

Abstract
Background
The benefits of erythropoiesis‐stimulating agents (ESA) for chronic kidney disease (CKD) patients have been previously demonstrated. However, the efficacy and safety of short‐acting epoetins administered at larger doses and reduced frequency as well as of new epoetins and biosimilars remains uncertain.
Objectives
This review aimed to evaluate the benefits and harms of different routes, frequencies and doses of epoetins (epoetin alpha, epoetin beta and other short‐acting epoetins) for anaemia in adults and children with CKD not receiving dialysis.
Search methods
We searched the Cochrane Kidney and Transplant Specialised Register to 12 September 2016 through contact with the Information Specialist using search terms relevant to this review. Studies contained in the Specialised Register are identified through search strategies specifically designed for CENTRAL, MEDLINE, and EMBASE; handsearching conference proceedings; and searching the International Clinical Trials Register (ICTRP) Search Portal and ClinicalTrials.gov.
Selection criteria
We included randomised control trials (RCTs) comparing different frequencies, routes, doses and types of short‐acting ESAs in CKD patients.
Data collection and analysis
Two authors independently assessed study eligibility and four authors assessed risk of bias and extracted data. Results were expressed as risk ratio (RR) or risk differences (RD) with 95% confidence intervals (CI) for dichotomous outcomes. For continuous outcomes the mean difference (MD) with 95% confidence intervals (CI) was used. Statistical analyses were performed using the random‐effects model.
Main results
We identified 14 RCTs (2616 participants); nine studies were multi‐centre and two studies involved children. The risk of bias was high in most studies; only three studies demonstrated adequate random sequence generation and only two studies were at low risk of bias for allocation concealment. Blinding of participants and personnel was at low risk of bias in one study. Blinding of outcome assessment was judged at low risk in 13 studies as the outcome measures were reported as laboratory results and therefore unlikely to be influenced by blinding. Attrition bias was at low risk of bias in eight studies while selective reporting was at low risk in six included studies.
Four interventions were compared: epoetin alpha or beta at different frequencies using the same total dose (six studies); epoetin alpha at the same frequency and different total doses (two studies); epoetin alpha administered intravenously versus subcutaneous administration (one study); epoetin alpha or beta versus other epoetins or biosimilars (five studies). One study compared both different frequencies of epoetin alpha at the same total dose and at the same frequency using different total doses.
Data from only 7/14 studies could be included in our meta‐analyses. There were no significant differences in final haemoglobin (Hb) levels when dosing every two weeks was compared with weekly dosing (4 studies, 785 participants: MD ‐0.20 g/dL, 95% CI ‐0.33 to ‐0.07), when four weekly dosing was compared with two weekly dosing (three studies, 671 participants: MD ‐0.16 g/dL, 95% CI ‐0.43 to 0.10) or when different total doses were administered at the same frequency (four weekly administration: one study, 144 participants: MD 0.17 g/dL 95% CI ‐0.19 to 0.53).
Five studies evaluated different interventions. One study compared epoetin theta with epoetin alpha and found no significant differences in Hb levels (288 participants: MD ‐0.02 g/dL, 95% CI ‐0.25 to 0.21). One study found significantly higher pain scores with subcutaneous epoetin alpha compared with epoetin beta. Two studies (165 participants) compared epoetin delta with epoetin alpha, with no results available since the pharmaceutical company withdrew epoetin delta for commercial reasons. The fifth study comparing the biosimilar HX575 with epoetin alpha was stopped after patients receiving HX575 subcutaneously developed anti‐epoetin antibodies and no results were available.
Adverse events were poorly reported in all studies and did not differ significantly within comparisons. Mortality was only detailed adequately in four studies and only one study included quality of life data.
Authors' conclusions
Epoetin alpha given at higher doses for extended intervals (two or four weekly) is non‐inferior to more frequent dosing intervals in maintaining final Hb levels with no significant differences in adverse effects in non‐dialysed CKD patients. However the data are of low methodological quality so that differences in efficacy and safety cannot be excluded. Further large, well designed, RCTs with patient‐centred outcomes are required to assess the safety and efficacy of large doses of the shorter acting ESAs, including biosimilars of epoetin alpha, administered less frequently compared with more frequent administration of smaller doses in children and adults with CKD not on dialysis.
Plain language summary
Short‐acting erythropoiesis agents in chronic kidney disease patients not requiring dialysis
What is the issue?
Anaemia due to reduced production by the kidneys of erythropoietin (a hormone which increases red cell production) is a major cause of tiredness and other problems experienced by people with chronic kidney disease requiring or not requiring dialysis.
Manufactured erythropoietins (epoetins) improve anaemia and are often prescribed for people with chronic kidney disease. Several different manufactured epoetins are now available.
What did we do?
We searched the Cochrane Kidney and Transplant Specialised Register to 12 September 2016 through contact with the Information Specialist using search terms relevant to this review. We included randomised control trials (RCTs) comparing different frequencies, routes, doses and types of short‐acting ESAs in patients with chronic kidney disease.
What did we find?
We examined the evidence from 14 studies with 2616 participants with CKD not receiving dialysis published before 12 September 2016 to determine whether differences in improvement in anaemia and in side effects existed between different short‐acting epoetins or between the same epoetins given at different frequencies. We did not find any studies using different frequencies of epoetins in children.
We found that the traditionally shorter acting epoetins given less often (two weekly to every four weeks) resulted in similar correction of anaemia compared with administration every week or every two weeks; there were no differences in side effects between the different comparisons. One study comparing subcutaneous administration of a newly manufactured HX575 epoetin alpha compared with epoetin alpha was discontinued after two patients developed anti‐erythropoietin antibodies. However more studies are required as most studies were small and poorly designed, which limits their application to the care of patients.
Summary of findings
Summary of findings for the main comparison. Epoetin alpha every 2 weeks versus to weekly for anaemia in CKD patients not receiving dialysis.
| Epoetin alpha every 2 weeks versus to weekly for anaemia in CKD patients not receiving dialysis | ||||||
| Patient or population: anaemia in predialysis patients Intervention: epoetin alpha every 2 weeks Comparison: weekly | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with weekly | Risk with Epoetin alpha every 2 weeks | |||||
| Change in Hb level | The mean change in Hb level was 0 g/dL | The mean change in Hb level in the intervention group was 0.19 g/dL lower (0.32 g/dL lower to 0.06 g/dL lower) | ‐ | 798 (4) | ⊕⊕⊝⊝ LOW 1 2 | downgraded for study limitations and indirectness |
| Number reaching target Hb | Study population | RR 0.96 (0.93 to 0.99) | 798 (4) | ⊕⊕⊝⊝ LOW 1 2 | downgraded for study limitations and indirectness | |
| 960 per 1000 | 922 per 1000 (893 to 951) | |||||
| Moderate | ||||||
| 947 per 1000 | 910 per 1000 (881 to 938) | |||||
| Number of deaths | Study population | RR 0.89 (0.38 to 2.07) | 838 (4) | ⊕⊕⊝⊝ LOW 1 3 | downgraded for study limitations and imprecision | |
| 28 per 1000 | 24 per 1000 (10 to 57) | |||||
| Moderate | ||||||
| 22 per 1000 | 20 per 1000 (9 to 46) | |||||
| Adverse events: RBC transfusions | Study population | RR 1.56 (0.71 to 3.45) | 580 (3) | ⊕⊕⊝⊝ LOW 3 4 | downgraded for imprecision and study limitations | |
| 33 per 1000 | 52 per 1000 (23 to 114) | |||||
| Moderate | ||||||
| 37 per 1000 | 58 per 1000 (26 to 128) | |||||
| Adverse events: hypertension | Study population | RR 0.85 (0.55 to 1.32) | 838 (4) | ⊕⊕⊕⊝ MODERATE 1 | downgraded for study limitations | |
| 100 per 1000 | 85 per 1000 (55 to 132) | |||||
| Moderate | ||||||
| 95 per 1000 | 81 per 1000 (52 to 126) | |||||
| Adverse events: thrombovascular events | Study population | RR 1.41 (0.67 to 3.00) | 838 (4) | ⊕⊕⊝⊝ LOW 1 3 | downgraded for study limitations and imprecision | |
| 28 per 1000 | 39 per 1000 (18 to 83) | |||||
| Moderate | ||||||
| 27 per 1000 | 38 per 1000 (18 to 80) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; Hb: haemoglobin; RBC: red blood cells | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 allocation concealment unclear in 3 of 4 studies
2 surrogate outcome
3 few studies with low numbers and wide confidence
4 allocation concealment unclear in 2 of 3 studies
Summary of findings 2. Epoetin alfa every four weeks versus with every two weeks in CKD patients not receiving dialysis.
| Epoetin alfa every four weeks versus with every two weeks in CKD patients not receiving dialysis | ||||||
| Patient or population: anaemia in predialysis patients Intervention: epoetin alpha every 4 weeks Comparison: every 2 weeks | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with every 2 weeks | Risk with Epoetin alpha every 4 weeks | |||||
| Change in Hb level | The mean change in Hb level was 0 | The mean change in Hb level in the intervention group was 0.15g/dL lower (0.41 g/dL lower to 0.1g/dL more) | ‐ | 671 (3) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | downgraded for study limitations, heterogeneity and indirectness |
| Number reaching target Hb | Study population | RR 0.95 (0.84 to 1.07) | 687 (3) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | downgraded for study limitations, heterogeneity and indirectness | |
| 916 per 1000 | 870 per 1000 (769 to 980) | |||||
| Moderate | ||||||
| 895 per 1000 | 850 per 1000 (752 to 957) | |||||
| Number of deaths | Study population | RR 0.95 (0.33 to 2.75) | 724 (3) | ⊕⊕⊝⊝ LOW 1 4 | downgraded for study limitations, imprecision | |
| 22 per 1000 | 21 per 1000 (7 to 62) | |||||
| Moderate | ||||||
| 26 per 1000 | 25 per 1000 (9 to 72) | |||||
| Adverse events: RBC transfusions | Study population | RR 1.26 (0.53 to 2.98) | 470 (2) | ⊕⊕⊝⊝ LOW 1 4 | downgraded for study limitations, imprecision | |
| 38 per 1000 | 48 per 1000 (20 to 114) | |||||
| Moderate | ||||||
| 35 per 1000 | 44 per 1000 (18 to 103) | |||||
| Adverse events: hypertension | Study population | RR 1.02 (0.62 to 1.69) | 724 (3) | ⊕⊕⊕⊝ MODERATE 1 | downgraded for study limitations | |
| 70 per 1000 | 72 per 1000 (44 to 119) | |||||
| Moderate | ||||||
| 62 per 1000 | 63 per 1000 (38 to 104) | |||||
| Adverse events: arteriovenous complications | Study population | RR 1.02 (0.39 to 2.68) | 724 (3) | ⊕⊕⊝⊝ LOW 1 4 | downgraded for study limitations, imprecision | |
| 26 per 1000 | 26 per 1000 (10 to 68) | |||||
| Moderate | ||||||
| 23 per 1000 | 24 per 1000 (9 to 62) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; Hb: haemoglobin; RBC: red blood cells | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 two of the three studies had unclear allocation concealment
2 surrogate outcome
3 unexplained heterogeneity
4 small numbers with wide confidence intervals
Summary of findings 3. Epoetin theta versus epoetin beta in CKD patients not receiving dialysis.
| Epoetin theta versus epoetin beta in CKD patients not receiving dialysis | ||||||
| Patient or population: anaemia in predialysis patients Intervention: epoetin theta Comparison: epoetin beta | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with epoetin beta | Risk with Epoetin theta | |||||
| Final Hb | The mean final Hb was 0 g/dL | The mean final Hb in the intervention group was 0.02 g/dL lower (0.25 g/dL lower to 0.21 g/dL higher) | ‐ | 288 (1) | ⊕⊕⊕⊝ MODERATE 1 | downgraded for indirectness ‐ surrogate outcomes |
| Mean weekly epoetin dose | The mean weekly epoetin dose was 0 units/week | The mean weekly epoetin dose in the intervention group was 0.4 units per week higher (5.68 units per week lower 6.48 units/week higher) | ‐ | 288 (1) | ⊕⊕⊝⊝ LOW 1 2 | downgraded for indirectness ‐ surrogate outcomes and imprecision |
| Deaths | Study population | RR 2.46 (0.29 to 20.77) | 288 (1) | ⊕⊕⊝⊝ LOW 2 | downgraded for imprecision | |
| 11 per 1000 | 26 per 1000 (3 to 219) | |||||
| Moderate | ||||||
| 11 per 1000 | 26 per 1000 (3 to 218) | |||||
| Adverse events: hypertension | Study population | RR 0.35 (0.11 to 1.08) | 288 (1) | ⊕⊕⊕⊝ MODERATE 2 | downgraded for imprecision | |
| 74 per 1000 | 26 per 1000 (8 to 80) | |||||
| Moderate | ||||||
| 74 per 1000 | 26 per 1000 (8 to 80) | |||||
| Adverse events: RBC transfusions | Study population | RR 1.48 (0.06 to 36.10) | 288 (1) | ⊕⊕⊝⊝ LOW 2 | downgraded for imprecision | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Adverse events: discontinuation of therapy | Study population | RR 1.77 (0.68 to 4.63) | 288 (1) | ⊕⊕⊝⊝ LOW 2 | downgraded for imprecision | |
| 53 per 1000 | 93 per 1000 (36 to 244) | |||||
| Moderate | ||||||
| 53 per 1000 | 93 per 1000 (36 to 244) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; RBC: red blood cells | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Surrogate outcome, not a patient‐centred outcome
2 Small numbers, wide confidence intervals
Background
Description of the condition
Anaemia is defined as haemoglobin (Hb) levels < 12.0 g/dL and 13.0 g/dL in adult females and males respectively based on the World Health Organization's definition of anaemia (KDIGO 2012; WHO 2011). Anaemia is diagnosed in children with chronic kidney disease (CKD) if Hb concentration is < 11.0 g/dL in children aged from six months to five years, < 11.5 g/dL in children aged five to 12 years, and < 12.0 g/dL in children from 12 to 15 years of age according to the Kidney Disease Improving Global Outcomes guidelines (KDIGO 2012). Anaemia is a known complication of CKD (Dmtrieva 2013) which develops as kidney function declines; prevalence increases as glomerular filtration rate (GFR) falls. CKD stage 3 to 5 is a predictor variable of decline in Hb (Dmtrieva 2013).
Anaemia prevalence ranges from 25% to 70% (Hsu 2002; Koch 1991), and most people with CKD stage 5 (GFR < 15 mL/min/1.73 m2) are anaemic (Astor 2002). Anaemia related to CKD results in significant morbidity, mortality and increased cardiovascular events, with symptoms including lack of energy, breathlessness, dizziness, angina, poor appetite and decreased exercise tolerance (Canadian EPO 1990; Lundin 1989).
The primary cause of anaemia in CKD is decreased production of the naturally‐occurring hormone, erythropoietin (EPO) in the kidney. Anaemia may be exacerbated by concurrent iron deficiency anaemia (KDIGO 2012).
Prior to the availability of recombinant human EPO (rHuEPO), anaemia was managed with blood transfusions together with iron and folate supplements. Cloning of the human gene for EPO was achieved in 1983 (Lin 1985) and production of rHuEPO followed. The efficacy of erythropoiesis‐stimulating agents (ESA) treatment in dialysis patients was demonstrated in 1986 (Winearls 1986) and several randomised controlled trials (RCTs) have documented a beneficial effect of ESA treatment in correcting the anaemia of CKD in non‐dialysis patients (Cody 2005; Stone 1988).
The increase in Hb levels following treatment with ESA leads to improved energy levels (Wolcott 1989), improved cardiac performance and increased ejection fraction (Pappas 2008) with normalisation of cardiac output and reduced left ventricular mass (Cannella 1990). The benefits of early treatment of anaemia with ESA in predialysis patients include increased exercise capacity, improved quality of life, improved cognitive function and a slower decline in kidney function (Ritz 2000; Roth 1994).
Description of the intervention
Administration of an ESA aims to replace endogenous EPO production, raise Hb levels and alleviate signs and symptoms of anaemia. Epoetin alpha has proven efficacy in treating anaemia in people with CKD (Eschbach 1987). Epoetin alpha has a relatively short half‐life and typically is administered twice or thrice weekly (Locatelli 2011). More recently new longer acting ESAs, which can be administered less frequently than short‐acting ESAs, have been developed allowing administration of ESA every one to four weeks depending on the preparation used and the individual patient response. Darbepoetin was the first ESA with a prolonged half‐life to enter the market enabling administration once a week to four weekly (Macdougall 1999). More recently, the use of the continuous EPO receptor activator (CERA), a pegylated epoetin, has extended dosing intervals to one dose every two to four weeks (Macdougall 2005). ESAs have to be administered intravenously or subcutaneously so the benefits of using ESAs in non‐dialysis CKD patients, who will generally receive subcutaneous injections in an outpatient setting, must be balanced against the inconvenience and/or discomfort of injections as well as potential harms of ESAs, which include hypertension, vascular access thrombosis and cardiovascular events. A significant concern in ESA therapy is the Hb target to be achieved. The CHOIR study reported a target Hb level of 13.5 g/dL compared with 11.3 g/dL was associated with increased mortality and cardiovascular risk and no considerable improvement in the quality of life. The study could not provide an explanation for poorer outcomes in patients with a higher target Hb (Singh 2006). Recommendations on when to commence ESA therapy are outlined in the KDIGO guidelines (KDIGO 2012). As the patents for epoetin alpha have expired, cheaper biosimilars of epoetin alpha have been developed. These biological products are highly similar though not identical to reference products and undergo a more limited appraisal before receiving marketing approval. As they are not generic versions of the reference products and could have differences particularly in adverse effects, these products should be submitted to rigorous assessment before marketing and to long term monitoring to ensure that adverse effects are recognised and attributed to the responsible biological preparation (Mikhail 2013; Schellekens 2009).
How the intervention might work
The primary cause of anaemia in CKD is the relative insufficiency of EPO which is mainly produced by peritubular fibroblasts in the kidney. EPO is part of a widespread system of hypoxia‐inducible gene expression mediated by hypoxia‐inducible transcription factors (HIFs).The factors associated with inadequate EPO production in progressive CKD remain unclear, though recent data indicate a deranged oxygen sensing, in addition to loss of EPO production, is involved (Bernhardt 2010). ESAs accelerate erythropoiesis, increase iron utilisation and raise Hb levels with clinical improvement in signs and symptoms of anaemia and avoidance of blood transfusions. ESA therapy aims to increase Hb levels slowly at a rate of 1 to 2 g/dL per month to correct anaemia. After correction of anaemia, dose adjustment may be necessary to maintain a stable Hb level. Anaemia is corrected slowly with ESA to avoid major side effects including hypertension and thrombotic events. ESA requirements are difficult to predict in individual patients, and may be increased in people with associated co‐morbidities including cardiovascular disease, diabetes, chronic inflammation and severe secondary hyperparathyroidism. ESA requirements are generally lower in patients not receiving dialysis. A major issue in ESA use relates to the Hb target to be achieved with increased cardiovascular risk noted with higher Hb targets (Drueke 2006; Singh 2006). Recent systematic reviews have suggested that aiming for Hb levels similar to those seen in healthy adults is associated with a significantly higher risk of mortality due to cardiovascular events, such as stroke and hypertension (Palmer 2010). The mechanisms for these treatment‐related harms are poorly understood though observational studies suggest treatment related toxicity secondary to impaired Hb responses and incremental erythropoietin dosing (Szczech 2008). KDIGO guidelines (KDIGO 2012) recommend that in general ESAs should not be used to raise Hb levels > 11.5 g/dL.
Why it is important to do this review
In a previous Cochrane systematic review, which included 15 RCTs, rHuEPO (epoetin alpha) significantly increased Hb (two studies) or haematocrit (HCT) levels (five studies) compared with placebo or no treatment and significantly reduced blood transfusion requirements (Cody 2005). Determining the ESA agent to be used should include assessment of the drug's pharmacodynamics, pharmacokinetics, route and frequency of administration, adverse effects, availability and any economic issues (KDIGO 2012). In most high income countries, the use of short‐acting ESAs (epoetin alpha, epoetin beta) in patients with CKD has been superseded by longer acting ESAs (darbepoetin, CERA) because of the reduced frequency of administration. In low income countries where newer longer acting ESAs are less likely to be accessible, clinicians may be limited to use short‐acting ESAs. The cost of using newer ESAs agents would have to be balanced against the costs and inconvenience of more frequent administration. Since the efficacy and safety of rHuEPO compared with placebo or no treatment has already been demonstrated (Cody 2005), this review aims to evaluate short‐acting ESAs (epoetin alpha, epoetin beta, other epoetins or epoetin biosimilars) in patients, both adults and children with CKD not on dialysis (CKD stages 2 to 5) with reference to route of administration (intravenous versus subcutaneous), frequency of administration, different doses and direct comparisons of different epoetins to provide additional information about the value of these agents for institutions where shorter acting ESAs are used.
This review will not evaluate studies comparing short‐acting with longer acting ESAs in CKD, different longer acting ESAs in CKD, different routes of administration in dialysis patients or kidney transplant recipients and different Hb targets as these are subject of other Cochrane published or planned systematic reviews (Hahn 2014; Palmer 2012; Palmer 2014a; Palmer 2014b; Strippoli 2006).
Objectives
This review aimed to evaluate the benefits and harms of different routes, frequencies and doses of epoetins (epoetin alpha, epoetin beta and other short‐acting epoetins) for anaemia in adults and children with CKD not receiving dialysis.
Methods
Criteria for considering studies for this review
Types of studies
All RCTs and quasi‐RCTs (RCTs in which allocation to treatment was obtained by alternation, use of alternate medical records, date of birth or other predictable methods) looking at epoetins (short‐acting ESAs) for treatment of anaemia in patients with CKD not on dialysis.
Types of participants
Inclusion criteria
Patients of any age (adults and children) with anaemia due to CKD (stages 2 to 5) of any severity, who were not receiving dialysis, were included. The definitions of CKD and anaemia used in individual studies were used in this review.
Exclusion criteria
Patients of any age receiving dialysis treatment. Patients receiving long‐acting ESAs or included in studies comparing shorter with longer acting ESAs were excluded. Kidney transplant recipients were also excluded.
Types of interventions
Short‐acting ESAs including epoetins alpha (Eprex®, Procrit®, Epogen®), beta (Recormin®), delta (Dynepo®), epoetin theta (Biopoin®) and biosimilars of epoetin alpha (HX575, EPO‐ hexal®, Abseamed®), epoetin zeta (Silapo®, Retacrit®, Epoetin Hospira®)
Short‐acting ESAs including epoetins with different routes of administration
Short‐acting ESAs including epoetins used at different frequencies of administration
Short‐acting ESAs including epoetins used at different doses
Head‐to‐head comparisons of different short‐acting ESAs.
Types of outcome measures
Primary outcomes
-
Death
All‐cause mortality
Mortality due to cardiac disease or cerebrovascular events
-
Measures of correction of anaemia
Values of Hb/HCT or change in Hb/HCT at the end of the study
Quality of life.
Secondary outcomes
-
Hypertension and blood pressure outcomes
Hypertension (number of patients presenting one or more episodes of hypertension)
Systolic blood pressure at end of treatment (mm Hg)
Diastolic blood pressure at end of treatment (mm Hg).
Cardiovascular morbidity
Cerebrovascular morbidity
-
Adverse effects
Number needing blood transfusion
Thrombotic events
Number ceasing ESA for adverse effects
Number of patients requiring hospitalisations for any cause
Number of patients developing antibody‐mediated pure red cell aplasia
Number of patients developing a malignancy.
Kidney function measures (GFR, serum creatinine (SCr), doubling of SCr) as reported by the authors of primary studies
Need for iron supplementation.
Search methods for identification of studies
Electronic searches
We searched the Cochrane Kidney and Transplant Specialised Register up to 12 September 2016 through contact with the Information Specialist using search terms relevant to this review. The Specialised Register contains studies identified from several sources.
Monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL)
Weekly searches of MEDLINE OVID SP
Handsearching of kidney‐related journals and the proceedings of major kidney conferences
Searching of the current year of EMBASE OVID SP
Weekly current awareness alerts for selected kidney journals
Searches of the International Clinical Trials Register (ICTRP) Search Portal and ClinicalTrials.gov.
Studies contained in the Specialised Register were identified through search strategies for CENTRAL, MEDLINE, and EMBASE based on the scope of Cochrane Kidney and Transplant. Details of these strategies, as well as a list of handsearched journals, conference proceedings and current awareness alerts, are available in the Specialised Register section of information about Cochrane Kidney and Transplant.
See Appendix 1 for search terms used in strategies for this review.
Searching other resources
Reference lists of review articles, relevant studies and clinical practice guidelines.
Letters seeking information about unpublished or incomplete studies to investigators known to be involved in previous studies.
Data collection and analysis
Selection of studies
The search strategy described was used to obtain titles and abstracts of studies that may be relevant to the review. The titles and abstracts were screened independently by two authors, who discarded studies that were not applicable; however studies and reviews that might have included relevant data or information on studies were retained initially. Two authors independently assessed the retrieved abstracts and, if necessary the full text, of these studies to determine which studies satisfied the inclusion criteria.
Data extraction and management
Data extraction was carried out independently by at least two authors using standard data extraction forms. Where there was more than one publication of a study, reports were grouped together and the publication with the most complete data was used in the analyses. Where relevant outcomes were only published in earlier versions these data were used. Any discrepancy between published versions was highlighted.
Assessment of risk of bias in included studies
The following items were independently assessed by two authors using the risk of bias assessment tool (Higgins 2011) (see Appendix 2).
Was there adequate sequence generation (selection bias)?
Was allocation adequately concealed (selection bias)?
-
Was knowledge of the allocated interventions adequately prevented during the study?
Participants and personnel (performance bias)
Outcome assessors (detection bias)
Were incomplete outcome data adequately addressed (attrition bias)?
Were reports of the study free of suggestion of selective outcome reporting (reporting bias)?
Was the study apparently free of other problems that could put it at a risk of bias?
Measures of treatment effect
For dichotomous outcomes (e.g. all‐cause mortality, adverse events, number needing transfusions) results were expressed as risk ratio (RR) with 95% confidence intervals (CI). Where continuous scales of measurement were used to assess the effects of treatment (e.g. final Hb/HCT or change in Hb/HCT, blood pressure, SCr), the mean difference (MD) was used, or the standardised mean difference (SMD) if different scales had been used.
Unit of analysis issues
We included only data from the first period of treatment in cross‐over studies (Higgins 2011). Data expressed in different metrics were analysed using SMD.
Dealing with missing data
Any further information required from the original author was requested by written correspondence (e.g. emailing corresponding authors) and any relevant information obtained in this manner was included in the review. We aimed to analyse available data in meta‐analyses using intention‐to‐treat (ITT) data. However, where only ITT data were available graphically or were not provided and additional information could not be obtained from the study authors, per‐protocol (PP) data was used in analyses.
We imputed a change‐from‐baseline standard deviation using an imputed correlation coefficient when sufficient data were available.
Assessment of heterogeneity
Heterogeneity was analysed using a Chi2 test on N‐1 degrees of freedom, with an alpha of 0.05 used for statistical significance and with the I2 test (Higgins 2003). I2 values of 25%, 50% and 75% correspond to low, medium and high levels of heterogeneity.
Assessment of reporting biases
The search strategy applied aimed to reduce publication bias caused by lack of publication of studies with negative results. We investigated for publication bias using funnel plots if there were sufficient studies of each comparison (Higgins 2011).
Data synthesis
Data were summarised using the random‐effects model but the fixed‐effect model was also used to ensure robustness of the model chosen. We qualitatively summarised data where insufficient data were available for meta‐analysis. Where there were multiple publications of the same study, all reports were reviewed to ensure that all details of methods and results were included. Qualitative review was conducted for adverse events and quality of life outcomes.
Subgroup analysis and investigation of heterogeneity
Subgroup analysis was used to explore possible sources of heterogeneity (participants, interventions and study quality). Heterogeneity among participants could be related to age (adult versus children) or stage of CKD. Heterogeneity in interventions could be related to dose, duration or frequency of rHuEPO treatment or to the route of administration. . Where possible, the risk ratio with 95% CI was calculated for each adverse effect, either compared to no treatment or to another agent.
Sensitivity analysis
Sensitivity analyses tested decisions where inclusion of a study may have altered the results of the meta‐analysis. In particular, sensitivity analysis may be used to test decisions where ITT and PP data were included in the same analyses.
'Summary of findings' tables
We presented the main results of the review in 'Summary of findings' tables. These tables present key information concerning the quality of the evidence, the magnitude of the effects of the interventions examined, and the sum of the available data for the main outcomes (Schünemann 2011a). The 'Summary of findings' tables also include an overall grading of the evidence related to each of the main outcomes using the GRADE (Grades of Recommendation, Assessment, Development and Evaluation) approach (GRADE 2008). The GRADE approach defines the quality of a body of evidence as the extent to which one can be confident that an estimate of effect or association is close to the true quantity of specific interest. The quality of a body of evidence involves consideration of within‐trial risk of bias (methodological quality), directness of evidence, heterogeneity, precision of effect estimates and risk of publication bias (Schünemann 2011b). We presented the following outcomes in the 'Summary of findings' tables.
-
Epoetin alpha every 2 weeks compared to weekly for anaemia in CKD patients not receiving dialysis (Table 1)
Final or change in Hb level (g/dL)
Number reaching target Hb
Number of deaths
Number with adverse events: red blood cell transfusions, hypertension, thrombovascular events
-
Epoetin alfa every four weeks compared with every two weeks in CKD patients not receiving dialysis (Table 2)
Final or change in Hb level (g/dL)
Number reaching target Hb
Number of deaths
Number with adverse events: red blood cell transfusions, hypertension, thrombovascular events
-
Epoetin theta versus epoetin beta (Table 3)
Final Hb (g/dL)
Mean weekly epoetin dose (U/kg)
Number of deaths
Number with adverse events: hypertension, red blood cell transfusions, discontinuation of therapy.
Results
Description of studies
Results of the search
Seventy‐three reports were identified from the search to 12 September 2016. After title and abstract screening 22 reports were excluded. Full‐text review was carried out the remaining 51 reports. Fourteen studies (30 reports) (Aggarwal 2002; Akiba 1992; Amon 1992; Frenken 1989; Gertz 2012; Haag‐Weber 2012; Knebel 2008; Kronborg 1994; Mignon 2000; Pergola 2009; Pergola 2010; PROMPT Study 2005; Sohmiya 1998; Spinowitz 2008) were included and 17 studies (19 reports) (Brown 1988; Clyne 1992; Duliege 2005; Furukawa 1992; Li 2004; Meloni 2003; NCT00240734; NCT00492427; NCT00563355; Patel 2012; Schwartz 1989; Shaheen 1983; Singh 1999; Teehan 1990; Teplan 1995; Yamazaki 1993; Zheng 1992) were excluded. Two recently completed studies (NCT01576341; NCT01693029) will be assessed in a future update of this review (Figure 1).
1.
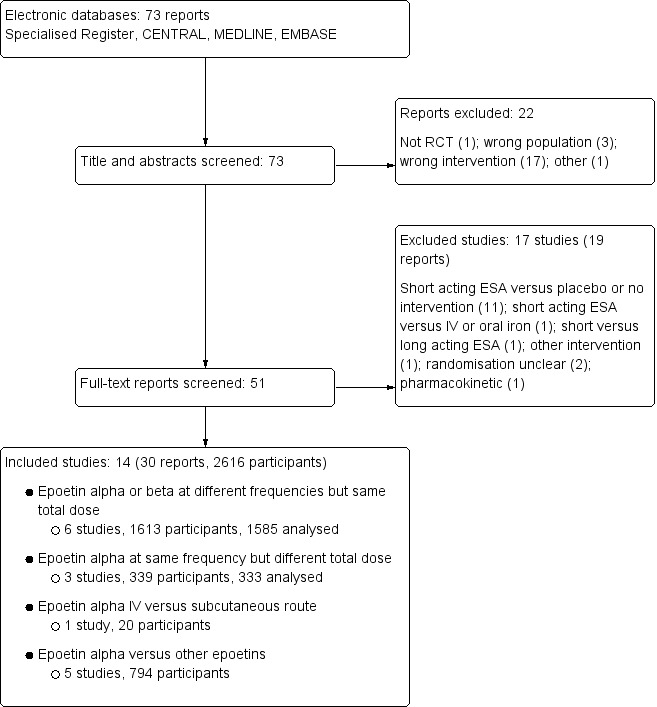
Study flow diagram.
Included studies
The 14 studies were divided into four treatment comparisons groups (Figure 1).
Epoetin alpha or beta administered at different frequencies using the same total dose
Six studies (Amon 1992; Pergola 2009; Pergola 2010; PROMPT Study 2005; Sohmiya 1998; Spinowitz 2008) (1613 enrolled/1585 evaluated participants) compared epoetin alpha or beta at different frequencies using the same total dose in each group.
Four studies (Pergola 2009; Pergola 2010; PROMPT Study 2005; Spinowitz 2008) with 840 enrolled (838 analysed) participants compared epoetin alpha administered at 10,000IU per week with 20,000IU given every two weeks.
Three studies (Pergola 2010; PROMPT Study 2005; Spinowitz 2008) with 724 analysed participants also compared epoetin alpha administered at 20,000 IU every two weeks with 40,000 IU every four weeks.
Amon 1992 (22 enrolled, 18 evaluated) compared subcutaneous epoetin alpha 50 IU/kg three times a week with 150 IU/kg given once weekly in children.
Sohmiya 1998 (5 enrolled, 5 evaluated) compared continuous infusion of epoetin beta with weekly subcutaneous injections for four weeks using the same total dose in each group in a cross over study.
Epoetin alpha administered at the same frequency using different total doses
Three studies (Akiba 1992; Frenken 1989; Spinowitz 2008) (339 enrolled/333 analysed participants) compared epoetin alpha at different doses but at the same frequencies.
Spinowitz 2008 (150 enrolled/144 evaluated) compared 20,000 IU given four weekly with 40 000IU given four weekly
Akiba 1992 (165 enrolled and evaluated) compared 3000 IU, 6000 IU and 12 000 IU given weekly to three groups
Frenken 1989 (24 enrolled and evaluated) compared 50 IU/Kg, 100 IU/kg and 150 IU/Kg given three times weekly.
Epoetin alpha intravenous versus subcutaneous administration
Aggarwal 2002 (20 participants enrolled and evaluated) compared subcutaneous with intravenous administration of epoetin alpha.
Epoetin alpha or beta versus other epoetins or biosimilars of epoetin alpha
Five studies Gertz 2012; Haag‐Weber 2012; Knebel 2008;Kronborg 1994; Mignon 2000) (794 participants) were included in this comparison.
Gertz 2012 (288 enrolled and evaluated) compared weekly subcutaneous epoetin theta with epoetin beta
Haag‐Weber 2012 (337 enrolled) compared a bio‐similar HX575 epoetin alpha with epoetin alpha (Eprex). This study was terminated due to the development of neutralising antibodies in two patients receiving subcutaneous HX575. Efficacy could not be assessed because the authors did not provide the number of patients, who contributed data to efficacy endpoints. Limited safety data were available.
Two studies (Mignon 2000, Knebel 2008) (140 enrolled) compared subcutaneous administration of epoetin delta with epoetin alpha. Both studies were terminated before completion when the pharmaceutical company ceased production of epoetin delta for commercial reasons and no data were available.
Kronborg 1994 (29 enrolled and evaluated) compared pain scores in participants treated with epoetin alpha or epoetin beta given subcutaneously using a visual analogue scale (VAS) and a verbal descriptive scale (VDS). As the results included a median with inter‐quartile ranges the data could not be included in a meta‐analysis.
Excluded studies
Seventeen studies were excluded. Eleven studies were ineligible as they compared a short‐acting ESA with placebo or no treatment (Brown 1988; Clyne 1992; Meloni 2003; NCT00240734; NCT00563355; Patel 2012; Schwartz 1989; Shaheen 1983; Singh 1999; Teehan 1990; Teplan 1995). NCT00492427 compared short‐acting ESA with the long acting ESA, darbepoetin and this study is included in another review (Palmer 2014b). Three studies assessed other interventions, or were pharmacokinetic studies (Duliege 2005; Furukawa 1992; Li 2004). Randomisation was unclear in two studies (Yamazaki 1993; Zheng 1992).
Risk of bias in included studies
The results of the risk of bias assessment are shown in Figure 2 and Figure 3.
2.
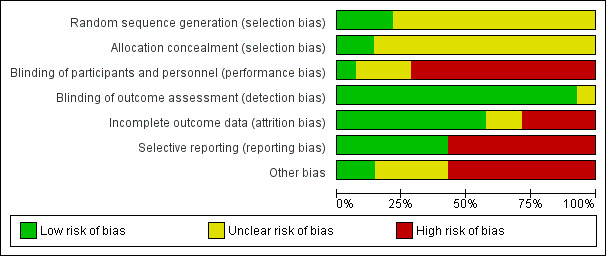
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
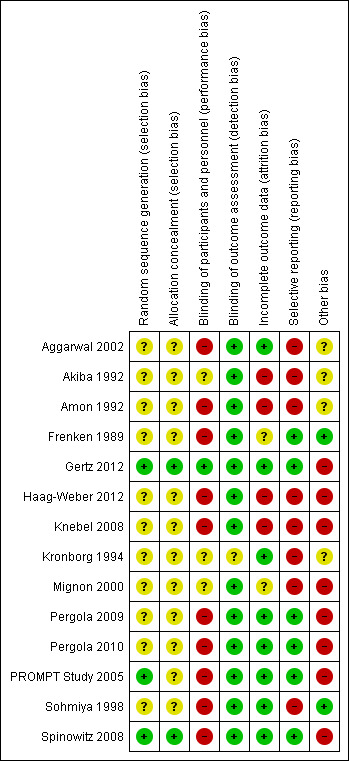
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Sequence generation was deemed at low risk of bias in three studies (Gertz 2012; PROMPT Study 2005; Spinowitz 2008) and unclear in the remaining eleven studies (Aggarwal 2002; Akiba 1992; Amon 1992; Frenken 1989; Haag‐Weber 2012; Knebel 2008; Kronborg 1994; Mignon 2000; Pergola 2009; Pergola 2010; Sohmiya 1998).
Allocation concealment was at low risk of bias in two studies (Gertz 2012; Spinowitz 2008) and unclear in the remaining twelve studies (Aggarwal 2002; Akiba 1992; Amon 1992; Frenken 1989; Haag‐Weber 2012; Knebel 2008; Kronborg 1994; Mignon 2000; Pergola 2009; Pergola 2010; PROMPT Study 2005; Sohmiya 1998).
Blinding
Only one study (Gertz 2012) was blinded and considered to be at low risk of bias for performance bias. Ten studies were not blinded and determined as high risk of performance bias (Aggarwal 2002; Amon 1992; Frenken 1989; Haag‐Weber 2012; Knebel 2008; Pergola 2009; Pergola 2010; PROMPT Study 2005; Sohmiya 1998; Spinowitz 2008). Blinding was unclear in the remaining three studies (Akiba 1992; Kronborg 1994; Mignon 2000).
As the primary outcomes (final Hb level or change in Hb level) in all studies were based on laboratory assessment, and therefore unlikely to be influenced by blinding, 13 studies were deemed to be at low risk of detection bias. The study by Kronborg 1994 was considered at unclear risk of detection bias; it assessed pain scores and was said to be double‐blinded though no information was provided as to how this was performed.
Incomplete outcome data
Eight studies were determined to be at low risk of attrition bias (Aggarwal 2002; Gertz 2012; Kronborg 1994; Pergola 2009; Pergola 2010; PROMPT Study 2005; Sohmiya 1998; Spinowitz 2008). Four studies were at high risk of bias because meta‐analyses could not be performed as total patient numbers were not provided (Akiba 1992; Haag‐Weber 2012; Knebel 2008) or because more than 10% patients were excluded from analysis (Amon 1992). Attrition bias was deemed unclear in the remaining two studies (Frenken 1989; Mignon 2000).
Selective reporting
Studies that did not provide data on final or change in Hb and on patient‐centred outcomes including adverse events such as blood transfusions, vascular access complications or all‐cause mortality were considered to be at high risk for selective reporting. Eight studies were considered at high risk of reporting bias (Aggarwal 2002; Akiba 1992; Amon 1992; Haag‐Weber 2012; Knebel 2008; Kronborg 1994; Mignon 2000; Sohmiya 1998). Six studies (Amon 1992; Gertz 2012; Pergola 2009; Pergola 2010; PROMPT Study 2005; Spinowitz 2008) were assessed at low risk for selective reporting.
Other potential sources of bias
Only two studies were assessed at free of other potential bias sources (Frenken 1989; Sohmiya 1998). Eight studies were industry funded and determined as at high risk of bias (Gertz 2012; Haag‐Weber 2012; Knebel 2008; Mignon 2000; Pergola 2009; Pergola 2010; PROMPT Study 2005; Spinowitz 2008). In the remaining four studies it was unclear whether the study was free of other potential sources of bias (Aggarwal 2002; Akiba 1992; Amon 1992; Kronborg 1994).
Effects of interventions
See: Table 1; Table 2; Table 3
Epoetin alpha administered at different frequencies using the same total dose
Six studies investigated this comparison (Amon 1992; Pergola 2009; Pergola 2010; PROMPT Study 2005; Sohmiya 1998; Spinowitz 2008).
Epoetin alpha weekly versus every two weeks using same total dose of epoetin
In meta‐analyses of four non‐inferiority studies (Pergola 2009; Pergola 2010; PROMPT Study 2005; Spinowitz 2008), final Hb levels (Analysis 1.1 (4 studies, 785 participants): MD ‐0.20 g/dL, 95% CI ‐0.33 to ‐0.07) and the number achieving target Hb were statistically significantly higher in patients receiving weekly doses compared with two weekly doses (Analysis 1.2 (4 studies, 798 participants): RR 0.96, 95% CI 0.93 to 0.99). The differences were not considered to be clinically significant. No significant heterogeneity was noted.
1.1. Analysis.
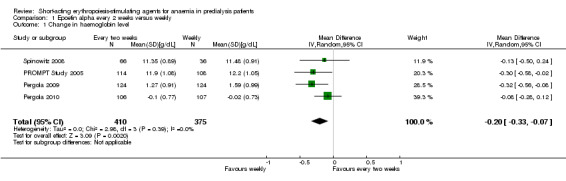
Comparison 1 Epoetin alpha every 2 weeks versus weekly, Outcome 1 Change in haemoglobin level.
1.2. Analysis.
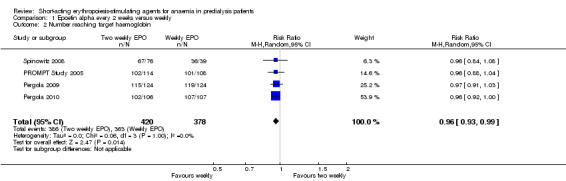
Comparison 1 Epoetin alpha every 2 weeks versus weekly, Outcome 2 Number reaching target haemoglobin.
There were no significant difference in all‐cause mortality (Analysis 1.3 (4 studies, 838 participants): RR 0.89, 95% CI 0.38 to 2.07), the number requiring transfusion (Analysis 1.4.1 (3 studies, 580 participants): RR 1.56, 95% CI 0.71 to 3.45), the number with hypertension (Analysis 1.4.2 (4 studies, 838 participants): RR 0.85, 95% CI 0.55 to 1.32), the number with thrombovascular complications Analysis 1.4.3 (4 studies, 838 participants): RR 1.41, 95% CI 0.67 to 3.00) or the number discontinuing therapy due to adverse effects (Analysis 1.4.4 (1 study, 258 participants): RR 0.98, 95% CI 0.20 to 4.79).
1.3. Analysis.
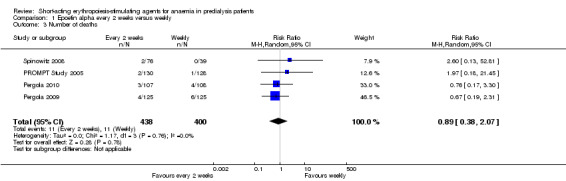
Comparison 1 Epoetin alpha every 2 weeks versus weekly, Outcome 3 Number of deaths.
1.4. Analysis.
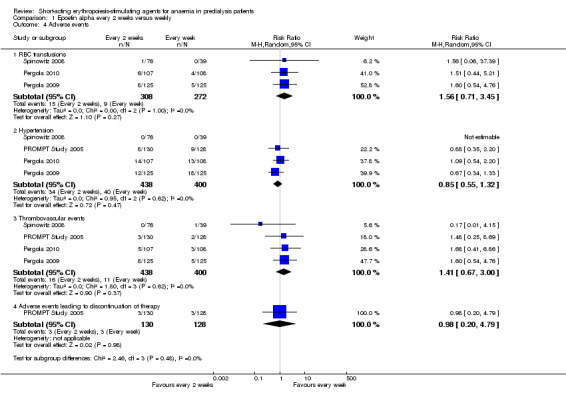
Comparison 1 Epoetin alpha every 2 weeks versus weekly, Outcome 4 Adverse events.
No neutralising antibodies were detected in study participants in Pergola 2009 and Pergola 2010. Most of the deaths were due to cardiovascular complications reflecting the underlying cardiovascular morbidity of the population studied. Only one study (PROMPT Study 2005) performed quality of life (QOL) assessments and reported no statistical differences in the final QOL scores between groups receiving epoetin once weekly or two weekly.
Epoetin alpha every two weeks versus every four weeks using same total dose of epoetin
In meta‐analyses of three non‐inferiority studies (Pergola 2010; PROMPT Study 2005; Spinowitz 2008), there were no significant differences in final Hb levels (Analysis 2.1 (3 studies, 671 participants): MD ‐0.16 g/dL, 95% CI ‐0.43 to 0.10; I2 = 63%) or in the number reaching target Hb levels (Analysis 2.2 (3 studies, 687 participants): RR 0.95, 95% CI 0.84 to 1.07) with dosing every two weeks compared with every four weeks. There was unexplained marked heterogeneity between these studies.
2.1. Analysis.
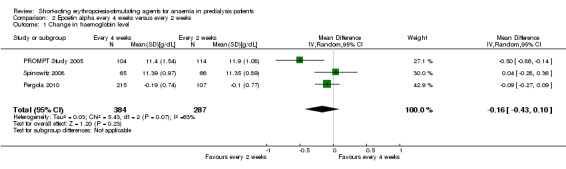
Comparison 2 Epoetin alpha every 4 weeks versus every 2 weeks, Outcome 1 Change in haemoglobin level.
2.2. Analysis.
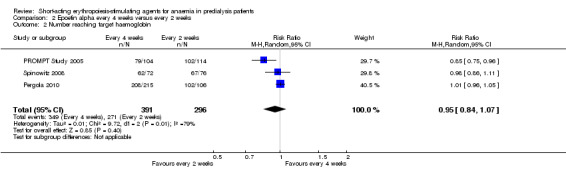
Comparison 2 Epoetin alpha every 4 weeks versus every 2 weeks, Outcome 2 Number reaching target haemoglobin.
There were no significant differences in all‐cause mortality (Analysis 2.3 (3 studies, 724 participants): RR 0.95, 95% CI 0.33 to 2.75), the number requiring transfusions (Analysis 2.4.I (2 studies, 470 participants): RR 1.26, 95% CI 0.53 to 2.98); the number with hypertension (Analysis 2.4.2 (3 studies, 724 participants): RR 1.02, 95% CI 0.62 to 1.69); and the number with thrombovascular complications (Analysis 2.4.3 (3 studies, 724 participants): RR 1.02, 95% CI 0.39 to 2.68). PROMPT Study 2005 noted no difference in final QOL scores between participants who received epoetins at two weekly or four weekly intervals.
2.3. Analysis.
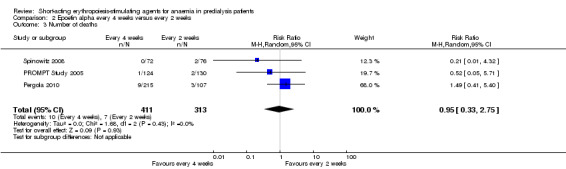
Comparison 2 Epoetin alpha every 4 weeks versus every 2 weeks, Outcome 3 Number of deaths.
2.4. Analysis.
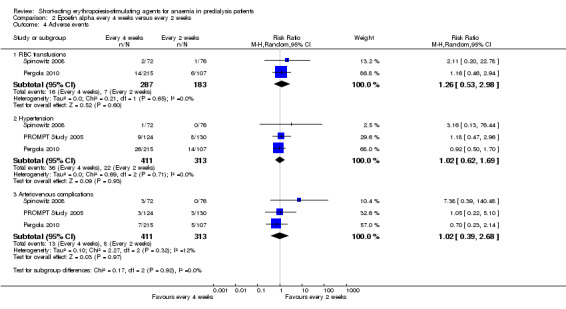
Comparison 2 Epoetin alpha every 4 weeks versus every 2 weeks, Outcome 4 Adverse events.
Other studies
Amon 1992 found that the time to reach a Hb level greater than 11.5 g/dL were significantly longer with weekly administration (15.6 weeks) compared with thrice weekly administration (9.3 weeks). Adverse effects were uncommon and did not differ between groups and there was no deterioration in glomerular filtration rate across the two groups. However there was no significant difference in mean dose/week to sustain Hb levels between different frequencies of administration.
Sohmiya 1998 in a cross‐over study found that continuous subcutaneous infusion of epoetin beta resulted in a significantly greater increase in Hb levels compared with weekly subcutaneous injections (2.56 ± 0.77 g/dL versus 0.28 ± 0.62 g/dL, P < 0.05)
Epoetin alpha administered at same frequency using different total doses
Three studies reported on this comparison (Akiba 1992; Frenken 1989; Spinowitz 2008)
Epoetin alpha different doses given three times weekly
Frenken 1989 reported no statistical significance in the final Hb in groups which received 100 IU/kg/dose (Analysis 3.1.1 (1 study, 16 participants): MD 0.70 g/dL, 95% CI ‐0.78 to 2.18) or 150 IU/kg/dose (Analysis 3.1.2 (1 study, 16 participants): MD 1.00 g/dL, 95% CI ‐0.18 to 2.18) compared with 50 IU/kg/dose. Final mean arterial blood pressures and serum creatinine levels did not differ between subgroups (Analysis 3.2; Analysis 3.3). No anti‐erythropoietin antibodies were detected in the study participants. The study reported overall improvement in well‐being in all participants receiving epoetin.
3.1. Analysis.
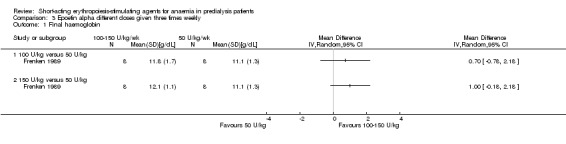
Comparison 3 Epoetin alpha different doses given three times weekly, Outcome 1 Final haemoglobin.
3.2. Analysis.
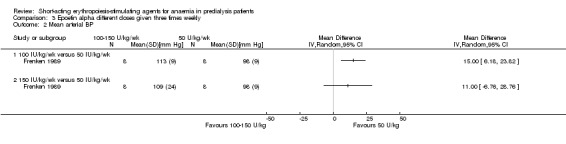
Comparison 3 Epoetin alpha different doses given three times weekly, Outcome 2 Mean arterial BP.
3.3. Analysis.
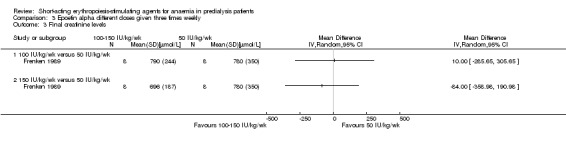
Comparison 3 Epoetin alpha different doses given three times weekly, Outcome 3 Final creatinine levels.
Epoetin alpha different doses given every four weeks
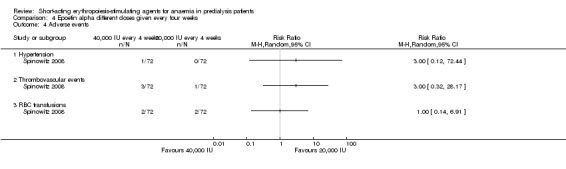
Spinowitz 2008 reported no significant difference in the final Hb level (Analysis 4.1 (1 study, 144 participants): MD 0.17 g/dL 95% CI ‐0.19 to 0.53) and the number reaching target Hb (Analysis 4.2 (1 study, 144 participants): RR 1.07, 95% CI 0.92 to 1.24) when epoetin alpha was administered at 20,000 U compared with 40,000 U every four weeks. There was no significant difference in all‐cause mortality (Analysis 4.3.1), the number with hypertension (Analysis 4.4.1), thrombovascular complications (Analysis 4.4.2) or number of patients requiring transfusions (Analysis 4.4.3).
4.1. Analysis.
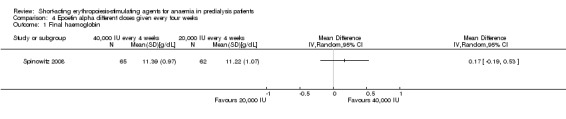
Comparison 4 Epoetin alpha different doses given every four weeks, Outcome 1 Final haemoglobin.
4.2. Analysis.

Comparison 4 Epoetin alpha different doses given every four weeks, Outcome 2 Number reaching target haemoglobin.
4.3. Analysis.
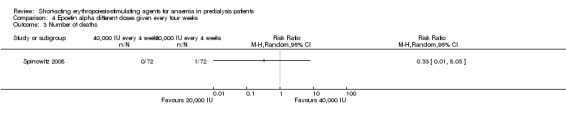
Comparison 4 Epoetin alpha different doses given every four weeks, Outcome 3 Number of deaths.
4.4. Analysis.
Comparison 4 Epoetin alpha different doses given every four weeks, Outcome 4 Adverse events.
Epoetin alpha different doses given every week
Akiba 1992 reported that 6000 IU and 12,000 IU given weekly increased HCT levels more than 3000 IU/week. No standard deviations were provided.
Epoetin alpha intravenous versus subcutaneous administration
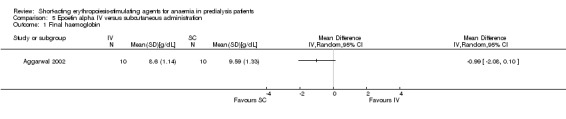
Aggarwal 2002 reported no significant difference in final Hb at 12 weeks (Analysis 5.1 (20 participants): MD ‐0.99 g/dL, 95% CI ‐2.08 to 0.10) between intravenous and subcutaneous administration of epoetin alpha.
5.1. Analysis.
Comparison 5 Epoetin alpha IV versus subcutaneous administration, Outcome 1 Final haemoglobin.
Epoetin alpha versus other epoetins or biosimilars
Five studies compared epoetin alpha with other epoetins or biosimilars.
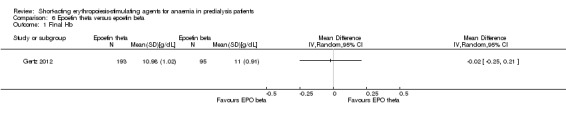
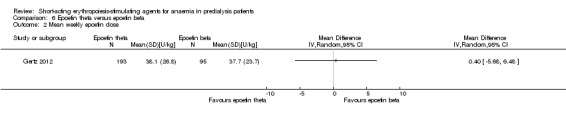
Gertz 2012 found no significant differences in final Hb (Analysis 6.1 (288 participants): MD ‐0.02 g/dL, 95% CI ‐0.25 to 0.21) and weekly epoetin doses (Analysis 6.2 (288 participants) MD 0.40 U/kg, 95% CI ‐5.68 to 6.48) between epoetin theta and epoetin beta. No significant differences were noted in all‐cause mortality (Analysis 6.3 (288 participants): RR 2.46, 95% CI 0.29 to 20.77), hypertension (Analysis 6.4.1 (288 participants): RR 0.35, 95% CI 0.11 to 1.08); transfusions (Analysis 6.4.2 (288 participants): RR 1.48, 95% CI 0.06 to 36.10) and discontinuation of therapy (Analysis 6.4.3 (288 participants): RR 1.77, 95% CI 0.68 to 4.63). No neutralising antibodies were noted in either intervention group. Most of the deaths were due to cardiovascular complications reflecting the population studied.
6.1. Analysis.
Comparison 6 Epoetin theta versus epoetin beta, Outcome 1 Final Hb.
6.2. Analysis.
Comparison 6 Epoetin theta versus epoetin beta, Outcome 2 Mean weekly epoetin dose.
6.3. Analysis.
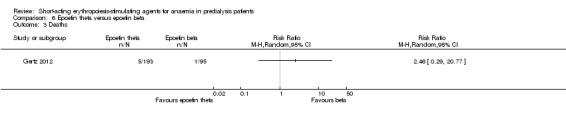
Comparison 6 Epoetin theta versus epoetin beta, Outcome 3 Deaths.
6.4. Analysis.
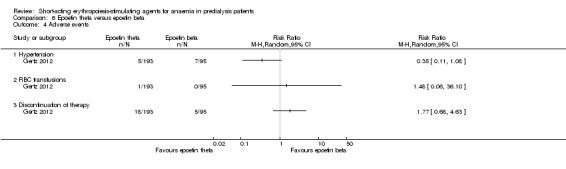
Comparison 6 Epoetin theta versus epoetin beta, Outcome 4 Adverse events.
The quality of this single study was assessed as moderate for the surrogate outcome of final Hb but as low for mean weekly epoetin dose because of imprecision (Table 3). The quality of evidence for patient‐centred outcomes was assessed as low for all‐cause mortality, need for blood transfusion and discontinuation of medications and moderate for hypertension because of imprecision due to small numbers of events
Haag‐Weber 2012 (337 participants) compared the biosimilar HX575 epoetin alpha with epoetin alpha; both medications were administered subcutaneously. The study was ceased when two patients receiving HX575 developed antibodies to epoetin and pure red cell aplasia and HX575 epoetin alpha was withdrawn for subcutaneous administration. The change in Hb from baseline at 13 weeks did not differ between groups (HX575 2.2 ± 0.9 g/dL; epoetin alpha 2.2 ± 1.0 g/dL) but the data could not be included in meta‐analyses since no denominators were provided and information could not be obtained from the authors.
Mignon 2000 (65 participants) found no significant differences in response between epoetin delta and epoetin alpha when both were given at the same dose (50 IU/kg/wk). Adverse events were similar between epoetin delta and epoetin alpha. No data were available from Knebel 2008 (60 participants). Since epoetin delta production was ceased for commercial reasons and no information could be obtained from the pharmaceutical company, no meta‐analyses were performed.
Kronborg 1994 (29 participants) found that pain scores were higher in participants treated with subcutaneous epoetin alpha compared with epoetin beta. The results were provided as median with inter‐quartile ranges so could not be included in a meta‐analysis.
Other outcomes
From the available studies, we were not able to analyse the outcomes of causes of death, cardiovascular and cerebrovascular morbidity, kidney function, number of hospitalisations, additional requirement for IV iron, serious infections, or de novo malignancies.
Discussion
Summary of main results
Fourteen studies (30 reports) with 2616 participants were included in this review, which evaluated the efficacy and safety of short‐acting ESAs in CKD patients, not requiring dialysis.
Six studies (1613 enrolled participants) compared epoetin alpha or epoetin beta at different frequencies using the same total dose in each group, with two studies having insufficient data for inclusion in meta‐analyses. Among four studies, no significant differences in end of study Hb, in the number of participants achieving target Hb, in all‐cause mortality or in adverse effects were identified when dosing every two weeks was compared with weekly dosing or when four weekly dosing was compared with two weekly dosing. These data suggest that larger doses of short‐acting epoetins given less frequently can be administered to CKD patients not requiring dialysis without a loss of efficacy or an increase in adverse effects. However this conclusion is based on low quality evidence (Table 1; Table 2). Three studies (including Spinowitz 2008, in which different frequencies were also evaluated) compared different total doses of epoetin alpha given at the same frequency though data from only two studies could be included in meta‐analyses. Both studies found no significant difference in final Hb or adverse effects with different total doses.
The remaining five studies evaluated different interventions. One study (288 participants) compared epoetin theta with epoetin alpha and found no significant differences in efficacy or adverse effects. Two studies (125 participants) compared epoetin delta with epoetin alpha. However no results were available since the pharmaceutical company withdrew epoetin delta for commercial reasons. The fourth study was terminated before completion after two patients receiving the biosimilar epoetin, HX575 epoetin alpha, developed anti‐epoetin antibodies and pure red cell aplasia. The fifth study found significantly higher pain scores with subcutaneous epoetin alpha compared with epoetin beta.
Overall completeness and applicability of evidence
For this review we were only able to identify 14 studies (2616 participants), which evaluated different frequencies, doses or routes of administration of the same epoetins or compared different epoetins in pre‐dialysis patients. Only seven studies contributed data to meta‐analyses with two of these studies enrolling only 20 and 24 patients respectively. Two studies evaluated children and neither had sufficient data for inclusion in meta‐analyses. Two studies were terminated before completion without available data when the pharmaceutical company ceased production of epoetin delta for commercial reasons. One study was terminated when two patients developed antibody mediated pure red cell aplasia with HX575 epoetin alpha, a biosimilar epoetin. No studies in pre‐dialysis patients were identified, which evaluated HX575 epoetin alpha, given intravenously, or of SB309 (epoetin zeta) given intravenously or subcutaneously. These biosimilar epoetins are approved for use by the European Medicines Agency. Preliminary information indicates that SB309 (epoetin zeta, Epoetin Hospira®) has not been approved for use in the USA (Big Molecule Watch Blog 2015).
Patient‐centred outcomes were generally poorly reported. Only one study reported on a quality of life assessment with no differences identified in end of study quality of life scores between different frequencies of epoetin alpha administration (PROMPT Study 2005). Six studies reported on all‐cause mortality. Data on hypertension and thrombovascular events, adverse effects known to be associated with epoetin administration, could be included in meta‐analyses from only five studies. Data from four studies on the number of participants, requiring blood transfusions, could be included in meta‐analyses. Anti‐erythropoietin antibodies, which can cause pure red cell aplasia, were assessed In only five studies.
Quality of the evidence
Of the fourteen studies included in this review, four studies were available in abstract format only.
Only three of 14 studies demonstrated adequate random sequence generation, with only two studies assessed as showing low risk of bias for allocation concealment. Blinding of participants and personnel was at low risk of bias in one study only. Blinding of outcome assessment was judged at low risk in 13 studies as the outcome measures were laboratory based. Attrition and reporting bias were at low risk of bias in eight and seven studies respectively (Figure 3).
Only five of 14 studies could be included in the Summary of Findings tables as other comparisons included single small studies only or data which could not be included in the meta‐analyses. Overall the quality of the studies (GRADE 2011a; GRADE 2011b) included in meta‐analyses was assessed as low indicating that our confidence in the results is significantly reduced because of poor study quality and the use of surrogate outcomes as the primary outcomes of the included studies.
The quality of the studies included in meta‐analyses comparing epoetin alpha every two weeks with weekly administration (Table 1) and in meta‐analyses comparing epoetin alpha every four weeks with two weekly administration (Table 2) for the efficacy outcomes of change in Hb level and number reaching target Hb were assessed as low or very low. These were down‐graded for indirectness on the GRADE profile, since these were surrogate outcomes and not patient‐centred outcomes. In the comparison of epoetin alpha given every four weeks compared with every two weeks, the results were further downgraded because of marked unexplained heterogeneity. In addition the quality of the evidence for efficacy was downgraded because of poor study design and/or reporting particularly of sequence generation and allocation concealment and for imprecision, where small numbers of events resulted in wide confidence intervals. Outcomes for adverse effects were downgraded because of poor study design and imprecision.
The quality of the single study included in meta‐analyses comparing epoetin theta with epoetin beta was assessed as moderate for the surrogate outcome of final Hb but as low for mean weekly epoetin dose because of imprecision (Table 3). The quality of evidence for patient‐centred outcomes was assessed as low for all‐cause mortality, need for blood transfusion and discontinuation of medications and moderate for hypertension because of imprecision due to small numbers of events.
Potential biases in the review process
For this review a comprehensive search of the Cochrane Kidney and Transplant's Specialised Register was performed, which reduced the likelihood that eligible published studies were omitted from the review. Eligible studies published after the last search date of 12 September 2016 or published in congress proceedings not routinely searched could have been missed. Four studies were only available in abstract form which provided limited information on study methods and results. Inclusion of these studies could be a source of bias.
The review was completed independently by at least three authors, who participated in all steps of the review. This limited the risk of errors in determining study eligibility, data extraction, risk of bias assessment and data synthesis.
Many of the earlier epoetin alpha studies were small with incomplete information on study methods and results. Further information could not be obtained about these studies from investigators or the literature.
Although six of the included studies were sponsored by pharmaceutical companies, these were multi‐centre studies and so were able to attract larger patient numbers. In two of these studies per‐protocol data were included in meta‐analyses for the primary outcomes of final Hb or change in Hb, since ITT data were only presented graphically. In both studies the authors reported that sensitivity analyses using ITT populations were consistent with those from the per‐protocol populations, thus increasing confidence in the findings.
Agreements and disagreements with other studies or reviews
No evidence was identified by the KDIGO working group to suggest that any given type of ESA was superior to another in terms of efficacy and safety (KDIGO 2012). The Working Group suggested ESA choice was dependent on patient and country specific issues including availability, cost and treatment setting. The NICE guidelines 2015 (NICE 2015) suggest the choice of ESA should be discussed with the patient with anaemia and CKD when initiating the patient on treatment, taking into consideration the route of administration and the local availability of ESA. There is no evidence to distinguish between ESAs in term of efficacy. The findings of this systematic review confirm these recommendations.
The findings of this review complement other Cochrane Kidney and Transplant reviews of ESA including an updated review comparing epoetin in pre‐dialysis patients with placebo or no specific treatment (Cody 2016), a review evaluating the benefits and harms of different Hb or HCT targets in CKD patients receiving ESA treatment for anaemia (Strippoli 2006), a review evaluating darbepoetin (Palmer 2014b) and a network meta‐analysis of studies of any ESA formulation (Palmer 2014a). While ESAs clearly reduce the need for blood transfusion, no systematic review to date has found clear evidence for the superiority of any ESA formulation over any other formulation based on available efficacy and safety data.
For consumers, clinicians and funders, considerations such as drug cost, availability and preferences for dosing frequency should be considered as the basis for individualising anaemia care due to lack of data for comparative differences in the clinical benefits and harms of different ESA preparations.
Authors' conclusions
Implications for practice.
A previous review identified that epoetin alpha was effective compared with placebo or no treatment in raising Hb levels without a significant reduction in GFR in patients with CKD not on dialysis (Cody 2016). Our review extends these observations to show that epoetin alpha given at higher doses for extended intervals is non‐inferior to more frequent dosing intervals. The benefits offered by the extended dosing intervals include convenience for the patients and healthcare providers and may also result in cost efficiency. This may be of benefit in countries with more limited resources and access to longer acting more costly ESAs. However the data are of low quality so that differences in efficacy and adverse effects cannot be completely excluded. We did not identify any studies which evaluated different frequencies of epoetin beta. In a single study, epoetin theta did not differ significantly from epoetin beta in haematological outcomes or adverse effects.
We only identified one study in predialysis patients comparing a biosimilar preparation of epoetin alpha (HX575 epoetin alpha, Binocrit®) in non‐dialysis patients and this study was terminated because of the development of pure red cell aplasia with neutralising antibodies (Haag‐Weber 2012). HX575 epoetin alpha and a second biosimilar of epoetin alpha (epoetin zeta, Retacrit®) received marketing authorization throughout the European Union in 2007; HX575 epoetin alpha is limited to intravenous use. Because the safety record of these compounds is limited compared with epoetin alpha and epoetin beta, the ERBP Work Group recommends stringent pharmacovigilance for biosimilars of epoetin alpha (ERBP 2009). Two clinical studies evaluating the biosimilar HX575 epoetin alpha in the USA have been completed (NCT01693029a, NCT01576341a) but the results are not yet available. Of these one (NCT01576341a) evaluated subcutaneous administration of HX575 epoetin alpha in predialysis patients in a single arm study aiming to determine efficacy, adverse effects and the incidence of anti‐epoetin antibodies. The uptake in the nephrology community of the biosimilar ESAs will ultimately depend on the balance between cost savings and residual concerns regarding safety (Mikhail 2013; Schellekens 2009).
Implications for research.
As noted in earlier ESA reviews (Hahn 2014; Strippoli 2006) the reporting of treatment effects of ESAs on potentially important patient outcomes is heterogeneous and poor, thereby limiting a good understanding of how ESA therapy affects the way patients feel and function. Currently decisions regarding different agents in clinical practice are dictated by physician and patient preference, drug cost and availability since we have inconclusive evidence of the effects of different short‐acting ESAs or of different frequencies of ESA administration on survival and quality of life. Data regarding effectiveness and safety when treating children with ESAs remains limited.
Therefore additional large, well designed, randomised studies are required in the following areas to compare larger doses of the shorter acting ESAs including new biosimilars of epoetin alpha administered less frequently with more frequent dosing in both children and adults with CKD not on dialysis. These studies should include patient‐centred outcomes including all‐cause mortality, cardiovascular mortality and morbidity, and quality of life assessment. Estimates of patient and carer satisfaction related to different frequencies of administration should be included. Studies of cost‐effectiveness of different frequencies of administration should also be undertaken.
Acknowledgements
We acknowledge the assistance of the Cochrane Kidney and Transplant editorial office for their assistance with this review. We would also like to thank the referees for their feedback and advice during the preparation of this review.
Appendices
Appendix 1. Electronic search strategies
| Database | Search terms |
| CENTRAL |
|
| MEDLINE |
|
| EMBASE |
|
Appendix 2. Risk of bias assessment tool
| Potential source of bias | Assessment criteria |
|
Random sequence generation Selection bias (biased allocation to interventions) due to inadequate generation of a randomised sequence |
Low risk of bias: Random number table; computer random number generator; coin tossing; shuffling cards or envelopes; throwing dice; drawing of lots; minimization (minimization may be implemented without a random element, and this is considered to be equivalent to being random). |
| High risk of bias: Sequence generated by odd or even date of birth; date (or day) of admission; sequence generated by hospital or clinic record number; allocation by judgement of the clinician; by preference of the participant; based on the results of a laboratory test or a series of tests; by availability of the intervention. | |
| Unclear: Insufficient information about the sequence generation process to permit judgement. | |
|
Allocation concealment Selection bias (biased allocation to interventions) due to inadequate concealment of allocations prior to assignment |
Low risk of bias: Randomisation method described that would not allow investigator/participant to know or influence intervention group before eligible participant entered in the study (e.g. central allocation, including telephone, web‐based, and pharmacy‐controlled, randomisation; sequentially numbered drug containers of identical appearance; sequentially numbered, opaque, sealed envelopes). |
| High risk of bias: Using an open random allocation schedule (e.g. a list of random numbers); assignment envelopes were used without appropriate safeguards (e.g. if envelopes were unsealed or non‐opaque or not sequentially numbered); alternation or rotation; date of birth; case record number; any other explicitly unconcealed procedure. | |
| Unclear: Randomisation stated but no information on method used is available. | |
|
Blinding of participants and personnel Performance bias due to knowledge of the allocated interventions by participants and personnel during the study |
Low risk of bias: No blinding or incomplete blinding, but the review authors judge that the outcome is not likely to be influenced by lack of blinding; blinding of participants and key study personnel ensured, and unlikely that the blinding could have been broken. |
| High risk of bias: No blinding or incomplete blinding, and the outcome is likely to be influenced by lack of blinding; blinding of key study participants and personnel attempted, but likely that the blinding could have been broken, and the outcome is likely to be influenced by lack of blinding. | |
| Unclear: Insufficient information to permit judgement | |
|
Blinding of outcome assessment Detection bias due to knowledge of the allocated interventions by outcome assessors. |
Low risk of bias: No blinding of outcome assessment, but the review authors judge that the outcome measurement is not likely to be influenced by lack of blinding; blinding of outcome assessment ensured, and unlikely that the blinding could have been broken. |
| High risk of bias: No blinding of outcome assessment, and the outcome measurement is likely to be influenced by lack of blinding; blinding of outcome assessment, but likely that the blinding could have been broken, and the outcome measurement is likely to be influenced by lack of blinding. | |
| Unclear: Insufficient information to permit judgement | |
|
Incomplete outcome data Attrition bias due to amount, nature or handling of incomplete outcome data. |
Low risk of bias: No missing outcome data; reasons for missing outcome data unlikely to be related to true outcome (for survival data, censoring unlikely to be introducing bias); missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk not enough to have a clinically relevant impact on the intervention effect estimate; for continuous outcome data, plausible effect size (difference in means or standardized difference in means) among missing outcomes not enough to have a clinically relevant impact on observed effect size; missing data have been imputed using appropriate methods. |
| High risk of bias: Reason for missing outcome data likely to be related to true outcome, with either imbalance in numbers or reasons for missing data across intervention groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk enough to induce clinically relevant bias in intervention effect estimate; for continuous outcome data, plausible effect size (difference in means or standardized difference in means) among missing outcomes enough to induce clinically relevant bias in observed effect size; ‘as‐treated’ analysis done with substantial departure of the intervention received from that assigned at randomisation; potentially inappropriate application of simple imputation. | |
| Unclear: Insufficient information to permit judgement | |
|
Selective reporting Reporting bias due to selective outcome reporting |
Low risk of bias: The study protocol is available and all of the study’s pre‐specified (primary and secondary) outcomes that are of interest in the review have been reported in the pre‐specified way; the study protocol is not available but it is clear that the published reports include all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon). |
| High risk of bias: Not all of the study’s pre‐specified primary outcomes have been reported; one or more primary outcomes is reported using measurements, analysis methods or subsets of the data (e.g. subscales) that were not pre‐specified; one or more reported primary outcomes were not pre‐specified (unless clear justification for their reporting is provided, such as an unexpected adverse effect); one or more outcomes of interest in the review are reported incompletely so that they cannot be entered in a meta‐analysis; the study report fails to include results for a key outcome that would be expected to have been reported for such a study. | |
| Unclear: Insufficient information to permit judgement | |
|
Other bias Bias due to problems not covered elsewhere in the table |
Low risk of bias: The study appears to be free of other sources of bias. |
| High risk of bias: Had a potential source of bias related to the specific study design used; stopped early due to some data‐dependent process (including a formal‐stopping rule); had extreme baseline imbalance; has been claimed to have been fraudulent; had some other problem. | |
| Unclear: Insufficient information to assess whether an important risk of bias exists; insufficient rationale or evidence that an identified problem will introduce bias. |
Data and analyses
Comparison 1. Epoetin alpha every 2 weeks versus weekly.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in haemoglobin level | 4 | 785 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.33, ‐0.07] |
| 2 Number reaching target haemoglobin | 4 | 798 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.93, 0.99] |
| 3 Number of deaths | 4 | 838 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.38, 2.07] |
| 4 Adverse events | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 RBC transfusions | 3 | 580 | Risk Ratio (M‐H, Random, 95% CI) | 1.56 [0.71, 3.45] |
| 4.2 Hypertension | 4 | 838 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.55, 1.32] |
| 4.3 Thrombovascular events | 4 | 838 | Risk Ratio (M‐H, Random, 95% CI) | 1.41 [0.67, 3.00] |
| 4.4 Adverse events leading to discontinuation of therapy | 1 | 258 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.20, 4.79] |
Comparison 2. Epoetin alpha every 4 weeks versus every 2 weeks.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in haemoglobin level | 3 | 671 | Mean Difference (IV, Random, 95% CI) | ‐0.16 [‐0.43, 0.10] |
| 2 Number reaching target haemoglobin | 3 | 687 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.84, 1.07] |
| 3 Number of deaths | 3 | 724 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.33, 2.75] |
| 4 Adverse events | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 RBC transfusions | 2 | 470 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.53, 2.98] |
| 4.2 Hypertension | 3 | 724 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.62, 1.69] |
| 4.3 Arteriovenous complications | 3 | 724 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.39, 2.68] |
Comparison 3. Epoetin alpha different doses given three times weekly.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Final haemoglobin | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 1.1 100 U/kg versus 50 U/kg | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 150 U/kg versus 50 U/kg | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Mean arterial BP | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2.1 100 IU/kg/wk versus 50 IU/kg/wk | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 150 IU/kg/wk versus 50 IU/kg/wk | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Final creatinine levels | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3.1 100 IU/kg/wk versus 50 IU/kg/wk | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 150 IU/kg/wk versus 50 IU/kg/wk | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 4. Epoetin alpha different doses given every four weeks.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Final haemoglobin | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Number reaching target haemoglobin | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Number of deaths | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Hypertension | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Thrombovascular events | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 RBC transfusions | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 5. Epoetin alpha IV versus subcutaneous administration.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Final haemoglobin | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 6. Epoetin theta versus epoetin beta.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Final Hb | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Mean weekly epoetin dose | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Deaths | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Hypertension | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 RBC transfusions | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Discontinuation of therapy | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Aggarwal 2002.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Lack of blinding could influence patient management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory based outcome and unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients included in assessment |
| Selective reporting (reporting bias) | High risk | No information on adverse effects |
| Other bias | Unclear risk | Insufficient information to permit judgement |
Akiba 1992.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment groups
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Said to be double blind, though insufficient information to permit judgement |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcome and blinding unlikely to influence |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No patient denominators supplied |
| Selective reporting (reporting bias) | High risk | Insufficient information to permit judgement (abstract only), however no full text publication by December 2016 |
| Other bias | Unclear risk | Insufficient information to permit judgement |
Amon 1992.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement ‐ says study is "prospective randomized multi‐centre study" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding and lack of blinding could influence management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcomes of interest are laboratory based and unlikely to be affected by knowledge of allocation |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol analysis was used, 18% of the children randomised to either treatment were excluded from analysis |
| Selective reporting (reporting bias) | High risk | All pre‐specified outcomes reported; however no full‐text publication by December 2016 |
| Other bias | Unclear risk | Insufficient information to permit judgement |
Frenken 1989.
| Methods |
|
|
| Participants |
|
|
| Interventions | Correction phase
Maintenance phase (at end of 8 weeks; non‐randomised)
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Lack of blinding could influence patient management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary outcome was laboratory based and unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes reported |
| Other bias | Low risk | Medication provided by Ortho Pharmaceutical Corporation (Raritan, New Jersey, USA) and Cilag B.V. (Herentals, Belgium) |
Gertz 2012.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Type of epoetin
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation via an interactive voice response system |
| Allocation concealment (selection bias) | Low risk | "randomised to treatment by using central randomisation via IVRS" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Similar syringes of intervention EPO Administration of intervention medication by a person who was not part of the study |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory‐based outcome and unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 89% were included of those randomised were included in the ITT population |
| Selective reporting (reporting bias) | Low risk | Studies pre‐specified outcomes reported |
| Other bias | High risk | Study and paper writing assistance was sponsored by a Pharma |
Haag‐Weber 2012.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group A
Treatment group B
Both groups
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "block randomisation, stratified by center" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Lack of blinding could influence patient management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory based outcome and unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Analysis done per‐protocol method. Study terminated & data in uncertain number of subjects only available to 13 weeks |
| Selective reporting (reporting bias) | High risk | Outcomes of interest for this review such as change in EPO dose, number reaching target Hb were presented in ways that could not be used in a meta‐analysis |
| Other bias | High risk | Study and paper writing assistance was sponsored by a Pharma |
Knebel 2008.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Phase III randomised, multi‐centre study. appropriate 3:1 ratio to epoetin delta or epoetin alfa |
| Allocation concealment (selection bias) | Unclear risk | No information provided other than that randomisation stratified by age and centre |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Lack of blinding could influence patient management |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary outcome was laboratory based and unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No data on outcomes provided |
| Selective reporting (reporting bias) | High risk | No data on outcomes provided |
| Other bias | High risk | Pharmaceutical study Analysis funded by Shire Pharmaceuticals Study terminated for commercial reasons |
Kronborg 1994.
| Methods |
|
|
| Participants |
|
|
| Interventions |
Treatment group 2
Control group
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Comparative double‐blind randomised placebo controlled study" but unclear how performed |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Said to double‐blind but unclear how this was achieved |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear who performed outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients received all injections and contributed to results |
| Selective reporting (reporting bias) | High risk | Insufficient information to permit judgement (abstract only), however no full text publication by December 2016 |
| Other bias | Unclear risk | Insufficient information to permit judgement |
Mignon 2000.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Treatment group 4
Control group
All groups
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "12 week multi‐centre, randomised, double blind parallel group study" but not details provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Said to be double blind but no information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary outcome was laboratory based and unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information provided. Abstracts only |
| Selective reporting (reporting bias) | High risk | Abstracts only, no full text publication by December 2016 |
| Other bias | High risk | Aventis Pharma/Shire PLC |
Pergola 2009.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Both groups
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "subjects ... were randomly assigned" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory based outcome and unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients accounts for and modified ITT data used |
| Selective reporting (reporting bias) | Low risk | Stated and important outcomes of interest were reported |
| Other bias | High risk | The study and its authorship were supported by pharmaceutical firm |
Pergola 2010.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Both groups
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Said that "subjects ... were randomly assigned" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory‐based outcome and unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients accounts for and modified ITT data used |
| Selective reporting (reporting bias) | Low risk | Pre‐specified outcomes mentioned |
| Other bias | High risk | The study and its authorship were supported by pharmaceutical firm |
PROMPT Study 2005.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Treatment group 4
All groups
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory‐based outcome and unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 90% of the patients who received epoetin were included in efficacy analysis |
| Selective reporting (reporting bias) | Low risk | Pre‐specified outcomes mentioned |
| Other bias | High risk | The study and its authorship were supported by pharmaceutical firm |
Sohmiya 1998.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Cross‐over study with 4‐week washout period |
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The cross‐over comparative study of two protocols was randomly performed for each patient with wash out period of 4 weeks" |
| Allocation concealment (selection bias) | Unclear risk | Unclear. No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding performed |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | End points were laboratory‐based |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data reported on all patients for each treatment |
| Selective reporting (reporting bias) | High risk | No report of adverse effects Data not provided in format that allowed results to be included in meta‐analyses |
| Other bias | Low risk | Grants from Japanese Government and research organisations |
Spinowitz 2008.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Treatment group 4
All groups
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation schedule |
| Allocation concealment (selection bias) | Low risk | Centrally generated using an interactive voice response system |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory‐based outcome and unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The primary end points were presented for over 98% of the study population using modified ITT analysis |
| Selective reporting (reporting bias) | Low risk | Pre‐specified outcomes mentioned |
| Other bias | High risk | The study and its authorship were supported by a pharmaceutical firm |
BUN ‐ blood urea nitrogen; CKD ‐ chronic kidney disease; EPO ‐ erythropoietin; ESA ‐ erythropoiesis‐stimulating agent/s; GFR ‐ glomerular filtration rate; Hb ‐ haemoglobin; HCT ‐ haematocrit; HTN ‐ hypertension; ITT ‐ intention‐to‐treat; IV ‐ intravenous; IVI ‐ IV infusion: M/F ‐ male/female; PRCA ‐ pure red cell aplasia; RBC ‐ red blood cell/s; RCT ‐ randomised controlled trial; SC ‐ subcutaneous; SCr ‐ serum creatinine; SD ‐ standard deviation; VAS ‐ visual analogue scale; VDS ‐ verbal descriptive scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Brown 1988 | Ineligible intervention; compares ESA with placebo |
| Clyne 1992 | Ineligible intervention; compares ESA with no treatment |
| Duliege 2005 | Phase 2 study of synthetic non‐epoetin agent |
| Furukawa 1992 | Pharmacokinetic study |
| Li 2004 | Ineligible intervention; compares Bushen Jianpi Recipe + ESA with ESA |
| Meloni 2003 | Ineligible intervention; compares short‐acting ESA with no ESA |
| NCT00240734 | Ineligible intervention; compares short‐acting ESA with placebo Study terminated because of slow enrolment and no results posted |
| NCT00492427 | Ineligible intervention; compares long acting ESA with short‐acting ESA |
| NCT00563355 | Ineligible intervention; compares short‐acting ESA with no treatment |
| Patel 2012 | Ineligible intervention; compares extended duration of short‐acting ESA with standard care for institution |
| Schwartz 1989 | Ineligible intervention; compares short‐acting ESA with placebo |
| Shaheen 1983 | Ineligible intervention; compares short‐acting ESA with no treatment |
| Singh 1999 | Ineligible intervention; compares short‐acting ESA with no treatment |
| Teehan 1990 | Ineligible intervention; compares short‐acting ESA with placebo |
| Teplan 1995 | Ineligible intervention; compares short‐acting ESA to no treatment |
| Yamazaki 1993 | Unclear whether this study is randomised |
| Zheng 1992 | Unclear whether this study is randomised |
ESA ‐ erythropoiesis‐stimulating agent/s
Characteristics of studies awaiting assessment [ordered by study ID]
NCT01576341.
| Methods |
|
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT01693029.
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
AE ‐ adverse events; CKD ‐ chronic kidney disease; EPO ‐ erythropoietin; ESA ‐ erythropoiesis‐stimulating agent/s; ESKD ‐ end‐stage kidney disease; Hb ‐ haemoglobin; HIV ‐ human immunodeficiency virus; IV ‐ intravenous; PRCA ‐ pure red cell aplasia; RCT ‐ randomised controlled trial; SC ‐ subcutaneous; SLE ‐ Systemic lupus erythematous
Differences between protocol and review
Removal of the comparison of short‐acting ESAs in evaluating the benefits and harms of different Hb or HCT targets in CKD patients receiving ESA treatment for anaemia as this is included in another systematic review (Strippoli 2006).
Contributions of authors
Draft the protocol: DH, EH
Study selection: DH, EH, NE
Extract data from studies: DH, EH, CE, NE
Enter data into RevMan: DH, CE, EH
Carry out the analysis: DH, EH, CE
Interpret the analysis: DH, EH
Draft the final review: DH, EH
Disagreement resolution: AW
Update the review: DH, EH
Declarations of interest
Deirdre Hahn: none known
Elisabeth M Hodson: none known
Angela C Webster: none known
Noha Elserafy: none known
Christopher Esezobor: none known
New
References
References to studies included in this review
Aggarwal 2002 {published data only}
- Aggarwal HK, Kaushik G, Singh S, Nand N, Singh M. Comparison of efficacy of subcutaneous versus intravenous erythropoeitin in anaemia of chronic renal failure [abstract no: 227]. Journal of the Association of Physicians of India 2001;49(Jan). [CENTRAL: CN‐01094842] [Google Scholar]
- Aggarwal HK, Nand N, Singh S, Singh M, Kaushik G. Comparative efficacy of subcutaneous versus intravenous dose of erythropoietin in pre‐dialysis patients of chronic renal failure. Journal of Indian Academy of Clinical Medicine 2002;3(1):46‐50. [CENTRAL: CN‐00689631] [Google Scholar]
Akiba 1992 {published data only}
- Akiba T, Koshikawa S, Mimura N, Maeda T, KRN5702 Pre‐Dialysis Study Group. Randomized double‐blind study of subcutaneous recombinant human erythropoietin in predialysis uraemic patients [abstract]. Nephrology Dialysis Transplantation 1992;7(7):765. [CENTRAL: CN‐00260735] [Google Scholar]
Amon 1992 {published data only}
- Amon O, Muller‐Wiefel DE, Daniel K, Eife R, Leititis J, Stolpe HJ. Is S.C. erythropoietin (rhEPO) treatment thrice per week superior to an application once per week? [abstract]. 9th Congress. International Pediatric Nephrology Association; 1992 Aug 30 ‐ Sep 4; Jerusalem, Israel. 1992:C143. [CENTRAL: CN‐00483004]
Frenken 1989 {published data only}
- Frenken LA, Verberckmoes R, Michielsen P, Koene RA. Efficacy and tolerance of treatment with recombinant‐human erythropoietin in chronic renal failure (pre‐dialysis) patients. Nephrology Dialysis Transplantation 1989;4(9):782‐6. [MEDLINE: ] [PubMed] [Google Scholar]
- Frenken LA, Verberckmoes R, Sluiter HE, Schrijver G, Michielsen P, Koene RA. An open study of the safety and efficacy of multiple doses of recombinant human erythropoietin in end‐stage renal disease (predialysis) patients [abstract]. Nephrology Dialysis Transplantation 1988;3(4):495. [CENTRAL: CN‐00260377] [Google Scholar]
- Frenken LA, Verberckmoes R, Sluiter HE, Schrijver G, Michielsen P, Koene RA. An open study of the safety and efficacy of multiple doses of recombinant‐human erythropoietin in end‐state renal disease (pre‐dialysis) patients [abstract]. Kidney International 1988;34(4):558. [CENTRAL: CN‐00644276] [Google Scholar]
- Koene R, Frenken L. Does treatment of predialysis patients with recombinant human erythropoietin compromise renal function?. Nefrologia 1990;10(Suppl 2):131‐6. [DOI] [PubMed] [Google Scholar]
- Koene RA, Frenken LA. Does treatment of predialysis patients with recombinant human erythropoietin compromise renal function?. Contributions to Nephrology 1990;87:105‐12. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Gertz 2012 {published data only}
- Gertz B, Kes P, Essaian A, Bias P, Buchner A, Zellner D. Epoetin theta: efficacy and safety of subcutaneous administration in anemic pre‐dialysis patients in the maintenance phase in comparison to epoetin beta. Current Medical Research & Opinion 2012;28(7):1101‐10. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Haag‐Weber 2012 {published data only}
- Haag‐Weber M, Eckardt KU, Horl WH, Roger SD, Vetter A, Roth K. Safety, immunogenicity and efficacy of subcutaneous biosimilar epoetin‐alpha (HX575) in non‐dialysis patients with renal anemia: a multi‐center, randomized, double‐blind study. Clinical Nephrology 2012;77(1):8‐17. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Knebel 2008 {published data only}
- Knebel W, Palmen M, Dowell JA, Gastonguay M. Population pharmacokinetic modeling of epoetin delta in pediatric patients with chronic kidney disease. Journal of Clinical Pharmacology 2008;48(7):837‐48. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kronborg 1994 {published data only}
- Kronborg J, Willassen Y, Stromsaether CE, Paulsen D. Pain after subcutaneous injection of erythropoietin (EPO). A comparison of two different preparations [abstract]. Journal of the American Society of Nephrology 1994;5(3):460. [Google Scholar]
Mignon 2000 {published data only}
- Mignon F, Andrassy K, Esnault VL, Kuhlmann MK, Sinnassamy P. Epoetin delta corrects and maintains haemoglobin in pre‐end stage renal disease [abstract]. 38th Congress. European Renal Association. European Dialysis and Transplantation Association; 2001 Jun 24‐27; Vienna, Austria. 2001:140. [CENTRAL: CN‐00461317]
- Mignon F, Andrassy K, Esnault VL, Kuhlmann MK, Sinnassamy P, European HMR4396 Study Group. Novel human erythropoietin corrects and maintains hemoglobin in pre‐end stage renal disease (ESRD) when administered twice weekly subcutaneously [abstract no: A0384]. Journal of the American Society of Nephrology 2000;11(Sept):71A. [MEDLINE: ] [Google Scholar]
- Pratt RD. Epoetin delta for the treatment of anemia in patients with CKD not requiring hemodialysis [abstract no: TH‐PO377]. Journal of the American Society of Nephrology 2006;17(Abstracts):187A. [CENTRAL: CN‐00765050] [Google Scholar]
Pergola 2009 {published data only}
- Pergola PE, Gartenberg G, Fu M, Bowers P. Extended dosing regimens of epoetin alfa (EPO) in EPO‐naive, pre‐dialysis subjects with anemia of chronic kidney disease (CKD): an open‐label randomized study [abstract no: F‐PO1833]. Journal of the American Society of Nephrology 2008;19(Abstracts Issue):523A. [CENTRAL: CN‐00747297] [Google Scholar]
- Pergola PE, Gartenberg G, Fu M, Wolfson M, Rao S, Bowers P. A randomized controlled study of weekly and biweekly dosing of epoetin alfa in CKD patients with anemia. Clinical Journal of the American Society of Nephrology: CJASN 2009;4(11):1731‐40. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pergola 2010 {published data only}
- Pergola P, Gartenberg G, Fu M, Bowers P, Sun S. Every 2 and 4 week dosing with epoetin alfa is non‐inferior to once weekly dosing in pre‐dialysis subjects with anemia of chronic kidney disease: an open‐label randomized study [abstract no: F‐PO1095]. Journal of the American Society of Nephrology 2009;20(Abstracts):364A. [Google Scholar]
- Pergola PE, Gartenberg G, Fu M, Sun S, Wolfson M, Bowers P. A randomized controlled study comparing once‐weekly to every‐2‐week and every‐4‐week dosing of epoetin alfa in CKD patients with anemia. Clinical Journal of the American Society of Nephrology: CJASN 2010;5(4):598‐606. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
PROMPT Study 2005 {published data only}
- Provenzano R, Bhaduri S, Singh AK, PROMPT Study Group. Extended epoetin alfa dosing as maintenance treatment for the anemia of chronic kidney disease: The PROMPT study. Clinical Nephrology 2005;64(2):113‐23. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Provenzano R, Bhaduri S, Tang KL, Klausner M, Singh A. Prospective, randomized evaluation of extended epoetin alfa dosing for treatment of anemia of chronic kidney disease (CKD): PROMPT interim analysis. [abstract no: SA‐PO729]. Journal of the American Society of Nephrology 2003;14(Nov):457A‐8A. [CENTRAL: CN‐00550618] [Google Scholar]
- Provenzano R, Bhaduri S, Tang L, Klausner M, Singh AK. Randomized, controlled trial of extended epoetin alfa dosing for the treatment of patients with the anemia of chronic kidney disease: final results from the PROMPT Study. [abstract no: SU‐PO055]. Journal of the American Society of Nephrology 2004;15(Oct):544A. [CENTRAL: CN‐00583874] [Google Scholar]
- Provenzano R, Singh AK. Hemoglobin maintenance with use of extended dosing of epoetin alfa in patients with diabetes and anemia of chronic kidney disease. Endocrine Practice 2007;13(3):251‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sohmiya 1998 {published data only}
- Sohmiya M, Kakiba T, Kato Y. Therapeutic use of continuous subcutaneous infusion of recombinant human erythropoietin in malnourished predialysis anemic patients with diabetic nephropathy. European Journal of Endocrinology 1998;139(4):367‐70. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Spinowitz 2008 {published data only}
- McGowan T, Beaver JS, Wolfson M. Pharmacokinetic and pharmacodynamic profiles of epoetin alfa (EPO) in anemic subjects with chronic kidney disease [abstract no: SU‐PO787]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):758A. [CENTRAL: CN‐00765903] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan T, Tang L, Wolfson M. Analysis of hemoglobin decline following hemoglobin increase to >12 g/dL when initiating epoetin alfa using extended dosing regimens [abstract no: SU‐PO786]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):758A. [CENTRAL: CN‐00763607] [Google Scholar]
- McGowan T, Vaccaro NM, Beaver JS, Massarella J, Wolfson M. Pharmacokinetic and pharmacodynamic profiles of extended dosing of epoetin alfa in anemic patients who have chronic kidney disease and are not on dialysis. Clinical Journal of the American Society of Nephrology: CJASN 2008;3(4):1006‐14. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinowitz B, Germain M, Benz R, Wolfson M, McGowan T, Tang KL, et al. A randomized study of extended dosing regimens for initiation of epoetin alfa treatment for anemia of chronic kidney disease. Clinical Journal of the American Society of Nephrology: CJASN 2008;3(4):1015‐21. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson M, Spinowitz B, Germain M, Benz R, Epoetin AEDSG. Extended dosing regimens for initiation of epoetin alfa for treatment of anemia of chronic kidney disease [abstract no: 238]. American Journal of Kidney Diseases 2007;49(4):B84. [CENTRAL: CN‐00644242] [Google Scholar]
References to studies excluded from this review
Brown 1988 {published data only}
- Brown CD, Kieran M, Dosunmu BV, Zhao Z, Larson RH, Friedman EA. Raised hematocrit (HCT) persists six‐weeks after stopping treatment with human recombinant erythropoietin (r‐HuEPO) in azotemic anemic patients [abstract]. Kidney International 1988;33(1):184. [CENTRAL: CN‐00602117] [Google Scholar]
Clyne 1992 {published data only}
- Clyne N, Jogestrand T. Effect of erythropoietin treatment on physical exercise capacity and on renal function in predialytic uremic patients. Nephron 1992;60(4):390‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Duliege 2005 {published data only}
- Duliege AM, Macdougall I, Duncan N, Wessels D, Iwashita J, Schatz P, et al. Hematide, a synthetic peptide‐based erythropoiesis stimulating agent (ESA), demonstrates erythropoietic activity in a phase 2 single dose, dose escalating study in patients with chronic kidney disease (CKD) [abstract no: 3532]. Blood 2005;106(11 Pt 1):986. [CENTRAL: CN‐00624867] [Google Scholar]
- Duliege AM, Macdougall IC, Goldsmith D, Wessels D, Iwashita J, Schatz P, et al. HematideTM, a synthetic peptide‐based erythropoiesis stimulating agent (ESA), demonstrates erythropoietic activity in single dose studies in normal healthy volunteers (HV) and in patients with chronic kidney disease (CKD) [abstract no: SP420]. Nephrology Dialysis Transplantation 2006;21(Suppl 4):iv155. [CENTRAL: CN‐00797702] [Google Scholar]
Furukawa 1992 {published data only}
- Furukawa A, Numata A, Imagawa A, Kaifu Y, Sumikura T, Miyake H, et al. Study of recombinant human erythropoietin treatment on the anemia of predialysis patients. Nippon Jinzo Gakkai Shi [Japanese Journal of Nephrology] 1992, (6):693‐700. [MEDLINE: ] [PubMed] [Google Scholar]
Li 2004 {published data only}
- Li QF, Ma QY, Zhu CF. Effect of combination of bushen jianpi recipe and erythropoietin on serum tumor necrosis factor alpha in patients with anemia. Zhongguo Zhongxiyi Jiehe Zazhi [Chinese Journal of Integrated Traditional & Western Medicine] 2004;24(2):106‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Meloni 2003 {published data only}
- Meloni C, Tozzo C, Rossi V, Borzi M, Flamini M, Grotta BD, et al. Early anaemia correction with EPO: one year effects on LVH and progression of chronic renal failure (CRF) in predialysis patients (PTS) [abstract]. Nephrology Dialysis Transplantation 2003;18(Suppl 4):157. [CENTRAL: CN‐00446732] [Google Scholar]
NCT00240734 {published data only}
- NCT00240734. A randomized, double‐blind, placebo‐controlled study evaluating weekly epoetin alfa (PROCRIT) administration on hemoglobin response and safety in diabetic subjects with the anemia of chronic kidney disease (CKD). www.clinicaltrials.gov/ct2/show/NCT00240734 (accessed 15 December 2016).
NCT00492427 {published data only}
- Baba T. Correction and maintenance study of subcutaneous injections of R744 to predialysis patients (phase III, comparative study in comparison with epoetin beta). www.clinicaltrials.gov/ct2/show/NCT00492427 (accessed 15 December 2016).
NCT00563355 {published data only}
- Wang AYM. A prospective randomised controlled trial to study the effects of recombinant human erythropoietin on the progression of atherosclerosis, cardiovascular function, nutrition and residual renal function in pre‐dialysis chronic renal failure patients. www.clinicaltrials.gov/ct2/show/NCT00563355 (accessed 15 December 2016).
Patel 2012 {published data only}
- Patel M, Thimons DG, Winston JL, Langholff W, McGowan T. An open‐label, randomized, multicenter, controlled study of epoetin alfa for the treatment of anemia of chronic kidney disease in the long term care setting. Journal of the American Medical Directors Association 2012;13(3):244‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schwartz 1989 {published data only}
- Prior JE, Terzian A, Schwartz AB, Kim KE, Mintz GS, Kahn SB. Prolonged RBC survival and hematopoietic response to recombinant human erythropoietin (rHuEPO) in chronic renal failure (CRF) [abstract]. Kidney International 1989;35(1):318. [CENTRAL: CN‐00766056] [Google Scholar]
- Schwartz AB, Mintz GS, Kim KE, Prior JE, Kahn SB. Recombinant human erythropoietin (rHuEPO) increases MAP, TPRI and systolic and diastolic dysfunction with increased impedance to LV ejection due to increased HCT and RBC mass in PTS with CRF [abstract]. Kidney International 1989;35(1):334. [CENTRAL: CN‐00766227] [Google Scholar]
Shaheen 1983 {published data only}
- Shaheen FA, Al‐Aqeil NA, Badawi L, Sheikh IA, Shalabi NM, Adiku W, et al. Correction of anemia by erythropoietin in pre‐dialysis patients. Saudi Kidney Diseases & Transplantation Bulletin 1983;4(3):215‐9. [CENTRAL: CN‐00525736] [Google Scholar]
Singh 1999 {published data only}
- Singh NP, Aggarwal L, Singh T, Anuradha S, Kohli R. Anaemia, iron studies and erythropoietin in patients of chronic renal failure. Journal of the Association of Physicians of India 1999;47(3):284‐90. [MEDLINE: ] [PubMed] [Google Scholar]
Teehan 1990 {published data only}
- Teehan BP, Benz RL, Sigler MH, Brown JM. Early intervention with recombinant human erythropoietin therapy. Seminars in Nephrology 1990;10(2 Suppl 1):28‐34. [MEDLINE: ] [PubMed] [Google Scholar]
Teplan 1995 {published data only}
- Teplan V, Schuck O, Mengerova O, Stollova M. Recombinant human erythropoietin (r‐hu EPO) and keto amino acids (KA) in predialysis: an accelerated metabolic effect [abstract]. ISN XIII International Congress of Nephrology; 1995 Jul 2‐6; Madrid, Spain. 1995:503. [CENTRAL: CN‐00509503]
Yamazaki 1993 {published data only}
- Yamazaki C, Watanabe Y, Sakamoto N. Pharmacokinetic study of recombinant human erythropoietin treatment in pre‐dialysis end stage renal disease patients. Nippon Jinzo Gakkai Shi [Japanese Journal of Nephrology] 1993;35(11):1233‐42. [MEDLINE: ] [PubMed] [Google Scholar]
Zheng 1992 {published data only}
- Zheng FL, Bi ZQ, Yang ZP, Li XW, Pu YF, Duan L. Effect of administration of low doses of rhuEPO in predialysis patients [abstract]. 9th Asian Colloquium in Nephrology; 1992 May 17‐21; Seoul, Korea. 1992:175. [CENTRAL: CN‐00462065]
References to studies awaiting assessment
NCT01576341 {published and unpublished data}
- NCT01576341. Open label, single arm, multicenter study to evaluate the safety and immunogenicity of HX575 epoetin alfa in the treatment of anemia associated with chronic kidney disease in pre‐dialysis and dialysis patients. www.clinicaltrials.gov/ct2/show/results/NCT01576341 (accessed 15 December 2016).
NCT01693029 {published data only}
- NCT01693029. Randomized, double‐blind, parallel group, multicenter study to evaluate the efficacy and safety of HX575 epoetin alfa vs. US licensed epoetin alfa (Epogen®/Procrit®) in the treatment of anemia associated with chronic kidney disease. www.clinicaltrials.gov/ct2/show/NCT01693029 (accessed 15 December 2016).
Additional references
Astor 2002
- Astor BC, Muntner P, Levin A, Eustace JA, Coresh J. Association of kidney function with anemia: the Third National Health and Nutrition Examination Survey (1988‐1994). Archives of Internal Medicine 2002;162(2):1401‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bernhardt 2010
- Bernhardt WM, Wiesener MS, Scigalla P, Chou J, Schmieder RE, Gunzler V, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. Journal of the American Society of Nephrology 2010;21(12):2151‐6. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Big Molecule Watch Blog 2015
- FDA rejects Hospira's Epoetin Biosimilar. www.bigmoleculewatch.com/2015/10/27/fda‐rejects‐hospiras‐epogen‐biosimilar/ 2015.
Canadian EPO 1990
- Anonymous. Association between recombinant human erythropoietin and quality of life and exercise capacity of patients receiving haemodialysis. Canadian Erythropoietin Study Group. BMJ 1990;300(6724):573‐8. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cannella 1990
- Cannella G, Canna G, Sandrini M, Gaggiotti M, Nordio G, Movilli E, et al. Renormalization of high cardiac output and of left ventricular size following long‐term recombinant human erythropoietin treatment of anemic dialyzed uremic patients. Clinical Nephrology 1990;34(6):272‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Cody 2005
- Cody JD, Daly C, Campbell MK, Khan I, Rabindranath KS, Vale L, et al. Recombinant human erythropoietin for chronic renal failure anaemia in pre‐dialysis patients. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD003266.pub2] [DOI] [PubMed] [Google Scholar]
Cody 2016
- Cody JD, Hodson EM. Recombinant human erythropoietin versus placebo or no treatment for the anaemia of chronic kidney disease in people not requiring dialysis. Cochrane Database of Systematic Reviews 2016, Issue 1. [DOI: 10.1002/14651858.CD003266.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dmtrieva 2013
- Dmitrieva O, Lusignan S, Macdougall IC, Gallagher H, Tomson C, Harris K, et al. Association of anaemia in primary care patients with chronic kidney disease: cross sectional study of quality improvement in chronic kidney disease (QICKD) trial data. BMC Nephrology 2013;14:24. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Drueke 2006
- Drueke TB, Locatelli F, Clyne N, Eckardt KU, Macdougall IC, Tsakiris D, et al. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. New England Journal of Medicine 2006;355(20):2071‐84. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
ERBP 2009
- Locatelli F, Covic A, Eckardt KU, Wiecek A, Vanholder R. ERA‐EDTA ERBP Advisory Board. Anaemia management in patients with chronic kidney disease: a position statement by the Anaemia Working Group of European Renal Best Practice (ERBP). Nephrology Dialysis Transplantation 2009;24(2):348‐54. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Eschbach 1987
- Eschbach JW, Egrie JC, Downing MR, Browne JK, Adamson JW. Correction of the anemia of end‐stage renal disease with recombinant human erythropoietin. Results of a combined phase I and II clinical trial. New England Journal of Medicine 1987;316(2):73‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
GRADE 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924‐6. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADE 2011a
- Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J, et al. GRADE Guidelines:1. Introduction‐GRADE evidence profiles and summary of findings tables. Journal of Clinical Epidemiology 2011;64(4):383‐94. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
GRADE 2011b
- Balshem H, Helfand M, Schunemann HJ, Oxman AD, Kunz R, Brozek J, et al. GRADE Guidelines: 3. Rating the quality of evidence. Journal of Clinical Epidemiology 2011;64(4):401‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Hahn 2014
- Hahn D, Cody JD, Hodson EM. Frequency of administration of erythropoiesis‐stimulating agents for the anaemia of end‐stage kidney disease in dialysis patients. Cochrane Database of Systematic Reviews 2014, Issue 5. [DOI: 10.1002/14651858.CD003895.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration. 2011. Available from www.cochrane‐handbook.org.
Hsu 2002
- Hsu CY, McCulloch CE, Curhan GC. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. Journal of the American Society of Nephrology 2002;13(2):504‐10. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
KDIGO 2012
- KDIGO. KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney International ‐ Supplement 2012;2(4):279‐335. [Google Scholar]
Koch 1991
- Koch KM, Frei U. Treatment of renal anemia, 1960‐1990. Advances in Nephrology From the Necker Hospital 1991;20:19‐30. [MEDLINE: ] [PubMed] [Google Scholar]
Lin 1985
- Lin FK, Suggs S, Lin CH, Browne JK, Smalling R, Egrie JC, et al. Cloning and expression of the human erythropoietin gene. Proceedings of the National Academy of Sciences of the United States of America 1985;82(22):7580‐4. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Locatelli 2011
- Locatelli F, Vecchio L. Erythropoiesis‐stimulating agents in renal medicine. Oncologist 2011;16 Suppl 3:19‐24. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lundin 1989
- Lundin AP. Quality of life: subjective and objective improvements with recombinant human erythropoietin therapy. Seminars in Nephrology 1989;9(1 Suppl 1):22‐9. [MEDLINE: ] [PubMed] [Google Scholar]
Macdougall 1999
- Macdougall IC, Gray SJ, Elston O, Breen C, Jenkins B, Browne J, et al. Pharmacokinetics of novel erythropoiesis stimulating protein compared with epoetin alfa in dialysis patients. Journal of American Society of Nephrology 1999;10(11):2392‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Macdougall 2005
- Macdougall IC. CERA (Continuous Erythropoietin Receptor Activator): a new erythropoiesis‐stimulating agent for the treatment of anemia. Current Hematology Reports 2005;4(6):436‐40. [MEDLINE: ] [PubMed] [Google Scholar]
Mikhail 2013
- Mikhail A, Farouk M. Epoetin biosimilars in Europe: five years on. Advances in Therapy 2013;30(1):28‐40. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
NICE 2015
- NICE Guideline. Chronic kidney disease: managing anaemia. Published: 3 June 2015. www.nice.org.uk/guidance/ng8 (accessed 15 December 2016).
Palmer 2010
- Palmer SC, Navaneethan SD, Craig JC, Johnson DW, Tonelli M, Garg AX, et al. Meta analysis: erythropoiesis stimulating agents in patients with chronic kidney disease. Annals of Internal Medicine 2010;153(1):23‐33. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Palmer 2012
- Palmer SC, Nand K, Dwi Nur Hidayati L, Munasinghe A, Nelson C, Khafaji MM, et al. Continuous erythropoiesis receptor activator (CERA) for the anaemia of chronic kidney disease. Cochrane Database of Systematic Reviews 2012, Issue 6. [DOI: 10.1002/14651858.CD009904] [DOI] [PMC free article] [PubMed] [Google Scholar]
Palmer 2014a
- Palmer SC, Saglimbene V, Mavridis D, Salanti G, Craig JC, Tonelli M, et al. Erythropoiesis‐stimulating agents for anaemia in adults with chronic kidney disease: a network meta‐analysis. Cochrane Database of Systematic Reviews 2014, Issue 12. [DOI: 10.1002/14651858.CD010590.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Palmer 2014b
- Palmer SC, Saglimbene V, Craig JC, Navaneethan SD, Strippolli GF. Darbepoetin for the anaemia of chronic kidney disease. Cochrane Database of Systematic Reviews 2014, Issue 3. [DOI: 10.1002/14651858.CD009297.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pappas 2008
- Pappas KD, Gouva CD, Katopodis KP, Nikolopoulos PM, Korantzopoulos PG, Michalis LK, et al. Correction of anemia with erythropoietin in chronic kidney disease (stage 3 or 4): effects on cardiac performance. Cardiovascular Drugs & Therapy 2008;22(1):37‐44. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ritz 2000
- Ritz E, Eisenhacht A. Early epoetin treatment in patients with renal insufficiency. Nephrology Dialysis Transplantation 2000;15 Suppl 3:40‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Roth 1994
- Roth D, Smith RD, Schulman G, Steinman TI, Hatch FE, Rudnick MR, et al. Effects of recombinant human erythropoietin on renal function in chronic renal failure predialysis patients. American Journal of Kidney Diseases 1994;24(5):777‐84. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schellekens 2009
- Schellekens H. Biosimilar therapeutics ‐ what do we need to consider?. NDT Plus 2009;2(Suppl 1):i27‐i36. [DOI: 10.1093/ndtplus/sfn177] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2011a
- Schünemann HJ, Oxman AD, Higgins JP, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and 'Summary of findings' tables. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Schünemann 2011b
- Schünemann HJ, Oxman AD, Higgins JP, Deeks JJ, Glasziou P, Guyatt GH. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Singh 2006
- Singh AK, Szczech L, Tang KL, Barnhart H, Sapp S, Wolfson M, et al. Correction of anemia with epoetin alfa in chronic kidney disease. New England Journal of Medicine 2006;355(20):2085‐98. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Stone 1988
- Stone WJ, Graber SE, Krantz SB, Dessypris EN, O'Neil VL, Olsen NJ, et al. Treatment of the anemia of predialysis patients with recombinant human erythropoietin: a randomized, placebo‐controlled trial. American Journal of the Medical Sciences 1988;296(3):171‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Strippoli 2006
- Strippoli GF, Navaneethan SD, Craig JC, Palmer SC. Haemoglobin and haematocrit targets for the anaemia of chronic kidney disease. Cochrane Database of Systematic Reviews 2006, Issue 4. [DOI: 10.1002/14651858.CD003967.pub2] [DOI] [PubMed] [Google Scholar]
Szczech 2008
- Szczech LA, Barnhart HX, Inrig JK, Reddan DN, Sapp S, Califf RM, et al. Secondary analysis of the CHOIR trial epoetin‐alpha dose and achieved hemoglobin outcomes. Kidney international 2008;74(6):791‐8. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2011
- World Health Organization. Haemoglobin concentrations for the diagnosis of anaemia and assessment of severity. Vitamin and Mineral Nutrition Information System. World Health Organization. 2011. www.who.int/vmnis/indicators/haemoglobin/en/ (accessed 15 December 2016). [WHO/NMH/NHD/MNM/11.1]
Winearls 1986
- Winearls CG, Oliver DO, Pippard MJ, Reid C, Downing MR, Cotes PM. Effect of human erythropoietin derived from recombinant DNA on the anaemia of patients maintained by chronic haemodialysis. Lancet 1986;2(8157):1175‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wolcott 1989
- Wolcott DL, Marsh JT, Rue A, Carr C, Nissenson AR. Recombinant human erythropoietin treatment may improve quality of life and cognitive function in chronic hemodialysis patients. American Journal of Kidney Diseases 1989;14(6):478‐85. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Hahn 2015
- Hahn D, Esezobor CI, Elserafy N, Webster AC, Hodson EM. Short‐acting erythropoiesis‐stimulating agents for anaemia in predialysis patients. Cochrane Database of Systematic Reviews 2015, Issue 5. [DOI: 10.1002/14651858.CD011690] [DOI] [PMC free article] [PubMed] [Google Scholar]