

Abstract
There is great interest in developing small molecules agents capable of reversing tumor immune escape to restore the body’s immune system. As an immunosuppressive enzyme, indoleamine 2,3-dioxygenase 1 (IDO-1) is considered a promising target for oncology immunotherapy. Currently, none of IDO-1 inhibitors have been launched for clinical practice yet. Thus, the discovery of new IDO-1 inhibitors is still in great demand. Herein, a series of diverse ortho-naphthaquinone containing natural product derivatives were synthesized as novel IDO-1 inhibitors. Among them, 1-ene-3-ketone-17-hydroxyl derivative 12 exhibited significantly improved enzymatic and cellular inhibitory activity against IDO-1 when compared to initial lead compounds. Besides, the molecular docking study disclosed that the two most potent compounds 11 and 12 have more interactions within the binding pocket of IDO-1 via hydrogen-bonding, which may account for their higher IDO-1 inhibitory activity.
Keywords: indoleamine 2,3-dioxygenase 1; immunosuppressive; natural product; ortho-naphthaquinone; molecular docking
1. Introduction
Although immune checkpoint blockades such as anti-PD-L1 (programmed cell death-ligand 1), anti-PD-1 (programmed cell death protein 1) [1], and anti-CTLA4 (cytotoxic T-lymphocyte-associated protein 4) [2] have demonstrated attractive therapeutic effects in multiple clinical trials, this new modality often suffers from a low response rate at least due to the immune escape developed by tumors [3]. Therefore, there is great interest in developing small molecule agents capable of reversing tumor immune escape to restore the body’s immune system.
Indoleamine 2,3-dioxygenase 1 (IDO-1) is a monomeric heme-containing enzyme found in nonhepatic human tissues [4]. It catalyzes the oxidative cleavage of the pyrrole ring of l-tryptophan (l-Trp) in the first and rate-limiting step of the kynurenine pathway to produce N-formylkynurenine [5]. This reaction not only leads to a local decrease of l-Trp concentration, but also generates a variety of catabolic products, which both account for the immunosuppressive effects of IDO-1 [6,7,8,9]. Thus, IDO-1 is considered as one of the key factors in the process of immune evasion in tumor microenvironment [10]. Overexpression of IDO-1 has been observed in a number of human malignancies, including ovarian, colorectal, and pancreatic cancers [11]. A growing body of clinical evidence indicated that the high expression of IDO-1 in tumors is directly correlated with a low survival rate [12]. In addition, IDO-1 inhibitors have demonstrated a significant cooperative effect with chemotherapy, radiotherapy, or cancer vaccines in preclinical models of cancer [13,14,15,16]. Accordingly, inhibition of IDO-1 would be a very useful strategy for the treatment of cancer. In the past few years, intense efforts have been devoted to developing IDO-1 inhibitors for the immuno-oncology therapy [17]. A number of structurally diverse natural products and synthetic IDO-1 inhibitors have been reported. Several IDO-1 inhibitors entered clinical trials such as indoximod (1) [13,18], epacadostat (2) [3], and NLG919 (3) (Figure 1) [19]. However, none of them have been launched for clinical practice yet. Thus, the discovery of new IDO-1 inhibitors is still in great demanded.
Figure 1.

Structures of known and our reported indoleamine 2,3-dioxygenase 1 (IDO-1) Inhibitors.
Natural products have long served as valuable starting points for drug discovery due to their unique molecular frameworks and novel mechanisms of actions. Danshen, a well-known traditional Chinese medicine (TCM) herb derived from the dried root or rhizome of Salvia miltiorrhiza Bunge, has long been used in Asian countries for multiple therapeutic remedies including cardiovascular and cerebrovascular disorders as well as inflammatory diseases [20,21,22]. Tanshinones such as Tan-IIA (6) (Figure 1), a group of lipophilic furano-o-naphthaquinone diterpenes isolated exclusively from this TCM herb, have demonstrated various pharmacological activities, such as cardio-protection, antibacterial, anti-inflammatory, antioxidant, anti-platelet aggregation, and anticancer properties [23,24].
As part of our drug discovery program towards identification of lead compounds from natural products bearing o-naphthaquinone scaffold [25,26,27,28,29,30,31], we screened our in-house compound library derived from o-naphthaquinones to pursue novel IDO-1 inhibitors, and found acyloxy derivatives of 6 possessing moderate IDO-1 inhibitory activity. When our project was ongoing, three o-naphthaquinone derivatives 4, 5, 9 were also identified as IDO-1 inhibitors by Wang and Xu et al. using machine-learning-based virtual screening (Figure 1) [32]. Inspired by these results, we further performed structural optimization based on the acyloxy derivatives to explore the chemical space of o-naphthaquinone scaffold for IDO-1 inhibition. Herein, we disclosed our effort on the chemical synthesis, biological evaluation, and molecular docking of o-naphthaquinone derivatives as novel IDO-1 inhibitors.
2. Results and Discussion
2.1. Chemistry
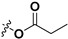
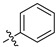
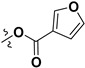
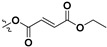
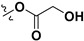
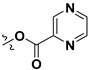
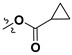
Following our previously established acyloxylation procedure [26,27,28], a series of diverse acyloxy derivatives 7a–m [27] were readily accessed by action of different carboxylic acids with 6 in 45%–95% yields (Table 1).
Table 1.
Structures of acyloxy derivatives 7a–m.
| Compound Number | R1 | R2 | Compound Number | R1 | R2 |
|---|---|---|---|---|---|
| 7a |
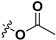
|
H | 7h |
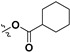
|
H |
| 7b |
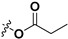
|
H | 7i |
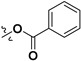
|
H |
| 7c |
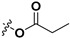
|
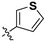
|
7j |
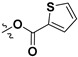
|
H |
| 7d |
|
|
7k |
|
H |
| 7e |
|
H | 7l |
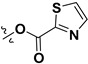
|
H |
| 7f |
|
H | 7m |
|
H |
| 7g |
|
H |
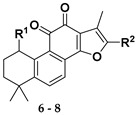
As outlined in Scheme 1, further hydrolysis of compound 7a with K2CO3 in methanol afforded 1-hydroxyl product 8 [33] in 99% yield, which further underwent an elimination reaction in the presence of pyridinium p–toluenesulfonate (PPTS) at 110 °C to give the 1-ene analogue 9 [33] in 85% yield. To introduce a hydroxyl group to the 3-position of the A-ring, an allylic oxidation was performed by treatment of 9 with selenium dioxide in refluxing 1,4-dioxane/H2O for 1.5 h to directly produce the Δ1-3-ketone (1-ene-3-ketone) derivative 11 [34,35] in 81% yield as sole product, instead of the 1-ene-3-hydroxyl derivative 10. Interestingly, prolonged reaction time not only provided 11, but also gave rise to 17-hydroxyl enone derivative 12 in 28% yield. It was believed that compounds 11 and 12 were produced by further oxidation of 10 although it was not observed in the reaction. Alternatively, reduction of 11 with NaBH4 provided allylic alcohol 10. In addition, treatment of compound 8 with 2-iodoxybenzoic acid in a mixture solvent of toluene and DMSO produced 1-ketone 13 [33] in 27% yield together with some minor enone derivative 14. Further oxidation of 13 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) could readily produce 1-ketone-Δ2 (1-ketone-2-ene) compound 14 in 70% yield. Subsequently, refluxing of 14 in dioxane/H2O using SeO2 as the oxidant afforded 17-hydroxy enone derivative 15 as the single product.
Scheme 1.

Synthesis of o-naphthaquinone derivatives 11–15. Reagents and conditions: (a) PPTS, toluene, 110 °C, 12 h; (b) SeO2, 1,4-dioxane, 100 °C; (c) NaBH4, MeOH, rt; (d) IBX, toluene/DMSO = 4:2, 60 °C, microwave, 3 h; (e) DDQ, toluene, 80 °C, 72 h; (f) SeO2, 1,4-dioxane, 100 °C, 36 h.
2.2. Biological Evaluation
All the synthesized derivatives were evaluated for their IDO-1 inhibitory activities using epacadostat as the positive control agent. The potent compounds were further assayed for their cellular activity in HEK 293 cells over-expressing human IDO-1. As shown in Table 2, a series of acyloxylated derivatives 7a–m was first evaluated. Compared to 6, acyclic acyloxylated derivatives 7a–b and 7e–f displayed significantly improved IDO-1 inhibitory activity with IC50 values ranging from 2 to 8 µM. However, installation of aryl or heteroaryl group at C-15 of the furan ring (7c–d) resulted in loss of the activity with IC50 value greater than 20 µM. Among cyclic acyloxylated derivatives 7g–m, furan compound 7k possessed the most potent activity against IDO-1 with an IC50 value of 4.60 µM. Most of the six-membered cyclic acyloxylated derivatives 7h–i exhibited relatively weak activity with IC50 values around 20 µM except pyrazine compound 7m. Hydrolysis of the acyloxyl into the hydroxyl (compound 8) displayed comparable inhibitory activity to that of precursor 7a. Compound 9 with 1-ene possessed 61.2% inhibitory rate at 20 µM, which is less potent than that reported by literature [32]. Compound 10 with hydroxyl group at the allylic position recovered the activity with an IC50 value of 3.10 µM. Compound 11 with 1-ene-3-ketone moiety exhibited significantly improved IDO-1 inhibitory activity with an IC50 value of 0.84 µM, and 17-hydroxyl derivative 12 also possessed more potent activity with 0.37 µM IC50 value. By contrast, both 1-ketone-2-ene compound 14 and its 17-hydroxyl analogue 15 demonstrated weak activities with IC50 values around 20 µM.
Table 2.
The IDO-1 inhibitory activity of o-naphthaquinone derivatives in enzyme- and cell-based assays.
| Compound Number | R1 | R2 | Enzymatic IC50 (μM) or Inhibitory Rate at 20 µM a | Cellular IC50 (μM) a | |
|---|---|---|---|---|---|
|
6 | H | H | 23.1 ± 1.6% | — b |
| 7a |
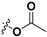
|
H | 3.72 ± 0.45 | — | |
| 7b |
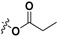
|
H | 4.72 ± 0.30 | — | |
| 7c |
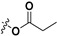
|
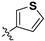
|
29.3 ± 2.2% | — | |
| 7d |
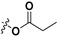
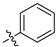
|
|
21.5 ± 1.1% | — | |
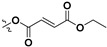
| 7e |
|
H | 7.47 ± 3.58 | — | |
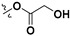
| 7f |
|
H | 2.74 ± 0.10 | — | |
| 7g |
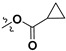
|
H | 7.05 ± 2.46 | — | |
| 7h |
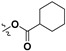
|
H | 48.0 ± 3.2% | — | |
| 7i |
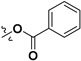
|
H | 54.5 ± 2.3% | — | |
| 7j |
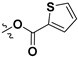
|
H | 60.4 ± 3.9% | — | |
| 7k |
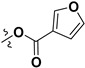
|
H | 4.60 ± 2.81 | — | |
| 7l |
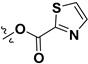
|
H | 5.71 ± 0.47 | — | |
| 7m |
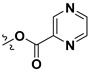
|
H | 4.96 ± 2.02 | — | |
| 8 | OH | H | 2.83 ± 0.12 | — | |
|
9 | H | 61.2 ± 2.1% | >10 | |
| 10 | OH | 3.10 ± 1.02 | >10 | ||
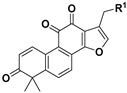
|
11 | H | 0.84 ± 0.26 | >10 | |
| 12 | OH | 0.37 ± 0.02 | 3.85 ± 0.86 | ||
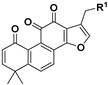
|
14 | H | 52.7 ± 2.5% | >10 | |
| 15 | OH | 68.0 ± 8.6% | — | ||
| Epacadostat | 0.086 ± 0.009 | 0.023 ± 0.003 | |||
a The values are the mean ± SE of two independent experiments; b Not determined.
Compounds 9–14 were further assayed for their activity to inhibit IDO-1 in HEK 293-hIDO-1 cells. Compared to their enzymatic activity, most of them displayed less potent inhibitory activity against IDO-1 probably due to their poor permeability. Among them, compound 12 exhibited the most potent cellular inhibitory activity with an IC50 value of 3.85 µM. The calculated cLogP values of 11 and 12 with Chemdraw were 3.90 and 2.36, respectively, which may account for their difference on the cellular potency.
2.3. Molecular Docking Study
To understand the binding modes of our compounds for IDO-1, compounds 11, 12, and 14 were docked into the binding pocket of an IDO-1 X-ray crystal structure (PDB code 5WHR). All of them could interact with residues Phe226, Thr379, Phe163, Tyr126, and Gly378 within hydrophobic pockets (Figure S17). The ortho-diketone moiety of 11 interacted with heme Fe2+ through two coordination bonds, and its 3-ketone had an additional hydrogen-bond interaction with Thr379 (Figure 2a). For compound 12, the ortho-diketone moiety not only formed two coordination bonds with heme Fe2+ (Figure 2b), but also had a hydrogen-bond interaction with the NH of Ala264 (Figure S17b). Besides, the 17-hydroxyl group of 12 also interacted with residue Ser167 via a hydrogen-bond (Figure S17b). The 1-ketone group of 14 formed only one coordination bond with Fe2+ of heme (Figure 2c). These docking results suggested that compounds 12 and 11 have more interactions with IDO-1 than 14, which may account for their more potent IDO-1 inhibitory activity.
Figure 2.

Docking models of compounds 11, 12, and 14 bound to IDO-1. (a) Computational modeling at the active site of IDO-1 for 11; (b) computational modeling at the active site of IDO-1 for 12; (c) computational modeling at the active site of IDO-1 for 14.
3. Materials and Methods
3.1. Chemistry
3.1.1. General Chemistry
All solvents and chemical reagents were obtained from commercial sources and used without further purifications. 1H- and 13C-NMR spectra were recorded on a Varian Mercury 400 NMR spectrometer (Varian, San Francisco, CA, USA) and Bruker avance III (Bruker, Karlsruhe, Germany). High-resolution mass spectrometry (HRMS) analysis was recorded at an ionizing voltage of 70 eV on a Agilent G6520 Q-TOF spectrometer (Agilent, San Francisco, CA, USA). Flash column chromatography on silica gel (200–300 mesh) was used for the routine purification of reaction products. All reactions were monitored by TLC on silica gel plates.
3.1.2. Procedure for the Preparation of Compounds
Synthesis of 1,6,6-trimethyl-6,7-dihydrophenanthro[1,2-b]furan-10,11-dione (9) [33]
To a solution of 8 (100 mg, 0.322 mmol) in toluene (3 mL) was added pyridinium p–toluenesulfonate (PPTS, 248 mg, 0.483 mmol). The reaction mixture was stirred at 110 °C for 12 h and then diluted with H2O and extracted with EtOAc (100 mL × 3). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was further purified by silica gel column, and elution with 15%–10% EtOAc in hexane afforded the desired product 9 (80 mg, 85%) as a red solid. 1H-NMR (400 MHz, CDCl3) δ 7.85 (dt, J = 10.2, 1.8 Hz, 1H), 7.55 (s, 2H), 7.23 (d, J = 1.3 Hz, 1H), 6.33 (dt, J = 10.1, 4.6 Hz, 1H), 2.29 (d, J = 1.8 Hz, 1H), 2.28 (d, J = 1.9 Hz, 1H), 2.27 (d, J = 1.3 Hz, 3H), 1.30 (s, 6H). 13C-NMR (126 MHz, CDCl3) δ 184.4, 175.9, 161.5, 148.5, 141.3, 137.4, 134.0, 130.3, 127.3, 124.5, 123.1, 121.5, 121.1, 120.0, 37.9, 34.2, 28.4, 8.8.
Synthesis of 1,6,6-trimethylphenanthro[1,2-b]furan-7,10,11(6H)-trione (11)
To a solution of 9 (35 mg, 0.120 mmol) in 1,4-dioxane (2 mL) was added selenium dioxide (13 mg, 0.120 mmol). The reaction mixture was stirred at 100 °C for 1.5 h and then diluted with H2O and extracted with EtOAc (50 mL × 3). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was further purified by silica gel column, and elution with 1% MeOH in CH2Cl2 afforded the desired product 11 (30 mg, 82%) as a red solid. m.p. 261.2–262.8 °C. 1H-NMR (400 MHz, CDCl3): δ 8.99 (d, J = 10.5 Hz, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.72 (d, J = 8.1 Hz, 1H), 7.30 (s, 1H), 6.41 (d, J = 10.5 Hz, 1H), 2.29 (s, 3H), 1.51 (s, 6H). 13C-NMR (126 MHz, CDCl3) δ 201.9, 184.2, 175.3, 160.5, 150.7, 142.0, 139.2, 132.3, 132.3, 128.6, 128.5, 125.3, 123.5, 121.5, 120.6, 47.9, 27.6, 8.8. MS (ESI, [M + Na]+) m/z 329.3. HRMS (ESI, [M + H]+) calcd for C19H15O4, 307.0965; found, 307.0973.
Synthesis of 1-(hydroxymethyl)-6,6-dimethylphenanthro[1,2-b]furan-7,10,11(6H)-trione (12)
To a solution of 9 (80 mg, 0.273 mmol) in 1,4-dioxane (2 mL) was added selenium dioxide (30 mg, 0.273 mmol). The reaction mixture was stirred at 100 °C for 24 h and then diluted with H2O and extracted with EtOAc (100 mL × 3). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was further purified by silica gel column, and elution with 1%–5% MeOH in CH2Cl2 afforded the desired product 11 (30 mg, 36%, Rf = 0.4 in 5% MeOH/CH2Cl2) and 12 (25 mg, 28%, Rf = 0.2 in 5% MeOH/CH2Cl2) as red solid. Compound 12: m.p. 213.4-216.5 °C. 1H-NMR (400 MHz, CDCl3) δ 8.99 (d, J = 10.5 Hz, 1H), 7.83 (d, J = 8.1 Hz, 1H), 7.75 (d, J = 8.3 Hz, 1H), 7.48 (s, 1H), 6.44 (d, J = 10.5 Hz, 1H), 4.71 (s, 2H), 1.52 (s, 6H). 13C-NMR (126 MHz, CDCl3) δ 201.6, 183.3, 175.5, 161.9, 151.6, 141.4, 138.8, 132.8, 132.5, 129.0, 128.0, 126.1, 125.2, 123.8, 120.1, 55.2, 48.1, 27.6. MS (ESI, [M + Na]+) m/z 345.2. HRMS (ESI, [M + H]+) calcd for C19H15O5, 323.0914; found, 323.0915.
Synthesis of 7-hydroxy-1,6,6-trimethyl-6,7-dihydrophenanthro[1,2-b]furan-10,11-dione (10) [33]
To a solution of 11 (30 mg, 0.098 mmol) in MeOH (2 mL) was added NaBH4 (11 mg, 0.300 mmol). The reaction mixture was stirred at rt for 1 h and then evaporated the solvent, diluted with H2O, and extracted with EtOAc (50 mL × 3). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was further purified by silica gel column, and elution with 1%–2% MeOH in CH2Cl2 afforded the desired product 10 (27 mg, 90%) as a red solid. m.p. 185.2-188.5 °C. 1H-NMR (400 MHz, CDCl3) δ 7.87 (d, J = 10.2 Hz, 1H), 7.64–7.55 (m, 2H), 7.24 (s, 1H), 6.39 (dd, J = 10.2, 4.4 Hz, 1H), 4.08 (d, J = 4.5 Hz, 1H), 2.26 (s, 3H), 1.39 (s, 3H), 1.28 (s, 3H). 13C-NMR (126 MHz, CDCl3) δ 184.2, 175.6, 161.2, 147.1, 141.5, 135.8, 135.5, 131.5, 127.7, 125.0, 123.6, 122.1, 121.2, 120.2, 72.5, 40.0, 26.3, 21.9, 8.8.
Synthesis of 1,6,6-trimethyl-7,8-dihydrophenanthro[1,2-b]furan-9,10,11(6H)-trione (13) and 1,6,6-trimethylphenanthro[1,2-b]furan-9,10,11(6H)-trione (14)
To a solution of 8 (200 mg, 0.644 mmol) in toluene (4 mL) and DMSO (2 mL) was added 2-Iodoxybenzoic acid (1.082 g, 3.864 mmol). The reaction mixture was stirred at 60 °C microwave for 3 h and then diluted with H2O and extracted with EtOAc (150 mL × 3). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was further purified by silica gel column, and elution with 30%–40% EtOAc in hexane afforded 13 (55 mg, 27%, Rf = 0.4 in 40% EtOAc/hexane) and 14 (38 mg, 19%, Rf = 0.2 in 40% EtOAc/hexane) as an orange solid. Compound 13: 1H-NMR (300 MHz, CDCl3) δ 7.72 (d, J = 8.2 Hz, 1H), 7.57 (d, J = 8.2 Hz, 1H), 7.26 (d, J = 1.4 Hz, 1H), 2.92 (t, J = 7.2 Hz, 2H), 2.26 (d, J = 1.3 Hz, 3H), 2.07 (t, J = 7.2 Hz, 2H), 1.34 (s, 6H). Compound 14: m.p. 158.3–160.8 °C.1H-NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.3 Hz, 1H), 7.72 (d, J = 8.3 Hz, 1H), 7.27 (s, 1H), 6.80 (d, J = 10.2 Hz, 1H), 6.48 (d, J = 10.2 Hz, 1H), 2.26 (s, 3H), 1.50 (s, 6H). 13C-NMR (126 MHz, CDCl3) δ 185.8, 183.4, 179.3, 159.6, 154.6, 151.8, 141.9, 135.3, 132.8, 131.6, 128.9, 127.4, 124.5, 121.3, 120.9, 38.3, 29.4, 8.7. MS (ESI, [M + H]+) m/z 307.4. HRMS (ESI) calcd for C19H15O4, 307.0965; found, 307.0967.
Synthesis of 1,6,6-trimethylphenanthro[1,2-b]furan-9,10,11(6H)-trione (14)
To a solution of 13 (44 mg, 0.14 mmol) in toluene (3 mL) was added 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 49 mg, 0.21 mmol). The reaction mixture was stirred at 80 °C for 36 h and then diluted with H2O and extracted with EtOAc (100 mL × 3). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was further purified by silica gel column, and elution with 40% EtOAc in hexane afforded 14 (31 mg, 70%).
Synthesis of 1-(hydroxymethyl)-6,6-dimethylphenanthro[1,2-b]furan-9,10,11(6H)-trione (15)
To a solution of 14 (30 mg, 0.098 mmol) in 1,4-dioxane (2 mL) was added selenium dioxide (44 mg, 0.392 mmol). The reaction mixture was stirred at 100 °C for 36 h and then diluted with H2O and extracted with EtOAc (50 mL × 3). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was further purified by silica gel column, and elution with 1%–2% MeOH in CH2Cl2 afforded the desired product 15 (5 mg, 16%) as an orange solid. m.p. 195.5–198.4 °C. 1H-NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.3 Hz, 1H), 7.75 (d, J = 8.3 Hz, 1H), 7.45 (s, 1H), 6.82 (d, J = 10.3 Hz, 1H), 6.47 (d, J = 10.2 Hz, 1H), 4.69 (s, 2H), 1.51 (s, 6H). 13C-NMR (126 MHz, CDCl3) δ 184.7, 183.3, 179.4, 161.1, 154.6, 152.5, 141.2, 135.7, 132.8, 131.8, 128.5, 127.4, 126.1, 124.7, 120.5, 55.2, 38.4, 29.5. HRMS (ESI, [M + H]+) calcd for C19H15O5, 323.0914; found, 323.0901.
3.2. Biology
hIDO-1 enzymatic assay. The hIDO-1 enzymatic assay was performed as described previously [36]. Briefly, a standard reaction mixture (30 μL) containing 100 mM potassium phosphate buffer (pH 6.5), 40 mmol/L ascorbic acid and 0.01% Triton X-100, 200 µg/mL catalase, 20 µmol/L methylene blue, and 0.05 µM rhIDO-1 was added to the solution (60 μL) containing the substrate l-tryptophan (250 µmol/L) and the test sample at a determined concentration. The reaction was carried out at 37 °C for 30 min and stopped by adding 45 µL of 30% (w/v) trichloroacetic acid. After being heated at 65 °C for 15 min, the reaction mixture was centrifuged at 12,000 rpm for 10 min. The supernatant (100 µL) was transferred into a well of a 96-well microplate and mixed with 100 µL of 2% (w/v) p-dimethylaminobenzaldehyde in acetic acid. The yellow pigment derived from kynurenine was measured at 492 nm using a Spectra Max Plus 384 microplate reader (Molecular Devices, Sunnyvale, CA, USA). IC50 values were calculated by using Graph Pad Prism 6 software (version 6.00, Graph Pad Software, San Diego, CA, USA).
Cell-based assay of IDO-1 activity. The cellular activity of IDO-1 was detected as described previously [36]. HEK 293 cells were seeded in a 6-well culture plate at a density of 5 × 105 cells/well and cultured overnight. After 24 h, HEK 293 cells were transfected with pcDNA3.1-hIDO-1 using Lipofectamine 2000 according to the manufacturer’s instructions. Cells were seeded in a 96-well culture plate at a density of 2.5 × 104 cells/well 24 h after transfection. A serial dilution of the tested compounds in 10 µL PBS was added to the cells. After an additional 12-h incubation, 200 µL of the supernatant per well was transferred to a new 96-well plate and mixed with 100 µL of 30% trichloroacetic acid in each well, and the plate was incubated at 65 °C for 15 min to hydrolyze N-formylkynurenine produced by the catalytic reaction of hIDO-1. The reaction mixture was then centrifuged for 10 min at 12,000 rpm to remove the sediments. Then, 100 µL of the supernatant per well were transferred to another 96-well plate and mixed with 100 µL of 2% (w/v) p-dimethylaminobenzaldehyde in acetic acid. The yellow color derived from kynurenine was measured at 492 nm using a Spectra Max Plus 384 microplate reader (Molecular Devices, Sunnyvale, CA, USA). IC50 values were calculated by using Graph Pad Prism 6 software (version 6.00, Graph Pad Software, San Diego, CA, USA).
3.3. Docking Study
Molecular modeling study of our compounds against IDO-1 was performed using the Schrödinger Suite 2015-4 (Schrödinger, LLC, New York, NY, USA). The X-ray crystal structure of IDO-1 was obtained from the Protein Data Bank. The IDO-1 protein preparation was revised using Protein Preparation Wizard. The receptor grid box was generated 25 × 25 × 25 Å cubic size. The compound was minimized using an OPLS_2005 force field with a dielectric constant value 80.0 in Macro Model.
4. Conclusions
In conclusion, we have designed and synthesized a series of furano-o-naphthaquinone derivatives as novel IDO-1 inhibitors. Among them, 1-ene-3-ketone-17-hydroxyl derivative 12 exhibited significantly improved enzymatic and cellular inhibitory activity against IDO-1 when compared to acyloxy derivatives. Besides, the molecular docking study also disclosed compounds 12 and 11 have more interactions with the binding pocket of IDO-1 via hydrogen-bond, which may account for their higher IDO-1 inhibitory activity. Although our synthesized derivatives displayed relative lower inhibitory activity than reported IDO-1 inhibitors, the o-naphthaquinone scaffold together with the novel binding model will open new avenues to develop better IDO-1 inhibitors for the immuno-oncology therapy.
Supplementary Materials
The following are available online.
Author Contributions
H.Z., P.S., W.G., and Y.W. performed the experiments; L.M., A.Z., and C.D. designed the experiment, analyzed the data, and wrote the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (21877120, 81430080) and the National Program on Key Basic Research Project of China (2015CB910603). Supporting from National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (Number: 2018ZX09711002-010-001), as well as grant from the ‘‘Personalized Medicines—Molecular Signature-based Drug Discovery and Development”, Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA12020320) are also highly appreciated.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 11, 12, 14 are available from the authors.
References
- 1.Robert C., Schachter J., Long G.V., Arance A., Grob J.J., Mortier L., Daud A., Carlino M.S., McNeil C., Lotem M., et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 2.Hodi F.S., O’Day S.J., McDermott D.F., Weber R.W., Sosman J.A., Haanen J.B., Gonzalez R., Robert C., Schadendorf D., Hassel J.C., et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yue E.W., Sparks R., Polam P., Modi D., Douty B., Wayland B., Glass B., Takvorian A., Glenn J., Zhu W., et al. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO-1) Inhibitor for Immuno-oncology. ACS Med. Chem. Lett. 2017;8:486–491. doi: 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Masanori S., Mark P.R., Eric D.C., John H.D. Heme-Containing Oxygenases. Chem. Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto S., Hayaishi O. Tryptophan pyrrolase of rabbit intestine. d- and l-tryptophan-cleaving enzyme or enzymes. J. Biol. Chem. 1967;242:5260–5266. [PubMed] [Google Scholar]
- 6.David H.M., Zhou M., John T.A., Igor B., Simon J.C., Brendan M., Corrie B., Andrew L.M. Prevention of Allogeneic Fetal Rejection by Tryptophan Catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 7.Hwu P., Du M.X., Lapointe R., Do M., Taylor M.W., Young H.A. Indoleamine 2,3-Dioxygenase Production by Human Dendritic Cells Results in the Inhibition of T Cell Proliferation. J. Immunol. 2000;164:3596–3599. doi: 10.4049/jimmunol.164.7.3596. [DOI] [PubMed] [Google Scholar]
- 8.Terness P., Bauer T.M., Röse L., Dufter C., Watzlik A., Simon H., Opelz G. Inhibition of Allogeneic T Cell Proliferation by Indoleamine 2,3-Dioxygenase–expressing Dendritic Cells. J. Exp. Med. 2002;196:447–457. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fallarino F., Grohmann U., You S., McGrath B.C., Cavener D.R., Vacca C., Orabona C., Bianchi R., Belladonna M.L., Volpi C., et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor-Chain and Induce a Regulatory Phenotype in Naive T Cells. J. Immunol. 2006;176:6752–6761. doi: 10.4049/jimmunol.176.11.6752. [DOI] [PubMed] [Google Scholar]
- 10.Munn D.H., Mellor A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Investig. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uyttenhove C., Pilotte L., Theate I., Stroobant V., Colau D., Parmentier N., Boon T., Van den Eynde B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 12.Okamoto A., Nikaido T., Ochiai K., Takakura S., Saito M., Aoki Y., Ishii N., Yanaihara N., Yamada K., Takikawa O., et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005;11:6030–6039. doi: 10.1158/1078-0432.CCR-04-2671. [DOI] [PubMed] [Google Scholar]
- 13.Hou D.Y., Muller A.J., Sharma M.D., DuHadaway J., Banerjee T., Johnson M., Mellor A.L., Prendergast G.C., Munn D.H. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 14.Muller A.J., DuHadaway J.B., Donover P.S., Sutanto-Ward E., Prendergast G.C. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005;11:312–319. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 15.Prendergast G.C., Malachowski W.P., DuHadaway J.B., Muller A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017;77:6795–6811. doi: 10.1158/0008-5472.CAN-17-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma M.D., Hou D.Y., Liu Y., Koni P.A., Metz R., Chandler P., Mellor A.L., He Y., Munn D.H. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood. 2009;113:6102–6111. doi: 10.1182/blood-2008-12-195354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rohrig U.F., Majjigapu S.R., Vogel P., Zoete V., Michielin O. Challenges in the Discovery of Indoleamine 2,3-Dioxygenase 1 (IDO-1) Inhibitors. J. Med. Chem. 2015;58:9421–9437. doi: 10.1021/acs.jmedchem.5b00326. [DOI] [PubMed] [Google Scholar]
- 18.Nakashima H., Uto Y., Nakata E., Nagasawa H., Ikkyu K., Hiraoka N., Nakashima K., Sasaki Y., Sugimoto H., Shiro Y., et al. Synthesis and biological activity of 1-methyl-tryptophan-tirapazamine hybrids as hypoxia-targeting indoleamine 2,3-dioxygenase inhibitors. Bioorg. Med. Chem. 2008;16:8661–8669. doi: 10.1016/j.bmc.2008.07.087. [DOI] [PubMed] [Google Scholar]
- 19.Mautino M., Kumar S., Waldo J., Jaipuri F., Kesharwani T. Fused Imidazole Derivatives Useful as IDO Inhibitors. WO2012/142237. U.S. Patent. 2012 Oct 18;
- 20.Nakao M., Fukushima T. On the chemical composition of Salvia miltiorrhiza (Chinese drug Tan-shen) J. Pharm. Soc. Jpn. 1934;54:154–162. doi: 10.1248/yakushi1881.54.9_844. [DOI] [Google Scholar]
- 21.Zhou L., Zuo Z., Chow M.S. Danshen: An overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. J. Clin. Pharmacol. 2005;45:1345–1359. doi: 10.1177/0091270005282630. [DOI] [PubMed] [Google Scholar]
- 22.Wang X.H., Morris-Natschke S.L., Lee K.H. New developments in the chemistry and biology of the bioactive constituents of Tanshen. Med. Res. Rev. 2007;27:133–148. doi: 10.1002/med.20077. [DOI] [PubMed] [Google Scholar]
- 23.Dong Y., Morris-Natschke S.L., Lee K.H. Biosynthesis, total syntheses, and antitumor activity of tanshinones and their analogs as potential therapeutic agents. Nat. Prod. Rep. 2011;28:529–542. doi: 10.1039/c0np00035c. [DOI] [PubMed] [Google Scholar]
- 24.Don M.J., Shen C.C., Syu W.J., Ding Y.H., Sun C.M. Cytotoxic and aromatic constituents from Salvia miltiorrhiza. Phytochemistry. 2006;67:497–503. doi: 10.1016/j.phytochem.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 25.Jiao M., Ding C., Zhang A. Facile construction of 3-hydroxyphenanthrene-1,4-diones Using a tandem three-step reaction sequence as key intermediates to tanshinone I and its 4-demethylated analogues. Tetrahedron. 2014;70:2976–2981. doi: 10.1016/j.tet.2014.03.019. [DOI] [Google Scholar]
- 26.Jiao M., Ding C., Zhang A. Preparation of 2-aryl derivatives of tanshinone I through a palladium-catalyzed Csp2–H activation/arylation approach. Tetrahedron Lett. 2015;56:2799–2802. doi: 10.1016/j.tetlet.2015.04.040. [DOI] [Google Scholar]
- 27.Ding C., Li J., Jiao M., Zhang A. Direct Catalyst-Free C(sp3)-H Acyloxylation of Tanshinone IIA: Regioselective Synthesis of C1-Acyloxyl Derivatives. J. Nat. Prod. 2016;79:2514–2520. doi: 10.1021/acs.jnatprod.6b00370. [DOI] [PubMed] [Google Scholar]
- 28.Liang B., Yu S., Li J., Wang F., Liang G., Zhang A., Ding C. Site-Selective Csp3-H Aryloxylation of Natural Product Tanshinone IIA and Its Analogues. Tetrahedron Lett. 2017;58:1822–1825. doi: 10.1016/j.tetlet.2017.03.078. [DOI] [Google Scholar]
- 29.Li J., Xue Y., Fan Z., Ding C., Zhang A. Dihydroxydifluorination of Tanshinones Analogues. J. Org. Chem. 2017;82:7388–7393. doi: 10.1021/acs.joc.7b01064. [DOI] [PubMed] [Google Scholar]
- 30.Ding C., Tian Q., Li J., Jiao M., Song S., Wang Y., Miao Z.-H., Zhang A. Structural Modification of Natural Product Tanshinone I Leading to Discovery of Novel Nitrogen-Enriched Derivatives with Enhanced Anticancer Profile and Improved Drug-Like Properties. J. Med. Chem. 2018;61:760–776. doi: 10.1021/acs.jmedchem.7b01259. [DOI] [PubMed] [Google Scholar]
- 31.Wang F., Yang H., Yu S., Xue Y., Fan Z., Liang G., Geng M., Zhang A., Ding C. Divergent Total Synthesis of (±)-Tanshinol B, (±)-Tanshindiol B, (±)-Tanshindiol C. and Tanshinone I. Org. Biomol. Chem. 2018;16:3376–3381. doi: 10.1039/C8OB00567B. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H., Liu W., Liu Z., Ju Y., Xu M., Zhang Y., Wu X., Gu Q., Wang Z., Xu J. Discovery of indoleamine 2,3-dioxygenase inhibitors using machine learning based virtual screening. Med. Chem. Commun. 2018;9:937–945. doi: 10.1039/C7MD00642J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding L., Wang J., Huang Z. Tanshinone IIA Derivative Useful in Treatment of Cancer and Its Preparation. CN 105,884,856. Patent. 2016 Aug 24;
- 34.Qin Y., Su M., Jin Q., Chen T., Wu X., Mao L. 1-Carbonyl Tanshinone IIA Sodium Sulfonate Analogue, Preparation Method and Application. CN 104,910,250. Patent. 2015 Sep 16;
- 35.Zhao Q., Leng Y., Deng X., Zhao Y., Shen Y., Luo X. Tanshinone Derivative, Its Pharmaceutical Composition, and Medical Application for Treating Diabetes Mellitus and Related Metabolic Diseases. CN 102,603,861. Patent. 2012 Jul 25;
- 36.Yu C.J., Zheng M.F., Kuang C.X., Huang W.D., Yang Q. Oren-gedoku-to and its constituents with therapeutic potential in Alzheimer’s disease inhibit indoleamine 2,3-dioxygenase activity in vitro. J. Alzheimers Dis. 2010;22:257–266. doi: 10.3233/JAD-2010-100684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.