

Abstract
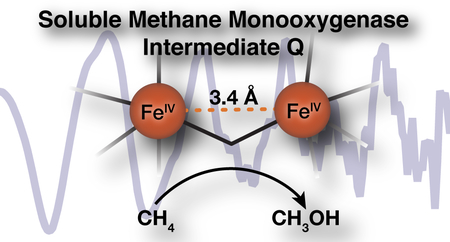
Despite decades of intense research, the core structure of the methane C-H bond breaking diiron(IV) intermediate, Q, of soluble methane monooxygenase (sMMO) remains controversial, with conflicting reports supporting either a ‘diamond’ diiron core structure or an open-core structure. Early EXAFS data assigned a short 2.46 Å Fe-Fe distance to Q (Shu, et al., Science 1997) that is inconsistent with several theoretical studies and in conflict with our recent high-resolution Fe K-edge X-ray absorption spectroscopy (XAS) studies (Castillo, et al., JACS, 2017). Herein, we revisit the EXAFS of Q using high-energy resolution fluorescence-detected extended X-ray absorption fine structure (HERFD-EXAFS) studies. The present data show no evidence for a short Fe-Fe distance, but rather a long 3.4 Å diiron distance, as observed in open-core synthetic model complexes. The previously reported 2.46 Å feature plausibly arises from a background metallic iron contribution from the experimental setup, which is eliminated in HERFD EXAFS due to the increased selectivity. Herein, we have explored the origin of the short diiron feature in partial-fluorescent yield EXAFS measurements and discuss the diagnostic features of background metallic scattering contribution to the EXAFS of dilute biological samples. Lastly, differences in sample preparation and resultant sample inhomogeneity in rapid-freeze quenched samples for EXAFS analysis are discussed. The presented approaches have broad implications for EXAFS studies of all dilute iron-containing samples.
Graphical Abstract
Introduction
Methanotrophic bacteria convert methane (CH4) to methanol (CH3OH) as the first step of their metabolic cycle utilizing methane monooxygenase enzymes (MMO).1 Two MMOs are found in nature, an iron-containing soluble MMO (sMMO) expressed under copper limiting conditions, and its copper-containing integral membrane protein counterpart (pMMO) expressed when copper is present at higher levels.2–7 Both soluble and particulate MMOs are able to activate O2 to subsequently break the strong, 104 kcal mol−1, C-H bond of methane and incorporate a single oxygen atom to form the methanol product.
The diiron active site of the hydroxylase protein (MMOH) of sMMO is transformed through several short-lived reaction cycle intermediates during the course of catalysis, two of which are shown in Figure 1.8–11 The dinuclear Fe(IV) MMOH-Q is the key intermediate that is able to cleave the C-H bond of methane.10–11 A detailed assignment of MMOH-Q’s structure is thus necessary to fully understand the process of biological methane oxidation. Numerous spectroscopic studies,8–9, 12–14 biomimetic synthetic efforts,15–19 and theoretical investigations20–23 have been dedicated to the elucidation of the structure of this critical intermediate.
Figure 1.

Abbreviated mechanism of sMMO with two proposed core structures of the MMOH-Q intermediate.
MMOH-Q from Methylosinus trichosporium OB3b (M.t. sMMO) is identified by a distinct electronic absorption band at 430 nm (ε430 = 7500 M−1 cm−1).8 Its two iron ions have equivalent environments based on their Mössbauer spectra,9 and a previous extended X-ray absorption fine structure (EXAFS) spectroscopy study of rapid-freeze-quenched (RFQ) samples of M.t. MMOH-Q assigned a short 2.46 Å Fe-Fe distance. Together, these findings supported the assignment of a closed, ‘diamond’-like, FeIV2(μ-O)2 core (Figure 1).13 Since the publication of this EXAFS spectrum two decades ago, the proposed structure has been called into question because neither computational21, 24 nor synthetic17, 25–26 efforts have provided support for such a dramatically short Fe-Fe distance in a high-valent diamond core. The consensus from these studies is that a distance of ~2.7–2.8 Å is more representative of a diamond core structure.27–28 Nevertheless, a recent continuous flow resonance Raman study of M.t. MMOHred (complexed with its regulatory protein component MMOB) reacting with O2 in solution (4 °C) reinforced the assignment of a diamond core structure for MMOH-Q.12 It was found that both atoms from O2(g) are inserted into the MMOH-Q core structure. The vibrational frequencies and the pattern of isotopic oxygen shifts observed for MMOH-Q made with 16O2, 18O2, or 16O18O were similar to those found for model compounds known to have a diamond core structure. In contrast, subsequent investigation of MMOH-Q by iron Kα High-Energy Resolution Fluorescence Detected (HERFD) X-ray Absorption Spectroscopy (XAS) provided evidence against a diamond core structure14 and, based on comparisons to relevant high-valent diiron model complexes,16, 26 supported an open core structure (Figure 1). In particular, the large pre-edge intensity observed in the HERFD-XAS spectrum of MMOH-Q could only be rationalized by a diiron core of significantly decreased symmetry and large covalent contributions, as might be provided by a terminal oxo ligand(s). Hence, multiple spectroscopic studies of MMOH-Q have yet to lead to a consensus assignment for its core structure. The key structural parameter that can distinguish between diamond and open core structures is the Fe-Fe distance, ~2.7–2.8 Å for diamond core versus ~3.4 Å for open core.26 In this context, it is important to revisit the EXAFS of MMOH-Q, which should be able to accurately reveal this distance.
Interestingly, the assignment of an anomalously short ~2.5 Å Fe-Fe distance based on EXAFS studies is not unique to MMOH-Q. Similar distances have been reported for samples of ferritin29 and ribonucleotide reductase (RNR)30–31 intermediates. In the case of RNR intermediate X, a subsequent EXAFS study by Dassama et al. (in which a 2.5 fold increase in protein concentration was achieved) showed no evidence for a ~2.5 Å Fe-Fe distance.32 The Fe-Fe distance was reevaluated to 2.8 Å, a value more consistent with both computational studies and synthetic models.32 Unfortunately, the authors did not identify the origin of the differences and simply concluded that their data were “inherently different, suggesting that they were obtained from inherently different samples.”32 The reexamination of RNR-X has set a precedent for the revaluation of similar diiron metallocenters with anomalously short distances.
Herein, we establish a new Fe-Fe distance for MMOH-Q by the HERFD-EXAFS technique. Additionally, newly acquired partial fluorescent yield (PFY)-EXAFS spectra of rapid-freeze quenched samples of MMOH-Q exhibit variable intensities of the previously observed 2.46 Å diiron feature. The possible origin and contributions to this feature observed only in the PFY-EXAFS are discussed in detail. The results of this study have important implications for the understanding of biological methane oxidation, as well as for the analysis of the EXAFS spectra of dilute biological iron-containing samples.
Materials and Methods
Preparation of freeze-quenched sMMO.
The sMMO proteins MMOH and MMOB were purified according to protocols described recently in the literature.33 The new purification scheme for MMOH provides protein with a significantly larger fraction of active MMOH,34 which enhances the yield of catalytic intermediates in the single turnover cycle. The procedure for preparing rapid freeze quench (RFQ) samples of MMOH-Q has been described previously in the HERFD XAS study on MMOH-Q.14 Each set of samples consisted of multiple replicates prepared by the same methodology. Some replicates employed 57Fe enriched MMOH to allow for Mössbauer quantitation of the yield of MMOH-Q. In the present study, two small modifications were made to this procedure to increase the yield of MMOH-Q; (i) the sMMO reaction was aged for 2.0 seconds before freeze quenching and (ii) the MMOH:MMOB protein concentration was reduced to 500 μM active sites (after mixing) in order to alleviate inefficient mixing of reduced enzyme and oxygen saturated buffer due to high sample viscosity. The total iron concentration for the MMOHred and MMOHox samples was 2.0 mM, while it was 1.0 mM for the intermediate MMOH-Q samples. The super-cooled liquid nitrogen used to prepare HERFD MMOH-RFQ-2 samples was generated by bubbling helium gas through liquid nitrogen for 5 min, at which point the temperature of liquid nitrogen dropped from −196 °C to −199 °C to decrease the boiling of the liquid.
Mössbauer Spectroscopy.
Zero-field Mössbauer spectra were recorded with a 57Co source in a Rh matrix using an alternating constant-acceleration Mössbauer spectrometer operated in transmission with a liquid helium cryostat (Oxford Instruments) for measurements at 80 K. The γ-source was kept at room temperature. All isomer shifts are quoted relative to iron metal at 300 K.
PFY X-ray spectroscopic measurements.
Iron K-edge XAS data were recorded on SSRL beamline 9–3 using a 100-element solid state Ge detector (Canberra) with a SPEAR storage ring current of ~500 mA at a power of 3.0 GeV as previously described.35 The incoming X-rays were selected using a Si(220) double-crystal monochromator and a Rh-coated mirror was utilized for harmonic rejection. Samples were maintained at ~20 K in a liquid helium flow cryostat. Data were calibrated by simultaneously measuring an iron foil, with the first inflection point set to 7111.2 eV.
HERFD X-ray spectroscopic measurements.
HERFD-XAS data were measured at the European Synchrotron Research Facility (ESRF) beamline ID-26 (6 GeV, 200 mA) equipped with a liquid helium cryostat and sample chamber operated at 20 K. Iron Kα X-ray emission detected XAS spectra were collected using a Si(311) double crystal monochromator upstream for energy selection and a 1 m radius Johann spectrometer for the measurement of X-ray emission. The crystal spectrometer was equipped with four Ge(440) crystal analyzers in a Rowland geometry. An energy selective Ketek detector, set to the Kα emission fluorescence region of interest, was employed to further increase S/N. All EXAFS and XANES spectra were collected at the maximum of the Kα emission energy (~ 6404 eV). HERFD-XAS spectra of metallic iron (α-Fe) were collected utilizing a 10 micron thick iron foil (>99.99% Fe, Goodfellow) within a layer of Kapton tape to mimic sample collection conditions. All HERFD-XAS data were calibrated to the first inflection point of an Fe reference foil set to an energy of 7111.2 eV during the initial calibration of the monochromator.
Radiation Damage.
Assessment of short XANES scans (5–60 sec for HERFD-XAS, and ~2 min for PFY-XAS) was used to maximize exposure times and assess radiation damage processes and determine dwell time limits. When necessary, the incident beam was attenuated by insertion of various number of aluminum foils into the beam path. Only scans that showed no evidence of radiation damage were included in the final analysis.
HERFD EXAFS Data Processing.
Multiple replicates of each sample (minimum 3) were used with >200 exposure spots per sample. The raw XAS spectra were initially averaged in Matlab 2014b and exported for further processing within Athena.36 All HERFD-EXAFS data were smoothed with a boxcar filter (kernel size 10) that exhibited no significant impact on the resultant Fourier transform of the EXAFS spectrum (See Supplemental Information ). A second order polynomial was fit to the pre-edge region and subtracted throughout the entire EXAFS spectrum. A three-region cubic spline (with the AUTOBK function within Athena) was employed to model the background function to a minimum of k = 14 Å−1 for all spectra. Fourier-transforms were performed over a windowed k range detailed in figure captions and all FT spectra are presented without a phase shift correction.
PFY EXAFS Data Processing.
Individual PFY-EXAFS scans were evaluated and processed in Athena prior to selected averaging as detailed in the Results. All processed PFY-EXAFS was treated the same as HERFD-EXAFS detailed above, with the exception of smoothing.
Digitalization of previously published Fourier transformed PFY-EXAFS spectra were performed with the aid of freely available WebPlotDigitizer ver. 3.12 (http://arohatgi.info/WebPlotDigitizer, June 2017).
EXAFS fitting.
See Supporting Information for description.
Results
HERFD EXAFS of sMMO
HERFD-EXAFS spectra were measured for three forms of M.t. MMOH in complex with MMOB: the FeII2 MMOHred, the FeIII2 MMOHox, and rapid-freeze quenched (RFQ) samples of the FeIV2 MMOH-Q intermediate. The Mössbauer spectra of MMOHred and MMOHox are consistent with those previously published (Figure S1, S2, and Table S1).9 The Fourier transformed (FT) HERFD-EXAFS spectra of M.t. MMOHred and MMOHox match well with previously published partial fluorescent yield (PFY) detected EXAFS spectra and those of the analogous Methylococcus capsulatus Bath MMOH (M.c. MMOH), Figure 2A, B.13, 37–38 Most critically, the outer shell scattering features of MMOHred and MMOHox yield very similar diiron radial shells at approximately R ~ 2.8 and 2.6 Å (non-phase shift corrected), respectively. This finding demonstrates the ability to directly compare more ‘traditional’ EXAFS collection methods, such as transmission mode or partial fluorescence yield (PFY) detection, with the HERFD-EXAFS methodology. This comparison also emphasizes the similarity of the core structures of MMOHred and MMOHox from Methylosinus trichosporium OB3b and Methylococcus capsulatus Bath.
Figure 2.

Non-phase shifted Fourier transform EXAFS spectra of MMOHred (A), MMOHox (B) and MMOH-RFQ (C) samples. HERFD-EXAFS (black) of M.t. MMOH is compared with the previously published PFY-EXAFS of M.t. MMOH (blue)13 and Mc (Bath) MMOH (orange). 37–38 For the FT-EXAFS from Shu, et al, a phase shift of 0.5 Å for MMOHred and 0.35 Å for MMOHox and MMOH-RFQ (see text) was subtracted from the previously published spectra to obtain a non-phase shifted FT-EXAFS for comparison. The published FT-EXAFS of Rudd et al, had no phase-shift previously applied. PFY-EXAFS of M.t. MMOH from Shu, L.; Nesheim, J. C.; Kauffmann, K.; Münck, E.; Lipscomb, J. D.; Que, L., Jr. Science 1997, 275 (5299), 515. Adapted with permission from AAAS. PFY-EXAFS of M.c. MMOH adapted with permission from Jackson Rudd, D.; Sazinsky, M. H.; Merkx, M.; Lippard, S. J.; Hedman, B.; Hodgson, K. O. Inorg. Chem. 2004, 43 (15), 4579 Rudd, D. J.; Sazinsky, M. H.; Lippard, S. J.; Hedman, B.; Hodgson, K. O. Inorg. Chem. 2005, 44 (13), 4546. Copyright 2004 and 2005 American Chemical Society.
For the HERFD-EXAFS of MMOH-Q, two methodologies were employed to trap the MMOH-Q intermediate: (i: HERFD MMOH-RFQ-1) a traditional RFQ trapping at the desired aging time (2 sec) onto liquid nitrogen cooled aluminum wheels and subsequent packing of the powder sample into sample cells33 or (ii: HERFD MMOH-RFQ-2) collection of the liquid reaction mixture into a pre-chilled sample cell followed by direct freezing after the desired aging time in helium infused (super-cooled) liquid nitrogen. The latter method is possible because the decay rate constant of MMOH-Q in the absence of a substrate is relatively slow at 4 °C (k = 0.03 s−1). This approach gives a uniformly frozen sample, but the freezing time is less precise. Both methods produced the characteristic yellow color and the appearance of a single Fe(IV) quadrupole doublet in the Mössbauer spectrum of the MMOH-Q intermediate Figure S3–4.33 The MMOH-RFQ samples contain three kinetically trapped species (MMOHred, MMOHox and MMOH-Q)33 that were quantified by Mössbauer spectroscopy, Table 1 and S1. The Kα HERFD-XAS spectrum of the MMOH-Q component of each MMOH-RFQ sample set (Figure S5) is in agreement with the recently reported HERFD-XAS spectrum.14
Table 1.
MMOH-RFQ Samples Presented
| Sample Name | % Q | Data Source |
|---|---|---|
| HERFD MMOH-RFQ-1 | 46 | This study |
| HERFD MMOH-RFQ-2 | 34 | This study |
| PFY MMOH-RFQ-1 | 61 | Shu et al. |
| PFY MMOH-RFQ-2 | 46 | Shu et al. |
| PFY MMOH-RFQ-3 | N/A | This study |
| PFY MMOH-RFQ-4 | 45* | This study |
Based on comparison of the FT-EXAFS spectrum with that of HERFD MMOH-RFQ-1 and 2.
While the FT spectra of both sets of HERFD-EXAFS of MMOH-RFQ samples were found to be qualitatively similar to each other, they differ significantly from the previously published PFY-EXAFS spectra Figure 2C.13 Most critically, there is the complete absence of the previously assigned short ‘Fe-Fe’ scattering shell at a radial distance of R = 2.15 Å in the spectra of both MMOH-RFQ samples measured by HERFD detection. The radial shell at 2.15 Å corresponds to the previously assigned short diiron scattering interaction of 2.46 Å (2.15 Å radial scattering + 0.35 Å phase shift = 2.45 Å atomic scattering distance). The absence of the previous ‘Fe-Fe’ scattering shell in the HERFD-EXAFS samples with comparable yields of MMOH-Q, while puzzling, conclusively indicates that the diiron distance of MMOH-Q is longer than 2.46 Å.
PFY vs. HERFD EXAFS
In order to understand the difference between the originally reported PFY-EXAFS of MMOH-Q vs. the present HERFD data, it is useful to briefly describe the differences in the experimental setups. The multi-element detectors typically employed in PFY-EXAFS measurements collect fluorescence events within a set energy range (~100–200 eV window), generally eliminating spectral contamination from other metal ions contained within the experiment, but not from potential background contamination of the metal being measured. As iron is the element of interest here, the potential for metallic iron (α-Fe) contained within the steel of the cryostat or other beam line components to contaminate the PFY-EXAFS spectrum is of clear concern, particularly more so for the measurement of dilute protein samples Figure 3.
Figure 3.

Basic schematic of cryostat and sample arrangement for PFY-EXAFS (A) and HERFD-EXAFS (B) measurements. The PFY detection methodology (A) collects all fluorescence emission events within the set detection energy window, including events from the sample (blue) and background contamination (red). The analyzer crystals of the HERFD-EXAFS setup (B), arranged in a Rowland geometry (C), selects fluorescent events arising from the sample (blue) to the spectrometer detector. The detection of all other fluorescent events (red) is greatly suppressed by the spectrometer as they do not satisfy Braggs’ law of the spectrometer geometry.
In a HERFD-XAS (EXAFS) experiment, the analyzer crystals and spectrometer’s detector are aligned to the desired origin of fluorescence (e.g. the sample).39–40 This Rowland circle geometric arrangement selects for fluorescence of a narrow bandwidth (< 1 eV) from the sample that satisfies Braggs’ angle at the spectrometer, specifically the angle between the sample, analyzer crystals and the detector, Figure 3C.41 Therefore, only fluorescent events from the sample at a selected energy (Fe Kα maximum, 6404 eV, Figure S6) are detected, while those arising from any position off the Rowland circle are greatly suppressed. Accordingly, background iron contamination signals from the cryostat or other components are dramatically reduced.
The non-phase shifted FT-EXAFS spectrum of metallic iron (α-Fe) exhibits a radial shell at the same position as the assigned diiron feature in the PFY-EXAFS spectrum of MMOH-Q, R = 2.15 Å (2.45 Å phase shifted) Figure 4.13 We hypothesize that background metallic iron in the previous PFY-EXAFS setup may have led to the distinct short Fe-Fe scattering feature.13 This can be illustrated clearly through a digital contamination of the HERFD-XAS spectrum with a stray metallic iron scatterer. The addition of a metallic iron component to MMOH-RFQ HERFD-XAS spectrum does not shift the rising-edge energy of the HERFD-XAS spectrum, but does significantly modulate the pre-edge region (Figure S7). As little as ~5% α-Fe contamination can be observed in the FT-EXAFS spectrum, Figure 4, and resembles the PFY-EXAFS previously published.13 This supports the contention that the short Fe-Fe scattering shell in the PFY-EXAFS measurement of MMOH-Q may have resulted from background metallic iron contamination.
Figure 4.

Non-phase shifted FT of k3-weighted HERFD-EXAFS of MMOH-RFQ - 1, black, compared with previously published PFY-EXAFS of MMOH-RFQ, blue.13 A 5% α-Fe EXAFS component was added to the HERFD-EXAFS of MMOH-RFQ-1 and Fourier-transformed to yield the digitally contaminated spectrum, red. FT HERFD-EXAFS of α-Fe, dashed grey line, is scaled by a factor of 0.1. Vertical dashed line at a radial distance of R ~ 2.15 Å is the previously assigned d(Fe-Fe) = 2.46 Å diiron scattering feature observed in the PFY-EXAFS data. The FT-EXAFS spectra are normalized to the intensity of the first shell of the HERFD-RFQ spectrum.
The digital contamination of the HERFD-EXAFS with metallic iron yields two prominent features in the FT of the EXAFS, the first at R ~ 2.15 Å, corresponding to the previously assigned 2.46 Å diiron distance, and a second feature at R ~ 4.3 Å. The latter feature is a combination of Fe-Fe single scattering at a distance of ~ 4.95 Å and strong forward and double forward multiple-iron scattering paths at a similar effective mean scattering path length.42 Both of these features should behave as clear indicators of metallic iron scattering contamination, even at small percent contributions (< 5%). Unfortunately, the previously published FT-EXAFS of MMOH-RFQ samples does not extend far enough to evaluate the presence of the R = 4.3 Å metallic iron radial shell. Therefore, PFY-EXAFS spectra of MMOH-RFQ samples were also remeasured employing standard collection practices.
PFY EXAFS of MMOH-Q
For samples MMOH-RFQ-3 and 4, prepared by the traditional RFQ methodology and measured by PFY-EXAFS, the average of all sample spots scans exhibited a radial shell at 2.1 Å, identical to that previously observed by Shu et al.13 However, the new data also shows a strong scattering interaction at R = 4.3 Å, consistent with the proposal of a metallic iron contribution.
Figure 5 shows the Fe K-edge XAS data (left) and the corresponding FT and EXAFS data (right) for two different MMOH-RFQ samples measured in PFY. Each sample was measured at multiple spots and the relative position on the sample is depicted graphically in the color-coded inset. For both samples, it is clear that by varying the spot the amount of metallic iron contamination also varies. This may be a result of the scatter contribution varying depending on the sample position and may also result from differences in sample packing (as will be discussed in more detail below). The background metallic iron contribution manifests itself most clearly in the FT of the EXAFS, where for certain sample spots intense FT features are observed at ~2.15 and 4.3 Å (non-phase shift corrected). These distinct scattering shells are in agreement with the two most prominent features of the FT-EXAFS of iron foil, Figure 4. The presence of metallic iron may also be correlated with more subtle changes in the Fe K- pre-edge XAS data. There is a correlated increase in intensity in the 7112–7118 eV region of the XAS spectrum with the ~2.15 and 4.3 Å features of the FT of the EXAFS. Therefore, the pre-edge may serve as a diagnostic for the extent of metallic background iron contributions. Using this approach for another duplicate sample of MMOH-RFQ (Figure 5C, D) it is shown that the metallic contributions to the FT can be greatly minimized by using the pre-edge region as an indicator for background iron contamination. Specifically, the sample spots with the least intense pre-edges in the 7112–7118 eV region were averaged to generate the grey spectra (Figure 5C), which correspondingly shows minimal background iron contributions in the FT (Figure 5D). This process yields an acceptable PFY-EXAFS spectrum, which is very similar to the HERFD-EXAFS spectra, Figure 6. These data thus provide strong evidence that the initial MMOH-Q data suffered from background iron contamination.
Figure 5.

PFY-XAS of MMOH-RFQ 3 and MMOH-RFQ 4. (A) XAS scans from three separate sample spots and (B) the corresponding k3-EXAFS and non-phase corrected FT spectra of each are exhibited in the top half of the figure. The bottom panels exhibit the selective averaging of different sample spots (maximum 8), as indicated by the corresponding color schematic in the inset (C). Corresponding k3-EXAFS and FT spectra of C are exhibited in panel D. Vertical dashed arrows indicate the correlation of the XAS pre-edge intensities in A and C with metallic iron scatting features at 2.15 and 4.3 Å in the FT-EXAFS spectra of B and D.
Figure 6.

Comparison of the non-phase shifted Fourier transform of the two HERFD MMOH-RFQ samples and a MMOH-RFQ sample measured by the PFY technique. The PFY EXAFS spectrum corresponds to the average of sample spots with minimal metallic iron scattering, as displayed in Figure 5. All FT EXAFS spectra are compared over a k-range of 2 to 11 Å-1.
It is important to note that the unacceptable PFY-EXAFS spectrum (from Figure 5D, blue) may be reproduced by digitally contaminating (as previously demonstrated in Figure 3) the acceptable PFY-EXAFS spectrum (Figure 5D, black) with a small component of a standard metallic iron EXAFS spectrum, Figure 7. This reaffirms the assignment of metallic iron as the background component in the PFY-EXAFS spectra.
Figure 7.

Non-phase shift corrected FT of k3-weighted PFY-EXAFS of MMOH-RFQ-4 with selective averaging of different sample spots, black and blue.13 A 5% α-Fe EXAFS component was added to the black spectrum of PFY-EXAFS MMOH-RFQ-4 and Fourier-transformed to yield the digitally contaminated spectrum, red. FT EXAFS of α-Fe, dashed grey line, is scaled by a factor of 0.1. Vertical dashed line at a radial distance of R ~ 2.15 Å is the previously assigned d(Fe-Fe) = 2.46 Å diiron scattering feature observed in the PFY-EXAFS data.
Sample Variation and Packing
The HERFD-EXAFS of MMOHred and MMOHox presented here are in excellent agreement with those samples of similar concentration studied by PFY-EXAFS. However, samples of MMOH-RFQ, also at comparable concentrations, exhibit dramatic differences between HERFD- and PFY-EXAFS spectra. Particularly, PFY-EXAFS of MMOH-RFQ samples appear to exhibit metallic iron contamination randomly over several sample spots. Samples of MMOHred and MMOHox are prepared as frozen solutions in this HERFD-EXAFS study and in previous PFY-EXAFS studies. However, rapid-freeze quenching of the MMOH-Q into liquid isopentane or onto liquid nitrogen cooled aluminum wheels yields a snow-like ‘powder’ sample that is challenging to pack uniformly. Therefore, the MMOH-RFQ samples are significantly more inhomogeneous in terms of the sample distribution within the cell compared to the frozen solutions of MMOHred and MMOHox. The homogenous frozen surface of MMOHred and MMOHox samples may enhance the absorption of the incident beam by the sample, reducing undesired scattering. In contrast, powder packed samples may present sample concentration differences, and even ‘holes’ in the sample, which allow the incident beam to pass through the sample without attenuation and scatter off other instrumental components.
To test the effects of sample preparation, MMOHox was prepared both as a frozen solution, and by RFQ to yield a powder-packed sample similar to the MMOH-RFQ samples. The powder-packed MMOHox sample has an XAS edge similar to that previously reported (Figure 8A), however, the FT-EXAFS is dramatically different from the PFY-EXAFS previously reported or the HERFD-EXAFS of MMOHox, Figure 8B. The distinct features of metallic iron scattering are observed in the FT of the PFY-EXAFS of the powder-packed MMOHox sample at R = 2.15 and 4.3 Å. The intensities of these features in the FT correlate with subtle modulations in pre-edge intensities of the XAS spectra, as also noted above for the MMOH-RFQ samples. Unfortunately, an ‘acceptable’ PFY-EXAFS spectrum could not be obtained by averaging of the XAS spectra with the lowest pre-edge intensities. XAS spectra of minimal pre-edge intensity still exhibited signs of metallic iron contamination.
Figure 8.

PFY-XAS of MMOHox as both a powder packed (A, B) and frozen solution (C, D) preparations. A shows the selective averaging of different sample spots (maximum 8), as indicated by the graphical color schematic shown in the inset. B exhibits the corresponding k3-EXAFS and FT spectra of A. The bottom panel (C, D) exhibits the average of four sample spots with the lowest pre-edge XAS intensity, and corresponding k3-EXAFS and FT spectra of C with minimal to vanishing background metallic iron scattering. The single selected sample spot in C, red, exhibited the highest pre-edge intensity of all sample spots and the corresponding metallic iron FT-EXAFS features. Vertical dashed arrows indicated the correlation of the XAS pre-edge intensity with metallic iron scatting features at 2.15 and 4.3 Å in the FT-EXAFS spectra. The omitted sample spots of C (white boxes) also possessed increased pre-edge XAS intensities, however, were omitted for clarity.
The frozen solution samples of MMOHox, in identical XAS sample cells, more closely resemble the HERFD-EXAFS. PFY-XAS spectra of the frozen solution of MMOHox appear to have no discernable metallic iron contributions in the FT of the EXAFS, Figure 8C,D. However, the PFY detected EXAFS of the frozen MMOHox is not immune to metallic iron scattering, as a single sample spot exhibiting an increased pre-edge XAS intensity (Figure 8C, red spectra) possesses the corresponding metallic iron FT EXAFS features, Figure 8D. Frozen sample preparation appears to increase the ratio of the desired sample EXAFS signal relative to undesired background contributions; however, it does not completely eliminate them and careful inspection of the data is required to identify these contributions.
Fitting of sMMO
Having now established that we have obtained reliable HERFD-detected EXAFS data for all three forms of MMOH, we now proceed to report the results of EXAFS fitting. The HERFD-EXAFS of MMOHred and MMOHox were each fit to EXAFS models with the minimal number of scattering paths to satisfactorily reproduce the experimental data. The resultant fit of MMOHox has a first coordination sphere of 5 O/N scatterers with an average mean distance of 2.06 Å and a fitted Debye-Waller like disorder fitting parameter of σ2 = 0.012 Å2. A single longer 2.48 Å Fe-O scattering interaction is also included in the fit. The large σ2-value of the 5 Fe-O/N is consistent with a large static disorder component of the first coordination shell, as this single scattering path has a five-fold degeneracy representing multiple Fe-ligand interactions at variable distances. The large σ2-value of the longer Fe-O interaction at 2.48 Å is in agreement with a disordered water molecule in the first coordination sphere as suggested by the X-ray crystal structure and the previous EXAFS analysis.37, 43 The fitted diiron distance of 3.05 Å is in excellent agreement with both the previously determined crystal structure and distances determined by EXAFS.13, 38, 43
The inclusion of a 3-fold degenerate long single Fe-C scattering path improves the fit of MMOHox. However, the fitting of this Fe-C is highly correlated with the Fe-Fe scattering path, as previously demonstrated.38 Rudd et al. have shown that the second radial-shell of MMOHox may be satisfactorily fit with either one Fe-Fe and degenerate single Fe-C scattering paths or one Fe-Fe and degenerate Fe-C-N multiple scattering paths. The fitted distances of the Fe-C and Fe-C-N scattering paths ranged from ~3.0 to 3.4 Å and exhibited high correlation with the Fe-Fe scattering path.38 Ultimately, it was clearly demonstrated by evaluation of the fitted Debye-Waller parameters that the Fe-Fe scattering interaction dominates the FT in this range of the spectrum. In the present fits, we also find that the Fe-Fe contribution dominates the outer shell. Inclusion of Fe-C single scattering paths visually improves the fit to the outer shell of the FT, but whether the Fe-C is included or not does not significantly perturb the fitted Fe-Fe distance (Figure S8, Table S2).
The non-phase shifted FT-EXAFS spectra measured by HERFD and the published PFY of MMOHred by Rudd et al. are in excellent agreement, Figure 2.37 The non-phase shifted FT HERFD-EXAFS of MMOHred compares well with the published FT PFY-EXAFS of Shu et al. after the subtraction of a 0.50 Å phase shift, Figure 2.13 We note that the 0.50 Å phase shift subtraction is larger than the 0.35 Å phase-shift that was reported. Subtraction of the reported 0.35 Å phase-shift results yields radial scattering shells of ~ 0.15 Å longer distance in the PFY-EXAFS of Shu et al. than both the PFY-EXAFS of Rudd et al. and the HERFD-EXAFS reported here, Figure S9. The phase shift discrepancy may be due to differences in data processing, including the background subtraction and/or the selection of the E0 value for the EXAFS reported in Shu et al.
The fitted diiron distance of the HERFD-EXAFS of MMOHred is 3.25 Å, in agreement with the EXAFS analysis of Rudd et al. In addition, the first shell is fit by 4 Fe-O/N scatters at ~2.04 Å, and a long ~2.54 Å Fe-O scattering interaction of modest disorder, as previously demonstrated.37 The fit of MMOHred is improved with the inclusion of degenerate Fe-C single-scattering contributions at approximately ~3 Å, corresponding to second-sphere interactions (Figure S10, Table S3). Unlike for MMOHox, the Fe-Fe and Fe-C radial shells are separable for MMOHred, as they appear as two distinct radial shells. Only fitting with a longer Fe-Fe distance and degenerate Fe-C scattering interactions at ~ 3.0 Å yields realistic σ2-values. Attempts to fit the reverse of these paths yields an unreasonably small σ2-value for the longer degenerate Fe-C path and a shorter Fe-Fe interaction with a large disorder.
The σ2-value for the Fe-Fe interaction of MMOHred is larger than that of MMOHox, again consistent with the active site structure previously observed.37 The larger σ2-value also appears consistent with the crystal structure of MMOHred, which shows variations in the Fe-Fe distances by as much as 0.15 Å in different protomers.43 As EXAFS measures the average of all the iron sites, the amount of static disorder will increase if each of the active sites do not possess the same diiron distances. Additionally, complexation of MMOH with MMOB, the regulatory protein of sMMO, induces a structural change at one iron atom of the reduced form of MMOH, potentially increasing the disorder.37, 43
Having shown that the HERFD EXAFS produces essentially identical results to previous PFY analyses, we now turn to the fits of MMOH-RFQ. Here, it is important to keep in mind that the EXAFS fits will reflect the average of all MMOH components in the samples. Based on Mössbauer spectroscopy, the MMOH-RFQ samples are comprised of 34–46% MMOH-Q, 39–54% MMOHred and 13–14% MMOHox. Hence, all three forms of MMOH will contribute to the EXAFS spectrum in varying ratios. Based on the relative scattering properties of Fe vs C, it is expected that the outer shells of the FT will be dominated by Fe-Fe contributions, as clearly demonstrated for MMOHox and also discussed above.38 From the analysis above, the diiron distances of MMOHox and MMOHred are 3.05 Å and 3.25 Å, respectively. In this simple fitting approach, we do not expect to separate the Fe-Fe of MMOH-Q from the other Fe-Fe contributions in the sample. The k = 2 to 11 Å−1 EXAFS range utilized for the Fourier transform yields a radial scattering shell resolution of ΔR ~ 0.17 Å. Therefore, if the diiron distance of MMOH-Q is within +/−0.17 Å of the Fe-Fe distance in MMOHox or MMOHred, it will not be separately resolvable. Rather, we will observe a change in the average Fe-Fe distances fitted in the MMOH-RFQ data. These fits should thus allow us to determine whether the data favor a shorter (~2.8 Å) or longer Fe-Fe contribution (~3.4 Å).
The k3-weighted HERFD-EXAFS of both the MMOH-RFQ samples sets are best fit with a 6-coordinate first shell, with a single resolved short Fe-O scattering interaction of ~1.75 Å likely corresponding to an Fe-O-Fe moiety, Figure 10, Table 3. The diiron scattering interactions observed in the MMOH-RFQ are the key interactions for the assignment of the MMOH-Q core structure. Both HERFD-EXAFS sample sets are best fit with two fractional Fe-Fe scattering interactions, one mean (N = 0.3) Fe-Fe interaction at 3.06–9 Å corresponding to the established Fe-Fe distance in MMOHox, and a longer (N = 0.7) Fe-Fe interaction at 3.30–3.35 Å. As these samples have three diiron scattering contributions with MMOHred and MMOH-Q being the largest components, the fitting of a diiron distance longer than MMOHred itself indicates that MMOH-Q possesses a diiron distance greater than 3.25 Å. The fractional coordination numbers are, within the error of EXAFS coordination number determination, in reasonable agreement with the percentages of the MMOHox and combined MMOHred and MMOH-Q components determined from Mössbauer. Importantly, by testing a range of diiron coordination number ratios (N = 0.2/0.8 to 0.5/0.5) for the two fitted diiron paths, it was determined that the fitted diiron distances in the MMOH-RFQ samples are quite stable. For instance, at N = 0.5 Fe-Fe for each scattering path, the distances vary a maximum of 0.03 Å from the Fe-Fe distances in Table 3. Additionally, attempts to include the Fe-C scattering contributions, as included in the individual fits of MMOHred and MMOHox, do not improve the fits of MMOH-RFQ. This likely results from the sample heterogeneity, which gives rise to a relatively large distribution of possible Fe-C scattering vectors, distributed over a range of distances, and resulting in relatively high disorder that cannot be well modeled. In contrast, the stronger scattering properties of the Fe-Fe contributions relative to the Fe-C contributions allow the metal-metal distances to be more reliably fit.38, 44
Figure 10.

Non-phase shifted Fourier transform of HERFD-EXAFS, black, of MMOH-RFQ HERFD-1 at 46% Q (A) and HERFD-2 at 34% Q (B). Raw k3-weighted HERFD-EXAFS, black, are included in the figure insets. Fits of each are in red.
Table 3.
HERFD-EXAFS Fit Results of MMOH-RFQ sample
| HERFD MMOH-RFQ – 1 | |||||
|---|---|---|---|---|---|
| N | Scattering Path | R (Å) | +/– | σ2 (×10−3 Å2) | +/– |
| 1 | Fe-O | 1.781 | 0.018 | 8.3 | 2.3 |
| 5 | Fe-O/N | 2.057 | 0.014 | 13.8 | 1.0 |
| 0.3 | Fe-Fe | 3.067 | 0.025 | 1.9 | 2.4 |
| 0.7 | Fe-Fe | 3.343 | 0.030 | 8.0 | 4.2 |
| ΔE0 = −1.454 eV | |||||
| HERFD MMOH-RFQ – 2 | |||||
| 1 | Fe-O | 1.758 | 0.011 | 7.6 | 1.5 |
| 5 | Fe-O/N | 2.035 | 0.009 | 12.1 | 0.6 |
| 0.3 | Fe-Fe | 3.053 | 0.019 | 1.2 | 1.8 |
| 0.7 | Fe-Fe | 3.303 | 0.015 | 3.7 | 1.6 |
| ΔE0 = −4.060 eV | |||||
Multiple attempts to fit a shorter diiron scattering interaction of approximately 2.8 Å, associated with diamond cores in model complexes17, 45 and RNR-X,32 fail to accurately reproduce the experimental data. As shown in Table S3 and Figure S10, when a 2.8 Å (N = 0.5) Fe-Fe scattering path is included in the model, the resultant fit yields an ~2.7 Å Fe-Fe vector with very large σ2-values of 0.014–0.017 Å2, which are unreasonable and unexpected for such a short, well-ordered, Fe-Fe distance typically possessing values of approximately 0.003 Å2. As the sample does consist of ~50% MMOHred, a Fe-Fe path at 3.25 Å is included in all fits of MMOH-RFQ, and when it is allowed to refine, it fits to a distance of ~3.34–3.39 Å, with reasonable Debye-Waller values. In summary, these fits further confirm that the Fe-Fe scattering contribution dominates the outer shell, and that the presence of MMOH-Q in the sample results in a longer average Fe-Fe distance relative to MMOHred.
The fitted long diiron distance supports an ‘open-core’-like structure model, as drawn in Figure 1. Such a model requires the presence of a terminal Fe=O unit with a shorter Fe-O distance (~1.65 Å) on one of the two iron atoms in the open core, as predicted by model chemistry.26 Assuming a 50% yield of MMOH-Q in the sample, such a scatterer would have a maximum coordination number of 0.25, which may be difficult to incorporate unambiguously into the fit. Short Fe-O/N scattering interactions are often difficult to accurately model, even at high yields, due to the proximity of such short vectors to the atomic background of EXAFS.46–49 We note that when modeling the spline function to extract EXAFS, one seeks to minimize low R components and such background removals can negatively impact the ability to determine short distances. Additionally, the resolution of separable interactions as determined by the EXAFS k-range (2 to 11 Å−1) yields a theoretical resolution of ΔR ~ 0.17 Å, placing the separation of a terminal Fe=O scatterer from the bridging oxo scatterer at the limit of the resolution.
Discussion and Outlook
Utilization of HERFD vs PFY
HERFD-XAS has been previously employed in the study of metalloproteins and biomimetic complexes to increase the resolution of the XAS spectrum, particularly in the pre-edge region, and also to obtain “site-selective” XAS separated by differences in metal spin state.14, 50–54 It is shown here that background iron contamination in EXAFS measurements can be eliminated by employing HERFD-EXAFS, a novel application of the HERFD technique. Using this new approach, the previously measured short 2.46 Å diiron distance13 for MMOH-Q is no longer observed. The origin of this feature in the older samples is assigned to background scattering fluorescence from metallic iron present in the EXAFS sample chamber, which can become dominant in dilute protein samples prepared by the rapid freeze quench technique. The distinct features of metallic iron contamination are easily diagnosed in the Fourier transform of the EXAFS spectrum. Modulations in the pre-edge region of the spectrum, which manifest in an intense “shelf-like” pre-edge in the 7112–7118 eV region, positively correlate with the presence of metallic iron features in the FT of the EXAFS spectrum, allowing for potential ‘real-time’ diagnosis of metallic iron contamination during data acquisition.
While the HERFD-EXAFS presented here demonstrates the ability of this technique to dramatically eliminate potential metallic scattering contributions, this study also provides insight and serves as a cautionary note for future studies of dilute metalloproteins and rapid-freeze quenched samples. It is feasible to collect satisfactory PFY-EXAFS of samples prepared by RFQ techniques, but additional data collection time may be necessary. Careful inspection of individual scans must be undertaken to recognize the presence of metallic scattering contributions and eliminate contaminated scans from final data averages. This is exemplified by the collection of a ‘background-free’ PFY-EXAFS spectrum with the same quality as HERFD-EXAFS detection, Figure 6. It is well-demonstrated through different EXAFS sample preparations of the better studied MMOHox that the physical preparation method may significantly influence background scattering contributions. The RFQ method results in ‘snow-like’ crystals that must be packed into a sample cell. These packed crystals may form an inhomogeneous scattering surface for EXAFS collection, allowing for increased background scattering when using PFY detection. In contrast, HERFD-EXAFS is effectively immune to this inhomogeneous packing problem, since the Bragg optic of the emission spectrometer rejects the undesired scattering events.
It is important to note, however, that there are other experimental considerations that should be taken into account when planning for HERFD- or PFY-detected EXAFS measurements of dilute metalloproteins. HERFD EXAFS on dilute metalloproteins requires a high flux insertion device beamline with a small focused beam size (for the present studies a 0.015 × 0.100 mm beam size was used) on the sample. In contrast, PFY EXAFS can be measured on either insertion device or bend magnet beamlines without the need for focusing (for the present PFY studies a 1 × 4 mm was used). Hence, the HERFD EXAFS is collected with 1–2 orders of magnitude more photon flux on the sample, so total sample exposure due to beam induced damage must be correspondingly reduced. This requires fast synchronized scanning of the monochromator and the insertion device in order to allow for a single HERFD-EXAFS scan to be acquired in as little as 5 seconds. This is in stark contrast to stepper monochromators, which typically require tens of minutes for an EXAFS scan. We note that HERFD detection is further limited by the use of the emission spectrometer, which significantly reduces the detected solid angle relative to large multi-element solid state detectors, employed for PFY EXAFS measurements. As a result, HERFD detection can require significantly more time to collect quality EXAFS. As an example, the PFY EXAFS of MMOHox required ~2 hours for usable EXAFS to k > 11 Å−1, while achieving similar results by HERFD detection required ~12 hours. Finally, we note that access to HERFD detection is also limited by the fact that there are relatively few beamlines with large array crystal spectrometers suitable for measurements on dilute biological systems. Hence, while HERFD EXAFS offers the ability to collect ‘background-free’ data of dilute samples, the technique at present cannot replace PFY EXAFS. We also would like to emphasize that through careful analysis of both the edge and EXAFS region of PFY-detected data, metal background contamination can be identified, and in some cases mitigated by optimizing sample preparation (e.g. maximizing absorber concentration and sample homogeneity), as is discussed further below. For this study of MMOH-Q, comparisons of HERFD and PFY-EXAFS have allowed for the clear assignment of background metallic contributions in PFY-EXAFS collection. Further development and improvement of PFY-EXAFS methodology may allow for similar ‘background-free’ EXAFS to be collected in the future without the need to turn to HERFD methodologies.
The metallic background contamination identified in the present study may also explain the erroneously short diiron distances observed in other biological systems examined over the past 20 years.29–31, 55 For instance, the first samples of the RNR-X intermediate for EXAFS characterization were prepared by rapid-freeze quenching of the intermediate into liquid isopentane and subsequent packing of the frozen powder into dual Mössbauer/X-ray sample cells.30 The EXAFS of RNR-X yielded a short diiron scattering distance of 2.5 Å, of similar character to the metallic scattering features presented here. Subsequent annealing of the RFQ-trapped sample to the oxidized form resulted in the disappearance of the short radial scattering shell assigned to the 2.5 Å distance of the intermediate. This was interpreted as validation of the RNR-X intermediate’s sample integrity. However, we hypothesize that the annealing process may have changed the physical morphology of the initially powder packed sample, resulting in a more uniform sample distribution better suited for EXAFS collection.
The subsequent reevaluation of the diiron distance of RNR-X to ~2.8 Å was achieved by using significantly more concentrated samples (6.2 mM vs ~ 2.0 mM Fe). While in the initial studies the concentrations of the samples were limited by the solubility of O2, the more recent study utilized chlorite dismutase to generate higher concentrations of O2 in solution, thus enabling the 3-fold increase in the concentration of the intermediate. The observation of a fundamentally different diiron distance in Dassama et al.’s study was attributed to the data being “obtained from inherently different samples (i.e., not from the same species).”32 In our view, a more likely interpretation is that the initial study suffered from background metallic iron contributions, which were eliminated in the subsequent study by significantly increasing the sample Fe concentration relative to potential background signals. Unfortunately, for many metalloproteins like sMMO, their intrinsic solubility may limit the practical realization of highly concentrated samples.32
The observation of erroneously short ~2.5 Å Fe scattering interactions29–31 appears to be a persistent issue, as EXAFS spectra measured more recently may continue to display this artifact. For example, the PFY-EXAFS of the hetero-bimetallic Mn-Fe RNR intermediate X55 showed a short metal scattering interaction only in the Fe-EXAFS spectrum but not within the Mn-EXAFS spectrum. The curious observation that the short ‘metal-metal’ scattering-like interaction is only observed in the Fe-EXAFS implies this feature is not from the Mn-Fe core itself, as it would then be present in both Mn and Fe-EXAFS spectra. However, this observation is consistent with background iron metal scattering that is only observed at the Fe K-edge. The HERFD-EXAFS technique applied here represents an important experimental advance for eliminating background contamination in studies of dilute biological samples. To our knowledge, this study is the first application of this method to eliminate confounding background artifacts.
MMOH-Q
Using HERFD-EXAFS, we have shown that MMOH-RFQ samples of Q possess a diiron distance longer than the diiron distances of MMOHox (3.05 Å) and MMOHred (3.27 Å). These fitted long diiron distances of ~3.30–3.34 Å represent the average of the inseparable MMOHred and MMOH-Q Fe-Fe interactions. The longer fitted distance than found for MMOHred clearly indicates that MMOH-Q is ‘pulling’ this diiron interaction to be longer than 3.27 Å. As we know the ratios of MMOHred and MMOH-Q in each of these samples from Mössbauer spectroscopy, and the Fe-Fe distance in MMOHred is uniquely determined from the MMOHred EXAFS fits, the distance of MMOH-Q can be estimated to be ~3.4 Å. The diiron distance of MMOHred and the estimated distance of MMOH-Q fall within the resolution limit of the present EXAFS (ΔR ~ 0.17 Å) and thus are not expected to be individually resolved. The present finding clearly contradicts the ‘diamond’ core structure for MMOH-Q initially proposed on the basis of the short 2.46 Å diiron distance measured by PFY-EXAFS.13 Synthetic model compounds with a Fe2(μ-O)2 diamond core,27–28, 45 as well as RNR-X with a Fe2(μ-O)(μ-OH)/Fe2(μ-O)(μ−1,1-carboxylate) diamond core,32 show Fe-Fe distances of 2.7 – 2.8 Å. On the other hand, a model compound with an open core (OH)Fe-μ(O)-Fe(O) structure shows an Fe-Fe distance of 3.3 Å.16, 26 Based upon this comparison, the long ~3.4 Å distance in MMOH-Q is suggestive of an open core structure, which agrees well with our recent analysis of the Fe Kα pre-edge data of MMOH-Q and a synthetic complex with an open core.14 However, the expected terminal Fe(IV)-oxo moiety characteristic of the open core structure cannot be definitively modeled in, nor clearly ruled out, due to the resolution of the EXAFS data presented here. This limitation makes it important to consider other models that would be consistent with all of the spectroscopic data available for MMOH-Q.
One possible, open core structure for MMOH-Q is shown in Figure 11A, but it fails to account for the observation of equivalent Fe(IV) sites by Mössbauer spectroscopy as well as the lack of a detectable terminal Fe(IV)-oxo feature in either the HERFD EXAFS or continuous flow resonance Raman spectra.12 An alternative shown in Figure 11B is suggested by the open core synthetic model in which both irons have oxygen ligands.16, 56 Such a structure might result from either cleavage of the proposed cis-μ−1,2 peroxo ligand of the preceding peroxo intermediate, MMOH-P, or opening of a precursor diamond core form of MMOH-Q to yield an terminal oxo at one iron followed by addition of a water-derived ligand to the other iron. Strong H-bonding within such a core in MMOH-Q could potentially shorten the Fe(IV)-hydroxo and lengthen the Fe(IV)-oxo bond to make the Mössbauer spectra of the iron atoms more equivalent and also make a discrete terminal Fe(IV)-oxo scatterer in the EXAFS spectrum more difficult to detect. However, DFT computations of the structure of the synthetic open-core model previously studied suggests that it has significant hydrogen bonding between the hydroxo- and oxo-ligands while retaining readily distinguishable iron sites in the Mössbauer spectrum and a detectible short Fe(IV)-oxo distance in the EXAFS spectrum.16, 56 These findings suggest that a much stronger hydrogen bond would be required in MMOH-Q in order to generate the observed spectroscopic features. Other spectroscopic features of MMOH-Q that must be accounted for include the uniquely low frequency of the resonance Raman feature at 690 cm−1 and the pattern of the frequency shift when using 18O2 or 18O16O. Although it may be possible for hydrogen bonding to decrease the frequency of the Fe-oxo vibration, such an effect has only recently been demonstrated for mononuclear Fe(III)-oxo complexes in the gas phase, in which 66 cm−1 downshifts can be observed.57 The observation of a resonance Raman vibration mode from MMOH-Q made with 18O16O halfway between that made with 16O2 or 18O2 requires coupling of the vibrational modes of the two O2-derived oxygens.12 Unfortunately, it has not been possible to measure the vibrational spectrum for the synthetic diiron(IV) open-core model. Therefore, the 690 cm−1 vibration for MMOH-Q associated in past studies with the tetra-atomic vibration of a diamond core should be further analyzed, preferably with experimental resonance Raman data of an appropriate open-core model. The high-valent irons in the open-core model described here are intermediate spin and ferromagnetically coupled, while those in MMOH-Q are high spin and antiferromagnetically coupled. These differences are likely to complicate direct comparisons of optical, vibrational, and Mössbauer spectra, suggesting additional synthetic models are required.
Figure 11.
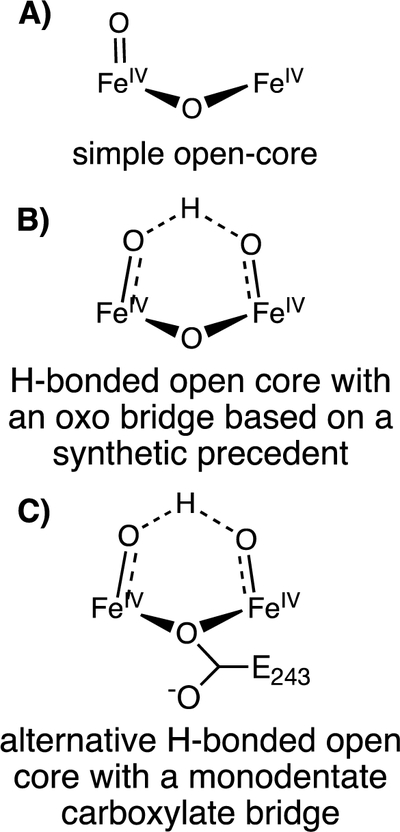
Speculative open-core models of MMOH-Q intermediate
The potential open-core model for MMOH-Q drawn in Figure 11B accounts for the required incorporation of both atoms of oxygen from O2 into the core of MMOH-Q, but the structure also contains an additional oxygen ligand (bridging or terminal) of unknown origin. It is possible that this extra oxygen derives either from the water known to be present as an iron ligand in MMOHred that is poised to initiate the catalytic cycle or from another water near the diiron cluster. Another possibility is that the μ−1,1-O(C(O)R) bridge found in MMOHred is maintained during the conversion of MMOH-P to MMOH-Q (Figure 11C). Indeed, a similar bridge is thought to be present in the RNR-X intermediate.60 This would allow for the formation of the core shown in Figure 11C with retention of both oxygens from O2 (both terminal). If the terminal oxygens are strongly hydrogen bonded, they might have similar electronic environments. However, one of the oxygens would have to shift from terminal to bridging upon reaction of MMOH-Q with substrate, because the resonance Raman spectrum shows that one oxygen from O2 is retained as a bridging oxo in the following intermediate T.12 It is unknown if the carboxylate bridge would possess weaker coupling of the iron sites leading to ferromagnetic coupling rather than the antiferromagnetic coupling characteristic of MMOH-Q.61 While carboxylate bridging leads to ferromagnetic coupling in MMOHred, other diiron iron sites in biology have demonstrated either ferro- and antiferromagnetic coupling depending on the active site metrics.26 Additionally, it is unknown whether this bridging mode would allow the vibrational modes of the terminal oxygens to couple, as required by the resonance Raman spectrum. In the future, advanced QM/MM modeling of the MMOH-Q structure will be necessary to shed light on the detailed physical and electron structure of MMOH-Q and its vibrational modes for comparison with structures such as those illustrated in Figure 11.
The present study provides the new key geometric constraint that the diiron distance is long (~3.4 Å), and explains previous observations of MMOH-Q. Further work to fully determine the exact character of MMOH-Q’s core structure must be done to ultimately understand nature’s most powerful oxidant.
Supplementary Material
Figure 9.

Non-phase shifted Fourier transform of HERFD-EXAFS, black, of MMOHox (A) and MMOHred (B). Raw k3-weighted HERFD-EXAFS, black, are included in the figure insets. Fits of each are in red.
Table 2.
HERFD-EXAFS Fit Results of MMOHox and MMOHred in complex with MMOB
| MMOHox | |||||
| N | Scattering Path | R (Å) | +/– | σ2 (×10−3 Å2) | +/– |
| 5 | Fe-O/N | 2.057 | 0.010 | 12.6 | 1.0 |
| 1 | Fe-O/N | 2.480 | 0.036 | 9.9 | 5.7 |
| 1 | Fe-Fe | 3.056 | 0.011 | 1.3 | 0.9 |
| 3 | Fe-C | 3.304 | 0.031 | 4.9 | 4.1 |
| ΔE0 = + 2.51 eV | |||||
| MMOHred | |||||
| N | Scattering Path | R (Å) | +/– | σ2 (×10−3 Å2) | +/– |
| 4 | Fe-O/N | 2.044 | 0.015 | 10.3 | 1.1 |
| 1 | Fe-O/N | 2.538 | 0.022 | 2.6 | 2.6 |
| 1 | Fe-Fe | 3.270 | 0.026 | 8.4 | 2.5 |
| 3 | Fe-C | 2.989 | 0.051 | 20.0 | 8.0 |
| ΔE0 = – 6.63 eV |
Acknowledgments:
Various members of the Inorganic Spectroscopy Department at MPI-CEC are thanked for helpful assistance with X-ray data collection. Bernd Meinert (MPI-CEC), Ruixi Fan and Dr. Yisong Guo (Carnegie Mellon University) for assistance in Mössbauer data collection. Financial support was provided by the Max Planck Society (S.D.). J.D.L. and L.Q. acknowledge the National Institutes of Health for funding (grants GM118030 to J.D.L and GM38767 to L.Q.) G.E.C. was supported by a fellowship from the Alexander von Humboldt Foundation.
The HERFD-XAS experiments were performed on beamline ID-26 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. We are grateful to Dr. Blanka Detlefs and Dr. Lucia Amidani at the ESRF for providing assistance in using beamline ID-26.
Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
Footnotes
Supporting Information:
The SI includes Mössbauer spectra, analysis and quantification, HERFD-XAS spectroscopy, Fe Kα emission spectroscopy, metallic iron XAS and EXAFS digital contamination studies, and additional EXAFS fittings. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hanson RS; Hanson TE, Methanotrophic bacteria. Microbiol. Rev 1996, 60 (2), 439–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sirajuddin S; Rosenzweig AC, Enzymatic oxidation of methane. Biochemistry 2015, 54 (14), 2283–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ross MO; Rosenzweig AC, A tale of two methane monooxygenases. J. Biol. Inorg. Chem 2017, 22 (2–3), 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tinberg CE; Lippard SJ, Dioxygen activation in soluble methane monooxygenase. Acc. Chem. Rev 2011, 44 (4), 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stanley SH; Prior SD; Leak DJ; Dalton H, Copper stress underlies the fundamental change in intracellular location of methane mono-oxygenase in methane-oxidizing organisms - studies in batch and continuous cultures. Biotechnol Lett 1983, 5 (7), 487–492. [Google Scholar]
- 6.Fox BG; Hendrich MP; Surerus KK; Andersson KK; Froland WA; Lipscomb JD; Münck E, Mössbauer EPR, and ENDOR studies of the hydroxylase and reductase components of methane monooxygenase from Methylosinus trichosporium OB3b. J. Am. Chem. Soc 1993, 115 (9), 3688––3701. [Google Scholar]
- 7.Wallar BJ; Lipscomb JD, Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem. Rev 1996, 96 (7), 2625–2658. [DOI] [PubMed] [Google Scholar]
- 8.Lee SK; Nesheim JC; Lipscomb JD, Transient intermediates of the methane monooxygenase catalytic cycle. J. Biol. Chem 1993, 268 (29), 21569–21577. [PubMed] [Google Scholar]
- 9.Lee SK; Fox BG; Froland WA; Lipscomb JD; Münck E, A transient intermediate of the methane monooxygenase catalytic cycle containing an FeIVFeIV cluster. J. Am. Chem. Soc 1993, 115 (14), 6450–6451. [Google Scholar]
- 10.Liu KE; Valentine AM; Wang DL; Huynh BH; Edmondson DE; Salifoglou A; Lippard SJ, Kinetic and spectroscopic characterization of intermediates and component interactions in reactions of methane monooxygenase from Methylococcus capsulatus (Bath). J. Am. Chem. Soc 1995, 117 (41), 10174–10185. [Google Scholar]
- 11.Liu KE; Wang DL; Huynh BH; Edmondson DE; Salifoglou A; Lippard SJ, Spectroscopic detection of intermediates in the reaction of dioxygen with the reduced methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath). J. Am. Chem. Soc 1994, 116 (16), 7465–7466. [Google Scholar]
- 12.Banerjee R; Proshlyakov Y; Lipscomb JD; Proshlyakov DA, Structure of the key species in the enzymatic oxidation of methane to methanol. Nature 2015, 518 (7539), 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shu L; Nesheim JC; Kauffmann K; Münck E; Lipscomb JD; Que L Jr., An Fe2IVO2 diamond core structure for the key intermediate Q of methane monooxygenase. Science 1997, 275 (5299), 515–8. [DOI] [PubMed] [Google Scholar]
- 14.Castillo RG; Banerjee R; Allpress CJ; Rohde GT; Bill E; Que L; Lipscomb JD; DeBeer S, High-energy-resolution fluorescence-detected X-ray absorption of the Q intermediate of soluble methane monooxygenase. J. Am. Chem. Soc 2017, 139 (49), 18024–18033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xue G; De Hont R; Münck E; Que L, Million-fold activation of the [Fe(2)(μ-O)(2)] diamond core for C-H bond cleavage. Nat. Chem 2010, 2 (5), 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue GQ; Fiedler AT; Martinho M; Münck E; Que L, Insights into the P-to-Q conversion in the catalytic cycle of methane monooxygenase from a synthetic model system. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (52), 20615–20620. [Google Scholar]
- 17.Que L Jr.; Tolman WB, Bis(μ-oxo)dimetal “diamond” cores in copper and iron complexes relevant to biocatalysis. Angew. Chem. Int. Ed 2002, 41 (7), 1114–1137. [DOI] [PubMed] [Google Scholar]
- 18.Tshuva EY; Lippard SJ, Synthetic models for non-heme carboxylate-bridged diiron metalloproteins: strategies and tactics. Chem. Rev 2004, 104 (2), 987–1012. [DOI] [PubMed] [Google Scholar]
- 19.Friedle S; Reisner E; Lippard SJ, Current challenges of modeling diiron enzyme active sites for dioxygen activation by biomimetic synthetic complexes. Chem. Soc. Rev 2010, 39 (8), 2768–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gherman BF; Baik M-H; Lippard SJ; Friesner RA, Dioxygen activation in methane monooxygenase: a theoretical study. J. Am. Chem. Soc 2004, 126 (9), 2978–2990. [DOI] [PubMed] [Google Scholar]
- 21.Huang SP; Shiota Y; Yoshizawa K, DFT study of the mechanism for methane hydroxylation by soluble methane monooxygenase (sMMO): effects of oxidation state, spin state, and coordination number. Dalton. Trans 2013, 42 (4), 1011–23. [DOI] [PubMed] [Google Scholar]
- 22.Yoshizawa K, Two-step concerted mechanism for methane hydroxylation on the diiron active site of soluble methane monooxygenase. J. Inorg. Biochem 2000, 78 (1), 23–34. [DOI] [PubMed] [Google Scholar]
- 23.Yoshizawa K; Yumura T, A non-radical mechanism for methane hydroxylation at the diiron active site of soluble methane monooxygenase. Chemistry 2003, 9 (10), 2347–58. [DOI] [PubMed] [Google Scholar]
- 24.Han W-G; Noodleman L, Structural model studies for the peroxo intermediate P and the reaction pathway from P → Q of methane monooxygenase using broken-symmetry density functional calculations. Inorg. Chem 2008, 47 (8), 2975–2986. [DOI] [PubMed] [Google Scholar]
- 25.Que L; Tolman WB, Biologically inspired oxidation catalysis. Nature 2008, 455 (7211), 333–340. [DOI] [PubMed] [Google Scholar]
- 26.Jasniewski AJ; Que L Jr., Dioxygen activation by nonheme diiron enzymes: diverse dioxygen adducts, high-valent intermediates, and related model complexes. Chem. Rev 2018, 118 (5), 2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zang Y; Dong Y; Que L; Kauffmann K; Münck E, The first bis(μ-oxo)diiron(III) complex. Structure and magnetic properties of [Fe2(μ-O)2(6TLA)2](ClO4)2. J. Am. Chem. Soc 1995, 117 (3), 1169–1170. [Google Scholar]
- 28.Hsu H-F; Dong Y; Shu L; Young VG; Que L, Crystal structure of a synthetic high-valent complex with an Fe2(μ-O)2 diamond core. Implications for the core structures of methane monooxygenase intermediate Q and ribonucleotide reductase intermediate X. J. Am. Chem. Soc 1999, 121 (22), 5230–5237. [Google Scholar]
- 29.Hwang J; Krebs C; Huynh BH; Edmondons DE; Theil EC; Penner-Hahn JE, A short Fe-Fe distance in peroxodiferric ferritin: Control of Fe substrate versus cofactor decay? Science 2000, 287 (5450), 122–125. [DOI] [PubMed] [Google Scholar]
- 30.Riggs-Gelasco PJ; Shu LJ; Chen SX; Burdi D; Huynh BH; Que L; Stubbe J, EXAFS characterization of the intermediate X generated during the assembly of the Escherichia coli ribonucleotide reductase R2 diferric tyrosyl radical cofactor. J. Am. Chem. Soc 1998, 120 (5), 849–860. [Google Scholar]
- 31.Baldwin J; Krebs C; Saleh L; Stelling M; Huynh BH; Bollinger JM; Riggs-Gelasco P, Structural characterization of the peroxodiiron(III) intermediate generated during oxygen activation by the W48A/D84E variant of ribonucleotide reductase protein R2 from Escherichia coli. Biochemistry 2003, 42 (45), 13269–13279. [DOI] [PubMed] [Google Scholar]
- 32.Dassama LMK; Silakov A; Krest CM; Calixto JC; Krebs C; Bollinger JM Jr.; Green MT, A 2.8 Å Fe–Fe Separation in the Fe2III/IV intermediate, X, from Escherichia coli ribonucleotide reductase. J. Am. Chem. Soc 2013, 135 (45), 16758–16761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banerjee R; Komor AJ; Lipscomb JD, Use of isotopes and isotope effects for investigations of diiron oxygenase mechanisms. Methods Enzymol 2017, 596, 239–290. [DOI] [PubMed] [Google Scholar]
- 34.Banerjee R; Meier KK; Münck E; Lipscomb JD, Intermediate P* from soluble methane monooxygenase contains a diferrous cluster. Biochemistry 2013, 52 (25), 4331–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strautmann JBH; George SD; Bothe E; Bill E; Weyhermüller T; Stammler A; Bögge H; Glaser T, Molecular and electronic structures of mononuclear iron complexes using strongly electron-donating ligands and their oxidized forms. Inorg. Chem 2008, 47 (15), 6804–6824. [DOI] [PubMed] [Google Scholar]
- 36.Ravel B; Newville M, ATHENA ARTEMIS,HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Sync. Rad 2005, 12 (4), 537–541. [DOI] [PubMed] [Google Scholar]
- 37.Rudd DJ; Sazinsky MH; Lippard SJ; Hedman B; Hodgson KO, X-ray absorption spectroscopic study of the reduced hydroxylases of methane monooxygenase and toluene/ o-xylene monooxygenase: Differences in active site structure and effects of the coupling proteins MMOB and ToMOD. Inorg. Chem 2005, 44 (13), 4546–4554. [DOI] [PubMed] [Google Scholar]
- 38.Jackson Rudd D; Sazinsky MH; Merkx M; Lippard SJ; Hedman B; Hodgson KO, Determination by X-ray absorption spectroscopy of the Fe−Fe separation in the oxidized form of the hydroxylase of methane monooxygenase alone and in the presence of MMOD. Inorg. Chem 2004, 43 (15), 4579–4589. [DOI] [PubMed] [Google Scholar]
- 39.Hamalainen K; Siddons DP; Hastings JB; Berman LE, Elimination of the inner-shell lifetime broadening in x-ray-absorption spectroscopy. Phys. Rev. Lett 1991, 67 (20), 2850–2853. [DOI] [PubMed] [Google Scholar]
- 40.Glatzel P; Bergmann U, High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes—electronic and structural information. Coord. Chem. Rev 2005, 249 (1–2), 65–95. [Google Scholar]
- 41.Bragg’s law (nλ=2d sinθ) sets the requirement that the fluorescent photon must satisfy a specific angular requirement (θ), dictated by the d spacing of the optic (analyzer crystals). Therefore, spectrometer has a strong dependence on the origin of the fluorescent photon, as it must satisfy this geometric requirement to be detected.
- 42.Ravel B, Quantitative EXAFS analysis In X-ray Absorption and X-ray Emission Spectroscopy: Theory and Applications, van Bokhoven JA; Lamberti C, Eds. John Wiley & Sons: West Sussex, United Kingdom, 2016; Vol. 1. [Google Scholar]
- 43.Rosenzweig AC; Nordlund P; Takahara PM; Frederick CA; Lippard SJ, Geometry of the soluble methane monooxygenase catalytic diiron center in two oxidation states. Chem. and Biol 1995, 2 (9), 409–418. [PubMed] [Google Scholar]
- 44.Beckwith MA; Ames W; Vila FD; Krewald V; Pantazis DA; Mantel C; Pcaut J; Gennari M; Duboc C; Collomb M-N; Yano J; Rehr JJ; Neese F; DeBeer S, How accurately can extended x-ray absorption spectra be predicted from first principles? Implications for modeling the oxygen-evolving complex in photosystem II. J. Am. Chem. Soc 2015, 137 (40), 12815––12834. [DOI] [PubMed] [Google Scholar]
- 45.Xue G; Wang D; De Hont R; Fiedler AT; Shan X; Münck E; Que L, A synthetic precedent for the FeIV2(μ-O)2 diamond core proposed for methane monooxygenase intermediate Q. Proc. Natl. Acad. Sci. U. S. A 2007, 104 (52), 20713––20718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lim MH; Rohde JU; Stubna A; Bukowski MR; Costas M; Ho RY; Münck E; Nam W; Que L Jr., An FeIV=O complex of a tetradentate tripodal nonheme ligand. Proc. Natl. Acad. Sci. U. S. A 2003, 100 (7), 3665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolter T; Meyer-Klaucke W; Müther M; Mandon D; Winkler H; Trautwein X, A.; Weiss R, Generation of oxoiron(IV) tetramesitylporphyrin π-cation radical complexes by m-CPBA oxidation of ferric tetramesitylporphyrin derivatives in butyronitrile at −78 °C. Evidence for the formation of six-coordinate oxoiron(IV) tetramesitylporphyrin π-cation radical complexes FeIV=O(tmp)X (X = Cl−, Br−), by Mössbauer and X-ray absorption spectroscopy. J. Inorg. Biochem 2000, 78 (2), 117–122. [DOI] [PubMed] [Google Scholar]
- 48.Riggs-Gelasco PJ; Price JC; Guyer RB; Brehm JH; Barr EW; Bollinger JM Jr.; Krebs C, EXAFS spectroscopic evidence for an Fe=O unit in the Fe(IV) intermediate observed during oxygen activation by taurine:α-ketoglutarate dioxygenase. J. Am. Chem. Soc 2004, 126 (26), 8108–9. [DOI] [PubMed] [Google Scholar]
- 49.Berry JF; Bill E; Bothe E; Debeer S; Mienert B; Neese F; Wieghardt K, An octahedral coordination complex of iron(VI) 2006, 312 (5782), 1937–1941. [DOI] [PubMed] [Google Scholar]
- 50.Kowalska JK; Lima FA; Pollock CJ; Rees JA; DeBeer S, A practical guide to high‐resolution x‐ray spectroscopic measurements and their applications in bioinorganic chemistry. Isr. J. Chem 2016, 56 (9–10), 803–815. [Google Scholar]
- 51.Rees JA; Bjornsson R; Kowalska JK; Lima FA; Schlesier J; Sippel D; Weyhermüller T; Einsle O; Kovacs JA; DeBeer S, Comparative electronic structures of nitrogenase FeMoco and FeVco. Dalton Trans 2017, 46 (8), 2445–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeBeer S, Advanced X-ray Spectroscopic methods for sudying iron-sulfur-containing proteins and model complexes. Method Enzymol 2018, 599, 427–450. [DOI] [PubMed] [Google Scholar]
- 53.Bauer M, HERFD-XAS and valence-to-core-XES: new tools to push the limits in research with hard X-rays? Phys. Chem. Chem. Phys 2014, 16 (27), 13827–13837. [DOI] [PubMed] [Google Scholar]
- 54.Lambertz C; Chernev P; Klingan K; Leidel N; Sigfridsson KGV; Happe T; Haumann M, Electronic and molecular structures of the active-site H-cluster in [FeFe]-hydrogenase determined by site-selective X-ray spectroscopy and quantum chemical calculations. Chem Sci 2014, 5 (3), 1187–1203. [Google Scholar]
- 55.Martinie RJ; Blaesi EJ; Krebs C; Bollinger JM; Silakov A; Pollock CJ, Evidence for a di-μ-oxo diamond core in the Mn(IV)/Fe(IV) activation intermediate of ribonucleotide reductase from Chlamydia trachomatis. J. Am. Chem. Soc 2017, 139 (5), 1950–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinho M; Xue G; Fiedler AT; Que L Jr.; Bominaar EL; Münck E, Mössbauer and DFT Study of the ferromagnetically coupled diiron(IV) precursor to a complex with an FeIV2O2 diamond core. J. Am. Chem. Soc 2009, 131 (16), 5823–5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andris E; Navrátil R; Jašík J; Puri M; Costas M; Que L Jr.; Roithová J, Trapping iron(III)–oxo species at the boundary of the “oxo wall”: Insights into the nature of the Fe(III)–O bond. J. Am. Chem. Soc 2018, 140, 14391–14400. [DOI] [PubMed] [Google Scholar]
- 58.Green MT, Application of Badger’s rule to heme and non-heme iron−oxygen bonds: An examination of ferryl protonation states. J. Am. Chem. Soc 2006, 128 (6), 1902–1906. [DOI] [PubMed] [Google Scholar]
- 59.Puri M; Que L Jr., Toward the synthesis of more reactive S = 2 non-heme oxoiron(IV) complexes. Acc. Chem. Rev 2015, 48 (8), 2443–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doan PE; Shanmugam M; Stubbe J; Hoffman BM, Composition and structure of the inorganic core of relaxed intermediate X(Y122F) of Escherichia coli ribonucleotide reductase. J. Am. Chem. Soc 2015, 137 (49), 15558–15566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mitic N; Clay MD; Saleh L; Bollinger JM; Solomon EI, Spectroscopic and electronic structure studies of intermediate X in ribonucleotide reductase R2 and two variants: A description of the FeIV-oxo bond in the FeIII-O-FeIV dimer. J. Am. Chem. Soc 2007, 129 (29), 9049–9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.