

Abstract
Export to the cytoplasm is a key regulatory junction for both protein-coding mRNAs and long noncoding RNAs (lncRNAs), and cytoplasmic enrichment varies dramatically both within and between those groups. We used a new computational approach and RNA-seq data from human and mouse cells to quantify the genome-wide association between cytoplasmic/nuclear ratios of both gene groups and various factors, including expression levels, splicing efficiency, gene architecture, chromatin marks, and sequence elements. Splicing efficiency emerged as the main predictive factor, explaining up to a third of the variability in localization. Combination with other features allowed predictive models that could explain up to 45% of the variance for protein-coding genes and up to 34% for lncRNAs. Factors associated with localization were similar between lncRNAs and mRNAs with some important differences. Readily accessible features can thus be used to predict RNA localization.
Keywords: long noncoding RNAs, nuclear export, RNA localization, post-transcriptional regulation, intron retention
INTRODUCTION
Subcellular localization, and particularly whether RNAs are exported to the cytoplasm or retained in the nucleus, plays a key role in the biology of long RNAs. Many long noncoding RNAs (lncRNAs) act in the nucleus, some of them while tethered to the chromatin (Ulitsky and Bartel 2013), and so their proper function requires pathways that ensure they are not exported. Messenger RNAs (mRNAs) of protein-coding genes (PCGs) are translated in the cytoplasm, and their retention in the nucleus can regulate the amount of protein produced from each mRNA, thus allowing tight temporal regulation of translation (Ninomiya et al. 2011; Mauger et al. 2016; Naro et al. 2017), or buffering of protein levels from bursty transcription (Bahar Halpern et al. 2015; Battich et al. 2015).
LncRNAs have been reported to be more nuclear on average than mRNAs (Derrien et al. 2012; Mukherjee et al. 2017), but the determinants of this difference are largely unknown. Since there are no known pathways for import of long RNAs, the cytoplasmic/nuclear (Cyto/Nuc) ratios of RNAs are likely dictated by a combination of the rate of their export and the stability of the RNA molecules in the different compartments. The decay of aberrant RNAs mostly occurs in the nucleus via quality control mechanisms (Bresson et al. 2015), whereas properly processed RNAs decay with varying rates in the cytoplasm (Garneau et al. 2007). How the nuclear export of long RNAs is regulated remains poorly understood. Specific sequences regulating nuclear retention have been identified in individual lncRNAs (Miyagawa et al. 2012; Zhang et al. 2014; Carlevaro-Fita et al. 2019), and more recently using massively parallel screens (Lubelsky and Ulitsky 2018; Shukla et al. 2018; Yin et al. 2018), but most RNAs retained in the nucleus do not contain any sequence elements associated with a known effect on nuclear export.
Intron retention (IR) is a widespread form of alternative splicing (Wang et al. 2008; Braunschweig et al. 2014), and it is regulated in various systems (Wong et al. 2013; Shalgi et al. 2014; Boutz et al. 2015; Dvinge and Bradley 2015; Mauger et al. 2016; Pimentel et al. 2016; Middleton et al. 2017). Retained introns have been associated with weaker splice sites, shorter length and higher G/C content (Galante et al. 2004; Sakabe and de Souza 2007; Yap et al. 2012; Braunschweig et al. 2014; Boutz et al. 2015; Mukherjee et al. 2017); higher intronic sequence conservation (Boutz et al. 2015); and alternative splicing of their flanking exons (Boutz et al. 2015; Mukherjee et al. 2017). A combination of such features can quite reliably predict which introns will undergo IR (Braunschweig et al. 2014; Mukherjee et al. 2017).
Protein-coding transcripts with retained introns that are exported to the cytoplasm can be subject to nonsense-mediated decay (NMD) (Chang et al. 2007), but only a minority of genes with IR appear to be NMD substrates (Braunschweig et al. 2014; Boutz et al. 2015), possibly because many potential targets do not reach the cytoplasm. Genes with IR were indeed reported to be enriched (or “detained”) in the nuclear fraction (Braunschweig et al. 2014; Boutz et al. 2015), but this phenomenon, the extent of nuclear enrichment of mRNAs and lncRNAs that can be explained by differences in splicing efficiency, and the relative contributions of other factors, have not been systematically evaluated.
The incompletely spliced transcripts can have various fates. Some accumulate in the nucleus and can be spliced and exported either slowly, or upon specific cues; others have been shown to be degraded by various pathways (Pendleton et al. 2018), involving hyperpolyadenylation and PABPN1 (Bresson and Conrad 2013; Bresson et al. 2015), or the exosome (Houseley et al. 2006). IR is also associated with lower expression level of the host gene and with increased accumulation of Pol2 on the intron (Braunschweig et al. 2014). Inhibition of transcription results in increased IR, supporting the connection between transcription efficiency and splicing (Braunschweig et al. 2014).
LncRNAs typically accumulate to levels substantially lower than mRNAs (Cabili et al. 2011; Mukherjee et al. 2017), are somewhat less stable (Clark et al. 2012), and are less efficiently spliced than mRNAs (Tilgner et al. 2012; Melé et al. 2017; Mukherjee et al. 2017), but the difference in splicing efficiency could not be explained by presence of exonic splicing enhancers (ESEs) or U1 binding sites, and was only mildly correlated with pyrimidine track and branch point sequences (Melé et al. 2017). Nuclear lncRNAs were also shown to be less stable than the ones enriched in the cytoplasm (Clark et al. 2012). A recent study revealed extensive alternative splicing of lncRNAs, with numerous alternative isoforms discovered at increasing sequencing depths, more so than in mRNAs (Deveson et al. 2018). Differences in splicing efficiency can thus explain some of the differences in subcellular localization between lncRNAs and PCGs.
Here we study RNA-seq data from cytoplasmic and nuclear fractions, and characterize the features that are associated with subcellular localization of lncRNAs and PCGs. We find that inefficient splicing, transcript length, sequence composition, and chromatin features all independently contribute to nuclear localization of subsets of lncRNAs and PCGs, and that their combination can be used to predict the subcellular localization of transcripts, with a substantially higher accuracy in PCGs. These features also contribute to lower expression levels of the inefficiently spliced transcripts, as those are subject to nuclear decay pathways. We further find that inefficient splicing is well conserved in evolution for PCGs, and that splicing and localization are strongly correlated also in mouse cells, and thus splicing efficiency impacts function through localization in both lncRNAs and PCGs.
RESULTS
Gene-level quantification of splicing efficiency and specificity
Quantification of IR using RNA-seq data is challenging, and can rely either on reads mapping to introns or on reads covering splice junctions (Vanichkina et al. 2017). The latter approach compares numbers of reads spanning exon–exon and intron–exon junctions and requires substantial sequencing depth, but does not suffer from the difficulties of uniquely mapping reads to repeat-rich intronic sequences (Vanichkina et al. 2017). We therefore opted for this scheme for quantifying splicing efficiencies in deeply sequenced data from human cell lines obtained by the ENCODE project (Tilgner et al. 2012).
Previous studies have considered IR on the level of individual introns (Braunschweig et al. 2014), or used just the longest transcript isoform of each gene (Melé et al. 2017), which appears suboptimal. Splicing and localization should ideally be studied on the level of all splicing isoforms of the gene, and then combined into gene-level metrics based on their relative abundances. Unfortunately, quantification of levels of individual transcripts is notoriously inaccurate and nonrobust when using short-read RNA-seq data (Merino et al. 2017). In our experience, subtle changes in read mapping between samples often result in substantial changes in relative isoform abundance estimates. It is therefore difficult to obtain robust isoform-specific expression and Cyto/Nuc ratio estimates. Further, as isoforms typically share most of their introns, computation of transcript-level splicing efficiency heavily relies on the accuracy of relative isoform abundance estimates, which is needed for “distributing” the splicing efficiencies of individual introns across the host isoforms. Another challenge is that the comprehensive GENCODE annotation contains many rarely spliced introns, and those can appear as commonly retained, skewing the splicing efficiency estimates of their host genes.
To address these challenges, we opted to develop a robust method for directly computing gene-level splicing efficiency (Fig. 1; Supplemental Fig. S1). Our approach (see Materials and Methods) starts with selecting a set of introns with confident support for their splicing when considering the full data set (whole-cell extract [WCE] RNA-seq from nine ENCODE cell lines in this study). We then count the reads overlapping exon–exon and exon–intron junctions to evaluate the splicing efficiency of each intron, defining splicing efficiency as in (Mukherjee et al. 2017), as the ratio between the exon–exon reads and the sum of the exon–exon and exon–intron reads. We consider two possible metrics for gene-level splicing efficiency—the average splicing efficiency across the confident introns, and the splicing efficiency of the intron with the worst efficiency, as splicing of that intron is presumably the rate-limiting step for full transcript maturation (Supplemental Data 1). We also used the consensus set of introns to compute gene-level splicing specificity, which is a measure of the extent of alternative splicing that reflects the frequency in which splicing events in the gene correspond to a single set of annotated and nonoverlapping introns (Supplemental Data 1, see Materials and Methods).
FIGURE 1.

Outline of the methodology for computing gene-level splicing efficiency and specificity. Data for the GAS5 lncRNA in ENCODE RNA-seq data for MCF7 cells are shown. All introns annotated in GENCODE were first considered and those poorly supported by spliced reads were discarded. Among the remaining introns, a nonoverlapping set of introns with the most confident support was selected and used for quantification. The method used for quantifying splicing efficiency and specificity at intron- and gene-level is illustrated at the bottom.
Splicing efficiency is prominently associated with localization of lncRNAs and PCGs
We quantified expression levels, Cyto/Nuc ratios, and splicing efficiency and specificity of 13,513 lncRNAs and 20,073 PCGs annotated in GENCODE v26 in nine cell lines profiled by the ENCODE project. Consistently with previous studies, we found that lncRNAs accumulate to lower levels (Fig. 2A; Supplemental Fig. S2A), are more enriched in the nucleus (Fig. 2B; Supplemental Fig. S2C), and exhibit substantially lower splicing efficiencies and specificities than mRNAs in all ENCODE cell lines (Fig. 2C,D; Supplemental Fig. S2D,E). Further, we found that splicing specificity, a measure of the prevalence of a dominant splicing pattern, was also significantly lower in lncRNAs compared to PCGs, that is, lncRNAs were substantially more alternatively spliced than mRNAs. Consistently with previous studies (Tilgner et al. 2012), splicing efficiencies were substantially lower in the nucleus than in the cytoplasm for both PCGs and lncRNAs (P < 10−50). Remarkably, splicing specificities were similar in the cytoplasmic and nuclear fractions for both gene classes (Fig. 2C,D; Supplemental Fig. S2D,E). This suggests that while IR plays a potentially prominent role in regulating nuclear export, alternative splicing rarely affects subcellular localization.
FIGURE 2.

Differences between PCGs and lncRNAs in K562 cells. (A–D) Distributions of expression levels (A), Cyto/Nuc ratios (B), splicing efficiencies (C), and specificities (D), for PCGs and lncRNAs. (*) P-value < 10−16 (Wilcoxon rank sum test).
Previous studies have shown that lncRNAs are shorter and contain fewer introns than PCGs (Cabili et al. 2011; Hezroni et al. 2015). LncRNAs also have shorter exons and slightly shorter introns as compared to PCGs (Supplemental Fig. S2B). These features may underlie some of the differences in localization and splicing between lncRNAs and PCGs. We therefore generated cell-type–specific sets of lncRNAs and PCGs matched for expression and exon number (Supplemental Fig. S2F; Materials and Methods). In this controlled setting, lncRNAs were still more enriched in the nucleus and less efficiently and less specifically spliced than mRNAs (Supplemental Fig. S2C–E). The vast majority of lncRNAs are classified by GENCODE as either “lincRNA” or “antisense,” based on their genomic positions, with a minority of lncRNAs labeled as “processed transcripts.” We evaluated length parameters, expression levels, splicing values and subcellular localization of these subgroups and found only minor differences between the two major classes in all cell lines, except for splicing efficiencies that were slightly higher for lincRNAs compared to antisense genes in most cell lines (Supplemental Fig. S3). Together, these results suggest that factors beyond gene architecture, genomic position and expression levels underlie the differences between PCGs and lncRNAs.
Strikingly, splicing efficiency was strongly associated with cytoplasmic localization of PCGs in all ENCODE cell lines (Figs. 3A, 4; Supplemental Fig. S4A), suggesting that splicing status substantially contributes to subcellular localization of protein-coding transcripts (though other explanations are also possible, see Discussion). This correlation was significantly weaker for lncRNAs (Fisher Z-transformation P-value < 2 × 10−16), despite their presumably similar processing and export mechanisms (Fig. 3B; Supplemental Fig. S4C, top). Splicing efficiency of the least efficiently spliced intron was correlated with localization better than the average efficiency across all introns, explaining up to ∼37% of the variance in Cyto/Nuc ratios for PCGs, but only ∼12% for lncRNAs (Fig. 3C,D; Supplemental Fig. S4B,C, bottom). Further, when comparing different cell lines, increased relative splicing efficiencies and, to a lesser extent, specificities were typically correlated with increased relative cytoplasmic enrichment for PCGs and in some cases also for lncRNAs (Fig. 3E; Supplemental Fig. S4D). These results suggest that regulation of splicing and particularly IR may underlie the transcriptome-wide differences in subcellular localization across different cell types. The milder yet significant correlation of splicing and subcellular localization of lncRNAs (Figs. 3B,D, 4; Supplemental Fig. S4C) and several stronger correlations of differential values between cell lines (Fig. 3E) suggest that at least some lncRNAs are subject to regulation of their localization state by splicing efficiency, similarly to PCGs.
FIGURE 3.

Association between splicing efficiency and RNA localization. (A,B) Correlation between splicing efficiency, averaged across all introns, and localization of PCGs (A) and lncRNAs (B) in HepG2 cells. Coloring indicates local point density. Regression line is shown in bold. (C,D) Correlation between the splicing efficiency of the least efficient intron and localization of PCGs (C) and lncRNAs (D) in HepG2 cells. Coloring indicates local point density. Regression line is shown in bold. (E) Correlations between difference in splicing efficiency and differences in localization when comparing the indicated pairs of cell lines for PCGs (top triangle) and lncRNAs (bottom triangle). Numbers indicate correlation coefficients. (*) P < 0.05. Correlation coefficients and P-values computed using Spearman's correlation.
FIGURE 4.

Association of different factors with localization of coding and noncoding RNAs in ENCODE cell lines. Correlation between the indicated parameters and Cyto/Nuc ratios in the indicated cell lines. Numbers indicate correlation coefficients. (*) P < 0.05. Correlation coefficients and P-values are computed using Spearman's correlation.
The association between localization, splicing, and Pol2 pausing is not explained by expression levels or gene architecture
We then looked at the correlation between Cyto/Nuc ratios and other factors and found that cytoplasmic localization was also consistently positively correlated with expression levels, splicing specificity, Pol2 occupancy on introns, and Pol2 pausing index for PCGs as well as for both major classes of lncRNAs (Fig. 4; Supplemental Fig. S5A). The association of localization with splicing remained significant also when controlling for expression levels and gene architecture (number of exons and exonic/intronic length, Supplemental Fig. S5B). In contrast, the association of localization with Pol2 occupancy on introns and pausing index had a variable and smaller effect for PCGs, but not for lncRNAs, where it remained significant after controlling for other factors (Supplemental Fig. S5B). Decreased Pol2 elongation rate is known to be associated with lower splicing efficiency (Kornblihtt 2006; Braunschweig et al. 2014), and here we found that promoter-proximal pausing is surprisingly associated with increased export in lncRNAs, in an expression level–independent way, perhaps because it allows for improved association of export factors with Pol2 (see Discussion). Pausing index was also significantly lower in lncRNAs compared to PCGs (Supplemental Fig. S5C). To better understand these results, we tested whether splicing-related sequence features may underlie the effect of Pol2 pausing on subcellular localization. To this end, we divided the lncRNAs expressed in HepG2 cells to equal-size subgroups, based on various sequence features of the splice sites in their first intron. Splitting lncRNAs based on splice-site strength measures, such as Senapathy and maxEnt scores, did not show any significant effect on the correlation (not shown). However, lncRNAs with highly conserved splice-site sequences exhibited significantly (Fisher Z-transformation P = 0.0021) lower correlation between Pol2 pausing and localization as compared to lncRNAs with low conservation scores, when considering either 5′ or 3′ splice sites. For the lncRNAs with highly conserved splice sites, the correlation between Pol2 pausing and localization resembled the correlation for PCGs (Supplemental Fig. S6). Together, these results suggest that Pol2 promoter-proximal pausing may play a role in modulating localization of lncRNAs with poorly conserved splice sites (which might also be less effective, but this difference does not appear to be captured by the splice-site scores that we tested). In contrast, pausing has a limited effect in lncRNAs and PCGs which bear highly conserved splice sites.
We also observed a weaker, yet consistent, negative correlation between cytoplasmic localization and exonic length, and variable correlations with number and lengths of introns—in PCGs longer transcription units with more exons were typically correlated with nuclear enrichment, whereas in lncRNAs such correlations were either absent or weaker (Fig. 4). Particularly long genes are expected to yield transcripts that spend a long time in the nucleus, as just the transcription of hundreds of kbs can take hours. The difference between PCGs and lncRNAs in the association between gene length and transcript localization presumably results from the scarcity of particularly long loci among lncRNAs—in our data set there were 2444 PCGs with loci longer than 100 kb and with >10 exons (12% of PCGs), compared to just 56 lncRNAs (0.4% of lncRNAs). Still, when the various factors related to gene architecture are considered, the strongest correlation with Cyto/Nuc ratios was observed for measures of splicing efficiency (Supplemental Fig. S5B).
Preference for C-rich hexamers is associated with nuclear enrichment
We recently reported that C-rich sequences in internal exons contribute to nuclear enrichment of lncRNAs and mRNAs through association with HNRNPK (Lubelsky and Ulitsky 2018), and C-rich motifs were also found as enriched in nuclear RNAs by others (Shukla et al. 2018). We therefore examined whether there is correlation between localization and the prevalence of hexamers enriched for each nucleotide (a hexamer was defined as enriched for base X if at least four of its six bases were X). To account for potential contribution of general G/C content, we also computed the “preference” for C-rich and A-rich hexamers (preference for C was the difference between densities of C-rich and of G-rich hexamers, and preference for A was the difference between densities of A-rich and of T-rich hexamers). For PCGs, we also computed the preference for a particular base in the third positions of codons, when accounting for overall codon usage (see Materials and Methods). Across these metrics, C-centric metrics universally significantly associated with nuclear enrichment in PCGs (Supplemental Fig. S7A), whereas A-centric metrics had a somewhat weaker and inverse effect. These effects were generally stronger in PCGs than in lncRNAs, perhaps because their exonic sequences are better defined, or because mRNAs are more likely to be found in regions of the nucleus where the relevant machinery is active (see Discussion). The more general correlation of export efficiency with G/C content was highly variable across cell lines, potentially reflecting differences in RNA-seq library quality, which can be affected by G/C content (Risso et al. 2011). Significant correlations were found between hexamer content and splicing efficiency (Supplemental Fig. S7B). The association of G/C content with splicing was much less variable across cell lines. The association between localization and preference for C-rich hexamers remained significant in PCGs when we controlled for splicing efficiency (Supplemental Fig. S7C), suggesting a splicing-independent contribution, and consistent with our previous report (Lubelsky and Ulitsky 2018).
Weak association between chromatin features and splicing efficiency and localization
As different chromatin features have been associated with splicing efficiency and with interactions with nuclear pores (Capelson et al. 2010; Luco et al. 2011), we next evaluated the correlation between chromatin marks, in the cell lines where those were measured, and localization and splicing, while controlling for expression levels. We considered separately the coverage of histone marks on the exon junctions and within introns (Supplemental Fig. S8). The observed trends in junctions and introns were similar with stronger correlations when considering the splice junctions. Here, in contrast to the general positive association of splicing efficiency and Cyto/Nuc ratios, we found that marks associated with active regulatory elements, H3K27 acetylation, and H3K4 di-/tri-methylation were positively correlated with cytoplasmic enrichment and negatively correlated with splicing efficiency. The presence of chromatin marks can be related to increased dwelling time of Pol2, which was also positively correlated with cytoplasmic enrichment (Fig. 4) and negatively correlated with splicing efficiency (see below).
Prediction of subcellular localization from genomic and splicing features
As different features were associated with subcellular localization to varying degrees, and potentially redundantly, we asked whether a combination of the features can be used to predict the gene-level subcellular localization. We first built a linear regression model based on 15 features of gene architecture, splicing, Pol2 occupancy, chromatin marks, and hexamer occurrences (Supplemental Data 6). This model could explain ∼45% of the variability in localization among PCGs, and 15%–30% of variability among lncRNAs (Fig. 5A; Supplemental Fig. S9A). We then evaluated the contribution of each feature and feature group to localization in the context of the model by comparing the regression coefficients and by considering the change in R2 when a group of features was omitted from the model (Fig. 5B; Supplemental Fig. S9B). Splicing-associated features had the strongest contributions in predicting localization of PCGs, whereas Pol2 pausing and chromatin marks had a more prominent and partially redundant contribution in predicting localization of lncRNAs. Gene architecture had a consistent effect for PCGs and lncRNAs, which was not redundant with expression or splicing features, as we observed prominent reduction in R2 when we excluded the three architectural features (number of exons and total exonic and intronic lengths). The contribution of hexamers was most variable across cell types, in agreement with our previous observations that the HNRNPK-mediated nuclear enrichment is more active in some cell types than others (and specifically more active in HepG2 over K562 cells) (Lubelsky and Ulitsky 2018).
FIGURE 5.

Predictive models for localization in HepG2 and K562 cells. (A) Coefficients of the indicated factors in the linear regression of localization. (*) P < 0.05, (**) P < 0.001, (***) P < 0.0001. Error bars represent standard errors of the coefficient estimates. Adjusted R2 is indicated for each cell line separately for PCGs and lncRNAs. (B) Changes in the adjusted R2 of the regression following omission of different factor groups: expression, Pol2 (Pol2 pausing and Pol2 over introns), histone marks (the four H3 modifications in A), gene structure (number of exons, exonic length, and intronic length), and hexamers (C-rich preference and A-rich preference). (C) AUC, precision and recall of random forest classifiers trained and tested with the indicated group of genes on data from the indicated cell line. Error bars indicate SD in a repeated 10-fold cross-validation analysis (see Materials and Methods).
To further evaluate the predictive ability of different features toward subcellular localization, we binned the genes into three groups based on their Cyto/Nuc ratios [using a threshold of log2 (Cyto/Nuc) = ±1] and trained Random Forest classifiers using the same set of 15 features. Our models showed very low predictive capacity for cytoplasmic enrichment of lncRNAs, which was not surprising given the scarcity of cytoplasmic lncRNAs (7%–17% in all cell lines), their relatively inefficient splicing, and low exon counts. We therefore grouped the “cytoplasmic” and “intermediate” classes together for lncRNAs and evaluated performance in a repeated 10-fold cross-validation setting separately for PCGs and lncRNAs (Fig. 5C; Supplemental Data 7A; see Materials and Methods). The classifier showed good predictive ability with typical precision and recall values of >60% for both PCGs and lncRNAs. Area under the curve (AUC) values for the cytoplasmic and nuclear classes (calculated separately in the case of PCGs) were higher than for the intermediate class, and typically close to 0.9, indicating better performance of the classifiers in more extreme cases. The nuclear class had a good precision but low recall in some of the cell lines, suggesting that additional features not captured by our model might account for nuclear retention of a substantial subset of RNAs (see below). Similarly good performance was observed for both PCGs and lncRNAs upon training the classifiers on data from one cell line and predicting localization in another (Supplemental Data 7B), suggesting that similar rules dictate most of the Cyto/Nuc localization variability across the cell lines profiled by ENCODE. In contrast, classifiers trained on one gene class (PCGs or lncRNAs) and tested on the other showed low predictive capacity (data not shown), consistent with the different contributions of features to classifier performance in the two gene classes, as described above.
Gene-level splicing efficiency and subcellular localization are highly conserved between human and mouse protein-coding genes
Features that are important for function are expected to be conserved in evolution. We therefore tested whether localization and splicing of PCGs and mRNAs are conserved between human and mouse. Since limited data on subcellular fractionations are available in mouse, we focused our comparison on the mouse liver (Cyto/Nuc RNA-seq data from Bahar Halpern et al. 2015 and WCE from ENCODE; Supplemental Data 8) and human liver carcinoma cell line HepG2 (ENCODE data). Splicing efficiency, specificity, and Cyto/Nuc ratios were significantly correlated for orthologous PCG pairs between the two species, and splicing efficiency was as conserved for lncRNAs as for PCGs (Fig. 6). However, splicing specificity and localization values for lncRNAs in human and mouse were not significantly correlated (Fig. 6B,C, right). It is possible that the difficulty in correctly assigning orthologs for lncRNAs, in which regions of sequence similarity are typically quite short (Hezroni et al. 2015), limits our ability to detect conservation in this context. Notably, when we examined the entire mouse liver data set, we observed similar correlations for PCGs and somewhat stronger correlations for lncRNAs (Fisher Z-transformation P = 0.017) between splicing efficiency and localization as compared to HepG2 (Supplemental Fig. S10). To test whether similar features are predictive of RNA localization in human and mouse, we trained our random forest classifiers on human data (either HepG2 or K562) and tested the performance on the mouse liver data set and vice versa (Supplemental Data 7C). Classifiers performed as well as for the different human cell lines (compare to Supplemental Data 7B), supporting the conservation of localization-controlling mechanisms. Notably, for lncRNAs the classifiers were not as successful when training on mouse data and testing on human data. Together, these results further suggest that efficiency of splicing plays important roles in modulating subcellular localization of PCGs and lncRNAs; however, the localization of the conserved lncRNAs subpopulation (and perhaps of some of the other lncRNAs) is mostly under control of other factors.
FIGURE 6.

Conservation of splicing and localization between human and mouse liver cells. (A–C) Correlation between splicing efficiency (A), splicing specificity (B), and localization (C) in mouse liver and human liver carcinoma cells HepG2. Regression line is shown in bold. Coloring indicates local point density. Indicated coefficient and P-values computed using Spearman's correlation.
Inefficiently spliced transcripts are targets of nuclear degradation pathways
We next evaluated whether the inefficiently spliced and nuclearly enriched transcripts are regulated by known decay pathways. We first compared the half-lives of genes in various groups using half-life data from HeLa (Ke et al. 2017) and MCF7 cells (Fig. 7A; Schueler et al. 2014). Genes enriched in the nucleus were generally less stable than other genes (consistently with previous studies on lncRNAs [Clark et al. 2012]), regardless of splicing efficiency.
FIGURE 7.

Susceptibility to cytoplasmic and nuclear decay factors. (A) Half-lives of genes in the indicated groups: efficiently spliced (“Eff.”, worst intron splicing efficiency >0.8) and cytoplasmic (“Cyt.”, Cyto/Nuc ratio >1); efficiently spliced and nuclear (“Nuc.”, Cyto/Nuc ratio <0.5); inefficiently spliced (“Ineff.”, worst intron splicing efficiency <0.5) and nuclear; and all other. (*) P-value = 6.4 × 10−5, (**) P-value < 2 × 10−16 (Wilcoxon rank sum test). (B) Expression changes of genes in the indicated group following KD of the indicated factors using siRNAs in HeLa cells, each compared to the nontargeting control from the same study. (*) P-value < 0.01, (**) P-value < 10−6 (Wilcoxon rank sum test). (C) Effect of KD of SMG6 and SMG7 versus KD of RRP40 on gene expression in HeLa cells. Coloring indicates local point density.
To evaluate which decay pathways act on transcripts enriched in the nucleus, we analyzed RNA-seq data sets obtained following siRNA knockdowns (KD) in HeLa cells of: (i) components of the NMD pathway SMG6, SMG7, and UPF1 (Colombo et al. 2017); (ii) common exosomal components RRP40, RRP6, and DIS3 (Tseng et al. 2015); (iii) components of the Nuclear EXosome Targeting (NEXT) complex ZCCHC8 and RBM7 (Meola et al. 2016); and (iv) components of the PolyA tail eXosome Targeting (PAXT) pathway PABPN1, PAP, and ZFC3H1 (Fig. 7B; Meola et al. 2016). Comparison of genes grouped by their splicing efficiency and localization revealed significant changes in their regulation. Subsets of both efficiently and inefficiently spliced nuclearly enriched transcripts were up-regulated following inhibition of NMD, but those subsets were generally small—only 66 of the 918 nuclearly enriched (>twofold) and inefficiently spliced genes were up-regulated by >twofold following either UPF1 or SMG6/7 KD. This suggests that for the vast majority of the nuclearly enriched transcripts, NMD is not the major cause for nuclear enrichment. Inefficiently spliced and nuclear transcripts were preferentially targeted by components of the exosome, and specifically the NEXT complex, and less so by the PAXT complex which was specifically linked to degradation of fully processed transcripts (Meola et al. 2016). As expected, there was no correlation between the susceptibility of transcripts to NMD (combined SMG6 and SMG7 KD) and the exosome (RRP40 KD) (Fig. 7C), suggesting that those pathways act on distinct groups of genes, determined at least in part by the efficiency of their splicing. We note that we could test a only a subset of the possible decay pathways, for which comparable data are available in HeLa cells, and it is possible that other pathways, including translation-related degradation (Carlevaro-Fita et al. 2016) preferentially affect subsets of lncRNAs that differ in their maturation status.
Nuclear-retained genes are enriched for signaling pathways and membrane proteins
Based on our observation that splicing efficiency differences are associated with differential localization when comparing pairs of the ENCODE cell lines (Fig. 3E), we were interested to characterize the biological processes that are preferentially affected by the splicing-localization pathway. GO term analysis revealed no significant enrichment of any particular biological process among inefficiently spliced genes (see Materials and Methods). However, signaling processes and cytoplasmic membrane transport pathways were enriched among nuclear-retained genes in most ENCODE cell lines (Fig. 8; Supplemental Data 10). Notably, many of the genes with strongest nuclear enrichment are characterized by low abundance, which does not allow reliable splicing quantification. Therefore, it is difficult to conclude whether nuclear retention of these particular genes is accompanied by inefficient splicing.
FIGURE 8.

GO enrichment of nuclear-retained genes. GO analysis of ranked K562 Cyto/Nuc localization values. Bars indicate −log10 FDR-corrected P-values for the respective GO-terms cluster (see Materials and Methods).
DISCUSSION
We describe here an attempt to use the existing information about the maturation level, chromatin marks, gene architecture and sequence features to predict the simplest dimension of subcellular localization of long RNAs in cells—nucleus versus cytoplasm. This attempt complements the recent development of machine learning approaches that attempt to predict subcellular localization using sequence features alone (Cao et al. 2018; Gudenas and Wang 2018; Su et al. 2018). Our study and others are based on ENCODE data, which include very high-quality RNA-seq on subcellular fractions, but is presently limited to human cancer cell lines. An important future prospect is to test and further develop the approach for primary cells and tissues, in which factors that influence localization might differ, though our preliminary analysis shows that a classifier used using human cancer cell lines works well when applied to mouse liver data (Supplemental Data 7C). Another present limitation is that we rely on gene models from GENCODE, which while being state-of-the-art in manual gene annotation, do suffer from occasional gene model incompleteness and potential errors in annotation of splice structures. Importantly, the new approach for calculating gene-level splicing that we introduced here helps address some of these challenges by first selecting for each gene a set of confident introns and then using only these introns for quantifying splicing efficiency and specificity.
Our results suggest a strong correlation between the efficiency of splicing and cytoplasmic localization. The two main underlying explanations for nuclear enrichment are slow nuclear export or cytoplasmic degradation (Bahar Halpern et al. 2015). The majority of inefficiently spliced genes do not appear to be sensitive to NMD that can recognize improperly spliced transcripts in the cytoplasm, and so the nuclear enrichment we observe for the inefficiently spliced genes is likely mostly due to inefficient export or nuclear degradation. Splicing was shown to dramatically improve export of model genes (Luo and Reed 1999; Valencia et al. 2008; Mor et al. 2010; Akef et al. 2015). It nevertheless remains uncertain if there is a direct genome-wide causal relationship between inefficient splicing and nuclear enrichment, that is, it is unclear how much of the nuclear enrichment across the transcriptome is caused by inefficient splicing. The canonical life cycle of an exported long RNA begins at the site of transcription on chromatin, continues to processing and maturation, that are believed to take place mostly in the nuclear speckles (Galganski et al. 2017), and then proceeds to the nuclear pore for export. Much of the splicing happens already on chromatin (Tilgner et al. 2012), perhaps more so for genes whose sites of transcription overlap the nuclear speckles (Galganski et al. 2017). The TREX export pathway was associated with transition of transcripts from the speckles to the nuclear pore (Dias et al. 2010), but other aspects of intra-nuclear transitions, and the points at which unprocessed transcripts are delayed or degraded are largely unknown.
The association between splicing and nuclear export can therefore result from various scenarios: (i) sequestration of RNA at chromatin, which may preclude its processing (indeed, highly insoluble transcripts are unspliced [Chujo et al. 2017]); (ii) limited spliceosome binding may prevent recruitment to the speckles (Dias et al. 2010); (iii) inefficient processing may increase time spent at the speckles; (iv) incompletely processed transcripts may be degraded following the release from speckles en route to or at the nuclear pore; or (v) nuclear pore may prevent export of incompletely processed transcript. It is probable that the global correlations we observe are due to a combination of signals from different transcript groups that are affected at different steps. It is possible to identify the relevant step for individual transcripts through reporter assays or genome editing, but classifying transcripts on a global level is challenging. Perturbations that affect splicing also affect cell viability, and so the perturbed cells can only be studied for short time windows, introducing transcript stability as a substantial confounding factor. Indeed, when we re-analyzed data from changes in Cyto/Nuc ratios following inhibition of splicing for 6 h using spliceostatin A (Yoshimoto et al. 2017), we found that transcript stability in unperturbed cells was strongly associated with changes in splicing efficiency, with the vast majority of affected transcripts having half-lives shorter than 5 h (Supplemental Fig. S11E–G). Transient metabolic labeling of just the newly produced transcripts may help overcome some of these issues (Meola et al. 2016; Wlotzka et al. 2017). Further difficulty in distinguishing between the models is that it is not possible to isolate RNAs found at specific subnuclear compartments, such as nuclear speckles or nuclear pores. The recently introduced methods for mapping of transcripts found in proximity to specific organelles, such as APEX-RIP (Kaewsapsak et al. 2017) have the potential of overcoming this difficulty, by labeling and sequencing RNAs found in proximity to proteins enriched in different compartments. Until such data become available, based on our analysis, it is tempting to speculate that nuclear enrichment of lncRNAs is driven more by features of their transcription (e.g., reduced association of Pol2 CTD with splicing and/or export factors), whereas PCGs are typically retained during maturation, as their nuclear enrichment was more associated with inefficient splicing, exonic structure and C-rich sequences (Fig. 4; Supplemental Figs. S5–S7). HNRNPK, which binds C-rich sequences, and some of its targets are enriched in the nuclear speckles, also contributes to nuclear sequestration of many transcripts in a splicing-independent manner (Lubelsky and Ulitsky 2018). However, our models for predicting nuclear localization, trained on all mentioned features and gene architecture exhibit relatively low recall for nuclear PCGs and lncRNAs (Fig. 5C; Supplemental Data 7), indicating that additional uncharacterized mechanisms act to prevent nuclear export.
The strong correlation between inefficient splicing and nuclear enrichment helps explain why lncRNAs, which are substantially less efficiently spliced than PCGs, are also more nuclear, but leaves open the question of why lncRNAs are substantially less spliced. When we evaluated the correlation between different genomic, sequence, and transcriptional features and splicing efficiency, we found that, as reported previously (Galante et al. 2004; Sakabe and de Souza 2007; Yap et al. 2012; Braunschweig et al. 2014; Boutz et al. 2015; Melé et al. 2017; Mukherjee et al. 2017), longer introns and splice-site sequences closer to the consensus and more highly conserved are associated with higher splicing efficiencies (Fig. 9). These features differ significantly between lncRNAs and PCGs, though the effect sizes of the difference for individual features are usually modest (Supplemental Figs. S2B, S11). Interestingly, expression levels were strongly correlated with better splicing in PCGs, but not in lncRNAs (Fig. 9), presumably because most abundant mRNAs need to be efficiently exported, whereas many abundant lncRNAs act in the nucleus. Consistently with the other analyses, increased Pol2 occupancy on introns was associated with reduced splicing efficiencies, in particular in lncRNAs (Fig. 9). Surprisingly, decreased Pol2 occupancy in promoter-proximal regions was associated with nuclear enrichment for lncRNAs (Fig. 4; Supplemental Figs. S5A,B, S6), suggesting that the effects of Pol2 dynamics on export of lncRNAs are likely not mediated by effects on splicing efficiency.
FIGURE 9.
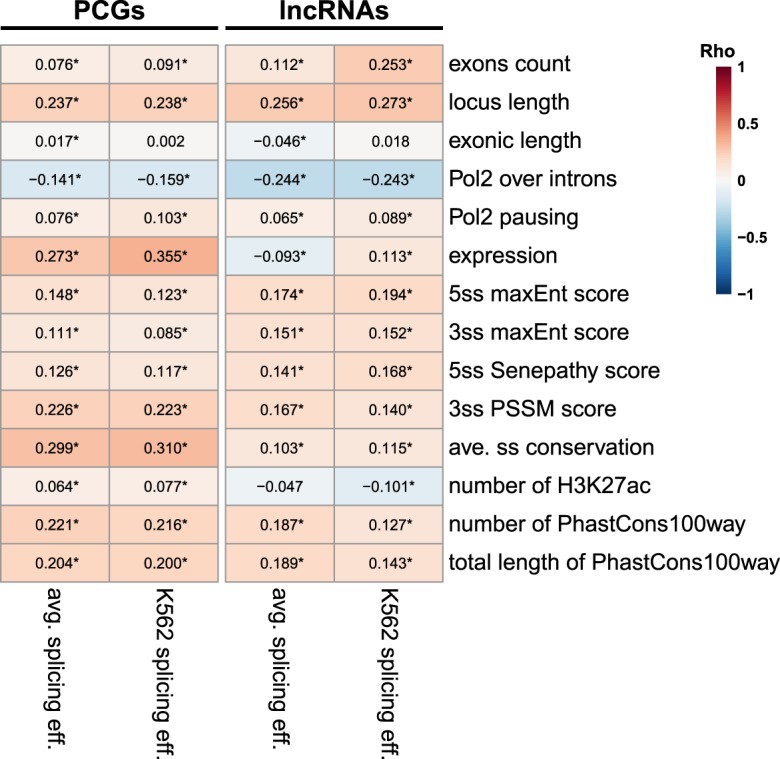
Factors associated with splicing efficiency. Correlation between the indicated features and splicing efficiency in PCGs and lncRNAs in K562 cells, or averaged across the nine cell lines (except for Pol2 occupancy, which is available in only six lines). Numbers indicate correlation coefficients. (*) P < 0.05. Correlation coefficients and FDR-adjusted P-values computed using Spearman's correlation.
The functions, if any, of the vast majority of lncRNAs remain unknown, but an increasing number of reports link lncRNAs to activity in the nucleus, which requires repression of their export to the cytoplasm. Inefficient splicing may help place lncRNAs at different subnuclear compartments and poise them for specific activities. Conversely, splicing itself is reported to be important for the functions of at least some lncRNAs (Engreitz et al. 2016; Gil and Ulitsky 2018; Tan et al. 2018). Regulated splicing can further assist in “releasing” the RNA from one compartment to another, allowing precise timing of its functional activity. For instance, a recent study has found that release from chromatin is essential for function of lncRNA A-ROD (Ntini et al. 2018). For PCGs, regulated nuclear retention through regulation of splicing has been shown to orchestrate translation and protein accumulation in several contexts (Ninomiya et al. 2011; Mauger et al. 2016; Naro et al. 2017). As complex regulatory networks for regulating splicing are in place in eukaryotic cells, the coupling of splicing with export has therefore significant regulatory potential for both coding and noncoding RNA.
MATERIALS AND METHODS
ENCODE RNA-seq data analysis
We downloaded publicly available RNA-seq data from nine ENCODE human cell lines [GSE30567; poly(A)+, WCE, cytosol and nucleus samples from GM12878, HeLa-S3, HepG2, HUVEC, IMR90, K562, MCF7, NHEK, and SK-N-SH]. Splicing analysis was based on mapping the reads to the human genome (hg19 assembly) using STAR (Dobin et al. 2013) and GENCODE v26 annotations. Expression levels in various fractions were quantified using RSEM (Li and Dewey 2011) and Bowtie2. We classified genes using the “gene_ type” field, and defined all genes with gene type “protein_coding” as “PCGs” and all genes with either of “lincRNA” (n = 7471), “antisense” (n = 5511), “processed_transcript” (n = 523), or “bidirectional_promoter_lncRNA” (n = 8) gene types as “lncRNAs.” Genes with a transcript_type containing “pseudogene” or “intronic” values were excluded. Cyto/Nuc ratios were computed using DESeq2 (Love et al. 2014) based on the RSEM quantifications. Average TPM values across replicates were used as the final expression values, and we considered in each cell line only genes with expression levels of >0.5 TPM.
To generate expression- and length-matched cell-type–specific sets of PCGs and lncRNAs, we classified all expressed lncRNAs into 12 groups of equal size based on their exon counts (two bins) and expression levels (WCE, six bins). PCGs were classified using the bin thresholds set for lncRNAs. For each bin, we randomly sampled the larger group (either PCGs or lncRNAs) to generate two groups of equal size that match in their expression and exon counts distributions. Total n numbers for all bins are as follows: Localization (Supplemental Fig. S2C): GM12878—1126; HeLa-S3—1101; HepG2—1030; HUVEC—816; IMR90—754; K562—1131; MCF-7—1077; NHEK—894; SK-N-SH—1038. Splicing (Supplemental Fig. S2D–E): GM12878—888; HeLa-S3—791; HepG2—787; HUVEC—568; IMR90—603; K562—815; MCF-7—838; NHEK—715; SK-N-SH—876 (see also Supplemental Fig. S2F).
Splicing quantification at gene level
In order to quantify splicing on the gene level, we used the following two-step algorithm that was applied separately to each multiexon gene in the GENCODE annotations. In the first step, we identified confidently supported introns by using all the BAM files from the ENCODE cell lines. We traversed the introns annotated for the gene, and identified (using the “M” and “N” CIGAR operators) reads that supported any of the splicing sites and those that supported the splicing of the specific intron, that is, reads that had consecutive segments mapping in the two flanking exons of the intron. Reads containing insertions or deletions were ignored. We discarded introns supported by less than three reads, and those supported by less than reads, where Nspliced is the total number of reads with splice sites; is the number of introns in the isoform of the gene that had the most introns; and K is a parameter aimed to exclude introns that had relatively poor support compared to other introns in the same gene, that is, we aim to ignore introns that have K-times less reads that the average intron of the same gene (we used K = 25 in this study).
The second phase was applied separately to each cell type, considered only introns that were kept in the first phase, and iterated over the reads overlapping the intron. For each intron we computed: Spliced (S)—the number of spliced reads supporting the intron; Unspliced (U)—the number of reads not containing a splicing event and overlapping one of the splice junctions; One-end (E)—number of spliced reads supported either the 5′ or the 3′ end of the intron; and Other (O)—number of spliced reads not overlapping either splice site (Supplemental Fig. S1). We then summed these counts over all the samples (e.g., nuclear, cytoplasmic, and WCE of the specific cell line), and considered further only introns with specificity S/(S + E + O) > 0.1. We then discarded introns that overlapped another intron that had better specificity or the same specificity but more reads. We then considered just the remaining introns and computed the efficiency of individual introns as (S + E + O)/(S + U + E + O) and the efficiency of the gene as Σ(S + E + O)/Σ (S + U + E + O) across all kept introns. Gene- and intron-level values for efficiency and specificity were averaged across replicates of corresponding samples (Supplemental Data 1 and 2).
To evaluate differences in splicing efficiency and specificity between cell lines (Fig. 3E), we re-analyzed all cell lines together as described above, to ensure that we quantify the same set of introns in all cell lines, thus making the efficiency and specificity values comparable.
Enrichment of hexamers
We first used all the coding sequences annotated in GENCODE to compute overall codon usage. We next considered for each gene only the isoform that had the most introns used in the splicing analysis. We then counted the normalized number of occurrences (“dense.” in Supplemental Fig. S7) of hexamers containing at least four X bases for X = A, C, G, and T, considering separately instances overlapping internal and terminal exons (Supplemental Data 5). In addition, we evaluated the preference of A-hexamers over T-hexamers and the preference of C-hexamers over G-hexamers by subtracting the density values (“A-rich pref.” and “C-rich pref.” respectively in Supplemental Fig. S7). For coding sequences, we counted the number of codons which ended with base X, and compared that number to the number expected given the amino acid sequence and the global codon usage, to compute the preference for usage of codons ending with X (“codon pref.” in Supplemental Fig. S7).
Pol2 and histone modifications ChIP-seq analysis
We downloaded available Pol2 ChIP-seq data for six ENCODE cell lines (GSE31477; bigWig files for GM12878, HeLa-S3, HepG2, HUVEC, IMR90, and K562) and applied the bigWigAverageOverBed tool with BED files containing regions of interest based on GENCODE v26 annotation.
We defined a Pol2 pausing (Supplemental Data 3) index as:
For coverage, we used the “sum” output of bigWigAverageOverBed. Pausing index was calculated only for loci longer than 2000 bp, and gene-level pausing index was defined as the maximum value across isoforms.
To assess Pol2 occupancy over introns (Supplemental Data 4), we used our splicing quantification tool to generate a unique, nonoverlapping list of introns supported by the RNA-seq data. For each cell line separately, we used bigWigAverageOverBed with those introns. To account for length differences, we used the “mean0” values (with noncovered bases counting as zeroes) for each intron and gene-level coverage was defined as mean across all introns of the gene.
To explore histone modifications over introns and exon–intron junctions, we downloaded available ChIP-seq data for seven ENCODE cell lines (GSE29611—Gm12878, HeLa-S3, HepG2, HUVEC, K562, and Nhek. Antibodies targeting H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me2, H3K4me3, H3K79me2, and H3K9me3; GSE31755—MCF-7. Antibodies targeting H3K27ac, H3K27me3, H3K36me3, and H3K9me3). Read coverage over introns was calculated like for Pol2 occupancy over introns. Histone marks coverage over exon–intron junction was calculated by generating a set of intervals file with ±50 bp sequence of all 5′ and 3′ splice sites defined by the cell-specific intron set and applying the bigWigAverageOverBed tool to it. Gene-level values were defined as mean of mean0 values of all splice sites.
Correlations with sequence parameters at gene level
To correlate gene-level localization and splicing values with length parameters and exon counts, we used maximal value among all gene isoforms for each of: locus length, number of exons, transcript length, total length of introns, mean exon length, and mean intron length. 5′ and 3′ Senapathy and PSSM scores were computed as in Schwartz et al. (2009) and MaxEnt scores as in Yeo and Burge (2004). PhyloP conservation scores for the 100-way vertebrate whole-genome alignment were obtained from the UCSC genome browser, and averaged for positions −3.. + 4 and −11.. + 1 for the 5′ and 3′ splice-site conservation scores, respectively. H3K27Ac regions were obtained from the ENCODE project (wgEncodeSydhHistoneMcf7H3k27acUcdPk).
All intron-level parameters were converted to gene-level using a list of unique, nonoverlapping introns generated by analyzing the RNA-seq data sets from all cell lines together. Considering only these introns, we defined gene-level maxEnt scores, 5′ss Senapathy score, 3′ss PSSM score and average ss conservation as mean across all introns for a given gene. Numbers and total lengths of H3K27ac peaks and of PhastCons100way peaks were aggregated into gene-level using sum of all intron-level values. For hexamer enrichment analysis, we selected the isoform that contains the maximal amount of supported introns generated by analyzing the RNA-seq data sets.
Combining the cell-specific splicing and localization values for Supplemental Fig. S8 was performed using the median value across cell lines for each gene (Supplemental Data 5).
Linear models and machine learning
For multiple regression analysis of localization determinants, we trained separate linear models for PCGs and lncRNAs using only expressed genes in each cell line. Genes with missing values for any of the parameters were omitted. The features used for training the models are listed in Supplemental Data 6.
Machine learning analysis to predict subcellular localization using the same set of parameters that we used for linear models was performed by discretization of the continuous Cyto/Nuc into three groups: cytoplasmic [log2(Cyto/Nuc) > 1], intermediate [−1 < log2(Cyto/Nuc) < 1], and nuclear [log2(Cyto/Nuc) < −1]. For lncRNAs, we merged the “intermediate” and “cytoplasmic” classes. We then trained a Random Forest classifier from the RWeka R package (Hornik et al. 2007) separately on PCGs and on lncRNAs, and tested it on the same data with a 10-fold cross validation using the evaluate_Weka_classifier function, which provides AUC, precision and recall values for each localization class (RWeka package). This analysis was repeated 100 times to evaluate the error in AUC, precision, and recall values. We also trained the random forest classifier on data from either HepG2 or K562 cells and tested it on all other cell lines separately for PCGs and lncRNAs using the predict function. Precision and recall were calculated manually from the confusion matrix, which was generated by the confusionMatrix function from the caret package.
All AUC, precision, and recall values are summarized in Supplemental Data 7.
Conservation analysis
To characterize the conservation of splicing efficiency and specificity and subcellular localization, we downloaded RNA-seq data sets of mouse liver WCE from ENCODE (GSE36025; adult, 8-wk-old mice), as well as RNA-seq data sets of cytosolic and nuclear fractions of mouse liver from (Bahar Halpern et al. 2015) (GSE73977). We analyzed these data sets the same way we did with human ENCODE cell lines, using WCE data for expression and splicing quantification and cytosol/nucleus data for subcellular localization (Supplemental Data 8). Orthologs of human and mouse PCGs were obtained from Ensembl Compara database (version 30). Orthologs of lncRNAs were obtained by applying the methods described in Hezroni et al. (2015) to the human and mouse GENCODE transcripts (versions 26 and M13, respectively), and considering pairs of human and mouse lncRNA genes supported by both sequence similarity and synteny. Orthologs available in Supplemental Data 9. Using these orthologs lists, we compared mouse liver data with human HepG2 data.
GO analysis
We evaluated enrichment for biological processes in our data using the ranked list option in GOrilla web tool (Eden et al. 2009). For cell-specific splicing, we ranked based on the splicing efficiency values of the worst intron for each gene. For localization, we ranked genes by cell-specific Cyto/Nuc ratios, smallest (most nuclear) first. The obtained FDR-corrected P-values were clustered by REVIGO (Supek et al. 2011). Splicing analysis revealed no significant enrichment (FDR < 0.01) for all cell lines. The results of localization analysis for all cell lines are presented in Supplemental Data 10.
Additional data sets
We obtained RNA-seq data from various perturbations in HeLa cells from GEO database, accession GSE84172, GSE86148, GSE73678, and GSE73776. These data sets were processed using the same RSEM/DESeq2 pipeline as the other data sets. Half-lives in HeLa and MCF-7 cells were obtained from GSE86336 and GSE49831 (Schueler et al. 2014; Ke et al. 2017), respectively.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Schraga Schwartz, Hadas Hezroni, and Yoav Lubelsky for comments on the manuscript, and members of the Ulitsky laboratory for helpful discussions. This work was supported by the Israeli Centers for Research Excellence (1796/12); Israel Science Foundation (1242/14 and 1984/14); Israeli Ministry of Health as part of the ERA-NET localMND; and German-Israel Foundation for Scientific Research and Development (GIF), grant number I-1455-417.13/2018. I.U. is an incumbent of the Sygnet Career Development Chair for Bioinformatics.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.068288.118.
REFERENCES
- Akef A, Lee ES, Palazzo AF. 2015. Splicing promotes the nuclear export of β-globin mRNA by overcoming nuclear retention elements. RNA 21: 1908–1920. 10.1261/rna.051987.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar Halpern K, Caspi I, Lemze D, Levy M, Landen S, Elinav E, Ulitsky I, Itzkovitz S. 2015. Nuclear retention of mRNA in mammalian tissues. Cell Rep 13: 2653–2662. 10.1016/j.celrep.2015.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battich N, Stoeger T, Pelkmans L. 2015. Control of transcript variability in single mammalian cells. Cell 163: 1596–1610. 10.1016/j.cell.2015.11.018 [DOI] [PubMed] [Google Scholar]
- Boutz PL, Bhutkar A, Sharp PA. 2015. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev 29: 63–80. 10.1101/gad.247361.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ. 2014. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res 24: 1774–1786. 10.1101/gr.177790.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresson SM, Conrad NK. 2013. The human nuclear poly(A)-binding protein promotes RNA hyperadenylation and decay. PLoS Genet 9: e1003893 10.1371/journal.pgen.1003893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresson SM, Hunter OV, Hunter AC, Conrad NK. 2015. Canonical poly(A) polymerase activity promotes the decay of a wide variety of mammalian nuclear RNAs. PLoS Genet 11: e1005610 10.1371/journal.pgen.1005610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. 2011. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25: 1915–1927. 10.1101/gad.17446611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Pan X, Yang Y, Huang Y, Shen HB. 2018. The lncLocator: a subcellular localization predictor for long non-coding RNAs based on a stacked ensemble classifier. Bioinformatics 34: 2185–2194. 10.1093/bioinformatics/bty085 [DOI] [PubMed] [Google Scholar]
- Capelson M, Liang Y, Schulte R, Mair W, Wagner U, Hetzer MW. 2010. Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell 140: 372–383. 10.1016/j.cell.2009.12.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlevaro-Fita J, Rahim A, Guigo R, Vardy LA, Johnson R. 2016. Cytoplasmic long noncoding RNAs are frequently bound to and degraded at ribosomes in human cells. RNA 22: 867–882. 10.1261/rna.053561.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlevaro-Fita J, Das M, Polidori T, Navarro C, Johnson R. 2019. Ancient exapted transposable elements drive nuclear localisation of lncRNAs. Genome Res 29: 208–222. 10.1101/gr.229922.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YF, Saadi Imam J, Wilkinson MF. 2007. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 76: 51–74. 10.1146/annurev.biochem.76.050106.093909 [DOI] [PubMed] [Google Scholar]
- Chujo T, Yamazaki T, Kawaguchi T, Kurosaka S, Takumi T, Nakagawa S, Hirose T. 2017. Unusual semi-extractability as a hallmark of nuclear body-associated architectural noncoding RNAs. EMBO J 36: 1447–1462. 10.15252/embj.201695848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MB, Johnston RL, Inostroza-Ponta M, Fox AH, Fortini E, Moscato P, Dinger ME, Mattick JS. 2012. Genome-wide analysis of long noncoding RNA stability. Genome Res 22: 885–898. 10.1101/gr.131037.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Karousis ED, Bourquin J, Bruggmann R, Mühlemann O. 2017. Transcriptome-wide identification of NMD-targeted human mRNAs reveals extensive redundancy between SMG6- and SMG7-mediated degradation pathways. RNA 23: 189–201. 10.1261/rna.059055.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. 2012. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22: 1775–1789. 10.1101/gr.132159.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveson IW, Brunck ME, Blackburn J, Tseng E, Hon T, Clark TA, Clark MB, Crawford J, Dinger ME, Nielsen LK, et al. 2018. Universal alternative splicing of noncoding exons. Cell Syst 6: 245–245.e5. 10.1016/j.cels.2017.12.005 [DOI] [PubMed] [Google Scholar]
- Dias AP, Dufu K, Lei H, Reed R. 2010. A role for TREX components in the release of spliced mRNA from nuclear speckle domains. Nat Commun 1: 97 10.1038/ncomms1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Bradley RK. 2015. Widespread intron retention diversifies most cancer transcriptomes. Genome Med 7: 45 10.1186/s13073-015-0168-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48 10.1186/1471-2105-10-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES. 2016. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539: 452–455. 10.1038/nature20149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galante PAF, Natanja Kirschbaum-Slager NJS, José de Souza S. 2004. Detection and evaluation of intron retention events in the human transcriptome. RNA 10: 757–765. 10.1261/rna.5123504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galganski L, Urbanek MO, Krzyzosiak WJ. 2017. Nuclear speckles: molecular organization, biological function and role in disease. Nucleic Acids Res 45: 10350–10368. 10.1093/nar/gkx759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. 2007. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 8: 113–126. 10.1038/nrm2104 [DOI] [PubMed] [Google Scholar]
- Gil N, Ulitsky I. 2018. Production of spliced long noncoding RNAs specifies regions with increased enhancer activity. Cell Syst 7: 537–547.e3. 10.1016/j.cels.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudenas BL, Wang L. 2018. Prediction of lncRNA subcellular localization with deep learning from sequence features. Sci Rep 8: 16385 10.1038/s41598-018-34708-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hezroni H, Koppstein D, Schwartz MG, Avrutin A, Bartel DP, Ulitsky I. 2015. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep 11: 1110–1122. 10.1016/j.celrep.2015.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornik K, Zeileis A, Hothorn T, Buchta C. 2007. RWeka: an R interface to Weka. R Package Version 03–04. http://CRAN.R-project.org/package=RWeka
- Houseley J, LaCava J, Tollervey D. 2006. RNA-quality control by the exosome. Nat Rev Mol Cell Biol 7: 529–539. 10.1038/nrm1964 [DOI] [PubMed] [Google Scholar]
- Kaewsapsak P, Shechner DM, Mallard W, Rinn JL, Ting AY. 2017. Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. eLife 6: e29224 10.7554/eLife.29224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vågbø CB, Geula S, Hanna JH, Black DL, Darnell JE Jr, Darnell RB. 2017. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 31: 990–1006. 10.1101/gad.301036.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblihtt AR. 2006. Chromatin, transcript elongation and alternative splicing. Nat Struct Mol Biol 13: 5–7. 10.1038/nsmb0106-5 [DOI] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M, Anders S, Huber W. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubelsky Y, Ulitsky I. 2018. Sequences enriched in Alu repeats drive nuclear localization of long RNAs in human cells. Nature 555: 107–111. 10.1038/nature25757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. 2011. Epigenetics in alternative pre-mRNA splicing. Cell 144: 16–26. 10.1016/j.cell.2010.11.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo MJ, Reed R. 1999. Splicing is required for rapid and efficient mRNA export in metazoans. Proc Natl Acad Sci 96: 14937–14942. 10.1073/pnas.96.26.14937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauger O, Lemoine F, Scheiffele P. 2016. Targeted intron retention and excision for rapid gene regulation in response to neuronal activity. Neuron 92: 1266–1278. 10.1016/j.neuron.2016.11.032 [DOI] [PubMed] [Google Scholar]
- Melé M, Mattioli K, Mallard W, Shechner DM, Gerhardinger C, Rinn JL. 2017. Chromatin environment, transcriptional regulation, and splicing distinguish lincRNAs and mRNAs. Genome Res 27: 27–37. 10.1101/gr.214205.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meola N, Domanski M, Karadoulama E, Chen Y, Gentil C, Pultz D, Vitting-Seerup K, Lykke-Andersen S, Andersen JS., Sandelin A, et al. 2016. Identification of a nuclear exosome decay pathway for processed transcripts. Mol Cell 64: 520–533. 10.1016/j.molcel.2016.09.025 [DOI] [PubMed] [Google Scholar]
- Merino GA, Conesa A, Fernández EA. 2017. A benchmarking of workflows for detecting differential splicing and differential expression at isoform level in human RNA-seq studies. Brief Bioinform 10.1093/bib/bbx122 [DOI] [PubMed] [Google Scholar]
- Middleton R, Gao D, Thomas A, Singh B, Au A, Wong JJ, Bomane A, Cosson B, Eyras E, Rasko JEJ, et al. 2017. IRFinder: assessing the impact of intron retention on mammalian gene expression. Genome Biol 18 10.1186/s13059-017-1184-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagawa R, Tano K, Mizuno R, Nakamura Y, Ijiri K, Rakwal R, Shibato J, Masuo Y, Mayeda A, Hirose T, et al. 2012. Identification of cis- and trans-acting factors involved in the localization of MALAT-1 noncoding RNA to nuclear speckles. RNA 18: 738–751. 10.1261/rna.028639.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor A, Suliman S, Ben-Yishay R, Yunger S, Brody Y, Shav-Tal Y. 2010. Dynamics of single mRNP nucleocytoplasmic transport and export through the nuclear pore in living cells. Nat Cell Biol 12: 543–552. 10.1038/ncb2056 [DOI] [PubMed] [Google Scholar]
- Mukherjee N, Calviello L, Hirsekorn A, de Pretis S, Pelizzola M, Ohler U. 2017. Integrative classification of human coding and noncoding genes through RNA metabolism profiles. Nat Struct Mol Biol 24: 86–96. 10.1038/nsmb.3325 [DOI] [PubMed] [Google Scholar]
- Naro C, Jolly A, Di Persio S, Bielli P, Setterblad N, Alberdi AJ, Vicini E, Geremia R, De la Grange P, Sette C. 2017. An orchestrated intron retention program in meiosis controls timely usage of transcripts during germ cell differentiation. Dev Cell 41: 82–93.e4. 10.1016/j.devcel.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya K, Kataoka N, Hagiwara M. 2011. Stress-responsive maturation of Clk1/4 pre-mRNAs promotes phosphorylation of SR splicing factor. J Cell Biol 195: 27–40. 10.1083/jcb.201107093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntini E, Liz J, Muino JM, Marsico A, Ørom UA. 2018. Chromatin-release of the long ncRNA A-ROD is required for transcriptional activation of its target gene DKK1. Nat Commun 9: 1636 10.1038/s41467-018-04100-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton KE, Park SK, Hunter OV, Bresson SM, Conrad NK. 2018. Balance between MAT2A intron detention and splicing is determined co-transcriptionally. RNA 24: 778–786. 10.1261/rna.064899.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel H, Parra M, Gee SL, Mohandas N, Pachter L, Conboy JG. 2016. A dynamic intron retention program enriched in RNA processing genes regulates gene expression during terminal erythropoiesis. Nucleic Acids Res 44: 838–851. 10.1093/nar/gkv1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Schwartz K, Sherlock G, Dudoit S. 2011. GC-content normalization for RNA-seq data. BMC Bioinformatics 12: 480 10.1186/1471-2105-12-480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe NJ, de Souza SJ. 2007. Sequence features responsible for intron retention in human. BMC Genomics 8: 59 10.1186/1471-2164-8-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueler M, Munschauer M, Gregersen LH, Finzel A, Loewer A, Chen W, Landthaler M, Dieterich C. 2014. Differential protein occupancy profiling of the mRNA transcriptome. Genome Biol 15: R15 10.1186/gb-2014-15-1-r15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Hall E, Ast G. 2009. SROOGLE: webserver for integrative, user-friendly visualization of splicing signals. Nucleic Acids Res 37: W189–W192. 10.1093/nar/gkp320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalgi R, Hurt JA, Lindquist S, Burge CB. 2014. Widespread inhibition of posttranscriptional splicing shapes the cellular transcriptome following heat shock. Cell Rep 7: 1362–1370. 10.1016/j.celrep.2014.04.044 [DOI] [PubMed] [Google Scholar]
- Shukla CJ, McCorkindale AL, Gerhardinger C, Korthauer KD, Cabili MN, Shechner DM, Irizarry RA, Maass PG, Rinn JL. 2018. High-throughput identification of RNA nuclear enrichment sequences. EMBO J 37: e98452 10.15252/embj.201798452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T. 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6: e21800 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su ZD, Huang Y, Zhang ZY, Zhao YW, Wang D, Chen W, Chou KC, Lin H. 2018. iLoc-lncRNA: predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics 34: 4196–4204. 10.1093/bioinformatics/bty508 [DOI] [PubMed] [Google Scholar]
- Tan JY, Biasini A, Young RS, Marques A. 2018. An unexpected contribution of lincRNA splicing to enhancer function. bioRxiv 10.1101/287706 [DOI] [Google Scholar]
- Tilgner H, Knowles DG, Johnson R, Davis CA, Chakrabortty S, Djebali S, Curado J, Snyder M, Gingeras TR, Guigo R. 2012. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res 22: 1616–1625. 10.1101/gr.134445.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng CK, Wang HF, Burns AM, Schroeder MR, Gaspari M, Baumann P. 2015. Human telomerase RNA processing and quality control. Cell Rep 13: 2232–2243. 10.1016/j.celrep.2015.10.075 [DOI] [PubMed] [Google Scholar]
- Ulitsky I, Bartel DP. 2013. lincRNAs: genomics, evolution, and mechanisms. Cell 154: 26–46. 10.1016/j.cell.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia P, Dias AP, Reed R. 2008. Splicing promotes rapid and efficient mRNA export in mammalian cells. Proc Natl Acad Sci 105: 3386–3391. 10.1073/pnas.0800250105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanichkina DP, Schmitz U, Wong JJ, Rasko JE. 2017. Challenges in defining the role of intron retention in normal biology and disease. Semin Cell Dev Biol 75: 40–49. 10.1016/j.semcdb.2017.07.030 [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476. 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlotzka W, von Haeseler A, Zuber J, Ameres SL. 2017. Thiol-linked alkylation of RNA to assess expression dynamics. Nat Methods 14: 1198–1204. 10.1038/nmeth.4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JJ, Ritchie W, Ebner OA, Selbach M, Wong JW, Huang Y, Gao D, Pinello N, Gonzalez M, Baidya K, et al. 2013. Orchestrated intron retention regulates normal granulocyte differentiation. Cell 154: 583–595. 10.1016/j.cell.2013.06.052 [DOI] [PubMed] [Google Scholar]
- Yap K, Lim ZQ, Khandelia P, Friedman B, Makeyev EV. 2012. Coordinated regulation of neuronal mRNA steady-state levels through developmentally controlled intron retention. Genes Dev 26: 1209–1223. 10.1101/gad.188037.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Burge CB. 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11: 377–394. 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
- Yin Y, Lu JY, Zhang X, Shao W, Xu Y, Li P, Hong Y, Zhang QS, Shen X. 2018. U1 snRNP regulates chromatin retention of noncoding RNAs. bioRxiv 10.1101/310433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto R, Kaida D, Furuno M, Maxwell Burroughs A, Noma S, Suzuki H, Kawamura Y, Hayashizaki Y, Mayeda A, Yoshida M. 2017. Global analysis of pre-mRNA subcellular localization following splicing inhibition by spliceostatin A. RNA 23: 47–57. 10.1261/rna.058065.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gunawardane L, Niazi F, Jahanbani F, Chen X, Valadkhan S. 2014. A novel RNA motif mediates the strict nuclear localization of a long noncoding RNA. Mol Cell Biol 34: 2318–2329. 10.1128/MCB.01673-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.