

Abstract
In this work the synthesis, photochemistry, and streptavidin interaction of new [Ru(tpy)(bpy)(SRR′)](PF6)2 complexes where the R′ group contains a free biotin ligand, are described. Two different ligands SRR′ were investigated: An asymmetric ligand 1 where the Ru‐bound thioether is a N‐acetylmethionine moiety linked to the free biotin fragment via a triethylene glycol spacer and a symmetrical ligand 2 containing two identical biotin moieties. The coordination of these two ligands to the precursor [Ru(tpy)(bpy)Cl]Cl was studied in water at 80 °C. In such conditions the coordination of the asymmetric ligand 1 occurred under thermodynamic control. After the reaction, a mononuclear and a binuclear complex were isolated. In the mononuclear complex, the ratio of methionine‐ {[6](PF6)2} vs. biotin‐bound {[7](PF6)2} regioisomer was 5.3 and the free biotin fragment of [6](PF6)2 allowed to purify it from its isomer [7](PF6)2 at small scales using avidin affinity chromatography. Coordination of the symmetrical ligand 2 afforded [Ru(tpy)(bpy)(2)](PF6)2 {[8](PF6)2} in synthetically useful scales (100 mg), good yield (82 %), and without traces of the binuclear impurity. In this complex, one of the biotin remains free whereas the second one is coordinated to ruthenium. Photochemical release of ligand 2 from [8](PF6)2 occurred upon blue light irradiation (465 nm) with a photosubstitution quantum yield of 0.011 that was independent of the binding of streptavidin to the free biotin ligand.
Keywords: Ruthenium, Streptavidin, Biotin, Photochemistry, Antitumor agents, Synthesis design, Thioethers
Introduction
One of the strongest known non‐covalent interactions in nature is the one between (strept)avidin and biotin (k d = 10–13 to 10 –15 L mol–1).1 Once the avidin‐biotin complex (ABC) is formed only extremely harsh conditions (6 m guanidine‐HCl, pH = 1.5) will lead to disassembly.2 This phenomenon, discovered 48 years ago, led to the emergence of the ABC field in biotechnology.1, 3 (Strept)avidin systems are utilized for bioconjugates, for theranostic purposes, but also as tumor‐targeting strategy,4 for which the high affinity of biotin for avidin is critical.3 Recently, e.g. an almost ten‐fold increase of cellular uptake of microspheres was reached by employing the ABC technique.5 However, for drug‐delivery strong binding also represent the major drawback, as it is difficult to obtain efficient release of the targeting moiety.6 Typically, targeting the cancer cells works well, but less cytotoxicity is observed, because the drug is not efficiently released from its targeting group and thus trapped in the endosomes for example.6 To solve this issue, strategies with pre‐determined “break junctions” have been developed. For example, the targeting fragment can be bound via linkers that are sensitive to intracellular triggers, such as pH, proteases, or glutathione concentration.6 These strategies, however, lack temporal and spatial control. Light activation would solve this issue by allowing to release the targeting moiety by an external trigger, i.e. photons.7 We reported earlier on a method to cleave methionine‐ and biotin‐based ligands from a ruthenium(II) polypyridyl complex using visible light.8 Following the steps of Lo et al.,9 Ward et al,10 and others,11 who showed that biotin‐tagged metal compounds could keep the biotin affinity for (strept)avidin, we embarked on investigating synthetically useful methods to functionalize photosubstitutionally active ruthenium polypyridyl complexes such as [Ru(tpy)(bpy)(Cl)]Cl with a free biotin fragment that can be removed by visible light, for future use in photochemotherapy.12, 13
Results and Discussion
Ligand Design and Synthesis
Thioether groups such as those found in N‐acetylmethionine or biotin are excellent ligands for ruthenium(II) polypyridyl complexes that can be cleaved by visible light irradiation.[8c] The dissymmetric ligand 1 that contains both sulfur donors separated by a short ethylene glycol spacer was hence designed first (Scheme 1). Based on the higher thermal stability of N‐acetylmethionine ruthenium complexes compared to biotin adducts,[8c] we hypothesized that coordination of the sterically less hindered methionine fragment of 1 may be preferred over that of biotin. In the symmetrical bis‐biotinylated ligand 2, on the contrary, one biotin should coordinate to ruthenium, while the second, identical fragment should remain free to interact with streptavidin.
Scheme 1.
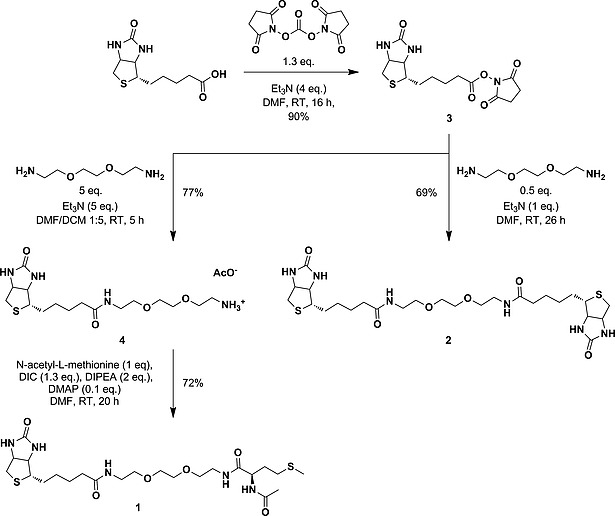
Overview of the synthetic routes used to obtain bis‐thioether ligands 1 and 2.
The first step in the synthesis of 1 (Scheme 1) involved the activation of biotin with disuccinimidyl carbonate to obtain the N‐hydroxysuccinimidyl ester of biotin, biotin‐NHS (3).14 In a second step, compound 3 was treated with 5 equiv. of the linker 2,2′‐(ethylenedioxy)bis(ethylamine) in DMF at room temp. which afforded the mono‐biotin (4) ligand. The final reaction of 4 with N‐acetyl‐l‐methionine (HAmet) afforded 1 as colorless, viscous oil in 72 % yield. Full characterization of ligand 1 was performed by NMR, ESI MS, and HRMS measurements (see Supporting Information). Ligand 2 was easily obtained from the bisamine spacer used for 1 and two equivalents of NHS‐biotin 3 (see Scheme 1 and Experimental Part).
Coordination of 1 and 2 to Ruthenium
For the synthesis of [Ru(tpy)(bpy)(1)](Cl)2 ([6]Cl2, Scheme 2), [Ru(tpy)(bpy)Cl]Cl was treated with 1 in water at 80 °C over three days. Under these conditions, the chloride precursor hydrolyses quickly[8c] to the aqua complex [Ru(tpy)(bpy)(OH2)]2+, which reacts with one or the other thioether moiety. After workup and column chromatography on silica, two different fractions were obtained. The 1H NMR (Figure S1) of the more polar fraction (R f = 0.1, acetone/H2O/1 m HCl [16:4:1]) showed two signals with the same integral between δ = 9.50–10.00 ppm, indicating two downfield‐shifted A6 protons on the bipyridine ligand instead of the expected unique doublet (see Scheme 2).[8c] Furthermore, the sum of integrals of the aromatic part was twice as high as that of the aliphatic part. Additionally, all methyl and methylene groups in close proximity to the sulfur‐atoms showed a significant downfield shift. This characteristic observation emerges from the shielding cone of the polypyridyl ligands when a monodentate ligand is coordinated to the ruthenium polypyridyl moiety.15 Taken together, NMR analysis indicated that this fraction contained a binuclear ruthenium complex [5]Cl4 (Scheme 2). This hypothesis was confirmed by mass spectrometry; a peak at m/z = 528.1 was found, which fits the calculated m/z value for [{Ru(tpy)(bpy)}2(1) + H2O + Cl]3+ (528.1). For the less polar fraction (R f = 0.3, acetone/H2O/1 m HCl [16:4:1]), mass analysis showed the expected peak at m/z = 519.2 for the mononuclear ruthenium complex [Ru(tpy)(bpy)(1)]2+ (calcd. m/z = 519.12). According to 1H‐NMR spectroscopy, the integral ratio between the characteristic signals of the aromatic part and that of the aliphatic part was close to one. Further analysis of the 1H NMR revealed significant up‐field shifts for the methyl signal of the methionine fragment (e.g. Δδ = –0.74 ppm for M1), which proved the formation of the desired mononuclear ruthenium complex [6]Cl2 coordinated via the sulfur of the N‐Acetylmethionine methionine as major isomer (Scheme 2). However, next to the characteristic A6 signal at δ = 9.75 ppm for [6]2+, a smaller A6 doublet at δ = 9.85 ppm (3 J = 5.5 Hz) was observed (Figure S2), suggesting a mixture of two isomers. The integral ratio between these two A6 peaks showed that [6]2+ was 83 % pure. The second, minor isomer is the biotin‐coordinated complex [7]2+, as shown by the identical mass spectrum but shifted chemical resonances of the biotin signals. Silica column chromatography enriched [6]2+ to some extent, however, a pure compound (> 99 % purity) could not be obtained under these conditions.
Scheme 2.
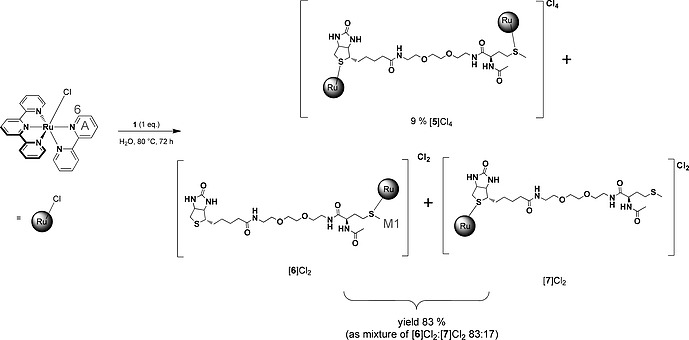
Coordination of 1 equiv. of 1 with [Ru(tpy)(bpy)Cl]Cl in water at 80 °C over three days yielded a mixture of a binuclear complex [5]Cl4 and two mononuclear isomer complexes [6]Cl2 and [7]Cl2.
In a next step, the synthetic conditions were varied to evaluate whether [6]2+ could be obtained regioselectively. By increasing the ligand‐to‐Ru ratio, we successfully suppressed the formation of the binuclear complex [5]4+. Changing the reaction times and using higher reaction temperatures showed an enrichment of the desired isomer [6]2+ and thus suggested that the selectivity of the reaction towards [6]2+ was not driven by kinetics, but the result of thermodynamic control. The equilibration process between 1, [Ru(tpy)(bpy)(OH2)]2+, [6]2+, and [7]2+ was hence followed in time by 1H NMR at 80 °C, as the reaction between the aqua complex and the sulfur donor is not observed below 50 °C. After the first 45 minutes [6]2+ and [7]2+ had formed in a ratio of 51:49, which showed that there was no measurable kinetic preference for one or the other isomer. The amount of [6]2+ in the reaction mixture quickly increased thereafter, to finally reach a plateau characterized by a [6]2+:[7]2+ ratio of 84:16 after 24 h (Figure 1). From these data, we concluded that the regioselectivity of the coordination to ruthenium of the methionine fragment in 1 vs. that of biotin, is due to the higher thermodynamic stability of the methionine‐coordinate isomer [6]2+, compared to [7]2+, rather than to a slower reaction of biotin. According to our data, the binding constant of [6]2+ is ca. 5 times higher than that of [7]2+, which is too low to drive the formation of analytically pure complex [6]2+. However, it was possible to purify [6]2+ from [7]2+ in small scales using avidin chromatography (see below).
Figure 1.
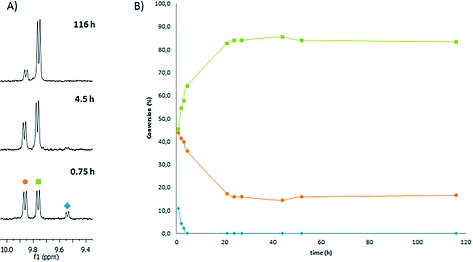
(A) Evolution of the 1H NMR spectra (300 MHz, D2O) during the coordination of ligand 1 to [Ru(tpy)(bpy)(OH2)](PF6)2 in D2O. (B) The [6]2+:[7]2+ ratio obtained from integration of the A6 proton peaks in the 10.0–9.4 ppm range, plotted vs. reaction time. Conditions: [Ru]0 = 4.2 mm, [1] = 42 mm, T = 353 K, in the dark. Blue diamond = [Ru(tpy)(bpy)(OH2)]2+ (δ = 9.54 ppm), green square = [6]2+(δ = 9.76 ppm), orange circle = [7]2+(δ = 9.86 ppm).
Analytically pure samples of a biotin‐functionalized ruthenium complex were obtained in preparative scale using the symmetrical ligand 2. Thermal reaction of an excess of 2 to [Ru(tpy)(bpy)(OH2)](PF6)2 in water at 80 °C, followed by silica gel chromatography, afforded the mononuclear adduct [8](PF6)2 (Scheme 3) in ca. 100 mg scale and 81 % yield. Coordination of only one of the two biotin moieties to ruthenium and structural information about the stereochemical orientation of the biotin ligand in [8]2+ was obtained by a combination of 2D NMR studies (NOESY, COSY, HMBC, HSQC), ESI MS, and elemental analysis. Based on these results (see Supporting Information), the 1,8‐diamino‐3,6‐dioxaoctane linker has no significant effect on the coordination mode of biotin to ruthenium, compared to the known crystal structure of the adduct [Ru(tpy)(bpy)(biotin methyl ester)]2+.[8c]
Scheme 3.
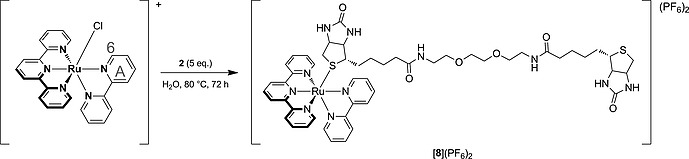
Coordination of 5 equiv. of 2 with [Ru(tpy)(bpy)Cl]Cl in water at 80 °C yielded [8](PF6)2.
Photochemical Investigation
Ruthenium polypyridyl complexes are known for their rich photochemistry.16 Excitation into the metal‐to‐ligand charge transfer (MLCT) absorption band leads to either emission, singlet oxygen production, or ligand photosubstitution. The concrete outcome depends on the ligand field splitting of the complex.16 For complexes of the [Ru(tpy)(bpy)(L)]2+ family, emission and singlet oxygen production are generally negligible as dissociative, metal‐centered triplet excited states (3MC) lie energetically close to the photochemically generated 3MLCT states.17 The photochemistry of compound [8](PF6)2 was studied under blue light irradiation (λ = 465 nm, Δλ 1/2 = 25 nm, photon flux 1.29 × 10–8 mol s–1) in PBS (pH = 7.03, I = 50 mm, [Ru]0 = 7.6 × 10–5 m) for 4 h at 298 K using a setup described earlier.[8c] An isosbestic point was observed at 445 nm, indicating that a single photochemical reaction occurred; the photostationary state was reached after 80 minutes (Figure S7). Mass spectrometry measurements after irradiation confirmed that photosubstitution of ligand 2 by water had occurred, as the initial peak at m/z = 545.4 for [8]2+ was replaced by a peak at m/z = 508.1 characteristic for [Ru(tpy)(bpy)(OH)]+ (calcd. m/z = 508.1). The photosubstitution rate constant k Φ = 7.1 × 10–4 s–1 was obtained from the slope of a plot of ln([RuS]/[Ru]0) vs. the irradiation time, where [RuS] represents the concentration of [8]2+ at time t, and [Ru]0 the total ruthenium concentration. The corresponding photosubstitution quantum yield was derived from the slope of a plot of the number of mol of thioether complex, n RuS, vs. the number of mol of photons, Q, absorbed since t = 0, according to the methods described in the experimental part (see also Figure S6). A photosubstitution quantum yield of 0.011 was found, which is higher, but about the same order of magnitude as the one found for the substitution of [Ru(typ)(bpy)(SChol)]2+ by water (ϕ = 0.0078).18
Binding of [8]2+ to Streptavidin and Photosubstitution
In order to check the suitability of [8]2+ for the ABC technology, a HABA [2‐(4‐hydroxyphenylazo)benzoic acid] displacement titration was applied to test the binding behavior of [8]2+ to streptavidin. HABA exhibits a characteristic absorption band with λ max at 506 nm when incorporated within streptavidin. Upon addition of four equivalents of biotin per streptavidin tetramer, the HABA is displaced leading to disappearance of the absorption band at 506 nm.19 Upon addition of more than four equivalents of biotin, the absorbance at 506 nm is constant. This behavior was also observed for [8]2+ and indicates an equally strong binding as biotin (Figure 2A). A slight increase in absorbance upon addition of more than four equivalents is due to the absorbance of [8]2+ itself. Irradiation of [8]2+ in absence and in presence of one equivalent tetrameric streptavidin in NaH2PO4 buffer (3.00 mL, 20 mm, pH 7.0) with blue light (λ = 465 nm, Δλ 1/2 = 25 nm) yielded the same spectroscopic changes as described in the previous section (Figure 2B). This observation meant that [8]2+ is furthermore a promising candidate for ABC‐based strategies, as the linker in 2 is long enough, so that the free biotin in complex [8]2+ still reaches the avidin binding pocket. In addition, the observed behavior of [8]2+ in the presence of streptavidin (Figure 2A) shows that the complex is kinetically stable under such conditions.
Figure 2.
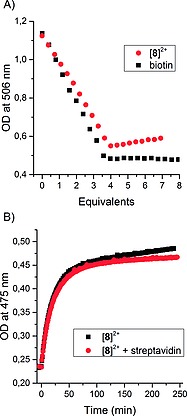
(A) Displacement titration of HABA within streptavidin by [8]2+ and biotin in NaH2PO4 buffer (2.7 mL, 20 mm, pH 7.0). (B) Evolution of the absorbance at 475 nm upon irradiation at λ = 465 nm (Δλ 1/2 = 25 nm) of 6.0 × 10–5 m [8]2+ in absence and in presence of one equivalent tetrameric streptavidin in NaH2PO4 buffer (3.00 mL, 20 mm, pH 7.0).
Application of the Selective Streptavidin Binding
The observation that the free biotin in [8]2+ interacted strongly with streptavidin suggested avidin‐based column chromatography as a potential method to separate the isomeric mixture of [6]2+ and [7]2+ mentioned above. An avidin column was hence incubated for 24 h with a mixture of [6]2+/[7]2+ in PBS. In a second step, the unbound complex mixture was washed from the column using buffer (PBS). Then, a biotin‐loaded PBS buffer was used to displace the non‐coordinated, avidin‐bound biotin moiety in [6]2+, with native biotin. This procedure yielded a mixture of [6]2+ and native biotin without traces of [7]2+, as shown by the unique A6 proton in the aromatic region of the 1H NMR spectrum of the purified sample (9.6–10.0 ppm, Figure 3). The low loading capacity and high price of commercially available avidin‐based purifications columns, however, was not suitable for a large scale separation of [6]2+, but it confirmed that the dangling biotin moiety in [6]2+ can still interact selectively with streptavidin.
Figure 3.
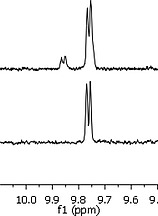
Comparison of the 1H NMR spectra (D2O, 300 MHz, zoom of A6 proton peaks) of the isomeric mixture [6]2+ and [7]2+ (top) and the isolated complex [6]2+ (bottom), before and after avidin affinity column chromatography, respectively.
Conclusions
In summary, the two different biotin‐containing thioether ligands 1 and 2, when coordinated to [Ru(tpy)(bpy)(OH2)]2+ in water, led to different results. The coordination of ligand 1 to [Ru(tpy)(bpy)Cl]Cl provided one of the rare regioselectivity study of the binding of thioethers to ruthenium(II). It showed that in the conditions of the experiment coordination is under thermodynamic control and that although biotin is more hindering than N‐acetylmethionine, the thermodynamic preference for [6]2+ vs. [7]2+ was not strong enough to lead to the regioselective preparation of [6]2+. Purification of [6]2+ from its regioisomer [7]2+ eventually succeeded in low scale via avidin‐affinity chromatography, proving that in [6]2+ the free biotin moiety allows for establishing strong interactions with (strept)avidin.
The symmetrical bis‐biotin ligand 2 offered a more straightforward and synthetically useful route towards the preparation of a ruthenium complex with a light‐cleavable, free biotin group. Complex [8](PF6)2 was obtained in ca. 100 mg scale with good yields (82 %). Meanwhile, photochemical studies with [8]2+ showed that, whether or not the dangling, non‐coordinated biotin moiety was bound to streptavidin, efficient photosubstitution of the coordinated biotin ligand occurred, thus providing a proof‐of‐concept that light‐cleavable free biotin moieties can be installed on photoactivated chemotherapeutic complexes to prepare streptavidin‐mediated targeted PACT systems. The synthetic strategy presented in this work would circumvent current problems of (strept)avidin‐biotin based cancer‐targeting methods, where the biologically active drug remains covalently bound to the biotin targeting group.
Experimental Section
Materials and Methods: Biotin was obtained from TCI Chemicals, N,N‐diisopropylethylamine (DIPEA) was obtained from Acros Organics. Disuccinimidyl carbonate, 1,8‐diamino‐3,6‐dioxaoctane, N,N′‐diisopropylcarbodiimide (DIC), 4‐dimethylaminopyridine (DMAP), Et3N and HABA [2‐(4‐hydroxyphenylazo)benzoic acid] were obtained from Sigma Aldrich. DSPE‐PEG‐2 K‐Biotin [385437‐57‐0] was obtained from Nanocs. [Ru(tpy)(bpy)(Cl)]Cl and [Ru(tpy)(bpy)(OH2)](PF6)2 were synthesized following literature procedures20 as well as wild type streptavidin.21 DMF was obtained from Alfa Aesar and dried with molecular sieves (3 Å). DCM was obtained from Alfa Aesar and dried using a PureSolve 400 solvent dispenser. Silica gel (60 Å, 230–400 mesh particle size, Ca ≈ 0.1 %) was used for column chromatography. Size exclusion chromatography was done over Sephadex LH‐20 prepared in MeOH. Avidin affinity column chromatography was done with a 2 mL Pierce Monomeric Avidin Agarose Kit from Thermo Scientific. 1H NMR and 13C NMR spectra were recorded with a Bruker DPX 300 spectrometer or a Bruker DMX 400 spectrometer. Chemical shifts are indicated in ppm relative to TMS. Numbering of the compounds and NMR spectra can be found in the Supporting Information. Mass spectra were recorded on a ThermoQuest Finnigan AQA spectrometer. UV/Vis spectra were obtained on a Varian Cary 50 UV/Visible spectrometer. The irradiation setup for UV/Vis experiments consisted of custom made LEDs directly placed on top of a 1 cm quartz cuvette, as described by Bahreman et al.22
Biotin‐NHS (3): Biotin (1.00 g, 4.10 mmol), disuccinimidyl carbonate (1.37 g, 5.33 mmol), and Et3N (2.30 mL, 16.4 mmol) were added to dry DMF (14 mL). The resulting suspension was stirred at room temperature under argon for 16 h. The solvent was removed by vacuum distillation at 323 K. The resulting solid was triturated in diethyl ether and ethyl acetate and subsequently filtered and washed with diethyl ether over a glass filter. The white powder was dried under air. Yield: 1.26 g (3.70 mmol, 90 %). R f = 0.68 (SiO2, BuOH/H2O/AcOH [3:1:1]). 1H NMR (300 MHz, [D]Chloroform): δ = (ppm): 4.60–4.49 (m, 1 H), 4.36 (dd, J = 7.9, 4.5 Hz, 1 H), 3.17 (td, J = 7.2, 4.5 Hz, 1 H), 2.94 (dd, J = 12.8, 5.0 Hz, 1 H), 2.89–2.81 (m, 4 H), 2.75 (d, 2 H), 2.65 (q, J = 7.0 Hz, 2 H), 1.93–1.65 (m, 6 H), 1.57 (q, J = 7.6 Hz, 2 H). 1H NMR spectroscopic data is in agreement with known literature.23
N‐Biotinyl‐8‐amino‐3,6‐dioxaoctane‐1‐amonium Acetate (4): Compound 3 (560 mg, 1.64 mmol) was dissolved in DMF (6 mL). The solution was added dropwise to a solution of 1,8‐diamino‐3,6‐dioxaoctane (1.20 mL, 8.20 mmol) and Et3N (1.14 mL, 8.20 mmol) in DCM (24 mL). The resulting solution was stirred at room temperature for 5 h, after which the solvents were removed in vacuo to give an oil. The crude product was purified twice by silica column chromatography using butanol/H2O/acetic acid (3:1:1) as the eluent. Excess of acetic acid was removed by co‐evaporation with toluene. Yield: 547 mg (1.26 mmol, 77 %). R f = 0.33 (SiO2, butanol/H2O/acetic acid [3:1:1]). 1H NMR (400 MHz, [D4]methanol): δ = (ppm): 4.50 (dd, J = 7.9, 4.8 Hz, 1 H, H2), 4.31 (dd, J = 7.8, 4.4 Hz, 1 H, H3), 3.70 (t, J = 4.9 Hz, 2 H, H13), 3.66 (s, 4 H, H11 + H12), 3.56 (t, J = 5.6 Hz, 2 H, H10), 3.37 (t, J = 5.6 Hz, 2 H, H9), 3.21 (ddd, J = 8.9, 5.8, 4.4 Hz, 1 H, H4), 3.11 (t, J = 4.7 Hz, 3 H, H14), 2.93 (dd, J = 12.7, J = 4.9 Hz, 1 H, H1B), 2.71 (d, J = 12.7 Hz, 1 H, H1A), 2.23 (t, J = 7.3 Hz, 2 H, H8), 1.91 (s, 3 H, CH3), 1.79–1.54 (m, 4 H, H5 + H7), 1.44 (p, J = 7.6 Hz, 2 H, H6). 13C NMR (75 MHz, [D4]methanol): δ =(ppm): 178.9 (C=O), 176.2 (Cb), 166.1 (Ca), 71.4 + 71.3 (C11 + C12), 70.7 (C10), 68.0 (C13), 63.4 (C3), 61.6 (C2), 57.0 (C4), 41.0 (C1), 40.6 (C14), 40.2 (C9), 36.7 (C8), 29.7 (C6), 29.5 (C5), 26.9 (C7), 23.1 (CH3). ESI MS: m/z (calc): 375.1 (375.2, [M – AcO]+).
N′′‐(Acetyl‐l‐methionine)‐N′‐biotinyl‐3,6‐dioxaoctane‐1,8‐diamine (1): N‐acetyl‐l‐methionine (512 mg, 2.68 mmol) and N,N′‐diisopropylcarbodiimide (DIC) (411 mg, 3.26 mmol) were dissolved in DMF (15 mL) and the resulting turbid solution was added dropwise to a suspension of compound 4 (1.12 mg, 2.57 mmol) in DMF (20 mL). DIPEA (0.885 mL, 5.35 mmol) and DMAP (37.1 mg, 0.30 mmol) were subsequently added. The resulting mixture was stirred under argon at room temperature for 20 h, after which DMF was removed by vacuum distillation at 323 K. The crude product was dissolved in H2O and the precipitated DIPEA was filtered off and washed with H2O. Water was removed by rotary evaporation and the crude oil was purified by silica column chromatography using DCM/MeOH (8:2) as eluent. Yield: 1.01 g (1.84 mmol, 72 %). R f = 0.7 (SiO2, DCM/MeOH [8:2]). 1H NMR (300 MHz, [D4]methanol): δ = (ppm): 8.24 (d, J = 7.6 Hz, 1 H, NH), 8.09 (t, J = 5.2 Hz, 1 H, NH), 8.01 (t, J = 5.5 Hz, 1 H, NH), 6.57 (s, 1 H, NH), 6.45 (s, 1 H, NH), 4.50 (dd, J = 7.8, 4.3 Hz, 1 H, H2), 4.44 (dd, J = 8.6, 5.5 Hz, 1 H, M4), 4.32 (dd, J = 7.9, 4.4 Hz, 1 H, H3), 3.62 (s, 4 H, H11 + H12), 3.55 (t, J = 5.4 Hz, 4 H, H10 + H13), 3.43–3.33 (m, 4 H, H9 + H14), 3.21 (ddd, J = 8.8, 5.8, 4.4 Hz, 1 H, H4), 2.93 (dd, J = 12.8, J = 4.9 Hz, 1 H, H1B), 2.71 (d, J = 12.7 Hz, 1 H, H1A), 2.63–2.42 (m, 2 H, M2), 2.23 (t, J = 7.3 Hz, 2 H, H8), 2.09 (s, 3 H, M1), 2.00 (s, 3 H, M5), 2.12–1.81 (m, 2 H, M3), 1.84–1.50 (m, 4 H, H5 + H7), 1.44 (p, J = 7.2 Hz, 2 H, H6). 13C NMR (75 MHz, [D4]methanol): δ =(ppm): 176.1 (Cb), 174.0 (Cc), 173.3 (Cd), 166.0 (Ca), 71.3 (C11 + C12), 70.6 + 70.4 (C10 + C13), 63.3 (C3), 61.6 (C2), 57.0 (C4), 54.0 (M4), 41.1 (C1), 40.3 (C9), 40.3 (C14), 36.7 (C8), 32.8 (M3), 31.1 (M2), 29.8 (C6), 29.5 (C5), 26.8 (C7), 22.6 (M5), 15.30 (M1). ESI MS: m/z (calc): 570.3 (570.2 [M + Na]+). HRMS: m/z (calc): 548.25717 (548.25710 [M + H]+).
Bis(N‐biotinyl)‐3,6‐dioxaoctane‐1,8‐diamine (2): A solution of 1,8‐diamino‐3,6‐dioxaoctane (110 mg, 0.74 mmol) and Et3N (152 mg, 1.50 mmol) in dry DMF (1.0 mL) was added to a solution of compound 3 (500 mg, 1.46 mmol) in dry DMF (15 mL). The resulting solution was stirred under argon for 26 h at room temperature, after which the DMF was removed by vacuum distillation at 323 K. The crude was purified twice by silica column chromatography, using BuOH/H2O/AcOH (3:1:1) and DCM/MeOH (78:22) as eluents, respectively. Yield: 308 mg (0.51 mmol, 69 %). R f = 0.38 (SiO2, BuOH/H2O/AcOH [3:1:1]). 1H NMR (300 MHz, [D4]methanol): δ = (ppm): 4.49 (dd, J = 7.8, 4.8 Hz, 2 H, H2), 4.31 (dd, J = 7.8, 4.4 Hz, 2 H, H3), 3.62 (s, 4 H, H11), 3.55 (t, J = 5.5 Hz, 4 H, H10), 3.37 (t, J = 5.5 Hz, 4 H, H9), 3.21 (ddd, J = 8.7, 5.6, 4.5 Hz, 2 H, H4), 2.93 (dd, J = 12.8, J = 4.9 Hz, 2 H, H1B), 2.71 (d, J = 12.7 Hz, 2 H, H1A), 2.23 (t, J = 7.3 Hz, 4 H, H8), 1.81–1.55 (m, 8 H, H5 + H7), 1.44 (p, J = 7.2 Hz, 4 H, H6). 13C NMR (75 MHz, [D4]methanol): δ = (ppm): 176.2 (Cb), 71.3 (C11), 70.6 (C10), 63.4 (C3), 61.6 (C2), 57.0 (C4), 41.1 (C1), 40.3 (C9), 36.8 (C8), 29.8 (C6), 29.5 (C5), 26.7 (C7). ESI MS: m/z (calc): 623.4 (623.3 [M + Na]+), 320.2 (320.1 [M + H + K]2+). HRMS: m/z (calc): 601.28364 (601.28365 [M + H]+).
[{Ru(tpy)(bpy)}2(1)]Cl4 and [Ru(tpy)(bpy)(1)]Cl2 (Compounds [5]Cl2 and [6]Cl2): A solution of ligand 1 (292 mg, 0.533 mmol) in H2O (15 mL) was deoxygenated with argon and [Ru(tpy)(bpy)Cl]Cl (299 mg, 0.533 mmol) was added. The resulting solution was stirred and heated to 353 K over 72 h in the dark under argon. Solvent was removed by rotary evaporation at 303 K and the crude product was purified by silica column chromatography using acetone/H2O/1 m HCl (16:4:1) as the eluent. Two orange fractions (R f = 0.3 and R f = 0.1, acetone/H2O/1 m HCl [16:4:1]) were collected. After removal of the solvent of the first fraction (R f = 0.30, acetone/H2O/1 m HCl [16:4:1]) by rotary evaporation at 303 K, the isomeric mixture [6]Cl2/[7]Cl2 (83:17) was obtained as an orange powder. Yield: 488 mg (0.440 mmol, 83 %). The binuclear complex [5]Cl4 was obtained as a second fraction (R f = 0.10, SiO2, acetone/H2O/1 m HCl [16:4:1]). The solvent was removed by rotary evaporation and [5]Cl4 was obtained as an orange powder. Yield: 81 mg (0.048 mmol, 9 %).
Characterization of [6]Cl2 (Contaminated with [7]Cl2 – See Supporting Information): R f = 0.30 (SiO2, acetone/H2O/1 m HCl [16:4:1]). 1H NMR (300 MHz, D2O): δ = (ppm): 9.75 (d, J = 5.5 Hz, 1 H, A6), 8.70–8.63 (m, 3 H, T3′ + A3), 8.49 (d, J = 7.8 Hz, 2 H, T3), 8.42 (d, J = 8.1 Hz, 1 H, B3), 8.36 (d, J = 8.0 Hz, 1 H, T4′), 8.39–8.27 (m, 1 H, A4), 8.02 (dd, J = 8.0, 7.6 Hz, 2 H, T4), 8.06–7.92 (m, 1 H, A5), 7.83 (dd, J = 7.8 Hz, 1 H, B4), 7.84–7.71 (m, 2 H, T6), 7.35 (dd, J = 7.5, 5.5 Hz, 2 H, T5), 7.23 (d, J = 5.6 Hz, 1 H, B6), 7.08 (dd, J = 7.5, 5.6 Hz, 1 H, B5), 4.54 (dd, J = 8.0, 4.7 Hz, 1 H, H2), 4.34 (dd, J = 8.1, 4.3 Hz, 1 H, H3), 4.14 (dd, J = 9.2, 4.5 Hz, 1 H, M4), 3.59–3.54 (m, 2 H, H10), 3.51 (t, J = 5.0 Hz, 2 H, H13), 3.32 (t, J = 5.1 Hz, 2 H, H9), 3.27 (t, J = 5.2 Hz, 2 H, H14), 3.24–3.15 (m, 1 H, H4), 2.88 (dd, J = 13.0, 4.8 Hz, 1 H, H1a), 2.65 (d, J = 13.0 Hz, 1 H, H1b), 2.15 (t, J = 7.7 Hz, 3 H, H8), 2.01–1.98 (m, 1 H, M2), 1.88 (s, 3 H, M5), 1.85–1.45 (m, 5 H, M3 + H5 + H7), 1.35 (s, 3 H, M1), 1.31–1.22 (m, 1 H, H6). ESI MS m/z (calc): 519.2 (519.2, [M – 2Cl]2+).
Characterization of [5]Cl4: R f = 0.10 (SiO2, acetone/H2O/1 m HCl [16:4:1]). 1H NMR (300 MHz, [D4]methanol): δ = (ppm): 9.86 (d, J = 5.4 Hz, 1 H, A6), 9.76 (d, J = 5.3 Hz, 1 H, A6*), 8.89–8.79 (m, 4 H, T3′), 8.81 (d, J = 8.3 Hz, 2 H, A3), 8.74–8.58 (m, 6 H, T3′′ + T3 + B3), 8.48–8.36 (m, 4 H, A4 + T4′), 8.18–8.08 (m, 4 H, T4′′ + T4), 8.09–8.04 (m, 2 H, A5), 8.00–7.88 (m, 4 H, B4 + T6′′), 7.84–7.74 (m, 2 H, T6), 7.53–7.42 (m, 4 H, T5′′ + T5), 7.32–7.20 (m, 4 H, B6 + B5), 4.28–4.21 (m, 1 H, M4), 4.21–4.12 (m, 2 H, H2 + H3), 3.58 (s, 4 H, H11 + H12), 3.52 (t, J = 5.9 Hz, 2 H, H10), 3.46 (t, J = 5.9 Hz, 2 H, H13), 3.40–3.31 (m, 2 H, H9), 3.30–3.23 (m, 2 H, H14), 2.33 (d, J = 11.7 Hz, 1 H, H4), 2.05 (t, J = 7.1 Hz, 2 H, H8), 1.95 (d, J = 12.2 Hz, 1 H, H1a), 1.89 (s, 3 H, M5), 1.76 (dd, J = 12.2, 4.3 Hz, 1 H, H1b), 1.83–1.65 (m, 2 H, M2), 1.60 (dd, J = 10.5, 5.4 Hz, 2 H, M3), 1.50–1.35 (m, 1 H, H5b), 1.38 (s, 3 H, M1), 1.28–1.14 (m, 2 H, H7), 1.14–1.00 (m, 1 H, H6a), 0.66–0.53 (m, 1 H, H6b), 0.52–0.38 (m, 1 H, H5a). 13C NMR (75 MHz, [D4]methanol): δ = (ppm): 175.7, 173.1 (d), 172.9, 159.6, 159.4, 158.7, 158.6, 158.5, 158.1, 158.1, 158.0, 154.8, 154.4, 154.3, 153.4 + 153.1 (A6), 150.8, 150.7, 140.4, 140.3, 139.8, 139.7, 139.6, 139.5, 138.6, 138.4, 129.9, 129.7, 129.2, 129.1, 128.6, 128.5, 126.7, 126.5, 126.4, 126.2, 126.0, 125.9, 125.6, 125.3, 125.2, 71.3 (C11 + 12), 70.5 (C10), 70.3 (C13), 60.2 (C3), 58.5 (C2), 57.5 (C4), 52.8 (M4), 40.5 (C1), 40.4 (C9), 40.2 (C14), 36.3 (C8), 31.3 (M3), 30.1 (M2), 28.0 (C6), 27.7 (C5), 26.3 (C7), 22.8 (M5), 14.2 (M1). ESI MS: m/z (calc): 528.1 (528.1, [M – 3Cl + H2O]3+), 405.1 (405.3, [M – 3Cl + Na + MeOH]4+). UV/Vis: λ max (ε in L mol–1 cm–1) in pure H2O: 447 nm (1.16 × 104).
Optimization Attempts for the Synthesis of [Ru(tpy)(bpy)(1)](PF6)2 {[6](PF6)2}: A solution of ligand 1 (52 mg, 0.095 mmol) in H2O (2.9 mL) was added to acetone (0.9 mL). The solution was deoxygenated with argon, after which [Ru(tpy)(bpy)(OH2)](PF6)2 (15 mg, 0.019 mmol) was added and the resulting solution was heated [different conditions (see Table S1): 333 K, 353 K, or reflux] under argon for either 2 or 48 h. The solvents were removed by rotary evaporation at 303 K. The crude was purified by silica column chromatography, using acetone/H2O/KPF6(sat) as the eluent, followed by size exclusion chromatography (Sephadex LH‐20) using MeOH as the eluent in order to remove the excess KPF6 salt. Analysis of the product composition in the different reaction conditions was performed by 1H NMR (see Figure S1).
[Ru(tpy)(bpy)(2)](PF6)2 {[8](PF6)2}: Compound 2 (250 mg, 0.416 mmol) and [Ru(tpy)(bpy)(OH2)](PF6)2 (66.5 mg, 83.2 µmol) were dissolved in degassed H2O (16 mL). The resulting solution was stirred and heated to 353 K under argon for 47 h. The solvent was removed by rotary evaporation at 303 K and the crude product was purified by silica column chromatography, using acetone/H2O/KPF6,sat. (12:4:1) as the eluent. Excess KPF6 was removed by size exclusion chromatography (Sephadex LH‐20) using MeOH as solvent. After evaporation of the solvent, an orange solid was obtained. Yield: 94 mg (0.068 mmol, 82 %). R f = 0.62 (SiO2, Acteon/H2O/HCl [100:10:1]). 1H NMR (400 MHz, [D4]methanol): δ = (ppm): 9.84 (d, J = 5.1 Hz, 1 H, A6), 8.84 (d, J = 8.1 Hz, 1 H, A3), 8.79 (d, J = 8.3 Hz, 2 H, T5′), 8.77 (d, J = 8.4 Hz, 1 H, T3′), 8.66 (d, J = 8.0 Hz, 1 H, T3′′), 8.62 (d, J = 8.0 Hz, 1 H, T3), 8.60 (d, J = 8.1 Hz, 1 H, B3), 8.41 (ddd, J = 7.9, 7.9, 1.5 Hz, 1 H, A4), 8.40 (dd, J = 8.2, 8.2 Hz, 1 H, T4′), 8.12 (ddd, J = 7.8, 7.8, 1.5 Hz, 1 H, T4′′), 8.10 (ddd, J = 7.8, 7.8, 1.6 Hz, 1 H, T4), 8.08 (ddd, J = 7.7, 5.6, 1.3 Hz, 1 H, A5), 7.94 (ddd, J = 8.2, 7.4, 1.6 Hz, 1 H, B4), 7.91 (dd, J = 5.5, 0.6 Hz, 1 H, T6′′), 7.78 (dd, J = 5.5, 0.8 Hz, 1 H, T6), 7.47 (ddd, J = 7.7, 5.5, 1.2 Hz, 1 H, T5′′), 7.44 (ddd, J = 7.6, 5.6, 1.3 Hz, 1 H, T5), 7.29–7.22 (m, 2 H, B6 + B5), 4.49 (dd, J = 7.8, 4.5 Hz, 1 H, H2), 4.31 (dd, J = 7.9, 4.4 Hz, 1 H, H3), 4.20 (dd, J = 8.4, 4.5 Hz, 1 H, H2′), 4.16 (dd, J = 8.4, 4.4 Hz, 1 H, H3′), 3.63 (s, 4 H, H11 + H11′), 3.60–3.51 (m, 4 H, H10 + H10′), 3.41–3.32 (m, 4 H, H9 + H9′), 3.21 (ddd, J = 8.8, 5.9, 4.4 Hz, 1 H, H4), 2.92 (dd, J = 12.8, 4.9 Hz, 1 H, H1b), 2.69 (d, J = 12.7 Hz, 1 H, H1A), 2.32 (ddd, J = 11.6, 4.4, 2.5 Hz, 1 H, H4′), 2.21 (t, J = 7.4 Hz, 2 H, H8), 2.06 (t, J = 7.1 Hz, 2 H, H8′), 1.93 (d, J = 12.2 Hz, 1 H, H1′a), 1.75 (dd, J = 12.2, 4.6 Hz, 1 H, H1′b), 1.72–1.51 (m, 4 H, H7 + H5), 1.51–1.36 (m, 3 H, H5′b + H6), 1.31–1.20 (m, 2 H, H7′), 1.15–1.02 (m, 1 H, H6′b), 0.65–0.53 (m, 1 H, H6′a), 0.53–0.43 (m, 1 H, H5′a). 13C NMR (101 MHz, [D4]methanol): δ = (ppm): 176.1 (b), 175.7 (b′), 165.2 (a′), 159.5, 159.3, 158.4, 158.1, 154.7 (T6′′), 154.3 (T6), 153.1 (A6), 150.6 (B6), 140.4 (T4 + T4′′), 139.8 (A4), 139.6 (B4), 138.7 (T4′), 129.8 (T5), 129.7 (T5′′), 129.1 (A5), 128.6 (B5), 126.6 (T3′′), 126.4 (B3), 126.2 (A3), 126.0 (T3′), 125.8 (T3′), 125.2 (T3), 71.2 (C11 + C11′), 70.5 (C10 + C10′), 63.3 (C3), 61.6 (C2), 60.2 (C3′), 58.5 (C2′), 57.4 (C4′), 57.0 (C4), 41.1 (C1), 40.5 (C1′), 40.3 (C9 + C9′), 36.8 (C8), 36.3 (C8′), 29.7 (C6), 29.5 (C5), 28.0 (C6′), 27.8 (C5′), 26.9 (C7), 26.3 (C7′). ESI MS: m/z (calc): 545.4 (545.7, [M]2+). Elemental analysis calcd. (%) for C51H63F12N11O6P2RuS2 + 1 H2O: C 43.78, H 4.68, N 11.01; found C 43.81, H 4.54, N 11.04. UV/Vis: λ max (ε in L mol–1 cm–1) in pure H2O: 448 nm (5.50 × 103).
Photochemistry
Determination of Extinction Coefficients: A stock solution of [8](PF6)2 (4.2 mg in 10.0 mL of H2O, 3.0 × 10–4 m) was prepared. Concentration series of 5 different concentrations were prepared by adding x mL of the stock solution (x = 0.40, 0.60, 0.80, 1.00, and 1.20 mL) to a quartz cuvette filled with 3.00‐x mL H2O or PBS buffer. Absorbance spectra were measured for each sample, and the extinction coefficient of both complexes was determined from the slope of a plot of the absorbance vs. concentration at each wavelength.
Kinetics of Photosubstitution of [8](PF6)2: A stock solution of [8](PF6)2 (0.6 mg in 2.5 mL of PBS solution, 1.7 × 10–4 m) was prepared. To investigate the photochemical properties of complex [8](PF6)2 x mL of stock solution (x = 1.30 mL) was added to a quartz cuvette filled with 3.00–x mL PBS solution. Final concentration of [8]2+ was 7.5 × 10–5 m. The sample was deoxygenated with argon for 20 min. Irradiation was performed using a custom 465 nm LED (Δλ 1/2 = 25 nm, 1.4 mW, 220 min, 19 J × cm–2), and the samples were stirred under argon and kept at 297 K for the duration of the experiment. During irradiation UV/Vis absorbance spectra were measured at variable time intervals, ranging from every 0.5 to 10 min. Mass spectrometry was used to determine the products presence in the solution after irradiation. Irradiation of 6.0 10–5 m [8](PF6)2 in absence and presence of one equivalent of streptavidin was performed in NaH2PO4 buffer (3.00 mL, 20 mm, pH 7.0) at 298 K. After deoxygenation with nitrogen irradiation occurred at 298 K under nitrogen flow with the above‐mentioned 465 nm LED at 3.30 W = 4.46 × 10–8 mol s–1.
HABA Displacement Titration: For each experiment streptavidin was added to a HABA solution (1.07 mm) in NaH2PO4 buffer (2.7 mL, 20 mm, pH 7.0) in a cuvette resulting in concentration of tetrameric streptavidin of 6.26 µm for the measurement with biotin and 7.27 µm with [8](PF6)2. The solution equilibrated for five minutes. UV/Vis spectra of this sample were recorded using the buffer as background. Aliquots (5 µL) of biotin in buffer (1.56 mm) or [8](PF6)2 in methanol (1.26 mm) were added and after each addition, a spectrum was recorded.
Supporting information
Supporting Information
Acknowledgements
The European Research Council is acknowledged for an ERC starting grant to S. B. NWO is acknowledged for a VIDI grant to S. B. A. P. gratefully acknowledges a postdoctoral fellowship from the Swiss National Science Foundation (SNSF), grant P2BSP2_175003. The authors thank Thomas Ward for a generous gift of mature streptavidin. The COST action CM1105 is acknowledged for stimulating scientific discussions. Prof. E. Bouwman is kindly acknowledged for support and scientific discussions.
References
- 1. Diamandis E. P. and Christopoulos T. K., Clin. Chem., 1991, 37, 625–636. [PubMed] [Google Scholar]
- 2. Cuatrecasas P. and Wilchek M., Biochem. Biophys. Res. Commun., 1968, 33, 235–239. [DOI] [PubMed] [Google Scholar]
- 3. Hermanson G. T. in Bioconjugate Techniques (Third edition), Academic Press, Boston, 2013, pp. 465–505. [Google Scholar]
- 4. Lesch H. P., Kaikkonen M. U., Pikkarainen J. T. and Ylä‐Herttuala S., Expert Opin. Drug Delivery, 2010, 7, 551–564. [DOI] [PubMed] [Google Scholar]
- 5. Bradley M., Alexander L. and Sanchez‐Martin R. M., J. Fluoresc., 2008, 18, 733–739. [DOI] [PubMed] [Google Scholar]
- 6. Soininen S. K., Lehtolainen‐Dalkilic P., Karppinen T., Puustinen T., Dragneva G., Kaikkonen M. U., Jauhiainen M., Allart B., Selwood D. L., Wirth T., Lesch H. P., Määttä A.‐M., Mönkkönen J., Ylä‐Herttuala S. and Ruponen M., Eur. J. Pharm. Sci., 2012, 47, 848–856. [DOI] [PubMed] [Google Scholar]
- 7.a) Arora K., White J. K., Sharma R., Mazumder S., Martin P. D., Schlegel H. B., Turro C. and Kodanko J. J., Inorg. Chem., 2016, 55, 6968–6979; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Joshi T., Pierroz V., Mari C., Gemperle L., Ferrari S. and Gasser G., Angew. Chem. Int. Ed., 2014, 53, 2960–2963; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2014, 126, 3004; [Google Scholar]; c) Li A., White J. K., Arora K., Herroon M. K., Martin P. D., Schlegel H. B., Podgorski I., Turro C. and Kodanko J. J., Inorg. Chem., 2016, 55, 10–12; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sharma R., Knoll J. D., Ancona N., Martin P. D., Turro C. and Kodanko J. J., Inorg. Chem., 2015, 54, 1901–1911; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Sharma R., Knoll J. D., Martin P. D., Podgorski I., Turro C. and Kodanko J. J., Inorg. Chem., 2014, 53, 3272–3274; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Rose M. J., Olmstead M. M. and Mascharak P. K., J. Am. Chem. Soc., 2007, 129, 5342–5343; [DOI] [PubMed] [Google Scholar]; g) Howerton B. S., Heidary D. K. and Glazer E. C., J. Am. Chem. Soc., 2012, 134, 8324–8327; [DOI] [PubMed] [Google Scholar]; h) Crespy D., Landfester K., Schubert U. S. and Schiller A., Chem. Commun., 2010, 46, 6651–6662; [DOI] [PubMed] [Google Scholar]; i) Presa A., Brissos R. F., Caballero A. B., Borilovic I., Korrodi‐Gregório L., Pérez‐Tomás R., Roubeau O. and Gamez P., Angew. Chem. Int. Ed., 2015, 54, 4561–4565; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2015, 127, 4644–4648 [Google Scholar]; j) Mari C., Pierroz V., Ferrari S. and Gasser G., Chem. Sci., 2015, 6, 2660–2686; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Farrer N. J., Salassa L. and Sadler P. J., Dalton Trans., 2009, 10690–10701; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Farrer N. J., Woods J. A., Salassa L., Zhao Y., Robinson K. S., Clarkson G., Mackay F. S. and Sadler P. J., Angew. Chem. Int. Ed., 2010, 49, 8905–8908; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2010, 122, 9089; [Google Scholar]; m) Schatzschneider U., Eur. J. Inorg. Chem., 2010, 1451–1467. [Google Scholar]
- 8.a) Askes S. H. C., Bahreman A. and Bonnet S., Angew. Chem. Int. Ed., 2014, 53, 1029–1033; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2014, 126, 1047; [Google Scholar]; b) Bahreman A., Limburg B., Siegler M. A., Koning R., Koster A. J. and Bonnet S., Chem. Eur. J., 2012, 18, 10271–10280; [DOI] [PubMed] [Google Scholar]; c) Goldbach R. E., Rodriguez‐Garcia I., van Lenthe J. H., Siegler M. A. and Bonnet S., Chem. Eur. J., 2011, 17, 9924–9929; [DOI] [PubMed] [Google Scholar]; d) van Rixel V. H. S., Busemann A., Göttle A. J. and Bonnet S., J. Inorg. Biochem., 2015, 150, 174–181. [DOI] [PubMed] [Google Scholar]
- 9. Lo K. K.‐W. and Lee T. K.‐M., Inorg. Chem., 2004, 43, 5275–5282. [DOI] [PubMed] [Google Scholar]
- 10.a) Loosli A., Rusbandi U. E., Gradinaru J., Bernauer K., Schlaepfer C. W., Meyer M., Mazurek S., Novic M. and Ward T. R., Inorg. Chem., 2006, 45, 660–668; [DOI] [PubMed] [Google Scholar]; b) Aacute M. H., Reuter R., Mallin H. and Ward T. R., Nat. Protocols, 2016, 11, 835–852; [DOI] [PubMed] [Google Scholar]; c) Heinisch T. and Ward T. R., Acc. Chem. Res., 2016, 49, 1711–1721. [DOI] [PubMed] [Google Scholar]
- 11. Zhang K. Y. and Lo K. K.‐W., Inorg. Chem., 2009, 48, 6011–6025. [DOI] [PubMed] [Google Scholar]
- 12. Siewert B., Langerman M., Hontani Y., Kennis J. T. M., van Rixel V. H. S., Limburg B., Siegler M. A., Saez V. Talens, Kieltyka R. E. and Bonnet S., Chem. Commun., 2017, 53, 11126–11129. [DOI] [PubMed] [Google Scholar]
- 13. Siewert B., van Rixel V. H., van Rooden E. J., Hopkins S. L., Moester M. J., Ariese F., Siegler M. A. and Bonnet S., Chem. Eur. J., 2016, 22, 10960–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kang S., Mou L., Lanman J., Velu S., Brouillette W. J. and Prevelige P. E., Rapid Commun. Mass Spectrom., 2009, 23, 1719–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Friebolin H., in: Ein‐ und zweidimensionale NMR‐Spektroskopie: Eine Einführung, Wiley VCH Verlag GmbH, 2013; [Google Scholar]; b) Ryan E. M., Wang R., Vos J. G., Hage R. and Haasnoot J. G., Inorg. Chim. Acta, 1993, 208, 49–58. [Google Scholar]
- 16.a) Juris A., Balzani V., Barigelletti F., Campagna S., Belser P. and von Zelewsky A., Coord. Chem. Rev., 1988, 84, 85–277; [Google Scholar]; b) Sauvage J. P., Collin J. P., Chambron J. C., Guillerez S., Coudret C., Balzani V., Barigelletti F., Decola L. and Flamigni L., Chem. Rev., 1994, 94, 993–1019. [Google Scholar]
- 17. Knoll J. D., Albani B. A. and Turro C., Acc. Chem. Res., 2015, 48, 2280–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bonnet S., Limburg B., Meeldijk J. D., Gebbink R. J. M. K. and Killian J. A., J. Am. Chem. Soc., 2011, 133, 252–261. [DOI] [PubMed] [Google Scholar]
- 19.a) Skander M., Humbert N., Collot J., Gradinaru J., Klein G., Loosli A., Sauser J., Zocchi A., Gilardoni F. and Ward T. R., J. Am. Chem. Soc., 2004, 126, 14411–14418; [DOI] [PubMed] [Google Scholar]; b) Keller S. G., Pannwitz A., Schwizer F., Klehr J., Wenger O. S. and Ward T. R., Org. Biomol. Chem., 2016, 14, 7197–7201. [DOI] [PubMed] [Google Scholar]
- 20. Wasylenko D. J., Ganesamoorthy C., Koivisto B. D., Henderson M. A. and Berlinguette C. P., Inorg. Chem., 2010, 49, 2202–2209. [DOI] [PubMed] [Google Scholar]
- 21. Jeschek M., Bahls M. O., Schneider V., Marliere P., Ward T. R. and Panke S., Metab. Eng., 2017, 40, 33–40. [DOI] [PubMed] [Google Scholar]
- 22. Bahreman A., Limburg B., Siegler M. A., Bouwman E. and Bonnet S., Inorg. Chem., 2013, 52, 9456–9469. [DOI] [PubMed] [Google Scholar]
- 23. Ma M. and Bong D., Org. Biomol. Chem., 2011, 9, 7296–7299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information