

Abstract
Background
Ustekinumab (CNTO 1275) and briakinumab (ABT‐874) are monoclonal antibodies that target the standard p40 subunit of the cytokines interleukin‐12 and interleukin‐23 (IL‐12/23p40), which are involved in the pathogenesis of Crohn's disease.
Objectives
The objectives of this review were to assess the efficacy and safety of anti‐IL‐12/23p40 antibodies for induction of remission in Crohn's disease.
Search methods
We searched the following databases from inception to 12 September 2016: PubMed, MEDLINE, EMBASE, and the Cochrane Library (CENTRAL). References and conference abstracts were searched to identify additional studies.
Selection criteria
Randomized controlled trials (RCTs) trials in which monoclonal antibodies against IL‐12/23p40 were compared to placebo or another active comparator in patients with active Crohn's disease were included.
Data collection and analysis
Two authors independently screened studies for inclusion and extracted data. Methodological quality was assessed using the Cochrane risk of bias tool. The primary outcome was failure to induce clinical remission, defined as a Crohn's disease activity index (CDAI) of < 150 points. Secondary outcomes included failure to induce clinical improvement, adverse events, serious adverse events, and withdrawals due to adverse events. Clinical improvement was defined as decreases of > 70 or > 100 points in the CDAI from baseline. We calculated the risk ratio (RR) and 95% confidence intervals (95% CI) for each outcome. Data were analyzed on an intention‐to‐treat basis. The overall quality of the evidence supporting the outcomes was evaluated using the GRADE criteria.
Main results
Six RCTs (n = 2324 patients) met the inclusion criteria. A low risk of bias was assigned to all studies. The two briakinumab trials were not pooled due to differences in doses and time points for analysis. In both studies there was no statistically significant difference in remission rates. One study (n = 79) compared doses of 1 mg/kg and 3 mg/kg to placebo. In the briakinumab group 70% (44/63) of patients failed to enter clinical remission at 6 or 9 weeks compared to 81% (13/16) of placebo patients (RR 0.86, 95% CI 0.65 to 1.14). Subgroup analysis revealed no significant differences by dose. The other briakinumab study (n = 230) compared intravenous doses of 200 mg, 400 mg and 700 mg with placebo. Eighty‐four per cent (154/184) of briakinumab patients failed to enter clinical remission at six weeks compared to 91% (42/46) of placebo patients (RR 0.92, 95% CI 0.83 to 1.03). Subgroup analysis revealed no significant differences by dose. GRADE analyses of the briakinumab studies rated the overall quality of the evidence for the outcome clinical remission as low. Based on the results of these two studies the manufacturers of briakinumab stopped production of this medication. The ustekinumab studies were pooled despite differences in intravenous doses (i.e. 1mg/kg, 3 mg/kg, 4.5 mg/kg, and 6 mg/kg), however the subcutaneous dose group was not included in the analysis, as it was unclear if subcutaneous was equivalent to intravenous dosing. There was a statistically significant difference in remission rates. At week six, 84% (764/914) of ustekinumab patients failed to enter remission compared to 90% (367/406) of placebo patients (RR 0.92, 95% CI 0.88 to 0.96; 3 studies; high‐quality evidence). Subgroup analysis showed a statistically significant difference for the 6.0 mg/kg dose group (moderate‐quality evidence). There were statistically significant differences in clinical improvement between ustekinumab and placebo‐treated patients. In the ustekinumab group, 55% (502/914) of patients failed to improve clinically (i.e. 70‐point decline in CDAI score), compared to 71% (287/406) of placebo patients (RR 0.78, 95% CI 0.71 to 0.85; 3 studies). Subgroup analysis revealed significant differences compared to placebo for the 1 mg/kg, 4.5 mg/kg and 6 mg/kg dosage subgroups. Similarly for a 100‐point decline in CDAI, 64% (588/914) of patients in the ustekinumab group failed to improve clinically compared to 78% (318/406) of placebo patients (RR 0.82, 95% CI 0.77 to 0.88; 3 studies; high‐quality evidence). Subgroup analysis showed a significant difference compared to placebo for the 4.5 mg/kg and 6.0 mg/kg (high‐quality evidence) dose groups. There were no statistically significant differences in the incidence of adverse events, serious adverse events or withdrawal due to adverse events. Sixty‐two per cent (860/1386) of ustekinumab patients developed at least one adverse event compared to 64% (407/637) of placebo patients (RR 0.97, 95% CI 0.90 to 1.04; 4 studies; high‐quality evidence). Five per cent (75/1386) of ustekinumab patients had a serious adverse event compared to 6% (41/637) of placebo patients (RR 0.83, 95% CI 0.58 to 1.20; 4 studies; moderate‐quality evidence). The most common adverse events in briakinumab patients were injection site reactions and infections. Infections were the most common adverse event in ustekinumab patients. Worsening of Crohn's disease and serious infections were the most common serious adverse events.
Authors' conclusions
High quality evidence suggests that ustekinumab is effective for induction of clinical remission and clinical improvement in patients with moderate to severe Crohn's disease. Moderate to high quality evidence suggests that the optimal dosage of ustekinumab is 6 mg/kg. Briakinumab and ustekinumab appear to be safe. Moderate quality evidence suggests no increased risk of serious adverse events. Future studies are required to determine the long‐term efficacy and safety of ustekinumab in patients with moderate to severe Crohn's disease.
Keywords: Humans; Antibodies, Monoclonal; Antibodies, Monoclonal/administration & dosage; Antibodies, Monoclonal/therapeutic use; Antibodies, Monoclonal, Humanized; Antibodies, Monoclonal, Humanized/administration & dosage; Antibodies, Monoclonal, Humanized/therapeutic use; Crohn Disease; Crohn Disease/therapy; Injections, Intravenous; Interleukin‐12; Interleukin‐12/antagonists & inhibitors; Interleukin‐12/immunology; Interleukin‐23; Interleukin‐23/antagonists & inhibitors; Interleukin‐23/immunology; Randomized Controlled Trials as Topic; Remission Induction; Remission Induction/methods; Ustekinumab; Ustekinumab/administration & dosage; Ustekinumab/therapeutic use
Plain language summary
Ustekinumab and briakinumab for the treatment of active Crohn's disease
What is Crohn's disease? Crohn's disease is a long‐term (chronic) inflammatory bowel disease that can affect any part of the gastrointestinal tract from mouth to anus. Symptoms include abdominal pain, non‐bloody diarrhoea, and weight loss.
What are ustekinumab and briakinumab? Ustekinumab and briakinumab are biologic medications. These medications can be injected under the skin using a syringe or directly infused into a vein (intravenous). Biologic therapies suppress the immune system and reduce the inflammation associated with Crohn's disease. When people with Crohn's disease are experiencing symptoms of the disease it is said to be ‘active’; periods when the symptoms stop are called ‘remission’.
What did the researchers investigate? The researchers investigated whether ustekinumab or briakinumab produce remission in people with active Crohn's disease; and whether these medications cause any harms (side effects). The researchers searched the medical literature up to 12 September 2016.
What did the researchers find? The researchers identified six studies that included a total of 2324 participants. Two studies compared briakinumab to placebo (a fake medicine) and four studies compared ustekinumab to placebo. All of the studies were high quality.
There was no difference in the proportion of briakinumab and placebo participants who achieved remission. An increase in side effects or severe side effects were not seen with briakinumab compared to placebo. The most common side effects in briakinumab participants were reactions at the site of injection and infections. Based on the results of these two studies the manufacturers of briakinumab stopped production of this medication.
High quality evidence suggests that ustekinumab is better than placebo for helping participants achieve remission and for reducing symptoms of active Crohn's disease. Different doses of ustekinumab were investigated and moderate to high quality suggests that 6.0 mg/kg is the most effective dose. An increase in side effects or serious side effects was not seen with ustekinumab compared to placebo. Infections were the most common adverse event in ustekinumab patients. Worsening of Crohn's disease and serious infections were the most common serious side effects in the ustekinumab studies. Ustekinumab is a promising as a therapy for inducing remission and improving symptoms in people with Crohn's disease. Further studies are required to determine the long‐term effectiveness and safety of ustekinumab in patients with moderate to severe Crohn's disease. The ideal dose of ustekinumab also needs to be determined.
Summary of findings
Summary of findings for the main comparison. Briakinumab compared to placebo for induction of remission in Crohn's disease.
| Briakinumab compared to placebo for induction of remission in Crohn's disease | ||||||
| Patient or population: induction of remission in Crohn's disease Settings: Intervention: Briakinumab Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| placebo | Briakinumab | |||||
|
Failure to induce clinical remission (Mannon 2004) CDAI (≤150 points) Follow‐up: 9 weeks |
812 per 10001 | 699 per 1000 (528 to 926) | RR 0.86 (0.65 to 1.14) | 79 (1 study) | ⊕⊕⊝⊝ low2,3 | |
|
Failure to induce clinical remission (Panaccione 2010) CDAI (≤150 points) Follow‐up: 6 weeks |
913 per 10001 |
840 per 1000 (758 to 940) |
RR 0.92 (0.83 to 1.03) |
230 (1 study) |
⊕⊕⊝⊝ low3,4 | |
|
Failure to induce clinical response
(Mannon 2004) CDAI ‐ (≥100 point reduction) Follow‐up: 9 weeks |
688 per 10001 | 447 per 1000 (289 to 681) | RR 0.65 (0.42 to 0.99) | 79 (1 study) | ⊕⊕⊝⊝ low5 | |
|
Failure to induce clinical response
(Panaccione 2010) CDAI ‐ (≥100 point reduction) Follow‐up: 6 weeks |
783 per 10001 |
642 per 1000 (525 to 775) |
RR 0.82 (0.67 to 0.99) |
230 (1 study) |
⊕⊕⊕⊝ moderate6 | |
|
Adverse events (Panaccione 2010) Follow‐up: 12 weeks |
783 per 10001 |
705 per 1000 (587 to 838) |
RR 0.90 (0.75 to 1.07) |
230 (1 study) |
⊕⊕⊕⊝ moderate7 | |
|
Serious adverse events (Panaccione 2010) Follow‐up: 12 weeks |
87 per 10001 | 45 per 1000 (15 to 140) | RR 0.52 (0.17 to 1.61) | 246 (1 study) | ⊕⊕⊝⊝ low8 | |
|
Withdrawals due to adverse event** (Pannaccione 2010) Follow‐up: 12 weeks |
44 per 10001 | 30 per 1000 (6 to 146) | RR 0.69 (0.14 to 3.31) | 246 (1 study) | ⊕⊕⊝⊝ low9 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). **Subject numbers are higher than those reported for the efficacy analyses of the individual studies due to the 200 mg, i.v. experimental group discontinuing enrolment during the induction phase due to poor patient enrolment (Panaccione, 2010). These patients were not included in the efficacy analyses, but were included in the safety analyses. CI: confidence interval; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Control group risk estimates come from control arm of meta‐analysis, based on included trials 2 Downgraded one level due to sparse data (57 events) 3 Downgraded on level because the 95% CI around the effect estimate includes both benefit and no effect 4 Downgraded one level due to sparse data (196 events) 5 Downgraded two levels due to very sparse data (39 events) 6 Downgraded one level due to sparse data (153 events) 7 Downgraded one level due to sparse data (177 events) 8 Downgraded two levels due to very sparse data (13 events) 9 Downgraded two levels due to very sparse data (8 events)
Summary of findings 2. Ustekinumab compared to placebo for induction of remission in Crohn's disease.
| Ustekinumab compared to placebo for induction of remission in Crohn's disease | ||||||
| Patient or population: patients with induction of remission in Crohn's disease Settings: Intervention: Ustekinumab Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| placebo | Ustekinumab | |||||
| Failure to induce clinical remission CDAI (≤ 150) Follow‐up: 6 weeks | 904 per 10001 | 832 per 1000 (795 to 868) | RR 0.92 (0.88 to 0.96) | 1320 (3 studies) | ⊕⊕⊕⊕ high | Sensitivity analysis excluding UNITI‐2 trial. These patients had more severe disease at study entry |
| Failure to induce clinical remission (6 mg/kg subgroup) CDAI (≤ 150) Follow‐up: 6 weeks | 907 per 10001 | 835 per 1000 (789 to 880) | RR 0.92 (0.87 to 0.97) | 916 (2 studies) | ⊕⊕⊕⊝ moderate2 | |
| Failure to induce clinical response CDAI (≥100 point reduction) Follow‐up: 6 weeks | 783 per 10001 | 642 per 1000 (603 to 689) | RR 0.82 (0.77 to 0.88) | 1320 (3 studies) | ⊕⊕⊕⊕ high | Sensitivity analysis excluding UNITI‐2 trial. These patients had more severe disease at study entry |
| Failure to induce clinical response (6 mg/kg subgroup) CDAI (≥100 point reduction) Follow‐up: 6 weeks | 780 per 10001 | 647 per 1000 (601 to 710) | RR 0.83 (0.77 to 0.91) | 916 (2 studies) | ⊕⊕⊕⊕ high | |
|
Adverse events Follow‐up: 8 weeks |
639 per 10001 | 620 per 1000 (575 to 664) | RR 0.97 (0.9 to 1.04) | 2023 (4 studies) | ⊕⊕⊕⊕ high | |
| Serious adverse events Follow‐up: 8 weeks | 64 per 10001 | 53 per 1000 (37 to 77) | RR 0.83 (0.58 to 1.2) | 2023 (4 studies) | ⊕⊕⊕⊝ moderate3 | |
| Withdrawals due to adverse event Follow‐up: 8 weeks | 54 per 10001 | 24 per 1000 (10 to 57) | RR 0.44 (0.18 to 1.05) | 657 (2 studies) | ⊕⊕⊝⊝ low4 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). **Subject numbers are higher than those reported for the efficacy analyses of the individual studies due to the omission of efficacy results for subjects receiving subcutaneous placebo and 90 mg ustekinumab, as well as subjects receiving 90mg s.c. and 4.5 mg/kg of ustekinumab in the open‐label arm of the study by Sandborn (2008). The results of these subjects were included in the safety analyses. CI: confidence interval; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Control group risk estimates come from control arm of meta‐analysis, based on included trials 2 Downgraded one level due to heterogeneity detected for 6 mg/kg subgroup (I2 = 39%) 3 Downgraded one level due to sparse data (116 events) 4 Downgraded two levels due to very sparse data (20 events)
Background
Description of the condition
Crohn's disease is a chronic inflammatory disease of the gastrointestinal tract that typically presents with abdominal pain, diarrhoea, and weight loss. In North America and Northern Europe, Crohn's disease affects up to 320 people per 100,000 (Molodecky 2012). Long‐term transmural inflammation can result in complications including stenoses and fistulas that require surgical management in up to a third of patients (Solberg 2007). Therapy is aimed at inducing and maintaining remission and preventing complications.
Corticosteroids were among the first available therapies for induction of remission in Crohn's disease (Summers 1979; Rutgeerts 1994). However, persistent remission (Faubion 2001) and mucosal healing (Modigliani 1990) have not been observed with corticosteroid therapy. Moreover, these drugs cause multiple adverse effects (Ford 2011; Yang 2002).
The immunosuppressives azathioprine, 6‐mercaptopurine and methotrexate have been widely used as corticosteroid‐sparing agents in the treatment of Crohn’s disease (McDonald 2012; Chande 2013). Although azathioprine is not effective for inducing remission (Chande 2013), it is beneficial when used in combination with infliximab therapy (Colombel 2010; Chande 2013). However, azathioprine requires careful monitoring for adverse events including pancreatitis, liver toxicity, and neutropenia (Raj 2010).
Due to the limited efficacy and potential toxicities of those drugs, monoclonal antibodies directed against tumor necrosis factor alpha (TNF‐α) were developed as more selective therapeutic agents. TNF‐α antagonists, which include infliximab, adalimumab, and certolizumab pegol, are effective for both induction and maintenance of remission in Crohn’s disease (Targan 1997; Hanauer 2002; Akobeng 2003; Hanauer 2006; Lawson 2006; Sandborn 2007; Schreiber 2007). However, concerns regarding infection (Colombel 2004; Colombel 2007; Schreiber 2007; Singh 2011) and a potentially increased risk of certain types of cancer such as non‐Hodgkin's lymphoma and non‐melanoma skin cancer exists (Burmester 2013; Hudesman 2013). Furthermore, primary and secondary failure of TNF‐α antagonists occurs in approximately 30 and 40% of patients, respectively (Targan 1997; Hanauer 2002; Hanauer 2006; Danese 2011). These limitations provided the impetus to investigate drugs that target different inflammatory pathways.
Description of the intervention
Ustekinumab (CNTO 1275) and briakinumab (ABT‐874) are fully human IgG1 monoclonal antibodies that target the common p40 subunit of the cytokines interleukin‐12 and interleukin‐23 (IL‐12/23p40). Ustekinumab and briakinumab are effective for the treatment of psoriasis (Papp 2008; Gottlieb 2009; Reich 2011), and early Phase I and II clinical trials of ustekinumab suggest a similar benefit in Crohn's disease (Mannon 2004; Sandborn 2008; Sandborn 2012).
How the intervention might work
Crohn's disease is characterized by dysregulated T‐cell activity. Interleukin‐12 (IL‐12) and IL‐23 are released by antigen presenting cells to trigger an acquired immune response (Watanabe 2004; Peluso 2006; Benson 2011; Duvallet 2011). Specifically, IL‐12 production stimulates a Th1 response, that culminates in the secretion of the pro‐inflammatory cytokines IFN‐ɣ and TNF‐α (Peluso 2006; Benson 2011; Cingoz 2011). Similarly, IL‐23 stimulates proliferation of Th17 lymphocytes which play a role in many chronic inflammatory conditions (Duvallet 2011). At a cellular level inhibition of IL‐12/23p40 leads to apoptosis of T cells in the gut mucosa (Fuss 1999). Furthermore, blockade IL‐12 and IL‐23, results in disease improvement in murine models of colitis (Neurath 1995). Collectively, these data suggest a possible therapeutic role for IL‐12/23p40 antibodies for the treatment of Crohn’s disease.
Why it is important to do this review
Ustekinumab, a humanized monoclonal antibody directed against the p40 subunit of IL‐12 and IL‐23, has been more thoroughly investigated in the treatment of psoriasis where it appears to be safe and effective (Gottlieb 2014; Papp 2013). This systematic review summarizes the current evidence regarding the use of anti IL‐12/23p40 antibodies for induction of remission in Crohn's disease.
Objectives
The objectives of this review were to determine the efficacy and safety of anti‐IL‐12/23p40 antibodies for induction of remission in Crohn's disease.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials (RCTs) that assessed the efficacy and safety of antibodies directed against the p40 subunit of IL‐12 and IL‐23 compared to placebo or other active treatments for induction of remission in Crohn's disease were considered for inclusion.
Types of participants
Participants were required to have active Crohn's disease defined by conventional clinical, radiological or endoscopic criteria. No age restrictions were applied.
Types of interventions
Interventions that involved the administration of an antibody directed against the p40 subunit of IL‐12 and IL‐23 were considered for inclusion. Interventions that altered IL‐12 or IL‐23 by another strategy, such as direct inhibition of transcription were not included (Sands 2010).
Types of outcome measures
Primary outcomes
The primary outcome measure was the proportion of patients who failed to enter clinical remission, as defined by the original studies (e.g. a Crohn's Disease Activity Index (CDAI) score of < 150)). An intention‐to‐treat analysis was used.
Secondary outcomes
Secondary outcome measures included:
clinical response;
endoscopic remission;
steroid withdrawal;
adverse events;
serious adverse events; and
withdrawal due to adverse events.
Search methods for identification of studies
Electronic searches
We searched PubMed, MEDLINE (OvidSP), EMBASE (Ovid SP), and the Cochrane Library (CENTRAL) from inception to 12 September 2016. The search strategies are listed in Appendix 1. Conference abstracts from Digestive Disease Week and the United European Gastoenterology Week were also searched to identify abstract publications.
Searching other resources
Reference lists
Manual searches of reference lists from potentially relevant papers were performed in order to identify additional studies that may have been missed using the computer‐assisted search strategy.
Correspondence
Leaders in the field (W. Strober, P. Mannon, W. Sandborn) were contacted to identify other studies, which may not be published. The manufacturers of briakinumab and ustekinumab (Abbott Laboratories, Abbott Park, IL, USA and Centocor, Horsham, PA, USA) were contacted for additional data.
Trial databases
The trial databases clinicaltrials.gov, clinicaltrials.ifpma.org and the meta register of controlled trials at controlled‐trials.com were searched to identify other studies that had not been reported.
Data collection and analysis
Selection of studies
All publications identified by the search strategy were assessed independently by two authors (TN and JKM), and relevant studies were selected according to the inclusion criteria. Disagreements were resolved by consensus.
Data extraction and management
Data were extracted independently by two authors (TN and JKM) using standardized extraction sheets. Information was extracted on data source; participants (age range, exclusion criteria, pre‐treatment medication, co‐medication, disease entity, activity); details of intervention (type of antibody, frequency and duration of treatment) and comparison treatment; outcomes studied and duration of follow up; study design features (including method of allocation, concealment, where applicable; intention‐to‐treat analysis); and results, including adverse events and loss to follow up. Discrepancies in the data extractions were resolved by consensus. Investigators were contacted for additional information when data were not available or unclear.
Assessment of risk of bias in included studies
We used the Cochrane risk of bias tool to assess the methodological quality of the included studies (HIggins 2011a). Two authors independently assessed the risk of bias (TN and JKM). The following study characteristics were assessed:
Randomization sequence generation;
Allocation concealment;
Blinding of participants, outcome assessors and investigators;
Incomplete outcome reporting (i.e. there was an acceptable method of dealing with attrition);
Selective outcome reporting (i.e. all outcomes described in the methods were included in the analysis); and
Other potential sources of bias.
Based on these characteristics, studies were judged to have a high, low or unclear risk of bias.
We used the GRADE approach to assess the overall quality of evidence for the primary outcome and secondary outcomes including clinical response, adverse events, serious adverse events and withdrawal due to adverse events. Outcomes from pooling of randomized trials start as high quality evidence, but may be downgraded due to: (1) risk of bias, (2) indirectness of evidence, (3) inconsistency (unexplained heterogeneity), (4) imprecision (sparse data), and (5) reporting bias (publication bias). The overall quality of evidence for each outcome was determined after considering each of these elements, and categorized as high quality (i.e. further research is very unlikely to change our confidence in the estimate of effect); moderate quality (i.e. further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate); low quality (i.e. further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate); or very low quality (i.e. we are very uncertain about the estimate) (Guyatt 2008; Schünemann 2011).
Measures of treatment effect
Data were analysed using Review Manager 5 (RevMan 5). Data were extracted from the original research articles and converted into 2×2 tables. The definitions of treatment success, remission and clinical improvement were set by the authors of each paper, and the data were combined for analysis only if these definitions were sufficiently similar (determined by consensus). All relevant outcomes in the identified studies were reported as dichotomous data so results were all pooled based on the relative risk (RR), with RR < 1.0 denoting superiority of the experimental treatment. Results were presented with 95% confidence intervals (CI). The 'Rule of Three' was used to estimate the 95% confidence interval for the frequency of rare, undetected adverse events, i.e. if no events were detected in n trial participants, the upper limit of the 95% confidence interval for the probability of the adverse event is 3/n (Loke 2007).
Unit of analysis issues
For three arm trials with a single placebo group and two treatment dose groups we split the placebo group in half to avoid a unit of analysis error (Higgins 2011b). In the case of an odd number of patients the groups were split in a way that led to a larger patient number in the placebo group for the lower dose arm in order to avoid overestimating effects in the higher dose arm, which was assumed to be more effective a priori. In order to avoid carry‐over effects we only included the first part of the study (i.e. before the cross‐over) for any cross‐over studies (Higgins 2011b).
Dealing with missing data
Missing outcome data were presented using the conservative intention‐to‐treat approach (i.e. all loss to follow up were considered treatment failures).
Assessment of heterogeneity
Statistical heterogeneity was examined by calculating the I2 statistic and the Chi2 test. Subgroup analysis (see below) was applied to explore clinical and methodological sources of heterogeneity.
Assessment of reporting biases
For future updates of this review, if there are a sufficient number of included studies (i.e. ≥ 10), we will investigate publication bias by means of a funnel plot (Egger 1997). Publication bias was not investigated for the current version of this review due to an insufficient number of studies.
Data synthesis
Results for the different antibodies were not combined. Also studies were not combined, if subgroup analysis suggested significant heterogeneity. A fixed‐effect model was applied when no heterogeneity was detected. Otherwise, a random‐effects model was calculated. Data were not pooled when I2 suggested considerable heterogeneity.
Subgroup analysis and investigation of heterogeneity
Preplanned subgroup analyses included:
studies investigating children and studies investigating adults;
different drug doses and/or routes of administration;
studies investigating patients with high disease activity; and
studies investigating only patients who failed anti‐TNF‐α therapy.
Due to the low number of studies identified subgroup analyses could only be performed for different drug doses.
Sensitivity analysis
Sensitivity analyses examined the impact of the following variables on the pooled effect:
random‐effects versus fixed‐effect modelling;
low risk of bias only versus unclear or high risk of bias; and
relevant loss to follow up (> 10%): Best‐case versus worst‐case scenario.
Sensitivity analysis could only be performed for random‐effects versus fixed‐effect modelling. No publication allowed for the simulation of best‐case and worst‐case scenarios.
Results
Description of studies
Results of the search
The literature search conducted on 12 September 2016 identified 183 records. After duplicates were removed, a total of 139 records were screened for inclusion. Of these, 16 studies were selected for full text review. Two studies were excluded leaving 27 reports of 6 trials (total 2324 patients) that met the pre‐defined inclusion criteria (See Figure 1). Leaders in the field and the manufacturers of briakinumab and ustekinumab were contacted and no additional studies were identified.
1.
Study flow diagram.
Included studies
All studies were multicenter randomized placebo‐controlled trials investigating patients with active Crohn's disease. The six studies investigated different anti‐IL12/23 antibodies. Mannon 2004 and Panaccione 2015 investigated briakinumab and four studies investigated ustekinumab (Sandborn 2008; Sandborn 2012; Feagan 2016 UNITI‐1; Feagan 2016 UNITI‐2). See Characteristics of included studies for further details.
Mannon 2004 investigated two different doses (1.0 mg and 3.0 mg/kg body weight) of briakinumab administered as 7 weekly subcutaneous injections against placebo. Each group was further randomized to uninterrupted therapy or a 4 week interval between the first and second doses. Follow‐up in this study was between 24 and 27 weeks. As the different dosing regimens differed only slightly, the treatment arms were combined for analysis. The two doses differed markedly with respect to effectiveness and were analysed as separate studies as described in the methods section.
Sandborn 2008 was a Phase IIa study of ustekinumab that compared 4 regimens of ustekinumab to placebo in 104 patients with active CD in both TNF‐α antagonist naive patients and those who had previously failed therapy with one or more of these agents. A sub‐study compared intravenous to subcutaneous therapy for the induction of clinical remission, however this portion of the trial was not included as it was not placebo‐controlled. Sandborn 2008 utilized a blinded cross‐over design that compared four groups: subcutaneous placebo at weeks 0–3, followed by 90 mg ustekinumab at weeks 8–11; subcutaneous 90 mg ustekinumab at weeks 0–3, then placebo at weeks 8–11; intravenous placebo at week 0, followed by 4.5 mg/kg ustekinumab at week 8; or intravenous 4.5 mg/kg ustekinumab at week 0, then placebo at week 8. Cross‐over to the alternate therapy occurred at week 8. The first part of the cross‐over trial was eligible for inclusion in this review. The subcutaneous doses were not included in the pooled analyses, as it was not clear that this was equivalent to intravenous dosing.
Panaccione 2015 was a double‐blind, placebo‐controlled Phase IIb trial in which 246 patients with moderate to severe Crohn’s disease were randomized (1:1:1:3) to placebo, 200 mg briakinumab, 400 mg briakinumab, or 700 mg briakinumab at weeks 0, 4, and 8 weeks. These patients were stratified by prior TNF‐α antagonist use and response to anti‐TNF‐α therapy. Patients that achieved clinical response at week 12 in the placebo and 400 mg briakinumab groups continued these treatment regimens at weeks 12, 16, 20 in the maintenance phase. Patients with clinical response in the 700 mg briakinumab group were re‐randomized (1:1:1) to placebo, 200 mg IV briakinumab, or 700 mg IV briakinumab for the maintenance phase. The maintenance data were not considered for this systematic review.
Sandborn 2012 was a double‐blind, placebo‐controlled Phase 2b trial of ustekinumab that randomized 526 patients, of which 524 had previously failed TNF‐α antagonist treatment (99.6%), to receive a single dose of 1, 3, or 6 mg/kg of intravenous ustekinumab or placebo. Patients that responded to ustekinumab were re‐randomized to receive maintenance therapy with 90 mg of subcutaneous ustekinumab or placebo at weeks 8 and 16. The maintenance data were not considered for this systematic review.
Feagan 2016 UNITI‐1 was a double‐blind, placebo‐controlled phase III trial of ustekinumab that randomized 741 patients with moderate to severe Crohn's disease, who had failed TNF‐α antagonist therapy, to a single dose of 130 mg or 6mg/kg of ustekinumab or placebo. Patients who responded to ustekinumab were randomized to receive maintenance therapy with 90 mg of subcutaneous ustekinumab (every 8 weeks or every 12 weeks) or placebo through week 40. The maintenance data were not considered for this systematic review.
Feagan 2016 UNITI‐2 was a double‐blind, placebo‐controlled phase III trial of ustekinumab that randomized 628 patients with moderate to severe Crohn's disease, who had failed corticosteroids or immunosuppressants or both, to a single dose of 130 mg or 6 mg/kg of ustekinumab or placebo. Patients who responded to ustekinumab were randomized to receive maintenance therapy with 90 mg of subcutaneous ustekinumab (every 8 weeks or every 12 weeks) or placebo through week 40. The maintenance data were not utilized for this systematic review.
Excluded studies
We excluded two studies (See Characteristics of excluded studies). Fasanmade 2008 was a pharmacokinetics study that compared intravenous ustekinumab at a dose of 4.5 mg/kg to subcutaneously administered ustekinumab at a dose of 90 mg. The active drug assessed in the Sands 2010 study was not an antibody directed against the p40 subunit of Il‐12 and IL‐23 (i.e. different mechanism of action than briakinumab and ustekinumab).
Risk of bias in included studies
The risk of bias analysis is summarized in Figure 2.
2.

Risk of bias summary: reviewers' judgements about each risk of bias item for each included study.
Allocation
All six studies utilized computer generated randomization and were rated as low risk of bias for random sequence generation. Five studies used a centralized randomization scheme and were rated as low risk of bias for allocation concealment (Feagan 2016 UNITI‐1; Feagan 2016 UNITI‐2; Panaccione 2015; Sandborn 2008; Sandborn 2012). The Mannon 2004 study allocated patients using a telephone based interactive voice response system and was rated as low risk of bias for allocation concealment.
Blinding
Five studies utilized an identical placebo and were rated as low risk of bias for blinding (Feagan 2016 UNITI‐1; Feagan 2016 UNITI‐2; Mannon 2004; Sandborn 2008; Sandborn 2012). Panaccione 2015 utilized a double‐blind design and reported that, "The study sponsor, site personnel, and patients were unaware of the treatment assignments throughout both the induction and maintenance phases." As a result we rated this study as low risk of bias for blinding.
Incomplete outcome data
All of the studies were rated as low risk for incomplete outcome data. Drop‐outs were generally balanced across treatment groups with similar reasons for withdrawal.
Selective reporting
Five studies were rated as low risk for selective reporting (Feagan 2016 UNITI‐1; Feagan 2016 UNITI‐2; Panaccione 2015; Sandborn 2008; Sandborn 2012). Mannon 2004 reported on pre‐specified primary and secondary outcomes (safety and efficacy) but only reported on histology and laboratory scores for the treatment group. This study was rated as unclear for selective reporting.
Other potential sources of bias
All of the studies appear to be free of other sources of bias and were rated as low risk of bias for this item.
Effects of interventions
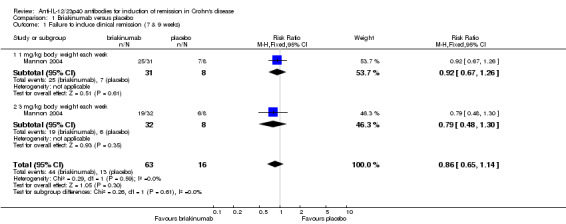
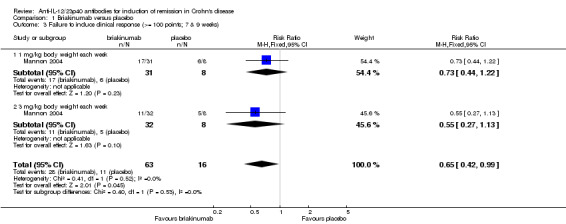
The two trials of briakinumab were not pooled due to differences in doses and time points for analysis (Mannon 2004; Panaccione 2015). However in both trials briakinumab was not statistically different than placebo for failure to induce remission. In the Mannon 2004 (N = 79) study 70% (44/63) of briakinumab patients failed to enter clinical remission at weeks 7 or 9 compared to 81% (13/16) of placebo patients (RR 0.86, 95% CI 0.65 to 1.14). Subgroup analysis showed no statistically significant differences by briakinumab dose (1 mg/kg or 3 mg/kg). No heterogeneity was detected for the dosage subgroup comparison (I2 = 0%). A GRADE analysis indicated that the quality of evidence supporting the primary outcome was low (See Table 1). There was a statistically significant difference in the proportion of patients who failed to have a clinical response (> 100 point improvement in CDAI from baseline) favouring briakinumab over placebo. Forty‐four per cent (28/63) briakinumab patients failed to respond to treatment compared to 69% (11/16) of placebo patients (RR 0.65, 95% CI 0.42 to 0.99). Subgroup analysis showed no statistically significant differences by briakinumab dose (1 mg/kg or 3 mg/kg). No heterogeneity was detected for this comparison (I2 = 0%). Sensitivity analyses utilizing a random‐effects model found similar results for induction of remission and clinical response (See Table 3). A GRADE analysis indicated that the quality of evidence supporting the outcome clinical response was low (See Table 1).
1. Sensitivity Analysis: Fixed Effects vs. Random Effects Modelling.
| Outcome | Fixed Effects Modelling | Random Effects Modelling |
| Briakinumab /Remission (Mannon 2004) | RR 0.86 [0.65, 1.14] | RR 0.88 [0.68, 1.15] |
| Briakinumab /Remission (Panaccione 2015) | RR 1.05 [0.90, 1.22] | RR 0.92 [0.83, 1.03] |
| Ustekinumab /Remission | RR 0.94 [0.88, 1.01] | RR 0.95 [0.89, 1.02] |
| Briakinumab /Response (Mannon 2004) | RR 0.65 [0.42, 0.99] | RR 0.66 [0.44, 1.01] |
| Briakinumab /Response (Panaccione 2015) | RR 0.82 [0,67, 0.99] | RR 0.81 [0.67, 0.99] |
| Ustekinumab /Response | RR 0.79 [0.71, 0.89] | RR 0.80 [0.72, 0.90] |
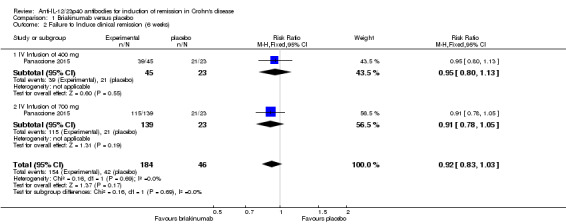
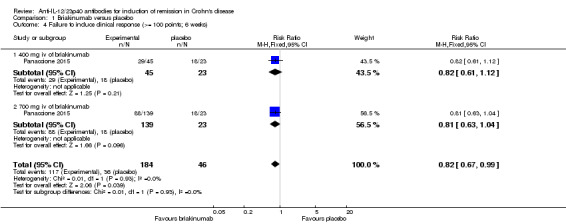
In the Panaccione 2015 (N = 230) study 84% (154/184) of briakinumab patients failed to enter clinical remission compared to 91% (42/46) of placebo patients (RR 0.92, 95% CI 0.83 to 1.03). Subgroup analysis showed no differences by briakinumab dose (400 mg or 700 mg). No heterogeneity was detected for the dosage subgroup comparison (I2 = 0%). A GRADE analysis indicated that the quality of evidence supporting the primary outcome was low (See Table 1). There was a difference in clinical response rates (> 100 point improvement in CDAI from baseline) between briakinumab and placebo patients at six weeks. Sixty‐four per cent (117/184) of briakinumab patients failed to respond compared to 78% of placebo patients (RR 0.82 95% CI 0.67 to 0.99). Subgroup analysis showed no differences by briakinumab dose (400 mg or 700 mg). No heterogeneity was detected for this comparison (I2 = 0%). A GRADE analysis indicated that the quality of evidence supporting this outcome was moderate (See Table 1). Although all patients completed the induction phase of this study, the sponsor ended the study during the maintenance phase because the primary endpoint was not achieved. Sensitivity analyses utilizing a random‐effects model found similar results for induction of remission and clinical response (See Table 3).
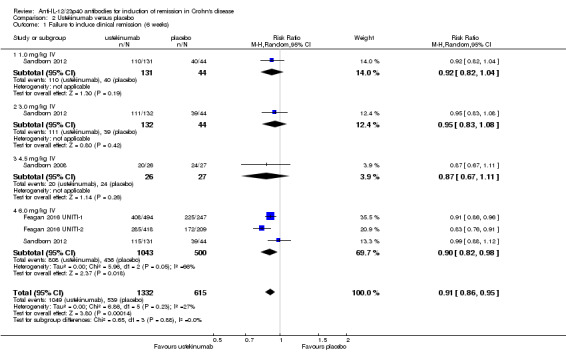
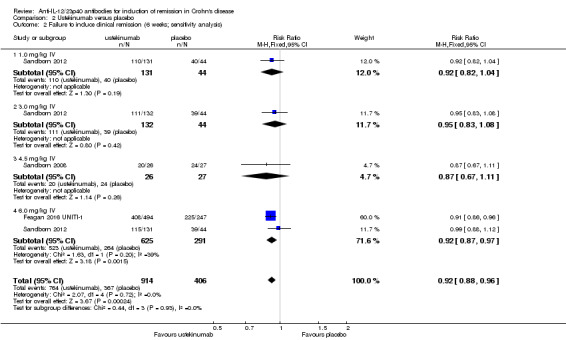
The ustekinumab trials (Feagan 2016 UNITI‐1; Feagan 2016 UNITI‐2; Sandborn 2008; Sandborn 2012), were pooled despite differences in intravenous doses (i.e. 1mg/kg, 3 mg/kg, 4.5 mg/kg, and 6 mg/kg), however the subcutaneous dose group was not included in the analysis, as it was unclear if subcutaneous was equivalent to intravenous dosing. In the pooled analysis (4 studies, 1947 patients), ustekinumab was statistically different than placebo for failure to induce remission. At week six, 79% (1049/1332) of ustekinumab patients failed to enter remission compared to 88% (539/615) of placebo patients (RR 0.91, 95% CI 0.86 to 0.95; I2 = 27%). Subgroup analysis showed a difference for the ustekinumab 6.0 mg/kg dose. However a moderate amount of heterogeneity was detected for this subgroup comparison (I2 = 66%). A visual inspection of the forest plot indicated that the heterogeneity appeared to be a result of the inclusion of the Feagan 2016 UNITI‐2 study in the pooled analysis. The Feagan 2016 UNITI‐2 study differed from the other studies in the pooled analysis because it required objective evidence of active Crohn's disease (e.g. elevated serum C‐reactive protein or fecal calprotectin concentrations or endoscopic ulcerations) for study entry whereas the other studies in the pooled analysis did not. This may be a plausible explanation for the higher response rates seen in the UNITI‐2 patients compared to the other studies in the pooled analysis. To investigate if this study was the source of the heterogeneity the analysis was repeated excluding this trial. The pooled analysis now included 3 studies and 1320 participants. No heterogeneity was detected for the overall comparison (P = 0.72; I2 = 0%) or for the 6.0 mg/kg subgroup (P 0.20; I2 = 39%). Eighty‐four per cent (764/914) of ustekinumab patients failed to enter remission at week 6 compared to 90% (367/406) of placebo patients (RR 0.92, 95% CI 0.88 to 0.96). A GRADE analysis indicated that the quality of evidence supporting the primary outcome was high (See Table 2). Subgroup analysis showed a difference in remission rates for the ustekinumab 6.0 mg/kg dose. Eighty‐four per cent (523/625) of ustekinumab 6.0 mg/kg patients failed to enter remission at week 6 compared to 91% (264/291) of placebo patients (RR 0.92, 95% CI 0.88 to 0.96).
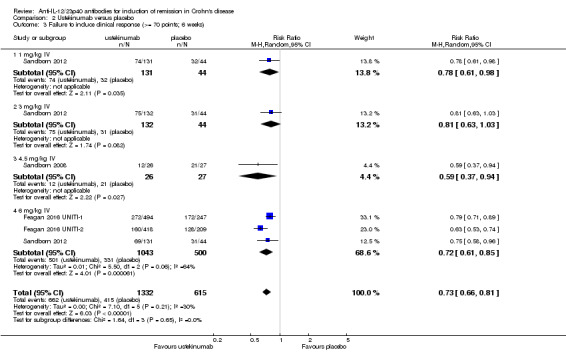
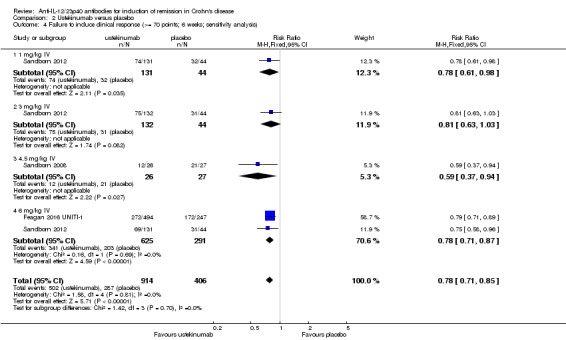
There were differences in clinical improvement between ustekinumab and placebo treated patients. In the ustekinumab group, 50% (662/1332) of patients failed to achieve a 70‐point decrease in CDAI score, compared to 68% (415/615) of placebo patients (RR 0.73, 95% CI 0.66 to 0.81; I2 = 30%). Subgroup analysis showed differences in failure to respond for the 1mg/kg, 4.5 mg/kg, and 6 mg/kg dosage subgroups. However, moderate heterogeneity was detected for the 6.0 mg/kg subgroup (I2 = 64%%). A visual inspection of the forest plot indicated that the heterogeneity appeared to be a result of the inclusion of the Feagan 2016 UNITI‐2 study in the pooled analysis. To investigate if this study was the source of the heterogeneity the analysis was repeated excluding this trial. The pooled analysis now included 3 studies and 1320 participants. No statistically significant heterogeneity was detected for the overall comparison (P = 0.81; I2 = 0%) or for the 6.0 mg/kg subgroup (P 0.69; I2 = 0%). Fifty‐five per cent (502/914) of ustekinumab patients failed to achieve a 70‐point decrease in CDAI score at week 6 compared to 71% (287/406) of placebo patients (RR 0.78, 95% CI 0.71 to 0.85). Subgroup analysis showed differences in failure to respond for the 1 mg/kg (RR 0.78, 95% CI 0.61 to 0.98), 4.5 mg/kg (RR 0.59, 95% CI 0.37 to 0.94), and 6 mg/kg (0.78, 95% CI 0.71 to 0.87) dosage subgroups. Similarly for a 100‐point decline in CDAI, 59% (782/1332) of patients in the ustekinumab group failed to have a 100‐point clinical response compared to 76% (467/615) of placebo patients (RR 0.77, 95% CI 0.69 to 0.87; I2 = 61%). Subgroup analysis showed a difference in failure to respond for the 4.5 mg/kg and 6.0 mg/kg dosage subgroups. However, a high degree of heterogeneity was detected for the 6.0 mg/kg subgroup comparison (I2 = 80%). A visual inspection of the forest plot indicated that the Feagan 2016 UNITI‐2 study could be the cause of this heterogeneity. To investigate if this study was the source of the heterogeneity the analysis was repeated excluding this trial. The pooled analysis now included 3 studies and 1320 participants. No heterogeneity was detected for the overall comparison (P = 0.57; I2 = 0%) or for the 6.0 mg/kg subgroup (P 0.72; I2 = 0%). Sixty‐four per cent (588/914) of ustekinumab patients failed to achieve a 100‐point decrease in CDAI score at week 6 compared to 78% (318/406) of placebo patients (RR 0.82, 95% CI 0.77 to 0.88). A GRADE analysis indicated that the quality of evidence supporting the outcome 100‐point clinical response was high (See Table 2). Subgroup analysis showed a differences in failure to respond for the 4.5 mg/kg (RR 0.59, 95% CI 0.39 to 0.89), and 6 mg/kg (RR 0.83, 95% CI 0.77 to 0.91) dosage subgroups.
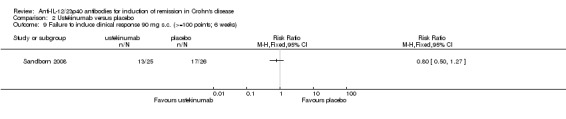
There were no differences in remission or clinical improvement rates in patients who received subcutaneous ustekinumab or placebo. Seventy‐two per cent (18/25) of patients in the subcutaneous ustekinumab group failed to enter remission at six weeks compared to 77% (20/26) of placebo patients (RR 0.94, 95% CI 0.68 to 1.29). Forty‐eight per cent (12/25) of patients in the subcutaneous ustekinumab group failed to achieve a 70‐point decrease in CDAI at six weeks compared to 62% (16/26) of placebo patients (RR 0.78, 95% CI 0.47 to 1.30). Fifty‐two per cent (13/25) of patients in the subcutaneous ustekinumab group failed to achieve a 100‐point decrease in CDAI at six weeks compared to 65% of placebo patients (RR 0.80, 95% CI 0.50 to 1.27). Sensitivity analyses utilizing a random‐effects model found similar results for induction of remission and clinical response.
Endoscopic remission was not assessed in any of the included studies. Steroid withdrawal following treatment was not assessed in any of the included induction studies. Sandborn 2012 evaluated corticosteroid‐free remission at 22 weeks during the maintenance phase.
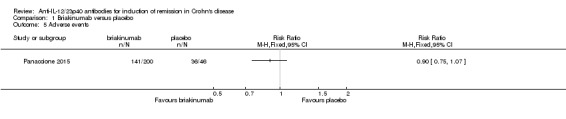
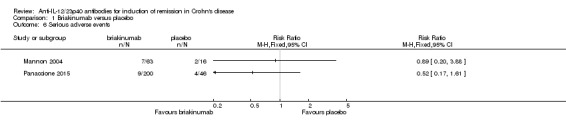
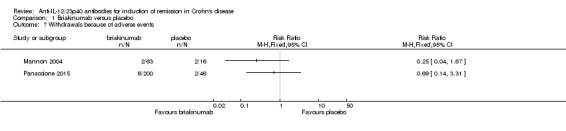
There was no difference in the proportion of patients who experienced at least one adverse event in the Panaccione 2015 study. Seventy‐one per cent (141/200) of briakinumab patients experienced at least one adverse event compared to 78% (36/46) of placebo patients (RR 0.90, 95% CI 0.75 to 1.07). A GRADE analysis indicated that the overall quality of the evidence supporting this outcome was moderate (See Table 1). Mannon 2004 did not report on the proportion of patients who had at least one adverse event. Mannon 2004 reported that injection site reactions were the most common adverse event. Injection site reactions occurred more frequently in the briakinumab groups (77% in the 1mg/kg group and 88% in the 3 mg/kg group) than the placebo group (25%). The majority of these injection site reactions were considered mild (88%). Panaccione 2015 reported infections as the most common adverse event. Thirty‐three per cent of briakinumab patients had an infection compared to 35% of placebo patients. There was no difference in either study in the proportion of patients who developed a serious adverse event or withdrew due to an adverse event. In the Mannon 2004 study, 11% (7/63) of briakinumab patients had a serious adverse event compared to 12% (2/16) of placebo patients (RR 0.89, 95% CI 0.20 to 3.88). In the Panaccione 2015 study 4% (9/200) of briakinumab patients had a serious adverse event compared to 9% (4/46) placebo patients (RR 0.52, 95% CI 0.17 to 1.61). Three per cent (2/63) of briakinumab patients in the Mannon 2004 study withdrew due to an adverse event compared to 12% (2/16) placebo patients (RR 0.25, 95% CI 0.04 to 1.67). Three per cent (6/200) of briakinumab patients in the Panaccione 2015 study withdrew due to an adverse event compared to 4% (2/46) of placebo patients (RR 0.69, 95% CI 0.14 to 3.31). GRADE analyses indicated that the overall quality of the evidence supporting the outcomes serious adverse events and withdrawal due to adverse events was low (See Table 1).
In the ustekinumab studies there was no difference in the proportion of patients who experienced at least one adverse event. Sixty‐two per cent (860/1386) of ustekinumab patients experienced at least one adverse event compared to 64% (407/637) of placebo patients (RR 0.97, 95% CI 0.90 to 1.04; participants = 2023; studies = 4; I2 = 0%). A GRADE analysis indicated that the overall quality of the evidence supporting this outcome was high (See Table 2). Infections were the most commonly reported adverse event in the ustekinumab studies. Other common adverse events included nasopharyngitis (Feagan 2016 UNITI‐1; Feagan 2016 UNITI‐2; Sandborn 2008; Sandborn 2012), abdominal pain (Feagan 2016 UNITI‐1; Feagan 2016 UNITI‐2; Sandborn 2008; Sandborn 2012), nausea (Feagan 2016 UNITI‐1; Feagan 2016 UNITI‐2; Sandborn 2008; Sandborn 2012), worsening Crohn's disease (Feagan 2016 UNITI‐1; Feagan 2016 UNITI‐2; Sandborn 2008; Sandborn 2012), arthralgia (Feagan 2016 UNITI‐1; Feagan 2016 UNITI‐2; Sandborn 2008; Sandborn 2012), headache (Feagan 2016 UNITI‐1; Feagan 2016 UNITI‐2; Sandborn 2008; Sandborn 2012), and dyspepsia (Sandborn 2008).
There was no difference in the incidence of serious adverse events. Serious adverse events occurred in 5% (75/1386) of ustekinumab patients compared to 6% (41/637) of patients in the placebo group (RR 0.83, 95% CI 0.58 to 1.20; participants = 2023; studies = 4; I2 = 0%). A GRADE analysis indicated that the overall quality of the evidence supporting this outcome was moderate (See Table 2). Worsening of CD and serious infections were the most common serious adverse events. Although the difference was not statistically significant, more placebo patients withdrew due to adverse events than ustekinumab patients. Five per cent (10/184) of placebo patients withdrew due to adverse events compared to 2% (10/473) of ustekinumab patients (RR 0.44, 95% CI 0.18 to 1.05; participants = 657; studies = 2; I2 = 0%).
Discussion
Summary of main results
Briakinumab (ABT‐874) and ustekinumab (CNTO 1275) are monoclonal antibodies that target the shared p40 subunit of IL‐12 and IL‐23, that have been studied for induction of remission in CD. Neither Mannon 2004 or Panaccione 2015 found briakinumab to be superior to placebo for induction of remission in CD. Grade analyses indicated that the overall quality of the evidence supporting this outcome was low due mostly to sparse data. Both Mannon 2004 and Panaccione 2015 found briakinumab to be superior to placebo for induction of clinical improvement. Grade analyses indicated that the overall quality of the evidence supporting this outcome was low due mostly to sparse data. Subgroup analysis did not identify any significant differences by dose. The Panaccione 2015 trial was terminated during the open‐label maintenance phase by the sponsor because the primary endpoint for induction of remission was not achieved, leaving to our knowledge ustekinumab as the only IL‐12/23p40 antagonist currently being investigated for the treatment of CD.
Ustekinumab patients were significantly more likely than placebo patients to achieve clinical remission and clinical improvement. Subgroup analysis identified a difference for the 6.0 mg/kg dose group for induction of remission. Clinical improvement results were consistent for both a 70 and 100 point reduction in CDAI scores. Subgroup analysis identified significant differences by dose for 70 (i.e. 1mg/kg, 4.5 mg/kg, and 6 mg/kg dosage subgroups) and 100 point reduction (i.e. 4.5 mg/kg and 6.0 mg/kg subgroups) in CDAI scores. The results of the GRADE analysis indicate that overall quality of the evidence supporting the outcomes clinical remission and 100‐point reduction in CDAI was high. Moderate to high quality evidence suggests that 6.0 mg/kg is the optimal dose for induction of remission and clinical response. Overall these results suggest a therapeutic benefit for ustekinumab in patients with moderate to severe CD.
With respect to safety, neither antibody led to an increase in adverse events or serious adverse events. Fewer patients that received anti‐IL‐12/23 monoclonal antibody treatment withdrew due to adverse events than placebo. However, these differences were not statistically significant. This may have been due to greater withdrawal of patients in the placebo group due to recurrence of CD symptoms. Although both drugs demonstrated an acceptable safety profile in these studies, the number of patients investigated did not allow for the assessment of rare adverse events. Studies with long term follow‐up are required to assess the risk of rare adverse events.
Overall completeness and applicability of evidence
The results of this review are applicable to patients with moderate to severe Crohn's disease and patients with moderate to severe Crohn's disease who have failed TNF‐alpha antagonists. High quality evidence suggests that ustekinumab is effective for induction of clinical remission and improvement in patients with moderate to severe Crohn's disease. Ustekinumab offers an out‐of‐class option for patients who fail currently available TNF‐α antagonists.
Quality of the evidence
All of the included studies were judged to be at low risk of bias. GRADE analyses indicated that the overall quality of the evidence supporting the primary and secondary outcomes from the briakinumab studies was moderate to low due mostly to sparse data. GRADE analyses indicated that the overall quality of evidence supporting the primary outcome from the ustekinumab studies was rated as high quality. The secondary outcome clinical response was rated as high quality. Moderate to high quality evidence suggests that 6.0 mg/kg is the optimal dose of ustekinumab for induction of remission and clinical improvement. The outcome adverse events was rated as high quality. The outcome serious adverse events was rated as moderate quality. The outcome withdrawal due to adverse events was rated as low quality due to very sparse data.
Potential biases in the review process
To reduce potential bias in the review process we performed a comprehensive literature search to identify all eligible studies. In addition, two review authors independently assessed studies for inclusion, extracted data and assessed study quality. There are several limitations to this review. The studies that investigated briakinumab were small, thus they were only able to detect large effects and frequent adverse events.
Agreements and disagreements with other studies or reviews
The results of our review agree with other published reviews on ustekinumab and briakinumab (Khanna 2013; Niederreiter 2013; Singh 2014). Our review is the only systematic review to include a pooled analysis for the ustekinumab studies.
Authors' conclusions
Implications for practice.
High quality evidence suggests that ustekinumab is effective for induction of clinical remission and improvement in patients with moderate to severe Crohn's disease. Moderate to high quality evidence suggests that the optimal dosage of ustekinumab is 6 mg/kg. Ustekinumab appears to be safe. Moderate quality evidence suggests no increased risk of serious adverse events in patients receiving ustekinumab. Production of briakinumab was stopped by the manufacturer because the primary endpoint for remission was not reached.
Implications for research.
Further studies are required to determine the long‐term efficacy and safety of ustekinumab in patients with moderate to severe CD.
What's new
| Date | Event | Description |
|---|---|---|
| 12 September 2016 | New citation required and conclusions have changed | Updated review with one new author. Conclusions changed. |
| 12 September 2016 | New search has been performed | New literature search performed on 12 September 2016. Two new studies were added. |
Acknowledgements
Partial funding for the Cochrane IBD Group (April 1, 2016 ‐ March 31, 2018) has been provided by Crohn's and Colitis Canada (CCC).
Appendices
Appendix 1. Search strategies
PubMed
8 Search (#6 AND #7)
7 Search (singl* OR doubl* OR tripl* OR trebl* OR blind* OR mask* OR placebo* OR single‐blind* OR double‐blind* OR triple‐blind* OR random* OR controlled clinical)
6 Search (#1 AND #5)
5 Search (#2 OR #3 OR #4)
4 Search anti‐IL‐12/23p40
3 Search ((IL‐12 OR interleukin 12 OR IL‐23 OR interleukin 23 OR p40) AND (antibod* OR anti‐IL*))
2 Search (abt‐874 OR ustekinumab OR "cnto 1275" OR briakinumab)
1 Search crohn* OR IBD OR "inflammatory bowel disease*"
EMBASE
1 random$.tw.
2 factorial$.tw.
3 (crossover$ or cross over$ or cross‐over$).tw.
4 placebo$.tw.
5 single blind.mp.
6 double blind.mp.
7 triple blind.mp.
8 (singl$ adj blind$).tw.
9 (double$ adj blind$).tw.
10 (tripl$ adj blind$).tw.
11 assign$.tw.
12 allocat$.tw.
13 crossover procedure/
14 double blind procedure/
15 single blind procedure/
16 triple blind procedure/
17 randomized controlled trial/
18 or/1‐17
19 (exp animal/ or animal.hw. or nonhuman/) not (exp human/ or human cell/ or (human or humans).ti.)
20 18 not 19
21 exp Crohn disease/ or crohn*.mp. or exp colon Crohn disease/
22 (inflammatory bowel disease* or IBD).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword]
23 21 or 22
24 ustekinumab.mp. or exp ustekinumab/
25 briakinumab.mp. or exp briakinumab/
26 (abt‐874 or "cnto 1275").mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword]
27 24 or 25 or 26
28 "interleukin 12".mp. or exp interleukin 12/
29 "interleukin 23".mp. or exp interleukin 23/
30 (IL‐12 or IL‐23 or p40).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword]
31 28 or 29 or 30
32 exp monoclonal antibody/ or exp antibody/ or antibod*.mp.
33 31 and 32
34 anti‐IL‐12 23p40.mp.
35 27 or 33 or 34
36 20 and 23 and 35
MEDLINE
1 random$.tw.
2 factorial$.tw.
3 (crossover$ or cross over$ or cross‐over$).tw.
4 placebo$.tw.
5 placebo$.tw.
6 double blind.mp.
7 triple blind.mp.
8 (singl$ adj blind$).tw.
9 (double$ adj blind$).tw.
10 (tripl$ adj blind$).tw.
11 assign$.tw.
12 allocat$.tw.
13 crossover procedure/
14 double blind procedure/
15 single blind procedure/
16 triple blind procedure/
17 randomized controlled trial/
18 or/1‐17
19 (exp animal/ or animal.hw. or nonhuman/) not (exp human/ or human cell/ or (human or humans).ti.)
20 18 not 19
21 exp Crohn Disease Activity Index/ or exp Crohn disease/ or crohn*.mp.
22 ("inflammatory bowel disease*" or IBD).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier]
23 21 or 22
24 (ustekinumab or briakinumab or "CNTO 1275" or ABT‐874).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier]
25 interleukin 12.mp. or exp Interleukin‐12/
26 interleukin 23.mp. or exp Interleukin‐23/
27 (IL‐12 or IL‐23 or p40).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier]
28 25 or 26 or 27
29 antibod*.mp. or exp Antibodies/ or exp Antibodies, Monoclonal/
30 28 and 29
31 IL‐12 23p40.mp.
32 24 or 30 or 31
33 20 and 23 and 32
Cochrane Library (CENTRAL)
1 ustekinumab or briakinumab or ABT‐874 or CNTO 1275
2 interleukin 12 or interleukin 23 or IL‐12 or il‐23 or p40
3 antibod*
4 #2 and #3 442
5 anti‐il‐12/23p40
6 #1 or #4 or #5
7 #6 and (Crohn* or IBD or "inflammatory bowel disease*")
Electronic databases of abstracts from Digestive Disease Week (DDW) (1994 to 2012)
(interleukin 12, interleukin‐12, IL‐12, interleukin 23, interleukin‐23, IL‐23, p40, ustekinumab, CNTO 1275, briakinumab and ABT‐874) were all searched with the search term: crohn*
Data and analyses
Comparison 1. Briakinumab versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to induce clinical remission (7 & 9 weeks) | 1 | 79 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.65, 1.14] |
| 1.1 1 mg/kg body weight each week | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.67, 1.26] |
| 1.2 3 mg/kg body weight each week | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.48, 1.30] |
| 2 Failure to Induce clinical remission (6 weeks) | 1 | 230 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.83, 1.03] |
| 2.1 IV Infusion of 400 mg | 1 | 68 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.80, 1.13] |
| 2.2 IV Infusion of 700 mg | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.78, 1.05] |
| 3 Failure to induce clinical response (>= 100 points; 7 & 9 weeks) | 1 | 79 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.42, 0.99] |
| 3.1 1 mg/kg body weight each week | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.44, 1.22] |
| 3.2 3 mg/kg body weight each week | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.27, 1.13] |
| 4 Failure to induce clinical response (>= 100 points; 6 weeks) | 1 | 230 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.67, 0.99] |
| 4.1 400 mg iv of briakinumab | 1 | 68 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.61, 1.12] |
| 4.2 700 mg iv of briakinumab | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.63, 1.04] |
| 5 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6 Serious adverse events | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7 Withdrawals because of adverse events | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
1.1. Analysis.
Comparison 1 Briakinumab versus placebo, Outcome 1 Failure to induce clinical remission (7 & 9 weeks).
1.2. Analysis.
Comparison 1 Briakinumab versus placebo, Outcome 2 Failure to Induce clinical remission (6 weeks).
1.3. Analysis.
Comparison 1 Briakinumab versus placebo, Outcome 3 Failure to induce clinical response (>= 100 points; 7 & 9 weeks).
1.4. Analysis.
Comparison 1 Briakinumab versus placebo, Outcome 4 Failure to induce clinical response (>= 100 points; 6 weeks).
1.5. Analysis.
Comparison 1 Briakinumab versus placebo, Outcome 5 Adverse events.
1.6. Analysis.
Comparison 1 Briakinumab versus placebo, Outcome 6 Serious adverse events.
1.7. Analysis.
Comparison 1 Briakinumab versus placebo, Outcome 7 Withdrawals because of adverse events.
Comparison 2. Ustekinumab versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to induce clinical remission (6 weeks) | 4 | 1947 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.86, 0.95] |
| 1.1 1.0 mg/kg IV | 1 | 175 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.82, 1.04] |
| 1.2 3.0 mg/kg IV | 1 | 176 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.83, 1.08] |
| 1.3 4.5 mg/kg IV | 1 | 53 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.67, 1.11] |
| 1.4 6.0 mg/kg IV | 3 | 1543 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.82, 0.98] |
| 2 Failure to induce clinical remission (6 weeks; sensitivity analysis) | 3 | 1320 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.88, 0.96] |
| 2.1 1.0 mg/kg IV | 1 | 175 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.82, 1.04] |
| 2.2 3.0 mg/kg IV | 1 | 176 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.83, 1.08] |
| 2.3 4.5 mg/kg IV | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.67, 1.11] |
| 2.4 6.0 mg/kg IV | 2 | 916 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.87, 0.97] |
| 3 Failure to induce clinical response (>= 70 points; 6 weeks) | 4 | 1947 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.66, 0.81] |
| 3.1 1 mg/kg IV | 1 | 175 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.61, 0.98] |
| 3.2 3 mg/kg IV | 1 | 176 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.63, 1.03] |
| 3.3 4.5 mg/kg IV | 1 | 53 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.37, 0.94] |
| 3.4 6 mg/kg IV | 3 | 1543 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.61, 0.85] |
| 4 Failure to induce clinical response (>= 70 points; 6 weeks; sensitivity analysis) | 3 | 1320 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.71, 0.85] |
| 4.1 1 mg/kg IV | 1 | 175 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.61, 0.98] |
| 4.2 3 mg/kg IV | 1 | 176 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.63, 1.03] |
| 4.3 4.5 mg/kg IV | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.37, 0.94] |
| 4.4 6 mg/kg IV | 2 | 916 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.71, 0.87] |
| 5 Failure to Induce clinical response (>=100 points; 6 weeks) | 4 | 1947 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.69, 0.87] |
| 5.1 1.0 mg/kg IV | 1 | 175 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.67, 1.01] |
| 5.2 3.0 mg/kg IV | 1 | 176 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.70, 1.04] |
| 5.3 4.5 mg/kg IV | 1 | 53 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.39, 0.89] |
| 5.4 6.0 mg/kg IV | 3 | 1543 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.63, 0.91] |
| 6 Failure to Induce clinical response (>=100 points; 6 weeks; sensitivity analysis) | 3 | 1320 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.77, 0.88] |
| 6.1 1.0 mg/kg IV | 1 | 175 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.67, 1.01] |
| 6.2 3.0 mg/kg IV | 1 | 176 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.70, 1.04] |
| 6.3 4.5 mg/kg IV | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.39, 0.89] |
| 6.4 6.0 mg/kg IV | 2 | 916 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.77, 0.91] |
| 7 Failure to induce clinical remission ‐ 90 mg, s.c. (6 weeks) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8 Failure to induce clinical response 90 mg s.c. (>= 70 points; 6 weeks) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9 Failure to induce clinical response 90 mg s.c. (>=100 points; 6 weeks) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 10 Adverse events | 4 | 2023 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.90, 1.04] |
| 11 Serious adverse events | 4 | 2023 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.58, 1.20] |
| 12 Withdrawals because of adverse events | 2 | 657 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.18, 1.05] |
2.1. Analysis.
Comparison 2 Ustekinumab versus placebo, Outcome 1 Failure to induce clinical remission (6 weeks).
2.2. Analysis.
Comparison 2 Ustekinumab versus placebo, Outcome 2 Failure to induce clinical remission (6 weeks; sensitivity analysis).
2.3. Analysis.
Comparison 2 Ustekinumab versus placebo, Outcome 3 Failure to induce clinical response (>= 70 points; 6 weeks).
2.4. Analysis.
Comparison 2 Ustekinumab versus placebo, Outcome 4 Failure to induce clinical response (>= 70 points; 6 weeks; sensitivity analysis).
2.5. Analysis.
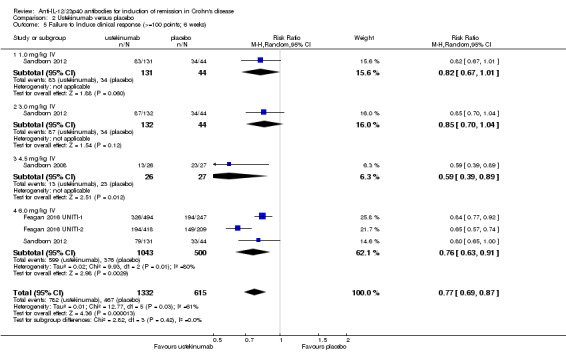
Comparison 2 Ustekinumab versus placebo, Outcome 5 Failure to Induce clinical response (>=100 points; 6 weeks).
2.6. Analysis.
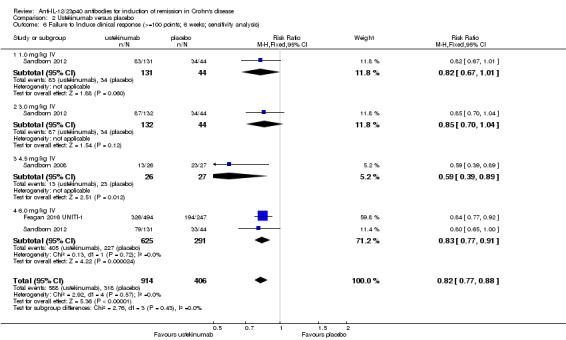
Comparison 2 Ustekinumab versus placebo, Outcome 6 Failure to Induce clinical response (>=100 points; 6 weeks; sensitivity analysis).
2.7. Analysis.
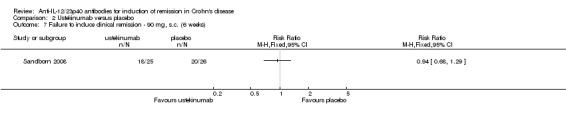
Comparison 2 Ustekinumab versus placebo, Outcome 7 Failure to induce clinical remission ‐ 90 mg, s.c. (6 weeks).
2.8. Analysis.
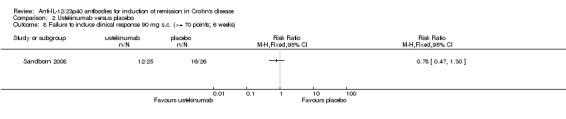
Comparison 2 Ustekinumab versus placebo, Outcome 8 Failure to induce clinical response 90 mg s.c. (>= 70 points; 6 weeks).
2.9. Analysis.
Comparison 2 Ustekinumab versus placebo, Outcome 9 Failure to induce clinical response 90 mg s.c. (>=100 points; 6 weeks).
2.10. Analysis.
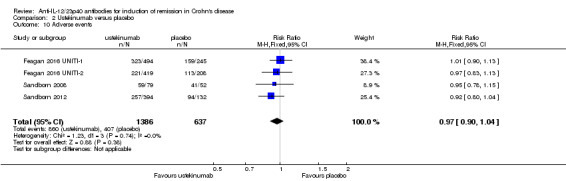
Comparison 2 Ustekinumab versus placebo, Outcome 10 Adverse events.
2.11. Analysis.
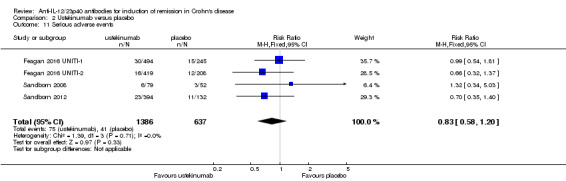
Comparison 2 Ustekinumab versus placebo, Outcome 11 Serious adverse events.
2.12. Analysis.
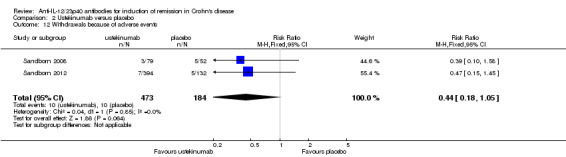
Comparison 2 Ustekinumab versus placebo, Outcome 12 Withdrawals because of adverse events.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Feagan 2016 UNITI‐1.
| Methods | Randomized, double‐blind, placebo‐controlled, multicenter trial | |
| Participants | Adult patients (> 18 years) with moderate to severe Crohn's disease who have failed TNF‐alpha antagonist therapy (N = 741) Patients must have received TNF‐alpha antagonists at approved doses and had documented primary non‐response, secondary non‐response or intolerance criteria |
|
| Interventions | Group 1: placebo; i.v. (n = 245) Group 2: 130 mg of ustekinumab, i.v. (n = 246) Group 3: 6 mg/kg of ustekinumab; i.v. (n = 249) |
|
| Outcomes | Primary outcome: Clinical response at week 6 (the number of patients in clinical response, as measured by a reduction in CDAI of > 100 points from baseline or CDAI < 150 at week 6) The primary outcome for the maintenance study was clinical remission at week 44 Secondary outcomes: 1. Clinical remission at week 8 (the number of patients in clinical remission, defined by CDAI < 150) 2. Clinical response at week 8 3. > 70 point CDAI decrease at weeks 3 and 6 4. Adverse events 5. Serious adverse events 6. Quality of Life 7. CRP |
|
| Duration of follow‐up | Induction phase: 8 weeks (20 weeks for patients not entering maintenance phase) Maintenance phase: 44 weeks |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated |
| Allocation concealment (selection bias) | Low risk | Centralized randomization using permuted blocks |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind Identical placebo |
| Incomplete outcome data (attrition bias) clinical relapse / remission | Low risk | Drop‐outs were balanced across interventions with similar reasons for withdrawal |
| Incomplete outcome data (attrition bias) adverse events | Low risk | Drop‐outs were balanced across interventions with similar reasons for withdrawal |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Feagan 2016 UNITI‐2.
| Methods | Randomized, double‐blind, placebo‐controlled, multicenter trial | |
| Participants | Adult patients (> 18 years) with moderate to severe Crohn's disease who have failed corticosteroids or immunosuppressants (azathioprine, 6‐mercaptopurine or methotrexate) or both (N = 628) Patients could have received TNF‐alpha antagonists without demonstration of inadequate response or intolerance |
|
| Interventions | Group 1: Placebo; i.v. 9 (n = 210) Group 2: 130 mg of Ustekinumab; i.v. (n = 209) Group 3: 6 mg/kg of body weight of Ustekinumab; i.v. (n = 209) |
|
| Outcomes | Primary outcome: Clinical response at week 6
Secondary outcomes: 1. Clinical remission at week 8 2. Clinical response through week 8 3. > 70 point CDAI decrease at weeks 3 and 6 4. Adverse events 5. Serious adverse events 6. Quality of Life 7. CRP |
|
| Duration of follow‐up | Induction phase: 8 weeks (20 weeks for patients not entering maintenance phase) Maintenance phase: 44 weeks |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated |
| Allocation concealment (selection bias) | Low risk | Centralized randomization using permuted blocks |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind Identical placebo |
| Incomplete outcome data (attrition bias) clinical relapse / remission | Low risk | Drop‐outs were balanced across interventions with similar reasons for withdrawal |
| Incomplete outcome data (attrition bias) adverse events | Low risk | Drop‐outs were balanced across interventions with similar reasons for withdrawal |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Mannon 2004.
| Methods | Randomized, double‐blind, placebo‐controlled, multi‐center trial in the USA, Germany, and the Netherlands Patients were randomized 1:2:2 to placebo or one of the two dosages of briakinumab |
|
| Participants | Participants with clinically active Crohn's disease (N = 79) Inclusion criteria: CDAI 220‐450, age ≥18 years Exclusion criteria: recently started Crohn's related medication, infections, history of malignancies, moderate to severe asthma, pregnancy/lactation, intestinal obstruction, stricture, ostomy, short bowel syndrome or probable operation in the near future |
|
| Interventions | Briakinumab was given at two different dosages: 1 mg/kg body weight subcutaneously (n = 31) or 3 mg/kg body weight subcutaneously (n = 32) Patients were enrolled into two cohorts with different dosing regimens (week 0, 4, 5, 6, 7, 8, 9 or week 0, 1, 2, 3, 4, 5, 6) with the first forty patients being enrolled in the former dosing regimen Each dosing cohort included a placebo group with 8 patients |
|
| Outcomes | Primary outcome: adverse events Secondary outcomes: clinical response (CDAI decrease ≥ 100), clinical remission (CDAI < 150), anti‐drug antibodies, histologic response (subgroup, modified D'Haens score, only partially reported), cytokine secretion by lamina propria mononuclear cells (subgroup, only partially reported) Endoscopic relapse / remission and quality of life were not assessed |
|
| Duration of follow‐up | ˜27 weeks after last injection (i.e. 5 to 6 months after randomisation) | |
| Notes | For the purpose of this review the two different dosing regimens were combined for each dosage of briakinumab Different dosages were assessed separately The respective control groups were split evenly |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated randomization (as per investigator) |
| Allocation concealment (selection bias) | Low risk | Telephone interactive voice response system for treatment allocation (as per investigator) |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Blinding of patients and investigators: The placebo was the same isotonic solution as the antibody |
| Incomplete outcome data (attrition bias) clinical relapse / remission | Low risk | Last observation carried forward for incomplete data Drop‐outs were balanced across interventions with similar reasons for withdrawal |
| Incomplete outcome data (attrition bias) adverse events | Low risk | Participants who discontinued the study were included in the safety analysis unless lost to follow‐up Drop‐outs were balanced across interventions with similar reasons for withdrawal |
| Selective reporting (reporting bias) | Unclear risk | Pre‐specified primary and secondary outcomes (safety and efficacy) were reported Histology and laboratory scores were only reported for the treatment group |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Panaccione 2015.
| Methods | Double‐blind, randomized, placebo‐controlled trial | |
| Participants | Inclusion criteria: Adult patients with a diagnosis of CD for > 4 months, and a Crohn’s Disease Activity Index (CDAI) score ≥ 220 and ≤ 450 (N = 246) Previous exposure to approved anti‐TNF agents (including adalimumab, certolizumab, etanercept, infiximab, certain investigational drugs, and tumor necrosis factor receptor [IgG1]) was permitted if discontinued at least 8 weeks prior to baseline Secondary non responders and primary non‐responders to prior anti‐TNF agents were eligible Patients were allowed to continue azathioprine, 6‐mercaptopurine (6‐MP), or methotrexate (MTX) provided they had received these medications for at least 12 weeks with stable doses for at least 4 weeks prior to entry Corticosteroids were permitted provided doses were stable (e.g. prednisolone ≤ 40 mg/day or equivalent, or budesonide ≤ 9 mg/day) for at least 2 weeks prior to entry |
|
| Interventions | I.V. infusions of briakinumab (200 mg, n = 16; 400 mg, n = 45; 700 mg, n = 139) or placebo (n = 46) administered at weeks 0, 4, and 8 and stratified at baseline (week 0) by prior TNF antagonist use (TNF antagonist naive vs TNF antagonist experienced) and TNF antagonist response (primary non‐response vs secondary loss of response or secondary non responders) | |
| Outcomes | Primary outcome: clinical remission (CDAI < 150) at week 6 Secondary outcomes: clinical remission at week 12, clinical response (CDAI decrease > 100) at week 6 and 12; IBDQ |
|
| Duration of follow‐up | Induction: 12 weeks Maintenance: 20 weeks (not reported in review) |
|
| Notes | 200 mg group was removed from efficacy analysis due to poor enrolment (included in safety analysis) Study was stopped by sponsor during the open‐label phase due to poor induction of remission results |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomization |
| Allocation concealment (selection bias) | Low risk | Centralized allocation |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind "The study sponsor, site personnel, and patients were unaware of the treatment assignments throughout both the induction and maintenance phases" |
| Incomplete outcome data (attrition bias) clinical relapse / remission | Low risk | Drop‐outs were balanced across interventions with similar reasons for withdrawal |
| Incomplete outcome data (attrition bias) adverse events | Low risk | Drop‐outs were balanced across interventions with similar reasons for withdrawal |
| Selective reporting (reporting bias) | Low risk | Expected outcomes were reported |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Sandborn 2008.
| Methods | Randomized, double‐blind, placebo‐controlled multi‐center trial with a cross‐over design in the USA Patients were randomized 1:1:1:1 to an intravenous or subcutaneous arm, each with a placebo group Within each arm patients crossed over to the treatment or control group after eight weeks |
|
| Participants | Participants with clinically active Crohn's disease (N = 104) Inclusion criteria: CDAI 220‐450, age ≥ 18 years. Patients had received at least one of the following in the past: 5‐ASA, antibiotics, corticosteroids, azathioprine, 6‐mercaptopurine, methotrexate, submaximal infliximab doses or regimens, or other anti‐TNF‐α‐agents Exclusion criteria: > 20 mg prednisolone, recent treatment with any investigational agent or an anti‐TNF‐α‐agents, infections, cancer, short‐bowel syndrome, ostomy, obstructive symptoms with strictures |
|
| Interventions | Ustekinumab was given at 90 mg s.c. per week over four weeks (n = 25) or 4.5 mg/kg body weight i.v. once (n = 26), each compared to s.c. placebo (n = 26) or i.v. placebo (n = 27) | |
| Outcomes | Primary outcome: Clinical response at week 8 (CDAI decrease ≥ 75 and ≥ 25%) Secondary outcomes: Clinical remission (CDAI < 150), laboratory results including CRP value, adverse events, anti‐drug antibodies, adherence to therapy. Endoscopic response / remission and quality of life were not assessed. |
|
| Duration of follow‐up | 8 weeks until cross‐over and 20 weeks thereafter | |
| Notes | Only the first part of the cross‐over design was evaluated for this review The placebo controlled study was accompanied by an unblinded study with participants who were non‐responsive to infliximab The unblinded sub‐study was not included in this review since it was not a placebo‐controlled or active comparator study |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated adaptive randomization stratified by investigative site |
| Allocation concealment (selection bias) | Low risk | Centralized randomization scheme |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Blinding of patients and investigators Identical placebo |
| Incomplete outcome data (attrition bias) clinical relapse / remission | Low risk | Intention‐to‐treat analysis with worst case assumption Drop‐outs were balanced across interventions with similar reasons for withdrawal |
| Incomplete outcome data (attrition bias) adverse events | Low risk | Drop‐outs were balanced across interventions with similar reasons for withdrawal |
| Selective reporting (reporting bias) | Low risk | All assessed outcomes were reported |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Sandborn 2012.
| Methods | Randomzed double‐blind, placebo‐controlled trial | |
| Participants | Inclusion criteria: Adults (> 18 years) at least a 3 month history of Crohn’s disease with a CDAI of 220 to 450 (N = 526) Patients had moderate to severe Crohn's disease that was resistant to TNF‐α antagonists Exclusion: Previous therapy specifically targeting interleukin‐12 or interleukin‐23 |
|
| Interventions | Intravenous ustekinumab 1 mg/kg (n = 131), 3 mg/kg (n=132), 6 mg/kg (n = 131) or placebo (n = 132) | |
| Outcomes | The primary outcome: clinical response (≥ 100 point decrease in CDAI) at week 6 Secondary outcomes: clinical remission (CDAI score, < 150 points) at week 6, clinical response at week 4, and clinical remission at week 22 among patients with a response to ustekinumab at week 6 |
|
| Duration of follow‐up | Induction phase: 8 weeks Maintenance phase: 36 weeks (not included) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated adaptive randomization stratified by investigative site and the initial response to a TNFalpha antagonist |
| Allocation concealment (selection bias) | Low risk | Centrally generated randomization scheme |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blind Identical placebo |
| Incomplete outcome data (attrition bias) clinical relapse / remission | Low risk | Intention‐to‐treat analysis and all subjects who began the study are accounted for in the results Drop‐outs were balanced across interventions with similar reasons for withdrawal |
| Incomplete outcome data (attrition bias) adverse events | Low risk | Drop‐outs were balanced across interventions with similar reasons for withdrawal |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | The study appears to be free of other sources of bias |
CDAI = Crohn's disease activity index
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Fasanmade 2008 | This pharmacokinetic study compared intravenous ustekinumab at a dose of 4.5 mg/kg to subcutaneously administered ustekinumab (90 mg) |
| Sands 2010 | Oral study drug (different route of administration) and drug is not a monoclonal antibody (different mechanism of action) |
Characteristics of ongoing studies [ordered by study ID]
NCT01369329.
| Trial name or title | A phase 3, randomized, double‐blind, placebo‐controlled, parallel‐group, multicenter study to evaluate the safety and efficacy of ustekinumab induction therapy in subjects with moderately to severely active Crohn's disease who have failed or are intolerant to TNF antagonist therapy (UNITI‐1) |
| Methods | Double‐blind, randomized, placebo‐controlled study; single i.v infusion of placebo or ustekinumab; safety and efficacy assessed through 8 weeks in subjects that had previously failed TNFa therapy |
| Participants | ˜ 703 subjects Inclusion Criteria:
|
| Interventions | Group 1: placebo; i.v. Group 2: 130 mg of ustekinumab, i.v. Group 3: 6 mg/kg of ustekinumab; i.v. |
| Outcomes | Primary outcome: Clinical response at week 6 (the number of patients in clinical response, as measured by reduction in CDAI from baseline)
Secondary outcomes: 1. Clinical remission at week 8 (the number of patients in clinical remission, defined by CDAI) 2. Clinical response through week 8 (the number of patients experiencing clinical response as measured by reduction in CDAI from baseline) |
| Starting date | July 2011 |
| Contact information | Janssen Research & Development, LLC |
| Notes | This study has been completed There are no published results as of 31 March 2015 |
NCT01369342.
| Trial name or title | A phase 3, randomized, double‐blind, placebo‐controlled, parallel‐group, multicenter study to evaluate the safety and efficacy of ustekinumab induction therapy in subjects with moderately to severely active Crohn's disease (UNITI‐2) |
| Methods | Double‐blind, randomized, placebo‐controlled study; single injection of ustekinumab or placebo; safety and efficacy assessed over an 8 week period |
| Participants | ˜ 612 subjects Inclusion Criteria:
|
| Interventions |
|
| Outcomes | Primary outcome: Clinical response at week 6
Secondary outcomes: 1. Clinical remission at week 8 2. Clinical response through week 8 |
| Starting date | July 2011 |
| Contact information | Janssen Research & Development, LLC |
| Notes | This study has been completed There are no published results as of 31 March 2015 |
NCT01369355.
| Trial name or title | A phase 3, randomized, double‐blind, placebo‐controlled, parallel‐group, multicenter study to evaluate the safety and efficacy of ustekinumab maintenance therapy in subjects with moderately to severely active Crohn's disease |
| Methods | Double‐blind, randomized, placebo‐controlled, parallel‐group design to assess whether additional subcutaneous (SC) ustekinumab treatment is beneficial in patients showing a clinical response to intravenous (IV) ustekinumab in the initial 2 induction studies |
| Participants | Estimated Enrolment is 1310 subjects Inclusion Criteria: Patients who received study agent at the start of study CNTO1275CRD3001 or CNTO1275CRD3002 and completed the Week 8 visit |
| Interventions | Group 1: Patients in response to IV ustekinumab will be randomized to receive either placebo, Group 2: Ustekinumab 90 mg SC every 12 weeks Group 3: Ustekinumab 90 mg SC every 8 weeks If patients in Groups 1 or 2 lose response, they will cross over to receive ustekinumab 90 mg every 8 weeks Other populations (nonresponders to prior IV ustekinumab or IV placebo) will receive ustekinumab at week 0 (either 90 mg SC or 130 mg IV, respectively) and continue SC ustekinumab if in response at week 8 |
| Outcomes | Primary outcome: Clinical remission at week 44 Secondary outcomes: 1. Remission in patient subgroups such as those in remission at Week 0 or those who previously failed TNF‐antagonists at week 44 2. Clinical response at week 44 3. Corticosteroid‐free remission at week 44 |
| Starting date | September 2011 |
| Contact information | |
| Notes | The estimated completion date is September 2018 |
Differences between protocol and review
We would like to acknowledge some differences between the protocol and review:
Primary and secondary outcomes: The primary and secondary outcomes were not well defined in the protocol. The primary outcome should have been defined as the proportion of patients who failed to enter clinical remission as defined by the included studies. The secondary efficacy outcomes should have been defined in a similar manner. The secondary outcome 'adverse events' was added after the protocol was published.
GRADE was added to the methods section. Please see MECIR C76.
We added a section on 'Unit of analysis issues' to the Methods section to explain how we would deal with these issues. This was not predefined in the protocol.
We added a section on 'Assessment of reporting biases' to the Methods section to explain how we would deal with issue for future updates of this review.This was not predefined in the protocol.
Contributions of authors
All authors contributed to planning the study, identification of relevant studies, assessment of methodological quality, data extraction, and manuscript preparation.
Sources of support
Internal sources
Charité ‐ Universitätsmedizin Berlin, Germany.
External sources
No sources of support supplied
Declarations of interest
John MacDonald: None known.
Tran Nguyen: None known
Reena Khanna has received honoraria from AbbVie, Jansen, Pfizer, Shire, and Takeda for consultancy. All of these activities are outside the submitted work.
Antje Timmer received grants (paid to institution) from Sanofi‐Aventis, Bayer, Takeda, Celgene, and Novartis for pharmacoepidemiological studies; and payment for lectures from The Falk Foundation, and MSD Sharp. All of these activities are outside the submitted work.
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Feagan 2016 UNITI‐1 {published data only}
- Adedokun OJ, Xu Z, Gasink C, Szapary P, Johanns J, Gao LL, et al. Pharmacokinetics and exposure‐response relationships of ustekinumab during IV induction and SC maintenance treatment of patients with Crohn's disease with ustekinumab: Results from the UNITI‐1, UNITI‐2, and IM‐UNITI studies. Gastroenterology 2016;150(4 Suppl 1):S408. [Google Scholar]
- Feagan BG, Sandborn WJ, Gasink C, Jacobstein D, Lang Y, Friedman JR, et al. Ustekinumab as induction and maintenance therapy for Crohn's disease. New England Journal of Medicine 2016. [DOI] [PubMed] [Google Scholar]
- Li K, Hayden K, Wadman E, Bhagat S, Emrich S, Friedman J, et al. Molecular response to ustekinumab in moderate‐to‐severe Crohn's disease by serum protein analysis: Results from uniti‐1 induction, uniti‐2 induction, and imuniti maintenance studies. Gastroenterology 2016;150(4 Suppl 1):S377. [Google Scholar]
- Sandborn W, Gasink C, Blank M, Lang Y, Johanns J, Gao LL, et al. A multicenter, double‐blind, placebo‐controlled phase 3 study of ustekinumab, a human IL‐12/23P40 mAB, in moderate to severe Crohn's disease refractory to anti‐TFNalpha: UNITI‐1. Inflammatory Bowel Diseases 2016;22(Suppl 1):S1. [Google Scholar]
- Sandborn W, Gasink C, Jacobstein D, Gao L, Johanns J, Targan S, et al. Assessment of serum C‐reactive protein, fecal lactoferrin, and fecal calprotectin in patients with moderate‐severely active Crohns disease: Results from the IM UNITI maintenance study. Gastroenterology 2016;150(4 Suppl 1):S982. [Google Scholar]
- Sands BE, Han C, Gasink C, Szapary P, Gao LL, Lang Y, et al. Ustekinumab improves general health status and disease‐specific health related quality of life of patients with moderate to severe Crohn's disease: Results from the uniti and IMUNITI phase 3 clinical trials. Gastroenterology 150(4 Suppl 1):S1004. [Google Scholar]
Feagan 2016 UNITI‐2 {published data only}
- Feagan B, Gasink C, Lang Y, Friedman J, Johanns J, Gao L, et al. A multicenter, double‐blind, placebo‐controlled pH3 study of ustekinumab, a human monoclonal antibody to IL‐12/23p40, in patients with moderately‐severely active Crohn's disease who are not naive or not refractory to anti‐TNFa: UNITI‐2. Canadian Journal of Gastroenterology and Hepatology 2016;2016. [Google Scholar]
- Feagan B, Gasink C, Lang Y, Friedman JR, Johanns J, Gao LL, et al. A multicenter, double‐blind, placebo‐controlled pH3 study of ustekinumab, a human monoclonal antibody to IL‐12/23p40, in patients with moderately‐severely active Crohn's disease who are not naive or not refractory to anti‐TNFa: UNITI‐2. United European Gastroenterology Journal 2015;3(6):563‐4. [Google Scholar]
Mannon 2004 {published data only}
- Mannon P, Fuss I, Mayer L, Elson CO, Sandborn WJ, Dolin B, et al. Anti‐interleukin‐12 treats active Crohn's disease. Gastroenterology 2004;126(4 Suppl 2):A22‐3. [Google Scholar]
- Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, et al. Anti‐interleukin‐12 antibody for active Crohn's disease. New England Journal of Medicine 2004;351(20):2069‐79. [DOI] [PubMed] [Google Scholar]
Panaccione 2015 {published and unpublished data}
- Panaccione R, Sandborn W, Gordon G, Lee S, Safdi A, Sedghi S, et al. Briakinumab (anti‐interleukin 12/23p40, ABT874) for treatment of Crohn's disease. American Journal of Gastroenterology 2010;105(Supp 1):S457‐8. [Google Scholar]
- Panaccione R, Sandborn WJ, Gordon GL, Lee SD, Safdi A, Sedghi S, et al. Briakinumab for treatment of Crohn's disease: results of a randomized trial. Inflammatory Bowel Diseases 2015;21(6):1329‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sandborn 2008 {published data only (unpublished sought but not used)}
- Sandborn W, Feagan B, Fedorak R, Scherl E, Fleisher M, Katz S, et al. A multicenter, randomized, phase 2A study of human monoclonal antibody to IL‐ 12/23P40 (CNTO 1275) in patients with moderately to severely active Crohn’s disease. Inflammatory Bowel Diseases 2008;14(Supp 1):S10. [Google Scholar]
- Sandborn WJ, Feagan BG, Fedorak R, Scherl E, Fleisher M, Katz S, et al. A multicenter, randomized, phase 2a study of human monoclonal antibody to IL‐12/23p40 (CNTO 1275) in patients with moderately to severely active Crohn's disease. Gastroenterology 2007;132(4 Suppl 1):A51. [Google Scholar]
- Sandborn WJ, Feagan BG, Fedorak RN, Scherl E, Fleisher MR, Katz S, et al. A randomized trial of ustekinumab, a human interleukin‐12/23 monoclonal antibody, in patients with moderate‐to‐severe Crohn's disease. Gastroenterology 2008;135(4):1130‐41. [DOI] [PubMed] [Google Scholar]
- Toedter GP, Blank M, Lang Y, Chen D, Sandborn WJ, Villiers WJ. Relationship of C‐reactive protein with clinical response after therapy with ustekinumab in Crohn's disease. American Journal of Gastroenterology 2009;104(11):2768‐73. [DOI] [PubMed] [Google Scholar]
Sandborn 2012 {published data only}
- Ding T, Telesco S, Monast C, Brodmerkel C, Yatsunenko T, Das A, et al. [The gut microbiome differentiates clinical phenotypes in moderate to severe Crohn's disease: Results from the certifi study]. Canadian Journal of Gastroenterology and Hepatology 2016;2016. [Google Scholar]
- Ding T, Telesco S, Monast CS, Brodmerkel C, Yatsunenko T, Das A, et al. [The gut microbiome differentiates clinical phenotypes in moderate to severe Crohn's disease: Results from the certifi study]. Gastroenterology 2015;148(4 Suppl 1):S713. [Google Scholar]
- Ding T, Telesco S, Monast CS, Brodmerkel C, Yatsunenko T, Das A, et al. [The gut microbiome differentiates clinical phenotypes in moderate to severe Crohn's disease: Results from the CERTIFI study]. United European Gastroenterology Journal 2015;3(5 Suppl 1):A133‐4. [Google Scholar]
- Feagan B, Gasink C, Gao L, Blank M, Johanns J, Guzzo C, et al. A multicenter, randomized, double‐blind, placebo‐controlled phase 2B study of ustekinumab, a human monoclonal antibody to IL‐12/23P40, in patients with moderately to severely active Crohn's disease: Results through week 36 from the CERTIFI trial. American Journal of Gastroenterology 2011;106(Supp 2s):S463. [Google Scholar]
- Feagan B, Gasink C, Gao LL, Blank M, Johanns J, Guzzo C, et al. Health related quality of life results through week 22 from the CERTIFI study, a multicenter, randomized, double‐blind, placebo‐controlled Phase2b study of ustekinumab in patients with moderately to severely active Crohn's disease. Journal of Crohn's and Colitis 2012;6(Supp 1):S129‐30. [Google Scholar]
- Gasink C, Chan D, Gao LL, Schenkel B, Han C. Assessment of sleep impairment in patients with Crohn's disease: Results from the ustekinumab CERTIFI study. Gastroenterology 2013;144(5 Suppl 1):S231. [Google Scholar]
- Gasink C, Friedman J, Gao L, Chan D, Sandborn W, Feagan B. Evaluation of an interim Crohn's disease outcome measure (PRO‐2) based on 2 patient‐reported components (stool frequency, abdominal pain) of the Crohn's disease activity index (CDAI) in the ustekinumab CERTIFI study. American Journal of Gastroenterology 2014;109(Suppl 2s):S497. [Google Scholar]
- Sandborn WJ, Gasink C, Gao LL, Blank M, Johanns J, Guzzo C, et al. A multicenter, randomized, double‐blind, placebo‐controlled Phase2b study of ustekinumab, a human monoclonal antibody to IL‐12/23p40, in patients with moderately to severely active Crohn's disease: Results through week 22 from the CERTIFI trial. Gastroenterology 2011;140(5 Suppl 1):S109. [Google Scholar]
- Sandborn WJ, Gasink C, Gao LL, Blank MA, Johanns J, Guzzo C, et al. Ustekinumab induction and maintenance therapy in refractory Crohn's disease. New England Journal of Medicine 2012;367(16):1519‐28. [DOI] [PubMed] [Google Scholar]
- Sands BE, Gasink C, Gao LL, Blank MA, Johanns J, Guzzo C, et al. Health related quality of life results through week 22 from the CERTIFI study, a multicenter, randomized, double‐blind, placebo‐controlled phase2b study ofUstekinumab in patients with moderately to severely active Crohn's disease. Inflammatory Bowel Diseases 2011;17(Supp 2):S24. [Google Scholar]
- Toedter G, Wu X, Gao LL, Gasink C. Reductions in fecal calprotectin and lactoferrin following ustekinumab induction therapy in patients with moderate to severe Crohn's disease who have previously failed or been intolerant of TNF antagonist therapies. Gastroenterology 2011;140(5 Suppl 1):S264. [Google Scholar]
References to studies excluded from this review
Fasanmade 2008 {unpublished data only}
- Fasanmade AA, Adedokun OJ, Johanns JR, Zhou H, Davis HM, Blank, M. Pharmacokinetics and exposure‐response relationship of ustekinumab, a human monoclonal antibody to interleukin 12/23 in patients with moderate‐to‐severe Crohn's disease. Gastroenterology 2008;134(4 Suppl 1):A‐490. [Google Scholar]
Sands 2010 {published data only}
- Sands BE, Jacobson EW, Sylwestrowicz T, Younes Z, Dryden G, Fedorak R, et al. Randomized, double‐blind, placebo‐controlled trial of the oral interleukin‐12/23 inhibitor apilimod mesylate for treatment of active Crohn's disease. Inflammatory Bowel Diseases 2010;16(7):1209‐18. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
NCT01369329 {published data only}
- NCT01369329. A phase 3, randomized, double‐blind, placebo‐controlled, parallel‐group, multicenter study to evaluate the safety and efficacy of ustekinumab induction therapy in subjects with moderately to severely active Crohn's disease who have failed or are intolerant to TNF antagonist therapy (UNITI‐1). clinicaltrials.gov/ct2/show/NCT01369329 (accessed March 31, 2015).
NCT01369342 {published data only}
- NCT01369342. A phase 3, randomized, double‐blind, placebo‐controlled, parallel‐group, multicenter study to evaluate the safety and efficacy of ustekinumab induction therapy in subjects with moderately to severely active Crohn's disease (UNITI‐2). clinicaltrials.gov/ct2/show/NCT01369342 (accessed 31 March 2015).
NCT01369355 {published data only}
- NCT01369355. A phase 3, randomized, double‐blind, placebo‐controlled, parallel‐group, multicenter study to evaluate the safety and efficacy of ustekinumab maintenance therapy in subjects with moderately to severely active Crohn's disease. clinicaltrials.gov/ct2/show/NCT01369355 (accessed 31 March 2015).
Additional references
Akobeng 2003
- Akobeng AK, Zachos M. Tumor necrosis factor‐alpha antibody for induction of remission in Crohn's disease. Cochrane Database of Systematic Reviews 2003, Issue 4. [DOI: 10.1002/14651858.CD003574.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Benson 2011
- Benson JM, Sachs CW, Treacy G, Zhou H, Pendley CE, Brodmerkel CM, et al. Therapeutic targeting of the IL‐12/23 pathways: generation and characterization of ustekinumab. Nature Biotechnology 2011;29(7):615‐24. [DOI] [PubMed] [Google Scholar]
Burmester 2013
- Burmester GR, Panaccione R, Gordon KB, McIlraith MJ, Lacerda AP. Adalimumab: long‐term safety in 23 458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn's disease. Annals of the Rheumatic Diseases 2013;72(4):517‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chande 2013
- Chande N, Tsoulis DJ, MacDonald JK. Azathioprine or 6‐mercaptopurine for induction of remission in Crohn's disease. Cochrane Database of Systematic Reviews 2013, Issue 4. [DOI: 10.1002/14651858.CD000545.pub4] [DOI] [PubMed] [Google Scholar]
Cingoz 2011
- Cingoz, O. Ustekinumab. Monoclonal Antibodies 2011;1(3):216‐221. [Google Scholar]
Colombel 2004
- Colombel JF, Loftus EV Jr, Tremaine WJ, Egan LJ, Harmsen WS, Schleck CD, et al. The safety profile of infliximab in patients with Crohn's disease: the Mayo clinic experience in 500 patients. Gastroenterology 2004;126(1):19‐31. [DOI] [PubMed] [Google Scholar]
Colombel 2007
- Colombel JF, Sandborn WJ, Rutgeerts P, Enns R, Hanauer SB, Panaccione R, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn's disease: the CHARM trial. Gastroenterology 2007;132(1):52‐65. [DOI] [PubMed] [Google Scholar]
Colombel 2010
- Colombel JF, Sandborn WJ, Reinisch W, Mantzaris GJ, Kornbluth A, Rachmilewitz D, et al. Infliximab, azathioprine, or combination therapy for Crohn's disease. New England Journal of Medicine 2010;362(15):1383‐95. [DOI] [PubMed] [Google Scholar]
Danese 2011
- Danese S, Fiorino G, Reinisch W. Review article: Causative factors and the clinical management of patients with Crohn's disease who lose response to anti‐TNF‐α therapy. Alimentary Pharmacology and Therapeutics 2011;34(1):1‐10. [DOI] [PubMed] [Google Scholar]
Duvallet 2011
- Duvallet E, Semerano L, Assier E, Falgarone G, Boissier MC. Interleukin‐23: a key cytokine in inflammatory diseases. Annals of Medicine 2011;43(7):503‐11. [DOI] [PubMed] [Google Scholar]
Faubion 2001
- Faubion WA Jr, Loftus EV Jr, Harmsen WS, Zinsmeister AR, Sandborn WJ. The natural history of corticosteroid therapy for inflammatory bowel disease: a population‐based study. Gastroenterology 2001;121(2):255‐60. [PUBMED: 11487534] [DOI] [PubMed] [Google Scholar]
Ford 2011
- Ford AC, Bernstein CN, Khan KJ, Abreu MT, Marshall JK, Talley NJ, et al. Glucocorticosteroid therapy in inflammatory bowel disease: systematic review and meta‐analysis. American Journal of Gastroenterology 2011;106(4):590‐9. [DOI] [PubMed] [Google Scholar]
Fuss 1999
- Fuss IJ, Marth T, Neurath MF, Pearlstein GR, Jain A, Strober W. Anti‐interleukin 12 treatment regulates apoptosis of Th1 T cells in experimental colitis in mice. Gastroenterology 1999;117(5):1078‐88. [PUBMED: 10535870] [DOI] [PubMed] [Google Scholar]
Gottlieb 2009
- Gottlieb A, Menter A, Mendelsohn A, Shen YK, Li S, Guzzo C, et al. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double‐blind, placebo‐controlled, crossover trial. Lancet 2009;373(9664):633‐40. [PUBMED: 19217154] [DOI] [PubMed] [Google Scholar]
Gottlieb 2014
- Gottlieb AB, Kalb RE, Langley RG, Krueger GG, Jong EM, Guenther L, et al. Safety observations in 12095 patients with psoriasis enrolled in an international registry (PSOLAR): experience with infliximab and other systemic and biologic therapies. Journal of Drugs in Dermatology 2014;13(12):1441‐8. [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hanauer 2002
- Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, et al. Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet 2002;359(9317):1541‐9. [DOI] [PubMed] [Google Scholar]
Hanauer 2006
- Hanauer SB, Sandborn WJ, Rutgeerts P, Fedorak RN, Lukas M, MacIntosh D, et al. Human anti‐tumor necrosis factor monoclonal antibody (adalimumab) in Crohn's disease: the CLASSIC‐I trial. Gastroenterology 2006;130(2):323‐33. [DOI] [PubMed] [Google Scholar]
HIggins 2011a
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. [Google Scholar]
Higgins 2011b
- Higgins JPT, Deeks JJ, Altman DG (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. [Google Scholar]
Hudesman 2013
- Hudesman D, Lichtiger S, Sands B. Risk of extraintestinal solid cancer with anti‐TNF therapy in adults with inflammatory bowel disease: review of the literature. Inflammatory Bowel Diseases 2013;19(3):644‐649. [DOI] [PubMed] [Google Scholar]
Khanna 2013
- Khanna R, Feagan BG. Ustekinumab for the treatment of Crohn's disease. Immunotherapy 2013;5(8):803‐15. [DOI] [PubMed] [Google Scholar]
Lawson 2006
- Lawson MM, Thomas AG, Akobeng AK. Tumour necrosis factor alpha blocking agents for induction of remission in ulcerative colitis. Cochrane Database of Systematic Reviews 2006, Issue 3. [DOI: 10.1002/14651858.CD005112.pub2] [DOI] [PubMed] [Google Scholar]
Loke 2007
- Loke YK, Price D, Herxheimer A, Cochrane Adverse Effects Methods Group. Systematic reviews of adverse effects: framework for a structured approach. BMC Medical Research Methodology 2007;7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
McDonald 2012
- McDonald JWD, Tsoulis DJ, MacDonald JK, Feagan BG. Methotrexate for induction of remission in refractory Crohn's disease. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.CD003459.pub3] [DOI] [PubMed] [Google Scholar]
Modigliani 1990
- ModiglianiR, Mary JY, Simon JF, Cortot A, Soule JC, Gendre JP, et al. Clinical, biological, and endoscopic picture of attacks of Crohn's disease. Evolution on prednisolone. Groupe d'Etude Thérapeutique des Affections Inflammatoires Digestives. Gastroenterology 1990;98(4):811‐8. [DOI] [PubMed] [Google Scholar]
Molodecky 2012
- Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012;142(1):46‐54.e42; quiz e30. [PUBMED: 22001864] [DOI] [PubMed] [Google Scholar]
Neurath 1995
- Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. Journal of Experimental Medicine 1995;182(5):1281‐90. [PUBMED: 7595199] [DOI] [PMC free article] [PubMed] [Google Scholar]
Niederreiter 2013
- Niederreiter L, Adolph TE, Kaser A. Anti‐IL‐12/23 in Crohn's disease: bench and bedside. Current Drug Targets 2013;14(12):1379‐84. [DOI] [PubMed] [Google Scholar]
Papp 2008
- Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, et al. Efficacy and safety of ustekinumab, a human interleukin‐12/23 monoclonal antibody, in patients with psoriasis: 52‐week results from a randomised, double‐blind, placebo‐controlled trial (PHOENIX 2). Lancet 2008;371(9625):1675‐84. [PUBMED: 18486740] [DOI] [PubMed] [Google Scholar]
Papp 2013
- Papp KA, Griffiths CE, Gordon K, Lebwohl M, Szapary PO, Wasfi Y, et al. Long‐term safety of ustekinumab in patients with moderate‐to‐severe psoriasis: final results from 5 years of follow‐up.. British Journal of Dermatology 2013;168(4):844‐54. [DOI] [PubMed] [Google Scholar]
Peluso 2006
- Peluso I, Pallone F, Monteleone G. Interleukin‐12 and Th1 immune response in Crohn's disease: pathogenetic relevance and therapeutic implication. World Journal of Gastroenterology 2006;12(35):5606‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Raj 2010
- Raj LS, Hawthorne AB. Optimising use of thiopurines in inflammatory bowel disease. Frontline Gastroenterology 2010;1:44‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Reich 2011
- Reich K, Langley RG, Papp KA, Ortonne JP, Unnebrink K, Kaul M, et al. A 52‐week trial comparing briakinumab with methotrexate in patients with psoriasis. New England Journal of Medicine 2011;365(17):1586‐96. [DOI] [PubMed] [Google Scholar]
RevMan 5 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Rutgeerts 1994
- Rutgeerts P, Löfberg R, Malchow H, Lamers C, Olaison G, Jewell D, et al. A comparison of budesonide with prednisolone for active Crohn's disease. New England Journal of Medicine 1994;331(13):842‐5. [DOI] [PubMed] [Google Scholar]
Sandborn 2007
- Sandborn WJ, Feagan BG, Stoinov S, Honiball PJ, Rutgeerts P, Mason D, et al. Certolizumab pegol for the treatment of Crohn's disease. New England Journal of Medicine 2007;357(3):228‐38. [DOI] [PubMed] [Google Scholar]
Schreiber 2007
- Schreiber S, Khaliq‐Kareemi M, Lawrance IC, Thomsen OØ, Hanauer SB, McColm J, et al. Maintenance therapy with certolizumab pegol for Crohn's disease. New England Journal of Medicine 2007;357(3):239‐50. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. [Google Scholar]
Singh 2011
- Singh JA, Wells GA, Christensen, Ghogomu ET, Maxwell L, MacDonald JK, et al. Adverse effects of biologics: a network meta‐analysis and Cochrane overview. Cochrane database of systematic reviews 2011;2:CD008794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Singh 2014
- Singh S, Garg SK, Pardi DS, Wang Z, Murad MH, Loftus EV Jr. Comparative efficacy of biologic therapy in biologic‐naive patients with Crohn disease: a systematic review and network meta‐analysis. Mayo Clinic Proceedings 2014;89(12):1621‐35. [DOI] [PubMed] [Google Scholar]
Solberg 2007
- Solberg IC, Vatn MH, Hoie O, Stray N, Sauar J, Jahnsen J, et al. Clinical course in Crohn's disease: results of a Norwegian population‐based ten‐year follow‐up study. Clinical Gastroenterology and Hepatology 2007;5(12):1430‐8. [DOI] [PubMed] [Google Scholar]
Summers 1979
- Summers RW, Switz DM, Sessions JT Jr, Becktel JM, Best WR, Kern F Jr, et al. National Cooperative Crohn's Disease Study: results of drug treatment. Gastroenterology 1979;77(4 Part 2):847‐69. [PubMed] [Google Scholar]
Targan 1997
- Targan SR, Hanauer SB, Deventer SJ, Mayer L, Present DH, Braakman T, et al. A short‐term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group.. New England Journal of Medicine 1997;337(15):1029‐35. [DOI] [PubMed] [Google Scholar]
Watanabe 2004
- Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll‐like receptor 2‐mediated T helper type 1 responses. Nature Immunology 2004;5(8):800‐8. [DOI] [PubMed] [Google Scholar]
Yang 2002
- Yang YX, Lichtenstein GR. Corticosteroids in Crohn's disease. American Journal of Gastroenterology 2002;97(4):803‐23. [DOI] [PubMed] [Google Scholar]