

Abstract
In a continuing effort to identify structural attributes required for strong binding and potent inhibition of human drug-metabolizing CYP3A4, we designed ten ritonavir-like analogs differing in the side-group stereochemistry, backbone atomic composition, and head-group spacing. All analogs had pyridine and tert-butyloxycarbonyl (Boc) as the heme-ligating and tail groups, respectively, phenyl side-groups, and either a methyl- or ethyl-pyridyl linker. Each linker subseries had S/R, R/S, R/R and S/S side-group conformers (4a-d and 4e-h, respectively), and one S/S stereoisomer with the backbone S-to-N-heteroatom substitution (6a and 6b). To elucidate structure-activity relationships, ligand-dependent changes in optical spectra, dissociation constant (Ks), inhibitory potency (IC50), thermostability, and heme ligation and reduction kinetics were analyzed. Comparison of the subseries and individual compounds showed that CYP3A4 only weakly discriminates between side-group configurations, associates more tightly with the pyridyl-ethyl-linker analogs, and strongly disfavors the N-containing backbone. Ks and IC50 for the pyridyl-ethyl R/R conformer, 4g, were the lowest and close to those for ritonavir: 0.04 and 0.31 µM versus 0.02 and 0.13 µM, respectively. Determination of the X-ray structures of the inhibitory complexes was critical for experimental data interpretation, especially for the uniquely oriented 4a and 4e. Based on structural analysis we conclude that, for this series of analogs, the ligand-mediated interactions near the heme are dominant and define the binding mode, and that fine-tuning of these interactions, as well as the backbone spacing could further improve the affinity and inhibitory strength.
Keywords: CYP3A4, ligand binding, inhibitor design, crystal structure, structure-activity relations
INTRODUCTION
Human cytochrome P450 3A4 (CYP3A4) is the major xenobiotic and drug metabolizing enzyme1–2 that preferentially binds and oxidizes hydrophobic molecules to facilitate their elimination. Some chemicals, including pharmaceuticals, can inhibit CYP3A4 via different mechanisms.3 This side effect is usually undesired but, in some cases, inhibition of CYP3A4 could be beneficial because it could improve therapeutic efficiency of the quickly metabolized drugs by increasing their plasma levels. This principle of pharmacoenhancement is currently exploited in the treatment of HIV and HCV infections, where ritonavir (Figure 1A) and its derivative cobicistat are used as boosters for anti-viral drugs.4–5 Neither marketed pharmacoenhancer was developed based on the CYP3A4 crystal structure. Therefore, it remains unclear what functionalities are required for potent inhibition, and whether the inhibitory potency of ritonavir-like molecules can be improved via rational structure-based inhibitor design.
Figure 1.
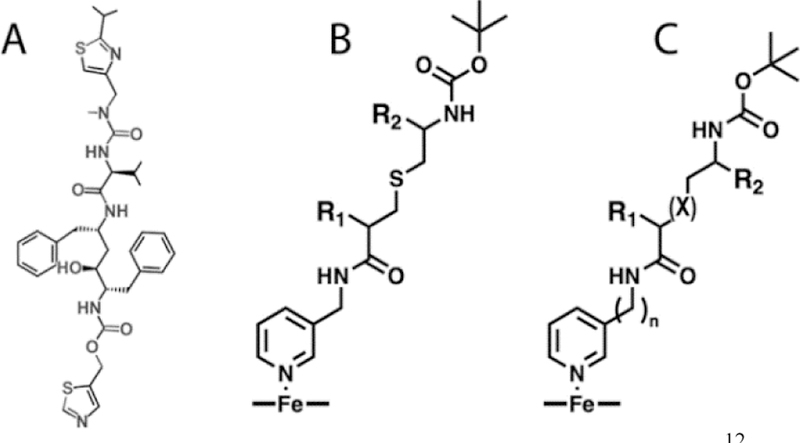
A, Ritonavir. B and C, The previously used12 and new scaffolds, respectively, where X = S or N, and n = 1 or 2. The pyridine nitrogen is the heme ligand; R1 and R2 are the side groups, and Boc is the protecting group serving as the terminal moiety.
Our ultimate goal is elucidation of the CYP3A4 ligand binding and inhibitory mechanism, which is critical for the development of safer and more effective drugs and pharmacoenhancers. To accurately map the active site and identify structural attributes required for strong binding and inhibition, we use a comparative approach and analyze structure-function relationships in series of rationally designed ritonavir-like molecules. Ritonavir was chosen for comparison because it remains the most potent inhibitor and its co-crystal structure with CYP3A4 had been determined.6–7
In our prior studies,6, 8–13 we developed a pharmacophore model for a CYP3A4-specific inhibitor, which was tested using two series of inhibitors with a scaffold shown in Figure 1B. The first series (series I) lacked the R1-side group and differed in the backbone length and R2 functionality,12 whereas the second series (series II) had the same backbone with various R1 and R1-R2 combinations.13 Structure/function analysis confirmed the importance of the backbone flexibility, H-bonding to the active site Ser119, and hydrophobic interactions mediated by the R1 and R2 moieties for the tight binding to CYP3A4. The latest study suggested also that optimization of the side-group stereochemistry and spacing between the functional groups may improve the affinity and inhibitory strength.13
To test this possibility, we utilized a different scaffold (Figure 1C) to design the third series (series III) of analogs with various stereochemical configurations of the R1/R2-phenyl functionalities, a one-carbon shorter R1-R2 distance (as in ritonavir), two varieties of the head-group spacer (methyl- or ethyl-pyridyl), and either a thioether or amino backbone linkage (Figure 2). As reported here, all three variables affect the binding and inhibitory strength, with the linker length and backbone composition being most critical.
Figure 2.
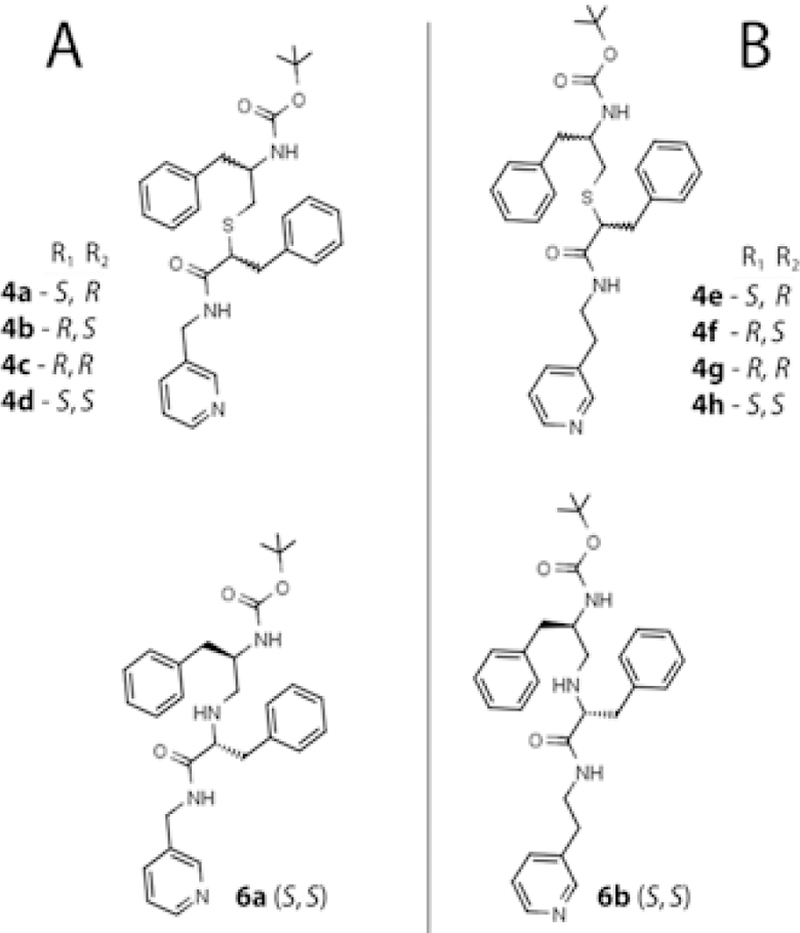
Methyl-pyridyl (A) and ethyl-pyridyl (B) linker subseries of compounds investigated in this study.
MATERIALS AND METHODS
Synthesis of CYP3A4 inhibitors, mass spectrometry and NMR data are included in the Supplemental Information section.
Protein Expression and Purification -
The codon-optimized full-length and Δ3–22 human CYP3A4 were produced as reported previously7 and used for assays and crystallization, respectively.
Spectral Binding Titrations –
Equilibrium ligand binding to CYP3A4 was monitored in a Cary 300 spectrophotometer at ambient temperature in 0.1M phosphate pH 7.4, containing 20% glycerol and 1 mM dithiothreitol. Inhibitors were dissolved in dimethyl sulfoxide (DMSO) and added to a 1.5–2 μM protein solution in small aliquots, with the final solvent concentration <2%. Spectral dissociation constants (Ks) were determined from the single-site hyperbolic or quadratic fits to titration curves.
Thermal Denaturation –
Thermal denaturation curves were recorded in 0.1 M phosphate, pH 7.4, in a Cary 300 spectrophotometer. Protein (1 μM) was mixed with a ligand or DMSO (20 μM and 2% final concentration, respectively) and incubated for 10 min at room temperature. Melting curves were recorded at 260 nm using a 0.2°C measurement step, 0.9°C/min ramp rate, and 50–75°C temperature range. A denaturation midpoint (melting temperature; Tm) was determined from non-linear fitting to a Boltzmann sigmoidal curve using the equation: Y = Amin + (Amax − Amin)/(1 + exp(Tm − X/Slope)), where Y - absorbance; X - temperature; Amin and Amax - absorbance at the lowest and highest temperature, respectively; Slope - steepness of the curve; and Tm - melting temperature.
Inhibitory Potency Assays –
Inhibitory potency for the 7-benzyloxy-4-(trifluoromethyl)coumarin (BFC) O-debenzylation activity of CYP3A4 was evaluated fluorometrically in an optimized soluble reconstituted system. The full-length CYP3A4 and rat cytochrome P450 reductase (40 μM and 60 μM, respectively) were preincubated at room temperature for 1 h before 20-fold dilution with the reaction buffer consisting of 0.1M potassium phosphate, catalase and superoxide dismutase (2 Units/ml each), and 0.0025% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate). Prior to measurement, 85 μl of the reaction buffer was mixed with 10 μl of the NADPH-regenerating system (10 mM glucose, 0.2 mM NADP+, and 2 Units/ml glucose-6-phosphate dehydrogenase), 5 μl of the protein mixture (0.1 μM final CYP3A4 concentration) and 2 μl of the inhibitor solution or DMSO. The mixture was incubated for 20 min at room temperature in the absence or presence of 70 μM NADPH. The reaction was initiated by adding 1 μl of 2 mM BFC and, when needed, 1 μl of 7 mM NADPH, and monitored for 2 min at room temperature in a Hitachi F400 fluorimeter (λex = 404 nm; λem = 500 nm). Within this time interval, the fluorescence change was linear. The reaction rates were calculated from linear fittings, and the average of three measurements was used to calculate the remaining activity using the fluorescence change in the presence of DMSO as a control. The IC50 values were derived from the [% activity] vs. [inhibitor] plots by fitting the data to a four-parameter logistic nonlinear regression equation: y = D + ((A-D)/(1+ 10(X-logC)•B)), where A and D are the maximal and minimal activity values, respectively, B - a slope factor, and C is IC50. The absolute rates of the product formation were measured only in the control samples using 7-hydroxy-4-trifluoromethyl coumarin (Santa Cruz Biotechnology) as a standard.
Kinetics of Ligand Binding and Heme Reduction –
Both kinetics were monitored in a SX.18MV stopped flow apparatus (Applied Photophysics, UK) at ambient temperatures in 0.1M phosphate, pH 7.4. To monitor the ligand binding reaction, 2 μM CYP3A4 was mixed with a 20 μM ligand solution and conversion of the heme iron to the low-spin form was followed at 427 nm. Kinetics of heme reduction in ligand-free and inhibitor-bound CYP3A4 was monitored at 417 and 443 nm, respectively, under anaerobic conditions in the presence of an oxygen scavenging system consisting of 10 mM glucose, glucose oxidase and catalase (5 Units/ml each). CYP3A4 (2 μM) was preincubated with 20 μM ligands for 10 min before mixing with 5 mM solution of sodium dithionite in a stopped flow apparatus purged with anaerobic buffer. Kinetic data were analyzed using manufacturer’s PROKIN software.
Determination of the X-ray Structures –
Δ3–22 CYP3A4 was co-crystallized with all ligands at room temperature by a microbatch method under oil. Protein (70–80 mg/ml) was incubated with a 5–10-fold ligand excess and centrifuged to remove the precipitate. The ligand-bound CYP3A4 (0.4–0.6 μl) was mixed with 0.4–0.6 μl of the crystallization solution (6–10% polyethylene glycol 3,350 and 60–90 mM sodium malonate, pH 6.0–7.0) and covered with paraffin oil. After harvesting, crystals were cryoprotected with Paratone-N and frozen in liquid nitrogen. X-ray diffraction data were collected at the Stanford Synchrotron Radiation Lightsource beamlines 9–2 and 12–2, and the Advanced Light Source beamline 8.2.2. The high resolution cutoffs were chosen based on the CC1/2 value according to the recommended guidelines.14 Crystal structures were solved by molecular replacement with PHASER15 and the 5VCC structure as a search model. Ligands were built with eLBOW16 and manually fit into the density with COOT.17 The initial models were rebuilt and refined with COOT and PHENIX.16 Data collection and refinement statistics are summarized in the Supplemental Information Tables S1 and S2. The atomic coordinates and structure factors for the 4a-, 4b-, 4c-, 4d-, 4e-, 4f-, 4g-, 4h-, 6a- and 6b-bound CYP3A4 were deposited in the Protein Data Bank with the ID codes 6DA2, 6DA3, 6DA5, 6DAA, 6DAB, 6DAC, 6DAG, 6DAJ, 6DA8 and 6DAL, respectively.
RESULTS AND DISCUSSION
Rationale for the New Series of Ritonavir Analogs
Our previous investigation on the series II inhibitors13 revealed that CYP3A4 selects from a racemic mixture and preferentially binds the S-configuration of R1-phenyl. The latter study also suggested that the series II compounds may have suboptimal pyridine-R1 and R1-R2 spacing, as neither analog could match the binding affinity and inhibitory potency of ritonavir. To test this possibility and gain deeper mechanistic and structural insights into the interaction of CYP3A4 with ritonavir-like molecules, we utilized a different scaffold (Figure 1C) to design the third series of analogs with the side-group functionalities and their spacing as in ritonavir (i.e., phenyl groups separated by two atoms). The pyridine-R1 distance was either the same (4 atoms; ethyl-pyridyl linker) or one-carbon shorter than in ritonavir (methyl-pyridyl linker). Each linker subseries contained four compounds with a thioether backbone and various R1/R2 configurations: S/R, R/S, R/R and S/S (4a-d and 4e-h, respectively; Figure 2). In addition, two S/S conformers with S-to-N substitution in the backbone (6a and 6b) were synthesized to elucidate if/how the heteroatom impacts the binding and inhibitory strength. To improve the accuracy and consistency of experimental data, all biochemical assays described herein were conducted with the full-length CYP3A4
Ligand-dependent Changes in the Absorbance Spectra and Binding Affinity
The maximum in the absolute spectra of inhibitor-ligated CYPs (λmax) is known to reflect electronic properties of the coordinating group, whereas the amplitude of the difference spectra (ΔAmax) is more sensitive to the steric properties and the environment distant from the heme: the larger ΔAmax, the more optimal orientation and stronger the coordination mode.18 One of our primary objectives was to identify spectral fingerprints for the weak and strong CYP3A4 binders. The earlier studies10, 12 suggested that one such feature could be the amplitude of the ferrous 442–443 nm band, found to be maximal in CYP3A4 complexed with the highly potent inhibitors, ritonavir and its analog GS3.6, 9 However, no correlation between IC50 and ferrous ΔAmax was observed for the series II analogs.13 Therefore, to further test the relationship between the ligand-induced spectral perturbations and inhibitory potency, we measured λmax and ΔAmax for both the ferric and ferrous ligand-bound forms (Figures 3 and S1; Table 1). Since the interaction of the full-length CYP3A4 with ritonavir has not been spectrally investigated, it was included in this study as well.
Figure 3.
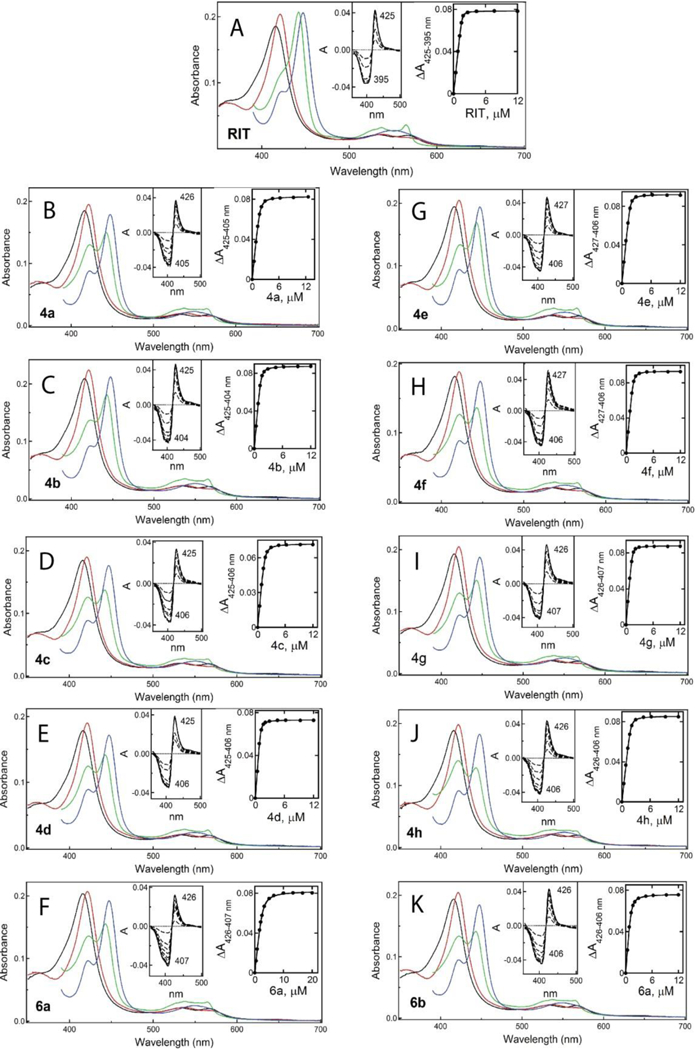
Spectral changes in CYP3A4 induced by ritonavir (A) and 4a-d (B-E), 4e-h (G-J) and 6a-b (F, K) analogs. Absorbance spectra of the oxidized ligand-free and ligand-bound CYP3A4 are in black and red, respectively. Spectra of the reduced and CO-adduct of the ligand-bound CYP3A4 are in green and blue, respectively. Left insets are difference spectra recorded during equilibrium titrations. Right insets are titration plots with hyperbolic or quadratic fittings, from which spectral dissociation constants (Ks) were derived (listed in Table 1).
Table 1.
Properties of the investigated compounds
| compound | λmax (nm) | ΔAmaxa | Ksb | IC50c | IC50/Ks | ΔTmd | ||
|---|---|---|---|---|---|---|---|---|
| ferric | ferrous | ferric | ferrous | μM | μM | °C | ||
| Ritonavir (R, R)e | 421 | 442 | 100% | 100% | 0.019± 0.002 | 0.13 ± 0.02 (0.14)f | 6.8 | +6.4 |
| pyridyl-methyl linker | ||||||||
| 4a(S, R) | 421 | 443 | 83% | 71% | 0.102±0.010 | 1.25±0.15 (1.30) | 12.3 | +4.1 |
| 4b(R, S) | 421 | 443 | 87% | 78% | 0.063±0.003 | 1.19± 0.06 (1.21) | 18.9 | +3.8 |
| 4c(R, R) | 421 | 442 | 81% | 67% | 0.070± 0.011 | 1.20± 0.05 (1.27) | 17.1 | +3.9 |
| 4d(S, S) | 421 | 443 | 86% | 71% | 0.042± 0.005 | 1.37± 0.11 (1.43) | 32.6 | +4.4 |
| 6a(S, S) | 421 | 443 | 86% | 69% | 0.340± 0.040 | 3.78± 0.29 (3.87) | 11.2 | +2.5 |
| pyridyl-ethyl linker | ||||||||
| 4e(S, R) | 422 | 443 | 99% | 79% | 0.042± 0.002 | 0.83± 0.07 (0.87) | 19.8 | +4.7 |
| 4f(R, S) | 422 | 443 | 91% | 66% | 0.045± 0.002 | 0.68± 0.05 (0.79) | 15.1 | +4.2 |
| 4g(R, R) | 421 | 443 | 94% | 71% | 0.040± 0.001 | 0.31± 0.04 (0.36) | 7.8 | +4.1 |
| 4h(S, S) | 421 | 443 | 94% | 58% | 0.055± 0.001 | 0.61± 0.02 (0.66) | 11.1 | +4.1 |
| 6b(S, S) | 421 | 443 | 95% | 70% | 0.122± 0.002 | 0.54± 0.05 (0.56) | 4.4 | +3.4 |
Maximal absorbance change in the ferric and ferrous ligand-bound CYP3A4 was calculated from the difference spectra (Figure S1) and expressed as a percentage of that induced by ritonavir. Prior to calculations, the difference spectra were normalized using 416 nm absorbance of the ligand-free form. The absorbance peak/trough at 425/394 nm and 443/412 nm was used to calculate ferric and ferrous ΔAmax, respectively, for ritonavir, and the respective values at 426/405–407 nm and 444/413 nm were used for the analogs.
The dissociation constant for the CYP3A4-inhibitor complex determined from the titration plots shown in Insets in Figure 3.
Determined for the BFC debenzylase activity of recombinant CYP3A4 in a reconstituted system with CPR.
Ligand-dependent change in the melting temperature relative to that of DMSO-bound CYP3A4 (Tm of 58.5°C).
Side-group stereochemistry if the backbone hydroxyl group was removed.
Values in the brackets are IC50s determined after preincubation of the inhibitor-bound CYP3A4 with NADPH.
As expected, all investigated compounds induced a red (type II) shift in the Soret band, indicative of the heme iron ligation to the nitrogen heteroatom (red spectra in Figure 3), whereas reduction of the ligand-bound forms with sodium dithionite led to formation of the 442–443 nm peak, characteristic for ritonavir-like inhibitors (green spectra in Figure 3). To ensure that there was no CYP3A4 inactivation during lengthy measurements, the ferrous CO-adduct spectra were recorded at the end of titrations. As evident from residual peaks at 420 nm, only a small fraction of ligand-bound CYP3A4 was unable to bind carbon monoxide either due to formation of an inactive P420 form or/and incomplete heme reduction.
Ferric and ferrous ΔAmax was calculated as a percentage of the absorbance change induced by ritonavir and compared to the spectral dissociation constants (Ks) derived from titration plots (right insets in Figure 3). Regardless of the redox state, both ΔAmax values were the highest for the CYP3A4-ritonavir complex, which correlated with the highest binding affinity (Ks of 0.019 μM). No clear correlation between ferric/ferrous ΔAmax and Ks was observed for other compounds. However, ferric ΔAmax was <90% for the methyl- pyridyl linker subseries and >90% for the elongated ethyl counterparts. With the exception of 4h, the latter subseries also had lower Ks values, and with a smaller spread (0.040–0.045 μM). Thus, in general, a one-carbon linker extension increases the binding affinity and leads to a larger spectral shift in ferric CYP3A4.
Regardless of the linker length, the binding affinity was markedly affected by the backbone S-to-N atom substitution: Ks for 6a and 6b was several-fold higher than for the sulfur-containing 4d and 4h (0.340 and 0.122 μM vs. 0.042 and 0.055 μM, respectively). Thus, all three variables in the ligands’ chemical structure affect the equilibrium binding to CYP3A4, with the pyridyl-R1 spacing and backbone composition being more critical than the side-group stereochemistry. The spectral data also demonstrate that linker elongation not only increases the affinity, but also partially compensates for the negative effect of the backbone heteroatom substitution.
Thermostability of CYP3A4
Although the relationship between binding affinity and melting temperature shift (ΔTm) is not a simple correlation,19 tighter-bound ligands usually increase the protein stability to a higher degree.20–21 Measurement of melting curves showed that neither analog could stabilize CYP3A4 as much as ritonavir, which increased Tm by 6.4°C (Table 1). Again, there was no strong relationship between the individual Ks and ΔTm values but, overall, compounds with an elongated linker had a more pronounced stabilizing effect. In each subseries, 4d and 4e had the largest impact and 6a-b the smallest (respective ΔTm of 4.4–4.7°C and 2.5–3.4°C). Thus, introduction of a backbone N-heteroatom not only lowers the binding affinity for CYP3A4, but also leads to protein destabilization.
Inhibitory Potency
The ability of new compounds to inhibit CYP3A4 was assessed by measuring their half inhibitory concentrations (IC50) for BFC debenzylation activity. Functional assays were conducted using an optimized lipid-free reconstituted system that, in addition to cytochrome P450 reductase, contained an NADPH-regeneration system and 0.0025% CHAPS. At this concentration, CHAPS did not form micelles and had no effect on the spin state of CYP3A4, but maximally stimulated the activity (by ~50%; Figure S2). One possible mechanism of its beneficial action could be a decrease in nonspecific protein aggregation and facilitation of the redox partner interaction, and/or an increase in solubility and availability of BFC. Importantly, the absolute rate of product formation in the soluble system was close to that for membranous CYP3A4: 0.33±0.02 min−1 versus 0.25–0.85 min−1,22 respectively.
Ritonavir is generally considered as a mechanism-based inhibitor of CYP3A4, mainly because its preincubation with microsomes and NADPH leads to an IC50 decrease.23–25 Thiazole and isopropylthiazole moieties of ritonavir are thought to be the sites of bioactivation, but the precise chemical nature of the reactive metabolite(s) remains unknown. Moreover, the latest detailed studies on ritonavir’s inhibitory mechanism were mutually exclusive, suggesting that only the heme or the apoprotein undergoes covalent modification.26–27 To determine if this mechanism-based inhibition occurs in our reconstituted system, the IC50 values were determined with and without 20 min preincubation with NADPH.
For all compounds, including ritonavir, preincubation with NADPH led to a small increase rather than decrease in IC50 (~2–16%), likely due to partial metabolism. There was no significant difference in IC50 for 4a-d, varying from ~1.2 to 1.4 μM (Table 1). The respective values for 4e-h were in the submicromolar range, with the lowest IC50 derived for 4g (0.31 μM). For the ethyl-pyridyl subseries, the heteroatom substitution had virtually no effect on the inhibitory potency but led to a 3-fold increase in IC50 for 6a. This implies once again that the negative impact of the amino-linked backbone on inhibitory complex formation can be compensated through a one-carbon-atom linker elongation. Pairwise comparison of Ks and IC50 also shows that tighter binding does not necessarily result in more potent inhibition (Table 1). No definitive conclusion could be drawn from the IC50/Ks ratios as well, as these values for the strongest inhibitors (4d, 4e and 4g) varied by several-fold.
Ligand Binding Kinetics
Two other factors that could affect IC50 are ligand binding and heme reduction rates. These parameters were measured by stopped flow spectrophotometry to better understand functional differences between the individual compounds. Ligand association to ferric CYP3A4 was monitored at 427 nm to follow heme iron conversion to a low-spin form. Under studied conditions, all reactions were four-phasic, with the initial burst followed by three slower phases (Figure S3). This complexity could arise from conformational heterogeneity and non-specific aggregation of the full-length CYP3A4,28 as well as the ability of ritonavir-like molecules to enter the active site with either the heme-ligating head- or tail-group. In the latter case, the bound inhibitor would have to reorient or re-enter the active site to allow for heme ligation, which would lead to the appearance of a slow kinetic phase(s).
For all investigated compounds, the percentage of the fast phase did not exceed 50%, and the amplitude of the total absorbance change at the end of the measurement (ΔA427 nm) was nearly the same (Figure S3; Table 2). The rate constants for the fast phase (kfast) were in the 5–14 s−1 range, with the highest value derived for 4f. The S/R stereoisomers (4a and 4c) were among the quickest binders, whereas 6a-b had the low binding rates. Ritonavir had the second lowest binding rate and the lowest percentage of the fast phase: 7.5 s−1 and 27%, respectively. For other inhibitors, however, no clear relationship between the kinetic parameters and IC50 could be derived. Thus, side-group stereochemistry and pyridyl-R1 spacing only modestly affect presteady-state kinetics, while the N-heteroatom imposes conformational restraints, likely by lowering the backbone flexibility.
Table 2.
Kinetic parameters for the ligand binding reaction and reduction of the ligand-bound CYP3A4 with sodium dithionite.
| ligand binding |
reduction with sodium dithionite |
||||||
|---|---|---|---|---|---|---|---|
| compound | kfasta | kslowb | ΔA427 nmc | kETfastd | kETslowe | ΔA443 nmf | |
| s−1 | s−1 | % | s−1 | s−1 | % | ||
| ligand-free | 4.6 (47%)g | 0.144 | |||||
| Ritonavir (R, R)h | 7.5 (27%)i | 1.13 | 0.23 | 100 | 0.015 (25%) | 0.005 | 100 |
| pyridyl-methyl linker | |||||||
| 4a(S, R) | 12.6 (44%) | 1.49 | 0.31 | 102 | 0.029 (55%) | 0.008 | 61 |
| 4b(R, S) | 9.5 (37%) | 1.57 | 0.28 | 97 | 0.035 (47%) | 0.007 | 60 |
| 4c(R, R) | 10.5 (40%) | 1.43 | 0.30 | 97 | 0.018 (39%) | 0.009 | 52 |
| 4d(S, S) | 10.4 (41%) | 1.42 | 0.29 | 96 | 0.026 (32%) | 0.006 | 56 |
| 6a(S, S) | 5.3 (29%) | 0.81 | 0.20 | 98 | 0.075 (53%) | 0.010 | 50 |
| pyridyl-ethyl linker | |||||||
| 4e(S, R) | 11.7 (32%) | 1.63 | 0.31 | 112 | 0.015 (75%) | 0.005 | 56 |
| 4f(R, S) | 13.8 (32%) | 1.98 | 0.35 | 108 | 0.022 (61%) | 0.008 | 45 |
| 4g(R, R) | 9.7 (42%) | 1.48 | 0.33 | 103 | 0.018 (53%) | 0.007 | 55 |
| 4h(S, S) | 11.7 (38%) | 1.62 | 0.30 | 106 | 0.022 (36%) | 0.007 | 40 |
| 6b(S, S) | 9.6 (38%) | 1.34 | 0.33 | 99 | 0.024 (56%) | 0.007 | 40 |
Rate constants for the fast and slow kinetic phase of the CYP3A4-ligand binding reaction determined at saturating ligand concentration.
Rate constants for the last reaction phase are given in Figure S3.
Percentage of the maximal absorbance change at 427 nm observed at the end of the reaction relative to that for ritonavir.
Rate constants for the fast and slow kinetic phase for the reduction of CYP3A4 with sodium dithionite.
Percentage of the absorbance change at 443 nm observed at the end of the reduction reaction relative to that for the ritonavir-bound form.
Percentage of the absorbance change taking place during the fast kinetic phase relative to those for ritonavir.
Side-group stereochemistry if the backbone hydroxyl group was removed.
Reduction of Ligand-Bound CYP3A4 with Sodium Dithionite
The amplitude of a characteristic 443 nm peak in ferrous inhibitor-bound forms (green spectra in Figure 3) could reflect not only the coordination geometry and strength but also the electron-accepting ability of CYP3A4, which is generally defined by the heme redox potential and accessibility of the heme iron to the reductant, sodium dithionite. Therefore, we measured kinetics of anaerobic reduction of ligand-free and ligand-bound CYP3A4 with sodium dithionite by monitoring absorbance changes at 417 and 443 nm, respectively. Within the studied time interval, all reactions were tri-phasic, with two major phases (fast and slow) and the residual third phase (<8% of total absorbance change; Figure S4). The ligand association led to a dramatic decrease in the heme reduction rates: 60-to-300-fold for the fast phase and 20-to-40-fold for the slow phase (kETfast and kETslow, respectively; Table 2). This supports our previous findings6 and implies that, upon ligation, ritonavir-like molecules markedly impede the electron transfer by lowering the redox potential of the heme and/or restiricting its access to the SO2- radical. As expected: (i) the ritonavir-bound form had one of the slowest reduction rates and the lowest percentage of the fast phase; (ii) the ethyl-pyridyl-linker compounds decreased kETfast more than the shorter counterparts; and (iii) in each subseries, analogs with the S-to-N backbone substitution had the lowest impact on the heme reduction.
Thus, according to the experimental data, all investigated parameters were affected by changes in analogs’ chemical structure, with alterations in the backbone length and composition having the largest impact. There was no strong correlation between parameters derived for the individual compounds but, in general, changes in Ks, ΔTm, kET and ferric ΔAmax coincide with changes in IC50. The sulfur-containing R/R and S/S conformers were amongst the strongest binders, inhibitors and stabilizers, whereas the S/R configuration and the amino-linked backbone were the least optimal for the association and/or inhibition of CYP3A4. Most importantly, despite the complexity of structure/activity relationships, it was possible to identify 4g as an outstanding leader, whose affinity and inhibitory potency for CYP3A4 were close to those of ritonavir.
Crystal Structures of the Inhibitory Complexes
To better understand structure-activity relationships for the series III analogs, each of them was co-crystallized with CYP3A4. The X-ray structures of the inhibitory complexes were determined to 2.15–2.80 Å resolution (Tables S1 and S2), and structural features are summarized and compared in Table 3.
Table 3.
Structural features of the CYP3A4-inhibitor complexes
| compound | Fe-N bond |
pyridine ring rotation (°)b | I-helix displacement (Å) c | H-bond with Ser119 (Å) d | pyridine-R2 ring angle and overlap | Phe304-R1 ring angle (°)and overlap | Boc-group conformation and contacts | |
|---|---|---|---|---|---|---|---|---|
| distance (Å) | angle (°)a | |||||||
| pyridyl-methyl linker | ||||||||
| 4a (S, R) | 2.38 | 10 | 17 | 0.67–0.81 | 3.10 | −25°; partiale | 90°; nonee | traceable; 106, 108, 215, 374 |
| 4b (R, S) | 2.26 | 3 | 25 | 1.39–0.88 | 2.54 | 20°; half | 0°; half | traceable; 57, 108, 220 |
| 4c (R, R) | 2.21 | 5 | 20 | 1.36–1.15 | 2.63 | 15°; half | 0°; half | traceable; 108, 220, 482 |
| 4d (S, S) | 2.25 | 3 | 28 | 0.95–1.06 | 2.55 | 15°; half | 15°; full | traceable; 108, 220 |
| 6a (S, S) | 2.40 | 0 | 30 | 0.79–1.24 | 2.87 | 32°; half | 12°; full | traceable; 108, 220 |
| pyridyl-ethyl linker | ||||||||
| 4e (S, R) | 2.22 | 5 | 47 | 2.16–2.36 | 3.16 | − 70°; partial | 87°; none | traceable; 108, 481 |
| 4f (R, S) | 2.29 | 2 | 20 | 1.25–0.85 | 3.24 | − 35°; half | 68°; none | disordered |
| 4g (R, R) | 2.25 | 5 | 33 | 0.97–1.37 | 2.93 | −45°; half | 25°; full | traceable, 105–108, 120 |
| 4h (S, S) | 2.31 | 5 | 20 | 0.54–0.71 | 2.99 | − 23°; half | 10°; half | traceable; 108, 220 |
| 6b (S, S) | 2.25 | 5 | 25 | 0.88–0.86 | 3.04 | −40°; half | 30°; half | disordered |
| Ritonavirf (R, R) | 2.16 | 8 | 45g | 1.99–2.18 | 2.92 (O24) | −65°; none g | 53°; half | |
| 2.84 (O41) | ||||||||
| 3.01 (N11) | ||||||||
Deviation from perpendicularity.
Angle between the planes passing through the pyridine ring and the NB-ND heme atoms
Distance between the Cα-atoms of Phe304 and Ala305 in the inhibitor-bound and ligand-free CYP3A4 (PDB ID 5VCC).
Hydrogen bond length between the inhibitor O21 atom and Ser119:OG. Ritonavir’s atoms H-bonded to Ser119 (5VC0 structure) are indicated in brackets.
In 4a, R1 and R2 side groups are placed near the heme-ligating pyridine and Phe304, respectively
Parameters derived from the 5VC0 structure. Side-group stereochemistry if the backbone hydroxyl group was removed.
Respective angle and overlap for the thiazole ring.
Structures of CYP3A4 bound to the methyl-pyridyl subseries of compounds -
Orientations of 4a-d and 6a relative to the adjacent I-helix and the omit electron density maps are displayed in Figure 4A–E. The central part of the I-helix in the superimposed water-bound CYP3A4 (4I3Q structure) is also shown to demonstrate the ligand-induced helix displacement and the interplay between the phenyl side-group and Phe304. As seen from the structural overlay (Figure 4F), 4a adapts a unique conformation with a reverse side-group orientation: R1 is placed near the heme-ligating pyridine, and R2 is near the hydrophobic pocket above the I-helix (designated as P2 and P1 sites, respectively, in our pharmacophore model).11 This binding mode allows only a partial overlap between the R1-phenyl and pyridine and none between the R2 and Phe304 rings, which are perpendicular to each other.
Figure 4.
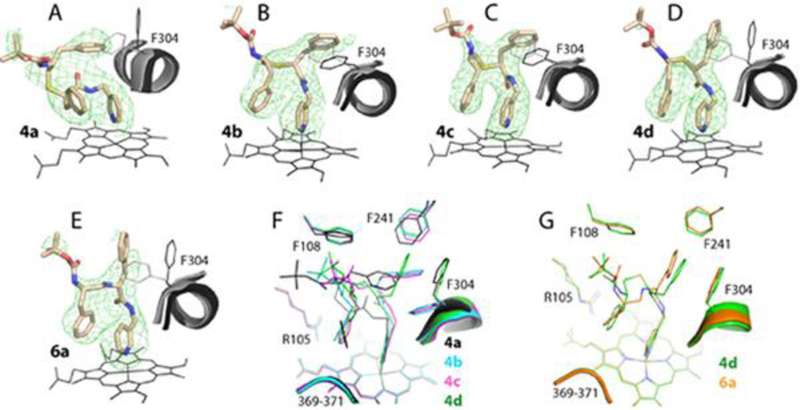
Crystal structures of CYP3A4 bound to compounds from the methyl-pyridyl subseries. A-E, The binding mode of 4a-d and 6a, respectively. The adjacent I-helix and Phe304 in the inhibitory complexes and in water-bound CYP3A4 (4I3Q structure) are shown in black and gray, respectively. Polder omit maps contoured at 4σ level are shown as green mesh. F and G, Structural overlays of 4a-d and the 4d-6a pair, respectively.
In contrast, 4b-d bind to CYP3A4 with a traditional R1/R2 orientation. Regardless of the side-group stereochemistry and the rotamer of Phe304, the R1-phenyls of 4b-d are parallel to and fully or partially overlap with the Phe304 ring. The R2-groups, in turn, are near parallel to and overlap with the heme-ligating pyridine (Figure 4F). Moreover, the R2-phenyls are closer to the Arg105 guanidine and tilt toward, rather than away from the pyridine, thereby enabling and strengthening π-π and cation-π stacking. The inability of 4a to establish stabilizing interactions at P2 site, as well as its longer Fe-N distance and H-bond with the active site Ser119 could explain why this analog is a weaker binder and a less potent inhibitor than the stereo counterparts (Tables 1 and 3).
Surprisingly, despite distinct curvatures of the backbone bearing the heteroatom, there was a high resemblance between the binding modes of 6a and 4d (Figure 4G). Nonetheless, 6a had a longer distance between the R1-phenyl and Phe304 rings (4.2 Å versus 3.6 Å in 4d), and the weaker Fe-N and O21-Ser119:OG bonds (Table 3), which could lower the affinity and inhibitory strength.
Structures of CYP3A4 bound to the ethyl-pyridyl subseries of compounds –
The active site positioning and structural overlays of 4e-h and 6b are shown in Figure 5. Except 4e, all analogs adapted a traditional orientation (R1 in P1 pocket and R2 at P2 site), meaning that this orientation is preferable but disallowed in the S/R conformers regardless of the linker length. Due to stereochemical restraints, the 4f-h backbones curve differently, leading to notable variations in the functional group positioning and the I-helix displacement (Figure 5E). Boc is completely disordered in 4f and 6b, but traceable in other compounds (Table 3). Notably, in 4e, the R2-phenyl orients perpendicularly to Phe304 and is adjacent but not inserted into the hydrophobic pocket. A near perpendicular orientation of R1 and Phe304, minimizing the aromatic ring overlap, is also observed in 4f. In contrast, the 4g/h R1 protrudes into the hydrophobic pocket deeper than in the methyl-pyridyl analogs and with a substantial R1-Phe304 overlap, maximized through Phe304 ring rotation. Another characteristic feature of this subseries is the outward tilted R2 (R1 in 4e), enabling stronger cation-π interactions with Arg105 and a larger overlap with the heme cofactor. In 4e, R1 is closer and near parallel to the heme, which could further strengthen hydrophobic/aromatic interactions at P2 site and compensate for the suboptimal R2 orientation.
Figure 5.
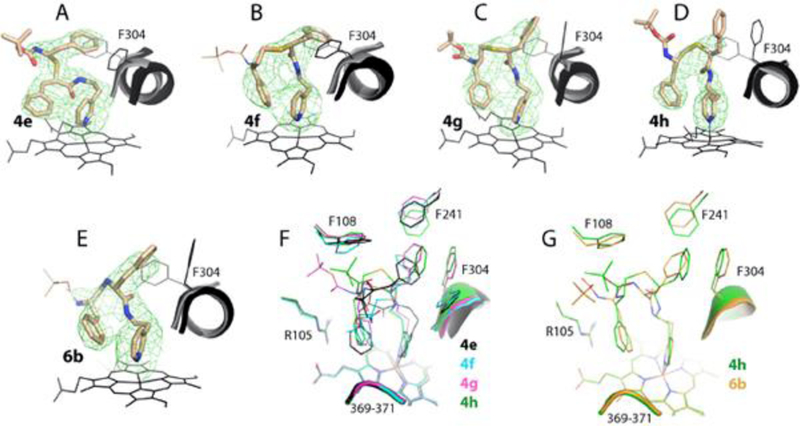
Crystal structures of CYP3A4 bound to compounds from the ethyl-pyridyl subseries. A-E, The binding mode of 4e-h and 6b, respectively. The adjacent I-helix and Phe304 in the inhibitory complexes and in water-bound CYP3A4 (4I3Q structure) are shown in black and gray, respectively. Polder omit maps contoured at 4σ level are shown as green mesh. In 4f and 6b, the Boc groups were disordered, and their arbitrary positions are shown in thin lines. F and G, Structural overlays of 4e-h and the 4h-6b pair, respectively.
The backbone heteroatom substitution led to notable changes in the ligand conformation (Figure 5G). Unlike the similarly oriented 4d-6a pair (Figure 4G), only the R2 functionalities coincide in 4h and 6b. Because of the distinctly curved backbone, the 6b R1 adapts a different rotamer to move closer to and maximize an overlap with Phe304. A smaller deviation in Ks and IC50 for the 4h-6b pair relative to 4d-6a (Table 1) implies that pyridine-R1 spacer elongation largely compensates for the restraints imposed by the amine-linked backbone.
Indeed, superposition of 6a-b (Figure 6A) shows that the longer analog adapts a more relaxed conformation and shifts sideways toward the wall of the active site cavity. This allows the 6b backbone to establish van der Waals contacts with the nearby residues (Ser119, Leu120 and the Arg105 side chain) and optimize the R2-mediated interactions. The resulting stabilization of the binding mode explains why 6b associates tighter and inhibits CYP3A4 considerably stronger than 6a (Table 1).
Figure 6.
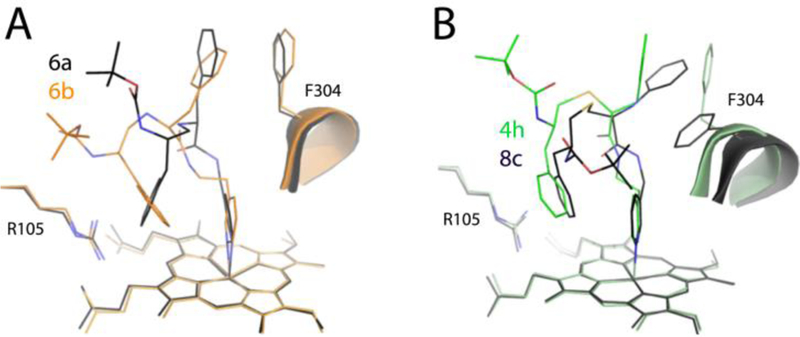
Superposition of the 6a-b (A) and 4h-8c pairs (B) showing how the ligand binding mode changes upon a one-carbon extension of the pyridyl-R1 and R1-R2 spacers, respectively. Compound 8c from the previous series12 has one extra carbon between the phenyl groups.
Impact of R1-R2 spacer shortening and the side-group configuration -
To better understand how the ligand orientation changes upon a one-carbon shortening of the R1-R2 spacer, 4h was superimposed with 8c, the series II compound.13 Although 8c was synthesized with the R1-phenyl in racemic mixture, CYP3A4 selected and co-crystallized with the S-conformer (6BDH structure). A C-to-N substitution in the 8c R1-phenyl could affect, to some degree, the R1 flexibility but not the overall ligand orientation. In contrast, the length of the R1-R2 spacer (2 atoms in 4h and 3 atoms in 8c) appears to be the major factor that impacts the ligand binding mode (Figure 6B). Except the heme-ligating pyridine, all structural elements of 4h are displaced: the backbone and R2 shift sideways toward the wall of the cavity, the R1 and Phe304 rings are at distinct positions and tilt angles, and the Boc group is pointing into an opposite direction. Given that 4h has a ~2-fold higher affinity and 14% lower inhibitory potency than 8c,13 it can be concluded that the shorter phenyl-group separation is more optimal for the interaction with CYP3A4.
To examine the impact of stereochemistry, a pairwise comparison of conformers from the methyl/ethyl-pyridyl subseries was conducted (Figure 7). Structural overlays demonstrate that analogs with the longer pyridyl-linker adapt a more relaxed conformation, with wider space between the head and tail parts. The largest difference in the pyridine and side-group orientations was observed only in the 4a-e pair (S/R conformers; Figure 7A). To optimize the side-group placement, the 4e pyridine tilts toward and shifts the I-helix by 2.4Å. This is the largest displacement observed thus far, and could be the reason for a significant increase in IC50.
Figure 7.
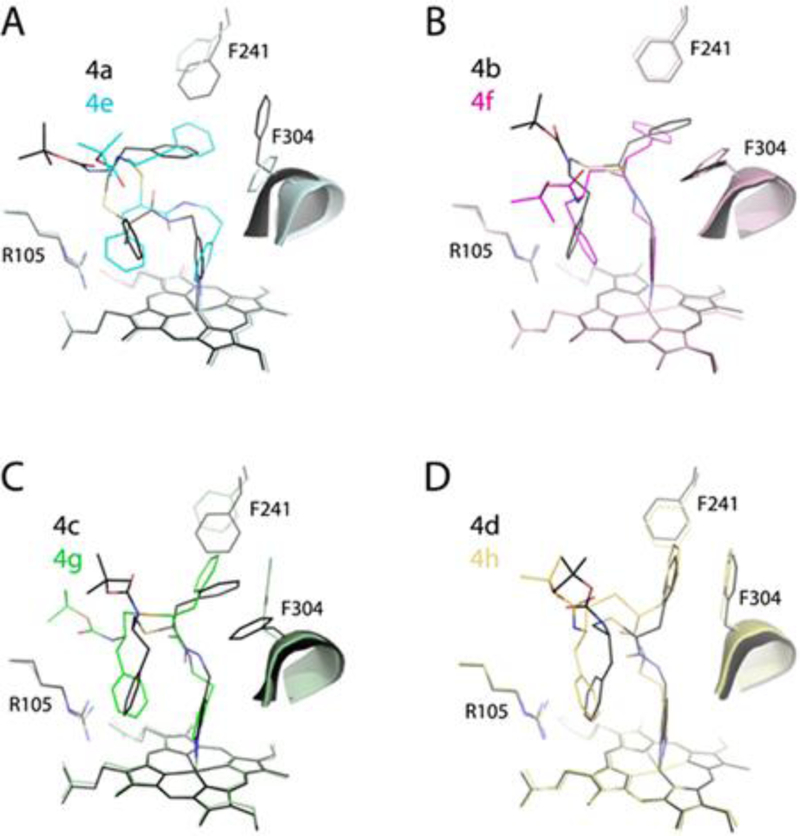
Pairwise comparison of the 4a-4e (A), 4b-4f (B), 4c-4g (C) and 4d-h (D) pairs showing that stereochemical restraints and the pyridyl-linker extension are compensated through changes in the backbone curvature and reorientation of the R1-phenyl, Phe241 and Phe304 rings.
In other pairs (Figure 7B–C), there were no changes in the pyridine orientation, as the binding modes were optimized primarily through the backbone curvature and via the R1, Phe241, and Phe304 interplay. The extent of conformational adjustments enabled by linker elongation was also quite similar and consistent with a small spread in Ks and IC50 (Table 1). Moreover, the relative orientation and larger R2-heme overlay in 4e-h analogs confirmed the notion that the hydrophobic and aromatic interactions at P2 site substantially contribute to the inhibitory potency.11–13 Finally, superposition of all investigated compounds (Figure 8) showed that, despite differences in the side-group stereochemistry, backbone length and atomic composition, the R2-phenyls occupy nearly the same spot. Thus, interactions established at P2 site are dominant and define the overall binding mode. This is in contrast to the series II compounds, where the R1-mediated interactions at P1 site appeared to play the leading role. 13
Figure 8.
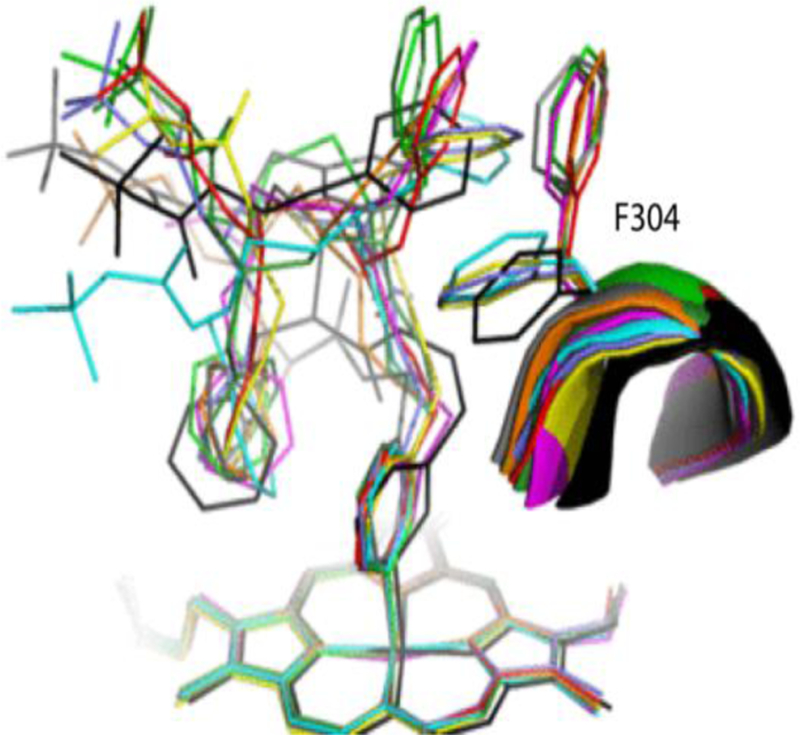
Structural overlay of all inhibitory complexes: 4a-d and 4e-h are in black, cyan, magenta, green, gray, lavender, yellow and red, respectively; 6a is in orange and 6b in dark green.
SUMMARY AND CONCLUSIONS
Human CYP3A4 is a major drug-metabolizing enzyme, whose inhibition could lead to undesired drug-drug interactions and toxicity, a major cause for late-stage clinical trial failures and withdrawal of marketed pharmaceuticals. Despite the central role of CYP3A4 in drug metabolism, there is limited understanding of how the protein adapts to such a wide range of substrates. Furthermore, there is no structure-based inhibitor pharmacophore that could assist in identification and early elimination of potential CYP3A4 inactivators during development of drugs and other chemicals relevant to public health.
We utilize a rational structure-based inhibitor design to address this issue and identify structural attributes required for strong binding and potent inhibition of CYP3A4. Having developed a pharmacophore model for a CYP3A4-speciic inhibitor,11 we undertook a build-from-sctratch approach for testing the relative importance of each pharmacophoric determinant. Starting with the pyridine ring as the heme-ligating moiety, the inhibitors were being built by gradually elongating the backbone and attaching various side-groups.
With the first two series of rationally designed analogs, we confirmed the importance of the backbone flexibility, H-bonding to the active site Ser119, and hydrophobic interactions mediated by the side-group moieties. The curent work was a logical continuation, designed to test if CYP3A4 has any preference for the side-group stereoconfiguration, side/head-group spacing, and backbone composition. For this purpose, ten novel compounds were synthesized, and biochemically and structurally characterized. It was found that:
Spectral properties, binding affinity, heme ligation/reduction rates, and the ability to stabilize and inhibit CYP3A4 were all affected by chemical changes, with alterations in the backbone length and composition having the largest impact.
CYP3A4 associates more tightly with the elongated compounds. Among the sulfur-containing conformers, R/R and S/S were stronger binders, inhibitors, and/or stabilizers, whereas the amino-linked backbone was the least favorable for association with CYP3A4.
The ethyl-pyridyl R/R conformer, 4g, was identified as an outstsnding leader in terms of binding affinity and inhibitory potency and, thus, it could be used as a template for analog synthesis for further evaluaton and optimization of the pharmacophore model.
No strong correlation was observed between parameters derived for the individual compounds but, collectively, Ks, ΔTm, kET and ferric ΔAmax could be used for the inhibitory potency predictions.
Under our experimental conditions, ritonavir and analogs exert their inhibitory action by strongly ligating to the heme and impeding its reduction. Since cytochrome b5 was not included in the assay system, it is possible that this or/and other microsomal proteins could mediate or directly bioactivate ritonavir.
Determination of the X-ray structures of the inhibitory complexes was essential for the experimental data interpretation. Moreover, without structural information, the unique oriention of 4a and 4e would be impossible to foresee.
The ethyl-pyridyl analogs adapt a more relaxed conformation, mainly through conformational adjustments in the backbone curvature and via the R1-Phe304 interplay.
Phe304 is a key residue, whose aromatic ring undergoes rotational and positional movements to adjust, improve and strengthen interactions with the phenyl side-group.
The nitrogen atom decreases the backbone flexibility, leading to an altered 6a-b binding mode. However, even in these analogs, conformational optimization could be partially achieved through pyridyl-linker elongation.
Regardless of the analogs’ chemical structure and backbone curvature, the phenyl-group closest to the heme occupies nearly the same space at P2 site, whereas orientation of the side-group at P1 site significantly varies. Thus, for the series III compounds, hydrophobic interactions at P2 site are dominant and define the binding mode.
Together, these findings suggest that there is still room for improvement and, prior to addition/optimization of the end-group, a further increase in the binding affinity and inhibitory strength could be achieved through an increase in R2 hydrophobicity and fine-tuning of the backbone length. Thus, this study not only provides novel insigts into structure-function relationships of ritonavir-like compounds and expands our knowledge on how CYP3A4 interacts with and adjusts to structurally diverse ligands, but also highlights strategies for for the next generation inhibitor design.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grant ES025767 and involves research carried out at the Stanford Synchrotron Radiation Lightsource and the Advanced Light Source. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Abbreviations
- CYP3A4
cytochrome P450 3A4
- BFC
7-benzyloxy-4-(trifluoromethyl)coumarin
- Boc
tert-butyloxycarbonyl
Footnotes
Accession ID
UniProtKB accession ID for human CYP3A4 is P08684.
Notes
The authors declare no competing financial interest.
Supplementary Information
Chemical synthesis, difference spectra for the ligand-bound forms, effect of CHAPS on the BFC debenzylase activity, ligand binding and heme reduction kinetics, X-ray data collection and refinement statistics, mass spectrometry and NMR data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Li AP, Kaminski DL, and Rasmussen A (1995) Substrates of human hepatic cytochrome P450 3A4. Toxicology 104, 1–8. [DOI] [PubMed] [Google Scholar]
- (2).Guengerich FP (1999) Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol 39, 1–17. [DOI] [PubMed] [Google Scholar]
- (3).Zhou SF (2008) Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr. Drug Metab 9, 310–322. [DOI] [PubMed] [Google Scholar]
- (4).Kempf DJ, Marsh KC, Kumar G, Rodrigues AD, Denissen JF, McDonald E, Kukulka MJ, Hsu A, Granneman GR, Baroldi PA, Sun E, Pizzuti D, Plattner JJ, Norbeck DW, and Leonard JM (1997) Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob. Agents Chemother 41, 654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Xu L, Liu H, Murray B, Callebaut C, Lee MS, Hong A, Strickley RG, Tsai L,K, Stray KM, Wang Y, Rhodes GR, and Desai MC (2010) Cobicistat (GS-9350): A potent and selective inhibitor of human CYP3A as a novel pharmacoenhancer. ACS Med. Chem. Lett 1, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sevrioukova IF, and Poulos TL (2010) Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. U S A 107, 18422–18427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sevrioukova IF (2017) High-level production and properties of the cysteine-depleted cytochrome P450 3A4. Biochemistry 56, 3058–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sevrioukova IF, and Poulos TL (2012) Interaction of human cytochrome P4503A4 with ritonavir analogs. Arch. Biochem. Biophys 520, 108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sevrioukova IF, and Poulos TL (2013) Pyridine-substituted desoxyritonavir is a more potent cytochrome P450 3A4 inhibitor than ritonavir. J. Med. Chem 56, 3733–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sevrioukova IF, and Poulos TL (2013) Dissecting cytochrome P450 3A4-ligand interactions using ritonavir analogues. Biochemistry 52, 4474–4481. [DOI] [PubMed] [Google Scholar]
- (11).Sevrioukova IF, and Poulos TL (2014) Ritonavir analogues as a probe for deciphering the cytochrome P450 3A4 inhibitory mechanism. Curr. Top. Med. Chem 14, 1348–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kaur P, Chamberlin AR, Poulos TL, and Sevrioukova IF (2016) Structure-based inhibitor design for evaluation of a CYP3A4 pharmacophore model. J. Med. Chem 59, 4210–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Samuels ER, and Sevrioukova IF (2018) Inhibition of human CYP3A4 by rationally designed ritonavir-like compounds: Impact and interplay of the side group functionalities. Mol. Pharm 15, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Karplus PA, and Diederichs K (2012) Linking crystallographic model and data quality. Science 336, 1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Section D 66, 213–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr. Section D 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Locuson CW, Hutzler JM, and Tracy TS (2007) Visible spectra of type II cytochrome P450-drug complexes: evidence that “incomplete” heme coordination is common. Drug Metab. Dispos 35, 614–622. [DOI] [PubMed] [Google Scholar]
- (19).Waldron TT, and Murphy KP (2003) Stabilization of proteins by ligand binding: application to drug screening and determination of unfolding energetics. Biochemistry 42, 5058–5064. [DOI] [PubMed] [Google Scholar]
- (20).Groftehauge MK, Hajizadeh NR, Swann MJ, and Pohl E (2015) Protein-ligand interactions investigated by thermal shift assays (TSA) and dual polarization interferometry (DPI). Acta Crystallogr. Section D 71, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Andreotti G, Monticelli M, and Cubellis MV (2015) Looking for protein stabilizing drugs with thermal shift assay. Drug Test. Anal 7, 831–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Davydov DR, Davydova NY, Sineva EV, Kufareva I, and Halpert JR (2013) Pivotal role of P450-P450 interactions in CYP3A4 allostery: the case of alpha-Naphthoflavone. Biochem. J 453, 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Koudriakova T, Iatsimirskaia E, Utkin I, Gangl E, Vouros P, Storozhuk E, Orza D, Marinina J, and Gerber N (1998) Metabolism of the human immunodeficiency virus protease inhibitors indinavir and ritonavir by human intestinal microsomes and expressed cytochrome P4503A4/3A5: mechanism-based inactivation of cytochrome P4503A by ritonavir. Drug Metab. Dispos 26, 552–561. [PubMed] [Google Scholar]
- (24).von Moltke LL, Durol AL, Duan SX, and Greenblatt DJ (2000) Potent mechanism-based inhibition of human CYP3A in vitro by amprenavir and ritonavir: comparison with ketoconazole. Eur. J. Clin. Pharmacol 56, 259–261. [DOI] [PubMed] [Google Scholar]
- (25).Obach RS, Walsky RL, and Venkatakrishnan K (2007) Mechanism-based inactivation of human cytochrome p450 enzymes and the prediction of drug-drug interactions. Drug Metab. Dispos 35, 246–55. [DOI] [PubMed] [Google Scholar]
- (26).Lin HL, D’Agostino J, Kenaan C, Calinski D, and Hollenberg PF (2013) The effect of ritonavir on human CYP2B6 catalytic activity: heme modification contributes to the mechanism-based inactivation of CYP2B6 and CYP3A4 by ritonavir. Drug Metab. Dispos 41, 1813–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rock BM, Hengel SM, Rock DA, Wienkers LC and Kunze KL (2014) Characterization of ritonavir-mediated inactivation of cytochrome P450 3A4. Mol. Pharmacol 86, 665–674. [DOI] [PubMed] [Google Scholar]
- (28).Sevrioukova IF, and Poulos TL (2015) Anion-dependent stimulation of CYP3A4 monooxygenase. Biochemistry 54, 4083–4096. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.