

Abstract
Oligomeric assemblies of amyloid-β (Aβ) peptide (Aβo) in the brains of individuals with Alzheimer's disease (AD) are toxic to neuronal synapses. More than a dozen Aβ receptor candidates have been suggested to be responsible for various aspects of the molecular pathology and memory impairment in mouse models of AD. A lack of consistent experimental design among previous studies of different receptor candidates limits evaluation of the relative roles of these candidates, producing some controversy within the field. Here, using cell-based assays with several Aβ species, including Aβo from AD brains obtained by autopsy, we directly compared the Aβ-binding capacity of multiple receptor candidates while accounting for variation in expression and confirming cell surface expression. In a survey of 15 reported Aβ receptors, only cellular prion protein (PrPC), Nogo receptor 1 (NgR1), and leukocyte immunoglobulin-like receptor subfamily B member 2 (LilrB2) exhibited direct binding to synaptotoxic assemblies of synthetic Aβ. Both PrPC and NgR1 preferentially bound synaptotoxic oligomers rather than nontoxic monomers, and the method of oligomer preparation did not significantly alter our binding results. Hippocampal neurons lacking both NgR1 and LilrB2 exhibited a partial reduction of Aβo binding, but this reduction was lower than in neurons lacking PrPC under the same conditions. Finally, binding studies with soluble Aβo from human AD brains revealed a strong affinity for PrPC, weak affinity for NgR1, and no detectable affinity for LilrB2. These findings clarify the relative contributions of previously reported Aβ receptors under controlled conditions and highlight the prominence of PrPC as an Aβ-binding site.
Keywords: Alzheimer disease; amyloid-β (Aβ); Aβ receptor; prion; receptor; oligomer; prion protein (PrPC); neurodegeneration; leukocyte immunoglobulin-like receptor subfamily B member 2 (LilrB2), Nogo receptor 1 (NgR1); EphA1; FcγRIIb; SorLA; sortilin; p75NTR; PGRMC1; NLGN1; RAGE; EphB2; FZD5; mGluR5; EphA4; nAchRa7; NR2B; NR1
Introduction
The pathophysiology responsible for the clinical signs and symptoms of AD2 has been studied since Alois Alzheimer first described the characteristic senile plaques and neurofibrillary tangles in the brain of a patient who suffered a deteriorating psychological condition that included sleep disturbances, memory impairment, and confusion (1). Today, AD is the leading cause of dementia worldwide and carries with it a tremendous economic burden projected to exceed 2 trillion United States dollars by the year 2030 (2). Despite more than a century of research, AD remains without a treatment that is capable of curing, preventing, or slowing the progression of disease. The role of senile plaques and neurofibrillary tangles in the etiology of Alzheimer's disease continues to be investigated and contested within the field. In 1998, Lambert et al. (3) described the self-assembly of synthetic β-amyloid monomers into soluble, multimeric, nonfibrillary aggregates dubbed Aβo. These oligomers were potently neurotoxic and capable of inducing cell death, and they inhibited long-term potentiation in organotypic hippocampal slices. Aβo are immunologically distinct from monomers or fibrils, induce synapse loss, and are correlated with disease progression (4–8). Similar species of Aβo were identified in brains from human AD patients in 2003 (9). The observations that synthetic and AD brain–derived Aβo bound to neurons in a trypsin-sensitive manner gave rise to the search for cell surface receptors capable of binding extracellular Aβo and transducing their neurotoxic signal intracellularly. More than a dozen proteins have been reported as responsible for mediating the deleterious effects of Aβo on neurons (10–25) (reviewed in Ref. 26). These studies have been highly disparate in both the quality and nature of evidence used to qualify a candidate as a receptor for Aβ (26). Variation in Aβ preparations, experimental design, and model systems have led to a call for a sharing of materials and validation of results between laboratories (26–28).
To address these discrepancies and better understand the relative contributions of each putative receptor to Aβo neurotoxicity, we compared the potential of each receptor to confer Aβ binding capacity to heterologous cells and neurons, the ability of each candidate to discriminate between nontoxic monomers and toxic oligomers, and the effect of different oligomer preparations on the binding profile. To determine whether synthetic preparations of Aβ faithfully recapitulate the binding profile of Aβ found in the brains of patients with AD, we also compared the ability of candidate receptors to bind soluble Aβ extracted from the brains of patients diagnosed with AD. These insights are critical to clarifying the roles of these receptors in AD pathogenesis and their therapeutic value to drug development. Preventing the interaction of neurotoxic Aβo with its receptors is an attractive drug target, and clinical trials targeting advanced glycosylation end product–specific receptor (RAGE), membrane-associated progesterone receptor component 1 (PGRMC1), and tumor necrosis factor receptor superfamily member 16 (p75NTR) are under way (NCT00141661, NCT00566397, NCT02916056, NCT02080364, NCT03522129, NCT03507790, and NCT03069014) (29–32).
Results
PrPc, LilrB2, and NgR1 bind oligomeric Aβ
Few descriptions of candidate receptors for Aβ have included a demonstration of sufficiency for conferring Aβ binding to live cells. To examine this attribute, we compiled a panel of putative receptors for Aβ and subcloned the cDNA of each into expression vectors encoding a Myc epitope at the cytoplasmic terminus of transmembrane proteins or at the mature N terminus of glycosylphosphatidylinositol-anchored proteins, which included PrPC and NgR1. These orientations were selected so as to leave the extracellular Aβ-binding domains undisturbed. The panel investigated here includes PrPC, LilrB2, NgR1, ephrin type-A receptor 1 (EphA1), low-affinity immunoglobulin γ Fc region receptor II-b (FcγRIIb), sortilin-related receptor (SorLA), sortilin, p75NTR, PGRMC1, neuroligin 1 (NLGN1), RAGE, ephrin type-B receptor 2 (EphB2), frizzled-5 (FZD5), metabotropic glutamate receptor 5 (mGluR5), and ephrin type-A receptor 4 (EphA4) (10–25). We also included neuropilin-1 (NRP1) as a negative control (Fig. S1). To our knowledge, this protein has not been implicated in binding Aβ.
We assessed the expression of each protein at the extracellular surface by cell surface biotinylation. Anti-Myc immunoprecipitation and immunoblotting confirmed that each protein was trafficked to the cell surface and of the expected molecular size (Fig. 1A). Furthermore, similar amounts of each receptor protein reached the plasma membrane and were subject to biotinylation by extracellular reagent. The Myc-tagged cytoplasmic protein collapsin response mediator protein 2 (CRMP2) was included as a control and demonstrated that only proteins localized to the cell surface were biotinylated.
Figure 1.

Myc-tagged candidates are expressed at similar levels and traffic to the cell surface. A, immunoblots of RIPA-soluble cell lysates following cell surface biotinylation and immunoprecipitation using anti-Myc-agarose beads. Candidate receptors were immunoblotted with an anti-Myc antibody (left) and fluorescently conjugated streptavidin (right). Empty, empty vector control. B, images of COS-7 cells transfected with the indicated candidate receptor and stained with anti-Myc antibody. Scale bar, 200 μm. C, quantification of B. All samples are normalized to hPrPC Myc signal. n = 32 experiments for hPrPC and 8–14 for all others. Individual data points indicate different experiments. Error bars, S.D.
For heterologous cell Aβ-binding experiments, we chose COS-7 cells, as they exhibit minimal basal Aβ binding. By staining transfected cells with an anti-Myc antibody and quantifying the average intensity, we found that receptor expression was similar across the panel (Fig. 1, B and C). Utilizing a common epitope tag for expression normalization across the panel is critical to making direct comparisons of Aβ binding. This allows us to account for any expression differences between constructs and experiments, without introducing variability from differences in the avidity of receptor-specific antibodies or potential interference with binding in the case of receptors whose Aβ binding domains have not been identified.
The concentration of oligomeric Aβ in AD patient brains has been reported to be ∼2.6 nm monomer equivalent (7). To examine the ability of candidate receptors to bind neurotoxic oligomers of Aβ, transfected cells were incubated with 1 μm monomer equivalent (∼5 nm oligomer) biotinylated Aβ oligomers (BAβo) at 4 °C to allow binding but minimize receptor internalization following binding. A high concentration was utilized so as to allow detection of Aβ binding even to low-affinity receptors. Following fixation and staining, the receptor expression–normalized Aβ binding was calculated and normalized to that of human PrPC (hPrPC). Fig. 2 (A and B) shows that PrPC, LilrB2, and NgR1 exhibit Aβo binding capacity. Among these, PrPC binds Aβo more than either LilrB2 or NgR1 (Fig. 2B). None of the other receptors demonstrated detectable Aβo binding with a lower limit of detection equal to 3% of hPrPC binding.
Figure 2.

Only PrPC, LilrB2, and NgR1 confer Aβo-binding capacity to cells. A, COS-7 cells expressing the Myc-tagged candidate receptor of interest and incubated with 1 μm biotin Aβo at 4 °C. Scale bar, 200 μm. B, quantification of biotin Aβo binding to cells in A, normalized to candidate receptor Myc signal. For each experiment, Myc-normalized biotin Aβo binding was expressed as a percentage of hPrPC in that experiment. n.s., not significant; ***, p < 0.001, one-sided t test comparing with an expected value of 100 (% hPrPC binding). Significance of a one-sided t test comparing with an expected value of 0 (no binding) is denoted by a number symbol below the x axis. #, p < 0.05; ##, p < 0.01. n = 4–7 experiments. Individual data points indicate different experiments. Error bars, S.D.
PrPC and NgR1 preferentially bind synaptotoxic Aβ species
An important characteristic of a receptor capable of transducing pathological signaling in response to Aβo is an ability to discriminate between neurotoxic oligomers and nontoxic monomeric Aβ. Aβ monomers spontaneously and rapidly assemble into high-molecular weight species in physiological buffers. Samples incubated overnight yield preparations enriched for oligomeric assemblies, which can be resolved using size-exclusion chromatography (SEC) (Fig. 3A). Analysis of the relative abundance of oligomeric Aβ in different preparations demonstrates that Aβ freshly prepared in Ham's F12 medium is 7% high-molecular weight (HMW) Aβo. Oligomeric preparations are 21% Aβo. Oligomerization can be inhibited by the use of nonphysiological buffers. When Aβ is dissolved in 0.1 n NaOH, only 3% of the preparation is HMW Aβo (Fig. 3B). Incubating transfected cells with freshly prepared Aβ in F12 is intended to provide a context in which monomeric Aβ is more readily available for binding; however, the preparation cannot be considered purely monomeric. Fig. 3C shows the expression-normalized monomeric Aβ signal for each receptor compared with oligomeric Aβ binding to hPrPC. None of the receptors bind this monomeric preparation of Aβ to a great extent (Fig. 3C). By comparing oligomeric and monomeric binding, we examined each receptor's ability to discriminate Aβ species. PrPC and NgR1 demonstrate an ability to distinguish between oligomeric Aβ and monomeric Aβ. LilrB2 binds oligomeric and monomeric Aβ to similar extents (Fig. 3D).
Figure 3.

PrPC and NgR1 preferentially bind neurotoxic Aβo. A, SEC trace of 50 μm BAβo showing absorbance at 280 nm. Alternate vertical columns indicate fractions collected for analysis. Yellow fraction, HMW Aβ. Elution times of various standards (kDa) are indicated by arrows. V0, void volume as determined by blue dextran, 2,000 kDa. B, detection and quantification of the effect of the preparative method on the distribution of BAβ in different SEC fractions by dot blot analysis. C, quantification of expression-normalized Aβ signal from COS-7 cells transfected with the indicated candidate receptor and incubated with 1 μm monomeric biotin Aβ at 4 °C. Values are normalized to oligomeric biotin Aβ binding to hPrPC. One-sided t test comparing with an expected value of 100. n = 3–5 experiments. D, binding of receptors to monomeric Aβ compared with Aβo from Fig. 2B. Shown are multiple t tests with Holm–Sidak correction for multiple comparisons. Individual data points indicate different experiments. Error bars, S.D. n.s., not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Neither temperature nor Aβ oligomerization method alters the binding profile of the receptor panel
Conducting binding assays at 4 °C affords convenience but decreases the fluidity of the cell membrane and impacts the thermodynamics of receptor–ligand interactions. To determine whether temperature influences the binding profile of the receptor panel, we incubated transfected cells with 1 μm monomer-equivalent BAβo at 37 °C. As with the 4 °C incubation, only PrPC, LilrB2, and NgR1 bind BAβo at 37 °C (Fig. 4A). In comparing binding at both temperatures, we found no effect of temperature on BAβo binding (Fig. 4B).
Figure 4.

Neither temperature nor oligomer preparation changes the binding profile of candidate receptors. A, quantification of 1 μm biotin Aβo binding to cells expressing candidate receptors at 37 °C. Binding is normalized to that of hPrPC at 4 °C. One-sided t test was used, comparing with an expected value of 100 (% hPrPC binding at 4 °C). n = 3–6 experiments. B, comparison of the capacity of a candidate receptor to bind biotin Aβo at 4 °C (Fig. 2B) versus 37 °C. Shown are multiple t tests with Holm–Sidak correction for multiple comparisons. C, atomic force microscopy images showing differences in Aβ generated by different preparations. Images are 200 × 200 nm. D, Myc-normalized binding of 1 μm Aβ prepared using the globulomer protocol. Data are expressed relative to oligomerized Aβ binding to hPrPC. One-sided t test was used, comparing with an expected value of 100 (% hPrPC binding); n = 3 experiments, 2 for PGRMC1. E, comparison of positive receptors' binding of oligomer (Fig. 2B) and globulomer Aβ. Shown are multiple t tests with Holm–Sidak correction for multiple comparisons. Individual data points indicate different experiments. Error bars, S.D. n.s., not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Multiple methods for the generation of oligomeric Aβ assemblies from monomeric synthetic peptides have been reported (10, 33, 34). Differences in preparation lead to unique profiles with regard to the types and relative abundance of multimeric Aβ assemblies as assessed under native conditions using SEC. Oligomers prepared in F12 culture medium, incubated overnight, and separated from insoluble fibrillary species by centrifugation generate larger species of oligomers with masses in the range of 100 kDa and larger (7, 10). To account for differences in oligomeric species generated, we utilized a second preparation of soluble multimeric Aβ termed globulomers (Aβg) that generates primarily a 60-kDa species by SEC (33). Atomic force microscopy confirms differences in sizes of soluble oligomers generated by the different preparations (Fig. 4C). Incubation of transfected cells with Aβg demonstrates a binding profile similar to that observed with the oligomeric preparations (Fig. 4D). When comparing binding of BAβo and Aβg to the binding-competent receptors, there is no effect of preparation on the binding (Fig. 4E).
NgR1 and LilrB2 are minor contributors to Aβo binding to neurons
Our survey of putative receptors has revealed that of those reported, only PrPC, LilrB2, and NgR1 are sufficient for binding to neurotoxic preparations of synthetic Aβ. PrPC has been demonstrated to be responsible for 50% of Aβo binding to hippocampal neurons (10). To examine the contribution of the two additional Aβo receptors confirmed here (11, 12), we examined the role of NgR1 and the murine homolog of LilrB2, Pirb, in double-knockout neurons under the same conditions used for PrPC (10). Loss of NgR1 and Pirb results in a 20% decrease in BAβo binding to double-knockout neurons (Fig. 5, A and B).
Figure 5.

Loss of NgR1 and the mouse LilrB2 homolog Pirb decreases Aβo binding to neurons. A, images showing punctate biotin Aβo binding to dendrites of hippocampal neurons. Scale bar, 50 μm. B, quantification of mean Aβo signal in A. n = 43–94 images from 5–7 dishes in four experiments. Unpaired t test was used. Error bars, S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
PrPC is the highest-affinity receptor for Aβo
To better understand the relative affinities of the positive receptors, we implemented plate-based assays of Aβ binding to purified protein. This assay is more sensitive and quantitative than the cell-based assays and allows for measurement of binding kinetics. We again utilized oligomeric, globulomeric, and monomeric Aβ. Fig. 6 (A–F) shows the binding curves and Scatchard plots for Aβo, Aβg, and Aβm, respectively. As measured in the cell-based assays, hPrPC, LilrB2, and NgR1 bound neurotoxic oligomers and globulomers (Fig. 6). The plate assay faithfully recapitulated effects of Aβ preparations on binding profiles. We observed highly similar binding of Aβo and Aβg to hPrPC and NgR1 (Fig. 6, G and H) and a preference of LilrB2 for Aβo over Aβg (Fig. 6I) as was also observed in Fig. 4E. Binding of Aβm to the receptors was minimal as was observed in the cell-based assay (Fig. 6, E and F). The dissociation constants and Bmax extracted from Fig. 6 are reported in Table 1. PrPC is the highest-affinity receptor for neurotoxic Aβo and Aβg, with affinities of 1.4 and 1.5 nm, respectively. The affinity of NgR1 for these preparations was ∼3–4-fold lower than that of hPrPC. LilrB2 demonstrated the lowest affinity, with KD of 42.3 and 207.2 nm. To quantify each receptor's ability to discriminate between neurotoxic and nontoxic preparations of Aβ, we calculated a discrimination factor by dividing each receptor's KD for Aβm by that of the indicated species. hPrPC was highly discriminate in its preference for oligomeric and globulomeric Aβ with discrimination factors of 68.5 and 63.9, respectively. NgR1 also discriminated in binding, although to a lesser degree than hPrPC. LilrB2 exhibited a modest preference for Aβo compared with monomers and bound Aβg with the same affinity as for Aβm (Table 1).
Figure 6.

hPrPC-, NgR1-, and LilrB2-coated plates recapitulate the cell-based assay. A, binding of Aβo to wells coated with hPrPC, NgR1, LilrB2, or Fc control protein. B, Scatchard plot of data in A. C, binding of Aβ globulomers to wells coated with hPrPC, NgR1, LilrB2, or Fc control protein. D, Scatchard plot of data in C. E, binding of Aβ monomers to wells coated with hPrPC, NgR1, LilrB2, or Fc control protein. F, Scatchard plot of data in E. G, comparison of the binding of Aβo, Aβg, and Aβm to hPrPC. H, comparison of the binding of Aβo, Aβg, and Aβm to NgR1. I, comparison of the binding of Aβo, Aβg, and Aβm to LilrB2. AU, arbitrary units. Error bars, S.D.
Table 1.
Binding affinities for positive receptor candidates and preparations of synthetic Aβ
The KD and Bmax of each of the receptors and the indicated Aβ preparation are shown. Concentrations are expressed as monomer equivalents. Discrimination factor is the quotient of a receptor's KD for monomeric Aβ divided by that of the indicated preparation. Greater values indicate a preference for binding to neurotoxic preparations. AU, arbitrary units.
| Parameter | Aβo | Aβg | Aβm |
|---|---|---|---|
| PrPC | |||
| KD (nm) | 1.4 ± 0.2 | 1.5 ± 0.2 | 95.9 ± 6 |
| Bmax (AU) | 6.27 × 106 | 5.9 × 106 | 6.46 × 106 |
| Discrimination factor | 68.5 | 63.9 | |
| LilrB2 | |||
| KD (nm) | 42.3 ± 4.4 | 207.2 ± 18.2 | 192.4 ± 12 |
| Bmax (AU) | 5.84 × 106 | 5.9 × 106 | 4.45 × 106 |
| Discrimination factor | 4.5 | 0.9 | |
| NgR1 | |||
| KD (nm) | 3.9 ± 0.4 | 6.6 ± 0.5 | 61.6 ± 5.3 |
| Bmax (AU) | 5.53 × 106 | 5.85 × 106 | 5.46 × 106 |
| Discrimination factor | 15.8 | 9.3 | |
PrPC and NgR1 bind Aβ present in the brains of patients with AD
Despite the ability of preparations of synthetic Aβ to generate neurotoxic species with masses similar to those found in the brains of patients with AD and to cause neurotoxicity in vitro, ex vivo, and in vivo, it is critical to examine each receptor's disease relevance using pathological Aβ present in disease (10, 33, 35–37). Having validated the concordance of our plate-based assay with the observations made in living cells, we quantified binding of Aβ present in TBS-soluble extracts of brains from 10 patients with autopsy-confirmed AD and compared it with signal from brains of 11 cognitively normal patients. The mean anti-Aβ signal from wells coated with hPrPC was highest and significantly greater than that from cognitively normal patients. LilrB2 exhibited no binding to Aβ present in brains of patients with AD and no difference between AD and control brains. NgR1 modestly bound Aβ present in AD patient brains, and this signal was significantly greater than that in brains from cognitively normal patients (Fig. 7).
Figure 7.
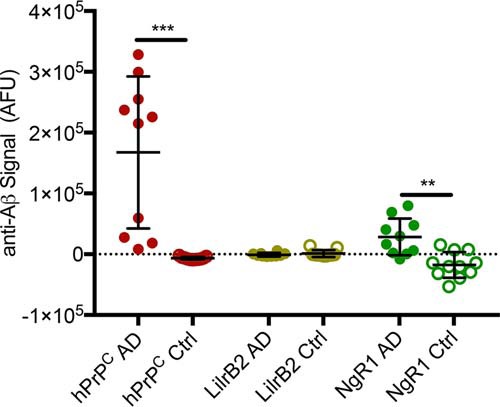
hPrPC and NgR1 bind soluble Aβ present in brains of AD patients. Shown is binding of Aβ in TBS-soluble fraction of brains of AD patients or control patients (Ctrl) to wells coated with hPrPC, NgR1, or LilrB2. Each dot represents an individual patient brain. Shown are multiple t tests with Holm-Sidak correction for multiple comparisons. Error bars, S.D. **, p < 0.01; ***, p < 0.001. AFU, arbitrary fluorescence unit.
Discussion
Here, a panel of putative Aβ receptors was expressed on the surface of nonneuronal cells and examined for sufficiency to mediate Aβ binding. By accounting for differences in receptor expression using a common epitope tag across the receptors, we made direct comparisons of Aβ affinity. Importantly, binding took place in living cells rather than cell-free systems. This allows for potential binding events to take place at the cell surface and in a physiological context with respect to the lipid and protein environment. Our experiments demonstrate that most of the reported receptors for Aβ are not sufficient to confer Aβ binding capacity to cells using three different preparations of synthetic Aβ. It is possible that those receptors that failed to bind Aβ in our experiments require a coreceptor that is not present in COS-7 cells. Whereas our cell surface biotinylation experiments demonstrate successful transport through the secretory system, which is typically associated with proper protein folding, they do not definitively exclude misfolding that could impact ligand binding. Whereas these negative proteins may have Aβ affinity under some experimental conditions, in a cellular context, their binding affinity is clearly much less than that of the three positive receptors. By including an analysis of binding to monomeric Aβ, we revealed that PrPC and NgR1 discriminate between pathological and nontoxic forms of Aβ. It should be noted that neither oligomeric nor monomeric preparations are exclusive to a single species but that these designations are meant to reflect the assemblies for which the preparation is enriched. It is possible that the reduced but persistent binding of monomer preparations to PrPC and NgR1 may reflect the decrease in the abundance of Aβo. It is clear, however, that upon enrichment for monomeric Aβ, none of the candidates that failed to bind Aβo showed an increase in binding. We also found that LilrB2 does not differentiate in binding these oligomeric and monomeric Aβ preparations. The promiscuity of LilrB2 may indicate that an epitope present in both species of synthetic Aβ mediates this interaction. Recently, Cao et al. demonstrated that LilrB2 can bind minimal peptides comprising Aβ amino acids 1–21, 15–35, and a tandem repeat of 16–21 without a specific oligomerization procedure (38). In contrast, PrPC has been shown to undergo structural changes specifically in the presence of oligomeric Aβg (39).
Neuronal acetylcholine receptor subunit α-7 (nAchRa7) and NMDA receptor subunits 1 and 2B have also been reported to bind Aβ (40–42). We sought to include these in our investigation; however, efficient transport to the cell surface as determined by cell surface biotinylation experiments was insufficient to make comparisons with the rest of the panel. Despite these data, and because we have observed that even very low expression levels of the high-affinity receptor PrPC can result in robust Aβo binding, nArchRα7 with and without the chaperone RIC-3 (resistance to inhibitors of cholinesterase 3) and GluN2B with and without GluN1 (glutamate receptor ionotropic NMDA 1) were included in our cell-binding experiments. No detectable Aβ-binding signal was observed in nArchRα7 or GluN2B experiments (data not shown).
Whereas the ability to bind Aβo is a crucial characteristic for a receptor, we found it important to interrogate the necessity of confirmed receptors for Aβo binding to neurons. Pirb is the mouse homolog of Lilrb2 and was shown to bind Aβo (11). By generating double-knockout mice deficient in NgR1 and Pirb, we found that up to 20% of binding of synthetic Aβo to neurons is mediated by these two receptors.
Preparations of synthetic Aβ peptide provide a convenient source of neurotoxic Aβ with species representative of those found in the brains of AD patients. When investigating the relevance of a potential receptor to human disease, the gold standard should be an ability to bind Aβ present in the brains of patients diagnosed with AD. We first validated our plate-based assay by replicating the experiments done in mammalian cells and found the two to be in close agreement. We determined that hPrPC is the highest-affinity receptor for Aβo (KD = 1.4), followed by NgR1 (KD = 3.9) and LilrB2 (KD = 42.3). We then utilized this assay for the detection of binding to soluble Aβ found in extracts of brains from patients diagnosed with AD. We found that hPrPC and NgR1 bound Aβ present in AD patient brains and that hPrPC did so to the greatest extent. Finally, we found that despite binding synthetic Aβ from multiple preparations and in cell-based and purified protein assays, LilrB2 did not detectibly bind the Aβ species present in AD patient brains.
The experiments described here provide much needed clarity to the field regarding the nature of the interaction of neurotoxic Aβo in the extracellular space with the cell surface. The observation made here that NgR1 and LilrB2 account for up to 20% of Aβo binding to hippocampal neurons, coupled with previous observations that PrPC is responsible for 50% of Aβo binding to neurons and the knowledge that Aβo binds neurons in a trypsin-sensitive manner, suggests that at least one additional receptor for Aβo remains to be identified (3, 10). Future characterizations of novel Aβo receptors should include experiments testing the necessity and sufficiency for cellular Aβo binding. It will also be necessary to examine the disease relevance of observations made with synthetic peptides and transgenic animals by using materials obtained from patients with AD.
Experimental procedures
Tissue culture
COS-7 or HEK293T cells were grown at 37 °C with 5% CO2 on 100-mm tissue culture dishes (Fisher, 08-772E) in Dulbecco's modified Eagle's medium (Gibco, 11965) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Gibco, 15140). For Aβ-binding experiments, COS-7 cells were seeded into 8-well chamber slides (Lab-Tek, 154941) and grown under the same conditions. Transfections were performed using Lipofectamine 3000 (Thermo Fisher Scientific, L3000015) diluted in Opti-MEM (Thermo Fisher Scientific, 31985).
Plasmids and cloning
cDNA encoding proteins of interest were obtained from Genecopoeia, Dharmacon, and Origene. cDNAs were subcloned into Myc-tagged expression vectors pcDNA3.1(+)/myc-His A (Thermo, V80020) or pSecTag2A (Thermo, V90020). Accession numbers for cDNA sequences are as follows: hPrP (NM_000311.3), mPrP (NM_011170.3), LilrB2 (XM_006726139.1), NgR1 (NM_023004.5), EphA1 (BC_130291.1), FcγRIIb (BC031992.1), SorLA (NM_003105.5), Sortilin (NM_002959.5), p75NTR (NM_002507.3), PGRMC1 (NM_006667.4), NLGN1 (NM_014932.3), RAGE (AB036432.1), EphB2 (BC146296.1), FZD5 (BC172518.1), mGluR5 (NM_017012.1), EphA4 (BC026327.1), CRMP2 (NM_001386). The construct encoding NRP1 has been described previously (43). Constructs were verified by DNA sequencing.
Cell surface biotinylation and immunoprecipitation
Dishes of transfected HEK293T cells were washed three times with ice-cold PBS, pH 8, and biotinylated with 10 ml of 0.48 mg/ml Sulfo-NHS-LC-LC-Biotin (Thermo Fisher 21338) in PBS, pH 8, and incubated at room temperature for 30 min. Cells were washed once with 50 mm Tris, pH 8, and then twice with ice-cold PBS, pH 8. Cells were harvested in 1 ml of ice-cold PBS and gently scraped to collect. Collected cells were transferred to a new tube and centrifuged at 500 × g for 3 min at 4 °C to pellet them. The cell pellet was resuspended in 1 ml of RIPA lysis buffer (Millipore 20-188) with PhosSTOP (Roche Diagnostics, 04906837001) and Complete Mini protease inhibitors (Roche Diagnostics, 11836170001) and sonicated. After sonication, samples were centrifuged at 100,000 × g, 4 °C for 30 min. Supernatants were transferred to c-Myc beads (Pierce 20169) and incubated at 4 °C with endo-over-end mixing for 1 h. Beads were washed three times with RIPA buffer. Proteins were eluted with 4× Laemmli sample buffer (Bio-Rad, 161-0747) and analyzed by immunoblotting using anti-Myc (Cell Signaling, catalog no. 2276, 1:1,000), donkey anti-mouse IRDye 680LT (LI-COR Biosciences, 926-68022, 1:20,000), and streptavidin IRDye 800CW (LI-COR Biosciences, 926-32230, 1:20,000) and imaged with the Odyssey infrared imaging system (LI-COR Biosciences).
Synthetic Aβ preparations
Lyophilized synthetic β-amyloid (residues 1–42) was purchased from The ERI Amyloid Laboratory, LLC. Vials of peptide were reconstituted in 1,1,1,3,3,3-hexafluoroisopropanol at 10 mg/ml, boiled at 70 °C for 1 h, aliquoted into microcentrifuge tubes at 0.5 mg/tube, and allowed to dry overnight at room temperature. The next day, the aliquots were further dehydrated in a SpeedVac for 1 h. Oligomers were prepared by dissolving an aliquot of Aβ in 40 μl of DMSO, separating into two aliquots, diluting in 1 ml of F12 medium (Atlanta Biologicals, M15350) to 55 μm, and incubating at room temperature overnight to allow oligomerization. The next day, samples were centrifuged in a tabletop centrifuge (Eppendorf 5430) at 20,817 × g for 15 min to precipitate fibrillary protein. The supernatant was treated as 55 μm and diluted to working concentrations in F12 medium. Monomeric Aβ was prepared by diluting the DMSO-dissolved peptide in F12 to working concentration as quickly as possible. Globulomer Aβ was generated according to the previously published protocol (33). HFIP-processed synthetic Aβ peptide films were resuspended in DMSO at 5 mm (22 μl of DMSO, 0.5 mg of Aβ). Then 250 μl of 1× PBS, pH 7.4, was added to each tube, immediately followed by 31 μl of 2% SDS. The resulting mix was incubated in the sealed tubes at 37 °C for 4–6 h and then diluted 4-fold with deionized water (resulting in 1.2 ml/tube final volume) and incubated at 37 °C overnight to yield mature globulomer Aβ. The preparations were then centrifuged for 10 min at 10,000 × g, and the supernatants were concentrated using 30-kDa cutoff Amicon filters. Concentrated Aβg (1 mm Aβ monomer) were then dialyzed against a 1000-fold excess of PBS diluted 4-fold with water (0.25 × PBS, pH 7.4) using 100 μl of 20-kDa cutoff Slide-A-Lyzer mini cassettes. Dialysis buffer was changed three times over a course of 36 h. Dialyzed Aβg was then centrifuged at 100,000 × g for 1 h at 4 °C, and supernatants were aliquoted and flash-frozen in liquid nitrogen. Typically, 70% of initial Aβ (dried HFIP film) was converted into Aβg as determined by absorbance at 280 nm using a ProtParam-derived extinction coefficient (1490 m−1 cm−1). For atomic force microscopy and SEC, NaOH monomer samples were generated by dissolving HFIP-treated Aβ in 440 μl of 0.1 m NaOH, resulting in a 250 μm stock, and processed for atomic force microscopy or SEC immediately.
Immunocytochemistry and heterologous cell-binding assays
COS-7 cells were cultured in 8-well chamber slides as described above. For Aβ binding experiments, the culture medium was removed from wells, leaving a volume sufficient to cover the cells, and washed with F12, and the indicated Aβ preparation was added. Slides were then incubated for 2 h at the indicated study temperature. Wells were washed three times with PBS and fixed with 3.7% formaldehyde (J.T. Baker, 2106) in PBS with 6% sucrose (AmericanBio, AB01900) for 30 min at room temperature. The fixative was then removed, and cells were permeabilized by incubation in 0.2% Triton X-100 (Fisher Scientific, 42235-5000) in PBS for 30 min. After permeabilization, cells were blocked by incubation with 5% normal donkey serum (Jackson ImmunoResearch, 017-000-121) for 1 h at room temperature. After blocking, cells were incubated with the indicated primary antibodies for anti-Myc (Cell Signaling, catalog no. 2276, 1:5,000) or anti-Aβ (Cell Signaling, catalog no. 8243, 1:1,000), diluted in 1% BSA (Sigma, catalog no. A9647) at 4 °C overnight. The next day, cells were washed three times with PBS with incubation periods of 0, 3, and 5 min. The indicated appropriate secondary antibodies diluted in 1% BSA in PBS (donkey anti-mouse Alexa Fluor 488, Life Technologies, catalog no. A21202, 1:500; donkey anti-rabbit Alexa Fluor 568, Life Technologies, catalog no. A10042, 1:500; Streptavidin Alexa Fluor 568, Life Technologies, S-11226, 1:500) were then added to wells and incubated for 1 h at room temperature. Wells were then washed four times with 0.02% Triton X-100 in PBS with incubation times of 0, 3, 5, and 15 min. After washing, slides were mounted using Vectashield with 4′,6-diamidino-2-phenylindole (Vector Laboratories, H-1200) and imaged on an Axio Imager M2. CellProfiler was used for quantification of Aβ binding to transfected cells (44). 4′,6-Diamidino-2-phenylindole and anti-Myc signal were used for the identification of transfected cells, and the intensity of Aβ and Myc inside of cell areas as well as background were measured in Aβ-treated and nontreated wells. Background signal was subtracted from cellular signal, and from that value, mean Aβ signal in cells transfected with cytoplasmic enhanced GFP was subtracted. The transfection-specific, background-subtracted Aβ signal was normalized by background-subtracted Myc signal and expressed as a percentage of that signal in hPrPC-transfected cells, which were included in every experiment for comparison. Limit of detection was calculated as the mean background-subtracted Aβ signal in cells transfected with cytoplasmic enhanced GFP, expressed as a percentage of that in hPrP-transfected cells, plus two S.D. values.
Size-exclusion chromatography
SEC was performed on the described preparations of biotinylated Aβ using a Superdex 200 Increase 10/300 GL column (GE Healthcare, 28990944) and Akta Pure 25 M1 (GE Healthcare) with an F9-C fraction collector. SEC was performed at 4 °C. 1.5-ml fractions were collected in 2-ml deep 96-well plates (USA Scientific, 1896-2110). For the representative trace, 50 μm BAβo in Ham's F12 medium was loaded into a 500-μl sample loop. For dot blot analysis, 1 μm samples were loaded. Samples prepared in Ham's F12 were run using Ham's F12 as the mobile phase. NaOH monomer samples were run in a mobile phase of 0.25× PBS, pH 7.4. Gel filtration standards used were blue dextran (Sigma, D4772), thyroglobulin, bovine γ-globulin, chicken ovalbumin, equine myoglobin, and vitamin B12 (Bio-Rad, 1511901). Fractions were concentrated using centrifugal filters with a 3,000 nominal molecular weight limit (Amicon UFC500324). Dot blot analysis was performed by applying 80% of concentrate volume to a nitrocellulose membrane using a Bio-Rad Bio-Dot apparatus. Samples were incubated on the membrane for 4 h at room temperature, washed twice with TBS, blocked for 1 h at room temperature with Rockland blocking buffer for fluorescent Western blotting (Rockland, MB070010TF), and probed overnight at 4 °C with streptavidin IRDye 800CW (LI-COR 926-32230, 1:1,000)
Atomic force microscopy
AFM samples were prepared by placing 10 μl of 0.25 mg/ml Aβ preparations on freshly cleaved mica and allowing it to adsorb for 2 min. The sample was washed twice with 200 μl of Milli-Q water and dried carefully. Images where taken in a Bruker Dimension Fastscan AFM in a tapping mode using silicon nitride cantilevers.
Mouse breeding and care
Mice were cared for by the Yale Animal Resource Center, and all experiments were approved by Yale's institutional animal care and use committee. NgR−/− (Rtn4r) and Pirb−/− mice have been described previously and were maintained on a C57/Bl6 background (45, 46). Pirb−/− mice were a generous gift of Dr. Toshiyuki Takai. NgR−/− and Pirb−/− were bred together to generate double-heterozygous and eventually double-knockout mice.
Neuron cultures and neuronal binding assays
Mouse hippocampal neurons from WT and RTN4R−/−, Pirb−/− double knockout animals were isolated from embryonic day 17 to postnatal day 0 pups and cultured on MatTek dishes (P35G-1.5-14-C) coated with poly-d-lysine (MP Bio, 0215017580) in Neurobasal medium (Gibco, 10888-022) supplemented with B-27 (Gibco, 17504-044), sodium pyruvate (Gibco, 11360-070), and Glutamax (Gibco, 35050-061), with 1% penicillin and streptomycin (Gibco, 15140). Binding experiments were performed at 18–20 days in vitro. After washing, neurons were incubated with 500 nm monomer-equivalent (∼2.5 nm oligomer) biotinylated Aβ for 1 h at 4 °C. Following fixation, permeabilization, and blocking, dendrites were visualized using anti-MAP2 (1:2,000, Millipore, AB5622) and Alexa Fluor 488 donkey anti-rabbit IgG (1:1,000, Life Technologies, Inc., A21206), and biotinylated Aβ was visualized with streptavidin Alexa Fluor 568 (1:500, Life Technologies, S11226). z-Stacks were collected at ×40 using a spinning disc confocal microscope. z-Stacks were then max-projected using National Institutes of Health ImageJ (47). Quantification was performed using CellProfiler (44). Integrated punctate Aβ signal was calculated after background subtraction and thresholding. Integrated signal was then normalized to the area positive for MAP2. When representing these data graphically, each data point represents a single region of interest from 5–7 dishes. The experiment was repeated four times.
Plate-based assay
384-well MaxiSorp plates (Thermo, 460372) were coated overnight with 20 μl/well of the indicated protein at 250 nm in 100 mm BupH carbonate-bicarbonate coating buffer (Thermo 28382) at 4 °C. Plates were washed twice with PBST (PBS, 0.05% Tween 20) and blocked with 25 μl of Protein-Free T20 PBS Blocking buffer (Pierce 37573) for 4 h at room temperature. After washing three times with PBST, 20 μl of either TBS fraction of human brain samples or synthetic Aβ was applied to wells and incubated overnight at 4 °C. Aβ binding was detected using D54D2 anti-Aβ antibody (Cell Signaling, catalog no. 8243, 1:2,000) in PBSTB (PBS, 0.05% Tween 20, 0.5% BSA) for 2 h. After four washes with PBST, 20 μl of Eu-N1 goat anti-rabbit IgG (PerkinElmer Life Sciences, AD105, 1:4,000) was diluted in DELFIA assay buffer (PerkinElmer Life Sciences) and incubated for 1 h at room temperature. After four washes with PBST, 20 μl of DELFIA Enhancement Solution (PerkinElmer Life Sciences) was applied, and time-resolved europium fluorescence was measured with a Victor 3V plate reader (PerkinElmer Life Sciences).
Recombinant hexahistadine-tagged hPrPC was generated using a method described previously (7). The extracellular domain of LilrB2 is encoded by amino acids 22–461. We subcloned cDNA encoding this region into pSecTag2A (Invitrogen) to include a C-terminal hexahistidine tag. HEK293T cells were transfected with this ecto-LilrB2 construct, and 24 h later, the medium was replaced with serum-free medium. The next day, the conditioned medium was collected and purified using the same method described for hPrPC. Human NgR-Fc decoy protein encodes amino acid residues 1–310 of human NgR1 with the C266A and C309A substitutions fused to the Fc domain of human IgG1. NgR(310)-Fc protein was produced as described previously and supplied by ReNetX Bio (48).
Human brain samples
Post-mortem human tissue was collected in accordance with institutional review board protocols approved by Yale University. Samples of brain tissue were microscopically analyzed to confirm the clinical diagnosis of AD (Braak Stage V or higher). Samples from neurologically healthy controls were required to have no or minimal histopathological signs of AD (Braak 0–II). Frozen prefrontal cortex was stored at −80 °C until used. Human brain was weighed and Dounce-homogenized in 3 volumes of TBS, pH 7.4 (Bio-Rad, catalog no. 170-6435) supplemented with Complete Mini Protease Inhibitors (Roche Diagnostics, 11836170001) and PhosStop phosphatase inhibitors (Roche Diagnostics, 04906837001). Samples were centrifuged at 100,000 × g at 4 °C for 1 h, and the supernatant was collected, flash-frozen in liquid nitrogen, and stored at −80 °C until assayed. The supernatant was referred to as the TBS-soluble fraction. Patient demographics for brains used are described in Table 2.
Table 2.
Characteristics of human brain tissue samples
Patient characteristics for human brain tissue used are described. Values are ± S.D.
| Sample size | Age | Sex (% female) | Post-mortem interval | |
|---|---|---|---|---|
| years | % | days | ||
| AD | 10 | 78 ± 12 | 50 | 13 ± 9 |
| Control | 11 | 71 ± 14 | 64 | 20 ± 7 |
Statistics
All results are presented are mean ± S.D. unless otherwise stated. Prism version 7 was used for statistical analysis, and specific tests used are indicated in the figure legends.
Author contributions
L. M. S. and S. M. S. conceptualization; L. M. S. data curation; L. M. S. formal analysis; L. M. S., M. A. K., and S. L. investigation; L. M. S., M. A. K., and S. L. methodology; L. M. S. writing-original draft; L. M. S., M. A. K., S. L., and S. M. S. writing-review and editing; S. M. S. supervision; S. M. S. funding acquisition; S. M. S. project administration.
Supplementary Material
Acknowledgments
We thank Stefano Sodi for help with mouse husbandry and Dr. Toshiyuki Takai for providing Pirb knockout mice.
This work was supported by National Institutes of Health Grants R01AG034924, P50AG047270, RF1AG05300, and R35NS097283 and the Falk Medical Research Trust (to S. M. S.). S. M. S. is the founder of ReNetX Bio, which seeks to develop therapeutics targeting NgR1, and is an inventor on a patent regarding PrPC antagonism for Alzheimer therapy. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- AD
- Alzheimer's disease
- Aβ
- amyloid-β
- Aβo
- amyloid-β oligomer(s)
- PrPC
- cellular prion protein
- NgR1
- Nogo receptor 1
- LilrB2
- leukocyte immunoglobulin-like receptor subfamily B member 2
- RAGE
- advanced glycosylation end product-specific receptor
- PGRMC1
- membrane-associated progesterone receptor component 1
- EphA1
- ephrin type-A receptor 1
- FcγRIIb
- low-affinity immunoglobulin γ Fc region receptor II-b
- SorLA
- sortilin-related receptor
- p75NTR
- tumor necrosis factor receptor superfamily member 16
- NLGN1
- neuroligin 1
- EphB2
- ephrin type-B receptor 2
- FZD5
- frizzled-5
- mGluR5
- metabotropic glutamate receptor 5
- EphA4
- ephrin type-A receptor 4
- CRMP2
- collapsin response mediator protein 2
- NRP1
- neuropilin-1
- BAβ
- biotin amyloid-β
- BAβo
- BAβ oligomers
- Aβg
- amyloid-β globulomer(s)
- Aβm
- amyloid-β monomer(s)
- Pirb
- paired immunoglobulin-like receptor b
- nArchRα7
- neuronal acetylcholine receptor subunit α-7
- GluN2B
- glutamate receptor ionotropic NMDA 2B
- hPrPC
- human PrPC
- SEC
- size-exclusion chromatography
- HMW
- high-molecular weight
- RIPA
- Radioimmunoprecipitation assay
- TBS
- Tris-buffered saline
- HFIP
- 1,1,1,3,3,3-hexafluoroisopropanol.
References
- 1. Alzheimer A. (1906) Uber einen eigenartigen schweren Erkrankungsprozeb der Himrinde. Neurol. Zentralbl. 23, 1129–1136 [Google Scholar]
- 2. Martin P. (2015) World Alzheimer Report 2015: The Global Impact of Dementia, Alzheimer's Disease International (ADI), London, UK [Google Scholar]
- 3. Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., Morgan T. E., Rozovsky I., Trommer B., Viola K. L., Wals P., Zhang C., Finch C. E., Krafft G. A., and Klein W. L. (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453 10.1073/pnas.95.11.6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., and Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 10.1126/science.1079469 [DOI] [PubMed] [Google Scholar]
- 5. Shankar G. M., Bloodgood B. L., Townsend M., Walsh D. M., Selkoe D. J., and Sabatini B. L. (2007) Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875 10.1523/JNEUROSCI.4970-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., and Ashe K. H. (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 10.1038/nature04533 [DOI] [PubMed] [Google Scholar]
- 7. Kostylev M. A., Kaufman A. C., Nygaard H. B., Patel P., Haas L. T., Gunther E. C., Vortmeyer A., and Strittmatter S. M. (2015) Prion-protein-interacting amyloid-β oligomers of high molecular weight are tightly correlated with memory impairment in multiple Alzheimer mouse models. J. Biol. Chem. 290, 17415–17438 10.1074/jbc.M115.643577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kayed R., Head E., Sarsoza F., Saing T., Cotman C. W., Necula M., Margol L., Wu J., Breydo L., Thompson J. L., Rasool S., Gurlo T., Butler P., and Glabe C. G. (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2, 18 10.1186/1750-1326-2-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gong Y., Chang L., Viola K. L., Lacor P. N., Lambert M. P., Finch C. E., Krafft G. A., and Klein W. L. (2003) Alzheimer's disease-affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U.S.A. 100, 10417–10422 10.1073/pnas.1834302100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Laurén J., Gimbel D. A., Nygaard H. B., Gilbert J. W., and Strittmatter S. M. (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132 10.1038/nature07761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim T., Vidal G. S., Djurisic M., William C. M., Birnbaum M. E., Garcia K. C., Hyman B. T., and Shatz C. J. (2013) Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer's model. Science 341, 1399–1404 10.1126/science.1242077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Park J. H., Gimbel D. A., GrandPre T., Lee J.-K., Kim J.-E., Li W., Lee D. H., and Strittmatter S. M. (2006) Alzheimer precursor protein interaction with the Nogo-66 receptor reduces amyloid-β plaque deposition. J. Neurosci. 26, 1386–1395 10.1523/JNEUROSCI.3291-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kam T.-I., Song S., Gwon Y., Park H., Yan J.-J., Im I., Choi J.-W., Choi T.-Y., Kim J., Song D.-K., Takai T., Kim Y. C., Kim K. S., Choi S. Y., Choi S., et al. (2013) FcγRIIb mediates amyloid-β neurotoxicity and memory impairment in Alzheimer's disease. J. Clin. Invest. 123, 2791–2802 10.1172/JCI66827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carlo A.-S., Gustafsen C., Mastrobuoni G., Nielsen M. S., Burgert T., Hartl D., Rohe M., Nykjaer A., Herz J., Heeren J., Kempa S., Petersen C. M., and Willnow T. E. (2013) The pro-neurotrophin receptor sortilin is a major neuronal apolipoprotein E receptor for catabolism of amyloid-β peptide in the brain. J. Neurosci. 33, 358–370 10.1523/JNEUROSCI.2425-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andersen O. M., Reiche J., Schmidt V., Gotthardt M., Spoelgen R., Behlke J., von Arnim C. A., Breiderhoff T., Jansen P., Wu X., Bales K. R., Cappai R., Masters C. L., Gliemann J., and Mufson E. J. (2005) Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 102, 13461–13466 10.1073/pnas.0503689102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andersen O. M., Schmidt V., Spoelgen R., Gliemann J., Behlke J., Galatis D., McKinstry W. J., Parker M. W., Masters C. L., Hyman B. T., Cappai R., and Willnow T. E. (2006) Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry 45, 2618–2628 10.1021/bi052120v [DOI] [PubMed] [Google Scholar]
- 17. Spoelgen R., von Arnim C. A., Thomas A. V., Peltan I. D., Koker M., Deng A., Irizarry M. C., Andersen O. M., Willnow T. E., and Hyman B. T. (2006) Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and β-secretase β-site APP-cleaving enzyme. J. Neurosci. 26, 418–428 10.1523/JNEUROSCI.3882-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yaar M., Zhai S., Pilch P. F., Doyle S. M., Eisenhauer P. B., Fine R. E., and Gilchrest B. A. (1997) Binding of β-amyloid to the p75 neurotrophin receptor induces apoptosis: a possible mechanism for Alzheimer's disease. J. Clin. Invest. 100, 2333–2340 10.1172/JCI119772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Izzo N. J., Xu J., Zeng C., Kirk M. J., Mozzoni K., Silky C., Rehak C., Yurko R., Look G., Rishton G., Safferstein H., Cruchaga C., Goate A., Cahill M. A., Arancio O., et al. (2014) Alzheimer's therapeutics targeting amyloid β1–42 oligomers II: σ-2/PGRMC1 receptors mediate Aβ42 oligomer binding and synaptotoxicity. PLoS One 9, e111899 10.1371/journal.pone.0111899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dinamarca M. C., Weinstein D., Monasterio O., and Inestrosa N. C. (2011) The synaptic protein neuroligin-1 interacts with the amyloid β-peptide: is there a role in Alzheimer's disease? Biochemistry 50, 8127–8137 10.1021/bi201246t [DOI] [PubMed] [Google Scholar]
- 21. Magdesian M. H., Carvalho M. M., Mendes F. A., Saraiva L. M., Juliano M. A., Juliano L., Garcia-Abreu J., and Ferreira S. T. (2008) Amyloid-β binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/β-catenin signaling. J. Biol. Chem. 283, 9359–9368 10.1074/jbc.M707108200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yan S. D., Chen X., Fu J., Chen M., Zhu H., Roher A., Slattery T., Zhao L., Nagashima M., Morser J., Migheli A., Nawroth P., Stern D., and Schmidt A. M. (1996) RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature 382, 685–691 10.1038/382685a0 [DOI] [PubMed] [Google Scholar]
- 23. Cissé M., Halabisky B., Harris J., Devidze N., Dubal D. B., Sun B., Orr A., Lotz G., Kim D. H., Hamto P., Ho K., Yu G. Q., and Mucke L. (2011) Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52 10.1038/nature09635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Renner M., Lacor P. N., Velasco P. T., Xu J., Contractor A., Klein W. L., and Triller A. (2010) Deleterious effects of amyloid β oligomers acting as an extracellular scaffold for mGluR5. Neuron 66, 739–754 10.1016/j.neuron.2010.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fu A. K., Hung K.-W., Huang H., Gu S., Shen Y., Cheng E. Y., Ip F. C., Huang X., Fu W.-Y., and Ip N. Y. (2014) Blockade of EphA4 signaling ameliorates hippocampal synaptic dysfunctions in mouse models of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 111, 9959–9964 10.1073/pnas.1405803111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith L. M., and Strittmatter S. M. (2017) Binding sites for amyloid-β oligomers and synaptic toxicity. Cold Spring Harb. Perspect. Med. 7, a024075 10.1101/cshperspect.a024075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benilova I., and De Strooper B. (2013) Promiscuous Alzheimer's amyloid: yet another partner. Science 341, 1354–1355 10.1126/science.1244166 [DOI] [PubMed] [Google Scholar]
- 28. Purro S. A., Nicoll A. J., and Collinge J. (2018) Prion protein as a toxic acceptor of amyloid-β oligomers. Biol. Psychiatry 83, 358–368 10.1016/j.biopsych.2017.11.020 [DOI] [PubMed] [Google Scholar]
- 29. Izzo N. J., Staniszewski A., To L., Fa M., Teich A. F., Saeed F., Wostein H., Walko T. 3rd, Vaswani A., Wardius M., Syed Z., Ravenscroft J., Mozzoni K., Silky C., Rehak C., et al. (2014) Alzheimer's therapeutics targeting amyloid β1–42 oligomers I: Aβ 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS One 9, e111898 10.1371/journal.pone.0111898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Galasko D., Bell J., Mancuso J. Y., Kupiec J. W., Sabbagh M. N., van Dyck C., Thomas R. G., Aisen P. S., and Alzheimer's Disease Cooperative Study (2014) Clinical trial of an inhibitor of RAGE-Aβ interactions in Alzheimer disease. Neurology 82, 1536–1542 10.1212/WNL.0000000000000364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burstein A. H., Sabbagh M., Andrews R., Valcarce C., Dunn I., and Altstiel L. (2018) Development of azeliragon, an oral small molecule antagonist of the receptor for advanced glycation endproducts, for the potential slowing of loss of cognition in mild Alzheimer's disease. J. Prev. Alzheimers Dis. 5, 149–154 [DOI] [PubMed] [Google Scholar]
- 32. Yang T., Knowles J. K., Lu Q., Zhang H., Arancio O., Moore L. A., Chang T., Wang Q., Andreasson K., and Rajadas J., Fuller G. G., Xie Y., Massa S. M., and Longo F. M. (2008) Small molecule, non-peptide p75NTR ligands inhibit Aβ-induced neurodegeneration and synaptic impairment. PLoS One 3, e3604 10.1371/journal.pone.0003604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barghorn S., Nimmrich V., Striebinger A., Krantz C., Keller P., Janson B., Bahr M., Schmidt M., Bitner R. S., Harlan J., Barlow E., Ebert U., and Hillen H. (2005) Globular amyloid β-peptide1–42 oligomer: a homogenous and stable neuropathological protein in Alzheimer's disease. J. Neurochem. 95, 834–847 10.1111/j.1471-4159.2005.03407.x [DOI] [PubMed] [Google Scholar]
- 34. Chromy B. A., Nowak R. J., Lambert M. P., Viola K. L., Chang L., Velasco P. T., Jones B. W., Fernandez S. J., Lacor P. N., Horowitz P., Finch C. E., Krafft G. A., and Klein W. L. (2003) Self-assembly of Aβ1–42 into globular neurotoxins. Biochemistry 42, 12749–12760 10.1021/bi030029q [DOI] [PubMed] [Google Scholar]
- 35. Cleary J. P., Walsh D. M., Hofmeister J. J., Shankar G. M., Kuskowski M. A., Selkoe D. J., and Ashe K. H. (2005) Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 8, 79–84 10.1038/nn1372 [DOI] [PubMed] [Google Scholar]
- 36. Li S., Hong S., Shepardson N. E., Walsh D. M., Shankar G. M., and Selkoe D. (2009) Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801 10.1016/j.neuron.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., Rowan M. J., and Selkoe D. J. (2002) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 10.1038/416535a [DOI] [PubMed] [Google Scholar]
- 38. Cao Q., Shin W. S., Chan H., Vuong C. K., Dubois B., Li B., Murray K. A., Sawaya M. R., Feigon J., Black D. L., Eisenberg D. S., and Jiang L. (2018) Inhibiting amyloid-β cytotoxicity through its interaction with the cell surface receptor LilrB2 by structure-based design. Nat. Chem. 10, 1213–1221 10.1038/s41557-018-0147-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kostylev M. A., Tuttle M. D., Lee S., Klein L. E., Takahashi H., Cox T. O., Gunther E. C., Zilm K. W., and Strittmatter S. M. (2018) Liquid and hydrogel phases of PrPC linked to conformation shifts and triggered by Alzheimer's amyloid-β oligomers. Mol. Cell 72, 426–443.e12 10.1016/j.molcel.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang H. Y., Lee D. H., Davis C. B., and Shank R. P. (2000) Amyloid peptide Aβ1–42 binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. J. Neurochem. 75, 1155–1161 [DOI] [PubMed] [Google Scholar]
- 41. Amar F., Sherman M. A., Rush T., Larson M., Boyle G., Chang L., Götz J., Buisson A., and Lesné S. E. (2017) The amyloid-β oligomer Aβ* 56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci. Signal. 10, eaal2021 10.1126/scisignal.aal2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lacor P. N., Buniel M. C., Chang L., Fernandez S. J., Gong Y., Viola K. L., Lambert M. P., Velasco P. T., Bigio E. H., Finch C. E., Krafft G. A., and Klein W. L. (2004) Synaptic targeting by Alzheimer's-related amyloid β oligomers. J. Neurosci. 24, 10191–10200 10.1523/JNEUROSCI.3432-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takahashi T., Nakamura F., Jin Z., Kalb R. G., and Strittmatter S. M. (1998) Semaphorins A and E act as antagonists of neuropilin-1 and agonists of neuropilin-2 receptors. Nat. Neurosci. 1, 487–493 10.1038/2203 [DOI] [PubMed] [Google Scholar]
- 44. Lamprecht M. R., Sabatini D. M., and Carpenter A. E. (2007) CellProfilerTM: free, versatile software for automated biological image analysis. BioTechniques 42, 71–75 10.2144/000112257 [DOI] [PubMed] [Google Scholar]
- 45. Kim J.-E., Liu B. P., Park J. H., and Strittmatter S. M. (2004) Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron 44, 439–451 10.1016/j.neuron.2004.10.015 [DOI] [PubMed] [Google Scholar]
- 46. Ujike A., Takeda K., Nakamura A., Ebihara S., Akiyama K., and Takai T. (2002) Impaired dendritic cell maturation and increased T H 2 responses in PIR-B−/− mice. Nat. Immunol. 3, 542–548 10.1038/ni801 [DOI] [PubMed] [Google Scholar]
- 47. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang X., Yigitkanli K., Kim C.-Y., Sekine-Komo T., Wirak D., Frieden E., Bhargava A., Maynard G., Cafferty W. B., and Strittmatter S. M. (2014) Human NgR-Fc decoy protein via lumbar intrathecal bolus administration enhances recovery from rat spinal cord contusion. J. Neurotrauma 31, 1955–1966 10.1089/neu.2014.3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.