

Abstract
Correct synthesis and maintenance of functional tRNA 3′-CCA-ends is a crucial prerequisite for aminoacylation and must be achieved by the phylogenetically diverse group of tRNA nucleotidyltransferases. While numerous reports on the in vitro characterization exist, robust analysis under in vivo conditions is lacking. Here, we utilize Escherichia coli RNase T, a tRNA-processing enzyme responsible for the tRNA-CCA-end turnover, to generate an in vivo system for the evaluation of A-adding activity. Expression of RNase T results in a prominent growth phenotype that renders the presence of a CCA- or A-adding enzyme essential for cell survival in an E. coli Δcca background. The distinct growth fitness allows for both complementation and selection of enzyme variants in a natural environment. We demonstrate the potential of our system via detection of altered catalytic efficiency and temperature sensitivity. Furthermore, we select functional enzyme variants out of a sequence pool carrying a randomized codon for a highly conserved position essential for catalysis. The presented E. coli-based approach opens up a wide field of future studies including the investigation of tRNA nucleotidyltransferases from all domains of life and the biological relevance of in vitro data concerning their functionality and mode of operation.
INTRODUCTION
Due to their essential role in translation, the correct synthesis and maintenance of functional tRNA is indispensable for cell survival. tRNAs are transcribed as precursor molecules that undergo a set of processing events, including the removal of leader and trailer sequences to generate mature 5′ and 3′ ends (1,2).
At the 3′ site, a functional tRNA end includes the invariant CCA triplet that is either already encoded in the tRNA gene or is posttranscriptionally added by ATP(CTP):tRNA nucleotidyltransferase (CCA-adding enzyme; 3–5). Using a single binding pocket, these enzymes have a unique mechanism of polymerization without the involvement of a nucleic acid-based template. Yet, the incorporation of both CTP and ATP occurs at high fidelity (6–10). Within the polymerase β superfamily, tRNA nucleotidyltransferases can be divided into class I (archaeal) and class II (eukaryotic and bacterial) enzymes, depending on the mode of action as well as on the structural organization (5,7,9). Both classes share a common signature motif containing two highly conserved carboxylates (DxD or DxE; 11,12).
Besides the almost error-free CCA-addition, tRNA nucleotidyltransferases show additional surprising features. In a considerable number of bacterial organisms, enzymes with partial activities collaborate to synthesize CCA-ends. One enzyme incorporates the first two CMP residues but fails to add the terminal adenosine (CC-adding enzymes), while the second one exclusively catalyzes the addition of the terminal AMP residue (A-adding enzyme; 13–17). Furthermore, CCA-adding enzymes are involved in tRNA quality control, where they catalyze two rounds of CCA addition on defect and structurally instable tRNAs. The resulting CCACCA stretch is recognized as a degradation tag, leading to a rapid elimination of nonfunctional transcripts from the tRNA pool (18–20). A second quality control mechanism is based on polymerization kinetics, as tRNAs carrying a nick in the sugar-phosphate backbone represent quite inefficient substrates for CCA-addition (21).
Due to the need for continuous CCA restoration as well as their contribution to tRNA quality control, CCA-adding enzymes are also of crucial importance in organisms with encoded CCA-ends (22–24). In Escherichia coli, all tRNA genes encode for a CCA sequence (25), yet the disruption of the cca gene leads to a mild growth phenotype due to the depleted repair activity of damaged tRNA 3′ ends (25,26). Such a damage can be caused by spontaneous or enzymatic hydrolysis. In E. coli, the prominent hydrolytic activity against CCA-ends is RNase T, a single-strand specific 3′ to 5′ exoribonuclease involved in both the final steps of short 3′ trailer trimming and tRNA end turnover/quality control (27–30,19,24).
Because of the astonishing functional and evolutionary diversity of tRNA nucleotidyltransferases, it is of great interest to search for novel ways to compare and characterize these enzymes, preferentially under conditions that are close to their native cellular environment. So far, tRNA nucleotidyltransferases have been in vivo complemented in an E. coli-based approach that is dependent on a multilayered reporter system involving an introduced suppressor tRNA gene and a chromosomal lacZ reporter gene containing the cognate amber-codon for suppression (14,31,32). tRNA nucleotidyltransferases may also be investigated in a temperature-sensitive yeast mutant. However, a background deprived of endogenous CCA-adding enzyme can only be introduced upon incubation at the non-permissive temperature (33,34).
Here, we present a straight-forward E. coli-based system that enables the characterization of a broad range of tRNA nucleotidyltransferases under in vivo conditions. Utilizing the well-characterized RNase T activity against the 3′ terminal AMP residue of E. coli tRNAs, we demonstrate that recombinant RNase T expression prevents E. coli growth in the absence of an A-adding activity. Furthermore, we show that the system is applicable for enzymes from all domains of life, including the evaluation of catalytic efficiency, fidelity, temperature sensitivity and protein evolution.
MATERIALS AND METHODS
Cloning of vector constructs
The pET28a(+) vector for RNase T overexpression has been previously described (19). In the pETDuet-1 constructs, the coding region of RNase T was inserted between the EcoRV and AvrII restriction sites (in MCS-2) of pETDuet-1 (Novagen). The autonomous expression platform for cca was generated via PCR including the native cca promotor and flanking restriction sites for EcoRI and NotI. The initial module comprising the coding region of E. coli CCA-adding enzyme was introduced in place of MCS-1 via megaprimer mutagenesis using HiFi polymerase (PCR Biosystems) and cloned into the original vector background between the EcoRI and NotI sites. Coding regions of further tRNA nucleotidyltransferases of interest were also introduced via megaprimer mutagenesis, site-specific mutations were introduced via site-directed mutagenesis. Further information on the constitution of the designated CCA gene module is provided in the supplementary section (Supplementary Figure S1).
Bacterial strains and knockout generation
The bacterial strains used in this study were E. coli BL21(DE3) (Novagen) and JM109(DE3) (Promega). Disruption of the cca gene (cca::cam, Δcca) was achieved using the Quick & Easy E. coli gene deletion kit #K006 (Genebridges). E. coli TOP10 or NEB 5-alpha were used for cloning and mutagenesis experiments.
Media and growth conditions
Escherichia coli cells were grown at 37°C in LB supplemented with antibiotics at the following concentrations: kanamycin (kan) 30 μg ml–1; chloramphenicol (cam) 34 μg ml–1; ampicillin (amp) 100 μg ml–1. For the analysis of tRNA nucleotidyltransferase complementation in the dual expression system, transformed cells from a 500 μl suspension were pelleted and 450 μl of the supernatant were withdrawn. Cells were resuspended in the remaining volume and 5 μl were streaked in sectors onto LB agar containing ampicillin. For growth curve measurements, overnight cultures were inoculated in 5 ml fresh LB containing kanamycin and incubated to an OD600 of approximately 0.5. Subsequently, a 50 ml culture was inoculated to a final OD600 of 0.005. Cells were grown at 37°C with agitation and the optical density was assessed at the indicated time points. To determine RNase T tolerance, cells were plated onto agar gradient plates prepared with two layers of sloped LB-amp agar (containing 0.0025 % 2,3,5-triphenyltetrazolium chloride (TTC) to visualize cell viability (35,36)) where only the bottom layer was supplemented with 0.2 mM IPTG for induction of RNase T expression.
Preparation of semi-randomized plasmid library and in vivo selection
A mutagenic pETDuet-1 vector library with a semi-randomization at the highly conserved position D23 in the catalytic DxD motif (D21x22D23; x = any amino acid) of E. coli CCA-adding enzyme was generated via site-directed mutagenesis starting from an inactive A23 variant (DxA) in 20 cycles using HiFi polymerase (PCR Biosystems) and the primer pair 5′-AAGACAGAGNNTGGGTGGTGGTCGGCAG-3′ and 5′-ACCACCCANNCTCTGTCTTTGACCGGTAGCC-3′ (HPLC-grade purity, biomers.net). Following DpnI digest, E. coli NEB 5-alpha cells (NEB) were transformed with the DNA sample. An overnight culture was prepared to isolate the plasmid library and to introduce the product mix into E. coli JM109(DE3) Δcca. Resulting transformants were plated onto LB agar containing ampicillin and sequences from single colonies were analyzed after an overnight incubation at 37°C.
Drop plate assay
For comparative growth studies on agar plates, the drop plate method was performed (37). 5 ml LB-amp/cam were inoculated with respective E. coli cells and incubated at 30°C for 2 days with agitation. OD600 was adjusted to 1.0 and 5 μl per drop of a 10-fold dilution series in LB were plated onto LB-amp agar using a multichannel pipette. Agar plates were incubated for two days at the indicated temperatures.
Preparation of in vitro tRNA substrates
Radioactively labelled yeast tRNAPhe(GAA) substrate was synthesized via in vitro transcription in the presence of α-32P ATP (Hartmann Analytic) and purified as described (38,39).
in vitro nucleotide incorporation assay
For qualitative analysis of crude extract activity, 2 ml of an overnight culture were pelleted. Cells were resuspended in 500 μl 30 mM HEPES/KOH pH 7.6, 30 mM KCl, 6 mM MgCl2, 1 mM DTT and disrupted using a FastPrep-24 homogenizer (MP Biomedicals). The crude extract was cleared from cell debris and kept on ice until further use.
In a total reaction volume of 20 μl, 20 % (v/v) crude extract were incubated at 37°C in 30 mM HEPES/KOH pH 7.6, 30 mM KCl, 6 mM MgCl2, 2 mM DTT in the presence of 1 mM of each NTP and 0.25 μM radioactively labeled yeast tRNAPhe(GAA) or tRNAPhe(GAA)-CC, respectively. Prior to addition to the reaction mix, the RNA was heated for 5 min at 65°C and slowly cooled down to room temperature. After 1 h incubation, reactions were ethanol-precipitated, redissolved in 8 μl RNA-loading dye (10 mM Tris–HCl pH 7.6, 80 % formamide, 0.25 % bromophenol blue, 0.25 % xylencyanol) and separated on a 10 % denaturing PAGE. RNA products were visualized on a PhosphorImager (GE Healthcare).
RESULTS
Overexpression of RNase T in E. coli Δcca impairs growth fitness
tRNA end turnover describes the opposing actions of E. coli RNase T and CCA-adding enzyme regarding the removal and re-addition of tRNA 3′-CCA-ends (19,25,26,28,40,41). In the absence of the CCA-adding enzyme, a mild growth defect based on the inability to repair damaged tRNA 3′ ends is observed. However, sporadic revertants can emerge that circumvent this detrimental effect by down-regulating the level of RNase T-mediated hydrolysis (28,42). To further elicit the cca knockout phenotype, we introduced a pET28a(+) vector for recombinant expression of RNase T into E. coli BL21(DE3) Δcca. The growth curve of the corresponding culture indicated a prominent inhibitory effect on E. coli BL21(DE3) Δcca (Figure 1). Even without induction, the basal expression of RNase T resulted in a clearly visible growth defect. Moreover, RNase T induction by IPTG (0.05 mM) led to complete growth impairment of E. coli Δcca, while the corresponding wild type strain was able to compensate for the increased tRNA end turnover to a certain extent.
Figure 1.
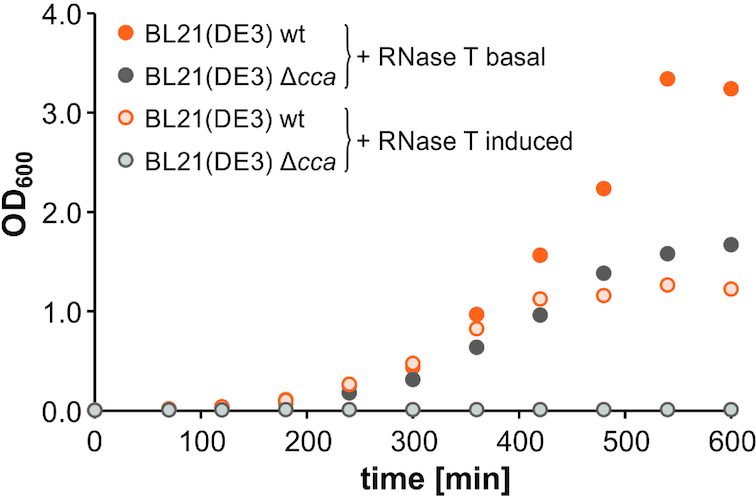
Effect of recombinantly expressed RNase T on growth behavior of E. coli. Growth was assessed for E. coli BL21(DE3) wild type (orange) and Δcca (gray) transformed with pET28a(+)_RNase T at basal (no IPTG) and induced (0.05 mM IPTG) RNase T expression. Growth curves are representative for experiments that were reproducible at least three times.
Survival under RNase T co-expression requires tRNA 3′ end repair activity
Next, we tested the impact on E. coli growth when both RNase T and tRNA nucleotidyltransferase are co-expressed from a single vector construct. The coding region of RNase T under the control of a T7 promotor was inserted into the multiple cloning site 2 (MCS-2) of pETDuet-1, while MCS-1 was replaced by an autonomous expression platform for tRNA nucleotidyltransferases under the control of the native E. coli cca promotor (CCA gene module, Figure 2A, see also Supplementary Figure S1). For further analysis, the strain JM109(DE3) was selected, as the ΔendA1 ΔrecA1 genotype leads to an improved DNA stability and overall plasmid quality. Transformation of E. coli JM109(DE3) Δcca with the pETDuet constructs resulted in a selective growth phenotype on agar plates (Figure 2B). In the absence of the two catalytically active carboxylates, E. coli CCA-adding enzyme (D21A/D23A, hereinafter referred to as AxA) is unable to repair tRNA 3′ ends which in turn prevented E. coli growth. Consistently, growth was not inhibited in a corresponding background of JM109(DE3) wild type, suggesting that the endogenous CCA-adding enzyme sufficiently compensated for the RNase T-mediated tRNA end hydrolysis. Similarly, the single replacement of aspartate at position 23 by alanine (DxA) prevented colony formation. Further, full growth complementation due to an efficient AMP incorporation was obtained by the introduction of the A-adding enzyme from Bacillus halodurans (Figure 2B). Intriguingly, the leaky basal expression of RNase T in the uninduced state was sufficient to generate the growth impairment in the absence of A-adding activity. In a corresponding in vitro nucleotide incorporation assay, all expected activities could be recovered from the respective crude extract preparations (Figure 2C).
Figure 2.

Dual expression of RNase T and tRNA nucleotidyltransferase in E. coli Δcca enforces selective growth phenotype. (A) Schematic presentation of the established in vivo system based on the dual expression of RNase T (orange) and tRNA nucleotidyltransferase (blue) from a pETDuet vector construct. In E. coli, both activities compete in the turnover of tRNA 3′ ends. (B) in vivo complementation requires efficient A-adding activity. Streak outs display E. coli JM109(DE3) Δcca (top) or wild type (bottom) transformed with the indicated pETDuet construct plated onto LB-amp. EcoCCA, E. coli CCA-adding enzyme; DxD, wild type enzyme with catalytically active carboxylates; AxA, inactive enzyme variant with catalytically active carboxylates replaced by alanine residues; DxA, inactive enzyme with catalytically active aspartate at position 23 replaced by alanine; BhaA, B. halodurans A-adding enzyme; Δcca, cca gene disruption; wt, wild type. (C) Radiolabeled yeast tRNAPhe(GAA) (top) or tRNAPhe(GAA)-CC (bottom), respectively, was incubated in the presence of 20 % (v/v) crude extract from different E. coli JM109(DE3) samples containing the indicated recombinantly expressed enzymes. Reaction products were resolved on a 10 % denaturing PAGE. Δcca, extract prepared from the strain carrying cca gene disruption; wt, extract from wild type strain; C, control incubation without extract, EcoCCA, extract from E. coli expressing recombinant CCA-adding enzyme; BhaA, E. coli extract expressing B. halodurans A-adding enzyme. (D) Utilization of dual expression as a selection system. A vector library with a semi-randomized GNN sequence at codon position 23 in the open reading frame for E. coli CCA-adding enzyme was generated. Introduction into E. coli JM109(DE3) Δcca resulted in colonies carrying vectors with GAT codons.
The dual expression method is suitable as an in vivo selection system to recapitulate protein evolution
The generation of a selective growth phenotype in E. coli Δcca allows to use the system for in vivo selection experiments. We performed a semi-randomization at the highly conserved position D23 in the E. coli CCA-adding enzyme. Together with D21, this position is involved in the coordination of the two metal ions essential for catalysis (43). We replaced the original aspartate codon by the partially randomized sequence ‘GNN’ in the open reading frame for this enzyme, leading to codons for aspartate, glutamate, glycine, valine or alanine, respectively. The randomization was performed on a catalytically inactive variant DxA to exclude selection of residual non-mutagenized original DxD sequences from the starting ORF. This pETDuet-based library was introduced into E. coli JM109(DE3) Δcca, and under the RNase T-based selection, all resulting colonies carried vectors with a GAT codon for aspartate at position 23, restoring the wild type activity (15 of 15 retrieved codons, Figure 2D, see also Supplementary Figure S2).
In vivo studies on enzyme variants with altered catalytic efficiency
During polymerization, CCA-adding enzymes switch their substrate specificity from CTP to ATP. For class II enzymes, a flexible loop region residing in the catalytic core acts as a lever and readjusts the nucleotide-recognizing amino acids within the nucleotide binding pocket (composition of the catalytic core motifs and the flexible loop element is shown in Supplementary Figure S3). This rearrangement of the templating residues switches the enzyme's specificity from CTP binding towards ATP (8,13,32,44,45). It was shown that the loop region from a phylogenetically closely related species can be readily exchanged with only minor impact on the enzyme's performance (45). Thus, replacing the loop of the E. coli CCA-adding enzyme by the corresponding region of the Wigglesworthia glossinidia enzyme (chimera EWEloop) sustained in vitro A-adding activity with only a slight reduction in the kcat value by 1.8-fold. In contrast, the replacement of conserved amino acids within the loop, like Y71 (in E. coli), by alanine reduced the kcat by 7.5-fold (45).
Based on these facts, we analyzed enzyme versions with different catalytic efficiencies in our in vivo approach. We introduced variants of the E. coli CCA-adding enzyme with the described alterations in the flexible loop region. On agar plates, both the tested EWEloop chimera as well as the E. coli CCA-adding enzyme variant Y71A can complement growth despite their varying catalytic properties (Figure 3A). For a more detailed estimation of the variant's impact on E. coli growth efficiency, we utilized the inducible RNase T expression system. The respective transformants were plated on agar containing an IPTG gradient from 0 to 0.2 mM (Figure 3A). The resulting gradient of expressed RNase T led to a gradual increase in the cellular stress level due to the induced tRNA 3′ end hydrolysis. Cells expressing wild type enzyme or EWEloop chimera exhibited a comparable growth behavior with a minimal inhibitory IPTG concentration of ∼0.1 mM. However, expression of the less efficient enzyme variant Y71A led to growth inhibition at ∼0.08 mM IPTG (Figure 3A). These findings are in good agreement with the reported turnover numbers (45) and thus illustrate that catalytic features of various enzyme variants can be easily displayed and compared in our complementation experiments.
Figure 3.

Growth defect depends on RNase T activity and CCA-adding enzyme fidelity. (A) RNase T expression level affects cell survival. Left: Streak outs of E. coli JM109(DE3) Δcca transformed with pETDuet constructs containing the coding regions of indicated enzymes. All introduced variants from E. coli CCA-adding enzyme show growth at conditions of RNase T basal expression. Right: E. coli growth was examined on LB-amp in an IPTG gradient from 0 to 0.2 mM, resulting in a gradual increase of RNase T-mediated tRNA end hydrolysis. The difference in RNase T tolerance is indicated by ‘Δ’. EcoCCA, E. coli CCA-adding enzyme; DxD, wild type enzyme with catalytically active carboxylates; AxA, inactive enzyme form with catalytically active carboxylates replaced by alanine residues; EWEloop, chimeric E. coli CCA-adding enzyme with flexible loop region from the corresponding W. glossinidia enzyme; wt, wild type E. coli CCA-adding enzyme with catalytically active DxD motif. (B) The described backup mechanism for CCA-addition by an error-prone enzyme is not sufficient for cell survival. Left: Streak outs of E. coli JM109(DE3) Δcca (top) or wild type (bottom) transformed with indicated constructs on LB-amp. JM109(DE3) Δcca expressing a CCA-adding enzyme with altered amino acid template EDxxA (EcoCCA R134A) does not survive in the presence of basally expressed RNase T. Right: Model for the interplay between an erroneous CCA-adding enzyme EcoCCA R134A and RNase T in quality control. The erroneous CCA-adding enzyme incorporates the terminal residue of the CCA-end at random, resulting in CCA, CCG, CCU and CCC ends. Due to the substrate preference of RNase T for purine ends, only those tRNAs are trimmed at their 3′ end, while tRNA-CCU and tRNA-CCC are not. The combination of RNase T activity and the erroneous nucleotide incorporation by EcoCCA R134A leads to an accumulation of non-functional tRNAs, resulting in a fatal situation for the cell.
As a prerequisite for the remarkable fidelity of CCA addition, the class II enzymes carry a set of highly conserved amino acids EDxxR (x, any residue) in the nucleotide binding pocket (Motif D, Supplementary Figure S3) that form Watson-Crick-like hydrogen bonds to the base moieties of CTP and ATP, respectively (8). The main interaction partner determining nucleotide specificity is the arginine residue, and a replacement by alanine leads to the incorporation of any of the four nucleotides (46,47). Yet, in vitro data show that the E. coli enzyme carrying the corresponding R134A alteration in the nucleotide binding pocket is able to synthesize proper CCA-ends, as only a tRNA primer carrying correctly added residues is further elongated by the enzyme, while primers ending in misincorporated residues are rejected. This contribution of the growing tRNA 3′ end to the fidelity of the polymerization resulted in a considerable amount of tRNA transcripts and was interpreted as a possible backup mechanism for CCA addition (47). Hence, it is a pertinent question as to whether this backup system for fidelity is also relevant in vivo. To address this for the terminal A incorporation, we introduced a plasmid expressing the E. coli R134A variant enzyme in our complementation system. However, even after prolonged incubation of the corresponding cells for up to 96 h, no colony growth was visible (Figure 3B). These data clearly indicate that the described backup mechanism is only traceable in vitro, while it does not contribute to cell survival in vivo.
tRNA nucleotidyltransferases from various organisms can complement in the in vivo system
To further estimate the applicability of the RNase T-based complementation system, we tested tRNA nucleotidyltransferases from all domains of life. Expression of the human CCA-adding enzyme also led to a growth restoration, although at a slightly lower rate (Figure 4A). Representing a member of class II tRNA nucleotidyltransferases, this enzyme has a catalytic mechanism of CCA polymerization very similar to that of the E. coli enzyme. Archaea, however, carry class I CCA-adding enzymes of very different shape and mode of action as well as nucleotide recognition, and it was unclear whether these highly different enzymes can complement for the terminal A-addition in an organism that usually carries a class II enzyme. Hence, we introduced the CCA-adding enzyme from the archaeon Methanosarcina barkeri in E. coli (Figure 4A). This very different enzyme form also led to similar growth behavior as the tested class II enzymes, indicating a successful complementation of the original activity in E. coli. Thus, our system has the potential for further investigating a broad phylogenetic range of tRNA nucleotidyltransferases.
Figure 4.

The E. coli system allows for the in vivo complementation of tRNA nucleotidyltransferases from all domains of life. (A) Streak outs of E. coli JM109(DE3) Δcca transformed with pETDuet constructs containing the coding regions of indicated enzymes. Both eukaryotic and archaeal enzymes can complement the lacking CCA-adding activity in the E. coli-based system. EcoCCA, E. coli CCA-adding enzyme; HsaCCA, Homo sapiens CCA-adding enzyme; MbaCCA, Methanosarcina barkeri CCA-adding enzyme; DxD, catalytically active carboxylates; AxA, catalytically active carboxylates replaced by alanines. (B) Drop plate assay to compare the growth of JM109(DE3) Δcca with pETDuet constructs containing coding regions of either wild type or L166S variant from human CCA-adding enzyme at indicated temperatures. Growth of cells with temperature-sensitive variant L166S is inhibited at increased temperatures. wt, wild type.
For the human CCA-adding enzyme, several mutations in the cca gene (TRNT1) resulting in partial loss-of-function were identified that correlate with certain hereditary diseases (24,34,48–52). For several of these disease-linked enzyme variants, an increased temperature sensitivity was observed, where L166S showed the most severe effect (Tm of 35.4°C compared to 42.4°C for the wild type enzyme; 53). To investigate whether the decreased thermal stability of this mutant enzyme form can be monitored in our in vivo system, temperature-dependent growth of E. coli expressing the human L166S variant was tested. In a drop plate approach, E. coli Δcca with both the wild type and the L166S version of the human CCA-adding enzyme exhibit a comparable growth behavior at the non-critical incubation temperatures 25 and 30°C (Figure 4B). In contrast and in accordance with reported in vitro data (53), the growth ability of E. coli Δcca expressing variant L166S is dramatically reduced at 37°C and completely abolished at 42°C (Figure 4B). Hence, our complementation system represents a useful sensor for thermostability of different CCA-adding enzymes within a natural environment, such as the evaluation of pathologically relevant variants from human tRNA nucleotidyltransferase.
DISCUSSION
Within the past decade, tRNA nucleotidyltransferases have emerged from sole processing enzymes that de novo synthesize and maintain a tRNA’s CCA-end to crucial scrutinizers in tRNA quality control for all domains of life (10,19,20,23,24,54). The detailed investigation of those highly versatile properties requires reliable assays to further elucidate and evaluate such surprising protein characteristics. In the present study, we established a simple procedure for the in vivo analysis of terminal AMP-incorporation based on an inducible, RNase T-mediated deactivation of E. coli tRNA 3′-CCA-ends. As RNase T action generates a tRNA-CC end with 3′ OH group, the system provides a tRNA substrate pool that is accessible to all tRNA nucleotidyltransferases of interest with A-adding activity.
Interestingly, tRNA nucleotidyltransferases from different organisms were able to complement for A-addition in our in vivo system, and it convincingly shows that the catalytic activity of a class I enzyme efficiently replaces the endogenous class II-mediated A-incorporation, indicating that activity and functionality of these substantially different nucleotidyltransferases are very similar and compatible. The system is also suitable for screening a randomized sequence pool for active enzyme variants, as it was shown for the catalytically important D23 position. Interestingly, we obtained only GAT codons in 15 selected colonies, while the second aspartate codon (GAC) was not observed. A possible explanation for this result is the codon usage in E. coli K12 that prefers GAT over GAC (65 % versus 35 %; see http://www.kazusa.or.jp/codon/). In addition, a screen of almost 200 tRNA nucleotidyltransferase sequences in seven genera of Enterobacteria revealed that the first catalytically active aspartate is encoded by GAC or GAT, while the second position (corresponding to D23 in E. coli), is exclusively represented by GAT. This indicates that the codon usage at the second aspartate site is not random but under selective pressure and seems to play an important role in translation so that the synonymous codons are not interchangeable at this position. Such a codon bias might be important for a correct cotranslational folding of the nascent protein, as it is frequently observed in many ORFs in all domains of life (55–57).
The complementation system can be further used to investigate in vitro findings concerning A-addition under the more stringent in vivo conditions, where the enzyme has to be active in the presence of additional and competing activities acting on tRNA. We addressed the relevance of the in vitro observed backup function in an E. coli CCA-adding enzyme where the nucleotide-recognizing arginine residue is replaced by alanine in the binding pocket (Supplementary Figure S3). In vitro, this variant has a high error rate (46), but is able to synthesize proper CCA-ends to a certain extent, as only primers with correctly added residues (C or CC) are accurately positioned and elongated (47). Our data show that the corresponding transformants are not viable, indicating that under in vivo conditions, this mechanism alone is not sufficient for a correct A incorporation (Figure 3B). In tRNA quality control, the terminal A residue is subjected to a frequent turnover, where RNase T removes this residue from the CCA-end (19,28,58). During restoration of this position, the compromised CCA-adding enzyme adds any of the four possible residues, so that at least a portion of the tRNAs should carry a functional CCA-end (46,47). However, RNase T has a strong substrate preference for RNA ending with purines, while it is almost completely inactive on pyrimidine ends, particularly on C residues (30). This enzymatic inhibition probably leads to an accumulation of nonfunctional deleterious tRNAs carrying CCC or CCU-ends, while the amount of tRNAs with correctly restored CCA-ends continuously decreases (Figure 3B). Hence, the RNase T-based 3′ end turnover and tRNA quality control requires a high fidelity CCA-adding enzyme that exclusively restores functional tRNA ends. Thus, although the in vitro data are very helpful to understand and clarify the mechanistic basis of proper CCA-addition and substrate selection, the biological relevance of such a backup mechanism can only be tested in a highly selective in vivo read-out system where such a limited enzymatic fidelity is not sufficient for tRNA quality control or repair and, consequently, cell survival.
Yet, one limitation of the E. coli-based assay is that the tested enzymes must be active within the temperature range of growth of the mesophilic gammaproteobacterium. While we were able to cover temperatures from 25 to 42°C, this range is not sufficient for thermophilic representatives, as the CCA-adding enzyme from Archaeoglobus fulgidus (thriving at 60–95°C; 59) did not lead to complementation (and growth) in E. coli (Supplementary Figure S4)—a fact consistent with the observation that the recombinant enzyme is not active at moderate temperatures in vitro. Hence, the particular applicability is defined by the temperature range of the host strain's growth ability.
Because of the essential function of the CCA-adding enzyme in eukaryotes (eukaryotic tRNA genes do not encode the CCA-end), it is not possible to generate complete cca knockouts for these organisms (33). Instead, a temperature-sensitive yeast mutant could be acquired as a host strain for complementation assays (33,34). Because of the necessity to grow the respective mutant cells at the non-permissive temperature (i.e. 37°C; 33), this approach is not suitable for the evaluation of temperature-dependent protein features. In the E. coli complementation system, however, temperature sensitivity can be readily detected. To our knowledge, the presented data demonstrate for the first time the in vivo relevance of in vitro findings regarding the thermal denaturation of human disease-associated enzyme variants (53). This easy-to-use detection method may prove useful as a rapid test for human disease manifestations associated with a cca mutation.
A great advantage of this dual expression system is the convenient and straight-forward procedure. A single transformation step is sufficient to simultaneously introduce both the tRNA nucleotidyltransferase of interest and the RNase T-mediated tRNA 3′ end-directed selective pressure. Moreover, it should be possible to replace the T7-based expression of RNase T by leaky or tunable endogenous E. coli promoters (like the lac or the ara promoters; 60,61), rendering the system applicable in almost every E. coli strain. In contrast, the previously described amber codon suppression system requires a particular E. coli strain CA244 that is equipped with a chromosomal lacZ mutant gene with amber codon and, additionally, the recombinant expression of an individual tRNATyrsu3+ (1,31). Moreover, the utilization of a suppressor tRNA does not exclude the potentially detrimental off-target effects caused by occasional read through of endogenous stop codons (1,62). Accordingly, the use of a single-copy plasmid vector is favored for suppressor tRNA transcription to minimize the occurrence of such effects (1,31). Further, the variation of codon suppression efficiency can vary (63), making it difficult to exclude unwanted false-positive events. In fact, previous studies report residual suppressor activity despite the disruption of the cca gene (31) which was explained by the side reactions of both poly(A) polymerase I and polynucleotide phosphorylase that are associated with residual tRNA end repair in the absence of CCA-adding enzyme (26,64). In the presented complementation system, however, such residual activities are not sufficient to overcome the increased hydrolysis of tRNA 3′ ends, as in principle the whole tRNA pool is affected by RNase T-mediated CCA degradation. Thus, the introduced selective pressure provides a robust and useful tool that allows for further investigations of tRNA nucleotidyltransferases, like the in vivo screening and selection for directed enzyme evolution via randomized, site-directed mutagenesis or the evaluation of pathogenic point mutations described for the human CCA-adding enzyme gene.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Tobias Friedrich, Bianca Lang and Isabelle Wachsmann for expert technical assistance and Zoya Ignatova for valuable discussion. We acknowledge support from the German Research Foundation (DFG) and Leipzig University within the program of Open Access Publishing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft (DFG) [MO 634/8-2] and [MO 634/12-1]. Funding for open access charge: DFG Open Access funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Reuven N.B., Deutscher M.P.. Multiple exoribonucleases are required for the 3′ processing of Escherichia coli tRNA precursors in vivo. FASEB J. 1993; 7:143–148. [DOI] [PubMed] [Google Scholar]
- 2. Hopper A.K. Transfer RNA post-transcriptional processing, turnover, and subcellular dynamics in the yeast Saccharomyces cerevisiae. Genetics. 2013; 194:43–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sprinzl M., Cramer F.. The -C-C-A end of tRNA and its role in protein biosynthesis. Prog. Nucleic Acid Res. Mol. Biol. 1979; 22:1–69. [DOI] [PubMed] [Google Scholar]
- 4. Deutscher M.P. Ribonucleases, tRNA nucleotidyltransferase, and the 3′ processing of tRNA. Prog. Nucleic Acid Res. Mol. Biol. 1990; 39:209–240. [DOI] [PubMed] [Google Scholar]
- 5. Betat H., Rammelt C., Mörl M.. tRNA nucleotidyltransferases: ancient catalysts with an unusual mechanism of polymerization. Cell. Mol. Life Sci. 2010; 67:1447–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deutscher M.P. Boyer PD. 7 tRNA nucleotidyltransferase. The Enzymes. Nucleic Acids Part B. 1982; NY: Academic Press; 183–215. [Google Scholar]
- 7. Yue D., Weiner A.M., Maizels N.. The CCA-adding enzyme has a single active site. J. Biol. Chem. 1998; 273:29693–29700. [DOI] [PubMed] [Google Scholar]
- 8. Li F., Xiong Y., Wang J., Cho H.D., Tomita K., Weiner A.M., Steitz T.A.. Crystal structures of the Bacillus stearothermophilus CCA-adding enzyme and its complexes with ATP or CTP. Cell. 2002; 111:815–824. [DOI] [PubMed] [Google Scholar]
- 9. Weiner A.M. tRNA maturation: RNA polymerization without a nucleic acid template. Curr. Biol. 2004; 14:R883–R885. [DOI] [PubMed] [Google Scholar]
- 10. Hou Y.-M. CCA addition to tRNA: implications for tRNA quality control. IUBMB Life. 2010; 62:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aravind L., Koonin E.V.. DNA polymerase beta-like nucleotidyltransferase superfamily. Identification of three new families, classification and evolutionary history. Nucleic Acids Res. 1999; 27:1609–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yue D., Maizels N., Weiner A.M.. CCA-adding enzymes and poly(A) polymerases are all members of the same nucleotidyltransferase superfamily: characterization of the CCA-adding enzyme from the archaeal hyperthermophile Sulfolobus shibatae. RNA. 1996; 2:895–908. [PMC free article] [PubMed] [Google Scholar]
- 13. Neuenfeldt A., Just A., Betat H., Mörl M.. Evolution of tRNA nucleotidyltransferases: a small deletion generated CC-adding enzymes. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:7953–7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomita K., Weiner A.M.. Collaboration between CC- and A-adding enzymes to build and repair the 3′-terminal CCA of tRNA in Aquifex aeolicus. Science. 2001; 294:1334–1336. [DOI] [PubMed] [Google Scholar]
- 15. Tomita K., Weiner A.M.. Closely related CC- and A-adding enzymes collaborate to construct and repair the 3′-terminal CCA of tRNA in Synechocystis sp. and Deinococcus radiodurans. J. Biol. Chem. 2002; 277:48192–48198. [DOI] [PubMed] [Google Scholar]
- 16. Tretbar S., Neuenfeldt A., Betat H., Mörl M.. An inhibitory C-terminal region dictates the specificity of A-adding enzymes. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:21040–21045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bralley P., Chang S.A., Jones G.H.. A phylogeny of bacterial RNA nucleotidyltransferases: Bacillus halodurans contains two tRNA nucleotidyltransferases. J. Bacteriol. 2005; 187:5927–5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuhn C.-D., Wilusz J.E., Zheng Y., Beal P.A., Joshua-Tor L.. On-enzyme refolding permits small RNA and tRNA surveillance by the CCA-adding enzyme. Cell. 2015; 160:644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wellner K., Czech A., Ignatova Z., Betat H., Mörl M.. Examining tRNA 3′-ends in Escherichia coli. Teamwork between CCA-adding enzyme, RNase T, and RNase R. RNA. 2018; 24:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilusz J.E., Whipple J.M., Phizicky E.M., Sharp P.A.. tRNAs marked with CCACCA are targeted for degradation. Science. 2011; 334:817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dupasquier M., Kim S., Halkidis K., Gamper H., Hou Y.-M.. tRNA integrity is a prerequisite for rapid CCA addition: implication for quality control. J. Mol. Biol. 2008; 379:579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hopper A.K., Huang H.-Y.. Quality control pathways for nucleus-encoded eukaryotic tRNA biosynthesis and subcellular trafficking. Mol. Cell Biol. 2015; 35:2052–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Betat H., Mörl M.. The CCA-adding enzyme: a central scrutinizer in tRNA quality control. Bioessays. 2015; 37:975–982. [DOI] [PubMed] [Google Scholar]
- 24. Wellner K., Betat H., Mörl M.. A tRNA’s fate is decided at its 3′ end: Collaborative actions of CCA-adding enzyme and RNases involved in tRNA processing and degradation. Biochim. Biophys. Acta. 2018; 1861:433–441. [DOI] [PubMed] [Google Scholar]
- 25. Zhu L., Deutscher M.P.. tRNA nucleotidyltransferase is not essential for Escherichia coli viability. EMBO J. 1987; 6:2473–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reuven N.B., Zhou Z., Deutscher M.P.. Functional overlap of tRNA nucleotidyltransferase, Poly(A) polymerase I, and polynucleotide phosphorylase. J. Biol. Chem. 1997; 272:33255–33259. [DOI] [PubMed] [Google Scholar]
- 27. Deutscher M.P., Marlor C.W.. Purification and characterization of Escherichia coli RNase T. J. Biol. Chem. 1985; 260:7067–7071. [PubMed] [Google Scholar]
- 28. Deutscher M.P., Marlor C.W., Zaniewski R.. RNase T is responsible for the end-turnover of tRNA in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1985; 82:6427–6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li Z., Pandit S., Deutscher M.P.. 3′ exoribonucleolytic trimming is a common feature of the maturation of small, stable RNAs in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:2856–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zuo Y., Deutscher M.P.. The physiological role of RNase T can be explained by its unusual substrate specificity. J. Biol. Chem. 2002; 277:29654–29661. [DOI] [PubMed] [Google Scholar]
- 31. Reuven N.B., Deutscher M.P.. Substitution of the 3′ terminal adenosine residue of transfer RNA in vivo. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:4350–4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Toh Y., Takeshita D., Numata T., Fukai S., Nureki O., Tomita K.. Mechanism for the definition of elongation and termination by the class II CCA-adding enzyme. EMBO J. 2009; 28:3353–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aebi M., Kirchner G., Chen J.Y., Vijayraghavan U., Jacobson A., Martin N.C., Abelson J.. Isolation of a temperature-sensitive mutant with an altered tRNA nucleotidyltransferase and cloning of the gene encoding tRNA nucleotidyltransferase in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1990; 265:16216–16220. [PubMed] [Google Scholar]
- 34. Chakraborty P.K., Schmitz-Abe K., Kennedy E.K., Mamady H., Naas T., Durie D., Campagna D.R., Lau A., Sendamarai A.K., Wiseman D.H. et al.. Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD). Blood. 2014; 124:2867–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bochner B.R., Savageau M.A.. Generalized indicator plate for genetic, metabolic, and taxonomic studies with microorganisms. Appl. Environ. Microbiol. 1977; 33:434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hurwitzx S.J., McCarthy T.J.. 2,3,5-Triphenyltetrazolium chloride as a novel tool in germicide dynamics. J. Pharm. Sci. 1986; 75:912–916. [DOI] [PubMed] [Google Scholar]
- 37. Chen C.-Y., Nace G.W., Irwin P.L.. A 6×6 drop plate method for simultaneous colony counting and MPN enumeration of Campylobacter jejuni, Listeria monocytogenes, and Escherichia coli. J. Microbiol. Methods. 2003; 55:475–479. [DOI] [PubMed] [Google Scholar]
- 38. Schürer H., Lang K., Schuster J., Mörl M.. A universal method to produce in vitro transcripts with homogeneous 3′ ends. Nucleic Acids Res. 2002; 30:e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mörl M., Lizano E., Willkomm D.K., Hartmann R.K.. Hartmann RK, Bindereif A., Schön A, Westhof E. Production of RNAs with Homogeneous 5′ and 3′ Ends. Handbook of RNA Biochemistry. 2005; Germany: Wiley-VCH, Weinheim; 22–35. [Google Scholar]
- 40. Deutscher M.P., Evans J.A.. Transfer RNA nucleotidyltransferase repairs all transfer RNAs randomly. J. Mol. Biol. 1977; 109:593–597. [DOI] [PubMed] [Google Scholar]
- 41. Padmanabha K.P., Deutscher M.P.. RNase T affects Escherichia coli growth and recovery from metabolic stress. J. Bacteriol. 1991; 173:1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deutscher M.P., Foulds J., McClain W.H.. Transfer ribonucleic acid nucleotidyl-transferase plays an essential role in the normal growth of Escherichia coli and in the biosynthesis of some bacteriophage T4 transfer ribonucleic acids. J. Biol. Chem. 1974; 249:6696–6699. [PubMed] [Google Scholar]
- 43. Steitz T.A., Jager J., Joyce C.M.. A unified polymerase mechanism for nonhomologous DNA and RNA polymerases. Science. 1994; 266:2022–2025. [DOI] [PubMed] [Google Scholar]
- 44. Just A., Butter F., Trenkmann M., Heitkam T., Mörl M., Betat H.. A comparative analysis of two conserved motifs in bacterial poly(A) polymerase and CCA-adding enzyme. Nucleic Acids Res. 2008; 36:5212–5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hoffmeier A., Betat H., Bluschke A., Günther R., Junghanns S., Hofmann H.-J., Mörl M.. Unusual evolution of a catalytic core element in CCA-adding enzymes. Nucleic Acids Res. 2010; 38:4436–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cho H.D., Verlinde C.L.M.J., Weiner A.M.. Reengineering CCA-adding enzymes to function as (U,G)- or dCdCdA-adding enzymes or poly(C,A) and poly(U,G) polymerases. Proc. Natl. Acad. Sci. U.S.A. 2006; 104:54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lizano E., Scheibe M., Rammelt C., Betat H., Mörl M.. A comparative analysis of CCA-adding enzymes from human and E. coli: differences in CCA addition and tRNA 3′-end repair. Biochimie. 2008; 90:762–772. [DOI] [PubMed] [Google Scholar]
- 48. Sasarman F., Thiffault I., Weraarpachai W., Salomon S., Maftei C., Gauthier J., Ellazam B., Webb N., Antonicka H., Janer A. et al.. The 3′ addition of CCA to mitochondrial tRNASer(AGY) is specifically impaired in patients with mutations in the tRNA nucleotidyl transferase TRNT1. Hum. Mol. Genet. 2015; 24:2841–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. DeLuca A.P., Whitmore S.S., Barnes J., Sharma T.P., Westfall T.A., Scott C.A., Weed M.C., Wiley J.S., Wiley L.A., Johnston R.M. et al.. Hypomorphic mutations in TRNT1 cause retinitis pigmentosa with erythrocytic microcytosis. Hum. Mol. Genet. 2016; 25:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hull S., Malik A.N.J., Arno G., Mackay D.S., Plagnol V., Michaelides M., Mansour S., Albanese A., Brown K.T., Holder G.E. et al.. Expanding the phenotype of TRNT1-Related immunodeficiency to include childhood cataract and inner retinal dysfunction. JAMA Ophthalmol. 2016; 134:1049–1053. [DOI] [PubMed] [Google Scholar]
- 51. Liwak-Muir U., Mamady H., Naas T., Wylie Q., McBride S., Lines M., Michaud J., Baird S.D., Chakraborty P.K., Holcik M.. Impaired activity of CCA-adding enzyme TRNT1 impacts OXPHOS complexes and cellular respiration in SIFD patient-derived fibroblasts. Orphanet. J. Rare Dis. 2016; 11:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Frans G., Moens L., Schaballie H., Wuyts G., Liston A., Poesen K., Janssens A., Rice G.I., Crow Y.J., Meyts I. et al.. Homozygous N-terminal missense mutation in TRNT1 leads to progressive B-cell immunodeficiency in adulthood. J. Allergy Clin. Immunol. 2017; 139:360–363. [DOI] [PubMed] [Google Scholar]
- 53. Leibovitch M., Hanic-Joyce P.J., Joyce P.B.M.. In vitro studies of disease-linked variants of human tRNA nucleotidyltransferase reveal decreased thermal stability and altered catalytic activity. Biochim. Biophys. Acta. 2018; 1866:527–540. [DOI] [PubMed] [Google Scholar]
- 54. Wilusz J.E. Controlling translation via modulation of tRNA levels. Wiley Interdiscip. Rev. RNA. 2015; 6:453–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Angov E. Codon usage: nature's roadmap to expression and folding of proteins. Biotechnol. J. 2011; 6:650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chaney J.L., Clark P.L.. Roles for synonymous codon usage in protein biogenesis. Annu. Rev. Biophys. 2015; 44:143–166. [DOI] [PubMed] [Google Scholar]
- 57. Komar A.A. A pause for thought along the co-translational folding pathway. Trends Biochem. Sci. 2009; 34:16–24. [DOI] [PubMed] [Google Scholar]
- 58. Deutscher M.P., Marlor C.W., Zaniewski R.. Ribonuclease T: new exoribonuclease possibly involved in end-turnover of tRNA. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:4290–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stetter K.O. Archaeoglobus fulgidus gen. nov., sp. nov.: a new taxon of extremely thermophilic archaebacteria. Syst. Appl. Microbiol. 1988; 10:172–173. [Google Scholar]
- 60. Rosano G.L., Ceccarelli E.A.. Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol. 2014; 5:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Marschall L., Sagmeister P., Herwig C.. Tunable recombinant protein expression in E. coli: promoter systems and genetic constraints. Appl. Microbiol. Biotechnol. 2016; 101:501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Herring C.D., Blattner F.R.. Global transcriptional effects of a suppressor tRNA and the inactivation of the regulator frmR. J. Bacteriol. 2004; 186:6714–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kleina L.G., Masson J.M., Normanly J., Abelson J., Miller J.H.. Construction of Escherichia coli amber suppressor tRNA genes. II. Synthesis of additional tRNA genes and improvement of suppressor efficiency. J. Mol. Biol. 1990; 213:705–717. [DOI] [PubMed] [Google Scholar]
- 64. Mohanty B.K., Kushner S.R.. Polynucleotide phosphorylase functions both as a 3′ right-arrow 5′ exonuclease and a poly(A) polymerase in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:11966–11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.