

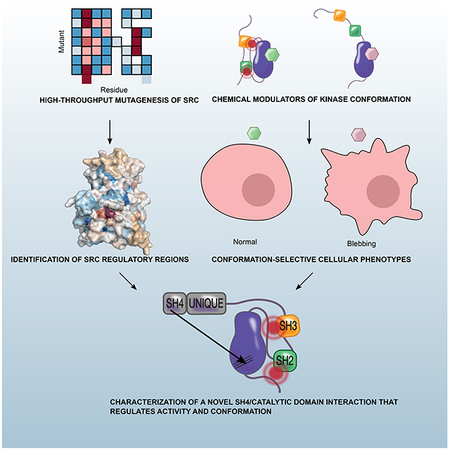
Summary
Multiple layers of regulation modulate the activity and localization of protein kinases. However, many details of kinase regulation remain incompletely understood. Here, we apply saturation mutagenesis and a chemical genetic method for allosterically modulating kinase global conformation to Src kinase, providing insight into known regulatory mechanisms and revealing a previously undiscovered interaction between Src’s SH4 and catalytic domains. Abrogation of this interaction increased phosphotransferase activity, promoted membrane association and provoked phosphotransferase-independent alterations in cell morphology. Thus, Src’s SH4 domain serves as an intramolecular regulator coupling catalytic activity, global conformation and localization, and mediating a phosphotransferase-independent function. Sequence conservation suggests that the SH4 domain regulatory interaction exists in other Src-family kinases. Our integrated approach’s ability to reveal a regulatory mechanism in one of the best-studied kinases suggests that it could be applied broadly to provide insight into kinase structure, regulation and function.
Graphical Abstract
eTOC
Ahler et al. combine saturation mutagenesis with chemical genetics to interrogate the regulatory mechanisms of Src kinase. They identify an intramolecular interaction between Src’s SH4 and catalytic domains that regulates kinase activity, membrane localization, and overall conformation of the protein.
Introduction
Eukaryotic protein kinases (EPKs) comprise a family of ~540 enzymes in humans that are integral mediators of signal transduction (Taylor et al., 2012). Dysregulation of EPK activity is associated with numerous human diseases (Lahiry et al., 2010). Regulation of EPK phosphotransferase activity is varied and complex, encompassing catalytic domain (CD)-intrinsic mechanisms contained completely within the CD (Figure 1A), interdomain mechanisms between the CD and other regions of the kinase, and extrinsic mechanisms that provide context-dependent control. Interdomain regulation is pervasive: half of all EPKs contain at least one other protein domain (Manning et al., 2002). Although these domains often directly regulate CD activity, they can also have important phosphotransferase-independent functions (Kung and Jura, 2016).
Figure 1. Dissection of intramolecular kinase regulation using a multidisciplinary approach.

A. Structural features of Src’s catalytic domain (CD) (PDB: 3DQW), showing the C-spine (Catalytic, green), R-spine (regulatory, blue), helix αC (yellow), and the activation (A, pink)-loop. B. Linear schematic of Src kinase (Unq = Unique domain). C. Src kinase in the closed, autoinhibited (left, PDB: 2SRC) and an open (right, PDB: 1Y57) global conformation. The SH3 domain is shown in orange, the SH2 domain in green and the CD in purple. D. Deep mutational scanning for the simultaneous measurement of the activity of nearly all possible single mutants of a kinase. E. Cysteine Installation for Modulating Allostery and Targeted Inhibition of Kinases (CystIMATIK), a method for selectively modulating intramolecular regulatory domain interactions and global conformation of a drug-sensitized kinase with conformation-selective, ATP-competitive probes.
One of the best-characterized families of multi-domain EPKs is the Src-family of kinases (SFKs). In addition to a C-terminal CD, SFKs contain a membrane-interacting SH4 domain, a Unique domain, and regulatory SH2 and SH3 domains (Figure 1B). Decades of research have elucidated the structure, mechanisms of regulation and biological functions of SFKs, with the roles of the SH2 and SH3 domains in regulating phosphotransferase activity best understood (Figure 1C) (Boggon and Eck, 2004). Despite this knowledge about SFKs in general and Src in particular, questions regarding regulation and phosphotransferase-independent functions remain. How do SFK domains coordinate to regulate CD activity? Do the less well understood SH4 and Unique domains play a role in directly regulating phosphotransferase activity? What are the phosphotransferase-independent roles of SFK regulatory domains? These questions are challenging to investigate—both for Src and for other multi-domain kinases—because of the dearth of tools for manipulating conformation, probing the importance of specific residues in regulating phosphotransferase activity and investigating phosphotransferase-independent functions.
To answer these questions, we combined CystIMATIK, a chemical genetic method for controlling kinase conformation, with comprehensive mutagenesis of Src’s CD (Figures 1D, 1E). This integrated approach revealed details of well-studied Src domains and uncovered an interaction between Src’s SH4 domain and a pocket in the C-lobe of Src’s CD with no previously known function. Abrogation of this SH4 domain/CD interaction increased phosphotransferase activity, disengaged the SH2 and SH3 regulatory domains, and increased association with membranes. Moreover, exposure of Src’s SH4 domain mediated phosphotransferase-independent alterations in cell morphology. Thus, Src’s SH4 domain serves as an intramolecular regulator coupling catalytic activity, global conformation and localization, and also mediating a phosphotransferase-independent cellular function. Analysis of SFK residue conservation in the light of comprehensive mutagenesis data, combined with additional biochemical experiments, strongly suggests that the SH4 regulatory interaction is conserved amongst SFKs.
Results
Parallel measurement of the activity of 3,506 Src mutants
To identify residues important for regulating Src’s activity, we used deep mutational scanning (DMS) (Fowler and Fields, 2014; Fowler et al., 2010), adapting an assay based on S. cerevisiae growth (Figure 2A) (Kritzer et al., 2018). Yeast provides a eukaryotic cellular environment to probe intrinsic regulation in the absence of extrinsic regulatory factors, with growth rates correlating to overall levels of cellular phospho-tyrosine (Figures 2B, 2C). We mutagenized the CD of full-length Src (Srcmyr, refer to Figure S1A and Table S1 for all constructs), transformed this library into yeast, collected samples during outgrowth and deeply sequenced each sample to quantify the frequency of all variants at every time point (Figures 2A, S1B). Then, we calculated activity scores for all 3,506 single amino acid variants in our library (Figures 2D, S1C, Table S2) (Rubin et al., 2017); classified each variant as either gain-of-function, loss-of-function, or WT-like (Figure 2E); and organized our large-scale Src variant activity scores into a sequence-function map covering ~70% of possible CD single mutants (Figure 2F). The map revealed expected patterns (Figures S1C, S1D) and identified 46 essential residues where >90% of mutations resulted in loss-of-function (Table S3). Finally, individually-determined growth rates were negatively correlated with large-scale variant activity scores (Figure 2G), and the activity scores accurately reflected yeast and human cellular phospho-tyrosine levels (Figures 2H, 2I, S1E, S1F).
Figure 2. Multiplex measurement of the activity of 3,506 single amino acid Src variants.

A. Schematic of a yeast growth-based deep mutational scan (DMS) of the Src catalytic domain. B. Individually-assessed growth curves for yeast expressing Srcmyr WT, K298M, T341I, or a control vector (n=3). C. Src and phospho-tyrosine (pTyr) immunoblots of yeast expressing Srcmyr variants for 24 hr. D. Scatterplot showing activity score correlations between two independent transformations of the Srcmyr variant library (Pearson’s R = 0.91). E. Activity scores for variants classified as gain-of-function (n=403, green), WT-like (n=1288, orange), or loss-of-function (n=1681, blue). F. Position-averaged activity scores mapped onto the Src catalytic domain (PDB: 3DQW). Nonsense mutants were excluded from the average score. Sequence-activity map of Src catalytic domain. Black dots in the map indicate the WT amino acid, gray tiles indicate missing data. Bar graph indicates relative evolutionary conservation at each position as determined by Kullback-Leibler entropy. Secondary structure and functional motif annotations were obtained from the ProKinO database. G. Dot plot of individually assessed growth rates compared to activity scores for a panel of Srcmyr variants (n=3; Pearson’s R = −.97). Growth rates for WT, K298M, and T341I from Figure 2B are shown for comparison. H. Src and phospho-tyrosine immunoblots of yeast expressing Srcmyr variants. I. Correlation between yeast DMS-derived activity scores and the ratio of phospho-tyrosine/Src levels in HEK293Ts for a panel of Srcmyr variants (n=3; Pearson’s R = 0.97; Figures S1E, S1F). Points represent individual measurements and the horizontal lines indicate the mean of all measurements. See also Figure S1, Tables S2 and S3.
Residue-scale mapping of Src regulatory interactions
Our large-scale mutagenesis data provide the first comprehensive functional view of Src intramolecular regulation at residue-scale resolution. We reasoned that residues in the CD that participate in autoinhibitory interactions would contain multiple gain-of-function mutations. To identify surfaces important for autoinhibition, we clustered the 27 residues in Src’s CD that had at least five gain-of-function mutations based on spatial proximity. Seven clusters of residues emerged, distributed across both lobes of the CD (Figure 3A).
Figure 3. Large-scale mutagenesis data reveal Src’s regulatory interfaces.

A. Hierarchical clustering of Src residues with at least five gain-of-function mutations based on spatial coordinates of the atomic centroids of each sidechain (right panel) projected onto the Src structure (left panel, PDB: 2SRC). B. Structural detail (left panels) and activity scores (right panels) for every variant at each residue comprising clusters that overlap with the SH2-CD (cluster 4, top), SH3/Linker-CD (cluster 7, middle) and αF pocket (cluster 2, bottom) interfaces (PDB: 2SRC). In the left panels, CD residues that are not part of a cluster are shown in white, SH2-linker residues in tan, SH2 residues in green, and SH3 residues in orange. C. Phosphotransferase activity of purified SrcFL WT, T293D, E381T, or I444K (n=3-6). D. Schematic of the SH3 pulldown assay. To detect global conformation, Src is incubated with an immobilized SH3 domain ligand. Closed, SH3-engaged Src is unable to bind to the resin, whereas open, SH3-disengaged Src binds. After washing, retained Src is eluted and quantified by western blot or in-gel fluorescence. E. Percent retained Src in the SH3 pulldown assay with purified SrcFL WT, T293D, or D368K (n=3-6). F. Sequence alignment of Src-family kinase αF pocket residues. G. Representative micrographs (left) and percent bleb quantification (right) for SYFs expressing either SrcGFP WT or SrcGFP E381T. Scale bars = 10 μm. Each point represents a replicate transfection with multiple cells imaged and scored in a double-blind fashion. Horizontal lines indicate the mean of all replicates. See Table S5 for the total number of replicates and cells analyzed. *p <0.05. See also Figure S2.
Two gain-of-function clusters, 4 and 7, coincided with autoinhibitory interactions between Src’s CD and its SH2 and SH3 domains. Autoinhibited Src adopts a closed global conformation, with the SH2 and SH3 domains forming an extensive interaction interface with the CD (Figure 1C, (Xu et al., 1997)). While the importance of the interaction between Src’s phosphorylated C-terminal tail and the SH2 domain in stabilizing the closed, autoinhibited global conformation is well understood (Young et al., 2001), the contributions of specific residues lining the SH2 domain/CD interface have not been determined. Thus, we mapped gain-of-function cluster 4 onto the CD’s SH2 domain interface (SH2-CD; Figure 3B, top). Every substitution at position D368 was activating, suggesting that D368’s interaction with R159 (Figure S2A) in the SH2 domain is important for stabilizing the closed, autoinhibited conformation. We purified a non-phosphorylated, full-length Src construct lacking an N-terminal myristoyl group (SrcFL), measured phosphotransferase activity (Figures 3C, S2B) and assessed intramolecular regulatory domain engagement using an immobilized SH3 domain ligand pulldown (Figure 3D). Consistent with D368K leading to increased activity by disrupting Src’s intramolecular autoinhibition, SrcFL D368K’s association with an immobilized SH3 ligand was increased −10-fold relative to SrcFL WT (Figures 3E, S2C). Therefore, the D368–K159 salt bridge is critical for stabilizing the closed, autoinhibited global conformation of Src, with other interface interactions making minimal contributions.
In autoinhibited Src, the SH3 domain binds to a surface composed of the CD’s N-terminal lobe and the linker connecting the CD to the SH2 domain (SH3/Linker-CD; Figure 3B, middle). Mutations that disrupt interactions between the SH3 domain, CD, and SH2-linker hyperactivate SFKs (Gonfloni et al., 1997; LaFevre-Bernt et al., 1998). We found that many of the residues lining the SH3 domain/CD/SH2-linker interface, which contains cluster 7, have gain-of-function mutations. Thus, unlike the SH2 domain/CD interface, the SH3 domain/CD/SH2-linker interface is much less tolerant of substitutions. SrcFL T293D, a gain-of-function mutant in this interface, had increased phosphotransferase activity and association with the immobilized SH3 domain ligand (Figures 3C, 3E). Our data support the canonical model of Src autoinhibition: the SH3 domain acts as a clamp that forms broadly distributed interactions with the SH2-linker and the CD, and the SH2 domain serves as a latch that stabilizes a closed global conformation (Xu et al., 1999).
Two of the largest gain-of-function clusters we identified do not overlap known intramolecular regulatory interfaces. The largest, cluster 2, is located at the intersection of the αF/αE/αI helices in the C-terminal lobe of Src’s CD and contains five of the fifteen most activating mutations (Figure 3B, bottom). This region, to the best of our knowledge, is not known to participate in regulatory interactions in any tyrosine kinase. However, surface geometry comparison analysis suggested that this region comprises an orphan binding pocket, called the “αF pocket”, for many kinases (Thompson et al., 2009). The αF pocket is highly conserved across SFKs (Figure 3F). Interestingly, two αF pocket Src mutants can transform cells (Levy et al., 1986) via an unknown mechanism (Bjorge et al., 1995; Seidel-Dugan et al., 1992).
Based on its large 1,361 Å2 surface area and the number of gain-of-function mutations, we hypothesized that the αF pocket serves as an intramolecular regulatory surface that stabilizes Src’s closed, autoinhibited global conformation. To test this hypothesis, we characterized the SrcFL αF pocket mutants E381T and I444K. In vitro, both mutants showed ~10-fold increases in kcat but little difference in Km for ATP relative to SrcFL WT (Figures 3C, S2B). Expression of Srcmyr E381T led to high levels of cellular phospho-tyrosine compared to WT in multiple mammalian cells lines (Figures 2I, S2D, S2E). Next, we examined the subcellular localization of GFP C-terminal fusions of Src in Src/Yes/Fyn (−/−/−) fibroblasts (SYFs) (Klinghoffer et al., 1999). SrcGFP E381T displayed enhanced localization to late endosomes and the perinuclear region compared to SrcGFP WT (Figures 3G, S2F). In addition, it promoted non-apoptotic membrane blebbing (Figure 3G), a phenotype consistent with localized disruption of the actin-myosin cortex (Charras, 2008). To the best of our knowledge, this blebbing phenotype has not previously been observed with any other hyperactive Src variants. Thus, we were curious whether blebbing derived from the E381T mutant’s increased phosphotransferase activity or through a phosphotransferase-independent mechanism such as exposure of an otherwise intramolecularly sequestered binding surface.
Chemical genetic control of kinase global conformation
Many protein kinases, including SFKs, possess phosphotransferase–independent cellular roles (Kung and Jura, 2016). Determining whether a phenotypic effect, like the one we observed for Src E381T, is caused by changes in phosphotransferase activity or via a phosphotransferase-independent mechanism is challenging as options for decoupling conformational state from activity are limited. Kinase inhibitors can modulate intramolecular regulatory domain interactions and global conformation by stabilizing different ATP-binding site conformations. For example, stabilization of SFKs in the helix αC-out conformation with inhibitors promotes a closed global conformation and reduces engagement of intermolecular binding partners (Krishnamurty et al., 2012; Leonard et al., 2014). Conversely, stabilization of an inactive conformation of the Asp/Phe/Gly (DFG)-motif at the base of the activation loop promotes an open global conformation (Skora et al., 2013). Thus, conformation-selective inhibitors that modulate SFK global conformation while simultaneously blocking phosphotransferase activity could be used to investigate phosphotransferase-independent phenotypic effects. However, this strategy requires a set of inhibitors selective for distinct conformations, each with specificity sufficient to ascribe observed phenotypes to modulation of the conformation of the targeted SFK.
To address this shortcoming, we developed Cysteine Installation for Modulating Allostery and Targeted Inhibition of Kinases (CystiMATiK). CystiMATiK is based on the introduction of a functionally neutral cysteine mutation into the active site of a target kinase, sensitizing it to a set of electrophile-containing CystiMATiK probes that stabilize different ATP-binding site conformations. Only the mutant kinase is sensitive to the CystiMATiK probes, which should not bind to other kinases or the WT targeted kinase. As a starting point for our probes, we used 1, a derivative of an inhibitor that only binds to the ATP-binding sites of kinases that contain both a Cys residue on the β2 strand of the N-terminal lobe and a Thr residue at the gatekeeper position (V284 and T341 in Src, respectively, (Figures 4A, 4B (Serafimova et al., 2012)). As expected, we found that WT Src and Hck, another SFK, are insensitive to 1, but that V284C variants are potently inhibited (Figures 4C, S3A).
Figure 4. Cysteine Installation for Modulating Allostery and Targeted Inhibition of Kinases (CystIMATIK).

A. Structure of the CD of Src (PDB: 2SRC) bound to AMP-PNP. Positions that are important for sensitivity to CystIMATIK probes are shown as sticks (T341 (red) and V284 (blue)). B. Sequence alignment of Src and Hck with various kinases and the C-terminal kinase domain of p90 ribosomal protein S6 kinase, CTD-RSK2. C. CystIMATIK probes 1-3. IC50 values (n=3, mean ± s.e.m), determined in the presence of 1 mM ATP, for Src3D WT, Src3D V284C, Hck3D WT, and Hck3D V284C are shown below each probe. Hck is numbered according to analogous Src residue. D. Crystal structures of the SrcCD V284C-1 (left; PDB:5SWH), SrcCD V284C-2 (middle; PDB:5TEH) and SrcCD V284C-3 (right; PDB:5SYS) complexes. Top panels: 1-3 are shown as sticks colored by atom. Bottom panels: Helix αC (yellow) and the Phe (pink) of the DFG motif are shown. C284, K298, and E313 are depicted as sticks. E. Percent retained Src in the SH3 pulldown assay for SrcFLAG V284C-expressing HEK293s treated with CystIMATIK probe 1, 2, or 3 (n=3). F. SH2 domain accessibility assay using Csk. Src3D V284C-CystIMATIK probe complexes are incubated with Csk and γ32P-ATP, and radioactive phosphate transfer to Src3D V284C is quantified. Closed, SH2-engaged Src cannot be phosphorylated by Csk whereas open, SH2-disengaged Src is efficiently phosphorylated by Csk. G. Quantification of Csk’s phosphorylation of the Src3D V284C-2 and Src3D V284C-3 complexes. H. Kinome profiling of CystIMATIK probes 1-3. Profiled kinases are represented by grey circles and interacting kinases by red circles. For interacting kinases, circle size scales with log2 SILAC ratio of DMSO control over 10 μM of each CystIMATIK probe (a mean log2 SILAC ratio >1 cut-off was applied; n=3). The heatmap shows all kinase targets for each CystIIMATIK probe. For 4E, 4G, points represent individual measurements and the horizontal lines indicate the mean of all measurements. See also Figure S3 and Table S7.
CystiMATiK probe 1 contains a substituent at the C-5 position of the pyrrolopyrimidine scaffold that should not affect the conformation of SFK ATP-binding sites, thus minimally perturbing global conformation. To generate conformation-selective versions of 1, we introduced substituents at the C-5 position to stabilize the helix αC-out (2) or DFG-out conformations (3). Like 1, probes 2 and 3 potently inhibited V284C mutants of Src and Hck but showed minimal inhibition of WT SFKs (Figure 4C). Crystal structures of 1-3 bound to SrcCD V284C confirmed that each probe stabilizes the expected ATP-binding site conformation (Figure 4D, Table 1). Thus, CystIMATIK probes that stabilize desired ATP-binding site conformations can be generated by varying the pyrrolopyrimidine scaffold’s C-5 substituent.
Table 1.
X-ray crystallography statistics for Src structures.
| 5SWH (VC 151) | 5TEH (VC 156) | 5SYS (VC 245) | |
|---|---|---|---|
| Space Group | P1 | P1 | P1 |
| Molecules | 1 | 1 | 1 |
| Cell dimensions | |||
| a | 41.82 | 41.79 | 41.81 |
| b | 64.14 | 63.59 | 63.86 |
| c | 74.91 | 76.07 | 75.63 |
| alpha | 77.94 | 76.73 | 77.87 |
| beta | 89.5 | 89.93 | 90.1 |
| gamma | 89.83 | 89.79 | 89.83 |
| Resolution | 73.2-2.5 | 74.0-3.0 | 73.9-2.8 |
| R merge | 0.186 | 0.168 | 0.176 |
| Rpim | 0.075 | 0.111 | 0.111 |
| <I/oI> | 8.5 | 6.0 | 5.7 |
| Completeness (%) | 80 (73) | 80 (74) | 87 (74) |
| Redundancy | 7.1 | 3.4 | 3.3 |
| No. Reflections | 19893 | 11499 | 15434 |
| R work/Rfree | 0.241/0.2871 | 0.3069/0.3659 | 0.2857/0.3339 |
| No. atoms | |||
| protein | 4111 | 4200 | 4128 |
| ligand/ion | 62 | 74 | 86 |
| water | 62 | 18 | 67 |
| B-factors | |||
| protein | 53.038 | 77.268 | 64.488 |
| ligand/ion | 74 | 52 | 87 |
| water | 30 | 52 | 27 |
| R.M.S. deviations | |||
| Bond lengths | 0.012 | 0.018 | 0.01 |
| Bond angles | 1.528 | 2.027 | 1.423 |
Next, we performed SH3 domain pulldown assays with CystIMATIK probe-SFK complexes to determine whether each probe promotes the expected SFK global conformation (Figures 3D, 4E) (Krishnamurty et al., 2012; Leonard et al., 2014). Helix αC-out-stabilizing inhibitor 2 led to minimal pulldown of SrcFLAG V284C, consistent with 2 stabilizing a closed global conformation. Conversely, the DFG-out-stabilizing inhibitor 3 greatly increased SH3 domain-accessible SrcFLAG V284C. 1, which minimally perturbs global conformation, yielded an intermediate level of SH3 domain accessibility. The same trend was observed with HckFLAG V284C in 1-3-treated cells (Figure S3B) and with purified Src lacking the SH4 and Unique domains (Src3D V284C) (Figure S3C). Furthermore, we found that the C-terminal tails of 2- and 3-bound Src3D V284C and Hck3D V284C demonstrated expected levels of accessibility to phosphorylation by C-terminal Src kinase (Csk; Figures 4F, 4G, S3D-F (Leonard et al., 2014)). Thus, CystIMATIK probes affect intramolecular engagement of SFK SH2 and SH3 domains and, consequently, global conformation as predicted based on the ATP-binding site conformations they stabilize.
To assess the specificity of our CystIMATIK probes, we performed comprehensive kinase profiling assays (Golkowski et al., 2017). Lysates from SILAC-labeled HEK293T cells, engineered to stably express HckFLAG V284C along with low levels of endogenous WT Hck, were incubated with 10 μM 1, 2, 3, or DMSO, and an immobilized matrix of non-selective inhibitors. Mass spectrometric determination of kinase enrichment in the presence of each CystIMATIK probe relative to DMSO allowed us to assess probe-kinase binding (Figure S3G). HckFLAG V284C was the only kinase among the ~220 enriched that was depleted in the presence of inhibitors 1 and 2. Inhibitor 3 was slightly less selective for HckFLAG V284C, with EPHA2 also showing significant depletion (Figures 4H, S3H). Therefore, CystIMATIK probes enable precise control of CD conformation in a highly selective manner, allowing us to decouple Src’s phosphotransferase activity from its global conformation.
Phenotypic effects of conformation-selective Src inhibition
We next used CystIMATIK to modulate Src’s global conformation in the absence of phosphotransferase activity. If Src E381T’s increased phosphotransferase activity drives the blebbing we observed (Figure 3G), all three CystIMATIK probes should abolish the effect. Conversely, if blebbing is a phosphotransferase-independent consequence of E381T exposing an otherwise intramolecularly sequestered binding surface, treatment with CystIMATIK probes 3 and, to a lesser extent, 1, should result in blebbing. Expression of SrcGFP V284C or SrcGFP WT in SYFs yielded a small number of blebbing cells, which was minimally affected by treatment with probe 2 (Figures 5A, S4A, S4B). However, treatment of SrcGFP V284C-expressing SYFs with probe 3 resulted in rapid (Figures 5B, S4C, Movies S1-2) and dose-dependent (Figure 5C) induction of blebbing. Blebbing appears to be a direct consequence of modulation of Src’s conformation because treatment of SYFs expressing SrcGFP WT, which is resistant to CystIMATIK probes, with 3 did not significantly increase membrane blebs (Figure S4D). Deletion of the GFP tag did not change the results (Figures S4E, S4F). To exclude artifacts arising from variability in probe cell permeability, we performed a pre-treatment competition experiment with inhibitor 2. Consistent with the ability of inhibitor 2 to rapidly permeate cells and occupy the ATP-binding site of Src, Src V284C-expressing cells pre-treated with 2 for 15 minutes prior to addition of 3 showed levels of blebbing comparable to cells treated with DMSO or 2 alone (Figures 5A, S4F). Thus, the open global conformation of Src, stabilized by 3, appears to promote blebbing in a phosphotransferase-independent fashion.
Figure 5. Conformational changes at the ATP-binding site drive phosphotransferase-independent cellular blebbing.

A. Representative micrographs (left) and percent bleb quantification (right) for SrcGFP V284C-expressing SYFs treated for 15 min with DMSO, CystIMATIK probe 1, 2, 3, or pre-treated with 2 for 15 min, followed by 3. B. Representative micrographs from time course experiments performed with SrcGFP V284C-expressing SYFs treated with CystIMATIK probe 2 or 3. Insets show membrane blebs. C. Percentage of SrcGFP V284C-expressing SYFs treated with 1, 3, or 10 μM 3 for 15 min showing blebs. Values for DMSO and 10 μM conditions were used previously in Figure 5A. D. Representative micrographs (left) and percent bleb quantification (right) for SrcGFP V284C-expressing SYFs pretreated with the Rock inhibitor GSK429286A, and then treated with DMSO or 3 for 15 minutes. E. Representative micrographs (left) and percent bleb quantification (right) for SrcGFP G2A/V284C-expressing SYFs treated with DMSO or 3 for 15 minutes. F. Structures of probes 4, 5, and 6 and IC50 values for SrcFL WT determined in the presence of 1 mM ATP (n=3, mean ± s.e.m). G. Quantification of Src immunoblots for co-sedimentation assays performed with apo SrcFLAG, SrcFLAG-4, SrcFLAG-5, or SrcFLAG-6, and lipsosomes composed of 1:1:1 phosphatidylcholine, phosphatidylserine and phosphatidylethanolamine (PC:PS:PE) or 2:1 PC and PS (2PC:PS) (Figures S4H, S4I). In all micrographs, scale bars = 10 μm. For 5A-5E, each point represents a replicate treatment with multiple cells imaged and scored in a double-blind fashion. See Table S5 for the total number of replicates and cells analyzed. Horizontal lines indicate the mean of all replicates. ***p < 0.001. See also Figure S4.
The blebbing observed in SrcGFP E381T- and SrcGFP V284C-expressing SYFs treated with 3 are similar in dynamics and scale to features that depend on Rho GTPase-mediated contractility (Charras, 2008). Indeed, pre-treating SrcGFP V284C-expressing SYFs or Srcmyr V284C-expressing HeLa cells with a selective inhibitor of the Rho effector kinase Rock prevented 3-promoted blebbing (Figures 5D, S4G). To explore whether Src membrane localization is important for blebbing, we tested whether 3 could promote blebbing in SYFs expressing SrcGFP G2A/V284C, which cannot be myristoylated at its N-terminus. This Src mutant demonstrates a decreased association with membranes because both the N-myristoyl group and the polybasic SH4 domain are required for high affinity membrane interaction (Sigal et al., 1994). Treatment of SrcGFP G2A/V284C-expressing SYFs with 3 did not produce blebbing (Figure 5E), suggesting that adoption of an open conformation alone is insufficient to induce blebbing and that high affinity membrane association is required.
Overexpression of SFK SH4 domain constructs also results in dynamic membrane blebbing through an incompletely defined mechanism (Tournaviti et al., 2007). Our discovery that 3 produces a similar phenotype suggests that Src’s membrane-interacting N-terminus can be allosterically modulated through Src’s ATP-binding site like its SH2 and SH3 domains. Thus, we performed co-sedimentation assays with purified, myristoylated WT Src (SrcFLAG), analogs of our CystIMATIK probes (inhibitors 4-6, Figure 5F), and liposomes (Figures 5G, S4H, S4I). The SrcFLAG-6 complex, stabilized in the DFG-out conformation, showed significantly increased liposome association compared to apo SrcFLAG and the SrcFLAG-4 complex, suggesting that the N-terminus is more accessible to membranes when intramolecular SH2 and SH3 domain engagement is disrupted. Furthermore, stabilization of the helix αC-out conformation with inhibitor 5 resulted in the lowest level of co-sedimentation, reinforcing a correlation between SH2/SH3 domain intramolecular engagement and N-terminus sequestration. Thus, stabilization of the DFG-out conformation, with concomitant adoption of an open global conformation, increases the accessibility of Src’s membrane-interacting N-terminus. Moreover, promoting Src’s open global conformation results in striking blebbing, which requires Src’s interactions with membranes but not its phosphotransferase activity.
Characterization of a N-terminal regulatory site in Src
Our co-sedimentation and cellular data are consistent with intramolecular sequestration of Src’s N-terminus in the closed global conformation, preventing membrane interaction. Disengagement of the SH2 and SH3 regulatory domains disrupts this sequestration and exposes the N-terminus in the open global conformation. Moreover, our large-scale mutagenesis and biochemical data support the importance of the αF pocket in regulating Src’s conformation. Thus, guided by structure-based modeling, we hypothesized that the αF pocket binds to and sequesters the N-terminus (Figures 6A, S5A). Our hypothesis predicts that αF pocket mutations should disrupt sequestration of the N-terminus, exposing it and increasing Src’s affinity for membranes. We performed co-sedimentation assays comparing membrane association of purified SrcFLAG WT and SrcFLAG E381T (Figures 6B, S5B). SrcFLAG E381T showed enhanced membrane association relative to SrcFLAG WT, implying that the E381T mutation results in a more membrane-accessible N-terminus.
Figure 6. Direct interaction of the SH4 and catalytic domain regulates Src activity and dictates cellular phenotype.

A. Proposed SH4 domain “fastener” model (left) and the Src constructs used for biochemical characterization (right). B. Quantification of Src immunoblots for co-sedimentation assays performed with SrcFLAG WT or SrcFLAG E381T, and liposomes composed of PC:PS:PE or 2PC:PS. C. Phosphotransferase activity of purified SrcFL, Src3D or SrcΔSH4 with either the WT or E381T sequence. (n=4-6). Values for SrcFL WT and E381T were used previously in Figure 3C. D. Autophosphorylation quantification of SrcFL or SrcΔSH4 with either the WT or E381T sequence at various time points after ATP addition (n=3). E. Percent retained Src in the SH3 pulldown assay with purified SrcFL or SrcΔSH4 with either the WT or E381T sequence (n=3-5). Values for SrcFL WT were used previously in Figure 3E. F. Model showing the global conformation of SrcFL K445C-5 or SrcFL K445C-6. G. Isotope-coded maleimide labeling of SrcFL K445C. An example mass spectrum of the light and heavy maleimide-labeled peptide containing K445C (αF peptide). The inset shows the location of the αF and control peptides. H. Peak intensity ratios of the maleimide-labeled αF and control peptides from the SrcFL K445C-5 or SrcFL K445C-6 complexes (n=4). Peak intensity ratios of the maleimide-labeled αF peptide from the Src3D K445C-5 or Src3D K445C-6 complexes (n=2) are also shown. I. SH4 pulldown assay schematic. Src variants are incubated with the immobilized SH4 domain (residues 1-18) of Src and the amount of retained Src is quantified after washing and elution. SrcCD variants with mutations outside (left) and within (right) the αF pocket are shown. J. Percent retained Src in the SH4 pulldown assay with purified SrcCD WT, E381T, I444K, T293D, or W285T (n=3). K. Percent retained Src in the SH4 pulldown assay with purified SrcCD WT or SrcFL WT (n=3). Points represent individual measurements and the horizontal lines indicate the mean of all measurements. See also Figure S5.
We also observed that the αF pocket mutations increase the phosphotransferase activity of full-length Src in yeast, mammalian cells, and in vitro. Moreover, deletion of the SH4 domain (Src3D or SrcΔSH4) resulted in a ~10-fold increase in phosphotransferase activity compared to SrcFL WT (Figures 6C, S5C-E). These observations suggest a model where the SH4 domain serves as a “fastener” promoting the intramolecular association of Src’s regulatory SH2 and SH3 domains through its interaction with the αF pocket, thereby decreasing phosphotransferase activity. If αF pocket mutations and SH4 domain deletions abrogate the same SH4 domain/aF pocket interaction, then combining the two should yield a minimal additional increase in phosphotransferase activity. Indeed, neither E381T nor I444K appreciably increased the phosphotransferase activity of Src3D or SrcΔSH4 (Figures 6C, S5C-E). In contrast, activating mutations located outside of the αF pocket further increased Src3D’s catalytic activity (Figure S5E). N-terminal myristoylation did not affect E381T activity, suggesting that E381T mainly disrupts interactions with the SH4 domain and not the N-terminal myristoyl group (Figure S5F). Finally, E381T increased purified SrcFL’s capacity for activation loop autophosphorylation, and, as for trans-phosphorylation, deletion of the SH4 domain did not increase autophosphorylation further (Figures 6D, S5G). Thus, mutations in the αF pocket and deletion of the SH4 domain are both activating in a variety of contexts, but combining the two does not further increase activity.
Consistent with the αF pocket/SH4 domain fastener model, the level of intramolecular SH3 domain engagement of constructs lacking an SH4 domain (SrcΔSH4 or Src3D WT) was lower than SrcFL WT in a pulldown assay (Figures 6E, S5H-J). While αF pocket mutations significantly decreased intramolecular SH3 domain engagement in SrcFL, they only minimally influenced the already increased level of SH3 domain disengagement in constructs lacking an SH4 domain (Figures 6E, S5H-J). Thus, mutations in the αF pocket appear to activate Src’s phosphotransferase activity by releasing intramolecular autoinhibition promoted by the SH4 domain fastener, although indirect disruption of the SH2/CD interface may also contribute. These results strongly suggest that the activity-modulating effects of the SH4 domain and the αF pocket operate through the same mechanism and that these two distal regions physically interact.
Next, we used chemical footprinting to determine whether the αF pocket is shielded when Src is in the closed global conformation (Figures 6F-H). We measured isotopically-coded maleimide labeling (Kahsai et al., 2014) of a purified Src construct (SrcFL K445C) containing a solvent accessible cysteine mutation. Isotopic coding allowed ratiometric, quantitative comparisons of maleimide labeling between SrcFL complexed to inhibitors 5 or 6, which promote closed or open global conformations, respectively. We found that K445C labeling was ~5-fold greater in the DFG-out, open conformation compared to the helix αC-out, closed conformation (Figure 6H). However, no change in labeling was observed at a control cysteine residue whose accessibility should not change between the closed and open conformations. Deletion of the SH4 and Unique domains abrogated global conformation-dependent differences in labeling of K445C, suggesting that the N-terminal region physically occludes the αF pocket.
We next evaluated the interaction in trans by performing pulldowns with an immobilized construct consisting of the non-myristoylated SH4 domain of Src (Figure 6I). A Src construct containing only the CD (SrcCD) exhibited efficient SH4 domain-mediated pulldown (Figures 6J, S5K). Moreover, E381T and I444K mutations abrogated pulldown in the context of SrcCD, while activating mutations located outside the αF pocket did not. SrcFL WT demonstrated less efficient pulldown than SrcCD WT, consistent with intramolecular SH4 domain engagement competing with SH4 pulldown in trans (Figure 6K). Moreover, pulldown of the SH2/SH3 domain-disengaged SrcFL V284C-3 complex was much greater than the SH2/SH3 domain-engaged SrcFL V284C-2 complex, showing that the conformation of Src’s regulatory SH2 and SH3 domains are coupled to the SH4 domain’s fastening interaction with the αF pocket (Figure S5L). Thus, a more open global conformation of full-length Src increases αF pocket accessibility and hence pulldown with an immobilized SH4 domain in trans.
Finally, the non-myristoylated SH4 domain construct used for pulldowns did not affect SrcCD autophosphorylation when added in trans, suggesting that the SH4 domain fastener does not directly influence the CD’s phosphotransferase activity (Figure S5M). However, the rate of SrcFL autophosphorylation increased in the presence of the non-myristoylated SH4 domain construct, consistent with disruption of intramolecular SH4 domain-mediated autoinhibition (Figure S5N). Autophosphorylation of I444K and E381T SrcFL were not affected by the addition of the non-myristoylated SH4 domain construct (Figure S5O), consistent with these mutations disrupting the interaction between the αF pocket and SH4 domain either in cis or in trans.
The SH4 domain regulatory interaction is conserved in other SFKs
We revealed a previously unknown, direct regulatory interaction between Src’s SH4 domain and its αF pocket and showed that the interaction regulates Src’s conformation, activity, localization and effect on cells. Src shares a common domain architecture with the seven other human SFKs, suggesting that the SH4 domain regulatory interaction might also be shared. The αF pocket is highly conserved amongst SFKs but varies considerably among the 12 human tyrosine kinases that have the same SH3/SH2/CD domain organization as the SFKs but lack SH4 domains (SH4-, Figures 7A, S6A). To better understand the meaning of these substitutions, we analyzed them in light of their effects on Src phosphotransferase activity (Figure 2F). Neither of the two SFK substitutions, relative to Src, were gain-of-function whereas 11 of the 48 SH4- kinase substitutions were gain-of-function. While the limited number of substitutions amongst the SFKs precludes meaningful statistical analysis, this trend suggests that αF pocket substitutions in SFKs preserve interaction with the SH4 domain, whereas substitutions in SH4- kinases disrupt it.
Figure 7. Functional characterization of the SH4 domain/αF pocket interaction.

A. Sequence alignment of SH4 domain-lacking human non-receptor tyrosine kinases (SH4-) at αF pocket residues. Colors indicate classification of the analogous mutation from the Src DMS. B. Phosphotransferase activity of purified FynFL WT or Fyn3D WT (n=3). C. Percent retained Fyn in the SH3 pulldown assay with purified FynFL WT or Fyn3D WT (n=3). D Representative micrographs (top panel) and percent bleb quantification (bottom panel) for Fynmyr-expressing SYFs treated with DMSO or CystIMATIK probe 3 for 15 min. E. DMS of Src’s SH4 domain. Dots indicate WT amino acid at that residue. F. Percent retained SrcTAMRA-3D in an SH4 pulldown assay with immobilized WT, P8N, or P8N/K5I SH4 domain (n=3). G. Proposed model of SH4 domain-mediated autoinhibition (myristoyl (green), unstructured SH4 and Unique (gray), SH3 (orange), SH2 (green), and CD (purple)). For 7D, each point represents a replicate treatment with multiple cells imaged and scored in a double-blind fashion. Horizontal lines indicate the mean of all replicates. See Table S5 for the total number of replicates and cells analyzed. *p <0.05; **p < 0.01, ***p < 0.001. See also Figure S6.
To provide additional support for the conservation of the SH4 domain/αF pocket interaction amongst SFKs, we investigated Fyn. As for Src, deletion of Fyn’s SH4 and Unique domains (Fyn3D) increased phosphotransferase activity and promoted an SH3 domain-accessible, open global conformation relative to FynFL (Figures 7B, S6B, 7C). Treatment of Fynmyr V284C-expressing SYFs with probe 3 resulted in a marked increase in blebbing, like for Src (Figures 7D, S6C). Thus, Fyn likely has an intramolecular SH4 domain interaction analogous to Src’s.
Unlike the αF pocket, SFK SH4 domain sequences are diverse, leading us to wonder which residues are important for the SH4 domain/αF pocket interaction. Therefore, we asked whether the phosphotransferase activity of SH4 domain variants could be measured in yeast. We worried that yeast activity measurements could be confounded by SH4 domain variant effects on myristoylation and localization. However, we found that two N-terminal Src truncations (SrcΔSH4 and Src3D), which are not myristoylated and cannot associate with membranes, strongly suppressed yeast growth, reflecting their enhanced in vitro activities (Figure S6D). Encouraged by these results, we performed a DMS of Src’s SH4 domain (residues 2-19) in the context of Srcmyr (Figure 7E). Unlike for Src’s CD, 76% of SH4 domain mutations were functionally neutral, with only 16% resulting in loss-of-function and 8% in gain-of-function (Table S4). Thus, the SH4 domain is under greatly reduced functional constraint, relative to the CD. Mutations at SH4 domain residues 2 through 9 had much larger effects, both positive and negative, than mutations at residues 10 through 19. In particular, mutations at residues Ser3 and Pro8 conferred greatly increased Src activity. Pulldown of Src3D with SH4 domain P8N, one of the most activating mutants, was substantially decreased compared to WT (Figures 7F, S6E). Introduction of a second gain-of-function mutant, K5I, further decreased Src3D pulldown. The importance of the N-terminal portion of the SH4 domain for interacting with the αF pocket helps to explain why disruption of this interaction alters Src’s capacity to interact with membranes in vitro and in cells because this region is also required for high affinity membrane interactions (McLaughlin and Aderem, 1995).
Discussion
Together, DMS and CystIMATIK revealed a regulatory interaction in Src, defined by the intramolecular engagement of the membrane-interacting, N-terminal SH4 domain with the αF pocket in the C-terminal lobe of the CD. We propose that the interaction between the SH4 domain and the αF pocket serves as a fastener that enhances autoinhibitory engagement of the core SH2/SH3 regulatory module with the CD (Figure 7G). By enhancing SH2/SH3 domain-mediated autoinhibition, the SH4 domain fastener ensures that Src possesses low phosphotransferase activity and minimal accessibility to intermolecular binding partners when the SH4 domain is not associated with membranes. Release of the SH4 domain fastener, through competitive association with membranes, could act as an initial step in Src activation by facilitating increased accessibility of Src’s SH2 and SH3 domains to other binding partners. Because the intramolecular engagement of the SH2, SH3, and SH4 domains of Src appear to be coupled, intermolecular binding partners that disrupt autoinhibitory SH2/SH3 domain engagement could also weaken the SH4 domain’s intramolecular interaction with the αF pocket and promote the enhanced membrane association of Src.
A recent report suggests that in the open global conformation, Src forms a homodimer that facilitates autophosphorylation via interaction of the N-terminus of one protomer with the CD domain of the other (Spassov et al., 2018). Intramolecular release of the SH4 domain fastener could enable the N-terminus to form an asymmetric dimer with the CD of a second Src protomer. Thus, this dimerization-dependent autophosphorylation mechanism is consistent with our SH4 domain fastener model, with the N-terminus of Src contributing to autoinhibition in a closed global conformation and autophosphorylation in an open global conformation.
A truncated construct comprising Src’s SH4, Unique and SH3 domains adopts a compacted structure with SH3 and SH4 interdomain contacts (Maffei et al., 2015). How this affects the SH4 domain’s interaction with the αF pocket is unclear, but additional structural studies with full length Src constructs should provide insight into whether these are competitive or complementary binding events. Another unresolved question is how the SH4 domain/αF pocket interaction affects autoinhibition through Src’s phosphorylated C-terminal tail. Our SH4 domain fastener model predicts that intramolecular engagement of Src’s SH4 domain should strengthen the interaction between the SH2 domain and the CD’s phosphorylated C-terminal tail and vice versa. Whether intramolecular engagement of the SH4 domain affects inhibitory C-terminal tail phosphorylation by Csk and dephosphorylation by phosphatases also remains to be determined.
The SH4 domain/αF pocket interaction likely plays a regulatory role in SFKs beyond Src. Deletion of Fyn’s N-terminus led to increased phosphotransferase activity and a more open global conformation, and CystIMATIK probes promoting an open global conformation of Fyn led to blebbing, consistent with release of SH4 domain sequestration. Moreover, the αF pocket sequence is highly conserved amongst SFKs and lacks any gain-of-function substitutions. By contrast, SFK SH4 domains are diverse and subjected to varying degrees of post-translational palmitoylation. What can explain this diversity? Our results show that the SH4 domain is under at least two distinct functional constraints: mediating binding to the αF pocket and binding to membranes. These two different constraints may help to explain the SH4 domain’s intrinsic disorder, which is a common strategy for binding to multiple partners (Maffei et al., 2015; Olsen et al., 2017). This intrinsic disorder is reflected in the relative mutational tolerance and low sequence conservation we observed, compared to the αF pocket. Nevertheless, our SH4 domain DMS results suggested that the N-terminal portion of Src’s SH4 domain, particularly residues Ser3 and Pro8, are important for interaction with the αF pocket.
In addition to Src, several other tyrosine kinase N-termini add a layer of regulation to a core SH3-SH2-linker-CD architecture (Devkota et al., 2017; Joseph et al., 2017; Wang et al., 2015). For example, Abl’s N-terminal myristate binds to its CD, reducing phosphotransferase activity (Hantschel et al., 2003). Like all SKFs, Src’s N-terminus is also myristoylated. However, our data, and a previous study, suggest that Src’s myristate does not function like Abl’s. (Patwardhan and Resh, 2010). The region of Src’s CD that overlaps with Abl’s myristate-binding pocket, which is ~20 Å away from the αF pocket, contains several sequence differences relative to Abl. Our DMS in yeast, where Src is likely myristoylated because yeast N-myristoyltransferase can efficiently modify Src’s N-terminus (Towler et al., 1988), shows almost no Src gain-of-function substitutions in this region (Figure S6F). The αF pocket could recognize Src’s myristate in addition to the SH4 domain, but the biochemical experiments we performed with myristoylated Src suggest that the αF pocket mainly interacts with the SH4 domain.
Like Src and Abl, Btk’s N-terminal PHTH domain is important for regulating CD phosphotransferase activity. The details of how the N-termini of Src, Abl, and Btk lead to autoinhibition vary, but they share common mechanistic themes. Like Src’s SH4 domain, Abl’s N-terminal myristate and Btk’s PHTH domain directly interact with the CD. All three N-termini strengthen autoinhibitory CD/SH3 and CD/SH2 domain interactions, further promoting a closed global conformation. The N-termini of all three kinases have an affinity for lipids, which intramolecular sequestration diminishes. Thus, coupling of membrane association to the release of autoinhibition via the N-terminus appears to be a recurrent theme amongst tyrosine kinases.
Interdomain regulation is a defining feature of multidomain kinases, and ~50% of kinases contain more than one domain. Our discovery of a intramolecular regulatory interaction in Src, one of the best-studied kinases, highlights the need to dissect kinase intramolecular regulation. In addition to modulating phosphotransferase activity, kinase regulatory domains often mediate important phosphotransferase-independent functions, as is the case for Src’s SH4 domain. We showed how two powerful tools, DMS and CystIMATIK, can be used to investigate intramolecular kinase regulation and probe phosphotransferase-independent function. Application of DMS and CystIMATIK to a wide range of kinases could reveal mechanisms of regulation and uncover biological functions.
Master STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dustin J. Maly (djmaly@uw.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
S. cerevisiae genetics and cell culture
Yeast experiments were performed in BY4741 Green Monster ((Suzuki et al., 2011)a generous gift from Dr. Fritz Roth, DMS experiments) or BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0, all other experiments). All Src constructs were cloned into the p415 GAL1 plasmid and transformants were selected for by plating on C-Leu media. All growth experiments were performed in C-Leu media to maintain plasmid.
Mammalian cell culture
Human HEK293s, HEK293Ts, HeLas and murine SYF cells were used for pulldowns and imaging experiments. Authenticity of human cell lines were confirmed by STR analysis, and SYF cells were purchased from ATCC. Cells were maintained in DMEM (Gibco) supplemented with 10% FBS (Gibco). Transient transfections were performed with a 3:1 ratio of μL turbofectin (Origene)/μg DNA.
METHOD DETAILS
All enzymes were purchased from New England Biolabs and all chemicals purchased from Sigma unless otherwise specified. All peptides were purchased from Genscript.
Cloning
All mutants were generated using either QuikChange Site Directed Mutagenesis (Agilent) or IVA cloning (García-Nafría et al., 2016) following standard protocols. Mutations were verified by Sanger sequencing. All subcloning was achieved using Gibson assembly or directional cloning following standard protocols and validated by Sanger sequencing.
Protein purification and expression
(SrcCD) For x-ray crystallography, SrcCD V284C (residues 261-536) was cloned into the bacterial expression plasmid pET-28a as a N-terminal His6-Tev tagged construct. The construct was co-transformed (Krishnamurty et al., 2012; Leonard et al., 2014) in E. coli with Yersinia pestis tyrosine phosphatase YopH (pCDF-Duet-1 vector) and chaperonin GroEL (pACYCDuet-1 vector) and plated on triple selective plates (Kanamycin/Chloramphenicol/Streptomycin). A single colony was picked and grown in an overnight culture of 50 mL of Terrific broth containing all three antibiotics. Three 2 L cultures were then inoculated with the starter culture, grown to an OD600 of 1.2, dropped to 18 °C and protein expression was induced with 0.2 M IPTG. The next day, cells were harvested through centrifugation, lysed by sonication in lysis buffer (50 mM HEPES, pH 8.0, 300 mM NaCl, 1 mM PMSF, 0.1% Triton-X, 20 mM imidazole), and centrifuged for 45 min at 10,000 rpm. Cleared lysates were incubated with 1.5 mL Ni-NTA resin (Thermo) for 1 h. The lysate was then discarded, and beads were washed with 20 mL lysis buffer before bound protein was eluted with lysis buffer containing 300 mM imidazole. Protein was dialyzed overnight at 4 °C in dialysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 5% glycerol, 1 mM DTT) with 1:25 ratio of Tev protease:eluted protein (wt:wt). The next day, the solution was filtered through a 0.22-μm filter then loaded onto a Ni-affinity column (GE Lifescience) and equilibrated with buffer A (50 mM Tris, pH 8.0, 5% glycerol, 1 mM DTT). The loaded column was washed with five column volumes of buffer A, then protein was eluted with a linear gradient of 0-35% of buffer B (50 mM Tris, pH 8.0, 1 M NaCl, 5% glycerol, 1 mM DTT). Peak fractions were analyzed by SDS-PAGE, and fractions containing Src were pooled, and used for crystallization. Relates to Figures S1A and 4D.
(SrcFL, SrcΔSH4, Src3D, SrcCD, Hck3D, FynFL, and Fyn3D) For in vitro biochemical assays, full length SFK (SFKfl; residues 1-536 for Src, residues 1-537 for Fyn), delta-SH4 (SrcδSH4; residues 19-536 for Src), three domain (3D) (SFK3D; residues 87-536 for Src, residues 85-537 for Fyn, residues 81-526 for Hck) or catalytic domain (SrcCD; residues 261-536) were cloned into the bacterial expression plasmid pMCSG7 as an N-terminal His6-SUMO tagged constructs (Hck lacked the SUMO tag). SFK constructs were co-transformed in E.coli with YopH/GroEL and plated on triple selective plates (Ampicilin/Chloramphenicol/Streptomycin). A single colony was picked and grown in an overnight culture of 15 mL of Terrific broth containing all three antibiotics. A 1 L culture was then inoculated with the starter culture, grown to an OD600 of 1.0, the temperature was then dropped to 18 °C and protein expression was induced with 0.4 M IPTG. Protein was expressed overnight at 18 °C. Ni-NTA was used to purify His6-SUMO-SFK after lysing cells in lysis buffer (50 mM HEPES, pH 8.0, 300 mM NaCl, 1 mM PMSF, 0.1% Triton-X, 20 mM imidazole) and eluted in purification buffer (50 mM HEPES, pH 8.0, 300 mM NaCl, 1 mM PMSF, 0.1% Triton-X, 10% glycerol, 0.2% BME, 100 mM imidazole). A 2 h dialysis in dialysis buffer (50 mM HEPES, pH 8.0, 150 mM NaCl, 1 mM DTT, 10% glycerol) was carried out before the first buffer exchange. 1:25 ratio of His6-ULP1 protease:eluted protein (wt:wt) was added into the dialysis cassette containing His6-SUMO-SFK and the mixture was allowed to cleave overnight in fresh dialysis buffer at 4 °C. Following cleavage, a second Ni-NTA purification was carried out to remove any non-cleaved SFK and His6-ULP1. Finally, an anion exchange column (Pierce) was used to remove YopH and GroEL to yield SFK at >95% purity. Relates to Figures S1A, 3C, 3E, S2B-C, 4C, 4E-G, S3A, S3C-E, 5F, 6A, 6C-E, S5G-K, S5M-O, 7B-C, and S6B.
(SrcTAMRA-FL and SrcTAMRA-3D) For N-terminal TAMRA labeling, Src was expressed as an N-terminal His6-SUMO construct with one extra glycine N-terminal to Gly2 (SrcTAMRA-FL) or two extra glycines added N-terminal to Thr87 (SrcTAMRA-3D). 5(6)-Carboxytetramethylrhodamine N-succinimydil ester was conjugated to ε-5-Caproic acid-LPYTGG (GenScript) to generate a rhodamine-labeled Sortase A substrate peptide (Theile et al., 2013). SFKs were expressed and the SUMO-tag was cleaved as described above. After the first dialysis, His6-Ulp1 was added to the dialysis cassette to cleave N-terminal His6-SUMO releasing N-terminal Gly-Gly motif. Anion exchange columns were then used to remove YopH and GroEL. Sortase A (Addgene, #64979), and rhodamine-labeled Sortase A substrate peptide were added to a purified SFK (10 (Sortase A): 10 (Sortase A substrate peptide): 1 (SFK)) and incubated in the dark for 2 h, at 4 °C in Sortase buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl). His6-Sortase and His6-Ulp1 were removed using Ni-NTA and excess peptide was removed using Zeba desalting columns (Thermo). Relates to Figures S1A, S3C, S5L, 7F, and S6E.
(purified SrcFLAG and HckFLAG) pcDNA 3.3 containing residues 1-536 of Src (residues 1-526 for Hck) with a C-terminal FLAG (SrcFLAG variants WT or E381T) was transiently transfected into HEK293Ts using 3:1 Turbofectin:μg DNA following manufacturer’s protocol. Cells were lysed after 24 h in Mod. RIPA buffer 3 (50 mM Tris, pH 7.8, 150 mM NaCl, 1 mM EDTA, 1x Pierce Protease Inhibitor Cocktail (Thermo), 1% IGEPAL CA-630), pelleted, and the supernatant was incubated with 50 nM of YopH and 0.5 mM BME at 4 °C for 2 h. BME was removed using Zeba Spin desalting column (Thermo) and the lysate was allowed to bind to Anti-Flag magnetic resin (Sigma) for 4 h at 4 °C. The magnetic bead was washed 3x with Flag purification buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 5% glycerol) and dephosphorylated myristoylated Src was eluted off the resin by treating resin with 3x Flag-peptide (Sigma) diluted in Flag elution buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 5% glycerol, 1mM DTT). Quantitative dephosphorylation was confirmed with a Src pTyr419 antibody (1:2500 Cell Signaling, #2101) and Tyr419 antibody (1:2500 Cell Signaling, #2102). Relates to Figures S1A, S3B, S3G, 5G, S4H-I, 6B, S5B, S5F.
(SH4SNAP) Residues 1-18 from Src were fused to the N-terminus of SNAP-tag through a LPETG linker (SH4SNAP). SH4SNAP (WT, P8N, K5I-P8N) constructs were cloned into the pMCSG7 vector and expressed in BL21 at an OD600 of 0.7 with 0.4 M IPTG. The protein was purified using Ni-NTA following the protocol described above using elusion buffer (50 mM HEPES, pH 7.6, 300 mM NaCl, 10% glycerol, 200 mM imidazole) and dialysed into storage buffer (50 mM HEPES, pH 7.6, 150 mM NaCl, 1mM DTT, 10% glycerol). Relates to Figures S1A, 6I-K, S5K, S5M-O, 7F, and S6E.
Csk was expressed and purified as a C-terminal GST tagged protein as described in (Leonard et al., 2014). Relates to Figures 4F-G and S3D-F.
Western blotting of yeast and mammalian cell lysates
For yeast protein prep, single colonies of each BY4741 strain were inoculated into 5 mL C-Leu with 2% glucose and grown overnight. The next day, each strain was back diluted into new C-Leu with 2% raffinose, grown for 3 h, then inoculated into C-Leu with 2% galactose. 24 h after induction, 3 OD600 units of each culture were collected, spun down, and the resultant pellet was lysed in 200 μL 1x SUMEB buffer (1% SDS, 8 M Urea, pH 6.8, 10 mM MOPS, 10 mM EDTA, 0.01% bromophenol blue, 5% 2-mercaptoethanol) with 1x Protease Inhibitor Cocktail and 200 μL acid washed beads. Each sample was vortexed for 3 min, heated at 65 °C for 10 min, and centrifuged at 17,000x g at 4 °C for 5 min. Protein was quantified using Qubit Protein Assay (Thermo). Samples were run on NuPAGE gels (Invitrogen) in 1x MOPS (Invitrogen) with 7 μL Spectra Multicolor Broad Protein Ladder (Thermo) at 200V for 1 h. Gels were transferred to a PVDF membrane in 1x Transfer buffer (Invitrogen) at 24V and 4 °C for 1 h. All subsequent wash steps were performed 3x in 1x TBST for 5 min while shaking. Membrane was washed and blocked in 5% BSA in 1x TBST for 1 h. Blocked membrane was washed and incubated with a Src antibody (Cell Signaling, #2110), anti-pTyr (Cell Signaling, #9411), or anti-Actin (Abcam, #8224) antibodies at 4 °C overnight. Probed membrane was washed and incubated with anti-Rabbit (1:10000) or anti-Mouse (1:10000) secondary (Invitrogen) for 1 h. After a final wash, blots were exposed using Super Signal West (Thermo) and developed. Blots were quantified using imageJ. Relates to Figures 2C and 2H.
HEK293T and SYF cells were transfected with the indicated constructs for 24 h, then washed once with 1x PBS and lysed in Mod RIPA buffer 1 (50 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1x Pierce Protease Inhibitor Cocktail (Thermo), 1% IGEPAL CA-630, 2 mM Sodium Orthovanadate, 30 mM Sodium Fluoride). Cells were pelleted at 13,000 g at 4 °C, then the supernatant was boiled with 3x SDS buffer (240 mM Tris, pH 6.8, 30% glycerol, 3% SDS, 0.01% bromophenol blue, 15% 2-mercaptoethanol) and separated by SDS-PAGE. Samples were visualized by western blotting with a Src antibody (Cell Signaling, #2110) and a Src pTyr419 antibody (Cell Signaling, #2101). After incubation with a secondary antibody, scanned blots were quantified with LI-COR Odyssey software to determine the percent phosphorylation. Relates to Figure 2I, S1E, S1F, S2D, S2F.
HeLa tRex cells were transfected with indicated constructs (in pcDNA5.0 under a doxycycline (Dox) inducible promoter) or blank using Turbofectin:DNA (1:1) for 24 h, and 1 μg/mL of Dox was added to respective wells. The wells were washed once with 1x PBS and lysed in Mod RIPA buffer 1 (50 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1x Pierce Protease Inhibitor Cocktail (Thermo), 1% IGEPAL CA-630, 2 mM Sodium Orthovanadate, 30 mM Sodium Fluoride). Cells were pelleted at 13,000x g at 4 °C, then the supernatant was boiled with 3x SDS buffer (240 mM Tris, pH 6.8, 30% glycerol, 3% SDS, 0.01% bromophenol blue, 15% 2-mercaptoethanol) and separated by SDS-PAGE. Samples were visualized by western blotting with a Src antibody (Cell Signaling, #2110) and a Src pTyr419 antibody (Cell Signaling, #2101). After incubation with a secondary antibody, scanned blots were quantified with LI-COR Odyssey software to determine the percent phosphorylation. Related to Figure S2E.
Inhibitor IC50 and Ki determination
For all IC50 determination experiments, a kinase titration was performed prior to inhibitor testing in order to ensure that the kinase concentration assayed was in the linear range.
(Src and Hck) Src3D WT (7.5 nM), Src3D V284C (7.5 nM), Hck3D WT (5 nM), Hck3D V284C (5 nM), SrcFL WT (8 nM), Abl3D WT (15 nM), or Abl3D V284C (24 nM) in assay buffer (76 mM HEPES, pH 7.5, 5 mM MgCl2, 150 mM NaCl, 3.8 mM EGTA, 0.2 mg/mL BSA, 150 uM Na3VO4, 1 mM BME) were incubated with inhibitors (initial concentration = 30 μM, 3-fold serial dilutions, 10 data points) and 1 mM ATP for 30 min in a 384-black assay plate (Corning, #3573). 20 μM of a fluorescent SFK peptide (EEEIYGE-(Dap-Pyrene)-EA) or 40 μM of an Abl peptide (AEAIYAA-(Dap-pyrene)-LA) (Wang et al., 2006) was then added to plate and incubated for 2 h. Raw fluorescence units were measured on an Envision plate reader (Perkin Elmer) with an excitation wavelength of 344 nm and an emission wavelength of 405 nm. Data was analyzed using GraphPad Prism software, and IC50 values were determined using “One-site fit logIC50” option. All IC50 values were then converted to Ki by implementing the Cheng-Prusoff equation using the ATP Km determined for each kinase and 1 mM ATP in case of SFKs; Ki is equal to IC50 for EphA2 and Rsk2. All assays were performed in triplicate. Relates to Figures 4C, S3H and 5F.
Kinobead profiling experiments
SILAC cell culture and generation of HEK293 lysate containing HckFLAG V284C
HckFLAG V284C was integrated into the Flp-In T-REx system (Thermo) as described in the product documentation and maintained with 50 μg/mL hygromycin after selection. Flp-In T-REx HEK293 cells stably expressing HckFLAG V284C were grown in custom -Lys/-Arg DMEM (Caisson Labs) supplemented with 10% dFBS (Sigma), 200 μg/ml proline and SILAC amino acids (0.2 mM natural isotope abundance Lys/Arg for light label, 0.2 mM Lys8/Arg10 for heavy label; Cambridge Isotope Labs). Cells were grown for at least 5 cell doublings in SILAC medium and harvested after reaching 90% confluency and 24 h induction with 0.5 mg/mL dox. For harvesting, cells were rinsed twice with ice cold PBS and lysed in 750 μL of mod. RIPA buffer 2 (50 mM Tris, pH 7.8, 150 mM NaCL, 10 mM NaF, 0.25% Na-deoxycholate, 1% NP-40 and 5% glycerol containing Halt Protease Inhibitor Cocktail (100x, Thermo Scientific), and Phosphatase inhibitor cocktail 2 and 3 (Sigma)). The ice-cold lysate was vortexed five times intermittently for 3 s and clarified by centrifugation at 21,000xg and 4 °C for 20 min. The protein content of the samples was determined using the Pierce 660 nm Assay Reagent (Thermo), then snap frozen in liquid nitrogen and stored at −80 °C until used (Golkowski et al., 2017). Relates to Figure 4H and S3G.
CystIMATIK probe profiling experiments
CystIMATIK probe profiling experiments were performed with a modified Kinobead profiling method (Golkowski et al., 2017). Briefly, 10 μL of a 50% Kinobead slurry (Kinobead matrix described in (Golkowski et al., 2017)) in 20% aqueous ethanol was pipetted into a 1.5 mL microtube and washed twice with 100 μL of mod. RIPA buffer 2. In parallel, 300 μg of cell extract in mod. RIPA buffer (ca. 2 mg/ml protein concentration) from either light or heavy labeled cells was pipetted into a 1.5 mL micro tube for each pulldown. Each CystIMATIK probe in DMSO (10 μM final concentration) or pure DMSO (control) was added to the corresponding SILAC lysate (1% DMSO final) and the tubes were agitated at 4 °C for 20 min on a rotator. For the SILAC label swap experiments, the addition of CystIMATIK probe to, for example, the light lysate was switched to heavy lysate, and addition of the DMSO control to the heavy lysate was switched to light lysate. After incubation, the pretreated lysate was pipetted individually to the tubes containing Kinobead affinity resin and the slurry was agitated on a rotator for 3 h at 4 °C. The supernatant was removed, and the beads were washed twice with 100 μL of ice cold mod. RIPA buffer. Then the beads were re-suspended in 100 μL of ice cold TBS (50 mM Tris-HCl, pH 7.8, 150 mM NaCl), the SILAC samples (beads) were combined pairwise (competition and control experiments) and the supernatant was removed. The beads were washed twice more with TBS and then re-suspended in 25 μL of 8 M tris buffered urea (8 M urea, 50 mM Tris-HCl, pH 7.8) containing 5 mM tris-(2-carboxyethyl)phosphine (TCEP) and 10 mM 2-chloroacetamide (CAM). The bead slurry was agitated for 30 min at 37 °C and 1400 rpm on a thermomixer. For digestion, the slurry was diluted two-fold with 100 mM aqueous triethylammonium bicarbonate solution (TEAB, urea concentration ≤4 M), the pH was adjusted to 9 with 1 N NaOH and 0.4 μg of LysC was added for every 5 uL of settled affinity resin that was added. The slurry was shaken for 2 h at 1400 rpm and 37 °C. Then the slurry was further diluted 2-fold with 100 mM TEAB (urea concentration ≤ 2 M) and 1 μg of trypsin per 5 μl of affinity resin was added. Samples were digested overnight on a shaker at 37 °C, diluted two-fold with 5% aqueous acetonitrile containing 0.1% TFA and acidified with formic acid (1% final). Peptides were extracted using StageTips (Rappsilber et al., 2007) and then analyzed in single nanoLC- MS/MS runs. Table S7 lists SILAC ratios and intensity values for proteins and kinases identified and quantified in competition experiments. Relates to Figures 4H and S3G.
LC-MS/MS and data analysis
Peptides were separated on a Thermo-Dionex RSLCNano UHPLC instrument with 10 cm fused silica capillary columns made in-house with a laser puller (Sutter) and packed with 3 μm 120 Å reversed phase C18 beads (a gift from Dr. Maisch, Ammerbuch, DE). The LC gradient was 90 min long with 10-35% Buffer B at 200 nL/min. LC solvent A was 0.1% acetic acid and LC solvent B was 0.1% acetic acid, 99.9% ACN. MS data was collected with a Thermo Orbitrap Elite spectrometer. Data-dependent analysis was applied using Top15 selection with CID fragmentation. Raw files were analyzed by MaxQuant/Andromeda (Golkowski et al., 2017) version 1.5.2.8 using protein, peptide and site FDRs of 0.01 and a score minimum of 40 for modified peptides, 0 for unmodified peptides; delta score minimum of 17 for modified peptides, 0 for unmodified peptides. MS/MS spectra were searched against the UniProt human database (updated July 22nd, 2015). MaxQuant search parameters: Variable modifications included Oxidation (M). Carbamidomethyl (C) was a fixed modification. Max. labeled amino acids was 3, max. missed cleavages was 2, enzyme was Trypsin/P, max charge was 7, multiplicity was either 1, 2 or 3, SILAC labels were Arg0/Lys0 (light), Arg6/Lys4 (medium), Arg10/Lys8 (heavy). The MaxQuant Re- Quantification feature was enabled. The initial search tolerance for FTMS scans was 20 ppm and 0.5 Da for ITMS MS/MS scans. Data was further processed using the Perseus software package (version 1.5.2.6), the R environment, Origin Pro 8.0 and Microsoft Excel. Significantly competed kinases were determined by applying a moderated two-tailed, two-sample t-test in Perseus with an FDR of 0.05. Relates to Figures 4H and S3G.
X-ray crystallography data acquisition and processing
A 20 mM stock solution of inhibitor 1, 2, or 3 (in 100% DMSO) was added to purified SrcCD V284C (5 mg/mL) to a final concentration of 75 μM. This mixture was incubated on ice for 1 h and spun down to remove insoluble compound prior to setting up the crystallization experiment. Crystals were grown at room temperature by vapor diffusion from a sitting drop containing 1 μL Src V284C-1, 2, or 3 solution (5 mg/mL protein, 100 mM NaCl, 5% glycerol, 1 mM DTT, 25 mM Tris, pH 8.0, 2 mM inhibitor, 10% DMSO) plus 1 μL crystallization buffer (1: 100mM MES, pH 6.0, 8% PEG 3350, 3% glycerol, 10 mM DTT, 10 mM NaOAc, 2: 100 mM MES, pH 6.3, 40 mM NaOAc, 8% PEG3350, 10% glycerol, 10 mM DTT, 3: 100 mM MES pH 6.0, 8% PEG 3350, 10 mM NaOAc, 14% glycerol, 10 mM DTT). Diffraction images were measured at SSRL Beamline BL9-2 at an X-ray wavelength of 0.9795 Å and processed using Mosflm and the CCP4 program suite. The initial structural models were taken from PDB entry 3UQF for Src-1, 5SWH for Src-2, and 3G6G for Src-3 and refined using alternating rounds of manual fitting in Coot and automated refinement in Refmac. Model geometry was validated using Molprobity. Crystallographic statistics are shown in Table 1. The final models have been deposited with the PDB as entry 5SWH (Src-1), 5TEH (Src-2), and 5SYS (Src-3). Relates to Figure 4D.
Yeast growth assay
To confirm that Src expression affects growth when expressed in S. cerevisiae, codon-optimized full-length human Srcmyr (Blue Heron Biotech) and indicated mutants were transformed into the S. cerevisiae BY4741 strain using standard LiAc transformation protocols (Gietz and Schiestl, 2007) and plated on C-Leu plates to select for successful transformants. Three independent colonies for each strain were collected and treated as biological replicates. Single colonies for each strain were grown overnight in 5 mL of 3% raffinose C-Leu to saturation. Strains were back diluted and allowed to double at least once, then 150 μL of each strain were plated in biological replicates and grown in a BioTek Synergy plate reader under constant shaking at 30 °C. OD600 was measured every 30 min over a 48 h time period. Growth rates of each strain were calculated using the GrowthRates software (Hall et al., 2013) using the BioTek plate readers option. Relates to Figures 2B, 2G, and S6D.
Src catalytic domain mutant library creation
All subsequent cleaning steps were performed using the Clean & Concentrate kit (Zymogen). To generate a mutant library of Src catalytic domain variants, we employed an inverse PCR saturation mutagenesis strategy (Jain et al. 2013). Briefly, we designed primer pairs corresponding to each of the 250 positions that comprise Src’s catalytic domain (residues 270-519, sequences labeled with SrcSSM in Table S6) in p415 GAL1 Src. For each position, the forward primer contained a degenerate ‘NNK’ codon corresponding to that position, where “N” represents any possible nucleotide while “K” represents a “G” or “T”. The reverse primer sits immediately upstream to the forward primer. Primer pairs were melting temperature and GC-content matched and ordered in 96-well format (Integrated DNA Technologies). For amplification of each position, 10 μL 2x KAPA HiFI HotStart ReadyMix (KAPA Biosystems), 2 μL of both 2.5 μM forward and reverse primers for that position, 5 μL H2O, and 1 μL 500 pg/μL p415 GAL1 Src. Thermocycler conditions used were: Initial denaturation at 95 °C for 3 min, followed by 20 cycles of 98 °C for 20 s, 60 °C for 15 s, and 72 °C for 8.5 min, and ending with a final extension at 72 °C for 10 min. ~90% of all positions were successfully amplified using these conditions, with the remaining positions requiring various PCR optimizations. 5 μL of each reaction was run on an agarose gel to verify correct amplification. Once confirmed, band intensities were quantified using the ImageJ software (https://imagej.nih.gov/ij/) and compared to a standard curve of intensities from known DNA concentrations. Based on measured intensities, each position was binned and pooled. After pooling, the linearized variant library was 5’phosphorylated by incubating with T4 Polynucleotide Kinase at 37 °C for 30 min, followed by a heat inactivation step at 65 °C for 20 min. This product was then ligated to re-circularize the vector with T4 DNA Ligase at 16 °C overnight, cleaned, and transformed by electroporation into One Shot TOP10F Electrocomp E. coli (Thermo) according to manufacturer’s protocol. Transformed cells were plated on LB + Ampicillin plates to determine library size and inoculated into LB + Ampicillin liquid and prepped using GenElute Midiprep Kit (Sigma). Relates to Figures 2F, S1B, and S1C.
Subassembly of Src variant library
Subassembly of variant library was performed following a previous protocol(Hiatt et al., 2010). The barcode oligo (EA01) was made double stranded by mixing 4.5 μL 25 μM EA01, 4.5 μL 25 μM EA02, 4 μL Buffer 3.1 and 27 μL H2O. This reaction was heated at 98 °C for 3 min, ramped down to 25 °C at a rate of 0.1 °C/s and then 1 μL of DNA Polymerase I, Large Fragment and 1.35 μL of 1 mM dNTPS (Qiagen) were added and incubated at 25 °C for 15 min. The now double stranded barcode was cleaned using and eluted in 10 μl H2O. In parallel, 10 μL of the plasmid-encoded Src variant library (268 ng/μL) was digested in 5 μL 10x Cutsmart Buffer, 1 μL Sall-HF, and 34 μL H2O overnight at 37 °C. The digested vector was treated with 5 μl 10x Antarctic Phosphatase Buffer and 1 μl Antarctic Phosphatase for 1 h at 37 °C, heat inactivated for 5 min at 70 °C, gel extracted (Qiagen), then cleaned and eluted in 10 μl H2O. 100 ng of vector and 150 ng of barcode were incubated with 2x Gibson Mastermix for 1 h at 50 °C, then cleaned and transformed into E. coli as above. Multiple gibsons were transformed in parallel to enable bottlenecking at a desired library size. A library corresponding to ~100,000 colonies was chosen for all future steps (henceforth referred to as Src CD library). A region encompassing Src’s catalytic domain and barcode was amplified using 25 μL 2x KAPA HiFI HotStart ReadyMix, 6 μL 2.5 μM EA03, 6 μL 2.5 μM EA04, 12 μL H2O, and 1 μL 10 ng/μL BC Src library, with 8 replicates for each of the three Bal-31 digestion conditions. Thermocycler conditions used were: Initial denaturation at 95 °C for 3 min, followed by 17 cycles of 98 °C for 20 s, 60 °C for 15 s, and 72 °C for 4.5 min. Eight replicates were pooled and cleaned. To generate fragments spanning the mutagenized region of Src’s catalytic domain, three different Bal-31 digestion conditions were performed: undigested, Bal-31 30 min, and ScaI/Bal-31 10 min. For the ScaI/Bal-31 10 min condition, 20 μL of amplicon, 5 μL 10x CutSmart buffer, 24 μL H2O, and 1 μL ScaI-HF was incubated overnight at 37 °C and subsequently cleaned. Each sample was digested using the endonuclease Bal-31 using 25 μL 2x Bal-31 Buffer, enough DNA for 40 ng/μL final concentration, and 1 μL of 1:5 Bal-31 enzyme at 25 °C for the indicated time. The digestion was stopped by adding 10 μL 100 mM EGTA and heating to 65 °C for 10 min. The digested DNA was cleaned and was run on a gel for visual conformation of fragment range. Each reaction was treated with END-IT Kit (Epicentre), cleaned, and A-tailed by adding the entire reaction to 5 μL 10x Taq Buffer, 8 μL H2O, 1 μL 10 mM ATP, and 1 μL GoTaq for 30 min at 72 °C then cooled down to 25 °C. Subassembly adapter was generated by mixing 10 μL of 100 μM EA05, 10 μL of 100 μM EA06, and 40 μL H2O, heating to 95 °C, then ramping down to 25 °C at 0.1°C/sec. Subassembly adapter was then ligated to cleaned DNA from above at a 3:1 Adapter:Amplicon ratio using T4 DNA ligase for 20 min at 25 °C, then heat inactivated for 10 min at 65 °C. Standard Illumina cluster generators were appended to ligated products by 25 μL KAPA HiFI HotStart ReadyMix, 1 μL 10 μM EA07, 1 μL 10 μM EA08, 21 μL H2O, and 2 μL 2 ng/μL respective template. Thermocycler conditions used were: Initial denaturation at 95 °C for 3 min, followed by 17 cycles of 98 °C for 20 s, 62.4 °C for 30 s, and 72 °C for 1 min. Cluster-forming amplicons were quantified using KAPA Library Quantification Kit (Kapa Biosystems). Paired end MiSeq 300bp kit (Illumina) using both the standard primers and a custom sequencing primer EA09 was used to sequence both the barcode and coding regions of each Src variant. All reads sharing a common barcode sequence were collapsed to form the consensus variant sequence, resulting in 72,822, barcodes, of which 31,728 contained “N”s, leaving 41,094 barcodes passing initial filter (Fowler et al., 2011; Starita et al., 2015). We then applied a quality filter to these remaining barcodes, wherein every base needed >=5x reads in agreement, leaving 25,390 high quality subassembled barcodes. These 25,390 barcodes represent 70% of all possible single amino acid mutants of the Src kinase domain, with an average of ~5 barcodes per variant. Relates to Figures 2F, S1B, and S1C.
Src library transformation into yeast
A single colony of freshly streaked BY4741 Green Monster was picked and grown in 5 mL of 2x YPAD (4% Peptone, 2% Bacto Dehydrated Yeast Extract, 0.008% Adenine sulfate, 2% glucose) overnight. The following morning, the saturated culture was backdiluted into 50 mL of 2x YPAD at an initial OD600 of 0.3 and grown until reaching an OD600 of ~2. The entire culture was centrifuged at 3,000 xg for 5 min and the resultant pellet was washed in 25 mL sterile H2O. This wash step was repeated twice. Pellet was resuspended in 1 mL sterile H2O, aliquoted equally into 10 eppendorf tubes, and spun down at 17,000G for 30 s to pellet. Each pellet was resuspended in transformation mix consisting of 240 μL 50% PEG, 50 μL 2 mg/mL Salmon Sperm (Invitrogen), 36 uL 1M LiAc. Eight of the samples had 500 ng BC Src library added, one had 250 ng BC Src library and 250 ng pRS411 added, and one received H2O vehicle. All samples were incubated at 42 °C for 45 min then washed in 1 mL sterile H2O. The eight 500 ng BC Src library tubes were pooled, added to 50 mL C-Leu 2% glucose and grown shaking at 30 °C. Serial dilutions of this culture were plated on C-Leu plates to enumerate library size, while dilutions of the BC Src/pRS411 condition were plated on C-Leu/Met plates to enumerate percent of double transformants. After 72 h of growth, aliquots of library were frozen in 20% glycerol at −80°C. Relates to Figures 2A, 2D, and 2F.
Selection & sequencing
Aliquots of frozen library were thawed and grown overnight in C-Leu 3% raffinose media. The next morning, cultures were back diluted to 0.5 OD600 in C-Leu 3% raffinose and grown for an additional doubling. Selection was initiated by inoculation at a final OD600 of 0.01 into 200 mL C-Leu 2% galactose media. Growth was monitored throughout the selection by measuring OD600. Selection 1 timepoints were OD600s: 0.164, 0.626, and 3.26. Selection 2 timepoints were OD600s: 0.239, 0.657, and 2.37. 4 OD600 units of each time point were harvested, spun down at 3,000G for 5 min, and immediately stored at −80 °C. Plasmids were extracted from frozen pellets using Yeast Plasmid Prep I (Zymogen) according to manufacturer’s protocol and resuspended in 15 μL H2O. To append Illumina cluster generators and append indices, 25 μL 2x KAPA2G Robust HotStart ReadyMix (KAPA Biosystems), 2 μL 10 μM EA10-EA12 (indexing primers), 2 μL 10 μM EA13-EA17 (indexing primers), 13.5 μL H2O, and 7.5 μL extracted plasmid. Thermocycler conditions used were: Initial denaturation at 95°C for 3 min, followed by 17 cycles of 95°C for 15s, 60°C for 15s, and 72°C for 15s. Each amplification was cleaned, quantified using KAPA Library Quantification Kit, then sequenced on NextSeq 500/550 High Output v2 kit (75 cycles) with standard Illumina sequencing primers and the custom primers EA18 and EA19. Relates to Figures 2D, 2F, S1B, and S1C.
Src variant library analysis
Raw Illumina reads were processed and demultiplexed using bcl2fastq and ea-utils. Functional scores for Src variants were calculated using Enrich2(Rubin et al., 2017) using Barcoded Variant SeqLib with “Weighted Least Squares” scoring option and “Wild Type” normalization. The OD600 readings of each sample (multiplied by 100, see “Selection & Sequencing”) were used as timepoints. The barcode map from subassembly step was used, with additional filtering of WT-associated barcodes (using 2x Std. Dev. of mean WT barcode activity score as a cutoff to filter out aberrantly-acting WT barcodes) and spiking in of barcoded variants Src K298M and T341I to ensure adequate representation of controls. Relates to Figures 2F, S1C, and S1D.
SH4 domain DMS
The SH4 domain mutant library was generated following a modified mutagenesis protocol(García-Nafría et al., 2016). For amplification of each position, 15 μL 2x KAPA HiFI HotStart ReadyMix, 0.9 μL of 10 μM mix of primers for that position (Table S6, sequences labeled with SH4), 10.1 μL H2O, and 4 μL 10 ng/ μL p415 GAL1 Src. Thermocycler conditions used were: Initial denaturation at 95 °C for 3 min, followed by 7 cycles of 98 °C for 20 s, 60 °C for 15 s, and 72 °C for 9 min, and ending with a final extension at 72 °C for 9 min. After amplification, 0.6 μL of DpnI was added and incubated at 37 °C for 1 h. Each product was cleaned and transformed into 10-beta electrocompetent cells (New England Biolabs) following manufacturer’s protocol. Transformed cells were plated on LB + Ampicillin plates to determine library size and inoculated into LB + Ampicillin liquid. This library (henceforth refered to as Src SH4 library) was transformed into yeast and grown as described for the Src CD library. Two independent transformations were performed, Selection 1 timepoints were OD600s: 0.156, 0.671, and 2.29. Selection 2 timepoints were OD600s: 0.171, 0.653, and 2.23. Plasmids were extracted and indexed as described for the Src CD library, except EA20 and EA21-EA28 were used. Amplicons were sequenced on NextSeq 500/550 High Output v2 kit (150 cycles) with custom primers EA29-EA32. Sequencing reads were processed and functional scores calculated as described for Src CD library, except the reads were paired using PEAR and the Basic SeqLib option was used. Relates to Figure 7E.
Rosetta modeling of N-tail conformation
We used the Rosetta Comparative Modeling protocol (Song et al., 2013) to model conformations of Src’s N-terminal tail (residues 2-86). A starting model of residues 87-536 created by running the Rosetta FastRelax protocol on the pdb structure 2SRC and threading the full sequence from residues 2-536 onto this structure. 3,798 models were generated using Rosetta@home (R@h; http://boinc.bakerlab.org/rosetta). Each final model was relaxed again using FastRelax to obtain the final score and conformation. Each output structure was analyzed using Pymol. Cα-Cα distances were calculated between tail residues (2-86) and all other residues. Cα-Cα distances < 8 Å are tabulated as a contact between residues. The Rosetta relaxed 2SRC structure was used to extract structural features of the kinase domain. Solvent accessible surface area of each residue was calculated using DSSP and the ratio of solvent accessible surface area to total surface area (rSASA) was calculated with respect to the theoretical surface areas(Tien et al., 2013). Relates to Figure S5A.
Phylogenetic analysis
Amino acid sequences for all aligned proteins were obtained from Uniprot and aligned using the MEGA software with the Muscle algorithm. Relates to Figures 3F and 7A.
Kinase activity assay
Activity of purified Src variants were measured using a self-reporting fluorescence SFK peptide (EEEIYGE-(DAP-Pyrene)-EA) in an in vitro kinase assay. Briefly, purified SrcFL, Src3D, or SrcΔSH4 constructs of each variant (WT, E381T, I444K) were diluted in kinase reaction buffer (76 mM HEPES, pH 7.5, 5 mM MgCl2, 150 mM NaCl, 3.8 mM EGTA, 0.2 mg/mL BSA, 150 uM Na3VO4) and incubated with 1 mM ATP at room temperature for 30 min to ensure full conversion of kinase to phosphorylated species. 60 nmoles of SFK peptide was pipetted to respective wells and raw fluorescence units were measured immediately on Envision (Perkin Elmer) with excitation wavelength of 344 nm and emission wavelength of 405 nm in real time for the first 10 min. Calculation of kinase activity in terms of pmole sec−1 of phosphorylated substrate is discussed below under “Calculation of Src kinase substrate phosphorylation rates”. Relates to Figures 3C, S2B, S3A, 6C, S5D-F, and 7B.
For S3A, relative fluorescence counts for kinase (Src3D WT or V284C or Hck3D WT or V284C) at concentrations 2.5 nM and 5 nM has been reported after kinase was incubated with SFK peptide and 1 mM ATP for 1 h.
SH3 domain pulldown assays
Generation of immobilized SH3 domain ligand: 20 μL of a 50% slurry of SNAP-capture pulldown resin was placed in a micro-centrifuge tube. The resin was washed (3x, 10 bead volumes) with pulldown buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM DTT and 0.2 mg/mL BSA). 8 μM of SNAP tag–polyproline peptide fusion (VSLARRPLPPLP) was loaded onto the resin at a final volume of 50 μL per 10 μL of bead in pulldown buffer (Leonard et al., 2014). The resin was rotated at room temperature for 1 h and then washed (3x, 10 bead volumes) prior to performing pulldown assays.
SH3 pull down assays performed with apo SFKs: apo SFK (100 nM) in 50 μL of pulldown buffer was incubated with 10 μL of the immobilized SH3 domain ligand. The resin was rotated at room temperature for 1 h. Post incubation, the flow-through was collected, and the resin was washed (3x, 10 bead volumes). To elute the retained kinase, 50 μL of 1x SDS loading buffer was added and the beads were boiled at 95 °C for 10 min. All samples were separated via SDS–PAGE and visualized using Western blotting using Src (1:2500; in case of Figures 3D-E, S2C, S2D, 6E, and S5H-J), or Tyr-419 (in case of 7C and S6B). (Cell Signaling, #2110 for SrcFL constructs or Cell Signaling, #2109 for Src3D constructs). The loaded amount of kinase was diluted to one fourth of the original concentration using 1x loading dye before running it on the gel as “I” (Input) along with the kinase eluted (“E”) off the resin. The scanned blots were quantified with ImageStudio Lite software and signal corresponding to input protein was scaled to original loaded kinase amount. The percentage of kinase retained on the resin (% retained) was then calculated on the basis of the loaded and eluted fraction based on curve fitting of immunoblot signal intensity to a Src titration.
SH3 pull down assays performed with purified inhibitor-SFKs complexes: Purified Src3D V284C (100 nM) was diluted in pulldown buffer. 3 or 5 μM of CystIMATIK probes 1, 2, or 3 was added to make a kinase-inhibitor complex. The mixture was incubated for 30 min at room temperature before being added to 10 μL of the immobilized SH3 domain ligand. Pulldown and quantification (with an anti-Src antibody) were performed as described above (Pull down assays with apo SFKs). Relates to Figures S3D. Inhibitor complexes of SrcTAMRA-3D V284C (50 nM, Figure S3D) were generated as described above and pulldowns were performed as described above. Quantification was performed with fluorescent gel scanning (using RITC filter on GE Typhoon FLA 900).
SH3 pull down assays performed with inhibitor-treated cells: HEK293Ts expressing SrcFLAG V284C or HckFLAG WT or V284C were treated with 10 μM CystIMATIK probes 1, 2, or 3 for 2 h before removal of media, 1x PBS wash, and lysis in 100 μL Mod. RIPA buffer (10 μM 1, 2, or 3, 50 mM Tris-HCL, pH 7.8 150 mM NaCl, 1% IGEPAL-630, 1 mM EDTA, 1x Pierce Protease Inhibitor Tablet (Pierce), and 1x Pierce Phosphatase Inhibitor tablet (Pierce) for 20 min on ice before clarification by centrifugation 17,000x g at 4 °C. Clarified lysate was added to 10 μL of the immobilized SH3 domain ligand and pulldown and quantification (with an anti-FLAG antibody) was performed as described above (Pull down assays with apo SFKs). Relates to Figures 4E and S3B.
SH4 domain pulldown assay
For data shown in Figure 6, 10 μL of a 50% slurry of SNAP-capture pulldown resin was placed in a micro-centrifuge tube. The resin was washed (3x, 10 bead volumes) with pulldown buffer (20 mM Tris, pH 7.0, 300 mM NaCl, 1mM BME, 0.2 mg/mL BSA). 5 μM (in case of Figure 6J) or 10 μM (in case of Figure 6K) of SH4SNAP was loaded onto the resin at a final volume of 50 μL per 10 μL bead volume in pulldown buffer. The resin was rotated at room temperature for 1 h and then washed (3x, 10 bead volumes) prior to performing pulldown assays. Next 75 nM of SrcCD (WT or mutants) was added to the resin to a final volume of 50 μL and incubated for 1 h at room temperature. The resin was then washed (3x, 10 bead volumes) with pulldown buffer described above and protein bound to the resin was eluted with 50 μL 1x loading dye and bead was boiled at 95 °C for 10 min. All samples were separated via SDS–PAGE and visualized using Western blotting using anti-Src (1:2500; Cell Signaling Technology #2109). For data shown in Figure S5I, pulldown was performed with 25 nM TAMRA-SrcFL V284C pre-treated with 5 μM of inhibitors 1, 2 or 3 for 30 min in buffer (20 mM Tris, pH 7.5, 100 mM NaCl, 1 mM 2-mercaptoethanol, 0.2 mg/mL BSA) before incubating kinase-inhibitor complex with 2.5 μM of SH4Snap protein conjugated to SNAP-capture pulldown resin. The resin was then washed (3x, 10 bead volumes) with pulldown buffer described above and protein bound to the resin was eluted with 50 μL 1x loading dye and bead was boiled at 95 °C for 10 min. All samples were separated via SDS–PAGE and visualized using RITC filter on GE Typhoon FLA 900. Relates to Figure 6I-K and S5K.
For data shown in Figure 7, 10 μL of a 50% slurry of SNAP-capture pulldown resin was placed in a micro-centrifuge tube. The resin was washed (3x, 10 bead volumes) with pulldown buffer (20 mM Tris, pH 7.5, 100 mM NaCl, 1 mM 2-mercaptoethanol, 0.2 mg/mL BSA). 2.5 μM of SH4SNAP (WT or P8N or P8N/K5I) was loaded onto the resin at a final volume of 50 μL per 10 μL bead volume in pulldown buffer. The resin was rotated at room temperature for 1 h and then washed (3x, 10 bead volumes) prior to performing pulldown assays with apo-SrcTAMRA-3D WT (50 nM) as described above (SH4 domain pull down assays performed with apo SFKs). Quantification was performed with fluorescent gel scanning (using RITC filter on GE Typhoon FLA 900). Relates to Figure 7F and S6E.
C-terminal Src kinase (Csk) accessibility assay
Csk was first profiled against CystIMATIK probes (2 and 3) using Csk peptide substrate (Csktide, KKKKEEIYFFFG) and γ32P-ATP. 25 nM of Csk was incubated with CystIMATIK probes (initial concentration = 10 μM, 3-fold serial dilutions, 10 data points) and 100 μM of ATP for 30 min in kinase reaction buffer (76 mM HEPES, pH 8.0, 5 mM MgCl2, 150 mM NaCl, 0.2 mg/mL BSA, 250 μM Na3VO4, and 1 mM DTT, 3.8 mM EGTA). 20 μM of Csktide and γ32P-ATP (0.08 μCi/well) were added and reaction was incubated for 1 h, then 4.6 μL of this reaction was spotted on a phosphocellulose membrane. Membranes were washed with 0.5% phosphoric acid (3x, 10 min each wash) and dried, and the radioactivity was determined by phosphor-imaging with a GE Typhoon FLA 9000 phosphor-scanner. The scanned membranes were quantified using ImageStudio Lite and plotted using GraphPad prism to determine % activity of Csk at different concentrations of inhibitors.
Titrations of SFK3D V284C starting at 1600 nM (2-fold serial dilutions) were incubated with 5 μM of 2 or 3 (SFK3D V284C was >98% inhibitor bound) with kinase reaction buffer. Following a 1 h incubation at room temperature, 25 nM Csk was added and phosphorylation was initiated by the addition of γ32P-ATP (0.08 μCi/well) and 100 μM of cold ATP at a final volume of 30 μL. The enzymatic reaction was run at room temperature for 1 h and then terminated by spotting 4.6 μL of the reaction mixture onto a phosphocellulose membrane. Membranes were washed with 0.05% phosphoric acid (3× 10 min wash) and air-dried, and the radioactivity was determined by phosphor-imaging with a GE Typhoon FLA 9000 phosphor-scanner. The scanned membranes were quantified with ImageStudioLite, and data were analyzed using GraphPad Prism and counts per minute was determined by dividing the raw counts over duration of the reaction. Relates to Figures 4F-G and S3D-F.
Co-Sedimentation assays
Generation of liposomes: For liposome co-sedimentation assay, sucrose loaded vesicles (1:1:1 phosphatidylcholine, phosphatidylserine and phosphatidylethanolamine (PC:PS:PE) or 2:1 PC and PS (2PC:PS)) were prepared as described previously (Giorgione and Newton, 2003).
Co-sedimentation of apo SFKs: SrcFLAG WT or E381T (150 nM) in co-sedimentation buffer (120 mM KCl, 6 mM MgCl2, 24 mM HEPES, pH 7.6, 1 mM DTT and 0.3 mg/mL BSA) was spun in a polycarbonate ultracentrifuge tube at 50,000 rpm at 4 °C for 10 min. Supernatant was removed and SrcFLAG was then incubated with 200 μM liposome at room temperature for 30 min before spinning the mixture at 100,000 rpm for 20 min. Supernatant was carefully removed and treated with 3x SDS loading dye, this served as the soluble fraction (“S”). The pelleted Src (“P”) was boiled with 1x SDS loading dye at 95 °C for 10 min. All samples were separated via SDS-PAGE and visualized by Western blotting using an anti-Src antibody (1:2500, Cell Signaling Technology #2110). The scanned blots were quantified with LI-COR Odyssey software to determine the percentage of kinase pelleted with liposomes on the basis of pelleted and soluble Src based on curve fitting of immunoblot signal intensity to a Src titration. Relates to Figures 6B and S5B.
Co-sedimentation of inhibitor SFK complexes: SrcFLAG WT (150 nM) in co-sedimentation buffer was spun in a polycarbonate ultracentrifuge tube at 50,000 rpm at 4 °C for 10 min. The soluble SrcFLAG WT was then incubated with DMSO or 5 μM inhibitors 4, 5, or 6 at room temperature for 30 min. The kinase-inhibitor complex was then added to 200 μM liposome and co-sedimentation assay was performed and quantified as described above (Co-sedimentation of apo SFKs). Relates to Figures 5F, 5G and S4H-I.
Microscopy
SYF cells were plated on 18 mm glass slides (Fisher) in a 12-well tissue culture plate at a density of 2×104. 24 h after plating, cells were transfected with a 3:1 ratio of μL turbofectin:μg DNA following manufacturer’s instructions. For untreated SYFs, 24 h after transfection the cells were washed once with PBS and fixed with a 4% paraformaldehyde mixture in PBS (Electron Microscopy Sciences) for 15 min. After fixing, cells were stained with NucBlue (Thermo) and Wheat-germ agglutinin conjugated to Alexafluor647 (Thermo) for 15 min. Relates to Figures 3G, and S2F.
SYF cells treated with CystIMATIK probes: SYFs were treated 24 h post-transfection with 10 μM 1 (Relates to Figure 5A), 3 μM 2 (Relates to Figure 5A, 5B, and S4E) or 10 μM 3 (Relates to Figure 5A-E, S4C-E, 7D, S6C) unless otherwise noted (Figure 5C: treated with 1, 3, or 10 μM of 3) for 15 min (except where otherwise noted, as in Figures 5B and S4C). For cells treated with both 2 and 3, cells were treated with 3 μM 2 for 15 min, then 10 μM 3 for 15 min (Relates to Figure 5A). Cells treated with Rock Inhibitor GSK429286A (Selleck Chem) were treated with 10 μM of the inhibitor for 2 h, then with 10 μM 3 for 15 min (Relates to Figure 5D). After treatment, cells were then washed once with PBS and fixed with a 4% paraformaldehyde mixture in PBS (Electron Microscopy Sciences) for 15 min. After fixing, cells were stained with NucBlue (Thermo) and Wheat-germ agglutinin conjugated to Alexafluor647 (Thermo) for 15 min.
HeLa cells treated with CystIMATIK probes: Flp-In T-Rex HeLa cells (gift from Stephen Taylor (Tighe et al., 2008)) stably expressing Srcmyr or Srcmyr V284C were plated on 18 mm glass slides (Fisher) in a 12-well tissue culture plate at a density of 2×104 with 1 μg/mL doxycycline. After 48 h, cells were treated with 5 μM 3b, an analog of 3 which differs by a methyl group (Relates to Figure S4F), 2 followed by 3b (same as described for SYFs, Relates to Figure S4F), or Rock inhibitor GSK429286A followed by 5 μM 3b (same as described for SYFs, relates to Figure S4G). After inhibitor treatment, cells were washed once with 1x PBS and fixed with a 4% paraformaldehyde mixture in 1x PBS for 15 min, followed by incubation with 250 μL of PBS containing NucBlue, Wheat-germ agglutinin conjugated to Alexafluor694 (Thermo) in the dark for 5 min.
For blebbing quantification of both SYF and HeLa fixed cells, slides were washed twice with 1x PBS, then mounted using Fluormount G (Southern Biotechnology) onto microscopy slides and sealed with nail polish. Images were obtained on a Leica SP8X, and images were processed using imageJ. Randomized images were scored by an unbiased scorer as either blebbing or not blebbing after viewing a training set of blebbing or non-blebbing cells for each condition. Cells were scored if they contained a single nucleus, had an intact and continuous membrane, and the majority of the cell was in the image. A cell was considered to be blebbing if it contained 3 or more rounded membrane protrusions. All images, as well as training sets and a master image list have been uploaded to Mendeley Data. Replicates consist of images from a single slide, and 2120 total cells were scored. All microscopy images were brightened for clarity. Relates to Figures 3G, 5A-E, S4B-G, 7D, and S6C.
Src autophosphorylation assay
SrcFL WT and E381T and SrcΔSH4 WT and E381T were purified in their dephosphorylated form. 160 μL kinase reaction mixture was prepared by adding Src constructs (final Src concentration = 50 nM) in kinase reaction buffer (76 mM HEPES, pH 7.5, 5 mM MgCl2, 150 mM NaCl, 3.8 mM EGTA, 0.2 mg/mL BSA, 150 uM Na3VO4). 32 μL of this was added to 3x loading dye for a 0 min timepoint sample. To initiate the autophosphorylation reaction, 30 μM ATP was then added to the rest 128 μL kinase reaction mixture and at various timepoints, the reaction was stopped by adding an aliquot of the reaction to 3x loading dye. Samples from 0, 5, 10 and 20 min were run on a SDS-PAGE gel and amount of phosphorylated and non-phosphorylated Src was visualized using anti-pTyr419 and anti-Tyr419 antibody (1:2500 dilution) respectively. Bands were quantified using ImageStudio Lite and percent autophosphorylated was calculated. Relates to Figures 6D and S5G.
Src autophosphorylation assay with the SH4 domain
SrcFL WT (50 nM), E381T (35 nM), I444K (35 nM) or SrcCD WT (30 nM) purified in their dephosphorylated form were incubated with 5 mM SNAP protein or 5 μM SH4SNAP protein for 30 min in kinase reaction buffer (76 mM HEPES, pH 7.5, 5 mM MgCl2, 150 mM NaCl, 3.8 mM EGTA, 0.2 mg/mL BSA, 150 uM Na3VO4). 30 μL of this reaction was added to 3x loading dye for a 0 min timepoint sample. To initiate the autophosphorylation reaction, 100 μM ATP was added and at the end of each timepoint, 30 μL of the reaction mixture was added to fresh 3x loading dye to terminate autophosphorylation. Samples from each timepoint were run on a SDS-PAGE gel and amount of phosphorylated Src and total Src was visualized using anti-pTyr419 and anti-Src antibody (1:2500 dilution), respectively. Bands were quantified using ImageStudio Lite and ratio of pTyr419 to total Src was determined. Relates to Figures S5M-O.
Maleimide labeling and mass spectrometry
Purified SrcFL or Src3D constructs K445C were diluted to 800 nM in mass spectrometry buffer (50 mM HEPES, pH 7.6, 150 mM NaCl, 5% glycerol and 0.02% (wt/vol) n-Dodecyl β-D- maltoside (DDM))(Kahsai et al., 2014) treated with 5 μM of inhibitors 5 or 6, and incubated at 25 °C for 30 min. The kinase-inhibitor complex was then treated with 1 mM heavy or light N-ethyl maleimide (NEM) in a LoBind 1.5 mL eppendorf tube at 25 °C for 30 min. The NEM labeling reaction was quenched with 10 mM of DTT in NH4HCO3 solution. 20 μL each of heavy and light maleimide-treated reactions were mixed and protein was precipitated using 0.02% deoxycholate and 10% trichloroacetic acid on ice for 10 min. The precipitated protein was pelleted by spinning at 10,000 rpm for 15 min. The pellet was dried with 10 μL acetone and re-suspended in peptide solubilization buffer (8 M urea, 200 mM Tris-HCl, pH 8.0, 2.4 mM iodoacetamide, 0.001% DDM) by vortexing briefly. The mixture was incubated in the dark for 30 min. Trypsin digestion solution (1 μg/mL of trypsin in 1 mM CaCl2, 200 mM Tris-HCl, pH 8.0) was added and the protein was digested overnight at 37 °C. Peptide was desalted using C18 ZipTips (Milipore) and each sample was run on the Finnigan LTQ Ion trap. [M+3H] peptide masses for both heavy and light NEM labeled peptides was analyzed using Xcalibur Quant software. Light and heavy NEM labeling reactions were swapped for each inhibitor and the above procedure was repeated. Relates to Figures 6F-H.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all statistical tests (unless otherwise noted), a two-tailed student’s t-test was used to compare means between two samples. A one-way ANOVA with post hoc Tukey’s HSD test was used to compare means between more than two samples. Significance is denoted as follows: * = p<0.05, ** = p<0.01, *** = p<0.001. Statistical tests were performed in R or GraphPad Prism.
Calculation of Ki, IC50 and Km
IC50 values were calculated in GraphPad Prism using the “One-Site Fit log IC50”. Km[ATP] values were determined using GraphPad Prism using “Plot Michaelis-Menten” option. Ki values for all SFKs were calculated using the Cheng-Prusoff equation at 1 mM ATP. Relates to Figures S2B, 4C, S3A, S3H, 5F, and S5C.
Calculation of log2 SILAC ratios
Data was analyzed using MaxQuant/Andromeda and processed with Perseus software package to compute log2 SILAC ratios. Our goal was to quantify kinases for secondary analysis (e.g. inhibitor profiling), we only consider proteins “identified” if MaxQuant is able to compute a protein intensity value; this is a more stringent criterion of protein identification as identifying peptides need to have good MS/MS spectra and quantifiable ion chromatograms. Significantly computed kinases were determined by applying a moderated two-tailed, two-sample t-test in Perseus with an FDR of 0.05. Relates to Figure 4H.
Classification of Src variants from activity scores
“Activity Scores” are the inverse of the Enrich2 output score function. To define “gain of function”, “functionally-neutral”, and “loss of function” activity classifications of Src variants we calculated ±2 standard deviations of the mean activity score of synonymous variants. All nonsynonymous variants with scores greater than the upper bound were classified as “gain of function”, all those with scores less than the lower bound were classified as “loss of function”, and all others were classified as “functionally-neutral”. Relates to Figures 2E, 3A-B, 3F, 7A and S6F.
Evolutionary conservation
Kullback-Leibler relative entropy was calculated with the pySCA software package (Rivoire et al., 2016) and multiple sequence alignments obtained from PFAM (http://pfam.xfam.org/). Relates to Figures 2F and S1D.
Calculation of amino acid centroids and hierarchical clustering of gain of function variants
The 2SRC structure was analyzed in R using the “bio3d” package and the centroid of each amino acid’s side chain was calculated using its atomic coordinates (backbone atoms were excluded). Hierarchical clustering was performed and visualized in R using the “dendextend” package. Relates to Figure 3A.
To define interdomain interfaces between catalytic domain (residues 270-519) and either the SH3 (residues 87-144) or SH2 (residues 149-239) domains, we calculated amino acid side chain centroids as above for all residues. Then, we imposed an 8 Å centroid-centroid distance cutoff to define residues in contact. Relates to Figure 3B.
Calculation of Src kinase substrate phosphorylation rates
Slopes were calculated from the linear portion of in vitro kinase enzyme assay to first obtain raw fluorescence count per sec. This was then divided by fluorescence change per picomoles of phosphorylated substrate (obtained from the slope of a standard curve of raw fluorescence versus phosphorylated substrate) to calculate phosphorylated substrate produced in pmoles sec−1. Relates to Figures 3C, 6C, S5D-F, and 7B
Synthesis of inhibitors
General Synthetic Procedures:
All chemicals purchased from commercial suppliers were used without further purification unless otherwise stated. Reactions were monitored with thin-layer chromatography (TLC) using silica gel 60 F254 coated glass plates (EM Sciences). Compound purification was performed with an IntelliFlash 280 automated flash chromatography system using pre-packed Varian SuperFlash silica gel columns (Hexane/EtOAc or CH2Cl2/MeOH gradient solvent). A Varian Dynamax Microsorb 100-5 C18 column (250 mm × 21.4 mm), eluting with H2O/CH3CN or H2O/ MeOH gradient solvent (+0.05% TFA), was used for preparatory HPLC purification. The purity of all final compounds was determined by analytical HPLC with an Agilent ZORBAX SB-C18 (2.1 mm × 150 mm) or Varian Microsorb-MV 100–5 C18 column (4.6 mm × 150 mm), eluting with either H2O/CH3CN or H2O/MeOH gradient solvent (+0.05% TFA). Elution was monitored by a UV detector at λ= 220 nm and λ= 254 nm, with all final compounds displaying >95% purity. Nuclear magnetic resonance (NMR) spectra were recorded on Bruker 300 or 500 MHz NMR spectrometers at ambient temperature. Chemical shifts were reported in parts per million (ppm) and coupling constants in hertz (Hz). 1H-NMR spectra were referenced to the residual solvent peaks as internal standards (7.26 ppm for CDCl3, 2.50 ppm for d6-DMSO, and 3.34 ppm for CD3OD). Mass spectra were recorded with a Bruker Esquire Liquid Chromatograph - Ion Trap Mass Spectrometer.
Synthesis and methods of purification for 4-chloro-5-iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine were described previously (Preparation of pyrrolo[2,3-d]pyrimidine derivatives as kinase inhibitors, WO 2014184069, Angiolini, Mauro; Buffa, Laura; Menichincheri, Maria; Motto, Ilaria; Polucci, Paolo; Traquandi, Gabriella; Zuccotto, Fabio). 4-methyl-3-((trimethylsilyl) ethynyl)aniline was synthesized according to previously reported publication (Röhrig et al., 2012). Synthesis and methods of purification for the compounds 1, 2, 3, 5 and 6 are described below.
4-chloro-5-iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde
In a round bottom flask, n-BuLi (540 mg, 3.6 mL, 2.5 M in Hexane, 8.4 mmol, 1.5 equiv.) was added dropwise to a solution of diisopropylamine (850 mg, 1.2 mL, 8.4 mmol, 1.5 equiv.) in THF (5 mL) under nitrogen at −78 °C. The solution was stirred at 0 °C for 1 hour and then cooled to −78 °C. A solution of 4-chloro-5-iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine1 (2000 mg, 5.8 mmol, 1.0 equiv.) in THF (1.25 mL) was added dropwise at −78 °C and the reaction was maintained at −78 °C for 1 hour. A solution of ethyl formate (658 mg, 8.9 mmol, 1.5 equiv.) in THF (1.5 mL) was then added dropwise at −78 °C. The reaction was allowed to warm up to room temperature overnight. Upon completion, the reaction was quenched with aqueous ammonium chloride solution (3.2 mL) at 0 °C. The aqueous layer was extracted with ethyl acetate (15 mL x3). Combined organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The solid residue was purified by flash chromatography using hexane/ethyl acetate gradient to afford 4-Chloro-5-iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde (600 mg, 28%). 1H-NMR (500 MHz, DMSO) δ = 9.63 (s, 1H), 8.49 (s, 1H), 4.57 (t, J = 5.50 Hz, 2H), 3.55 (t, J = 5.50 Hz, 2H), 3.16 (s, 3H); 13C-NMR (125 MHz, DMSO) δ = 185.28, 155.23, 153.60, 152.52, 132.80, 116.05, 71.10, 70.10, 57.95, 42.69; MS (ESI, m/z) calculated for C10H9CIIN3O2 364.9, [M+H]+ found 366.5.
4-amino-5-iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde

4-Chloro-5-iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde (200 mg, 0.54 mmol) and ammonium hydroxide solution (1.5 mL, 28.0 %- 30.0 % NH3 in water) were dissolved in 1,4-dioxane (1.5 mL) in a pressure tube and then heated to 60 °C for 3 to 5 hours. Upon completion, 1,4-dioxane was removed in vacuo. The solid residue was then resuspended in water (2 mL) and filtered. The filtrate was used in further steps without purification (95 mg, 50%). 1H-NMR (500 MHz, DMSO) δ= 9.75 (s, 1H), 8.27 (s, 1H), 4.65 (t, J = 5.56 Hz, 2H), 3.59 (t, J = 5.56 Hz, 2H), 3.19 (s, 3H); 13C-NMR (125 MHz, DMSO) δ= 192.92, 168.80, 165.12, 162.12, 138.40, 114.14, 82.14, 80.08, 67.66, 51.41; MS (ESI, m/z) calculated for C10H11IN4O2 346.0, [M+H]+ found 347.2.
4-Amino-5-(4-chlorophenyl)-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde
4-Amino-5-iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde (100 mg, 0.28 mmol, 1.0 equiv.), potassium phosphate (118 mg, 0.56 mmol, 2.0 equiv.), bis(triphenylphosphine) palladium(N) dichloride (11 mg, 0.014 mmol, 0.05 equiv.) and 4-chlorophenylboronic acid (54 mg, 0.34 mmol, 1.2 equiv.) were dissolved in a mixture of 1,4-dioxane (4 mL) and water (1 mL) in a 20-mL microwave vial. The resulting reaction mixture was then heated in a microwave reactor at 85 °C for one hour. After cooling, the reaction was diluted with ethyl acetate (10 mL) and quenched with water (5 mL). The organic phase was washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was then purified by flash chromatography on silica gel, eluting with CH2Cl2/MeOH gradient, afforded 4-amino-5-(4-chlorophenyl)-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde (38 mg, 40%). 1H-NMR (500 MHz, CDCl3) δ= 9.62 (s, 1H), 8.44 (s, 1H), 7.57-7.43 (m, 4H), 5.23 (broad s, 2H), 4.86 (t, J = 5.59 Hz, 2H), 3.78 (t, J = 5.59 Hz, 2H), 3.35 (s, 3H); 13C-NMR (124.75 MHz, CDCl3) δ= 181.71, 159.04, 156.0, 152.18, 135.43, 131.70, 129.69, 129.21, 129.10, 128.82, 101.60, 71.37, 58.76, 42.71; MS (ESI, m/z) calculated for C16H15CIN4O2 330.1, [M+H]+ found 331.4.
(E)- & (Z)- 3-(4-Amino-5-(4-chlorophenyl)-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl)-2-cyano-N-isopropylacrylamide (1):
To a solution of 4-amino-5-(4-chlorophenyl)-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde (28 mg, 0.084 mmol, 1.0 equiv.) and 2-cyano-N-isopropylacetamide (11 mg, 0.093 mmol, 1.0 equiv.) in tetrahydrofuran (5 mL) was added 1,8-diazabicyclo[5.4.0]undec-7-ene (8 μL, 0.048 mmol, 1.0 equiv). The reaction mixture was stirred at ambient temperature for 24 to 48 hours. Upon completion, the reaction was quenched by aqueous ammonium chloride solution and diluted with ethyl acetate. The organic layer was washed with brine (20 mL), dried over Na2SO4, and concentrated in vacuo. The crude product was purified by flash chromatography using a dichloromethane/methanol solvent gradient to afford a mixture of (E) and (Z) diastereomers (1:1) of 3-(4-amino-5-(4-chlorophenyl)-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl)-2-cyano-N-isopropylacrylamide (6 mg, 16 %). 1H-NMR (300 MHz, CD3OD) δ= 8.36 (s, 1H), 8.21 (s, 1H), 8.18 (s, 1H), 7.81 (s, 1H), 7.56-7.45 (m, 4H), 7.44-7.38 (m, 2H), 7.36-7.30 (m, 2H), 4.55-4.40 (m, 4H), 4.10 (m, 1H), 3.76-3.65 (m, 4H), 3.54 (m, 1H), 3.34 (s, 3H), 3.32 (s, 3H), 1.19 (d, J = 6.63 Hz, 6H), 0.95 (d, J = 6.63 Hz, 6H); 13C-NMR (124.75MHz, CD3OD) δ= 160.42, 160.33, 158.25, 157.83, 153.28, 152.75, 152.33, 151.54, 150.96, 140.83, 139.09, 131.73, 131.57, 131.56, 129.75, 129.58, 119.94, 117.83, 116.51, 114.57, 114.27, 113.67, 113.51, 111.28, 101.94, 101.61, 71.37, 71.28, 58.13, 58.11, 48.70, 48.35, 48.18, 43.16, 42.79, 42.33, 20.99, 20.99; MS (ESI, m/z) calculated for C22H23CIN6O2 438.2, [M+H]+ found 439.2; HPLC purity: 95%.
4-Amino-7-(2-methoxyethyl)-5-(4-phenoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidine-6-Carbaldehyde
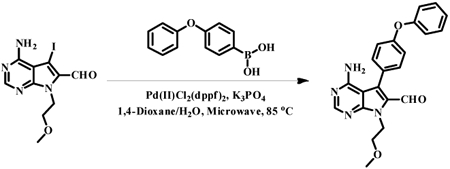
4-Amino-5-iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde (50 mg, 0.14 mmol, 1 equiv.), potassium phosphate (59 mg, 0.28 mmol, 2.0 equiv.), bis(triphenylphosphine) palladium(II) dichloride (5.6 mg, 0.007 mmol, 0.05 equiv.) and 4-phenoxyphenylboronic acid (37 mg, 0.17 mmol, 1.2 equiv.) were dissolved in a mixture of 1,4-dioxane (4 mL) and water (1 mL) in a 20-mL microwave vial. The reaction mixture was heated in a microwave reactor at 85 °C for one hour. After cooling, ethyl acetate (10 mL) and water (5 mL) were added and the organic phase was separated. The combined organic phase was washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was then purified with flash chromatography on silica gel, eluting with CH2Cl2/MeOH gradient, afforded 4-amino-7-(2-methoxyethyl)-5-(4-phenoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde (19 mg, 35%). 1H-NMR (500 MHz, CDCl3) δ= 9.66 (s, 1H), 8.43 (s, 1H), 7.48-7.38 (m, 4H), 7.21 (m, 1H), 7.16-7.08 (m, 4H), 5.32 (br s, 2H), 4.86 (t, J = 5.59 Hz, 2H), 3.78 (t, J = 5.59 Hz, 2H), 3.36 (s, 3H); MS (ESI, m/z) calculated for C22H20N4O3 388.2, [M+H]+ found 389.2.
(E)- & (Z)- 3-(4-Amino-7-(2-methoxyethyl)-5-(4-phenoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl)-2-cyano-N-isopropylacrylamide (2):

To a solution of 4-amino-7-(2-methoxyethyl)-5-(4-phenoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde (20 mg, 0.051 mmol, 1.0 equiv.) and 2-cyano-N-isopropylacetamide (6.40 mg, 0.051 mmol, 1.0 equiv) in tetrahydrofuran (5 mL) was added 1,8-Diazabicyclo[5.4.0]undec-7-ene (9.3 μL, 0.061 mmol, 1.2 equiv.). The reaction mixture was stirred at ambient temperature for 24 to 48 hours and monitored by TLC. Upon completion, the reaction was quenched with aqueous ammonium chloride solution, and diluted with ethyl acetate. The organic phase was washed with brine (20 mL), dried over Na2SO4, and concentrated in vacuo. The crude product was purified by flash chromatography using a dichloromethane/methanol solvent gradient to afford a mixture of (E) and (Z) diastereomers (8:2) of 3-(4-amino-7-(2-methoxyethyl)-5-(4-phenoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl)-2-cyano-N-isopropylacrylamide (10 mg, 15%). 1H-NMR (300 MHz, CD3OD) δ= 8.36 (s, 1H), 8.22 (s, 1H), 8.19 (s, 1H), 7.80 (s, 1H), 7.46-7.29 (m, 8H), 7.23-7.06 (m, 10H), 4.45-4.41 (m, 4H), 4.15 (m, 1H), 3.79-3.67 (m, 4H), 3.66-3.56 (m, 1H), 3.35 (s, 3H), 3.30 (s, 3H), 1.22 (d, J = 6.63 Hz, 6H), 0.99 (d, J = 6.42 Hz, 6H); 13C-NMR (124.75MHz, CD3OD) δ= 162.07, 161.84, 159.69, 159.33, 159.29, 159.26, 158.56, 158.50, 158.45, 158.31, 154.57, 153.98, 152.84, 152.19, 142.13, 140.22, 132.89, 131.35, 131.19, 129.62, 129.10, 128.90, 128.71, 125.29, 125.08, 125.04, 124.91, 122.43, 120.97, 120.72, 120.59, 120.49, 120.02, 119.72, 117.98, 116.02, 115.40, 111.84, 103.49, 103.11, 72.76, 72.69, 59.48, 59.44, 44.60, 44.38, 44.14, 43.67, 22.40, 22.20; MS (ESI, m/z) calculated for C28H28N6O3 496.2, [M+H]+ found 497.4; HPLC purity: 95%.
5-Iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine
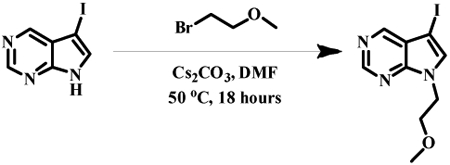
5-iodo-7H-pyrrolo[2,3-d]pyrimidine (2000 mg, 8.2 mmol, 1.0 equiv.) and 1-bromo-2-methoxyethane (1700 mg, 1.2 mL, 12 mmol, 1.5 equiv.) were dissolved in dry DMF (20 mL). To the solution was added cesium carbonate (5300 mg, 16 mmol, 2.0 equiv.). The reaction mixture was then stirred at 50 °C for 18 hours and then fil tered. The filtrate was rinsed with EtOAc (200 mL) and organic layer was collected. The combined organic layer was washed with water (30 mL), brine (30 mL) and dried over anhydrous Na2SO4. Purification by flash chromatography on silica gel (MeOH: DCM= 0:100 to MeOH: DCM= 10:90) afforded 5-iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine as a pale yellow solid (1.7 g, 69%); 1H NMR (300 MHz, CDCl3) δ= 8.85 (s, 1H), 8.70 (s, 1H), 7.41 (s, 1H), 4.41 (t, J = 5.0 Hz, 2H), 3.68 (t, J = 5.0 Hz, 2H), 3.30 (s, 3H); 13C NMR (75 MHz, CDCl3) δ= 152.25, 150.83, 150.08, 134.19, 121.38, 59.06, 52.18, 44.45; MS (ESI, m/z) calculated for C9H10IN3O 303.1, [M+H]+ found 303.8.
5-Iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde
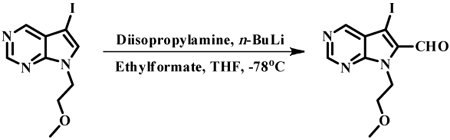
Diisopropylamine (340 mg, 470 μL, 3.3 mmol, 1.5 equiv.) was dissolved in dry THF (8 mL) and cooled to −78°C. n-BuLi solution (2.5 M in hexane, 1.3 mL, 3.3 mmol, 1.5 equiv.) was added dropwise to the above solution. The resulting reaction mixture was stirred at 0 °C for 1 hour and then cooled to −78 °C. A solution of 5-iodo-7-(2-me thoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine (700 mg, 2.3 mmol, 1.0 equiv., dissolved in 3 mL dry THF) was added dropwise and the reaction was then maintained at −78 °C for 1 hour. Ethyl formate (302 mg, 330 μL, 4.0 mmol, 1.8 equiv.) was then added dropwise at −78°C. The reaction mixture was allowed to warm to room temperature overnight. The reaction was quenched by saturated aqueous ammonium chloride solution (2 mL) and diluted with ethyl acetate (200 mL). The organic layer was washed with saturated aqueous NaHCO3 solution, brine and dried over anhydrous Na2SO4. Purification by flash chromatography on silica gel (EtOAc: Hexane= 0:100 to EtOAc: Hexane= 60:40) afforded 5-iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde as a bright yellow powder (514 mg, 67%). 1H-NMR (500 MHz, CDCl3) δ= 10.04 (s, 1H), 9.10 (s, 1H), 8.98 (s, 1H), 4.91 (t, 2H), 3.75 (t, 2H), 3.29 (s, 3H); 13C-NMR (126 MHz, CDCl3) δ= 184.56, 156.31, 154.12, 153.45, 132.11, 121.15, 71.55, 71.35, 59.04, 43.03; MS (ESI, m/z) calculated for C10H10IN3O2 331.0, [M+H]+ found 332.3.
N-(3-ethynyl-4-methylphenyl)-3-(trifluoromethyl)-benzamide
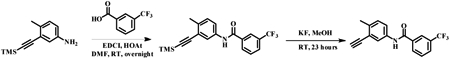
4-Methyl-3-((trimethylsilyl)ethynyl)aniline (1000 mg, 4.9 mmol, 1.0 equiv.), 3-trifluomethyl-benzoic acid (1400 mg, 7.4 mmol, 1.5 equiv.) and HOAt (1.0 g, 7.4 mmol, 1.5 equiv.) were dissolved in dry DMF (20 mL). The solution was cooled to 0 °0 and to it was added 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (1400 mg, 7.4 mmol, 1.5 equiv.). The reaction mixture was then allowed to warm to room temperature overnight. The solvent was removed in vacuo and the solid residue was dissolved in EtOAc (200 mL). The organic layer was washed with saturated aqueous KH2PO4 solution (30 mL), saturated aqueous NaHCO3 solution (30 mL), brine (30 mL) and dried over anhydrous Na2SO4. The solvent was removed in vacuo and the solid residue was dissolved in dry MeOH (30 mL). Potassium fluoride (1300 mg, 22 mmol, 4.5 equiv.) was added and the resulting mixture was stirred at room temperature for 23 hours. The reaction mixture was then filtered and concentrated in vacuo. The solid residue was dissolved in EtOAc (200 mL) and washed with water (20 mL), brine (30 mL) and dried over anhydrous Na2SO4. Purification by flash chromatography with silica gel afforded N-(3-ethynyl-4-methylphenyl)-3-(trifluoromethyl)-benzamide as a pale brown solid (977 mg, 66% for two steps). 1H-NMR (500 MHz, CDCl3) δ= 8.10 (s, 1H), 8.03 (d, J = 7.7 Hz, 1H), 7.90 (s, 1H), 7.80 (d, J = 7.8 Hz, 1H), 7.69 (s, 1H), 7.61 (t, J = 7.8 Hz, 1H), 7.56 (d, J = 8.2 Hz, 1H), 7.20 (d, J = 8.3 Hz, 1H), 3.28 (s, 1H), 2.43 (s, 3H); 13C NMR (126 MHz, CDCl3) δ= 164.44, 137.73, 135.68, 135.10, 131.44, 130.50, 130.28, 129.61, 128.61, 124.40, 124.13, 123.75, 122.73, 121.30, 81.98, 81.62, 20.22; MS (ESI, m/z) calculated for C17H12F3NO 303.09, [M+H]+ found 304.1.
N-(3-((6-formyl-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d] pyrimidin-5-yl)ethynyl)-4-methylphenyl)-3-(trifluoromethyl)benzamide
5-Iodo-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde (150 mg, 0.45 mmol, 1.0 equiv.) was dissolved in dry DMF (6 mL). The solution was flushed with a moderate stream of nitrogen gas for 10 minutes. Triethylamine (190 mg, 250 μL, 1.8 mmol, 4.0 equiv.), N-(3-ethynyl-4-methylphenyl)-3-(trifluoromethyl)benzamide (210 mg, 0.68 mmol, 1.5 equiv.), bis(triphenylphosphine)palladium(II) dichloride (16 mg, 0.023 mmol, 0.05 equiv.) and copper(I) iodide (8.6 mg, 0.045 mmol, 0.10 equiv.) were added sequentially. The resulting mixture was stirred at room temperature for 30 minutes and then maintained at 50 °C for 14 hrs. The solvent was removed in vacuo and the solid residue was dissolved in EtOAc (300 mL). The organic layer was washed with saturated aqueous NH4Cl, saturated aqueous NaHCO3, brine and then dried over anhydrous Na2SO4. Purification by flash chromatography on silica gel (EtOAc: Hexane= 0:100 to EtOAc: Hexane= 60:40) afforded N-(3-((6-formyl-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d] pyrimidin-5-yl)ethynyl)-4-methylphenyl)-3-(trifluoromethyl)benzamide as a pale yellow solid (189 mg, 80%). 1H-NMR (300 MHz, CDCl3) δ= 10.37 (s, 1H), 9.29 (s, 1H), 9.10 (s, 1H), 8.16 (s, 1H), 8.11 (d, J = 7.8 Hz, 1H), 8.03 (s, 1H), 7.94 (s, 1H), 7.82 (d, J = 7.7 Hz, 1H), 7.72 – 7.54 (m, 2H), 7.29 (d, J = 8.3 Hz, 1H), 4.92 (t, J = 5.2 Hz, 2H), 3.77 (t, J = 5.2 Hz, 2H), 3.30 (s, 3H), 2.55 (s, 3H); 13C-NMR (126 MHz, CDCl3) δ= 181.91, 164.40, 156.03, 152.49, 152.16, 137.08, 135.73, 135.63, 135.51, 131.14, 130.68, 130.47, 129.55, 128.64, 124.19, 123.97, 123.72, 122.55, 121.84, 118.45, 110.33, 96.83, 82.47, 71.26, 59.01, 43.15, 20.52; MS (ESI, m/z) calculated for C27H21F3N4O3 506.16, [M+H]+ found 508.0.
(E)- and (Z)- N-(3-((6-(2-cyano-3-(methylamino)-3-oxoprop-1-en-1-yl)-7-(2-methoxy ethyl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)ethynyl)-4-methylphenyl)-3-(trifluoromethyl)benzamide (3a)
N-(3-((6-formyl-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)ethynyl)-4-methylphenyl)-3-(trifluoromethyl)benzamide (30 mg, 0.059 mmol, 1.0 equiv.) and 2-cyano-N-methylacetamide (8.7 mg, 0.089 mmol, 1.5 equiv.) were dissolved in dry THF (2.2 mL). 1,8-Diazabicyclo[5.4.0]undec-7-ene (18 mg, 18 μL, 0.12 mmol, 2.0 equiv.) was added and the resulting solution was stirred at 23 °C for 2 hours. The reaction mixture was then diluted with EtOAc (20 mL) and washed with saturated aqueous NH4Cl solution, brine and dried over anhydrous Na2SO4. Flash chromatography on silica gel (EtOAc: Hexane= 0:100 to EtOAc: Hexane= 90:10) afforded a mixture of (E) or (Z) diastereomers (53:47) of N-(3-((6-(2-cyano-3-(methylamino)-3-oxoprop-1-en-1-yl)-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)ethynyl)-4-methylphenyl)-3-(trifluoromethyl)benzamide as a bright yellow solid powder (14 mg, 40%). 1H-NMR (500 MHz, MeOD) δ= 10.26 (s, 1H), 10.25 (s, 1H), 9.03 (s, 1H), 8.94 (s, 1H), 8.80 (s, 1H), 8.78 (s, 1H), 8.44 (s, 1H), 8.39 (d, J = 4.5 Hz, 1H), 8.23 – 8.04 (m, 2H), 7.96 (s, 1H), 7.87 (s, 1H), 7.82 – 7.72 (m, 3H), 7.66 – 7.54 (m, 3H), 7.45 (d, J = 8.2 Hz, 1H), 7.22 – 7.08 (m, 1H), 4.50 – 4.40 (m, 4H), 3.65 (t, J = 4.9 Hz, 2H), 3.60 (t, J = 4.8 Hz, 2H), 3.21 (s, 3H), 3.15 (s, 3H), 2.81 (d, J = 4.4 Hz, 3H), 2.55 (d, J = 4.6 Hz, 3H), 2.40 (s, 3H), 2.38 (s, 3H); 13C-NMR (126 MHz, CDCl3) δ= 164.28, 160.33, 154.29, 151.47, 150.77, 139.75, 137.55, 136.71, 136.33, 135.65, 135.56, 130.53, 130.53, 130.25, 129.55, 128.57, 124.26, 123.83, 123.27, 120.89, 119.43, 116.11, 111.86, 101.65, 99.85, 85.67, 71.16, 59.26, 43.56, 27.55, 20.39; MS (ESI, m/z) calculated for C31H25F3N6O3 586.2, [M+H]+ found 587.7; HPLC purity: > 99%.
(E)- and (Z)- N-(3-(2-(6-(3-amino-2-cyano-3-oxoprop-1-enyl)-7-(2-methoxyethyl)-7H-pyrrolo [2,3-d]pyrimidin-5-yl)ethynyl)-4-methylphenyl)-3-(trifluoromethyl)benzamide (3b)
N-(3-((6-formyl-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)ethynyl)-4-methylphenyl)-3-(trifluoromethyl)benzamide (30 mg, 0.059 mmol, 1.0 equiv.) and 2-cyanoacetamide (7.5 mg, 0.089 mmol, 1.5 equiv.) were dissolved in dry THF (2.2 mL). 1,8-Diazabicyclo[5.4.0]undec-7-ene (18 mg, 18 μL, 0.12 mmol, 2.0 equiv.) was added and the resulting solution was stirred at 23 °C for 45 minutes. The reaction mixture was then diluted with EtOAc (20 mL) and washed with saturated aqueous NH4Cl solution, brine and dried over anhydrous Na2SO4. Flash chromatography on silica gel (MeOH:DCM =0:100 to MeOH:DCM = 10:90) afforded a mixture of (E) or (Z) diastereomers (75:25) of (E)- and (Z)- N-(3-(2-(6-(3-amino-2-cyano-3-oxoprop-1-enyl)-7-(2-methoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)ethynyl)-4-methylphenyl)-3-(trifluoromethyl) benzamide as a bright yellow solid powder (25 mg, 74%). 1H-NMR (500 MHz, MeOD) δ= 9.09 (s, 1H), 8.98 (s, 1H), 8.86 (s, 1H), 8.82 (s, 1H), 8.48 (s, 1H), 8.18 (s, 1H), 8.13 (d, J= 7.7 Hz, 1H), 7.98 (d, J= 2.1 Hz, 1H), 7.92 (s, 1H), 7.80 (d, J=6.7 Hz, 1H), 7.71-7.58 (m, 2H), 7.54 (dd, J= 8.3, 2.1 Hz, 1H), 7.21 (d, J= 8.3 Hz, 2H), 4.58-4.43 (m, 3H), 3.73-3.58 (m, J= 18.5, 5.0 Hz, 3H), 3.19 (s, 5H), 2.46 (s, 3H); MS (ESI, m/z) calculated for C30H23F3N6O3 572.2, [M+H]+ found 573.7; HPLC purity: > 99%.
1-Cyclopentyl-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (5)
1-Cyclopentyl-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine (100 mg, 0.304 mmol, 1.00 equiv.) and 4-Phenoxyphenylboronic acid (97.5 mg, 0.456 mmol, 1.50 equiv.) were dissolved in a mixture of 1,4-Dioxane (3.5 mL) and water (0.88 mL). Potassium phosphate (162 mg, 0.760 mmol, 2.50 equiv.) and bis(triphenylphosphine) palladium(II) dichloride (24.8 mg, 0.0304 mmol, 10 %) were added and the resulting suspension was heated at 85 °C for 2 hours in a microwave reactor. After cooling, the reaction was diluted with ethyl acetate (40 mL) and quenched with saturated aqueous NH4Cl solution (10 mL). The organic phase was separated and washed with brine, dried over anhydrous Na2SO4. Flash chromatography on silica gel (eluted with a gradient of EtOAc:Hexane= 0:100) to EtOAc:Hexane= 90:10) afforded 1-cyclopentyl-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine as a pale yellow solid (92.7 mg, 82%). 1H NMR (500 MHz, CDCl3) δ= 8.25 (s, 1H), 7.59 (d, J = 8.6 Hz, 2H), 7.44 – 7.39 (m, 1H), 7.28 – 7.20 (m, 2H), 7.17 (d, J = 8.5 Hz, 2H), 7.10 (d, J = 7.7 Hz, 2H), 5.36 – 5.27 (m, 1H), 2.26 – 2.12 (m, 4H), 2.10 – 1.93 (m, 4H); MS (ESI, m/z) calculated for C22H21N5O 371.2, [M+H]+ found 372.2; HPLC purity: > 99%.
N-(3-((4-amino-1-cyclopentyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)ethynyl)-4-methylphenyl)-3-(trifluoromethyl)benzamide (6)
1-Cyclopentyl-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine (100 mg, 0.304 mmol, 1.00 equiv.) was dissolved in anhydrous DMF (4 mL) and the solution was flushed with a moderate stream of nitrogen for 5 minutes. Triethylamine (124 mg, 1.22 mmol, 4.00 equiv., 167 uL), N-(3-ethynyl-4-methylphenyl)-3-(trifluoromethyl)-benzamide (138 mg, 0.456 mmol, 1.50 equiv), bis(triphenylphosphine)palladium(II) dichloride (10.7 mg, 0.0152 mmol, 5.0%) and copper(I) iodide (5.8 mg, 0.0304 mmol, 10%) were added sequentially. The resulting mixture was heated at 50 °C for 14 hours and then diluted with EtOAc (40 mL). The organic phase was washed with saturated aqueous NH4Cl solution (10 mL), brine, dried over anhydrous Na2SO4 and concentrated in vacuo. Purification with Flash chromatography using a gradient of EtOAc:Hexane= 0:100 to EtOAc:Hexane= 90:10) afforded N-(3-((4-amino-1-cyclopentyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)ethynyl)-4-methylphenyl)-3-(trifluoromethyl) benzamide as a pale yellow solid (120 mg, 78%). 1H NMR (500 MHz, CDCl3) δ= 8.72 (s, 1H), 8.26 (s, 1H), 8.23 (d, J = 7.7 Hz, 1H), 8.05 (s, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 7.4 Hz, 1H), 7.60 (t, J = 7.6 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 5.34 – 5.23 (m, 1H), 2.49 (s, 3H), 2.28 – 2.09 (m, 4H), 2.09 – 1.97 (m, 2H), 1.85 – 1.72 (m, 2H); MS (ESI, m/z) calculated for C27H23F3N6O 504.2, [M+H]+ found 505.6; HPLC purity: > 99%.
DATA AND SOFTWARE AVAILABILITY
Sequencing reads were deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through accession number GSE114098. Microscopy images and Rosetta models are accessible in Mendeley Data DOI: 10.17632/4wcrr6vswv.1.
Supplementary Material
Movie S1. Live-cell imaging of SrcGFP V284C treated with 2 (3 μM). Relates to Figure 5B.
Movie S2. Live-cell imaging of SrcGFP V284C treated with 3 (10 μM). Relates to Figure 5B.
Table S2. Scores and classifications of Src CD variants. Relates to Figure 2F.
Table S4. Scores and classifications of Src SH4 variants. Relates to Figure 7E.
Table S6. Primers used in study. Relates to all Figures.
Table S7. Log2 SILAC ratios from Kinobead profiling. Relates to Figure 4H.
Scheme 1:

a) Diisopropylamine, n-BuLi, ethyl formate, THF, −78 °C; b) Ammonium hydroxide, 1,4-Dioxane, 60 °C; c) K3PO4, PdCl2(dppf)2. 4-Chlorophenylboronic acid, 1, 4-Dioxane/H2O, 85 °C (microwave); d) K3PO4, PdCl2(dppf)2. 4-Phenoxyphenyl boronic acid, 1, 4-Dioxane/H2O, 85 °C (microwave); e) 2-cyano-N-isopropylacetamide, DB U, THF, RT.
Scheme 2:

a) 2-Bromoethyl methyl ether, CS2CO3, DMF, 50 °C; b) Diisopropylamine, n-BuLi, ethyl formate, THF, −78 °C to RT; c) 3-(Trifluorome thyl)benzoic acid, EDCI, HOAt, DMF, RT; d) KF, MeOH, RT; e) PdCl2(PPh3)2, CuI, Et3N, DMF, 50 °C; f) 2-Cyano-N-methylacetamide, DBU, THF, RT.
Highlights.
Functional characterization of ~3,500 mutants of Src kinase
Development of compounds for precise control of Src conformation in mammalian cells
Discovery of regulatory role for Src's SH4 domain
Acknowledgements
We thank Stan Fields, Christine Queitsch and Lea Starita for guidance. We thank Nathaniel Peters at the W.M. Keck Center for Advanced Studies in Neural Signaling for microscopy assistance and Martin Sadilek for mass spectrometry assistance. This work was supported by the National Institute of General Medical Sciences (R01GM109110 to D.M.F. and R01GM086858 to D.J.M.). D.M.F. is a CIFAR Azrieli Global Scholar. E.A. and E.M.D. were supported by a National Science Foundation Graduate Research Fellowship. E.A. was supported by the Ruth L. Kirschstein National Research Service Award T32 from the NHGRI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Bjorge JD, Bellagamba C, Cheng H-C, Tanaka A, Wang JH, and Fujita DJ (1995). Characterization of Two Activated Mutants of Human pp60c-srcThat Escape c-Src Kinase Regulation by Distinct Mechanisms. Journal of Biological Chemistry 270, 24222–24228. [DOI] [PubMed] [Google Scholar]
- Boggon TJ, and Eck MJ (2004). Structure and regulation of Src family kinases. Oncogene 23, 7918–7927. [DOI] [PubMed] [Google Scholar]
- Charras GT (2008). A short history of blebbing. J Microsc 231, 466–478. [DOI] [PubMed] [Google Scholar]
- Devkota S, Joseph RE, Boyken SE, Fulton DB, and Andreotti AH (2017). An Autoinhibitory Role for the Pleckstrin Homology Domain of Interleukin-2-Inducible Tyrosine Kinase and Its Interplay with Canonical Phospholipid Recognition. Biochemistry 56, 2938–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M, Wilson LK, Trager JB, Thorner J, and Martin GS (1994). Aberrant protein phosphorylation at tyrosine is responsible for the growth-inhibitory action of pp60v-src expressed in the yeast Saccharomyces cerevisiae. Molecular Biology of the Cell 5, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler DM, and Fields S (2014). Deep mutational scanning: a new style of protein science. Nat Meth 11, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, and Fields S (2010). High-resolution mapping of protein sequence-function relationships. Nat Meth 7, 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler DM, Araya CL, Gerard W, and Fields S (2011). Enrich: software for analysis of protein function by enrichment and depletion of variants. Bioinformatics 27, 3430–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Nafría J, Watson JF, and Greger IH (2016). IVA cloning: A single-tube universal cloning system exploiting bacterial In Vivo Assembly. Nature Publishing Group; 6, 27459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, and Schiestl RH (2007). High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nature Protocols 2, 31–34. [DOI] [PubMed] [Google Scholar]
- Giorgione J, and Newton AC (2003). Measuring the binding of protein kinase C to sucrose-loaded vesicles. Methods Mol. Biol. 233, 105–113. [DOI] [PubMed] [Google Scholar]
- Golkowski M, Vidadala RSR, Lombard CK, Suh HW, Maly DJ, and Ong S-E (2017). Kinobead and Single-Shot LC-MS Profiling Identifies Selective PKD Inhibitors. J. Proteome Res. 16, 1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonfloni S, Williams JC, Hattula K, Weijland A, Wierenga RK, and Superti-Furga G (1997). The role of the linker between the SH2 domain and catalytic domain in the regulation and function of Src. Embo J. 16, 7261–7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BG, Acar H, Nandipati A, and Barlow M (2013). Growth Rates Made Easy. Molecular Biology and Evolution 31, 232–238. [DOI] [PubMed] [Google Scholar]
- Hantschel O, Nagar B, Guettler S, Kretzschmar J, Dorey K, Kuriyan J, and Superti-Furga G (2003). A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 112, 845–857. [DOI] [PubMed] [Google Scholar]
- Hiatt JB, Patwardhan RP, Turner EH, Lee C, and Shendure J (2010). Parallel, tag-directed assembly of locally derived short sequence reads. Nat Meth 7, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph RE, Wales TE, Fulton DB, Engen JR, and Andreotti AH (2017). Achieving a Graded Immune Response: BTK Adopts a Range of Active/Inactive Conformations Dictated by Multiple Interdomain Contacts. Structure 25, 1481–1494.e1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahsai AW, Rajagopal S, Sun J, and Xiao K (2014). Monitoring protein conformational changes and dynamics using stable-isotope labeling and mass spectrometry. Nat Protoc 9, 1301–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinghoffer RA, Sachsenmaier C, Cooper JA, and Soriano P (1999). Src family kinases are required for integrin but not PDGFR signal transduction. Embo J. 18, 2459–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurty R, Brigham JL, Leonard SE, Ranjitkar P, Larson ET, Dale EJ, Merritt EA, and Maly DJ (2012). Active site profiling reveals coupling between domains in SRC-family kinases. Nat Chem Biol 9, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritzer JA, Freyzon Y, and Lindquist S (2018). Yeast can accommodate phosphotyrosine: v-Src toxicity in yeast arises from a single disrupted pathway. FEMS Yeast Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung JE, and Jura N (2016). Structural Basis for the Non-catalytic Functions of Protein Kinases. Structure 24, 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFevre-Bernt M, Sicheri F, Pico A, Porter M, Kuriyan J, and Miller WT (1998). Intramolecular regulatory interactions in the Src family kinase Hck probed by mutagenesis of a conserved tryptophan residue. Journal of Biological Chemistry 273, 32129–32134. [DOI] [PubMed] [Google Scholar]
- Lahiry P, Torkamani A, Schork NJ, and Hegele RA (2010). Kinase mutations in human disease: interpreting genotype–phenotype relationships. Nature Reviews Genetics 11, 60–74. [DOI] [PubMed] [Google Scholar]
- Leonard SE, Register AC, Krishnamurty R, Brighty GJ, and Maly DJ (2014). Divergent Modulation of Src-Family Kinase Regulatory Interactions with ATP-Competitive Inhibitors. ACS Chem. Biol 9, 1894–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JB, Iba H, and Hanafusa H (1986). Activation of the transforming potential of p60c-src by a single amino acid change. Proc. Natl. Acad. Sci. U.S.a. 83, 4228–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei M, Arbesú M, Le Roux A-L, Amata I, Roche S, and Pons M (2015). The SH3 Domain Acts as a Scaffold for the N-Terminal Intrinsically Disordered Regions of c-Src. Structure 23, 893–902. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, and Sudarsanam S (2002). The protein kinase complement of the human genome. Science 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- McLaughlin S, and Aderem A (1995). The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends in Biochemical Sciences 20, 272–276. [DOI] [PubMed] [Google Scholar]
- Olsen JG, Teilum K, and Kragelund BB (2017). Behaviour of intrinsically disordered proteins in protein-protein complexes with an emphasis on fuzziness. Cellular and Molecular Life Sciences 74, 3175–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan P, and Resh MD (2010). Myristoylation and Membrane Binding Regulate c-Src Stability and Kinase Activity. Mol. Cell. Biol. 30, 4094–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Rivoire O, Reynolds KA, and Ranganathan R (2016). Evolution-Based Functional Decomposition of Proteins. PLoS Comput Biol 12, e1004817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röhrig UF, Majjigapu SR, Grosdidier A, Bron S, Stroobant V, Pilotte L, Colau D, Vogel P, Van den Eynde BJ, Zoete V, et al. (2012). Rational Design of 4-Aryl-1,2,3-Triazoles for Indoleamine 2,3-Dioxygenase 1 Inhibition. J. Med. Chem. 55, 5270–5290. [DOI] [PubMed] [Google Scholar]
- Rubin AF, Gelman H, Lucas N, Bajjalieh SM, Papenfuss AT, Speed TP, and Fowler DM (2017). A statistical framework for analyzing deep mutational scanning data. Genome Biol. 18, 741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel-Dugan C, Meyer BE, Thomas SM, and Brugge JS (1992). Effects of SH2 and SH3 deletions on the functional activities of wild-type and transforming variants of c-Src. Mol. Cell. Biol. 12, 1835–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frodin M, and Taunton J (2012). Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat Chem Biol 8, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal CT, Zhou W, Buser CA, McLaughlin S, and Resh MD (1994). Amino-terminal basic residues of Src mediate membrane binding through electrostatic interaction with acidic phospholipids. Proc. Natl. Acad. Sci. U.S.a. 91, 12253–12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skora L, Mestan J, Fabbro D, Jahnke W, and Grzesiek S (2013). NMR reveals the allosteric opening and closing of Abelson tyrosine kinase by ATP-site and myristoyl pocket inhibitors. Proc. Natl. Acad. Sci. U.S.a. 110, E4437–E4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, DiMaio F, Wang RY-R, Kim D, Miles C, Brunette TJ, Thompson J, and Baker D (2013). High-Resolution Comparative Modeling with RosettaCM. Structure 21, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassov DS, Ruiz-Saenz A, Piple A, and Moasser MM (2018). A Dimerization Function in the Intrinsically Disordered N-Terminal Region of Src. Cell Reports 25, 449–463.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starita LM, Young DL, Islam M, Kitzman JO, Gullingsrud J, Hause RJ, Fowler DM, Parvin JD, Shendure J, and Fields S (2015). Massively Parallel Functional Analysis of BRCA1 RING Domain Variants. Genetics 200, 413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Onge RPS, Mani R, King OD, Heilbut A, Labunskyy VM, Chen W, Pham L, Zhang LV, Tong AHY, et al. (2011). Knocking out multigene redundancies via cycles of sexual assortment and fluorescence selection. Nat Meth 8, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Keshwani MM, Steichen JM, and Kornev AP (2012). Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philosophical Transactions of the Royal Society B: Biological Sciences 367, 2517–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile CS, Witte MD, Blom AEM, Kundrat L, Ploegh HL, and Guimaraes CP (2013). Site-specific N-terminal labeling of proteins using sortase-mediated reactions. Nature Protocols 8, 1800–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson EE, Kornev AP, Kannan N, Kim C, Eyck, Ten LF, and Taylor SS (2009). Comparative surface geometry of the protein kinase family. Protein Science 18, 2016–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien MZ, Meyer AG, Sydykova DK, Spielman SJ, and Wilke CO (2013). Maximum Allowed Solvent Accessibilites of Residues in Proteins. PLoS ONE 8, e80635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tighe A, Staples O, and Taylor S (2008). Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. The Journal of Cell Biology 181, 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournaviti S, Hannemann S, Terjung S, Kitzing TM, Stegmayer C, Ritzerfeld J, Walther P, Grosse R, Nickel W, and Fackler OT (2007). SH4-domain-induced plasma membrane dynamization promotes bleb-associated cell motility. Journal of Cell Science 120, 3820–3829. [DOI] [PubMed] [Google Scholar]
- Towler DA, Adams SP, Eubanks SR, Towery DS, Jackson-Machelski E, Glaser L, and Gordon JI (1988). Myristoyl CoA:protein N-myristoyltransferase activities from rat liver and yeast possess overlapping yet distinct peptide substrate specificities. Journal of Biological Chemistry 263, 1784–1790. [PubMed] [Google Scholar]
- Wang Q, Vogan EM, Nocka LM, Rosen CE, Zorn JA, Harrison SC, and Kuriyan J (2015). Autoinhibition of Bruton's tyrosine kinase (Btk) and activation by soluble inositol hexakisphosphate. Elife 4, e06074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Cahill SM, Blumenstein M, and Lawrence DS (2006). Self-Reporting Fluorescent Substrates of Protein Tyrosine Kinases. J. Am. Chem. Soc. 128, 1808–1809. [DOI] [PubMed] [Google Scholar]
- Xu W, Doshi A, Lei M, Eck MJ, and Harrison SC (1999). Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell 3, 629–638. [DOI] [PubMed] [Google Scholar]
- Xu W, Harrison SC, and Eck MJ (1997). Three-dimensional structure of the tyrosine kinase c-Src. Nature 385, 595–602. [DOI] [PubMed] [Google Scholar]
- Young MA, Gonfloni S, Superti-Furga G, Roux B, and Kuriyan J (2001). Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell 105, 115–126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Live-cell imaging of SrcGFP V284C treated with 2 (3 μM). Relates to Figure 5B.
Movie S2. Live-cell imaging of SrcGFP V284C treated with 3 (10 μM). Relates to Figure 5B.
Table S2. Scores and classifications of Src CD variants. Relates to Figure 2F.
Table S4. Scores and classifications of Src SH4 variants. Relates to Figure 7E.
Table S6. Primers used in study. Relates to all Figures.
Table S7. Log2 SILAC ratios from Kinobead profiling. Relates to Figure 4H.
Data Availability Statement
Sequencing reads were deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through accession number GSE114098. Microscopy images and Rosetta models are accessible in Mendeley Data DOI: 10.17632/4wcrr6vswv.1.