

Abstract
The direct enantioselective addition of water to unactivated alkenes could simplify the synthesis of chiral alcohols and solve a long‐standing challenge in catalysis. Here we report that an engineered fatty acid hydratase can catalyze the asymmetric hydration of various terminal and internal alkenes. In the presence of a carboxylic acid decoy molecule for activation of the oleate hydratase from E. meningoseptica, asymmetric hydration of unactivated alkenes was achieved with up to 93 % conversion, excellent selectivity (>99 % ee, >95 % regioselectivity), and on a preparative scale.
Keywords: alkenes, asymmetric hydration, biocatalysis, decoy molecules, rational enzyme design
Carbon–carbon double bonds are one of the most important functional groups in organic chemistry and allow the synthesis of complex organic molecules. Their widespread availability and reactivity has made alkene‐functionalization reactions essential to organic synthesis. A long‐standing challenge in the field is the asymmetric addition of water across a carbon–carbon double bond.1, 2 Considerable effort has been devoted to this topic and many catalysts have been developed, including acids, metal oxides, zeolites, as well as clays. Notably, the hydration of alkenes to produce the corresponding alcohols is carried out industrially on a large scale.3 However, these applied catalysts do not offer stereocontrol for this chemical transformation. The synthesis of chiral alcohols from alkenes depends currently on two‐step oxidation–reduction sequences (Scheme 1) or resolution of racemic alcohols (Figure S1 in the Supporting Information).4, 5 The few reported catalysts for the asymmetric addition of water across alkenes show often only moderate stereocontrol and are limited to activated alkenes such as enones and hydroxystyrenes (Figure S2).6, 7 Catalysts for the asymmetric hydration of unactivated, aliphatic alkenes (e.g., 1‐alkenes) to produce higher‐value chiral alcohols have not been reported to date. These molecules are challenging substrates because of the inert nature of aliphatic carbon–carbon double bonds, their limited steric and electronic bias, as well as their high degree of conformational flexibility.8 Therefore, the direct addition of water to an alkene in a stereoselective way under mild reaction conditions is certainly “a synthetic chemists dream”5, 9 as it generates valuable chiral alcohols in a reaction that displays both atom economy10 and redox neutrality.11
Scheme 1.
Methods for the hydration of unactivated aliphatic alkenes to chiral alcohols.
While asymmetric alkene hydration has proven difficult to achieve with synthetic catalysts, enzymes perform this reaction with high activity and selectivity. Hydratases catalyze the addition of water to activated alkenes such as α,β‐unsaturated carbonyl groups as well as unactivated carbon–carbon double bonds.9, 12, 13, 14 The advantage of enzymes for the asymmetric hydration of alkenes is the precise positioning of a water molecule in the active site. Several hydratase enzymes for the regio‐ and stereoselective hydration of unactivated alkenes have been reported, including carotenoid‐1,2‐hydratases,15, 16, 17, 18, 19 the linalool dehydratase isomerase,20, 21, 22, 23 and fatty acid hydratases (Figure S3).12, 24, 25, 26, 27 These enzymes, however, are limited to a specific substrate motif. Carotenoid‐1,2‐hydratases are specific to isoprene units in long‐chain terpenes12 and the linalool dehydratase isomerase is limited to isoprenoid dienes.20, 22 In contrast, fatty acid hydratases, including oleic acid hydratases (OAHs), convert isolated carbon–carbon double bonds of unsaturated fatty acids to the corresponding chiral alcohols. This is achieved by alkene protonation in combination with water activation by an active‐site Brønsted base (Figure 1 A). The strict requirement for carboxylic acid substrates, however, limits the synthetic application of OAHs.27, 28, 29, 30, 31, 32
Figure 1.
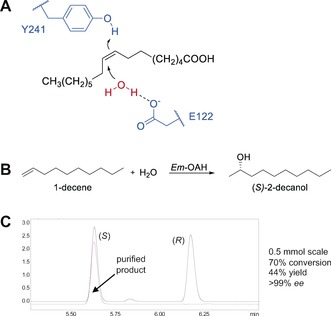
Asymmetric hydration of aliphatic alkenes using Em‐OAH. A) Postulated reaction mechanism of the Em‐OAH‐catalyzed hydration of oleic acid. The protonation of the carbon–carbon double bond at the C9 atom of oleic acid is initiated by Y241, followed by a nucleophilic attack of a water molecule that is activated by E122.32 B) Enantioselective hydration of 1‐decene by Em‐OAH wildtype enzyme using heptanoic acid as a decoy molecule. C) GC analysis on a chiral support, and results for the preparative‐scale reaction of 1‐decene (red line in GC trace). Upscaling of 1‐decene hydration by Em‐OAH wildtype was performed with 70 mg starting material (2 mm 1‐decene) using a whole‐cell catalyst (100 mg mL−1) and heptanoic acid as a decoy molecule.
We have recently found first evidence that this substrate limitation can be overcome by studying the OAH from Elizabethkingia meningoseptica (Em‐OAH) in combination with carboxylic acid decoy molecules (Figure S4).33 Decoy molecules interact with active site substrate binding motifs and, thus, activate enzymes for catalysis on alternative substrates.34, 35, 36 Herein, we describe the selective asymmetric hydration of a wide range of unactivated alkenes by combining decoy molecules and protein engineering of Em‐OAH.
In a first set of experiments, we investigated the influence of the carboxylic acid decoy molecule on alkene hydration. 1‐Decene was selected as a model substrate for the development of a selective hydration protocol. While Em‐OAH wildtype shows only modest activity with 1‐decene as substrate, the addition of hexanoic acid as decoy molecule activated Em‐OAH for more efficient alkene hydration. Moreover, we found that an equimolar ratio of 1‐decene and decoy molecule showed highest activity in 1‐decene hydration (Figure S5). Highest 1‐decene conversion was observed with heptanoic acid as a decoy molecule. Carboxylic acids with different chain lengths (C5–C8) and functionalized derivatives such as amides, esters, and alcohols did not further increase 1‐decene hydration activity (Figures S6 and S7).
Next, we screened different substrate concentrations (0.5 to 4.0 mm) to find conditions for high substrate conversion and product formation (Table S1). Strikingly, we found reaction conditions (0.5 mm 1‐decene) that enabled up to 93 % conversion, while generating the chiral alcohol product with excellent enantioselectivity (>99 % ee). To demonstrate that the asymmetric hydration of 1‐decene can also be performed on preparative scale (0.5 mmol), (S)‐2‐decanol was synthesized using conditions with a trade‐off in conversion and product formation (2 mm 1‐decene). The results are shown in Figures 1 B and C. (S)‐2‐Decanol was obtained with high conversion (70 %), 44 % yield of isolated product, and excellent enantioselectivity (>99 % ee).
We further examined the scope of asymmetric hydration towards short‐chain 1‐alkenes using wildtype Em‐OAH. Interestingly, we observed highly reduced conversion of 1‐octene compared to 1‐decene using our optimized hydration protocol. It should be noted that even after four days of incubation, the wildtype Em‐OAH showed only modest product formation. As 1‐octene is two carbon atoms shorter than 1‐decene, we assumed that 1‐octene might not bind effectively into the large substrate binding pocket of Em‐OAH. Therefore, a rational mutagenesis approach was applied aiming to activate Em‐OAH for efficient hydration of short‐chain 1‐alkenes (C5–C8).
We harnessed the diversity of fatty acid hydratase sequences that cluster in a superfamily to identify active site hotspot positions for mutagenesis. While catalytically relevant positions are conserved in all fatty acid hydratases, substrate specificity‐determining positions are expected to be conserved within functional subfamilies, but are different between them.37 To identify subfamily‐specific positions in fatty acid hydratases, multiple sequence alignments were performed using the Hydratase Engineering Database (HyED, https://hyed.biocatnet.de).26 Position 248 attracted our attention because this active‐site amino acid is not conserved in the fatty acid hydratase superfamily while at the same time showed conservation within homologous families. A more detailed analysis of the Em‐OAH crystal structure (PDB code: 4UIR)32 further supports the functional relevance of position 248 for substrate binding as it is located at the end of the alkyl binding pocket and points towards the terminal carbon atom of the natural substrate oleic acid (Figure 2 A). As a result, we hypothesized that position 248 might control the substrate specificity of fatty acid hydratases and selected alanine 248 of Em‐OAH for site‐saturation mutagenesis.
Figure 2.
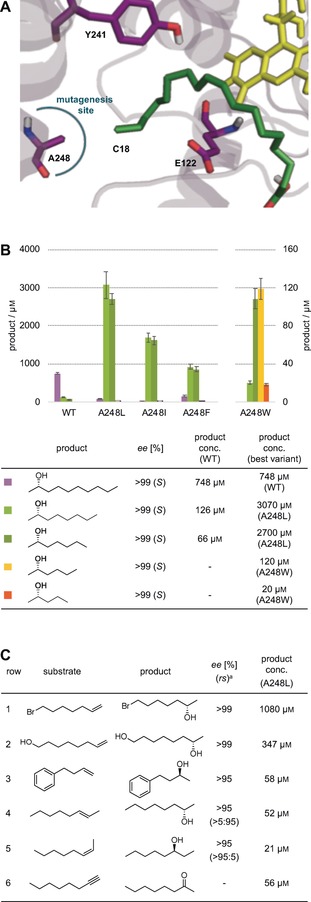
Rational enzyme engineering and substrate scope. A) Substrate binding pocket of Em‐OAH with bound FAD (yellow) and the docked oleic acid substrate (green). Alanine 248 (highlighted with the blue circle) was identified as potential specifity‐determining position and selected for site‐saturation mutagenesis. The terminal carbon atom of oleic acid points towards A248 (4 Å distance) and is labelled as C18. The proposed catalytic residues Y241 and E122 are shown as purple sticks. B) Asymmetric hydrations of 1‐decene, 1‐octene, 1‐heptene, 1‐hexene, and 1‐pentene by Em‐OAH wildtype enzyme and best variants. Reactions were performed using the two‐phase system. C) Substrate scope studies using functionalized alkenes (row 1–3), internal alkenes (row 4 and 5), and alkyne (row 6). Reactions were performed using the two‐phase system (row 1 and 4) as well as standard whole‐cell biotransformation conditions (rows 2, 3, 5, and 6). See Supporting Information for details. [a] rs describes the regioselectivity for internal alkene hydration as ratio of the 3‐octanol to the 2‐octanol product.
Various 1‐alkenes (C5–C8 and C10) were chosen for characterization of the different mutant enzymes with heptanoic acid as a decoy molecule. Results of the biotransformation are shown in Figure 2 B. The generated variants enabled asymmetric hydration of all tested 1‐alkenes. Furthermore, a closer comparison permitted a correlation of the substrate specificity with the size of the introduced amino acid residue at position 248. Selective hydration of short‐chain 1‐alkenes was either very low (1‐octene and 1‐heptene) or not detectable (1‐hexene and 1‐pentene) using the Em‐OAH wildtype. This indicates a non‐optimal fit of these substrates in the wildtype enzyme bearing an alanine at position 248. In contrast, variant A248L demonstrated increased product formation in the hydration of 1‐octene and 1‐heptene by a factor of 24 and 41, respectively. Moreover, the introduction of a tryptophan residue (Em‐OAH A248W) resulted in highest activity towards 1‐hexene and even enabled 1‐pentene hydration. In order to compare the activity of the different variants with various 1‐alkenes as substrate, we quantified the product formation in the asymmetric hydration. Product concentrations of up to 3 mm 2‐octanol, 2.7 mm 2‐heptanol, 120 μm 2‐hexanol, and 20 μm 2‐pentanol were obtained using the engineered Em‐OAH variants (Figure 2 B). The presence of a larger residue at position 248 most probably shifts the alkene double bond closer to the nucleophilic water molecule, which could account for the observed increment in product formation.
It is noteworthy that mutations at position 248 did not affect the selectivity of the enzyme; all alcohol products were formed with very good enantioselectivity (>99 % ee). As the asymmetric hydration of unactivated alkenes is a particular challenge in catalyst design, the excellent selectivities obtained in our experiments emphasize the high degree of control that enzymes offer in stereoselective catalysis. Even for the short‐chain 1‐pentene substrate, the engineered Em‐OAH precisely positioned a water molecule for the selective attack from one side of the prochiral carbon‐carbon double bond, producing (S)‐2‐pentanol with very good stereoselectivity (>99 % ee; see Figure S8).
Next, we were interested to explore the substrate scope towards functionalized and internal alkenes. Since single enzymes do typically not show high activity on an extremely broad substrate scope,38 we aimed to confirm initial activity on a diverse set of substrates which may serve as starting point for further protein engineering. Overall, we have tested a set of 23 alkenes using enzyme variants shown in Figure 2 B and different decoy molecules (see Figure S9 for more details). Six alkenes showed significant conversion while control experiments using cells containing an empty vector did typically not show any activity. These six alkenes were further characterized using the best variant (Em‐AOH A248L) and hexanoic acid as decoy molecule. The functionalized alkenes 7‐bromohept‐1‐ene and 7‐octen‐1‐ol were converted with good activity and excellent enantioselectivity (>99 % ee, rows 1 and 2 in Figure 2 C and Figure S10). Surprisingly, we could even confirm asymmetric hydration of the bulky 4‐phenyl‐1‐butene, generating the chiral alcohol with low activity and good enantioselectivity (>95 % ee, row 3 in Figure 2 C and Figure S10). Please note that in the case of 4‐phenyl‐1‐butene as substrate, control experiments (whole cells containing an empty vector) revealed a very low level of background hydration generating the racemic alcohol product. Strikingly, we could also identify activities for internal alkenes (rows 4 and 5 in Figure 2 C). A248L converted trans‐2‐octene to (S)‐2‐octanol and cis‐2‐octene to (S)‐3‐octanol. Both reactions revealed very good enantio‐ (>95 %) and regioselectivities (>95 %) in the asymmetric alkene hydration (see Figure S11). To the best of our knowledge, this is the first example of high regio‐ and enantiocontrol in the catalytic hydration of internal alkenes. In addition, we have found moderate activity for the hydration of 1‐octyne to yield 2‐octanone (row 6 in Figure 2 C). The only example of enzymatic hydration of alkynes reported to date is that of acetylene by a tungsten‐dependent, oxygen sensitive enzyme.39
Finally, to further prove that this reaction can be accomplished on a preparative scale (0.5 mmol), we performed the asymmetric hydration of 1‐octene with Em‐OAH A248L under optimized conditions. Figure 3 shows the production of (S)‐2‐octanol with 50 % conversion, 22 % isolated yield and an enantiomeric excess of >99 % (Figure 3).
Figure 3.
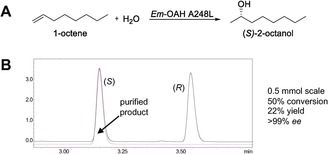
Asymmetric hydration of 1‐octene on preparative scale. A) Enantioselective hydration of 1‐octene by Em‐OAH A248L using hexanoic acid as a decoy molecule. B) Results of 1‐octene preparative‐scale reaction (red line in GC trace). Upscaling of 1‐octene hydration by Em‐OAH A248L was performed with 56 mg starting material (1 mm 1‐octene) using a whole‐cell catalyst (100 mg mL−1) and hexanoic acid as a decoy molecule.
In conclusion, we demonstrated the first catalytic asymmetric hydration of unactivated, aliphatic alkenes. In addition to high enantioselectivity (>99 % ee) and in some cases high conversion (up to 93 %), we could further reveal high regioselectivity in the hydration of internal alkenes. Key to this catalytic activity is the precise orientation of alkene substrates and water molecules in a protein macromolecular catalyst. Increased enzyme activity was achieved by using a carboxylic acid decoy molecule that activates the fatty acid hydratase for alkene hydration in combination with rational enzyme engineering to expand the substrate scope. The preparative‐scale reactions prove the practical utility of this procedure. It should be noted that we did not observe side reactions or decoy molecule conversion during the reaction time. Considering the environmental, quality, and cost benefits that enzymes bring to synthetic organic chemistry,40, 41 enzymatic alkene hydration may enable sustainable, efficient, and stereoselective synthesis of chiral alcohols from cheap starting materials. Further enzyme characterization will show whether more efficient hydratases can be found in nature or can even be engineered in the laboratory to perform asymmetric alkene hydration on a broad class of terminal, internal, and branched alkenes. Because Em‐OAH shows initial activity with 1‐decene in the absence of the carboxylic acid decoy molecule, engineering of fatty acid hydratases might be a good strategy to yield biocatalysts that function without decoy molecule activation. As fatty acid hydratases catalyze alkene hydration with (R)‐ and (S)‐selectivity in nature,24, 27, 32, 42, 43, 44 this class of enzymes might provide a significant source of catalysts for the challenging asymmetric hydration of unactivated alkenes.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This project has received funding from the European Union's Horizon 2020 Research and Innovation Programme under Grant agreement No 635536. We thank Samuel Kleehammer for support in generating the enzyme variants, Dr. Bernd Nebel for assistance in chiral GC analysis and Max‐Philipp Fischer as well as Jens Schmid for fruitful discussions.
R. M. Demming, S. C. Hammer, B. M. Nestl, S. Gergel, S. Fademrecht, J. Pleiss, B. Hauer, Angew. Chem. Int. Ed. 2019, 58, 173.
References
- 1. Beller M., Seayad J., Tillack A., Jiao H., Angew. Chem. Int. Ed. 2004, 43, 3368–3398; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3448–3479. [Google Scholar]
- 2. Hintermann L., Top. Organomet. Chem. 2010, 31, 123–155. [Google Scholar]
- 3. Wiseman P., An Introduction to Industrial Chemistry, Applied Science, London, 1979. [Google Scholar]
- 4. Breuer M., Ditrich K., Habicher T., Hauer B., Kesseler M., Stürmer R., Zelinski T., Angew. Chem. Int. Ed. 2004, 43, 788–824; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 806–843. [Google Scholar]
- 5. Sato H., Hummel W., Gröger H., Angew. Chem. Int. Ed. 2015, 54, 4488–4492; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4570–4574. [Google Scholar]
- 6. Boersma A. J., Coquiére D., Geerdink D., Rosati F., Feringa B. L., Roelfes G., Nat. Chem. 2010, 2, 991–995. [DOI] [PubMed] [Google Scholar]
- 7. Wuensch C., Gross J., Steinkellner G., Gruber K., Glueck S. M., Faber K., Angew. Chem. Int. Ed. 2013, 52, 2293–2297; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2349–2353. [Google Scholar]
- 8. Coombs J. R., Morken J. P., Angew. Chem. Int. Ed. 2016, 55, 2636–2649; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2682–2696. [Google Scholar]
- 9. Jin J., Hanefeld U., Chem. Commun. 2011, 47, 2502–2510. [DOI] [PubMed] [Google Scholar]
- 10. Trost B. M., Science 1991, 254, 1471–1477. [DOI] [PubMed] [Google Scholar]
- 11. Burns N. Z., Baran P. S., Hoffmann R. W., Angew. Chem. Int. Ed. 2009, 48, 2854–2867; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2896–2910. [Google Scholar]
- 12. Hiseni A., Arends I. W. C. E., Otten L. G., ChemCatChem 2015, 7, 29–37. [Google Scholar]
- 13. Demming R. M., Fischer M.-P., Schmid J., Hauer B., Curr. Opin. Chem. Biol. 2018, 43, 43–50. [DOI] [PubMed] [Google Scholar]
- 14. Resch V., Hanefeld U., Catal. Sci. Technol. 2015, 5, 1385–1399. [Google Scholar]
- 15. Steiger S., Mazet A., Sandmann G., Arch. Biochem. Biophys. 2003, 414, 51–58. [DOI] [PubMed] [Google Scholar]
- 16. Maresca J. A., Graham J. E., Bryant D. A., Photosynth. Res. 2008, 97, 121–140. [DOI] [PubMed] [Google Scholar]
- 17. Sun Z., Shen S., Wang C., Wang H., Hu Y., Jiao J., Ma T., Tian B., Hua Y., Microbiology 2009, 155, 2775–2783. [DOI] [PubMed] [Google Scholar]
- 18. Hiseni A., Arends I. W. C. E., Otten L. G., Appl. Microbiol. Biotechnol. 2011, 91, 1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hiseni A., Otten L. G., Arends I. W. C. E., Appl. Microbiol. Biotechnol. 2016, 100, 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brodkorb D., Gottschall M., Marmulla R., Lüddeke F., Harder J., J. Biol. Chem. 2010, 285, 30436–30442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lüddeke F., Harder J., Z. Naturforsch. C 2011, 66, 409–412. [DOI] [PubMed] [Google Scholar]
- 22. Nestl B. M., Geinitz C., Popa S., Rizek S., Haselbeck R. J., Stephen R., Noble M. A., Fischer M.-P., Ralph E. C., Hau H. T., et al., Nat. Chem. Biol. 2017, 13, 275–281. [DOI] [PubMed] [Google Scholar]
- 23. Weidenweber S., Marmulla R., Ermler U., Harder J., FEBS Lett. 2016, 590, 1375–1383. [DOI] [PubMed] [Google Scholar]
- 24. Kim K.-R., Oh H.-J., Park C.-S., Hong S.-H., Park J.-Y., Oh D.-K., Biotechnol. Bioeng. 2015, 112, 2206–2213. [DOI] [PubMed] [Google Scholar]
- 25. Lorenzen J., Janke R., Waldow A., Qoura F., Loll B., Brück T., ChemCatChem 2018, 10, 407–414. [Google Scholar]
- 26. Schmid J., Steiner L., Fademrecht S., Pleiss J., Otte K. B., Hauer B., J. Mol. Catal. B 2016, 133, S243–S249. [Google Scholar]
- 27. Hirata A., Kishino S., Park S.-B., Takeuchi M., Kitamura N., Ogawa J., J. Lipid Res. 2015, 56, 1340–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rosberg-Cody E., Liavonchanka A., Göbel C., Ross R. P., O'Sullivan O., Fitzgerald G. F., Feussner I., Stanton C., BMC Biochem. 2011, 12, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Volkov A., Khoshnevis S., Neumann P., Herrfurth C., Wohlwend D., Ficner R., Feussner I., Acta Crystallogr. Sect. D 2013, 69, 648–657. [DOI] [PubMed] [Google Scholar]
- 30. Volkov A., Liavonchanka A., Kamneva O., Fiedler T., Goebel C., Kreikemeyer B., Feussner I., J. Biol. Chem. 2010, 285, 10353–10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takeuchi M., Kishino S., Hirata A., Park S.-B., Kitamura N., Ogawa J., J. Biosci. Bioeng. 2015, 119, 636–641. [DOI] [PubMed] [Google Scholar]
- 32. Engleder M., Pavkov-Keller T., Emmerstorfer A., Hromic A., Schrempf S., Steinkellner G., Wriessnegger T., Leitner E., Strohmeier G. A., Kaluzna I., et al., ChemBioChem 2015, 16, 1730–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Demming R. M., Otte K. B., Nestl B. M., Hauer B., ChemCatChem 2017, 9, 758–766. [Google Scholar]
- 34. Shoji O., Yanagisawa S., Stanfield J. K., Suzuki K., Cong Z., Sugimoto H., Shiro Y., Watanabe Y., Angew. Chem. Int. Ed. 2017, 56, 10324–10329; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10460–10465. [Google Scholar]
- 35. Shoji O., Watanabe Y., Isr. J. Chem. 2015, 55, 32–39. [Google Scholar]
- 36. Zilly F. E., Acevedo J. P., Augustyniak W., Deege A., Häusig U. W., Reetz M. T., Angew. Chem. 2011, 123, 2772–2776. [DOI] [PubMed] [Google Scholar]
- 37. Suplatov D., Kirilin E., Takhaveev V., Švedas V., J. Biomol. Struct. Dyn. 2014, 32, 1752–1758. [DOI] [PubMed] [Google Scholar]
- 38. Reetz M. T., Chem. Rec. 2016, 16, 2449–2459. [DOI] [PubMed] [Google Scholar]
- 39. Seiffert G. B., Ullmann G. M., Messerschmidt A., Schink B., Kroneck P. M. H., Einsle O., Proc. Natl. Acad. Sci. USA 2007, 104, 3073–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bornscheuer U. T., Huisman G. W., Kazlauskas R. J., Lutz S., Moore J. C., Robins K., Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- 41. Nestl B. M., Hammer S. C., Nebel B. A., Hauer B., Angew. Chem. Int. Ed. 2014, 53, 3070–3095; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3132–3158. [Google Scholar]
- 42. Takeuchi M., Kishino S., Park S.-B., Hirata A., Kitamura N., Saika A., Ogawa J., J. Appl. Microbiol. 2016, 120, 1282–1288. [DOI] [PubMed] [Google Scholar]
- 43. Kang W.-R., Park C.-S., Shin K.-C., Kim K.-R., Oh D.-K., J. Am. Oil Chem. Soc. 2016, 93, 649–655. [Google Scholar]
- 44. Park J.-Y., Lee S.-H., Kim K.-R., Park J.-B., Oh D.-K., J. Biotechnol. 2015, 208, 1–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary