

SUMMARY
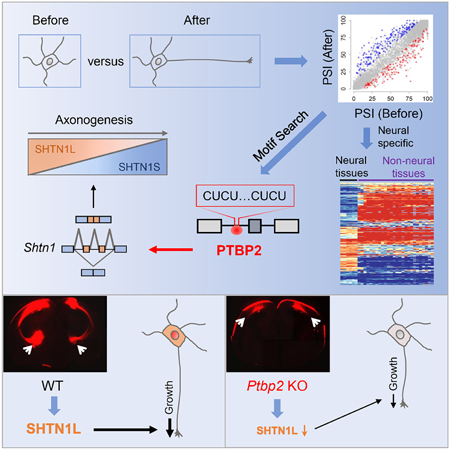
How a neuron acquires an axon is a fundamental question. Piecemeal identification of many axonogenesis-related genes has been done, but coordinated regulation is unknown. Through unbiased transcriptome profiling of immature primary cortical neurons during early axon formation, we discovered an association between axonogenesis and neuron-specific alternative splicing. Known axonogenesis genes exhibit little expression alternation but widespread splicing changes. Axonogenesis-associated splicing is governed by RNA binding protein PTBP2, which is enriched in neurons and peaks around axonogenesis in the brain. Cortical depletion of PTBP2 prematurely induces axonogenesis-associated splicing, causes imbalanced expression of axonogenesis-associated isoforms, and specifically affects axon formation in vitro and in vivo. PTBP2-controlled axongenesis-associated Shtn1 splicing determines SHTN1’s capacity to regulate actin interaction, polymerization, and axon growth. Precocious Shtn1 isoform switch contributes to disorganized axon formation of Ptbp2−/− neurons. We conclude that PTBP2-orchestrated alternative splicing programming is required for robust generation of a single axon in mammals.
Keywords: axon formation, neurite, polarity, polarization, RNA binding protein, polypyrimidine-tract, PTB, nPTB, Shootin1, SHOT1
Graphical Abstract
eTOC
Zhang et al. discover a PTBP2-orchestrated neural specific alternative splicing program essential for robust axon formation. These findings could shed light on axon pathophysiology in diseases and how axonogenesis happens only in neurons but not in any other cells.
INTRODUCTION
The directional propagation of information in the mammalian brain requires morphological and functional asymmetry of neurons. The foremost step in establishing neuronal polarity is the compartmentalization of distinct axonal and somatodendritic domains, which is a prerequisite for subsequent synaptogenesis, myelination, synaptic transmission and circuit wiring. How a neuron acquires its one and only axon is one of the most fundamental questions in neurobiology.
The exact determinants and precise hierarchy of molecular events leading to axon formation have been difficult to dissect (Barnes and Polleux, 2009; Cheng and Poo, 2012; de la Torre-Ubieta and Bonni, 2011). The success in differentiating embryonic or induced pluripotent stem cells into functional neurons in the absence of a niche microenvironment strengthened the argument for the intrinsic capability of neurons to form an axon. If axonogenesis is exclusive to neurons, neurons are probably uniquely equipped with a set of special molecules and/or regulatory rules for their ability to form an axon, which have not been clearly defined.
Genes controlling axon formation have been identified and studied piecemeal. Their presumed coordination to fulfill robust axon formation, however, has not been reported. These molecules include transcription factors and growth factors, polarity genes believed to be symmetry-breaking signals, enzymes as signal transducers remodeling the cytoskeleton, molecular motors, and scaffolding proteins establishing the axonal compartment (van Beuningen et al., 2015; Chen et al., 2013; McNeely et al., 2017; Namba et al., 2014; Neukirchen and Bradke, 2011; Rao et al., 2017; Tahirovic et al., 2010; de la Torre-Ubieta and Bonni, 2011; Zollinger et al., 2015). Most studies relied on overexpression of proteins in cultured neurons and recently on RNAi-mediated knockdown. Only a few analyzed physiological functions of genes in vivo using knockout animals (Barnes et al., 2007; Garvalov et al., 2007; Kishi et al., 2005; Yi et al., 2010). In vivo studies of axon formation and analyzing single axons are challenging, because axonogenesis occurs within a short time window and most axons have narrow diameters (< 1 µm).
Neurons make extensive use of splicing regulation to generate specialized protein isoforms, presumably in keeping with their morphological and functional complexity. In many cases, alternative splicing is a tightly controlled mRNA processing step embedded in the genetically hardwired regulatory program of tissue development. Studies have shown alternative splicing is important for neurogenesis, synaptogenesis, and synaptic functions (Darnell, 2013; Raj and Blencowe, 2015; Vuong et al., 2016a; Zheng and Black, 2013). The contribution of alternative splicing to axonogenesis, however, has not been systematically examined.
Alternative splicing is governed by specialized RNA binding proteins (RBPs). With their ability to expand RNA and proteome diversity, RBPs could be molecular orchestrators of neuronal differentiation. RBPs known to regulate alternative splicing in the brain include polypyrimidine tract binding protein 2 (PTBP2). The roles of PTBP2 in the brain has been difficult to dissect. Neural stem cells and progenitors express PTBP1, a close paralog of PTBP2, until they exit mitosis, when PTBP1 is sharply downregulated and PTBP2 is induced (Boutz et al., 2007; Linares et al., 2015). The functional significance of such a paralog switch is still a mystery, but PTBP2 is mostly irreplaceable by PTBP1 during brain development (Vuong et al., 2016b). PTBP2 expression is then maintained for days before drastically reduced prior to synapse formation (Zheng et al., 2012). These phasic expression patterns of PTBP1 and PTBP2 are conserved between human and mouse (Zheng, 2016a). Ectopic PTBP2 expression in mature neurons inhibits synapse formation (Zheng et al., 2012). However, Ptbp2−/− neurons do not precociously generate synapses (Li et al., 2014), suggesting additional unknown PTBP2 functions in differentiating neurons before synapse formation.
Seeking molecular determinants of cortical axonogenesis, we uncovered a surprisingly predominant association between alternative splicing programming and axonogenesis. Motif analysis predicted PTBP2 as a major regulator. Using comprehensive multidisciplinary approaches, we showed that PTBP2 directly influences early axon formation.
RESULTS
The transcriptome landscape during early axonogenesis
Comparing transcriptomes before and after an axon is specified can be informative to reveal regulatory rules that ensure production of a single axon (Figure 1A). Developing cortical excitatory neurons display a multipolar shape in the subventricular and intermediate zones, and subsequently a bipolar shape in the cortical plate (Figure S1A). Axons emerge during the multipolar-to-bipolar transition (Hand and Polleux, 2011; Hatanaka and Yamauchi, 2013). It is currently infeasible to directly purify neurons right before and after axon emergence from the brain. Therefore, we cultured immature cortical neurons from embryonic day 14 neocortices, which predominantly consist of neural stem cells and progenitors (Chen et al., 2005; Dehay and Kennedy, 2007; Fan et al., 2008; Klingler, 2017; Zimmer et al., 2004). Primary neurons undergo stereotypical morphological transformation, mimicking in vivo transitions. At 1 day in vitro (1 DIV), these neurons extend multiple indistinguishable processes, like pre-axonogenesis neurons in vivo. Around 2–3 DIV, one of the neurites accelerates its growth to become the axon (Figure S1B).
Figure 1. The transcriptome landscape during early axonogenesis.
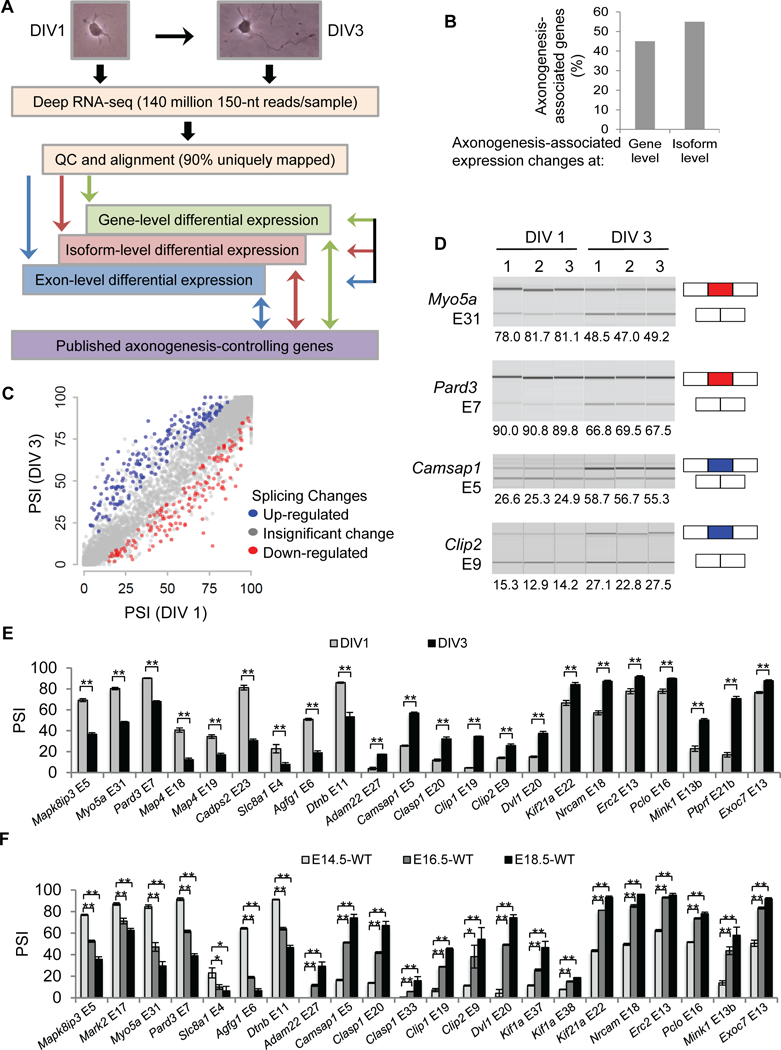
(A) Transcriptome profiling design and analysis. (B) Distribution of genes that exhibit differential expression at the gene level vs at the isoform level alone during early axon formation. (C) PSI of alternative exons in DIV 1 neurons vs DIV 3 neurons. Each dot represents a cassette exon passing the coverage criteria. (D) Virtual gels of quantitative capillary electrophoresis show splicing changes of Myo5a, Pard3, Camsap1, and Clip2 in three biological replicates during early axonogenesis. PSIs are stated below the images. Red: down-regulated exons. Blue: up-regulated exons. PSIs of axonogenesis-associated exons in cortical cultures (E) at DIV 1 and DIV 3 and in neocortices (F) at E14.5, E16.5, and E18.5. Data are represented as mean ± SEM of 3–4 (E) and 2–3 (F) animals of different litters. **, P-value < 0.01; *, P-value < 0.05; Student’s t test. See also Figures S1, S2.
We performed unbiased deep RNA sequencing (RNA-Seq) of DIV 1 and DIV 3 neurons and identified differentially expressed genes (DEGs). Transcriptome profiling was highly reproducible for biological replicates but showed larger differences before and after axons are generated (Figure S2A-F). From DIV 1 to DIV 3, 287 and 147 genes are up- and down-regulated, respectively (fold change ≥ 2, P-value ≤ 0.001, the corresponding FDR=0.003), representing 0.87% of total (49,671) examined genes (Table S1).
Gene-level analysis underappreciates the complexity of transcriptome regulation. We determined differentially expressed isoforms (DEIs) and found about half of the transcriptome changes occurred at the isoform level (Figure 1B). Specifically, we applied the same algorithm (RSEM) and the same criteria for calling differential expression of DEGs and DEIs, thereby preventing biases of using different pipelines. DEGs should exhibit DEIs: 252 of 434 DEGs did and herein referred to “axonogenesis-associated gene-level changes”. For the other 182 DEGs, their isoform abundances were below the expression cutoff. We uncovered an additional 304 genes with DEIs that were not DEGs (Table S2). We termed these changes “axonogensis-associated isoform-level changes”.
The parsimonious explanation for isoform- but not gene-level changes is differential alternative splicing. We directly inferred alternative splicing level and assigned each exon a “percent spliced in” (PSI) value using the GeneSplice pipeline (Vuong et al., 2016b). We found 181 activated (PSIDIV3 – PSIDIV1 > 10) and 150 repressed (PSIDIV3 – PSIDIV1 < −10) cassette exons (P-values ≤ 0.001, the corresponding FDR=0.02), representing 2.2% of all examined cassette exons (Figure 1C, Table S3). We called them “axonogenesis-associated splicing changes” (corresponding to 271 genes).
To validate the RNA-seq findings we performed RT-PCR followed by quantitative capillary electrophoresis analysis. Among 28 randomly selected axonogenesis-associated exons, all showed statistically significant ∆PSI between DIV 1 and DIV 3, and 22 exhibited |∆PSI| > 10 (Figure 1D-E). These genes contain a wide range of functions associated with axonogenesis. For example, Pard3 is a key component of the Par polarity complex (PAR3/PAR6/aPKC) essential to polarity of many cell types including neurons (Hapak et al., 2018). Myo5a and Kif21a are molecular motors responsible for cargo transports. Clip1, Clip2, Camsap1, Clasp1, Map4, and Pclo interact with cytoskeleton molecules and/or influence cytoskeleton dynamics.
We also validated the results in vivo. Developing neocortices contain an increasing proportion of axon-generating neurons over time during embryonic development, so splicing differences before and after axonogenesis should be reflected by splicing differences between early (e.g., E14) and late (e.g., E18) embryonic cortices. We tested 24 of 28 aforementioned exons and 23 exhibited significant ∆PSI between E14.5 and E18.5 neocortices: 22 showed |∆PSI| > 10 (Figure 1F). The |∆PSIE16.5-E14.5| were generally larger than |∆PSIE18.5-E16.5|, agreeing with the notion that the splicing changes we identified reflected programming of early axonogenesis.
Differential splicing is strongly associated with characteristics of axonogenesis
We next asked whether differential gene expression and differential splicing contributed to different aspects of axonogenesis. To systemically describe biological characteristics of axon formation, we curated 159 protein-coding genes previously reported to regulate axon formation (Table S4), and determined their collective features (e.g., gene ontology, protein domains, and molecular pathways). Published axonogenesis-controlling genes are enriched for terms like axonogenesis, cytoskeleton, and protein binding. We tested which terms were enriched in DEGs versus in differentially spliced genes.
Enriched terms of differentially spliced genes significantly overlapped with those of the published axonogenesis genes (P-value = 3.42×10−18, hypergeometric test; Figure 2A, blue dots). The overlap of enriched terms between DEGs and published axonogenesis genes was less significant (P-value = 0.013, hypergeometric test; Figure 2A, grey dots). Differentially spliced genes had a very small overlap with DEGs (Figure 2B, P value = 0.21, hypergeometric test), further indicative of two independent regulatory modes.
Figure 2. Differential splicing is associated with characteristics of axonogenesis.
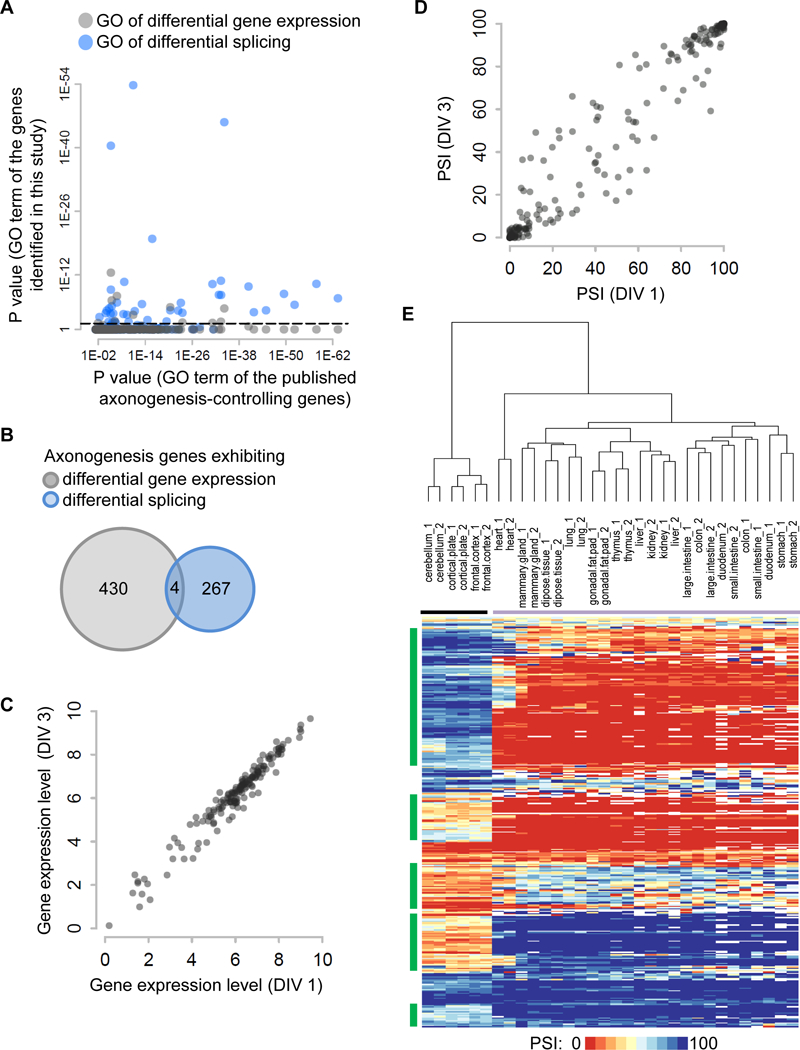
(A) A scatter plot shows the Bonferroni-corrected P values of enriched Gene Ontology terms for genes exhibiting differential gene expression (grey dots) and differential splicing (blue dots) on the y axis vs previously published axonogenesis-controlling genes on the x axis. Dashed lines demark P value equal to 0.05 on the y axis. (B) An insignificant overlap of genes exhibiting differential gene expression and those exhibiting differential splicing during axon formation. (C) Gene expression levels (log2 (TPM +1)) of published axonogenesis-controlling genes at DIV 1 and DIV 3. Each dot represents an axonogenesis gene. (D) PSI of published axonogenesis-controlling genes at DIV 1 and DIV 3. Each dot represents a cassette exon. (E) Heatmap of PSI values of axonogenesis-associated exons in adult tissues profiled in the ENCODE project. These include neural tissues (cerebellum, cortical plate, frontal cortex labeled by black horizontal line) and non-neural tissues (heart, mammary gland, subcutaneous adipose tissue, lung, gonadal fat pad, thymus, liver, kidney, large intestine, colon, duodenum, small intestine, and stomach labeled by purple horizontal line). Green vertical lines indicate exon clusters that exhibit distinct PSI values between neural and non-neural tissues. See also Figure S2.
Differentially spliced genes and DEGs were identified from different algorithms using different metrics (∆PSI vs fold change). To reassess, we compared axonogenesis-associated gene-level changes (group 1, 252 genes) versus axonogenesis-associated isoform-level changes (group 2, 304 genes), both derived from the same RSEM algorithm with the same cutoffs. Only group 2 genes were enriched for the same terms as published axonogenesis genes (P-value = 2.93×10−14, hypergeometric test; Figure S2G, pink dots), whereas group 1 had no significant overlap (P-value = 0.15, hypergeometric test; Figure S2G, grey dots), consistent with our prior observation.
Our data show that axonogenesis coincides with extensive changes in alternative exon usage. Differential splicing is more associated with key characteristics of axonogenesis than differential gene expression, hence is probably a critical driver of axon formation. Consistent with this notion, almost all published axonogenesis genes exhibit insignificant expression changes between DIV 1 and DIV 3 neurons (Figure 2C). In contrast, many of these genes exhibited marked splicing alteration (Figure 2D) with a much wider dispersion between DIV 1 and DIV 3 neurons (dispersion values: 0.02 for gene expression changes vs 0.04 for splicing changes, P-value = 0.00014, Student’s t test).
Axon formation happens only in neurons and not in any other cells. We asked whether axonogenesis-associated exons show distinct splicing between neural and non-neural tissues. Using the mouse RNA-seq data from the ENCODE project (ENCODE Project Consortium, 2012), we determined the PSI values of all axonogenesis-associated exons in adult tissues and performed two-way hierarchical clustering (Figure 2E). All brain tissues, including cerebellum, cortical plates, and frontal cortex, were clustered in one group, and all non-neural tissues were clustered in the other group. The distance between the two groups was large. The difference between neural and non-neural groups was not driven by the differences of a subset of tissues. Rather, the PSI values were quite similar within each group, suggesting a binary separation (e.g., green exons in Figure 2E). Therefore, most axonogenesis-associated exons exhibit neural-specific splicing, which might reflect axonogenesis as a neural-specific phenomenon.
Although most exons encode amino acids of unknown motifs, we identified examples of interest to axonogenesis. For instance, CLIP2 knockdown decreases axon growth (Neukirchen and Bradke, 2011) and its gene deletion causes Williams-Beuren Syndrome. The axonogenesis-associated exon encodes amino acids within the first coiled-coil domain (Figure S2H), which may be necessary for CLIP2 to interact with its binding partners and to stabilize microtubules. CAMSAP1 mutations induced overextension of neurites and impaired axon regeneration (Chuang et al., 2014; Marcette et al., 2014). Inclusion of the axonogenesis-associated exon splits CAMSAP1’s Calponin-homology domain and may affect its activity of protecting microtubules from depolymerization (Figure S2I). As a target of ABL-family nonreceptor tyrosine kinase, ABI2 conveys extracellular signals to affect dynamics of actin cytoskeleton (Chen et al., 2014; Courtney et al., 2000). The ABI2 axonogenesis-associated exon encodes a stretch of proline residues at the heart of the proline-rich domain that mediates ABI2’s interaction with c-ABL (Figure S2J) (Dai and Pendergast, 1995). Therefore, skipping this exon may diminish or abolish ABI2’s role as a signal transducer. PPP3CA is the catalytic subunit of Calcineurin, a calcium and calmodulin dependent serine/threonine protein phosphatase. Inhibition of Calcineurin promoted axon growth (Wang et al., 2016). The amino acids encoded by the axonogenesis-associated exon are within its autoinhibitory domain (Figure S2K) and likely modulates Calcineurin’s activity.
Shtn1 switches isoform expression during early axonogenesis
We prioritized known axonogenesis genes displaying an obvious isoform switch but insignificant gene-level change, and therefore focused on Shtn1 (Shootin1). Originally discovered as a neuronal polarization-induced gene (Toriyama et al., 2006), Shtn1 knockout mice display agenesis of multiple axonal tracts (Baba et al., 2018). The originally identified SHTN1 protein is the short isoform (SHTN1S), which skips exon 15 and 16. Inclusion of these exons shifts the reading frame and alters the C terminus of the protein, resulting in an uncharacterized longer isoform Shtn1L (Figure 3A). The functional differences between the two isoforms have not been reported. The mechanism of SHTN1S upregulation during neuronal polarization has not been described either.
Figure 3. Shtn1 switches isoform expression during early axonogenesis.
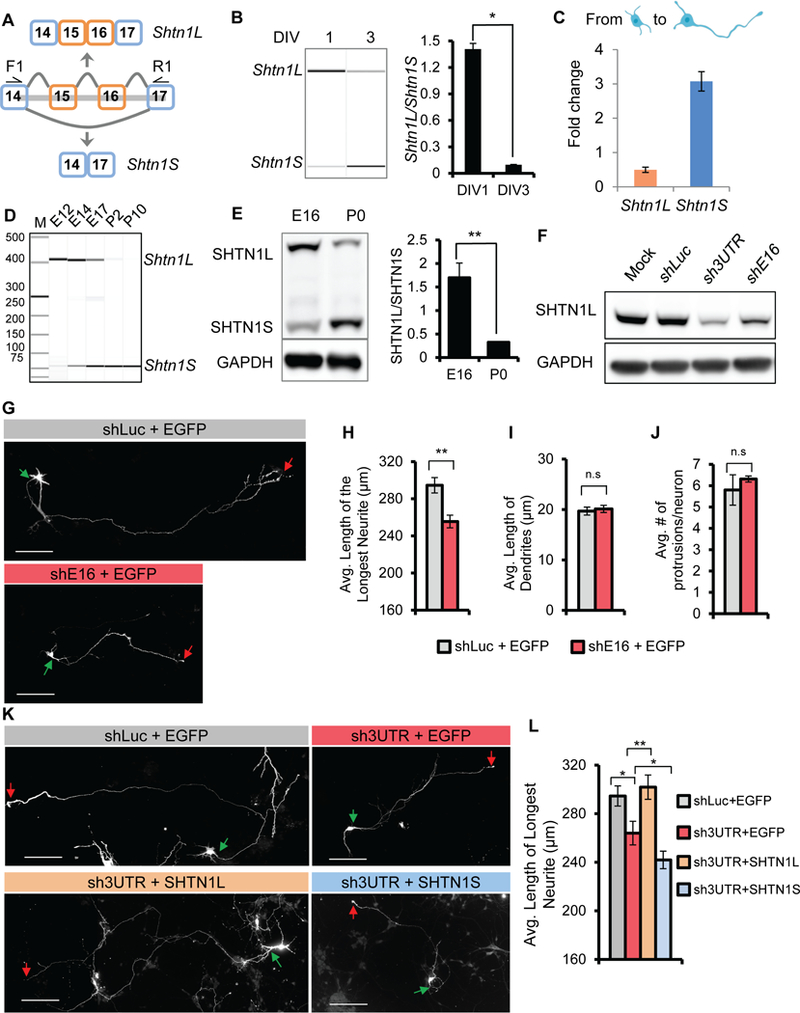
(A) Schematic of Shtn1 alternative splicing. F1 and R1 depict the location of primers for RT-PCR analysis of Shtn1 alternative splicing. Exclusion of exons 15 and 16 generates a frame-shift and results in an early STOP codon in exon 17. (B) A virtual gel image shows Shtn1 switches its mRNA isoform expression from DIV 1 to DIV 3. Bar graph summarizes the ratio of the expressed isoforms. Data represent mean ± SD. * p< 0.05. (C) TPM values in DIV 3 neurons relative to the TPM values in DIV 1 neurons (Fold change) of Shtn1L and Shtn1S mRNA isoforms. (D) Shtn1 switches its mRNA isoform expression in neocortices from Shtn1L at E12 to Shtn1S at P2. (E) Isoform switch of SHTN1 proteins from E16 to P0. Bar graph summarizes the ratio of the expressed isoforms. Data represent mean ± SD from 3 independent experiments. ** p< 0.01. (F) Western blot shows SHTN1L knockdown by small hairpin RNAs targeting exon 16 (shE16), and 3’ UTR (sh3UTR) of endogenous Shtn1 mRNA, in N2a cells. (G-H) Knockdown of Shtn1L isoform in mouse primary cortical neurons using shE16 leads to shorter primary axons at DIV5 without affecting minor neurite growth or neurite number. N = 242 and 256 neurons for shLuc and shE16, respectively from 3 animals of 3 litters. Data are represented as mean ± SEM. (K-L) The shortened axons upon sh3UTR transfection are rescued by overexpression of Shtn1L but not Shtn1S. N = 242, 133, 149, 156 neurons for shLuc, sh3UTR+EGFP, sh3UTR+ Shtn1L, sh3UTR+ Shtn1S, respectively from 2–3 animals of 3 litters. Data are represented as mean ± SEM. Student’s t-test, 2-tailed, *p<0.05, **p<0.01. Green and red arrows indicate starting and ending points of primary axons, respectively. Scale bars: 50µm.
We found a noticeable transition from Shtn1L to Shtn1S during early axonogenesis (Figure 3B). This was primarily due to alternative splicing, as shown by an independent analysis using isoform-level quantitation in which Shtn1S was up-regulated by threefold, whereas Shtn1L was down-regulated by twofold (Figure 3C).
We found that Shtn1 splicing is developmentally regulated in neocortices (Figure 3D). The E12 neocortex expresses Shtn1L almost exclusively, whereas at postnatal day 2 (P2) when primary axon formation is completed, the neocortex expresses only Shtn1S. Therefore, the transition from Shtn1L to Shtn1S is concurrent with axonogenesis in vitro and in vivo. Consistently, the Shtn1L-to-Shtn1S switch was observed at the protein level in mouse neocortices (Figure 3E). Therefore, axonogenesis-associated Shtn1 expression is regulated by its isoform switch, which explains polarization-induced SHTN1S expression.
Shtn1L and Shtn1S isoforms exhibit different activity during early axonogenesis
The robust switch from Shtn1L to Shtn1S suggested that the two Shtn1 isoforms may exert different roles during axon formation. Inagaki’s group reported that Shtn1S overexpression induced multiple short axons (Kubo et al., 2015; Toriyama et al., 2006). We found overexpressing Shtn1L did not lead to an increase in axon number, suggesting the two isoforms have differential abilities in specifying axons.
To investigate Shtn1L’s function, we designed Shtn1L isoform-specific shRNA targeting exon 16 (shE16), which effectively decreased SHTN1L protein (Figure 3F). Depletion of Shtn1L in neurons decreased axon length without affecting dendrite length or neurite number (Figure 3G-J). The impact of SHTN1S on axon length has not been clearly documented in literature. To test whether Shtn1L and Shtn1S were equivalent in promoting axon growth, we knocked down total Shtn1 proteins using a shRNA targeting the common 3’ UTR region in exon 17 (sh3UTR) and compensated with cDNA expression of individual isoforms. Transfection of sh3UTR led to shorter axon as shE16 did (Figure 3K-L), further confirming Shtn1’s role in axon growth. This defect was rescued by the Shtn1L isoform, but not the Shtn1S isoform, suggesting that SHTN1L is the major force accounting for SHTN1’s activity in promoting axon growth. In summary, the two Shtn1 isoforms are expressed sequentially via alternative splicing to fulfill different aspects of axonogenesis. Shtn1L promotes axon growth, determining axon length. Shtn1S appears to promote axon specification, determining axon fate.
SHTN1L isoform directly interacts with actin and promotes actin polymerization
To understand the differential biochemical activities of two SHTN1 isoforms, we investigated their possible interactions with cytoskeleton molecules. Due to lack of isoform-specific antibodies we expressed N-terminal EGFP-tagged SHTN1 proteins and performed co-immunoprecipitation using anti-GFP antibody. We found that actin co-precipitated with SHTN1L but not with SHTN1S proteins (Figure 4A-B).
Figure 4. SHTN1L but not SHTN1S binds actin and enhances actin polymerization.
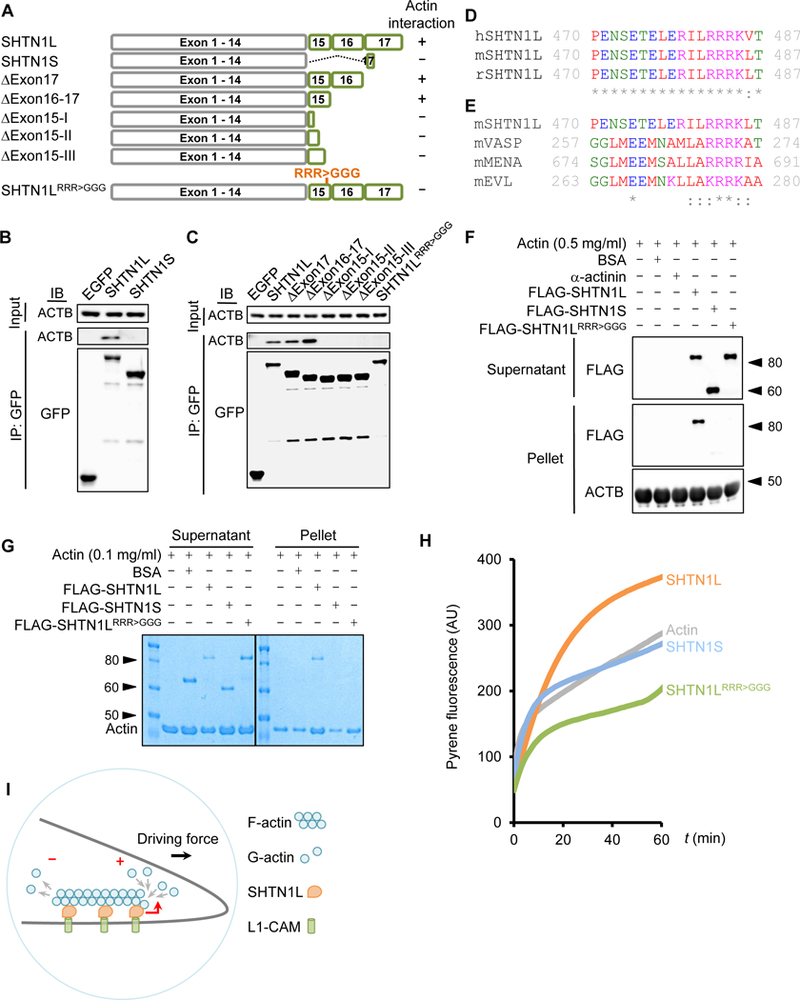
(A) Schematic of SHTN1 isoforms and mutant constructs, as well as their actin-binding abilities. (B) Actin is co-immunoprecipitated with SHTN1L but not SHTN1S. EGFP-tagged SHTN1L and SHNT1S, and EGFP were expressed in N2a cells for anti-GFP co-immunoprecipitation. (C) Immunoprecipitation of EGFP-tagged SHTN1L and various mutants for detecting the coimmunoprecipitated actin molecules. (D) Clustal Omega multiple sequence alignment of the human (h), mouse (m) and rat (r) SHTN1L peptide sequence within exon 15. Asterisk: a conserved residue. Colon: a conserved substitution. Red: small and hydrophobic. Blue: acidic. Magenta: basic. Green: hydroxyl or sulhydryl or amine or G. (E) Comparison of F-actin interaction sites of mouse SHTN1L, VASP, MENA and EVL proteins. Numbers depict the positions of the F-actin interaction sites within each protein. (F) Actin cosedimentation assay assesses FLAG-SHTN1 binding to F-actin. Pellet and supernatant fractions were resolved and immunoblotted with anti-FLAG antibody. (G) SHTN1L promotes actin polymerization. A representative image from the cosedimentation assay performed under a buffer condition refractory to actin self-polymerization (STAR Methods). Pellet and supernatant fractions were resolved by SDS/PAGE and visualized by Coomassie blue staining. (H) A representative image of pyrene-actin assay showing that SHTN1L promotes actin polymerization. G-actin monomers (2 µM, 12.5% pyrene labeled; primed with 1 mM EGTA and 0.1 mM MgCl2) were simultaneously assembled in the absence or presence of each FLAG-SHTN1 variants (275 nM). Experiments were repeated independently three times with similar results. (I) Schematic illustration of the proposed function of SHTN1L in growth cones. See also Figure S3.
Shtn1L and Shtn1S encode the same N-terminal sequence but different C-termini (Figure 4A). SHTN1S’s unique C-terminus is only three residues (ASQ) whereas SHTN1L’s is 178 amino acids long. Therefore, SHTN1L C-terminus likely contains an actin-binding motif. Deletion of exon 17, or exons 16 and 17, did not impact SHTN1L’s interaction with actin, indicating that the actin-binding motif likely resides in exon 15. Exon 15 encodes 40 amino acids. We sequentially removed 10 additional amino acids. Deletion of exons 16, 17, and the last 10 amino acids of exon 15 (∆Exon15-III) completely abolished SHTN1L binding to actin (Figure 4C). Therefore, amino acids 31–40 encoded by exon 15 harbor or overlap a sequence motif necessary for actin interaction. Human and rodent SHTN1L are largely identical around this region (Figure 4D).
Many actin-binding proteins interact with actin through positively-charged amino acids. We identified a conserved basic patch, RRRK, within SHTN1L exon 15, which is similar to those of F-actin-binding regions of VASP, MENA, and EVL (Figure 4E) (Breitsprecher et al., 2011; Chereau and Dominguez, 2006). When the consecutive arginine residues were mutated to glycine, SHNT1L(RRR>GGG) mutant no longer interacted with actin (Figure 4C), demonstrating essential roles of RRR for SHTN1L-actin interaction.
We next tested whether SHTN1L directly interacts with actin polymers (F-actin). We first purified FLAG-tagged SHTN1L, SHTN1S, and SHNT1L(RRR>GGG) proteins (Figure S3A). F-actin co-sedimentation is a common assay to examine protein interaction to F-actin. We incubated different SHTN1 proteins with in vitro preassembled F-actin molecules. Only SHTN1L but not SHTN1S or SHNT1L(RRR>GGG) mutant co-precipitated with F-actin (Figure 4F). These data showed that SHTN1L directly interacts with F-actin via the RRR motif.
To understand the functional outcome of SHTN1L‒F-actin interaction, we tested whether SHTN1L affected actin polymerization. Actin monomers (G-actin) polymerize spontaneously but slowly in vitro under refractory conditions (low salt and low ATP). We incubated purified SHTN1L protein with G-actin in such a condition for 1 hour. At the end of reactions, F-actin was precipitated and separated from G-actin in the solution via ultracentrifugation. SHTN1L proteins clearly augmented the F-actin pellet, an indication of promoting actin polymerization, while SHTN1S or SHTN1L(RRR>GGG) mutant did not (Figure 4G). The polymerization of actin was proportional to SHTN1L concentrations, as more G-actin molecules were depleted from the supernatant and incorporated into F-actin in the pellet in the presence of increasing SHTN1L protein (Figure S3B). In contrast, even at a high concentration, the RRR>GGG mutant did not promote actin polymerization (Figure S3C). To further evaluate SHTN1L’s activity in promoting actin polymerization, we measured actin filament formation with a pyrene actin polymerization assay. We found that SHTN1S barely influenced the kinetics of de novo actin assembly. Addition of SHNT1L, but not the SHTN1L(RRR>GGG) mutant, increased the production of actin filament (Figure 4H).
Taken together, we identified SHTN1L as a novel actin-binding molecule and the RRR-containing motif distinguishes SHTN1L from SHTN1S in actin binding and polymerization. Actin polymerization is critical in force generation and axonal growth, and actin-remodeling molecules are often found to configure axon formation. SHTN1S has been shown to serve as a clutch molecule creating a driving force for axon growth (Kubo et al., 2015). Our results suggest that SHTN1L propels actin polymerization to increase the force, resulting in enhanced axonal growth (Figure 4I). These data collectively demonstrate alternative splicing is a regulatory switch influencing SHTN1’s role in axon formation.
PTBP2 coordinates axonogenesis-regulated alternative splicing
To identify trans splicing factors of axonogenesis, we searched de novo for overrepresented sequence motifs surrounding axonogenesis-associated exons (Figure 5A). We found five cis elements (E value < 10−4) for the up-regulated exons and one for down-regulated exons (Figure 5B, Figure S4A). All were flanking the exons, suggesting that most regulatory actions occurred via intron interaction. The most significant motif is a sequence of alternating C and U nucleotides, which is reminiscent of PTBP binding sites.
Figure 5. PTBP2 coordinates axonogenesis-associated alternative splicing.
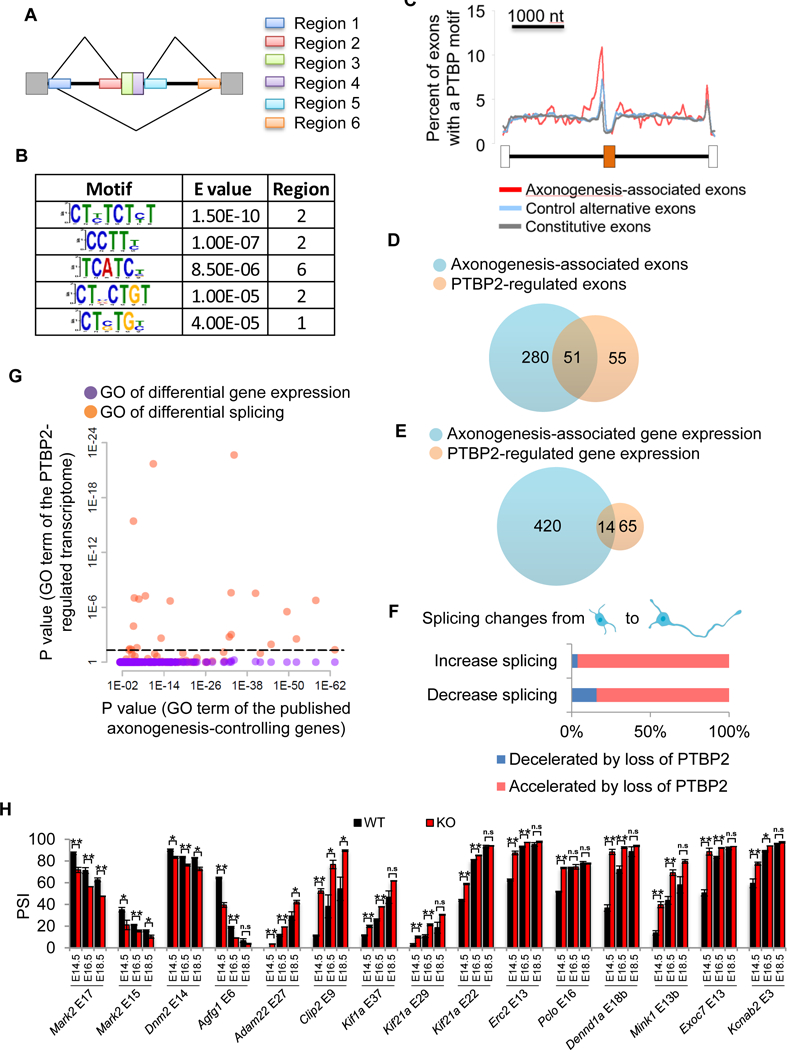
(A) Six regions of the axonogenesis-associated exons are selected to search for enriched sequence motifs. (B) All identified motifs (E value <0.0001) for the up-regulated exons. (C) Percent of exons containing previously characterized PTBP motifs at each position surrounding the axongenesis-associated up-regulated exons, control non-axonongenesis-associated cassette exons, and constitutive exons. (D) PTBP2 direct target exons significantly overlap with axonogenesis-associated exons. P value = 1.68 × 10−56, hypergeometric test. (E) A small overlap of differentially expressed genes during early axonogenesis and those dysregulated in Ptbp2−/− neurons. P value = 9.1×10−15, hypergeometric test. (F) Splicing changes (increase or decrease) from DIV1 neurons to DIV3 neurons are mostly accelerated by loss of PTBP2. (G) A scatter plot shows the Bonferroni-corrected P values of enriched Gene Ontology terms for genes exhibiting differential gene expression (purple dots) and differential splicing (orange dots) in Ptbp2−/− neurons on the y axis vs previously published polarity-controlling genes on the x axis. Dashed lines demark P value equal to 0.05 on the y axis. (H) RT-PCR validation of PTBP2-regulated cassette exons in E14.5/16.5/18.5 Ptbp2-Emx-Cre KO cortices and wild-type littermates. Data are represented as mean ± SEM of 2 (E18.5) or 3 (E14.5/E16.5) animals. Student’s t-test with *p<0.05, **p<0.01. See also Figures S4, S5, S6, S7.
We took a reverse approach to investigate the well-characterized PTBP motifs across axonogenesis-associated exons, control non-regulated cassette exons, and constitutive exons. The PTBP motifs, CTCT[CT][CT] and [ACT][CT]TTT[CT]T, were obtained from a published database (http://rmaps.cecsresearch.org/Help/RNABindingProtein). Axonogenesis-associated alternative exons, particularly the up-regulated exons, contained the PTBP motif more frequently than control alternative exons and constitutive exons (Figure 5C). Additionally, the PTBP motifs were further distant from the 3’ splice sites of axonogenesis-associated exons. This data corroborated the de novo motif discovery supporting a regulatory role for PTBP during axonogenesis. Since PTBP1 expression is sharply diminished when neural stem cells exit the cell cycle, and PTBP2 is abundantly expressed in immature neurons, we focused our study on PTBP2.
PTBP2 is expressed much higher in the brain than in peripheral tissues, suggesting its specialized role(s) in the nervous system (Figure S5). Furthermore, PTBP2 expression in the brain is the highest during embryonic development and downregulated at birth, concurrent with axonogenesis. This temporal regulation implies a unique activity for PTBP2 on axon formation. Further supporting PTBP2’s regulatory role was the crosslinking immunoprecipitation (CLIP-Seq) experiment, which profiled transcriptome binding of PTBP2 in embryonic brains (Licatalosi et al., 2012). Axonogenesis-associated exons were more bound by PTBP2 than control non-regulated alternative exons and constitutive exons (Figure S4B).
To test whether PTBP2 truly regulates axonogenesis-associated alternative splicing, we compared the transcriptomes of Ptbp2−/− and wildtype cortical neurons at DIV 1 by deep RNA-seq. Biological replicates of RNA-seq were highly reproducible (Pearson correlation 0.997). PTBP2 depletion up-regulated 122 exons and down-regulated 29 exons (|PSIPtbp2−/− – PSIWT| > 10, P-value ≤ 0.001, Table S5). A significant proportion of these exons, 106 of 151 (70.2%), contained strong PTBP2 CLIP-Seq signals in their flanking introns (enrichment P value = 4.01 × 10−48 compared to non-PTBP2-regualted exons, one-sided Fisher’s exact test), and thus were likely direct PTBP2 targets. These include Clip2 exon 9, Map4 exon 18, Ppp3ca exon 13, and Plekha5 exon 10, etc. (Figure S6).
To further confirm PTBP2 regulation, we inserted target candidates and their flanking introns into a minigene reporter (pflareA, Figure S7). The minigene reporter produces GFP and RFP from the exon-skipping and exon-including isoforms, respectively (Zheng, 2016b; Zheng et al., 2013). We built minigenes for Clip2 exon 9 and Plekha5 exon 10, both repressed by PTBP2. We also constructed their mutant versions deleting the PTBP2 binding sites based on the CLIP-Seq signal. We then transfected these minigenes separately into cortical neurons. Because transfection efficiency is low, we imaged individual transfected neurons with the same acquisition parameters.
In WT neurons, both GFP and RFP of Clip2 minigene were readily detectable (Figure S7B). In Ptbp2−/− neurons, RFP became brighter and GFP was often barely detected, indicating an increase in exon inclusion, consistent with loss of PTBP2 repression (Figure S7C). Similarly, the Clip2 minigene mutant displayed a preferential expression of RFP in most transfected neurons (Figure S7D). We might not have deleted all PTBP2 regulatory elements. As a result, the change in RFP/GFP of the mutant minigene was not as dramatic as the one observed in Ptbp2−/−neurons.
Plekha5 exon 10 minigene predominantly expressed GFP in WT neurons (Figure S7F). When expressed in Ptbp2−/− neurons, GFP was much weaker and RFP became stronger (Figure S7G), because PTBP2 inhibition was removed and exon splicing was enhanced. Deletion of PTBP2 regulatory sequences substantially boosted RFP expression and diminished GFP in WT neurons, consistent with increased exon inclusion (Figure S7H). These minigene experiments show that Clip2 exon 9 and Plekha5 exon 10 are true PTBP2 direct targets.
PTBP2 direct targets and axonogenesis-associated exons significantly overlapped (Figure 5D, P value = 1.68 × 10−56, hypergeometric test). The overlap was still highly significant if all 151 PTBP2 general targets were considered (Figure S4C, P value = 4.4 × 10−78, hypergeometric test). In contrast, PTBP2 knockout showed lesser effects on axonogenesis-associated gene expression change (Figure 5E). Gene-level dysregulation in Ptbp2−/− neurons was low in number and did not exhibit any enriched GO terms (Figure 5G, purple dots), whereas PTBP2-controlled exons were enriched in gene ontology terms shared by the published axonogenesis genes (Figure 5G, orange dots).
Splicing shifts in both directions upon PTBP2 depletion and during axonogenesis, but the overlapping exons were not randomly distributed (Figure S4D). Instead, 67 of 71 overlapping exons belonged to only two scenarios, suggesting that PTBP2 does not randomly influence axonogenesis-associated exons. We found that loss of PTBP2 mostly accelerated developmental changes of axonogenesis-associated exons. This was seen for exons increasing, and decreasing, splicing during axonogenesis (Figure. 5F). Such nearly unidirectional activity of PTBP2 showed PTBP2 coordinated axonogenesis-associated splicing changes.
We asked whether gain of PTBP2 function, via lentivirus transduction, would come to a similar conclusion. We compared transcriptomes of PTBP2-expressing neurons versus control GFP-expressing neurons by deep RNA-Seq. Differentially-spliced exons upon PTBP2 overexpression significantly overlapped with axonogenesis-associated exons (Figure S4E, P value = 3.23×10−82, hypergeometric test). Considering the direction of splicing changes, axonogenesis-associated splicing was decelerated by gain of PTBP2 function (Figure S4F).
To confirm that PTBP2 depletion precociously induced the switches of axonogenesis-associated exons, we extended the validation analysis of PTBP2-controlled axonogenesis-associated exons from DIV 1 and 3 to DIV 5 (Figure S4G). By comparing PSI values at these three stages, we determined the developmental courses of splicing changes. Except for Dnm2 exon 14, where developmental changes were minimal, the splicing of all other exons were altered by PTBP2 knockout in the same directions as their developmental trajectories. To further test this idea in vivo, we analyzed splicing changes in wildtype and Ptbp2−/− neocortices at E14.5, E16.5, and E18.5 (Figure 5H). Indeed, PTBP2 depletion prematurely induced axonogenesis-associated splicing changes for developmentally up-regulated and down-regulated exons. With all these data, we concluded that PTBP2 coordinates axonogenesis-associated alternative splicing.
Axonogenesis of Ptbp2−/− neurons is disorganized
We compared primary neurons from Ptbp2−/− neocortices and wild-type control littermates. Using [polarization index (the ratio of the length of the longest protrusion to that of the second longest protrusion) ≥ 2] as the criterion for determining polarized neurons, we found that 36% of Ptbp2−/−neurons were unpolarized in comparison to 16% in WT littermates (Figure 6A). This defect was primarily attributed to a decrease in the length of the longest neuronal protrusion (Figure 6B). We used the classical criterion (length >50 µm) to define an axon and found that at 5 DIV most WT neurons had already extended their presumptive axons. Only 5.5% WT neurons, but 21.8% Ptbp2−/− neurons, lacked an obvious axon (Figure 6C).
Figure 6. Axonogenesis of Ptbp2−/− neurons is disorganized in vitro.
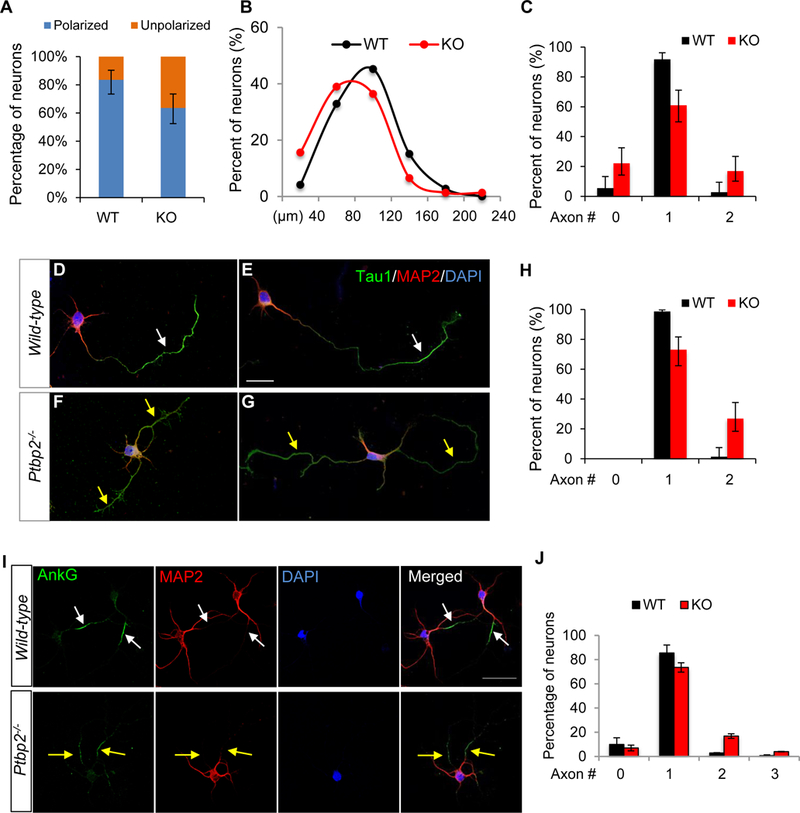
(A) A larger percentage of Ptbp2−/− (KO) neurons than WT neurons are unpolarized. The polarity index, i.e., the length of the longest neurite in relative to the length of the 2nd longest neurite, classifies polarized neurons (index ≥ 2) and unpolarized neurons (index < 2). N = 73 (WT) and 77 (KO) neurons. P value = 0.0047, Fisher’s exact test. Error bars represent 95% confidence intervals. (B) Density plot shows the longest protrusions in KO are shorter than those in WT neurons. P = 0.00259, Wilcoxon test. (C) More KO neurons than WT neurons display two axon-like processes (P value = 0.0034) and zero axon (P value = 0.0029), Fisher’s exact test. Neurites longer than 50 µm are classified as axons. Error bars represent 95% confidence intervals. (D-G) Immunostaining of axon marker Tau1 and dendrite marker MAP2 in WT (D-E) and Ptbp2−/− (F-G) neurons. Arrows point to Tau1+ MAP2- axons. (H) More KO neurons display two Tau1+ MAP2- axon-like processes. Error bars represent 95% confidence intervals. P value <0.0001, Fisher’s exact test. (I) Immunostaining of AIS marker AnkG and dendrite marker MAP2 in WT and Ptbp2−/− DIV 5 neurons. Arrows point to AnkG+ MAP2- axons. White: single axon. Yellow: surplus axons. (J) Quantification of axon numbers based on AnkG+ MAP2- staining. N = 247 (WT) and 228 (KO). Data are represented as mean ± SEM of 3 animals of 3 liters. P = 0.0009 for 2-axons and 0.0054 for 3-axons, Student’s t-test. See also Figure S8.
To ensure the phenotype originated from loss of PTBP2, we performed a rescue experiment expressing PTBP2 in Ptbp2−/− neurons by lentivirus transduction. The PTBP2 lentivirus co-expressed PTBP2 and GFP, whereas the control virus expressed only GFP. We used GFP as the indicator of an infected neuron and to measure the length of its longest neurite (axon). Infecting Ptbp2 null neurons with the PTBP2 virus allowed the knockout neurons to grow longer axons, reaching a length comparable to normal WT neurons (Figure S8). The rescue of axon length, however, depended on the amount of virus used for infection (MOI: 938–1,875), indicating that a proper level of PTBP2 expression was critical.
We found some Ptbp2 null neurons generated surplus axons longer than 50 µm (Figure 6C). To confirm the disorganized axonogenesis in Ptbp2−/−, we used classical molecular markers Tau1 and MAP2 as independent criteria for defining axons. A typical axon is Tau1 positive and MAP2 negative in its distal part (Figure 6D-E). The immunostaining confirmed the Ptbp2−/− neuron’s capacity to produce Tau1+ MAP2− axons. However, Ptbp2−/− Tau1+ MAP2− protrusions were generally shorter than WT counterparts (Figure 6F-G). Furthermore, a greater number of Ptbp2−/− neurons displayed two or more Tau1+ MAP2− protrusions than WT neurons (Figure 6H).
We examined whether a Ptbp2−/− axon could develop to obtain an axon initiation segment (AIS), a unique structure demarcating the boundary between axonal and somatodendritic compartments. Ankyrin G (AnkG) is a key scaffolding protein of AIS and AnkG clustering along the proximal axon is often used as another molecular marker of axon (or axon maturation). We found Ptbp2 null neurons were as competent as WT neurons in generating AIS, as the percentages of AnkG negative neurons were similar between Ptbp2 KO and WT (Figure 6I-J). Most strikingly, the supernumerary axons in Ptbp2−/− neurons were positive for AnkG staining, indicating that the second Tau1+ axon was not an artifact. Based on these criteria, loss of PTBP2 impedes axon growth while stimulating axon specification. As a result, Ptbp2−/− neurons are not as robust as WT neurons in generating one and only one axon.
Cell-autonomous axonal defects of Ptbp2−/− neurons in vivo
To examine axonogenesis of individual Ptbp2−/− neurons in vivo, we in utero electroporated (IUE) CAG-EGFP plasmid to E13.5 neocortex and dissected the brain at E17.5 to examine GFP+ neurons. WT neurons in cortical layers exhibited a typical bipolar morphology with one leading process (dendrite) and one trailing process (axon). By contrast, about 25% of Ptbp2−/− neurons displayed two or more trailing processes from the soma (the primary axon) toward the intermediate zone (Figure S9A-B). In cases where a trailing process stopped within rather than travelling out of the brain section, a careful assessment showed Ptbp2−/− axons were shorter in general and some failed to reach the IZ (Figure S9C-D). This explains why Ptbp2−/− axonal fibers originating from cortical neurons were thinner in the IZ (see below).
To test whether the axonogenesis phenotype was cell autonomous to loss of PTBP2, we used IUE on Ptbp2loxp/loxp neocortices with a mixture of Cre-expressing plasmids and Cre-dependent EGFP expression (LSL-GFP) reporter plasmids. This ensured that only Cre-expressing electroporated neurons in Ptbp2loxp/loxp cortices were depleted of PTBP2 and simultaneously gained GFP expression (aka, GFP+ cell is Ptbp2−/−), allowing examination of Ptbp2−/− neurons by GFP in an otherwise WT dark background. Four days following IUE, co-staining of GFP and PTBP2 showed only GFP+ neurons were deficient in PTBP2 expression (Figure 7A-D). The same IUE procedure in WT littermates (from Ptbp2loxp/+ x Ptbp2loxp/+ breeding) was used as a control. Again, significantly more GFP+ neurons exhibiting multiple, and often short, trailing processes in the Ptbp2loxp/loxp cortices than in the control (Figure 7E-G). The percentage in KO could be underestimated because only clearly separated trailing processes were included in the analyses (e.g., Figure 7F1 and F4) and microscopically unresolved processes were not considered (e.g., Figure 7F2 and F3). We conclude the axon growth defect and the multi-axon phenotype are intrinsic to PTBP2 deficiency.
Figure 7. Axonal defects of Ptbp2−/− neurons in vivo.
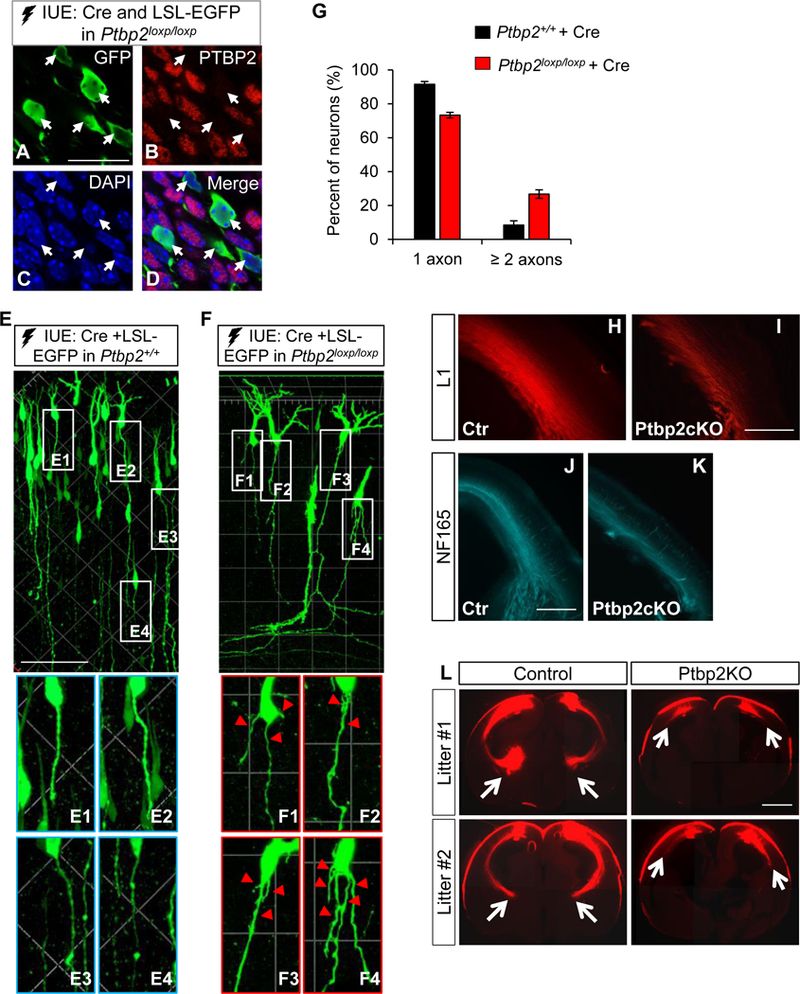
(A-D) Cre and LSL-EGFP plasmid IUE of the Ptbp2loxp/loxp neocortex effectively depletes PTBP2 expression in Cre-expressing GFP+ neurons (arrows). Scale bar: 20 µm. (E-F) Representative images showing morphology of GFP+ neurons four days after Cre and LSL-EGFP IUE of the Ptbp2+/+ and Ptbp2loxp/loxp neocortices. Scale bar: 80 µm. Arrows point to Ptbp2−/− neurons exhibiting supernumerary trailing processes (axons). (G) Ptbp2−/− cortical neurons display a higher percentage of surplus axons than WT neurons. Data are represented as mean ± SEM of 7 WT (647 neurons) and 11 KO (902 neurons) brains. P < 0.0001, Student’s t-test, 2-tailed. (H-K) Emx1-Ptbp2cKO brains exhibit weaker and narrower staining of axonal markers L1 and NF165 at E17.5. Scale bar: 500 µm. (L) Dil tracing at E18.5 shows a substantial defect of axonal growth from Ptbp2−/− cortical neurons. Arrows indicate the terminal of axonal tract. Scale bar: 1000 µm. See also Figure S9, S10.
To examine the aggregate effect of defective axonal growth and specification, we conducted extensive phenotypic analysis of prenatal Emx1-Ptbp2cKO (Ptbp2loxp/loxp, Emx1-Cre) and WT (Ptbp2loxp/+ or Ptbp2loxp/loxp) brains. Stained with axonal markers L1 and NF165, axonal bundles in the intermediate zone (IZ) of E17.5 Emx1-Ptbp2cKO were weaker and thinner than WT (Figure 7H-K). Consistently, the gap between the cortical plate and the ventricle zone was narrower in Emx1-Ptbp2cKO (Figure S10). Axonal loss could be due to neuronal loss. However, using various markers, we found no difference in subtype specification, neuron numbers or cell death between WT and Ptbp2cKO at E17.5 (Figure S10).
We deposited DiI crystals in the dorsal cortical plate to trace axonal tracts. In control brains, the cortical-thalamic projections grew through the internal capsule and reached the thalamus, whereas the DiI-labeled Ptbp2-KO projections were shorter: most failed to exit the neocortex (Figure 7L). These data further proved the growth defects of Ptbp2−/− axons.
PTBP2 controls the developmental switch of Shtn1 isoforms
To understand the mechanism by which PTBP2 loss leads to a deficiency in axonal growth, we focused on Shtn1. As shown by PTBP2 CLIP-Seq, PTBP2 interacts with at least five polypyrimidine tracts within the region from intron 14 to intron 16 (Figure 8A). These polypyrimidine tracts, all in introns, run from 21 to 126 nucleotides, of considerable length to confer PTBP2 regulation. The longest polypyrimidine sequence, consisting of 98% C and U, localizes between exon 15 and 16.
Figure 8. PTBP2-regulated Shtn1L rescues Ptbp2−/− axonal length.
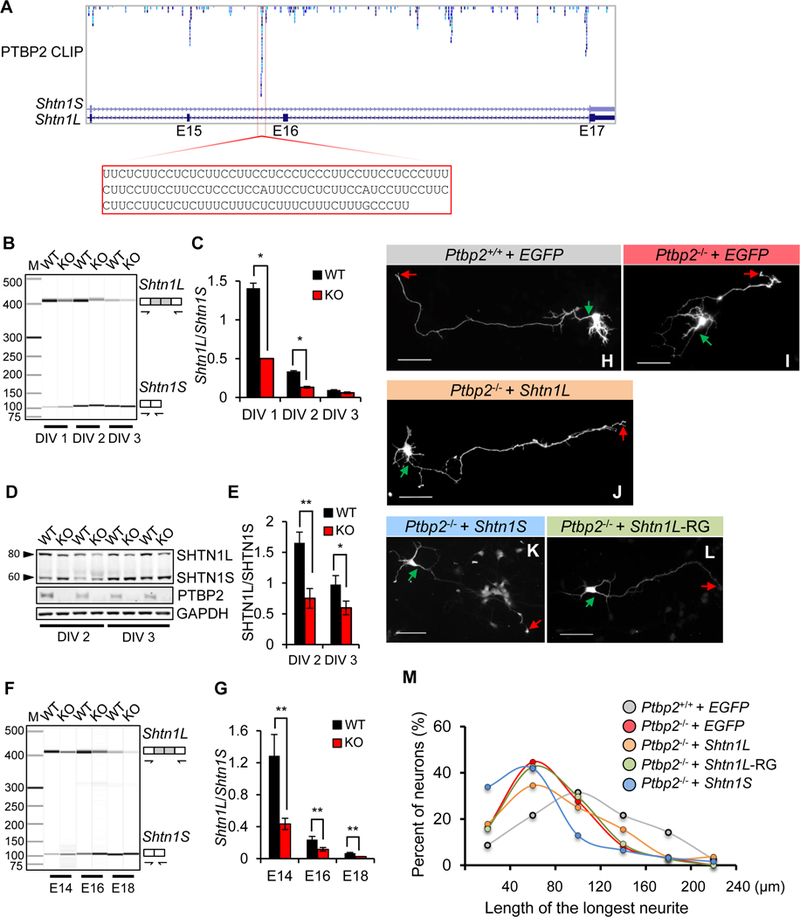
(A) Browser track of PTBP2 CLIP-Seq shows multiple PTBP2 binding sites in Shtn1 introns 14, 15, and 16. One prominent binding site is highlighted. (B-C) Alternative splicing of Shtn1 mRNA in WT and KO primary cortical neurons at DIV 1, 2, and 3. Data are represented as the mean ± SD from three independent experiments. * p< 0.05, Student’s t-test. (D-E) A representative immunoblot demonstrating downregulation of SHTN1L protein in Ptbp2−/− primary cortical neurons. Data are represented as the mean ± SD. * p< 0.05, ** p< 0.01, Student’s t-test. (F-G) Representative virtual gel image of Shtn1 mRNA splicing in the WT and Ptbp2-KO neocortices at E14.5, E16.5 and E18.5. Data are represented as the mean ± SD. ** p< 0.01, Student’s t-test. (H-L) Representative images of Ptbp2+/+ neurons, Ptbp2−/− neurons, and Ptbp2−/− neurons transfected with Shtn1L-, Shtn1S-, or Shtn1L(RRR>GGG)-expressing plasmids. Shtn1L-RG indicates Shtn1L(RRR>GGG). Neurons were transfected at DIV1 and visualized by transfected GFP at DIV3. Green arrows indicate the starting sites of axons and red arrows the axonal tips. Scale bars: 25µm. (M) Density plot of Fig.8H-I shows the length distribution of the longest neurites (axons). Shtn1L, not Shtn1S or Shtn1L(RRR>GGG), partially rescues the defect of axonal length in Ptbp2−/− neurons. N=162 (Ptbp2+/+), 159 (Ptbp2−/−), 84 (Ptbp2−/− + Shtn1L), 108 (Ptbp2−/− + Shtn1L-RG) and 62 (Ptbp2−/− + Shtn1S) neurons from 4–7 biological replicates.
PTBP binding to RNA does not necessarily result in alternative splicing regulation (Vuong et al., 2016b). We tested whether PTBP2 controlled Shtn1 splicing and found PTBP2 inhibited the Shtn1L-to-Shtn1S developmental switch. The Shtn1L/Shtn1S mRNA ratio was significantly decreased in Ptbp2−/− neurons at DIV 1 and 2 (Figure 8B-C). The SHTN1L/SHTN1S protein ratio was smaller in Ptbp2−/− neurons at DIV 2 and 3 (Figure 8D-E). We also found a reduced Shtn1L/Shtn1S mRNA ratio in Ptbp2−/− brains at E14 and E16 (Figure 8F-G). In all cases, the differences between WT and KO were the largest during early development, consistent with PTBP2 affecting early axonogenesis.
Since SHTN1L promotes axon growth, we hypothesized that reduced SHTN1L expression in Ptbp2−/− cultured neurons caused a decrease in axon length. To test this, we overexpressed SHTN1L, SHNT1L(RRR>GGG) mutant, and SHTN1S separately in Ptbp2−/− neurons at DIV 1 and measured axon length at DIV 3 (Figure 8H-L). SHTN1L, but not the RRR>GGG mutant or SHTN1S, partially rescued the axon length defect in Ptbp2−/− neurons (Figure 8M). The rescue was most obvious for axons of medium length (60–140 µm). Shorter axons (< 60 µm) were not rescued and SHTN1L overexpression rarely induced axon growth over 180 µm, suggesting limitations to SHNT1L ability to promote the growth of Ptbp2−/− axons. This rescue experiment showed that reduced SHTN1L isoform expression contributes to the decreased axon length in Ptbp2−/− cortical neurons.
DISCUSSION
In this study, we show that alternative splicing programming is critical to the robust axonogenesis of mammalian cortical neurons. Unbiased transcriptome profiling revealed axonogenesis-associated alternative splicing is overtly enriched for features of axon formation. Since axon formation is a neuron-specific phenotype, the neuron-specific expression pattern of axonogenesis-associated splicing may be part of the intrinsic program underlying neurons’ unique capability to generate an axon. Interestingly, genes known to regulate axonogenesis exhibit changes in splicing but not in transcription during early axon formation. These genes have been studied individually but are presumably coordinated to guarantee axon generation. We now show many of them are orchestrated by alternative splicing.
Axonogenesis-associated splicing is governed by PTBP2. Concurring with axonogenesis, PTBP2 expression in neocortices is the highest during embryonic development. The tissue-specific and temporal-specific expression patterns of PTBP2 indicate that specialized regulation takes place to ensure precise programming of axonogenesis-associated splicing and that PTBP2 has a unique role in axon formation. Indeed, knockout of Ptbp2 results in a phenotype specific to axonogenesis.
PTBP2 depletion precociously induced switches of many axonogenesis-associated alternative exons. This led us to hypothesize that loss of PTBP2 would promote or accelerate axon formation. However, we observed more complicated phenotypes. While some Ptbp2−/− neurons fail to extend axons, others display surplus axons. In either case, axons are generally shorter. This defect is likely due to imbalanced expression of isoforms, because gene-level changes are modest.
We propose a probabilistic model to explain the Ptbp2−/− phenotype. The precise choreography of axon formation likely involves sequential interconnected molecular cellular events leading to neuritogenesis, rapid axon growth, stabilization, specification, and maturation. The unusual Ptbp2−/− phenotype reflects a shortage of some isoforms for initial axon growth and an oversupply of other isoforms for axon specification. Impaired growth increases the probability of a Ptbp2−/− neuron generating a short axon. Excessive axon-specifying molecules increase the probability of specifying an extra axon. Robust production of a single axon requires minimizing both probabilities, which in turn demands balanced expression of isoforms.
One example supporting this model is Shtn1. The long isoform (Shtn1L) specialized for axon growth is reduced in Ptbp2−/− neurons, whereas the short isoform (Shtn1S), previously shown to affect axon number, is increased. We show that the phasic expression of SHNT1L and SHTN1S is developmentally programmed and the two isoforms exhibit different roles in axon formation. We further found that SHTN1L promotes actin polymerization thanks to its distinct C-terminus. Actin polymerization is unique to SHNT1L, providing novel insights to distinguishing the two isoforms. The role of SHTN1 in axon formation proposed by Inagaki’s group rests on its ability to create mechanical forces on adhesive substrates for growth cone migration. Molecularly, SHTN1 (or more precisely SHTN1S) interacts with L1-CAM and Cortactin, and Cortactin binds to F-actin, thereby SHTN1 couples cell adhesion molecules and F-actin to generate the driving force (Kubo et al., 2015; Shimada et al., 2008). These molecular interactions are mediated by protein sequences shared by both isoforms, so the model should apply to SHNT1L as to SHTN1S. In the above model the critical protein(s) responsible for actin polymerization has not yet been specified. Our data show that SHTN1L (but not SHTN1S) can play this role (Figure 4I). SHTN1L presents an advantage over SHTN1S by augmenting actin polymerization and thereby the driving force. These observations do not mean SHTN1S is not engaged in the coupling or generation of the driving force; it only suggests that the SHTN1S driving force (in cortical neurons) has not translated into substantial growth cone movement and may be counterbalanced by other molecules.
We found SHTN1L is a novel actin-binding protein. This suggests that SHTN1L may not require Cortactin to couple L1-CAM and F-actin. Excluding Cortactin as a limiting factor would give SHTN1L additional latitude to promote axon growth, which might be critical during early axonogenesis. Because actin binding and actin polymerization are both abolished in the SHTN1L (RRR>GGG) mutant, SHTN1L’s actin polymerization activity may depend on its actin binding ability. This would explain why SHTN1S does not promote actin polymerization, since SHTN1S does not directly interact with F-actin. In any case, these isoform differences originate from the distinct C-terminus of SHTN1L. Therefore, alternative splicing alters SHTN1’s activity during development and is important for SHTN1-mediated axonogenesis.
To date, only a few genetic knockout mice showed a significant effect on axonogenesis. In each case, axon formation is repressed (Barnes et al., 2007; Kishi et al., 2005; Yi et al., 2010), so it is difficult to distinguish and ascribe a precise cause for axon growth vs. axon specification. The two terms are often used interchangeably in literature, as axon fate cannot be determined without axon growth. It is not known whether axon growth leads to axon specification or vice versa. When axon growth is impeded in Ptbp2 null neurons, axons still express canonical axonal markers. On the other hand, when axons are specified in Ptbp2 null neurons, and sometimes surplus axons are produced, axon growth can appear subpar. These observations demonstrate a break in strict correlation between axon growth and specification. Therefore, the Ptbp2−/− mouse presents a phenotype of its own in which the retarded axon growth, coexisting with surplus axons, indicates that axon specification and axon growth might be distinguishable.
Our findings raise the hypothesis that neurons alter the functions of a wide range of genes, to a previously unappreciated degree, to initiate and establish axons. Shtn1 is one example. Another example is AnkG (Jacko et al., 2018). Jacko et al. showed that a 33-nt cassette exon was down-regulated in maturing motor neurons. Skipping this exon was necessary to generate effective AIS. This AnkG splicing switch occurred during maturation rather than early axonogenesis, suggesting multiple waves of splicing regulation account for different stages of axon formation. Many other isoform switches warrant future investigation to fully understand axon formation.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Sika Zheng (sika.zheng@ucr.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Maintenance
All mice were maintained and used for experiments according to the requirements of the Institutional Animal Care and Use Committees (IACUC) at the University of California, Riverside. Under routinely monitoring and checking of veterinary and mouse room staff, all animals used in this study were kept healthy in a mouse room with a stable temperature at 22±2°C and a 12h light/dark cycle. Ventilated cages and clean bedding were changed every two weeks. Food and water were provided ad libitum. No more than 5 mice were allowed in each cage. Mice of both genders were used. Embryonic day 14 (E14) embryos were collected for primary neuron cultures. E13-P0 animals were used for histological and morphological studies. The mouse lines are:
C57BL/6J mice purchased from the Jackson Laboratory and bred in house for timed pregnancy.
Timed pregnant CD-1 (ICR) mice ordered from Charles River Hollister.
Ptbp2+/− mice inbred to generate Ptbp2−/− and wildtype littermate controls.
Ptbp2loxp/loxp mice used in IUE experiment or bred with Ptbp2loxp/+;Emx-Cre+/− to generate Ptbp2 conditional KO.
Emx-Cre+/− mice bred with Ptbp2loxp/loxp mice to generate Ptbp2loxp/+;Emx-Cre+/− breeders.
Mouse Cortical Primary Neuron Culture
Primary cortical neurons were cultured as previously reported (Zheng et al., 2010). Briefly, E14 mouse cerebral neocortices were dissected out and treated with 0.25% trypsin (Thermo Fisher Scientific) and 1mM EDTA in HBSS at 37°C for 8–10 minutes. After trypsin was removed, neurons were dissociated in warm plating media (20% heat inactivated horse serum, 25mM glucose, 1x GlutaMax in MEM), centrifuged, resuspended in fresh plating media and plated with different cell densities according to experiments. Media were changed to feeding media (1x GlutaMax, 1x B-27 in Neurobasal) 6 hours after plating. For regular maintenance, half of the feeding media was refreshed every 3–4 days.
METHOD DETAILS
RNA Sequencing libraries
One million mouse cortical neurons (E14) were plated in a 35mm dish coated with 0.1mg/ml poly L-lysine (Sigma-Aldrich) and maintained in feeding media until use. Total RNA was extracted using TRIzol reagent and purified with TURBO DNase (Thermo Fisher Scientific). RINs were determined by Bioanalyzer (Agilent). RNA-seq libraries were constructed using Illumina TruSeq Stranded Total RNA LT Sample Prep kits. RNA-seq samples were derived from different embryonic mice (cortices). One embryonic mouse was used per replicate.
RNA-Seq analysis
We quantified gene expression using RSEM (Li and Dewey, 2011) and corrected sequencing biases to determine differentially expressed genes (DEGs) using a model (below) based on GPSeq (Srivastava and Chen, 2010). Gencode annotation (version M12) was used for the quantification (49,761 genes and 123,063 transcripts). We focused on genes with length (number of nonredundant exonic positions) >500bp and transcripts whose corresponding genes satisfy the length requirement. The differential expression (DE) analysis for genes and transcripts was performed by this GP-based approach with bias correction. We limited the DE analysis on genes and transcripts with bias-corrected TPM > 5.0 for both conditions. The P value threshold for differential expression (of genes or isoforms) is ≤ 0.001 (the corresponding FDR <0.005) and the fold change is ≥ 2.0.
We conservatively focused on Gencode cassette exons, because the independent Gencode evidence for alternative splicing enhanced the validity of the analytical results. We applied sequence-read filters to further obtain cassette exons with high quality sequencing profiling for both developmental stages. The Gencode annotation contains 14,931 cassette exons. The quantification of alternative splicing was performed using the GeneSplice pipeline (Vuong et al., 2016b) and based on junction read counts summarized by STAR (Dobin et al., 2013). To pass the coverage criteria, a cassette exon is required to have ≥ 10 exclusive junction reads or ≥ 20 inclusive junction reads (upstream inclusive junction reads plus downstream inclusive junction reads). To detect differentially spliced cassette exons, we focus on cassette exons passing the coverage criteria under both the experimental and control conditions. The Fisher’s exact test was applied on the two-by-two table for the exclusive and the upstream inclusive junction counts as well as the table for the exclusive and the downstream inclusive junction counts. The corresponding p-value on both tests should be ≤ 0.001 (the corresponding FDR<0.05). And we additionally require a minimum change at the splicing ratio level (|∆PSI| > 10). To increase the analysis sensitivity, replicated samples were pooled together to increase the sequencing depth.
Mouse adult tissue RNA-seq data from ENCODE (generated by Gingeras’ group) were used to study the tissue-specific splicing of axonogenesis-associated cassette exons (ENCODE Project Consortium, 2012). We quantified splicing ratios (PSI) of cassette exons as described above. Hierarchical clustering with complete linkage was performed for different tissues or for different cassette exons based on the euclidean distances among splicing ratios. To show the expression levels of Ptbp2 across different tissues, we additionally considered the fetus brain RNA-seq data in ENCODE (generated by Wold’s group). There are two biological replicates for each of ENCODE tissues.
To determine the dispersion values of gene expression differences or splicing differences for published axonogenesis genes, we scatter-plotted the expression or splicing values of each gene in pre-axonogenesis neurons vs those in axon-generating neurons, and calculated the average distance of the points to the identity line Y=X. For the gene expression values (log(TPM+1) as shown in the figure), we divided them by the maximum gene expression value so that the range was 0 to 1. Note that the range for splicing ratio (PSI/100) was also 0 to 1.
The de novo motif discovery was performed using DREME (MEME Suite version 5.0.2) with the default setting for RNA sequences (Bailey, 2011). The regions around cassette exons considered for motif discovery include: (1) downstream 500bp-intronic region of the upstream 5’ splicing sites, (2) upstream 500bp-intronic region of the upstream 3’ splicing site, (3) 5’ 50bp-exonic region, (4) 3’ 50bp-exonic region, (5) downstream 500bp-intronic region of the downstream 5’ splicing site, (6) upstream 500bp-intronic region of the downstream 3’ splicing site. The control exon set for the motif analysis included 4,799 alternative exons passing coverage criteria and whose P values for differential splicing were larger than 0.1 and |∆PSI| between DIV 3 and DIV 1 neurons < 0.01. The two known motifs for PTBPs (curated at http://rmaps.cecsresearch.org/Help/RNABindingProtein) were used to scan the above six regions around axonogenesis-associated cassette exons, aforementioned control cassette exons, and constitutive exons (i.e. middle exons appearing in every transcript of a multi-transcript gene in Gencode) to determine position-dependent motif frequency. PTBP2 CLIP-Seq of embryonic brains were obtained from Licatalosi et al. 2012 (Licatalosi et al., 2012). We calculated the percent of exons with enriched PTBP2 CLIP signal after first determining the number of CLIP tags for each position of the above six regions around cassette exons, control cassette exons, and constitutive exons. To claim direct PTBP2 targets by overlapping the CLIP-Seq data and RNA-seq data of Ptbp2−/− neurons, we first calculated the average CLIP coverage (density) across the considered 2100 nts (regions 1 to 6). We used a stringent density cutoff (>0.1) to qualify a direct PTBP2 target.
Gene ontology analysis was performed using DAVID (https://david.ncifcrf.gov/). We focused on terms with at least three genes in our lists. Significant terms are those with the Bonferroni-corrected P value ≤ 0.05.
GP-based models for expression quantification and differential expression analysis
For a considered gene g, for each sample r, we obtain position-level read counts: based on the STAR alignment and the feature Counts summary for each exonic position of the gene, where is the effective gene length from RSEM. We assume
where is the bias parameter and can be estimated as is the average position-level read count and is the variance of the position-level read counts, and is the expression parameter. We write , where represents the normalization factor across all samples. Note that is the biased corrected molecular amount (similar to the denominator calculation in TPM). We have subsequently omitted subscript g for brevity. Based on the property of GP distributions, for gene g from sample r, we have
The observed , where is the gene-level read count. Thus, position-level read counts were not required for the following analysis. We perform the following transformation:
so , transforming our observed . We assume our transformed variable . To compare under condition 1 with under condition 2, we can pool replicates as:
Correspondingly,
According to the property of Poisson distribution, we have:
Under the null: . Then . We calculate the following probabilities:
The p-value for DE detection is . The bias corrected molecular amount for the considered gene, based on pooled replicates, is therefore: We normalized it by the total bias corrected molecular amount to achieve a bias-corrected tpm The quantification and DE analysis for transcripts were similar and we assigned gene-level to transcripts since the position-level read count along the same transcript is not available. Note that position-level read counts are not required for other steps of our model.
RNA extraction, RT-PCR and QIAxcel quantitative capillary electrophoresis analysis
Total RNA from embryonic cortex tissues or 1.0×106 cultured primary neurons per sample were extracted using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. DNase I (Thermo Fisher Scientific) was used to remove DNA contamination from RNA samples prior to cDNA synthesis. First-strand cDNAs were synthesized using M-MLV reverse transcriptase (Promega) with random hexamers and a total of 1 µg RNA input as previously described (Chen and Zheng, 2008, 2009). Subsequently, cDNA was diluted 1:10 and 6 µl used per PCR reaction. For QIAxcel analysis cDNA was amplified with Taq DNA polymerase (New England Biolabs) as per manufacturer’s instructions and each primer at a final concentration of 0.25 or 0.5 µM. Cycling parameters were as follows: 95°C at 3 min for 1 cycle; 95°C 30 s, 63°C 30 s, 72°C 30 s for 27–30 cycles; 72°C at 5 min for 1 cycle. Primer sequences used to detect PTBP2-targeted exons are listed in Table S6. The RT-PCR amplicons were subjected to capillary electrophoresis analysis on the QIAxcel System by loading onto a QIAxcel DNA cartridge (Qiagen) and run next to a 50–500 bp DNA size marker (Qiagen) as per manufacturer’s instructions. Visualization of RT-PCR products was performed with QIAxcel ScreenGel software (Qiagen), and statistical analyses of calculated concentrations of targets were carried out using Microsoft Excel. At least three independent biological replicates of the samples were included in the analysis. Differences were considered statistically significant at p<0.05.
Cell culture and transient expression
Mouse Neuro-2a cells (Cat#CCL-131; ATCC) were cultured in DMEM including high glucose, 10% FBS (Thermo Fisher Scientific) and 2 mM GlutaMAX (Thermo Fisher Scientific) according to the ATCC’s instructions. Neuro-2a cells were plated at the density of 1×105 per cm2 and transfected with cDNA expression or shRNA plasmids using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. Forty-eight hours after transfection, cells were harvested for immunoprecipitation, immunoblotting, or protein purification according to experiments.
DNA constructs
In this study, pEGFP-C1, psiRNA-W[H1.4], pFlareA and pCAGIG were used as host vectors. All constructs were sequenced prior to transfections to ensure no mutations were introduced during molecular cloning. All the plasmids were propagated in DH5α E. coli cells and plasmids were prepared using Qiagen miniprep kits. Briefly, recombinant plasmids for cDNA expression of mouse Shtn1 long and Shtn1 short isoforms, which were inserted between XhoI and TspMI (New England Biolabs) sites in pEGFP-C1 mammalian expression vector. Primer sequences used for the generation of pEGFP-C1 constructs are shown in Table S6. pCAGIG plasmids encoding for FLAG-SHTN1L and FLAG-SHTN1S proteins were generated by PCR and inserts were subcloned into pCAGIG vector linearized by XhoI enzyme via In-Fusion HD enzyme (Takara), which fuses PCR-generated amplicons and linearized vector precisely by recognizing a 15 bp overlap at their ends. Oligonucleotide primer sequences used for the pCAGIG constructs are shown in Table S6.
Short hairpin RNAs (shRNAs) targeting exon 16, 3UTR of mouse Shtn1 mRNA and luciferase mRNA (negative control) were designed according to the previous instructions (Brummelkamp et al., 2002). The hairpin constructs were generated by annealing two synthetic oligonucleotides and cloned into the BbsI site of psiRNA-W [H1.4] host vector. To increase the stability of the hairpin structures, C>T and/or A>G nucleotide substitutions were introduced to oligos resulting in G/U wobble pairings. The oligos used to make the hairpins are listed in Table S6, and nucleotide substitutions are underlined.
Minigene constructs pFlareA-Clip2 and pFlareA-Plekha5 were created using standard cloning techniques. Clip2 and Plekha5 genomic DNA, which include the exon 9 and exon 10 along with flanking introns, respectively, were PCR amplified and cloned in EcoRI and BamHI enzyme sites in pFlare9A. To generate mutant minigene constructs, dsDNA fragments were synthesized (gBlock, IDT) that were lack of the PTBP2-binding sites in flanking introns and cloned in EcoRI and BamHI enzyme sites in pFlareA. The genomic sequences used to make WT and mutant minigene constructs are shown in Table S6.
Protein extraction and immunoblotting
Mouse cortices, primary cortical cultures, and Neuro-2a cells were lysed in RIPA buffer containing 10 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 1 mM PMSF and protease inhibitor cocktail as previously described (Li et al., 2017). Protein extracts were cleared by centrifugation at 14,000 rpm for 15 minutes at 4 °C to separate insoluble fractions. To determine protein concentration of the samples, BCA assay was performed using BSA dilutions as standard. Samples were boiled with Bio-Rad’s 4X Laemmli buffer containing 10% β-mercaptoethanol at 95°C for 10 min. Proteins were separated on 4–10% SDS-PAGE gels and transferred to Immobilon-FL PVDF membrane (Millipore) and probed for proteins of interests and fluorescence conjugated secondary antibodies. The membrane was blocked in TBST plus 3% BSA and then incubated overnight with the following primary antibodies: rabbit polyclonal anti-Shootin1 (clone B627; Ab for both isoform; 1:1000), rabbit polyclonal anti-PTBP2 (1:1000), and mouse monoclonal anti-GAPDH (clone 6C5 Thermo Fisher Scientific Cat#AM4300; 1:2000). Primary antibodies were detected by appropriate Alexa Fluor conjugated secondary antibodies (Thermo Fisher Scientific; 1:2000). Blots were visualized by Typhoon FLA9000 and band intensities were quantified by the ImageJ software (NIH). Statistical analyses were carried out using a Microsoft Excel. Differences were considered statistically significant at P<0.05.
Site-directed mutagenesis
QuickChange approach with non-overlapping primers was carried out to generate site-directed mutagenesis on host vectors. A series of deletion and conversion mutants were made by PCR amplification of pEGFP-C1-SHTN1L or pCAGIG-FLAG-SHTN1L plasmids in vitro to incorporate mutagenic oligonucleotides by Phusion HotStart II DNA polymerase (New England Biolabs). Cycling parameters for mutanegenesis were as follows, 98°C 3 min for 1 cycle then 98°C 15 s, 69°C 30 s, 72°C 4 min for 20 cycles then 72°C 10 min for 1 cycle. Template plasmids (10 ng) were subjected to DpnI (New England Biolabs) digestion to eliminate non-mutated templates. The resulting plasmids were recovered by transformation into DH5α E. coli competent bacteria. Mutated constructs were confirmed by DNA sequencing. The oligonucleotides used for site-directed mutagenesis are shown in Table S6.
Protein expression and purification
pCAGIG-FLAG-SHTN1L, pCAGIG-FLAG-SHTN1S and pCAGIG-FLAG-SHTN1LRRR>GGG constructs were individually transfected into Neuro-2a cells cultured in 15-cm dishes. Forty-eight hours after transfection, cells overexpressing the FLAG-tagged target proteins were washed with ice-cold PBS, scraped off and lysed in 5 ml buffer containing 50 mM Tris.HCl pH 7.4, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, Protease/Phosphatase inhibitor cocktails (Roche), 1 mM PMSF, and 50U/ml Benzonase (Novagen). For affinity purification, 250 µl slurry of anti-FLAG M2 magnetic beads (Sigma-Aldrich) equilibrated in lysis buffer were added into cleared cell lysate and incubated for 3 hrs at 4°C. Then collected beads on magnetic rack were washed three times with 2.5 ml lysis buffer. Bound proteins were eluted two times for 15 min at RT with 1.25 ml elution buffer containing 10 mM Tris.HCl pH 7.4, 100 mM NaCl and 200 µg/ml 3X FLAG-peptide (Apex Bio). Elutions were concentrated and washed with TBS buffer by Amicon Ultra centrifugal filters MWCO 30 kDa. Concentrated proteins and BSA standards were analyzed by polyacrylamide gel electrophoresis stained with Coomassie brilliant blue G-250 dye (Teknova) to calculate molar concentration of target molecules.
Coimmunoprecipitation
To detect interaction partners of SHTN1 molecules by coimmunoprecipitation, Neuro2a cells transiently expressing GFP fused full-length wild type and truncated/mutated SHTN1 proteins were lysed in 1 ml buffer containing 50 mM Tris.HCl pH 7.4, 150 mM NaCl, 1mM EDTA, 0.5% Triton X-100, Protease/Phosphatase inhibitor cocktails (Roche), 1 mM PMSF, and 50U/ml Benzonase (Novagen). Cell extracts were incubated with 10 µl GFP-Trap magnetic beads (Chromotek) for 1 h at 4°C with gentle rotation. Beads were washed three times with 1 ml stringent buffer containing 50 mM Tris.HCl pH 7.4, 300 mM NaCl, 0.5% Triton X-100, 1 mM PMSF. Bead-bound proteins were denatured by boiling in Laemmli sample buffer before SDS-PAGE. The total protein lysate (input) and the IP fractions were analyzed by immunoblotting. Proteins were resolved on polyacrylamide gels, transferred to Immobilon-FL PVDF membrane, and probed with primary antibodies against mouse monoclonal β-Actin antibody (1:500, sc-47778, Santa Cruz Biotecnology), chicken polyclonal GFP antibody (1:2500, GFP-1020, AvesLab).
Actin co-sedimentation assays
Human platelet actin protein (Cytoskeleton Inc.) was resuspended in general actin buffer (5 mM Tris.HCl pH 8.0, 0.2 mM CaCl2) at 10 mg/ml concentration and incubated for 1 h on ice. Actin oligomers were removed by centrifuge at 14,000 rpm for 30 min at 4°C. Actin filaments were produced by adding 1/10 volume of actin polymerization buffer (10X; 500 mM KCl, 20 mM MgCl2 and 10 mM ATP) and incubating at RT for 1 h. Recombinant target molecules used at their highest concentrations (FLAG-SHTN1L 240 nM, FLAG-SHTN1L 403 nM, and FLAG-SHTN1LRRR>GGG 294 nM in 5 µl volume) were incubated with prepolymerized actin filaments for 30 min at RT and sedimented at 70,000 rpm (TLA100 rotor; Beckman Coulter) for 1 h in ultracentrifuge tube with adapter (Beckman Coulter). G-actin (supernatant, unbound targets) and F-actin (pellet, bound targets) fractions were individually mixed with 4X Laemmli sample buffer in equal volume, boiled, run on SDS-PAGE and analyzed by Coomassie blue staining or immunoblotting with anti-FLAG antibody to increase visibility of the pelleted targets.
To test whether SHTN1L could involve in actin dynamics by polymerizing the filament, we utilized cosedimentation assays performed under stringent conditions to restrict self-polymerization of G-actin monomers. Actin was prepared as above mentioned except that actin polymerization buffer was not added to generate actin filaments before addition of target proteins. Cosedimentation experiments were carried out using 0.1 mg/ml (2 µM) actin alone or with 344 nM FLAG-SHTN1L, 320 nM FLAG-SHTN1S, and 344 nM FLAG-SHTN1LRRR>GGG. Purified proteins were normalized to 10 µl total volume in TBS buffer prior to mixing. After ultracentrifugation at 70,000 rpm for 1 h, pellet and supernatant fractions were resolved by SDS-PAGE and visualized by Coomassie blue-stained gel. BSA (200 nM) was used as negative control. Lastly, the effect of triple arginine mutations (R482G, R483G, and R484G; SHTN1LRRR>GGG mutant) was assessed by high-speed cosedimentation assay. Experimental steps were followed as mentioned above, using 0.15 mg/ml (3 µM) actin alone or with 1:2 serial dilutions of purified FLAG-SHTN1L and FLAG-SHTN1LRRR>GGG proteins starting from 286 nM for both.
Pyrene-actin polymerization assay
Pyrenyl F-actin emits 7–10 times greater fluorescence than monomeric pyrenyl G-actin, and the increase in fluorescence intensity is proportional to the amount of pyrenyl G-actin incorporated into F-actin (Cooper et al., 1983). Pyrene-labeled actin assays were performed using 1:1 mixture of nonmuscle actin and pyrene-labeled muscle actin (Cytoskeleton Inc. #AP05) resuspended in general actin buffer containing 0.2 mM ATP, 1 mM DTT, 1 mM EGTA, 0.1 mM MgCl2 to obtain 4 µM, 25% pyrenyl actin stock according to the manufacturer’s recommendations. Purified proteins (of equal molar concentrations in a range from 100 nM to 350 nM) were normalized to 10 µl total volume in TBS buffer and, all reaction components except the actin were first mixed together in 90 µl 1X actin polymerization buffer (50 mM KCl, 2 mM MgCl2, 1 mM EGTA and 1 mM ATP). Samples were preincubated for 15 min on ice. The reaction was started by adding 100 µl mixture of actin and pyrene-actin to the 100 µl preincubated protein mix in 96-well black plate (Greiner Bio-One, Fluotrac 600) and fluorescence changes were measured with excitation at 365 nm and emission at 385 nm in a fluorescence spectrophotometer (GloMax®-Multi+ Detection System, Promega, Inc.). Data analyses were carried out using a Microsoft Excel.
Mouse Brain Sectioning and Immunofluorescence
For layer markers, axonal markers and IUE brain analysis, embryonic mouse brains were dissected out and fixed in 4% PFA at 4°C for 6–10 h ours, washed three times with pH7.4 PBS, embedded in 3% agarose in PBS and then sectioned to 100–150 µm using a Vibratome LEICA VT1000 S (Leica). Brain sections were rinsed three time with PBS, permeabilized in 0.5% Triton X-100 in PBS for 30 min, incubated in blocking buffer (10% donkey serum, 2% BSA, 0.3% Triton X-100 in pH7.4 PBS) for 60 min. After blocking, sections were incubated with primary antibodies for SATB2 (Abcam, ab51502, 1:200), TBR1 (Abcam, ab31940, 1:400), CUX1/CDP (Santa Cruz, sc-13024, 1:1000), Ctip2 (Abcam, ab18465, 1:200), L1 (Millipore, MAB5272, 1:200), GFP (Aves Labs, GFP-1020, 1:1000) or NF165 (DSHB, 2H3, 1:120) in blocking buffer at 4°C overnight. On the next day, sections were thric e rinsed with 0.3% Triton X-100 in PBS and incubated in appropriate Alexa Fluor secondary antibodies (Life Technologies, 1:1000) in blocking buffer at 4°C overnight. On the third day, after staining with DAPI in PBS (1:500) for 30 min at room temperature, sections were washed three times with PBS and then mounted with ProLong Gold Antifade Mountant (Thermo Fisher Scientific). Images for layer makers were taken on LSM800 (Zeiss). Images for axonal markers were captured on Nikon Eclipse Ci microscope.
DiI Labeling
E18.5 mouse brains were dissected out and fixed in 4% PFA overnight in 4°C. On the second day, DiI crystals were embedded into the cerebral cortex using a 28.5G needle. The brains were returned to 4% PFA and kept at 37°C for 11 weeks to let DiI diffuse from soma to axonal tips. The brains were sectioned to 200 µm using a LEICA VT1000 S (Leica) and mounted with ProLong Gold Antifade Mountant (Thermo Fisher Scientific). Sections were imaged on a Nikon Eclipse Ci microscope with Nikon DS-Qi2 camera, PRIOR Lumen 200 light source and NIS-Elements BR4.5 software and reconstructed using Photoshop CS4.
Transfection, Infection, and Immunofluorescence Staining of Primary Cortical Neurons
Mouse cortical primary neurons were plated on glass coverslips (Thermo Fisher Scientific, 50949009) pre-treated with coating media (0.1mg/ml poly L-lysine, 5µg/ml Laminin in H2O), and maintained in feeding media. For Ptbp2−/− neuron morphological analysis and PTBP2 lentivirus transduction, 4 ×104 cells were plated per coverslip in 24-well plates; for Shtn1 experiments, 7.5 ×104 cells were plated per coverslip; and for mini gene transfection, 4 ×105 cells were plated. Plasmids were delivered into neurons by transfection (Lipofectamine 2000, Life Tech) or lentiviral transduction. In transfection experiments, for Shtn1 knockdown, 0.3 µg shRNA plasmids with 0.1 µg pCAG-GFP were co-transfected; for Shtn1 overexpression, 0.4 µg pEGFP-C1, pEGFP-Shtn1L, pEGFP-Shtn1S or pEGFP-Shtn1L -RG was transfected; for mini gene expression, 0.5 µg plasmid was transfected into neurons 6 hours after plating. Lentiviral particles (in the FUCGW backbone) expressing GFP (final concentration 1 µl/ml) or coexpressing PTBP2 and GFP (final concentration 3–6 µl/ml) together with 8 µg/ml protamine sulfate in feeding media were transduced into neurons 6 hours after plating and collected 2 days later. At the desired time point, cells were washed twice with cold PBS, fixed with 4% PFA for 15 min at room temperature and then rinsed three times with PBS. For immunofluorescent staining, cells were washed three times with PBS, permeabilized in 0.3% Triton X-100 in PBS for 10 min and incubated in blocking buffer (5% Donkey serum, 2% BSA, 0.1% Triton X-100 in pH7.4 PBS) for 60 min. After that, cells were incubated with primary antibodies for MAP2 (Abcam, ab5392, 1:4000), Tau1 (Millipore, MAB3420, 1:1000), GFP (Aves Labs, GFP-1020, 1:2000) in blocking buffer at 4°C overnight. On the second day, cells w ere rinsed with 0.1% Triton X-100 in PBS for three times, incubated with appropriate Alexa Fluor secondary antibodies (Life Technologies, 1:1000) in PBST for 60 min at room temperature. After staining with DAPI in PBS (1:500) for 10 min at room temperature, cells were washed three times with PBS and mounted with ProLong Gold Antifade Mountant (Thermo Fisher Scientific). Images were taken on a Nikon Eclipse Ci microscope with Nikon DS-Qi2 camera, PRIOR Lumen 200 light source and NIS-Elements BR4.5 software, or on LSM800 (Zeiss).
Image Processing, and Analysis
Images for primary neuron culture acquired on Nikon Eclipse Ci microscope or Zeiss LSM800 were analyzed with NIS-Elements BR4.5 software or ZEN software. Axons were defined and lengths measured according to Tau1 and MAP2 fluorescent signals or GFP fluorescence according to experiments. The presented images of cultured neurons with long axons were reconstructed by two original images using Photoshop CS4. Images for mouse brain sections captured on Zeiss LSM 800 were quantified or processed with ZEN software. To monitor the morphology of individual cortical neurons in IUE brains, the sections were imaged under Plan Apo 63x/1.40 oil lens for a 100–130µm Z-stack. Axon length was measured based on 3D reconstructed images from soma to the visible end of the trailing process before it merged into the axonal track. Only trailing processes longer than 10 µm are considered.
In Utero Electroporation
In utero electroporation (IUE) was performed according to the protocol described previously (Saito and Nakatsuji, 2001). Briefly, a timed-pregnant mouse (E13.5) was anesthetized with a mix of vaporized isoflurane and air (2:1) in the chamber and transferred to the 37℃ heat pad with its head under the mask for continuous isoflurane. Carprofen (5 mg/kg) was administered before the first incision. One side of the embryos was taken out of the abdominal cavity and immersed in warm saline with 1% Pen/Strep, and ~1 µl of Endo-free plasmid DNA (1–1.5 µg/ µl) containing 0.05% Fast Green was microinjected into the lateral ventricles of brain via glass micropipettes (VWR International). Electric pulses with forceps-type electrodes were delivered every second, with a duration of 50 ms per pulse in total five times at the voltage 32–35 V using an electroporator (BTX, ECM 830). Afterwards, this side of embryos were replaced in the abdominal cavity and this procedure was replicated on the other side. Finally, warm saline with 1% Pen/Strep was added into the abdominal cavity and the wound was surgically sutured. The whole process was performed under sterile conditions and the mice were monitored on the heat pad after surgery until they recovered completely.
QUANTIFICATION AND STATISTICAL ANALYSIS
We follow common practices of applying statistical analysis in biological data. For data presented as counts in a 2×2 table, we used Fisher’s exact test. For data expected to follow a normal distribution approximately, we used Student’s t-test. For data not expected to follow a (normal) distribution, we used non-parametric Wilcoxon test. R or Microsoft Excel (v.14) were used to carry out the aforementioned statistical tests. Significance threshold was set as 0.05 and presented as: p> 0.05, no significant “ns.”; p< 0.05, “*”; p< 0.01, “**”. Experimental numbers “N” and what they represent can be found in figure legends. More specifically, for analyses of proteins (Western Blot) and RNA (RT-PCR), “N” in figure legends indicates animal number or biological repeats of cell line experiments. For morphological analysis, “N” in figure legends represents total cell number from 2–11 animals of different liters. Data were blindly analyzed without considering genotype. For IUE experiments, only trailing processes longer than 10 µm were considered as axon and included for analysis. mRNA expression of the target amplicons was identified and quantified on QIAxcel Advanced capillary electrophoresis system. Densitometric quantification of the blots was carried out by ImageJ software (NIH). Band intensities obtained from the individual lanes was used to calculate fold change in protein expression following normalization to their internal loading control intensities.
Supplementary Material
Table S1. Axonogenesis-associated gene-level changes. Related to Figure 1.
Table S2. Axonogenesis-associated isoform-level changes. Related to Figure 1.
Table S3. Axonogenesis-associated splicing changes. Related to Figure 1.
Table S4. Known axonogenesis-controlling genes. Related to Figure 2.
Table S5. Alternative exons mis-regulated in Ptbp2-/-. Related to Figure 5.
HIGHLIGHTS.
Axon formation is orchestrated by neural-specific alternative splicing programming.
Early axonogenesis-associated splicing changes are governed by PTBP2.
Splicing-dependent functional changes of SHTN1 in actin binding and polymerization.
PTBP2 depletion impedes axon growth while stimulating axon specification.
ACKNOWLEDGEMENTS
We thank Dr. Orly Reiner (Weizmann Institute of Science, Israel) for the SHTN1 antibody and Dr. Douglas Black (University of California, Los Angeles) for the PTBP2 antibody. We thank Dr. Martin Riccomagno (University of California Riverside) for technical assistance with the DiI experiment. We thank Dr. Ayala Rao (University of California Riverside) for sharing the ultracentrifuge. We thank other members in the Zheng lab for various technical helps. V.E. was supported for 1-year by International Postdoctoral Scholarship Program (BIDEB-2219; 1059B191501385) from TUBITAK and Erzincan University. This work was supported by the NIH grants R01GM097230 (to L.C.), R01NS104041, and R01MH116220 (to S.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
ACCESSSION NUMBERS
The RNA-seq data are deposited in GEO (GSE124554).
REFERENCES
- Baba K, Yoshida W, Toriyama M, Shimada T, Manning CF, Saito M, Kohno K, Trimmer JS, Watanabe R, and Inagaki N (2018). Gradient-reading and mechano-effector machinery for netrin-1-induced axon guidance. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL (2011). DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27, 1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes AP, and Polleux F (2009). Establishment of axon-dendrite polarity in developing neurons. Annu. Rev. Neurosci 32, 347–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, Sanes JR, and Polleux F (2007). LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell 129, 549–563. [DOI] [PubMed] [Google Scholar]
- van Beuningen SFB, Will L, Harterink M, Chazeau A, van Battum EY, Frias CP, Franker MAM, Katrukha EA, Stucchi R, Vocking K, et al. (2015). TRIM46 Controls Neuronal Polarity and Axon Specification by Driving the Formation of Parallel Microtubule Arrays. Neuron 88, 1208–1226. [DOI] [PubMed] [Google Scholar]
- Boutz PL, Stoilov P, Li Q, Lin C-H, Chawla G, Ostrow K, Shiue L, Ares M Jr, and Black DL (2007). A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev 21, 1636–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitsprecher D, Kiesewetter AK, Linkner J, Vinzenz M, Stradal TEB, Small JV, Curth U, Dickinson RB, and Faix J (2011). Molecular mechanism of Ena/VASP-mediated actin-filament elongation. EMBO J 30, 456–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, and Agami R (2002). A system for stable expression of short interfering RNAs in mammalian cells. Science 296, 550–553. [DOI] [PubMed] [Google Scholar]
- Chen L, and Zheng S (2008). Identify alternative splicing events based on position-specific evolutionary conservation. PLoS One 3, e2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, and Zheng S (2009). Studying alternative splicing regulatory networks through partial correlation analysis. Genome Biol 10, R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Brinkmann K, Chen Z, Pak CW, Liao Y, Shi S, Henry L, Grishin NV, Bogdan S, and Rosen MK (2014). The WAVE regulatory complex links diverse receptors to the actin cytoskeleton. Cell 156, 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J-G, Rasin M-R, Kwan KY, and Sestan N (2005). Zfp312 is required for subcortical axonal projections and dendritic morphology of deep-layer pyramidal neurons of the cerebral cortex. Proc. Natl. Acad. Sci. U. S. A 102, 17792–17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Chen J, Shi H, Wei M, Castaneda-Castellanos DR, Bultje RS, Pei X, Kriegstein AR, Zhang M, and Shi S-H (2013). Regulation of microtubule stability and organization by mammalian Par3 in specifying neuronal polarity. Dev. Cell 24, 26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng P-L, and Poo M-M (2012). Early events in axon/dendrite polarization. Annu. Rev. Neurosci 35, 181–201. [DOI] [PubMed] [Google Scholar]
- Chereau D, and Dominguez R (2006). Understanding the role of the G-actin-binding domain of Ena/VASP in actin assembly. J. Struct. Biol 155, 195–201. [DOI] [PubMed] [Google Scholar]
- Chuang M, Goncharov A, Wang S, Oegema K, Jin Y, and Chisholm AD (2014). The microtubule minus-end-binding protein patronin/PTRN-1 is required for axon regeneration in C. elegans. Cell Rep 9, 874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA, Walker SB, and Pollard TD (1983). Pyrene actin: documentation of the validity of a sensitive assay for actin polymerization. J. Muscle Res. Cell Motil 4, 253–262. [DOI] [PubMed] [Google Scholar]
- Courtney KD, Grove M, Vandongen H, Vandongen A, LaMantia AS, and Pendergast AM (2000). Localization and phosphorylation of Abl-interactor proteins, Abi-1 and Abi-2, in the developing nervous system. Mol. Cell. Neurosci 16, 244–257. [DOI] [PubMed] [Google Scholar]
- Dai Z, and Pendergast AM (1995). Abi-2, a novel SH3-containing protein interacts with the c-Abl tyrosine kinase and modulates c-Abl transforming activity. Genes Dev 9, 2569–2582. [DOI] [PubMed] [Google Scholar]
- Darnell RB (2013). RNA protein interaction in neurons. Annu. Rev. Neurosci 36, 243–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay C, and Kennedy H (2007). Cell-cycle control and cortical development. Nat. Rev. Neurosci 8, 438–450. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Kim H-J, Bouton D, Warner M, and Gustafsson J-Å (2008). Expression of liver X receptor β is essential for formation of superficial cortical layers and migration of later-born neurons. Proc. Natl. Acad. Sci. U. S. A 105, 13445–13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvalov BK, Flynn KC, Neukirchen D, Meyn L, Teusch N, Wu X, Brakebusch C, Bamburg JR, and Bradke F (2007). Cdc42 regulates cofilin during the establishment of neuronal polarity. J. Neurosci 27, 13117–13129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand R, and Polleux F (2011). Neurogenin2 regulates the initial axon guidance of cortical pyramidal neurons projecting medially to the corpus callosum. Neural Dev 6, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hapak SM, Rothlin CV, and Ghosh S (2018). PAR3-PAR6-atypical PKC polarity complex proteins in neuronal polarization. Cell. Mol. Life Sci 75, 2735–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatanaka Y, and Yamauchi K (2013). Excitatory cortical neurons with multipolar shape establish neuronal polarity by forming a tangentially oriented axon in the intermediate zone. Cereb. Cortex 23, 105–113. [DOI] [PubMed] [Google Scholar]
- Jacko M, Weyn-Vanhentenryck SM, Smerdon JW, Yan R, Feng H, Williams DJ, Pai J, Xu K, Wichterle H, and Zhang C (2018). Rbfox Splicing Factors Promote Neuronal Maturation and Axon Initial Segment Assembly. Neuron 97, 853–868.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi M, Pan YA, Crump JG, and Sanes JR (2005). Mammalian SAD kinases are required for neuronal polarization. Science 307, 929–932. [DOI] [PubMed] [Google Scholar]
- Klingler E (2017). Development and Organization of the Evolutionarily Conserved Three-Layered Olfactory Cortex. eNeuro 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y, Baba K, Toriyama M, Minegishi T, Sugiura T, Kozawa S, Ikeda K, and Inagaki N (2015). Shootin1-cortactin interaction mediates signal-force transduction for axon outgrowth. J. Cell Biol 210, 663–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Zheng S, Han A, Lin C-H, Stoilov P, Fu X-D, and Black DL (2014). The splicing regulator PTBP2 controls a program of embryonic splicing required for neuronal maturation. Elife 3, e01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Vuong JK, Zhang M, Stork C, and Zheng S (2017). Inhibition of nonsense-mediated RNA decay by ER stress. RNA 23, 378–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Yano M, Fak JJ, Mele A, Grabinski SE, Zhang C, and Darnell RB (2012). Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev 26, 1626–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares AJ, Lin C-H, Damianov A, Adams KL, Novitch BG, and Black DL (2015). The splicing regulator PTBP1 controls the activity of the transcription factor Pbx1 during neuronal differentiation. Elife 4, e09268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcette JD, Chen JJ, and Nonet ML (2014). The Caenorhabditis elegans microtubule minus-end binding homolog PTRN-1 stabilizes synapses and neurites. Elife 3, e01637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeely KC, Cupp TD, Little JN, Janisch KM, Shrestha A, and Dwyer ND (2017). Mutation of Kinesin-6 Kif20b causes defects in cortical neuron polarization and morphogenesis. Neural Dev 12, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba T, Kibe Y, Funahashi Y, Nakamuta S, Takano T, Ueno T, Shimada A, Kozawa S, Okamoto M, Shimoda Y, et al. (2014). Pioneering axons regulate neuronal polarization in the developing cerebral cortex. Neuron 81, 814–829. [DOI] [PubMed] [Google Scholar]
- Neukirchen D, and Bradke F (2011). Cytoplasmic linker proteins regulate neuronal polarization through microtubule and growth cone dynamics. J. Neurosci 31, 1528–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B, and Blencowe BJ (2015). Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 87, 14–27. [DOI] [PubMed] [Google Scholar]
- Rao AN, Patil A, Black MM, Craig EM, Myers KA, Yeung HT, and Baas PW (2017). Cytoplasmic Dynein Transports Axonal Microtubules in a Polarity-Sorting Manner. Cell Rep 19, 2210–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, and Nakatsuji N (2001). Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol 240, 237–246. [DOI] [PubMed] [Google Scholar]
- Sharma S, Falick AM, Black DL (2005). Polypyrimidine Tract Binding Protein Blocks the 5′ Splice Site-Dependent Assembly of U2AF and the Prespliceosomal E Complex. Mol. Cell 19, 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Toriyama M, Uemura K, Kamiguchi H, Sugiura T, Watanabe N, and Inagaki N (2008). Shootin1 interacts with actin retrograde flow and L1-CAM to promote axon outgrowth. J. Cell Biol 181, 817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, and Chen L (2010). A two-parameter generalized Poisson model to improve the analysis of RNA-seq data. Nucleic Acids Res 38, e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahirovic S, Hellal F, Neukirchen D, Hindges R, Garvalov BK, Flynn KC, Stradal TE, Chrostek-Grashoff A, Brakebusch C, and Bradke F (2010). Rac1 regulates neuronal polarization through the WAVE complex. J. Neurosci 30, 6930–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriyama M, Shimada T, Kim KB, Mitsuba M, Nomura E, Katsuta K, Sakumura Y, Roepstorff P, and Inagaki N (2006). Shootin1: A protein involved in the organization of an asymmetric signal for neuronal polarization. J. Cell Biol 175, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, and Bonni A (2011). Transcriptional regulation of neuronal polarity and morphogenesis in the mammalian brain. Neuron 72, 22–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong CK, Black DL, and Zheng S (2016a). The neurogenetics of alternative splicing. Nat. Rev. Neurosci 17, 265–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong JK, Lin C-H, Zhang M, Chen L, Black DL, and Zheng S (2016b). PTBP1 and PTBP2 Serve Both Specific and Redundant Functions in Neuronal Pre-mRNA Splicing. Cell Rep 17, 2766–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Rai A, Hur E-M, Smilansky Z, Chang KT, and Min K-T (2016). DSCR1 is required for both axonal growth cone extension and steering. J. Cell Biol 213, 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Barnes AP, Hand R, Polleux F, and Ehlers MD (2010). TGF-β Signaling Specifies Axons during Brain Development. Cell 142, 144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S (2016a). Alternative splicing and nonsense-mediated mRNA decay enforce neural specific gene expression. Int. J. Dev. Neurosci [DOI] [PMC free article] [PubMed]
- Zheng S (2016b). IRAS: High-Throughput Identification of Novel Alternative Splicing Regulators. Methods Enzymol 572, 269–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, and Black DL (2013). Alternative pre-mRNA splicing in neurons: growing up and extending its reach. Trends Genet 29, 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Eacker SM, Hong SJ, Gronostajski RM, Dawson TM, and Dawson VL (2010). NMDA-induced neuronal survival is mediated through nuclear factor I-A in mice. J. Clin. Invest 120, 2446–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Gray EE, Chawla G, Porse BT, O’Dell TJ, and Black DL (2012). PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nat. Neurosci 15, 381–388, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Damoiseaux R, Chen L, and Black DL (2013). A broadly applicable high-throughput screening strategy identifies new regulators of Dlg4 (Psd-95) alternative splicing. Genome Res 23, 998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer C, Tiveron M-C, Bodmer R, and Cremer H (2004). Dynamics of Cux2 expression suggests that an early pool of SVZ precursors is fated to become upper cortical layer neurons. Cereb. Cortex 14, 1408–1420. [DOI] [PubMed] [Google Scholar]
- Zollinger DR, Baalman KL, and Rasband MN (2015). The ins and outs of polarized axonal domains. Annu. Rev. Cell Dev. Biol 31, 647–667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Axonogenesis-associated gene-level changes. Related to Figure 1.
Table S2. Axonogenesis-associated isoform-level changes. Related to Figure 1.
Table S3. Axonogenesis-associated splicing changes. Related to Figure 1.
Table S4. Known axonogenesis-controlling genes. Related to Figure 2.
Table S5. Alternative exons mis-regulated in Ptbp2-/-. Related to Figure 5.