

Abstract
We report an unusual 3-substituted pyridine polyketide, onydecalin A (1), which was obtained along with 2 as a major constituent from the fungus Aioliomyces pyridodomos (Order: Onygenales) following a two-month fermentation. Feeding studies demonstrated that the pyridine subunit originates via an unprecedented biosynthetic process in comparison to other polyketide-linked pyridines or derivatives such as pyridones. The slow growth of the fungus led us to perform a one-year fermentation, leading to production of compounds 2-4 as the major constituents. These compounds showed modest but selective inhibition against a variety of transient receptor potential channels, as well as against the human pathogenic fungus, Histoplasma capsulatum.
Graphical Abstract
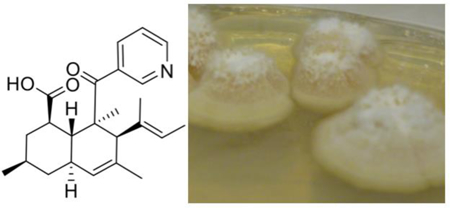
Fungal genomes typically encode a large number of orphan secondary metabolite biosynthetic gene clusters, leading to an explosion in genome mining methods.1 Another approach is to focus on the chemical potential found in novel or previously uncultivated fungal taxa.2 In particular, much of fungal biodiversity has yet to be assessed by natural products chemists. In our program to isolate microbes associated with marine invertebrates, we serendipitously obtained a novel fungus from the order Onygenales, strain 110162,3 with the proposed name Aioliomyces pyridodomos. Onygenales is a diverse fungal group including human and animal pathogens such as Coccidioides and Histoplasma species, as well as a variety of non-pathogenic strains.4 Despite this importance, it has received only modest attention in natural products chemistry.5 We thus took the opportunity to investigate this marine-derived strain for its chemical potential. In a previous study, Aioliomyces pyridodomos was grown for 25 days, leading to oxazinin A as a major component.3 Here, by growing the strain for 60 days, we obtained a series of decalin derivatives that exhibit antagonism to transient receptor potential (TRP) channels and antifungal activity.
RESULTS AND DISCUSSION
A. pyridodomus was grown for two months and extracted with organic solvents. Fractions were screened against a panel of TRP channels expressed in cultivated mammalian cells, leading to isolation of onydecalin A (1), which was partly responsible for observed antagonism to TRPV1 and TRPV4.
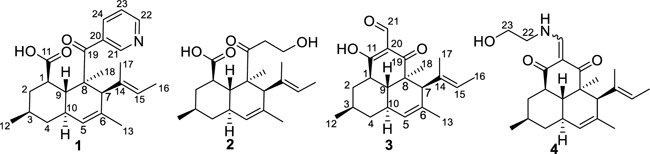
Compound 1 was obtained as a pale yellow solid and assigned the molecular formula C24H31NO3 based on HRFTMS analysis and NMR experiments (Table 1; Figure 1). The free base contained minor impurities, which could be removed by repurification with TFA to afford the salt. For both the free base and the salt, 1H and 2D NMR spectra were readily obtained, but due to molecular properties most 13C signals were visible only in HSQC or HMBC spectra, and not in the 1D 13C spectrum. Using these data, 24 13C signals were readily observed, and an odd number of nitrogen atoms was inferred from the m/z. These spectra revealed two olefins, one aromatic ring, a ketone, a carboxylic acid, and five methyl groups.
Table 1.
NMR data for compounds 1 and 2 in DMSO-d6.
| No. | 1 |
2 |
||||
|---|---|---|---|---|---|---|
| 13C NMR | 1H NMR (J in Hz) | ROESY | 13C NMR | 1H NMR (J in Hz) | ROESY | |
| 1 | 45.9 CH | 2.04 dd (10.7, 10.7) | 45.3 CH | 2.00 m | ||
| 2 | 39.7 CH2 | 1.7 brd (13.0), 1.29 q (13.0) | 39.5 CH2 | 1.70 m, 1.23 m | ||
| 3 | 32.2 CH | 1.56 m | H-1, H-10 | 32.0 CH | 1.53 m | H-10 |
| 4 | 42.7 CH2 | 1.81 d (12.8), 0.90 m | 42.6 CH2 | 1.80 m, 0.83 m | ||
| 5 | 127.1 CH | 5.30 brs | 126.7 CH | 5.26 brs | ||
| 6 | 132.0 C | - | 130.9 C | - | ||
| 7 | 61.1 CH | 2.93 brs | H-18 | 61.0 CH | 2.55 brs | H-18 |
| 8 | 54.7 C | - | 53.1 C | - | ||
| 9 | 45.3 CH | 2.28 dd (10.3, 9.1) | H-17, H-12 | 43.7 CH | 2.02 m | H-17 |
| 10 | 39.7 CH | 1.87 dd (11.7, 10.3) | 39.3 CH | 1.80 m | ||
| 11 | 177.3 C | - | 176.2 C | - | ||
| 11-OH | - | 11.81 brs | ||||
| 12 | 22.3 CH3 | 0.91 d (6.7) | 22.5 CH3 | 0.90 d (6.7) | ||
| 13 | 22.8 CH3 | 1.49 s | 22.8 CH3 | 1.44 s | ||
| 14 | 136.8 C | - | 135.9 C | - | ||
| 15 | 124.2 CH | 5.19 q (6.7) | 123.6 CH | 5.29 q (6.7) | ||
| 16 | 13.9 CH3 | 1.37 d (6.7) | 13.8 CH3 | 1.45 s | ||
| 17 | 13.9 CH3 | 1.37 s | 12.6 CH3 | 1.27 s | ||
| 18 | 20.2 CH3 | 1.44 s | H-1, H-10, H-7 | 18.5 CH3 | 1.35 s | H-1, H-10, H-7 |
| 19 | 204.9 C | - | 210.7 C | - | ||
| 20 | 136.6 C | - | 42.8 CH2 | 2.70 m, 2.54 m | H-18, H-7, H-15 | |
| 21 | 150.5 CH | 8.77 brs | H-18, H-7, H-15 | 57.1 CH2 | 3.58 m, 3.33 m | H-18, H-7, H-15 |
| 22 | 151.4 CH | 8.61 dd (4.9, 1.5) | ||||
| 23 | 123.3 CH | 7.40 dd (8.0, 4.9) | ||||
| 24 | 137.2 CH | 8.08 brd (6.5) | H-18, H-7, H-15 | |||
Figure 1.

Key HMBC, 1H-1H COSY and ADEQUATE correlations.
The vinyl proton signal at δ 5.19 ppm (H-15) was correlated by COSY to a vinylic methyl group (H-16). Further, vinylic methyl groups H-16 and H-17 exhibited HMBC correlations to C-15 and C-14, and to C-14 and C-15, respectively. Taken together, these data indicated the presence of a trisubstituted double bond. A second trisubstituted double bond at C-5 to C-6 was evident from HMBC correlations between H-13 and C-5 and C-6. A spin system of from H-1 to H-9, including H-12 and H-5, was deduced from COSY correlations (Figure 1), indicating the presence of a cyclohexane moiety in the molecule. The two double bonds and the cyclohexane ring could be joined based upon HMBC correlations from singlet methyl protons H-18 (to C-8, C-7, C-9 and C-19) and from a singlet methine proton H-7 (to C-8, C-14, and C-6). These data revealed a decalin substructure, which is tetramethyl-substituted at C-3, C-6, C-8 and C-17. Furthermore, an HMBC correlation from H-1 to C-11 (δC 177.3, δH 11.81 ppm) indicated a carboxylic acid group substituted at C-1 of the decalin ring. This established a core structure bearing substantial similarity in chemical shift data to the large family of decalin-bearing polyketides from fungi.
In addition to the signals of the decalin ring system, signals for four aromatic protons at δ 8.77 ppm (H-21), 8.61 ppm (H-22), 7.40 ppm (H-23), 8.08 ppm (H-24) were observed in the 1H NMR spectrum. COSY correlations between H-23/H-22, H-23/H-24 indicated that both H-24 and H-22 are ortho to H-23. Meta-coupling between H-24/H-21 was observed in the COSY spectrum. HMBC correlations from H-23 to C-20, from H-21 to C-20 and C-22, as well as the chemical shifts of C-21 (δ 150.5) and C-22 (δ 152.4 ppm), indicated the presence of a 3-substituted pyridine ring substructure. Although no HMBC correlation was observed between the ketone carbonyl C-19 (δ 204.9) of the decalin moiety and the protons in the pyridine moiety, the connection between them could be established by the ROESY correlations (Figure 3) from both H-21 and H-24 to H- 18, H-7 and H-15. Further, the chemical shift of C-19 was consistent with a conjugated aromatic ketone carbonyl.
Figure 3.

Experimental and calculated CD spectra for 1, 3 and 4. Blue: experimental; red: calculated.
With these data in hand, the absence of certain 13C signals in 1D spectra, but not in 2D spectra, could be potentially explained. We believe that this effect was most likely caused by a slow rotation of the pyridine ring in the NMR timescale. This was reflected both in the ROESY data, in which correlations corresponding to two major conformers of the pyridine ring were observed, and in molecular modeling, in which two major conformers were present (Figure 2). In particular, ROESY crosspeaks between pyridine protons H-21 and H-22 with decalin substituents H-7, H-15, and H-18 (Figure 2A) reflect two different orientations of the pyridine ring (Figures 2B and 2c) in comparison to the decalin.
Figure 2.

A) Key ROESY correlations of compound 1, B,C) Two major optimized conformations of compound 1 obtained using B3LYP/6–31G(d,p) in Gaussian09.
The configuration of 1 was determined based on NMR and CD data. ROESY correlations (H-7/H-18/H-10/H-1/H-3 and H-12/H-9/H-17) (Figure 2 and Table 1) indicated the relative configuration of the decalin ring. The absolute configuration was determined by comparing the calculated and experimental CD spectra. In the CD spectrum of 1, we observed a negative Cotton effect at 345 nm (n→π*) and a positive Cotton effect at 265 nm (π →π*). Compound 1 was modeled using an arbitrary isomer (1R, 3R, 7S, 8R, 9R, 10R). Initial structures were obtained using the Merck molecular force field (MMFF).6 Subsequently, these starting minima were re-optimized using time-dependent density-functional theory (TDDFT), resulting in 5 optimized conformers for which ECD was calculated using SpecDis (Figure 3).7 Because the experimental and calculated data provided a relatively good match, it is likely that the absolute configuration is as shown in 1. This is identical to the reported C-8 configuration of eupenicinicol C, a somewhat related compound with known configuration.8
Onydecalin B (2) was isolated as a colorless solid with the molecular formula C21H32O4 based on the analysis of HRFTMS and NMR data (Table 1). The 1D NMR data (Table 1) of 2 were very similar to those of 1, clearly indicating the presence of an identical decalin ring. The only obvious difference was the absence of aromatic NMR signals found in 1. Instead, an ethyl alcohol group (δC 42.8, δH 2.70, 2.54 and δC 57.1, δH 3.58, 3.33 ppm) was observed in the NMR spectra of 2. The chemical shift of C-19 (δ 210.7 ppm) was downfield in comparison to C-19 in 1, further indicating the loss of the aromatic group. NOESY correlations (Table 1) between H-20, H-21 and H-18, H-7, H-15 established the connection between the decalin moiety and the ethyl alcohol moiety through the C-19-C-20 bond. NOESY correlations confirmed that the decalin ring of 2 has the same relative configuration as 1. The standard modeling procedure failed to provide unambiguous ECD data for 2: both a positive and a negative Cotton effect at 280 nm could be obtained depending upon the details of the method. We tentatively propose that 2 has the same absolute configuration of 1 based upon their probable common biosynthetic origin. Notably, the functional group arrangement and the resulting chemical shifts are very close to those found in the betaenone series of fungal polyketides.9–10
We attempted a long-term fermentation of the fungus to determine whether new derivatives would be produced. After 365 days, the extract contained barely detectable 1, with 2 as the major compound. In addition, new congeners onydecalins C (3) and D (4) were obtained. The fungus continued to grow and produce new mycelia over this period, indicating slow growth.
Compound 3 was isolated as pale yellow solid. the molecular formula was determined as C21H28O3 based on the analysis of HRESIMS and NMR data (Table 2). In chromatographic separations, a well-resolved peak corresponding to 3 was observed only under acidic conditions, suggesting a potential low pKa. The 1H NMR spectrum of 3 showed two sets of signals with the ratio 1.6:1, indicating the presence of two exchangeable tautomeric forms. The NMR data showed that 3 contained an identical ring to that found in 1 and 2. In addition, in the NMR spectra of 3, an aldehyde group was observed at δC 195.6 and δH 9.64 ppm, with the minor tautomeric form at δC 195.0 and δH 9.55 ppm. HMBC correlations from H-21 to C-19, from H-19 to C-19, from H-1 to C-11 for the major tautomer, along with the HMBC correlations from H-21 to C-11, from H-18 to C-19 for the minor tautomer, suggested a 2-formycyclohexane-1,3-dione substructure. Thus, the tautomerization of 3 was due to the rotation around the C20-C21 bond in the NMR time scale.
Table 2.
NMR data for compounds 3 and 4.
| No. | 3 |
4 |
||||||
|---|---|---|---|---|---|---|---|---|
| 13C NMRa | 13C NMRb | 1H NMR (J in Hz)a | 1H NMR (J in Hz)b | 13C NMRa | 13C NMRb | 1H NMR (J in Hz)a | 1H NMR (J in Hz)b | |
| 1 | 43.1 CH | 39.6 CH | 2.15 m | 2.47 m | 43.3 CH | 43.0 CH | 2.22 m | 2.16 m |
| 2 | 34.8 CH2 | 34.8 CH2 | 2.35 m 0.88 m | 2.40 m 0.97 m | 36.0 CH2 | 36.0 CH2 | 2.39 m 0.88 m | 2.39 m 0.88 m |
| 3 | 31.7 CH | 31.7 CH | 1.54 m | 1.54 m | 31.9 CH | 31.9 CH | 1.56 m | 1.56 m |
| 4 | 40.7 CH2 | 40.7 CH2 | 1.84 m 0.78 m | 1.84 m 0.78 m | 41.3 CH2 | 41.3 CH2 | 1.82 m 0.78 m | 1.82 m 0.78 m |
| 5 | 125.2 CH | 124.2 CH | 5.29 brs | 5.22 brs | 124.8 CH | 125.2 CH | 5.24 m | 5.26 m |
| 6 | 133.6 C | 134.9 C | - | - | 135.3 C | 135.0 C | - | - |
| 7 | 57.1 CH | 56.9 CH | 2.84 m | 2.82 m | 57.9 CH | 57.9 CH | 2.83 m | 2.82 m |
| 8 | 44.6 C | 47.3 C | - | - | 47.1 C | 47.1 CH | - | - |
| 9 | 40.1 CH | 40.8 CH | 1.80 m | 1.83 m | 40.0 CH | 40.0 CH | 1.82 m | 1.82 m |
| 10 | 37.4 CH | 37.4 CH | 1.94 m | 1.94 m | 38.2 CH | 38.2 CH | 1.94 m | 1.94 m |
| 11 | 196.5 C | 195.7 C | - | - | 199.9 C | 197.5 C | - | - |
| 12 | 22.3 CH3 | 22.3 CH3 | 0.99 d (6.7) | 1.01 d (6.7) | 22.6 CH3 | 22.6 CH3 | 1.00 d (6.7) | 1.00 d (6.7) |
| 13 | 22.3 CH3 | 22.3 CH3 | 1.56 s | 1.56 s | 22.9 CH3 | 22.9 CH3 | 1.55 brs | 1.55 brs |
| 14 | 134.8 C | 135.6 C | - | - | 134.6 C | 134.3 C | - | - |
| 15 | 126.1 CH | 124.8 CH | 5.40 m | 5.30 m | 124.0 CH | 124.3 CH | 5.32 m | 5.33 m |
| 16 | 13.9 CH3 | 13.9 CH3 | 1.53 s | 1.55 s | 14.1 CH3 | 14.0 CH3 | 1.53 brs | 1.53 brs |
| 17 | 18.0 CH3 | 18.9 CH3 | 1.58 s | 1.62 s | 18.8 CH3 | 18.6 CH3 | 1.57 brs | 1.61 brs |
| 18 | 20.2 CH3 | 19.7 CH3 | 1.31 s | 1.11 s | 20.4 CH3 | 20.6 CH3 | 1.14 s | 1.18 s |
| 19 | 199.6 C | 199.2 C | - | - | 2005 C | 203.7 C | - | - |
| 20 | 112.9 C | 112.9 C | - | - | 108.2 C | 107.9 C | - | - |
| 21 | 195.6 CH | 195.0 CH | 9.64 s | 9.55 s | 160.4 CH | 160.4 CH | 8.10 d (14.1) | 8.17 d (14.1) |
| 21-NH | - | - | - | - | - | - | 11.4 brs | 11.4 brs |
| 22 | 52.4 CH2 | 52.4 CH2 | 3.55 m | 3.55 m | ||||
| 23 | 61.12 CH2 | 61.12 CH2 | 3.82 m | 3.82 m | ||||
major isomer
minor isomer
Compound 4 was isolated as pale yellow solid. The molecular formula was determined as C23H33NO3 based on the analysis of HRESIMS and NMR data (Table 2). The NMR spectra of 4 also showed two sets of signals, indicating that it was also a mixture of two tautomers. The NMR spectra of 4 were very similar to those of 3, except for the absence of the aldehyde group. Instead, a doublet methine proton at δ 8.10 ppm (H-21) was observed, with a corresponding carbon at δ 160.4 ppm (C-21). This finding indicated the presence of a 2-(aminomethylene)cyclohexane-1,3-dione moiety in 4 instead of the 2-formycyclohexane-1,3-dione moiety in 3. HMBC correlations from H-21 to C-11, C-19, from H-1 to C-11, and from H-18 to C-8, C-7, C-9, C-19 confirmed this substructure (Figure 1). Additionally, COSY correlations linking H-2½1-NH/H-22/H-23 indicated a side chain. The chemical shifts of C-22 (δ 52.4 ppm) and C-23 (δ 61.2 ppm) were consistent with an amine functional group at C-22 and a terminal free alcohol functional group at C-23, respectively.
The calculated and measured ECD spectra of 3 and 4 matched relatively well, leading to the same absolute configurational assignment as for compound 1.
To determine the biochemical origin of 1, feeding with U-13C-glucose was used. This method was selected because of the slow growth and production rate. After a two-month fermentation with U-13C-glucose, 13C-enriched 1 was purified and analyzed by 1H, 13C, and ADEQUATE NMR spectroscopy. The 1H spectrum of 13C-1 showed that all signals, with the exceptions of H-22, H-23, and H-24, were robustly coupled to 13C. Using ADEQUATE, we determined that carbons C-1 through C-21 originated in polyketide metabolism (Scheme S1). Units arising from acetate showed clear 13C-13C coupling, with the exception of C-8 to C19, which could not be observed in the ADEQUATE because of a lack of attached protons. Because the type of methyl group observed in onydecalins is known to originate from S-adenosylmethionine in fungal polyketides,11 the lack of ADEQUATE coupling and the strong 1H-13C coupling in the 1H spectrum for C-12, C-13, and C-17 further support the polyketide origin of 1. Incorporation of 13C into carbons C-1 through C-21 was estimated as 37% on the basis of the integration of H-7, which provided labeled and unlabeled signals without overlap in the 1H NMR spectrum. Overall, this result showed that the decalin core of the compound, and two carbons in the pyridine ring, originated from relatively standard fungal polyketide metabolism.
By contrast, no 13C incorporation was seen for C-22, C-23, or C-24. Not only did we fail to observe 1H-13C coupling for those signals in the 1H spectrum, but also no ADEQUATE coupling was observed for C-22, C-23, or C-24. Although unlabeled 1 provided poor 13C NMR spectra, the 13C NMR spectrum of 13C-1 was clear except for C-22, C-23, and C-24, where no signals were visible even after 4000 scans. This result shows that, the nitrogen-bearing unit in 1 is not made from glucose metabolism, but instead arises from an unknown component in the complex medium, ISP2 (such as an amino acid from malt extract or yeast extract). Therefore, the pyridine moiety is synthesized de novo following a polyketide synthase step. This result is unique in fungal polyketide metabolism, to the best of our knowledge.
3-substituted pyridines and related derivatives are found in many natural products.12–13 In known cases of polyketide biosynthesis, nicotinate is incorporated as a starter, with the carboxylate of nicotine incorporated into the chain.14–15 By contrast, in 1 the feeding study demonstrates that the carbonyl originates in the polyketide chain, and the labeling pattern is inconsistent with nicotinate biosynthesis. Pyridones are also common substituents of fungal polyketides, but result from a biochemical mechanism that does not pertain to 1.16–17 In addition, compounds 2-4 bear some resemblance to the known natural and rearrangement products in the betaenone series, the biosynthesis of which has recently been elucidated.19 On that basis, we have proposed a hypothetical biogenesis relating compounds 1-4 (Scheme S1).
Activation and inhibition of TRP ion channels was tested using crude organic extract fractions of the A. pyridodomos cultures, leading to the isolation of 1 as a modestly potent human TRPV1 and human TRPV4 inhibitor of capsaicin (TRPV1)- and GSK1016790A (TRPV4)-induced calcium flux in BEAS-2B (TRPV1) and HEK-293 (TRPV4) cells overexpressing these ion channels. IC50 values of ~81.6 and 45.9 μM were observed, respectively (Table S2 and Figure S36). Compounds 1-4 were further tested as agonists and antagonists of human TRPA1, M8, V1, V3, and V4 (Table S2). Using icilin as an agonist, it was found that 2 and 4 were modestly potent inhibitors of the human TRPM8 channel, exhibiting IC50 values of 18.5 and 61.6 μM respectively (Table S2).
TRP ion channels are integral in sensory physiology. They play central roles in human pathologies and agonists and antagonists have been shown to have therapeutic potential, particularly TRPV1 and TRPM8 in pain.20–22 TRPV4 has also been associated with numerous pathologies, including glaucoma, where TRPV4 inhibition has been shown to prevent ocular damage caused by elevated intraocular pressure.23 Results on TRPM8 inhibition by 2 are encouraging as the potency is comparable to existing agonists suggesting potential uses for this novel compound, or further optimized variants of 2 in TRPM8 research and/or therapeutics. TRP channel proteins are also widely distributed in the fungal kingdom.24
Compounds 3 and 4 arrested the growth of H. capsulatum at MICs of 2 µg/mL and 16 µg/mL respectively (Table 3). Compounds 1 and 2 did not alter the growth of H. capsulatum at the highest concentration (64 µg/mL) tested (Figure S37). None of the compounds restricted the growth of C. albicans or A. fumigatus, while itraconazole and amphotericin B showed broad inhibition of the growth of fungi tested, with MICs of 0.001 µg/mL and 0.5 µg/mL against H. capsulatum, respectively. Thus, compound 3 in particular shows some potency and has a different spectrum of action than other compounds that kill H. capsulatum but broadly target fungi.
Table 3.
Antifungal activity of compounds 1–4, itraconazole and amphotericin B against H. capsulatum
| Compound | MIC (µg/mL) |
|---|---|
| 1 | > 64 |
| 2 | > 64 |
| 3 | 2 |
| 4 | 16 |
| Amphotericin B | 0.5 |
| Itraconazole | 0.001 |
EXPERIMENTAL SECTION
General Experimental Procedures
Unless stated otherwise, all reagents and solvents were purchased from commercial suppliers. Yields were calculated by HPLC chromatography or 1H NMR spectroscopy. Circular dichroism spectra were obtained on a Jasco J720A spectropolarograph. NMR data were collected using either a Varian INOVA 500 (1H 500 MHz, 13C 126 MHz) NMR spectrometer with a 3 mm Nalorac MDBG probe or a Varian INOVA 600 (1H 600 MHz, 13C 150 MHz) NMR spectrometer equipped with a 5 mm 1H[13C,15N] triple resonance cold probe with a z-axis gradient, utilizing residual solvent signals for referencing. High-resolution mass spectra (HRMS) were obtained using a Bruker (Billerica, MA) APEXII FTICR mass spectrometer equipped with an actively shielded 9.4 T superconducting magnet (Magnex Scientific Ltd., UK), an external Bruker APOLLO ESI source, and a Synrad 50W CO2 CW laser. Supelco Discover HS (4.6 × 150 mm) and semipreparative (10 × 150 mm) C18 (5 μm) columns were used for analytical and semipreparative HPLC, respectively, as conducted on a Hitachi Elite Lachrom System equipped with a Diode Array L-2455 detector.
Fermentation and Extraction
A. pyridodomos was cultured at 30 °C with 4×4 L KIMAX glass flasks (NO.26500) each containing ISP2 medium (2 L; 0.4% yeast extract, 1% malt extract, 0.4% glucose, 2% NaCl) in a shaker at a speed of 200 rpm. After 60 days, the broth was centrifuged and the supernatant was extracted with HP-20 resin for 4 h. The resin was filtered through cheesecloth and washed with H2O to remove salts. The filtered resin was eluted with MeOH. The mycelium was then extracted by acetone 3 times. The acetone and MeOH-filtered resin extracts were combined to yield extract A (350 mg). Subsequently, the strain was allowed to grow statically at room temperature (~21 °C) with 4 × 4 L KIMAX glass flasks (NO.26500) containing ISP2. After 365 days (September 18, 2013 to September 18, 2014), different extraction method was used for the static culture. EtOAc (200 mL) was added to each of the flasks to sterilize them. Then, the mycelium was filtered through cheesecloth and extracted by acetone 3 times to yield fraction B (600 mg). The broth was extracted with EtOAc 3 times to yield fraction C (350 mg).
Purification
Extract A was separated into 7 fractions (A-Fr1-A-Fr8) on a C18 column using step-gradient elution of MeOH in H2O (20%, 40%, 60%, 70%, 80%, 90%, and 100%). Fraction A-Fr6 was further subjected to a C18 HPLC (75% CH3CN in H2O isocratic) to yield compound 1 (4.1 mg) and a subfraction A-Fr6–3, which was further purified by C18 HPLC (65% CH3CN in H2O isocratic) to yield compound 2 (3.2 mg). Extract B was fractionated with the same method. Fraction B-Fr7 was further purified by C18 HPLC (90% CH3CN in H2O with 0.1% TFA) to yield compound 3 (3.2 mg). Fraction B-Fr6 was further fractionated by C18 HPLC (65% CH3CN in H2O), one of the sub-fractions B-Fr6–7 was purified by fractionated by C18 HPLC (60% CH3CN in H2O) to yield compound 4 (3 mg). The 60-day fermentation was repeated on 4 separate occasions, yielding 2 4/4 times and 1 ¾ times, indicating good reproducibility. Due to the long duration, the one-year fermentation was performed only a single time.
Onydecalin A (1):
colorless solid; UV (MeOH) λmax 263 nm; 1H and 13C NMR (see Table 1); HRFTMS m/z 382.2382 [M+H]+ (calcd for C24H31NO3, 382.2377).
Onydecalin B (2):
colorless solid; UV (MeOH) λmax 210 nm; 1H and 13C NMR (see Table 1); HRFTMS m/z 349.2379 [M+H]+ (calcd for C21H32NO4, 349.2373).
Onydecalin C (3):
colorless solid; UV (MeOH) λmax 234, 281 nm; 1H and 13C NMR (see Table 2); HRESIMS m/z 329.2111 [M+H]+ (calcd for C21H28O3, 329.2111).
Onydecalin D (4):
colorless solid; UV (MeOH) λmax 256, 306 nm; 1H and 13C NMR (see Table 2); HRESIMS m/z 372.2534 [M+H]+ (calcd for C23H33NO3, 372.2533).
U-13C-glucose Feeding
Mycelia were inoculated into ISP2 medium (150 mL) in a 500 mL flask. After 28 days culture at 30 °C, U-13C-glucose (500 mg) was added in the flask. The culture was continued for another 25 days. Acetone (150 mL) was added, and the resulting culture broth was stirred overnight. Mycelia were removed by filtration through paper. The broth was dried in vacuo to remove acetone, and the resulting aqueous residue was extracted with ethyl acetate (150 mL) three times. The combined organic layers were treated as described above to yield 13C-enriched 1.
ECD Calculation
Conformational searches were performed by the Spartan 14 package using MMFF molecular mechanics force field. The geometries with Boltzmann distribution >5% were optimized with DFT calculations at the B3LYP/6–31G(d,p) level using the Gaussian09 program. Calculations of ECD spectra were performed at the TDDFT level of theory using CAM-B3LYP/SVP. Theoretical ECD spectra were obtained as weighted averages of Boltzmann populations using the program SpecDis.
TRP Channel Activity Screening
All cells were maintained using standard cell culture conditions. HEK-293 cells (ATCC, Manassas, VA) engineered to stably overexpress human TRPA1, TRPM8, TRPV3, and TRPV4.25 were grown in DMEM:F12 media supplemented with 5% FBS and 300 μg/mL Geneticin®. Prior to experiments, the cells were plated in 96-well plates pre-coated with 1% gelatin and grown to confluence. Cells were loaded with the calcium indicator Fluo 4-AM, using the Fluo-4 Direct assay kit (Invitrogen), diluted 2X with LHC-9 media for 60 min at 37 °C in the dark. BEAS-2B cells (ATCC) engineered to stably over-express human TRPV126 were also used, but loading with Fluo-4 was performed at room temperature; these cells were grown in LHC-9 media in plates pre-coated with LHC-basal media. For both HEK-293 and BEAS-2B TRP-overexpressing cells, thirty minutes prior to activity analysis, the loading solution was replaced with LHC-9 media containing 1 mM water soluble probenecid and 0.75 mM trypan red (ATT Bioquest); for inhibition assays the test compounds were diluted in this solution and applied to the cells at this time. For agonist assays the, test compounds were prepared as a 3X concentrate in LHC-9 medium and arrayed in a separate 96-well plate. Treatment-induced changes in cellular fluorescence were quantified using a NOVOStar fluorescence plate reader (BMG Labtech; Offenberg, Germany) using the plate to plate transfer feature to deliver the test compounds and/or prototypical receptor agonists. Data were normalized by subtracting the negative control response (LHC-9 only) and then to the maximum change in fluorescence elicited by a prototypical agonist for each TRP receptor. The agonists used were: TRPA1 (2,4-ditert butylphenol; 250 μM), TRPM8 (icilin; 50 μM), TRPV1 (nonivamide; 5 μM), TRPV3 (carvacrol; 300 μM), and TRPV4 (GSK1016790A; 0.03 μM). IC50 values were calculated from ≥8-point concentration-response curves using an n=2–3 at each concentration, and fit using the dose response-inhibition log(inhibitor) vs. normalized response equation in GraphPad 7.0 Software (GraphPad Software, La Jolla, CA).
Antifungal Activity Assays
Fungal strains Histoplasma capsulatum G217B strain was grown in Brain-Heart infusion medium for 3 days at 37 °C with 5%CO2 in the yeast form and Candida albicans strain SC5314 were grown overnight in YPD media at 30 °C in the yeast form. Cell density was measured by direct counts using hemocytometer and adjusted to 1 X 104 cells/mL in RPMI medium for H. capsulatum and in YPD for C. albicans. In addition, dry spores of Aspergillus fumigatus Af293 strain were resuspended in RPMI medium supplemented with 0.1% Tween-80 to avoid clumping of the spores. Spore density was enumarated by direct counts under hemocytometer and adjusted to 1 X 104 spores/mL in RPMI medium.
For antifungal activity assays with xompounds 1–4, each compound was dissolved in DMSO at the concentration of 10 mg/mL. The stocks were serially diluted in RPMI (H. capsulatum and A. fumigatus) or YPD (for C. albicans) further to obtain a series of concentrations including 128 µg/mL, 64 µg/mL, 32 µg/mL, 16 µg/mL, 8 µg/mL, 4 µg/mL, 2 µg/mL, 1 µg/mL, 0.5 µg/mL, and 0.25 µg/mL. Each compound was tested separately against each fungal species. Briefly, 100 µL of a fungal culture at 2X cell density (2 X 104 cells or spores/ml) were dispensed in individual wells of a 96-well plate containing 100 µL of a xompound 1–4 solution (128 to 0.25 ug/ml). Thus, the final fungal culture density was 1 X 104 cells or spores/ml and the final compound concentrations were 64 µg/mL, 32 µg/mL, 16 µg/mL, 8 µg/mL, 4 µg/mL, 2 µg/mL, 1 µg/mL, 0.5 µg/mL, 0.25 µg/mL and 0.125 µg/mL Control wells with no DMSO and no compound (DMSO only) were also prepared similarly. The plates containing C. albicans were incubated at 30 oC for 48 hours, A. fumigatus were incubated at 37 oC for 48 hours, and H. capsulatum were incubated at 37 oC with 5% CO2 for 10–14 days. MIC testing of itraconazole and amphotericin B against H. capsulatum was carried out similarly: Briefly, itraconazole stock was prepared in DMSO at a concentration of 1.25 mg/mL, and amphotericin B stock was prepared in dH2O at a concentration of 250 μg/mL. These stocks were used to generate two-fold dilution series for itraconazole (16 µg/mL to 0.002 ng/mL) and amphotericin B (16 µg/mL to 0.008 µg/mL), and co-incubated with H. capsulatum (1 X 104 cells/ml) at 37oC with 5%CO2 for 10–14 days. Control wells with no vehicle, dH2O and DMSO were prepared and incubated similarly. Fungal growth was monitored each day visually and microscopically. Experiments were repeated at least three independent times.
Supplementary Material
ACKNOWLEDGMENT
This work was funded by NIH R01GM107557 and R35GM122521 to EWS and CR; and NIH R00AI112691 and R01AI137418 to SB. Computer time was graciously provided by the University of Utah Center for High Performance Computing.
REFERENCES
- 1.Brakhage AA; Schroeckh V, Fungal secondary metabolites - strategies to activate silent gene clusters. Fungal Genet Biol 2011, 48 (1), 15–22. [DOI] [PubMed] [Google Scholar]
- 2.Hawksworth DL; Lucking R, Fungal Diversity Revisited: 2.2 to 3.8 Million Species. Microbiol Spectr 2017, 5 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin Z; Koch M; Abdel Aziz MH; Galindo-Murillo R; Tianero MD; Cheatham TE; Barrows LR; Reilly CA; Schmidt EW, Oxazinin A, a pseudodimeric natural product of mixed biosynthetic origin from a filamentous fungus. Org Lett 2014, 16 (18), 4774–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dukik K; Munoz JF; Jiang Y; Feng P; Sigler L; Stielow JB; Freeke J; Jamalian A; Gerrits van den Ende B; McEwen JG; Clay OK; Schwartz IS; Govender NP; Maphanga TG; Cuomo CA; Moreno LF; Kenyon C; Borman AM; de Hoog S, Novel taxa of thermally dimorphic systemic pathogens in the Ajellomycetaceae (Onygenales). Mycoses 2017, 60 (5), 296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin WB; Chooi YH; Smith AR; Cacho RA; Hu Y; White TC; Tang Y, Discovery of cryptic polyketide metabolites from dermatophytes using heterologous expression in Aspergillus nidulans. ACS Synth Biol 2013, 2 (11), 629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halgren TA, Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comp. Chem 1996, 17, 490–519. [Google Scholar]
- 7.Bruhn T; Schaumloffel A; Hemberger Y; Bringmann G, SpecDis: quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25 (4), 243–9. [DOI] [PubMed] [Google Scholar]
- 8.Li G; Kusari S; Golz C; Laatsch H; Strohmann C; Spiteller M, Epigenetic Modulation of Endophytic Eupenicillium sp. LG41 by a Histone Deacetylase Inhibitor for Production of Decalin-Containing Compounds. J Nat Prod 2017, 80 (4), 983–988. [DOI] [PubMed] [Google Scholar]
- 9.Ichihara A; Oikawa H; Hayashi K; Sakamura S; Furusaki A; Matsumoto T, Structures of betaenones A and B, novel phytotoxins from Phoma betae Fr. J. Am. Chem. Soc 1983, 105, 2907–2908. [Google Scholar]
- 10.Ichihara A; Oikawa H; Hashimoto M; Sakamura S; Haraguchi T; Nagano H, A phytotoxin betaenone C, and its related metabolites of Phoma betae Fr. Agric. Biol. Chem 1983, 47, 2965–2967. [Google Scholar]
- 11.Chooi YH; Tang Y, Navigating the fungal polyketide chemical space: from genes to molecules. J Org Chem 2012, 77 (22), 9933–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li C; Gloer JB; Wicklow DT; Dowd PF, Antiinsectan decaturin and oxalicine analogues from Penicillium thiersii. J Nat Prod 2005, 68 (3), 319–22. [DOI] [PubMed] [Google Scholar]
- 13.Sepcic K, Bioactive alkylpyridinium compounds from marine sponges. J Toxicol-Toxin Rev 2000, 19 (2), 139–160. [Google Scholar]
- 14.Cutignano A; Tramice A; De Caro S; Villani G; Cimino G; Fontana A, Biogenesis of 3-alkylpyridine alkaloids in the marine mollusc Haminoea orbignyana. Angew Chem Int Ed Engl 2003, 42 (23), 2633–6. [DOI] [PubMed] [Google Scholar]
- 15.Fukuda T; Miller ED; Clark BR; Alnauman A; Murphy CD; Jensen PR; Fenical W, Structures and biosynthesis of the pyridinopyrones, polyenepyrones from a marine-derived Streptomyces species. J Nat Prod 2011, 74 (8), 1773–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halo LM; Heneghan MN; Yakasai AA; Song Z; Williams K; Bailey AM; Cox RJ; Lazarus CM; Simpson TJ, Late stage oxidations during the biosynthesis of the 2-pyridone tenellin in the entomopathogenic fungus Beauveria bassiana. J Am Chem Soc 2008, 130 (52), 17988–96. [DOI] [PubMed] [Google Scholar]
- 17.Eley KL; Halo LM; Song Z; Powles H; Cox RJ; Bailey AM; Lazarus CM; Simpson TJ, Biosynthesis of the 2-pyridone tenellin in the insect pathogenic fungus Beauveria bassiana. Chembiochem 2007, 8 (3), 289–97. [DOI] [PubMed] [Google Scholar]
- 18.Li G; Kusari S; Lamshoft M; Schuffler A; Laatsch H; Spiteller M, Antibacterial secondary metabolites from an endophytic fungus, Eupenicillium sp. LG41. J Nat Prod 2014, 77 (11), 2335–41. [DOI] [PubMed] [Google Scholar]
- 19.Ugai T; Minami A; Fujii R; Tanaka M; Oguri H; Gomi K; Oikawa H, Heterologous expression of highly reducing polyketide synthase involved in betaenone biosynthesis. Chem Commun (Camb) 2015, 51 (10), 1878–81. [DOI] [PubMed] [Google Scholar]
- 20.Julius D, TRP channels and pain. Annu Rev Cell Dev Biol 2013, 29, 355–84. [DOI] [PubMed] [Google Scholar]
- 21.Sousa-Valente J; Andreou AP; Urban L; Nagy I, Transient receptor potential ion channels in primary sensory neurons as targets for novel analgesics. Br J Pharmacol 2014, 171 (10), 2508–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perez de Vega MJ; Gomez-Monterrey I; Ferrer-Montiel A; Gonzalez-Muniz R, Transient Receptor Potential Melastatin 8 Channel (TRPM8) Modulation: Cool Entryway for Treating Pain and Cancer. J Med Chem 2016, 59 (22), 10006–10029. [DOI] [PubMed] [Google Scholar]
- 23.Ryskamp DA; Frye AM; Phuong TT; Yarishkin O; Jo AO; Xu Y; Lakk M; Iuso A; Redmon SN; Ambati B; Hageman G; Prestwich GD; Torrejon KY; Krizaj D, TRPV4 regulates calcium homeostasis, cytoskeletal remodeling, conventional outflow and intraocular pressure in the mammalian eye. Sci Rep 2016, 6, 30583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saimi Y; Zhou X; Loukin SH; Haynes WJ; Kung C, Microbial TRP channels and their mechanosensitivity. Curr Topics Membranes 2007, 58, 311–327. [Google Scholar]
- 25.Deering-Rice CE; Romero EG; Shapiro D; Hughen RW; Light AR; Yost GS; Veranth JM; Reilly CA, Electrophilic components of diesel exhaust particles (DEP) activate transient receptor potential ankyrin-1 (TRPA1): a probable mechanism of acute pulmonary toxicity for DEP. Chem Res Toxicol 2011, 24 (6), 950–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reilly CA; Taylor JL; Lanza DL; Carr BA; Crouch DJ; Yost GS, Capsaicinoids cause inflammation and epithelial cell death through activation of vanilloid receptors. Toxicol Sci 2003, 73 (1), 170–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.