

Abstract
Nuclear receptor tyrosine kinases (nRTKs) are aberrantly upregulated in many types of cancers, but the regulation of nRTK remains unclear. We previously showed androgen deprivation therapy (ADT) induces nMET in castration resistant prostate cancer (CRPC) specimens. Through gene expression microarray profiles reanalysis, we identified that nMET signaling requires ARF in CRPC in vivo Arf deficiency in Pten/Trp53 conditional knockout mouse model. Accordingly, aberrant MET/nMET elevation correlates with ARF in human prostate cancer (PCa) specimens. Mechanistically, ARF elevates nMET through binding to MET cytoplasmic domain to stabilize MET. Furthermore, carbon nanodots resensitize cancer cells to MET inhibitors through DNA damage response. The inhibition of phosphorylation by carbon nanodots was identified through binding to phosphate group of phosphory-tyrosine via computational calculation and experimental assay. Thus nMET is essential to precision therapy of MET inhibitor. Our findings reveal for the first time that targeting nMET axis by carbon nanodots can be a novel avenue for overcoming drug resistance in cancers especially prostate cancer.
Keywords: nuclear MET, ARF, CRPC, MET inhibitor, carbon nanodots
Introduction
Prostate cancer (PCa) is the second leading cause of cancer-related deaths among men in Western countries. Androgen deprivation therapy (ADT) is a standard therapy for the patients who have PCa at advanced stages including metastasis. However, relapse is frequently observed in patients as the castration resistant forms of PCa (CRPC), metastatic CRPC, and multiple drug resistance may develop after ADT (1–4). Clonal growth as a stem trait can be elevated in drug resistance during cancer evolution, suggesting a cause of the cancerous stem and the expansion of individual cancer cells (5). The signaling pathways that promote the clonal resistant stem cells to evade the drug treatment is still unclear, even there are some reports on the stem signaling crosstalk with drug efflux pumps such as ABC transporters (4, 6, 7).
Numerous studies revealed that aberrant activation of receptor tyrosine kinases (RTKs) regulates CRPC growth and drug resistance (8). For example, MET, especially nuclear form of MET (nMET) is associated with CRPC through cancer stem-like signaling including β-catenin (9). As a receptor tyrosine kinase, the canonical MET, namely membrane MET (mMET or c-MET) is activated by HGF ligand which results in autophosphoylation at Y1234/Y1235 that provides a docking platform to recruit kinases, adaptors to further activate downstream signaling pathways such as AKT, β-catenin (10, 11). It has been shown that the nMET is increased in breast cancer cells upon cleavage (12), but the regulation of nMET is largely unknown. We recently found that nMET expression is correlated with CRPC growth in human PCa specimens (13). A few MET inhibitors such as crizotinib (14) have introduced to clinical trials but acquired drug resistance frequently develop in patients (15, 16). Therefore, targeting MET/nMET activators or crosstalk pathways would be more efficient to treat advanced PCa or CRPC.
ARF named as human p14ARF (or ARF) and mouse p19Arf, is an alternative transcript of ARF-INK4a locus on human chromosome 9q21 (17) with the canonical function of tumor suppression. However, ARF is frequently upregulated by oncogenic stimuli such as PTEN or TP53 mutation (18). Our previous work and some recent discovery indicate ARF may potentiate promoter function in tumor. For examples, ARF correlates with PTEN loss in human PCa specimens (18), promotes epithelial–mesenchymal transition (EMT) and microenvironmental invasiveness in PCa (19, 20), inhibits cell death (21), and functions as a tumor promoter upon nutrition depletion (22). In PCa, ARF is also elevated in many cancer cell lines (20). However, molecular mechanism of ARF in CRPC remains elusive.
Carbon nanodots (c-dots) derived from differential resources have been widely studied in imaging, drug delivery but less studied in intrinsic enzyme (nanozyme) activity, and mechanisms underlying the potential as anti-cancer agents (23). For example, food-derived c-dots have been found to be able to inhibit tumor growth in xenograft model (23). Previously, we found tea-derived c-dots can bind certain amino acids on ARF and colocalize with ARF in nucleus which sensitizes cancer cells to rapamycin (24,25). As the tea leaves derived c-dots are believed mainly contain carbohydrate along with some functional groups such as carbonyl or carboxylic acid, or both, other carbon sources should also be developed into carbon dots and the combination with facial modification through doping may bring some special functions. Meanwhile, computational approaches have been introduced to simulate the interactions between nanomaterials and biomolecular systems (26). Dynamic simulation, although well represents the realities of the system, sacrifices some atomistic and/or molecular details, not to mention the high computation and time cost (27–28). Some computational simulations are aiming at the accurate estimation of the toxicity of nanomaterial or the electrostatic interaction between biomolecular systems and nanoparticles within a macroscopic dimension, wherein, the detailed such as the shape, size, surface distribution of functional groups about nanoparticles is rather important (29–30), whereas, in the study of drug delivery or other biomolecular mechanisms, the quantum mechanical model is based on the scaffold of the functional groups that well represent the chemical reactivity of the involved systems(31). In the present work, we reported soybean-derived carbon nanodots that inhibit ARF/MET pathways, and provided explanations on novel mechanisms of the combination therapy by performing quantum mechanical simulation. Our model revealed that the functional groups from both c-dots and phosphorylated tyrosine on nMET are critical for the enzyme activity of carbon nanodots through binding to phosphor groups.
Results
1. Array profiles re-analysis identified that p19Arf is required for nMET in castration resistant prostate tumor growth
Previously we showed that in genetic engineered mouse model of prostate cancer, generated by Pten/Trp53 double-mutant (referred to as Pten/Trp53), MET amplification is ranked as top elevated genomic changes (11). To further identify upstream pathways that regulate MET signaling, we applied gene expression profiles of microarray re-analysis using Pten/Trp53/p19Arf triple-mutant mice as this type of mice dramatically deceased phenotype of tumorigenesis compared to Pten/Trp53 mice (20). We found many genes expressed differentially are related to MET signaling such as Muc20, Mapk13 (Figure 1A). The data suggest that ARF may crosstalk with MET pathways.
Figure 1. Array analysis and genetically engineering mouse model identified that Met requires p19Arf in CRPC.
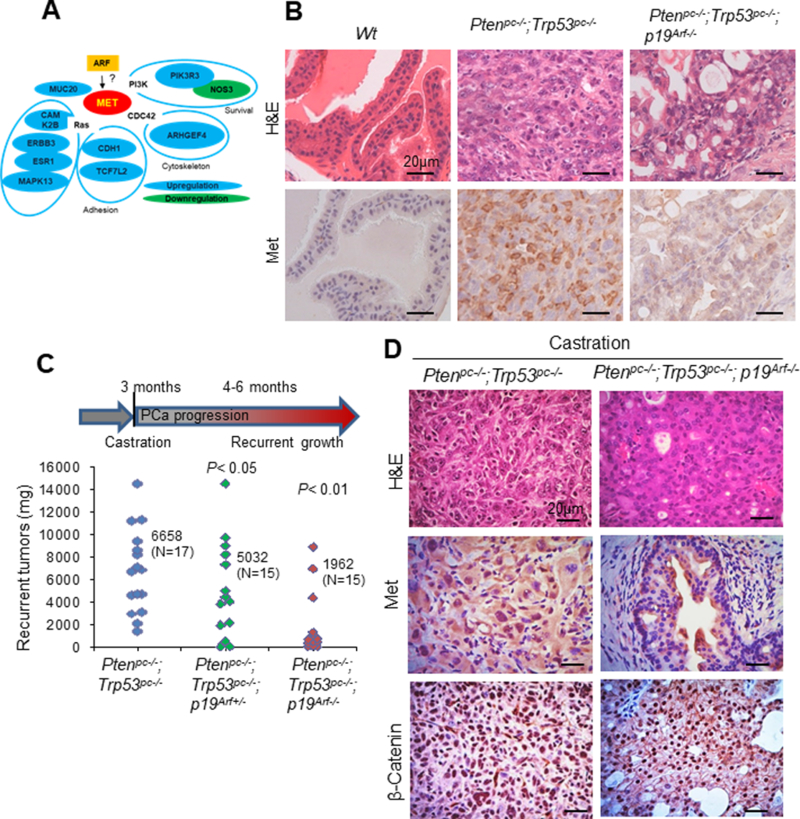
(A) Microarray reanalysis identified that ARF regulates MET pathway. (B-D) IHC analysis of Met protein expression in mouse prostate tissues (B) or recurrent prostate tumors (C-D) In Pten/Trp53 mutant mice, p19Arf deletion decreases the recurrent growth of prostate tumors of castrated Pten/Trp53 mutant mice at 4–6 months of age (C). Data are indicated by individual dots with analysis of p value. Nuclear MET and nuclear β-Catenin expression decreases upon p19Arf deletion (D).
Then we tested whether Met involves in the p19Arf deficiency mediated tumor restriction. As shown in Figure 1B and S1, Met protein is highly expressed in prostate tumors of Pten/Trp53 mice but not p19Arf deficient Pten/Trp53 mice. To be note, Met expression predominately localizes in plasma membrane in tumor cells of Pten/Trp53 mice. This is consistent with our previous findings (13, 19). Thus our data suggest that ARF may also regulate MET expression for tumor progression. We are attempted to further investigate whether ARF also contributes to CRPC in vivo. We examined the effects of p19Arf deficiency on the recurrent growth of prostate tumors in mice (Figure 1C). As shown, a complete loss of p19Arf decreased the recurrent growth of prostate tumors significantly in castrated Pten/Trp53 mice (Figure 1C). Moreover, IHC analysis revealed that p19Arf loss resulted in a decrease in the nucleus localization of Met in CRPC mice with elevated nuclear β-catenin (Figure 1D). Therefore, ARF correlates with CRPC and MET/nuclear MET (nMET)/ β-catenin signaling in vivo.
2. ARF associates with nMET through cytoplasmic domain and correlates with nMET in human PCa specimens
Given the important roles of both ARF and MET in CRPC, we examined the association and correlation of ARF with nMET in both PCa cell lines and human specimens. To test whether ARF and MET interacts in nucleus, we first performed IF staining and found that ARF is co-localized with MET in nucleus of PC3 and DU145 cells (Figure 2A and S2A). Immunoprecipitation (IP) analysis further showed that ARF physically binds with MET in nuclear extract of PC3 cells (Figure 2B). Finally, ARF or MET knockdown decreased PC3 cell growth and co-knockdown both ARF and MET dramatically decreased cell growth (Figure 2C and S2B). Thus our data suggest that ARF interacts with MET, increases MET levels and promotes androgen insensitive PCa cell growth.
Figure 2. MET interacts with ARF in androgen insensitive PCa cells and correlates with ARF in human PCa specimens.
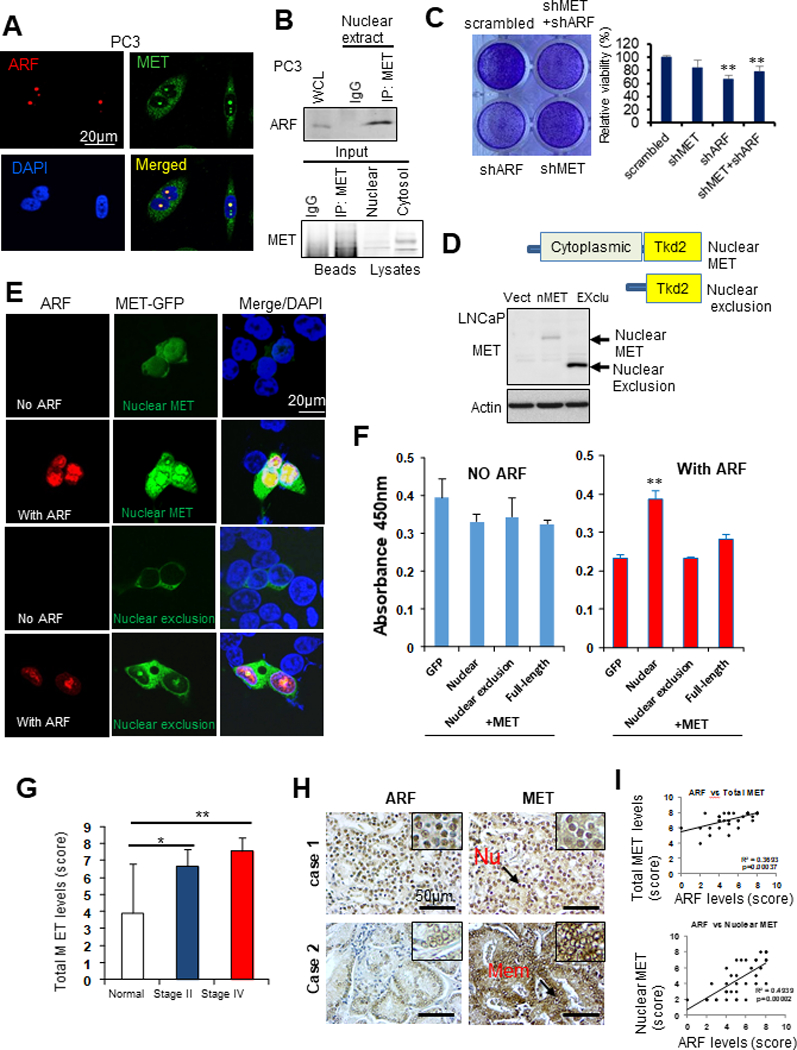
(A) IF images show co-localizations of ARF and MET in PC3 cells. (B) Co-IP assay shows ARF associates with MET in nuclear extracts from PC3 cells. (C) Concurrent knockdown of ARF and MET decreases PC3 cell growth. (D) A diagram shows the plasmid construct contains different domains. (E) IF images show MET deletion mutant translocases into nucleus upon ARF overexpression in androgen insensitive C4–2B cells. (F) MET deletion mutant promotes C4–2B cells proliferation upon ARF overexpression. C4–2B cells were viral infected by construct expressing different genes as indicated and cell proliferation analysis was performed by WST-1 assay. (G) Elevated expression of ARF in different stages of tumors compared to normal prostate tissues. (H) IHC staining on ARF and MET expression in PCa samples. (I) ARF correlates with MET expression in PCa samples. Data are representative of averages+ SD (standard deviation).
To further explore the binding mechanism between ARF and nMET, we applied truncated mutants to investigate the domains that mediated this event. We co-transfected cells with ARF and MET-GFP fusion mutant plasmids. As expected, without ARF, two distinct mutants either localize to nucleus or not nucleus (nucleus exclusion) (Figure 2D). We found ARF overexpression promotes nMET levels (Figure 2E). For nucleus- exclusion mutant, ARF elevation showed very limited effect on nMET levels (Figure 2E). Based on the mutant domain structures, it is quite clear that the nucleus translocation by ARF is predominately mediated by cytoplasmic domain. Consistent with the nMET levels enhanced by ARF, C4–2B cell growth was promoted by nMET only upon ARF overexpression (Figure 2F). Hence ARF promotes nMET expression and cell proliferation not through kinase domain but through cytoplasmic domain.
Next, in prostate tissue microarrays analysis, it was observed that MET expression levels were significantly higher in both stage II and stage IV PCa samples, compared to normal tissues (Figure 2G-2I). Our results indicated that MET expression was positively correlated with the severity of advanced prostate cancer. The combined scores of IHC staining showed that ARF levels are positively correlated with MET especially nMET (Figure 2I). It was suggested that ARF and MET/nMET may crosstalk in driving advanced PCa.
3. MET requires ARF for turnover and recruitment of β-Catenin
First we found that acute ARF inactivation decreased MET expression upon a transient ARF knockdown in PC3 and DU145 PCa cells, two hormone insensitive cell lines (Figure 3A). Then we used Urea to disrupt the protein folding and found ARF knockdown decreased maturation of MET (Figure 3B). Further protein synthesis chase assay demonstrated ARF knockdown decreased MET half-life from 6hrs to 3hrs (Figure S3). Consistent with the protein degradation assay, MET Ubiquitination was reduced by ARF overexpression or enhanced by ARF knockdown (Figure 3C-D).
Figure 3. MET requires ARF for stabilization and recruitment of β-Catenin.
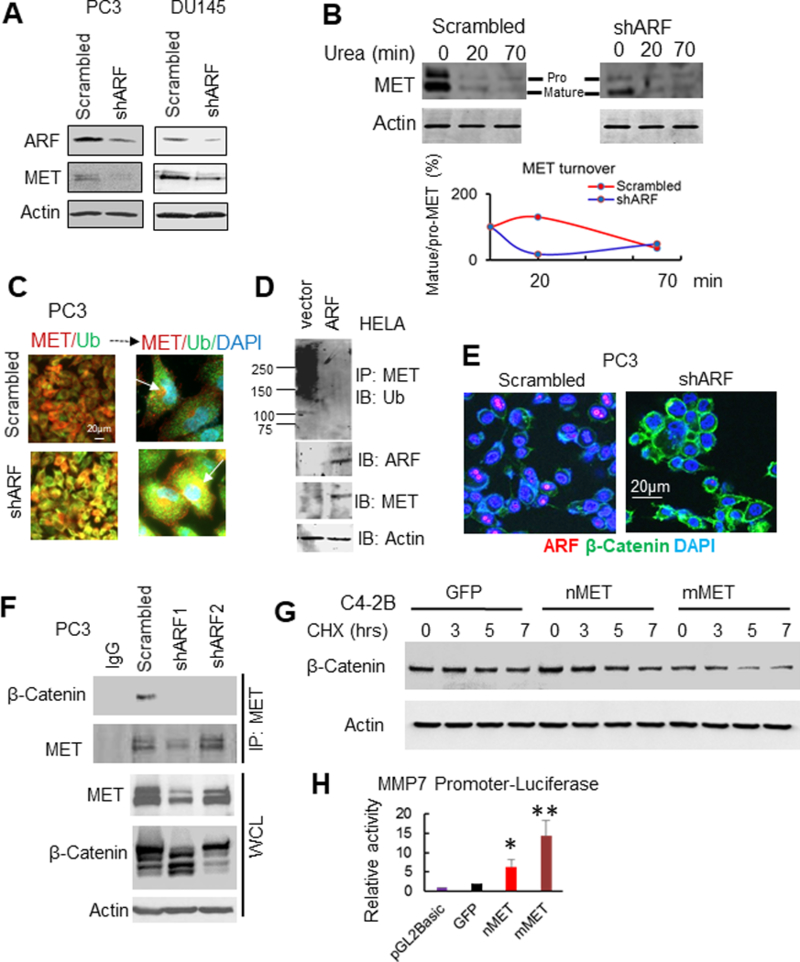
(A) knockdown of ARF decreases MET levels in PC3 and DU145 cells. (B) ARF knockdown decreases MET folding and turnover. (C) ARF knockdown increases colocalization of MET and Ubiquitin (Ub). (D) ARF overexpression decreases ubiquitination of MET. (E) IF images show ARF knockdown increases membrane localization of β-Catenin but decreases nuclear β-Catenin. (F) Co-IP assay shows ARF knockdown disrupts MET and β-Catenin association to decrease MET and β-Catenin expression. (G) MET increases β-Catenin degradation in stably expressing nMET and mMET C4–2B cells which we described previously (13). (H) nMET induces transcription of β-Catenin target gene MMP7. Data are representative of averages+ SD (standard deviation).
Previously we found nMET activates nuclear β-Catenin. In the present work, we found ARF knockdown enhanced membrane but reduced nuclear accumulation of β-Catenin (Figure 3E). IP results convinced that ARF knockdown disrupted the recruitment of β-Catenin to MET (Figure 3F). To be noted, ARF knockdown led to a decrease of both MET and β-Catenin expression in PC3 cells (Figure 3F). As nuclear β-Catenin is transcriptional active and fast destructive, therefore β-Catenin nuclear accumulation promoted by ARF may contribute to ARF/MET signaling activation. Indeed, nMET destructs β-Catenin faster by protein synthesis chase assay (Figure 3G). In addition, MET/nuclear MET activates transcription of β-catenin target gene MMP7 (Figure 3H) and AR (13). Overall, our data suggest that ARF promotes recruitment of MET/nMET/β-Catenin for transcriptional regulation of downstream signaling targets genes.
4. nMET mediates drug resistance through DNA damage response
First, nMET accumulation was commonly observed in clonal tumors cells treated with MET inhibitor Crizotinib in Pten/Trp53 mouse tumors (data not shown). We then discovered that the PC3 cells treated by Crizotinib though experienced membrane MET loss, remained sustained or even elevated nMET with co-upregulation of ARF tested by different c-MET antibodies of C28 or D1C2. This suggests nMET mediates MET inhibitor drug resistance (Figure 4B). To exam whether nMET induction is related to DNA damage response with ARF, the DNA damage agent doxorubicin (DOX) was applied and the results showed that DOX also induced nMET depending on ARF and HSP90 suggesting that nMET induction is through folding and turnover (Figure 4C) which is consistent with above finding (Figure 3B). Further tests indicated that ARF and MET co-knockdown enhanced the DNA damage which results in inhibition of cell growth (Figure 4 D). After all, MET requires ARF in insensitization to DNA damage drug. Moreover, the fact that ARF knockdown inhibited nMET accumulation upon Crizotinib treatment once again, emphasized ARF-dependence of nMET.
Figure 4. nMET requires ARF in drug resistance.
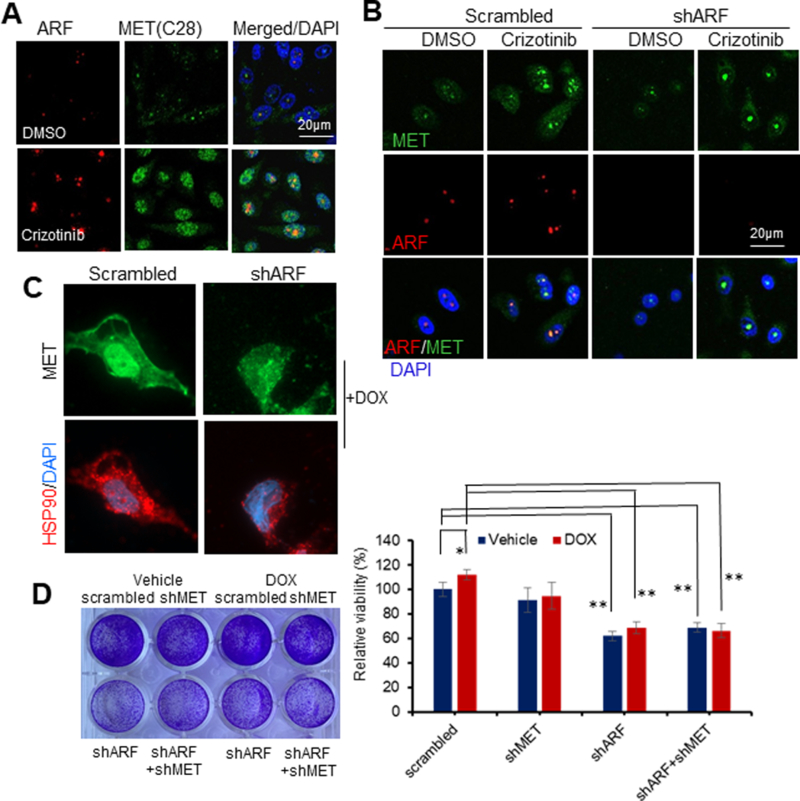
(A-C) IF images show MET inhibitor Crizotinib (A,B) or doxorubicin (DOX) treatment induces MET nuclear accumulation which depends on HSP90 (C) and ARF (B). PC3 cells with knockdown of ARF or control were treated with Crizotinib at 100nM or DOX at 1μM for 24hrs followed by Immunofluorescence Microscopy. (D). ARF and MET knockdown induces DNA damage response and decrease cell resistance to DOX. Data are representative of averages+ SD (standard deviation).
5. Androgen receptor interrupts ARF/MET axis
As AR plays essential roles in PCa and CRPC, we then attempted to exam the correlation among ARF, AR, and MET. It was discovered that MET correlates with ARF positively, but negatively with AR in PCa cell lines investigated (Figure 5A). Then androgen depletion was performed in LAPC4, an androgen sensitive and AR positive cell line, by treatment of charcoal striped FBS (cFBS). We found the androgen depletion led to the elevation of both ARF and MET, with nMET target SOX9 and β-catenin which are essential nMET targets (Figure 5B). Since nMET level is less in AR positive sphere cells but sustained with AR at extremely low level in non-sphere cells, the interaction and crosstalk between nMET and AR is likely weak (Figure 5C). It was noticed that ARF knockdown reduces cytosolic MET, SOX9 and β-catenin, that is to say, ARF does not directly promote nuclear translocation but rather do that through indirect cytosolic stabilization (Figure 5D). Moreover, activation of AR abolished the ARF knockdown effect on MET downregulation, suggesting AR interferes the ARF/MET axis (Figure 5E). This is consistent with the observation where AR overexpression in PC3 cells abolishes the interaction between ARF and MET (Figure 5F). AR also abolished ARF mediated androgen-independent cell growth in androgen insensitive, AR positive C4–2B cells (Figure 5G). Moreover, AR promoted mMET but not nMET mediated cell migration (Figure 5H). Further experiments indicated that AR correlates with DNA repair protein of nuclear BRCA1, however, ARF opposes nuclear BRCA1(Figure 5I-J). In addition, ARF mediates ATM mediated DNA damage and PARP mediates DNA repair, while AR dysregulates the ARF mediated DNA damage response (Figure 5M). In conclusion, ARF, AR, and MET may exert distinct signaling in CRPC by interfere or crosstalk.
Figure 5. Androgen receptor interrupts ARF/MET axis.

(A) AR inversely correlates with MET in prostate cancer cell lines. ARF levels were used as reference control based on our previous report (20). (B) ARF, MET and its signaling proteins β-Catenin, SOX9 co-upregulation by androgen depletion in LAPC4 cells. LAPC4 cells were cultured in cFBS to deplete androgen for 6 days and regular FBS as control. (C) AR inversely correlates with MET in the prostate sphere. (D) ARF knockdown mediated MET downregulation is cytosolic MET-dependent. C, cytosolic lysates; N, nuclear lysates. (E) ARF knockdown mediated MET downregulation is androgen-dependent and is interrupted by AR activation. (F) AR overexpression and activation disrupts ARF and MET interactions. PC3 cells were transfected by AR and stimulated by DHT at 15nM for 24hrs. (G) ARF inducible expression in LNCaP cells increases androgen independent cell growth. GFP fusion ARF expression is detected by GFP expression by imaging (left panel) and cell growth were detected by crystal violet staining (right panel). (H) Nuclear MET stimulates LNCaP cells migration independent on androgen but mMET dependents on androgen. (I) AR correlates with DNA repair protein BRCA1 in nucleus. (J) ARF knockdown increases BRCA1 nuclear localization. (K) AR, nMET, and ARF regulate ATM and PARP but not caspase 3 investigated by western blot. Data are representative of averages+ SD (standard deviation).
6. nMET phosphorylation is sustained by ARF and is abolished by carbon nanodots
Complementary overexpression or knockdown of ARF effect on cell growth and invasion suggest that ARF cooperates with nMET but not significantly with mMET in cancer cells (Figure 6A-B). Moreover, nMET is a phosphorylated protein, the elevation of its phosphorylation depends on ARF, as ARF knockdown prevents MET phosphorylation in nucleus (Figure 6C-E). A novel treatment was then developed in the present work to prevent nMET phosphorylation. The carbon nanoparticles, with an average size of ~10 nm, derived from soybean, showed some impressive inhibition against nMET and its phosphorylation through direct binding to phosphorylated amino acid tyrosine (Figure 6F-G). The carbon nanodots derived from soybean inhibiting nMET and its phosphorylated form is then seen as a promising future cancer therapy.
Figure 6. nMET phosphorylation is sustained by ARF and is abolished by carbon nanodots.
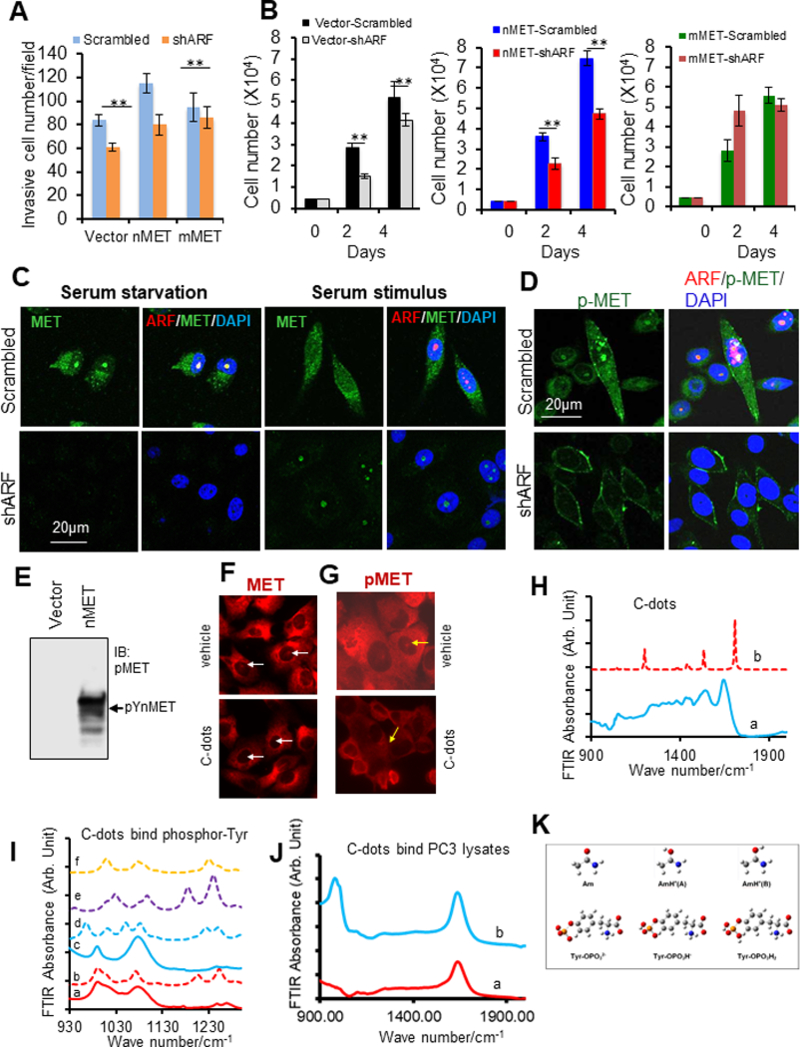
(A-B) Stable ARF knockdown decreases nMET-mediated cell invasion and cell proliferation. (C-D) Stable ARF knockdown decreases nMET expression and phosphorylation. (E) nMET is phosphorylated protein. (F-G) C-dots abolish nMET and phosphorylated nMET levels in PC3 cells. (H) FT-IR spectra of c-dots. (a): experimental FT-IR spectrum of c-dots. (b): Calculated IR spectrum of protonated acetamide based on geometry in Figure 8 AnH+(B); (I): FT-IR spectra of phosphorylated tyrosine before and after addition of c-dots (a): FT-IR spectrum of phosphorylated tyrosine (b): calculated IR spectrum of phosphorylated tyrosine (c): FT-IR spectrum of phosphorylated tyrosine with the addition of c-dots (d): calculated IR spectrum of phosphorylated tyrosine and acetamide complex in Figure 8A (e) calculated IR spectrum of phosphorylated tyrosine and acetamide complex in Figure 8B (f) calculated IR spectrum of phosphorylated tyrosine and acetamide complex in Figure 8C (g) calculated IR spectrum of phosphorylated tyrosine and acetamide complex in Figure 8D; (J) FT-IR spectra of total protein extracted from PC3 cell (a) FT-IR spectra of total protein extracted from PC3 cell without c-dots (b) FT-IR spectra of total protein extracted from PC3 cell with the addition of c-dots. (K) Optimized structure for acetamide w/o protonated and phosphorylated tyrosine at different degrees of protonation. Legend of colours: white (H), grey (C), blue (N), red (O), and orange (P). Data are representative of averages+ SD (standard deviation).
7. Interaction between c-dots and phosphate anion
Atomic force microscopy result suggested that the synthesized carbon dots have an average size of 9 ± 2 nm. The FT-IR spectra of the C-dot is presented in the wavenumber range of 900 cm−1 to 1900 cm−1, where nearly all the typical absorption bands for amide are emerged, among which we observed the peak at 1637 cm−1 representing the C=O stretching vibration in amide, the peak at 1540 cm−1 representing N-H bending vibration, and the peak at 1440 cm−1 representing C-N vibration. The bands at 2960 cm−1 and 3004 cm−1 representing N-H stretching were also observed. The DNA gel electrophoresis test indicated the synthesized c-dots carry positive charge (data not shown) then we studied its binding to phosphor-group (Figure 6H-K). In the present work, we first confirmed its binding to phosphate acid (Figure 6H) then we tested the extracted the total protein from PC3 cells, by adding c-dots. The FTIR spectra in Figure 6J showed the addition of c-dots led to a suppression of the entire band centered at 980 cm-1.
In order to specify the interaction nature between c-dots and phosphate group, we synthesized phosphorylated tyrosine. The FT-IR spectra of phosphorylated tyrosine before and after adding c-dots were collected and compared as in Figure 6I. On both spectra, there are mainly three bands located at 1632 cm−1, 1072 cm−1, and 990 cm−1. The band at 1632 cm−1 generally represents the C=O stretching vibration, whereas the other two bands can be assigned as out of phase O=P=O stretching mode ѵa(PO) (1072 cm−1) and in phase O=P=O stretching mode ѵs (PO) (990 cm−1) (32). It may be noticed the band at 990 cm−1 is very broad with a shoulder representing P-OH-H bending mode 𝛿 (POHH) for the phosphorylated tyrosine. This band is slimmed down with the disappearance of the shoulder upon addition of c-dots.
8. Carbon nanodots sensitize PCa cells to MET inhibitor
To further explore the potential treatment avenues using soybean c-dots targeting both ARF and MET, the c-dots effect on expression of ARF and MET levels was examined. The results showed that the application of c-dots to the system led to a decreased protein levels (data not shown). It was further discovered that c-dots induced DNA damage markers of γ-H2AX and phosphor-ATR, DNA comet tails, as well as the ROS generation at dose of 0.1mg/ml (Figure 7A-C). It was assumed that c-dots may target phosphor-nMET and inhibit its phosphorylation. Since MET inhibitor has less efficiency on nMET, we then combined c-dots with MET inhibitor and higher efficiency was observed on DNA tails, ROS generation, the reduction of MET levels, and more cleaved PARP. The DNA damaged repair deficiency and cell growth inhibition in both PC3 and DU145 cells were achieved by drug combination through ubiquitination (Figure 7A-G). Figure 7H-I indicates DOX promoted the efficiency of MET inhibitor leading to the suppression of cancer cell growth. This implies that DNA damaging potentially crosstalk with MET inhibition pathways. Further analysis of DNA damage showed that the combination with c-dots enhanced MET inhibitor induced DNA damage and the repair response detected by markers of γ-H2AX and ATR (Figure 7I). More importantly, c-dots enhanced MET inhibitor sensitization requires the cooperation of ARF, as ARF overexpression can block the effect (Figure S4). Thus, targeting ARF/MET/nMET axis using unconventional green nanomaterials of carbon dots would be promising in prevention of drug resistance and CRPC cells growth simultaneously.
Figure 7. Carbon nanodots sensitize PCa cells to MET inhibitor.
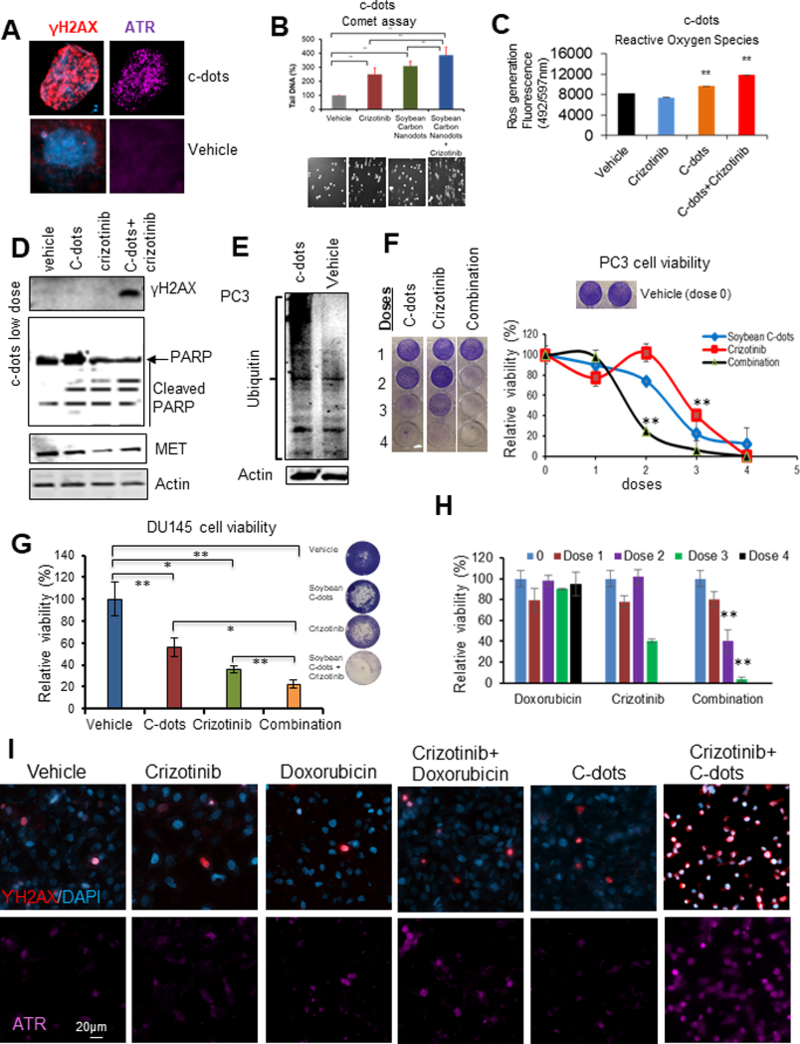
(A) C-dots induce DNA damage response in DU145 cells at dose of 0.1mg/ml. (B-C) C-dots induce more DNA damage response and ROS generation in PC3 cells with MET inhibitor crizotinib. Cells were treated by c-dots at 0.1mg/ml with or without 10μM crizotinib followed by comet or ROS assay. (D) Western blot showing C-dots at low dose of 0.025 mg/ml enhance DNA damage response, cleaved PARP, and decreased MET levels in PC3 cells with MET inhibitor crizotinib. (E) C-dots induce high molecular weight protein ubiquitination in PC3 cells. (F-H) C-dot or DOX increases efficacy of MET inhibitor on PCa cell growth. (I) C-dot or DOX increases efficacy of MET inhibitor on DNA damage response. Data are representative of averages+ SD (standard deviation).
9. The calculated energies along with the vibrational spectra using a system made of protonated acetamide and the phosphorylated tyrosine
A quantum mechanical calculation was performed to simulate the interaction between carbon dots and phosphorylated tyrosine. The FT-IR spectrum of c-dots indicates the major functional group distributed on the surface of the carbon dots is amide group. Considering the fact c-dots carry positive charge, we use a protonated acetamide to represent c-dots. The calculated IR spectrum based on carbonyl oxygen protonated acetamide with a structure shown in Figure 6 K as AmH+(B) exhibits correspondence among quite a few bands, such as H-N-H bending vibration dominated band at 1637 cm−1, C-N-H bending dominated absorption at 1540 cm−1, and C-N stretching involved band at 1245 cm-1. The highly consistency between the FT-IR spectrum of c-dots and the calculated IR spectrum of protonated acetamide implies that protonated acetamide is representative at least with respect to the chemical reactivity of the c-dots.
The FT-IR spectrum of phosphorylated tyrosine represents a vibrational pattern of HPO42-, in other word, the phosphate group is monoprotonated (denoted as Tyr-OPO3H-) whose structure is displayed in Figure 8. The calculations on energetics were performed based on four fundamental structures established between (A). non-protonated acetamide and Tyr- Tyr-OPO3H- in Figure8 A; (B).protonated acetamide and Tyr-OPO3H-, where the involved oxygens on Tyr-OPO3H- are both not protonated(Figure 8B); (C). protonated acetamide and phosphorylated tyrosine, where the protonated C=O on acetamide interacts as a proton donor with the protonated P-O on Tyr-OPO3H- (Figure 8C); (D). protonated acetamide and Tyr-OPO3H-, where the protonated C=O on acetamide interacts as an electron lone pair donor with the protonated P-O on Tyr-OPO3H- (Figure 8D). Among the various interactions between actamide w/o protonation and Tyr-OPO3H-, structure in Figure 8B shows the most negative enthalpy change. The IR spectra corresponding to different interacting complexes are displayed in Figure 6H as the dished lines. Overall, the calculated vibrational patterns for the interacting complex 8B, 8C, 8D show certain consistency with the experimental spectrum of phosphorylated tyrosine with the addition of c-dots in terms of in phase/ out of phase O=P=O stretching modes.
Figure 8. Optimized structures for the complexes formed between acetamide w/o protonated and phosphorylated tyrosine.
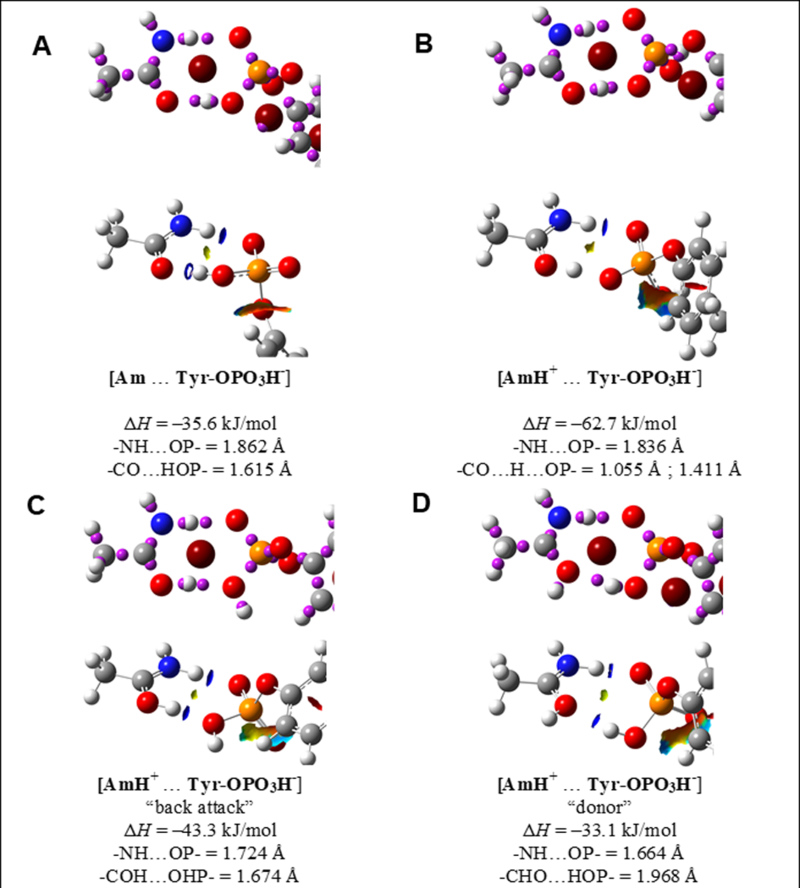
(A) non-protonated acetamide and Tyr- Tyr-OPO3H-. (B) Protonated acetamide and Tyr-OPO3H-, where the involved oxygens on Tyr-OPO3H- are both not protonated. (C) Protonated acetamide and phosphorylated tyrosine, where the protonated C=O on acetamide interacts as a proton donor with the protonated P-O on Tyr-OPO3H-. (D) Protonated acetamide and Tyr-OPO3H-, where the protonated C=O on acetamide interacts as an electron lone pair donor with the protonated P-O on Tyr-OPO3H-.
Most importantly, based on the calculation, we further found the carbon nanodots have intrinsic phosphatase-like activity by in vitro assay (Figure S6). Thus, our data suggest that soybean-derived carbon nanodots may function as a phosphatase or kinase inhibitor and through combination with specific kinase inhibitors, they may more specifically target kinases induced tumor growth especially resistant tumors.
Discussion
In this study, we elucidated for the first time that androgen deprivation activates ARF/MET//nMET/β-Catenin pathway both in vitro and in vivo and a novel mechanism in which MET requires ARF to sustain nuclear oncogenic functions for PCa progression, at least in the context of PTEN/p53 loss. Nano carbon dots may be a potential avenue to disrupt the ARF/MET signaling and enhance the efficacy of MET inhibitor.
One of the important discoveries in this study is that MET in nucleus is sustained by ARF but disrupted by AR. Many studies suggested that MET in nucleus may play pivotal roles in oncogenic functions. Early studies showed that MET in nucleus promotes the cell proliferation in breast cancer (12) and nuclear reprogramming in PCa (13). Here we further observed that nuclear MET correlates with ARF in human PCa specimens and is stabilized by ARF, but is disrupted by AR through distinct DNA damage repair systems from ARF or AR. Given that both ARF and AR regulate each other’s expression or activity in PCa cells (26, 33), the feedback loop may be formed among ARF/MET/nMET/β-Catenin/AR. Nevertheless, whether, and why reactivated AR may in turn negatively regulates MET remains unclear. One of the possible mechanisms is the intrinsic protection to prevent further aggressive cancer development initiated by cell stress response. Recent studies showed that AR positively regulates ARF thus the dual positive feedback elevation of MET by both ARF and AR may be complementary or competitive inhibition. The detailed mechanisms is left for further investigation to elucidate the correlation of ARF/MET/nMET under low androgen condition.
In addition to its tumor suppressor function, emerging evidence revealed that the novel functions of ARF in cells and in mouse tissues depend solely on metabolisms and signaling pathways, but are independent of Mdm2-p53 pathway. It has been shown that ARF has the pro-proliferative or oncogenic-promoting roles in some cells under various contexts of physiological and genetic alterations such as nutrition depletion, the viability of meiotic germ cells, or PTEN loss (2, 20–22, 25). Similarly, the uncoupling scenario of p19Arf-p53 upon oncogenic insults was found in Pten-deficient prostate tumorigenesis (2). These studies provided lines of genetic evidence that the activation of intact p53 signaling pathway plays critical roles in biological functions of ARF. Our findings demonstrated that ARF regulates MET in both mouse prostate tumors and PCa cells null for PTEN and p53 genes, a frequent event at the advanced stages of human prostate malignancy (34). Differential to its tumor suppressive role in glioma and melanoma (35), ARF regulates functions and transcriptional activation of oncogenic signaling events in nucleus of cancer cells through multiple mechanisms (36). Thus the details in ARF transition from tumor suppressor to promoter deserve further investigation which may link aging to cancer transformation (37).
The interaction between phosphorylated tyrosine and c-dots becomes evident considering both the FT-IR experiment and quantum mechanical calculation results. In the FT-IR spectrum, the broad shoulder representing P-OH-H bending mode (POHH) on Tyr-OPO3H- disappeared once the c-dots were added in the system. On the other hand, the quantum mechanical calculation indicates the protonated acetamide (the protonation site is on C=O group) tends to have stronger interaction with Tyr-OPO3H- than the acetamide without protonation. It confirms the necessity of the positive charge on c-dots. The comparison of the experimental and calculated IR spectra implies the complexes formed between c-dots and phosphorylated tyrosine is likely a mixture of the structures shown in Figure 8B, 8C, and 8D. This leads us to a realization that positive charged carbon nanodots can bind phosphorylated tyrosine and inhibit phosphorylation of nMET, and further promote MET inhibitor sensitization. Thus, c-dots can be used as targeting therapy as both a deliver/carrier and an anti-cancer drug. The dual role of c-dots is benefited from its higher toxicity than most types of nanoparticles. In the future, the conjugation of kinase inhibitors and c-dots holds huge potential in targeted inhibition of kinases.
Given that drug resistance frequently occur in cancer through multiple mechanisms, nanoparticles are the best drug delivery carrier as non-substrate of drug transporters, reversing resistance by nanoparticles have been acknowledged in preclinical or clinical studies (38). For example, milk-derived carbon nanoparticles with fabrication of doxorubicin through electrostatic interactions have been found to delivery DOX to nucleus and showed increased efficacy of DOX in cancer cells but less in normal fibroblast cells (39). Moreover, BYL719, a PI3Kα inhibitor conjugated with P-selectin-targeted nanoparticles, showed higher delivery efficiency to head and neck tumor (40). Compared to other nanoparticles, due to water soluble, biocompatibility, the potential of c-dots in drug delivery is promising if targeted into tumor cells.
However, if not targeted into ideal tumor tissues, the limitation of c-dots on drug delivery is obvious so that side effect may appear. As carbon materials are green materials to the microenvironment, compared to other metal related nanoparticles, carbon dots showed significant mild side effect on tissues. In cell level, based on our results and personal communication, food-derived c-dots can enter in to nucleus to abolish nuclear drug targets such as epigenetic factors, transcriptional events, DNA, nuclear RTK/MET kinase or other nuclear targets of prostate cancer such as YAP, AR (25). Therefore, more precision delivery should be designed as far as c-dots mediated drug delivery is concerned.
Most importantly, we provided experimental, conceptual and promising novel treatment avenue that using c-dots to enhance RTK inhibitor efficiency, which is the first report in the green carbon-drug field, though there is a report using gold nanoparticles conjugated with FLT3 kinase inhibitor in leukemia (41). Thus green carbon nanoparticles would provide a more ecologically benefit treatment for combinatorial therapy or drug delivery using RTK inhibitors.
In summary, it is the first report on how nuclear RTKs can be sustained by a tumor suppressor for drug resistance. Our novel finding that androgen deprivation mediated ARF/MET/nMET/AR axis would provide the guidance for precision treatment. Moreover, ARF might be a pharma-marker for individual treatment with MET inhibitor. Finally, we provided a novel avenue using food-derived nanomaterials for CRPC treatment by targeting ARF/MET axis and overcoming drug resistance to MET inhibitor. Emerging data have shown that nMET is widely found in many types of cancer, thus c-dots based novel therapy might be broadly attractive for targeted precision cancer therapy (38–39,42–43). Moreover, in a broad concept, drug resistance to many kinase inhibitors often occurs through multiple mechanisms such as reactivation of crosstalk pathways or self-induction (44–45). For example, HGF ligand can activate MET inhibitor resistance, and MET elevation can resistant to EGFR inhibitor (44–45). Based on our finding, c-dots can bind phosphor-tyrosine and may inhibit phosphorylation by physical occupying through electric statics which is a good events for protecting additional molecular binding or phosphorylation occurring. The carbon nanodots would have great application potential in preventing RTK inhibitor resistance. Thus conjugated carbon nanodots-RTK inhibitor compound would provide a novel avenue for targeted therapy in cancer for preventing drug resistance.
Materials and methods
1. Mutant mice, tumor analysis and microarray based pathway reanalysis
In accordance with and approved by IACUC guidelines at Meharry Medical College ( MMC), the C57BL/6 knockout mice were bred and maintained at MMC animal core facility. Pten and Trp53 conditional knockout in prostates are controlled by Probasin-Cre recombination (18). All male, aged at 3months or 6 months of wild type (Wt), p19Arf knockout mice (p19Arf−/−), Pten/Trp53 double-mutant (referred to as Pten/Trp53), and Pten/Trp53/p19Arf triple-mutant mice (referred to as Pten/Trp53/p19Arf) were generated and genotyped as described previously (18, 19). Sample numbers are indicated in figures. Sample sizes are collection of all mice which were generated in this specific experiment at the similar experimental period and without exclusion, without randomization, without blinding during the analysis were used. Tissues were fixed and processed and 5 μm thickness of tissue sections were applied for H&E and immunohistochemically (IHC) staining. For IHC analysis, sections were stained with antibodies: MET (C28, Santa Cruz), β-Catenin (1:100, Cell Signaling).
2. ARF knockdown by small hairpin RNA (shRNA), cell proliferation, drug response and cell invasion assays
p14ARF stable knockdown and scrambled control cell lines were established as described previously (19,46). For transient knockdown, shRNA constructs were transfected into cells, and cell lysates were collected and subjected to immunoblotting analysis after 48–72 hrs. For cell proliferation, cells were replated in 96-well plates at a density of 3000 cells per well. Cell numbers were determined at 2, 4, and 6 days by counting cell number or by WST-1 assay based on commercial instructions (Roche). Alternatively, for drug response of cell growth assay, cells were seeded in 24-well plates at a density of 5×105 per well and 24hrs post-transfection, cells were treated with drugs indicated in figure legends, fixed, stained with 0.3% crystal violet and counted through absorbance. For shRNA mediated cell growth inhibition assay, cells were transfected with scrambled or shARF plasmid using Lipofectamine 2000 or Lipofectamine 3000 (Life Technologies) or infected by lentivirus containing shRNA plasmid. For cytotoxicity assay, drugs including doxorubicin (DOX) or nanodots were used for treatment 24hrs post plating of cells and 3 days later, cells were fixed, stained with 0.3% crystal violet and counted through absorbance. For invasion assay, cells were cultured in 60mm Petri dishes until confluence. After starved for 16 hrs, cells were seeded at a density of 5×104 per well into the upper chamber with 8 μm polyethylene terephthalate membrane filters (Becton-Dickinson). Finally cells were fixed, stained with 0.3% crystal violet and counted.
3. Immunofluorescence (IF), immunoblotting (IB) and co-immunoprecipitation (IP) assays
For IF analysis, cells were seeded on coverslips for 2 days and then fixed in 4% paraformaldehyde for 15 min. Antibodies for IF were: p14ARF monoclonal antibody (14P02, 1:100, NeoMarkers, Catalog No.MS850P1 of Thermo Scientific™, as this item has been discontinued by the manufacturer, new Thermo Scientific™ 14P02 (DCS-240), Catalog # MA5–14260), MET polyclonal antibody (1:50, Met (D1C2) XP® Rabbit mAb #8198, Cell Signaling), Phospho-Met (Tyr1234/1235) (1:100, (D26) XP® Rabbit mAb #3077 Cell Signaling), MET(1:100, Met (C-28): sc-161, Santa Cruz), γH2AX (1:100, Phospho-Histone H2A.X (Ser139) (20E3) Rabbit mAb 9718, Cell signaling), ATR (1:100, clone C-1, sc-515173, Santa Cruz), AR (1:100, N-20, sc-816, Santa Cruz), BRCA1(1:100 (D-9): sc-6954, Santa Cruz), Ubiquitin (1:200, P4D1, sc-8017, Santa Cruz), HSP90 (1:100, 4F10, sc-69703) and β-Catenin (1:100, β-Catenin (D10A8) XP® Rabbit mAb #8480, Cell Signaling). For IB, cell lysates were prepared in NP40 cell lysis buffer (ThermoFisher), and subjected to a standard procedure of SDS-PAGE and antibody detection (1:1000, except special note). Additional antibodies list except IF, used for Western blotting were: mouse monoclonal anti-β-actin (AC-74, 1:5000 dilution, Catalog# A5316, Sigma Sigma), p14ARF (14P02, 1:750, new Thermo Scientific™ 14P02 (DCS-240), Catalog # MA5–14260), PARP (1:1000, #9542, Cell Signaling), Caspase 3 (1:1000, E-8, sc-7272, Santa Cruz), and MET (D1C2, 1:1000, #8198, Cell Signaling). For IP assay, cellular proteins were extracted by IP lysis buffer (Thermo Fisher), followed by immunoprecipitation with antibody of MET (C28, sc-161, Santa Cruz), and subjected to immunoblotting.
4. Signaling pathway reanalysis of microarray data
The gene expression profiles dataset of Pten/Trp53/p19Arf triple knockout mice (19) were used for reanalysis of ARF regulated signaling pathways as described previously (20) and genes expressed differently were analyzed by pathways using DAVID tools (https://david.ncifcrf.gov/), with Mus musculus as the species background (47–50).
5. Prostate tissue microarrays (TMA)
Human prostate tissue microarrays were performed using paraffin-embedded normal prostate tissue (n=5) and PCa samples (n=35) (US Biomax) with information on the stage, Gleason’s score and grade (40 cases/80 cores, Catalog# PR808, informed consent was managed by Biomax.) which was approved by institutional committee with the aid of morphology core facility at Meharry Medical College. Tissue sections were stained with antibodies against p14ARF (4C6/4, 1:50, p14ARF (4C6/4, 1:50, Catalog #2407, Cell Signaling) and MET (C28, sc-161, 1:500, Santa Cruz). The intensity of the staining was investigated blindly and graded as reported previously (51). The proportion and intensity scores were combined (0–8 range) (51). The correlation between MET and ARF levels were analyzed based on their combined scores.
6. Statistical analysis
All data were evaluated using two-tailed Student’s t-test unless otherwise stated, and values of p≤0.05, or p<0.01 were considered statistically significant assuming similar variation but calculated and indicated in figures. For statistics in figure legend, **, p<0.01; *, p≤0.05. Experiments were performed with three repeats or triplicates with reproducible repeat.
7. C-dots synthesis, characterization, and calculation of binding to phosphory-Tyrosine
The mixture of soybean and ethylenediamine (1:4) (Sigma Aldrich®) with water were heated at 200 oC for 5h. After centrifuge, the supernatant was collected by evaporating the water in vacuo using rotavap, followed by wash with methanol. The final solid product was obtained by heating in oven at 80oC for 24 h.
A SmartSPM 100 scanning probe microscope (AIST-NT) in non-contact mode was used to characterize the morphology and particle size of the carbon dots using high accuracy HA-NC “etalon” probes (cantilever frequency 140 kHz) at a scan rate of 1 Hz. The functional groups distributed on the surface of c-dots were characterized using a FTIR spectrometer. The FTIR absorbance was recorded in the range of 400 cm−1 to 4000 cm-1. Calculation of C-dots binding to phosphory-Tyrosine was performed as described in details in supplemental methods (32).
8. Comet, ROS and Luciferase reporter assay
The PC3 cells were treated by carbon nanodots at 0.1 mg/ml for 2hrs followed by the comet and ROS assay. For comet assay, cells were subjected to treatment as protocol of the following (https://www.sigmaaldrich.com/life-science/cell-biology/cancer-research/learning-center/cancer-research-protocols/comet-assay.html; http://www.cometassayindia.org). Briefly, trypsinized cells went through neutralization with DMEM medium and were diluted to 0.5×106 cells/ml with 1xPBS. Next, 0.5% low-melting agarose was melted, cooled for 20 min at room temperature and then kept in the Thermoblocker at 37°C followed by mixing with diluted cells (50μl low-melting agarose: 20μl diluted cells) at 37°C. The solution was immediately transferred to anti-frost glass slides followed by covering by 1% agarose on top. The slides were finally put into the chamber protected from light with lysate buffer for 15–20 hrs at 4°C. Following steps include electrophoresis in running buffer (pH 11.0, 300 mM NaOH / 1 mM EDTA) for 50 min (20 min without running and 30 min at 24V, 100mA), washing with neutralization buffer for 5 min, washing by ddH2O for 1 min, and staining with ethidium bromide [2μg/ml] for 30 sec. Samples were kept under low light conditions. In addition, to avoid background noise running buffer solutions were freshly prepared. Finally, samples were visualized under Fluorescent microscope and analyzed using Comet Score software. ROS assay was performed based on the protocol of the manufacture using Invitrogen™ Total Reactive Oxygen Species (ROS) Assay Kit 520 nm (Invitrogen). Cells were seeded on a black 96-well plate with clear bottom (1000cells/100μl/well) in phenol red free medium for overnight. CM-H2DCFDA dye was used for staining cells and the plate with samples were analyzed on a Cytation 5 (BioTek) imaging and Synergy H1 microplate reader. The Dual-Luciferase® Reporter (DLR™) Assay System kit (E1910, Promega) was used for MMP7 promoter-reporter assay. Cells were transfected as reported previously and assay was performed based on the protocol of manufacture (https://worldwide.promega.com/resources/protocols/technical-manuals/0/dual-luciferase-reporter-assay-system-protocol/ )
9. Cell line and cell culture
Cells were cultured in medium of RPMI1640 or DMEM with 10% FBS and antibiotics. Most cell lines were purchased from ATCC within 1–2 years. C4–2B cells were purchased from MD Anderson Cancer center with authenticated report. LAPC4 cell line was a gift provided by Memorial Sloan Kettering Cancer Center. Others are mentioned in Acknowledgement part.
Supplementary Material
Acknowledgements
We would like to thank Drs. Simon W. Hayward and LaMonica Stewart for cell lines, Lynn M. Matrisian for providing MMP7-Luc plasmid. This work was supported in part by NIH grants MD004038, MD007586, CA091408 and CA163069, UL1 RR024975–01 and UL1 TR000445–06. IF and IHC analysis was performed by Meharry Medical College Morphology Core which is supported in part by NIH grants U54 MD007593, G12 MD007586, R24 DA036420, and S10RR0254970. We would like to thank Dr. Guoliang Li, Zhanna Alexeyeva, Aigerim Kabulova, Zhibek Keneskhanova, Akerke Altaikyzy for the kind help or assistant on research. Quantum mechanical calculations were performed on the clusters of the Siberian Supercomputer Center (http://www2.sscc.ru) of the Institute of Computational Mathematics and Mathematical Geophysics of the Siberian Branch of the Russian Academy of Sciences.
Footnotes
Conflict of Interest Statement
There is no conflict of interest.
References
- 1.Huggins C, and Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin1972; 22: 232–240. [DOI] [PubMed] [Google Scholar]
- 2.Chen WY, Tsai YC, Yeh HL, Suau F, Jiang KC, Shao AN et al. Loss of SPDEF and gain of TGFBI activity after androgen deprivation therapy promote EMTand bone metastasis of prostate cancer. Sci Signal 2017; 10: 492. [DOI] [PubMed] [Google Scholar]
- 3.Petrylak DP, Tangen CM, Hussain MH, Lara PNJ, Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004; 351: 1513–1520. [DOI] [PubMed] [Google Scholar]
- 4.Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012; 22:373–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greaves M, Maley CC. Clonal Evolution in cancer. Nature 2012; 481(7381): 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie Y, Xu K, Linn DE, Yang X, Guo Z, Shimelis H, et al. The 44-kDa Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J Biol Chem 2008; 283: 3349–3356. [DOI] [PubMed] [Google Scholar]
- 7.Xie Y, Burcu M, Linn DE, Qiu Y, and Baer MR. Pim-1 kinase protects P-glycoprotein from degradation and enables its glycosylation and cell surface expression. Mol Pharmacol 2010; 78: 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 2006; 10: 309–319. [DOI] [PubMed] [Google Scholar]
- 9.Verras M, Lee J, Xue H, Li TH, Wang Y, Sun Z. The androgen receptor negatively regulates the expression of c-Met: implications for a novel mechanism of prostate cancer progression. Cancer Res 2007; 67: 967–975. [DOI] [PubMed] [Google Scholar]
- 10.Gherardi E, Birchmeier W, Birchmeier C, Vande WG. Targeting MET in cancer: rationale and progress. Nat Rev Cancer 2012; 12: 89–103. [DOI] [PubMed] [Google Scholar]
- 11.Wanjala J, Taylor BS, Chapinski C, Hieronymus H, Wongvipat J, Chen Y, et al. Identifying Actionable Targets through Integrative Analyses of GEM Model and Human Prostate Cancer Genomic Profiling. Mol Cancer Ther 2015; 14: 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pozner-Moulis S, Pappas DJ, and Rimm DL. Met, the hepatocyte growth factor receptor, localizes to the nucleus in cells at low density. Cancer Res 2006; 66: 7976–7982. [DOI] [PubMed] [Google Scholar]
- 13.Xie Y, Lu W, Liu S, Yang Q, Carver BS, Li E, et al. Crosstalk between nuclear MET and SOX9/beta-catenin correlates with castration-resistant prostate cancer. Mol Endocrinol 2014; 28: 1629–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tu WH, Zhu C, Clark C, Christensen JG, and Sun Z. Efficacy of c-Met inhibitor for advanced prostate cancer. BMC Cancer 2010; 10: 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012; 4: 120ra–117.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song A, Kim TM, Kim DW, Kim S, Keam B, Lee SH, et al. Molecular changes associated with acquired resistance to crizotinib in ROS1-rearranged non-small cell lung cancer (NSCLC). Clin Cancer Res 2015; 21(10):2379–87. [DOI] [PubMed] [Google Scholar]
- 17.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell 2006; 127: 265–275. [DOI] [PubMed] [Google Scholar]
- 18.Chen Z, Carracedo A, Lin HK, Koutcher JA, Behrendt N, Egia A, et al. Differential p53-independent outcomes of p19(Arf) loss in oncogenesis. Sci Signal 2009; 2: ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie Y, Liu S, Lu W, Yang Q, Williams KD, Binhazim AA, et al. Slug regulates E-cadherin repression via p19Arf in prostate tumorigenesis. Mol Oncol 2014; 8: 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie Y, Lu W, Liu S, Yang Q, Goodwin JS, Sathyanarayana SA et al. MMP7 interacts with ARF in nucleus to potentiate tumor microenvironments for prostate cancer progression in vivo. Oncotarget 2016; 7(30): 47609–47619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vivo M, Fontana R, Ranieri M, Capasso G, Angrisano T, Pollice A, et al. p14ARF interacts with the focal adhesion kinase and protects cells from anoikis. Oncogene 2017; 36(34): 4913–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Humbey O, Pimkina J, Zilfou JT, Jarnik M, Dominguez-Brauer C, Burgess DJ, et al. The ARF tumor suppressor can promote the progression of some tumors. Cancer Res 2008; 68: 9608–9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li CL, Ou CM, Wu WC, Chen YP, Lin TE, Ho LC, et al. Carbon dots prepared from ginger exhibiting efficient inhibition of human hepatocellular carcinoma cells. J Mater Chem B 2014; 2: 4564–4571. [DOI] [PubMed] [Google Scholar]
- 24.Xie Y, Filchakova O, Yang Q, Yesbolatov Y, Tursynkhan D, Kassymbek A, et al. Inhibition of Cancer Cell Proliferation by Carbon Dots Derived from Date Pits at Low Dose. ChemistrySelect 2017; 2: 4079–4083. [Google Scholar]
- 25.Xie Y, Sun Q, Nurkesh AA, Lu J, Kauanova S, Feng J, et al. Dysregulation of YAP by ARF Stimulated with Tea-derived Carbon Nanodots. Sci Rep 2017; 7(1): 16577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding HM, Ma YQ. Computational approaches to cell-nanomaterial interactions: keeping balance between therapeutic efficiency and cytotoxicity. Nanoscale Horiz 2018; 3: 6–27. [DOI] [PubMed] [Google Scholar]
- 27.Liu S, Meng XY, Perez-Aguilar JM, Zhou R. An In Silico study of TiO(2)nanoparticles interaction with twenty standard amino acids in aqueous solution. Sci Rep 2016; 6: 37761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem 2005; 16:1781–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Likos CN. Effective interactions in soft condensed matter physics. Phys Rep 2001; 348: 267–439. [Google Scholar]
- 30.Vácha R, Martinez-Veracoechea FJ, Frenkel D. Receptor-mediated endocytosis of nanoparticles of various shapes. Nano Lett 2011; 11: 5391–5395. [DOI] [PubMed] [Google Scholar]
- 31.Kaur D, Khanna S. Intermolecular hydrogen bonding interactions of furan, isoxazole and oxazole with water. Computational and Theoretical Chemistry 2011; 963: 71–75. [Google Scholar]
- 32.Klähn M, Mathias G, Kötting C, Nonella M, Schlitter J, Gerwert K, et al. IR Spectra of Phosphate Ions in Aqueous Solution: Predictions of a DFT/MM Approach Compared with Observations. J of Phy Chem A 2004; 108 (29): 6186–6194. [Google Scholar]
- 33.Mudryj M, Siddiqui S, Libertini SJ, Lombard AP, Mooso B, D’Abronzo L, Melgoza F, Borowsky A, Drake C, Qi L, Ghosh PM. Androgen receptor-mediated regulation of p14ARF transcription in prostate tumor cells. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res. 2015;75(15 Suppl): Abstract nr 5051. doi: 10.1158/1538-7445.AM2015-5051. [DOI] [Google Scholar]
- 34.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005; 436: 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lachat Y, Diserens AC, Nozaki M, Kobayashi H, Hamou MF, Godard S, et al. INK4a/Arf is required for suppression of EGFR/DeltaEGFR(2–7)-dependent ERK activation in mouse astrocytes and glioma. Oncogene 2004; 23: 6854–6863. [DOI] [PubMed] [Google Scholar]
- 36.Yu J, Zhang SS, Saito K, Williams S, Arimura Y, Ma Y, et al. PTEN regulation by Akt-EGR1-ARF-PTEN axis. EMBO 2009; 28: 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khoo CM, Carrasco DR, Bosenberg MW, Paik JH, Depinho RA. Ink4a/Arf tumor suppressor does not modulate the degenerative conditions or tumor spectrum of the telomerase-deficient mouse. Proc Natl Acad Sci U S A 2007; 104: 3931–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang M, Liu E, Cui Y, Huang Y. Nanotechnology-based combination therapy for overcoming multidrug-resistant cancer. Cancer Biol Med.2017; 14(3): 212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan Y, Guo B, Hao L, Liu N, Lin Y, Guo W, Li X, Gu B. Doxorubicin-loadedenvironmentally friendly carbon dots as a novel drug delivery system for nucleus targeted cancer therapy. Colloids Surf B Biointerfaces 2017; 159: 349–359. [DOI] [PubMed] [Google Scholar]
- 40.Mizrachi A, Shamay Y, Shah J, Brook S, Soong J, Rajasekhar VK, et al. Tumour-specific PI3K inhibition via nanoparticle-targeted delivery in head and neck squamous cell carcinoma. Nat Commun 2017; 8: 14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petrushev B, Boca S, Simon T, Berce C, Frinc I, Dima D, et al. Gold nanoparticles enhance the effect of tyrosine kinase inhibitors in acute myeloid leukemia therapy. Int J Nanomedicine 2016; 11: 641–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie Y, Istayeva S, Chen Z, Tokay T, Zhumadilov Z, Wu D, et al. nMET, A New Target in Recurrent Cancer. Curr Cancer Drug Targets 2016; 16(7): 572–8. [DOI] [PubMed] [Google Scholar]
- 43.Yuan Y, Guo B, Hao L, Liu N, Lin Y, Guo W et al. Doxorubicin-loaded environmentally friendly carbon dots as a novel drug delivery system for nucleus targeted cancer therapy. Colloids Surf B Biointerfaces. 2017; 159: 349–359. [DOI] [PubMed] [Google Scholar]
- 44.Kim SM, Kim H, Yun MR, Kang HN, Pyo KH, Park HJ et al. Activation of the Met kinase confers acquired drug resistance in FGFR-targeted lung cancer therapy. Oncogenesis 2016; 5(7): e241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahn SY, Kim J, Kim MA, Choi J, Kim WH. Increased HGF Expression Induces Resistance to c-MET Tyrosine Kinase Inhibitors in Gastric Cancer. Anticancer Res 2017;37(3):1127–1138. [DOI] [PubMed] [Google Scholar]
- 46.Lu W, Xie Y, Ma Y, Matusik RJ, Chen Z. ARF represses androgen receptor transactivation in prostate cancer. Mol Endocrinol 2013; 27: 635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017; 45: D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang DW, Sherman B T, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols 2008; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 49.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016; 44: D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanehisa M, Goto S KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 2000; 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ji H, Wang J, Nika H, Hawke D, Keezer S, Ge Q et al. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Mol Cell 2009; 36: 547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.