

Abstract
The conjugation of antibodies with cytotoxic drugs can alter their in vivo pharmacokinetics. As a result, the careful assessment of the in vivo behavior — and specifically the tumor-targeting properties — of antibody-drug conjugates represents a crucial step in their development. In order to facilitate this process, we have created a methodology that facilitates the dual labeling of an antibody with both a toxin and a radionuclide for positron emission tomography (PET). To minimize the impact of these modifications, this chemoenzymatic approach leverages strain-promoted azide-alkyne click chemistry to graft both cargoes to the heavy chain glycans of the immuoglobulin’s Fc domain. As a proof-of-concept, a HER2-targeting trastuzumab immunoconjugate was created bearing both a monomethyl auristatin E (MMAE) toxin as well as the long-lived positron-emitting radiometal 89Zr (t1/2 ~ 3.3 days). Both the tumor targeting and therapeutic efficacy of the 89Zr-trastuzumab-MMAE immunoconjugate were validated in vivo using a murine model of HER2-expressing breast cancer. The site-specifically dual-labeled construct enabled the clear visualization of tumor tissue via PET imaging, producing tumoral uptake of ~70 %ID/g. Furthermore, a longitudinal therapy study revealed that the immunoconjugate exerts significant anti-tumor activity, leading to a >90% reduction in tumor volume over the course of 20 days.
Keywords: Antibody-drug conjugate, ADC, positron emission tomography, PET, site-specific bioconjugation, heavy chain glycans, click chemistry, strain-promoted azide-alkyne click chemistry
Graphical Abstract
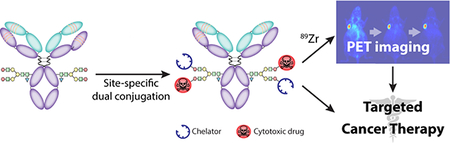
INTRODUCTION
The idea of conjugating toxins to antibodies in order to enhance the tumor-specific delivery of chemotherapeutics dates back to the middle of the 20th century. This field has largely been driven by two parallel trends: (1) the discovery of non-selective yet extremely powerful cytotoxic agents and (2) the development of immunoglobulins with extraordinarily high affinity and selectivity for cancer biomarkers. Over the years, the study of these immunoconjugates — now ubiquitously known as antibody-drug conjugates (ADCs) — has exploded. To wit, as of 2017, there are a remarkable 108 different reviews containing “antibody-drug conjugate” in their title.1
A sizeable contingent of ADCs has been synthesized via the random conjugation of toxins to the amino acids of the antibody, almost certainly due to the relative ease of this approach. For example, one of the two ADCs currently approved by the United States FDA — KADCYLA® — is synthesized through the random conjugation of emtansine to the lysines of the HER2-targeting antibody trastuzumab.2 However, recent years have witnessed a dramatic shift in the field toward ADCs synthesized using site-specific conjugation methods. This pivot toward more well-defined and homogeneous ADCs has been fueled in large part by several studies demonstrating the superior in vivo performance of site-specifically modified immunoconjugates as well as the exigencies of the regulatory review process.3–7 Yet it is steadily becoming apparent that even the site-specific conjugation of payloads to biomolecules may not be as benign as previously thought. Recent studies have clearly demonstrated that the attachment of cargoes to biomolecular vectors can dramatically alter the pharmacokinetic profiles of the bioconjugates and even impede their ability to reach their target in vivo.8,9
In light of this data, assessing the tumor targeting and in vivo behavior of ADCs using data obtained for the parent antibody is clearly a shortcut rife with problems. Admittedly, reliable methods do exist for directly determining the concentration of ADCs in blood and serum; however, techniques for quantifying drug concentrations in other tissues remain imperfect. The latter, for example, has typically been performed by analyzing tissues from organs harvested during necropsy, hardly a viable approach with human patients undergoing therapy.
Clearly, the optimal solution is a targeted drug delivery system that can be tracked in vivo using quantitative non-invasive imaging. Non-invasive imaging modalities —primarily magnetic resonance imaging (MRI) or positron emission tomography (PET) — have already been used to facilitate the visualization and quantification of nanomedicine. Grange et al., for example, coated doxorubicin-loaded liposomes with gadolinium complexes and used MRI to verify the tumoral delivery of the liposomes in mice bearing Kaposi’s sarcoma.10 More recently, a group from King’s College (UK) labeled doxorubicin- and alendronate-containing liposomes with 89Zr and used PET imaging to study the biodistribution of the particles in mice bearing mammary carcinoma.11
Yet somewhat surprisingly, this approach has been applied to ADCs remarkably sparingly, especially considering the advent of immunoPET over the last two decades. Indeed, to the best of our knowledge, only two reports exists detailing ADCs that have also been labeled with a radionuclide for nuclear imaging. In this first, Cohen, et al. used a dual radiolabeling approach to create a 89Zr-labeled variant of trastuzumab that also bears a 131I-labeled tubulysin A analogue. The presence of two different radionuclides allowed the authors to track the biodistributions of the antibody and the toxin independently and quantify their delivery to both target and non-target tissues.12 In the second, Boswell, et al. radiolabeled an anti-TENB2 ThioMab conjugated with monomethyl auristatin E (MMAE) with 111In.13 The in vivo tracking of the 111In-labeled ADC via SPECT imaging allowed the authors to assess the effect of the pre-injection of unconjugated antibody on the ability of the ADC to reach TENB2-expressing xenografts.
The limits of traditional bioconjugation strategies almost certainly play a role in this dearth of imaging-enabled antibody-drug conjugates (IEADCs). If, as we have noted, the random conjugation of a single payload can create problems, the random attachment of two different moieties only multiplies potential complications, including heterogeneity, impaired immunoreactivity, and suboptimal in vivo performance. In the investigation at hand, we have circumvented this issue by creating what is — to the best of our knowledge — the first immunoconjugate that has been site-specifically labeled with both a toxin and a positron-emitting radiometal. More specifically, we have used a chemoenzymatic approach to create DFO:MMAE-sstrastuzumab, an IEADC in which both monomethyl auristatin A (MMAE) and the radiometal chelator desferrioxamine (DFO) have been conjugated to the heavy chain glycans of the HER2-targeting antibody trastuzumab. Using a murine model of HER2-expressing breast cancer, we clearly illustrate that this IEADC is an effective therapeutic agent and can be tracked noninvasively using PET imaging when labeled with the long-lived positron-emitting isotope 89Zr (t1/2 ~3.3 d). Ultimately we envision that dual-labeled radioADCs such as 89Zr:MMAE-sstrastuzumab could play important roles in clinic, both as more accurate scout imaging agents prior to ADC-based therapy and as imaging agents capable of providing real-time dose information during treatment.
EXPERIMENTAL PROCEDURES
Modification of Trastuzumab
5.0 mg of trastuzumab in 750 μL of buffer (50 mM Bis-Tris, 100 mM NaCl, pH 6.0) were incubated with 40 μL of β-(1,4)-galactosidase (2.0 U/mL) at 37°C overnight. A solution of 60 μL of 1 M Tris, 12 μL of 1M solution of MnCl2, 100 μL of GalT, 30 μL of a 40 mM solution of UDP-GalNAz, and 300 μL of deionized water was then added to the antibody solution, and the resultant mixture was incubated at 30 °C overnight. The excess of UDP-GalNAz was removed by washing the functionalized antibody 4 times with TBS (50 mM Tris.HCl, 150 mM NaCl, pH 7.6) using a 2 mL Amicon Centrifugal Filter with a 50K molecular weight cut-off. The solution of antibody was diluted in TBS to reach a concentration of 2g/L (2200 μL), and the DIBO precursors (in solution in DMSO at 8mM) were added in order to reach a final DIBO concentration of 0.3 mM (DFO-sstrastuzumab: 82.5 μL of DIBO-DFO; DFO:MMAE-sstrastuzumab: 41.3 μL of DIBO-DFO, 41.3 μL of DIBO-MMAE) The mixture was incubated overnight at room temperature before purification via size-exclusion chromatography (Sephadex G-25 M, PD-10 column, GE Healthcare; dead volume = 2.5 mL, eluted with 2 × 1 mL fractions of TBS, pH 7.6) and concentration using a 2 mL Amicon Centrifugal Filter with a 50K molecular weight cut-off, ultimately producing the immunoconjugates in a global yield of 70%.
Radiolabeling of Immunoconjugates
For each antibody construct, 500 μg of immunoconjugate solution was diluted to 400μL with PBS, pH 7.4. [89Zr]Zr-oxalate (1500 μCi) in 150 μL of 1.0 M oxalic acid was adjusted to pH 7.0–7.5 with 1.0 M Na2CO3. After the bubbling of CO2 stopped, the 89Zr solution was added to the antibody solution, and the resulting mixture was incubated at room temperature for 1h. The reaction progress was then assayed using iTLC and an eluent of 50mM EDTA (pH 5). Subsequently, the reaction was quenched with 13 μL of 50mM of EDTA (pH=5), and the antibody construct was purified using size exclusion chromatography (Sephadex G-25 M, PD-10 column, GE Healthcare; dead volume = 2.5 mL, eluted with 500 μL fractions of PBS, pH 7.4) and if necessary concentrated via centrifugal filtration units with a 50,000 molecular weight cut off (Amicon™ Ultra 4 Centrifugal Filtration Units, Millipore Corp. Billerica, MA). The radiochemical purity of the final radiolabeled bioconjugate was assayed by radio-TLC again using 50mM EDTA (pH 5) as an eluent. In the iTLC experiments, free 89Zr4+ cations and [89Zr]-EDTA elute with solvent front, while radiolabeled antibody construct remains at the baseline.
PET imaging
PET imaging experiments were conducted on an Inveon microPET-CT (Siemens). Athymic nude mice bearing subcutaneous BT474 xenografts (left shoulder, 60–120 mm3, 25–30 days after inoculation) were administrated with the radioimmunoconjugates (150 μCi, 60–65μg, in solution in 200 μL of saline) via tail vein injection. Approximately 5 min before PET imaging, mice were anesthetized by inhalation of a 2% isoflurane (Baxter Healthcare):oxygen gas mixture and placed on the scanner bed. Anesthesia was maintained using a 1% isoflurane mixture. PET data for each mice were recorded via static scans at 24 h, 72 h and 120 h p.i.
Therapy study
Athymic nude mice bearing subcutaneous BT474 xenografts (left flank; 60–120 mm3) were randomized in 5 groups (n=10) before the study and were administrated with DFO:MMAE-sstrastuzumab at 2.5 or 10 mg/kg, or with controls (saline solution, 10 mg/kg native trastuzumab, or 0.1 mg/kg free MMAE) via tail vein injection. Following injection, the length (L) and width (W) of the tumors were measured every 2–3 days using calipers, and the tumor volume (V) was assessed from these measured parameters using the formula V=1/2*L*W2.
RESULTS AND DISCUSSION
Design and Synthesis
For the synthesis of our site-specifically labeled IEADC, we used a reliable and modular approach that our laboratories have developed over the last 5 years: the chemoenzymatic modification of the heavy chain glycans. This strategy — originally inspired by the work of Qasba and Hseih-Wilson — relies on two sequential enzymatic reactions to append terminal azide-bearing monosaccharides to the pair of biantennary sugar chains on the CH2 domain of the antibody’s FC region.14–16 Subsequently, a strain-promoted azide-alkyne cycloaddition (SPAAC) reaction between the azide-bearing antibody and cyclooctyne-modified payloads facilitates the site-specific grafting of cargoes to the immunoglobulin. This approach seemed ideally suited for our application for two reasons. First, we have already successfully demonstrated the utility of this strategy for dual-modification and, just as importantly, demonstrated that the ratio of the two payloads can be altered by modifying the initial ratio of DIBO-bearing precursors in the reaction mixture.17,18 Second, non-radiolabeled ADCs have recently been synthesized using analog site-specific glycan modification methods and exhibited unimpaired therapeutic potency.19
The design choices for this proof-of-concept study are straightforward. Trastuzumab was chosen as the targeting vector because the antibody is robust, extraordinarily well characterized, and has been the basis of a variety of different ADCs.2,20–22 The tubulin inhibitor monomethyl auristatin E (MMAE) was selected as the toxin because ADCs bearing MMAE have been used widely in cancer therapy, with several of these agents currently in clinical trials.28 Finally, 89Zr and its gold standard chelator desferrioxamine (DFO) were enlisted due to the advantageous match between the radionuclidic half-life of 89Zr (t1/2 ~3.3 days) and the biological half-life of immunoglobulins as well as the clear clinical utility demonstrated by 89Zr-immunoPET.29
The chemoenzymatic modification of trastuzumab was performed according to established procedures via the incubation of the antibody with β−1,4-galactosidase and then the promiscuous galactosyltransferase GalT(Y289L) in conjunction with the azide-modified sugar GalNAz (Fig. 1A). Subsequently, this N3-sstrastuzumab intermediate was reacted with cyclooctyne-bearing variants of both the drug and chelator — MMAE-DIBO and DFO-DIBO — in mixture in a 1:1 molar ratio to form the completed IEADC: DFO:MMAE-sstrastuzumab (Fig. 1B). The immunoconjugate was prepared with a global yield of 70% over three steps.
Figure 1.

(A) The chemoenzymatic approach used to create the IEADC; (B) Schematic structures of MMAE-DIBO and DFO-DIBO.
Chemical and In Vitro Characterization
The site-specific nature of the bioconjugation reaction was verified via both SDS-PAGE and mass spectrometry. The denaturing electrophoresis gel experiment displayed a clear upward shift in the molecular weight of the heavy chain following the ligation of the payloads to the glycans (Fig. 2). In contrast, no shift was observed for the light chain, demonstrating the site-specific character of the conjugation. To complement this data, mass spectrometry analysis was also performed on the DFO:MMAE-sstrastuzumab immunoconjugate. After Fc-F(ab´)2 cleavage, the mass spectrometry shows no modification in the Fab region compared to unmodified trastuzumab (Supp. Fig. S1A). However, a mass increase is observed between the Fc domain of the azide modified trastuzumab – N3-sstrastuzumab – and the Fc domain of DFO:MMAE-sstrastuzumab (Supp. Fig. S1B). These data clearly illustrate that the conjugation occurs site-specifically on the CH2 domain of the Fc region.
Figure 2.

Denaturing polyacrylamide gel electrophoresis of (1) native trastuzumab, (2) DFO-sstrastuzumab, and (3) DFO:MMAE-sstrastuzumab, demonstrating the site-specific conjugation on the heavy chain. HC: heavy chains, LC: light chains.
The mass spectrometry of the whole antibody reveals similar results and allowed us to discriminate the different species constituting DFO:MMAE-sstrastuzumab (Supp. Fig. S1C). As expected, the predominant species bears 2 DFO and 2 MMAE. However, other species containing different combinations of payloads – ranging from 2 DFO:1 MMAE to 4 MMAE – are also present in the mixture. Overall, the average degree of conjugation for each of the payloads is 2.1 ± 0.2 DFO/mAb and 1.7 ± 0.2 MMAE/mAb. It is important to note that while these data clearly illustrate that DFO:MMAE-sstrastuzumab is not perfectly homogenous, it is far more homogenous than an analogous randomly conjugated IEADC. In all cases, the payloads are site-specifically appended to the biantennary glycans chains of the FC domain glycans chains, far from the antigen-binding complementarity determining regions (CDRs). This distance between the modification site and the CDRs stands as one of the primary advantages of this bioconjugation approach compared to traditional, non-site-specific bioconjugation methods. In the latter, lysines within the CDRs can be modified, detrimentally affecting — or even completely abrogating — the ability of the antibody to bind to its antigen.
Following this chemical characterization, the in vitro HER2-binding properties of DFO:MMAE-sstrastuzumab were investigated via flow cytometry and surface plasmon resonance. Not surprisingly, flow cytometry experiments with HER2-expressing BT474 human breast cancer cells revealed that DFO:MMAE-sstrastuzumab exhibited identical immunoreactivity to both native trastuzumab and a variant of trastuzumab that had been site-specifically modified with DFO alone (DFO-sstrastuzumab) (Supp. Fig. S2). In addition, surface plasmon resonance experiments revealed that both unmodified trastuzumab and the immunoconjugates demonstrated robust binding with purified HER2. All the three constructs yielded nanomolar KD values, which were well within range of each other (~ 5 nM) (Supp. Fig. S3). These data clearly indicate the immunoconjugates suffer no loss of immunoreactivity for HER2 in vitro compared to the parent antibody.
Radiochemistry and In Vivo PET Imaging
The IEADC DFO:MMAE-sstrastuzumab and its toxin-lacking cousin DFO-sstrastuzumab were labeled with 89Zr in preparation for in vivo PET imaging. Both immunoconjugates were radiolabeled and purified according to standard published protocols, yielding 89Zr:MMAE-sstrastuzumab and 89Zr-sstrastuzumab in >99% radiochemical purity and with specific activities of 2.7 ± 0.1 and 2.5 ± 0.1 mCi/mg, respectively. Subsequent stability assays revealed both radioimmunoconjugates to be >96% intact after 7 days in human serum at 37 °C (Supp. Fig. S4). The immunoreactive fractions of the radioimmunoconjugates with HER2-positive BT474 human breast cancer cell line were similar for both constructs: 0.92 ± 0.02 and 0.95 ± 0.01 for 89Zr-sstrastuzumab and 89Zr:MMAE-sstrastuzumab, respectively. This confirms that the immunoreactivity of the constructs was not adversely affected by the conjugation or radiolabeling procedures.
The in vivo behavior of 89Zr:MMAE-sstrastuzumab and 89Zr-sstrastuzumab was investigated in athymic nude mice bearing subcutaneous HER2-positive BT474 xenografts. To this end, the mice were administered ~150 μCi of each radioimmunoconjugate and imaged at 24, 72 and 120 h post-injection (Fig. 3). It is important to note up front that we elected not to perform control experiments using a HER2-negative cell line or a blocking dose of trastuzumab. This decision was born not of carelessness but rather a desire to avoid treading on well-trod scientific soil. Indeed, the specificity of trastuzumab-based radiotracers for HER2-positive and HER2-negative xenografts is very well-established.23–27 In this case, the radioADC and 89Zr-sstrastuzumab exhibited very similar behavior. At 24 h p.i., some tumoral uptake is observed, but the activity concentration in the blood pool also remains relatively high. Over time, however, the radiotracers leave the blood compartment and concentrate overwhelmingly within the tumor, allowing for extremely clear visualization of the tumor by 72 and 120 h after injection. The biodistribution data from 120 h p.i. tell a very similar story (Fig 4. and Supp. Table S5). At this time point, both 89Zr:MMAE-sstrastuzumab and 89Zr-sstrastuzumab display relatively low activity concentrations — less than 8 %ID/g — in the blood while concomitantly producing high activity concentrations in the tumor: 76 ± 13 and 70 ± 7 %ID/g, respectively. Uptake in non-target tissues was also low, with the highest activity concentrations observed in the liver, lungs, kidneys, and bone: each tissue displayed slightly above 3 %ID/g with each of the conjugates. Interestingly, in this case, the presence of the MMAE toxin seems to make very little difference on the in vivo behavior of the two constructs. Finally, autoradiography of tumors excised 120 after the administration of 89Zr-sstrastuzumab revealed significant correlation between the localization of the radiotracer and vascular staining with Hoechst 33342 (Supp. Fig. S6)
Figure 3.

Planar (left) and maximum intensity projection (right) PET images of athymic nude mice bearing HER2-expressing BT474 breast cancer xenografts (white arrow) following the injection of 89Zr:MMAE-sstrastuzumab and 89Zr-sstrastuzumab (150 μCi, 60–65μg). The coronal slices intersect the center of the tumors.
Figure 4.

Biodistribution data collected 120 h after the administration of 89Zr:MMAE-sstrastuzumab and 89Zr-sstrastuzumab (150 μCi; 60–65 μg) to athymic mice bearing HER2-expressing, subcutaneous BT474 human breast cancer xenografts. Stomach, small intestine, and large intestine values include contents.
In Vivo Therapeutic Efficacy
After confirming the tumor targeting properties of the 89Zr-IEADC via PET, we next investigated the therapeutic efficacy of the IEADC. To this end, a longitudinal treatment study was performed using athymic nude mice bearing subcutaneous HER2-expressing BT474 xenografts. Based on previous studies, doses of 2.5 and 10 mg/kg DFO:MMAE-sstrastuzumab were chosen for assessment.19,30,31 The unlabeled IEADC was used for logistical reasons: in order to simplify the experiment and reduce the radiation dose associated with repeated measurements of tumor volume. Moreover, in this context, it is critical to remember that in formulation of the 89Zr-labeled IEADC — as with any 89Zr-labeled antibody — the radionuclide is just a statistical label: >99% of the biomolecules are actually the DFO:MMAE-sstrastuzumab precursor. Due to the high energies of the γ-photons resulting from its decay — 511 and 909 keV from positron annihilation and the decay of 89mY, respectively — 89Zr has very limited cytotoxic effect at short range. Therefore, the presence or absence of 89Zr on the IEADC should have no impact on the therapeutic effect of the conjugate. Control cohorts were studied that received doses of vehicle alone, 10 mg/kg of native trastuzumab, and 0.1 mg/kg of MMAE (a dose which provides slightly more MMAE than that contained in a 10 mg/kg dose of DFO:MMAE-sstrastuzumab). Prior to the experiment, the mice were randomized into cohorts of n = 10. Subsequently, the mice of each cohort received single injections of each agent at day 0 of the therapy study. The tumor volume was monitored via caliper for the next 20 days, an endpoint determined by the last remaining day of efficacy of the estrogen pellets needed to grow the BT474 xenografts.
The mice treated with 0.1 mg/kg MMAE or 10 mg/kg native trastuzumab failed to display a significant reduction in tumor volume over the course of 20 days after injection compared to the vehicle-only control cohort (Fig 5. and Supp. Fig. S7). Importantly, however, the administration of 10 mg/kg DFO:MMAE-sstrastuzumab resulted in a marked, statistically significant reduction in tumor volume compared to the three control groups. Moreover, this therapeutic effect appears to be dose dependent. To wit, while the administration of 2.5 mg/kg of DFO:MMAE-sstrastuzumab led to a decrease of tumor size to 50% of its initial volume 20 days after injection, the administration of 10 mg/kg of the unlabeled IEADC led to an average reduction of the tumors >90% after the same period of time.
Figure 5.

Relative tumor volume of athymic nude mice (n = 10 per group) bearing subcutaneous HER2-expressing BT474 xenografts following the injection (t = 0) of saline (blue), 10 mg/kg native trastuzumab (red), a 0.1 mg/kg MMAE (grey), 2.5 mg/kg DFO:MMAE-sstrastuzumab (green), and 10 mg/kg DFO:MMAE-sstrastuzumab (purple). The solid lines are the moving averages.
CONCLUSION
The chemoenzymatic modification of the heavy chain glycans represents a powerful tool for the construction of site-specifically modified antibodies. In this investigation, we have detailed the synthesis, characterization, and in vivo evaluation of 89Zr:MMAE-sstrastuzumab, a site-specifically modified immunoconjuate bearing both a toxin and a positron-emitting radiometal. Chemical and in vitro characterization assays clearly demonstrated the site-specific nature of the bioconjugation reaction as well as the unimpaired ability of the dual-labeled construct to bind HER2-expressing cells. Subsequently, PET imaging and biodistribution experiments study in mice bearing HER2-expressing BT474 human breast cancer xenografts confirmed that the dual-labeled radioADC produces very high activity concentrations in target tissues and exhibits nearly identical in vivo behavior compared to a variant labeled only with 89Zr. Even more importantly, a longitudinal therapy study in the same murine model confirmed the dose-dependent therapeutic efficacy of the dual labeled conjugate. Indeed, treatment with 10 mg/kg DFO:MMAE-sstrastuzumab reduced tumor volumes by >90% over the course of just 20 days. Even beyond facilitating the construction of well-defined, homogeneous, and highly immunoreactive constructs, one of the main advantages of this chemoenzymatic approach is its modularity. This strategy can be used with any antibody and any set of two toxin and reporter cargoes. Moreover, the ratio of the reporter and toxin can be altered in a straightforward manner to create IEADCs bearing a higher DOL of toxin.
In the end, the principal advantage of this approach is that it produces an all-but-identical theranostic pair. Practically speaking, we envision these radioADCs can be employed in three different settings: (1) as probes to study in vivo biodistribution and pharmacokinetics in preclinical investigations, (2) as scout imaging agents administered prior to therapy in the clinic, and (3) as companion imaging agents that can provide real-time voxel-by-voxel drug-dose information during ADC-based therapy. Ultimately, it is our hope that dual-labeled constructs such as these help usher in an era in which true theranostics — constructs that can perform both therapy and imaging — become valuable preclinical and clinical tools.
Supplementary Material
ACKNOWLEDGEMENTS
Services provided by the MSKCC Small-Animal Imaging Core Facility were supported in part by NIH grants R24 CA83084 and P30 CA08748. The authors are also grateful for the generous financial support of the National Institutes of Health (4R00CA178205-02 and R01204167), the TeamConnor Childhood Cancer Foundation, the National Institute on Minority Health and Health Disparities (G12MD007599).
ABBREVIATIONS
- ADC
antibody-drug conjugate
- IEADC
imaging-enabled antibody-drug conjugate
- DFO
desferrioxamine
- MMAE
monomethyl auristatin E
- PET
positron emission tomography
Footnotes
SUPPORTING INFORMATION
Reagents and general procedures; cell culture; xenograft models; SDS-PAGE; flow cytometry; radiolabeled antibody stability assays; biodistribution protocols, statistical analysis protocols, supplementary figures and tables.
REFERENCES
- (1).https://www.ncbi.nlm.nih.gov/pubmed/?term=antibody-drug+conjugates (2017, January 19) antibody-drug conjugates - PubMed - NCBI.
- (2).Phillips GDL, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blättler WA, Lambert JM, Chari RVJ, Lutz RJ, et al. (2008) Targeting HER2-Positive Breast Cancer with Trastuzumab-DM1, an Antibody–Cytotoxic Drug Conjugate. Cancer Res. 68, 9280–9290. [DOI] [PubMed] [Google Scholar]
- (3).Panowski S, Bhakta S, Raab H, Polakis P, and Junutula JR (2013) Site-specific antibody drug conjugates for cancer therapy. mAbs 6, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Behrens CR, and Liu B (2014) Methods for site-specific drug conjugation to antibodies. mAbs 6, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Merten H, Brandl F, Plückthun A, and Zangemeister-Wittke U (2015) Antibody–Drug Conjugates for Tumor Targeting—Novel Conjugation Chemistries and the Promise of non-IgG Binding Proteins. Bioconjug. Chem. 26, 2176–2185. [DOI] [PubMed] [Google Scholar]
- (6).Agarwal P, and Bertozzi CR (2015) Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjug. Chem 26, 176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Adumeau P, Sharma SK, Brent C, and Zeglis BM (2016) Site-Specifically Labeled Immunoconjugates for Molecular Imaging—Part 1: Cysteine Residues and Glycans. Mol. Imaging Biol 18, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dearling JLJ, Paterson BM, Akurathi V, Betanzos-Lara S, Treves ST, Voss SD, White JM, Huston JS, Smith SV, Donnelly PS, et al. (2015) The Ionic Charge of Copper-64 Complexes Conjugated to an Engineered Antibody Affects Biodistribution. Bioconjug. Chem 26, 707–717. [DOI] [PubMed] [Google Scholar]
- (9).Janzer M, Larbig G, Kübelbeck A, Wischnjow A, Haberkorn U, and Mier W (2016) Drug Conjugation Affects Pharmacokinetics and Specificity of Kidney-Targeted Peptide Carriers. Bioconjug. Chem 27, 2441–2449. [DOI] [PubMed] [Google Scholar]
- (10).Grange C, Geninatti-Crich S, Esposito G, Alberti D, Tei L, Bussolati B, Aime S, and Camussi G (2010) Combined Delivery and Magnetic Resonance Imaging of Neural Cell Adhesion Molecule–Targeted Doxorubicin-Containing Liposomes in Experimentally Induced Kaposi’s Sarcoma. Cancer Res. 70, 2180–2190. [DOI] [PubMed] [Google Scholar]
- (11).Edmonds S, Volpe A, Shmeeda H, Parente-Pereira AC, Radia R, Baguña-Torres J, Szanda I, Severin GW, Livieratos L, Blower PJ, et al. (2016) Exploiting the Metal-Chelating Properties of the Drug Cargo for In Vivo Positron Emission Tomography Imaging of Liposomal Nanomedicines. ACS Nano 10, 10294–10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cohen R, Vugts DJ, Visser GWM, Stigter-van Walsum M, Bolijn M, Spiga M, Lazzari P, Shankar S, Sani M, Zanda M, et al. (2014) Development of novel ADCs: conjugation of tubulysin analogues to trastuzumab monitored by dual radiolabeling. Cancer Res. 74, 5700–5710. [DOI] [PubMed] [Google Scholar]
- (13).Boswell CA, Mundo EE, Zhang C, Stainton SL, Yu S-F, Lacap JA, Mao W, Kozak KR, Fourie A, Polakis P, et al. (2012) Differential Effects of Predosing on Tumor and Tissue Uptake of an 111In-Labeled Anti-TENB2 Antibody–Drug Conjugate. J. Nucl. Med 53, 1454–1461. [DOI] [PubMed] [Google Scholar]
- (14).Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, Agnew BJ, and Hsieh-Wilson LC (2008) Direct In-Gel Fluorescence Detection and Cellular Imaging of O-GlcNAc-Modified Proteins. J. Am. Chem. Soc 130, 11576–11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Boeggeman E, Ramakrishnan B, Pasek M, Manzoni M, Puri A, Loomis KH, Waybright TJ, and Qasba PK (2009) Site Specific Conjugation of Fluoroprobes to the Remodeled Fc N-Glycans of Monoclonal Antibodies Using Mutant Glycosyltransferases: Application for Cell Surface Antigen Detection. Bioconjug. Chem 20, 1228–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zeglis BM, Davis CB, Aggeler R, Kang HC, Chen A, Agnew BJ, and Lewis JS (2013) Enzyme-Mediated Methodology for the Site-Specific Radiolabeling of Antibodies Based on Catalyst-Free Click Chemistry. Bioconjug. Chem 24, 1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zeglis BM, Davis CB, Abdel-Atti D, Carlin SD, Chen A, Aggeler R, Agnew BJ, and Lewis JS (2014) Chemoenzymatic Strategy for the Synthesis of Site-Specifically Labeled Immunoconjugates for Multimodal PET and Optical Imaging. Bioconjug. Chem 25, 2123–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Houghton JL, Zeglis BM, Abdel-Atti D, Aggeler R, Sawada R, Agnew BJ, Scholz WW, and Lewis JS (2015) Site-specifically labeled CA19.9-targeted immunoconjugates for the PET, NIRF, and multimodal PET/NIRF imaging of pancreatic cancer. Proc. Natl. Acad. Sci 112, 15850–15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).van Geel R, Wijdeven MA, Heesbeen R, Verkade JMM, Wasiel AA, van Berkel SS, and van Delft FL (2015) Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody–Drug Conjugates. Bioconjug. Chem 26, 2233–2242. [DOI] [PubMed] [Google Scholar]
- (20).Pinto AC, Ades F, de Azambuja E, and Piccart-Gebhart M (2013) Trastuzumab for patients with HER2 positive breast cancer: Delivery, duration and combination therapies. The Breast 22, Supplement 2, S152–S155. [DOI] [PubMed] [Google Scholar]
- (21).Larsen PB, Kümler I, and Nielsen DL (2013) A systematic review of trastuzumab and lapatinib in the treatment of women with brain metastases from HER2-positive breast cancer. Cancer Treat. Rev 39, 720–727. [DOI] [PubMed] [Google Scholar]
- (22).Ahmed S, Sami A, and Xiang J (2015) HER2-directed therapy: current treatment options for HER2-positive breast cancer. Breast Cancer 22, 101–116. [DOI] [PubMed] [Google Scholar]
- (23).Lub-de Hooge MN, Kosterink JGW, Perik PJ, Nijnuis H, Tran L, Bart J, Suurmeijer AJH, de Jong S, Jager PL, and de Vries EGE (2004) Preclinical characterisation of 111In-DTPA-trastuzumab. Br. J. Pharmacol 143, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).McLarty K, Cornelissen B, Scollard DA, Done SJ, Chun K, and Reilly RM (2009) Associations between the uptake of 111In-DTPA-trastuzumab, HER2 density and response to trastuzumab (Herceptin) in athymic mice bearing subcutaneous human tumour xenografts. Eur. J. Nucl. Med. Mol. Imaging 36, 81–93. [DOI] [PubMed] [Google Scholar]
- (25).Dijkers ECF, Kosterink JGW, Rademaker AP, Perk LR, Dongen G. A. M. S. van, Bart J, Jong J. R. de, Vries E. G. E. de, and Hooge MNL (2009) Development and Characterization of Clinical-Grade 89Zr-Trastuzumab for HER2/neu ImmunoPET Imaging. J. Nucl. Med 50, 974–981. [DOI] [PubMed] [Google Scholar]
- (26).Holland JP, Caldas-Lopes E, Divilov V, Longo VA, Taldone T, Zatorska D, Chiosis G, and Lewis JS (2010) Measuring the Pharmacodynamic Effects of a Novel Hsp90 Inhibitor on HER2/neu Expression in Mice Using 89Zr-DFO-Trastuzumab. PLOS ONE 5, e8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zeglis BM, Mohindra P, Weissmann GI, Divilov V, Hilderbrand SA, Weissleder R, and Lewis JS (2011) Modular Strategy for the Construction of Radiometalated Antibodies for Positron Emission Tomography Based on Inverse Electron Demand Diels–Alder Click Chemistry. Bioconjug. Chem 22, 2048–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Rostami S, Qazi I, and Sikorski R (2014) The Clinical Landscape of Antibody-drug Conjugates. ADC Rev. [Google Scholar]
- (29).Jauw YWS, Oordt M. der H. van, Willemien C, Hoekstra OS, Hendrikse NH, Vugts DJ, Zijlstra JM, Huisman MC, Dongen V, and S GAM (2016) Immuno-Positron Emission Tomography with Zirconium-89-Labeled Monoclonal Antibodies in Oncology: What Can We Learn from Initial Clinical Trials? Front. Pharmacol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yao X, Jiang J, Wang X, Huang C, Li D, Xie K, Xu Q, Li H, Li Z, Lou L, et al. (2015) A novel humanized anti-HER2 antibody conjugated with MMAE exerts potent anti-tumor activity. Breast Cancer Res. Treat 153, 123–133. [DOI] [PubMed] [Google Scholar]
- (31).Li H, Yu C, Jiang J, Huang C, Yao X, Xu Q, Yu F, Lou L, and Fang J (2016) An anti-HER2 antibody conjugated with monomethyl auristatin E is highly effective in HER2-positive human gastric cancer. Cancer Biol. Ther 17, 346–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.