

Abstract
The damage to liver mitochondria is universally observed in both humans and animal models after excessive alcohol consumption. Acute alcohol treatment has been shown to stimulate calcium (Ca2+) release from internal stores in hepatocytes. The resultant increase in cytosolic Ca2+ is expected to be accumulated by neighboring mitochondria, which could potentially lead to mitochondrial Ca2+ overload and injury. Our data indicate that total and free mitochondrial matrix Ca2+ levels are, indeed, elevated in hepatocytes isolated from alcohol-fed rats compared with their pair-fed control littermates. In permeabilized hepatocytes, the rates of mitochondrial Ca2+ uptake were substantially increased after chronic alcohol feeding, whereas those of mitochondrial Ca2+ efflux were decreased. The changes in mitochondrial Ca2+ handling could be explained by an up-regulation of the mitochondrial Ca2+ uniporter and loss of a cyclosporin A-sensitive Ca2+ transport pathway. In intact cells, hormone-induced increases in mitochondrial Ca2+ declined at slower rates leading to more prolonged elevations of matrix Ca2+ in the alcohol-fed group compared with controls. Moreover, treatment with submaximal concentrations of Ca2+-mobilizing hormones markedly increased the levels of mitochondrial reactive oxygen species (ROS) in hepatocytes from alcohol-fed rats, but did not affect ROS levels in controls. The changes in mitochondrial Ca2+ handling are expected to buffer and attenuate cytosolic Ca2+ increases induced by acute alcohol exposure or hormone stimulation. However, these alterations in mitochondrial Ca2+ handling may also lead to Ca2+ overload during cytosolic Ca2+ increases, which may stimulate the production of mitochondrial ROS, and thus contribute to alcohol-induced liver injury.
Introduction
Previous studies have provided ample evidence linking excessive alcohol consumption with abnormalities in liver mitochondria morphology and function in animal models [1,2] as well as in both alcoholic [3,4] and nonalcoholic human volunteers [5]. Mitochondrial elongation and swelling is an early manifestation of alcohol-induced liver injury, which precedes or is concomitant with alcoholic steatosis [6–8]. Moreover, the abundance and activity of mitochondrial respiratory complexes (I, III and IV) are decreased by chronic ethanol feeding resulting in lower rates of oxidative phosphorylation and lower levels of cellular ATP production [1,2,9–16].
The mechanism of alcohol-induced mitochondrial dysfunction and liver damage has not been fully elucidated. Ethyl alcohol can perturb multiple signaling pathways in hepatocytes including the mobilization of internal Ca2+ stores by activating receptor-coupled phospholipase C (PLC) and increasing the formation of inositol 1,4–5-trisphophate (InsP3) [17–19]. Frequent and prolonged ethanol-induced increases in cytosolic Ca2+ concentration ([Ca2+]i) are expected to increase the risk of mitochondrial damage, as the subsequent accumulation of Ca2+ into the matrix could stimulate the formation of mitochondrial ROS [20]. Prior exposure to aberrant Ca2+ increases during chronic alcohol feeding could explain why mitochondria isolated from these animals are more sensitive to Ca2+-induced swelling and injury [21,22].
Mitochondria accumulate Ca2+ electrophoretically through the Ca2+ uniporter, which is a multiprotein signaling complex containing the mitochondrial Ca2+ uniporter (MCU) channel and accessory regulatory subunits [23–25]. The rapid transfer of Ca2+ increases from the cytosol to the mitochondria plays an important role in regulating hepatic energy metabolism [26–28]. However, high sustained levels of mitochondrial Ca2+ coupled to increased production of ROS can also activate a high conductance channel in the inner membrane known as the mitochondrial permeability transition pore (mPTP). The opening of mPTP disrupts the integrity of the mitochondrial inner membrane leading to depolarization of the mitochondrial membrane potential (ΔΨm), matrix swelling, reorganization of cristae membranes, and ultimately irreversible mitochondrial damage and cell death [29,30]. The mitochondrial swelling observed after excessive alcohol consumption suggests that mitochondrial Ca2+ homeostasis and/or the threshold for inducing mitochondrial permeability transition may be altered by sustained exposure to ethanol.
In the present study, we investigated the effects of long-term alcohol feeding on mitochondrial Ca2+ handling in rat hepatocytes. Our data indicate that the rates of mitochondrial Ca2+ accumulation are increased after chronic alcohol feeding, whereas those of mitochondrial Ca2+ efflux are impaired. The changes in mitochondrial Ca2+ handling could be attributed to increased levels of MCU protein and the loss of a cyclosporin A (CsA)-sensitive mitochondrial Ca2+ transport pathway in hepatocytes from alcohol-fed animals. As expected, the resting levels of both total and free mitochondrial matrix Ca2+ levels were elevated after chronic alcohol feeding. Moreover, hormone-induced mitochondrial Ca2+ spikes decayed at significantly slower rates, which led to more prolonged increases of matrix Ca2+ in the alcohol-fed group compared with controls. We propose that the changes in mitochondrial Ca2+ handling could be a response to alcohol induced increases in cytosolic Ca2+. Increasing Ca2+ uptake and retention in the mitochondria may protect the hepatocyte from cytoplasmic Ca2+ overload and cytotoxicity. However, this protective mechanism may lead to matrix Ca2+ overload and higher rates of mitochondrial ROS formation that could contribute to alcohol-induced mitochondrial injury.
Experimental
Materials
Chemicals were purchased from BDH chemicals or Sigma-Aldrich, unless otherwise specified. Gamitrinib-triphenylphosphonium (G-TPP) was a gift from Dr Altieri (The Wistar Institute, Philadelphia), mitochondrially targeted circularly permuted yellow fluorescent protein (mito-cpYFP) [31] was a gift from Drs W. Wang (University of Washington) and P. Cheng (Peking University, China) and mitochondrially targeted cameleon, 4mtD3cpv [32], was a gift from Dr A. Palmer (University of Colorado Boulder). GCaMP3 was obtained from Loren Looger through Addgene (Plasmid # 22692). GCaMP3 was subcloned into a pShooter mitochondrial targeting vector. Mitochondrially targeted HyPer was purchased from Evrogen.
Alcohol feeding model
The Lieber-DeCarli pair-feeding model for chronic alcohol feeding was used in this study [33]. All experiments were carried out using pairs of 3- to 4-week-old male Sprague-Dawley rat littermates (Harlan Laboratories). Rats were acclimated to the liquid diets and then the ‘alcoholic’ animals were maintained on an ethanol-containing (36% of total calories) liquid diet for 2–4 months, while the littermate controls received an equivalent amount of an isocaloric diet with carbohydrate substituted for ethanol. The average daily alcohol consumption ranged between 12 and 18 g/kg body weight and rats maintained on the alcohol-containing liquid diet routinely developed steatotic livers. The Lieber-DeCarli ′82 alcohol (F1258SP) and control liquid (F1259SP) diets were purchased from Bioserv, Inc., Frenchtown, New Jersey. Animal studies were approved by the Institutional Animal Care and Use Committee at Rutgers, New Jersey Medical School.
Hepatocyte isolation and culture
Hepatocytes were isolated by a two-step collagenase perfusion of livers as previously described [26,34]. Cell suspensions were maintained on ice in a HEPES-buffered Krebs-Ringers bicarbonate (KRB) until used. KRB is composed of (in mM) 121 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 24 NaHCO3, 1 CaCl2, 11 glucose, 1 glutamine and 12.5 HEPES, pH 7.4, at 37°C. Cell viability was determined by Trypan blue exclusion and typically ranged between 85 and 95%. Hepatocytes (7 × 105) were plated on collagen-coated glass coverslips in William’s E Medium (WEM, Invitrogen) supplemented with 5% (v/v) fetal bovine serum (Atlanta Biologicals), 2 mM glutamine, 10 units/ml penicillin, 10 μg/ml streptomycin and 50 μg/ml gentamycin. Cells were maintained in short-term culture for 1–3 h in the absence of insulin or cultured 24–36 h in the presence of insulin (14 nM, Gibco) In some experiments, hepatocytes were transfected by electroporation using an Amaxa Rat/Mouse Hepatocyte Nucleofector Kit according to the manufacturer’s instructions (Lonza).
Mitochondrial Ca2+ uptake and matrix swelling in permeabilized cells
Hepatocyte suspensions were washed once with PBS containing 1 mM EGTA plus 1 mM EDTA to remove extracellular Ca2+ and then twice with intracellular-like medium (ICM) to remove the chelators. Cells were stored on ice in ICM containing 5 mM glutamate and 1 mM pyruvate until used. ICM is composed of (in mM) 135 KCl, 15 NaCl, 1.2 MgSO4, 5 KH2PO4, 10 HEPES and 10 MES, pH 7.2, at 37°C. Hepatocytes (3–5 mg protein) were added to 2 ml of prewarmed ICM containing 5 mM glutamate, 1 mM pyruvate, 2 mM ATP, 1 μM thapsigargin, protease inhibitor cocktail (Calbiochem IV), 40 μg/ml digitonin and either 2 μM fura-2 FF free acid (Kd = 25 μM, TEFLABS) or 2 μM fura-6F (Kd = 5μM, Molecular Probes). An ATP-regenerating system composed of 5 mM creatine phosphate and 5 units/ml creatine phosphokinase was included in the buffer when indicated in the figure legends.
Hepatocytes were digitonin-permeabilized in thermostatically regulated cuvettes (37°C) with continuous stirring for 5–10 min prior to data acquisition. Extra-mitochondrial Ca2+ concentration and light scattering intensities were recorded simultaneously using a high-speed, DeltaRAM spectrofluorometer (Photon Technology International) with dual monochromators and company written software routines. Fura-2 FF (or fura-6F) fluorescence intensities were acquired by alternating excitation at 340, 380 and 510 nm emission. Light scattering was determined at a 90° angle using 630 nm excitation and emission. A decrease in light scattering is a relative measurement of mitochondrial swelling. The light scattering intensities were normalized to the fluorescence obtained after treatment with the K+ ionophore, valinomycin. The normalization protocol was used to compare the extent of mitochondrial swelling between different hepatocyte preparations. Time constants (τ) were calculated from the exponential fits using GraphPad Prism software.
Digitonin fractionation and enzyme marker assays
Hepatocytes (5 mg protein) were incubated with digitonin (40 μg/ml) for 5 min in ICM. Cells were collected by centrifugation and a half volume aliquot of the supernatant was transferred to a new tube. Supernatant and cell pellets fractions were treated with 2% (v/v) Triton-X 100 and then stored in liquid nitrogen until assayed. The activities of lactate dehydrogenase (LDH) and L-glutamate dehydrogenase (GDH) were used as enzyme markers for the cytosolic and mitochondrial fractions, respectively. Lactate dehydrogenase activity was determined spectrophotometrically at 340 nm by following the oxidation of NADH with pyruvate [35]. The activity of GDH is measured as described by Fisher [36] following the oxidation of NADPH with α-ketoglutarate and ammonia. Citrate synthase (CS) activity was determined with a kit from Sigma-Aldrich. The assay is based on the chemical coupling of CoA-SH with Ellman’s reagent, 5,5ʹ-dithiobis-(2-nitrobenzoic acid). The resultant product, 5-thio-2-nitrobenzoic acid, was measured at 412 nm. The efficacy of digitonin to permeabilize the plasma membrane was the same in both preparations. More importantly, >98% of the mitochondrial marker remained with the cell pellets after digitonin treatment, indicating that protocol did not rupture the mitochondria in either control or alcohol groups (Table 1).
Table 1. Digitonin permeabilization.
Freshly isolated hepatocytes were incubated with digitonin (40 μg/ml) for 5 min in ICM and then centrifuged to separate the cytosolic constituents from the cell pellet. The activity of LDH or GDH in the supernatant or cell pellet fractions was normalized to the total enzymatic activity measured in the samples (% total). Data shown are mean ± SEM of samples from three pairs of alcohol-fed rats and their pair-fed littermate controls. Abbreviations: ns, not significant; paired t-test.
| LDH (% total) |
GDH (% total) |
|||
|---|---|---|---|---|
| Control | Alcohol-fed | Control | Alcohol-fed | |
| Supernatant | 95.01 ±6.21 | 97.28 ± 2.83 | 1.20 ± 0.67 | 1.29 ± 0.03 |
| Pellet | 4.99 ±6.21 | 2.72 ± 2.83ns | 98.80 ± 0.67 | 98.71 ±0.03ns |
Mitochondrial mass measurement by flow cytometry
Hepatocyte suspensions were loaded with 200 nM MitoTracker® Deep Red (Molecular Probes) in WEM for 30 min at 37°C. Cells were washed once with WEM without dye and collected by centrifugation. Fluorescence intensity of individual cells was quantified by a flow cytometry with the 635 nm laser and the emission of 661 ± 16 nm.
Total and free matrix Ca2+ measurements
Hepatocyte cultures were loaded with fura-2 by incubation with 5 μM fura-2/AM in a HEPES-buffered physiological saline solution (HBSS) supplemented with 0.02% (w/v) Pluronic® F-127 and an organic anion transport inhibitor bromsulphthalein (100 μM) for 30 min at 37°C. HBSS is composed of (in mM) 121 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 5 NaHCO3, 1.3 CaCl2, 25 HEPES, 5 glutamate, 1 pyruvate and 0.25% (w/v) BSA, pH 7.4, at 37°C. Fura-2-loaded hepatocytes were transferred to a thermostatically regulated microscope chamber (37°C) and then washed into a Ca2+-free HBSS just prior to data acquisition. Fura-2 fluorescence images (excitation, 340 and 380 nm, and emission 420–600 nm) were acquired at 5 s intervals with a cooled charge-coupled device (CCD) camera attached to an epifluorescent microscope, as previously described [37]. FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone, 5 μM) and oligomycin (5 μg/ml) were added to collapse ΔΨm and release mitochondrial matrix Ca2+.
Hepatocyte suspensions (5 mg protein) were permeabilized with digitonin in ICM for 5 min as described above. The release of mitochondrial matrix calcium (Ca2+) was induced by the addition of 1 μM CCCP (carbonyl cyanide-m-chlorophenyl hydrazone) and changes in the fura-2 FF ratio were monitored with a DeltaRAM spectrofluorometer as described above. The amount of Ca2+ released was calibrated at the end of each run by the addition of ionomycin (5 μM) followed by 20 nmol of Ca2+.
Mitochondrial-free Ca2+ was determined in cells expressing mitochondrially targeted 4mtD3cpv cameleon. Fluorescent images were acquired with 436 nm excitation and the emitted fluorescence split into donor and acceptor images with a 515 nm dichroic mirror followed by 480 ± 30 nm (CFP) or 535 ±40nm (cpVenus) emission filters [37].
Fura-2 and mitochondrial-GCaMP3 fluorescence images were collected with alternating excitation at 340, 380 and 485 nm as described above. The emitted fluorescence was collected using a 495 nm long band-pass dichroic beam splitter and a 500 nm long band-pass emission filter.
Mitochondrial ROS measurements
Confocal images of mito-cpYFP fluorescence (excitation, 488 nm and emission >500 nm) are acquired with a Bio-Rad Radiance 2002-MP confocal system using the manufacture’s software. Fluorescence images of mitochondrial HyPer (mito-HyPer) were acquired with an EMCCD camera (Hamamatsu) under computer control (Metafluor) using alternating excitation at 485 ± 20 and 405 ± 20 nm. The emitted fluorescence was collected using a 495 nm long band-pass dichroic and a 500 nm long band-pass emission filter. Mito-HyPer excitation ratios were normalized to the peak ratios obtained after treatment with maximal hydrogen peroxide (H2O2) concentrations (>100 μM).
Hepatocytes (50 000 cells/cm2) were plated onto 96-well plates and maintained in WEM plus insulin for 16–18 h. Cultures were washed once then incubated in Hank’s buffer supplemented with 3 mM HEPES and 0.2% BSA, pH 7.4. Cultures were treated with 1 nM vasopressin (VP), 10 ng/ml tumor necrosis factor α (TNFα, PeproTech) or both reagents for 120 min. Cultures were loaded with MitoSOX Red (1 μM) during the last 30 min of the treatment protocol. Cells were washed once with Hank’s buffer and then counterstained with Hoechst 33342 (0.5 μg/ml) for 5 min just prior to measurement. MitoSOX Red (excitation 531 nm and emission 572 nm) and Hoechst (excitation 340 nm and emission 460 nm) fluorescence intensities were measured on a Victor3 plate reader (PerkinElmer). MitoSOX Red fluorescent intensities were normalized to the Hoechst fluorescence to account for differences in cell density.
ATP content
Freshly isolated hepatocytes (3 mg protein) were suspended in 2 ml of HBSS supplemented with 5 mM glutamate and 1 mM pyruvate and then incubated for 10 min in thermostatically regulated cuvettes (37°C) with continuous stirring. Aliquots of cells (250 μl) were denatured with an equal volume of trichloroacetic acid (0.2 M). Samples were centrifuged (30 s at 16 000 ×g) and the supernatant (400 μl) was collected and neutralized with 250 μl of 0.2 M KOH. The extracted ATP was quantified with the kit purchased from Promega.
Triglyceride measurements
The analysis of triglyceride levels was determined by saponification of isolated hepatocytes in ethanolic KOH as described previously [38]. The extract was neutralized with MgCl2 and glycerol content was determined enzymatically with the Glycerol Free Reagent kit (F6428, Sigma).
Western blot analysis
Whole cell lysates were prepared in a buffer comprised of (in mM) 100 Tris-HCl, 2 EDTA, 2 EGTA, 1 PMSF, 1 benzamidine, 1 dithiothreitol, 1 Na3VO4, 20 NaF, 10 Na2MoO4, 10 Na-pyrophosphate and 50 β-glycerol phosphate and supplemented with a GBioscience PhosphataseArrest™ III inhibitor cocktail, Roche complete proteinase inhibitor tablet, 10 μg/ml pepstatin A and 4% (w/v) SDS (pH 7.2). Lysates were resolved by SDS-PAGE on a 10 or 12% polyacrylamide gel and transferred to PVDF or nitrocellulose membranes. Membranes were blocked for 1 h in Tris-buffered saline ( pH 7.5) containing 3% (w/v) BSA and 0.1% (v/v) Tween-20. The membranes were incubated with primary antibodies overnight at 4°C. Rabbit polyclonal antibody against MCU/CCDC109A was obtained from Sigma, rabbit polyclonal antibody against voltage-dependent anion channel (VDAC) was from Cell Signaling, mouse monoclonal antibody against NDUFA9 was from Molecular Probes, mouse monoclonal antibodies against CypD/CypF and mitochondrial respiratory chain polypeptides were from Abcam and rabbit monoclonal antibodies against Hsp10 and Hsp60 were from Epitomics. Protein loading was determined by stripping the membranes and reprobing with anti-α-tubulin or anti-β-actin antibodies (Cell Signaling). The levels of α-tubulin or β-actin did not change with alcohol feeding and either protein was used as a loading control.
Protein determination
Protein content was determined by the Pedersen modified Lowry assay [39].
Statistical analysis
Data are presented as means ± SEM for the number of pairs or measurements indicated in the figure legend. Statistical differences between experimental group means were examined by ANOVA or by Student’s t-test when comparing just two groups. A value of P < 0.05 was considered statistically significant.
Results
Chronic alcohol feeding and hepatocyte injury
Sprague-Dawley rats were maintained on a Lieber-DeCarli alcohol-containing liquid diet for 60 ± 5 days prior to isolation of hepatocytes. This length of ethanol feeding has been shown to induce alcoholic fatty liver disease and mitochondrial dysfunction including decreases in the protein abundance of all complexes involved in oxidative phosphorylation except complex II [2,14–16]. The data shown in Figure 1 validate that the hepatocytes from alcohol-fed rats used in this study also show signs of steatosis and mitochondrial injury. The confocal images show paraformaldehyde-fixed, primary cultured hepatocytes stained with 1,6-diphenyl-1,3,5-hexatriene to visualize neutral lipids [40]. As expected, chronic alcohol feeding significantly increased the number and size of lipid droplets in hepatocytes compared with cells isolated from pair-fed littermates (compare Figure 1A with B). The average number of lipid droplets per cell increased from 4 ± 3.0 per cell in the controls to 33 ± 4.0 per cell after alcohol feeding (n > 90 cells/condition; P < 0.05). In addition, biochemical measurement of lipid content showed a significant increase in triglyceride levels. Hepatic triglycerides increased from 0.15 ± 0.05 mg per 1 × 106 cells in control hepatocytes to 0.4 ±0.07 mg per 1 × 106 cells after alcohol feeding (Figure 1C; P < 0.05). Long-term alcohol consumption decreased the levels of nuclear-encoded polypeptide subunits involved in the assembly of mitochondrial complex I (NDUFB8 and NDUFA9) and a mitochondrially encoded core subunit of complex IV (MTCO1, Figure 1D,E and Table 2). Alcohol-dependent decreases in these subunits has been reported in previous studies [15,16]. The loss of these polypeptide chains was associated with an approximately 40% decrease in cellular ATP levels (Figure 1F). Cellular ATP levels were 33.40 ± 5.0 vs. 18.8 ± 2.9 nmol/ mg protein in littermate controls vs. hepatocytes from alcohol-fed animals, respectively (P <0.05). We next determined if chronic alcohol feeding had any effect on the expression of the VDAC, a protein involved in the transport of nucleotides and metabolites across the outer mitochondrial membrane. The data show a significant increase in the protein levels of VDAC in the alcohol-fed group compared with controls (Figure 1G and Table 2). Finally, previous studies have provided evidence linking alcohol-induced mitochondrial injury with the activation of stress responses including the induction of mitochondrial-specific chaperones, e.g. Hsp60 [15]. Increases in mitochondrial chaperone expression are thought to be a protective response to elevated levels of misfolded and oxidatively damaged matrix proteins. Western blot analysis for Hsp10 and Hsp60 revealed increases in these proteins after alcohol feeding (Figure 1H and Table 2).
Figure 1. Chronic alcohol feeding causes mitochondrial dysfunction.
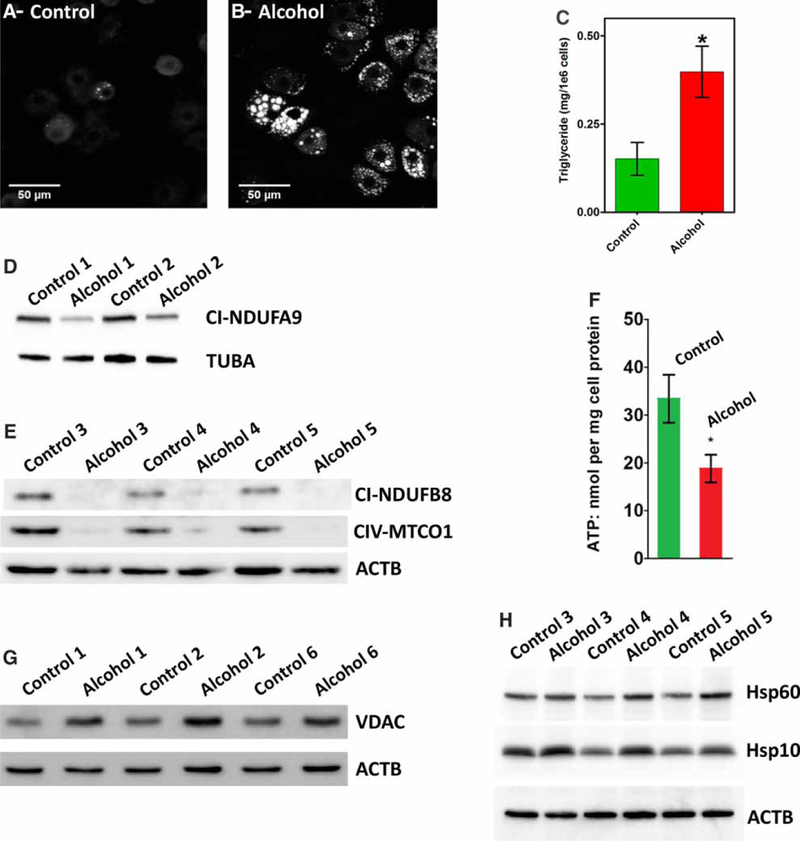
(A and B) Representative confocal images showing the lipid droplets stained with 1,6-diphenyl-1,3,5-hexatriene in hepatocytes isolated from control (A) or alcohol-fed rats (B) and maintained in primary culture for 1 h. (C) Biochemical determination of triglyceride levels in hepatocytes from control and alcohol-fed rats. Data are the means ± SEM, n = 4 pairs; *P< 0.05. (D and E) Immunoblot bands of mitochondrial respiratory chain polypeptides: (D) subunit NDUFA9 of complex I and (E) subunit NDUFB8 of complex I and subunit MTCO1 of complex IV. (F) Total ATP levels determined in hepatocytes from control and alcohol-fed rats incubated in the presence of 5 mM glutamate and 1 mM pyruvate. Data are the means ± SEM, n = 5 pairs; *P < 0.05. Immunoblot bands of VDAC (G) or Hsp10 and Hsp60 (H) in hepatocytes isolated from control and alcohol-fed rats. The numbers following the control and alcohol labels refer to a specific pairs of animals, i.e. the whole cell lysates used in E and H were from the same animals. Densitometric analysis of immunoblot bands are shown in Table 2.
Table 2. Fold changes in the levels of mitochondrial proteins after chronic alcohol feeding.
Protein levels of the indicated mitochondrial proteins were determined in whole cell lysates prepared from hepatocytes isolated from control and alcohol-fed animals. Immunoblot bands were quantified by densitometry analysis using Image J (NIH). Representative examples of immunoblots are shown in Figure 1. Data shown are the means ± SEM from 3 to 6 pairs of alcohol-fed rats and their pair-fed littermate controls. Proteins levels are normalized to TUBA or ACTB as indicated.
| Protein | Control | Alcohol-fed |
|---|---|---|
| CI-NDUFA9:TUBA | 1.00 ± 0.07 | 0.59 ± 0.07* |
| CI-NDUFB8:ACTB | 1.00 ± 0.15 | 0.14 ±0.02* |
| CIV-MTCO1:ACTB | 1.00 ±0.24 | 0.13 ± 0.02* |
| VDAC:ACTB | 1.00±0.10 | 1.60 ± 0.27* |
| Hsp10:ACTB | 1.00 ± 0.20 | 1.48 ± 0.06* |
| Hsp60:ACTB | 1.00±0.10 | 1.52 ±0.17* |
P <0.05
Taken together, these data confirm that our animal model reproduces some of the key features of experimentally induced alcoholic fatty liver disease including fat accumulation, decreases in polypeptide subunits that are assembled into mitochondrial respiratory chain complexes I and IV [15,16], compromised capacity to synthesize ATP [9,13] and activation of mitochondrial stress responses [1]. The increase in VDAC protein was unexpected as previous studies had not detected significant changes in the levels of VDAC protein after alcohol feeding [16,41]. We do not have an explanation for this discrepancy. An increase in VDAC protein could potentially have significantly effects on mitochondrial Ca2+ homeostasis. VDAC has been proposed to form a large signaling complex with the type 1 InsP3 receptor through the chaperone, grp75. This signaling complex acts to increase Ca2+ accumulation into the mitochondria during hormone stimulation [42,43].
Long-term ethanol consumption increases mitochondrial Ca2+ levels
Excessive accumulation of Ca2+ ions into the mitochondria is known to cause matrix swelling and increase ROS formation [20] eventually resulting in irreversible injury to the organelle. Dysregulation in matrix Ca2+ homeostasis could be an underlying mechanism involved in alcohol-induced mitochondrial damage. To test this possibility, we determined the effects of chronic alcohol feeding on mitochondrial Ca2+ content and Ca2+ handling pathways.
Cultured hepatocytes were loaded with fura-2/AM and then washed into a Ca2+-free buffer for 5 min prior to the addition of the FCCP plus oligomycin to collapse mitochondrial proton motive force (PMF). The dissipation of mitochondrial PMF stimulates the release of matrix Ca2+ and the resultant rise in [Ca2+]i is an indirect measurement of the amount of Ca2+ in the mitochondria. The traces in Figure 2A are the mean FCCP-stimulated increases in [Ca2+]i from cells isolated from alcohol-fed rats (red) or their pair-fed control littermates (green). A summary of the peak increases in the fura-2 ratio (ΔR) for each hepatocyte is shown in Figure 2B. The data indicate that treatment with FCCP induces a larger rise in [Ca2+]i in hepatocytes from alcohol-fed animals compared with littermate controls. Similar results were also observed in hepatocytes maintained in primary culture overnight in the absence of alcohol. The mean FCCP-induced increase in the fura-2 ratio was 0.47 ±0.08 vs. 0.21 ±0.03 in overnight cultured hepatocytes from control and alcohol-fed animals, respectively (mean ± SEM; n = 200–275 cells isolated from four pairs of alcohol-fed rats and their littermate controls, P < 0.05).
Figure 2. Chronic alcohol feeding increases mitochondrial matrix Ca2+ levels in hepatocytes.
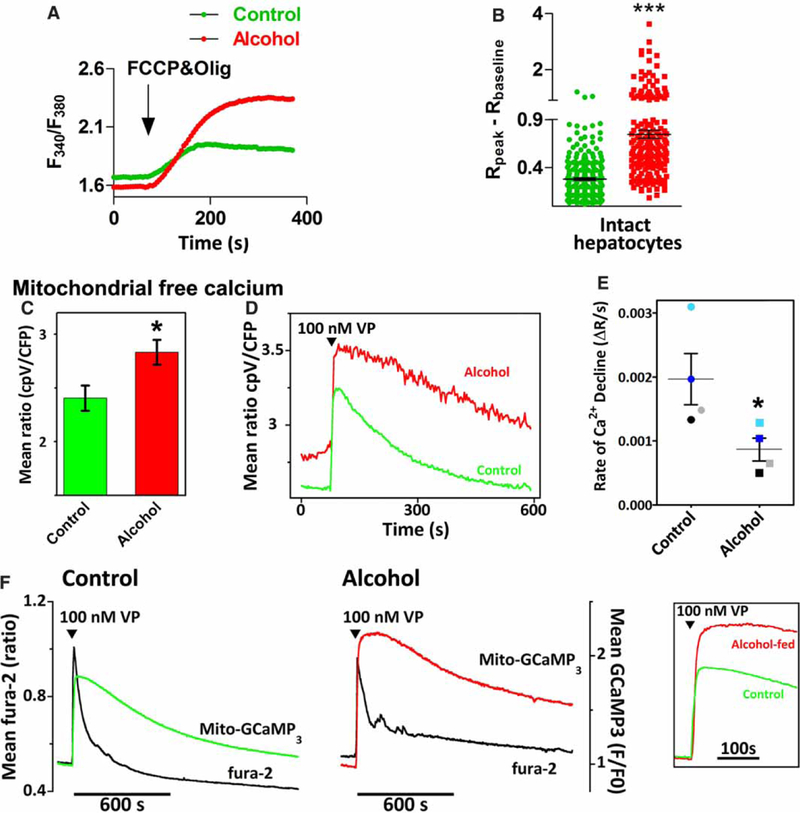
(A) The traces are the mean FCCP-evoked increases in cytosolic Ca2+ measured in fura-2/AM-loaded hepatocytes from alcohol-fed rats (red) or their pair-fed littermates (green); n = 4 pairs, 30–100 cells/pair. Cells were maintained in culture for 1 h prior to experimentation. FCCP (5 μM) plus oligomycin (5 μg/ml) are added at the arrow. (B) Summary data for the peak FCCP-induced increase in the fura-2 ratio (Rpeak) - baseline ratio (Rbaseline) calculated for each cell shown in A; ***P< 0.001 compared with littermate controls. (C-E) Hepatocytes from control and alcohol-fed animals were transfected with mitochondrially targeted 4mtD3cpv. (C) Basal mitochondrial 4mtD3cpv emission ratio. Data are the means ± SEM determined from 7–9 separate measurements using cells isolated from three separate pairs of control and alcohol-fed livers. (D) VP-evoked mitochondrial Ca2+ increases in the absence of extracellular Ca2+. Traces are the mean responses from all 4mtD3cpv expressing cells (n = 5–10 cells) in the field of view. (E) The initial rates of Ca2+ decline were determined by linear regression curve fits. Data points are the means calculated from 3–5 replicate measurements carried out in hepatocytes isolated from four pairs of alcohol-fed rats and littermate controls. Symbols with alike colors are from the same pair. *P <0.05 compared with controls. (F) Hepatocytes from control and alcohol-fed rats were transfected with mitochondrially targeted GCaMP3. Cells were loaded with fura-2/AM washed into Ca2+-free buffer then treated with 100 nM VP. Traces are the mean cytosolic (black traces) and mitochondrial Ca2+ increases (red and green traces) recorded in all GCaMP3-expressing cells within the field of view (n = 17–25 cells). Insert shows VP-induced mito-GCaMP3 increases on expanded timescale.
We next tested if chronic alcohol feeding had any effect on matrix-free Ca2+. These studies were carried out in hepatocytes transfected with mitochondrially targeted cameleon, 4mtD3cpv [32], and maintained in culture for 20–24 h in the absence of alcohol. The data show that the resting emission ratio was significantly higher in hepatocytes from alcohol-fed animals compared with controls, indicating an increase in mitochondrial-free Ca2+ (Figure 2C). In some experiments, the cultures were washed into a Ca2+-free buffer then challenged with maximal vasopressin (VP) concentrations. The traces shown in Figure 2D are the time courses for the mean increases in the 4mtD3cpv emission ratio from hepatocytes isolated from control and alcohol-fed animals. The amplitude of the VP-evoked mitochondrial Ca2+ increases was slightly higher after alcohol feeding, but did not reach statistical significance (3.1 ±0.3 vs. 3.5 ±0.3 in control and alcohol-fed, respectively; P >0.1). The falling phase of the mitochondrial Ca2+ spike displayed the largest difference between the control and alcohol groups. In control cells, the mitochondrial Ca2+ spike declined exponentially, while the Ca2+ decay rates could not be adequately described by exponential curve fits after alcohol feeding. Therefore, linear regression analysis on the first 20–120 s after the mitochondrial Ca2+ peak was carried out to assess the rates of Ca2+ decay. The data indicate that the initial rates of mitochondrial Ca2+ decline are significantly slower after alcohol feeding (Figure 2E). Consequently, hormone-induced mitochondrial Ca2+ increases recovered more rapidly to baseline levels in control cells compared with the alcohol group. The 4mtD3cpv emission ratio recovered 93 ± 6% from the hormone-induced peak within 10 min in control hepatocytes, while only 60 ± 9% in hepatocytes from alcohol-fed animals (P < 0.05).
The 4mtD3cpv measurements were confirmed in hepatocytes transfected with mitochondrially targeted GCaMP3 (mito-GCaMP3). This Ca2+-sensitive biosensor has a high signal-to-noise ratio and is spectrally compatible with measurements of cytosolic Ca2+ using fura-2, but has the disadvantage of being a single wavelength probe and having a lower Kd for Ca2+ compared with 4mtD3cpv (350 vs. 600 nM). Mito-GCaMP3-transfected cultures were treated with maximal VP doses in the absence of extracellular Ca2+ while simultaneously monitoring both mitochondrial and cytosolic Ca2+ responses (Figure 2F). Hormone stimulation induced a rapid and large increase in cytosolic Ca2+ (black traces) that was transitory due to the absence of Ca2+ influx and depletion of internal Ca2+ stores. There were no measureable differences in the amplitude of the hormone-induced Ca2+ increases or the size of thapsigargin-sensitive Ca2+ stores between the control and alcohol-fed groups (Bartlett J. P, Antony N. A, Agarwal A, Hilly M, Prince L. V, Combettes L, Hoek B. J and Gaspers D. L, unpublished work). The peak VP-induced increases in mitochondrial-GCaMP3 fluorescence were not markedly different between the control (green trace) and alcohol-fed groups (red trace). However, mitochondrial Ca2+ increases remained elevated at a plateau for a longer time before starting to slowly decline in the alcohol-fed group compared with controls (Figure 2F, insert). Taken together, these data suggest that alcohol feeding may increase the transfer of Ca2+ from the cytosol to the mitochondria during hormone stimulation while also prolonging the falling phase of the mitochondrial Ca2+ spike leading to longer lasting Ca2+ increases in the matrix.
To ensure that culturing the cells did not affect our mitochondrial Ca2+ measurements, total matrix Ca2+ was also determined in freshly isolated hepatocytes. Cell suspensions were rapidly washed to remove extracellular Ca2+ and then suspended in an ICM containing thapsigargin to block Ca2+ uptake into the endoplasmic reticulum (ER) and fura-2 free acid to monitor changes in media-free Ca2+ concentration. Digitonin was added to permeabilize the plasma membrane and then mitochondrial Ca2+ was released by the addition of a proton-phore, CCCP plus oligomycin. CCCP was used in the cell suspension studies because it is less fluorescent than FCCP. The data show that treatment with CCCP releases more Ca2+ in hepatocytes from alcohol-fed animals compared with their pair-fed controls (Figure 3A,B). The increase in mitochondrial Ca2+ levels could not be explained by changes in mitochondrial mass, as the activities of CS and GDH were not significantly altered after alcohol feeding. Hepatic CS activity was 1.0 ±0.1 vs. 1.2 ±0.1 μmol/min/mg protein and GDH activity was 4.8 ± 0.3 vs. 4.0 ± 0.3 μmol/min/mg protein in hepatocytes isolated from control and alcohol-fed rats, respectively (P >0.1). In addition, flow cytometric analysis of MitoTracker® Deep Red stained hepatocytes showed similar levels of labeling in both cell populations (Figure 3C,D). Moreover, our previous studies in paraformaldehyde-fixed livers did not show an increase in the number of mitochondria per cell after alcohol feeding [6]. Taken together, these data indicate that chronic ethanol feeding increases total and free mitochondrial matrix Ca2+ levels, but does not alter mitochondrial mass.
Figure 3. Effect of alcohol feeding on mitochondrial total Ca2+ content and mass in freshly isolated hepatocytes.
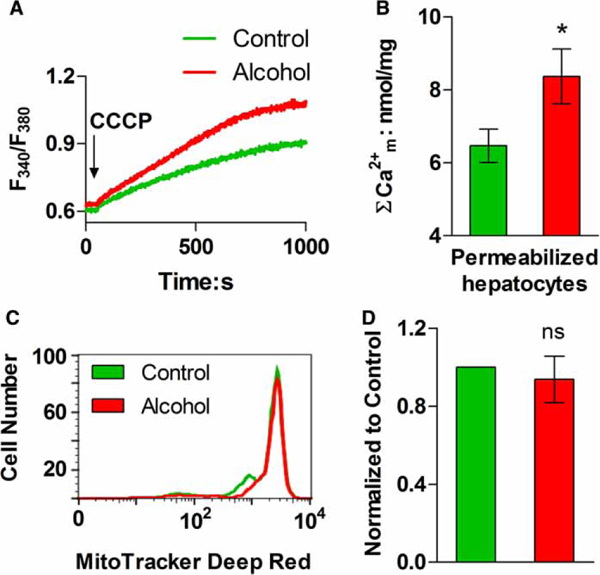
(A) Representative traces of CCCP-induced Ca2+ release in digitonin-permeabilized hepatocyte suspensions. (B) Data shown are the mean (±SEM) CCCP-induced Ca2+ release from experiments similar to those shown in A and calibrated with a reference Ca2+ pulse; n = 3 pairs; *P < 0.05 compared with littermate controls. (C and D) Isolated hepatocytes were stained with MitoTracker® Deep Red FM and the fluorescence intensity of each cell was determined by flow cytometry. (C) The distribution of fluorescent intensity in cells isolated from an alcohol-fed rat (red) and corresponding littermate control (green). (D) Summary data for the average cellular fluorescence in hepatocytes isolated from control and alcohol-fed animals, n = 3 pairs; ns: not significant.
The effect of alcohol feeding on mitochondrial Ca2+ transport
The rates of mitochondrial Ca2+ uptake were determined in digitonin-permeabilized hepatocytes suspended in ICM supplemented with an ATP-regenerating system, fura-6F free acid and thapsigargin. Cell suspensions were incubated for 5 min at 37°C in a stirred and thermostatically regulated cuvette and then treated with a series of CaCl2 pulses (300 nmol/pulse). Representative traces for the first three Ca2+ pulses are shown in Figure 4A. Summary data for the calculated half-times (τ) for mitochondrial Ca2+ uptake for each Ca2+ pulse are shown in Figure 4B. The data indicated that the rates of mitochondrial Ca2+ uptake are faster in hepatocytes from alcohol-fed animals compared with controls. Mitochondrial Ca2+ accumulation was completely blocked by pretreatment with ruthenium red (not shown), suggesting that activity of the MCU Ca2+ channel was solely responsible for Ca2+ uptake. The protein levels of MCU were determined by Western blot analysis of whole cell lysates prepared from freshly isolated hepatocytes from control and alcohol-fed animals. The results indicate that MCU protein levels are increased 2- to 3-fold after chronic alcohol feeding, which could explain the faster rates of Ca2+ uptake (Figure 4C,D).
Figure 4. The rates of mitochondrial Ca2+ uptake are increased by chronic alcohol consumption.
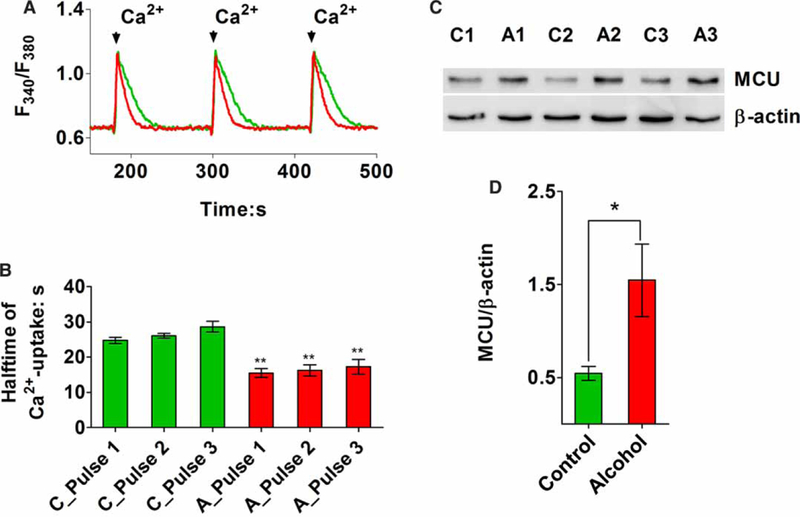
Hepatocytes (4 × 106 cells/ml) were digitonin-permeabilized in ICM containing 5 mM glutamate, 1 mM pyruvate, 2 mM ATP, 5 mM creatine phosphate, 5 units/ml creatine phosphokinase, 2 μM fura-6F free acid, 1 μM thapsigargin and a protease inhibitor cocktail. (A) Data shown are representative traces for the first three Ca2+ pulses (300 nmol/pulse) in alcohol-fed (red) and their pair-fed littermate controls (green). Mitochondrial Ca2+ uptake is indicated by a decrease in the excitation ratio. (B) Summary results are shown for the calculated half-times (τ) of mitochondrial Ca2+ uptake for the first three Ca2+ pulses in hepatocytes from control (C) and alcohol-fed animals (A). Data are means ± SEM from 4 pairs and 12 separate measurements; **P <0.01 vs. parallel control time constant. (C) Western blot showing the protein levels of MCU in hepatocytes isolated from alcohol-fed rats (A) and their pair-fed controls (C). (D) Summary data for MCU expression normalized to β-actin in cells from control and alcohol-fed animals; n = 6 pairs; *P <0.05.
In the next series of experiments, we tested the effects of cyclosporin A (CsA) on mitochondrial Ca2+ transport. We have previously demonstrated that CsA increases rates of mitochondrial Ca2+ uptake in hepatocytes, while also suppressing the rates of Ca2+ efflux [44]. The traces in Figure 5A show that the rates of mitochondrial Ca2+ uptake in control hepatocytes are significantly increased by CsA treatment. In contrast, CsA treatment had minimal effects on the rates of Ca2+ uptake in hepatocytes from alcohol-fed animals (Figure 5B). Summary data are shown in Figure 5C. The addition of CsA normalized the differences in the rates of Ca2+ uptake between control and alcohol groups (Figure 5C, compare blue and black bars). The data indicate that the rates of mitochondrial Ca2+ uptake in hepatocytes from alcohol-fed animals were already maximal in the absence of CsA, which is most likely due to the up-regulation of the MCU.
Figure 5. The effects of CsA on mitochondrial Ca2+ handling in hepatocytes from control and alcohol-fed animals.
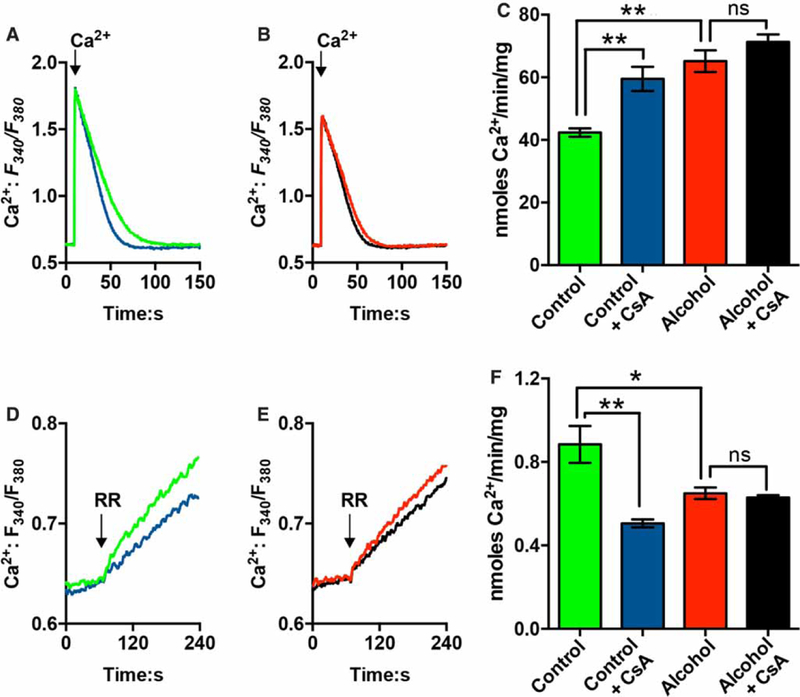
Mitochondrial Ca2+ uptake (A-C) and Ca2+ efflux (D and E) in permeabilized hepatocytes isolated from control (A and D) or alcohol-fed rats (B and E). Hepatocytes (2.5 mg protein/ml) were digitonin-permeabilized in ICM containing 5 mM glutamate, 1 mM pyruvate, 2 mM ATP, 2 μM fura-2 FF, 1 μM thapsigargin and a protease inhibitor cocktail. Where indicated, CsA (1 μM) was added 5 min prior to data acquisition. (A and B) Representative traces showing mitochondrial Ca2+ uptake in the absence (green and red traces) or presence of CsA (blue and black traces). The arrow shows the addition of 120 nmol Ca2+/mg protein. (C) Summary data for the rates of mitochondrial Ca2+ uptake for the indicated conditions. Data are means ± SEM, n = 5 pairs of control and alcohol-fed rats. (D and E) Cell suspensions were treated with ruthenium red (1 μM, RR) once the Ca2+ pulse (660 nmol) was completely accumulated. This inhibits Ca2+ uptake allowing Ca2+ efflux to be measured. Representative traces showing mitochondrial Ca2+ efflux in hepatocytes from control (D) or alcohol-fed animals (E). CsA was present in the blue and black traces. (F) Summary data for the calculated initial rates of mitochondrial Ca2+ egress. Data are means ± SEM, n = 4 pairs of control and alcohol-fed rats. *P <0.05, **P<0.01.
After mitochondrial Ca2+ uptake was complete, cell suspensions were treated with ruthenium red (RR, μM) to block Ca2+ uptake and unmask Ca2+ efflux from the mitochondria (Figure 5D-F). The rates of mitochondrial Ca2+ egress were significantly slower in hepatocytes from alcohol-fed animals compared with controls (Figure 5D-F). Pretreatment with CsA decreased the rates of mitochondrial Ca2+ egress in control hepatocytes, but had no effect on Ca2+ efflux in hepatocytes from alcohol-fed animals (Figure 5F). These results indicate that the activity of a CsA-sensitive channel or carrier contributes to the rates of Ca2+ efflux from the mitochondrial matrix in control cells, but this Ca2+ release pathway is absent after chronic alcohol feeding. These data could help to explain the slower recovery rates of hormone-induced mitochondrial Ca2+ increases in hepatocytes from alcohol-fed rats (Figure 2D,E).
The effect of chronic alcohol consumption on Ca2+-induced mitochondrial swelling
The volume of the mitochondrial matrix was qualitatively followed by monitoring changes in light scattering at F630 nm [45,46]. Light scattering intensities were normalized to the fluorescence values obtained after treatment with the K+ ionophore, valinomycin, which induces maximal swelling of the mitochondrial matrix. The addition of a Ca2+ bolus to permeabilized hepatocyte suspensions induced a transient decrease in light scattering consistent with Ca2+-induced matrix swelling (Figure 6A, orange trace). Note that these measurements were carried out in a potassium-based buffer that allows for the recovery of the matrix volume, which is not possible in sucrose-based media. The prior addition of CsA completely blocked the Ca2+-induced decrease in light scattering, indicating that the response is due to a transient opening of mPTP in a fraction of the mitochondrial pool (Figure 6A, blue trace).
Figure 6. The effect of chronic ethanol feeding on Ca2+-induced mitochondrial swelling in permeabilized cells.
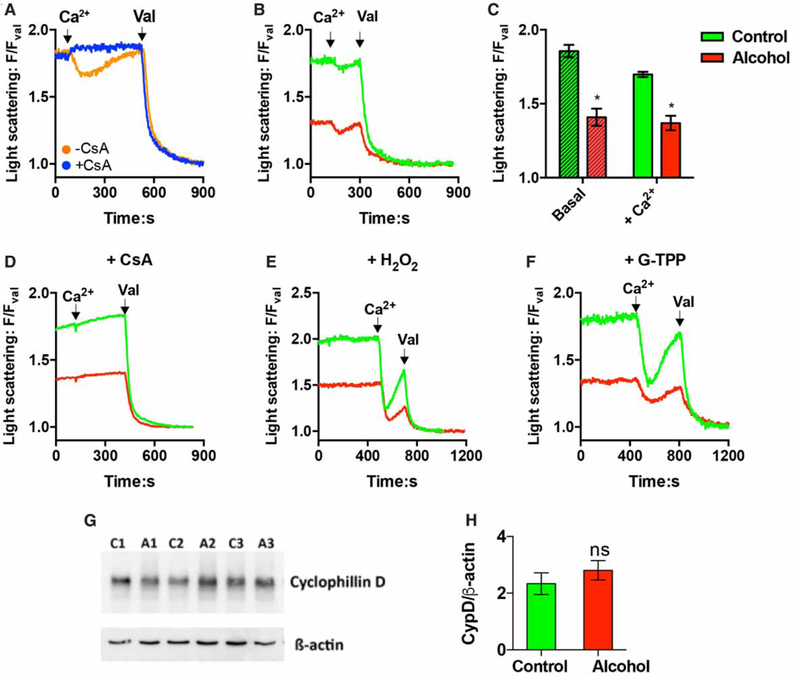
(A-F) Digitonin-permeabilized hepatocytes (2.5 mg protein/ml) were incubated as described in Figure 5. The addition of CaCl2 (120 nmol/mg) and valinomycin (Val, 2 μM) are indicated by the arrows. The relative change in light scattering intensities was monitored at F630 nm and normalized to values obtained after treatment with Val. (A) Cell suspensions were treated with CsA (1 μM, CsA) or DMSO (0.1% v/v) for 5 min prior to the addition of Ca2+ and Val. (B) Representative light scattering traces showing the Ca2+-induced changes in F630/FVal ratio in hepatocytes from alcohol-fed (red) and control (green) animals. (C) Summary data are shown for Ca2+-induced swelling in hepatocytes from control and alcohol-fed animals. Data are means ± SEM; *P < 0.05 significantly different from control; n = 4 pairs. (D-F) Hepatocyte suspensions were pretreated with CsA (1 μM, 5 min), H2O2 (500 μM, 5 min) or G-TPP (20 μM, 20 min) prior to the addition of Ca2+. (G and H) Western blot showing the protein levels of cyclophilin D (CypD) in hepatocytes isolated from alcohol-fed rats (A) and their pair-fed controls (C). Summary data are shown in H; ns, not significant.
The magnitude of the initial light scattering ratios was significantly lower in permeabilized hepatocytes from alcohol-fed rats compared with controls, suggesting that alcohol feeding causes some matrix swelling (Figure 6B,C). The F630/FVal ratios were 1.9 ±0.0 vs. 1.4 ±0.1 in hepatocytes from control and alcohol-fed animals, respectively (P < 0.05, n = 4 pairs). The addition of Ca2+ caused a transient decline in F630/FVal ratio that recovered to baseline values in both cell preparations. The Ca2+-induced decrease in the F630/FVal ratio was larger in hepatocytes from control littermates compared with alcohol-fed animals, but this response did not reach statistical significance. The ΔF630/FVal ratio was −0.16 ±0.05 vs. −0.04 ±0.01 in control and alcohol groups, respectively (P > 0.05). We next tested whether constitutive activation of the mPTP could explain the lower initial F630/FVal ratios in hepatocytes from alcohol-fed animals. Pretreatment with CsA did not alter light scattering ratios in either cell preparation, indicating that the mPTP was not active (Figure 6D). The F630/FVal ratio was 1.8 ± 0.04 vs. 1.4 ± 0.09 after CsA treatment in control and alcohol groups, respectively (P >0.05, n = 3 pairs). The addition of CsA effectively blocked Ca2+-induced swelling in both cell preparations confirming the potency of drug treatment to inhibit the mPTP (Figure 6D). The apparent increase in mitochondrial matrix volume after alcohol feeding could not be explained by differences in CypD expression as no significant changes were detected by Western blot analysis of whole cell lysates (Figure 6G,H).
Cell suspensions were next treated with H2O2 to oxidize protein thiols, which sensitize the mPTP to Ca2+ (Figure 6E). Alternatively, cells were preincubated with a mitochondrially targeted geldanamycin analog, G-TPP (Figure 6F), to inhibit matrix Hsp90 activity. Treatment with G-TPP has also been shown to potentiate the opening of mPTP [47,48]. The results indicate that Ca2+ treatment induces a larger mitochondrial swelling response after either drug treatment with the F630 nm/FVal ratio declining to similar values in both control and alcoholic hepatocytes (Figure 6E,F). The addition of Ca2+ transiently decreased the F630/FVal ratio to 1.2 ±0.02 vs. 1.1 ±0.02 in H2O2-treated (n = 2) and 1.3 ±0.05 vs. 1.2 ±0.06 in G-TPP-treated (n = 2) control and alcohol groups, respectively. The addition of the non-targeted parental compound, 17-(allylamino)-17-demethoxy-geldanamycin (17-AAG, Cayman Chemical), had no effect on the magnitude of Ca2+-induced swelling (not shown). These data suggest that mitochondria in control hepatocytes are in a condensed state and can be induced to undergo large Ca2+-dependent decreases in light scattering, whereas, potentiating the opening of the mPTP in hepatocytes from alcohol-fed animals results in smaller Ca2+-induced decreases in light scattering presumably because the mitochondrial pool is already partially swollen.
Hormone-induced mitochondrial ROS formation
Hepatocytes from control or alcohol-fed rats were transfected with a mito-cpYFP (Figure 7A) to determine if the elevated levels of matrix Ca2+ are associated with an increase in ROS production. Previous studies have reported spontaneous and transient spikes in mito-cpYFP fluorescence intensity called mito-flashes. These flash events have been interpreted as bursts of superoxide anion production in single mitochondria [31,49–51]. However, this interpretation is controversial with other studies, suggesting that changes in matrix pH can also contribute [52–54]. Real-time confocal imaging readily detected mito-cpYFP spikes in hepatocytes from alcohol-fed animals under basal conditions, whereas these flash events were rare or below the detection limits of the biosensor in cells from pair-fed controls (Figure 7B,C). Superoxide produced in the matrix is rapidly dis-mutated by Mn-superoxide dismutase producing H2O2. Thus, the levels of mitochondrial H2O2 were determined in cultured hepatocytes expressing a peroxide-sensitive fluorescent protein, HyPer, in the mitochondrial matrix [55]. Mito-HyPer excitation ratios were normalized to the peak ratio values obtained after the addition of maximal H2O2 concentrations (>100 μM). The data in Figure 7D indicate that alcohol feeding induces a small increase in the basal levels of mitochondrial H2O2 compared with controls (≈7%). The difference between the control and alcohol groups was statistically significant; nevertheless, the small nature of the increase makes it unclear what effect these changes in ROS will have on mitochondrial function. Acute alcohol exposure (30 mM, EtOH) induced a significant increase in the mito-HyPer excitation ratio within 5−10 min in the alcohol group, but did not markedly increase mitochondrial H2O2 levels in the control group over this time. These data are consistent with a previous study, showing that chronic alcohol feeding exacerbates ROS generation in hepatocytes exposed to acute ethanol treatment [56].
Figure 7. The effect of Ca2+-mobilizing hormones on mitochondrial ROS formation in hepatocytes from control and alcohol-fed animals.
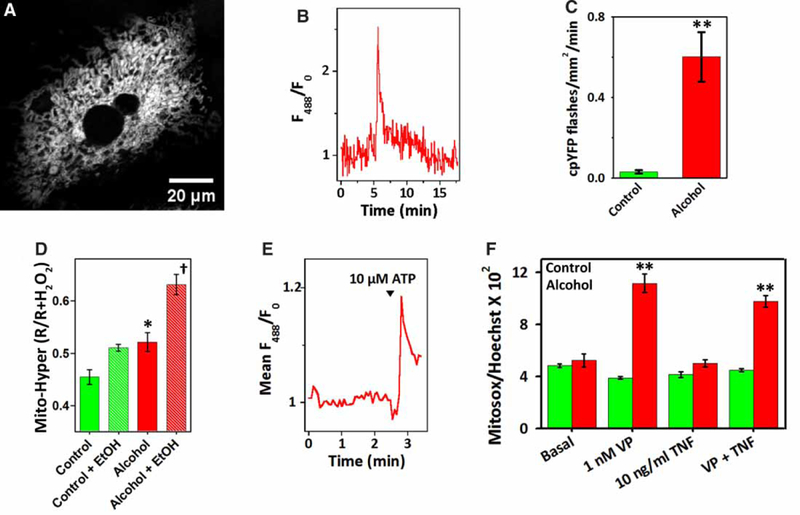
Hepatocytes were transfected with mitochondrially targeted cpYFP (A-C and E), mitochondrially targeted Hyper (mito-Hyper, D) or loaded with MitoSOX Red™ (F). (A) Confocal image of a hepatocyte-expressing mito-cpYFP. (B) Trace shows a spontaneous mito-cpYFP increase or flash in a hepatocyte from an alcohol-fed animal. (C) Summary data for the frequency of mito-cpYFP spikes. Data are the mean ± SEM, n = 5−12 cells isolated from three pairs of alcohol-fed rats (red) and their pair-fed controls (green). (D) Data are the mito-Hyper excitation ratios measured before and after acute ethanol exposure (30 mM, +EtOH). Excitation ratios were normalized to the peak ratio values obtained after the addition of maximal H2O2 concentrations (≥100 μM). Data shown are the means ± SEM from five pairs of control (green) and alcohol-fed (red) animals, n = 30–60 cells per pair. (E) Trace shows the mean ATP-induced increase in mito-cpYFP fluorescence in three hepatocytes from an alcohol-fed animal. (F) Hepatocytes cultured on 96-well plates were treated with VP, TNFα or both reagents for 120 min. Cells were loaded with MitoSOX Red™ (1 μM) during the last 30 min of the treatment and then counterstained with Hoechst 33342 (0.5 μg/ml) just prior to measurement. MitoSOX Red fluorescence was normalized to the Hoechst fluorescence intensities. Data shown are means ± SEM, n = 6 measurements. *P<0.05 vs. corresponding control group; **P<0.01 vs. corresponding control group; †P <0.01 vs. other groups.
We next determined if elevating matrix Ca2+ levels stimulated an increase in mitochondrial ROS formation. We have previously reported that maximal concentrations of Ca2+-mobilizing hormones that evoke sustained increases in [Ca2+]i produce a slow rise in mitochondrial ROS production, presumably mediated by matrix Ca2+ overload [57]. The trace in Figure 7E shows the mean mito-cpYFP increase in hepatocytes from an alcohol-fed rat challenged with 10 μM ATP, a dose that induces a sustained rise in [Ca2+]i. The data indicate that hormone stimulation causes a rapid increase in the production of mitochondrial ROS after ethanol feeding. These findings were also confirmed with the pH-insensitive indicator dye MitoSOX™ Red. Hepatocytes from control and alcohol-fed animals were cultured on 96-well plates and then stimulated for 2 h with an EC50 concentration of VP (1 nM, [58]), TNFα or VP plus TNFα. The cultures were loaded with MitoSOX™ Red for the last 30 min to determine mitochondrial levels. The data indicate that the levels of MitoSOX™ Red fluorescence intensity were similar in the control and alcohol groups under basal conditions (Figure 7F, left bars), which is consistent with the small differences detected with mito-Hyper. The addition of VP induced a significant increase in mitochondrial production in hepatocytes from alcohol-fed animals, but had no effect on ROS formation in pair-fed controls. Treatment with TNFα had little effect on MitoSOX™ Red fluorescence intensities in either cell type over this time period and did not synergize with VP to potentiate ROS formation (Figure 7F). Taken together, these data suggest that the basal rates of mitochondrial H2O2 production are not markedly elevated in cultured hepatocytes after chronic alcohol feeding, whereas, hormone stimulation potentiated the formation of mitochondrial in alcohol-injured hepatocytes but not in control cells. We speculate that the more prolonged hormone-induced increases in mitochondrial Ca2+ may contribute to the enhanced rates of ROS formation in the alcohol group.
Discussion
The impetus for the present investigation was based on previous studies, demonstrating that acute alcohol treatment stimulates receptor-coupled PLC activity in isolated hepatocytes leading to increases in InsP3 and mobilization of internal Ca2+ stores [17,19] and that mitochondria isolated from chronically ethanol-fed rats are more sensitive to Ca2+-induced swelling compared with their pair-fed control littermates [21,22]. We hypothesized that chronic alcohol feeding or excessive alcohol intake may cause aberrant and persistent increases in cytosolic Ca2+ in the liver eventually leading to mitochondrial Ca2+ overload, matrix swelling and higher rates of ROS production.
We demonstrate that chronic alcohol feeding is associated with elevated levels of matrix Ca2+ in freshly isolated hepatocytes as well as in hepatocytes cultured overnight in the absence of alcohol. These data point to stable changes in the pathways regulating mitochondrial Ca2+ homeostasis. We provide evidence that the protein levels of the mitochondrial Ca2+ uniporter and VDAC are significantly increased after alcohol feeding and these changes are the mostly likely explanation for the faster rates of mitochondrial Ca2+ uptake. These results are unanticipated. First, the loss of mitochondrial electron transport chain components observed in hepatocytes from alcohol-fed animals are expected to impair the formation of ΔΨm and slow the rates of mitochondrial Ca2+ uptake. Obviously, the level of mitochondrial injury achieved in our alcohol feeding paradigm was not sufficient to decrease the magnitude of the ΔΨm and limit Ca2+ uptake. Secondly, previous studies have clearly shown that liver mitochondria from alcohol-fed animals are more sensitive to Ca2+-induced permeability transition compared with littermate controls [21,22]. The opening of mPTP would depolarize the ΔΨm and slow the rates of mitochondrial Ca2+ accumulation. Previous morphometric studies of fixed liver samples indicate that a fraction of the mitochondrial pool is swollen after chronic alcohol feeding [6] and this could explain the lower light transmission values recorded in this study (Figure 6B). The addition of Ca2+ decreased light scattering by a similar magnitude in both control and alcohol groups, suggesting that the potency of Ca2+ to trigger the permeability transition is similar in both cell preparations. It should be pointed out, however, that the relationship between light scattering and mitochondrial volume is indirect and could be influenced by other subcellular structures in permeabilized hepatocytes, such as lipid droplets. We presume that our normalization protocol would account for these other light scattering structures, but perhaps it is not possible to directly compare changes in light scattering between hepatocytes from control and alcohol-fed animals. Irrespective of these potential issues, increasing the open time of the mPTP is not consistent with higher matrix Ca2+ levels or faster rates of Ca2+ accumulation in the alcohol group.
Our data also indicate that alcohol feeding suppresses the rates of mitochondrial Ca2+ egress and this appears to be due to a loss of a CsA-sensitive Ca2+ efflux pathway. We propose that the loss of this CsA-sensitive Ca2+ efflux pathway along with increased Ca2+ accumulation by the MCU contributes to the slower falling phase of the mitochondrial Ca2+ spike in intact cells (Figure 2D,E). This conjecture is based on previous studies, showing that genetic silencing or pharmacological inhibition of mitochondrial Na+/Ca2+ exchanger (NCLX) also significantly decreases the rates of mitochondrial Ca2+ efflux, but does not alter the amplitude of agonist-induced mitochondrial Ca2+ spikes [59–61]. Moreover, transient overexpression of MCU has been shown to significantly increase the amplitude of agonist-induced mitochondrial Ca2+ increases, but did not alter the rates of mitochondrial Ca2+ efflux nor significantly prolong the duration of the mitochondrial Ca2+ spike [23,62]. These data imply that mitochondrial Ca2+ efflux pathways can efficiently remove large Ca2+ loads as long as these efflux pathways are not impaired. Therefore, it is probably the amalgamation of larger MCU-dependent Ca2+ uptake and the loss of the CsA-sensitive Ca2+ efflux, which combine to markedly prolong the falling phase of the mitochondrial Ca2+ spike.
The transit opening of the mPTP is thought to provide an alternative pathway for Ca2+ egress particularly when mitochondrial Ca2+ levels are high enough to saturate the capacity of the H+/Ca2+ and Na+/Ca2+ exchangers [63]. This hypothesis is supported by studies showing that treatment with CsA inhibits mitochondrial Ca2+ efflux [44,64–66] and genetic depletion of CypD results in a substantial increase in basal mitochondrial matrix Ca2+ levels [67]. Moreover, mitochondria accumulate and retain more Ca2+ during stimulus-evoked Ca2+ increases in neurons isolated from CypD knockout mice [68]. The increase in mitochondrial basal Ca2+ levels, the slower rates of mitochondrial Ca2+ decline after hormone stimulation and the loss of a CsA-sensitive Ca2+ efflux pathway after chronic alcohol feeding could be interpreted as inhibition of Ca2+ efflux through the mPTP.
The hypothesis that chronic alcohol feeding may alter Ca2+ efflux through the mPTP is predicated on the presumption that the only pharmacological target of CsA is the pore. However, CypD binds to multiple matrix proteins and also interacts with mitochondrial chaperone networks [69]. The displacement of CypD from these proteins by CsA or off-target effects of the drug could directly or indirectly regulate the rates of mitochondrial Ca2+ egress. Indeed, Wei et al. [70] have demonstrated that micromolar concentrations of CsA inhibit the activity of Na+/Ca2+ exchanger by 50% in heart mitochondria. These data indicate that CsA cannot be used to unequivocally establish a role for mPTP in mitochondrial Ca2+ efflux. Moreover, it has also been reported that CsA treatment does not alter the rates of mitochondrial Ca2+ egress in HeLa cells, indicating that the pore does not participate in mitochondrial Ca2+ extrusion in this cell type [62].
In our permeabilized hepatocyte studies, mitochondrial Ca2+ efflux should be predominately Na+-independent since the physiological levels of Mg2+ used in our buffer have been shown to strongly inhibit Na+/Ca2+ exchange activity in isolated mitochondria [71,72]. Nevertheless, there are still measurable rates of Na+-dependent mitochondrial Ca2+ efflux in our experimental model (not shown). Thus, it is still plausible that some of the effects of CsA on mitochondrial Ca2+ egress could be mediated at the level of the Na+/Ca2+ exchanger. It is currently unknown if CsA has any effects on the putative mitochondrial H+/Ca2+ antiporter. Taken together, we can only conclude that alcohol feeding inhibits a CsA-sensitive mitochondrial Ca2+ efflux pathway. Further work is required to determine if chronic alcohol consumption alters the rates of Na+-dependent or Na+-independent Ca2+ extrusion from the mitochondria and if the mPTP contributes to Na+-independent Ca2+ efflux in hepatocytes. Carrying out chronic alcohol studies in the CypD knockout mouse model would help to clarify the role of the mPTP in mitochondrial Ca2+ handling.
Previous studies have reported that long-term alcohol feeding increases the basal rates of mitochondrial H2O2 production by several fold [56,73] not a few percent as reported here. The small changes in matrix H2O2 levels may reflect the possibility that hepatocytes cultured in the absence of alcohol partially recover from alcohol-induced injury or it may reflect the sensitivity of the biosensors. The peroxidase moiety in HyPer reacts with peroxide about a 100-fold slower than the endogenous mitochondrial peroxiredoxins, which may prohibit the biosensor from accurately detecting basal levels of H2O2 [74]. It is also possible that previous studies may have overestimated the basal rates of mitochondrial H2O2 production. The fluorescent dyes used in previous studies can either undergo auto-oxidation through redox-cycling (e.g. DCF [75]) or can be metabolized by mitochondrial enzymes (e.g. Amplex Red [76]) into fluorescent products and increase the apparent rates of ROS formation. More importantly, our data indicate that Ca2+-mobilizing hormones stimulate a significant increase in mitochondrial levels in the alcohol-fed group, but not in the controls. These data suggest that hormone-induced increases in matrix Ca2+ can exacerbate ROS formation in the alcohol-injured mitochondria. The hormone-induced increases in mitochondrial ROS could potentially feedback onto the InsP3 receptors and stimulate additional Ca2+ release from the ER. Hajnóczky and collaborators have recently reported that Ca2+-mobilizing hormones can induce H2O2 release from the mitochondrial matrix into the cleft between the ER and mitochondria, which are sufficient in size and duration to sensitize InsP3 receptors and modulate Ca2+ release [77].
In summary, our data demonstrate that chronic alcohol consumption up-regulates the MCU Ca2+ channel and VDAC while suppressing the activity of a CsA-sensitive mitochondrial Ca2+ efflux pathway leading to the accumulation and retention of Ca2+ in the mitochondrial matrix. The changes in mitochondrial Ca2+ handling in hepatocytes could be the liver’s response to alcohol-induced increases in [Ca2+]i. The faster rates of mitochondrial Ca2+ uptake should locally suppress Ca2+-induced Ca2+-release at the level of InsP3 receptors and potentially counteract the stimulatory actions of alcohol on receptor-coupled PLC activity. In parallel, the inhibition of the CsA-sensitive Ca2+ efflux pathway could increase Ca2+ retention in the mitochondria and more effectively buffer the ethanol-induced rises in [Ca2+]i. These mitochondrial responses to alcohol exposure may initially protect the hepatocytes from cytoplasmic Ca2+ overload and cytotoxicity. However, this protective mechanism could still contribute to cell injury by causing matrix Ca2+ overload, particularly during hormone stimulation. The prolonged increases in mitochondrial Ca2+ levels could lead to mitochondrial injury by stimulating the production of mitochondrial ROS.
Acknowledgements
We thank Drs Paula Bartlett, Natalia Shirokova, Lai-Hua Xie, Jan Hoek and Andrew P. Thomas for helpful comments. We also thank Anil Noronha for technical assistance with isolating hepatocytes.
Funding
This work was supported, in whole or in part, by the National Institutes of Health Grant [AA017752] (to L.D.G.).
Abbreviations
- Ca2+
calcium
- AM
acetoxymethyl
- CFP
cyan fluorescent protein
- DCF
dihydrodichlorofluorescein
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid
- InsP3
inositol 1,4–5-trisphosphate
- PLC
phospholipase C
- mPTP
mitochondrial permeability transition pore
- CsA
cyclosporin A
- MCU
mitochondrial Ca2+ uniporter
- RR
ruthenium red
- ΔΨm
mitochondrial membrane potential
- CypD
cyclophilin D
- ICM
intracellular-like medium
- ROS
reactive oxygen species
- CCCP
carbonyl cyanide m-chlorophenyl hydrazine
- FCCP
carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- G-TPP
Gamitrinib-triphenylphosphonium
- KRB
Krebs-Ringers bicarbonate
- WEM
William’s E Medium
- mito-cpYFP
mitochondrially targeted circularly permuted yellow fluorescent protein
- mito-HyPer
mitochondrial HyPer
- VP
vasopressin
- PMF
proton motive force
- mito-GCaMP3
mitochondrially targeted GCaMP3
- ER
endoplasmic reticulum
- CS
citrate synthase
- H2O2
hydrogen peroxide
- NCLX
mitochondrial Na+/Ca2+ exchanger
Footnotes
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Hoek JB, Cahill A and Pastorino JG (2002) Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology 122, 2049–2063 doi: 10.1053/gast.2002.33613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoek JB (1994) Mitochondrial energy metabolism in chronic alcoholism. Curr. Top. Bioenerg. 17, 197–241 doi: 10.1016/B978-0-12-152517-0.50012-8 [DOI] [Google Scholar]
- 3.Porta EA, Bergman BJ and Stein AA (1965) Acute alcoholic hepatitis. Am. J. Pathol. 46, 657–689 PMID: [PMC free article] [PubMed] [Google Scholar]
- 4.Rubin E and Lieber CS (1967) Early fine structural changes in the human liver induced by alcohol. Gastroenterology 52, 1–13 PMID: [PubMed] [Google Scholar]
- 5.Rubin E and Lieber CS (1968) Alcohol-induced hepatic injury in nonalcoholic volunteers. N. Engl. J. Med. 278, 869–876 doi: 10.1056/NEJM196804182781602 [DOI] [PubMed] [Google Scholar]
- 6.Das S, Hajnoczky N, Antony AN, Csordas G, Gaspers LD, Clemens DL et al. (2012) Mitochondrial morphology and dynamics in hepatocytes from normal and ethanol-fed rats. Pflug. Arch. Eur. J. Phys. 464, 101–109 doi: 10.1007/s00424-012-1100-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iseri OA, Lieber CS and Gottlieb LS (1966) The ultrastructure of fatty liver induced by prolonged ethanol ingestion. Am. J. Pathol. 48, 535–555 PMCID: [PMC free article] [PubMed] [Google Scholar]
- 8.Lieber CS, Jones DP and Decarli LM (1965) Effects of prolonged ethanol intake: production of fatty liver despite adequate diets. J. Clin. Invest. 44, 1009–1021 doi: 10.1172/JCI105200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cunningham CC, Coleman WB and Spach PI (1990) The effects of chronic ethanol consumption on hepatic mitochondrial energy metabolism. Alcohol Alcohol 25, 127–136 PMID: [DOI] [PubMed] [Google Scholar]
- 10.Coleman WB and Cunningham CC (1991) Effect of chronic ethanol consumption on hepatic mitochondrial transcription and translation. Biochim. Biophys. Acta Bioenergetics 1058, 178–186 doi: 10.1016/S0005-2728(05)80235-X [DOI] [PubMed] [Google Scholar]
- 11.Thayer WS and Rubin E (1979) Effects of chronic ethanol intoxication on oxidative phosphorylation in rat liver submitochondrial particles. J. Biol. Chem. 254, 7717–7723 PMID: [PubMed] [Google Scholar]
- 12.Thayer WS and Rubin E (1980) Effects of chronic ethanol consumption on the respiratory chain of rat liver submitochondrial particles. Adv. Exp. Med. Biol. 132, 385–392 PMID: [DOI] [PubMed] [Google Scholar]
- 13.Young TA, Bailey SM, Van Horn CG and Cunningham CC (2006) Chronic ethanol consumption decreases mitochondrial and glycolytic production of ATP in liver. Alcohol Alcohol. 41, 254–260 doi: 10.1093/alcalc/agl017 [DOI] [PubMed] [Google Scholar]
- 14.Venkatraman A, Landar A, Davis AJ, Chamlee L, Sanderson T, Kim H et al. (2004) Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. J. Biol. Chem. 279, 22092–22101 doi: 10.1074/jbc.M402245200 [DOI] [PubMed] [Google Scholar]
- 15.Sun Q, Zhong W, Zhang W and Zhou Z (2016) Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: role of zinc deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G205–G214 doi: 10.1152/ajpgi.00270.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiser B, Gonye G, Sykora P, Crumm S and Cahill A (2011) Chronic ethanol feeding causes depression of mitochondrial elongation factor Tu in the rat liver: implications for the mitochondrial ribosome. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G815–G822 doi: 10.1152/ajpgi.00108.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoek JB, Thomas AP, Rubin R and Rubin E (1987) Ethanol-induced mobilization of calcium by activation of phosphoinositide-specific phospholipase C in intact hepatocytes. J. Biol. Chem. 262, 682–691 PMID: [PubMed] [Google Scholar]
- 18.Hoek JB, Thomas AP, Rooney TA, Higashi K and Rubin E (1992) Ethanol and signal transduction in the liver. Faseb J. 6, 2386–2396 PMID: [PubMed] [Google Scholar]
- 19.Thomas AP, Hoek JB, Rubin R and Rubin E (1989) Activation of the inositol-1,4,5-trisphosphate signaling system by acute ethanol treatment of rat hepatocytes In Cell Calcium Metabolism (Fiskum G, ed.), pp. 169–177, Springer US [Google Scholar]
- 20.Brookes PS, Yoon Y, Robotham JL, Anders MW and Sheu SS (2004) Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287, C817–C833 doi: 10.1152/ajpcell.00139.2004 [DOI] [PubMed] [Google Scholar]
- 21.Pastorino JG, Marcineviciute A, Cahill A and Hoek JB (1999) Potentiation by chronic ethanol treatment of the mitochondrial permeability transition. Biochem. Biophys. Res. Commun. 265, 405–409 doi: 10.1006/bbrc.1999.1696 [DOI] [PubMed] [Google Scholar]
- 22.King AL, Swain TM, Dickinson DA, Lesort MJ and Bailey SM (2010) Chronic ethanol consumption enhances sensitivity to Ca2+-mediated opening of the mitochondrial permeability transition pore and increases cyclophilin D in liver. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G954–G966 doi: 10.1152/ajpgi.00246.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Stefani D, Raffaello A, Teardo E, Szabo I and Rizzuto R (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 doi: 10.1038/nature10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y et al. (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345 doi: 10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchi S and Pinton P (2014) The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J. Physiol. 592, 829–839 doi: 10.1113/jphysiol.2013.268235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R and Thomas AP (1998) Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 17, 4987–5000 doi: 10.1093/emboj/17.17.4987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hajnoczky G, Robb-Gaspers LD, Seitz MB and Thomas AP (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82, 415–424 doi: 10.1016/0092-8674(95)90430-1 [DOI] [PubMed] [Google Scholar]
- 28.McCormack JG, Halestrap AP and Denton RM (1990) Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 70, 391–425 PMID: [DOI] [PubMed] [Google Scholar]
- 29.Halestrap AP (2009) What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 46, 821–831 doi: 10.1016/j.yjmcc.2009.02.021 [DOI] [PubMed] [Google Scholar]
- 30.Rasola A and Bernardi P (2011) Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 50, 222–233 doi: 10.1016/j.ceca.2011.04.007 [DOI] [PubMed] [Google Scholar]
- 31.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J et al. (2008) Superoxide flashes in single mitochondria. Cell 134, 279–290 doi: 10.1016/j.cell.2008.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palmer AE, Giacomello M, Kortemme T, Hires SA, Lev-Ram V, Baker D et al. (2006) Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem. Biol. 13, 521–530 doi: 10.1016/j.chembiol.2006.03.007 [DOI] [PubMed] [Google Scholar]
- 33.Lieber CS and DeCarli LM (1989) Liquid diet technique of ethanol administration: 1989 update. Alcohol Alcohol. 24, 197–211 PMID: [PubMed] [Google Scholar]
- 34.Rooney TA, Sass EJ and Thomas AP (1989) Characterization of cytosolic calcium oscillations induced by phenylephrine and vasopressin in single fura-2-loaded hepatocytes. J. Biol. Chem. 264, 17131–17141 PMID: [PubMed] [Google Scholar]
- 35.McNeela PM and Dehn PF (1993) An automated kinetic microassay for lactate dehydrogenase using a microplate reader. J. Tissue Cult. Methods 15, 19–22 doi: 10.1007/BF02387285 [DOI] [Google Scholar]
- 36.Fisher HF (1985) [3] L-Glutamate dehydrogenase from bovine liver. Methods Enzymol. 113, 16–27 doi: 10.1016/S0076-6879(85)13006-5 [DOI] [PubMed] [Google Scholar]
- 37.Gaspers LD and Thomas AP (2008) Calcium-dependent activation of mitochondrial metabolism in mammalian cells. Methods 46, 224–232 doi: 10.1016/j.ymeth.2008.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salmon DM and Flatt JP (1985) Effect of dietary fat content on the incidence of obesity among ad libitum fed mice. Int. J. Obes. 9, 443–449 PMID: [PubMed] [Google Scholar]
- 39.Peterson GL (1983) Determination of total protein. Methods Enzymol. 91, 95–119 doi: 10.1016/S0076-6879(83)91014-5 [DOI] [PubMed] [Google Scholar]
- 40.Ranall MV, Gabrielli BG and Gonda TJ (2011) High-content imaging of neutral lipid droplets with 1,6-diphenylhexatriene. BioTechniques 51, 35–36, 38–42 doi: 10.2144/000113702 [DOI] [PubMed] [Google Scholar]
- 41.Zelickson BR, Benavides GA, Johnson MS, Chacko BK, Venkatraman A, Landar A et al. (2011) Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochim. Biophys. Acta Bioenergetics 1807, 1573–1582 doi: 10.1016/j.bbabio.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D et al. (2006) Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911 doi: 10.1083/jcb.200608073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G et al. (2002) Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159, 613–624 doi: 10.1083/jcb.200205091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smaili SS, Stellato KA, Burnett P, Thomas AP and Gaspers LD (2001) Cyclosporin A inhibits inositol 1,4,5-trisphosphate-dependent Ca2+ signals by enhancing Ca2+ uptake into the endoplasmic reticulum and mitochondria. J. Biol. Chem. 276, 23329–23340 doi: 10.1074/jbc.M100989200 [DOI] [PubMed] [Google Scholar]
- 45.Quinlan PT, Thomas AP, Armston AE and Halestrap AP (1983) Measurement of the intramitochondrial volume in hepatocytes without cell disruption and its elevation by hormones and valinomycin. Biochem. J. 214, 395–404 doi: 10.1042/bj2140395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunter DR and Haworth RA (1979) The Ca2+-induced membrane transition in mitochondria: I. The protective mechanisms. Arch. Biochem. Biophys. 195, 453–459 doi: 10.1016/0003-9861(79)90371-0 [DOI] [PubMed] [Google Scholar]
- 47.Altieri DC, Stein GS, Lian JB and Languino LR (2012) TRAP-1, the mitochondrial Hsp90. Biochim. Biophys. Acta Mol. Cell Res. 1823, 767–773 doi: 10.1016/j.bbamcr.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang BH, Plescia J, Song HY, Meli M, Colombo G, Beebe K et al. (2009) Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J. Clin. Invest. 119, 454–464 doi: 10.1172/JCI37613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei-LaPierre L, Gong G, Gerstner BJ, Ducreux S, Yule DI, Pouvreau S et al. (2013) Respective contribution of mitochondrial superoxide and pH to mitochondria-targeted circularly permuted yellow fluorescent protein (mt-cpYFP) flash activity. J. Biol. Chem. 288, 10567–10577 doi: 10.1074/jbc.M113.455709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quatresous E, Legrand C and Pouvreau S (2012) Mitochondria-targeted cpYFP: pH or superoxide sensor? J. Gen. Physiol. 140, 567–570 doi: 10.1085/jgp.201210863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang X, Huang Z, Hou T, Xu J, Wang Y, Shang W et al. (2013) Superoxide constitutes a major signal of mitochondrial superoxide flash. Life Sci. 93, 178–186 doi: 10.1016/j.lfs.2013.06.012 [DOI] [PubMed] [Google Scholar]
- 52.Schwarzländer M, Wagner S, Ermakova YG, Belousov VV, Radi R, Beckman JS et al. (2014) The ‘mitoflash’ probe cpYFP does not respond to superoxide. Nature 514, E12–E14 doi: 10.1038/nature13858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwarzländer M, Logan DC, Fricker MD and Sweetlove LJ (2011) The circularly permuted yellow fluorescent protein cpYFP that has been used as a superoxide probe is highly responsive to pH but not superoxide in mitochondria: implications for the existence of superoxide ‘flashes’. Biochem. J. 437, 381–387 doi: 10.1042/BJ20110883 [DOI] [PubMed] [Google Scholar]
- 54.Santo-Domingo J and Demaurex N (2012) Perspectives on: SGP symposium on mitochondrial physiology and medicine: the renaissance of mitochondrial pH. J. Gen. Physiol. 139, 415–423 doi: 10.1085/jgp.201110767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, Terskikh AV et al. (2006) Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 3, 281–286 doi: 10.1038/nmeth866 [DOI] [PubMed] [Google Scholar]
- 56.Bailey SM and Cunningham CC (1998) Acute and chronic ethanol increases reactive oxygen species generation and decreases viability in fresh, isolated rat hepatocytes. Hepatology 28, 1318–1326 doi: 10.1002/hep.510280521 [DOI] [PubMed] [Google Scholar]
- 57.Gaspers LD, Mémin E and Thomas AP (2012) Calcium-dependent physiologic and pathologic stimulus-metabolic response coupling in hepatocytes. Cell Calcium 52, 93–102 doi: 10.1016/j.ceca.2012.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel S, Gaspers LD, Boucherie S, Memin E, Stellato KA, Guillon G et al. (2002) Inducible nitric-oxide synthase attenuates vasopressin-dependent Ca2+ signaling in rat hepatocytes. J. Biol. Chem. 277, 33776–33782 doi: 10.1074/jbc.M201904200 [DOI] [PubMed] [Google Scholar]
- 59.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J et al. (2010) NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl Acad. Sci. USA 107, 436–441 doi: 10.1073/pnas.0908099107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim B, Takeuchi A, Koga O, Hikida M and Matsuoka S (2012) Pivotal role of mitochondrial Na+-Ca2+ exchange in antigen receptor mediated Ca2+ signalling in DT40 and A20 B lymphocytes. J. Physiol. 590, 459–474 doi: 10.1113/jphysiol.2011.222927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A and Demaurex N (2014) NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 289, 20377–20385 doi: 10.1074/jbc.M113.540898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Marchi E, Bonora M, Giorgi C and Pinton P (2014) The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium 56, 1–13 doi: 10.1016/j.ceca.2014.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bernardi P and von Stockum S (2012) The permeability transition pore as a Ca2+ release channel: new answers to an old question. Cell Calcium 52, 22–27 doi: 10.1016/j.ceca.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Altschuld RA, Hohl CM, Castillo LC, Garleb AA, Starling RC and Brierley GP (1992) Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am. J. Physiol. 262, H1699–H1704 PMID: [DOI] [PubMed] [Google Scholar]
- 65.Ichas F, Jouaville LS and Mazat J-P (1997) Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89, 1145–1153 doi: 10.1016/S0092-8674(00)80301-3 [DOI] [PubMed] [Google Scholar]
- 66.Nicchitta CV, Kamoun M and Williamson JR (1985) Cyclosporine augments receptor-mediated cellular Ca2+ fluxes in isolated hepatocytes. J. Biol. Chem. 260, 13613–13618 PMID: [PubMed] [Google Scholar]
- 67.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA et al. (2010) Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 120, 3680–3687 doi: 10.1172/JCI43171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barsukova A, Komarov A, Hajnoczky G, Bernardi P, Bourdette D and Forte M (2011) Activation of the mitochondrial permeability transition pore modulates Ca2+ responses to physiological stimuli in adult neurons. Eur. J. Neurosci. 33, 831–842 doi: 10.1111/j.1460-9568.2010.07576.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alam MR, Baetz D and Ovize M (2015) Cyclophilin D and myocardial ischemia-reperfusion injury: a fresh perspective. J. Mol. Cell. Cardiol. 78, 80–89 doi: 10.1016/j.yjmcc.2014.09.026 [DOI] [PubMed] [Google Scholar]
- 70.Wei A-C, Liu T, Cortassa S, Winslow RL and O’Rourke B (2011) Mitochondrial Ca2+ influx and efflux rates in Guinea pig cardiac mitochondria: low and high affinity effects of cyclosporine A. Biochim. Biophys. Acta Mol. Cell Res. 1813, 1373–1381 doi: 10.1016/j.bbamcr.2011.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rizzuto R, Bernardi P, Favaron M and Azzone GF (1987) Pathways for Ca2+ efflux in heart and liver mitochondria. Biochem. J. 246, 271–277 doi: 10.1042/bj2460271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wingrove DE and Gunter TE (1986) Kinetics of mitochondrial calcium transport. I. Characteristics of the sodium-independent calcium efflux mechanism of liver mitochondria. J. Biol. Chem. 261, 15159–15165 PMID: [PubMed] [Google Scholar]
- 73.Song BJ, Moon K-H, Olsson NU and Salem N Jr (2008) Prevention of alcoholic fatty liver and mitochondrial dysfunction in the rat by long-chain polyunsaturated fatty acids. J. Hepatol. 49, 262–273 doi: 10.1016/j.jhep.2008.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morgan B, Van Laer K, Owusu TNE, Ezerina D, Pastor-Flores D, Amponsah PS et al. (2016) Real-time monitoring of basal H2O2 levels with peroxiredoxin-based probes. Nat. Chem. Biol. 12, 437–443 doi: 10.1038/nchembio.2067 [DOI] [PubMed] [Google Scholar]
- 75.Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB et al. (2012) Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 52, 1–6 doi: 10.1016/j.freeradbiomed.2011.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miwa S, Treumann A, Bell A, Vistoli G, Nelson G, Hay S et al. (2016) Carboxylesterase converts amplex red to resorufin: implications for mitochondrial H2O2 release assays. Free Radic. Biol. Med. 90, 173–183 doi: 10.1016/j.freeradbiomed.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Booth DM, Enyedi B, Geiszt M, Várnai P and Hajnoczky G (2016) Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol. Cell 63, 240–248 doi: 10.1016/j.molcel.2016.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]