

Abstract
The functional evolution of proteins advances through gene duplication followed by functional drift, whereas molecular evolution occurs through random mutational events. Over time, protein active-site structures or functional epitopes remain highly conserved, which enables relationships to be inferred between distant orthologs or paralogs. In this study, we present the first functional clustering and evolutionary analysis of the RCSB Protein Data Bank (RCSB PDB) based on similarities between active-site structures. All of the ligand-bound proteins within the RCSB PDB were scored using our Comparison of Protein Active-site Structures (CPASS) software and database (http://cpass.unl.edu/). Principal component analysis was then used to identify 4,431 representative structures to construct a phylogenetic tree based on the CPASS comparative scores (http://itol.embl.de/shared/jcatazaro). The resulting phylogenetic tree identified a sequential, step-wise evolution of protein active-sites and provides novel insights into the emergence of protein function or changes in substrate specificity based on subtle changes in geometry and amino acid composition.
Keywords: Functional Evolution, Proteins, CPASS, Protein Active-sites
Introduction
A functional clustering of ligand-defined active-sites in the RCSB Protein Data Bank (RCSB PDB)1 was undertaken to infer an evolutionary lineage of enzymatic function. Conversely, sequence based phylogenetic methods are typically utilized to produce evolutionary trees originating from a common ancestor.2 The resulting phylogenetic tree can be used to infer an evolutionary relationship between species, to predict protein functions, and to reconstruct the sequence of ancestral proteins.3 This is possible because molecular evolution occurs through random mutations at a constant rate.4 Importantly, molecular evolution assumes the sequence alignment is based on homologous proteins derived from a common ancestor in which function has been maintained. Alternatively, the goal of functional evolution is to increase the diversity of protein functions, which may not occur through a common ancestor. In fact, there are likely multiple origin events leading to the same protein function, which may occur through either convergent or divergent evolution.5 While molecular evolution and functional evolution both require random mutations to occur, functional evolution also requires a gene duplication event to occur in order to enable functional diversification.6 Without this gene duplication event, random mutations would only exchange one protein function for another. There would be no evolutionary path for the diversity of protein functions currently realized. Additionally, while survivability and fitness are important factors for the selection of mutations, functional evolution involves other mechanisms of natural selection: neofunctionalization, subfunctionalization, and selection for gene dosage.6 In effect, molecular evolution and functional evolution result from innately different environmental pressures. Molecular evolution strives to maintain and enhance existing beneficial traits, while functional evolution can be viewed as the development of new traits in response to needs, stressors or competition.
The reliability of sequence-based evolutionary measurements becomes suspect when protein sequence homology enters the “twilight-zone” and falls below 25% sequence identity.7 In fact, the sequence alignment of proteins with less than 25% sequence identity results in over 95% of the proteins having distinct structures and function. Accordingly, low sequence identity raises serious concerns about the aligned proteins – are they really homologous proteins with the same function and from the same ancestor? Simply, the accuracy of a phylogenetic tree is directly dependent on the accuracy of the sequence alignment, which becomes undependable at low sequence identity.8,9 Therefore, due to the large sequential dissimilarity for the entirety of proteins deposited in the RCSB PDB, sequence-based evolutionary methods are not easily or reliably employed across an all-inclusive set of protein functional classes. Conversely, sequence alignments and sequence-based database searches are intended to identify proteins that share the same function. In fact, advanced sequence alignment techniques rely on multiple sequence alignments of presumed homologous proteins and such features as Hidden Markov models (HMM profiles) or genetic algorithms in order to maximize an underlying similarity between the aligned proteins.
Structure-based alignment is an alternative to sequence based alignments, especially considering the tremendous reduction in structure space relative to sequence space. Recent estimates suggest that only a few-thousand distinct protein folds exists,10,11 which is consistent with the 1391 protein topologies currently identified by CATH.12 Nevertheless, the alignment of protein structures is even more challenging than sequence alignment, and fails for completely dissimilar structures.11 Like sequence, the arrangement of tertiary structures is extremely evolutionarily labile when considering the entirety of known protein functions. While global protein sequence and structure may drift without detrimental consequences, dramatic changes to an active-site or functional epitope of a protein may negatively impact the survivability of an organism. Instead, functional evolution progresses slowly through gene duplication and functional drift to avoid negative influence on cellular fitness.13,14 This occurs because even minor changes in the spatial orientation or amino acid composition within an active-site may lead to dramatic changes in substrate and reaction specificity. Consequently, protein active-sites mutate at a much slower rate relative to other structural elements and remain highly conserved over time.15 In effect, a similarity in protein active-sites may remain even though the overall sequence or structure of a protein has completely diverged. Thus, it may be possible to infer an evolutionary functional relationship based on similarities in protein active-sites in situations when global sequence or structure similarities no longer exist. Again, a global sequence or structure alignment of functionally dissimilar proteins is very likely to fail. There is simply too much noise (e.g., large regions of sequence and structure differences) that would mask any residual signal (e.g., functional epitope or ligand-binding site). Instead, by focusing only on the active site/ligand binding site we can effectively remove or reduce the noise and enhance the signal.
Several methods and databases have been previously published describing the clustering of proteins from the RCSB PDB. These include sequence,16 structure,17 ligand conformation,18 atomic properties,19 and putative cavity20 based approaches. Similarly, evolutionary analyses are possible on large and divergent superfamilies using structure-function relationships21 or a combination of sequence, structure, and reaction mechanism data.22 However, a clustering and subsequent phylogenetic analysis based on ligand-defined active-sites has not been done. The Comparison of Protein Active-site Structures (CPASS) software and database compares the geometry and amino acid similarity between pairs of experimentally determined ligand-defined active-sites. CPASS is distinctly different from protein cavity approaches because it focuses on known binding sites rather than putative pocket detection. Further, substrate conformation is only used in the determination of active-site residues and not in the CPASS scoring function. Consequently, the evolutionary analysis of protein functions in the RCSB PDB based on active-site similarity is a novel approach.
We previously demonstrated the utility of CPASS to decipher the functional evolution (not molecular evolution) of proteins by comparing the active-sites of 204 PLP-dependent enzymes.23 We produced the first-ever phylogenetic tree that contained all four families or fold-types (I to IV) for PLP-dependent enzymes. The resulting phylogenetic tree correctly distinguished between the four individual folds and further sorted the enzymes by substrate specificity and function. Critically, no functional information was utilized to produce the phylogenetic tree of PLP-dependent enzymes, yet the enzymes were clustered perfectly based on EC number (i.e., branches were comprised of enzymes with the same EC number). Furthermore, examining individual branches of the phylogenetic tree illustrates the step-wise evolution of function through a series of single amino-acid substitutions. In effect, nearest neighbors in the CPASS derived phylogenetic tree identified subtle differences in active-site sequences and structures that led to changes in enzymatic activity and substrate specificity. It is important to note that the nearest neighbors in the CPASS derived phylogenetic tree do not necessarily share a common ancestor nor do nearest neighbors infer an evolutionary relationship between species. The CPASS derived phylogenetic tree captures functional evolution not molecular evolution. Nevertheless, we were still able to produce a phylogenetic tree for the PLP-dependent enzymes despite sequence identity well-below 20% and poor structural alignments between folds (TM-align24 score of ~ 0.3).
Based on this prior success, we expanded upon the phylogenetic tree of PLP-dependent enzymes by using CPASS to functionally cluster all ligand-containing proteins present in the RCSB PDB. In essence, CPASS was used to produce an unrooted phylogenetic tree containing essentially all protein functional classes present in the RCSB PDB. CPASS was used to make a pair-wise comparison between all of the ligand-defined binding sites within the RCSB PDB to produce an all-versus-all CPASS similarity score matrix. The proteins were then clustered by the identity of the bound ligand. Principal component analysis of the CPASS scores was employed to identify a representative structure for each functional class (i.e., same ligand and EC number) in order to reduce the overall size of the dataset. The representative structure for each functional class was then successfully modeled into a single unrooted phylogenetic tree based on the CPASS similarity score matrix. The resulting unrooted phylogenetic tree demonstrates the functional evolution across all of the protein functional classes within the RCSB PDB. Again, to be clear, since CPASS does not utilize global sequence or structure similarity the resulting unrooted phylogenetic tree does not describe molecular evolution from a common ancestor. Instead, the CPASS phylogenetic tree highlights the large-number of distinct origin events that have led to the diversity of known protein functions. To further illustrate the effectiveness of our approach, we also highlight two specific regions of the phylogenetic tree that demonstrate the stepwise substrate and enzymatic evolution of fructose-6-phosphate (F6P) bound active-sites.
Materials and Methods
Active-site Structure Comparison
Protein structures with ligand defined active-sites were collected from the Protein Data Bank 25 and subjected to an all versus all comparison using CPASS.26,27 It is important to note, that some protein structures contain more than one bound ligand. In these cases, each unique bound ligand was treated as a separate and distinct ligand-binding site and was included in the all versus all comparison. Unique ligands are defined as being different chemical compounds or sharing less than 80% sequence identity in the ligand-defined active sites. The primary goal of these exclusion criteria was to remove redundant ligand-binding sites from X-ray structures that contain multiple identical copies of the protein structure within the unit cell.
CPASS scores were subsequently converted to relative distances by subtracting from 100% CPASS similarity. The distance matrix, due to size and computational constraints, was divided into smaller matrices based on bound ligand. Principal component analysis was applied to the smaller ligand defined matrices using MVAPACK28 where functional clusters were generated based on Enzyme Commission (EC) number.29 For each PCA scores plot, only the first two principal components, which capture the highest and second highest amount of variance in the datasets, were chosen. For each EC number cluster, a 95% confidence ellipse was calculated which was used to find the representative active-site with the shortest Euclidean distance to its center. Ligands that appear only once or a few times in the PDB were not amenable to this PCA analysis. Instead, a representative active site was randomly selected from these small membered (e.g., singleton) classes only if the EC number was not previously identified from the prior PCA analysis. A total of 4431 representative active-sites were identified and then utilized to produce the CPASS phylogenetic tree.
Phylogenetic Analysis of Representative Active-sites
The CPASS distance matrix for the representative active-sites was input into FastME for tree generation using the Neighbor-join algorithm.30 Briefly, the neighbor-join algorithm joins the two closest taxa or nodes in the distance matrix and creates a new node, which has recalculated distances to the remaining taxa and nodes. Multiple iterations of this process build the unrooted tree until only a pair of nodes remains. Identification and investigation of the resulting unrooted tree structure was accomplished through visual inspection using the Interactive Tree of Life online tool.31 The unrooted tree, available at http://itol.embl.de/shared/jcatazaro, is searchable and has also been shared on our website at https://www.bionmr.unl.edu. Leaves are labeled by PDB ID, colored by EC function, and contain popup windows with links to the respective PDB entry, EC function, and bound ligand. A complete table with the unique, non-redundant mappings for each PDB ID to their corresponding representative PDB ID can be found in the supplemental information (Figure S1).
Active-site Overlays
From the CPASS representative dataset and tree, 9 enzymes were selected for additional investigation. Structural and sequential differences between the active-sites of the enzymes (PDB IDs: 1H83, 3BI5, 1SEZ, 3M5P, 2O2D, 2P3V, 1LBY, 1JP4, 1KA1) were elucidated by visual inspection using Chimera.32 In each case, residues were considered to be in the active-site based on their relative proximity to the bound ligand in their respective crystal structure (6Å). The orientation of the active-sites relative to one another was also determined by CPASS, as a standard 3D overlay of the tertiary structures would result in misalignment of the active-sites.
Results
Functional Clustering and Principal Component Analysis of Ligand Defined Active-sites.
The Comparison of Protein Active-site Structures (CPASS) software and database (http://cpass.unl.edu/) was used to compare all protein active-sites from the RCSB PDB that contained a bound ligand. Please note, some protein structures contain more than one bound ligand. In these cases, all of the unique ligand binding sites (e.g., different compound and location) were used in the CPASS analysis. CPASS performs a pairwise comparison between two protein active-sites, where active-site residues were determined based on a defined distance to the bound ligand (6Å). CPASS similarity scores are determined by similarities in both amino acid composition and by the relative amino acid positions between the two compared active-site. An “all versus all” distance matrix derived from CPASS similarity scores was initially calculated for all of the ligand defined active-sites in the RCSB PDB.
The protein structures were then clustered based on the identity of the bound ligand in order to create function specific protein groupings and to reduce the size of the dataset. A total of 169 protein function groups were created based on a shared identity of the bound ligand. Consequently, a total of 169 principal component analyses (PCA) were then performed using our MVAPACK28 software for each of these ligand defined protein groups. Group membership within the PCA scores plot was further defined by Enzyme Commission (EC)29 number and demarcated by a 95% confidence ellipse. A representative example of a PCA scores plot for the collection of fructose-6-phosphate (F6P) bound active-sites is shown in Figure 1. There are 8 different enzymatic functional classes (EC numbers: 1.2.1.9, 2.7.1.105, 2.7.1.11, 3.1.3.11, 3.1.3.25, 3.5.1.25, 5.3.1.8, and 5.3.1.9) and one unannotated group in the PCA scores plot.
Figure 1.
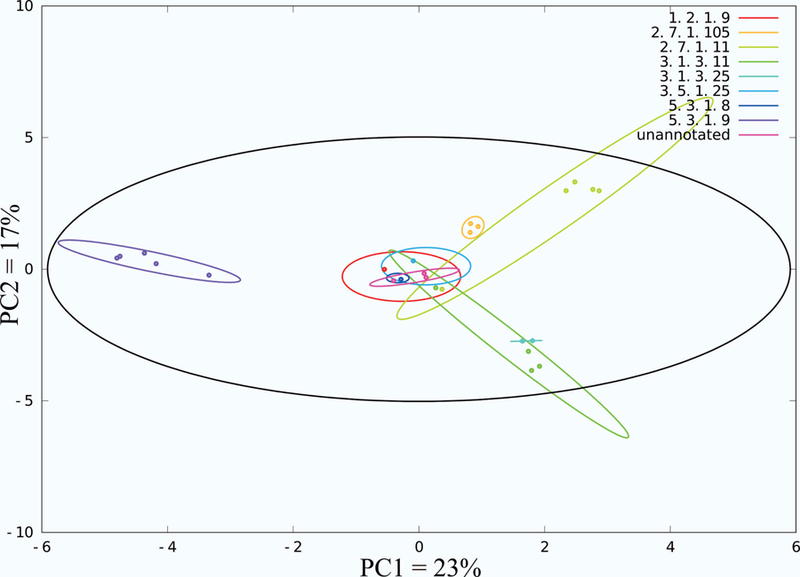
The PCA scores plot of a CPASS distance matrix for fructose-6-phosphate bound proteins. Active-sites are clustered by Enzyme Commission number, which refers to a specific function. Ellipses correspond to the 95% confidence intervals for each of the functional clusters (colored) and the dataset (black).
Structural Representatives of Functional Classes.
The PCA scores plots were leveraged to find a representative protein structure for each functional class based on EC number and the type of bound ligand. For each functional class in the PCA scores plot, the protein active-site with the shortest Euclidean distance to the center of the 95% confidence ellipse was chosen as a representative structure. Again, the 95% confidence ellipse defines the membership for a given functional class. Accordingly, the selected protein active-site should have a high CPASS similarity score or a small variance relative to the other protein active-sites in the functional class. In effect, the selected protein active site is expected to serve as a structural “average” for the functional class. This is supported by the histogram plots of the CPASS similarity scores shown in Figure 2A and 2B. The CPASS similarity scores between members of a given functional class (e.g., same bound ligand and EC number) are significantly larger (Figure 2B) than the CPASS similarity scores between members of different functional classes (Figure 2A). The relatively flat distribution of lower CPASS scores in Figure 2B is attributed to members of unannotated groups that presumably have different functions despite binding the same ligand. In total, the 169 PCA score plots identified a representative structure for 4431 EC functional classes. A complete table with the unique, non-redundant mappings for each RCSB PDB structure to their corresponding representative structure can be found in the supplemental information (Figure S1).
Figure 2.
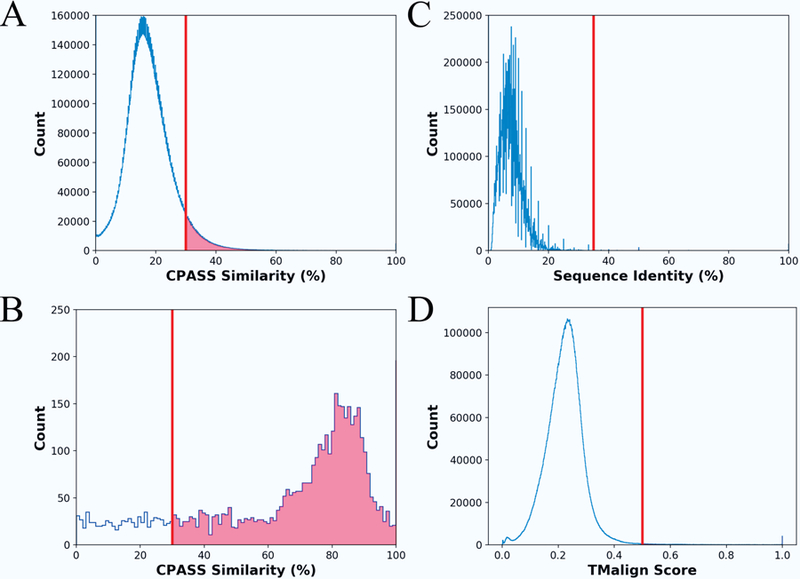
Histogram plots illustrating the distribution of (A) CPASS similarity scores (blue line), (B) the CPASS similarity scores between the representative active-site for each functional class and the other members of the group (i.e., same EC number and bound ligand), (C) percent sequence identity, and (D) TMalign similarity scores for the pair-wise comparison of the 4431 representative active-sites used to generate the phylogenetic tree in Figure 3. The vertical line in each histogram plot identifies the lower score that defines a significant level of similarity.
Phylogenetic Analysis.
A phylogenetic analysis was conducted using a distance matrix based on CPASS similarity scores for the 4431 protein active-sites. The phylogenetic analysis used the neighbor-join algorithm and the resulting unrooted phylogenetic tree is shown in Figure 3. An annotated, interactive and searchable version of the phylogenetic tree is hosted by the Interactive Tree Of Life (iTOL)31 and is located at http://itol.embl.de/shared/jcatazaro, and has also been shared on our website at https://www.bionmr.unl.edu. The unrooted phylogenetic tree is shown in a circular display with leaves colored according to the function defined by the first EC number [oxidoreductases (red), transferases (blue), hydrolases (yellow), lyases (green), isomerases (purple), ligases (orange), not annotated (black)]. Importantly, the functional classification was not used as part of the phylogenetic analysis. Instead, the resulting phylogenetic tree was simply annotated with the known functional classifications. A full, linear unrooted tree with annotated leaves is provided in the supplemental information (Figure S2). Existing tools provided within iTOL enable searching the tree by PDB ID, modifying the tree display (circular, linear, etc.), as well as exporting high resolution images. Additionally, pop up boxes have been implemented for each representative active-site, which contains the EC number, the bound ligand, the 3D structure of the protein, and links to the RCSB PDB1 and KEGG33 databases. Notably, the link to the RCSB also provides the CATH12 and SCOP34 classification for each protein structure. The tree structure can be downloaded directly from iTOL and the raw distance matrix can be provided upon request.
Figure 3.

A phylogenetic tree of 4431 representative CPASS active sites from the RCSB PDB is presented. The phylogenetic tree highlights the functional evolutionary relationships between protein active-site structures. Leaves are colored according to the first EC number of the annotated active-site (1: oxidoreductases, red; 2: transferases, blue; 3: hydrolases, yellow; 4: lyases, green; 5: isomerases, purple; 6: ligases, orange; not annotated: black). An annotated, searchable and interactive phylogenetic tree is located at the iTOL 31 website http://itol.embl.de/shared/jcatazaro.
Active Site Similarity versus Sequence or Structural Alignment.
The entire sequence and the complete structure for the 4431 representative proteins were subjected to a multiple sequence alignment with Clustal Omega or a three-dimensional (3D) structural alignment with TMalign.24,35 Histograms of CPASS similarity scores, percent sequence identities, and TMalign similarity scores are shown in Figure 2. A CPASS similarity score of ~30% is considered reliable and indicates conserved features between the two active sites. As evident by the histogram plot (Figure 2A), a significant number of the pair-wise comparisons of active sites fall in the significant >30% range. Conversely, a sequence identity less than 20% or a TMalign similarity score below 0.5 are considered insignificant and suggest the proteins are not homologous.7,24 Accordingly, the histograms displaying the distribution of sequence (Figure 2C) and structure (Figure 2D) similarity scores suggest minimal or non-existent similarities between the 4431 representative proteins. Specifically, of the approximately 10 million pairwise sequence and structure comparison only 3304 (0.03%) homologous pairs (>35% sequence identity, >0.5 TMalign score) were identified.
Discussion
Herein, we report the first functional clustering and evolutionary analysis of the entirety of proteins deposited in the RCSB PDB with a bound ligand. A functional evolution (not molecular evolution) was based on active-site similarities determined by our CPASS software and database. Protein active-sites were first divided into functional classes based on the type of bound ligand. PCA of the CPASS similarity scores was then used to visualize the relative similarities of the functional class membership. The resulting PCA scores plot was then annotated with EC numbers and the 95% confidence ellipses (Figure 1) were used to define the membership of each functional class within the scores plot.
PCA has been extensively used in chemometrics and various ‘omics’ fields for fingerprint analysis.36 In this study, PCA was used to reduce the variance within each functional class while also reducing the size of the dataset used for the phylogenetic analysis. The PCA scores plot for the collection of F6P bound active-sites (Figure 1) yielded several important observations. First, a number of the 95% confidence ellipses partially overlap in the PCA scores plot. This suggests that there are structural elements that remain consistent within the active-site even though the enzymatic functions vary considerably. Second, a complete separation of two functional classes would indicate that the active-sites have either diverged significantly over time or have converged to act upon the same substrate. An evolutionary functional drift is also apparent when considering the shape and positions of the ellipses in the scores plot. The various clusters appear to drift away from the center of the scores plot. Assuming the center of the PCA scores plot is the structural average of all active-sites bound to a ligand, the movement of an ellipse or active-site toward or away from the center would indicate convergent or divergent evolution, respectively. In effect, the substrate specificity and/or enzymatic activity is diverging as the enzyme moves away from the center of the scores plot or converging as it moves towards its center.
In this study, PCA was primarily utilized to identify a representative protein structure for each functional class. Simply, the protein structure closest to the center of each ellipse was identified as the representative active-site for the functional class. For example, a total of eight protein structures were identified from the PCA scores plot of the F6P bound active sites shown in Figure 1. This corresponds to one protein representative for each of the seven EC functional classes and one protein for the single unannotated class. In this manner, PCA allowed for a drastic reduction in the size of the dataset to about 10% of its initial size. A representative active-site was randomly selected from low-populated EC functional classes (e.g., singletons) that were not amenable to this PCA analysis. This only occurred if the EC functional class was not already present in the list of active-sites identified from the PCA analysis. Importantly, the resulting set of proteins achieved a maximal variety of functional classes with little sequence (< 20% identity) or structural (< 0.4 TMalign score) similarity between each member of the set (Figure 2). This also indicates that the data set is mostly comprised of non-homologous proteins since likely homology is inferred from a sequence identity > 35% or from a similar structure (> 0.5 TMalign score).7,24 A distance matrix was then generated from an all-vs-all comparison of the 4431 representative protein active-sites from each functional class. The matrix of CPASS similarity scores were then subjected to the neighbor-join algorithm for a phylogenetic analysis (Figures 3 and S2). The resulting unrooted phylogenetic tree captures the stepwise functional evolution of essentially all of the protein functional classes present in the RCSB PDB.
Protein active-sites were paired together in the tree according to enzymatic function, which was also seen in our previous study of PLP-dependent enzymes.23 Consistent with this trend was the observation that 66% of the limited number of homologous pairs were found on nearby branches of the CPASS tree. The remaining homologous pairs were found on distant branches. The separation of homologous pairs is a result of proteins containing multiple ligand-binding sites, where these alternative ligand binding sites are not related to their EC classification. For example, the RNAse enzyme 1AFL has two bound ligands (2’-monophosphoadenosine-5’-diphosphate and citrate) where 2’-monophosphoadenosine-5’-diphosphate is relevant to its EC classification (EC 3.1.27.5). But, 1AFL was placed into the CPASS tree based on its citrate binding site, which is likely not related to its EC classification. Accordingly, nearest neighbors to 1AFL on the CPASS tree may be distinct from its EC classification because of the unique ligand-binding site.
The structure and amino-acid composition of a protein active-site is typically highly conserved in order to maintain function and retain cellular fitness. Thus, functional evolution of a protein progresses slowly and likely follows a step-wise process of single-amino substitutions that also involves a prior gene duplication event. The process proceeds until a new function or substrate specificity is achieved. Importantly, this step-wise evolution of function is clearly evident in our phylogenetic tree of protein active-sites. Nearest-neighbors, even those from different organisms, have very subtle differences in active-site structures and/or sequence. Simply, as an active-site progresses towards the next node, a change in substrate specificity or enzymatic activity may result from a few amino-acid substitutions and/or minor conformational change (Figures 4 and 5). Importantly, since the CPASS phylogenetic tree describes functional evolution, and not molecular evolution, nearest neighbors do not necessarily share a common ancestor. Nearest neighbors may be orthologs or paralogs that result from divergent or convergent evolution. In fact, nearest neighbors may actually be from species very far apart on the evolutionary tree. Simply put, nearest neighbors are functionally, not evolutionarily related.
Figure 4.
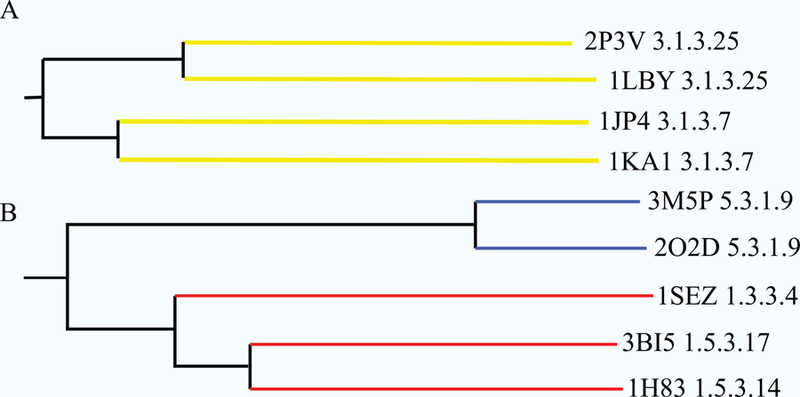
Two regions of the phylogenetic tree from Figure 3 were selected for further detailed analysis. (A) The protein active-sites in this branch of the phylogenetic tree illustrate protein functional evolution that results in changes in substrate specificity. (B) The protein active-sites in this branch of the phylogenetic tree illustrate protein functional evolution that results in changes in both enzymatic activity and substrate specificity. Proteins are listed by their PDB IDs and EC functions (3.1.3.25: inositol-phosphate phosphatase; 3.1.3.7: 3'(2'), 3’ phosphoadenosine-5’-phosphate phosphatase; 5.3.1.9: glucose-6-phosphate isomerase; 1.3.3.4: protoporphyrinogen oxidase; 1.5.3.17: non-specific polyamine oxidase; 1.5.3.14: polyamine oxidase (propane-1,3-diamine-forming)).
Figure 5.
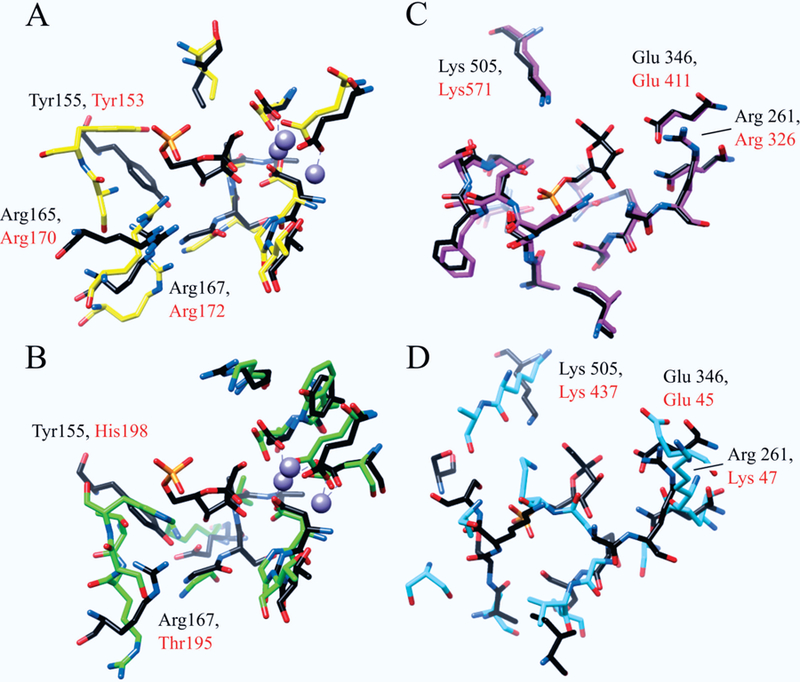
Structural overlays of the active-sites for (A) 1LBY (black) and 2P3V (yellow), and (B) 1LBY (black) and 1JP4 (green). Residues are labeled by type and sequence position with those from 1LBY in black and those from 2P3V and 1JP4 in red. Overlays are oriented relative to the bound F6P in 1LBY with the coordinated magnesium ions displayed in purple. Structural overlays of the active-sites for (C) 3M5P (black) and 2O2D (pink), and (D) 3M5P (black) and 1SEZ (cyan). Residues are labeled by type and sequence position with those from 3M5P in black and those from 2O2D and 1SEZ in red. Overlays are oriented relative to the bound F6P in 3M5P. Residues were chosen for the comparative analysis if they were within 6Å of the bound ligand and were used in the CPASS similarity scoring.
Interestingly, while nearest neighbors share similar function, an overall view of the functional distribution throughout the entire phylogenetic tree is more complex and diverse. This is apparent from the relatively random distribution of colors throughout the phylogenetic tree, where leaves are colored according to the first EC number for each representative protein. The phylogenetic tree is not uniformly divided or colored into six contiguous functional classes. Instead, there are many small pairings and subgroupings of similar functional classes that are evenly distributed throughout the tree. This mixing of function is likely due to multiple ancestral active-site scaffolds that have evolutionarily diverged and then expanded their biological roles. In effect, there is not one active site template for all hydrolases, not one template for all transferases, or not one template for all ligases. Furthermore, the lack of homologous proteins, based on low sequence and structure similarity (Figures 2C, D), also implies, by definition, that the 4431 proteins evolved from multiple ancestors. It is important to note that some of the apparent randomness in the distribution of protein function may be explained by proteins having multiple distinct ligand binding sites, but only a single EC classification. Accordingly, some proteins may be positioned into the tree based on these secondary ligand binding sites that may not be related to their defined function. In this regards, the functional color-labeling may not be correct.
While not assumed, our analysis provides strong evidence that all known active-sites did not emerge from a single ancestor nor did each EC class emerge from a single unique ancestor. Instead, the known diversity of protein function evolved from multiple random and independent origin events. In other words, there are multiple functional ancestors that have produced the diversity of protein functions. Proteins may share a similar active-site, but this function may be positioned on completely different structural scaffolds that evolved from distinct sets of ancestors. Accordingly, the organization of the phylogenetic tree is also consistent with convergent evolution where distant active-site architectures have slowly mutated toward the same enzymatic function. In essence, the dramatic dispersion of color throughout the phylogenetic tree is further evidence of the multitude of divergent and convergent events that have occurred in the evolution of protein functions. Additionally, our analysis considers each ligand defined active site within a particular protein independently. One protein may have multiple ligand binding sites and, thus, each site would have a unique representative in the tree. This method is in stark contrast to sequence and structure tools where the entire primary sequence or 3D model is used for comparison and subsequent phylogenetic analyses. Active site comparisons are not bound by these constraints and, therefore, the CPASS representative tree may capture the functional evolution of two or more ligand binding sites associated with one protein. This further explains the dispersion of color throughout the CPASS phylogenetic tree.
Two representative regions of the phylogenetic tree have been highlighted to further illustrate the effectiveness of an evolutionary clustering of function based on CPASS similarity scores (Figure 4). It is important to note that two branches highlighted in Figure 4 come from distinct regions of the phylogenetic tree. Nevertheless, both branches contain a protein active-site bound to fructose-6-phosphate (F6P), where two proteins (1LBY 3.1.3.25, 3M5P 5.3.1.9)37 were representative structures identified from the PCA scores plot displayed in Figure 1. Using these two proteins as arbitrary starting points, a step-wise evolution of substrate specificity and enzymatic activity is easily observed. The active-site of 1LBY has inositol-phosphate phosphatase activity and was found to be most similar to 2P3V,38 which shares the same function as 1LBY (Figure 4A). An overlay of the 1LBY and 2P3V CPASS determined active-sites reveals an almost identical match in terms of both amino acid identity and geometry (Figure 5A). This is to be expected as nearest neighbors have the closest distance (highest CPASS similarity). Furthermore, the primary difference between the two active-sites is the identity of the bound ligand. 2P3V is bound to S,R meso-tartaric acid instead of F6P, which was simply a result of the crystallization conditions. This outcome also demonstrates an important feature, the robustness of CPASS to identify highly similar active-sites independent of the identity of the bound ligand.
The next nearest node to 1LBY in Figure 4A includes two protein active-sites (1JP439 and 1KA140) with a similar function (identical for the first three EC numbers), but that act on different substrates. The CPASS determined active-sites for 1JP4 and 1KA1 are quite similar (not shown), where the primary difference is the identity of the bound ligand (adenosine monophosphate vs. adenosine-3’−5’-diphosphate). In effect, these two nearest-neighbor nodes (Figure 4A) contain a pair of proteins with similar functional classification (3.1.3.25 or 3.1.3.7), but with different bound ligands in the experimental structures deposited in the RCSB PDB. A comparison of the 1LBY active site, an inositol-phosphate phosphatase, with 1JP4, a 3’ phosphoadenosine-5’-phosphate phosphatase, reveals minor structural and amino acid differences between the two active sites (Figure 5B). Since the active-sites have a similar function but different substrate specificity, the observed changes in amino acid composition and active-site geometry are most likely related to substrate binding.
The four proteins (1LBY, 2P3V, 1JP4, 1KA1) comprising this node are magnesium dependent phosphatases, which have an evolutionarily conserved active-site and coordinate 2 to 3 metal ions.40 The metal ions specifically enable the catalytic dephosphorylation of bound substrates and are essential to enzyme activity. Interestingly, the metal coordination sites are strictly conserved even though the metal ions do not participate in substrate binding. Critical to our study, the sequence identity for members of the Mg2+-dependent superfamily is below 25%,39 which makes sequence-based evolutionary analysis extremely challenging and further highlights the benefits of our CPASS approach. In fact, the CPASS analysis further confirms the high conservation of the metal coordination site. This is apparent in the structural overlays in Figure 4. The active-site residues identified by CPASS around the coordination sites deviate very little in position while sequence identity is absolutely maintained. Conversely, the residues opposite the metal coordination sites, which do change between Figures 5A to 5B, are involved in substrate recognition.
Since the active-sites for 1LBY and 2P3V have the same function and act upon the same substrate, the tyrosine (1LBY:Tyr155, 2P3V:Tyr153) and arginine (1LBY:Arg165, Arg167, 2P3V: Arg170, Arg172) residues on the distal side of the active-site relative to the metal ions are conserved. These residues assist in the coordination of the substrate sugar and phosphate moieties, respectively. For 1JP4, the substrate is changed to 3’-phosphoadenosine 5’-phosphate (PAP), which induces spatial changes and amino-acid substitutions in the active-site (Figure 5B). Specifically a tyrosine is replaced by a histidine (1JP4: His198) and an arginine is replaced by a threonine (1JP4: Thr195). These amino-acid substitutions form a new hydrogen bond network around the PAP 5’-phosphate moiety.39 A reorientation of the side chain of the remaining arginine creates space to accommodate the increase in size of the PAP ligand. Interestingly, the PAP phosphatase maintains some of its inositol 1-phosphate/fructose-1,6-bisphosphate phosphatase activity.39 Considering how close the active-sites are to one another in the phylogenetic tree and the similarity of the active-site structures, the residual enzymatic activity is understandable.
A similar comparative analysis of protein functional evolution is illustrated by examining another branch from the phylogenetic tree (Figure 4B). Unlike the first illustrated example that lead to an evolution of substrate specificity (Figure 4A), this branch leads to the proteins adopting new functions in addition to changes to substrate specificities. The focus of this branch is the active-site of 3M5P, which is a glucose-6-phosphate (G6P) isomerase and was identified as a representative structure from the PCA scores plot in Figure 1.
The active site of 3M5P was found to be most similar to 2O2D,41 which has the same function as 3M5P (EC 5.3.1.9, G6P isomerases). An overlay of the two CPASS active-site structures in Figure 5C indicates near identity in regards to both amino-acid composition and structure. Again, the only difference in these two active-site structures is the nature of the bound ligand. 3M5P is bound to F6P; whereas, 2O2D is bound to citrate. This difference is likely just a result of differences in the crystallization buffers. Critical residues in the active-sites of 3M5P and 2O2D that are directly responsible for enzymatic activity are a lysine (3M5P: Lys505, 2O2D: Lys571), a glutamate (3M5P: Glu346, 2O2D: Glu411), and an arginine (3M5P: Arg261, 2O2D: Arg326). These residues are positioned directly around the substrate sugar moiety,41 facilitate proton transfer (lysine, glutamate), and stabilize the intermediate structure.
The next nearest node to 3M5P includes three protein active-sites (1SEZ, 3BI5, 1H83)42,43 with a similar function (oxidoreductase activity), but act on different substrates. However, the active-sites are no longer conserved when comparing 3M5P, a G6P isomerase, to 1SEZ, a protoporphyrinogen oxidase (PPO) (Figure 5D). The lysine, glutamate, and arginine residues, which are important to enzymatic activity remain in 3M5P, but now occupy different positions within the active-site. Of particularly note, the importance of these residues to the enzymatic activity of 1SEZ has been diminished. Now, the residues likely only assist in hydrogen bonding to the substrate rather than serving a more integral role in the enzymatic activity of the protein. Moreover, the critical arginine in the G6P isomerase active site is no longer strictly conserved in the CPASS determined active-site for PPO. Simply, there is no longer a reaction intermediate in PPO that requires stabilization by an arginine. As a result, the arginine is not conserved and the space it occupied has been better utilized. In essence, the overlay of active sites in Figures 5C and 5D demonstrates the stepwise evolution from a glucose-6-phosphate isomerase to a PPO enzyme. A similar result is obtained when 3M5P is compared to 3BI5 or 1H83, in which a G6P isomerase is converted into a polyamine oxidase. Comparing these active-site structures provides a clear understanding of the slow, step-wise evolution of protein function that is essential to the survivability and adaptability of a cell or organism.
Each nearest-neighbor in the phylogenetic tree represents a functional relationship that may be further explored and studied in detail. Nearest-neighbor pairs may demonstrate active site rearrangements or mutations that occur to change substrate specificity, function, or both. Therefore, the potential information that may be extracted from the phylogenetic tree is enormous and beyond the scope of this study. For example, there are substantial drug discovery and therapeutic opportunities that may be realized from studying the active site structures of cytochrome P450 enzymes, or proteins that bind ATP, NAD or chemical analogs.
Conclusion
Our CPASS phylogenetic tree (http://itol.embl.de/shared/jcatazaro, https://www.bionmr.unl.edu) depicts the functional similarity of 4431 protein active-sites from the RCSB PDB. In this manner the step-wise transformation of enzymatic activity and substrate specificity is easily visualized through a comparison of nearest neighbors. Simply, nearest neighbors’ share a similar function while functional diversity ensues as proteins move further apart along the tree. In essence, our CPASS phylogenetic tree provides a visual map of protein functional space. It is important to appreciate that our CPASS phylogenetic tree does not depict the traditional hierarchal evolution in time from a common ancestor. Instead, we have simply employed a special graph sub-type, an unrooted tree, to visually cluster protein active–sites based on their relative similarity in shape and amino-acid composition. In this regards, nearest neighbors on the CPASS phylogenetic tree do not necessarily share a common ancestor, and, importantly, our analysis does not suggest that all protein active-sites share a common ancestor. To the contrary, our CPASS phylogenetic tree clearly demonstrates that a multitude of independent, random, evolutionary events have occurred, which has produced multiple functional ancestors. In effect, nature is constantly “re-inventing the wheel” when it comes to protein function.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Allergy and Infectious Diseases (R21AI081154), the Redox Biology Center (P30 GM103335, NIGMS); and the Nebraska Center for Integrated Biomolecular Communication (P20 GM113126, NIGMS). The research was performed in facilities renovated with support from the National Institutes of Health (RR015468–01). This work was completed utilizing the Holland Computing Center of the University of Nebraska.
Footnotes
Conflict of Interests
The authors declare no conflict of interests.
References
- 1.Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res 2000;28(1):235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Todd AE, Orengo CA, Thornton JM. Evolution of Function in Protein Superfamilies, from a Structural Perspective. Journal of Molecular Biology 2001;307:1113–1143. [DOI] [PubMed] [Google Scholar]
- 3.Gabaldon T Evolution of proteins and proteomes: A phylogenetics approach. Evol Bioinf Online 2005;1:51–61. [PMC free article] [PubMed] [Google Scholar]
- 4.Fay JC, Wu C-i. Sequence divergence, functional constraint, and selection in protein evolution. Annu Rev Genomics Hum Genet 2003;4:213–235. [DOI] [PubMed] [Google Scholar]
- 5.Ponting CP, Russell RR. The natural history of protein domains. Annu Rev Biophys Biomol Struct 2002;31:45–71. [DOI] [PubMed] [Google Scholar]
- 6.Conant GC, Wolfe KH. Turning a hobby into a job: How duplicated genes find new functions. Nat Rev Genet 2008;9(12):938–950. [DOI] [PubMed] [Google Scholar]
- 7.Rost B Twilight zone of protein sequence alignments. Protein Eng 1999;12(2):85–94. [DOI] [PubMed] [Google Scholar]
- 8.Cantarel BL, Morrison HG, Pearson W. Exploring the relationship between sequence similarity and accurate phylogenetic trees. Mol Biol Evol 2006;23(11):2090–2100. [DOI] [PubMed] [Google Scholar]
- 9.Liu K, Linder CR, Warnow T. Multiple sequence alignment: a major challenge to large-scale phylogenetics. PLoS Curr 2010;2:RRN1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schaeffer RD, Daggett V. Protein folds and protein folding. Protein Eng Des Sel 2011;24(1–2):11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolodny R, Pereyaslavets L, Samson AO, Levitt M. On the universe of protein folds. Annu Rev Biophys 2013;42:559–582. [DOI] [PubMed] [Google Scholar]
- 12.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH - a hierarchic classification of protein domain structures. Structure (London) 1997;5(8):1093–1108. [DOI] [PubMed] [Google Scholar]
- 13.Hughes AL. The evolution of functionally novel proteins after gene duplication. Proc R Soc London, Ser B 1994;256(1346):119–124. [DOI] [PubMed] [Google Scholar]
- 14.Prince VE, Pickett FB. Splitting pairs: The diverging fates of duplicated genes. Nat Rev Genet 2002;3(11):827–837. [DOI] [PubMed] [Google Scholar]
- 15.Martincorena I, Seshasayee ASN, Luscombe NM. Evidence of non-random mutation rates suggests an evolutionary risk management strategy. Nature (London, U K) 2012;485(7396):95–98. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006;22(13):1658–1659. [DOI] [PubMed] [Google Scholar]
- 17.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics 2008;24(23):2780–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shin JM, Cho DH. PDB-Ligand: a ligand database based on PDB for the automated and customized classification of ligand-binding structures. Nucleic Acids Res 2005;33(Database issue):D238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Totrov M Ligand binding site superposition and comparison base on Atomic Property Fields: identification of distant homologues, convergent evolution, and PDB-wide clustering of binding sites. BMC Bioinformatics 2011;12(Suppl 1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leinweber M, Fober T, Strickert M, et al. CavSimBase: A Database for Large Scale Comparison of Protein Binding Sites. IEEE Transactions on Knowledge and Data Engineering 2016;28(6):1423–1434. [Google Scholar]
- 21.Akiva E, Brown S, Almonacid DE, et al. The Structure-Function Linkage Database. Nucleic Acids Res 2014;42(D1):D521–D530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furnham N, Sillitoe I, Holliday GL, et al. FunTree: a resource for exploring the functional evolution of structurally defined enzyme superfamilies. Nucleic Acids Res 2012;40(Database issue):D776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Catazaro J, Caprez A, Guru A, Swanson D, Powers R. Functional Evolution of PLP-Dependent Enzymes Based on Active Site Structural Similarities Proteins: Struct, Funct, Bioinf 2014;82:2597–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 2005;33(7):2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res 2000;28(1):235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Powers R, Copeland JC, Germer K, Mercier KA, Ramanathan V, Revesz P. Comparison of protein active site structures for functional annotation of proteins and drug design. Proteins 2006;65(1):124–135. [DOI] [PubMed] [Google Scholar]
- 27.Powers R, Copeland JC, Stark JL, Caprez A, Guru A, Swanson D. Searching the protein structure database for ligand-binding site similarities using CPASS v.2. BMC Research Notes 2011;4(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Worley B, Powers R. MVAPACK: a complete data handling package for NMR metabolomics. ACS Chem Biol 2014;9(5):1138–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cornish-Bowden A Current IUBMB recommendations on enzyme nomenclature and kinetics. Perspectives in Science 2014;1(1–6):74–87. [Google Scholar]
- 30.Lefort V, Desper R, Gascuel O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program. Mol Biol Evol 2015;32(10):2798–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 2016;44(W1):W242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera-A Visualization System for Exploratory Research and Analysis. Journal of Computational Chemistry 2004;25(13):1605–1612. [DOI] [PubMed] [Google Scholar]
- 33.Kanehisa M, Goto S, Kawashima S, Nakaya A. The KEGG databases at GenomeNet. Nucleic Acids Res 2002;30(1):42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 1995;247(4):536–540. [DOI] [PubMed] [Google Scholar]
- 35.Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 2011;7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Worley B, Powers R. Multivariate Analysis in Metabolomics. Current Metabolomics 2013;1:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stieglitz KA, Johnson KA, Yang H, et al. Crystal structure of a dual activity IMPase/FBPase (AF2372) from Archaeoglobus fulgidus. The story of a mobile loop. J Biol Chem 2002;277(25):22863–22874. [DOI] [PubMed] [Google Scholar]
- 38.Stieglitz KA, Roberts MF, Li W, Stec B. Crystal Structure of the Tetrameric Inositol 1-phosphate phosphatase (TM1415) from the Hyperthermophile, Thermotoga maritima. The FEBS journal 2007;274:2461–2469. [DOI] [PubMed] [Google Scholar]
- 39.Patel S, Yenush L, Rodriguez PL, Serrano R, Blundell TL. Crystal structure of an enzyme displaying both inositol-polyphosphate-1-phosphatase and 3’-phosphoadenosine-5’-phosphate phosphatase activities: a novel target of lithium therapy. J Mol Biol 2002;315(4):677–685. [DOI] [PubMed] [Google Scholar]
- 40.Patel S, Martínez-Ripoll M, Blundell TL, Albert A. Structural Enzymology of Li+-sensitive/Mg2+-dependent Phosphatases. Journal of Molecular Biology 2002;320(5):1087–1094. [DOI] [PubMed] [Google Scholar]
- 41.Arsenieva D, Appavu BL, Mazock GH, Jeffery CJ. Crystal structure of phosphoglucose isomerase from Trypanosoma brucei complexed with glucose-6-phosphate at 1.6 A resolution. Proteins: Struct, Funct, Bioinf 2009;74(1):72–80. [DOI] [PubMed] [Google Scholar]
- 42.Koch M, Breithaupt C, Kiefersauer R, Freigang J, Huber R, Messerschmidt A. Crystal structure of protoporphyrinogen IX oxidase: a key enzyme in haem and chlorophyll biosynthesis. The EMBO journal 2004;23(8):1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Binda C, Angelini R, Federico R, Ascenzi P, Mattevi A. Structural Bases for Inhibitor Binding and Catalysis in Polyamine Oxidase. Biochemistry 2001;40(9):2766–2776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.