

Abstract
T-cell receptors (TCR) have considerable potential as therapeutics and antibody-like reagents to monitor disease progression and vaccine efficacy. Whereas antibodies recognize only secreted and surface-bound proteins, TCRs recognize otherwise inaccessible disease-associated intracellular proteins when they are presented as processed peptides bound to major histocompatibility complexes (pMHC). TCRs have been primarily explored for cancer therapy applications but could also target infectious diseases such as cytomegalovirus (CMV). However, TCRs are more difficult to express and engineer than antibodies, and advanced methods are needed to enable their widespread use. Here, we engineered the human CMV–specific TCR RA14 for high-affinity and robust soluble expression. To achieve this, we adapted our previously reported mammalian display system to present TCR extracellular domains and used this to screen CDR3 libraries for clones with increased pMHC affinity. After three rounds of selection, characterized clones retained peptide specificity and activation when expressed on the surface of human Jurkat T cells. We obtained high yields of soluble, monomeric protein by fusing the TCR extracellular domains to antibody hinge and Fc constant regions, adding a stabilizing disulfide bond between the constant domains and disrupting predicted glycosylation sites. One variant exhibited 50 nm affinity for its cognate pMHC, as measured by surface plasmon resonance, and specifically stained cells presenting this pMHC. Our work has identified a human TCR with high affinity for the immunodominant CMV peptide and offers a new strategy to rapidly engineer soluble TCRs for biomedical applications.
Keywords: glycosylation, antibody engineering, protein engineering, recombinant protein expression, immunoglobulin fold, affinity, antiviral treatment, cytomegalovirus, T-cell receptor
Introduction
Antibodies represent a large and growing class of successful therapeutics, by virtue of their abilities to tightly bind antigens that are secreted or expressed on a target cell surface. The structurally analogous T-cell receptor (TCR)3 provides access to a much wider array of intracellular and extracellular antigens that are presented on a cell surface as proteolyzed peptides bound to major histocompatibility complexes (MHC). Exhibiting clear structural homology to antibodies, TCR-binding sites are formed by six complementarity determining regions (CDRs) on the α- and β-variable domains, with the CDR3 loops dominating peptide interactions (1). When a peptide–MHC complex is recognized by the TCR expressed on a T cell, activation, cytokine release, and cell killing can follow.
There is growing interest in using TCRs as therapeutics and reagents to monitor the presence of disease-related peptides. For example, engineered TCRs can be used in adoptive T cell therapies to re-direct patient T cells to recognize a chosen target (2), whereas soluble TCRs can be used as antibody-like reagents to bind specific peptide MHC complexes presented on a cell surface (3). Proof-of-concept for TCR therapeutic applications has been demonstrated by TCRs targeting the immunodominant Gag epitope SL9 from HIV. When transduced into patient T cells, high-affinity TCRs were able to control viral replication (4). When expressed as a soluble TCR linked to a CD3-specific single–chain antibody, the chimeric protein was able to redirect polyclonal CD8+ T cells to kill CD4+ T cells harboring reactivated HIV (5).
Viral infection by cytomegalovirus (CMV) is also controlled by cytotoxic T cells in healthy individuals but causes disease in the very young, very old, and immunocompromised, with no vaccine candidate yet nearing licensure (6). CMV-specific cytotoxic T cells primarily target peptides from the pp65 tegument protein, with the immunodominant peptide residing between residues 495 and 503 (sequence NLVPMVATV, hereafter called NLV) (7, 8). This peptide is restricted to HLA-A*02 (hereafter called A2), the most common allele in North America (9). Adoptive transfer of NLV-specific T cells is sufficient to control infection in allogeneic hematopoietic stem cell transplantation patients with CMV infection (10). Notably, NLV is presented on the infected cell surface early after infection, prior to de novo protein synthesis and in the presence of therapeutics blocking viral replication (11).
Identification of a validated, CMV-specific peptide–MHC complex suggests opportunities to monitor NLV-presenting cells, if an appropriate peptide-specific TCR is available. Although hundreds of TCRs can recognize an immunodominant peptide, the NLV/A2 response is dominated by “public” clones whose CDR3α and/or CDR3β sequences are shared among unrelated individuals (12, 13). One of these, RA14, emerged as the dominant clone after rounds of immunosuppression and viral reactivation in a rheumatoid arthritis patient with asymptomatic CMV infection (12). RA14 contains the two most common public features observed in NLV-reactive TCRs: CDR3α sequence XnGNQF (where Xn indicates a variable number of residues), observed in 14% of all sequences obtained from multiple donors; and CDR3β sequence SXnTGXnGY, observed in 13% of sequences (13). The RA14 TCR has been crystallized in complex with its ligand NLV/A2, revealing a typical TCR–pMHC binding interface characterized by high structural complementarity for the entire peptide (14).
Although RA14 appears to be an excellent candidate to monitor NLV/A2 presence and may be suitable for adoptive therapy applications, there are several limitations to using TCRs as soluble, antibody-like reagents. First, TCR ligand–binding affinities are much weaker than antibodies: RA14 has been reported to have a 6–30 μm equilibrium binding affinity for NLV/A2 (14, 15), whereas antibody–ligand affinities are typically >1000-fold stronger (1–10 nm Kd). Second, soluble expression of TCRs continues to present challenges, with no generally successful strategies identified.
To address these shortcomings, TCRs have been engineered for increased stability, expression level, and affinity. This has been achieved using a single-chain format and yeast display (16–18), but engineering of each unique TCR appears required to incorporate properly folded protein into these formats (19). Phage display of the TCR extracellular regions with an engineered disulfide bond (20) has also been used to identify several very high–affinity human TCR variants (4, 21, 22). While successful, phage display lacks the eukaryotic protein folding machinery that is likely required to allow expression of a greater range of TCRs and is not compatible with efficient FACS-based selection strategies. To produce soluble protein, single-chain or extracellular two-chain TCRs are most commonly refolded from bacterial inclusion bodies with varying levels of success (19). Some TCR sequences are amenable to soluble expression in bacteria with chaperone co-expression (23–25), in yeast (16) or in mammalian cells (26–28), but these represent only a fraction of TCRs of interest. Taken together, considerable effort is required to convert a TCR into a soluble protein that can be used in biochemical assays.
Here, we aimed to develop a better platform for TCR engineering and soluble expression and to validate this platform by engineering the RA14 TCR for use as an antibody-like reagent to monitor peptide presentation on cells. We first modified our previously-described CHO-cell display system (29) to display RA14, and we then designed a library in CDR3α and CDRβ and screened for better binders. We reformatted the selected clones as TCR-Fc fusion proteins and identified an optimal backbone for improved soluble expression. One variant, RA14 α2.β8, had a 50 nm Kd and was able to detect pMHC on the surface of cells at physiologically-relevant peptide concentrations. This protein could be used to monitor NLV presentation after vaccination with novel CMV vaccines such as the NLV–peptide vaccine (30) or to replace the cumbersome pp65 antigenemia assay used to detect active infection in organ transplant recipients (31).
Results
Display of pp65 NLV-specific TCR RA14 on the CHO cell surface
To first determine the level of recombinant TCR display on the CHO cell surface, we cloned the truncated extracellular α- and β-chains of the human RA14 TCR into a pcDNA3-based plasmid with a CMV promoter, mouse IgΗ leader sequence, one TCR chain, and T2A peptide sequence followed by the second TCR chain fused in-frame to a platelet-derived growth factor receptor (PDGFR)-transmembrane region (TM, Fig. 1a). As only the second chain is fused to the transmembrane region and chain order can impact yields (24), we cloned the chains in both the α/β-TM and β/α-TM orientations. Similarly, since moving the terminal inter-chain disulfide bond to the Vα:T84C and Vβ:S79C position (IMGT numbering used throughout) and removing the free cysteine at position Vβ85.1 have been reported to improve expression of soluble and phage/yeast-displayed TCRs (20, 29), these modifications were also tested in each chain orientation.
Figure 1.
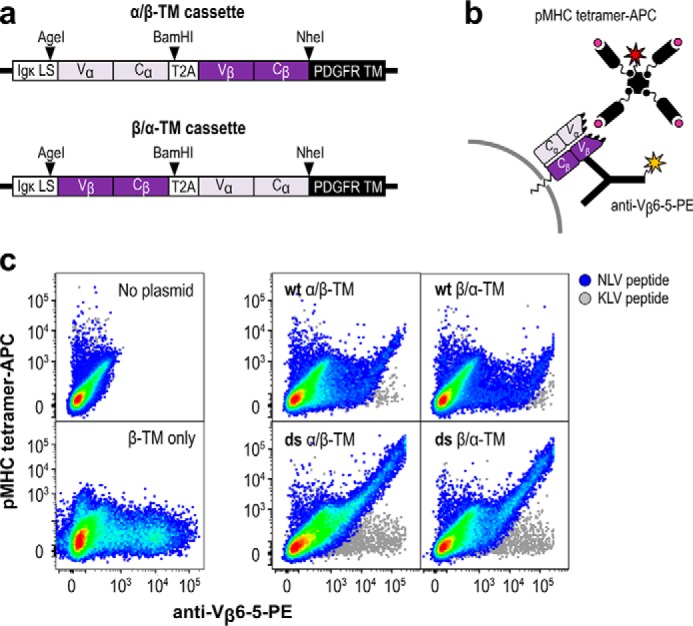
NLV-specific human TCR RA14 displays on the surface of CHO-K1 cells. a, RA14 variable and constant regions were cloned in-frame with the mouse IgH leader sequence (LS), a T2A peptide for cleavage, and the PDGFR transmembrane domain (TM) with either the α-chain (α/β-TM) or the β-chain (β/α-TM) in the first position. The cassettes were then cloned into a pcDNA3 mammalian expression vector. b, display of functional RA14 TCR was detected with a dual-staining approach, in which an anti-Vβ6-5 antibody-PE conjugate was used to detect expression of the TCR β-chain, whereas a peptide/A2 tetramer conjugated to APC was used to assess ligand binding. c, plasmids encoding the TCR in both chain orientations and with the wildtype (wt) or engineered disulfide (ds) constant regions were transfected, stained 2 days later, and assayed for APC and PE signal via flow cytometry. Rainbow dots depict staining using tetramer presenting the NLV peptide from the CMV pp65 protein, and the gray dots depict staining with tetramer presenting the control peptide KLV. Control transfections without plasmid and with a plasmid lacking the α-chain are also shown.
After cloning and sequence confirmation, midi-prepped plasmid DNA was transiently transfected into CHO-T cells, and TCR surface display was assessed by flow cytometry 2 days later. The presence of TCR on the cell surface was monitored by an antibody binding the human variable β-chain (Vβ6-5-PE), whereas NLV/A2 tetramers conjugated to APC were used to assess ligand-binding activity. A tetramer presenting an unrelated peptide from hepatitis C virus (HCV1406–1415 sequence KLVALGINAV; hereafter called KLV) complexed with A2 was used to evaluate peptide specificity (Fig. 1b).
Flow cytometry showed varying expression patterns for each vector design, with the PE- and APC-positive population indicative of cells binding tetramer and displaying TCR (Fig. 1c). Cells transfected with empty vector showed minimal binding to either reagent, whereas a β-TM construct only bound the Vβ antibody, indicating that unpaired TCRβ chains can be displayed but will not be detected by tetramer binding. By contrast, cells transfected with constructs containing both TCR chains presented a diagonal double-positive population, indicative of the expected correlation between tetramer staining and surface display and a wide range of expression levels. No staining was observed with the negative control KLV tetramer, indicating that the displayed TCRs retained peptide specificity. All samples included a population of unstained, nonexpressing cells, which is expected for eukaryotes with unsynchronized growth cycles. Although inclusion of the modified disulfide bond greatly increased the specific tetramer-binding activity (tetramer binding/TCR display ratio), chain order had minimal impact. Accordingly, we chose one design, the α/β-TM configuration with the modified disulfide bond, for further use.
Design of CDR3α and CDR3β libraries
Analysis of the RA14-NLV/A2 crystal structure revealed that RA14 engages nearly all solvent-exposed peptide residues and forms hot spots with peptide residues P4:Pro, P5:Met, and P8:Thr (14). In the α-chain, CDR3α:N114 forms a key hydrogen bond with P5:Met, which is also present in the structure of the related TCR C7 with NLV/A2 (32), which has a nearly identical CDR3α. In the β-chain, CDR3β:T110 forms multiple hydrogen bonds with P8:Pro. To identify high-affinity RA14 variants, we designed two separate CDR3α and CDR3β libraries. This allowed us to generate libraries with more residues randomized per CDR than if we had screened simultaneous CDR3α and CDR3β libraries, yet include every possible sequence in the library. Three anchor residues (Vα:N114, Vβ:T110, and Vβ:Y114) were retained while the remaining peptide-contacting residues plus one flanking residue on either side were randomized to optimize the TCR–pMHC interface (Fig. 2).
Figure 2.
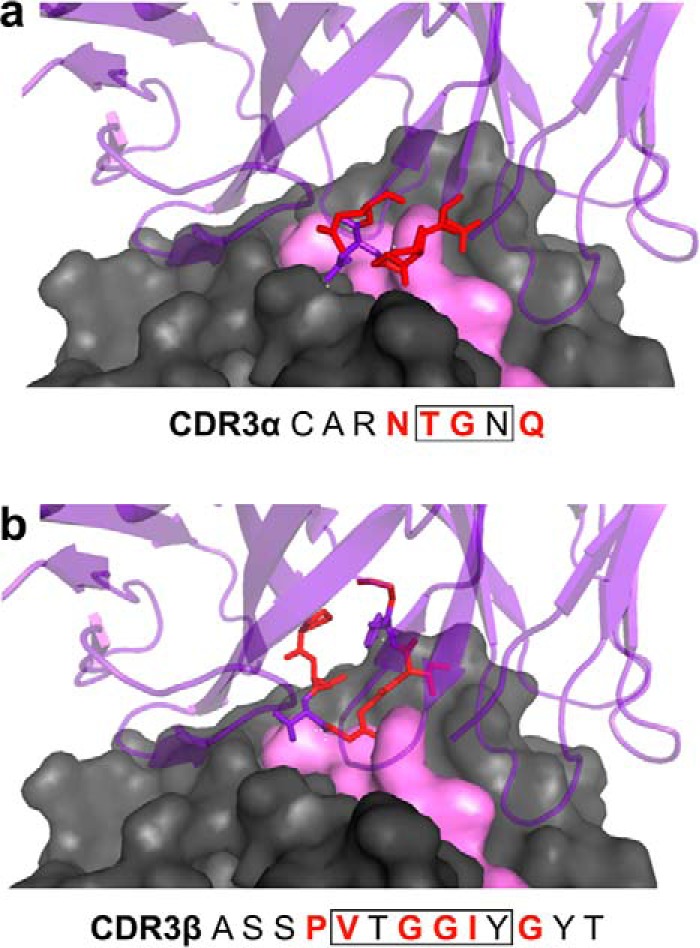
Structural interactions between RA14 CDR3 loops and NLV peptide/A2. The crystal structure of the RA14 TCR complexed with NLV/A2 (PDB code 3GSN) was used to guide library design. The A2 surface is shown in gray space-fill, the NLV peptide surface in pink space-fill, and the RA14 structure in purple ribbon. The residues comprising the CDR3α (a) and CDR3β (b) loops are listed, with those targeted for mutagenesis highlighted in red in the text and in the structure. Boxed residues form direct pMHC contacts in the WT crystal as reported previously (14).
To create each library, primers incorporating degenerate codons were designed to maximize amino acid diversity while keeping the theoretical library sizes (1 × 106 for CDR3α and 4 × 106 for CDR3β) near ∼106, a limitation determined by mammalian cell culture volume constraints. Mutagenized cassettes were generated using overlap PCR with these primers, followed by overlap extension PCR to produce full-length inserts. These were digested and ligated into the pPyEBV vector, which includes the polyoma virus origin of replication, Epstein-Barr virus nuclear antigen and OriP that allow for plasmid retention and amplification in CHO-T cells that stably express the polyoma virus large T antigen (33). After transformation into Escherichia coli, actual library sizes were estimated as 4 × 105 for the CDR3α library and 1 × 106 for the CDR3β library, with diversity confirmed by DNA sequencing.
Selection of RA14 variants with improved tetramer binding
Pooled library plasmids were diluted with a 1:4 molar ratio of carrier plasmid to ensure each cell acquired at most one library clone (29) and transfected into CHO-T cells. After 2 weeks of growth under antibiotic selection to eliminate cells lacking the pPyEBV plasmid, cells were stained with AlexaFluor-647 (AF647)-labeled NLV/A2 tetramers to detect ligand binding and anti-Vβ6-5-PE to detect surface TCR display. Each library was sorted by FACS to collect the 1–3% of cells with the highest ratio of tetramer binding to TCR display. After sorting, each library was expanded for 1 week before being sorted again, for a total of three rounds. Comparison of the libraries at each step demonstrated enrichment of clones with improved specific tetramer binding (Fig. 3, a and b). The CDR3α and CDR3β libraries each showed a 5–10-fold increase in the number of cells falling within the gated area per round. Based on the percent of the library that was collected each round, we expect only a handful of clones to remain after three rounds (e.g. collecting 2% of a one million member library over three rounds would result in ∼8 clones = 2%*2%*2%* 106 clones). To retain a diverse collection of clones for analysis, we did not pursue further sorting rounds.
Figure 3.

RA14 variants with improved tetramer binding can be isolated by CHO display. The TCR display cassette optimized in Fig. 1 was mutagenized to create two libraries following the strategy in Fig. 2 and cloned into a pPy vector to allow for episomal maintenance in CHO-T cells. CDR3α (a) and CDR3β (b) libraries were separately transfected into CHO-T cells, stained, and sorted over three rounds to enrich for improved tetramer binding. Untransfected cells and cells displaying Fab hu1B7 (39) are shown as controls. The gate drawn is representative of the sorting gate used in round three, with the percentage of cells falling into the gate noted in red to facilitate comparisons. Individual clones selected during round three of the CDR3α (c) and CDR3β (d) libraries were re-transfected, stained, and assessed for specific tetramer binding relative to the WT RA14. Specific tetramer binding is the ratio of the AF647 signal (tetramer binding) to the PE signal (anti-TCRβ display) calculated on a per-cell basis. The median fluorescence intensity for this new variable was then normalized to the value for cells expressing WT RA14.
TCR sequences were recovered from pooled cells after round three by PCR amplification from total cellular DNA, followed by re-cloning into the pPyEBV plasmid for sequencing. Analysis of 20–35 colonies revealed seven unique CDR3α and 10 unique CDR3β sequences (Table S1). The WT residues were largely retained at positions Vβ:109 and Vβ:111 within the CDR3β library, whereas positions Vβ:112 and Vβ:113 were highly variable. RA14 covers an unusually high percentage of the exposed peptide upon binding, and CDR3β contacts in particular are mostly backbone-mediated, which may explain the high variation observed in these residues (14). Finally, larger residues were frequently found in the flanking sites, which may have been preferentially enriched if they were able to introduce additional peptide contacts.
Characterization of RA14 variants with improved tetramer binding
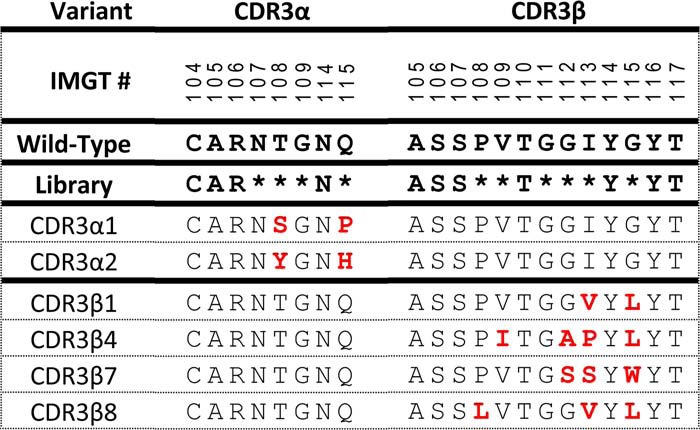
All identified CDR variants were transfected into fresh CHO-T cells in combination with the complementary WT chain and analyzed for TCR display level and tetramer binding as single clones. To compensate for TCR expression level differences, we compared the specific tetramer-binding activity (tetramer-AF647 signal/anti-TCR–PE signal) on a per cell basis for each clone. The median of this distribution was then normalized to the median value obtained for WT RA14 to report a fold-increase as compared with RA14. A similar process was performed after staining with the control KLV tetramers, revealing that peptide restriction was retained for all selected variants. Only two CDR3α variants (α1 and α2) showed binding similar to or improved over WT, with the best variant (α2) having a 2.3-fold increase in normalized binding (Fig. 3c and Fig. S1). In contrast, all 10 of the CDR3β variants showed significant improvements over WT, with 1.8–3.5-fold improved specific tetramer binding (Fig. 3d and Fig. S2). The variants with the greatest specific tetramer-binding activities (α1, α2, β1, β4, β7, and β8) were selected for further analysis.
We wished to determine whether the best α- and β-variants could be combined to further improve tetramer binding, as reported in other studies (21). The WT and selected α- (α1, α2) and β- (β1, β4, β7, β8) variants were combined pairwise, transfected, and analyzed as before. The combinations generally showed further improved specific tetramer binding (Fig. 4a), with some variation in TCR display levels among clones (Fig. S3). As surface display levels can be a useful predictor of soluble expression yields (16), we selected the α2 and β8 combination as the lead candidate. Variant α2 contains the Vα:T108Y and Vα:Q115H substitutions, whereas β8 contains Vβ:P108L and the frequently-observed Vβ:I113V and Vβ:G115L mutations (Table 1).
Figure 4.
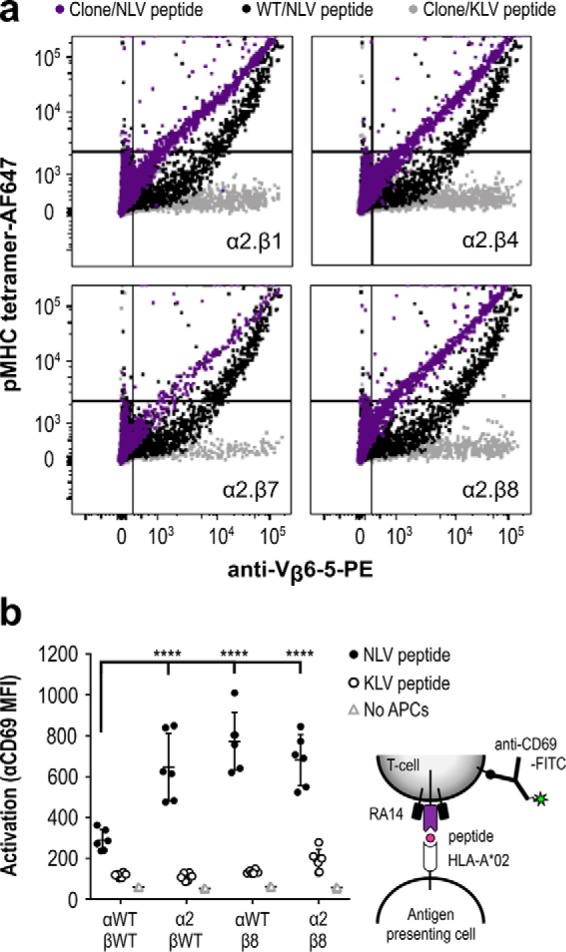
Combining selected CDR3 variants further improves tetramer binding and TCR activation. a, the most improved α-chain variant (α2) was transfected into CHO-T cells in pairwise combinations with the WT and selected β-chain variants (β1, β4, β7, and β8). After 2 days of expression, cells were stained and analyzed by flow cytometry, as in Fig. 3. b, activation of human Jurkat T cells expressing RA14 and selected TCR variants was measured by CD69 up-regulation. Selected TCR variable regions were cloned into expression vectors with mouse constant domains, native TCR transmembrane, and signaling sequences and transfected into Jurkat cells. After 24 h of co-culture with peptide-pulsed human T2 antigen-presenting cells, TCR-positive cells (NLV-tetramer-binding and Vβ-positive) were further monitored for CD69 up-regulation using an anti-CD69-FITC antibody. Data shown are the results, average and standard deviation of three independent experiments, each performed in duplicate for every treatment condition. Analysis of variance was used to compare the anti-CD69 mean fluorescence intensity for each clone combination (****, p < 0.001).
Table 1.
Amino acid sequences of charaterized RA14 CDR3 variants
To better understand the impact of the selected CDR changes on TCR function, we evaluated TCR activation after transfection into human Jurkat T cells. The native RA14 and selected α2 and β8 variable regions were appended with murine constant regions followed by human TCR transmembrane regions to allow TCR signaling and prevent mispairing with the endogenous human TCR expressed in Jurkat cells (34). After electroporation, Jurkat cells were co-cultured with human T2 antigen-presenting cells preincubated with 0.1 μm NLV or control KLV peptide, with activation measured by CD69–up-regulation using flow cytometry 24 h later (Fig. 4b). All TCRs tested showed activation in the presence of NLV but not KLV-pulsed T2 cells. Moreover, the engineered TCRs showed significantly improved activation (2.2–2.7-fold) as compared with the WT RA14. Although our selection strategy was based upon TCR display levels and tetramer binding, these data suggest that features required for peptide-specific TCR activation were also retained (35), which is not always the case for affinity-matured TCRs (36, 37).
Production of RA14 TCR as a soluble Fc-fusion protein
We next set out to express these variants as soluble proteins for further characterization. TCRs are notoriously difficult to produce, with no generally successful strategies yet identified (24, 25). Because TCRs are naturally produced by mammalian cells and because fusion of a poorly expressed protein to an antibody Fc domain generally increases expression level, increases avidity through bivalency, and provides a convenient detection handle for purification and immunoassays, we selected a TCR-Fc format for production in CHO cells. We modified the pcDNA display plasmid to express the WT RA14 variable and constant domains (with engineered disulfide bond) with either the α- or β-chain fused to the human IgG1 Fc domain (named TCRds-huFc) with the two chains separated by a T2A linker. In a second generation, the chains were encoded on separate plasmids. This two-plasmid system, with the α-chain fused to the Fc, resulted in the highest expression levels in a small-scale transfection test. Finally, we further optimized this format by adding a second disulfide bond joining the base of the TCR constant domains, encoded by the antibody upper hinge sequence (named TCR2ds–huFc).
To compare RA14 and the engineered variants in the TCR2ds–huFc format, the α2 and β8 domains were cloned into the two-plasmid expression system and transfected pairwise with plasmids encoding the WT RA14 chains to produce four variants: WT RA14 (αWT.βWT), α2.βWT, αWT.β8, and α2.β8. After protein A purification, size-exclusion chromatography (SEC) was performed to isolate the dominant peak (eluting at ∼10.5 ml) containing protein with the highest tetramer-binding activity and appearing to correspond to intact bivalent protein. SDS-PAGE analysis showed similar high levels of purity for all variants (Fig. 5a). Protein stability was compared by thermal unfolding, using differential scanning fluorimetry and using the inflection point of the first unfolding event to compare TCR domain stabilities. The β8 changes are mildly destabilizing, inducing a >4 °C decrease in melting temperature as compared with RA14. In contrast, the α2 changes increase thermal stability >2 °C and partially compensate for the presence of the less stable β8 in the combined α2.β8 variant (Table 2).
Figure 5.

Engineered RA14 TCR2ds–huFc variants show increased affinity for NLV/A2. a, TCR2ds–huFc formats of each variant were purified by protein A and size-exclusion chromatography to isolate only the intact protein. Protein purity was evaluated by nonreduced and reduced 4–20% gradient SDS-polyacrylamide gel (3 μg of protein per lane). b, tetramer-binding activities of purified RA14 variants were compared by ELISA. Plates were coated with NLV/A2 tetramer, followed by TCR2ds–huFc and goat anti-human Fc-HRP. Data shown are the average and range of duplicate series for a representative experiment; this was repeated several times with similar results. c, pMHC binding kinetics were measured by SPR. Each TCR2ds–huFc variant was immobilized on a CM5 chip at 2000–5000 RUs, after which monomeric NLV/A2 was injected at six concentrations between 3.9 and 500 nm. An in-line blank flow cell was used to assess background binding. Peptide specificity was evaluated with injections of monomeric KLV/A2 at the maximum concentration used for NLV/A2 for each variant. All injections were performed in duplicate; shown are the data and fits with the numerical values reported in Table 2.
Table 2.
Binding kinetics for NLV/A2 and thermal stabilities of RA14 TCR2ds-huFc variants
| Variant | Tm, glycosylated | Tm, deglycosylated | ka × 105 | kd | χ2 | Kd = kd/ka | Equilibrium Kd |
|---|---|---|---|---|---|---|---|
| °C | °C | m−1 s−1 | s−1 | nM | nm | ||
| αWT.βWT | 67.9 ± 0.4 | 68.8 ± 0.3 | 0.95 ± 0.1 | 0.29 ± 0.2 | 1.7 ± 0.6 | 3092 ± 400 | 1480 ± 60 |
| α2.βWT | 70.2 ± 0.5 | 71.3 ± 0.1 | 3.1 ± 0.8 | 0.15 ± 1 | 14 ± 2 | 514 ± 100 | 570 ± 13 |
| αWT.β8 | 63.7 ± 0.7 | 64.1 ± 0.7 | 1.3 ± 0.02 | 0.032 ± 0.1 | 6.1 ± 0.6 | 243 ± 7 | 224 ± 7 |
| α2.β8 | 65.2 ± 0.1 | 67.0 ± 0.5 | 3.5 ± 0.02 | 0.018 ± 0.1 | 1.4 ± 0.1 | 53 ± 1 | 62 ± 1 |
Ligand-binding affinity of engineered variants as soluble TCR2ds–huFc fusion proteins
As an initial assessment of NLV/A2 binding, we compared the ability of purified TCR2ds–huFc variants to bind immobilized pMHC in an ELISA. The NLV/A2 or control KLV/A2 tetramer was coated onto an ELISA plate, and the TCR2ds–huFc protein was titrated and detected by anti-human–Fc-HRP (Fig. 5b). No binding was detected for any variant to the control surface, whereas RA14 provided a clear dose-response curve on NLV tetramer–coated wells, with a detection limit near 0.1 μg/ml. Both variants including the β8-chain showed a distinct increase in binding, with the detection limit ∼30-fold lower than observed for the other variants. In contrast, the α2.βWT showed improved detection but a much shallower slope than observed for RA14.
We used surface plasmon resonance to rigorously quantify the binding kinetics of the four RA14 variants to NLV/A2. SEC-purified TCR2ds–huFc was coupled to the sensor surface, with varying concentrations of the NLV/A2 or KLV/A2 monomer passed over the surface. The WT RA14 exhibited the slow on-rates (9.5 × 104 m−1 s−1) and fast off-rates (0.29 s−1) typical of TCRs, resulting in a calculated Kd of 3.1 μm for the bivalent TCR2ds–huFc format. This compares well to the 6.3 μm Kd previously reported for monovalent RA14 using an immobilized pMHC orientation but is tighter than the 27.7 μm reported using immobilized TCR (14, 15). The α2 CDR changes primarily affected the on-rate, with a 3-fold increase, whereas the β8 CDR changes primarily impacted the off-rate, with a 10-fold decrease (Fig. 5c). When combined, these changes were additive, conferring a 50 nm Kd for α2.β8, which is a 60-fold improved affinity over RA14. Equilibrium binding analyses of these data yielded similar results (Table 2 and Fig. S4). No variant exhibited detectable binding to the control KLV/HLA-A2 monomer at the highest concentrations used (Fig. 5c). Because a bivalent TCR-like antibody with 300 nm Kd was able to detect the NLV/A2 complex presented by CMV-infected primary human fibroblasts (38), this TCR-Fc with 50 nm Kd seemed sufficient for use as an antibody-like reagent.
Enhanced TCR-Fc expression by removal of N-linked glycosylation sites
Although we were able to obtain relatively pure preparations of TCR2ds–huFc for SPR, we incurred considerable protein losses during SEC purification to collect properly assembled protein. Moreover, SDS-PAGE analysis showed doublets near the expected protein molecular weight, suggestive of multiple glycoforms (Fig. 5a). Inspection of the RA14 sequence revealed five predicted N-linked glycosylation sites, at positions Vα:N20, Cα:N90, Cα:N109, βV:N77, and βC:N85.6. Several of these are near domain interfaces and could sterically inhibit proper TCR-Fc assembly. Treatment of purified TCR2ds–huFc protein with peptide:N-glycosidase resulted in more homogeneous bands on SDS-PAGE, supporting this hypothesis (data not shown). We therefore designed a third TCR-Fc format in which these glycosylation motifs were disrupted by Asn-to-Gln amino acid substitutions (named TCR2dsΔgly-huFc; Fig. 6a).
Figure 6.

Expression and stability of the WT RA14 TCR as a soluble Fc fusion protein. a, several iterations of the TCR-Fc fusion protein were designed. In all scaffolds, the TCR α-chain was fused to a human IgG1 core/lower hinge and Fc. Open circles represent native glycosylation sites. Additional modifications include a second disulfide bond resulting from inclusion of the upper hinge sequence (ds, green) in the TCR2ds–huFc format and the removal of predicted N-linked glycosylation sites (Δgly, blue crosses) in the TCR2dsΔgly-huFc format. b, purity of each protein A purified design was evaluated via reducing and nonreducing 4–20% gradient SDS-PAGE, with 3 μg loaded per lane. c, protein homogeneity was analyzed by size-exclusion chromatography. The arrow indicates the major peak collected for experiments using a glycosylated scaffold. Triangles indicate elution volumes for molecular weight standards: thyroglobulin with a 669-kDa size eluted at 8.7 ml; ferritin 440 kDa at 10.5 ml; aldolase 158 kDa at 12.8 ml; conalbumin 75 kDa at 14.3 ml; ovalbumin 44 kDa at 15.2 ml; carbonic anhydrase 29 kDa at 16.7 ml. Representative data are shown for each design. d, melting temperature of each design was analyzed by differential scanning fluorimetry. An average value for two separate normalized curves is shown for each scaffold from a representative experiment.
We then expressed, purified, and characterized RA14 in all three formats (TCRds-huFc, TCR2ds–huFc, and TCR2dsΔgly-huFc). SDS-PAGE analysis shows that genetic de-glycosylation resulted in very sharp protein bands indicative of a single major species (Fig. 6b). The second disulfide bond apparently improved α–β pairing, as it eliminated the minor SEC peak eluting at ∼13.5 ml, which is expected to be free β-chain (Fig. 6c). The deglycosylated format eliminated the second minor peak at ∼12.5 ml, resulting in a single, monodisperse product on SEC. To evaluate the potential impact of these changes on protein stability, we monitored the thermal stability of each version, as above (Fig. 6d). The melting point of the RA14 TCRds-huFc increased from 66.3 to 67.9 °C with the addition of the second disulfide bond, and it further increased to 68.8 °C after deglycosylation (Table 2), which is in the lower range of values typically reported for antibody Fab domains (67–79 °C) (39). Comparison of the RA14 variants in the TCR2ds–huFc versus TCR2dsΔ-huFc formats showed that all three exhibited similar increases in monodispersity (Fig. S5) while retaining NLV/A2 binding affinity and specificity (Fig. S6).
Staining of pMHC-displaying cells using engineered TCRs
To demonstrate the potential utility of high-affinity TCR-Fc fusion proteins to detect CMV-positive cells, we assessed their abilities to stain peptide-pulsed antigen-presenting cells. To prevent background staining due to binding between the human Fc on the TCR2ds–huFc proteins and human Fcγ receptors on the T2 cells, we replaced the human IgG1 hinge and Fc domains with their murine IgG2a counterparts to create TCR2ds–mFc constructs, which achieved similar yields and purity as the TCR2ds–huFc format (Fig. 7a). Human T2 antigen-presenting cells were incubated overnight with 100 μm purified NLV or KLV peptide, stained with 1 μm purified TCR2ds–mFc, followed by anti-mouse Fc-Alexa647 and analyzed by flow cytometry. Cells incubated with the KLV control peptide showed no shift in fluorescence as compared with cells incubated only with the anti-mouse Fc-AF647. A clear correlation between fluorescence shift and TCR affinity was observed, which increased from no detectable staining for WT RA14 to a distinct population for α2.β8 (Fig. 7b). Finally, to determine the peptide detection sensitivity, the peptide dose was serially diluted before incubation with T2 cells and staining with 1 μm α2.β8 TCR2dsΔgly-mFc. Signals did not saturate at high peptide concentrations, as is expected for these TAP-deficient cells that up-regulate MHC levels in the presence of peptide, and signals were detectable down to 0.5 μm peptide (Fig. 7c).
Figure 7.

High-affinity TCR-mFc proteins bind peptide-pulsed antigen-presenting cells. a, wildtype (WT) RA14 and improved variants were expressed as TCR2ds–mFc proteins with mouse IgG2a hinge and Fc domains to reduce binding to human Fc receptors expressed on T2 cells. SDS-PAGE was used to assess protein size and purity (3 μg/lane). b, human T2 antigen-presenting cells were pulsed overnight with 100 μm NLV or KLV peptide and stained with 1 μm TCR2ds–mFc followed by 1:500 anti-mouse Fc-AF647 before flow cytometric analysis. This experiment was performed twice with similar results; representative data from one experiment are shown. c, human T2 antigen-presenting cells were pulsed overnight with NLV at 0, 0.5, 1, 5, 10, 25, 50, 100, 250, and 500 μm KLV peptide at 500 μm and stained with 1 μm α2.β8 in the TCR2dsΔgly-mFc format and 1:500 anti-mouse Fc-AF647 before flow cytometric analysis; 20,000 events were collected per condition to calculate a geometric mean fluorescence intensity (GMFI). Data shown are the average and range of two independent experiments performed with separate cells and protein preparations on different days.
Discussion
Soluble TCRs have potential as therapeutics and reagents to monitor disease progression or vaccine efficacy. In particular, T cells recognizing the immunodominant NLV peptide from CMV are sufficient to maintain clinical latency, suggesting NLV is a useful marker to monitor disease or vaccination status. Here, we identified variants of the human CMV-specific TCR RA14 with nanomolar affinity for the cognate NLV/A2 complex by selection on the CHO cell surface. These variants retained peptide selectivity and activity when expressed on the surface of human Jurkat T cells. Moreover, we observed high-level production of homogeneous protein when the TCR domains were fused to an antibody Fc domain to create an antibody-like targeting molecule. This construct specifically detected NLV/A2 complexes on the surface of human antigen-presenting cells at low display levels and provided proof-of-concept for a new TCR engineering strategy.
Because TCR engineering continues to present challenges for phage and yeast display platforms, we reasoned that TCR expression on the near-native eukaryotic membrane might offer a more straightforward approach. We were able to display high levels of active TCR using a PDGFR transmembrane domain, modified only by inclusion of a previously described non-native disulfide bond between the TCR constant domains (20). Specific pMHC binding activity was measured as the ratio of NLV/A2 tetramer binding to TCR display level, with the WT RA14 showing a clear population of cells with high specific activity (Fig. 1). In contrast, phage or yeast display of single-chain TCRs or paired TCR extracellular domains is restricted to well-behaved germline segments (e.g. mouse TRBV13 and human TRAV12) (19, 21, 24) or requires extensive engineering to achieve display of active protein, including identification of specific mutations and co-expression of chaperones (24, 25). Even so, variants with partially suppressed stop codons have been isolated, suggesting that expression of these proteins harms their bacterial hosts (21).
Eukaryotic display has been explored previously for TCR engineering. Importantly, these prior reports used completely native TCRs, with TCR extracellular, transmembrane, and intracellular signaling domains expressed on T cells (40–42). In contrast, our use of the PDGFR transmembrane domain fused to just one TCR chain eliminated TCR dependence on CD3 co-expression, provided selection pressure for TCR α/β heterodimerization, and allowed the use of CHO cells. An episomal plasmid allowed us to avoid the more cumbersome retroviral transfection system used in the earlier studies yet maintained TCR expression for ∼2 months, whereas inclusion of a 2A peptide ensured equimolar expression of the α- and β-chains. Because most therapeutic proteins are produced in CHO cells, this system allows for selection of TCRs with characteristics that are expected to be more predictive of the soluble protein. Importantly, this includes glycosylation sites that may negatively impact ligand binding (29) or TCR assembly (Fig. 6).
Eukaryotic display platforms have constrained library sizes due to their lower transfection efficiencies and larger culture volumes as compared with bacteria and yeast. Despite this, the three previously reported eukaryotic TCR libraries all produced interesting clones (40–42). These strategies retained the native TCR transmembrane domains that couple TCR display level to CD3 level (40) and may present challenges during selection, especially if the cells become activated and down-regulate CD3 (43). Kessels et al. (40) randomized seven codons in CDR3β of the flu-specific F5 TCR to generate a library of ∼3 × 104 unique clones. After four rounds of flow cytometric sorting with labeled tetramer, they isolated a variant with physiologic affinity and newly acquired binding to an altered peptide ligand containing two residue changes (40). Similarly, Chervin et al. (41) altered five codons in the mouse 2C TCR CDR3α to yield a library of ∼103–104 clones. After two rounds of flow cytometry, they isolated variant m100 with an estimated 1900 nm affinity, representing a 4–15-fold improvement over the WT TCR. By contrast, an even higher affinity 2C variant, m33 with an ∼32 nm affinity, was isolated from a similarly designed, but larger (5 × 105), yeast display library (36).
The libraries reported here, while still of modest size (4 × 105 for CDR3α and 1 × 106 for CDR3β), were larger than those previously reported for eukaryotic TCR display systems. Taking into account an ∼50% transfection efficiency, cell death and dilution with blank plasmid, a single T-150 flask with adherent CHO cells can reasonably yield 5-fold coverage of a 5 × 105 member library. With shake flasks and suspension cells to facilitate scale up, a library of 107 should be achievable. Although this remains smaller than many bacterial or yeast-display libraries, limiting libraries to include only the peptide-specific contacts mostly found in CDR3 can result in targeted mutagenesis libraries rich in higher affinity variants.
From these libraries, we isolated variants with increased on-rates and decreased off-rates (Table 2). Importantly, the changes in specific tetramer binding observed during library screening anticipated the affinity improvements measured by SPR (Fig. 3). When the two selected chains were combined to create variant α2.β8, the benefits were roughly additive, resulting in an overall ∼60-fold improved NLV/A2 affinity as compared with the WT RA14. Specificity for the NLV peptide was retained, as binding to a control HCV/A2 complex was not detected even at 500 nm (Fig. 5). Analysis of previously engineered TCRs indicates that affinity improvements are typically due to large decreases in off-rate and small increases in on-rate, as was observed for RA14. Structurally, this has been mediated by increases in overall shape complementarity and formation of new contacts between the TCR and peptide residues that retain the native TCR/pMHC binding angle (44, 45). For example, four residue changes in CDR3β of the A6 TCR formed 26 new peptide contacts that were sufficient to increase the Kd for the tax/A2 complex from 3.2 μm to 4 nm (45).
As expected, based on the significantly lower affinities present in endogenous TCR repertoires, the sequences identified here have not been reported in human sequencing studies of NLV/A2-binding T cells (12, 13). In CDR3α, two residues from the XnGNQF motif were altered; the conserved glycine was not always observed in position α:109, and the enhanced on-rate α2 variant replaced the canonical α:Q115 with a histidine. Regardless, these modest sequence and affinity changes support the idea that this common public CDR3α sequence is relatively optimized for NLV/A2 binding. Furthermore, the structure of the related TCR C7 (CDR3α sequence: ITGNQF) in complex with NLV/A2 demonstrates the ability of this CDR3α to preserve a similar peptide-binding interaction while making small adjustments to accommodate a different CDR3β (32). Changes to the CDR3β motif SXnTGXnYGY were more dramatic despite revealing Vβ:G111 to be absolutely conserved in all sequences recovered in this work; notably, residue Vβ:G111 was anticipated as crucial for binding from the initial analysis of the RA14-NLV/A2 crystal structure (14). Among other CDR3β residues, the motif residue Vβ:G115 was not always retained but was preferentially replaced with larger hydrophobic residues, with five of 10 clones using leucine. The other randomized CDR3β positions all showed evidence of structural plasticity, with the slow off-rate β8 variant preferring hydrophobic residues in all three modified positions (Table 1 and Table S1).
High-level expression of soluble TCR proteins continues to present challenges due to their low expression levels and weak heterodimerization properties. Common expression strategies include generation of single-chain TCRs, which typically requires identification of specific residue changes that support folding and expression in this format (18, 19, 24); refolding of intact extracellular domains (46), often with a modified disulfide bond supporting constant domain heterodimerization introduced at residues Cα:T84C and Cβ:S79C (20); and TCR-antibody chimeras expressed in eukaryotes. For this latter approach, a variety of designs has been evaluated, including single-chain TCRs fused to a constant β domain and then antibody heavy chain domains 1–3 (26), complete TCR extracellular domains appended with constant κ domains (47), or an intact antibody fused to the TCR C terminus (48, 49) and simply replacing the antibody variable regions with TCR variable regions (48).
Our approach was to use the simplest antibody-like design that supported expression of active TCR material. This strategy was guided by the desire to employ established antibody purification processes and minimize the risks of proteolysis and immunogenicity in the resulting protein. After evaluation of several designs, substitution of the antibody Fab domains with the TCR extracellular domains emerged as the best approach. Specifically, the extracellular TCR α-chain was fused to the antibody Fc domain with the β-chain expressed in trans. Subsequent replacement of the human Fc for the corresponding mouse Fc domains allowed for detection of NLV/A2 complexes on peptide-pulsed antigen-presenting cells (Fig. 7).
Additional modifications were introduced to support proper assembly of the TCR-Fc chimera. In addition to the previously described engineered disulfide bond between the TCR constant domains (20), the human IgG1 upper and core hinge region introduced a second disulfide bond joining the TCR constant domains and two disulfide bonds stabilizing the Fc homodimer, while the free cysteine at position 85.1 was replaced with an alanine. Finally, two predicted n-linked glycosylation sites in the variable domains and three in the human constant TCR domains were eliminated by Asn-to-Gln substitutions. Together, this allowed for production of 0.75–1 mg of purified TCR2dsΔgly–huFc protein from 50 ml of media, matching typical antibody yields in our lab-scale transient CHO cell expression system. This strategy has yielded similar production levels for three additional TCRs (data not shown).
Prior attempts to replace the antibody Fab with the TCR extracellular domains were less successful (48, 50), but these did not include the upper antibody hinge region responsible for the second inter-chain disulfide bond and did not remove glycosylation sites then thought to increase protein solubility. Although endogenous TCRs are highly glycosylated on the T-cell surface, analysis of TCR affinity and structure are typically performed on bacterially expressed TCR protein, which is aglycosylated. The expression system developed here allows for direct comparison of identical TCR protein with and without glycosylation at specific sites. Removal of predicted N-linked glycosylation sites dramatically increased protein homogeneity as measured by size-exclusion chromatography and SDS-PAGE (Fig. 6), with no negative impact on yield or thermal stability. Further investigation could provide insights into the impact of glycosylation on TCR function, as removal of glycosylation sites has been shown to increase the functional avidity of TCRs expressed on T cells (51).
The ability of TCRs to detect pMHC on the target cell surface is inextricably linked to not just the TCR–pMHC affinity but also the peptide display level and pMHC clustering on the cell surface. Peptide display level in turn reflects several factors, including antigen expression level, proteolysis sensitivity, and peptide–MHC affinity. For TCRs expressed with their native signaling machinery on T cells, a Kd of 1–5 μm seems sufficient to confer maximal signaling responses (52), while retaining the ability to respond to as few as 1–10 pMHC complexes per cell (53, 54). For soluble TCRs, which lack the elaborate array of co-receptors that support cellular TCR–pMHC recognition, high affinity is crucial to allow detection of low levels of target pMHC antigens. In this work, the ability of RA14 TCR variants to detect NLV/A2 correlated strongly with affinity for human T2 cells pulsed with 100 μm NLV peptide (Fig. 7b). A subsequent peptide dosing experiment demonstrated that the α2.β8 variant could detect NLV/A2 complexes after pulsing T2 cells with as little as 0.5 μm peptide (Fig. 7c).
The sensitivity exhibited by α2.β8 is relevant for detection in clinical settings. The NLV peptide was previously reported to present ∼100 molecules/cell when primary human fibroblasts were infected with an AD169 strain that retains the ability to suppress MHC display (38). A bivalent TCR-like antibody with 300 nm Kd detected the NLV/A2 complex after infection of primary human fibroblasts with CMV, but it has not been further developed (38). The higher 50 nm Kd of our TCR suggests that the α2.β8 clone could be used to track pMHC display and demonstrates the feasibility of our TCR engineering approach. Moreover, flow cytometer sensitivity can be increased with an enzymatic amplification step (55) or a single molecule fluorescence assay (56).
There is growing interest in using TCRs to monitor the presence of disease-related peptides and a need to detect the NLV/A2 complex to support CMV diagnostics and therapeutics development. High-affinity soluble TCRs directed at cancer antigens are currently under clinical evaluation as part of a bispecific molecule (57). As compared with cancer antigens, infectious disease-associated antigens are more likely to be unique to diseased tissue, thereby reducing the risks of toxicity. The high-affinity TCRs reported here are currently under evaluation for their abilities to detect NLV-positive cells in vaccine and infection-related settings.
Experimental procedures
Display of recombinant RA14 variants on the CHO cell surface
The amino acid sequences of the extracellular α- and β-chains of the human TCR RA14 were obtained from the Protein Data Bank (PDB code 3GSN) (14), with the constant regions including the native-PESSC and -C on the α- and β-chain C termini, respectively. Minor changes were introduced into this sequence: Vα:1I and Vβ:M2A were added to match the germline TRAV24 and TRBV6-5 sequences (IMGT), and αA78V (PDB residue 152) substitution was made to match the germline TRAC gene (UniProt). These sequences were then reverted to DNA with CHO cell-optimized codons and synthesized as a gblock (IDT). These were assembled into a cassette with a murine IgΗ leader sequence (58) followed by one TCR chain, a T2A cleavage site with furin cut site (bold), and GSG linker (underlined; sequence: RRKRGSGEGRGSLLTCGDVEENPGP), then the second TCR chain fused to a PDGFR transmembrane region (Fig. 1a) (59). The TCR chains were cloned in both orientations, α-T2A-β and β-T2A-α. The constant regions were further modified to create a disulfide variant (ds) by introducing the amino acid substitutions Vα:T84C, Vβ:S79C, and Vβ:C85.1A to move the terminal disulfide to a more central position and remove a free cysteine (20). The cassette was cloned into a pcDNA3 (Invitrogen) backbone for transient expression and moved into a pPyEBV backbone (Acyte Biotech) (33) for semi-stable replication used during library sorting.
CHO cell transfection for TCR display analysis
CHO-T cells (Acyte Biotech) (33) were grown in CHO-S–SFM II media (Gibco) supplemented with 2× GlutaMax and penicillin/streptomycin. For transfection, cells were spun and resuspended at a concentration of 1.5 × 106 cells/ml, with 2 ml plated per well in a 6-well plate. For each well, 250 μl of OptiMEM (ThermoFisher Scientific) was mixed with 10 μl of Lipofectamine 2000 (ThermoFisher Scientific) and added to another tube with 250 μl of OptiMEM and 4 μg of DNA. The solution was mixed and allowed to equilibrate for 30 min at room temperature, before adding the solution to the appropriate well. The next day, cells were fed an additional 1 ml of media.
TCR library design and cloning
Two separate libraries were generated, one targeting the CDR3α and the other targeting CDR3β with saturation mutagenesis. The targeted region was defined as the single continuous stretch of residues in each CDR3 having direct contact with the pMHC (14), as well as an additional residue on either side to confer additional loop flexibility (Vα:107–115 and Vβ:108–115; Fig. 2). To limit library size and retain ligand binding, three residues forming hydrogen bonds with the peptide (Vα:114, Vβ:110, and Vβ:114) were not randomized.
Libraries were generated using overlap PCR with degenerate codons (CDR3α: NNS–NNS–NNS–AAC–NNS and CDR3β: NBS–VBC–ACC–VBC–VBC–VBC–TAC–NBS) and Q5 hot-start master mix (New England Biolabs). The PCR insert and pPyEBV backbone were both digested with restriction enzymes flanking each targeted region (for CDR3α, AgeI and NheI; for CDR3β, BamHI and NheI, New England Biolabs), gel-extracted, and desalted. For each library, ∼1 μg of vector was ligated with insert at a 3:1 (CDR3α) or 6:1 (CDR3β) ratio overnight using T4 ligase (New England Biolabs). The following day, ligations were desalted and transformed into fresh NEB10β electrocompetent cells. After 1 h of recovery, dilutions were plated and incubated, and colony counts were used to estimate the library size. The library was grown to an OD600 of 2 in liquid culture, which was then used to make frozen stocks and inoculate a new flask for overnight growth. Library DNA was prepared using a Maxiprep kit (Qiagen).
Tetramer preparation
Biotinylated HLA-A*0201 monomer loaded with the pp65495–503 NLV peptide from human CMV or a control HCV peptide (Biolegend and the National Institutes of Health tetramer facility) at 100 μg/ml was combined in a 4:1 molar ratio with streptavidin-conjugated to APC or AF647 (ThermoFisher Scientific). Streptavidin was added slowly over 1 h on ice to favor the formation of tetramer. Biotin (30 μm) was then added to block any unfilled biotin-binding sites, and the tetramers were incubated overnight at 4 °C.
Flow cytometry and analysis
Transfected CHO-K1 or CHO-T cells (∼1 × 106) were centrifuged (250 × g for 10 min) and resuspended in 100 μl of PBS plus 2% FBS (Sigma) with 2 μg/ml NLV or HCV tetramer and a 1:50 dilution of anti-Vβ6-5-PE (Beckman Coulter). Cells were stained on ice for 1 h, then rinsed and resuspended in 0.5 ml of PBS plus 2% FBS. Samples were run on a Fortessa cytometer (BD Biosciences). A forward- and side-scatter gate was used to select live cells, with TCR-positive cells defined as having a PE signal greater than or equal to 103. To compare binding activity of TCR variants, NLV tetramer binding was normalized by TCR display level by dividing the AF647 signal by the PE signal on a per-cell basis for the TCR-positive population. All analyses were performed with FlowJo software.
Library transfection and sorting
Confluent CHO-T cells (1 or 2× T-150 flasks) were transfected as above, with plasmid quantities scaled accordingly. Library DNA was diluted 1:4 with an inert yeast plasmid as carrier DNA (pCTCON) (60) so that each CHO cell received at most one TCR expressing pPy plasmid. Flasks were also transfected with the WT pPy_RA14 as a positive control and pPy_hu4D5 expressing the anti-HER2 Fab as a negative control. Two days later, cells were scanned for TCR surface display using the anti-Vb6–5 antibody to evaluate transfection efficiency, and media were replaced with half-strength selective media (CHO-S-SFM II plus 2× GlutaMax plus 150 μg/ml hygromycin). Four or 5 days after transfection, cells were expanded and transferred into full-strength selective media with 300 μg/ml hygromycin. Cells were maintained in selective media until reaching confluence in a T-150 flask (∼2 weeks).
For sorting, ∼1 × 107 live cells were centrifuged (250 × g, 5 min) and resuspended in OptiMEM plus 1% BSA and stained with anti-TCR Vβ6–5 and NLV tetramer as described above. Cells were sorted using a FACSAria, with the sort gate drawn to collect the most fluorescent 1–2% of cells in the population (∼1 × 105 total cells), biased toward cells with a high tetramer binding to TCR display ratio. The sorted cells were expanded for ∼1 week, and the process was repeated two times. To recover plasmid from sorted CHO cells, genomic DNA was prepared from the pooled population 1 week after each sort. The region of interest (CDR3α or CDR3β, depending on the library) was PCR-amplified using flanking primers and 500 ng of template DNA. PCR product was ligated into a TOPO vector (Invitrogen/ThermoFisher Scientific) or digested and ligated into empty pPy backbone. After transformation, 10–40 individual colonies were sequenced from each round of sorting.
Expression of soluble TCR-Fc fusion proteins
To express soluble TCR-Fc fusion proteins, DNA encoding the RA14 extracellular α- and β-chains was cloned into pcDNA3.0 plasmids downstream of a mouse IgG heavy chain leader sequence. Both the native human TCR constant domains and versions with the additional inter-chain disulfide introduced were used. The Cα domain was followed by the upper hinge sequence of human IgG1 (VEPKSC), the core and lower hinge, and then the Fc domains. The native IgG1 light-chain cysteine was inserted at the C terminus of Cβ to pair with the upper hinge cysteine and further stabilize the TCR heterodimerization. Additional modifications included the removal of five N-linked glycosylation sites predicted by NetNGlyc 1.0 Server (DTU Bioinformatics; http://www.cbs.dtu.dk/services/NetNGlyc/)4 by introducing Vα:N20Q, Cα:N90Q, α:N109Q, Vβ:N77Q, and Cβ:N85.6Q substitutions (Fig. 6a).
In one version, the construct consisted of α-T2A-β–huIgG1 hinge and CH2 and CH3 domains, and in another version, these two chains were encoded on separate pcDNA3.0-based plasmids, with either α- or β-chain fused to the Fc. Both human IgG1 and mouse IgG2a hinge–Fc sequences were used (Uniprot accession numbers P01857 and P01863, respectively). Constructs containing the mouse IgG2a Fc retained the human IgG1 VEPKSC before the mouse hinge-Fc sequence. Cloning was performed using Q5 hot-start polymerase (New England Biolabs) and either traditional digestion/ligation or Gibson assembly methods and confirmed by Sanger sequencing.
For large-scale expression, two T-150 flasks of adherent CHO-K1 cells (ATCC CCL-61) were grown to confluency and transfected using the same cell/media/reagent ratio described above for each variant. Cells were grown in high-glucose Dulbecco's modified Eagle's medium (Sigma) with 10% low-IgG FBS (ThermoFisher Scientific) and no antibiotics at 37 °C with 5% CO2. Media were replaced the day after transfection, and cells were transferred to a 32 °C, 5% CO2 incubator for 1 week. Media were harvested and loaded onto a protein A column using an FPLC (ÅKTAPure, GE Healthcare) using 100 mm phosphate, 150 mm NaCl, pH 7.2, and eluted with 100 mm glycine, pH 2.5. The eluate was immediately neutralized with 1 m Tris, pH 8, and buffer-exchanged into PBS, pH 7.4, using a 50,000 MWCO Amicon centrifugal filter.
Protein biophysical characterization
Purified TCR-Fc proteins (3 μg each) were prepared in reducing or nonreducing 6× SDS loading buffer and incubated for 5 min at 80 or 42 °C, respectively. Samples were separated on a 4–20% gradient gel (Bio-Rad) and stained with GelCode Blue (ThermoFisher Scientific). Analytical size-exclusion chromatography was performed with 100 μg of purified protein in 100 μl using a Superdex S200 column and Åkta FPLC with PBS as the running buffer and gel filtration calibration kit high- and low-molecular-weight standards (GE Healthcare). For affinity variants, the peak eluting at ∼10.5 ml containing properly assembled, bivalent TCR-Fc was collected and concentrated. To monitor thermal stability, protein was prepared at 200 μg/ml and diluted with protein thermal shift dye (ThermoFisher Scientific) following the recommended protocol. Samples were heated at 1 °C/min on a RT-PCR machine measuring fluorescence.
Protein–protein binding assays
For ELISA analyses, high-protein binding plates (Costar) were coated with 1 μg/ml NLV/A2, HCV/A2 tetramer, or nothing in PBS overnight at 4 °C, before blocking with 5% milk in PBS with 0.05% Tween 20 (PBS-T) for 1 h at room temperature. The plate was washed three times with PBS-T, and purified TCR-Fc was titrated in 1:5 dilution steps from 10 μg/ml and incubated for 1 h. After washing again, 1:1000 dilution of goat anti-human Fc-HRP conjugate (Southern Biotech) was added to the plate for 1 h. After a final wash, the plate was developed with TMB (ThermoFisher Scientific), quenched with 1 n HCl, and absorbance measured at 450 nm on a Molecular Devices Spectramax. All plated volumes were 50 μl. Data were analyzed with Graphpad Prism 5.
Dynamic and equilibrium binding kinetics were obtained using a BIAcore 3000 instrument. The purified TCR-Fc was immobilized on a CM5 sensor chip (GE Healthcare) via EDC/NHS coupling using a sodium acetate buffer at pH 4.0 for a total of 2000–5000 response units, with a blank flow cell used as the reference channel. Monomeric NLV/A2 was injected at concentrations ranging from 4 to 1000 nm at 30 μl/min for 2 min and allowed to dissociate for 6 min, which resulted in a return to baseline without regeneration. The negative control HCV/A2 monomer was injected at the highest concentration for all variants. All data were measured at 25 °C. On-rate, off-rate, and equilibrium binding analyses were performed using BIAEvaluation 3.0 software and fit using the 1:1 Langmuir binding model. All injections were performed twice, and final kinetic values reported are the average and standard deviation for the entire dataset.
Staining peptide-pulsed antigen-presenting cells
Human TAP-deficient T2 lymphoblasts (174 × CEM.T2; ATCC no. CRL-1992) expressing empty HLA-A2 were cultured in Iscove's modified Dulbecco's medium supplemented with 4 mm glutamine at 37 °C and 5% CO2. The pp65495–503 peptide NLVPMVATV and control HCV1406–1415 peptide KLVALGINAV were produced by solid-phase synthesis (Peptide 2.0) and dissolved in DMSO for a final peptide stock of 50 mm. T2 cells in a six-well plate (2 ml/well) containing 106 cells/ml were adjusted to 100 μm or the indicated peptide concentration and incubated at 37 °C. After 24 h, 5 × 105 cells per sample were stained on ice for 1 h using 2 μm purified TCR-mFc in PBS with 1% FBS (PBS-F) with 50 μg/ml human Fc block (BD Biosciences) in a 50-μl volume. Cells were washed twice with PBS-F and bound TCR-mFc detected with a 1:500 dilution of goat anti-mouse Fc-AF647 (Jackson Immunobiology) for another hour. After a final wash, cells were resuspended and assayed for AF647 signal using a flow cytometer (Fortessa, BD Biosciences).
Activation of human Jurkat T cells expressing RA14 variants
A pcDNA3.1-derived plasmid with a CMV promoter was modified to support transient expression of a signaling-competent TCR composed of the RA14 variable regions and mouse constant/transmembrane regions. First, a Kozak sequence with an optimal ribosome-binding site (sequence CCACCATGG), multiple cloning site, and stop codon followed by a HindIII site and terminal SV40 poly(A) tail signal were added to pcDNA3.1. Next, separate plasmids containing the TCR α- and β-chains were assembled. The pRA14α α-chain plasmid includes a murine α-chain TCR signal sequence from IMGT TRAV5D-4 (amino acid sequence MKTYAPTLFMFLWLQLDGMSQ) in-frame with the human TCR α variable region, both flanked by restriction sites EcoRI and AflII, the murine TCR α constant region (TRAC*01), and α transmembrane domain between restriction sites AflII and HindIII. The pRA14β β-chain plasmid was constructed similarly but with the murine β-chain signal sequence from IMGT TRBV13-2 (MGSRLFFVLSSLLCSKHM) and human variable β domain flanked by EcoRI and AflII sites, mouse β constant region (TRBC*02), and β transmembrane domain between restriction sites AflII and HindIII. Signal sequences were encoded by oligonucleotides, and constant regions were amplified from mouse DO11.10 hybridoma mRNA by RT-PCR. Engineered RA14 variable regions were introduced by PCR amplification followed by digestion/ligation into the EcoRI and AflII sites.
Human Jurkat T-cells, clone E6-1 (ATCC no. TIB-152), were grown in RPMI 1640 media with 10% FBS and 100 units/ml penicillin-streptomycin (Sigma) and transfected as described previously (61). Briefly, 106 cells per transfection were centrifuged at 250 × g for 5 min, resuspended in 5 ml of OptiMEM, and incubated at room temperature for 8 min. Cells were centrifuged as before and resuspended in 400 μl of OptiMEM. Cells were then mixed with 7.5 μg of each α- and β-plasmid in a 4-mm electroporation cuvette (ThermoFisher Scientific) and incubated for 8 min before pulsing exponentially with 250 V, 950 microfarads, and ∞ ohms on a Bio-Rad GenePulser. After an 8-min recovery period, cells were rescued with 7 ml of RPMI 1640 medium (supplemented with 10% FBS and antibiotics) in a T-25 flask at 37 °C and 5% CO2. After 18–24 h, recombinant TCR expression was monitored by flow cytometry, with the RA14 display level monitored by the anti-TRBV6-5 antibody-PE, and binding activity monitored by NLV/A2 tetramer-AF647, as described above.
The ability of transfected RA14 variants to activate Jurkat cells was monitored by CD69 up-regulation after incubation with peptide-pulsed T2 cells. T2 cells (105) were pulsed with NLV and HCV peptides at 0.1 μm for 4 h. T2 cells were washed once in RPMI medium to remove excess peptide. Transfected Jurkat cells (105) were co-cultured with pulsed T2 cells at a TCR-positive effector/target ratio of 1:1. After 24 h, cells were collected and incubated with 50 μg/ml human Fc block (BD Biosciences) for 10 min before adding anti-TRBV6-5-PE, NLV/A2 tetramer-AF647, and anti-CD69-FITC (Biolegend; 1 μl of each antibody and 5 nm tetramer per 50 μl of staining volume) for 1 h on ice. Cells were scanned on a Fortessa cytometer (BD Biosciences) and gated for display of RA14 variants (PE signal ≥500) to exclude T2 cells. All analyses were performed with FlowJo.
Author contributions
E. K. W., A. N. Q., and J. A. M. conceptualization; E. K. W. and A. N. Q. formal analysis; E. K. W., A. N. Q., C. A. S., A. W. N., and G. D. investigation; E. K. W., A. N. Q., C. A. S., A. W. N., G. D., and J. A. M. methodology; E. K. W. and J. A. M. writing-original draft; E. K. W., A. N. Q., C. A. S., A. W. N., G. D., and J. A. M. writing-review and editing; A. N. Q. data curation; A. W. N. and J. A. M. supervision; J. A. M. funding acquisition; J. A. M. project administration.
Supplementary Material
Acknowledgments
The National Institutes of Health tetramer facility is supported by Contract HHSN272201300006C from NIAID. We thank Zachary P. Frye for support in cloning for Jurkat display.
This work was supported by Welch Foundation Grant F-1767 (to J. A. M.), the Clayton Foundation for Biomedical Research (to G. D.), National Institutes of Health NRSA Grant F32GM111018 (to A. W. N.), and National Science Foundation GRFP fellowships (to E. K. W. and A. N. Q.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S6 and Table S1.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- TCR
- T-cell receptor
- A2
- human HLA-A*02 MHC allele
- CDR
- complementarity determining region
- CHO
- Chinese hamster ovary
- CMV
- cytomegalovirus
- Fc
- antibody crystallizable fragment
- KLV
- peptide fragment between residues 1406–1415 of the hepatitis C virus protein
- NLV
- peptide fragment between residues 495–503 of the pp65 CMV protein
- PDGFR
- platelet-derived growth factor receptor
- pMHC
- peptide-major histocompatibility complex
- TM
- transmembrane
- FBS
- fetal bovine serum
- PE
- phycoerythrin
- APC
- allophycocyanin
- PDB
- Protein Data Bank
- SEC
- size exclusion chromatography
- SPR
- surface plasmon resonance.
References
- 1. Rossjohn J., Gras S., Miles J. J., Turner S. J., Godfrey D. I., and McCluskey J. (2015) T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 33, 169–200 10.1146/annurev-immunol-032414-112334 [DOI] [PubMed] [Google Scholar]
- 2. Harris D. T., and Kranz D. M. (2016) Adoptive T cell therapies: a comparison of T-cell receptors and chimeric antigen receptors. Trends Pharmacol. Sci. 37, 220–230 10.1016/j.tips.2015.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oates J., Hassan N. J., and Jakobsen B. K. (2015) ImmTACs for targeted cancer therapy: why, what, how, and which. Mol. Immunol. 67, 67–74 10.1016/j.molimm.2015.01.024 [DOI] [PubMed] [Google Scholar]
- 4. Varela-Rohena A., Molloy P. E., Dunn S. M., Li Y., Suhoski M. M., Carroll R. G., Milicic A., Mahon T., Sutton D. H., Laugel B., Moysey R., Cameron B. J., Vuidepot A., Purbhoo M. A., Cole D. K., et al. (2008) Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat. Med. 14, 1390–1395 10.1038/nm.1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang H., Buisson S., Bossi G., Wallace Z., Hancock G., So C., Ashfield R., Vuidepot A., Mahon T., Molloy P., Oates J., Paston S. J., Aleksic M., Hassan N. J., Jakobsen B. K., and Dorrell L. (2016) Elimination of latently HIV-infected cells from antiretroviral therapy-suppressed subjects by engineered immune-mobilizing T-cell receptors. Mol. Ther. 24, 1913–1925 10.1038/mt.2016.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Plotkin S. A., and Boppana S. B. (2018) Vaccination against the human cytomegalovirus. Vaccine 2018, S0264–410X(18)30288–3 10.1016/j.vaccine.2018.02.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wills M. R., Carmichael A. J., Mynard K., Jin X., Weekes M. P., Plachter B., and Sissons J. G. (1996) The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J. Virol. 70, 7569–7579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saulquin X., Ibisch C., Peyrat M. A., Scotet E., Hourmant M., Vie H., Bonneville M., and Houssaint E. (2000) A global appraisal of immunodominant CD8 T cell responses to Epstein-Barr virus and cytomegalovirus by bulk screening. Eur. J. Immunol. 30, 2531–2539 10.1002/1521-4141(200009)30:9<2531::AID-IMMU2531>3.0.CO;2-O [DOI] [PubMed] [Google Scholar]
- 9. Gonzalez-Galarza F. F., McCabe A., Melo Dos Santos E. J., Takeshita L., Ghattaoraya G., Jones A. R., and Middleton D. (2018) Allele frequency net database. Methods Mol. Biol. 1802, 49–62 10.1007/978-1-4939-8546-3_4 [DOI] [PubMed] [Google Scholar]
- 10. Feuchtinger T., Opherk K., Bethge W. A., Topp M. S., Schuster F. R., Weissinger E. M., Mohty M., Or R., Maschan M., Schumm M., Hamprecht K., Handgretinger R., Lang P., and Einsele H. (2010) Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 116, 4360–4367 10.1182/blood-2010-01-262089 [DOI] [PubMed] [Google Scholar]
- 11. Riddell S. R., Rabin M., Geballe A. P., Britt W. J., and Greenberg P. D. (1991) Class I MHC-restricted cytotoxic T lymphocyte recognition of cells infected with human cytomegalovirus does not require endogenous viral gene expression. J. Immunol. 146, 2795–2804 [PubMed] [Google Scholar]
- 12. Trautmann L., Rimbert M., Echasserieau K., Saulquin X., Neveu B., Dechanet J., Cerundolo V., and Bonneville M. (2005) Selection of T cell clones expressing high-affinity public TCRs within human cytomegalovirus-specific CD8 T cell responses. J. Immunol. 175, 6123–6132 10.4049/jimmunol.175.9.6123 [DOI] [PubMed] [Google Scholar]
- 13. Wang G. C., Dash P., McCullers J. A., Doherty P. C., and Thomas P. G. (2012) T-cell receptor αβ diversity inversely correlates with pathogen-specific antibody levels in human cytomegalovirus infection. Sci. Transl. Med. 4, 128 10.1126/scitranslmed.3003647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gras S., Saulquin X., Reiser J. B., Debeaupuis E., Echasserieau K., Kissenpfennig A., Legoux F., Chouquet A., Le Gorrec M., Machillot P., Neveu B., Thielens N., Malissen B., Bonneville M., and Housset D. (2009) Structural bases for the affinity-driven selection of a public TCR against a dominant human cytomegalovirus epitope. J. Immunol. 183, 430–437 10.4049/jimmunol.0900556 [DOI] [PubMed] [Google Scholar]
- 15. Gakamsky D. M., Lewitzki E., Grell E., Saulquin X., Malissen B., Montero-Julian F., Bonneville M., and Pecht I. (2007) Kinetic evidence for a ligand-binding-induced conformational transition in the T-cell receptor. Proc. Natl. Acad. Sci. U.S.A. 104, 16639–16644 10.1073/pnas.0707061104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shusta E. V., Kieke M. C., Parke E., Kranz D. M., and Wittrup K. D. (1999) Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J. Mol. Biol. 292, 949–956 10.1006/jmbi.1999.3130 [DOI] [PubMed] [Google Scholar]
- 17. Holler P. D., Holman P. O., Shusta E. V., O'Herrin S., Wittrup K. D., and Kranz D. M. (2000) In vitro evolution of a T-cell receptor with high affinity for peptide/MHC. Proc. Natl. Acad. Sci. U.S.A. 97, 5387–5392 10.1073/pnas.080078297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shusta E. V., Holler P. D., Kieke M. C., Kranz D. M., and Wittrup K. D. (2000) Directed evolution of a stable scaffold for T-cell receptor engineering. Nat. Biotechnol. 18, 754–759 10.1038/77325 [DOI] [PubMed] [Google Scholar]
- 19. Aggen D. H., Chervin A. S., Insaidoo F. K., Piepenbrink K. H., Baker B. M., and Kranz D. M. (2011) Identification and engineering of human variable regions that allow expression of stable single-chain T-cell receptors. Protein Eng. Des. Sel. 24, 361–372 10.1093/protein/gzq113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boulter J. M., Glick M., Todorov P. T., Baston E., Sami M., Rizkallah P., and Jakobsen B. K. (2003) Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 16, 707–711 10.1093/protein/gzg087 [DOI] [PubMed] [Google Scholar]
- 21. Li Y., Moysey R., Molloy P. E., Vuidepot A. L., Mahon T., Baston E., Dunn S., Liddy N., Jacob J., Jakobsen B. K., and Boulter J. M. (2005) Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 23, 349–354 10.1038/nbt1070 [DOI] [PubMed] [Google Scholar]
- 22. Liddy N., Bossi G., Adams K. J., Lissina A., Mahon T. M., Hassan N. J., Gavarret J., Bianchi F. C., Pumphrey N. J., Ladell K., Gostick E., Sewell A. K., Lissin N. M., Harwood N. E., Molloy P. E., et al. (2012) Monoclonal TCR-redirected tumor cell killing. Nat. Med. 18, 980–987 10.1038/nm.2764 [DOI] [PubMed] [Google Scholar]
- 23. Wülfing C., and Plückthun A. (1994) Correctly folded T-cell receptor fragments in the periplasm of Escherichia coli. Influence of folding catalysts. J. Mol. Biol. 242, 655–669 10.1006/jmbi.1994.1615 [DOI] [PubMed] [Google Scholar]
- 24. Maynard J., Adams E. J., Krogsgaard M., Petersson K., Liu C. W., and Garcia K. C. (2005) High-level bacterial secretion of single-chain αβ T-cell receptors. J. Immunol. Methods 306, 51–67 10.1016/j.jim.2005.07.022 [DOI] [PubMed] [Google Scholar]
- 25. Gunnarsen K. S., Kristinsson S. G., Justesen S., Frigstad T., Buus S., Bogen B., Sandlie I., and Loset G. Å. (2013) Chaperone-assisted thermostability engineering of a soluble T-cell receptor using phage display. Sci. Rep. 3, 1162 10.1038/srep01162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mosquera L. A., Card K. F., Price-Schiavi S. A., Belmont H. J., Liu B., Builes J., Zhu X., Chavaillaz P. A., Lee H. I., Jiao J. A., Francis J. L., Amirkhosravi A., Wong R. L., and Wong H. C. (2005) In vitro and in vivo characterization of a novel antibody-like single-chain TCR human IgG1 fusion protein. J. Immunol. 174, 4381–4388 10.4049/jimmunol.174.7.4381 [DOI] [PubMed] [Google Scholar]
- 27. Walseng E., Wälchli S., Fallang L. E., Yang W., Vefferstad A., Areffard A., and Olweus J. (2015) Soluble T-cell receptors produced in human cells for targeted delivery. PLoS ONE 10, e0119559 10.1371/journal.pone.0119559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Foss S., Grevys A., Sand K. M. K., Bern M., Blundell P., Michaelsen T. E., Pleass R. J., Sandlie I., and Andersen J. T. (2016) Enhanced FcRn-dependent transepithelial delivery of IgG by Fc-engineering and polymerization. J. Control. Release 223, 42–52 10.1016/j.jconrel.2015.12.033 [DOI] [PubMed] [Google Scholar]
- 29. Nguyen A. W., Le K. C., and Maynard J. A. (2018) Identification of high affinity HER2 binding antibodies using CHO Fab surface display. Protein Eng. Des. Sel. 31, 91–101 10.1093/protein/gzy004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang X., Wong C. W., Urak R., Mardiros A., Budde L. E., Chang W. C., Thomas S. H., Brown C. E., La Rosa C., Diamond D. J., Jensen M. C., Nakamura R., Zaia J. A., and Forman S. J. (2015) CMVpp65 vaccine enhances the antitumor efficacy of adoptively transferred CD19-redirected CMV-specific T cells. Clin. Cancer Res. 21, 2993–3002 10.1158/1078-0432.CCR-14-2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ho S. K., Lo C. Y., Cheng I. K., and Chan T. M. (1998) Rapid cytomegalovirus pp65 antigenemia assay by direct erythrocyte lysis and immunofluorescence staining. J. Clin. Microbiol. 36, 638–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang X., Gao M., Chen G., Pierce B. G., Lu J., Weng N. P., and Mariuzza R. A. (2015) Structural basis for clonal diversity of the public T cell response to a dominant human cytomegalovirus epitope. J. Biol. Chem. 290, 29106–29119 10.1074/jbc.M115.691311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kunaparaju R., Liao M., and Sunstrom N. A. (2005) Epi-CHO, an episomal expression system for recombinant protein production in CHO cells. Biotechnol. Bioeng. 91, 670–677 10.1002/bit.20534 [DOI] [PubMed] [Google Scholar]
- 34. Cohen C. J., Zhao Y., Zheng Z., Rosenberg S. A., and Morgan R. A. (2006) Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 66, 8878–8886 10.1158/0008-5472.CAN-06-1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sibener L. V., Fernandes R. A., Kolawole E. M., Carbone C. B., Liu F., McAffee D., Birnbaum M. E., Yang X., Su L. F., Yu W., Dong S., Gee M. H., Jude K. M., Davis M. M., Groves J. T., et al. (2018) Isolation of a structural mechanism for uncoupling T-cell receptor signaling from peptide–MHC binding. Cell 174, 672–687.e27 10.1016/j.cell.2018.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holler P. D., Chlewicki L. K., and Kranz D. M. (2003) TCRs with high affinity for foreign pMHC show self-reactivity. Nat. Immunol. 4, 55–62 10.1038/ni863 [DOI] [PubMed] [Google Scholar]
- 37. Zhao Y., Bennett A. D., Zheng Z., Wang Q. J., Robbins P. F., Yu L. Y., Li Y., Molloy P. E., Dunn S. M., Jakobsen B. K., Rosenberg S. A., and Morgan R. A. (2007) High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J. Immunol. 179, 5845–5854 10.4049/jimmunol.179.9.5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Makler O., Oved K., Netzer N., Wolf D., and Reiter Y. (2010) Direct visualization of the dynamics of antigen presentation in human cells infected with cytomegalovirus revealed by antibodies mimicking TCR specificity. Eur. J. Immunol. 40, 1552–1565 10.1002/eji.200939875 [DOI] [PubMed] [Google Scholar]
- 39. Nguyen A. W., Wagner E. K., Laber J. R., Goodfield L. L., Smallridge W. E., Harvill E. T., Papin J. F., Wolf R. F., Padlan E. A., Bristol A., Kaleko M., and Maynard J. A. (2015) A cocktail of humanized anti-pertussis toxin antibodies limits disease in murine and baboon models of whooping cough. Sci. Transl. Med. 7, 316ra195 10.1126/scitranslmed.aad0966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kessels H. W., van Den Boom M. D., Spits H., Hooijberg E., and Schumacher T. N. (2000) Changing T cell specificity by retroviral T-cell receptor display. Proc. Natl. Acad. Sci. U.S.A. 97, 14578–14583 10.1073/pnas.97.26.14578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chervin A. S., Aggen D. H., Raseman J. M., and Kranz D. M. (2008) Engineering higher affinity T-cell receptors using a T cell display system. J. Immunol. Methods 339, 175–184 10.1016/j.jim.2008.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Malecek K., Zhong S., McGary K., Yu C., Huang K., Johnson L. A., Rosenberg S. A., and Krogsgaard M. (2013) Engineering improved T-cell receptors using an alanine-scan guided T cell display selection system. J. Immunol. Methods 392, 1–11 10.1016/j.jim.2013.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Borroto A., Lama J., Niedergang F., Dautry-Varsat A., Alarcón B., and Alcover A. (1999) The CD3 ϵ subunit of the TCR contains endocytosis signals. J. Immunol. 163, 25–31 [PubMed] [Google Scholar]
- 44. Sami M., Rizkallah P. J., Dunn S., Molloy P., Moysey R., Vuidepot A., Baston E., Todorov P., Li Y., Gao F., Boulter J. M., and Jakobsen B. K. (2007) Crystal structures of high affinity human T-cell receptors bound to peptide major histocompatibility complex reveal native diagonal binding geometry. Protein Eng. Des. Sel. 20, 397–403 10.1093/protein/gzm033 [DOI] [PubMed] [Google Scholar]
- 45. Cole D. K., Sami M., Scott D. R., Rizkallah P. J., Borbulevych O. Y., Todorov P. T., Moysey R. K., Jakobsen B. K., Boulter J. M., Baker B. M., and Yi L. (2013) Increased peptide contacts govern high affinity binding of a modified TCR whilst maintaining a native pMHC docking mode. Front. Immunol. 4, 168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. van Boxel G. I., Stewart-Jones G., Holmes S., Sainsbury S., Shepherd D., Gillespie G. M., Harlos K., Stuart D. I., Owens R., and Jones E. Y. (2009) Some lessons from the systematic production and structural analysis of soluble (α)(β) T-cell receptors. J. Immunol. Methods 350, 14–21 10.1016/j.jim.2009.08.008 [DOI] [PubMed] [Google Scholar]
- 47. Grégoire C., Rebaï N., Schweisguth F., Necker A., Mazza G., Auphan N., Millward A., Schmitt-Verhulst A. M., and Malissen B. (1991) Engineered secreted T-cell receptor αβ heterodimers. Proc. Natl. Acad. Sci. U.S.A. 88, 8077–8081 10.1073/pnas.88.18.8077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lunde E., Loset G. Å., Bogen B., and Sandlie I. (2010) Stabilizing mutations increase secretion of functional soluble TCR-Ig fusion proteins. BMC Biotechnol. 10, 61 10.1186/1472-6750-10-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lebowitz M. S., O'Herrin S. M., Hamad A. R., Fahmy T., Marguet D., Barnes N. C., Pardoll D., Bieler J. G., and Schneck J. P. (1999) Soluble, high-affinity dimers of T-cell receptors and class II major histocompatibility complexes: biochemical probes for analysis and modulation of immune responses. Cell Immunol. 192, 175–184 10.1006/cimm.1999.1441 [DOI] [PubMed] [Google Scholar]
- 50. Ozawa T., Horii M., Kobayashi E., Jin A., Kishi H., and Muraguchi A. (2012) The binding affinity of a soluble TCR-Fc fusion protein is significantly improved by cross-linkage with an anti-Cβ antibody. Biochem. Biophys. Res. Commun. 422, 245–249 10.1016/j.bbrc.2012.04.134 [DOI] [PubMed] [Google Scholar]
- 51. Kuball J., Hauptrock B., Malina V., Antunes E., Voss R. H., Wolfl M., Strong R., Theobald M., and Greenberg P. D. (2009) Increasing functional avidity of TCR-redirected T cells by removing defined N-glycosylation sites in the TCR constant domain. J. Exp. Med. 206, 463–475 10.1084/jem.20082487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Irving M., Zoete V., Hebeisen M., Schmid D., Baumgartner P., Guillaume P., Romero P., Speiser D., Luescher I., Rufer N., and Michielin O. (2012) Interplay between T-cell receptor binding kinetics and the level of cognate peptide presented by major histocompatibility complexes governs CD8+ T cell responsiveness. J. Biol. Chem. 287, 23068–23078 10.1074/jbc.M112.357673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kageyama S., Tsomides T. J., Sykulev Y., and Eisen H. N. (1995) Variations in the number of peptide–MHC class I complexes required to activate cytotoxic T cell responses. J. Immunol. 154, 567–576 [PubMed] [Google Scholar]
- 54. Sykulev Y., Joo M., Vturina I., Tsomides T. J., and Eisen H. N. (1996) Evidence that a single peptide–MHC complex on a target cell can elicit a cytolytic T cell response. Immunity 4, 565–571 10.1016/S1074-7613(00)80483-5 [DOI] [PubMed] [Google Scholar]
- 55. Anikeeva N., Mareeva T., Liu W., and Sykulev Y. (2009) Can oligomeric T-cell receptor be used as a tool to detect viral peptide epitopes on infected cells? Clin. Immunol. 130, 98–109 10.1016/j.clim.2008.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bossi G., Gerry A. B., Paston S. J., Sutton D. H., Hassan N. J., and Jakobsen B. K. (2013) Examining the presentation of tumor-associated antigens on peptide-pulsed T2 cells. Oncoimmunology 2, e26840 10.4161/onci.26840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bossi G., Buisson S., Oates J., Jakobsen B. K., and Hassan N. J. (2014) ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol. Immunother. 63, 437–448 10.1007/s00262-014-1525-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Smith K., Garman L., Wrammert J., Zheng N. Y., Capra J. D., Ahmed R., and Wilson P. C. (2009) Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat. Protoc. 4, 372–384 10.1038/nprot.2009.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chng J., Wang T., Nian R., Lau A., Hoi K. M., Ho S. C., Gagnon P., Bi X., and Yang Y. (2015) Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. MAbs 7, 403–412 10.1080/19420862.2015.1008351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Colby D. W., Kellogg B. A., Graff C. P., Yeung Y. A., Swers J. S., and Wittrup K. D. (2004) Engineering antibody affinity by yeast surface display. Methods Enzymol. 388, 348–358 10.1016/S0076-6879(04)88027-3 [DOI] [PubMed] [Google Scholar]
- 61. Goyarts E. C., Vegh Z., Kalergis A. M., Hörig H., Papadopoulos N. J., Young A. C., Thomson C. T., Chang H. C., Joyce S., and Nathenson S. G. (1998) Point mutations in the β-chain CDR3 can alter the T-cell receptor recognition pattern on an MHC class I/peptide complex over a broad interface area. Mol. Immunol. 35, 593–607 10.1016/S0161-5890(98)00056-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.