

Abstract
The anthracycline–DNA complex, which is a potent agent for cancer chemotherapy, has a unique intercalating molecular structure with preference to the GC bases of DNA, as shown by Rich's group in studies of single-crystal x-ray diffraction. Understanding cytotoxicity and its photoenhancement requires the unraveling of the dynamics under the solution-phase, physiological condition. Here we report our first study of the primary processes of drug function. In a series of experiments involving the drug (daunomycin and adriamycin) in water, the drug–DNA complexes, the complexes with the four nucleotides (dGTP, dATP, dCTP, and dTTP), and the drug-apo riboflavin-binding protein, we show the direct involvement of molecular oxygen and DNA base-drug charge-separation—the rates for the reduction of the drug and dioxygen indicate the crucial role of drug/base/O2 in the efficient and catalytic redox cycling. These dynamical steps, and the subsequent reactions of the superoxide product(s), can account for the photoenhanced function of the drug in cells, and potentially for the cell death.
Drug molecules that target a particular DNA sequence have shown selectivity to inhibit or modulate gene expression and are valuable for a variety of chemotherapeutic strategies (1, 2). Daunomycin (DM), one of the anthracycline antibiotics, is among the most effective drugs for cancer chemotherapy. Previous studies of single-crystal molecular structures (3, 4) and solution-phase binding thermodynamics and kinetics (5, 6) have demonstrated that DM molecules have preference for the GC bases of DNA and intercalate into GC base pairs (Fig. 1). It is also known that the cytotoxicity of DM is enhanced by photoactivation (7, 8). The understanding of the recognition process and drug action requires not only the important information about static molecular structures, but also knowledge of the elementary steps of dynamics. For a rational design of reactivity (9), unraveling the dynamics under physiological conditions of hydration and oxygenation is essential to the function.
Figure 1.

Crystal structure of DM-DNA complex. DM, shown as space-filled, intercalates into GC base pairs while the amino sugar ring, daunosamine, fits in the minor groove. (Upper) The crystal structure obtained from x-ray diffraction by Rich and his group (3). (Lower) The DNA double strand—in our case, a GC homopolymer.
In this contribution, we report direct observation of the primary processes involved in DM and adriamycin (AM) drug action. It is found that the selective recognition of GC enhances charge separation with the net transfer of electron to the drug. Furthermore, we observe a striking effect of dioxygen on the drug–dGTP nucleotide in water—the presence of O2 depletes the drug radical population, leading to the formation of superoxide anion radicals (see below), whereas its absence leads to charge recombination to reform the initial state. Because these reactions were found to be ultrafast in nature, all subsequent reactions involving dioxygen occur as a result of charge separation. These processes can account for the photoenhancement of the drug action in cells, cytotoxicity, and potentially as precursors for the cell death. We discuss the relationship of this effect of enhanced cell damage to the less efficient thermal reaction involving a similar transition state. We also report results for DM-apo riboflavin-binding protein complex, which show the analogy with DNA hydrophobic recognition, but with redox cycling due to tryptophans.
Methodology
For thermal reactions, charge separation is a slow process because of energy-barrier crossing. We used femtosecond (fs) excitation to induce (over the barrier) electron transfer from the base to the intercalated (DNA) or complexed (nucleotide) drug. We then probed, with fs resolution, the evolution of the charge-transfer state and its subsequent decay, either by charge recombination recovery or by reaction with O2. We have invoked an elaborate laser setup to study these systems by using transient absorption and fluorescence up-conversion methods, as described (10). Briefly, a fs laser pulse at 480 nm or 510 nm (controlled, 0.1–0.4 μJ) was used to excite the drug molecules. A second fs pulse at ≈800 nm (80 fs and 40 μJ) was used to probe the fluorescence emission by mixing it in a 0.2-mm BBO crystal in the up-conversion experiment. For the transient absorption experiment a fs probe pulse in the wavelength range 465–700 nm or at ≈400 nm was used to measure the absorption transient induced by the excitation pulse. The polarization of the pump and probe pulses were set to magic angle conditions (54.7°) to avoid contributions from orientational motion. The experiments were performed at room temperature.
Sample Preparation
Mononucleotides (dGTP, dATP, dCTP, and dTTP) and anticancer drugs (DM and AM, 98%) were from Sigma. DNA double helix is GC homopolymers obtained from Amersham Pharmacia. The DNA sample was prepared and purified as described (6). Before further use, the DNA was dialyzed against a buffer consisting of 6 mM Na2HPO4, 2 mM NaH2PO4, 1 mM Na2EDTA, and 0.185 M NaCl at pH 7.0 for 24 h. Ambient oxygen, as well oxygen from a cylinder, was used in experiments involving the effect on the observed dynamics.
Results and Discussion
The spectroscopy of DM is known (unpublished data; see also ref. 11): the absorption peak is at 480 nm and the emission “mirrors” the absorption with a peak at 590 nm. The time-resolved spectra of DM in water are detailed elsewhere: because the emission bands do not shift to longer wavelength with increasing polarity, transient solvation of DM by water is minor. In water, our measured vibrational relaxation time is ≈200 fs and up to 1 picosecond (ps) and the fluorescence lifetime is 1 ns.
Fig. 2 shows the fs-resolved fluorescence of DM in water, in the presence of DNA (GC homopolymer), and also with guanine nucleotide, 2′-deoxyguanosine 5′-triphosphate (dGTP); not shown are the other nucleotides studied: dATP, dCTP, and dTTP and the data for AM. The dramatic change in rates is evident: DM in water (1 ns), DM-DNA (290 fs; 1.7 ps), and DM-dGTP (600 fs; 3 ps). The intercalation of DM in DNA is certain (3–6). This intercalation leads to a dramatic shortening of the decay time of DM, by 3 orders of magnitude, from 1 ns to 290 fs. This huge increase in rates is clearly the result of complexation of the DNA base with the drug and is due to electron transfer from the base to DM. The major part of the transfer (63%) occurs in 290 fs; the 1.7-ps component may reflect the other conformations of unprivileged structures. NMR studies and theoretical calculations (12, 13) indicate the existence of two conformations of DM, and the observed time constant for the electron transfer is consistent with at least two structurally different complexes. However, charge separation may involve a distribution of rates due to vibration relaxation (see above) and also orientation relaxation.
Figure 2.

Fluorescence up-conversion decays for drug DM (10 μM) in water (610 nm); with DNA (base pairs 1 mM); and with nucleotide dGTP (90 mM). GC homopolymer was prepared as described in ref. 6. The buffer used consisted of 6 mM Na2HPO4, 2 mM NaH2PO4, 1 mM Na2EDTA, and 0.185 M NaCl at pH 7.0 for all samples. Details of the experimental apparatus are given in ref. 10. The measured decay time constants are given for the three cases (see text).
For DM-DNA complexes, the fluorescence signal decays almost to zero in a few ps. The remaining nanosecond component accounts for less than 1% and is due to free DM molecules in water (14). This large ratio shows that almost all DM molecules were intercalated into the hydrophobic environment of GC base pairs at our concentration ratio of 100:1; [base pairs] = 1 mM, [DM] = 10 μM. The equilibrium constant has recently been measured (6) and it is 2.6 × 105 M−1, indicating that larger than 99% of complexes of DM-DNA are formed under our conditions. For the nucleotides, the equilibrium constant is significantly smaller and from concentration-dependence of the time-resolved fluorescence, we estimated it to be 160 M−1; the percentage of complexes formed is 97% at the stated conditions. For nucleotides, the concentration ratio typically is 9,000:1. Clearly the concentration of free drugs and their dimers is less than 1% for DNA and 3% for nucleotides.
The evidence for charge separation comes from several observations. First, the decay of DM in DNA is similar to that observed with the nucleotide dGTP, but vastly different from that of the free drug in water. Second, the decay rate of DM in the presence of the four different nucleotides exhibits a trend consistent with the driving force ΔG0 of the reaction for electron transfer: the easier the oxidation of the nucleotide, the faster the rate of electron transfer. Electrochemistry studies of DM give the reduction potential [DM/DM−⋅ is −0.38 ± 0.08 V vs. NHE Standard (ref. 15 and references therein)] and knowing the oxidation potential of the base [G/G+⋅ ≈ +1.4 V vs. NHE (16–18)] and the energy of 0, 0 transition (2.3 eV; 1 eV = 1.602 × 10−19 J), we obtained a net ΔG0 of −0.5 eV. Similarly, we obtained ΔG0 for the other bases; the rate is largest for dGTP, then for dATP (reaction time, 18 ps and 60 ps), and the rates for dTTP and dCTP are slower than (1 ns)−1. We also investigated the involvement of other possible processes. From our results the following processes can be excluded: proton transfer, resonance energy transfer, internal conversion, and intersystem crossing. We observed no noticeable change with D2O/H2O solution and with pH 5–7; the drug lifetime in different solvents is still on the nanosecond scale, and the absorption of bases does not overlap with the drug emission. There is also a direct evidence; namely, the observation of the charge-separated state—its formation, and recombination and reaction.
In Fig. 3, we display the striking change in the ground-state transient absorption (λext = 510 nm; λprobe = 480 nm) of DM-dGTP in the presence and absence of dioxygen. In both cases, we first observe negative absorption. At room temperature, the concentration of O2 is ≈1 mM in water. On purging with argon to remove O2, the observed signal shows a rise from negative to positive. In the presence of oxygen, either in the water solution or by bubbling it through the sample, the positive absorption disappears. It is important to note that we have repeated the fluorescence decay measurements with and without O2 for DM-dGTP and observed no change (see Fig. 3), indicating that the forward rate for charge separation is unaffected by oxygen. Also, note that the observed negative absorbance is not due to stimulated emission because our pump pulse (510 nm) was at lower energy than the probe pulse (480 nm) and we only monitored the change in absorption of the probe (480 nm).
Figure 3.

Transient absorption decays of DM-dGTP nucleotide in the absence and presence of dioxygen; the ratio of nucleotide to drug concentration is 900. The best theoretical fits are shown as solid lines. Note the positive absorbance when O2 is absent. (Inset) The states described by the steps in Eq. (1). (Upper Inset) The fluorescence decays (using up-conversion) with and without oxygen (no change).
The above observations elucidate the elementary steps involved in the mechanism:
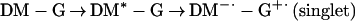 |
1a |
 |
1b |
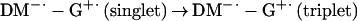 |
1c |
 |
1d |
Fig. 3 contains the analysis of the population flow described by Eqs. 1a–d. With oxygen, the rise of the negative absorption of DM-dGTP was observed in ≈600 fs and 3 ps, and certainly the signal was back to almost zero in at most 10 ps. This indicates that the signal is due to the formation and recombination to the ground state of the singlet charge transfer state and that the rate of this recovery is not larger than (10 ps)−1. In the absence of oxygen, the absorption rises from negative to positive values, then decays in ≈35 ps, which may imply a recovery with this time constant. However, careful measurements of the positive absorption as a function of pump laser power (and using cw vs. pulsed lasers), cell thickness, and stirring of the solution indicated that this signal is produced as a result of a long-lived triplet species, with lifetime shorter than a millisecond, as determined by our repetition rate; it quenches effectively with O2. The minor product contribution (resulting from the triplet state) is monitored by its absorption at 510 nm, and the ≈35-ps decay is from the probe signal at 480 nm; the product is formed by a diffusion-controlled bimolecular reaction, likely to form complexes of reduced DM dimers (or with other moeities) that have absorption similar to DM but red shifted, as we observed experimentally. It should be emphasized that such fs–ps transient behavior was absent for samples of the nucleotide or the drug alone in solution—in fact we observed only the coherence spike for the former and the negative absorption (ns) for the latter. Accordingly, we have observed channel (Eq. 1d) with a long-lived charge separated state capable of producing the superoxide.
These results show the direct involvement of oxygen. In water and at room temperature, a diffusion-controlled process will give a decay time close to a microsecond for a bimolecular encounter with O2, faster than the lifetime on the millisecond or longer scale. The redox property of DM and O2 supports this picture: the potential for DM/DM−⋅ is ≈−0.4 V (ref. 15 and references therein) and for O2/O2− is ≈−0.16 V in water (19), thus the driving force is favorable. From the steady-state redox chemistry of anthracyclines, it has been reported that redox recycling, which begins with the formation of the DM−⋅ radical and the following reaction with oxygen to generate superoxide, is an important aspect of drug cytotoxicity (ref. 15, and references therein, and ref. 20). In our studies, the key is the direct, enhanced reduction of the drug and the clocking of subsequent reactions.
With AM, we have obtained similar results for the time scales involved in charge separation, indicating that the primary processes for both drugs are of a similar nature. The molecular structures of the drugs (Scheme S1) differs only in one of the terminal substituents, suggesting that subsequent reactions involving O2− that account for the difference in cytotoxicity may involve the hydrogen migration through enediol tautomerization. The experiments on DNA–drug complexes show a faster time for charge separation, reflecting the ≈3-Å short separation between GC and the drug in the confined intercalation site, in contrast with the less confined nucleotide complexes. The positive absorption was not observed for the DNA case, but the recovery of the negative absorption was still occurring in ≈5 ps. This finding is consistent with G+⋅-hole migration to the neighboring site(s) DM−⋅-G-C-G+⋅; the latter is known to be on the ps time scale (10, 21) and can result in DNA cleavage (22).
Scheme 1.

Molecular structure of DM and AM drugs.
As with DNA, many proteins have hydrophobic domains. To test for the applicability of the above mechanism in proteins we also carried out a series of experiments for DM in the apo riboflavin-binding protein (chicken egg white). The crystal structure is known (23) and there are several tryptophan residues in the hydrophobic pocket. These residues, similar to G, have the ability to donate electrons (tryptophan/tryptophan+⋅ ≈ +1.15 V (24); net ΔG0 = −0.75 eV). We observed similar dynamical behavior in the DNA and protein systems, with and without oxygen, except for a change by a factor of 3 in the electron transfer rates—the drug–DNA complex is more efficient, likely due to a more perfect chelation. In the protein, the rates decrease as we increase the degree of unfolding by changing the concentration of the denaturing agent (Fig. 4). The drug DM behaves as in water (see Fig. 2) when the concentration of the agent reaches 2 M, entirely consistent with the effect of binding and charge separation in the confined environment.
Figure 4.

Fluorescence up-conversion decays (610 nm) of DM (50 μM) bound to the protein, chicken egg white apo riboflavin-binding protein (0.4 mM). Shown is the dependence on the concentration of denaturing agent, guanidine hydrochloride, from 0–2 M.
In living cells, previous studies have shown that DM cytotoxicity is significantly enhanced by laser photoactivation (7, 8). However, the cause of the enhancement was not clear. With the mechanism we report here, we can elucidate the drug function. The photoexcitation enhances the rate of charge separation over that of the thermal reaction (barrier crossing) and triggers a redox cycling with a much higher efficiency, thus resulting in enhanced cell killing by the anticancer drug. The reaction with oxygen guarantees the success of trajectories forming the superoxide even for the slow encounter characteristics of thermal reactions. Our ultrafast rate of charge separation is for excitation at λ = 510 or 480 nm, and it is remarkable that the photoactivated cytotoxicity was found efficient at 514 nm. Moreover, the product for in vitro drug solutions, excited at 438 nm, was spin-trapped and found to be O2−, which decays to give hydroxyl radicals (25).
In general, it has been reported that this class of drugs can stimulate topoisomerase II-mediated DNA cleavage by inducing structural alterations of the DNA intercalation sites; ternary complex of topo II/drug/DNA have been isolated (ref. 15, and references therein, and ref. 26). This process can cause deletions, insertions, and chromosomal aberrations and trigger lethal pathways in damaged cells and finally kill cancerous cells (27, 28). Our results of ultrafast electron transfer between guanine (smaller rate for A, C, and T) and the drug and the direct role of molecular oxygen suggest that this redox activation can trigger damage and may stimulate topoisomerase II poisons, which can ultimately lead to cell death (Scheme S2).
Scheme 2.

Schematic of the involvement of drug and molecular oxygen in the dynamics of the nucleotide and DNA activation. Also shown is the mechanism of topoisomerase II enzyme-mediated DNA cleavage. The associated O2 is only for illustration.
The enhanced effect induced by light is thus due to the energetically and kinetically favored reaction over the barrier. Both the thermal and photoactivated reactions are likely to have similar transition states, as dictated by state correlations involving ionic potentials (29, 30). The extension of our studies to other drug/DNA systems is needed before generalization can be made. However, the results reported here clearly elucidate the role of charge separation and the biologically reactive molecular oxygen. The intercalation with DNA is important for equilibrium properties (equilibrium constant and ΔG0), but the primary function of the drug cannot be fully revealed without knowledge of the dynamics that control the subsequent steps for the disruption of DNA and RNA synthesis. Currently we are studying different types of DNA in the hope of generalizing the above picture for the molecular basis of clinical observations.
Acknowledgments
We wish to thank Dr. Spencer Baskin for his devoted effort during many hours of experiments and discussions. This work is supported by the National Science Foundation. H.-C.B. is grateful for financial support from The Wenner-Gren Foundations.
Abbreviations
- DM
daunomycin
- AM
adriamycin
References
- 1.Chaires J B. Curr Opin Struct Biol. 1998;8:314–320. doi: 10.1016/s0959-440x(98)80064-x. [DOI] [PubMed] [Google Scholar]
- 2.Foote-Zewail M, Hurley L H. Anticancer Drug Des. 1999;14:1–9. [PubMed] [Google Scholar]
- 3.Wang A H, Ughetto G, Quigley G J, Rich A. Biochemistry. 1987;26:1152–1163. doi: 10.1021/bi00378a025. [DOI] [PubMed] [Google Scholar]
- 4.Frederick C A, Williams L D, Ughetto G, van der Marel G A, van Boom J H, Rich A, Wang A H. Biochemistry. 1990;29:2538–2549. [PubMed] [Google Scholar]
- 5.Chaires J B, Dattagupta N, Crothers D M. Biochemistry. 1985;24:260–267. doi: 10.1021/bi00323a004. [DOI] [PubMed] [Google Scholar]
- 6.Qu X, Trent J O, Fokt I, Priebe W, Chaires J B. Proc Natl Acad Sci USA. 2000;97:12032–12037. doi: 10.1073/pnas.200221397. . (First Published October 10, 2000; 10.1073/pnas.200221397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paiva M B, Saxton R E, Graeber I P, Jongewaard N, Eshraghi A A, Suh M J, Paek W H, Castro D J. Lasers Surg Med. 1998;23:33–39. doi: 10.1002/(sici)1096-9101(1998)23:1<33::aid-lsm5>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 8.Andreoni A, Colasanti A, Malatesta V, Roberti G. Radiat Res. 1991;127:24–29. [PubMed] [Google Scholar]
- 9.Lerner R A, Benkovic S J, Schultz P G. Science. 1991;252:659–667. doi: 10.1126/science.2024118. [DOI] [PubMed] [Google Scholar]
- 10.Fiebig T, Wan C, Kelley S O, Barton J K, Zewail A H. Proc Natl Acad Sci USA. 1999;96:1187–1192. doi: 10.1073/pnas.96.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaires J B, Dattagupta N, Crothers D M. Biochemistry. 1982;21:3933–3940. doi: 10.1021/bi00260a005. [DOI] [PubMed] [Google Scholar]
- 12.Penco S, Angelucci F, Ballabio M, Barchielli G, Suarato A, Vanotti E, Vigevani A, Arcamone F. Heterocycles. 1984;21:21–28. [Google Scholar]
- 13.Tosi C, Fusco R, Ranghino G, Malatesta V. J Mol Struct Theochem. 1986;134:341–350. [Google Scholar]
- 14.Malatesta V, Andreoni A. Photochem Photobiol. 1988;48:409–415. doi: 10.1111/j.1751-1097.1988.tb02838.x. [DOI] [PubMed] [Google Scholar]
- 15.Priebe W, editor. Anthracycline Antibiotics, New Analogues, Methods of Delivery & Mechanisms of Actions. Washington, DC: Am. Chem. Soc.; 1995. [Google Scholar]
- 16.Hush N S, Cheung A S. Chem Phys Lett. 1975;34:11–13. [Google Scholar]
- 17.Seidel C A M, Schultz A, Sauer M H M. J Phys Chem. 1996;100:5541–5553. [Google Scholar]
- 18.Steenken S, Jovanovic S. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 19.Sawyer D T, editor. Oxygen Chemistry. New York: Oxford Univ. Press; 1991. [Google Scholar]
- 20.Taatjes D J, Gaudiano G, Resing K, Koch T H. J Med Chem. 1997;40:1276–1286. doi: 10.1021/jm960835d. [DOI] [PubMed] [Google Scholar]
- 21.Wan C, Fiebig T, Kelley S O, Treadway C R, Barton J K, Zewail A H. Proc Natl Acad Sci USA. 2000;97:14052–14055. doi: 10.1073/pnas.250483297. . (First Published December 5, 2000; 10.1073/pnas.250483297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gray P J, Phillips D R, Wedd A G. Photochem Photobiol. 1982;36:49–57. doi: 10.1111/j.1751-1097.1982.tb04339.x. [DOI] [PubMed] [Google Scholar]
- 23.Monaco H L. EMBO J. 1997;16:1475–1483. doi: 10.1093/emboj/16.7.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeFelippis M R, Murthy C P, Broitman F, Weinraub D, Faraggi M, Klaffer M H. J Phys Chem. 1991;95:3416–3419. [Google Scholar]
- 25.Carmichael A J, Mossoba M M, Riesz P. FEBS Lett. 1983;164:401–405. doi: 10.1016/0014-5793(83)80325-1. [DOI] [PubMed] [Google Scholar]
- 26.Bodley A, Liu L F, Israel M, Seshadri R, Koseki Y, Giuliani F C, Kirschenbaum S, Silber R, Potmesil M. Cancer Res. 1989;49:5969–5978. [PubMed] [Google Scholar]
- 27.Kingma P S, Corbett A H, Burcham P C, Marnett L J, Osheroff N. J Biol Chem. 1995;270:21441–21444. doi: 10.1074/jbc.270.37.21441. [DOI] [PubMed] [Google Scholar]
- 28.Chen A Y, Liu L F. Annu Rev Pharmacol Toxicol. 1994;34:191–218. doi: 10.1146/annurev.pa.34.040194.001203. [DOI] [PubMed] [Google Scholar]
- 29.Steiger B, Baskin J S, Anson F C, Zewail A H. Angew Chem Int Ed. 2000;39:257–260. [PubMed] [Google Scholar]
- 30.Donahue N M, Clarke J S, Anderson J G. J Phys Chem A. 1998;102:3923–3933. [Google Scholar]