

Abstract
α-Melanocyte–stimulating hormone (α-MSH) has been shown to be involved in nociception, but the underlying molecular mechanisms remain largely unknown. In this study, we report that α-MSH suppresses the transient outward A-type K+ current (IA) in trigeminal ganglion (TG) neurons and thereby modulates neuronal excitability and peripheral pain sensitivity in rats. Exposing small-diameter TG neurons to α-MSH concentration-dependently decreased IA. This α-MSH–induced IA decrease was dependent on the melanocortin type 4 receptor (MC4R) and associated with a hyperpolarizing shift in the voltage dependence of A-type K+ channel inactivation. Chemical inhibition of phosphatidylinositol 3-kinase (PI3K) with wortmannin or of class I PI3Ks with the selective inhibitor CH5132799 prevented the MC4R-mediated IA response. Blocking Gi/o-protein signaling with pertussis toxin or by dialysis of TG neurons with the Gβγ-blocking synthetic peptide QEHA abolished the α-MSH–mediated decrease in IA. Further, α-MSH increased the expression levels of phospho-p38 mitogen-activated protein kinase, and pharmacological or genetic inhibition of p38α abrogated the α-MSH–induced IA response. Additionally, α-MSH significantly increased the action potential firing rate of TG neurons and increased the sensitivity of rats to mechanical stimuli applied to the buccal pad area, and both effects were abrogated by IA blockade. Taken together, our findings suggest that α-MSH suppresses IA by activating MC4R, which is coupled sequentially to the Gβγ complex of the Gi/o-protein and downstream class I PI3K-dependent p38α signaling, thereby increasing TG neuronal excitability and mechanical pain sensitivity in rats.
Keywords: electrophysiology, patch clamp, potassium channel, signal transduction, G-protein-coupled receptor (GPCR), ion channel, pain, cell signaling, A-type K+ current, alpha-melanocyte-stimulating hormone, melanocortin type 4 receptor (MC4R), neuronal excitability, trigeminal ganglion neurons, pain perception, nociception, action potential
Introduction
The α-melanocyte–stimulating hormone (α-MSH)4 is primarily a pigmentary hormone derived from proopiomelanocortin (POMC), a common precursor protein of all melanocortin peptides, which predominantly expresses in the pituitary gland (1). Five G-protein–coupled melanocortin receptors, named MC1R–MC5R, have been characterized as the endogenous receptors of α-MSH (2). These receptors differ in their tissue distribution and in their ability to recognize various melanocortins. Consistent with the distribution of MCRs in the nervous system (mainly MC3R and MC4R) and peripheral organs (3), α-MSH exerts a large array of behavioral effects and regulates a variety of physiological functions, including blood pressure regulation, grooming, immunity, and body weight homeostasis (4, 5). Additionally, in vivo studies have revealed that α-MSH, as well as the melanocortin receptor agonist melanotan II, causes hyperalgesia in various pain models (6–8). Further support for this hypothesis came from the observation that antagonism of the MC3R and MC4R can effectively alleviate chronic constriction injury (CCI)-induced allodynia (9, 10). Nevertheless, the contributing mechanisms underlying α-MSH hyperalgesic actions are not well-understood.
Alterations in the excitability of nociceptive sensory neurons can directly affect allodynia and hyperalgesia, the two common symptoms of pain (11). Voltage-gated ion channels, including K+ channels (Kv), are pivotal determinants of membrane excitability in peripheral nociceptive neurons (12) and are classified into two major categories, A-type and delayed rectifier channels, which mediate currents of IA and IDR, respectively (13, 14). IA currents are defined by their sensitivity to 4-aminopyridine and their rapidly activating and quick inactivating characteristics (15). These channels play critical roles in determining the intrinsic membrane properties and the excitability of nociceptive neurons (12, 13), and they have been widely implicated in pain plasticity (15). For instance, peripheral nerve injury was shown to induce the down-regulation of IA, resulting in increased excitability of sensory neurons, which may increase the responsiveness to nociceptive stimulation (16). In addition, genetic and functional analyses have firmly established an important role of A-type channels in amplifying nociceptive signals in the periphery and in contributing to central sensitization in the spinal dorsal horn (15, 17, 18). Manipulation of A-type channels, therefore, is expected to modulate intrinsic neuronal excitability and subsequent nociceptive transmission, which is considered useful for pain therapy.
In the present study, we determined the role of α-MSH in regulating IA and elucidated the detailed mechanisms underlying the actions of α-MSH in small-diameter (<30 μm) TG neurons. Our findings demonstrated that α-MSH decreased IA via a Gβγ-dependent class I PI3K and the downstream p38α signaling pathway. This response occurred through the MC4R activation and contributed to the neuronal hyperexcitability of TG neurons and pain hypersensitivity in rats.
Results
α-MSH selectively decreases IA in small-sized TG neurons
Different subtypes of peripheral sensory neurons are usually examined in in vitro studies of nociceptive and other forms of sensory processing (19–23). In this study, we sorted adult rat TG neurons into small-sized (<30 μm in soma diameter) and medium-sized (30–45 μm in soma diameter) groups and limited the whole-cell recording to the small ones, as these neurons played pivotal roles in the nociceptive processing (20, 24). Kv currents in the small-sized nociceptive neurons are functionally characterized into two main types, including a transient outward, A-type K+ current (IA) and a delayed rectifier K+ current (IDR) (13, 25). Therefore, we first separated the two kinetically different whole-cell currents in our patch clamp recordings. A whole-cell outward K+ current was elicited by a command potential of +40 mV from a holding potential of −80 mV in TG neurons (Fig. 1A). The typical current profile observed in these neurons exhibited a component characteristic of fast inactivation and a following sustained portion. After inactivating the transient outward K+ currents by a 150-ms-long prepulse, only the IDR was left. Further off-line subtraction of IDR from the total outward current yielded IA (Fig. 1A). The application of 5 mm 4-aminopyridine (4-AP) to TG neurons dramatically inhibited this IA (decrease of 81.9 ± 5.1% at +40 mV, n = 9; Fig. 1B), indicating the effective separation of IA. Bath application of α-MSH (0.1 μm) significantly reduced the peak current amplitude of IA by 33.2 ± 2.8% in small-sized TG neurons (n = 8), whereas IDR was not significantly affected (decrease of 2.1 ± 1.6%, n = 8) (Fig. 1, C and D). The inhibition of IA was partially reversible within 5 min of washout of α-MSH (Fig. 1D). Further investigation showed that the α-MSH–induced IA suppression was concentration-dependent and had a median effective concentration (EC50) of 36.9 nm (Fig. 1E).
Figure 1.

α-MSH decreases IA in TG neurons. A, representative current traces from small-sized TG neurons before (top) and after (bottom) application of 5 mm 4-AP. Left, IA was isolated using a two-step voltage protocol. Right, IA was obtained after off-line subtraction of the noninactivating component. Insets in the top panel show the stimulation waveform, which was used for the IA isolation indicated in all figures. B, current-voltage (I/V) relationships of IA current density versus test potential for 5 mm 4-AP effects (n = 9). C and D, representative traces (C) and summary data of current density (D) (n = 8) demonstrating that 0.1 μm α-MSH selectively decreases IA. E, concentration–response curve plotting the inhibitory effects of α-MSH on IA. The curves were fitted with a sigmoidal Hill function (I/Icontrol = 1/(1 + 10(logEC50 − log(α-MSH))nH) to compute the effective concentration at which the half-maximum effect occurs (EC50). The number of cells tested at each concentration of α-MSH is shown in brackets. F and G, representative current traces (F) and I/V curves (G) (n = 8) indicating the inhibitory effects of 0.1 μm α-MSH on the current density of IA. The holding potential was −80 mV, and currents were stimulated with voltage protocols from −70 to +70 mV with +10-mV increments. H and I, application of 0.1 μm α-MSH to TG neurons had no significant effect on the voltage dependence of activation potential (H) (n = 12) but leftward-shifted the half-potential of the inactivation curve (I) (n = 12). To measure the voltage dependence of activation, voltage stimuli lasting 400 ms were delivered with voltage steps ranging from −70 to +70 mV. To determine the steady-state inactivation, prepulses ranging from −120 to +20 mV were applied with 10-mV increments, followed by a 500-ms voltage step to +40 mV. J, bar graph showing the effects of 0.1 μm α-MSH on Vhalf of the activation and inactivation curves. *, p < 0.05; **, p < 0.01 versus control. Error bars, S.E.
We then investigated whether the biophysical properties of A-type channels was also affected by α-MSH. The current–voltage relationships (I/V curves) indicated that at each test potential above −20 mV, 0.1 μm α-MSH significantly reduced the peak current amplitude of IA (Fig. 1, F and G), and at +40 mV, the peak current density of IA decreased from 134.7 ± 21.7 to 78.3 ± 15.8 pA/picofarad (n = 8; Fig. 1G). Next, we analyzed the voltage dependence of activation and inactivation potentials following the α-MSH application. Whereas the potential of voltage-dependent activation did not change significantly (Vhalf from 8.9 ± 2.4 to 9.7 ± 3.3 mV; n = 12; Fig. 1, H and J), application of 0.1 μm α-MSH to TG neurons significantly shifted the inactivation curve toward the hyperpolarizing direction by ∼10.7 mV (Vhalf from −52.6 ± 1.9 to −63.3 ± 1.4 mV; n = 12; Fig. 1, I and J). These results reveal that the increased proportion of A-type channels in the steady-state inactivation might contribute to the α-MSH-induced IA decrease.
MC4R mediates the α-MSH–induced IA decrease
Five types of melanocortin receptors in the mammalian genome have been identified as the endogenous receptors for α-MSH, among which only MC3R and MC4R prominently expressed in the spinal cord and dorsal root ganglia (3, 7, 26, 27). We determined the participation of these two receptors in α-MSH–induced IA decrease by first examining the expression profile of MC3R and MC4R. RT-PCR analysis demonstrated that only the MC4R transcripts (the predicted size is 923 bp), and not the MC3R (the predicted size is 307 bp), were detected in rat TG tissues (Fig. 2A). MC4R protein expression in TGs was confirmed by Western blotting (Fig. 2B). Next, we analyzed the subcellular localization of MC4R protein by immunofluorescent staining. In rat TG sections, 32.5 ± 6.3% of MC4R-positive neurons were co-stained with IB4, and 43.9 ± 5.2% of MC4R-positive neurons exhibited immunoreactivity for CGRP (Fig. 2, C and D). In contrast, only 7.5 ± 1.7% of MC4R-positive neurons were positive for neurofilament 200, a specific marker for neurons with myelinated fibers (Fig. 2, C and D). The expression pattern of MC4R suggests that it might be involved in the α-MSH–mediated IA response in small-sized TG neurons. Indeed, the pretreatment of cells with 0.5 μm HS024, a selective MC4R antagonist, completely abolished the 0.1 μm α-MSH–mediated decrease in IA (decrease of 2.1 ± 0.9%, n = 9; Fig. 2, E and F), whereas the application of HS024 (0.5 μm) alone had no effects on IA (Fig. 2, E and F). Next, we investigated whether selective activation of MC4R mimics the inhibitory effect of α-MSH. Indeed, bath application of 0.2 μm Ro 27-3225, a specific MC4R agonist, dramatically decreased the peak amplitude of IA (27.7 ± 1.9%, n = 6; Fig. 2, G and H), further supporting the involvement of MC4R in α-MSH–induced IA decrease.
Figure 2.

MC4R mediates the α-MSH–induced IA decrease. A, determination of MC3R and MC4R transcripts in rat TGs. The negative control without reverse transcriptase (−RT) did not show any signal. B, Western blot analysis indicating the protein expression profile of MC4R (predicted band size 37 kDa). The GAPDH level served as a loading control. The blots shown are representative of three independent experiments. C, colocalization of MC4R (red) with three markers (green) (IB4, CGRP, and neurofilament 200) in naive rat TG sections. Arrows, colocalization. Scale bars, 40 μm. D, quantification of MCR4 colocalization in TGs. E and F, time course of IA changes (E) and summary of results (F) (n = 9) showing that pre-incubation of neurons with HS024 (0.5 μm) prevented the 0.1 μm α-MSH–induced IA decrease. The letters on the plot indicate which points were utilized for sample traces. G and H, representative current traces (G) and summary of results of current density (H) (n = 6) indicating the effect of 0.2 μm Ro 27-3225 on IA. Insets, representative current traces. *, p < 0.05 versus control; ***, p < 0.001 versus 0.1 μm α-MSH. Error bars, S.E.
MC4R mediates IA decrease via Gβγ-dependent PI3K signaling
The signaling of MC4R activation in a variety of primary cells and tissues is known to act via heterotrimeric Gs-proteins (28). Pre-incubating TG neurons with 0.5 μg/ml cholera toxin (CTX), which effectively inactivated by Gαs (24, 29), did not affect the α-MSH–induced IA response (decrease of 29.1 ± 3.7%, n = 10; Fig. 3, A and C). Contrastingly, inhibition of Gαi/o by pretreatment with 0.2 μg/ml pertussis toxin (PTX) for 16 h abolished the inhibitory effect of α-MSH on IA (decrease of 1.2 ± 3.9%, n = 8; Fig. 3, B and C). Dialysis of small-sized TG neurons with 10 μm QEHA, a synthetic peptide that competitively binds Gβγ and blocks the Gβγ-mediated signaling (30), prevented the α-MSH–induced response in IA (decrease of 2.1 ± 1.8%, n = 8; Fig. 3D), whereas intracellular application of SKEE (10 μm), the scrambled peptide of QEHA, did not elicit such effects (decrease of 34.1 ± 4.6%, n = 11; Fig. 3D). These results suggest that the Gβγ complex of Gi/o-protein participates in the MC4R-induced IA decrease. Previous studies showed that protein kinase A (PKA) is downstream of Gβγ activation (31); however, dialyzing small TG neurons with 1 μm PKI 6-22, a synthetic peptide inhibitor of PKA, did not alter the ability of α-MSH to decrease IA (decrease of 31.2 ± 3.9%, n = 9; Fig. 3D). The PKI 6-22 at 1 μm was effective in this assay because the intracellular administration of PKI 6-22 resulted in a nearly complete blockade of 20 μm forskolin-induced IA response (decrease of 1.5 ± 3.1%, n = 7; Fig. 3E). PI3K activation has been shown to modulate IA and act as a downstream effector of Gβγ activation (32). Application of either 0.1 μm α-MSH or the selective MC4R agonist Ro 27-3225 at 0.2 μm significantly enhanced the PI3K activity, and the pretreatment of TG neurons with the selective MC4R antagonist HS024 at 0.5 μm completely abolished these effects (Fig. 3F). Further, pretreating TG neurons with 0.5 μm wortmannin, a PI3K inhibitor (decrease of 3.1 ± 2.7%, n = 8; Fig. 3G), or 1 μm CH5132799, a selective inhibitor of class I PI3Ks (decrease of 1.1 ± 3.9%, n = 7; Fig. 3G), completely abolished the α-MSH–mediated decrease in IA, indicating the requirement of class I PI3K in the MC4R-mediated IA response.
Figure 3.

The α-MSH–induced IA decrease requires the Gβγ-dependent PI3K. A and B, time course of changes in IA amplitude mediated by 0.1 μm α-MSH in CTX (0.5 μg/ml for 16-h pretreatment, n = 10) (A) and PTX (0.2 μg/ml for 16-h pretreatment, n = 8) (B), respectively. Insets, representative current traces. The letters on the plot indicate which points were used for sample traces. C, summary data showing the effect of 0.1 μm α-MSH on IA in cells pretreated with CTX and PTX, respectively, indicated in A and B. D, summary of results showing the effects of 0.1 μm α-MSH on IA in the presence of QEHA (10 μm, intracellular application, n = 8), SKEE (10 μm, intracellular application, n = 11), and PKI 6-22 (1 μm, intracellular application, n = 9), respectively. E, representative current traces (left) and summary of results (right) depicting that dialysis with 1 μm PKI 6-22 prevented 20 μm forskolin-induced IA response (n = 7). F, bar graph indicating that pre-incubation of cells with HS024 abolished the increase in PI3K activity induced by 0.1 μm α-MSH or 0.2 μm Ro 27-3225. All experiments were performed in triplicate with similar results. G, summary data indicating that treatment of TG cells with wortmannin (0.5 μm, n = 8) or CH5132799 (1 μm, n = 7) prevented the 0.1 μm α-MSH–induced IA decrease. ***, p < 0.001 versus control. #, p < 0.05; ##, p < 0.01 versus vehicle. @, p < 0.05 versus α-MSH. $, p < 0.05 versus Ro 27-3225. Error bars, S.E.
The α-MSH–induced IA decrease requires p38 MAPK
Akt is a common and major downstream target of PI3K. Therefore, we examined whether the α-MSH action in TG neurons was through the Akt activation. Although the phosphorylated Akt (p-Akt) level was significantly increased following the α-MSH treatment (Fig. 4A), the inhibition of Akt activity by 10 μm Akt inhibitor III showed no influence on 0.1 μm α-MSH–induced IA decrease (decrease of 31.9 ± 7.6%, n = 11; Fig. 4B). This finding suggests that the α-MSH–induced IA response is independent of Akt. It has been shown that mitogen-activated protein kinase (MAPK) cascades play pivotal roles in pain sensation (33), and the MAPK family molecules are involved in the regulation of IA (34). Therefore, subsequently, we investigated in TG neurons whether MAPK participated in the MC4R-mediated decrease in IA. Immunoblot analysis indicated that exposure of TG cells to 0.1 μm α-MSH markedly increased phosphorylated p38 (p-p38), whereas the total p38 (t-p38), as well as phospho-JNK and phospho-ERK (p-ERK) expression levels, remained unaffected (Fig. 4C). Pre-incubation of TG cells with the PI3K antagonist wortmannin at 0.5 μm eliminated the α-MSH–induced p38 activation (Fig. 4D), indicating the involvement of PI3K-dependent p38 MAPK activation. Furthermore, the pretreatment of TG neurons with the p38 MAPK inhibitor SB203580 at 10 μm abrogated the α-MSH–induced decrease in IA (decrease of 2.9 ± 2.1%, n = 9; Fig. 4, E and G). Contrastingly, the negative control compound SB202474, which is structurally related to SB203580 but does not inhibit p38 MAPK activity, elicited no such effects at the same concentration (decrease of 30.9 ± 4.9%, n = 7; Fig. 4, F and G). Thus, the Akt-independent p38 MAPK activation is required for the α-MSH–induced IA decrease in TG neurons.
Figure 4.

α-MSH attenuates IA through activation of p38 MAPK. A, α-MSH at 0.1 μm induced a significant increase in the expression levels of phosphorylated Akt (p-Akt) in TG cells. This response was prevented by pretreatment with the Akt inhibitor III (Akt inhibitor; 10 μm). The blots shown are representative of three independent experiments. B, time course of IA changes (left) and summary of results (right) showing that pre-incubation of cells with 10 μm Akt inhibitor did not affect the 0.1 μm α-MSH–induced IA response (n = 11). The letters on the plot indicate which points were utilized for sample traces. Insets, representative current traces. C, α-MSH at 0.1 μm significantly increased the protein expression levels of phosphorylated p38 (p-p38) but did not affect the levels of phospho-JNK (p-JNK) and phospho-ERK (p-ERK) in TG cells. All blots are representative of three independent experiments. D, pretreating TG cells with 0.5 μm wortmannin (wort.) abolished the 0.1 μm α-MSH–induced p38 activation. The blots shown are representative of three independent experiments. E and F, time course of changes in IA amplitude mediated by 0.1 μm α-MSH in cells pretreated with 10 μm SB203580 (n = 9) (E) or SB202474 (10 μm, n = 7) (F). G, summary data showing that pretreating cells with 10 μm SB203580 prevented the 0.1 μm α-MSH–induced IA decrease. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus control. Error bars, S.E.
p38α mediates the α-MSH–induced IA response
In the nervous system, such as in rat spinal cord, two major isoforms of p38 exist, p38α and p38β (Fig. 5A). We further dissected the exact p38 isoform involved in the α-MSH–induced IA decrease in rat TG neurons. Western blot analysis revealed that only p38α, and not p38β, was endogenously expressed in adult rat TGs (Fig. 5A). Pretreating TG neurons with 50 nm JX-401, a potent inhibitor specific to the p38α isoform, completely abolished the 0.1 μm α-MSH–mediated decrease in IA (decrease of 2.9 ± 3.5%, n = 9; Fig. 5B). To further confirm this specific p38α-mediated signaling pathway, we utilized an adenovirus-based shRNA approach to knock down p38α in TG cells. In contrast to the substantial expression of p38α in the cells transduced with negative control shRNA (NC-shRNA), the protein expression level of p38α was markedly reduced in TG cells transduced with the p38α-specific shRNA (p38α-shRNA; Fig. 5C). Knockdown of p38α in TG neurons led to the attenuation of the α-MSH–mediated decrease in IA (decrease of 2.3 ± 2.1%, n = 17; Fig. 5D), indicating that p38α is specifically involved in the α-MSH–induced IA decrease.
Figure 5.

The p38α mediates the α-MSH–induced IA response. A, Western blot analysis showing that p38α, but not p38β, was endogenously expressed in rat TGs. Rat spinal cord was used as a positive control. The blots shown are representative of three independent experiments. B, time course of changes in IA amplitude (left) and summary data (right) indicating the effect of α-MSH (0.1 μm) on IA in cells pretreated with JX-401 (50 nm, n = 9). C, immunoblot analysis showed that the protein expression level of p38α was significantly reduced in p38α-shRNA–transducing groups. Depicted immunoblots are representative of three different experiments. D, exemplary current traces (left) and summary data (right) demonstrating that the treatment of TG neurons with p38α-shRNA prevented the 0.1 μm α-MSH–induced decrease in IA (n = 17). *, p < 0.05; ***, p < 0.001 versus control; ##, p < 0.01 versus NC-shRNA. Error bars, S.E.
α-MSH enhances TG neuronal excitability through IA modulation
IA encoded by Kv channels plays pivotal roles in modulating membrane excitability in multiple excitable cell types, including TG neurons (15, 23). To determine the functional roles of the IA decrease mediated by MC4R activation, we investigated whether α-MSH application might affect the neuronal excitability. The application of 0.1 μm α-MSH to small-sized TG neurons had no effect on the whole-cell Nav currents (decrease of −2.7 ± 1.6% at −10 mV, n = 10; Fig. 6A). It has been shown that the activation of MC4R inhibits presynaptic N-type Ca2+ channels in amygdaloid complex neurons (35). To exclude the possible influence of α-MSH regulation of N-type as well as other types of Ca2+ channels in TG neurons, we applied CdCl2 (100 μm) in the external solution to block voltage-gated Ca2+ channels during current clamp recordings. We observed that the application of α-MSH at 0.1 μm significantly increased action potential (AP) firing frequency by 61.8 ± 5.3% (n = 15; Fig. 6, B and C). Additionally, 0.1 μm α-MSH significantly shortened first spike latency and increased AP amplitude (n = 15; Fig. 6D). Other membrane properties of neuronal excitability, such as resting membrane potential and input resistance, were not significantly changed by the α-MSH application (not shown). Pretreating TG neurons with the MC4R antagonist HS024 (0.5 μm) prevented the increased AP firing rate induced by 0.1 μm α-MSH, indicating the MC4R involvement (n = 11; Fig. 6E). To further verify that the MC4R-mediated neuronal hyperexcitability occurred through the decrease of IA, we applied 4-AP in the external solution to block IA and found that application of 5 mm 4-AP mimicked the MC4R-mediated neuronal hyperexcitability (n = 10; Fig. 6, F and G). Notably, application of α-MSH during the 4-AP–induced response failed to produce any further increase in firing frequency (Fig. 6G). These findings suggest that the MC4R-mediated IA decrease contributes to the α-MSH–induced TG neuronal hyperexcitability.
Figure 6.

α-MSH enhances TG neuronal excitability. A, I/V curves (left) and summary of results (right) depicting the effect of 0.1 μm α-MSH on the whole-cell Nav currents (INa, n = 10). INa was elicited by a 40-ms depolarizing step pulse from −80 to +60 mV with the holding potential at −60 mV. B and C, exemplary traces of AP firing (B) and summary of results (C) depicting the change of AP firing rate before versus after 0.1 μm α-MSH application (n = 15). Current injections of +130 pA into the soma are shown in the top panels. D, bar graph showing that α-MSH at 0.1 μm significantly shortened first-spike latency and increased the AP amplitude in small TG neurons (n = 15). E, summary of results depicting that pretreating TG neurons with 0.5 μm HS024 abolished the increase of AP firing rate in response to 0.1 μm α-MSH (n = 11). F and G, representative traces of AP firing (F) and summary of results (G) depicting that pre-incubation of TG neurons with 5 mm 4-AP prevented the 0.1 μm α-MSH–induced neuronal hyperexcitability (n = 10). Current injections of +80 pA into the soma are shown in the top panels. *, p < 0.05; **, p < 0.01 versus control. Error bars, S.E.
α-MSH–mediated IA decrease induces pain hypersensitivity in vivo
To further determine the functional implications of MC4R at the behavioral level, we investigated whether α-MSH affects peripheral pain sensitivity in rats. The escape threshold to mechanical facial nociception was determined by an ascending series of von Frey filaments, which were applied to the buccal pad region. Intra-TG injection of α-MSH at 2 nmol induced a marked decrease in the escape threshold 1–2 h post-administration (Fig. 7A), and this effect recovered after 3 h. The α-MSH–mediated mechanical pain hypersensitivity was abrogated by intra-TG injection of the MC4R inhibitor HS024 at 5 nmol (Fig. 7B). The participation of IA in the α-MSH–mediated mechanical hypersensitivity was further examined using the specific A-type channel blocker 4-AP. Intra-TG application of 4-AP at 25 nmol exhibited a significant increase in mechanical sensitivity as compared with the control animals that received a saline injection (Fig. 7C). Sensitivity assessed after intra-TG injection of α-MSH showed that α-MSH had no additive effect to 4-AP on mechanical sensitivity in rats (Fig. 7C), which suggests that the effect of α-MSH and 4-AP can be mediated through the same signaling pathway in vivo. Collectively, our data provide evidence that the α-MSH–induced IA decrease contributes to the MC4R-mediated mechanical pain hypersensitivity in rats.
Figure 7.
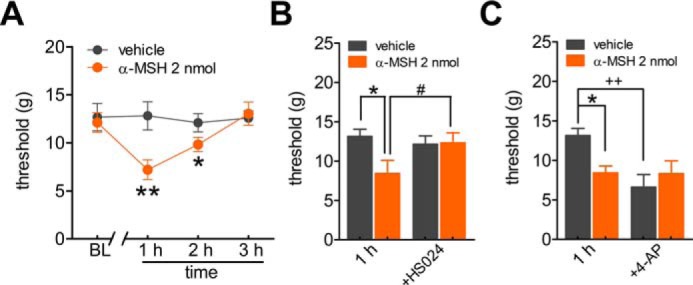
Involvement of peripheral MC4R in pain hypersensitivity. A, intra-TG injection of α-MSH (2 nmol), but not vehicle, induced a markedly mechanical pain hypersensitivity. *, p < 0.05; **, p < 0.01, α-MSH versus vehicle, one-way ANOVA. BL, baseline. B, pretreatment of HS024 (5 nmol) prevented the α-MSH–induced mechanical hypersensitivity. *, p < 0.05, α-MSH versus vehicle; #, p < 0.05, α-MSH + HS024 versus α-MSH at 1 h, one-way ANOVA. Intra-TG injection of 5 nmol of HS024 did not affect the basal escape threshold of normal rats. C, intra-TG pre-injection of 4-AP (25 nmol) occluded the mechanical hypersensitivity induced by 2 nmol of α-MSH. *, p < 0.05, α-MSH versus vehicle; ++, p < 0.01, 4-AP injection versus vehicle at 1 h. For all animal behavior data, n ≥ 7 rats. Error bars, S.E.
Discussion
This study reveals a novel functional role of α-MSH in the regulation of IA in rat TG neurons. We identify a signaling pathway by through α-MSH attenuates IA through stimulation of MC4R that couples to a Gβγ-dependent class I PI3K and the downstream p38α signaling (Fig. 8). One of the immediate outcomes is the induction of sensory neuronal hyperexcitability and mechanical pain hypersensitivity.
Figure 8.
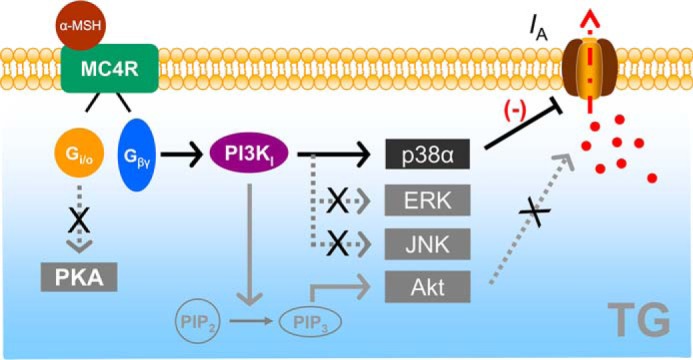
Scheme of the proposed mechanisms of MC4R on IA. α-MSH acts through MC4R, which is coupled to the G-protein Gi/o, causing it to release the Gβγ subunits. The released Gβγ causes an increase in class I PI3K activity in TG neurons. Stimulation of PI3K signaling may phosphorylate p38α to regulate IA and induces TG neuronal hyperexcitability and pain hypersensitivity. PI3K can catalyze the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 4,5-trisphosphate (PIP3), which serves as a second messenger that helps to activate Akt. However, neither Akt nor PKA is necessary for the MC4R-mediated decrease of IA. Determination of whether p38α in small-sized TG neurons directly phosphorylates IA channel subtypes or acts through some intermediate signal molecules requires further investigation.
Studies examining the PKA-dependent modulation of IA lead to conflicting conclusions. In hippocampal pyramidal neurons, activation of PKA by 8-bromo-cAMP significantly decreased IA (36). Similarly, 3-isobutyl-1-methylxanthine stimulation of PKA down-regulated the peak current density of IA (37). In contrast, PKA inducer mimicked the response of serotonin 1D receptor activator and was shown to stimulate IA in TG neurons (38). In the current study, the decrease of IA induced by α-MSH was not affected by the PKA inhibitor PKI 6-22, suggesting that other mechanisms rather than PKA participated in the MC4R-mediated IA response. We observed that in small-sized TG neurons, the selective inhibition of IA by MC4R activation was mediated by the class I PI3K and then relayed to the downstream p38-dependent signaling. Our data are consistent with previous studies conducted in early hippocampal neurons, which demonstrate that the PI3K/Akt signaling mediated the inhibition of IA induced by amyloid-β (39). Similarly, Kv currents, including IA, recorded from pancreatic β cells decrease in response to PI3K activation (40). Interestingly, the activation of PI3K has been shown to increase IA in cultured rat cerebellar granule cells (41), and PI3K-induced activation of the Kv4.3 channel through glucocorticoid-inducible kinase-1 has also been identified (42). Although the discrepancy requires further investigation, the regulatory effects of different PI3K subtypes, including class I through class III, might be variable in distinct tissue/cell types expressing Kv channels.
Akt is the most common component downstream of PI3K. Previous studies suggested that Akt may regulate the activity of Kv4 channels in a cell type– and signal–specific manner. For instance, in cultured cerebellar granule cells of rats, Akt has been shown to down-regulate Kv4.2 channels (43), whereas the Akt-dependent signaling stimulated Kv4 in the arcuate nucleus (44). In the present study, the MC4R-mediated IA decrease appears not to be mediated by Akt in small TG neurons. Accumulating evidence has suggested that the extracellular signal–regulated kinase-1 and -2 (ERK1/2) signaling is involved in the modulation of pain (45). Additionally, ERK was shown to directly phosphorylate the pore-forming subunit of Kv4.2 channels (46), and activation of ERK attenuated IA in neurons in superficial laminae of the spinal dorsal horn (34). In contrast, ERK signaling mediating the dopamine-induced IA increase in lateral pyloric neurons was also reported (47). However, in this study, the stimulation of MC4R in small TG neurons resulted in an elevated level of phospho-p38, but not phospho-ERK or phospho-JNK, excluding the possible involvement of ERK in the MC4R-induced IA response. Our findings are in line with previous studies conducted in dorsal root ganglion neurons and heart cells, in which the p38 activation led to a decrease of Kv currents (48) by phosphorylation of the residues Tyr-124 of Kv channels (49). On the contrary, p38 has been shown to stimulate Kv currents in transfected Chinese hamster ovary cells (50). This discrepancy could be explained by distinct p38α phosphorylation sites, by different Kv subtypes encoding IA in TG neurons and in modified Chinese hamster ovary cells (51), or by distinct splice variants of Kv channel–interacting protein (52).
Changes of peripheral neuronal excitability can directly influence painful conditions such as hyperalgesia and allodynia in vivo (11, 12). A-type channels, the key components influencing neuronal excitability, have been implicated in controlling both the delay of first-spike latency and the decrease in spike frequency (15, 53), which are two important determinants in the release time course of neurotransmitter and hence the nociceptive transmission (54). An important consequence of peripheral A-type channel modulation is to influence somatic and visceral nociceptive inputs, and the decrease of IA results in significant nociception in a variety of animal neuropathic pain models (17). In the current study, consistent with the α-MSH–induced decrease in IA in small-sized TG neurons, activation of MC4R significantly increased TG neuronal excitability along with the shorter first-spike latency and increased firing frequency; blockade of IA by 4-AP prevented this effect. Moreover, the acute mechanical hypersensitivity induced by α-MSH was occluded by the A-type channel blocker application. As such, it is reasonable to infer that the nociceptive effects of MC4R activation are mediated, partially if not completely, through its inhibition of IA. Indeed, our present results are in line with previous in vivo studies indicating that antagonism of MC4R reduces the CCI-induced allodynia in rodents (9, 55, 56), whereas activation of MC4R causes hyperalgesia in various pain models (6–8). Nevertheless, potential channel targets other than A-type channels can also be activated by the MC4R pathway. It has been shown that the activation of MC4R with its agonist melanotan II inhibits presynaptic N-type Ca2+ channels in amygdaloid complex neurons (35). In addition, MC4R constitutive activity inhibits Cav1.2 channel currents in transiently transfected HEK293 cells (57).
In summary, we present new insights underlying the effect of α-MSH on IA in small-sized TG neurons. Our findings indicated that α-MSH decreases IA through stimulation of a Gi/o-protein–coupled MC4R and downstream class I PI3K-mediated p38α signaling. The identified signaling pathway mediates the functional effects of α-MSH on induced sensory neuronal hyperexcitability and mechanical pain hypersensitivity. It would be interesting to further examine the effects of MC4R signaling on other types of pain (e.g. pain caused by thermal injuries) in future work. Moreover, previous studies have revealed that the expression of p38α is up-regulated in ipsilateral TGs in an orofacial inflammatory pain model induced by complete Freund's adjuvant (58) and that pharmacological blockade of p38 activation reduces the CCI-induced thermal hyperalgesia (56). Therefore, the present findings regarding the MC4R-mediated p38α pathway in TG neurons may be reproduced in other sensory neurons, including dorsal root ganglia, and may pave the way for MC4R to be developed as a potential therapeutic target for the clinical management of chronic pain.
Experimental procedures
Dissociation of TG neurons
Rats were maintained in specific pathogen-free facilities on a 12-h/12-h light/dark cycle at a room temperature (22 ± 1 °C) and housed in cages with access to food and water ad libitum. All procedures in the animal studies were approved by the Animal Studies Committee of Soochow University and were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. TG neurons were dissociated from Sprague-Dawley rats (220–250 g, regardless of gender) according to the established protocol (38, 39). Briefly, the TGs were dissected out bilaterally, and the connective tissue was removed. After they were minced into 8–10 pieces, the tissues were enzymatically digested first with 1.5 mg/ml collagenase II (Worthington) for 30 min and then with 20 units/ml papain for 20 min (Worthington). After papain treatment, the tissue was mechanically dissociated by trituration with sterile fire-polished glass pipettes. After centrifugation, the pellet was resuspended in minimum essential medium (Invitrogen) containing 10% fetal bovine serum (Hyclone, GE Healthcare), 2% B27 supplements (Invitrogen), 1% GlutaMAX, and 1% penicillin/streptomycin (Invitrogen) and then plated onto glass coverslips coated with Matrigel. The cells were maintained at 37 °C in humidified incubators with 5% CO2 and 95% air. Electrophysiological recordings were performed 2–6 h after plating.
Whole-cell patch clamp recordings
Electrophysiological procedures were performed at room temperature (23 ± 1 °C) as described previously (22, 24). We chose only small-sized TG neurons that were less than 30 μm in soma diameter for patch clamp experiments. Pipettes (Sutter Instruments) had 3–5-megaohm resistance when filled with a pipette solution containing the following: 140 mm KCl, 10 mm HEPES, 1 mm MgCl2, 0.5 mm CaCl2, 5 mm EGTA, 0.5 mm Na2GTP, and 3 mm Mg-ATP, with pH of 7.4 and osmolarity of 295 mosmol/kg H2O. The recording chamber was superfused continuously with the external solution containing the following: 150 mm choline-Cl, 10 mm HEPES, 1 mm MgCl2, 0.03 mm CaCl2, 5 mm KCl, and 10 mm glucose, with pH of 7.4 and osmolarity of 310 mosmol/kg H2O. Series resistance was compensated by at least 75% in voltage clamp mode. Currents were filtered at 1 kHz and recorded using a MultiClamp 700B amplifier (Molecular Devices). The whole-cell current clamp was used to record changes in action potential firing of TG neurons. In whole-cell current clamp experiments and Nav current recordings, the internal solution of electrodes contained 10 mm NaCl, 110 mm KCl, 2 mm EGTA, 25 mm HEPES, 0.5 mm Na2GTP, and 4 mm Mg-ATP, with pH of 7.3 and osmolarity of 295 mosmol/kg H2O. The bath solution contained 2 mm KCl, 128 mm NaCl, 2 mm CaCl2, 2 mm MgCl2, 30 mm glucose, and 25 mm HEPES, with pH of 7.4 and osmolarity of 305 mosmol/kg H2O. During current clamp recordings, we applied 100 μm CdCl2 in the external solution to block voltage-gated Ca2+ channels. In experiments in which neurons were dialyzed with compounds, patch pipette resistance ranged from 2 to 3 megaohms for infusion, and currents were measured at least 5 min after breaking into the whole-cell configuration.
Detection of gene expression
RT-PCR was performed as described previously (30). Briefly, total RNA from rat entire TG tissues was extracted using an RNeasy kit (Qiagen, Germantown, MD) according to the manufacturer's protocol. RNA was treated with DNase (Promega Corp., Madison, WI) and then reverse-transcribed with SuperScriptTMII (Thermo Fisher Scientific). The primers, designed in Primer version 5.0 software (Premier Biosoft International, Palo Alto, CA), were derived from a partial genomic sequence: MC3R, accession number NM_001025270.3, 5′-TCTGCTGTGCTGTGGGGGTG-3′ (forward) and 5′-CGGTTAGGCGGGTCGGGATC-3′ (reverse); MC4R, accession number NM_013099.3, 5′-CCACAAGAGAAGCACCTAGA-3′ (forward) and 5′-GTTGCCGTTCCTCACCACAG-3′ (reverse). Experiments included non-reverse-transcribed DNase-treated RNA samples as negative controls. PCR of these RT control groups never showed amplification, indicating that the RNA was genomic DNA-free. The assays were carried out in duplicate using the same samples to confirm the reproducibility of the results.
Western blot analysis
Western blot analyses were performed following a procedure described previously (22, 24). In brief, 25 μg of extracted proteins were separated in 7.5% SDS-polyacrylamide gels and electroblotted onto polyvinylidene difluoride membranes (Merck Millipore). Membranes were blocked with 5% nonfat dry milk in TBST (0.05% (w/v) Tween in TBS) and were incubated overnight at 4 °C with primary antibodies. The primary antibodies utilized included MC4R (rabbit, 1:1000; Abcam), phospho-Akt (rabbit, 1:1000; Cell Signaling Technology), Akt (rabbit, 1:2000; Cell Signaling Technology), phospho-p38 (rabbit, 1:600; Cell Signaling Technology), p38 (rabbit, 1:1000; Cell Signaling Technology), p38α (rabbit, 1:500; Santa Cruz Biotechnology), p38β (rabbit, 1:1000; Santa Cruz Biotechnology), phospho-ERK1/2 (rabbit, 1:1000; Cell Signaling Technology), ERK1/2 (rabbit, 1:6000; Cell Signaling Technology), phospho-SAPK/JNK (rabbit, 1:500; Cell Signaling Technology), SAPK/JNK (rabbit, 1:1000; Cell Signaling Technology), and GAPDH (rabbit, 1: 3000, Abcam). After extensive washing, the membranes were incubated with appropriate horseradish peroxidase–conjugated secondary antibodies (Cell Signaling Technology) and then reacted with enhanced chemiluminescence substrate (Pierce). Western blotting signals were visualized using the ChemiDoc XRS system (Bio-Rad). The Quantity One software (Bio-Rad) was used for chemiluminescence measurements and quantification of the immunoblot data.
Immunofluorescence
The standard procedure of immunohistochemistry was performed as described in our previous studies (22). Briefly, after sectioning (15 μm) in a cryostat (CM 1950; Leica), TG sections were blocked with 5% normal goat serum in PBS plus 0.2% Triton X-100 for 1 h and then incubated overnight in the primary antibody against MC4R (rabbit, 1:500; Abcam), antibody against NF-200 (mouse, 1:1000; Abcam), or antibody against CGRP (mouse, 1:1000; Abcam). The sections were washed with PBS, followed by Cy3-conjugated goat anti-rabbit IgG (1:500; Abcam), FITC-conjugated goat anti-mouse IgG (1:400; Abcam), or IB4-FITC (5 μg/ml; Sigma-Aldrich) for 2 h at room temperature. Slices were viewed under an upright fluorescence microscope (BX-51, Olympus), and images were taken using a CCD camera (DP70, Olympus). Negative controls incubated with secondary antibody only did not display any positive staining (not shown).
Measurement of PI3K activity
PI3K activity was determined as described previously (32). In brief, after stimulation of cells with α-MSH (0.1 μm) or Ro 27-3225 (0.2 μm) for 20 min, PI3K activity in homogenates was determined using a PI3K HTRFTM assay kit (Millipore Corp., Bedford, MA) according to the manufacturer's protocol. HTRF was then measured with an excitation wavelength of 335 nm and emission wavelengths of 620 and 665 nm with a spectrofluorometer (Tecan, Infinite M1000, Salzburg, Austria).
Adenovirus transduction
Adenovirus-mediated gene silencing was performed following a procedure described previously (22, 32). Three candidate shRNAs targeting p38α (GenBankTM accession number NM_031020.2) were designed, and the shRNA sequence (5′-GGACCTCCTTATAGACGAATG-3′) with the best knockdown efficiency were selected. The nonsense sequence was designed as the negative control (NC-shRNA, 5′-ACCTGACTGTGTCAGGAATCA-3′). The pAdTrack-CMV-GFP vector carrying p38α shRNA (p38α-shRNA) plasmid was packaged by Genechem Co., Ltd. (Shanghai, China). After 48 h of infection, the efficiency of shRNA knockdown was determined by Western blot analysis as described above. For electrophysiological analysis, small TG neurons expressing GFP were subjected to whole-cell recordings.
Escape threshold for mechanical stimulation
Animals (Sprague-Dawley rats, 220–250 g, regardless of gender) were housed under standard conditions (22 ± 1 °C, 50–70% humidity, 12-h/12-h light/dark cycle, with ad libitum access to food and water) and were allowed to habituate to laboratory conditions for at least 3 days prior to the experiments. The escape threshold to mechanical stimulation was determined by an ascending series of von Frey filaments (Ugo Basile) as described previously (59, 60). Von Frey stimuli were applied to the buccal pad region and were applied three times in each series of trials. α-MSH (2 nmol), HS024 (5 nmol), or 4-AP (25 nmol) was intra-TG–injected into the trigeminal ganglia with a 30-gauge needle inserted through the infraorbital foramen, infraorbital canal, and foramen rotundum. The needle tip was positioned in the medial part of the ganglia, and the treatment agent was slowly delivered in a volume of 5 μl.
Drug application
CdCl2, 4-aminopyridine, cholera toxin, and pertussis toxin were purchased from Sigma-Aldrich; α-MSH was from Abcam; PKI 6-22 was from Tocris Bioscience; and Akt inhibitor III was from Santa Cruz Biotechnology, all of which were prepared in distilled deionized water as stock solutions. Forskolin, wortmannin, Ro 27-3225, CH5132799, and SB203580 were obtained from Sigma-Aldrich; HS024 was from Tocris Bioscience; SB202474 was from Merck Millipore; and JX-401 was from R&D systems, all of which were prepared as concentrated stock solutions in DMSO. The final concentration of DMSO in the bath solution was less than 0.01%, and this compound had no functional effects on IA (not shown).
Data analysis
Data acquisition and analysis were done using Clampfit version 10.2 software (Molecular Devices) and GraphPad Prism version 5.0 (GraphPad Software). Data are expressed as original traces or presented as means ± S.E. The amplitude of IA was measured at the peak. The voltage dependence of activation and inactivation of IA was fitted with standard Boltzmann equations. A paired or two-sample t test was used when comparisons were restricted to two means. Treatment effects were statistically analyzed using one-way analysis of variance (ANOVA) followed by Dunnett's test or two-way ANOVA followed by Bonferroni's test. Error probabilities of p < 0.05 were considered statistically significant.
Author contributions
Z. Q., X. J., and J. T. conceived the idea, planned the experiments, and wrote the manuscript. Y. Z., D. J., H. L., and J. T. performed the experiments, wrote the manuscript, and analyzed the data. Y. S. contributed to designing experimental procedures. S. G. provided technical assistance. All authors reviewed the manuscript.
This work was supported by National Natural Science Foundation of China Grants 81873731, 81771187, 81622014, 81671080, 81571063, and 81371229; the Innovation Project of Jiangsu Province Qing-Lan Project (to J. T.); Six Talent Peaks Project of Jiangsu Province Grant JY-065; Jiangsu Key Laboratory of Neuropsychiatric Diseases Grant BM2013003; and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions. The authors declare that they have no conflicts of interest with the contents of this article.
- α-MSH
- α-melanocyte–stimulating hormone
- MC4R
- melanocortin type 4 receptor
- TG
- trigeminal ganglion
- Kv
- voltage-gated K+ channel(s)
- IA
- transient outward K+ channel current(s)
- IDR
- sustained delayed-rectifier K+ channel current(s)
- PI3K
- phosphatidylinositol 3-kinase
- PKA
- protein kinase A
- MAPK
- mitogen-activated protein kinase
- CGRP
- calcitonin gene–related peptide
- 4-AP
- 4-aminopyridine
- POMC
- proopiomelanocortin
- CCI
- chronic constriction injury
- CTX
- cholera toxin
- PTX
- pertussis toxin
- NC
- negative control
- AP
- action potential
- ERK
- extracellular signal–regulated kinase
- JNK
- c-Jun N-terminal kinase
- SAPK
- stress-activated protein kinase
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- ANOVA
- analysis of variance.
References
- 1. Mountjoy K. G., Caron A., Hubbard K., Shome A., Grey A. C., Sun B., Bould S., Middleditch M., Pontré B., McGregor A., Harris P. W. R., Kowalczyk R., Brimble M. A., Botha R., Tan K. M. L., et al. (2018) Desacetyl-α-melanocyte stimulating hormone and α-melanocyte stimulating hormone are required to regulate energy balance. Mol. Metab. 9, 207–216 10.1016/j.molmet.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang Y., and Harmon C. M. (2017) Molecular signatures of human melanocortin receptors for ligand binding and signaling. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 2436–2447 10.1016/j.bbadis.2017.04.025 [DOI] [PubMed] [Google Scholar]
- 3. Adan R. A., and Gispen W. H. (1997) Brain melanocortin receptors: from cloning to function. Peptides 18, 1279–1287 10.1016/S0196-9781(97)00078-8 [DOI] [PubMed] [Google Scholar]
- 4. Brzoska T., Luger T. A., Maaser C., Abels C., and Böhm M. (2008) α-Melanocyte-stimulating hormone and related tripeptides: biochemistry, antiinflammatory and protective effects in vitro and in vivo, and future perspectives for the treatment of immune-mediated inflammatory diseases. Endocr. Rev. 29, 581–602 10.1210/er.2007-0027 [DOI] [PubMed] [Google Scholar]
- 5. Bertolini A., Tacchi R., and Vergoni A. V. (2009) Brain effects of melanocortins. Pharmacol. Res. 59, 13–47 10.1016/j.phrs.2008.10.005 [DOI] [PubMed] [Google Scholar]
- 6. Sandman C. A., and Kastin A. J. (1981) Intraventricular administration of MSH induces hyperalgesia in rats. Peptides 2, 231–233 10.1016/S0196-9781(81)80040-X [DOI] [PubMed] [Google Scholar]
- 7. Beltramo M., Campanella M., Tarozzo G., Fredduzzi S., Corradini L., Forlani A., Bertorelli R., and Reggiani A. (2003) Gene expression profiling of melanocortin system in neuropathic rats supports a role in nociception. Brain Res. Mol. Brain Res. 118, 111–118 10.1016/j.molbrainres.2003.08.001 [DOI] [PubMed] [Google Scholar]
- 8. Klusa V., Germane S., Svirskis S., Opmane B., and Wikberg J. E. (2001) The γ(2)-MSH peptide mediates a central analgesic effect via a GABA-ergic mechanism that is independent from activation of melanocortin receptors. Neuropeptides 35, 50–57 10.1054/npep.2000.0843 [DOI] [PubMed] [Google Scholar]
- 9. Vrinten D. H., Adan R. A., Groen G. J., and Gispen W. H. (2001) Chronic blockade of melanocortin receptors alleviates allodynia in rats with neuropathic pain. Anesth. Analg. 93, 1572–1577, table of contents 10.1097/00000539-200112000-00052 [DOI] [PubMed] [Google Scholar]
- 10. Vrinten D. H., Gispen W. H., Groen G. J., and Adan R. A. (2000) Antagonism of the melanocortin system reduces cold and mechanical allodynia in mononeuropathic rats. J. Neurosci. 20, 8131–8137 10.1523/JNEUROSCI.20-21-08131.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Julius D., and Basbaum A. I. (2001) Molecular mechanisms of nociception. Nature 413, 203–210 10.1038/35093019 [DOI] [PubMed] [Google Scholar]
- 12. Waxman S. G., and Zamponi G. W. (2014) Regulating excitability of peripheral afferents: emerging ion channel targets. Nat. Neurosci. 17, 153–163 10.1038/nn.3602 [DOI] [PubMed] [Google Scholar]
- 13. Rasband M. N., Park E. W., Vanderah T. W., Lai J., Porreca F., and Trimmer J. S. (2001) Distinct potassium channels on pain-sensing neurons. Proc. Natl. Acad. Sci. U.S.A. 98, 13373–13378 10.1073/pnas.231376298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo J. L., Qin H. Y., Wong C. K., Tsang S. Y., Huang Y., and Bian Z. X. (2011) Enhanced excitability and down-regulated voltage-gated potassium channels in colonic drg neurons from neonatal maternal separation rats. J. Pain 12, 600–609 10.1016/j.jpain.2010.11.005 [DOI] [PubMed] [Google Scholar]
- 15. Hu H. J., Carrasquillo Y., Karim F., Jung W. E., Nerbonne J. M., Schwarz T. L., and Gereau R. W. 4th (2006) The kv4.2 potassium channel subunit is required for pain plasticity. Neuron 50, 89–100 10.1016/j.neuron.2006.03.010 [DOI] [PubMed] [Google Scholar]
- 16. Takeda M., Tsuboi Y., Kitagawa J., Nakagawa K., Iwata K., and Matsumoto S. (2011) Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol. Pain 7, 5 10.1186/1744-8069-7-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duan K. Z., Xu Q., Zhang X. M., Zhao Z. Q., Mei Y. A., and Zhang Y. Q. (2012) Targeting A-type K+ channels in primary sensory neurons for bone cancer pain in a rat model. Pain 153, 562–574 10.1016/j.pain.2011.11.020 [DOI] [PubMed] [Google Scholar]
- 18. Chien L. Y., Cheng J. K., Chu D., Cheng C. F., and Tsaur M. L. (2007) Reduced expression of A-type potassium channels in primary sensory neurons induces mechanical hypersensitivity. J. Neurosci. 27, 9855–9865 10.1523/JNEUROSCI.0604-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen Y., Williams S. H., McNulty A. L., Hong J. H., Lee S. H., Rothfusz N. E., Parekh P. K., Moore C., Gereau R. W. 4th, Taylor A. B., Wang F., Guilak F., and Liedtke W. (2013) Temporomandibular joint pain: a critical role for Trpv4 in the trigeminal ganglion. Pain 154, 1295–1304 10.1016/j.pain.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tao J., Liu P., Xiao Z., Zhao H., Gerber B. R., and Cao Y. Q. (2012) Effects of familial hemiplegic migraine type 1 mutation T666M on voltage-gated calcium channel activities in trigeminal ganglion neurons. J. Neurophysiol. 107, 1666–1680 10.1152/jn.00551.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Orestes P., Osuru H. P., McIntire W. E., Jacus M. O., Salajegheh R., Jagodic M. M., Choe W., Lee J., Lee S. S., Rose K. E., Poiro N., Digruccio M. R., Krishnan K., Covey D. F., Lee J. H., et al. (2013) Reversal of neuropathic pain in diabetes by targeting glycosylation of Ca(V)3.2 T-type calcium channels. Diabetes 62, 3828–3838 10.2337/db13-0813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Y., Ji H., Wang J., Sun Y., Qian Z., Jiang X., Snutch T. P., Sun Y., and Tao J. (2018) Melatonin-mediated inhibition of Cav3.2 T-type Ca2+ channels induces sensory neuronal hypoexcitability through the novel protein kinase C-η isoform. J. Pineal. Res. 64, e12476 10.1111/jpi.12476 [DOI] [PubMed] [Google Scholar]
- 23. Zhang Y., Wang H., Ke J., Wei Y., Ji H., Qian Z., Liu L., and Tao J. (2018) Inhibition of A-gype K+ channels by urotensin-II induces sensory neuronal hyperexcitability through the PKCα-ERK pathway. Endocrinology 159, 2253–2263 10.1210/en.2018-00108 [DOI] [PubMed] [Google Scholar]
- 24. Zhang Y., Qin W., Qian Z., Liu X., Wang H., Gong S., Sun Y. G., Snutch T. P., Jiang X., and Tao J. (2014) Peripheral pain is enhanced by insulin-like growth factor 1 through a G protein-mediated stimulation of T-type calcium channels. Sci. Signal. 7, ra94 10.1126/scisignal.2005283 [DOI] [PubMed] [Google Scholar]
- 25. Winkelman D. L., Beck C. L., Ypey D. L., and O'Leary M. E. (2005) Inhibition of the A-type K+ channels of dorsal root ganglion neurons by the long-duration anesthetic butamben. J. Pharmacol. Exp. Ther. 314, 1177–1186 10.1124/jpet.105.087759 [DOI] [PubMed] [Google Scholar]
- 26. van der Kraan M., Tatro J. B., Entwistle M. L., Brakkee J. H., Burbach J. P., Adan R. A., and Gispen W. H. (1999) Expression of melanocortin receptors and pro-opiomelanocortin in the rat spinal cord in relation to neurotrophic effects of melanocortins. Brain Res. Mol. Brain Res. 63, 276–286 10.1016/S0169-328X(98)00291-5 [DOI] [PubMed] [Google Scholar]
- 27. Tanabe K., Gamo K., Aoki S., Wada K., and Kiyama H. (2007) Melanocortin receptor 4 is induced in nerve-injured motor and sensory neurons of mouse. J. Neurochem. 101, 1145–1152 10.1111/j.1471-4159.2006.04432.x [DOI] [PubMed] [Google Scholar]
- 28. Adan R. A., and Gispen W. H. (2000) Melanocortins and the brain: from effects via receptors to drug targets. Eur. J. Pharmacol. 405, 13–24 10.1016/S0014-2999(00)00537-9 [DOI] [PubMed] [Google Scholar]
- 29. Yue J., Zhang Y., Li X., Gong S., Tao J., and Jiang X. (2014) Activation of G-protein-coupled receptor 30 increases T-type calcium currents in trigeminal ganglion neurons via the cholera toxin-sensitive protein kinase A pathway. Pharmazie 69, 804–808 [PubMed] [Google Scholar]
- 30. Tao J., Hildebrand M. E., Liao P., Liang M. C., Tan G., Li S., Snutch T. P., and Soong T. W. (2008) Activation of corticotropin-releasing factor receptor 1 selectively inhibits CaV3.2 T-type calcium channels. Mol. Pharmacol. 73, 1596–1609 10.1124/mol.107.043612 [DOI] [PubMed] [Google Scholar]
- 31. Hu C., Depuy S. D., Yao J., McIntire W. E., and Barrett P. Q. (2009) Protein kinase A activity controls the regulation of T-type CaV3.2 channels by Gβγ dimers. J. Biol. Chem. 284, 7465–7473 10.1074/jbc.M808049200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y., Ying J., Jiang D., Chang Z., Li H., Zhang G., Gong S., Jiang X., and Tao J. (2015) Urotensin-II receptor stimulation of cardiac L-type Ca2+ channels requires the βγ subunits of Gi/o-protein and phosphatidylinositol 3-kinase-dependent protein kinase C β1 isoform. J. Biol. Chem. 290, 8644–8655 10.1074/jbc.M114.615021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ji R. R., Gereau R. W. 4th, Malcangio M., and Strichartz G. R. (2009) MAP kinase and pain. Brain Res. Rev. 60, 135–148 10.1016/j.brainresrev.2008.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu H. J., Glauner K. S., and Gereau R. W. 4th (2003) ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. I. Modulation of A-type K+ currents. J. Neurophysiol. 90, 1671–1679 10.1152/jn.00340.2003 [DOI] [PubMed] [Google Scholar]
- 35. Agosti F., López Soto E. J., Cabral A., Castrogiovanni D., Schioth H. B., Perelló M., and Raingo J. (2014) Melanocortin 4 receptor activation inhibits presynaptic N-type calcium channels in amygdaloid complex neurons. Eur. J. Neurosci. 40, 2755–2765 10.1111/ejn.12650 [DOI] [PubMed] [Google Scholar]
- 36. Hoffman D. A., and Johnston D. (1998) Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J. Neurosci. 18, 3521–3528 10.1523/JNEUROSCI.18-10-03521.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Coutts C. A., Balt L. N., and Ali D. W. (2009) Protein kinase A modulates A-type potassium currents of larval zebrafish (Danio rerio) white muscle fibres. Acta Physiol. (Oxf.) 195, 259–272 10.1111/j.1748-1716.2008.01889.x [DOI] [PubMed] [Google Scholar]
- 38. Zhao X., Zhang Y., Qin W., Cao J., Zhang Y., Ni J., Sun Y., Jiang X., and Tao J. (2016) Serotonin type-1D receptor stimulation of A-type K+ channel decreases membrane excitability through the protein kinase A- and B-Raf-dependent p38 MAPK pathways in mouse trigeminal ganglion neurons. Cell. Signal. 28, 979–988 10.1016/j.cellsig.2016.05.004 [DOI] [PubMed] [Google Scholar]
- 39. Wang H., Qin J., Gong S., Feng B., Zhang Y., and Tao J. (2014) Insulin-like growth factor-1 receptor-mediated inhibition of A-type K+ current induces sensory neuronal hyperexcitability through the phosphatidylinositol 3-kinase and extracellular signal-regulated kinase 1/2 pathways, independently of Akt. Endocrinology 155, 168–179 10.1210/en.2013-1559 [DOI] [PubMed] [Google Scholar]
- 40. Xu Y., Chiamvimonvat N., Vázquez A. E., Akunuru S., Ratner N., and Yamoah E. N. (2002) Gene-targeted deletion of neurofibromin enhances the expression of a transient outward K+ current in Schwann cells: a protein kinase A-mediated mechanism. J. Neurosci. 22, 9194–9202 10.1523/JNEUROSCI.22-21-09194.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu R. L., Butler D. M., and Barish M. E. (1998) Potassium current development and its linkage to membrane expansion during growth of cultured embryonic mouse hippocampal neurons: sensitivity to inhibitors of phosphatidylinositol 3-kinase and other protein kinases. J. Neurosci. 18, 6261–6278 10.1523/JNEUROSCI.18-16-06261.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lang F., and Shumilina E. (2013) Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. FASEB J. 27, 3–12 10.1096/fj.12-218230 [DOI] [PubMed] [Google Scholar]
- 43. Pieri M., Amadoro G., Carunchio I., Ciotti M. T., Quaresima S., Florenzano F., Calissano P., Possenti R., Zona C., and Severini C. (2010) SP protects cerebellar granule cells against beta-amyloid-induced apoptosis by down-regulation and reduced activity of Kv4 potassium channels. Neuropharmacology 58, 268–276 10.1016/j.neuropharm.2009.06.029 [DOI] [PubMed] [Google Scholar]
- 44. Roepke T. A., Malyala A., Bosch M. A., Kelly M. J., and Rønnekleiv O. K. (2007) Estrogen regulation of genes important for K+ channel signaling in the arcuate nucleus. Endocrinology 148, 4937–4951 10.1210/en.2007-0605 [DOI] [PubMed] [Google Scholar]
- 45. Popiolek-Barczyk K., Makuch W., Rojewska E., Pilat D., and Mika J. (2014) Inhibition of intracellular signaling pathways NF-κB and MEK1/2 attenuates neuropathic pain development and enhances morphine analgesia. Pharmacol. Rep. 66, 845–851 10.1016/j.pharep.2014.05.001 [DOI] [PubMed] [Google Scholar]
- 46. Schrader L. A., Birnbaum S. G., Nadin B. M., Ren Y., Bui D., Anderson A. E., and Sweatt J. D. (2006) ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am. J. Physiol. Cell Physiol. 290, C852–C861 10.1152/ajpcell.00358.2005 [DOI] [PubMed] [Google Scholar]
- 47. Rodgers E. W., Krenz W. D., Jiang X., Li L., and Baro D. J. (2013) Dopaminergic tone regulates transient potassium current maximal conductance through a translational mechanism requiring D1Rs, cAMP/PKA, Erk and mTOR. BMC Neurosci. 14, 143 10.1186/1471-2202-14-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hagenacker T., Hillebrand I., Büsselberg D., and Schäfers M. (2010) Myricetin reduces voltage activated potassium channel currents in DRG neurons by a p38 dependent mechanism. Brain Res. Bull. 83, 292–296 10.1016/j.brainresbull.2010.07.010 [DOI] [PubMed] [Google Scholar]
- 49. Norris C. A., He K., Springer M. G., Hartnett K. A., Horn J. P., and Aizenman E. (2012) Regulation of neuronal proapoptotic potassium currents by the hepatitis C virus nonstructural protein 5A. J. Neurosci. 32, 8865–8870 10.1523/JNEUROSCI.0937-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Redman P. T., Hartnett K. A., Aras M. A., Levitan E. S., and Aizenman E. (2009) Regulation of apoptotic potassium currents by coordinated zinc-dependent signalling. J. Physiol. 587, 4393–4404 10.1113/jphysiol.2009.176321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li N., Lu Z. Y., Yu L. H., Burnstock G., Deng X. M., and Ma B. (2014) Inhibition of G protein-coupled P2Y2 receptor induced analgesia in a rat model of trigeminal neuropathic pain. Mol. Pain 10, 21 10.1186/1744-8069-10-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Van Hoorick D., Raes A., Keysers W., Mayeur E., and Snyders D. J. (2003) Differential modulation of Kv4 kinetics by KCHIP1 splice variants. Mol. Cell Neurosci. 24, 357–366 10.1016/S1044-7431(03)00174-X [DOI] [PubMed] [Google Scholar]
- 53. Vydyanathan A., Wu Z. Z., Chen S. R., and Pan H. L. (2005) A-type voltage-gated K+ currents influence firing properties of isolectin B4-positive but not isolectin B4-negative primary sensory neurons. J. Neurophysiol. 93, 3401–3409 10.1152/jn.01267.2004 [DOI] [PubMed] [Google Scholar]
- 54. Tolhurst D. J., Smyth D., and Thompson I. D. (2009) The sparseness of neuronal responses in ferret primary visual cortex. J. Neurosci. 29, 2355–2370 10.1523/JNEUROSCI.3869-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Starowicz K., Mousa S. A., Obara I., Chocyk A., Przewłocki R., Wedzony K., Machelska H., and Przewłocka B. (2009) Peripheral antinociceptive effects of MC4 receptor antagonists in a rat model of neuropathic pain: a biochemical and behavioral study. Pharmacol. Rep. 61, 1086–1095 10.1016/S1734-1140(09)70171-9 [DOI] [PubMed] [Google Scholar]
- 56. Chu H., Xia J., Yang Z., and Gao J. (2012) Melanocortin 4 receptor induces hyperalgesia and allodynia after chronic constriction injury by activation of p38 MAPK in DRG. Int. J. Neurosci. 122, 74–81 10.3109/00207454.2011.630542 [DOI] [PubMed] [Google Scholar]
- 57. Agosti F., Cordisco Gonzalez S., Martinez Damonte V., Tolosa M. J., Di Siervi N., Schioth H. B., Davio C., Perello M., and Raingo J. (2017) Melanocortin 4 receptor constitutive activity inhibits L-type voltage-gated calcium channels in neurons. Neuroscience 346, 102–112 10.1016/j.neuroscience.2017.01.007 [DOI] [PubMed] [Google Scholar]
- 58. Dong Y., Li P., Ni Y., Zhao J., and Liu Z. (2014) Decreased microRNA-125a-3p contributes to upregulation of p38 MAPK in rat trigeminal ganglions with orofacial inflammatory pain. PLoS One 9, e111594 10.1371/journal.pone.0111594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang Q., Cao D. L., Zhang Z. J., Jiang B. C., and Gao Y. J. (2016) Chemokine CXCL13 mediates orofacial neuropathic pain via CXCR5/ERK pathway in the trigeminal ganglion of mice. J. Neuroinflammation 13, 183 10.1186/s12974-016-0652-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dixon W. J. (1980) Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 20, 441–462 10.1146/annurev.pa.20.040180.002301 [DOI] [PubMed] [Google Scholar]