

Abstract
In this study, we combined photo‐organo redox catalysis and biocatalysis to achieve asymmetric C–H bond functionalization of simple alkane starting materials. The photo‐organo catalyst anthraquinone sulfate (SAS) was employed to oxyfunctionalise alkanes to aldehydes and ketones. We coupled this light‐driven reaction with asymmetric enzymatic functionalisations to yield chiral hydroxynitriles, amines, acyloins and α‐chiral ketones with up to 99 % ee. In addition, we demonstrate functional group interconversion to alcohols, esters and carboxylic acids. The transformations can be performed as concurrent tandem reactions. We identified the degradation of substrates and inhibition of the biocatalysts as limiting factors affecting compatibility, due to reactive oxygen species generated in the photocatalytic step. These incompatibilities were addressed by reaction engineering, such as applying a two‐phase system or temporal and spatial separation of the catalysts. Using a selection of eleven starting alkanes, one photo‐organo catalyst and 8 diverse biocatalysts, we synthesized 26 products and report for the model compounds benzoin and mandelonitrile > 97 % ee at gram scale.
Keywords: Photocatalysis, Chemoenzymatic cascades, Photo‐organo catalyst, Asymmetric synthesis, Enzymes
Introduction
Combining several catalytic steps to conduct a precisely arranged sequence of chemical transformations in a single reaction vessel exhibits an enormous potential for more economically and ecologically efficient synthesis routes, and thus, the development of one‐pot (cascade) reactions is a growing research field.1 Cascades are attractive from the perspective to reduce effort and waste, which usually arises from intermittent work‐up steps. Secondly, they also allow minimizing production time, if all catalysts and reagents are present from the beginning of the reaction (this type of reaction is referred to as concurrent cascade or tandem reaction). As a further advantage, tandem reactions facilitate steps that generate unstable or toxic intermediates, or that are thermodynamically challenging and thus would not be beneficial if performed as a single reaction.2 Cascade reactions have been developed within the fields of homogeneous‐,3 heterogeneous‐,[1c], [1d], 4 organo‐,5 photo‐, and biocatalysis.[1a], 6 On the contrary, combining catalysts from different catalysis fields (“worlds”) is sometimes more challenging[1e], [1f] for compatibility reasons: Whereas there are many reactions in homogeneous and organocatalysis operating in organic solvents, enzyme catalysis, for example, requires aqueous conditions in most cases, which is inconceivable for many transition metal‐catalysed reactions. Solving these compatibility challenges, in return, opens the possibility to combine reaction steps relying on different chemistries und thus to attain products that would not be accessible in cascades including reactions from one catalysis domain alone.
Fruitful interdomain combinations employ manifold solutions to tackle compatibility issues,[1e], [1f], [4b] such as compartmentalization by two‐phase systems, nanoparticles or membrane systems. Especially photocatalysts have been combined with various types of other catalysts yielding synergistic dual catalytic systems, where two types of catalysts participate in one catalytic cycle.8 The combination of photocatalytic and biocatalytic steps for organic synthesis, however, are not systematically explored until now. Most examples of combining photo‐ and biocatalysis focus on photocatalytic in situ regeneration of redox enzymes7 (Figure 1A). This approach, similar to Nature's photosynthesis, uses the light energy to feed electrons directly into the enzyme, or to provide stoichiometric redox equivalents such as NADPH or hydrogen peroxide for the enzymatic reaction step. Thus, light is used indirectly to fuel chemical transformations.[7c], 9 On the contrary, in this study, we aim to couple photoorganocatalytic reactions that use light energy directly to drive small molecule conversions with further enzymatic functionalization steps to develop photobiocatalytic one‐pot (tandem) reactions (Figure 1B). Only two examples of photobiocatalytic cascades combining metal photocatalysts with enzymes have been reported very recently.10 A very recent example couples an organo‐photocatalytic E/Z isomerization with an enzymatic reduction of the double bond,11 thereby increasing the conversion of the enzymatic reaction from 50 to 95 %. Beyond E/Z isomerization, other photocatalytic reactions have been developed also facilitating bond‐cleaving and bond‐forming reactions, which are very useful for the functionalization of simple starting materials.12
Figure 1.
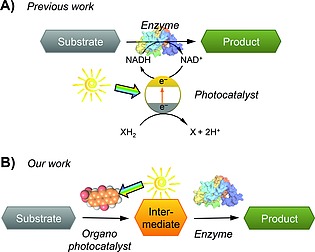
Cascade reactions combine catalysts from the same or different fields of catalysts. A) Literature‐reported photobiocatalytic approaches utilize photocatalysts to provide redox co‐substrates.7 B) In this study, we combine photocatalytic transformations with enzymatic reactions steps.
In this study, we explored the potential of photocatalytic oxidation/oxyfunctionalisation reactions generating aldehydes and ketones from simple alkene starting materials followed by biocatalytic asymmetric transformations yielding a range of chiral, high‐value added products. We demonstrate the general feasibility of this approach and also identify limitations. This asymmetric photo‐chemo‐enzymatic functionalization of a range of hydrocarbons substantially extends available chemoenzymatic approaches.
Results and Discussion
We chose the photocatalytic oxyfunctionalisation of alkanes 1–11 (Scheme 1) catalysed by sodium anthraquinone sulfonate (SAS) as a model reaction for the photocatalytic step. SAS is a quinoid photo‐organo catalyst well‐known for the light‐driven oxidation of alcohols. It also catalyses oxyfunctionalisation of alkanes in benzylic and allylic positions with moderate efficiency, which are the by far more attractive reactions.13 Converting (non)activated C‐H‐bonds by SAS into aldehydes 12–15 or ketones 16–22 does not introduce chirality into the respective compounds. We thus envisioned cascade reactions combining the SAS‐catalysed oxyfunctionalisation reactions with enzymatic reactions to generate further functionalized products 23–41 and to introduce the desired chirality. Due to the sulfonate substituent, SAS dissolves well in an aqueous reaction media, allowing to easily combine SAS‐catalysed reactions with various enzymatic reaction steps to explore the potential for different two‐step cascades.
Scheme 1.
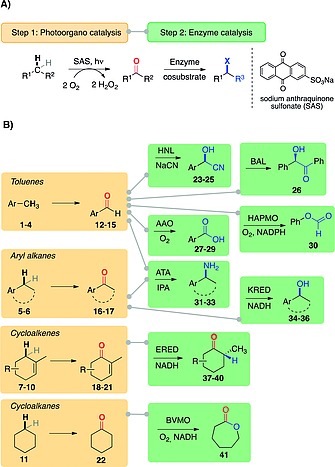
A) General principle of C–H functionalization by a photo‐enzymatic cascade. B) Modular combinations of SAS‐catalysed oxyfunctionalisation reactions and enzymatic transformations explored in this study. HNL: hydroxynitrile lyase; BAL: benzaldehyde lyase; AAO: aryl alcohol oxidase; HAPMO: 4‐hydroxyacetophenone monooxygenase; ATA: amine transaminase; KRED: keto reductase; ERED: ene‐reductase; CHMO: cyclohexanone monooxygenase.
Ideally, the reactions are taking place simultaneously in one reaction vessel (concurrent cascade). To prove that simultaneous catalysis by enzymes and SAS is possible, we first investigated a simple reaction system to produce functionalized but achiral products. We used benzyl alcohol as starting material, which can be readily oxidised by SAS, and added 4‐hydroxyacetophenone monooxygenase (HAPMO) to the reaction mixture, a Baeyer–Villiger monooxygenase (BVMO) known to catalyse ketone oxidation. Indeed, when both catalysts are present in the reaction mixture, phenyl formate 30 was formed (Table 1, entry 1). Similarly, cyclohexanol could be converted with cyclohexanone monooxygenase (CHMO) to ε‐caprolactone 41, although with lower conversion (Table 1, entry 5). When starting with the alkanes toluene 1 and cyclohexane 11 [20 mm dissolved in 10 % (v/v) acetonitrile in reaction buffer], no conversion could be detected. Interestingly, this changed if 1 was applied in excess, resulting in a two‐phase system (Table 1, entries 3 and 4). The improved conversion might be due to protecting the BVMOs from inhibition, as these enzymes are sensitive to product and substrate inhibitions. The non‐activated cyclohexane, however, could not be converted into 41.
Table 1.
Concurrent cascades of photocatalytic oxidation and enzymatic functionalization. Standard reaction conditions: 20 mm substrate, 10 % (v/v) acetonitrile in aqueous reaction buffer, 1–2 mg/mL enzyme (lyophilisate), 30 °C, 6–24 h. Details of buffer compositions, co‐factors, co‐substrates and additives are given in the Supporting Information
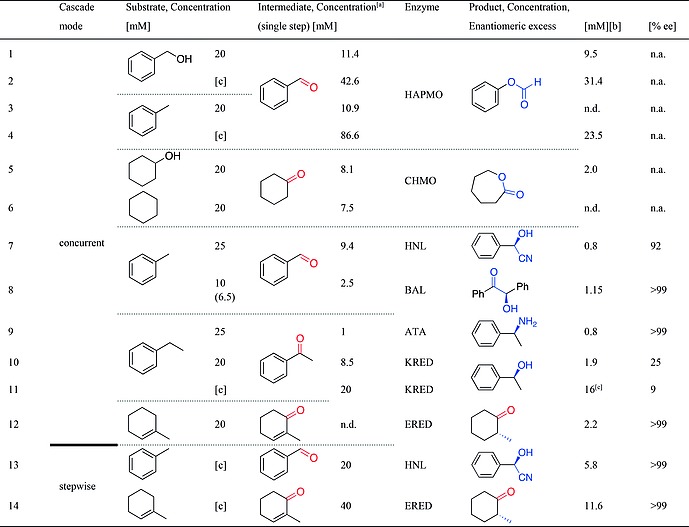
Maximum formed concentration of the intermediate if the photocatalytic reaction is given only. Determined by GC; for details see Supporting Information.
Concentration of product in the cascade reaction.
Substrate was used as solvent in a two‐phase system, volume ratio 1:1 (v/v). n.a. – not applicable; n.d. – not determined.
Next, we drew our attention on reactions directly starting from toluene, phenylethane and cyclohexene derivatives 1–10 (Scheme 1, see SI for a complete list of substrates and products) in the presence of biocatalysts chosen from three enzyme classes.
Benzaldehyde lyase (BAL), the nicotinamidedinucleotide‐dependent oxidoreductases ene‐reductase (ERED) and ketoreductase (KRED), as well the C–N–bond forming amine transaminase (ATA) are well‐known to convert carbonyl substrates with high to excellent enantioselectivity to functionalized products (Scheme 1). The photo‐oxidation step generated carbonyl intermediates in a concentration range of 2–45 mm depending on the used alkane (Table 1, entries 7–12). Pleasingly, in all cases the expected functionalized products 23–40 were detected when theses enzymes were included in the reaction mixtures (see Scheme 2 for structures of all products), although the conversions of the carbonyl intermediates in the enzymatic step were relatively low in most cases, ranging from 5–25 %. The reaction involving BAL is a notable exception: an equal amount of benzoin 26 formed corresponding to the consumed two equivalents of benzaldehyde.
Scheme 2.
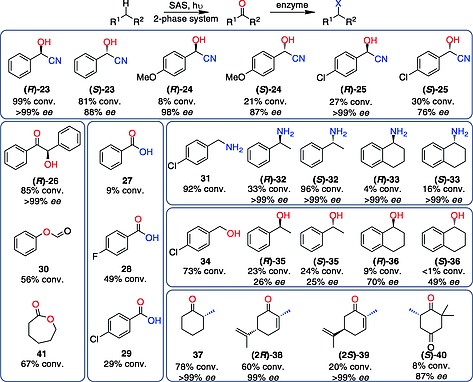
Scope of the photo‐chemo‐enzymatic approach for C–H bond transformation. The first step is performed as two phase reaction, where the substrate is used as organic phase. Conversions refer to consumption of the intermediate (aldehyde/ketone) in the enzymatic step.
For the optimization of cascade reactions, inhibition or deactivation of the catalysts by reaction participants or (co)‐solvents often play a role, but also cross‐reactivities leading to side reactions of reaction participants might occur.14 Furthermore, the equilibrium constant of the biocatalytic step might require actions to shift the equilibrium, especially in ATA and HNL‐catalysed reactions. In the modular reaction system, we identified the following issues that have to be addressed: (i) In the presence of SAS and light, substrates that are prone to oxidation such as HCN and NADH decomposed over time (see Figure S3), as observed in the HNL‐catalysed reactions. (ii) As reactive oxygen species and other radical intermediates are formed in the photocatalytic reaction, it was not surprising to note that also enzyme deactivation occurred over time: product formation often declined after a few hours, but SAS‐mediated alkane oxidation still continued over time. (iii) Incompatibility of catalysts with necessary co‐solvents occurred in case of the transaminase reactions. With our studied ATAs we had to replace acetonitrile by DMSO due to rapid inactivation of the transaminases.
DMSO is often employed in transaminase reactions and well‐tolerated by many enzymes, but unfortunately it decreased SAS activity by 25‐fold: Only ca. 1 mm of benzaldehyde and ketones formed when DMSO was present (compare entries 9 and 10, Table 1). (iv) Most enzymes maintained their excellent stereoselectivities, only KRED‐catalysed reactions yielded alcohols with a low enantiomeric excess of 25 % ee. This might be attributed to a radical‐mediated racemization of the alcohol products.
After having identified several incompatibility issues, we aimed at improving the conversions especially of the enzymatic step of the cascades. Reactive oxygen species such as H2O2, which are produced also by other photocatalysts under an oxidative reaction regime, are a general challenge for enzyme stability. When we added catalase to the reaction mixture of the ERED reaction to decompose H2O2, however, no beneficial effect on conversion was detected. Inspired by the success of a two‐phase approach with BVMO, we therefore investigated this approach for ATA and KRED. The low concentration of alcohol in the KRED reaction is very likely due to the facile oxidation of alcohols by SAS. In a two‐phase system, the organic phase provides a product sink, thereby protecting the alcohol product 34 from back oxidation. Therefore, the two‐phase system increased product concentrations by 10‐fold for the KRED reaction (entries 10 and 11), but had no impact on the ATA reaction due to the higher solubility of the amine in the aqueous phase. Spacial and temporal separation of catalysts is an often‐applied solution if incompatibility of reaction conditions cannot be avoided.[1e], 14 Therefore we turned our attention to a sequential reaction mode: after the photocatalytic step, we added the appropriate enzyme and protected the reactions from light to decrease oxidative damage. This resulted in a 3–4‐fold increased formation of the final product: mandelonitrile 23 concentration in the HNL reaction increased sixfold, and notably also an excellent enantiomeric excess of > 99 % ee was obtained (entry 13). Especially if the alcohols were employed as starting material, HAPMO and ERED achieved 74 % and > 98 % conversions of the formed intermediates to yield 30 and 38 (Table 1, entry 14 and Table S6). Despite the increased product formation in the enzymatic step, conversion was incomplete in most cases and we observed that reactions stagnated after a few hours. To exclude inhibitory effects of side‐products from the photocatalytic reactions, we modified the step‐wise reaction as follows: a two‐phase SAS‐catalysed oxidation resulted in the accumulation of the intermediate in the organic phase. We then discarded the SAS‐containing aqueous phase and provided reagents and enzymes dissolved in aqueous reaction buffer to start the second reaction. In this way, we could further increase conversions in the enzymatic step. To show the generality of this approach, we explored different alkane substrates and also employed ATA, HNL or KRED enzymes with opposite stereo‐preferences to produce both product enantiomers (Scheme 2, Figure S4–S17). In total, this approach generated 26 products with varying conversion, but at least one example with good to quantitative conversions of the formed intermediate was obtained with most of the investigated enzymes. Finally, these photo‐chemo‐enzymatic transformations were performed on preparative scale for (R)‐mandelonitrile and (R)‐benzoin synthesis, respectively. For the (R)‐HNL catalysed hydrocyanation of benzaldehyde, we performed a micro‐aqueous biocatalytic reaction: 1.4 g of essentially pure product (97.7 % ee) was obtained in 85 % isolated yield (in the enzymatic step). The synthesis of (R)‐benzoin resulted in 1.8 g of product with > 99 % ee in 97 % isolated yield (see SI for details).
Conclusions
In conclusion, the studied combination of photo‐ and enzyme catalysis enabled C–H bond transformations to six different functional groups using cheap alkanes as starting material, e.g. toluene and cyclohexene derivatives. Amines, cyanohydrins, the acyloin benzoin, and α‐chiral ketones are obtained with excellent optical purities, which is a key advantage of the photo‐chemo‐enzymatic cascade. A one‐pot reaction is especially suitable for enzymes that do not require redox cofactors, such as lyases or transferases. As a compromise, a stepwise reaction can be alternatively carried out to overcome the observed incompatibility between photo‐organo catalyst and biocatalyst. We envision that the efficiency of reactions with redox cofactor‐depending enzymes will be further increased in the future, when photocatalytic water oxidation has been established as a generally applicable method for cofactor regeneration. Furthermore, protein engineering can be used to improve enzyme resistance to oxidative stress and photo‐degradation to further increase catalyst compatibility in photo‐biocatalytic cascades.
Experimental Section
Supporting Information (see footnote on the first page of this article): All experimental details are given in the Supporting information.
Supporting information
Supporting Information
Acknowledgements
We gratefully acknowledge Prof. Wolfgang Kroutil for providing us the vectors encoding the KREDs. FH thanks for the financial support by the European Research Council (ERC Consolidator Grant No. 648026) and the Netherlands Organisation for Scientific Research (VICI grant, No. 724.014.003). RW and LLM acknowledge funding from the European Union's Horizon 2020 MSCA ITN‐EID program under grant agreement No. 634200 (Project BIOCASCADES). MH has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 759262). SS has received funding from the European Union's Horizon 2020 MSCA ITN‐EJD program under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 764920, Project PhotoBioCat).
Contributor Information
Matthias Höhne, Email: Matthias.hoehne@uni-greifswald.de, www.protein‐biochemistry.de.
Sandy Schmidt, Email: s.schmidt@tugraz.at, https://www.tugraz.at/arbeitsgruppen/bpe/forschung/.
References
- 1.a) Schrittwieser J. H., Velikogne S., Hall M. and Kroutil W., Chem. Rev., 2018, 118, 270–348; [DOI] [PubMed] [Google Scholar]; b) Muschiol J., Peters C., Oberleitner N., Mihovilovic M. D., Bornscheuer U. T. and Rudroff F., Chem. Commun., 2015, 51, 5798–5811; [DOI] [PubMed] [Google Scholar]; c) Climent M. J., Corma A., Iborra S. and Sabater M. J., ACS Catal., 2014, 4, 870–891; [Google Scholar]; d) Climent M. J., Corma A. and Iborra S., RSC Adv., 2012, 2, 16–58; [Google Scholar]; e) Rudroff F., Mihovilovic M. D., Gröger H., Snajdrova R., Iding H. and Bornscheuer U. T., Nat. Catal., 2018, 1, 12–22; [Google Scholar]; f) Lohr T. L. and Marks T. J., Nat. Chem., 2015, 7, 477–482. [DOI] [PubMed] [Google Scholar]
- 2.a) Li Z., Assary R. S., Atesin A. C., Curtiss L. A. and Marks T. J., J. Am. Chem. Soc., 2014, 136, 104–107; [DOI] [PubMed] [Google Scholar]; b) Wang X. Y. and Rinaldi R., Angew. Chem. Int. Ed., 2013, 52, 11499–11503; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2013, 125, 11713–11717. [Google Scholar]
- 3.a) Galván A., Fañanás F. J. and Rodríguez F., Eur. J. Org. Chem., 2016, 2016, 1306–1313; [Google Scholar]; b) Huff C. A. and Sanford M. S., J. Am. Chem. Soc., 2011, 133, 18122–18125; [DOI] [PubMed] [Google Scholar]; c) Perez‐Temprano M. H., Casares J. A. and Espinet P., Chem. Eur. J., 2012, 18, 1864–1884. [DOI] [PubMed] [Google Scholar]
- 4.a) Guo Z., Liu B., Zhang Q. H., Deng W. P., Wang Y. and Yang Y. H., Chem. Soc. Rev., 2014, 43, 3480–3524; [DOI] [PubMed] [Google Scholar]; b) Szőllősi G., Catal. Sci. Technol., 2018, 8, 389–422. [Google Scholar]
- 5. Chanda T. and Zhao J. C. G., Adv. Synth. Catal., 2018, 360, 2–79. [Google Scholar]
- 6.a) Zhang Y. F. and Hess H., ACS Catal., 2017, 7, 6018–6027; [Google Scholar]; b) Quin M. B., Wallin K. K., Zhang G. and Schmidt‐Dannert C., Org. Biomol. Chem., 2017, 15, 4260–4271; [DOI] [PubMed] [Google Scholar]; c) Köhler V. and Turner N. J., Chem. Commun., 2015, 51, 450–464. [DOI] [PubMed] [Google Scholar]
- 7.a) Choudhury S., Baeg J.‐O., Park N.‐J. and Yadav R. K., Green Chem., 2014, 16, 4389–4400; [Google Scholar]; b) Macia‐Agullo J. A., Corma A. and Garcia H., Chem. Eur. J., 2015, 21, 10940–10959; [DOI] [PubMed] [Google Scholar]; c) Lee S. H., Choi D. S., Kuk S. K. and Park C. B., Angew. Chem. Int. Ed., 2018, 57, 7958–7985; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2018, 130, 8086. [Google Scholar]
- 8.a) Fabry D. C. and Rueping M., Acc. Chem. Res., 2016, 49, 1969–1979; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hoffmann N., ChemCatChem, 2015, 7, 393–394; [Google Scholar]; c) Hopkinson M. N., Tlahuext‐Aca A. and Glorius F., Acc. Chem. Res., 2016, 49, 2261–2272; [DOI] [PubMed] [Google Scholar]; d) Lerch S., Unkel L. N. and Brasholz M., Angew. Chem. Int. Ed., 2014, 53, 6558–6562; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2014, 126, 6676–6680; [Google Scholar]; e) Levin M. D., Kim S. and Toste F. D., ACS Cent. Sci., 2016, 2, 293–301; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Silvi M., Arceo E., Jurberg I. D., Cassani C. and Melchiorre P., J. Am. Chem. Soc., 2015, 137, 6120–6123; [DOI] [PubMed] [Google Scholar]; g) Skubi K. L., Blum T. R. and Yoon T. P., Chem. Rev., 2016, 116, 10035–10074; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Wozniak L., Magagnano G. and Melchiorre P., Angew. Chem. Int. Ed., 2018, 57, 1068–1072; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem., 2018, 130, 1080; [Google Scholar]; i) Yoon T. P., Acc. Chem. Res., 2016, 49, 2307–2315; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Chen K., Berg N., Gschwind R. and König B., J. Am. Chem. Soc., 2017, 139, 18444–18447. [DOI] [PubMed] [Google Scholar]
- 9.a) Seel C. J., Králík A., Hacker M., Frank A., König B. and Gulder T., ChemCatChem, 2018, 10, 3960–3963; [Google Scholar]; b) Zhang W., Fernandez‐Fueyo E., Ni Y., van Schie M., Gacs J., Renirie R., Wever R., Mutti F. G., Rother D., Alcalde M. and Hollmann F., Nat. Catal., 2018, 1, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Guo X., Okamoto Y., Schreier M. R., Ward T. R. and Wenger O. S., Chem. Sci., 2018, 9, 5052–5056; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lauder K., Toscani A., Qi Y., Lim J., Charnock S. J., Korah K. and Castagnolo D., Angew. Chem. Int. Ed., 2018, 57, 5803–5807; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2018, 130, 5905. [Google Scholar]
- 11. Litman Z. C., Wang Y., Zhao H. and Hartwig J. F., Nature, 2018, 560, 355–359. [DOI] [PubMed] [Google Scholar]
- 12.a) Marzo L., Pagire S. K., Reiser O. and König B., Angew. Chem. Int. Ed., 2018, 57, 10034–10072; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2018, 130, 10188; [Google Scholar]; b) Ravelli D., Protti S. and Fagnoni M., Chem. Rev., 2016, 116, 9850–9913; [DOI] [PubMed] [Google Scholar]; c) Roslin S. and Odell L. R., Eur. J. Org. Chem., 2017, 2017, 1993–2007. [Google Scholar]
- 13. Zhang W., Gacs J., Arends I. W. C. E. and Hollmann F., ChemCatChem, 2017, 9, 3821–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmidt S., Castiglione K. and Kourist R., Chem. Eur. J., 2018, 24, 1755–1768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information