

Pulmonary arterial hypertension (PAH) is a deleterious disease of the lung vasculature characterized by endothelial dysfunction, medial hypertrophy and proliferation, and eventual occlusion of the pulmonary arterioles. Although a number of genetic and environmental triggers have been described as causative, the specific molecular origins of this vascular disease are still unknown.
Multiple lines of evidence now converge to connect immune dysregulation to PAH in humans and animal models. Previous reports suggest that altered inflammatory dynamics, such as hematopoietic and myeloid expansion, B- and T-cell dysfunction, increased cytokine production, circulating autoantibodies, and tertiary lymphoid tissue neogenesis, may initiate or drive PAH.1 Clinically, a well-established causative link exists between autoimmune connective tissue disorders and PAH. Furthermore, infectious agents, such as Schistosoma mansoni and human immunodeficiency virus (HIV), are known to alter the immune cell repertoire and predispose to the development of PAH. However, a mechanistic understanding of the exogenous or endogenous factors that drive immune dysfunction remains largely undefined.
Prior studies have aimed to characterize the role of exogenous viruses in PAH. For example, HIV-associated PAH has been attributed to decreased CD4+ T-cell function, indicating a role for T-cell-mediated immune dysregulation.2 Separately, HIV-encoded proteins such as Nef have been reported to be pleiotropic meditators promoting inflammation and endothelial dysfunction in PAH, independent of vascular infection. Human herpesvirus 8 infection, often observed coincident with HIV, is the causative agent for Kaposi sarcoma—tumors characterized by endothelial hyperproliferation with histology similar to that observed in plexiform lesions of end-stage PAH. A role for humanherpes virus 8 in PAH was proposed by demonstrating upregulation of human herpes virus 8 viral protein and genomic expression in PAH lung tissue.3 However, these results have not been observed in separate, larger patient cohorts.4 Additionally, connections have been proposed between PAH and chronic infection of Epstein-Barr virus and hepatitis C virus.
In this issue of Circulation, Saito et al5 present evidence supporting a novel model of pathogenesis, implicating an endogenous, rather than an exogenous, retrovirus in the inflammatory processes of PAH. Human endogenous retroviruses (HERVs) are ancestral proviruses classified as mobile genetic elements. Although these viruses no longer replicate, they can be transmitted vertically and are activated by infection or other stressors. Activation may lead to transcription or mobilization of HERV genes, resulting in functional abnormalities and chromosomal instability.6 HERV-K sequences are transcriptionally active and have been associated with chronic inflammatory conditions such as cancer,6 neurological disorders, and other autoimmune diseases.
Saito and colleagues5 utilized a comprehensive approach combining confocal microscopy with mass spectrometry to identify SAMHD1, an innate immune factor associated with autoimmunity and HIV infection, localized to lung immune complexes in human PAH. It is important to note that an unbiased metagenomic viral screen revealed a significant increase in HERV, specifically HERV-K, in PAH lung tissue, whereas no increase occurred in exogenous viruses. The authors discovered that HERV-K components, HERV-K (II) envelope and HERV-K dUTPase mRNA, are upregulated in PAH lungs, notably in pulmonary macrophages and circulating monocytes. Mechanistically, they found that recombinant HERV-K dUTPase can induce SAMDH1 expression in pulmonary arterial endothelial cells and monocytes, driving endothelial apoptosis and monocytic expression of inflammatory cytokines. In vivo, injections of recombinant dUTPase in rats resulted in hemodynamic, histological, and inflammatory alterations consistent with PAH.
Collectively, these findings represent an innovative and substantial step forward in our understanding of viral pathogenesis in vascular inflammation and PAH (Figure). However, we are just beginning to obtain comprehensive, cause-and-effect proof of this pathobiology. Historically, the generation of rigorous scientific evidence for a direct pathogenic role for any endogenous retrovirus has been challenging. In part, this stems from the undefined fundamental biology and modes of activation of these genetic sequences. Their nearly ubiquitous prevalence in the general population, with multiple insertion sites as well as complete and incomplete open reading frames, also make straightforward comparative studies difficult. Additionally, it is not known how HERV-K is upregulated in PAH. Saito and colleagues5 found that HERV-K mRNA expression increased in monocytes after genotoxic lipopolysaccharide exposure. In contrast, inducible pluripotent stem cells from patients with PAH exhibited higher HERV-K dUPTase mRNA levels without any inflammatory stimulation. Although the definitive molecular driver(s) of HERV-K dUTPase expression remains undefined, at the least, these data suggest a more complex cell type-and perhaps context-specificity of this regulation. It is also not clear whether other endogenous retroviruses may display the same activity as HERV-K in PAH or whether HERV-K carries unique pathogenic activity for the pulmonary vasculature. It would be interesting to search for endogenous retrovirus activity in PAH in other mammals, particularly those with known genetic predisposition, such as brisket disease in cattle.7 It is also notable that many human diseases associated with HERV-K have not been linked to PAH, such as neuropsychiatric illnesses, thus providing an opportunity for future studies to search for a molecular explanation for that discrepancy. Finally, given the historical difficulties of proving a critical role for specific exogenous viruses in PAH, it will be imperative to corroborate these findings in a larger cohort of patients, hopefully with stratification of different subtypes of PAH and pulmonary hypertension in general.
Figure. HERV-K may initiate or propagate the inflammation that defines pulmonary arterial hypertension.
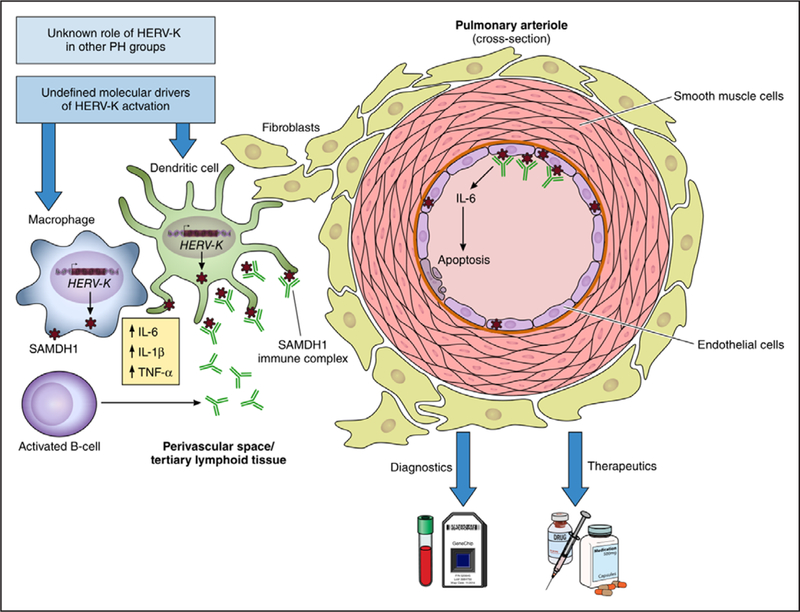
Saito and colleagues propose a role for HERV-K in the pathogenesis of PAH. Specifically, HERV-K components induce SAMDH1 expression, resulting in cytokine release, endothelial apoptosis, and B-cell activation. How HERV-K is activated or its diagnostic and therapeutic implications are still undefined. HERV-K indicates human endogenous retrovirus K; IL, interleukin; PAH, pulmonary arterial hypertension; and TNF, tumor necrosis factor.
Overall, this publication, which is built on prior work in both chronic inflammation and viral pathogenesis in PAH, highlights an intriguing new direction of discovery regarding the pathobiology of endogenous retroviruses. If relevant molecular mechanisms can be established in future work, substantial opportunity is available to design new diagnostic markers of PAH by tracking the activation of HERV-K. Furthermore, given the rapid advances of CRISPR-Cas9 gene editing tools in vivo for endogenous retroviruses in mammals,8 HERV-K may represent an exceptionally novel therapeutic target for PAH in the not-so-distant future.
Acknowledgments
SOURCES OF FUNDING
This work was supported by National Institutes of Health grants F30 HL139017–01 (M.K. Culley) and R01 HL124021, HL 122596, HL 138437, and UH2 TR002073 (S.Y. Chan).
Footnotes
DISCLOSURES
Dr Chan has served as a consultant for Actelion (significant), Gilead, Pfizer, and Vivus (modest).
M.K. Culley declares no conflicts of interest.
FOOTNOTES
Circulation is available at http://circ.ahajournals.org
The opinions expressed in this article are not necessarily those of the editors or of the American Heart Association.
REFERENCES
- 1.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Degano B, Guillaume M, Savale L, Montani D, Jaïs X, Yaici A, Le Pavec J, Humbert M, Simonneau G, Sitbon O. HIV-associated pulmonary arterial hypertension: survival and prognostic factors in the modern therapeutic era. AIDS. 2010;24:67–75. doi: 10.1097/QAD.0b013e328331c65e. [DOI] [PubMed] [Google Scholar]
- 3.Cool CD, Rai PR, Yeager ME, Hernandez-Saavedra D, Serls AE, Bull TM, Geraci MW, Brown KK, Routes JM, Tuder RM, Voelkel NF. Expression of human herpesvirus 8 in primary pulmonary hypertension. N Engl J Med. 2003;349:1113–1122. doi: 10.1056/NEJMoa035115. [DOI] [PubMed] [Google Scholar]
- 4.Henke-Gendo C, Mengel M, Hoeper MM, Alkharsah K, Schulz TF. Absence of Kaposi’s sarcoma-associated herpesvirus in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2005;172:1581–1585. doi: 10.1164/rccm.200504-546OC. [DOI] [PubMed] [Google Scholar]
- 5.Saito T, Miyagawa K, Chen S-Y, Tamosiuniene R, Wang L, Sharpe O, Samayoa E, Harada D, Moonen J- RAJ, Cao A, Chen P- I, Hennigs JK, Gu M, Li CG, Leib RD, Li D, Adams CM, del Rosario PA, Bill M, Haddad F, Montoya JG, Robinson WH, Fantl WJ, Nolan GP, Zamanian RT, Nicolls MR, Chiu CY, Ariza ME, Rabinovitch M. Upregulation of human endogenous retrovirus-K is linked to immunity and inflammation in pulmonary arterial hypertension. Circulation. 2017;136:1920–1935. doi: 10.1161/CIRCULATIONAHA.117.027589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoye JP. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol. 2012;10:395–406. doi: 10.1038/nrmicro2783. [DOI] [PubMed] [Google Scholar]
- 7.Newman JH, Holt TN, Cogan JD, Womack B, Phillips JA III, Li C, Kendall Z, Stenmark KR, Thomas MG, Brown RD, Riddle SR, West JD, Hamid R. Increased prevalence of EPAS1 variant in cattle with high-altitude pulmonary hypertension. Nat Commun. 2015;6:6863. doi: 10.1038/ncomms7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niu D, Wei HJ, Lin L, George H, Wang T, Lee IH, Zhao HY, Wang Y, Kan Y, Shrock E, Lesha E, Wang G, Luo Y, Qing Y, Jiao D, Zhao H, Zhou X, Wang S, Wei H, Güell M, Church GM, Yang L. Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9. Science. 2017;357:1303–1307. doi: 10.1126/science.aan4187. [DOI] [PMC free article] [PubMed] [Google Scholar]