

A protein complex key to the global nitrogen cycle has an unprecedented electron transport network of 192 heme groups.
Abstract
Anaerobic ammonium oxidation (anammox) is a major process in the biogeochemical nitrogen cycle in which nitrite and ammonium are converted to dinitrogen gas and water through the highly reactive intermediate hydrazine. So far, it is unknown how anammox organisms convert the toxic hydrazine into nitrogen and harvest the extremely low potential electrons (−750 mV) released in this process. We report the crystal structure and cryo electron microscopy structures of the responsible enzyme, hydrazine dehydrogenase, which is a 1.7 MDa multiprotein complex containing an extended electron transfer network of 192 heme groups spanning the entire complex. This unique molecular arrangement suggests a way in which the protein stores and releases the electrons obtained from hydrazine conversion, the final step in the globally important anammox process.
INTRODUCTION
The biological nitrogen cycle consists of a set of biochemical reactions performed by microorganisms that convert nitrogen from one chemical form into another. Starting at the extremely inert atmospheric dinitrogen (N2), the first step is the reduction of N2 to the far more reactive ammonium () by specialized nitrogen-fixing bacteria. The ammonium is then assimilated into biochemical compounds or oxidized to nitrite () by nitrifying bacteria. In the classical picture of the nitrogen cycle, nitrite is, in turn, reduced by denitrifying bacteria to nitric oxide (NO), nitrous oxide (N2O), and finally back to N2, closing the cycle. In the 1990s, however, another bacterial process, called anaerobic ammonium oxidation (anammox), was discovered (1), which is now believed to be responsible for up to 30 to 70% of the yearly nitrogen removal from the oceans (2). In this process, nitrite reacts with ammonium, yielding dinitrogen and water, as well as energy for the organism (reaction 1). The anammox process takes place in a specialized intracellular compartment, the anammoxosome (3), and starts with the one-electron reduction of to NO (reaction 2). A three-electron reduction reaction then condenses NO with ammonium to form the extremely reactive intermediate hydrazine (N2H4, reaction 3). Last, a four-electron oxidation converts hydrazine to dinitrogen gas (reaction 4).
| (1) |
| (2) |
| (3) |
| (4) |
Redox reactions 2 to 4 drive an electron transport chain that creates a proton gradient across the anammoxosome membrane driving adenosine 5′-triphosphate (ATP) synthesis (4). This use of hydrazine as a metabolic fuel by anammox bacteria is unique in nature. At −750 mV, its midpoint potential is far outside the typical range of biological redox potentials, and hydrazine is highly toxic. At present, while a mechanism for hydrazine synthesis by anammox organisms has been proposed (5), little is known about how these bacteria harness the energy released by hydrazine oxidation.
The enzyme responsible for hydrazine oxidation (reaction 4), hydrazine dehydrogenase (HDH), is a soluble multiheme c-type cytochrome complex that is not spatially associated with the anammoxosome membrane (6). Therefore, the generated electrons must be transported to the quinone pool in the membrane over a large distance. This is in stark contrast to the situation in, e.g., mitochondria, where the high-energy electrons at the start of the electron transport chain are generated inside an integral membrane protein from where they can be directly fed into the quinone pool. Moreover, care must be taken that the electrons are not inadvertently fed back to the electron-consuming enzymes that produce hydrazine in a futile electron cycle, as these enzymes are also soluble complexes residing in the anammoxosome (6). Thus, HDH should be able to safely store the low-potential electrons obtained from hydrazine oxidation until they can be passed on to the right electron transport protein with high specificity. To investigate how this is achieved, we performed biochemical investigations and determined the 2.8 Å resolution crystal structure as well as single-particle cryo electron microscopy (cryo-EM) reconstructions of HDH.
RESULTS AND DISCUSSION
Consistent with our previous studies (7), purified HDH from the anammox organisms Kuenenia stuttgartiensis and Brocadia fulgida shows a range of oligomers depending on ionic strength, with large particles showing four- or fivefold symmetry predominating at high salt concentrations (Fig. 1, A to D). Taking this information into account, we grew crystals of K. stuttgartiensis HDH at high ionic strength. These contained a very large, cube-shaped, ~150 × 150 × 150 Å3 assembly in the asymmetric unit. This assembly contains 24 copies of the HDH monomer arranged as an octamer of trimers (α3)8 (Fig. 1E). Cryo-EM reconstructions confirm that this assembly corresponds to the fourfold symmetric particles observed by negative-stain EM (figs. S1 and S2 and Supplementary Text). During model building, additional electron density was observed for 12 copies of an unexpected, ~10-kDa protein located at the vertices of the cube. Using Edman degradation, this polypeptide was identified as Kustc1130, a protein that is conserved in various anammox genera and is highly expressed in K. stuttgartiensis (Fig. 1E, fig. S3, and Supplementary Text). When K. stuttgartiensis HDH was supplemented with Kustc1130 (Fig. 1, F and G), only the fourfold symmetrical particles corresponding to the cubic assembly were observed, even at low ionic strength, indicating that Kustc1130 acts as an assembly factor. Similar results were obtained using the HDH from B. fulgida and the corresponding homolog of Kustc1130, Broful2728 (Fig. 1, H and I), suggesting that the observed complex is a general feature of HDHs. Thus, given the observation of the cubic particles in HDHs from two different organisms, the high expression level of the assembly factor (in approximately stoichiometric amounts, see fig. S3C), and the fact that in the presence of the assembly factor only the cubic assemblies are formed, we conclude that the observed cube-shaped, 1.7-MDa (α3)8β12 complex probably represents the functionally relevant oligomeric state of HDH.
Fig. 1. HDH complex.
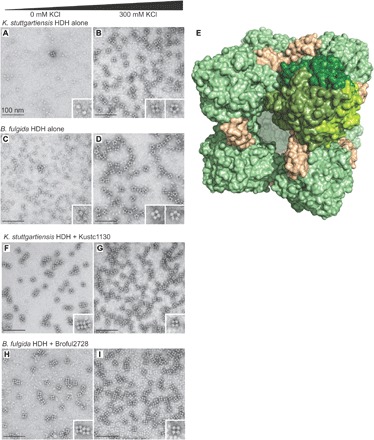
(A and B) Negative-stain electron micrographs of K. stuttgartiensis HDH alone in the absence and presence (300 mM) of KCl. (C and D) B. fulgida HDH alone, without salt, and with 300 mM KCl. (E) Crystal structure of the K. stuttgartiensis HDH/Kustc1130 assembly. HDH trimers are shown in green, with the monomers of one trimer shown in different shades of green. Assembly factor molecules are shown in beige. (F and G) K. stuttgartiensis HDH supplemented with the assembly factor Kustc1130 without salt and with 300 mM KCl. (H and I) B. fulgida HDH supplemented with the assembly factor Broful2728 without salt and with 300 mM KCl.
HDH is a member of the hydroxylamine oxidoreductase (HAO)–like enzyme family. HAO-like enzymes typically occur as cone-shaped trimers in which the eight heme groups (named hemes 1 to 8) of the three monomers are combined into a multiheme, ring-shaped relay system for electrons between the active sites at the heme 4 groups inside the trimer and binding sites for redox partners at the heme 1 groups located at the outside of the trimer (Fig. 2, A and B) (8–10). In the HDH complex, however, the situation is notably different, with the HDH trimers interacting at the heme 1 moieties, burying these and bringing them in sufficiently close proximity [7.7 Å edge-to-edge distance (11)] with each other (Fig. 2C) to allow for efficient electron transfer (12). This would result in the formation of an extended system of 192 heme groups spanning the entire assembly (Fig. 2D and movie S1), which could allow electrons generated at any of the active sites to be relayed to and stored at any other part of the complex (Fig. 2, D and E). Multiheme systems for electron transport and storage have been studied in a number of proteins, such as trimeric HAO-like proteins (13) and multiheme proteins involved in bacterial nanowire formation and dissimilatory metal reduction (14–16). However, a system with 192 hemes within one protein complex, spanning distances of >100 Å, is unprecedented.
Fig. 2. Electron transfer network in HDH.
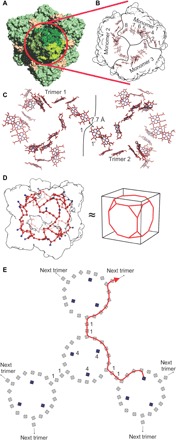
(A) Entire HDH assembly, with one trimer (front, red circle) shown in different shades of green. (B) Heme network in a trimer of HDH. The eight heme groups of each monomer form a ring-like relay system for electrons, connecting the active site heme 4 moieties to the exit sites for electrons at heme 1 as in a typical HAO-like enzyme. (C) Heme networks of two individual trimers in the HDH complex. Heme 1 of the one trimer is in close proximity to a heme 1 of the other trimer, likely allowing efficient electron transfer. Their edge-to-edge distance is indicated. (D) Proposed network of heme groups in the HDH complex. Each heme group is represented by its iron atom, shown as a red sphere or a blue sphere in case of an active site heme 4 iron. The surface of the HDH complex is shown as a black outline. The heme network approximates a truncated cube. (E). Schematic of part of the heme network in the HDH complex. Active site hemes (labeled “4”) are shown in solid blue, and other heme groups are shown as open black symbols. A possible path for electrons from one active site to a distant trimer in the complex is indicated by the red line.
The active-site heme 4 groups are each accessible from the outside of the complex through a 20 Å long tunnel. At the tunnel entrance, the protein surface is negatively charged (fig. S4). This may serve to lead hydrazine, which is likely to be protonated () at pH 6 in the anammoxosome (17), into the tunnel. The N terminus of an adjacent monomer forms part of the tunnel wall, bringing the N-terminal residue (after cleavage of the signal peptide), Val33′, into the active-site pocket located at the distal side of heme 4 (Fig. 3A). The N-terminal amino group is held in place by an N-aromatic interaction with the side chain of Trp204. Heme 4 itself is covalently bound with its C4 and C5 atoms to the Oη and Cε1 atoms of Tyr462″ from yet another monomer, resulting in pronounced perturbation of the heme known as “ruffling” (18, 19), as in other oxidizing HAO-like proteins (9, 10). Moreover, in addition to the covalent bonds between the heme’s vinyl groups and the two cysteines of the heme c binding motif, an unexpected additional covalent bond is present between one of the heme 4 methyl groups and the Sγ atom of the conserved Cys202 (Fig. 3A). Such an extra bond, which was also observed in the active site of the Kuenenia hydrazine synthase γ subunit (5), likely contributes to the heme ruffling and so may contribute to the low redox potential of this heme (7, 20–23). The active-site pocket itself is distinctly amphipathic, with one side being dominated by a His/Asp pair conserved in HAO-like proteins, whereas the other side is predominantly hydrophobic, being made up of the side chains of Met319, Trp204, and the N-terminal Val33′. The conserved His/Asp pair is perfectly positioned to abstract a proton from each of the two nitrogens of a hydrazine molecule bound to the heme 4 iron, which, with concomitant removal of two electrons, would result in a diazene intermediate. The removal of another two electrons, together with the abstraction of the remaining protons by the conserved His/Asp pair, would complete the reaction, resulting in the formation of dinitrogen. The four abstracted protons may be transferred to the large water-filled cavity present in the center of each α3 trimer, which lies directly behind the loop containing the His/Asp pair (fig. S5), as in other HAO-like enzymes (10). The four electrons, however, must be stored until they can be passed on to a redox partner and fed into the anammoxosomal electron transport chain through an exit site on the surface of the complex.
Fig. 3. Details of the HDH complex structure.
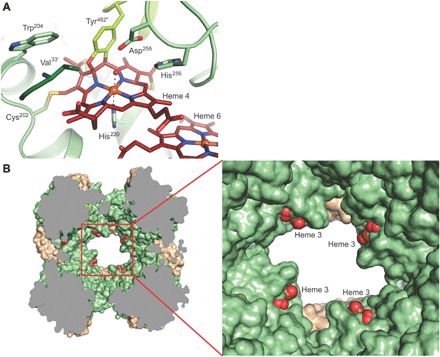
(A) Close-up of the active site. Three monomers, shown in different colors, contribute side chains to the active-site cavity, such as the conserved Asp255/His256 pair from one monomer and the N-terminal Val33′ from another. The active-site heme 4 is bound covalently to Tyr462″ from a third monomer, as well as to the conserved Cys202 in addition to the two cysteines of the heme-binding motif (in the background). (B) Sliced view of the HDH complex. Heme groups 3 of each monomer are solvent-exposed just inside the holes, leading to the central cavity in the complex.
However, since heme 1, which is the canonical exit site for electrons in HAO-like proteins, is buried in the HDH complex, another exit site is required. This can only be heme 3, which is solvent-exposed in HDH, in contrast to the situation in other HAO-like enzymes. In HDH, heme 3 binds to the protein through a rare CX4CH heme binding motif, which is considered to be a diagnostic feature for HDHs (7, 24, 25). The heme 3 moieties are located on the inside of the holes in the sides of the HDH assembly (Fig. 3B), making them accessible only to acceptors able to enter these holes. The ~30 × 50 Å holes may thus act as a filter conferring selectivity for the correct electron acceptor. Two copies of Kustc1130 flank each of the holes, which may aid selection of the correct acceptor. While the identity of the electron acceptor is currently unknown, it has been suggested that a small, single-heme cytochrome could fulfill this role (26). A ~10- to 15-kDa cytochrome c–like protein would be small enough to access the electron exit sites. Direct electron transfer to a large complex such as hydrazine synthase (5) or the proposed nitrite reductase (4), however, would likely be made impossible by the selectivity filter, which would help to prevent futile electron cycling.
In conclusion, as may be expected for an enzyme generating as many as four electrons per substrate molecule, the structure of the HDH assembly appears geared toward the storage and distribution of electrons. The extended heme system would enable several electrons to be stored far apart from each other, which is favorable for electrostatic reasons. Moreover, by allowing these electrons to be removed from the heme network through all 24 exit sites, the assembly likely permits very efficient transfer to the acceptor proteins, by increasing the chance that an acceptor is available for electron abstraction. Furthermore, the selectivity filter implied by the holes in the assembly would help to avoid accidental transfer of the low-redox potential electrons to the wrong acceptor. Thus, the HDH assembly structure presented in this study appears to offer distinct advantages for carrying out the central role of hydrazine oxidation in anammox metabolism.
MATERIALS AND METHODS
K. stuttgartiensis HDH was prepared as described previously (7) and concentrated to 20 mg/ml in 25 mM Hepes-KOH (pH 7.5) and 25 mM KCl. B. fulgida HDH was purified from a community culture by ion-exchange, hydroxyapatite, and size exclusion chromatography, as will be described elsewhere. The genes coding for the small binding proteins Kustc1130 and Broful02728 with their endogenous signal sequences were amplified from genomic DNA by polymerase chain reaction (PCR), using the following primers: 5′-CAACTACTTCACGTTGGCGAAGGTCATCTCGAGCACC-3′ (forward) and 5′-GGTGCTCGAGGTGGCTCCCTGAAGGATGAC-3′ (reverse) for Kustc1130, and 5′-GAAGTACCATGGCATTTAAGAAGATATGTGCAGG-3′ (forward) and 5′-GGTGCTCGAGGTGATGTCCGCTTGGATGAAG-3′ (reverse) for Broful02728, and were cloned into pET24d vectors with C-terminal hexa-histidine tags using the Nco I and Xho I restriction sites. The proteins were expressed in BL21 (DE3) and purified using Ni-affinity chromatography as follows. For each construct, 3 × 2–liter LB medium supplemented with kanamycin (50 μg/ml) was inoculated with 1% precultures and grown at 37°C while shaking at 120 rpm. Protein expression was induced at an OD600 (optical density at 600 nm) of 0.8 to 1.0 by the addition of 500 μM isopropyl-β-d-thiogalactopyranoside (IPTG), after which incubation was continued at 20°C, 120 rpm for 18 to 20 hours.
The bacterial cells were harvested by centrifugation at 8000 × g, 10 min, and 4°C. The cell pellets were resuspended in wash buffer (WB) [50 mM tris-HCl (pH 8.0), 300 mM NaCl, and 10 mM imidazole]. The cells were disrupted using a microfluidizer (Microfluidics, Westwood, USA) operated at a pressure of 0.7 MPa, and the lysate was clarified by centrifugation at 43,456 × g for 20 min at 4°C. The cleared lysate was loaded onto a column containing 2 ml of Ni-NTA (nickel–nitrilotriacetic acid) agarose pre-equilibrated with buffer WB. The column was further washed with 20 ml of buffer WB. The individual proteins were eluted using elution buffer (EB) [same as buffer WB but with 250 mM imidazole (pH 8.0)] and collected in 1-ml fractions. Fractions containing the desired and nearly pure proteins were pooled and concentrated using 10-kDa cutoff Amicon concentrators (Millipore Bioscience, Schwalbach, Germany). The concentrated proteins were further purified using gel filtration chromatography on a Superdex 75 (10/300 GL) column (GE Healthcare, Chicago, USA) in gel filtration buffer (GFB) [50 mM Hepes-KOH (pH 7.5) and 150 mM NaCl]. Both proteins were concentrated and buffer-exchanged to storage buffer (SB) [25 mM Hepes-KOH (pH 7.5) and 25 mM KCl] using 10-kDa cutoff Amicon concentrators up to 8 mg/ml. Both proteins were flash-frozen in small aliquots in liquid nitrogen and stored at −80°C until further use.
Crystallization and crystal structure determination
K. stuttgartiensis HDH crystals were grown in 1 + 1 μl of hanging drops against a reservoir solution containing 8 to 9% (w/v) polyethylene glycol 4000 and 0.25 M (NH4)2SO4. Hexagonal needles of 200 to 400 μm length grew in 3 to 4 days and were cryoprotected in mother liquor with 25% (v/v) glycerol before flash-cooling in liquid nitrogen. 2.8 Å resolution diffraction data were collected at the X10A beamline of the Swiss Light Source, at a wavelength of 0.95376 Å. The data were phased by molecular replacement using the KsHOX structure (10) as the search model, taking care to include the heme irons in the search. The resulting electron density map was subjected to 24-fold averaging for the HDH subunits and 12-fold averaging for the Kustc1130 subunits using DM (27), resulting in a map of excellent quality into which the model was built. The final model was refined using a test set defined in thin resolution shells because of the high noncrystallographic symmetry. It has excellent geometry, with 96.92% of residues in the most favored regions of the Ramachandran plot, 3.07% in allowed regions, and only 0.01% Ramachandran plot outliers. The quality of the electron density map is shown in fig. S6, and data collection and refinement statistics are given in table S1. Electrostatic analyses were performed using APBS (28) and PDB2PQR (29). Electrostatic parameters for the heme groups were adapted from the PARSE force field (30).
Identification of the assembly factor
The crystallographic electron density maps of K. stuttgartiensis HDH showed electron density for 12 copies of an unknown small, ~10-kDa protein. Purified HDH showed a prominent band with a mobility corresponding to ~10 kDa on 15% SDS-PAGE (polyacrylamide gel electrophoresis). This band was transferred by semi-dry blotting onto a polyvinylidene difluoride membrane using a transfer buffer consisting of 20% ethanol in 1× SDS-PAGE running buffer and a current of 40 mA during 1.5 hours. The membrane was then stained using a solution containing 0.1% Coomassie brilliant blue R250 (SERVA GmbH, Heidelberg, Germany), 10% acetic acid, and 40% methanol. The ~10-kDa band was cut out, dried, and sent for N-terminal sequencing using Edman degradation at the Proteome Factory (Germany), which showed the N-terminal amino acids to be YDVKPAKLXV, where the “X” at the ninth position in the sequence represents an amino acid whose identity could not be unambiguously determined. A BLAST search against the genome of K. stuttgartiensis then revealed the identity of the protein to be the product of kustc1130.
Negative-stain electron microscopy
HDH samples were diluted in 25 mM Hepes-KOH (pH 7.5), with or without 300 mM KCl. The final concentration of HDH was 3 μM. The assembly factors were added to the respective HDHs at a final concentration of 17 μM and incubated for 30 min. The samples were stained with 1% uranyl acetate (pH 4.2 to 4.5) on glow-discharged copper grids with carbon-coated formvar (Plano GmbH, Wetzlar, Germany). Electron micrographs were recorded on an FEI Tecnai G2 T20 transmission electron microscope (FEI NanoPort, Eindhoven, The Netherlands) running at 200 kV equipped with an FEI Eagle 4k HS, 200-kV charge-coupled device camera.
Cryo electron microscopy
Large K. stuttgartiensis HDH particles were obtained either by simple incubation at high salt concentration (300 mM KCl) or by cross-linking under high-salt conditions, as described in (7). Briefly, KsHDH was dispersed in 150 mM KCl, 50 mM Hepes-KOH (pH 7.5) at an A2801 cm of 0.5. Then, glutaraldehyde was added from a freshly diluted stock [final concentration, 0.1% (v/v)], and the mixture was incubated at 37°C for 30 min. The cross-linking reaction was quenched by adding Tris-HCl (pH 8.0) to a final concentration of 167 mM. The sample was then concentrated using a 100-kDa MWCO Amicon ultrafiltration device (Millipore, Schwalbach, Germany) and subjected to gel filtration on a Superose 6 (10/300) column (GE Healthcare, Uppsala, Sweden) in 150 mM KCl and 50 mM Hepes-KOH (pH 7.5). The peak eluting at 11 ml was pooled, and the protein was concentrated while exchanging the buffer to 25 mM HEPES-KOH and 25 mM KCl by ultrafiltration as before. The final concentration corresponded to an A2801 cm of ~3. Cryo-EM samples were prepared by placing cross-linked or non–cross-linked K. stuttgartiensis HDH on freshly glow-discharged Quantifoil R2/2 holey carbon grids (Quantifoil Micro Tools, Jena, Germany) and blotted automatically with Vitrobot (FEI NanoPort, Eindhoven, The Netherlands) using a blot time of 4 s, drain and wait times of 0 s, and a blot force of 4 a.u. (arbitrary units). Data were recorded on an FEI Tecnai Polara transmission electron microscope operating at 300 kV equipped with a Gatan K2 back-thinned direct electron detector operating in counting mode, resulting in datasets of 452 and 151 micrographs for the cross-linked and non–cross-linked samples, respectively. Images were collected manually at a nominal magnification of ×200,000 with a calibrated pixel size of 1.09 Å pixel−1. Videos were collected for 6 s with a total of 30 frames with a calibrated dose of about 1.5 e−/Å2 per frame, at defocus values between −0.75 and −2.75 μm. The micrographs were corrected using the algorithm described by Li et al. (31). Particles were automatically picked using RELION (32) and the semiautomatic procedure of EMAN (33). The contrast transfer functions were determined using gctf (34). The initial datasets contained 10,332 particle images for the cross-linked (cubic) 24- and 30-mer assembly reconstructions and 5866 for the non–cross-linked particles, extracted using a box of 350 pixels. The particle images were subjected to an initial reference-free two-dimensional (2D) classification in RELION2.1, after which visual selection of particles with interpretable features resulted in datasets of 4235 and 2071 particles, respectively, for the cross-linked 24- and 30-mer assemblies and 3246 for the non–cross-linked particles, which were used for 3D consensus refinement. Maps of the initial 3D classes were low pass–filtered to 50 Å and used as initial models for 3D gold standard refinement in RELION2.1. The final maps were sharpened by applying a negative B-factor in RELION (32), using a B-factor value estimated by the postprocessing procedure in that program. All resolutions were estimated on the basis of the gold standard Fourier shell correlation 0.143 criterion (35) between two independently refined half maps (36). Local map resolution was calculated using ResMap (37). The atomic model of the 30-mer was prepared by manually placing HDH trimers excised from the crystal structure into the density in COOT followed by rigid body refinement in PHENIX (38). In none of the EM maps, the assembly factor (Kustc1130) could be observed. This may be due to two factors: First, a highly purified fraction of HDH was used, in which only small amounts of the assembly factor may have been present. Second, given the fact that the interactions between HDH and Kustc1130 are mainly electrostatic, the high ionic strength used to prepare samples may have inhibited binding of the assembly factor.
Supplementary Material
Acknowledgments
We thank the staff at the PXII beamline of the Swiss Light Source (Paul Scherrer Institute, Villigen, Switzerland) for their help and facilities. We also thank I. Schlichting for expert crystallographic data collection and critical reading of the manuscript, and C. Roome for excellent computing support. We thank D. Mills for help and advice during cryo-EM data collection. T.R.M.B. and K.P. are very grateful to I. Schlichting and W. Kühlbrandt for continuous support. Funding: This work was funded by the Max Planck Society. B.K. was funded by ERC grant 640422. Author contributions: M.A., A.D., and T.R.M.B. designed experiments. M.A. crystallized HDH and performed biochemical experiments. M.A. and T.R.M.B. determined the crystal structure. M.A. and U.M. prepared cryo-EM and negative-stain EM samples and performed negative-stain EM. S.P. and K.P. performed cryo-EM data collection and processing. M.A., W.M., C.F., and N.M.d.A. purified proteins. M.A. and T.R.M.B. wrote the paper with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The crystal structure of K. stuttgartiensis HDH and the structure factor amplitudes have been deposited in the Protein Data Bank under accession code 6HIF. Cryo-EM maps have been deposited in the EMDB under accession codes EMD-0205, EMD-0206, and EMD-0207.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaav4310/DC1
Supplementary Text
Fig. S1. Single-particle cryo-EM map quality.
Fig. S2. Single-particle cryo-EM of HDH.
Fig. S3. Details of the assembly factor Kustc1130.
Fig. S4. Electrostatics of HDH complex.
Fig. S5. Central cavity in HDH trimers.
Fig. S6. Stereo figure showing the quality of the crystallographic electron density map.
Fig. S7. Structure of 30-mer HDH particles.
Fig. S8. Interactions in HDH assemblies.
Table S1. Data collection and refinement statistics.
Movie S1. Heme network in the HDH complex.
REFERENCES AND NOTES
- 1.Mulder A., van de Graaf A. A., Robertson L. A., Kuenen J. G., Anaerobic ammonium oxidation discovered in a denitrifying fluidized bed reactor. FEMS Microbiol. Ecol. 16, 177–183 (1995). [Google Scholar]
- 2.Arrigo K. R., Marine microorganisms and global nutrient cycles. Nature 437, 349–355 (2005). [DOI] [PubMed] [Google Scholar]
- 3.van Niftrik L. A., Fuerst J. A., Damsté J. S. S., Kuenen J. G., Jetten M. S. M., Strous M., The anammoxosome: An intracytoplasmic compartment in anammox bacteria. FEMS Microbiol. Lett. 233, 7–13 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Kartal B., de Almeida N. M., Maalcke W. J., Op den Camp H. J. M., Jetten M. S. M., Keltjens J. T., How to make a living from anaerobic ammonium oxidation. FEMS Microbiol. Rev. 37, 428–461 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Dietl A., Ferousi C., Maalcke W. J., Menzel A., de Vries S., Keltjens J. T., Jetten M. S. M., Kartal B., Barends T. R. M., The inner workings of the hydrazine synthase multiprotein complex. Nature 527, 394–397 (2015). [DOI] [PubMed] [Google Scholar]
- 6.de Almeida N. M., Neumann S., Mesman R. J., Ferousi C., Keltjens J. T., Jetten M. S. M., Kartal B., van Niftrik L., Immunogold localization of key metabolic enzymes in the anammoxosome and on the tubule-like structures of Kuenenia stuttgartiensis. J. Bacteriol. 197, 2432–2441 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maalcke W. J., Reimann J., de Vries S., Butt J. N., Dietl A., Kip N., Mersdorf U., Barends T. R. M., Jetten M. S. M., Keltjens J. T., Kartal B., Characterization of anammox hydrazine dehydrogenase, a key N2-producing enzyme in the global nitrogen cycle. J. Biol. Chem. 291, 17077–17092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Igarashi N., Moriyama H., Fujiwara T., Fukumori Y., Tanaka N., The 2.8 Å structure of hydroxylamine oxidoreductase from a nitrifying chemoautotrophic bacterium, Nitrosomonas europaea. Nat. Struct. Biol. 4, 276–284 (1997). [DOI] [PubMed] [Google Scholar]
- 9.Cedervall P., Hooper A. B., Wilmot C. M., Structural studies of hydroxylamine oxidoreductase reveal a unique heme cofactor and a previously unidentified interaction partner. Biochemistry 52, 6211–6218 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Maalcke W. J., Dietl A., Marritt S. J., Butt J. N., Jetten M. S. M., Keltjens J. T., Barends T. R. M., Kartal B., Structural basis of biological NO generation by octaheme oxidoreductases. J. Biol. Chem. 289, 1228–1242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moser C. C., Chobot S. E., Page C. C., Dutton P. L., Distance metrics for heme protein electron tunneling. Biochim. Biophys. Acta Bioenerg. 1777, 1032–1037 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moser C. C., Anderson J. L. R., Dutton P. L., Guidelines for tunneling in enzymes. Biochim. Biophys. Acta Bioenerg. 1797, 1573–1586 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurnikov I. V., Ratner M. A., Pacheco A. A., Redox equilibria in hydroxylamine oxidoreductase. Electrostatic control of electron redistribution in multielectron oxidative processes. Biochemistry 44, 1856–1863 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Clarke T. A., Edwards M. J., Gates A. J., Hall A., White G. F., Bradley J., Reardon C. L., Shi L., Beliaev A. S., Marshall M. J., Wang Z., Watmough N. J., Fredrickson J. K., Zachara J. M., Butt J. N., Richardson D. J., Structure of a bacterial cell surface decaheme electron conduit. Proc. Natl. Acad. Sci. U.S.A. 108, 9384–9389 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Breuer M., Rosso K. M., Blumberger J., Electron flow in multiheme bacterial cytochromes is a balancing act between heme electronic interaction and redox potentials. Proc. Natl. Acad. Sci. U.S.A. 111, 611–616 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Firer-Sherwood M., Pulcu G. S., Elliott S. J., Electrochemical interrogations of the Mtr cytochromes from Shewanella: Opening a potential window. J. Biol. Inorg. Chem. 13, 849–854 (2008). [DOI] [PubMed] [Google Scholar]
- 17.van der Star W. R. L., Dijkema C., de Waard P., Picioreanu C., Strous M., van Loosdrecht M. C. M., An intracellular pH gradient in the anammox bacterium Kuenenia stuttgartiensis as evaluated by 31P NMR. Appl. Microbiol. Biotechnol. 86, 311–317 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jentzen W., Song X.-Z., Shelnutt J. A., Structural characterization of synthetic and protein-bound porphyrins in terms of the lowest-frequency normal coordinates of the macrocycle. J. Phys. Chem. B 101, 1684–1699 (1997). [Google Scholar]
- 19.Shelnutt J. A., Song X.-Z., Ma J.-G., Jia S.-L., Jentzen W., Medforth C. J., Medforth C. J., Nonplanar porphyrins and their significance in proteins. Chem. Soc. Rev. 27, 31–42 (1998). [Google Scholar]
- 20.Kleingardner J. G., Bren K. L., Biological significance and applications of heme c proteins and peptides. Acc. Chem. Res. 48, 1845–1852 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Liptak M. D., Wen X., Bren K. L., NMR and DFT investigation of heme ruffling: Functional implications for cytochrome c. J. Am. Chem. Soc. 132, 9753–9763 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleingardner J., Levin B. D., Zoppellaro G., Andersson K. K., Elliott S. J., Bren K. L., Influence of heme c attachment on heme conformation and potential. J. Biol. Inorg. Chem. 23, 1073–1083 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Liu J., Chakraborty S., Hosseinzadeh P., Yu Y., Tian S., Petrik I., Bhagi A., Lu Y., Metalloproteins containing cytochrome, iron–sulfur, or copper redox centers. Chem. Rev. 114, 4366–4469 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimamura M., Nishiyama T., Shigetomo H., Toyomoto T., Kawahara Y., Furukawa K., Fujii T., Isolation of a multiheme protein with features of a hydrazine-oxidizing enzyme from an anaerobic ammonium-oxidizing enrichment culture. Appl. Environ. Microbiol. 73, 1065–1072 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oshiki M., Mizuto K., Kimura Z.-i., Kindaichi T., Satoh H., Okabe S., Genetic diversity of marine anaerobic ammonium-oxidizing bacteria as revealed by genomic and proteomic analyses of ‘Candidatus Scalindua japonica’. Environ. Microbiol. Rep. 9, 550–561 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Kartal B., Keltjens J. T., Anammox biochemistry: A tale of heme c proteins. Trends Biochem. Sci. 41, 998–1011 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Cowtan K., ‘dm’: An automated procedure for phase improvement by density modification. Joint CCP4 ESF-EACBM Newsletter Protein Crystallogr. 31, 34–38 (1994). [Google Scholar]
- 28.Jurrus E., Engel D., Star K., Monson K., Brandi J., Felberg L. E., Brookes D. H., Wilson L., Chen J., Liles K., Chun M., Li P., Gohara D. W., Dolinsky T., Konecny R., Koes D. R., Nielsen J. E., Head-Gordon T., Geng W., Krasny R., Wei G.-W., Holst M. J., McCammon J. A., Baker N. A., Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dolinsky T. J., Czodrowski P., Li H., Nielsen J. E., Jensen J. H., Klebe G., Baker N. A., PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 35, W522–W525 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sitkoff D., Sharp K. A., Honig B., Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 98, 1978–1988 (1994). [Google Scholar]
- 31.Li X., Mooney P., Zheng S., Booth C. R., Braunfeld M. B., Gubbens S., Agard D. A., Cheng Y., Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheres S. H. W., RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ludtke S. J., Baldwin P. R., Chiu W., EMAN: Semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Zhang K., Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenthal P. B., Henderson R., Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Scheres S. H. W., Chen S. X., Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kucukelbir A., Sigworth F. J., Tagare H. D., Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Afonine P. V., Poon B. K., Read R. J., Sobolev O. V., Terwilliger T. C., Urzhumtsev A., Adams P. D., Real-space refinement in PHENIX for cryo-EM and crystallography. Acta. Crystallogr. D Struct. Biol. 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holm L., Rosenström P., Dali server: Conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomis-Ruth F. X., Companys V., Qian Y., Fricker L. D., Vendrell J., Avilés F. X., Coll M., Crystal structure of avian carboxypeptidase D domain II: A prototype for the regulatory metallocarboxypeptidase subfamily. EMBO J. 18, 5817–5826 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reverter D., Maskos K., Tan F., Skidgel R. A., Bode W., Crystal structure of human carboxypeptidase M, a membrane-bound enzyme that regulates peptide hormone activity. J. Mol. Biol. 338, 257–269 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Budzik J. M., Marraffini L. A., Schneewind O., Assembly of pili on the surface of Bacillus cereus vegetative cells. Mol. Microbiol. 66, 495–510 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Budzik J. M., Poor C. B., Faull K. F., Whitelegge J. P., He C., Schneewind O., Intramolecular amide bonds stabilize pili on the surface of bacilli. Proc. Natl. Acad. Sci. U.S.A. 106, 19992–19997 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amelung S., Nerlich A., Rohde M., Spellerberg B., Cole J. N., Nizet V., Chhatwal G. S., Talay S. R., The FbaB-type fibronectin-binding protein of Streptococcus pyogenes promotes specific invasion into endothelial cells. Cell. Microbiol. 13, 1200–1211 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Messerschmidt A., Niessen H., Abt D., Einsle O., Schink B., Kroneck P. M. H., Crystal structure of pyrogallol–phloroglucinol transhydroxylase, an Mo enzyme capable of intermolecular hydroxyl transfer between phenols. Proc. Natl. Acad. Sci. U.S.A. 101, 11571–11576 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caspar D. L. D., Klug A., Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 27, 1–24 (1962). [DOI] [PubMed] [Google Scholar]
- 47.Kartal B., Maalcke W. J., de Almeida N. M., Cirpus I., Gloerich J., Geerts W., Op den Camp H. J. M., Harhangi H. R., Janssen-Megens E. M., Francoijs K.-J., Stunnenberg H. G., Keltjens J. T., Jetten M. S. M., Strous M., Molecular mechanism of anaerobic ammonium oxidation. Nature 479, 127–130 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Read R. J., Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. 42, 140–149 (1986). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaav4310/DC1
Supplementary Text
Fig. S1. Single-particle cryo-EM map quality.
Fig. S2. Single-particle cryo-EM of HDH.
Fig. S3. Details of the assembly factor Kustc1130.
Fig. S4. Electrostatics of HDH complex.
Fig. S5. Central cavity in HDH trimers.
Fig. S6. Stereo figure showing the quality of the crystallographic electron density map.
Fig. S7. Structure of 30-mer HDH particles.
Fig. S8. Interactions in HDH assemblies.
Table S1. Data collection and refinement statistics.
Movie S1. Heme network in the HDH complex.