

Abstract
The interchange between different repair mechanisms in human cells has long been a subject of interest. Here, we provide a direct demonstration that the oxidatively generated guanine lesions spiroiminodihydantoin (Sp) and 5-guanidinohydantoin (Gh) embedded in double-stranded DNA, are substrates of both BER and NER mechanisms in intact human cells. Site-specifically modified, 32P-internally labeled double-stranded DNA substrates were transfected into fibroblasts or HeLa cells and the BER and/or NER mono- and dual incision products were quantitatively recovered after 2–8 hour incubation periods and lysis of the cells. DNA duplexes bearing single benzo[a]pyrene-derived guanine adduct were employed as positive controls of NER. The NER activities, but not the BER activities, were abolished in XPA−/− cells, while the BER yields were strongly reduced in NEIL1−/− cells. Co-transfecting different concentrations of analogous DNA sequences bearing the BER substrates 5-hydroxyuracil, diminish the BER yields of Sp lesions and enhanced the yields of NER products. These results are consistent with a model based on the local availability of BER and NER factors in human cells and their competitive binding to the same Sp or Gh BER/NER substrates.
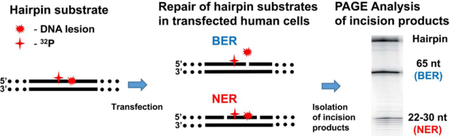
Graphical Abstract
INTRODUCTION
Existing paradigms suggest that non-bulky oxidatively generated DNA lesions are removed by base excision repair (BER) mechanisms, while bulky DNA double helix-distorting lesions are excised by the nucleotide excision repair (NER) pathway.1–4 For example, DNA lesions such as oxidatively generated 8-oxo-7,8-dihydroguanine (8-oxoG) are known to be substrates of BER only.5–7 The 8-oxoG lesions are more easily oxidized than the parent base guanine,8 and their oxidation can lead to the formation of stereoisomeric spiroiminodihydantoin (Sp) and 5-guanidinohydantoin (Gh) lesions.9–11 The accumulation of Sp lesions was detected in Nei-deficient E. coli cells treated with chromate,12 and were also detected in both the liver and colon tissues of Rag2−/− mice at levels ~100 times lower than those of 8-oxoG.13 The low levels of the hydantoin lesions in cellular environment can be associated with the competitive formation of other oxidation products,14 including crosslinks between 8-oxoG and histones in nucleosomes in relatively high yields.15 Although the cellular levels of hydantoin lesions are low, they can contribute to the malignant transformation of cells because they are at least one order of magnitude more mutagenic than 8-oxoG.16
Recent observations suggest that the hydantoin lesions are not only excellent BER substrates,17–21 but are also excised by the NER pathway in human cell-free extract.22 It remained unclear how these two repair pathways can cooperate to excise the same DNA lesions and whether they compete with one another in the biologically more relevant environment of intact human cells.
In cell extracts, the BER proteins and the DNA damage-sensing NER factor XPC-RAD23B locate their target substrates by a free diffusion mechanism.23 The yields of BER and NER incision products depend on the relative concentrations of these proteins in cell extracts, thus suggesting that these two repair pathways indeed can compete with one another.22 These observations are consistent with the notion that the NER mechanism serves as a backup for the BER pathway. However, it is not known whether similar phenomena can occur within the confines of intact cells where the protein concentrations and diffusion characteristics of proteins are different than in cell extracts. To answer this question, we used 32P-internally labeled DNA hairpin double-stranded DNA substrates24, 25 containing single, site specifically inserted Gh or Sp lesions. The 5’- and 3’-ends of these hairpins were capped with biotin and dideoxycytidine (ddC) to protect these templates from non-specific degradation by exonucleases, as described by Shen et al.24 As a positive control of NER, bulky benzo[a]pyrene-derived DNA lesions were substituted at the same sites in the DNA substrates (Figure 1). Experiments using varying concentrations of 5-hydroxyuracil lesions (5-OHU), which are classical substrates of BER proteins,26, 27 were employed to test the BER vs. NER competition hypothesis.
Figure 1.

Structures of Sp (S absolute configuration), Gh, 5-OHU, (+)-cis-B[a]P-dG, and (+)-trans-B[a]P-dG lesions (X) site-specifically positioned in the DNA hairpin double-stranded substrates.
In this work, we demonstrate the feasibility of quantitatvely recovering 32P-labeled DNA repair products that arise from the removal of single, structurally defined lesions embedded in DNA constructs transfected into human cells. In the case of the bulky (+)-cis- or (+)-trans-B[a]P-dG adducts that are known to be substrates of NER, but not BER mechanisms, only the characteristic ladders of dual incision NER products are recovered after transfection and incubation. However, in the case of single Gh or Sp lesions positioned in the same DNA sequences, both single incision BER products and NER dual incision products are observed. The interplay of BER and NER mechanisms in the repair of Gh and Sp hydantoin lesions was confirmed using human fibroblasts with a deficient NER pathway lacking the protein XPA (XPA−/−),3, 24 or lacking the DNA glycosylase NEIL1 (NEIL1−/−).2 The XPA protein is one of the critical NER factors,3 while the base excision repair of hydantoin lesions is known to be suppressed in NEIL1−/− cells19 because these lesions are classical substrates of the DNA glycosylase NEIL1.17–21 Control experiments in these genetically modified, NER inactive XPA−/− cells, or BER deficient NEIL1−/− cells, confirm the cooperation of these two repair systems in excising Gh and Sp lesions. It is shown that the BER and NER pathways compete with one another in intact human cells and can incise the same Gh and Sp DNA lesions with efficiencies that depend on the relative concentrations of BER and NER proteins.
EXPERIMENTAL PROCEDURES
Construction of Hairpin Duplexes.
The 2’-oligodeoxynucleotides were purchased from Integrated DNA Technologies (Coralville, IA, USA) and were purified by denaturing polyacrylamide gel electrophoresis (PAGE). The 5′-CCATC[5-OHU]CTACC sequence was purchased from Trilink Biotechnologies (San Diego, CA, USA). The 11-mer oligonucleotide adducts containing the S-Sp or R-Sp diastereomers were generated by the site-selective oxidation of guanine in the 5′-CCATCGCTACC sequence using photochemically generated carbonate radical anions at pH 7.5 – 8.0 to oxidize the guanine residue as described earlier;28–30 the modified oligonucleotides were isolated by anion-exchange HPLC methods using DNAPac PA-100 columns (Dionex, Sunnyvale, CA, USA).31, 32 The Gh-modified oligonucleotides were prepared by the oxidation of 5’-CCATC[8-oxoG]CTACC sequences with (NH4)2IrCl6 complex at pH 6.0.31, 33 The 11-mer bulky adducts with 10R (+)-cis-anti-B[a]PDE-N2-dG and 10R (+)-trans-anti-B[a]PDE-N2-dG adducts were generated by reacting the racemic (±)-anti-7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene diol epoxide with the 5’-CCTACGCTACC sequence. The (+)-cis- and (+)-trans-products were separated from one another and from the other stereoisomeric products and purified by reversed-phase HPLC as described earlier34 and by denaturing gel electrophoresis.
The DNA hairpin constructs containing single site-specifically inserted lesions were designed as described by Shen et al.24 The hairpins contained guanine lesions positioned at the 66th nucleotide from the 5’-end were generated by ligating the 5’−32P-endlabeled 11-mer 5′-CCATCXCTACC (X = S-Sp, Gh or B[a]P-dG) to 5’-and 3’-flanking 60- and 108-mer sequences.22 The flanking sequences were capped with biotin in the 60-mer 5’-biotin-GCACGTCAGGCACGGCGTCGGTACCAGCTGCGGCAAGGCCGGATCCAGACCTCGTCACCA sequence, and with dideoxycytidine (ddC) in the 108-mer strand 5’-ACCTCAGTGGTCCGCTCGAGACAC CGAAAACGGTGTCTCGAGC GGACCACTGAGGTGGTAGCGATGGTGGTGACGAGGTCTGGATCCGGCCTTGCCGCAGCTGGT-AC[ddC]. The end-capping was introduced in order to reduce degradation of these strands by cellular non-specific exonucleases.24 The hairpin strands obtained by this approach were purified by denaturing PAGE and annealed by heating at 90 °C for 5 min, followed by a slow cooling to room temperature overnight to form the hairpin duplexes used in the subsequent experiments.
DNA Repair Assays in Cultured Human Cells.
The SV40-transformed human fibroblast cell lines (GM00637) and fibroblasts from a xeroderma pigmentosum (XP) patients with mutant XPA alleles (GM04429) were purchased from the Coriell Institute for Medical Research (Camden, NJ, USA). The HeLa S3 cells were purchased from American Type Culture Collection (Manassas, VA, USA). The NEIL1 knockout human embryonic kidney 293T cell line (NEIL1−/−) created by CRISPR technology was purchased from EdiGene (Cambridge, MA, USA). All cells were maintained according to the manufacturer’s protocols.
Human fibroblasts and HeLa S3 cells were transfected with the hairpin duplexes employing a classical calcium-phosphate (CaP) method.24, 35 The 32P-internally labeled hairpins (~ 0.1 pmol) in 250 μL H2O containing 200 mM CaCl2 were added drop-by-drop (with gentle mixing) to 250 μL 2× HBS buffer (1.5 mM Na2HPO4, 50 mM HEPES, 10 mM KCl, 280 mM NaCl, 12 mM Dextrose, pH 7.04). The reaction mix was incubated at room temperature for 1 min to form the DNA-CaP precipitate,35 the latter was added to the cells to initiate transfection. In these experiments, ~4×106 cells in the exponential growth phase36 were seeded into 75 cm2 flasks the day before the transfection. One hour before the DNA-CaP precipitate solution (500 μL) was added to the cells, the medium was replaced with fresh medium (3 mL). The cells in the medium (3.5 mL) containing ~ 30 pM hairpins were then incubated for 2 – 8 h at 37 °C in a 5% CO2 humidified incubator.
After incubation for fixed periods of time, the cells were washed twice with cold phosphate buffer saline (PBS) and harvested as pellets. The latter were re-suspended in a 10× packed cell volume (PCV) of lysis buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 1.2% SDS and 100 μg/mL RNase A) and incubated for 15 min at room temperature.37 After addition of one-fourth volume of 5 M NaCl with gentle mixing, the samples were incubated on ice overnight. Insoluble material containing genomic DNA was removed by centrifugation at 14,000 × g at 4 °C for 1 h. The supernatant was transferred to a new tube, treated with proteinase K (0.1 mg/mL) for 30 min at 37 °C, and extracted with phenol/chloroform. The low molecular weight DNA was ethanol-precipitated and subjected to denaturing 12% PAGE. The dry gels were analysed using Molecular Dynamics Storage Phosphor Screens scanned with a Typhoon FLA 9000 laser scanner, and analysed using the ImageQuant software.
The relative efficiencies of recovery of the BER incision and NER excision products leading to longer (65 nt) and shorter (24 – 30 nt) oligonucleotide products, respectively, were assessed. In these experiments, human cells were transfected with mixtures of 32P-labeled oligonucleotides 65 nt and 28 nt in length. Following a typical incubation period of 2 h, the oligonucleotides were recovered as described in the above paragraph and the relative proportions of 65 and 28 nt sequences were assessed by denaturing 12% PAGE and analysis of the autoradiographs (representative results obtained with HeLa cells are summarized in Figure S1 in Supporting Information). These control experiments indicate that the recovery method used for isolation of low molecular weight DNA37 provides a reasonable estimate of the yields of BER and NER products because the above difference is close to the experimental reproducibility of such experiments (Figure 7).
Figure 7.

(A) Competitive repair of the spiroiminodihydantoin and 5-hydroxyuridine lesions by the BER and NER mechanisms in intact HeLa cells. The mixtures containing the 32P-internally labeled S-Sp hairpins (0.1 pM) and increasing quantities (0, 0.3, 1, 3 and 10 pM) of non-radioactive 5-OHU hairpins that correspond to 5-OHU/Sp molar ratios of 0, 3, 10, 30, or 100, were transfected into BER/NER-fully competent HeLa cells. (B) The 5-hydroxyuracil lesions are excellent substrates of BER only in intact cells. The 32P-internally labeled DNA hairpins harboring 5-OHU lesions were transfected into intact HeLa cells. The incubation times in all experiments were 2 h. Lane M: oligonucleotide size markers. Lane C: Control experiment; the 179 nucleotide-long hairpin in buffer solution (not transfected into the cells, but subjected to the same treatment otherwise). Lane 1: the full hairpin with 5-OHU lesions after transfection into intact HeLa cells and full digestion to the 66-nucleotide-long single-incision BER product. (C, D) The BER and NER yields were calculated by integration of the histograms derived from the denaturing gel autoradiographs at each particular molecular ratio of the 5-OHU and S-Sp hairpins used for transfection and incubation time of 2 h. The results of three independent experiments and their standard deviations are shown.
The complete repair of the hairpins by the full BER mechanism associated with polymerase β and ligase activities24 would lead to an underestimation of the initial BER-induced incision yields based on the quantitation of the 65 nt-long intermediate products (Figures 4 and 5). We therefore tested the hypothesis that the intact hairpin duplexes represented by the upper bands in Figure 5 are a result of a limited BER DNA repair capacity of the cells. To assess this possibility, the concentration of the hairpin substrates transfected into the cells was reduced from ~ 30 pM (as in the experiment shown in Figure 5), to ~10 pM. These experiments showed that after a 2 – 4 h incubation period, all of the initially transfected hairpins were completely cleaved by BER and NER mechanisms (Figure S2 in Supporting Information). Thus, under our experimental conditions, the subsequent BER-mediated repair steps that follow the initial incision appear to be slow, and the limited DNA repair capacity of the cells can account for the residual intact hairpins that are observed when the initial hairpin concentration is ~30 pM.
Figure 4.

The 5-guanidinohydantoin lesions are substrates of both NER and BER pathways in intact human cells. The 32P-internally labelled DNA hairpins harboring Gh or (+)-cis-B[a]P-dG lesions were transfected either into HeLa cells, fully NER-competent (XPA+/+) or NER-deficient (XPA−/−) human fibroblasts and incubated for the time intervals indicated. Lanes M: oligonucleotide size markers. Lanes C: Control experiments; the hairpins are not transfected into the cells, but are otherwise subjected to the same post-incubation treatments. The panel shown is a composite of the autoradiographs of two gels
Figure 5.

The spiroiminodihydantoin lesions are substrates of BER and NER in intact cells. The 32P-internally labeled DNA hairpins harbouring Sp lesions were transfected either into fully BER/NER-competent HeLa cells (lanes 1–4), or into NEIL1−/− cells that lack the glycosylase NEIIL1 (lanes 5–8) that is known to excise Sp lesions in double-stranded DNA.19, 20 The incubation times (t) varied between 2 and 8 hours. Lanes M: oligonucleotide size markers (We note that the intensity of the synthetic maker Lane M only, was diminished separately from the rest of the gel because it was oversaturated). Lanes C: Control experiments; the hairpins were not transfected into the cells, but were subjected to the same treatment otherwise. The panel shown is a composite of autoradiographs of two gels.
RESULTS
Excision Repair of DNA Lesions in Intact Human Cells and Recovery of the Products.
A hallmark of successful NER activity is the appearance of the oligonucleotide dual incision products ~24 – 30 nucleotides in lengths.38, 39 In order to detect repair activities in intact cells by monitoring NER excision products, we adopted the simple hairpin design described earlier24 that contains a single site-specifically inserted lesion X (Figure 1). Briefly, the hairpin was 179 nucleotides (nt) in length and was capped with biotin and dideoxycytidine (ddC) to protect these templates from potential non-specific degradation by exonucleases. The DNA sequences containing the lesions were synthesized separately as described.22, 28, 32, 34 The 11-mer sequences 5’…CCATCXCTACC… that contained the guanine lesions X = Gh, Sp (S absolute configuration), X = 5 -OHU, (+)-cis-B[a]P-dG, or (+)-trans-B[a]P-dG], were inserted into the 77 base pair-long double stranded part of the hairpin construct. The lesions were embedded at the 66-th nucleotide counted from the 5’-side of the hairpin.
The feasibility of recovering the NER dual incision 32P-labeled oligonucleotide products 24 – 30 nucleotides in length after incubation of the hairpins in intact cells, was tested with a known NER substrate derived from a metabolite of the environmental carcinogen benzo[a]pyrene.40 The metabolic activation of polycyclic aromatic hydrocarbons like benzo[a]pyrene (B[a]P) by cytochrome P450 in mammalian cells gives rise to a number of oxygenated metabolites including the highly reactive (+)-7R,8S-dihydrodiol, 9S,10R-epoxy-tetrahydrobenzo[a]pyrene enantiomer (B[a]PDE). This diol epoxide derivative reacts with the exocyclic amino groups of guanine in DNA by cis- or trans-addition to form the stereoisomeric DNA adducts abbreviated as (+)-cis-B[a]P-dG, or (+)-trans-B[a]P-dG, respectively41 (Figure 1). In human cell extracts, the (+)-cis-adducts are better substrates of NER than the (+)-trans adducts42 by a factor of ~ 5 in a double-stranded DNA ,,..CG*C... sequence with G* designating the B[a]P-dG adducts.43 Therefore, these stereoisomeric bulky adducts are ideal not only for testing the feasibility of recovering the NER excision products from cells, but also to test whether the difference in NER incision efficiencies observed in human cell extracts42, 43 are maintained in intact cells.
Typical results of the DNA repair assays in HeLa cells transfected with the 32P-internally labeled hairpins harboring (+)-cis-B[a]P-dG, and (+)-trans-B[a]P-dG] bulky lesions are depicted in the autoradiograph of a denaturing polyacrylamide gel shown in Figure 2. It has been shown earlier that the (+)-cis-B[a]P-dG lesion is as good a substrate of NER as the 6–4 UV photoproduct (6,4 pyrimidine-pyrimidone) in human cell extracts.44 After incubation of HeLa cells transfected with hairpins harboring the two stereoisomeric B[a]P-dG adducts for 2 – 8 h, the characteristic ladders of NER dual incision products of the 32P-labeled oligonucleotide fragments ~ 26 – 32 nt in lengths are clearly visible (Figure 2, lanes 5 – 8 and 9 – 12). However, analogous bands are not observed in the case of unmodified hairpins used as negative controls (Figure 2, lanes 1 – 4). These observations demonstrate that HeLa cells transfected with hairpins harboring bulky adducts are NER-active in the intact human cells selected for this study.
Figure 2.

The appearance of dual incision NER products in HeLa cells transfected with 32P-internally labeled DNA hairpins harboring (+)-cis-B[a]P-dG or (+)-trans-B[a]P-dG] lesions as a function of incubation time. The (+)-cis-B[a]P-dG or (+)-trans-B[a]P-dG] lesions are NER substrates in intact human cells. The unmodified hairpins with guanine instead of the lesion were used as negative controls of NER activity. Lanes M: oligonucleotide size markers. Lanes C: Control experiments; the DNA hairpins were not transfected into the cells, but were otherwise subjected to the same post-incubation treatment. The panel shown is a composite of the autoradiographs of three gels.
Examples of the NER kinetics in HeLa cells transfected with hairpins containing (+)-cis-B[a]P-dG or (+)-trans-B[a]P-dG] lesions are depicted in Figure 3. In this figure, the yields of dual incision products calculated from the gel autoradiographs (Figure 2) are plotted as a function of incubation time (Figure 3). These yields increase as a function of time and reach a maximum level of about 6 % of the original hairpin DNA substrates present in the cells within ~ 4 – 5 hours after the start of the transfection. However, the dual incision kinetics are significantly slower than in human cell extracts. As in HeLa cell extracts,43 the yield of NER products associated with the (+)-cis-B[a]P-dG adducts is larger by a factor 3.7 ± 1 than the yield derived from the stereoisomeric (+)-trans-B[a]P-dG adducts (Figure 3). This value is close to the ratio of ~ 5 observed in HeLa cell extracts43. In the case of the two stereoisomeric B[a]P-dG adducts studied, similar differences in NER efficiencies are found in HeLa cell extracts and in intact HeLa cells.
Figure 3.

Kinetics of appearance of NER dual incision products in HeLa cells transfected with the 32P-internally labelled DNA hairpins harboring (+)-cis-B[a]P-dG, or (+)-trans-B[a]P-dG] lesions. The NER yields were calculated by integration of the histograms derived from the denaturing gel autoradiographs at each particular incubation time (2, 4, 6 or 8 h). The results of three independent experiments and their standard deviations are shown. Other details are described in Methods.
These results demonstrate that the recovery of NER excision products and the quantitative analysis of relative repair yields are feasible.
BER and NER of Oxidatively Generated Guanine Lesions in Human Cells.
The Sp and Gh lesions are excellent substrates of BER in vitro.17–21 Incisions of these lesions by BER proteins are expected to generate the 32P-labeled 65-mer oligonucleotide fragments, while successful NER dual incisions should generate the usual ladder of shorter oligonucleotides in the range of 24 – 30 nucleotides in lengths.
Typical results of DNA repair assays in NER-competent human fibroblasts (XPA+/+) and HeLa cells with hairpins harboring (+)-cis-B[a]P-dG or Gh are shown in Figure 4. Hairpins with B[a]P-dG adducts were included as positive controls of NER activity, and negative control of BER activity. The same hairpin samples were also transfected into NER-inactive XPA−/− fibroblasts. Incisions at the sites of the lesions and the appearance of 32P-labeled 65-mer oligonucleotide fragments are evident in the case of the Gh hairpin DNA substrates and are attributed to the BER mechanism (Figure 4, lanes 7 – 12). There is no evidence of any BER activity at the site of (+)-cis-B[a]P-dG adducts (Figure 4, lanes 1 – 6). The ladders of the 32P-labeled dual incision products (Figure 4, lanes 1–4) demonstrate that the Gh lesions are substrates of not only BER, but also of NER in human fibroblasts (XPA+/+) and HeLa cells that are both NER-competent (Figure 2).
When (+)-cis-B[a]P-dG or Gh hairpins are incubated into NER-deficient XPA−/− fibroblasts, the typical ladder of NER excision products is strongly suppressed (Figure 4, lanes 5,6 and 11,12, respectively). These results are consistent with those published by Shen et al.24 who showed that the NER activities in XPA-deficient fibroblasts transfected with DNA hairpins are reduced by factor of ~ 4 relative to normal fibroblasts. However, the Gh BER incision products are clearly visible (Figure 4, lanes 11 and 12). Analogous results of DNA repair assays in human cells transfected with 32P-internally labelled DNA hairpins harboring Sp lesions are depicted in Figure 5. In addition to the BER incision products, the ladder of 32P-labeled NER dual incision products is also visible in intact HeLa cells (Figure 5, lanes 1 – 4).
DNA Repair Assays in Intact BER-Deficient (NEIL1−/−) or NER-Deficient (XPA−/−) Cells.
The hydantoin lesions (Sp and Gh) are well-known substrates of the bifunctional DNA glycosylase NEIL1.17–21 In order to verify that the single incision bands are indeed the result of BER activity in HeLa cells, the same DNA sequences with Sp lesions were transfected into genetically altered NEIL1−/− cells. Typical results of DNA repair assays in these cells with hairpins harboring Sp lesions are shown in Figure 5. The intensities of the single incision bands of 65 nt products in lanes 5 and 6 are significantly weaker than they are in the BER-competent HeLa cells (lanes 7 and 8, Figure 5), while NER ladders of dual incision products are still evident (lanes 5,6 vs. lanes 7,8, Figure 5).
To summarize, the existence of parallel NER and BER activities that have been documented in HeLa cell extracts,22 also manifest themselves at the level of intact HeLa cells and NER-competent fibroblasts.
Kinetics of Single BER Incisions and NER Dual Incisions.
Examples of the repair kinetics in HeLa cells transfected with the hairpins containing S-Sp and Gh lesions are depicted in Figure 6.
Figure 6.

Kinetics (A) and (B) maximum averaged yields of BER and NER incision products in HeLa cells transfected with DNA hairpins that contain the single S-Sp and Gh lesions. The BER and NER yields were calculated by integration of the histograms derived from the denaturing gel autoradiographs at each particular incubation time (2, 4, 6 or 8 h). The results of four independent experiments and their standard deviations are shown.
The yields of BER and NER incision products are plotted as a function of incubation time after transfection of the DNA hairpins into intact HeLa cells. The average BER and NER yields increase as a function of incubation time and approach relatively constant values after a 4 h incubation period (Figure 6A). However, the BER and NER kinetics are significantly slower than in human cell extracts.22 The repair of the transfected hairpins occurs in parallel with transfection initiated by addition of the DNA-CaP precipitate, which is also time-dependent.35 Thus, the DNA repair kinetics shown in Figures 3 and 6 reflect a superposition of the transfection and repair processes, and are suitable for monitoring the relative efficiencies of the BER and NER mechanisms.
The maximum average yields of NER products for hairpins containing Sp and Gh lesions are close to one another, while the BER yields for Sp modified hairpins are greater by a factor of ~2 than in the case of the Gh-modified hairpins (Figure 6B). The ratios of BER/NER activities in these examples for Sp and Gh lesions are 9.5 ± 3.1 and 5.0±1.9, respectively. In HeLa cell extract experiments the BER/NER ratios were in the range of 1 – 2, and thus significantly smaller than in intact cells; furthermore, the levels of NER dual incision yields in HeLa cell extracts varied from preparation to preparation with NER yields in the range of 5 – 25% after 30 min incubation periods,22 depending on the particular cell extract. In the intact cell examples shown, the NER yields for the same lesions are at level of 4 ± 1% after a 4 h incubation time period (Figure 6). The BER and NER yields in intact cells tend to level off after a ~ 2 hour incubation period after transfection, indicating that ~ 50% or more of the lesions remain unrepaired in these particular experiments.
Experiments shown in Supporting Information (Figure S1) indicate that the recovery of BER and NER excision products of different lengths is quantitative. In these experiments, HeLa cells were transfected with mixtures of 32P-labeled 65 and 28 nt oligonucleotides without lesions, and incubated for 2 h. After recovery of the oligonucleotides, the relative proportions of 65 and 28 nt sequences were assessed by denaturing 12% PAGE (Figure S1). We found that the ratio of the 65 and 28 nt bands recovered in these control experiments were within 10 ± 7% of the correct initial value before transfection, which is within the experimental error bars observed in typical experiments (Figure 6). These control experiments thus demonstrate that the recovery method used provides a reasonable estimate of the relative yields of BER and NER products.
The Incisions of DNA Hairpins with Sp Lesions in Human Cells Occur by Competitive BER and NER Pathways.
In these experiments, the 32P-internally labeled Sp hairpins were transfected into HeLa cells together with varying quantities of the same, but unlabelled hairpins containing the well-known BER substrates 5-OHU (Figure 7A, lanes 1 – 4).
The co-transfection of unlabelled DNA hairpins containing single 5-OHU lesions significantly suppressed the yields of the 32P-labeled BER products excised from the radioactively labelled hairpins with single Sp lesions. For instance, increasing the hairpin molar ratio (5-OHU/Sp) from 0 to 100 results in a significant reduction of BER yields from Sp hairpins from 75±3% to 3±2% (Figure 7C). By contrast, the yields of the 32P-labeled NER dual incision products (the ladder of bands ~ 24 – 30 nucleotides in lengths) are enhanced by factor of ~2, from 4.5±1.5% in the absence of, to 9.5±2.5% in the presence of the 5-OHU-containing hairpins (Figure 7D). Control experiments with the 32P-internally labeled 5-OHU hairpins clearly demonstrate that these lesions are repaired exclusively by the BER pathways, while NER dual incision products are entirely absent (Figure 7B, lane 1).
In summary, these experiments clearly demonstrate that the NER machinery (most likely the initial DNA damage-sensing NER factor XPC-RAD23B) competes with BER proteins for the Sp lesions, which are substrates of both DNA repair pathways, BER and NER, in intact human cells as well as in cell extracts.
DISCUSSION
Employing 32P-internally labelled DNA sequences harboring site-specifically positioned guanine lesions, we have shown that Sp and Gh are substrates of two different repair pathways, BER and NER, in intact human cells. Interestingly, Sp, Gh and other hydantoin lesions are also substrates of the prokaryotic UvrABC NER system in vitro.45 The approach described here, is based on the direct recovery of the 32P-internally labeled BER and NER incision products that does not require lesion-specific antibodies as in immunoprecipitation approaches,38 or biotin- or 32P-postlabeling of the dual incision products.37 The transfection of DNA hairpins, in which the ends are protected from non-specific degradation by exonucleases, have been successfully used for monitoring full repair processes in human cells (from the detection and removal of the lesion to the complete restoration of damaged DNA by polymerase and ligase activities) by NER24 and BER25 pathways. This so-called Oligonucleotide Retrieval Assay (ORA) involves the PCR amplification of the hairpin products recovered by biotin binding to streptavidin. Our approach does not require these two additional steps and allows for the simultaneous monitoring of the primary events initiated by BER and NER mechanisms. We note that in both NER and BER processes only the primary steps (from recognition of lesions to their excision) depend on the structure of the DNA lesion that is crucial for achieving a better understanding of the structure-function relationships of the excision stages of DNA lesions. The further processing required for the completion of the repair and restoration of the undamaged DNA by polymerase and ligase activities do not depend on the structural features of the DNA lesions.
The hypothesis was tested that in intact human cells the relative yields of NER and BER products depends on the competitive binding of BER and NER DNA damage-sensing proteins (DNA glycosylases and XPC-RAD23B, respectively) to the same DNA lesions. In cell-free extracts, the concentrations of BER and NER proteins are of the order of ~0.1 nM and the binding to their NER substrates depends on free diffusion mechanisms. In intact cells however, the concentrations of repair proteins are of the order of ~0.1 μM,23, 46 three orders of magnitude higher than in cell-free extracts. Once the glycosylases bind to their substrate, the removal of the DNA lesion occurs within seconds in vitro, although dissociation of BER enzymes from the product complexes can take tens of minutes.20, 47 In turn, the binding of the dimeric NER factor XPC-RAD23B to its substrate is followed within minutes by the recruitment of the other, subsequent NER factors23 that lead to the dual incisions and excision of the 24 – 32 nucleotide-long NER products. The irreversibility of these initial binding events supports the hypothesis that the ultimate yields of BER and NER products depend on the competition between DNA glycosylases and XPC-RAD23B to form the initial complexes with Sp and Gh.22 Experiments conducted with NEIL−/− cells demonstrate that the DNA glycosylase NEIL1 plays an important role in excising the Gh and Sp lesions (Figure 5). Thus, the BER vs. NER competition is likely to depend on the ratios of the local concentrations of NEIL1 and XPC in intact human cells, as well as the relative binding affinities of these proteins to their DNA substrates.
The repair of oxidatively generated DNA lesions is a crucial factor for maintaining genomic stability during oxidative stress. Small DNA lesions such as the hydantoin lesions Gh and Sp generated by reactive oxygen species are generally efficiently recognized and excised by DNA glycosylases. Using radioactively labeled DNA sequences harboring Gh and Sp lesions, it is shown that these lesions are substrates of both BER and NER mechanisms. The relative contribution of either process in intact cells depends on the local availability of the primary NER and BER factors that recognize and bind to the same lesions in a competitive fashion. While BER yields of Sp up to 80% have been observed, the maximum NER yields were less than ~ 10% even when BER was suppressed by unlabeled BER (but not NER) substrates, or when the excision of bulky benzo[a]pyrene-derived guanine adducts that are substrates of NER only, was examined. These results suggest that the NER yields are limited by the local cellular concentrations of at least one of the NER factors that are critical for successful NER dual incisions, while the BER proteins are ~ 8 times more abundant (Figure 7). The experimental approaches described are useful for assessing the DNA repair capacities intact human cells up to the incision stages of repair.
Supplementary Material
Acknowledgments
Funding
This work was supported by the National Institute of Environmental Health Sciences grant R01 ES-027059 to V.S. Components of this work were conducted in the Shared Instrumentation Facility at New York University that was constructed with support from Research Facilities Improvement grant C06 RR-16572 from the National Center for Research Resources, National Institutes of Health (USA).
ABBREVIATIONS
- 8-oxoG
8-oxo-7,8-dihydroguanine
- Sp
spiroiminodihydantoin
- Gh
5-guanidinohydantoin
- B[a]PDE
(+)-7R,8S-dihydrodiol, 9S,10R-epoxy-tetrahydrobenzo[a]pyrene enantiomer
- (+)-cis-B[a]P-dG
10R (+)-cis-anti-B[a]PDE-N2-dG adduct
- (+)-trans-B[a]P-dG
10R (+)-trans-anti-B[a]PDE-N2-dG adduct
- ddC
dideoxycytidine
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Autoradiographs of denaturing gels (Figures S1–S2) (PDF).
REFERENCES
- (1).Lindahl T, and Wood RD (1999) Quality control by DNA repair. Science 286, 1897–1905. [DOI] [PubMed] [Google Scholar]
- (2).Wallace SS (2014) Base excision repair: a critical player in many games. DNA Repair (Amst) 19, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Marteijn JA, Lans H, Vermeulen W, and Hoeijmakers JH (2014) Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell. Biol 15, 465–481. [DOI] [PubMed] [Google Scholar]
- (4).Yu Y, Cui Y, Niedernhofer LJ, and Wang Y (2016) Occurrence, Biological Consequences, and Human Health Relevance of Oxidative Stress-Induced DNA Damage. Chem. Res. Toxicol 29, 2008–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bruner SD, Norman DP, and Verdine GL (2000) Structural basis for recognition and repair of the endogeneous mutagen 8-oxoguanine in DNA. Nature 403, 859–866. [DOI] [PubMed] [Google Scholar]
- (6).David SS, O’Shea VL, and Kundu S (2007) Base-excision repair of oxidative DNA damage. Nature 447, 941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wallace SS, Murphy DL, and Sweasy JB (2012) Base excision repair and cancer. Cancer Lett 327, 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Steenken S, and Jovanovic SV (1997) How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc 119, 617–618. [Google Scholar]
- (9).Luo W, Muller JG, Rachlin EM, and Burrows CJ (2000) Characterization of spiroiminodihydantoin as a product of one-electron oxidation of 8-oxo-7,8-dihydroguanosine. Org. Lett 2, 613–616. [DOI] [PubMed] [Google Scholar]
- (10).Niles JC, Wishnok JS, and Tannenbaum SR (2001) Spiroiminodihydantoin is the major product of the 8-oxo-7,8-dihydroguanosine reaction with peroxynitrite in the presence of thiols and guanosine photooxidation by methylene Blue. Org. Lett 3, 963–966. [PubMed] [Google Scholar]
- (11).Sugden KD, Campo CK, and Martin BD (2001) Direct oxidation of guanine and 7,8-dihydro-8-oxoguanine in DNA by a high-valent chromium complex: a possible mechanism for chromate genotoxicity. Chem. Res. Toxicol 14, 1315–1322. [DOI] [PubMed] [Google Scholar]
- (12).Hailer MK, Slade PG, Martin BD, and Sugden KD (2005) Nei deficient Escherichia coli are sensitive to chromate and accumulate the oxidized guanine lesion spiroiminodihydantoin. Chem. Res. Toxicol 18, 1378–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mangerich A, Knutson CG, Parry NM, Muthupalani S, Ye W, Prestwich E, Cui L, McFaline JL, Mobley M, Ge Z, Taghizadeh K, Wishnok JS, Wogan GN, Fox JG, Tannenbaum SR, and Dedon PC (2012) Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci. U S A 109, E1820–E1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cadet J, Douki T, Gasparutto D, and Ravanat JL (2003) Oxidative damage to DNA: formation, measurement and biochemical features. Mutat. Res 531, 5–23. [DOI] [PubMed] [Google Scholar]
- (15).Bai J, Zhang Y, Xi Z, Greenberg MM, and Zhou C (2018) Oxidation of 8-Oxo-7,8-dihydro-2′-deoxyguanosine Leads to Substantial DNA-Histone Cross-Links within Nucleosome Core Particles. Chem. Res. Toxicol 31, 1364–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, and Essigmann JM (2003) The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry 42, 9257–9262. [DOI] [PubMed] [Google Scholar]
- (17).Leipold MD, Muller JG, Burrows CJ, and David SS (2000) Removal of hydantoin products of 8-oxoguanine oxidation by the escherichia coli DNA repair enzyme, FPG. Biochemistry 39, 14984–14992. [DOI] [PubMed] [Google Scholar]
- (18).Hazra TK, Muller JG, Manuel RC, Burrows CJ, Lloyd RS, and Mitra S (2001) Repair of hydantoins, one electron oxidation product of 8-oxoguanine, by DNA glycosylases of Escherichia coli. Nucleic Acids Res 29, 1967–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hailer MK, Slade PG, Martin BD, Rosenquist TA, and Sugden KD (2005) Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair (Amst) 4, 41–50. [DOI] [PubMed] [Google Scholar]
- (20).Krishnamurthy N, Zhao X, Burrows CJ, and David SS (2008) Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry 47, 7137–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhao X, Krishnamurthy N, Burrows CJ, and David SS (2010) Mutation versus repair: NEIL1 removal of hydantoin lesions in single-stranded, bulge, bubble, and duplex DNA contexts. Biochemistry 49, 1658–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shafirovich V, Kropachev K, Anderson T, Liu Z, Kolbanovskiy M, Martin BD, Sugden K, Shim Y, Chen X, Min JH, and Geacintov NE (2016) Base and Nucleotide Excision Repair of Oxidatively Generated Guanine Lesions in DNA. J Biol. Chem 291, 5309–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Luijsterburg MS, von Bornstaedt G, Gourdin AM, Politi AZ, Mone MJ, Warmerdam DO, Goedhart J, Vermeulen W, van Driel R, and Hofer T (2010) Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. J. Cell. Biol 189, 445–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Shen JC, Fox EJ, Ahn EH, and Loeb LA (2014) A rapid assay for measuring nucleotide excision repair by oligonucleotide retrieval. Sci. Rep 4, 4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Golato T, Brenerman B, McNeill DR, Li J, Sobol RW, and Wilson DM 3rd. (2017) Development of a Cell-Based Assay for Measuring Base Excision Repair Responses. Sci. Rep 7, 13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bandaru V, Sunkara S, Wallace SS, and Bond JP (2002) A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst) 1, 517–529. [DOI] [PubMed] [Google Scholar]
- (27).Takao M, Kanno S, Shiromoto T, Hasegawa R, Ide H, Ikeda S, Sarker AH, Seki S, Xing JZ, Le XC, Weinfeld M, Kobayashi K, Miyazaki J, Muijtjens M, Hoeijmakers JH, van der Horst G, and Yasui A (2002) Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. Embo J 21, 3486–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Joffe A, Geacintov NE, and Shafirovich V (2003) DNA lesions derived from the site-selective oxidation of guanine by carbonate radical anions. Chem. Res. Toxicol 16, 1528–1538. [DOI] [PubMed] [Google Scholar]
- (29).Crean C, Uvaydov Y, Geacintov NE, and Shafirovich V (2008) Oxidation of single-stranded oligonucleotides by carbonate radical anions: generating intrastrand cross-links between guanine and thymine bases separated by cytosines. Nucleic Acids Res 36, 742–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rokhlenko Y, Geacintov NE, and Shafirovich V (2012) Lifetimes and reaction pathways of guanine radical cations and neutral guanine radicals in an oligonucleotide in aqueous solutions. J. Am. Chem. Soc 134, 4955–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kornyushyna O, Berges AM, Muller JG, and Burrows CJ (2002) In vitro nucleotide misinsertion opposite the oxidized guanosine lesions spiroiminodihydantoin and guanidinohydantoin and DNA synthesis past the lesions using Escherichia coli DNA polymerase I (Klenow fragment). Biochemistry 41, 15304–15314. [DOI] [PubMed] [Google Scholar]
- (32).Khutsishvili I, Zhang N, Marky LA, Crean C, Patel DJ, Geacintov NE, and Shafirovich V (2013) Thermodynamic profiles and nuclear magnetic resonance studies of oligonucleotide duplexes containing single diastereomeric spiroiminodihydantoin lesions. Biochemistry 52, 1354–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Fleming AM, Muller JG, Dlouhy AC, and Burrows CJ (2012) Structural context effects in the oxidation of 8-oxo-7,8-dihydro-2′-deoxyguanosine to hydantoin products: electrostatics, base stacking, and base pairing. J. Am. Chem. Soc 134, 15091–15102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mao B, Xu J, Li B, Margulis LA, Smirnov S, Ya NQ, Courtney SH, and Geacintov NE (1995) Synthesis and characterization of covalent adducts derived from the binding of benzo[a]pyrene diol expoxide to a -GGG-sequence in a deoxyoligonucleotide. Carcinogenesis 16, 357–365. [DOI] [PubMed] [Google Scholar]
- (35).Jordan M, Schallhorn A, and Wurm FM (1996) Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res 24, 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Masters JR, and Stacey GN (2007) Changing medium and passaging cell lines. Nat Protoc 2, 2276–2284. [DOI] [PubMed] [Google Scholar]
- (37).Choi JH, Gaddameedhi S, Kim SY, Hu J, Kemp MG, and Sancar A (2014) Highly specific and sensitive method for measuring nucleotide excision repair kinetics of ultraviolet photoproducts in human cells. Nucleic Acid. Res 42, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hu J, Choi JH, Gaddameedhi S, Kemp MG, Reardon JT, and Sancar A (2013) Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J. Biol. Chem 288, 20918–20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Huang JC, Svoboda DL, Reardon JT, and Sancar A (1992) Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc. Natl. Acad. Sci. U S A 89, 3664–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Conney AH (1982) Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons: G. H. A. Clowes Memorial Lecture. Cancer Res 42, 4875–4917. [PubMed] [Google Scholar]
- (41).Cosman M, Ibanez V, Geacintov NE, and Harvey RG (1990) Preparation and isolation of adducts in high yield derived from the binding of two benzo[a]pyrene-7,8-dihydroxy-9,10-oxide stereoisomers to the oligonucleotide d(ATATGTATA). Carcinogenesis 11, 1667–1672. [DOI] [PubMed] [Google Scholar]
- (42).Hess MT, Gunz D, Luneva N, Geacintov NE, and Naegeli H (1997) Base pair conformation-dependent excision of benzo[a]pyrene diol epoxide-guanine adducts by human nucleotide excision repair enzymes. Mol. Cell. Biol 17, 7069–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Mocquet V, Kropachev K, Kolbanovskiy M, Kolbanovskiy A, Tapias A, Cai Y, Broyde S, Geacintov NE, and Egly JM (2007) The human DNA repair factor XPC-HR23B distinguishes stereoisomeric benzo[a]pyrenyl-DNA lesions. EMBO J 26, 2923–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kropachev K, Kolbanovskii M, Cai Y, Rodriguez F, Kolbanovskii A, Liu Y, Zhang L, Amin S, Patel D, Broyde S, and Geacintov NE (2009) The sequence dependence of human nucleotide excision repair efficiencies of benzo[a]pyrene-derived DNA lesions: insights into the structural factors that favor dual incisions. J. Mol. Biol 386, 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).McKibbin PL, Fleming AM, Towheed MA, Van Houten B, Burrows CJ, and David SS (2013) Repair of hydantoin lesions and their amine adducts in DNA by base and nucleotide excision repair. J. Am. Chem. Soc 135, 13851–13861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Odell ID, Newick K, Heintz NH, Wallace SS, and Pederson DS (2010) Non-specific DNA binding interferes with the efficient excision of oxidative lesions from chromatin by the human DNA glycosylase, NEIL1. DNA Repair (Amst) 9, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Porello SL, Leyes AE, and David SS (1998) Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry 37, 14756–14764. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.