

ABSTRACT
Trypanosoma (Herpetosoma) lewisi is a globally distributed rat trypanosome, currently considered as a zoonotic pathogen; however, a detailed understanding of the morphological events occurring during the cell cycle is lacking. This study aimed to investigate the cell cycle morphology and cleavage events of Trypanosoma lewisi (T. lewisi) during in vitro cultivation. By establishing in vitro cultivation of T. lewisi at 37°C, various cell morphologies and stages could be observed. We have provided a quantitative analysis of the morphological events during T. lewisi proliferation. We confirmed a generation time of 12.14 ± 0.79 hours, which is similar to that in vivo (12.21 ± 0.14 hours). We also found that there are two distinct cell cycles, with a two-way transformation connection in the developmental status of this parasite, which was contrasted with the previous model of multiple division patterns seen in T. lewisi. We quantified the timing of cell cycle phases (G1n, 0.56 U; Sn, 0.14 U; G2n, 0.16 U; M, 0.06 U; C, 0.08 U; G1k, 0.65 U; Sk, 0.10 U; G2k, 0.17 U; D, 0.03 U; A, 0.05 U) and their morphological characteristics, particularly with respect to the position of kinetoplast(s) and nucleus/nuclei. Interestingly, we found that both nuclear synthesis initiation and segregation in T. lewisi occurred prior to kinetoplast, different to the order of replication found in Trypanosoma brucei and Trypanosoma cruzi, implicating a distinct cell cycle control mechanism in T. lewisi. We characterized the morphological events during the T. lewisi cell cycle and presented evidence to support the existence of two distinct cell cycles with two-way transformation between them. These results provide insights into the differentiation and evolution of this parasite and its related species.
KEYWORDS: Trypanosoma lewisi, cell cycle, In vitro, multiplication division, zoonotic pathogen
Introduction
Trypanosoma lewisi (T. lewisi) is a globally distributed parasitic protozoan found in rats (Rattus spp.). It is transmitted by rat-fleas, e.g. Nosopsyllus fasciatus, and has a complex life cycle creating a variety of morphologies [1]. Traditionally, it has been considered a rat-specific trypanosome found only in rats. However, occasional human infection cases are reported from tropical regions and it has been suggested as a neglected zoonotic pathogen due to its resistance to lysis by normal human serum [2,3]. As with the infection found in its natural host, the rat, a high parasitaemia was also reported in cases of human infections, particularly in infants [4–9]. Death due to T. lewisi infection in humans has also been recorded [10].
T. lewisi exhibits various life cycle forms in the bloodstream of infected rats and these include typical slender forms as well as multiple rosette forms [1,11]. In contrast, few dividing cells have been observed from infected individuals. In addition, little is known about the cell cycle of this mysterious trypanosome. Several investigators have cultured Herpetosoma trypanosomes in association with various mammalian cell lines [12–15]. In the early studies, T. lewisi had been maintained continuously [11] and morphological polymorphism was observed during in vitro cultivation [16–18]. However, there has been no systematic analysis, to date, that resolves these morphological cell types and relates them to the corresponding cell cycle events.
Multiplication (including cytokinesis) is one of the most important characteristics of all living cells including protists. It is precisely controlled by very complex mechanisms found in a wide range of cell types from single-celled eukaryotes to metazoa. Multiplication is constrained within complex cell cycle events which results in division into two identical daughter cells. The eukaryotic cell cycle events generally include nuclear/cytosolic activities described in five phases. S-phase is defined as the nuclear DNA synthesis period, separating two preparatory periods, known as the gap 1 (G1) and gap 2 (G2) phases. These are followed by nuclear division (mitosis, M-phase) and cytoplasmic division (cytokinesis, C-phase). However, the classical cell cycle descriptions do not include the events required for replication of the extra-nuclear genetic material, such as mitochondrial and chloroplast DNAs, due to the presence of multiple copies of these organelles. In some early branching eukaryotes, such as the Kinetoplastida, there is only one mitochondrion (More detailed could be found in recent reviews [19,20]). This group includes many human and animal pathogens, such as Trypanosoma brucei, T. cruzi, T. evansi and Leishmania spp. In these species, the mitochondrial DNA is packaged into a compact disc-shaped kinetoplast (K), allowing easy detection and identification of K duplication events together with nuclear duplication stages. Therefore, it is important to consider all of the events surrounding the cell cycle in the Kinetoplastida (K changes, kinetoplast G1-phase, S-phase, G2-phase, division phase (D-phase) and the post-division phase (A-phase)). The presence of two DNA-containing organelles in the cell cycle also raises important questions regarding the co-ordination of DNA replication during the cell cycle events.
The events surrounding the changes in kinetoplast and nucleus configurations during the cell cycle of T. brucei have been described using an asynchronous cell culture condition [21] or a synchronous model [22]. According to those descriptions, the kinetoplast DNA replication and division in T. brucei occurs prior to the nuclear DNA replication and division during the cell cycle. Cells with two kinetoplasts and one nucleus (2K1N) are commonly observed in an asynchronous population because of this order of activity. A similar situation was also observed in T. cruzi [23,24]. Therefore, it has been considered a classical model, in trypanosomes, that kDNA replication and division occurs prior to nuclear division. To our surprise, we found that in contrast to T. cruzi and T. brucei, we could not find any 2K1N cells in T. lewisi but instead found cells with one kinetoplast and two nuclei (1K2N), suggesting a different cell cycle process.
In this study, we have modified the in vitro cultivation system of T. lewisi described by Behr et al. [11] and provided the first detailed description, with a comprehensive and quantitative analysis, of the morphological events occurring during the cell division cycle. This includes a description of a novel two-way transformation connection between two distinct cell cycles in the developmental events in T. lewisi. These provide an alternative explanation to the multiple morphological states observed in the T. lewisi cell cycle. Furthermore, we have also calculated the timing of key cell cycle events within the developmental cycle, including nuclear DNA replication, mitosis, kinetoplast duplication, flagellum duplication and segregation, providing a further understanding of the proliferative cell cycle of T. lewisi.
Materials and methods
Growth characteristics of T. lewisi in vitro and in vivo
Trypanosoma lewisi (CPO02), isolated from a field rat in Guangzhou, China [3], were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS, Genetimes Technology Inc.) and a feeder layer of Sprague Dawley (SD) rat primary embryo fibroblasts (REF) as modified from Behr et al. [11]. Growth curves and generation times were determined by counting the parasites under a light microscope at 12-hour intervals. Doubling times were calculated from log-transformed growth curve data derived from the logarithmic phase of growth and using trendline analysis. All values are expressed as mean ± standard error of 3 biological repeats. In vivo, T. lewisi (2 ×105 cells) were injected intraperitoneally (i.p.) into SD rats (n = 3) and the parasitemia was monitored twice a day. Blood smears were stained with Giemsa (Sigma) for 40 min at room temperature and doubling times were calculated from log-transformed growth curve data. All rodents used in this work were treated in strict accordance with protocols approved by the Laboratory Animal Use and Care Committee of Sun Yat-Sen University under the license for grant Number 31672276.
Measurement of cell morphological parameters
For measurement of cell morphological parameters, cell samples were prepared as described by Wheeler and colleagues [25]. Briefly, mid-log phase cells (2 ×106 cells/ml) were settled onto glass slides, fixed, and stained with a DNA intercalating dye, DAPI (4ʹ,6-diamidino-2-phenylindole). Cell body length and flagellum length were quantified by Image J [26], as well as the DNA intensity of the nucleus and kinetoplast, which were quantitated by their dye-signal intensity.
Flow cytometry and analysis of DNA content
Flow cytometry was performed as described in Signorell et al. [27]. Briefly, trypanosome cells (2 ×106) were fixed in 0.25% formaldehyde and subsequently in 10 volumes of ice-cold 70% ethanol. Cells were then re-suspended in 4 ml of phosphate-buffered saline (PBS, pH7.4) containing 40 μg/ml propidium iodide (PI) and 10 μg/ml RNase A, and incubated at 37°C for 30 min in the dark. The DNA contents of PI-stained cells were analyzed with a flow cytometer. A total of 10,000 events were recorded. The percentage of cells with different DNA contents were determined by Flowjo [28].
EdU labeling and immunofluorescence microscopy
A Cell-light EdU Apollo567 in Vitro Kit (Ribobio) was used to determine the timing of initiation and the duration of nuclear and kinetoplast S-phase in T. lewisi. Briefly, exponential cells (1 ×106 cells/ml) were incubated with 25 μM 5-ethynyl-2-deoxy-uridine (EdU). At half-hourly intervals over a period of 4 hours, 0.5 ml samples were collected and cells were then harvested by centrifugation at 2000 g for 5 min. Following this, they were washed once in cold PBS (pH7.4) and were then adhered onto glass slides, fixed, stained and washed according to the manufacturer’s manual (Ribobio). Mouse monoclonal antibody L8C4, which recognizes the T. brucei para-flagella rod protein PFR2, was used to label the flagellum as previously described [29].
Measurement of timing of events in the cell cycle
Using an asynchronous logarithmic culture, the timing of cell cycle events, which gives rise to different morphologies, can be determined by the frequency of cells with each particular morphology. The duration of mitosis and cytokinesis was calculated based on DAPI staining using the Equation (1) below: [25]
Where x is the cumulative time within the cycle required to reach the end of the stage in question, y is the cumulative percentage of cells up to and including the stage in question, expressed as a fraction of one unit, and t is the generation time.
The duration of the S phase of either the nucleus or kinetoplast was calculated individually by (2):
Where = In 2/T and Z = (duration of G2) + (duration of mitosis) + (duration of cytokinesis) of either the nucleus or the kinetoplast, T is the generation time, t is the duration of the EdU labeling period, and L is the proportion of cells exhibiting labelled nuclei or labelled kinetoplasts [30].
Scanning electron microscopy (SEM) for morphological observation
Samples for scanning electron microscopy were prepared as previously described [31]. Trypanosome cells (107) were washed with PBS and settled onto coverslips. Cells were fixed with 2.5% glutaraldehyde in 100 mM phosphate buffer (pH7.4) overnight, washed three times with PBS and dehydrated through an ethanol series [30%, 50%, 70%, 80%, 90%, and 100% (vol/vol)] for 10 min each. The coverslips were critical-point dried and coated with 8 nm gold nanoparticles (Pt:Pd 80:20, Ted Pella Inc) using a sputter-coater (Cressington Sputter 208 HR, Ted Pella Inc). Images were captured using an electron microscope (S-3400N, Hitachi), with a scanning work distance of 5 mm and a high voltage of 8 kV.
Time-lapse video microscopy for cell division monitoring
Each rosette cell cluster was placed into a 35 mm petri dish at 37°C in a 5% CO2 humid atmosphere using the ECU-HOC STEV2 system (WPI). Proliferation of multiple kinetoplasts and multiple nuclei (xKxN) rosette cells were monitored under an inverted microscope Vert A1 (Zeiss) for ~3 hours. When the number of individuals (i) in rosette clusters were clarified by observation and continuous observations were made until i increases to i +1 and i +2. The period of time that elapsed from i +1 to i +2 was defined as the duplication time for the rosettes. We also followed the rosette from i +2 when it reduced back to i +1 when an individual was released, and this period was defined as the segmentation time.
Results
Logarithmic culture of T. lewisi and the progress of the cell cycle
To understand the events during the cell cycle, T. lewisi bloodstream forms were cultured in vitro in an asynchronous manner. The cells maintained a logarithmic growth between cell density of 2 ×105 cells/ml to 4.5 ×106 cells/ml, and entered a stationary phase at 5 ×106 cells/ml followed by a declining phase (Figure 1(a)). The logarithmic growth phase of T. lewisi was confirmed using a log plot of growth over this period (60 hours) and a doubling time of 12.14 ± 0.79 h (log2/0.0248) was determined (Figure 1(b)). Logarithmical growth of the repeated sub-culture of this parasite maintained the cell density between 5.3 (log 2 ×105 cells/ml) and 6.5 (log 3.5 ×106 cells/ml) at 50 hrs intervals (Figure 1(c)). In in vivo experiments, the parasitemia in rat blood was monitored twice daily, and growth curves were constructed and transformed into a logarithmic scale (Figure S1). Analysis of the trend line indicated that the generation time was 12.21 ± 0.14 h.
Figure 1.
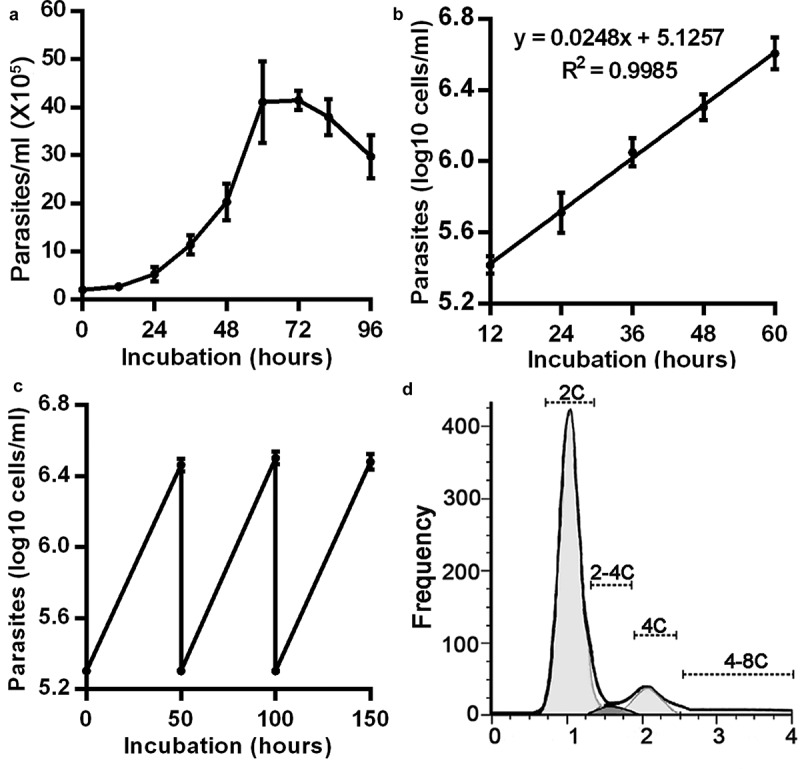
Growth curves and properties of Kinetoplast and Nuclear (K/N) configurations of T. lewisi in in vitro bloodstream cultures. (a) A typical growth curve of T. lewisi bloodstream forms during in vitro cultivation. Parasites (2 × 105) were inoculated in RPMI-1640 with 10% FBS, supplemented with a feeder layer of rat embryo fibroblasts cells, and were continually monitored for 96 hours. (Values indicate the mean ± standard error of the mean of three replicates). (b) The growth curve using the first 60 hours of data (from (a)) transformed to a log scale. Trend line analysis shows the doubling time as 12.14 hours (calculated from log2/0.0248). (c) Growth curve of T. lewisi where cells were repeatedly subcultured at 50-hour intervals (covering the logarithmic growth phase) to confirm the doubling time of 12.14 hours. (d) Flow cytometry analysis was conducted, using the propidium iodide (PI) DNA stain, and a histogram of DNA content was analyzed to determine the proportions of cells in each phase. Based on 10,000 events, proportions were recorded as follows: G1-phase (2C, 81.16%), S-phase (2C-4C, 3.04%) and G2-phase (4C, 10.44%) and others (4C-8C, 5.36%).
To further investigate the cell division cycle, flow cytometry showed that in an asynchronous T. lewisi culture, most cells were present as diploid (G1-phase, 81.16%) or tetraploid (G2-phase, mitosis phase or cytokinesis phase, 10.44%). The remaining portion was DNA synthesis (S) phase cells (3.04%) and other aneuploid cells (5.36%) (Figure 1(d)).
To better resolve the kinetoplast configuration, DAPI staining was applied to the logarithmic phase cells. Various K/N configurations were found in the culture (Figure 2). The majority of the cells (~91%) contained only one nucleus and one kinetoplast (1K1N) (Figure 2(a,b,h). A small proportion of cells contained two nuclei and one kinetoplast (1K2N) (~2%; Figure 2(c,h), two nuclei and two kinetoplasts (2K2N) (~2%; Figure 2(d,e,h), or multiple nuclei and multiple kinetoplasts (xKxN) (~4%; Figure 2(f,h).
Figure 2.
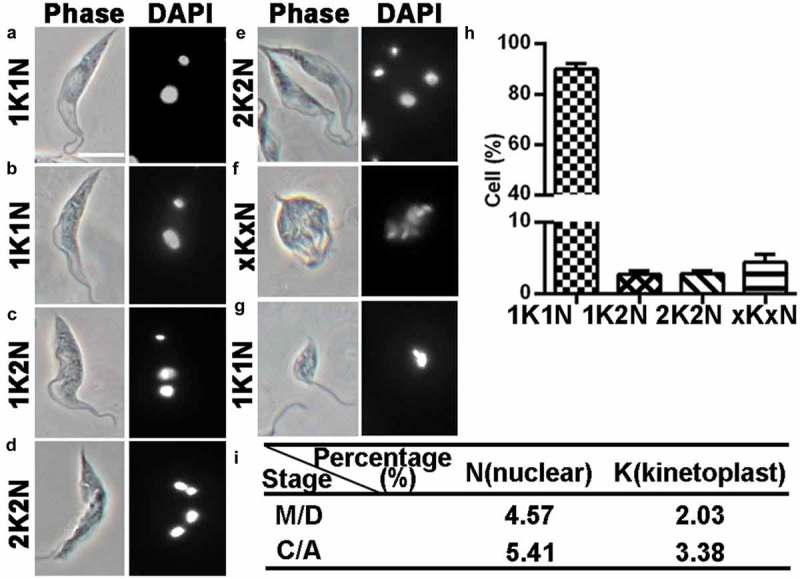
DAPI staining of T. lewisi cells to determine the distribution of kinetoplasts and nuclei. A logarithmic growing culture of 5 × 106 cells were fixed in 4% paraformaldehyde, permeabilized and stained with DAPI and the cells characterized. (a) Trypomastigote with 1K1N (G1/2, S-phase), (b) with 1K2N (M-phase), (c) with 1K2N (C-phase), (d and e) with 2K2N (C-phase), (f) with xKxN, (g) epimastigote form with 1K1N, (h) Proportion of the cells with different number of nucleus (N) and kinetoplast (K) configurations in a logarithmic growth and (i) proportion of M/D, C/A cells. n = 562, mean value ±sem. Scale bar represents 10 μm.
This initial analysis established a basic order and timing of the cell cycle unit, as previously defined [32]. The total time of a cell cycle was calculated to be 12.14 h (shown as a unit, “U”) and the proportion of M/D (4.57%/2.03%), C/A (5.41%/3.38%) cells were calculated (Figure 2(i). Through Equation (1) (Materials and methods), the duration of M-phase was calculated to be 0.75 h, taking only 6% of a general cell cycle time (0.06 U), C-phase time 0.92 h (0.08 U), D-phase time 0.34 h (0.03 U) and A-phase time 0.58 h (0.05 U).
Subsequently, the duration of G2 period was determined by measuring the progressive appearance of EdU-labeled DNA in dividing nuclei or kinetoplasts. Initially, cultures were checked to see whether those treated with 25 μM EdU grew at the same rate as the EdU-free controls (Figure S2). Superimposed curves from EdU and non-EdU treated cultures (n =3) demonstrate that this is indeed the case. Samples were collected every 30 min for over 4 hours and EdU incorporation was assessed by immunofluorescence microscopy (Figure 3). Cells without any EdU incorporation in either the nucleus or the kinetoplast were predominant from the beginning of the experiment (Figure 3(a)). As time progressed, cells were observed where only the nucleus was labelled (Figure 3(b)) or where the nucleus was strongly labelled and the kinetoplast was weakly labelled (Figure 3(c)). After just one-hour incubation with EdU, cells were detected with both nuclei and kinetoplasts strongly labelled (Figure 3(d)).
Figure 3.
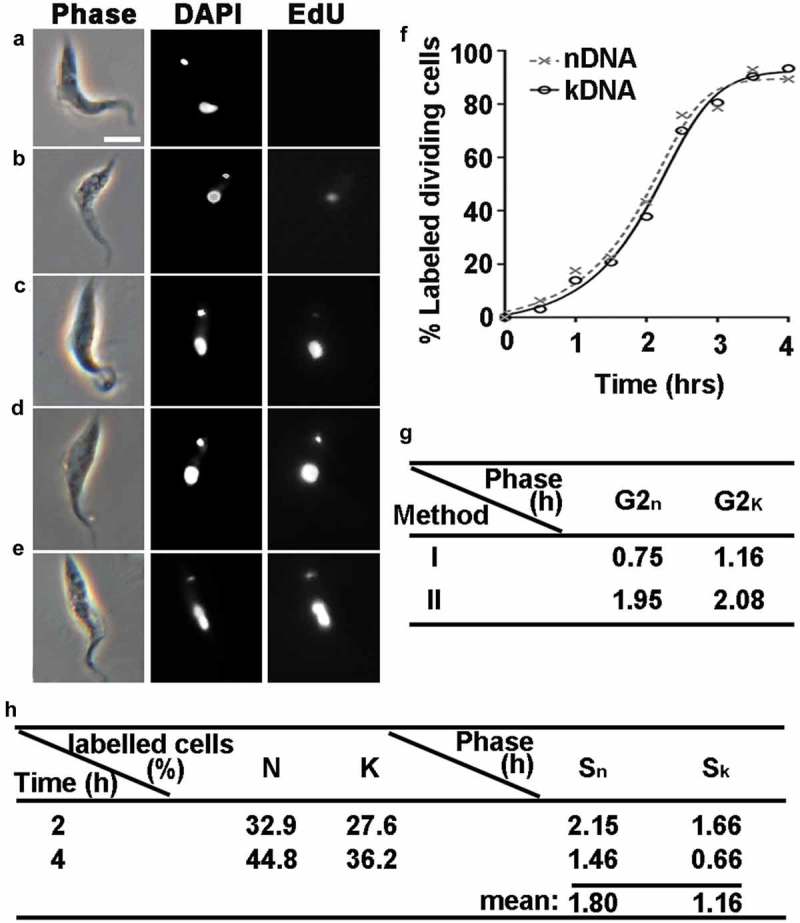
Determination of S/G2-Phase durations and newly synthesized DNA in T. lewisi using staining with EdU. Exponentially growing cells (1 × 106 cells/ml) were incubated with 25 μM EdU, and 0.5 ml samples were taken at half-hourly intervals for a period of 4 hours and then immunofluorescence signals from each cell was detected. (a) Cells with no EdU incorporation in either the nucleus or the kinetoplast. (b) Labelled signal was only found in nucleus but not in the kinetoplast. (c) The nucleus in the cell was labelled more strongly than the kinetoplast. (d) A strongly labelled signal was detected in both the nucleus and kinetoplast. (e) Cells labelled with two nuclei and one kinetoplast. The scale bar represents 10 μm. (f) Proportion of the cells labelled with the nucleus (solid line, closed circles) or the kinetoplast (dotted line, cross). The time at which 50% of dividing nuclei (or kinetoplasts) were labelled was defined as the average G2 period: nucleus (1.95 h, 0.16 U); kinetoplast (2.08 h, 0.17 U). n = 800. (g) Determinates G2n and G2k phase with two methods. Method I considers the incubation time that appears the first cell labeled with EdU dye in the kinetoplast/nucleus equals to the duration time of G2 + M/D phases. G2 phase, is determined by a subtraction of M/D-phase. Method II only counts cells with dividing organelles. When 50% of them were labelled the corresponding the incubation time is considered as the average duration of G2-phase. (h) Evaluation of S phase. After incubation of EdU for 2/4 hrs, cells with labelled organelles were determined, and values of S phase were calculated by applying Equation (2).
To estimate the duration of nuclear/kinetoplast G2 phase (G2n & G2k), T. lewisi cells were maintained in the presence of EdU until cells containing two EdU-labeled nuclei/kinetoplasts were observed (Figure 3(e)). The results showed that both 1K2N and 2K2N cells were first detected 1.5 h after EdU incorporation, meaning that cells at the end of S phase took 1.5 h to go through G2n/G2k phase and mitosis (Figure 3(g), method I), which was performed in Trypanosomatids previously [24,33]. Considering this finding and that the duration of M and D phase in T. lewisi trypanomastigotes are 0.75 h, we could estimate the duration of G2n as 0.75 h (0.06 U) and G2k as 1.16 h (0.1 U) (Figure 3(g), method I). Alternatively, the time taken for 50% of the dividing organelles to become labelled was taken as the average duration of G2 (Figure 3(f)), which is 1.95 h (0.16U) for G2n and 2.08 h (0.17 U) for G2k (Figure 3(g), method II).
Considering that method II determines the mean time of G2 phase in a T. lewisi population, this value was used to calculate S phase. The S-phase durations were determined using Equation (2) [30], we have performed this analysis in cells with inoculation for two periods (2 and 4 hrs), the proportions of cells exhibiting either labelled nuclei or kinetoplasts were recorded (Figure 3(h)). The mean time S-phase of the nuclear cycle (Sn) was 1.80 h or 0.14 U and that of the kinetoplast cycle (Sk) was 1.16 h or 0.10 U (Figure 3(h)).
The calculations of the G1 period for the nucleus (G1n) and kinetoplast (G1k) were achieved by subtracting all of the above calculated periods. The overall order and duration of each of the T. lewisi cell cycle periods, G1n/k, Sn/k, G2n/k, M/D, and C/A are summarized in Figure 8.
Figure 8.
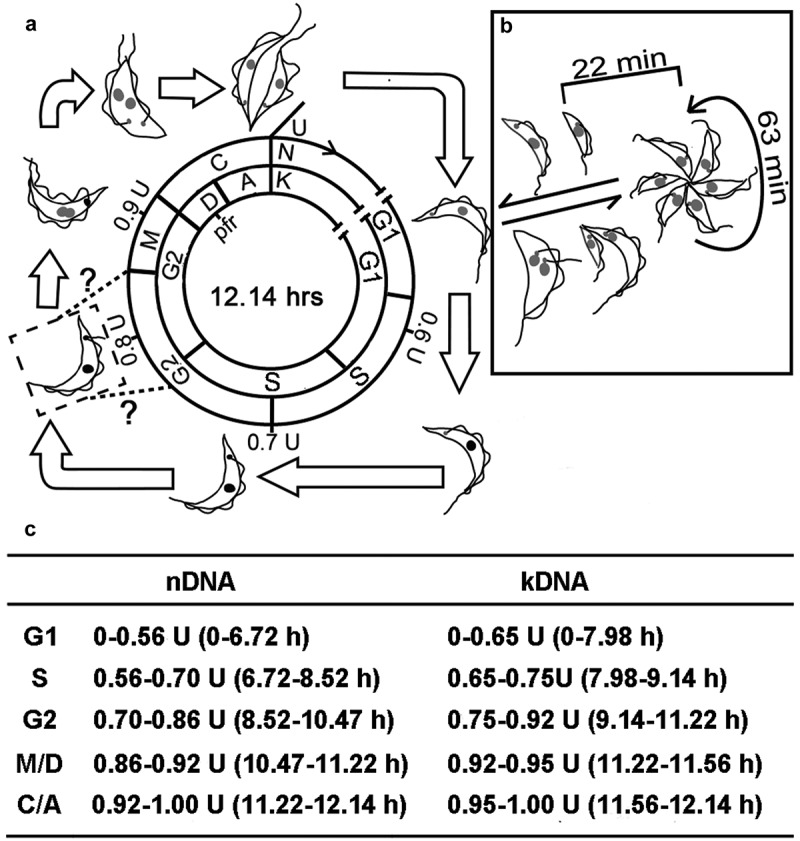
Schematic representation of the morphological alterations and the duration of the phases of the T. lewisi cell cycle. (a) Binary fission comprises the following stages: G1n/k (gap1), Sn/k (DNA synthesis period), G2n/k (gap2), M (mitosis) or D (division phase), and C (cytokinesis) or A (post-division phase) were indicated. Two equal length flagella were observed at M-phase, but the initial assembly time point for the new flagellum might occur in either Sn/k or G2n/k phase, according to related studies in T. brucei [21] or T. cruzi [23]. The duration of the cell cycle phases was listed in the table below. (b) The rosette stage undergoes multiple division and individuals can develop to either form a new rosette or to transform into transitional trypomastigotes and finally to return to fully developed trypomastigotes which can undergo binary fission. On the other hand, trypomastigotes may transform to rosette forms by means of unequal division events. (c). The duration of the cell cycle phases with respect to the nucleus and the kinetoplast. The duration of the phases of the cell cycle were calculated as described (methods, results). The phases of the nuclear events are G1n, Sn, G2n, M and C-phase, while the phases of the kinetoplast events are G1k, Sk, G2k, D and A-phase. The overall generation time is 12.14 h.
Based on the observations of the K/N configuration changes and the order of the cell cycle events, nucleus synthesis initiation and division were found to occur prior to that of the kinetoplast. To corroborate these findings further, we developed an independent approach. The DNA contents of individual nuclei and kinetoplasts were quantified using the quantification programme, ImageJ, after PI and DAPI staining, respectively. Using PI staining, a broad range of nuclear DNA contents was detected for those cells which are 1K1N (majority of G1n cells, and some in Sn and G2n phases) while for the 1K2N and 2K2N cells, the nuclear DNA content mainly was around two-fold intense of that in the 1K1N cells. For example, 1K2N, 2.50 times; 2K2N, 1.91 times, as the nuclear DNA in 1K2N and 2K2N cells had replicated completely before M-phase (Figure 4(a)). Meanwhile, using DAPI staining, the kinetoplast DNA (kDNA) contents were detected for 1K1N (majority of G1k cells, and some in Sn phase) and 1K2N cells (majority of Sn and G2n phases), and a higher kDNA intensity was found in 2K2N cells (2.22 times 1K1N and 1.78 times, 1K2N) (Figure 4(b)). This was due to the completion of kDNA replication before D-phase.
Figure 4.
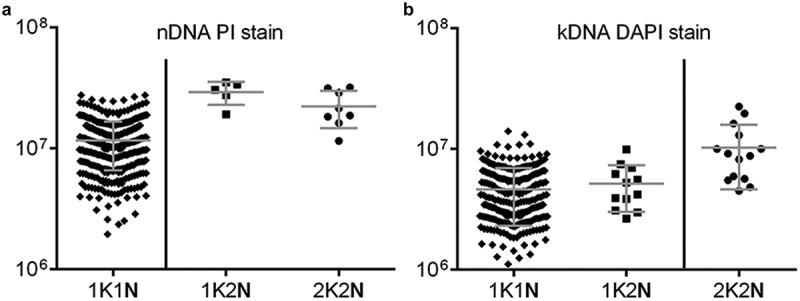
Replication of nDNA prior to kDNA could be verified based on DNA intensity analysis using PI and DAPI stains. The cells were fixed and stained with DAPI/PI and the detection of DNA intensity in the exponentially growing cells was measured by software programme Image J (mean ± standard deviation) for 1K1N, 1K2N, 2K2N arrangements of the nuclei and kinetoplasts, respectively. (a) For the nucleus, the mean of the nDNA intensity in a single cell of 1K2N (n = 5) and 2K2N (n = 8) cells are two-fold higher than 1K1N (n = 380) cells, due to the nDNA duplicating completely in cells containing 2 nuclei. (b) For the kinetoplast, the mean intensity of the kDNA in a single cell of 2K2N (n = 12) cells is two-fold higher than that in the 1K1N (n = 494) and 1K2N (n = 14) cells, due to the nDNA being duplicated completely in cells with 2 kinetoplasts.
Morphometric analysis revealed two distinct cell types
In additional to cells following the classical binary fission cell cycle, we also found rosette-like cells, which possessed multiple kinetoplasts, multiple nuclei and multiple flagella (xKxNxF) (Figure 2(f)). Scanning electron microscopy (SEM) and paraflagellar rod (PFR) immunofluorescence confirmed that they had flagella facing outwards from a central position, forming a rosette (Figures 5(f–h) and 7(h,i)), as compared with the standard arrangement (Figure 5(a–c)). However, DAPI staining revealed a close positional association between the kinetoplast and the nucleus, suggesting that these cells are not trypomastigotes, but epimastigotes (Figure 2(f,g); Figure 7(h,i); Figure S1(c)) [1]. Using live cell imaging, we were able to observe the proliferation of these xKxN rosette cells. As shown in Figure 5(j), a rosette cell contained four individuals that are undergoing incomplete fission. Thirty three minutes later, a fifth individual was generated and remained attached to the xKxN population of cells, resulting in a five-individual rosette (i = 5). After a further period of 48 min, the number of individuals increased to six (i = 6), and 25 min later, one individual was released from the rosette (segmentation) and became a free daughter cell. A more detailed division progress is shown in Figure S3, which includes a continual record for about 3 hrs. According to these observations on xKxN rosette proliferation, the average time for individual duplication was calculated to be around 63 min (52– 74 min, n =3). We were able to observe the full progression onto separation in three cases of segmetation, which took about 22 min (19– 25 min, n =3).
Figure 5.
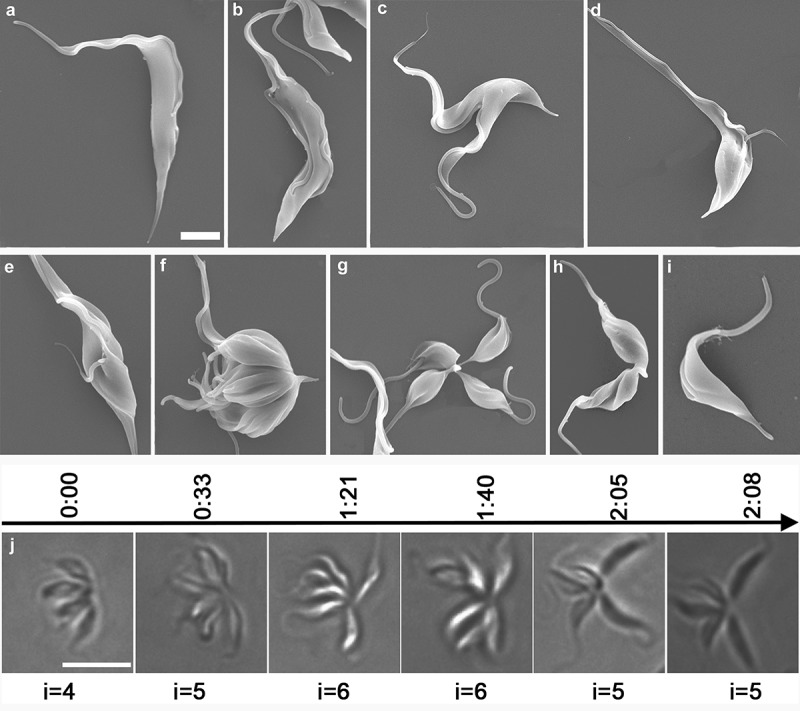
Investigation of the progression of T. lewisi and time-lapse observations of a single rosette cell progressing through the cell cycle. Trypanosomes (107) were fixed overnight, washed three times with PBS and dehydrated through a series of concentration of ethanol. (a) T. lewisi trypomastigote with a long flagellum, (b) trypomastigote with two equal flagella, (c) trypomastigote during the cytokinesis phase, (d,e) cells in a different cell cycle route found in cells developing from trypomastigotes to dividing epimastigotes, (f–h) dividing epimastigotes, (i) transitional form found in cells transforming from epimastigotes to trypomastigotes. Scale bar represents 2 μm. (j) Cells were trapped in 0.5% agarose to assist observation. A series images of the same rosette were shown in a timeline (hours) whilst undergoing proliferation. The number of individuals (i) is indicated, and these increased after incomplete fission and decreased if segmentation occured. Scale bar represents 10 μm.
Figure 7.
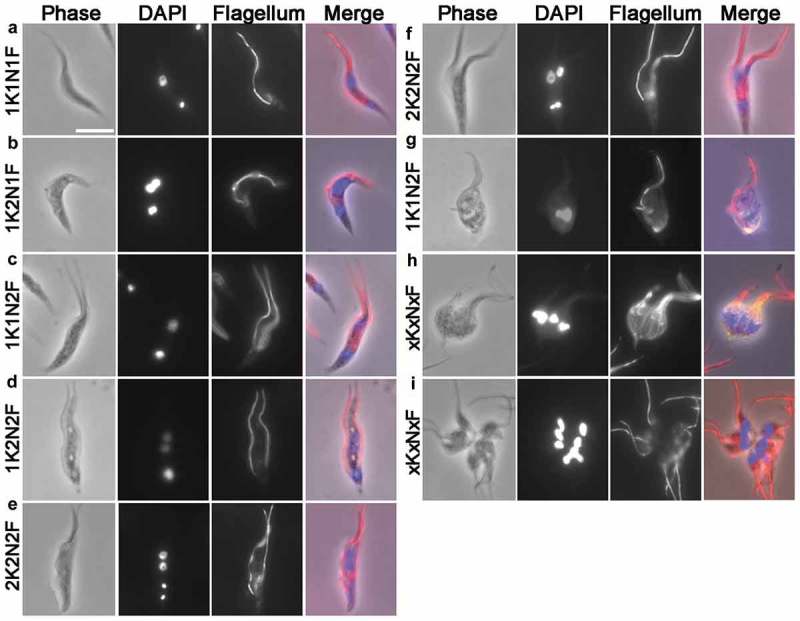
Immunofluorescence staining of T. lewisi cells using anti-paraflagella rod antibodies. Trypanosomes were settled onto glass microscope slides, fixed in methanol at −20°C, and a mouse monoclonal antibody (mAb) against the T. brucei flagellum L8C4 was used as primary antibody, followed by secondary antibody (goat anti-mouse IgG, Invitrogen). DNA was stained by DAPI and cells were categorized according to their number of nuclei (N), kinetoplasts (K) and flagella (F). (a) 1K1N1F, (b) 1K2N1F, (c) 1K1N2F, (d) 1K2N2F, (e and f) 2K2N2F, (g) 1K1N2F (trypomastigote form in transition towards a dividing epimastigote), (h,i) xKxNxF. In the merged images, the red staining represents the flagellum while the blue staining highlights the nuclei and kinetoplasts. Scale bar represents 10 μm.
By serial dilution we successfully picked out trypomastigotes (three wells, each containing 5–10 cells). One day later, transformation to rosettes was found in all wells. On the other hand, when picking rosettes (two wells, each containing three rosettes), trypomastigotes were observed in both wells on the second day. Both experiments suggested that two-way transformations have occurred (Figure S4). Using SEM, we confirmed the existence of various cell types found by light microscopy (Figure 5). Figure 5(d,e); Figure 7(g) and Figure S1(i) show a cell probably undergoing transformation from trypomastigote to epimastigote, due to its long cell body (trypomastigote origin) and short (newly assembled epimastigote) flagella. Figures 5(d) and Figure 7(g) show the cells that are likely to be synthesizing their new flagellum, as the flagella tip comes out from the flagella pocket, forming a cell with multiple K and multiple N (pre-rosette stage). Figure 5(e) shows another cell also with multiple K and multiple N (pre-rosette stage), while Figure 5(f–h) shows a regular rosette cell with several individuals.
Based on these observations, we attempted to distinguish this rosette proliferation cell cycle from the classical binary fission cell cycle. To do so, we went through the DAPI stained images of xKxN rosette cells, and found clear and consistent association of the kinetoplasts with the nuclei (Figure 2(f); Figure 7(h,i). Thus, these rosettes were actually epimastigotes and constituted ~4% of the total population of cells (Figure 2(f)). These epimastigotes also had a shorter cell body length than the trypomastigotes generated by binary fission. Therefore, by conducting scatter plot analysis of the length of the cell body (PA) and the distance between the nucleus and kinetoplast (K-N), we were able to cluster the dividing epimastigotes and trypomastigotes into two separate groups (Figure 6). The cells that fell between these two groups were probably in a transition stage (Figure 5(i)). This division cycle is summarized in Figure 8(b) and proportions of each of these cell types were listed in table below (Figure 8).
Figure 6.
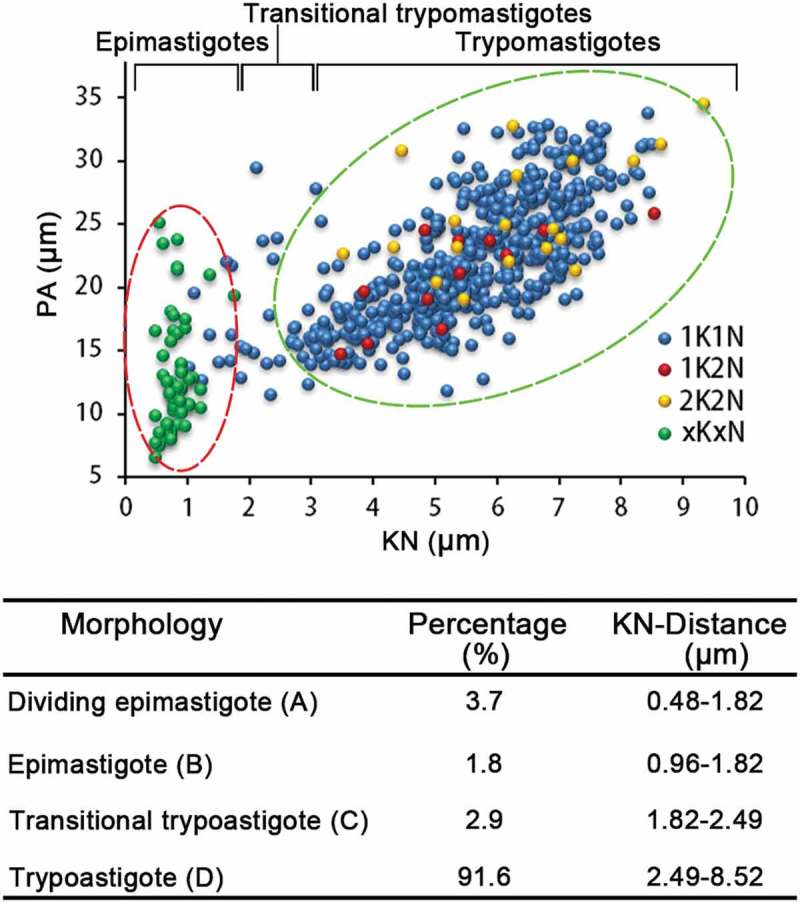
Scatter plot analysis of the two cell cycles of T. lewisi based on morphological characteristics. Cell body (PA) length and distance between the Kinetoplast and Nucleus (KN) was measured in a heterogeneous range of cells, by Photoshop (Adobe), and resolved in a scatter plot. The division of epimastigotes (multiplication division) and trypomastigotes (generated by binary fission) were grouped in circles of red and green dotted lines, respectively. 1K1N cells within each of the two circles were classified as epimastigotes and trypomastigotes, while the rest that fell between the two circles were defined as transitional trypomastigotes. n = 597. Percentage of cells within the population exhibiting in the table below.
Since we have identified two distinct proliferation cycles in the T. lewisi cultivation, we estimated, using the individual duplication time of 63 min and segmentation time of 22 min as the generation time of epimastigotes, that the daughter cells required for maintaining a rosette population were ~4% and the rest of them transformed into trypomastigotes. Therefore, we were able to estimate a corrected time for the classical binary fission route, by subtracting the epimastigotes, the transition cells and their resultant trypomastigotes from the population. A complete cycle of the classical binary fission cell cycle was determined as 18.13 hours and each phase was corrected and presented in Table S1.
Formation of the new flagellum during the cell cycle
The growth of the new flagellum in trypomastigote cells was investigated by confocal immunofluorescence microscopy using anti-paraflagellar rod antibodies. The trypomastigote population of 1K1N1F, 1K2N2F and 2K2N2F cells can be detected (Figure 7). Both 1K2N1F and 1K1N2F cells were detected, although 1K2N1F cells (2.4%, 14/581) were more abundant than 1K1N2F cells (0.5%, 3/581). Overall the cells with two flagella make up to ~4% of total cells (n =581). Surprisingly, we only observed trypomastigote cells with either one flagellum or two flagella of similar length, but not cells with one short flagellum and one long flagellum. All the cell types observed with light-microscopy were also identified under SEM (Figure 5). Taking advantage of the ability to clearly visualize flagella using SEM, we were able to examine over 3600 cells and identified only 4 of them possessing one short flagellum and one long flagellum (Figure 5(d,e)). However, they all seemed to be epimastigotes, as judged by the position of the flagellum. In addition, the tip of the short new flagellum was not attached to the old flagellum and no flagella connector was observed. Overall, using the data on numbers of biflagellate cells, we are able to propose that flagellum duplication completes at 0.962 U (Figure 8(a)).
Discussion
Trypanosoma lewisi, a globally distributed rat trypanosome, has long been considered as non-pathogenic to humans [1]. However, occasionally human infected cases are reported [2,34] and it has been suggested as a neglected zoonotic pathogen [3]. We know little about the cell cycle and the division mechanisms of this parasite. It has long been thought that T. lewisi undergoes an atypical cell cycle in the rat bloodstream. Epimastigote stages with multiple divisions forming rosette shaped structures were described in early studies [13,16,17], while the expected proliferation in trypomastigotes was rarely observed and it was suggested that they may require transformation to epimastigotes [1]. In a later study, proliferation of trypomastigotes was observed when it was cultured in vitro [18]. However, detailed information regarding the cell cycle events was not provided. Here, we have established in vitro cultivation of T. lewisi trypomastigotes at 37°C, and observed the proliferation phases of the trypomastigote cell cycle, which demonstrate that the trypomastigote is one of the typical reproductive stages as observed in other kinetoplastid parasites. In addition, using flow cytometry, we showed that most T. lewisi cells (~94%) underwent standard binary fission rather than entering multiplication through the rosette stages. However, rosette multiplication events were also observed using live cell imaging. Furthermore, we showed the occurrence of inter-conversion events between epimastigotes and trypomastigotes, which demonstrates that these two distinct cell cycles are connected. The reasons for these fast and frequent switches in cell morphology, between trypomastigote and epimastigote forms, currently remain unclear. However, a potential underlying mechanism has been recently revealed in Trypanosoma brucei, which involves the assembly and disassembly of the FAZ structure[35]. In past studies, such transitional trypomastigotes had been recognized, although little evidence was provided to confirm their identity [16,17]. We have also calculated the time of each proliferation phase during trypomastigote division (summarized in Figure 8). The measured doubling time of T. lewisi in our culture system was 12.14 hours, which very closely matches the value calculated from in vivo infection of rats (12.21 hours). This was convincing evidence that our culture system replicated the biological reality. Having recognized, from the culture system, that that two independent duplication cycles are occurring in parallel, we set out to calculate the proliferation times of each of those cycles. The rosette stage took 63 min to generate a new individual and 22 min to segment an individual from the rosette. On the other hand, once corrected for the removal of the rosette cycle, the classical binary fission route took 18.1 hours to complete the cycle. This suggests that there are both long and short proliferation cycles and supports a notion that rosette multiplication is essential for maintenance of the population of T. lewisi particularly during the early stages of infection [36,37]. It has been suggested that the rosette multiplication might reside in the small vessels of the internal organs, especially the kidneys [1]. They have also been observed in the bloodstream of a monkey during infection [38].
It is interesting to speculate on the evolution and role of these rosette stages. If they help an early infection to become established as proposed in the early studies described above [36,37], they might reside as a reservoir of dividing cells in tissues at the site of infection (the skin). In this context, interestingly, recent studies have demonstrated that the skin and dermal layers can act as a reservoir for T. b. gambiense in human infections [39,40]. In fact, visualization of these reservoirs by SEM [40] (e.g. in their Figure 4) shows that aggregates of trypanosomes become trapped in collagen networks. While in T. brucei these aggregates are not necessarily rosettes, perhaps the rosette stages of T. lewisi are better adapted for becoming trapped in interstitial tissues and thereby establishing localized reservoirs.
Rosette and transitional forms have been observed in in vitro cultivation of other Herpetosoma trypanosomes, e.g T. musculi and T. grosi [16]. However, this phenomenon has not been observed in T. rangeli, in which division was not found in the bloodstream forms but instead it multiplies inside host hepatic parenchyma cells as amastigotes similar to members of the subgenus Schizotrypanum, e.g. Trypanosoma cruzi [41]. The dramatic differences in the ways which the bloodstream forms divide when comparing T. rangeli and other Herpetosoma members support the suggestion of reclassification of T. rangeli from the subgenus Herpetosoma into Schizotrypanum as suggested from studies based on molecular data [42,43].
Although there are no published studies on rosette multiplication of T. lewisi in the rat bloodstream so far, our work shows that short epimastigotes are easily found and transitional forms can be observed in rats from 3–5 days after infection. We show that the rosette cell cycle takes 22 min for individual parasites to segment from the rosette, while cytokinesis takes 28.3 min to complete during binary fission. These durations are consistent with those found for T. brucei (24.5 min) [31].
The doubling time of 12.14 hours in T. lewisi logarithmic growth culture is consistent with the doubling time of this parasite in vivo (12.21 hours). It is much longer than many other related species that have been investigated, such as T. brucei (8.67 h) [21], L. mexicana (7.1 h) [25], and Crithidia fasciculata (6.2 h) [44], but it is much shorter than that found in T. cruzi (24 h) [23]. According to the suggestion by Elias [23], the time of reproduction may positively correlated with the amount of DNA, because the more DNA there is, the more prolonged is the time needed for replication to form two identical copies of DNA. As the amount of DNA and G1n/k-phase is known in T. brucei (26 Mb) [45], Leishmania species (32 Mb) [46], T. rangeli (24 Mb) [47] and T. cruzi (60 Mb) [48], it is possible to predict that the genome size of T. lewisi may be between 30–60 Mb given the duration of G1n of 7.52 h (0.62 U) and G1k of 7.73 h (0.64 U), assuming all other parameters are similar.
The general structure (cell length, nuclear apparatus) of T. lewisi (adult form) was described in 1909 in a pioneering study from Minchin [49]. T. lewisi contains two copies of genetic material located in the nucleus and the kinetoplast, both of which require replication and division in the cell cycle. Traditionally, replication and division of kinetoplast DNA were recognized as occurring prior to nuclear DNA division both in the African trypanosome T. brucei and the American trypanosome T. cruzi [21,23]. In contrast to T. brucei and T. cruzi, nuclear DNA replication and division in T. lewisi occurs prior to the replication and division of kDNA during the cell cycle. This reverse order has also been observed in other members of the Kinetoplastida such as L. mexicana [25], and C. fasciculata [44], when they were maintained at room temperature. It is interesting to consider that when C. fasciculata is cultured at 32°C, it undergoes a cell cycle with the kDNA replication phase occurring prior to the nDNA phase, suggesting that kDNA polymerase (Pol β) in C. fasciculata was more temperature-sensitive than the nDNA polymerase (Pol A, B) [50,51]. More interestingly, the situation is complex in the genus Leishmania. In contrast to L. mexicana (N prior to K) [25], the order of K/N replication and division is found as “K prior to N” in L. major [52] and L. tarentolae [53], and these even co-exist within the population of a culture in other Leishmania species (e.g. in L. amazonensis and L. donovani) [33,54]. Although the underlying mechanisms remain unclear, we consider the complicated situation in the order of K/N replication and division in kinetoplastida may be due to the complex organization of the kDNA network and its replication system. Actually, we have also observed 1K2N and 2K2N forms in blood-smears from rats infected with T. lewisi (Figure S1), which rules out the possibility that these cell types are artifacts of in vitro cultivation. Therefore, we propose that as the division of the nucleus occurs prior to kinetoplast in T. lewisi, this is a unique characteristic of the cell cycle which indicates that there are two separate mechanisms of nuclear and kinetoplast segregation among the species within the Kinetoplastida. Compared to mammalian cells, in which the centrosome is used for nuclear DNA segregation, the centrosome (basal body) in trypanosomes is responsible for kinetoplast DNA segregation. The centrosome and nuclear chromosomes in mammalian cells share some common subunits of the cohesin complex [55], which are responsible for synchronization of these two organelles. However, in trypanosomes the cohesin complex subunits are used in the nucleus, but not in the basal bodies [56]. Therefore, the shift in the order of nuclear and kinetoplast replication may indicate a dissociation of both processes and a cohesin-independent mechanism probably exists.
The flagella of cells were quantified and those with 1K2N1F were found 4 times more abundant than those with 1K1N2F. This indicated that the formation of new flagella was likely to be synchronized with the separation of the nuclei. However, it is still not possible to assign a fixed order. In over 3600 cells examined, those trypomastigotes with two flagella, all had flagella of equal length. Therefore, it suggests that there is a short time requirement for new flagellar elongation and that there is a high flagellar elongation speed. Alternatively, it might be due to close proximity of the newly-synthesized flagellum to the existing one. In order to distinguish these two possibilities, scanning electron microscopy was helpful. However, very few cells were found with one long flagellum and one short flagellum, therefore, we favor the high-speed possibility, given the existence of a fast cell cycle (~1 hour only) in rosette forms of T. lewisi. New flagellum assembly in T. lewisi may be extremely fast, compared to the known three species below. Firstly, T. brucei, whose flagellar assembly initiates in G2k-phase and is completed at the onset of C-phase, lasts about 0.3 U 21. Secondly, T. cruzi, whose flagellar assembly initiates in G2n/k-phase and may be completed in C-phase, lasts over 0.3 U [23] . Thirdly, L. mexicana, initiates in G2n/k-phase and may be completed in the following cell cycle [25].
In this paper, we have provided a quantitative characterization of the morphological events during T. lewisi proliferation and provided evidence to support the existence of two distinct cell cycles with two-way transformation between them. This raises interesting questions as to how the parasite controls the decision-making processes as to whether to enter the epimastigote rosette phase and also the evolutionary role of this alternative cell cycle. Our data may provide a platform for further studies on the identification of the cell cycle regulation machinery in this zoonotic pathogen as well as in other species of Kinetoplastida.
Funding Statement
The work was supported by the grants from the National Science Foundation of China (#31672276, #31772445 and #31720103918), the Natural Science Foundation of Guangdong Province (#2016A030306048), Guangzhou Science Technology and Innovation Commission (#201506010011).
Author contributions
Xuan Zhang, Zhao-Rong Lun and De-Hua Lai designed experiments; Xuan Zhang and Su-Jin Li performed the experiments; De-Hua Lai, Xuan Zhang, Zhao-Rong Lun and Ziyin Li analyzed experimental results and wrote the manuscript; Cynthia Y. He and Geoff Hide revised the manuscript.
Conflict interest
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary material for this article can be accessed here.
References
- [1].Hoare CA. The Trypanosome of mammals. A zoological monograph. Oxford and Edinburgh: Blackwell Scientific Publications; 1972. [Google Scholar]
- [2].Truc P, Buscher P, Cuny G, et al. Atypical human infections by animal trypanosomes. PLoS Negl Trop Dis. 2013;7:e2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lun ZR, Wen YZ, Uzureau P, et al. Resistance to normal human serum reveals Trypanosoma lewisi as an underestimated human pathogen. Mol Biochem Parasitol. 2015;199:58–61. [DOI] [PubMed] [Google Scholar]
- [4].Johnson PD. A case of infection by Trypanosoma lewisi in a child. Trans R Soc Trop Med Hyg. 1933;26:467–468. [Google Scholar]
- [5].Kaur R, Gupta VK, Dhariwal AC, et al. A rare case of trypanosomiasis in a two month old infant in Mumbai, India. J Commun Dis. 2007;39:71–74. [PubMed] [Google Scholar]
- [6].Shrivastava KK, Shrivastava GP. Two cases of Trypanosoma (Herpetosoma) species infection of man in India. Trans R Soc Trop Med Hyg. 1974;68:143–144. [DOI] [PubMed] [Google Scholar]
- [7].Sarataphan N, Vongpakorn M, Nuansrichay B, et al. Diagnosis of a Trypanosoma lewisi-like (Herpetosoma) infection in a sick infant from Thailand. J Med Microbiol. 2007;56:1118–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Verma A, Manchanda S, Kumar N, et al. Trypanosoma lewisi or T. lewisi-like infection in a 37-day-old Indian infant. Am J Trop Med Hyg. 2011;85:221–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bharodiya D, Singhal T, Kasodariya GS, et al. Trypanosomiasis in a young infant from rural Gujarat, India. Indian Pediatr. 2018;55:69–70. [PubMed] [Google Scholar]
- [10].Doke PP, Kar A. A fatal case of Trypanosoma lewisi in Maharashtra, India. Ann Trop Med Public Health. 2011;4:91–95. [Google Scholar]
- [11].Behr MA, Mathews SA, D’Alesandro PA. A medium for the continuous cultivation of bloodstream forms of Trypanosoma lewisi at 37°C. J Parasitol. 1990;76:711–716. [PubMed] [Google Scholar]
- [12].Hommel M, Robertson E. In vitro attachment of trypanosomes to plastic. Experientia. 1976;32:464–466. [DOI] [PubMed] [Google Scholar]
- [13].Mohamed HA, Maraghi S, Wallbanks KR, et al. In vitro cultivation of Herpetosoma trypanosomes on embryonic fibroblasts and in semidefined cell-free medium. J Parasitol. 1988;74:421–426. [PubMed] [Google Scholar]
- [14].Albright JW, Albright JF. Growth of Trypanosoma musculi in cultures of murine spleen cells and analysis of the requirement for supportive spleen cells. Infect Immun. 1978;22:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vincendeau P, Guillemain B, Daulouede S, et al. In vitro growth of Trypanosoma musculi: requirements of cells and serum free culture medium. Int J Parasitol. 1986;16:387–390. [DOI] [PubMed] [Google Scholar]
- [16].Mohamed HA, Molyneux DH. In vitro cultivation of Herpetosoma trypanosomes in insect cell tissue culture media. Parasitol Res. 1987;73:9–14. [DOI] [PubMed] [Google Scholar]
- [17].Proulx C, Olivier M, Mora L, et al. Infectivity and route of penetration in rats after oral and intraperitoneal inoculations of bloodstream and in vitro-cultured metacyclic forms of Trypanosoma lewisi. J Parasitol. 1989;75:964–969. [PubMed] [Google Scholar]
- [18].Ashraf M, Nesbitt RA, Humphrey PA, et al. Comparative positions of kinetoplasts in Trypanosoma musculi and Trypanosoma lewisi during development in vitro. Cell Prolif. 2002;35:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lukes J, Butenko A, Hashimi H, et al. Trypanosomatids are much more than just trypanosomes: clues from the expanded family tree. Trends Parasitol. 2018;34:466–480. [DOI] [PubMed] [Google Scholar]
- [20].Maslov DA, Opperdoes FR, Kostygov AY, et al. Recent advances in trypanosomatid research: genome organization, expression, metabolism, taxonomy and evolution. Parasitology. 2018;146;1–27. [DOI] [PubMed] [Google Scholar]
- [21].Woodward R, Gull K. Timing of nuclear and kinetoplast DNA replication and early morphological events in the cell cycle of Trypanosoma brucei. J Cell Sci. 1990;95(Pt 1):49–57. [DOI] [PubMed] [Google Scholar]
- [22].Benz C, Dondelinger F, McKean PG, et al. Cell cycle synchronisation of Trypanosoma brucei by centrifugal counter-flow elutriation reveals the timing of nuclear and kinetoplast DNA replication. Sci Rep. 2017;7:17599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Elias MC, Da Cunha JP, de Faria FP, et al. Morphological events during the Trypanosoma cruzi cell cycle. Protist. 2007;158:147–157. [DOI] [PubMed] [Google Scholar]
- [24].Da Silva MS, Munoz PAM, Armelin HA, et al. Differences in the detection of BrdU/EdU incorporation assays alter the calculation for G1, S, and G2 phases of the cell cycle in Trypanosomatids. J Eukaryot Microbiol. 2017;64:756–770. [DOI] [PubMed] [Google Scholar]
- [25].Wheeler RJ, Gluenz E, Gull K. The cell cycle of Leishmania: morphogenetic events and their implications for parasite biology. Mol Microbiol. 2011;79:647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Collins TJ. ImageJ for microscopy. BioTechniques. 2007;43:S25–S30. [DOI] [PubMed] [Google Scholar]
- [27].Signorell A, Gluenz E, Rettig J, et al. Perturbation of phosphatidylethanolamine synthesis affects mitochondrial morphology and cell-cycle progression in procyclic-form Trypanosoma brucei. Mol Microbiol. 2009;72:1068–1079. [DOI] [PubMed] [Google Scholar]
- [28].Gosink JJ, Means GD, Rees WA, et al. Bridging the divide between manual gating and bioinformatics with the bioconductor package flowFlowJo. Adv Bioinformatics. 2009;2009:809469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kohl L, Sherwin T, Gull K. Assembly of the paraflagellar rod and the flagellum attachment zone complex during the Trypanosoma brucei cell cycle. J Eukaryot Microbiol. 1999;46:105–109. [DOI] [PubMed] [Google Scholar]
- [30].Stanners CP, Till JE. DNA synthesis in individual L-strain mouse cells. Biochem Biophys Acta. 1960;37:406–419. [DOI] [PubMed] [Google Scholar]
- [31].Wheeler RJ, Scheumann N, Wickstead B, et al. Cytokinesis in Trypanosoma brucei differs between bloodstream and tsetse trypomastigote forms: implications for microtubule-based morphogenesis and mutant analysis. Mol Microbiol. 2013;90:1339–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Williams FM. Dynamics of microbial population In: Patten B, editor. Systems analysis and simulation ecology. Vol. 1 New York (NY): Academic Press; 1971. p. 247–262. [Google Scholar]
- [33].Da Silva MS, Monteiro JP, Nunes VS, et al. Leishmania amazonensis promastigotes present two distinct modes of nucleus and kinetoplast segregation during cell cycle. PloS one. 2013;8:e81397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lun ZR, Reid SA, Lai DH, et al. Atypical human trypanosomiasis: a neglected disease or just an unlucky accident? Trends Parasitol. 2009;25:107–108. [DOI] [PubMed] [Google Scholar]
- [35].Sunter JD, Gull K. The flagellum attachment zone: ‘The cellular ruler’ of trypanosome morphology. Trends Parasitol. 2016;32:309–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Davis BS. Studies on the trypanosomes of some california mammals. Univ California Pub Zool; 1952;57:145–250. [Google Scholar]
- [37].Ormerod WE. The initial stages of infection with Trypanosoma lewisi; control of parasitaemia by the host. In: Gamham, PCC, Pierce AE, Roitt I,editor.Immunology to protozoa A symposium of the Brit Soc for Immunology. Oxford: Blackwell; 1963, p. 213–227. [Google Scholar]
- [38].Maia Da Silva F, Marcili A, Ortiz PA, et al. Phylogenetic, morphological and behavioural analyses support host switching of Trypanosoma (Herpetosoma) lewisi from domestic rats to primates. Infect Genet Evol. 2010;10:522–529. [DOI] [PubMed] [Google Scholar]
- [39].Capewell P, Cren-Travaille C, Marchesi F, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5:e17716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Caljon G, Van Reet N, De Trez C, et al. The dermis as a delivery site of Trypanosoma brucei for tsetse flies. PLoS Pathog. 2016;12:e1005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Urdaneta-Morales S, Tejero F. Trypanosoma (Herpetosoma) rangeli Tejera, 1920. Intracellular amastigote stages of reproduction in white mice. Rev Inst Med Trop Sao Paulo. 1986;28:166–169. [DOI] [PubMed] [Google Scholar]
- [42].Stevens JR, Teixeira MM, Bingle LE, et al. The taxonomic position and evolutionary relationships of Trypanosoma rangeli. Int J Parasitol. 1999;29:749–757. [DOI] [PubMed] [Google Scholar]
- [43].Lin RH, Lai DH, Zheng LL, et al. Analysis of the mitochondrial maxicircle of Trypanosoma lewisi, a neglected human pathogen. Parasit Vectors. 2015;8:665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cosgrove WB, Skeen MJ. The cell cycle in Crithidia fasciculata. Temporal relationships between synthesis of deoxyribonucleic acid in the nucleus and in the kinetoplast. J Protozool. 1970;17:172–177. [DOI] [PubMed] [Google Scholar]
- [45].Berriman M, Ghedin E, Hertz-Fowler C, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. [DOI] [PubMed] [Google Scholar]
- [46].Ivens AC, Peacock CS, Worthey EA, et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Stoco PH, Wagner G, Talavera-Lopez C, et al. Genome of the avirulent human-infective trypanosome- Trypanosoma rangeli. PLoS Negl Trop Dis. 2014;8:e3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].El-Sayed NM, Myler PJ, Bartholomeu DC, et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309:409–415. [DOI] [PubMed] [Google Scholar]
- [49].Minchin EA. The strcuture of Trypanosoma lewisi in relation to microscopical technique. J Cell Sci. 1909;53:755. [Google Scholar]
- [50].Holmes AM, Cheriathundam E, Kalinski A, et al. Isolation and partial characterization of DNA polymerases from Crithidia fasciculata. Mol Biochem Parasitol. 1984;10:195–205. [DOI] [PubMed] [Google Scholar]
- [51].Torri AF, Kunkel TA, Englund PT. A beta-like DNA polymerase from the mitochondrion of the trypanosomatid Crithidia fasciculata. J Biol Chem. 1994;269:8165–8171. [PubMed] [Google Scholar]
- [52].Ambit A, Woods KL, Cull B, et al. Morphological events during the cell cycle of Leishmania major. Eukaryot Cell. 2011;10:1429–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Simpson L, Braly P. Synchronization of Leishmania tarentolae by hydroxyurea. J Protozool. 1970;17:511–517. [DOI] [PubMed] [Google Scholar]
- [54].Minocha N, Kumar D, Rajanala K, et al. Kinetoplast morphology and segregation pattern as a marker for cell cycle progression in Leishmania donovani. J Eukaryot Microbiol. 2011;58:249–253. [DOI] [PubMed] [Google Scholar]
- [55].Piel M, Meyer P, Khodjakov A, et al. The respective contributions of the mother and daughter centrioles to centrosome activity and behavior in vertebrate cells. J Cell Biol. 2000;149:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bessat M, Ersfeld K. Functional characterization of cohesin SMC3 and separase and their roles in the segregation of large and minichromosomes in Trypanosoma brucei. Mol Microbiol. 2009;71:1371–1385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.