Ed 54, 7003–7006 (2015). [DOI] [PubMed] [Google Scholar]
- 260.Friedrich J et al. Titanocene catalyzed 4-exo cyclizations: mechanism, experiment, catalyst design. J. Am. Chem. Soc 130, 1788–1796 (2008). [DOI] [PubMed] [Google Scholar]
- 261.Gansauer A, Shi L & Otte M Catalyticenantioselective radical cyclization via regiodivergent epoxide opening. J. Am. Chem. Soc 132, 11858–11859 (2010). [DOI] [PubMed] [Google Scholar]
- 262.Hildebrandt S & Gansauer A Synthesis of dihydropyrrolizine and tetrahydroindolizine scaffolds from pyrroles by titanocene(III) catalysis. Angew.Chem. Int. Ed 55, 9719–9722 (2016). [DOI] [PubMed] [Google Scholar]
- 263.Gansauer A, Lauterbach T, Bluhm H &Noltemeyer M A catalytic enantioselective electron transfer reaction: titanocene-catalyzed enantioselective formation of radicals from meso-epoxides. Angew. Chem. Int. Ed 38, 2909–2910 (1999). [DOI] [PubMed] [Google Scholar]
- 264.Streuff J A titanium(III)-catalyzed redox umpolung reaction for the reductive cross-coupling of enones with acrylonitriles. Chem. Eur. J 17, 5507–5510 (2011). [DOI] [PubMed] [Google Scholar]
- 265.Bichovski P, Haas TM, Keller M & Streuff J Direct conjugate alkylation of a, p-unsaturated carbonyls by TiIII-catalysed reductive umpolung of simple activated alkenes. Org. Biomol. Chem 14, 5673–5682 (2016). [DOI] [PubMed] [Google Scholar]
- 266.Leijendekker LH, Weweler J, Leuther TM & Streuff J Catalytic reductive synthesis and direct derivatization of unprotected aminoindoles, aminopyrroles, and iminoindolines. Angew. Chem. Int. Ed 56, 6103–6106 (2017). [DOI] [PubMed] [Google Scholar]
- 267.Streuff J, Feurer M, Bichovski P, Frey G &Gellrich U Enantioselective titanium(III)-catalyzed reductive cyclization of ketonitriles. Angew. Chem. Int. Ed 51, 8661–8664 (2012). [DOI] [PubMed] [Google Scholar]
- 268.Frey G, Luu H-T, Bichovski P, Feurer M & Streuff J Convenient titanium(III)-catalyzed synthesis of cyclic aminoketones and pyrrolidinones-development of a formal [4 + 1] cycloaddition. Angew. Chem. Int. Ed 52, 7131–7134 (2013). [DOI] [PubMed] [Google Scholar]
- 269.Feurer M, Frey G, Luu H-T, Kratzert D& Streuff J The cross-selective titanium(III) catalysed acyloin reaction. Chem. Commun 50, 5370–5372 (2014) [DOI] [PubMed] [Google Scholar]
- 270.Streuff J Reductive umpolung reactions with low-valent titanium catalysts. Chem. Rec 14, 1100–1113 (2014). [DOI] [PubMed] [Google Scholar]
- 271.Bichovski P, Haas TM, Kratzert D & Streuff J Synthesis of bridged benzazocines and benzoxocines by a titanium-catalyzed double-reductive umpolung strategy. Chem. Eur. J 21,2339–2342 (2015). [DOI] [PubMed] [Google Scholar]
- 272.Luu H-T, Wiesler S, Frey G & Streuff J A titanium(III)- catalyzed reductive umpolung reaction for the synthesis of1,1-disubstituted tetrahydroisoquinolines. Org. Lett 17, 2478–2481 (2015). [DOI] [PubMed] [Google Scholar]
- 273.Streuff J & Gansauer A Metal-catalyzed β-functionalization of Michael acceptors through reductive radical addition reactions. Angew. Chem. Int. Ed 54, 14232–14242 (2015).This review highlights strategies for accessing catalytic single-electron chemistry with early transition metal complexes and their subsequent applications in Michael-type chemistry.
- 274.Streuff J et al. Mechanism of the TiIII-catalyzed acyloin-type umpolung: a catalyst-controlled radical reaction. J. Am. Chem. Soc 137, 14396–14405 (2015)This report details a bimolecular mechanism for the acyloin-type umpolung reaction that avoids free radical intermediates from open-shell catalysts.
- 275.Gansauer A & Hildebrandt S in Topics in Heterocyclic Chemistry Vol. 54 (ed. Landais Y) 253–283 (Springer, 2018). [Google Scholar]
- 276.Zheng X et al. Umpolung of hemiaminals: titanocene-catalyzed dehydroxylative radical coupling reactions with activated alkenes. Angew. Chem. Int. Ed 52, 3494–3498 (2013). [DOI] [PubMed] [Google Scholar]
- 277.Estevez RE et al. Stereocontrolled coupling between aldehydes and conjugated alkenals mediated by TiIII/H2O. Org. Lett 8, 5433–5436 (2006). [DOI] [PubMed] [Google Scholar]
- 278.Kosal AD & Ashfeld BL Titanocene-catalyzed conjugate reduction of a, p-unsaturated carbonyl derivatives. Org. Lett 12, 44–47 (2010). [DOI] [PubMed] [Google Scholar]
- 279.Hao W et al. Radical redox-relay catalysis: formal [3+2] cycloaddition of N-acylaziridines and alkenes. J. Am. Chem. Soc 139, 12141–12144 (2016) [DOI] [PubMed] [Google Scholar]
- 280.Zhang Y-Q, Vogelsang E, Qu Z-W, Grimme S & Gansauer A Titanocene-catalyzed radical opening of N-acylated aziridines. Angew. Chem. Int. Ed 56, 12654–12657 (2017). [DOI] [PubMed] [Google Scholar]
- 281.Hao W, Harenberg JH, Wu X, MacMillan SN & Lin S Diastereo- and enantioselective formal [3 + 2] cycloaddition of cyclopropyl ketones and alkenes via Ti-catalyzed radical redox relay. J. Am. Chem. Soc 140, 3514–3517 (2018). [DOI] [PubMed] [Google Scholar]
- 282.Alvarado J, Herrmann AT & Zakarian A Stereoselective a-fluorination of N-acyloxazolidinones at room temperature within 1 h. J. Org. Chem 79, 6206–6220 (2014). [DOI] [PubMed] [Google Scholar]
- 283.Mabe PJ & Zakarian A Asymmetric radical addition of TEMPO to titanium enolates. Org. Lett 16, 516–519 (2014). [DOI] [PubMed] [Google Scholar]
- 284.Herrmann AT, Smith LL & Zakarian A A simple method for asymmetric trifluoromethylation of N-acyl oxazolidinones via Ru-catalyzed radical addition to zirconium enolates. J. Am. Chem. Soc 134, 6976–6979 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Gu Z, Herrmann AT & Zakarian A Dual Ti-Ru catalysis in the direct radical haloalkylation of N-acyl oxazolidinones. Angew. Chem. Int. Ed 50, 7136–7139 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Ciez D, Patasz A & Trzewik B Titanium enolate chemistry at the beginning of the 21st century. Eur. J. Org. Chem 2016, 1476–1493 (2016). [Google Scholar]
- 287.Beaumont S, Ilardi EA, Monroe LR & Zakarian A Valence tautomerism in titanium enolates: catalytic radical haloalkylation and application in the total synthesis of heodysidenin. J. Am. Chem. Soc 132, 1482–1483 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Moreira I & de PR et al. Unconventional biradical character of titanium enolates. J. Am. Chem. Soc 130, 3242–3243 (2008). [DOI] [PubMed] [Google Scholar]
- 289.Dieguez HR et al. Weakening C-O bonds: Ti(III), a new reagent for alcohol deoxygenation and carbonyl coupling olefination. J. Am. Chem. Soc 132, 254–259 (2010). [DOI] [PubMed] [Google Scholar]
- 290.Pappas I & Chirik PJ Ammonia synthesis by hydrogenolysis of titanium-nitrogen bonds using proton coupled electron transfer. J. Am. Chem. Soc 137, 3498–3501 (2015). [DOI] [PubMed] [Google Scholar]
- 291.Pappas I & Chirik PJ Catalytic proton coupled electron transfer from metal hydrides to titanocene amides, hydrazides and imides: determination of thermodynamic parameters relevant to nitrogen fixation. J. Am. Chem. Soc 138, 13379–13389 (2015)This report highlights the large impact that coordination to an early transition metal can have on the N-H BDFE in NH3.
- 292.Cuerva JM et al. Water: the ideal hydrogen-atom source in free-radical chemistry mediated by Tilll and other single-electron-transfer metals? Angew. Chem. Int. Ed 45, 5522–5526 (2006). [DOI] [PubMed] [Google Scholar]
- 293.Paradas M et al. Understanding the exceptional hydrogen-atom donor characteristics of water in TiIII-mediated free-radical chemistry. J. Am. Chem. Soc 132, 12748–12756 (2010). [DOI] [PubMed] [Google Scholar]
- 294.Gansauer A et al. H2O activation for hydrogen-atom transfer: correct structures and revised mechanisms. Angew. Chem. Int. Ed 51,3266–3270 (2012). [DOI] [PubMed] [Google Scholar]
- 295.Kessler M et al. Photoassisted Ti-O activation in a decamethyltitanocene dihydroxido complex: insights into the elemental steps of water splitting. Angew. Chem. Int. Ed 51,6272–6275 (2012). [DOI] [PubMed] [Google Scholar]
- 296.Godemann C et al. Highly selective visible light-induced Ti-O bond splitting in an ansa-titanocene dihydroxido complex. Chem. Commun 51, 3065–3068 (2015). [DOI] [PubMed] [Google Scholar]
- 297.Godemann C et al. A model of a closed cycle of water splitting using ansa-titanocene(III/IV) triflate complexes. J. Am. Chem. Soc 137, 16187–16195 (2015). [DOI] [PubMed] [Google Scholar]
- 298.Harrigan RW, Hammond GS & Gray HB Photochemistry of titanocene(IV) derivatives. J. Organomet. Chem 81, 79–85 (1974). [Google Scholar]
- 299.Kessler M et al. Synthesis of Cp*2Ti(OTf) and its reaction with water. Eur. J. Inorg. Chem 2011, 627–631 (2011). [Google Scholar]
- 300.Zhang Y, Petersen JL & Milsmann C A luminescent zirconium(IV) complex as a molecular photosensitizer for visible light photoredox catalysis. J. Am. Chem. Soc 138, 13115–13118 (2016). [DOI] [PubMed] [Google Scholar]
- 301.Zhang Y, Lee TS, Petersen JL & Milsmann C A zirconium photosensitizer with a long-lived excited state: mechanistic insight into photoinduced single-slectron transfer. J. Am. Chem. Soc 140, 5934–5947 (2018).This study shows that early transition metals can act as photosensitizers, taking advantage of LMCT processes as opposed to the classic MLCT processes used for Ru(bpy)32+ and similar complexes.
- 302.Roizen JL, Harvey ME & Du Bois J Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C-H bonds. Acc. Chem. Res 45, 911–922 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Qin C & Davies HML Role of sterically demanding chiral dirhodium catalysts in site-selective C-H functionalization of activated primary C-H bonds. J. Am. Chem. Soc 136, 9792–9796 (2014). [DOI] [PubMed] [Google Scholar]
- 304.Iovan DA, Wilding MJT, Baek Y, Hennessy ET & Betley TA Diastereoselective C-H bond amination for disubstituted pyrrolidines. Angew. Chem. Int. Ed 56, 15599–15602 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Basuli F, Wicker B, Huffman JC & Mindiola DJ Understanding the role of an easy-to-prepare aldimine-alkyne carboamination catalyst, [Ti(NMe2)3(NHMe2)][B(C5F5)4j. J. Organomet. Chem 696, 235–243 (2011). [Google Scholar]
- 306.Fantauzzi S et al. Origin of the deactivation in styrene aziridination by aryl azides, catalyzed by ruthenium porphyrin complexes. Structural characterization of a A2–1,2,3-triazoline RuII(TPP)CO complex. Organometallics 24, 4710–4713 (2005). [Google Scholar]
- 307.King ER, Hennessy ET & Betley TA Catalytic C-H bond amination from high-spin iron imido complexes. J. Am. Chem. Soc 133, 4917–4923 (2011). [DOI] [PubMed] [Google Scholar]
- 308.Cramer SA & Jenkins DM Synthesis of aziridines from alkenes and aryl azides with a reusable macrocyclic tetracarbene iron catalyst. J. Am. Chem. Soc 133, 19342–19345 (2011). [DOI] [PubMed] [Google Scholar]
- 309.Heyduk AF, Zarkesh RA & Nguyen AI Designing catalysts for nitrene transfer using early transition metals and redox-active ligands. Inorg. Chem 50, 9849–9863 (2011). [DOI] [PubMed] [Google Scholar]
- 310.Zarkesh RA, Ziller JW & Heyduk AF Four-electron oxidative formation of aryl diazenes using a tantalum redox-active ligand complex. Angew. Chem. Int. Ed 47, 4715–4718 (2008). [DOI] [PubMed] [Google Scholar]
- 311.Zarkesh RA & Heyduk AF Reactivity of organometallic tantalum complexes containing a bis(phenoxy)amide (ONO)3- ligand with aryl azides and 1,2-diphenylhydrazine. Organometallics 30, 4890–4898 (2011). [Google Scholar]
- 312.Blackmore KJ, Lal N, Ziller JW & Heyduk AF Catalytic reactivity of a zirconium(IV) redox-active ligand complex with 1,2-diphenylhydrazine. J. Am. Chem. Soc 130, 2728–2729 (2008). [DOI] [PubMed] [Google Scholar]
- 313.Nguyen AI, Zarkesh RA, Lacy DC, Thorson MK & Heyduk AF Catalytic nitrene transfer by a zirconium(IV) redox-active ligand complex. Chem. Sci 2, 166–169 (2011). [Google Scholar]
- 314.Gilbert ZW, Hue RJ & Tonks IA Catalytic formal [2 + 2 + 1] synthesis of pyrroles from alkynes and diazenes via TiII/TiIV redox catalysis. Nat. Chem 8, 63–68 (2016).This study provides an example of Ti redox catalysis and group transfer in the absence of redox non-innocent ligands, highlighting the role that π-accepting ligands can play in promoting catalysis with low-valent early transition metal complexes.
- 315.Chiu H & Tonks IA Trimethylsilyl-protected alkynes as selective cross-coupling partners in titanium-catalyzed [2 + 2 + 1] pyrrole synthesis. Angew. Chem. Int. Ed 57, 6090–6094 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Davis-Gilbert ZW, Yao LJ & Tonks IA Ti-catalyzed multicomponent oxidative carboamination of alkynes with alkenes and diazenes. J. Am. Chem. Soc 138, 14570–14573 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Pearce AJ, See XY & Tonks IA Oxidative nitrene transfer from azides to alkynes via Ti(II)/Ti(IV) redox catalysis: formal [2 + 2 + 1] synthesis of pyrroles. Chem. Commun 54, 6891–6894 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Davis-Gilbert ZW, Wen X, Goodpaster JD & Tonks IA Mechanism of Ti-catalyzed oxidative nitrene transfer in [2 + 2 + 1] pyrrole synthesis from alkynes and azobenzene. J. Am. Chem. Soc 140, 7267–7281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Guo J et al. Differences between the elimination of early and late transition metals: DFT mechanistic insights into the titanium-catalyzed synthesis of pyrroles from alkynes and diazenes. Chem. Sci 8, 2413–2425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Guo J et al. Strong preference of the redox-neutral mechanism over the redox mechanism for the TiIV catalysis involved in the carboamination of alkyne with alkene and diazene. Chem. Eur. J 24, 7010–7025 (2018). [DOI] [PubMed] [Google Scholar]
- 321.Tomson NC, Arnold J & Bergman RG Halo, alkyl, aryl, and bis(imido) complexes of niobium supported by the (3-diketiminato ligand. Organometallics 29, 2926–2942 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Kriegel BM, Bergman RG & Arnold J Nitrene metathesis and catalytic nitrene transfer promoted by niobium bis(imido) complexes. J. Am. Chem. Soc 138, 52–55 (2016). [DOI] [PubMed] [Google Scholar]
- 323.Schmidt M et al. Transfer reagent for bonding isomers of iron complexes. J. Am. Chem. Soc 139, 13981–13984 (2017). [DOI] [PubMed] [Google Scholar]
- 324.Corbey JF, Fang M, Ziller JW & Evans WJ Cocrystallization of (^-S2)2- and (n-S)2- and formation of an [r|2-S3N(SiMe3)2] ligand from chalcogen reduction by (N2)2- in a bimetallic yttrium amide complex. Inorg. Chem 54, 801–807 (2015). [DOI] [PubMed] [Google Scholar]
- 325.Fieser ME et al. Dinitrogen reduction, sulfur reduction, and isoprene polymerization via photochemical activation of trivalent bis(cyclopentadienyl) rare-earth-metal allyl complexes. Organometallics 34, 4387–4393 (2015). [Google Scholar]
- 326.Camp C, Maron L, Bergman RG & Arnold J Activation of white phosphorus by low-valent group 5 complexes: formation and reactivity of cyclo-P4 inverted sandwich compounds. J. Am. Chem. Soc 136, 17652–17661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Seitz AE et al. Pnictogen-silicon analogues of benzene. J. Am. Chem. Soc 138, 10433–10436 (2016). [DOI] [PubMed] [Google Scholar]
- 328.Vogel U et al. Access to phosphorus-rich zirconium complexes. Angew. Chem. Int. Ed 50, 8982–8985 (2011). [DOI] [PubMed] [Google Scholar]
- 329.Cossairt BM, Piro NA & Cummins CC Early transition metal mediated activation and transformation of white phosphorus. Chem. Rev 110, 4164–4177 (2010). [DOI] [PubMed] [Google Scholar]
- 330.Cossairt BM & Cummins CC A niobium-mediated cycle producing phosphorus-rich organic molecules from white phosphorus (P4) through activation, functionalization, and transfer reactions. Angew.Chem. Int. Ed 47, 8863–8866 (2008). [DOI] [PubMed] [Google Scholar]
- 331.Silvia JS & Cummins CC Ligand-based reduction of CO2 to CO mediated by an anionic niobium nitride complex. J. Am. Chem. Soc 132, 2169–2171 (2010). [DOI] [PubMed] [Google Scholar]