

Abstract
Based principally on the cancer incidence found in survivors of the atomic bombs dropped in Hiroshima and Nagasaki, the International Commission on Radiation Protection (ICRP) and the United States National Council on Radiation Protection and Measurements (NCRP) have recommended that estimates of cancer risk for low dose exposure be extrapolated from higher doses by using a linear, no-threshold model. This recommendation is based on the dogma that the DNA of the nucleus is the main target for radiation-induced genotoxicity and, as fewer cells are directly damaged, the deleterious effects of radiation proportionally decline. In this paper, we used a precision microbeam to target an exact fraction (either 100% or ≤20%) of the cells in a confluent population and irradiated their nuclei with exactly one α particle each. We found that the frequencies of induced mutations and chromosomal changes in populations where some known fractions of nuclei were hit are consistent with non-hit cells contributing significantly to the response. In fact, irradiation of 10% of a confluent mammalian cell population with a single α particle per cell results in a mutant yield similar to that observed when all of the cells in the population are irradiated. This effect was significantly eliminated in cells pretreated with a 1 mM dose of octanol, which inhibits gap junction-mediated intercellular communication, or in cells carrying a dominant negative connexin 43 vector. The data imply that the relevant target for radiation mutagenesis is larger than an individual cell and suggest a need to reconsider the validity of the linear extrapolation in making risk estimates for low dose, high linear-energy-transfer (LET) radiation exposure.
Radiation can cause as well as cure cancer. The risk of developing radiation-induced cancer has traditionally been estimated from cancer incidence among survivors of the atomic bombs dropped in Hiroshima and Nagasaki in 1945. These data provide the best estimate of human cancer risk over the dose range from 20 to 250 cGy for low linear energy transfer radiation such as X- or γ-rays. The cancer risk at doses below 20 cGy, however, is uncertain and has been the subject of controversy for decades. Both the International Commission on Radiation Protection and the U.S. National Council on Radiation Protection and Measurements have recommended using a linear no-threshold extrapolation from higher doses where more accurate risk estimates are available (1, 2). However, this approach has drawn criticisms for being too strict on the one hand and too conservative on the other (3). A better understanding of the mechanisms of radiobiological effects at low doses would shed light on the validity of the currently used model and provide a rationale for the best estimates of risk.
Ever since X-rays were shown to induce mutation in Drosophila and maize, it has been accepted dogma that the deleterious effects of radiation, such as mutation and carcinogenesis, were due mainly to direct damage to DNA. Evidence is now emerging that extranuclear or extracellular targets are extremely important in mediating the genotoxic effects of radiation (4–16). We showed, for example, that irradiation of just the cellular cytoplasm could induce mutation in the nucleus of the target cells by a process involving oxyradicals (11). Furthermore, very low doses of α particles induced significantly higher levels of p53 in populations of human fibroblasts than expected from the number of cells that had actually been hit by an α particle (5). The excess in the fraction of responding cells, which received no radiation exposure, were termed “bystanders.” It has been difficult to measure the induction of mutations in populations of mammalian cells where only a small fraction were traversed by an exact number of α particles. Here, we used a precision charged particle beam to deliver exactly one α particle through the nuclei of a known proportion of human-hamster hybrid AL cells to clearly ascertain the magnitude of this bystander mutagenic effect. We found that cells irradiated with a single α particle can induce bystander mutagenic response in nonirradiated neighboring cells, and that gap junction cell–cell communication plays a critical role in mediating that bystander mutagenesis. Furthermore, irradiation of 10% of a population resulted in a mutagenic yield that was similar to when all of the cells in the population were hit. These results are of considerable importance in reassessing the potential genotoxic effect of low dose radiation and suggest that the assumption of direct proportionality in radiation risk assessment is seriously in error.
Materials and Methods
Cell and Culture Conditions.
The human–hamster hybrid AL cells containing a standard set of Chinese hamster ovary-K1 (CHO K1) chromosomes plus a single copy of human chromosome 11 were used in this study. Chromosome 11 encodes cell surface antigens (CD59) that render AL cells sensitive to killing by specific monoclonal antibody E7.1 in the presence of complement. Rabbit serum complement was from HRP (Denver, PA). Antibody specific to the CD59 antigen was produced from hybridoma cultures as described (17, 18). Cells were maintained in Ham's F-12 medium supplemented with 8% heat-inactivated FBS, 25 μg/ml gentamycin, and 2 × 10−4 M glycine at 37°C in a humidified 5% CO2 incubator, and passaged as described (19, 20).
Irradiation Procedure.
Approximately 500 exponentially growing AL cells in 0.5 μl volume were inoculated into each of a series of microbeam dishes constructed by drilling a 1/4 inch hole in the center of 60-mm diameter non-tissue-culture dishes as described (11, 13, 19). A 3.8-μm-thick polypropylene film was epoxied over the bottom of the hole, creating a miniwell that was then coated with Cel-Tak (BD Biosciences, Bedford, MA) to enhance cell attachment. Two days after plating, when the number of attached cells reached an average of 2,000 per dish with ≈70% of the attached cells in contact with neighboring cells, the nuclei of attached cells were stained with a 50 nM solution of Hoechst 33342 dye for 30 min. The image analysis system then located the centroid of each nucleus and irradiated some or all of them randomly, one at a time, with an exact number of α particles. After irradiation, cells were maintained in the dishes for 3 days before being removed by trypsinization and replated into culture flasks. After culture for 4–5 days, the cells were trypsinized and replated to measure the mutant fraction as described (19, 20).
Cytotoxicity of a Single α Particle Traversal Through the Nucleus.
Conditions for assessing the clonogenic survival of cells irradiated with a single α particle have previously been described (19). Briefly, irradiated and control cells in a series of miniwells were trypsinized and replated into 60-mm-diameter Petri dishes for colony formation. After incubating for 7–9 days, cultures were fixed with formaldehyde and stained with Giemsa. The number of colonies was counted to determine the surviving fraction as described (11, 13, 19).
Quantification of CD59− Mutants.
Determination of the mutant fraction was carried out as described (11, 17–20). Briefly, 5 × 104 cells were plated into each of six 60-mm dishes in 2 ml of growth medium, and the cultures were incubated for 2 h to allow for cell attachment, after which 0.3% CD59 antiserum (E7.1) and 1.5% (vol/vol) freshly thawed complement were added to each dish as described (19, 20). The cultures were further incubated for 7–8 days for colony formation. At this time, the cells were fixed and stained, and the number of CD59− mutant colonies was scored. Controls included sets of dishes containing antiserum alone, complement alone, or neither agent. Mutant yields in the cultures derived from each radiation group were determined for two consecutive weeks to ensure full expression of the mutations. The mutant fraction at each dose was calculated as the number of surviving colonies divided by the total number of cells plated after correction for any nonspecific killing because of complement alone and was expressed as the number of mutants per 105 clonogenically viable cells.
Prediction of the Mutant Yields.
Predictions of the yield of mutants where a known fraction of the cells was irradiated through the nucleus with exactly one α particle were made based on the assumption of no bystander mutagenic effect. Mathematically, we can predict the mutant fraction in a culture where a known fraction of cells has been irradiated as follows.
The number of cells that were irradiated, survived, and formed mutants is given as: F × N × P.E.IR × MIR where F is the fraction of cells irradiated with exactly one α particle, N is the total number of cells in the population, P.E.IR and MIR are the plating efficiency and mutant fraction where 100% of cells have been irradiated with exactly one α particle, respectively.
The number of cells that were not irradiated, that were attached, and that produced mutants is given as: (1 − F) × N × P.E.c × Mc where P.E.c, and Mc are the plating efficiency and mutant fraction of the controls, respectively.
The expected mutant fraction in population where a known fraction (F) of cells was irradiated by a single α particle is therefore:
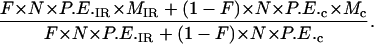 |
Cancel out N and divide all terms by P.E.c, then the formation becomes
 |
where S.F.IR is the survival fraction where 100% of the cells have been irradiated with exactly one α particle.
Treatment with Octanol.
Octanol, an effective inhibitor of gap junction communication (21), was used to investigate the role of gap junction-mediated cell–cell communication in bystander mutagenesis. Cells were treated with a 1-mM dose of octanol 2 h before and maintained until 3 days after the irradiation. After treatment, cultures were washed, trypsinized, and replated for survival and mutagenesis as described above.
Bystander Mutagenesis in Cells Genetically Deficient in Gap Junction-Mediated Cell–Cell Communication.
To further investigate the role of cell–cell communication in bystander mutagenesis, we transfected AH1-9 cells (a variant of AL cells containing a hygromycin resistant marker on chromosome 11) with either a dominant negative connexin 43 vector or with connexin 43 expressing vector and repeated the bystander mutagenic studies. Connexin 43 is the principal protein component of gap junctions (22). There is good evidence that connexin of itself (assembled in a lipid bilayer) is sufficient and necessary for the generation of gap junction channels (23, 24). The scrape-loading assay (25) was used to test the existence of gap junction-mediated intercellular communication in the AH1-9-transfected cells. Briefly, confluent, density-inhibited cells were scraped with a scalpel blade, exposed to Lucifer yellow (0.05%) and Rhodamine dextran solution (0.05%) for 3 min, and washed with PBS three times, and the distance traveled by the migrating dye was determined under a fluorescent microscope.
Detection of Chromosomal Damage.
The use of premature chromosome condensation to analyze the frequency of chromatid break as an index of chromosomal damage has been described (26). We chose to use a Calyculin A-induced G2 phase premature chromosome condensation (G2 PCC) assay to detect chromatid damage instead of the conventional metaphase spread because of its higher sensitivity. Immediately after irradiation, cells were treated with Calyculin A at a final concentration of 50 nM for 30 min at 37°C. The PCC samples were prepared according to conventional cytogenetic procedure (26, 27). Briefly, cells were treated with 75 mM KCl for 20 min at 37°C and fixed in methanol/acetic acid (3:1). The cell suspension was dropped on ethanol-cleaned slides, air-dried, stained with 5% Giemsa solution, and scored under a microscope. Chromatid-type breaks, which included chromatid breaks and acentric fragments, were scored from a minimum of 50 G2 PCC samples per experiment. The estimated chromatid-type breaks per cell, assuming no interaction between irradiated and nonirradiated cells, were similarly calculated as described above. To assess the role of gap junctions in mediating the bystander process, lindane (40 μM) or octanol (1 mM) was added to the cultures 2 h before irradiation as described (13).
Statistical Analysis.
Data were calculated as means and standard deviations. Comparisons of surviving fractions and induced mutant fractions between treated groups and controls were made by Student's t test. A P value of 0.05 or less between groups was considered to be significant.
Results
Bystander Mutagenesis in AL Cells Induced by a Single α Particle Through the Nucleus.
Consistent with our previous finding, traversal of the nucleus with a single α particle was only slightly cytotoxic to AL cells, resulting in a surviving fraction ≈0.79 ± 0.05 (19). The yield of CD59− mutants induced in populations of AL cells in which 5, 10, 20, or 100% of the cells had received exactly one α particle through the nucleus is shown in the upper curve of Fig. 1. The mutant fractions (MF) predicted, assuming no bystander interaction between the irradiated and nonirradiated cells, are shown in the lower curve. The experimental curve is significantly different from that expected. For example, the mutant fraction when 5% of the cells had been irradiated was 58% of that when all of the cells were irradiated (induced mutant fractions were 57 and 98 per 105 survivors, respectively). It is of interest to note that there was little change in the yield of mutants when the fraction of irradiated cells increased from 10 to 100%. This result could be a reflection that the percentage of irradiated cells in the population that were in direct contact with non-hit cells in mediating the bystander response had reached a plateau at 10% level and that further increases in the proportion of irradiated cells would not enhance the bystander response. Because the range of secondary electrons from α particles of this energy is ≈0.25 μm (28), it is highly unlikely that direct radiation damage to the nontargeted cells by secondary electrons contributes to the bystander effect.
Figure 1.

Induced CD59− mutant fractions per 105 survivors obtained from populations of AL cells in which 0, 5, 10, 20, or 100% had been irradiated with exactly one α particle through its nucleus. Induced mutant fraction = total mutant fraction minus background incidence, which was 46 ± 10 mutants per 105 clonogenic survivors in AL cells used in these experiments. Data are pooled from three to seven independent experiments. Error bars represent ± SD. The calculated curve deviates slightly from a straight line fitting because of the slight cytotoxic effect of single particle traversal among the irradiated cells.
Involvement of Gap Junction-Mediated Cell–Cell Communication in Bystander Mutagenesis.
Because a high cell density implies cell–cell contact in the process, we investigated the relationship between gap junctional activity and α particle-induced bystander mutagenic effect in two ways: (i) the use of octanol to inhibit gap junction-mediated intercellular communication (21) and (ii) the use of genetically engineered cells that lack gap junctions. In our first set of studies, we treated AL cells with a nontoxic and largely nonmutagenic dose of octanol (1 mM) beginning 2 h before and until 3 days after irradiation. As shown in Fig. 2, octanol reduced the yield of induced CD59− mutants from 92 ± 35 to 16 ± 3 per 105 survivors. Treatment of octanol alone resulted in an induced mutant fraction of ≈10 ± 4 per 105 survivors. Although this result indicates a role of gap junctions in the bystander mutagenic response, the effects of octanol are not limited to gap junctions but can affect other cellular structures and functions, including membrane fluidity (29). Therefore, to investigate more specifically the role of gap junction-mediated cell–cell communication with α particle-induced bystander mutagenicity, we used cells in which gap junctional activity was suppressed by a dominant negative connexin construct.
Figure 2.

Effect of octanol treatment (1 mM, 2 h before and maintained until 3 days after irradiation) on mutant fractions of AL cell population of which 20% had been irradiated with a single α particle through the nucleus. Data are from three independent experiments. Error bars represent ± SD.
AL-AH1-9 Cells Genetically Deficient in Connexin 43 Demonstrate No Bystander Mutagenesis.
We transfected AH1-9 cells with either a connexin 43 overexpressing or a dominant negative construct. By using the standard scrape-loading test as a measure of gap junction activity (22), we found that the migration of Lucifer yellow was completely blocked in cells carrying the dominant negative connexin 43 vector (Fig. 3B, DN6). In contrast, the dye was found to migrate many cell layers in distance among cells carrying connexin 43 overexpressing construct (Fig. 3A, CX10). Parental AL cells and AH1-9 cells showed a moderate migration of Lucifer yellow (data not shown). Significantly, AH1-9 cells containing the connexin 43 expressing vector showed a higher bystander mutagenic yield than that of vector control (Fig. 4, Table 1). In contrast, there was little, if any, bystander effect among AH1-9 cells carrying the dominant negative vector (Table 1). These data clearly show that gap junction intercellular communication is critical in mediating the bystander mutagenesis, although the nature of the signaling molecules involved in the communication between α particle-traversed and nontraversed cells remains to be established.
Figure 3.

The scrape-loading assay (20) was used to evaluate levels of gap junction-mediated intercellular communication in AL-AH1-9-transfected cells. In connexin 43 overexpressing (CX10) cells (A), Lucifer yellow migrated a distance of several cell layers away from the scrape. In contrast, in cells transfected with the dominant negative vector (DN6) cells (B), there was no migration of the dye (×200).
Figure 4.

Mutation fraction (MF) from population of AL-AH1-9 cells transfected with connexin 43 overexpressing vector (CX10), a dominant negative connexin 43 vector (DN6), or vector alone (CXV2). Data are from three to four independent experiments. Error bars represent ± SD. The populations of AH1-9 cells used in these experiments have higher mutant induction as well as background mutant level than the parental AL cells.
Table 1.
Comparison of mutant fractions (MF) in population of AL-AH1-9 cells transfected with either connexin 43 overexpressing vector (CX10), dominant negative vector (DN6), or with vector alone (CXV2)
| Cell line | Level of GJCC* | Induced MF† | Predicted MF | MF because of bystander‡ |
|---|---|---|---|---|
| CX10 | High | 467 | 97 | 370 |
| CXV2 | Moderate | 346 | 123 | 223 |
| DN6 | None | 149 | 141 | 8 |
GJCC, gap junction-mediated cell–cell communication.
Mutant fraction, number of CD59− mutants per 105 survivors.
Bystander mutagenic yield = induced MF minus predicted MF, assuming no interaction between irradiated and nonirradiated cells.
Using Chromatid Breakage as an Endpoint To Assess the Bystander Effect.
In addition to gene mutations, chromosomal aberrations are an important class of DNA damages induced by α particles. Therefore, we further compared the incidence of chromatid-type breaks induced in AL cells where a single α particle was delivered to the nucleus in either 20 or 100% of the cultures. As shown in Fig. 5B, in population where every cell had been irradiated, 93% of the cells contained three or more chromatid breaks. This result was in sharp contrast to the control where only 10% of the cells contained one break (Fig. 5A). When 20% of the cells in a population were irradiated with a single particle, 75% of the cells were expected to contain no breaks if one assumed there was no interaction between the irradiated and nonirradiated cells (Fig. 5C). In actuality, only 36% of the cells in this population showed no chromatid breaks (Fig. 5D). Furthermore, the profile of chromatid breaks was very different from that in which 100% of the cells in the population were hit. Addition of octanol (1 mM) or lindane (40 μM) to the 20% irradiated culture likewise obliterated the increase in chromatid breaks resulting from the bystander effects to a profile similar to that shown in Fig. 5C (data not shown).
Figure 5.

Induction of chromatid-type breaks per cell from populations of AL cells in which 0, 20 or 100% of cells were traversed by exactly one α particle through the nucleus. The data are from three to four independent experiments. Bars represent ± SD.
Discussion
Both epidemiological and experimental animal studies have indicated an association between exposure to radon (α particles) and the incidence of lung cancer (see ref. 30 for review). Although the mechanisms of radiation carcinogenesis have not been elucidated, there is good evidence that radiation-induced genetic changes such as chromosomal aberrations, gene mutations, and genomic instability play a critical role in the process. Because direct epidemiological studies on indoor radon exposure and lung cancer are equivocal, risk for low level of exposure received by the general population have been based on extrapolation from higher exposures in studies of underground miners assuming a linear, no-threshold dose-response relationship. This model for cancer estimation, however, has been a subject of controversy for decades because there is insufficient observational basis to confirm the model (3, 31). Moreover, application of this model as a basis of radioprotection and risk assessment by no means signifies its validity, merely a precautionary necessity.
It has always been accepted that most of the deleterious effects of ionizing radiation including α particles are attributable to direct nuclear hits. Recent evidence, however, indicates that extranuclear or extracellular events are also important in mediating the genotoxic effects of α particles. Early investigations of the radiation-induced bystander effect measured the frequency of sister chromatid exchanges (SCE) in populations of cells exposed to low fluencies of α particles. It was found that SCE levels were significantly higher than expected from target theory calculations of the number of cells that had actually been hit by an α particle (4, 6). Furthermore, such biological effects as induction of micronuclei (10), gene mutation (12, 13), and expression of stress-related genes (5, 9, 15) can occur in a significantly higher proportion of cells than in those traversed by an α particle. There is also evidence that bystander effects are involved in malignant transformation of mammalian cells in vitro (16).
By using a precision charged-particle microbeam, we reported that cells that had been lethally irradiated with α particles could induce mutagenesis in neighboring cells not directly hit by the particles, and that mutant induction depended on cell–cell communication (13). However, exposure to high dose of α particles is an unlikely scenario in environmental exposures to radon. To extend this observation, we show here that a single α particle traversal of a small fraction of AL cells (10–20%) induces a mutagenic response similar to that occurring when 100% of the cells in the population are hit, and that gap junction-mediated cell–cell communication plays an important role in the process. Although it is not clear whether directly irradiated cells are equally responsive to the bystander effect observed in nonirradiated cells, it is likely that, when cells are directly hit, they initiate a series of self-preservation mechanisms including DNA repair and a cell-signaling process that diminish their ability to respond to bystander signaling. In other words, irradiated cells behave differently from bystander, nonirradiated cells in their collective response to mutagenic signals.
Two important questions need to be addressed: (i) What are the mechanism(s) of the bystander mutagenic process, and (ii) what is the implication of the present findings to low dose radiation risk assessment? Based on the literature, it is likely that at least two pathways are involved in mediating radiation-induced bystander effects (see ref. 32 for review). In sparsely populated cultures, any induction of a bystander response clearly requires the presence of oxyradicals or other soluble mediators (10). In contrast, studies (including ours) with confluent monolayers have implicated gap junctional activities (12–15). These latter findings are consistent with our result that certain cytotoxic factor(s), such as cytokines and reactive oxygen or nitrogen species released into the culture medium from irradiated cells, have little, if any, effect on bystander mutagenesis (33). Furthermore, pretreatment of cells with the intracellular radical scavenger, N-acetyl cysteine (10 mM) had little effect on bystander mutagenic yield (data not shown). However, there is evidence that, among confluent human fibroblast cultures, secretion of cytokines or other growth-promoting factors by irradiated cells leads to enhanced production of reactive oxygen species in bystander cells (6, 7). These two observations among confluent cultures are not necessarily mutually exclusive because there is evidence that radiation induces long-lived organic radicals that persist for hours (34).
We also found that the gap junction inhibitor octanol significantly decreased the mutant yield. These results were further confirmed in transfected cells carrying a dominant negative connexin that reduced the mutation frequency of the cell populations to the level expected assuming no bystander effect. The gap junction channels have an apparent selectivity based principally on molecular size, allowing the movement of molecules smaller than 1,000 Da, such as cAMP, but preventing the movement of proteins or nucleic acids. These findings show that gap junction intercellular communication plays a critical role in bystander mutagenesis when cells are in close contact, although the nature of the signaling molecules involved in the communication between α particle-traversed and nontraversed cells remains to be established.
Our G2 PCC studies indicated that bystander effect could also be demonstrated at the chromosomal level. Compared with the traditional mitotic preparation based on metaphase spread, the use of chemically induced PCC provides a more sensitive and easier approach to score chromatid damage in mammalian cells (26). For example, in Calyculin A-treated cells, the G2 PCC index was found to be seven times higher than the mitotic index after a comparable treatment with colcemid (26). It should be noted that the profile of chromatid breaks found in populations where 100% of cells were hit was very different from that in which only 20% of cells received a hit. The findings are consistent with the observations that (i) high linear-energy-transfer radiation produces multiple damaged sites in the nucleus of hit cells (35) and (ii) the type of mutations induced as a result of the bystander effect is qualitatively different from that of direct nuclear hit (12, 13). The findings that, in the presence of octanol or lindane, the profile of chromatid breaks in this latter population resembled that found in controls suggest that gap junctions are indeed involved in the bystander phenomenon.
Our studies provide clear evidence that a single α particle can induce mutations and chromosome aberrations in cells that received no direct radiation exposure to their DNA. These findings imply that the target for radiation-induced genetic damage is larger than an individual cell. The observation is important in formulating risk assessment models because, for α particles, a cell cannot receive a dose lower than a single traversal and these hit cells are a minority population in lung tissue exposed to environmental radon. The observation that irradiation of as few as 10% of a cell population results in a mutagenic yield similar to that when all of the cells in the population are hit indicates that low dose α particle irradiation can induce a huge bystander mutagenic response in neighboring cells not directly traversed by α particles. The genotoxic risk at such a low dose region, therefore, may be significantly underestimated based on current practice. Because radiation-induced bystander response (mainly cell killing and genomic instability) has been demonstrated with low linear-energy-transfer radiation such as X- or γ-rays (8, 32), our findings may not be inconsistent with the recent study of radiation-related cancer risk among A-bomb survivors at the dose range of 0.15–0.30 Sv (36). Results of our present studies cast doubt on the dose at which dose linearity would be expected, a strong indication that the models presently used in predicting radiation risk at low doses are inadequate and need to be reexamined.
Acknowledgments
We thank Drs. Eric Hall and Charles Geard for helpful discussion and Dr. Hiroshi Yamasaki for providing the dominant negative connexin 43 plasmids. This work was supported by National Cancer Institute Grants CA75384, 49062, and 36447 and by National Cancer Institute Research Resource Grant RR 11623.
Abbreviations
- CX10
AH1-9 cells carrying connexin 43 overexpressing construct
- G2 PCC
G2 phase premature chromosome condensation
References
- 1.International Commission on Radiological Protection. Recommendations. New York: Pergamon; 1991. , Report no. 60. [Google Scholar]
- 2.National Council on Radiation Protection and Measurements. Report 116. Bethesda: Natl. Counc. Radiat. Prot. Meas.; 1993. [Google Scholar]
- 3.Strom D J, Cameron J R, Cohen B L. Med Phys. 1998;25:273–278. doi: 10.1118/1.598207. [DOI] [PubMed] [Google Scholar]
- 4.Nagasawa H, Little J B. Cancer Res. 1992;52:6394–6396. [PubMed] [Google Scholar]
- 5.Hickman A W, Jaramillo R J, Lechner J F, Johnson N F. Cancer Res. 1994;54:5797–5800. [PubMed] [Google Scholar]
- 6.Deshpande A, Goodwin E H, Bailey S M, Marrone B L, Lehnert B E. Radiat Res. 1996;145:260–267. [PubMed] [Google Scholar]
- 7.Narayanan P K, Goodwin E H, Lehnert B E. Cancer Res. 1997;57:3963–3971. [PubMed] [Google Scholar]
- 8.Mothersill C, Seymour C B. Radiat Res. 1998;149:256–262. [PubMed] [Google Scholar]
- 9.Azzam E I, de Toledo S M, Gooding T, Little J B. Radiat Res. 1998;150:497–504. [PubMed] [Google Scholar]
- 10.Prise K M, Belyakov O V, Folkard M, Michael B D. Int J Radiat Biol. 1998;74:793–798. doi: 10.1080/095530098141087. [DOI] [PubMed] [Google Scholar]
- 11.Wu L J, Randers-Pehrson G, Xu A, Waldren C A, Geard C R, Yu Z, Hei T K. Proc Natl Acad Sci USA. 1999;96:4959–4964. doi: 10.1073/pnas.96.9.4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagasawa H, Little J B. Radiat Res. 1999;152:552–557. [PubMed] [Google Scholar]
- 13.Zhou H, Randers-Pehrson G, Waldren C A, Vannais D, Hall E J, Hei T K. Proc Natl Acad Sci USA. 2000;97:2099–2104. doi: 10.1073/pnas.030420797. . (First Published February 11, 2000; 10.1073/pnas.030420797) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bishayee A, Hill H Z, Stein D, Rao D V, Howell R W. Radiat Res. 2001;155:335–344. doi: 10.1667/0033-7587(2001)155[0335:friagj]2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Azzam E I, de Toledo S M, Little J B. Proc Natl Acad Sci USA. 2001;98:473–478. doi: 10.1073/pnas.011417098. . (First Published January 9, 2001; 10.1073/pnas.011417098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawant S G, Randers-Pehrson G, Geard C R, Brenner D J, Hall E J. Radiat Res. 2001;155:397–401. doi: 10.1667/0033-7587(2001)155[0397:tbeiro]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 17.Waldren C A, Jones C, Puck T T. Proc Natl Acad Sci USA. 1979;76:1358–1362. doi: 10.1073/pnas.76.3.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waldren C A, Correll L, Sognier M A, Puck T T. Proc Natl Acad Sci USA. 1986;83:4839–4843. doi: 10.1073/pnas.83.13.4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hei T K, Wu L J, Liu S X, Vannais D, Waldren C A. Proc Natl Acad Sci USA. 1997;94:3765–3770. doi: 10.1073/pnas.94.8.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hei T K, Piao C Q, He Z Y, Vannais D, Waldren C A. Cancer Res. 1992;52:6305–6309. [PubMed] [Google Scholar]
- 21.Princen F, Robe P, Lechanteur C, Mesnil M, Rigo J, Gielen J, Merville M, Bours V. Clin Cancer Res. 1999;5:3639–3643. [PubMed] [Google Scholar]
- 22.Bruzzone R, Meda P. Eur J Clin Invest. 1998;18:13–21. doi: 10.1111/j.1365-2362.1988.tb01038.x. [DOI] [PubMed] [Google Scholar]
- 23.Bruzzone R, White T W, Goodenough D A. BioEssays. 1996;18:709–718. doi: 10.1002/bies.950180906. [DOI] [PubMed] [Google Scholar]
- 24.Trosko J E. Environ Health Perspect. 1998;106, Suppl. 1:331–339. doi: 10.1289/ehp.98106s1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Fouly M H, Trosko J E, Chang C C. Exp Cell Res. 1987;168:422–430. doi: 10.1016/0014-4827(87)90014-0. [DOI] [PubMed] [Google Scholar]
- 26.Masao S, Piao C, Hall E J, Hei T K. Radiat Res. 2001;155:432–439. doi: 10.1667/0033-7587(2001)155[0432:ckacdi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 27.Durante M, Furusawa Y, Majima J, Kawata T, Gotoh E. Radiat Res. 1999;151:670–676. [PubMed] [Google Scholar]
- 28.Magee J, Chatterjee A. J Phys Chem. 1980;84:3529–3536. [Google Scholar]
- 29.Sikkema J, de Bont J A, Poolman B. J Biol Chem. 1994;269:8022–8028. [PubMed] [Google Scholar]
- 30.National Research Council Committee on Health Risk of Exposure to Radon (BEIR VI) Health Effects of Exposure to Radon: Time for Reassessment. Washington, DC: Natl. Acad. Press; 1994. [Google Scholar]
- 31.Kellerer A M. Radiat Environ Biophys. 2000;39:17–24. doi: 10.1007/pl00007679. [DOI] [PubMed] [Google Scholar]
- 32.Mothersill C, Seymour C. Radiat Res. 2001;155:759–767. doi: 10.1667/0033-7587(2001)155[0759:ribeph]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 33.Zhou, H., Suzuki, M., Geard, C. & Hei, T. K. (2002) Mutat. Res., in press. [DOI] [PMC free article] [PubMed]
- 34.Koyama S, Kodama S, Suzuki K, Matsumoto T, Miyazaki T, Watanabe M. Mutat Res. 1998;421:45–54. doi: 10.1016/s0027-5107(98)00153-5. [DOI] [PubMed] [Google Scholar]
- 35.Goodhead D T. Int J Radiat Biol. 1994;65:7–17. doi: 10.1080/09553009414550021. [DOI] [PubMed] [Google Scholar]
- 36.Pierce D A, Preston D L. Radiat Res. 2000;154:178–186. doi: 10.1667/0033-7587(2000)154[0178:rrcral]2.0.co;2. [DOI] [PubMed] [Google Scholar]