

Abstract
Background
Members of the regulator of G‐protein signaling (RGS) family inhibit G‐protein coupled receptor signaling by modulating G‐protein activity. In platelets, there are 3 different RGS isoforms that are expressed at the protein level, including RGS16. Recently, we have shown that CXCL12 regulates platelet function via RGS16. However, the role of RGS16 in platelet function and thrombus formation is poorly defined.
Methods and Results
We used a genetic knockout mouse model approach to examine the role(s) of RGS16 in platelet activation by using a host of in vitro and in vivo assays. We observed that agonist‐induced platelet aggregation, secretion, and integrin activation were much more pronounced in platelets from the RGS16 knockout (Rgs16 −/−) mice relative to their wild type (Rgs16 +/+) littermates. Furthermore, the Rgs16 −/− mice had a markedly shortened bleeding time and were more susceptible to vascular injury–associated thrombus formation than the controls.
Conclusions
These findings support a critical role for RGS16 in regulating hemostatic and thrombotic functions of platelets in mice. Hence, RGS16 represents a potential therapeutic target for modulating platelet function.
Keywords: hemostasis, platelet, regulator of G‐protein signaling, RGS16, signal transduction, thrombosis
Subject Categories: Basic Science Research, Cell Signalling/Signal Transduction, Platelets, Vascular Biology
Clinical Perspective
What Is New?
This is the first study to demonstrate that regulator of G‐protein signaling 16 negatively regulates platelet function and consequently their hemostatic and thrombotic functions in mice.
What Are the Clinical Implications?
Regulator of G‐protein signaling 16 represents a potential therapeutic target for modulating platelet function.
Platelets are nonnucleated blood cells that are activated at sites of vascular injury1, 2 and are essential for normal hemostasis. Platelets also have the capacity to contribute to pathology when activated excessively, including in ischemic disorders such as stroke and myocardial infarction.1, 2 Under normal physiological conditions, patrolling platelets stay in a quiescent, nonadhesive state, which occurs through balanced inhibitory and stimulatory signaling.3 In response to a vascular insult, platelets rapidly bind to damaged blood vessels and aggregate to form thrombi in order to prevent excessive bleeding.3 When platelet dysfunction occurs through breakdowns in inhibitory signaling, inappropriate activation and excessive response to injury can occur.2, 4
Activated platelets secrete a host of diffusible locally acting mediators or agonists (eg, thrombin,5 adenosine diphosphate,6 or thromboxane A2,)3 which serve to recruit and activate additional platelets at the site of injury. For example, adenosine diphosphate potentiates the aggregatory effects of other stimuli such as thrombin and thromboxane A2, thereby contributing to stable thrombus formation. These mediators/agonists activate platelets by binding to their respective receptors on the platelet membrane, most of which belong to the G‐protein coupled receptor (GPCR) family.7 GPCR activation engages and initiates the activation of the coupled G proteins (GPs) through the exchange of GTP for GDP.8 Activation of the GP in turn results in several molecular changes, including inhibition of adenylyl cyclase and reduction in intracellular cyclic AMP levels (via the Gαi subunit), and activation of phospholipase C, which increases intracellular calcium levels (via the Gαq subunit), among others.7, 8, 9, 10 These molecular changes induce a host of cellular responses resulting in platelet aggregation, secretion, etc.7
Once the Gα/GP/GPCR signaling pathway has fulfilled its function, it must undergo a rapid turnoff to restore the “G protein cycle” by the GP's own intrinsic capacity to hydrolyze GTP, thereby returning to its quiescent (GDP‐bound) state,9 through re‐formation of a trimeric complex and termination of the signal. Thus, the duration in which Gα is in its GTP‐bound state serves as a major determining factor of the magnitude of GPCR signaling. Regulators of G protein signaling (RGS) proteins are GTPase‐accelerating proteins for Gα subunits, which increase the rate of GPCR signal termination.11 The RGS superfamily has an abundance of over 30 members in mammalian cells, which negatively regulate G protein activity.12, 13 RGS10, 16, and 18 are known to be expressed in platelets at the protein level.14, 15, 16
We have previously shown that RGS10 and 18 play a direct and prominent role in regulating platelet activation.14, 15 RGS16, which is the next most abundant RGS protein in platelets,17, 18 is a negative regulator of chemokine receptor CXCR4 signaling in megakaryocytes13 and its ligand CXCL12 in platelets.16 In addition, RGS16 was found to determine the signaling specificity of Gq‐coupled GPCRs,19 which is consistent with receptor‐specific regulation by other RGS proteins reported elsewhere. However, whether RGS16 plays a direct and/or more universal role in regulating platelet function and thrombogenesis remains to be determined.
The present study investigates the function of RGS16 in platelets by utilizing Rgs16 −/− mice. We found that platelets isolated from Rgs16 −/− mice exhibited enhanced aggregation, secretion, integrin activation, and phosphatidylserine exposure, when compared with Rgs16 +/+ littermates. We also observed that Rgs16 −/− mice exhibited an enhanced hemostasis response (shortened tail bleeding time) and an increased risk for thrombosis. Collectively, our findings indicate that RGS16 plays an essential negative regulatory role in platelet‐dependent hemostasis and thrombogenesis.
Materials and Methods
Reagents and Materials
Thrombin, stir bars, and other disposables were from Chrono‐Log (Havertown, PA). The protease‐activated receptor 4 agonist (TRAP4 peptide) was from Peptides International (Louisville, KY). Apyrase was obtained from Sigma Aldrich (St. Louis, MO). FITC‐conjugated CD62P and Annexin V antibodies were obtained from BD Biosciences (San Jose, CA). PE‐conjugated rat anti‐mouse Integrin αIIbβ3 (active form) JonA antibody was purchased from Emfret Analytics (Eibelstadt, Germany). RGS16 antibody was from Santa Cruz Biotechnology (sc‐166083, Dallas, TX). Other reagents were of analytical grade.
Animals and Genotyping
Rgs16 −/− mice were generated and genotyped as described previously.16, 20 Mice were housed in groups of 1 to 4 at 24°C, under 12/12 h light/dark cycles, with access to water and food ad libitum.
Methods
All experiments involving animals were performed in compliance with the institutional guidelines approved by the Institutional Animal Care and Use Committee.
Blood withdrawal was conducted as per our approved studies by the Institutional Review Board, and after the subjects gave informed consent.
The authors declare that all supporting data are available within the article data. Requests for detailed analytic methods will be addressed by the corresponding author. As for requests for the RGS16 deletion mice, these will be addressed on a case‐by‐case basis, and will be referred by the corresponding author to Dr. Kirk Druey.
Immunoblotting Studies
Human platelet proteins were separated on SDS‐PAGE gels and electrophoretically transferred to Immobilon‐P PVDF membranes, as previously described.16, 21 The blots were incubated with anti‐RGS16. Following washing, the blots were incubated with HRP‐labeled anti‐rabbit IgG. Signal was detected using enhanced chemiluminescence substrate (Thermo Scientific, Rockford, IL). Images were obtained with ChemiDoc MP Imaging System (Bio‐Rad, Hercules, CA).
Platelets Preparation
Platelets were prepared as previously described.14, 15, 22 Mouse blood was collected from a ventricle and the citrated (0.38%) blood was mixed with phosphate‐buffered saline, pH 7.4, and before incubation with prostaglandin I2 (10 ng/mL; 5 minutes) and centrifugation at 237g for 10 minutes at room temperature. Platelet‐rich plasma (PRP) was recovered and platelets were pelleted at 483g for 10 minutes at room temperature. The pellets were resuspended in HEPES/Tyrode buffer (20 mmol/L HEPES/KOH, pH 6.5, 128 mmol/L NaCl, 2.8 mmol/L KCl, 1 mmol/L MgCl2, 0.4 mmol/L NaH2PO4, 12 mmol/L NaHCO3, 5 mmol/L D‐glucose) supplemented with 1 mmol/L EGTA, 0.37 U/mL apyrase, and 10 ng/mL epoprostenol. Platelets were then washed and resuspended in HEPES/Tyrodes (pH 7.4) without EGTA, apyrase, or epoprostenol. Platelets were counted with an automated hematology analyzer (Drew Scientific, Dallas, TX) and adjusted to the indicated concentrations.
In Vitro Platelet Aggregation
The platelet‐rich plasma from Rgs16 +/+ and Rgs16 −/− mice was stimulated with 80 μmol/L TRAP4, 0.05 U/mL thrombin, or 1.25 μg/mL collagen. Note, collagen studies were performed with or without aspirin (1 μmol/L) or apyrase (0.03 U/mL). Platelet aggregation was measured by the turbidometric method using models 490 or 700 aggregometry systems (Chrono‐Log Corporation). Each experiment was repeated at least 3 times and blood was pooled from at least 3 separate groups of 8 mice.
ATP Release
This assay was performed as we described before.14, 16 Platelets were prepared as described above (250 μL; 7×107/mL) before being placed into siliconized cuvettes and stirred for 5 minutes at 37°C. The luciferase substrate/luciferase mixture (12.5 μL, Chrono‐Log) was then added, followed by the addition of the agonists 80 μmol/L TRAP4, 0.05 U/mL thrombin, or 1.25 μg/mL collagen. Note, collagen studies were performed with or without aspirin (1 μmol/L) or apyrase (0.03 U/mL). Each experiment was repeated 3 times, each time using blood pooled from 8 mice per group.
Flow Cytometric Analysis
Platelets from Rgs16 +/+ and Rgs16 −/− mice were stimulated with the 80 μmol/L TRAP4 or 0.05 U/mL thrombin for 3 minutes at room temperature. The reaction was stopped by fixing cells with 2% paraformaldehyde in PBS. Fixed platelets were labeled with PE‐conjugated rat anti‐mouse Integrin αIIbβ3 (active form) JonA antibody or FITC‐conjugated CD62P and Annexin V antibodies. Samples (105 platelets/100 μL) were then analyzed using an ACURI C6 flow cytometer as previously described.14, 15, 16, 22
Tail Bleeding Time
Hemostasis was examined using the tail transection technique.14, 15, 22 Briefly, mice were anesthetized with isoflurane and placed on a 37°C homeothermic blanket and their tails were transected 5 mm from the tip. The tail was placed in saline at 37°C and the time to bleeding cessation was measured. Bleeding stoppage was not considered complete until bleeding had stopped for 1 minute. When required, measurements were terminated at 10 minutes to prevent excessive blood loss.
In Vivo FeCl3‐Induced Carotid Artery Injury Model
These studies were performed as described previously.14, 15, 16, 22 Briefly, Rgs16 +/+ and Rgs16 −/− mice (8–10 weeks old) were anesthetized with 2.5% tribromoethanol (Avertin). The left carotid artery was exposed and cleaned, and baseline carotid artery blood flow was measured with a Transonic micro‐flowprobe (0.5 mm; Transonic Systems Inc, Ithaca, NY). After stabilization of blood flow, 7.5% ferric chloride (FeCl3) was applied to a filter paper disc (1‐mm diameter) that was immediately placed on top of the artery for 3 minutes. Blood flow was continuously monitored for 45 minutes, or until blood flow reached stable occlusion (zero blood flow for 2 minutes). Time to vessel occlusion was calculated as the difference in time between stable occlusion and removal of the filter paper (with FeCl3). An occlusion time of 10 minutes was considered as the cut‐off time for the purpose of statistical analysis.
Statistical Analysis
All experiments were performed at least 3 times. Analysis of the data was performed using GraphPad PRISM statistical software (San Diego, CA) and presented as mean±SEM, unless otherwise stated. The Mann–Whitney test was used for the evaluation of differences in mean bleeding and occlusion times, but similar results were observed using the t test. The flow cytometry data were analyzed (mean fluorescence intensity) using the t test. Significance was accepted at P<0.05, unless stated otherwise.
Results
Rgs16 Gene Deletion Does Not Affect Platelet or Peripheral Blood Cell Counts
We have previously shown that mouse platelets express several RGS proteins including RGS16.16 In order to extend these findings to humans, we isolated platelets from blood samples obtained by venipuncture and assessed RGS16 expression by immunoblotting. We detected RGS16 expression in lysates of human platelets (Figure 1; n=3). Given these results, we investigated the role of RGS16 in platelet development and function using WT and Rgs16 −/− mice. As we reported previously,16 the Rgs16 knockout strain showed no gross physical differences from their wild‐type counterparts, and both strains produced viable and healthy offspring. Platelet counts in Rgs16 +/+ and Rgs16 −/− mice were comparable (Table). All other blood parameters analyzed, including red blood cells and total white blood cells, were also normal in Rgs16 −/− mice. These data suggest that RGS16 deficiency does not affect megakaryopoiesis or hematopoiesis in general. Of note, the genotype of our RGS16 mice was verified via PCR analysis of genomic DNA, as previously reported.16
Figure 1.
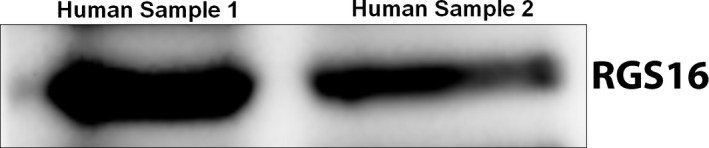
RGS16 is expressed in human platelets. RGS16 expression was analyzed in lysates of human platelets by immunoblotting (2×108/mL sample). RGS16 indicates regulator of G protein signaling 16.
Table 1.
Peripheral Blood Cell Counts in Rgs16 +/+ and Rgs16 −/− Mice
| Rgs16+/+ | Rgs16−/− | P Value | |
|---|---|---|---|
| Platelets | 1212.72±30.5 | 1296.68±30.0 | NS |
| MPV | 5.64±2.64 | 5.75±3.78 | NS |
| Red blood cells | 9.65±1.25 | 8.95±1.78 | NS |
| Lymphocytes | 5.31±1.34 | 5.95±1.69 | NS |
| Monocytes | 0.40±0.012 | 0.35±0.016 | NS |
| Granulocytes | 2.78±0.27 | 2.54±0.26 | NS |
Blood was collected from the heart and was counted as mentioned in the Methods section. All counts are thousands per microliter, except for red blood cells, which are millions per microliter. Data are represented as mean±SD. MPV indicates mean platelet volume; NS, not significant.
Deletion of Rgs16 Alters Agonist‐Induced Platelet Aggregation and Dense Granule Secretion
Platelets are hyperactivated in various pathological conditions, which can ultimately lead to heart attacks or stroke. To determine whether loss of RGS16 has an impact on platelet functions, we first examined the platelet aggregation response in WT or Rgs16 −/− mice. While Rgs16 gene deletion did not induce spontaneous aggregation, treatment of platelets from Rgs16 −/− mice with protease‐activated receptor 4 (PAR4) peptide (TRAP4) resulted in significantly increased aggregation compared with the responses of WT counterparts (Figure 2A; n=3). Similar results were observed with thrombin stimulation (Figure 2C; n=3). Platelet secretion of ATP from dense granules is a critical event in platelet activation, which amplifies its physiological responses. Here we found that platelets from Rgs16 −/− mice exhibit enhanced dense granule release, as measured by ATP secretion, compared with platelets from Rgs16 +/+ mice in response to either TRAP4 (Figure 2B; n=3) or thrombin (Figure 2D; n=3). To determine whether RGS16 regulates GPCR‐independent aggregation and secretion, we stimulated platelets with collagen, which activates platelets through glycoprotein VI (GPVI) receptors.23 Surprisingly, collagen‐induced aggregation and secretion were also increased in platelets from RGS16‐deficent mice compared with WT (Figure 2E and 2F; n=3). Collagen/GPVI‐mediated platelet activation is thought to involve feedback pathways such as thromboxane A2 and adenosine diphosphate, which in turn can act in an autocrine/paracrine manner on their cognate GPCRs to amplify aggregation and secretion.24 In order to determine whether the effects of RGS16 on collagen‐induced platelet function depend on this mechanism, we inhibited these mediators by pretreating platelets with aspirin or apyrase, respectively.24 We found that neither of these pretreatments significantly affected the response of platelets from Rgs16 −/− mice to collagen (Figure 3A through 3H; n=3). Thus, RGS16 regulates both GPCR‐dependent and ‐independent pathways in platelets.
Figure 2.
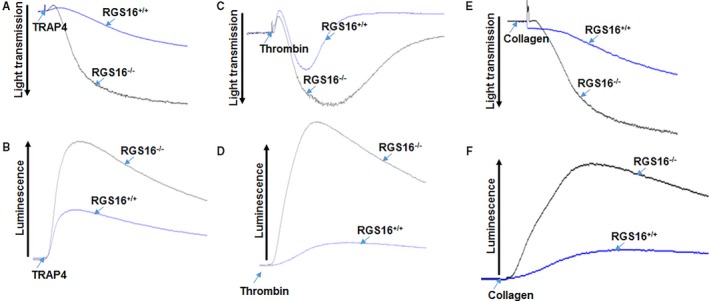
Deletion of RGS16 alters agonist‐induced platelet activation and dense granule secretion. Platelets from Rgs16 +/+ and Rgs16 −/− mice were prepared and aliquots (2.5×108/mL) were stimulated with (A and B) 80 μmol/L TRAP4, (C and D) 0.05 U/mL thrombin, or (E and F) 1.25 μg/mL collagen. Platelet aggregation (A, C, and E) and ATP release (B, D, and F) were measured with constant stirring in a lumiaggregometer. Each experiment was repeated at least 3 times, each with blood pooled from a group of 8 mice++. RGS16 indicates regulator of G protein signaling 16; TRAP4, protease‐activated receptor 4.
Figure 3.
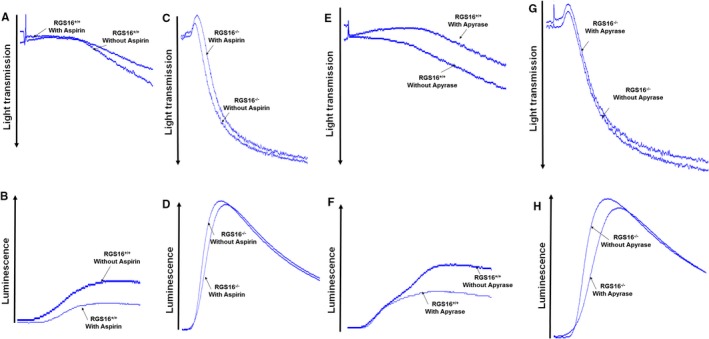
Deletion of RGS16 alters collagen‐induced platelet activation and dense granule secretion in aspirin and apyrase‐treated platelets. Platelets from Rgs16 +/+ and Rgs16 −/− mice were prepared and aliquots (2.5×108/mL) pretreated with or without aspirin or apyrase were stimulated with 1.25 μg/mL collagen. Platelet aggregation of the Rgs16 +/+ (A and E) and Rgs16 −/− (C and G) as well as ATP release of the Rgs16 +/+ (B and F) and Rgs16 −/− (D and H) were measured with constant stirring in a lumiaggregometer. Each experiment was repeated at least 3 times, each with blood pooled from a group of 8 mice. RGS16 indicates regulator of G protein signaling 16.
Loss of Rgs16 Causes Enhanced P‐Selectin Expression in Stimulated Platelets
Upon activation, platelets express P‐selectin, which is mobilized from α‐granules to the platelet surface,25 which is a measure of their α‐granule response. To examine the role of RGS16 in α‐granule release, we measured the marker P‐selectin using flow cytometry. While resting platelets from Rgs16 +/+ or Rgs16 −/− mice exhibited comparable surface expression of P‐selectin, stimulation with either TRAP4 or thrombin increased surface P‐selectin expression in platelets from Rgs16 −/− mice relative to controls, suggesting exaggerated α‐granule release (Figure 4A and 4B; n=3). Thus, RGS16 also regulates α‐granule release in response to PAR stimulation.
Figure 4.
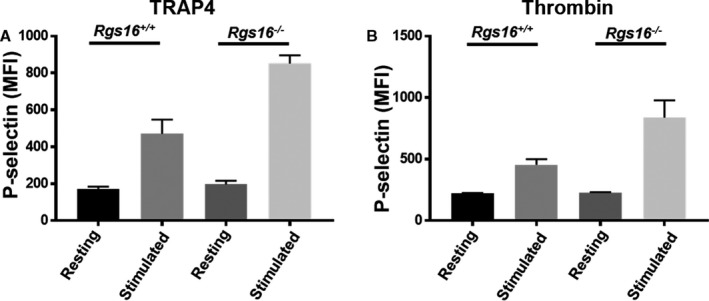
Deletion of RGS16 protein causes enhanced P‐selectin expression in stimulated platelets. Platelets from Rgs16 +/+ or Rgs16 −/− mice platelets (105 platelets/100 μL) were stimulated with (A) 80 μmol/L TRAP4 (P<0.00001; ANOVA) and (B) 0.05 U/mL thrombin (P<0.00001; ANOVA) for 3 minutes, fixed, and labeled with CD62P antibody. Samples were analyzed using a flow cytometer. Average mean fluorescence intensities are shown. Experiment was repeated at least 3 times, with blood pooled from a group of 8 mice each time. MFI indicates mean fluorescence intensity; RGS16, regulator of G protein signaling 16; TRAP4, protease‐activated receptor 4.
Deletion of RGS16 Causes Enhanced αIIbβ3 Activation in Stimulated Platelets
Upon stimulation, the platelet surface integrin αIIbβ3 becomes activated and undergoes a conformational change leading to a cascade of events that is required for platelet aggregation.26, 27, 28, 29 To assess whether RGS16 regulates integrin αIIbβ3, we measured cell surface expression of activated αIIbβ3 by flow cytometry. The surface intensity of the active form of the integrin was found to be significantly increased in RGS16‐deficient platelets following stimulation with either TRAP4 or thrombin, when compared with Rgs16 +/+ (Figure 5A and 5B; n=3), whereas the resting expression of activated αIIbβ3 in platelets from Rgs16 +/+ and Rgs16 −/− mice was indistinguishable from one another. Taken together, these results indicate that platelets from Rgs16 −/− mice aggregate excessively in response to PAR stimulation compared with WT counterparts, which occurs through an upregulation of dense and α‐granule release and through integrin αIIbβ3 activation.
Figure 5.
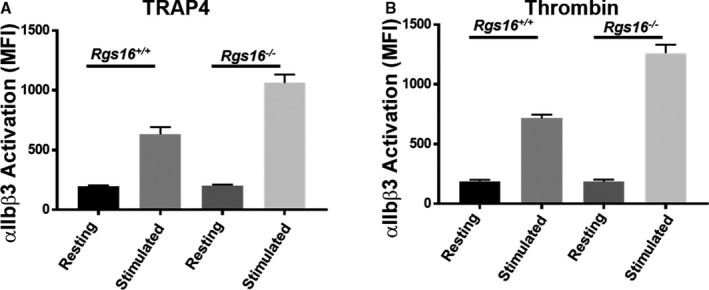
Deletion of RGS16 protein causes enhanced αIIbβ3 activation in stimulated platelets. Platelets from Rgs16 +/+ or Rgs16 −/− mice platelets (105 platelets/100 μL) were stimulated with (A) 80 μmol/L TRAP4 (P<0.00001; ANOVA) and (B) 0.05 U/mL thrombin (P<0.00001; ANOVA) for 3 minutes, fixed, and labeled with JON/A antibody. Samples were analyzed using a flow cytometer. Average mean fluorescence intensities are shown. Experiment was repeated at least 3 times, with blood pooled from a group of 8 mice each time. MFI indicates mean fluorescence intensity; RGS16, regulator of G protein signaling 16; TRAP4, protease‐activated receptor 4.
Increased Phosphatidylserine Expression in Stimulated Platelets from Rgs16 −/− Mice
Negatively charged, procoagulant phospholipid phosphatidylserine shifts from the inner surface of the plasma membrane to the outer surface upon agonist stimulation, thereby facilitating platelet aggregation and coagulation.30, 31 Maintenance of this asymmetry requires energy and is of major importance for platelet function since the appearance of phosphatidylserine at the cell surface is associated with several physiologic and pathologic phenomena.32, 33, 34, 35 Therefore, we examined phosphatidylserine expression under resting and activated conditions by flow cytometry. Whereas phosphatidylserine expression in platelets from WT and Rgs16 −/− mice was comparable at baseline, stimulation with either TRAP4 or thrombin increased cell surface phosphatidylserine expression, and the response was increased in platelets from Rgs16 −/− mice relative to controls (Figure 6A and 6B; n=3). Thus, RGS16 regulates phosphatidylserine exposure induced by PAR stimulation.
Figure 6.
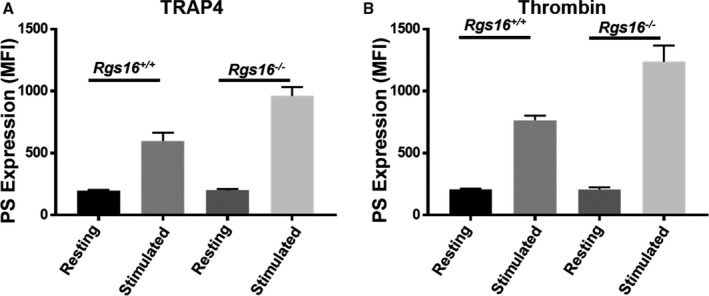
Deletion of RGS16 protein causes enhanced phosphatidylserine expression in stimulated platelets. Platelets from Rgs16 +/+ or Rgs16 −/− mice platelets (105 platelets/100 μL) were stimulated with (A) 80 μmol/L TRAP4 (P<0.00001; ANOVA) and (B) 0.05 U/mL thrombin (P<0.00001; ANOVA) for 3 minutes, fixed, and labeled with Annexin V antibody. Samples were analyzed using a flow cytometer. Average MFIs are shown. Experiment was repeated at least 3 times, with blood pooled from a group of 8 mice each time. MFI indicates mean fluorescence intensity; PS, phosphatidylserine; RGS16, regulator of G protein signaling 16; TRAP4, protease‐activated receptor 4.
Deletion of Rgs16 Enhances Hemostasis and Predisposes to Thrombosis
Previous work by our laboratory and others indicated that several RGS proteins (RGS10 and RGS18) prevent excessive responses to vascular injury through negative regulation of platelet activation.14, 15, 16 To determine whether the hyperactivity of platelets from Rgs16 −/− mice also translated to aberrant hemostasis in vivo, we measured tail bleeding times. We observed a significantly shortened bleeding time in Rgs16 −/− mice following tail transection (≈59 s±13.7) compared with WT littermates (≈260 s±33.4; Figure 7A; n=8 each; P=0.0002). This finding indicates that RGS16 negatively regulates the physiological hemostatic function of platelets in vivo. Platelet hyperactivity is also known to promote thrombogenesis, which can lead to a host of deleterious health problems, such as heart attack and stroke. We hypothesized that Rgs16 −/− mice would be more prone to thrombotic disorders. To test this, we applied FeCl3 to the carotid artery, which is an established method to induce vascular injury–mediated thrombus formation and artery occlusion. Rgs16 −/− mice had significantly shortened occlusion times, averaging ≈40 s (±10.6) compared with WT littermates of ≈420 s (±54.8) (Figure 7B; n=5 each; P=0.0079). These results demonstrate that RGS16 inhibits thrombus formation in vivo.
Figure 7.
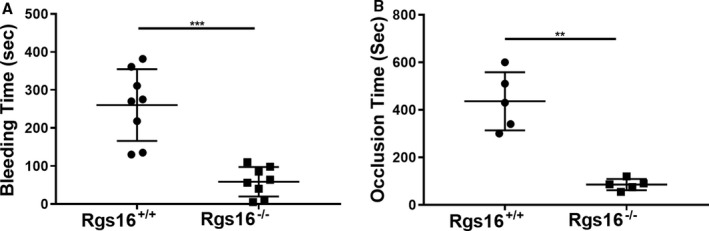
Deletion of RGS16 protein alters physiological hemostasis and development of thrombosis. (A) Bleeding times were measured in Rgs16 +/+ (n=8) or Rgs16 −/− (n=8) mice following venisection. Each point represents the bleeding time of a single animal (P=0.0002; Mann–Whitney test). (B) Thrombosis was induced in Rgs16 +/+ (n=5) or Rgs16 −/− (n=5) mice using chemical injury (FeCl3). Each point represents an occlusion time of a single animal (P=0.0079; Mann–Whitney test). ***P<0.0001, whereas **P<0.001. RGS16 indicates regulator of G protein signaling 16.
Discussion
RGS16 and other closely related RGS proteins in the R4 family negatively regulate G‐protein activity,36 and thus represent a feedback mechanism to inhibit Gi/Gq signaling a variety of cell systems.19, 37, 38 Previous studies have indicated that RGS16 has GAP activity toward different Gi and/or Gq class α subunits but not GαS or Gα12. 39, 40, 41, 42, 43, 44 RGS16 regulates a wide variety of physiological as well as pathophysiological conditions, in cells and tissues, such as proto‐oncogene expression in neuronal cells, respiratory and cardiovascular functions, and immune responses.16, 20, 45, 46, 47, 48, 49 Although the full spectrum of RGS expression and function in platelets has not been fully defined, we and others have recently shown that RGS10 and 18 regulate GPCR‐dependent platelet function.14, 15, 50, 51 Moreover, stimulated platelets release various growth factors and chemokines into their microenvironment including CXCL12,52 which we recently showed to be sensitive to regulation by RGS16.16
Here we found that PAR‐mediated platelet aggregation and activation (as measured by dense and α granule secretion, integrin αIIbβ3 activation, and phosphatidylserine exposure) of platelets was enhanced by RGS16 deficiency, which translated to a hypercoagulable state in vivo. These findings suggest a negative regulatory function for RGS16 in platelets through GAP activity on Gi and Gq.19 Published work has indicated that although RGS16 lacks GAP activity for G13, it also has the capacity to negatively regulate G13‐mediated cancer cell proliferation and morphological changes of neurons by blocking G protein‐effector interactions.53 Aside from Gq, PAR1, PAR4, and thromboxane receptors couple to G13, 7, 54, 55 raising the possibility that RGS16 might also regulate G13‐dependent signaling through specific GPCRs in platelets.
Unexpectedly, we found that RGS16 modifies the platelet response to collagen, which acts through GPVI, an inhibitory tyrosine activation motif (ITAM)‐containing receptor that acts through multiple pathways including PI3 kinase and phospholipase Cγ, but independently of G proteins.56 Noncanonical roles for RGS proteins in various signaling pathways have been documented.57, 58, 59, 60 For example, several RGS proteins including RGS16 interact directly with the p85 regulatory subunit of PI3K and modulate PI3K activity.61 In breast cancer cells, RGS16 inhibited formation of a PI3K‐Gab1‐epidermal growth factor receptor complex that promotes cell proliferation.62 RGS16‐depleted cell lines proliferated more than controls in response to epidermal growth factor stimulation, suggesting that RGS16 negatively regulates PI3K‐dependent proliferative signaling. In our study, enhanced platelet responses to collagen in the absence of RGS16 persisted even when collagen‐induced mediators (GPCR agonists adenosine diphosphate and thromboxane A2) were inhibited, suggesting that RGS16 regulates GPVI signaling directly. Since PI3K activation appears to be critical for GPVI‐mediated activation of platelets,56, 63 RGS16 might regulate this pathway through its interaction with PI3K.
We and others have shown that RGS proteins are rather promiscuous with respect to their capacity to modulate various GPCR‐G‐protein pathways.13, 14, 15, 16, 51, 64, 65, 66 For instance, RGS4, binds to and serves as a GTPase‐activating protein for Gαi1, Gαi2, Gαi3, Gαo, Gαt, Gαz, and Gαq. 39, 40, 67 Similarly, RGS16 binds to Gαi2, Gαi3, Gαo, Gαt, 41, 68 and Gαq. Interestingly, investigations of RGS function in mammalian cells indicate that individual family members seem to be selective in the regulation of specific G protein–linked signaling pathways. RGS1, RGS2, RGS3, and RGS4 were found to differ in their ability to impair interleukin‐8 receptor signaling (ERK activation), with RGS4 showing the greatest potency followed by RGS3, RGS1, and RGS2 in overexpression systems.69 In a separate study, RGS3, but not RGS1, RGS2, or RGS4, suppressed the IP3 responses induced by the gonadotropin‐releasing hormone.70 RGS16 may selectively regulate platelet GPCRs‐G‐proteins in a similar manner, exhibit functional specificity in interacting with GPs, or in its cross‐talk with other signaling pathways. In addition, it is likely that other factors such as tissue and cell type distribution, temporal expression, and posttranslational modification act in concert to dictate the specificity of RGS function.
One potential challenge to investigating RGS protein function in living animals is the potential for functional redundancy and compensatory changes in RGS protein expression resulting from loss of a single protein. However, given the phenotypes we observed/reported with 3 RGS knockout mice, it does not seem that there is functional redundancy, at least in the platelet functions we analyzed, and our previous work revealed no changes in the expression levels of RGS10 and RGS18, as a result of Rgs16 deletion.16
Unlike in the case of RGS18, which was found to regulate platelet generation,65 our findings revealed no impairment in platelet formation in the absence of RGS16 in mice. Thus, although RGS16 and RGS18 are both expressed in megakaryocytes, RGS16 does not appear to regulate megakaryopoiesis in the bone marrow.13, 71, 72 Moreover, because deletion of RGS16 did not affect other blood cell counts, our results suggest that RGS16 does not modulate hematopoiesis in general. In contrast to RGS18, RGS16 was shown to act as a negative regulator of chemokine (CXCR4) signaling in megakaryocytes,13 which was considered to be the result of its GAP activity.73 These findings suggest that RGS proteins could play different roles in platelet biology. However, the present results, coupled with our studies showing that RGS10 and RGS18 also regulate platelet function in vitro and in vivo,14, 15 indicate that platelet‐associated RGS proteins play distinct and indispensable roles in platelet activation. The mechanism(s) underlying these distinct roles will be the scope of future investigations.
In conclusion, RGS16 plays a critical role in fine‐tuning platelet function and may serve as a potential therapeutic target for managing several thrombotic disorders. Future studies of the full spectrum of GPCR pathways regulated by RGS16 in platelets, as well as their signaling mechanisms, are warranted in order to investigate differential regulation of RGS16‐GPCRs in platelet function.
Sources of Funding
This research was supported, in part, by funds provided by the American Heart Association, grant number 16GRNT31350032 (to Khasawneh) and by the Intramural Research Program, NIAID/NIH (to Druey).
Disclosures
None.
(J Am Heart Assoc. 2019;8:e011273 DOI: 10.1161/JAHA.118.011273.)
References
- 1. Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359:938–949. [DOI] [PubMed] [Google Scholar]
- 2. Jackson SP. Arterial thrombosis—insidious, unpredictable and deadly. Nat Med. 2011;17:1423–1436. [DOI] [PubMed] [Google Scholar]
- 3. Paez Espinosa EV, Murad JP, Khasawneh FT. Aspirin: pharmacology and clinical applications. Thrombosis. 2012;2012:173124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nieswandt B, Pleines I, Bender M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J Thromb Haemost. 2011;9(suppl 1):92–104. [DOI] [PubMed] [Google Scholar]
- 5. Wiisanen ME, Moliterno DJ. Platelet protease‐activated receptor antagonism in cardiovascular medicine. Coron Artery Dis. 2012;23:375–379. [DOI] [PubMed] [Google Scholar]
- 6. Gurbel PA, Kuliopulos A, Tantry US. G‐protein‐coupled receptors signaling pathways in new antiplatelet drug development. Arterioscler Thromb Vasc Biol. 2015;35:500–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Offermanns S. Activation of platelet function through g protein‐coupled receptors. Circ Res. 2006;99:1293–1304. [DOI] [PubMed] [Google Scholar]
- 8. McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS. G‐protein signaling: back to the future. Cell Mol Life Sci. 2005;62:551–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dohlman HG. Thematic minireview series: cell biology of g protein signaling. J Biol Chem. 2015;290:6679–6680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wettschureck N, Offermanns S. Mammalian g proteins and their cell type specific functions. Physiol Rev. 2005;85:1159–1204. [DOI] [PubMed] [Google Scholar]
- 11. Siderovski DP, Willard FS. The GAPs, GEFs, and GDIs of heterotrimeric G‐protein alpha subunits. Int J Biol Sci. 2005;1:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bansal G, Druey KM, Xie Z. R4 RGS proteins: regulation of G‐protein signaling and beyond. Pharmacol Ther. 2007;116:473–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berthebaud M, Riviere C, Jarrier P, Foudi A, Zhang Y, Compagno D, Galy A, Vainchenker W, Louache F. RGS16 is a negative regulator of SDF‐1‐CXCR4 signaling in megakaryocytes. Blood. 2005;106:2962–2968. [DOI] [PubMed] [Google Scholar]
- 14. Alshbool FZ, Karim ZA, Vemana HP, Conlon C, Lin OA, Khasawneh FT. The regulator of G‐protein signaling 18 regulates platelet aggregation, hemostasis and thrombosis. Biochem Biophys Res Commun. 2015;462:378–382. [DOI] [PubMed] [Google Scholar]
- 15. Hensch NR, Karim ZA, Druey KM, Tansey MG, Khasawneh FT. RGS10 negatively regulates platelet activation and thrombogenesis. PLoS One. 2016;11:e0165984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karim ZA, Alshbool FZ, Vemana HP, Conlon C, Druey KM, Khasawneh FT. CXCL12 regulates platelet activation via the regulator of G‐protein signaling 16. Biochim Biophys Acta. 2016;1863:314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gagnon AW, Murray DL, Leadley RJ. Cloning and characterization of a novel regulator of g protein signalling in human platelets. Cell Signal. 2002;14:595–606. [DOI] [PubMed] [Google Scholar]
- 18. Louwette S, Van Geet C, Freson K. Regulators of G protein signaling: role in hematopoiesis, megakaryopoiesis and platelet function. J Thromb Haemost. 2012;10:2215–2222. [DOI] [PubMed] [Google Scholar]
- 19. Xu X, Zeng W, Popov S, Berman DM, Davignon I, Yu K, Yowe D, Offermanns S, Muallem S, Wilkie TM. RGS proteins determine signaling specificity of Gq‐coupled receptors. J Biol Chem. 1999;274:3549–3556. [DOI] [PubMed] [Google Scholar]
- 20. Shankar SP, Wilson MS, DiVietro JA, Mentink‐Kane MM, Xie Z, Wynn TA, Druey KM. RGS16 attenuates pulmonary Th2/Th17 inflammatory responses. J Immunol. 2012;188:6347–6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karim ZA, Hensch NR, Qasim H, Alshbool FZ, Khasawneh FT. Role of ikappab kinase beta in regulating the remodeling of the CARMA1‐Bcl10‐MALT1 complex. Biochem Biophys Res Commun. 2018;500:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hensch NR, Karim ZA, Pineda J, Mercado N, Alshbool FZ, Khasawneh FT. P2Y12 antibody inhibits platelet activity and protects against thrombogenesis. Biochem Biophys Res Commun. 2017;493:1069–1074. [DOI] [PubMed] [Google Scholar]
- 23. Liu Y, Liu T, Ding K, Liu Z, Li Y, He T, Zhang W, Fan Y, Ma W, Cui L, Song X. Phospholipase Cgamma2 signaling cascade contribute to the antiplatelet effect of notoginsenoside Fc. Front Pharmacol. 2018;9:1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roger S, Pawlowski M, Habib A, Jandrot‐Perrus M, Rosa JP, Bryckaert M. Costimulation of the Gi‐coupled ADP receptor and the Gq‐coupled TXA2 receptor is required for ERK2 activation in collagen‐induced platelet aggregation. FEBS Lett. 2004;556:227–235. [DOI] [PubMed] [Google Scholar]
- 25. Mumford AD, Frelinger AL III, Gachet C, Gresele P, Noris P, Harrison P, Mezzano D. A review of platelet secretion assays for the diagnosis of inherited platelet secretion disorders. Thromb Haemost. 2015;114:14–25. [DOI] [PubMed] [Google Scholar]
- 26. Fong KP, Zhu H, Span LM, Moore DT, Yoon K, Tamura R, Yin H, DeGrado WF, Bennett JS. Directly activating the integrin alphaiibbeta3 initiates outside‐in signaling by causing alphaiibbeta3 clustering. J Biol Chem. 2016;291:11706–11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hodivala‐Dilke KM, McHugh KP, Tsakiris DA, Rayburn H, Crowley D, Ullman‐Cullere M, Ross FP, Coller BS, Teitelbaum S, Hynes RO. Beta3‐integrin‐deficient mice are a model for glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest. 1999;103:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma YQ, Qin J, Plow EF. Platelet integrin alpha(iib)beta(3): activation mechanisms. J Thromb Haemost. 2007;5:1345–1352. [DOI] [PubMed] [Google Scholar]
- 29. Mehrbod M, Trisno S, Mofrad MR. On the activation of integrin alphaiibbeta3: outside‐in and inside‐out pathways. Biophys J. 2013;105:1304–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bevers EM, Comfurius P, van Rijn JL, Hemker HC, Zwaal RF. Generation of prothrombin‐converting activity and the exposure of phosphatidylserine at the outer surface of platelets. Eur J Biochem. 1982;122:429–436. [DOI] [PubMed] [Google Scholar]
- 31. Bevers EM, Comfurius P, Zwaal RF. Changes in membrane phospholipid distribution during platelet activation. Biochim Biophys Acta. 1983;736:57–66. [DOI] [PubMed] [Google Scholar]
- 32. Zwaal RF, Schroit AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89:1121–1132. [PubMed] [Google Scholar]
- 33. Brzoska T, Suzuki Y, Mogami H, Sano H, Urano T. Binding of thrombin‐activated platelets to a fibrin scaffold through alpha(iib)beta(3) evokes phosphatidylserine exposure on their cell surface. PLoS One. 2013;8:e55466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fukuda DA, Caporrino MC, Barbaro KC, Della‐Casa MS, Faquim‐Mauro EL, Magalhaes GS. Recombinant phospholipase d from loxosceles gaucho binds to platelets and promotes phosphatidylserine exposure. Toxins (Basel). 2017;9:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pang A, Cui Y, Chen Y, Cheng N, Delaney MK, Gu M, Stojanovic‐Terpo A, Zhu C, Du X. Shear‐induced integrin signaling in platelet phosphatidylserine exposure, microvesicle release and coagulation. Blood. 2018;132:533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ross EM, Wilkie TM. GTPase‐activating proteins for heterotrimeric g proteins: regulators of G protein signaling (RGS) and RGS‐like proteins. Annu Rev Biochem. 2000;69:795–827. [DOI] [PubMed] [Google Scholar]
- 37. Luo X, Popov S, Bera AK, Wilkie TM, Muallem S. RGS proteins provide biochemical control of agonist‐evoked [ca2+]i oscillations. Mol Cell. 2001;7:651–660. [DOI] [PubMed] [Google Scholar]
- 38. Zeng W, Xu X, Popov S, Mukhopadhyay S, Chidiac P, Swistok J, Danho W, Yagaloff KA, Fisher SL, Ross EM, Muallem S, Wilkie TM. The n‐terminal domain of RGS4 confers receptor‐selective inhibition of G protein signaling. J Biol Chem. 1998;273:34687–34690. [DOI] [PubMed] [Google Scholar]
- 39. Berman DM, Kozasa T, Gilman AG. The GTPase‐activating protein RGS4 stabilizes the transition state for nucleotide hydrolysis. J Biol Chem. 1996;271:27209–27212. [DOI] [PubMed] [Google Scholar]
- 40. Berman DM, Wilkie TM, Gilman AG. GAIP and RGS4 are GTPase‐activating proteins for the Gi subfamily of G protein alpha subunits. Cell. 1996;86:445–452. [DOI] [PubMed] [Google Scholar]
- 41. Chen C, Zheng B, Han J, Lin SC. Characterization of a novel mammalian RGS protein that binds to Galpha proteins and inhibits pheromone signaling in yeast. J Biol Chem. 1997;272:8679–8685. [DOI] [PubMed] [Google Scholar]
- 42. Heximer SP, Cristillo AD, Forsdyke DR. Comparison of mRNA expression of two regulators of G‐protein signaling, RGS1/BL34/1R20 and RGS2/G0S8, in cultured human blood mononuclear cells. DNA Cell Biol. 1997;16:589–598. [DOI] [PubMed] [Google Scholar]
- 43. Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. D‐AKAP2, a novel protein kinase A anchoring protein with a putative RGS domain. Proc Natl Acad Sci USA. 1997;94:11184–11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yan Y, Chi PP, Bourne HR. RGS4 inhibits Gq‐mediated activation of mitogen‐activated protein kinase and phosphoinositide synthesis. J Biol Chem. 1997;272:11924–11927. [DOI] [PubMed] [Google Scholar]
- 45. Hoshi Y, Endo K, Shirakihara T, Fukagawa A, Miyazawa K, Saitoh M. The potential role of regulator of G‐protein signaling 16 in cell motility mediated by deltaef1 family proteins. FEBS Lett. 2016;590:270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Patten M, Bunemann J, Thoma B, Kramer E, Thoenes M, Stube S, Mittmann C, Wieland T. Endotoxin induces desensitization of cardiac endothelin‐1 receptor signaling by increased expression of RGS4 and RGS16. Cardiovasc Res. 2002;53:156–164. [DOI] [PubMed] [Google Scholar]
- 47. Stuebe S, Wieland T, Kraemer E, Stritzky A, Schroeder D, Seekamp S, Vogt A, Chen CK, Patten M. Sphingosine‐1‐phosphate and endothelin‐1 induce the expression of RGS16 protein in cardiac myocytes by transcriptional activation of the RGS16 gene. Naunyn Schmiedebergs Arch Pharmacol. 2008;376:363–373. [DOI] [PubMed] [Google Scholar]
- 48. Suurvali J, Pahtma M, Saar R, Paalme V, Nutt A, Tiivel T, Saaremae M, Fitting C, Cavaillon JM, Ruutel Boudinot S. RGS16 restricts the pro‐inflammatory response of monocytes. Scand J Immunol. 2015;81:23–30. [DOI] [PubMed] [Google Scholar]
- 49. Vivot K, Moulle VS, Zarrouki B, Tremblay C, Mancini AD, Maachi H, Ghislain J, Poitout V. The regulator of G‐protein signaling RGS16 promotes insulin secretion and beta‐cell proliferation in rodent and human islets. Mol Metab. 2016;5:988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ma P, Cierniewska A, Signarvic R, Cieslak M, Kong H, Sinnamon AJ, Neubig RR, Newman DK, Stalker TJ, Brass LF. A newly identified complex of spinophilin and the tyrosine phosphatase, SHP‐1, modulates platelet activation by regulating G protein‐dependent signaling. Blood. 2012;119:1935–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Signarvic RS, Cierniewska A, Stalker TJ, Fong KP, Chatterjee MS, Hess PR, Ma P, Diamond SL, Neubig RR, Brass LF. RGS/Gi2alpha interactions modulate platelet accumulation and thrombus formation at sites of vascular injury. Blood. 2010;116:6092–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jonnalagadda D, Izu LT, Whiteheart SW. Platelet secretion is kinetically heterogeneous in an agonist‐responsive manner. Blood. 2012;120:5209–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Johnson EN, Seasholtz TM, Waheed AA, Kreutz B, Suzuki N, Kozasa T, Jones TL, Brown JH, Druey KM. RGS16 inhibits signalling through the G alpha 13‐Rho axis. Nat Cell Biol. 2003;5:1095–1103. [DOI] [PubMed] [Google Scholar]
- 54. French SL, Hamilton JR. Protease‐activated receptor 4: from structure to function and back again. Br J Pharmacol. 2016;173:2952–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moers A, Wettschureck N, Offermanns S. G13‐mediated signaling as a potential target for antiplatelet drugs. Drug News Perspect. 2004;17:493–498. [DOI] [PubMed] [Google Scholar]
- 56. Moroi AJ, Watson SP. Impact of the PI3‐kinase/Akt pathway on ITAM and hemITAM receptors: haemostasis, platelet activation and antithrombotic therapy. Biochem Pharmacol. 2015;94:186–194. [DOI] [PubMed] [Google Scholar]
- 57. Derrien A, Druey KM. RGS16 function is regulated by epidermal growth factor receptor‐mediated tyrosine phosphorylation. J Biol Chem. 2001;276:48532–48538. [DOI] [PubMed] [Google Scholar]
- 58. Hunt TW, Fields TA, Casey PJ, Peralta EG. RGS10 is a selective activator of G alpha i GTPase activity. Nature. 1996;383:175–177. [DOI] [PubMed] [Google Scholar]
- 59. Sethakorn N, Yau DM, Dulin NO. Non‐canonical functions of RGS proteins. Cell Signal. 2010;22:1274–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ghavami A, Hunt RA, Olsen MA, Zhang J, Smith DL, Kalgaonkar S, Rahman Z, Young KH. Differential effects of regulator of G protein signaling (RGS) proteins on serotonin 5‐HT1a, 5‐HT2a, and dopamine D2 receptor‐mediated signaling and adenylyl cyclase activity. Cell Signal. 2004;16:711–721. [DOI] [PubMed] [Google Scholar]
- 61. Bansal G, Xie Z, Rao S, Nocka KH, Druey KM. Suppression of immunoglobulin E‐mediated allergic responses by regulator of G protein signaling 13. Nat Immunol. 2008;9:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liang G, Bansal G, Xie Z, Druey KM. RGS16 inhibits breast cancer cell growth by mitigating phosphatidylinositol 3‐kinase signaling. J Biol Chem. 2009;284:21719–21727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Manganaro D, Consonni A, Guidetti GF, Canobbio I, Visconte C, Kim S, Okigaki M, Falasca M, Hirsch E, Kunapuli SP, Torti M. Activation of phosphatidylinositol 3‐kinase beta by the platelet collagen receptors integrin alpha2beta1 and GPVI: the role of Pyk2 and c‐Cbl. Biochim Biophys Acta. 2015;1853:1879–1888. [DOI] [PubMed] [Google Scholar]
- 64. Anger T, Klintworth N, Stumpf C, Daniel WG, Mende U, Garlichs CD. RGS protein specificity towards Gq‐ and Gi/o‐mediated ERK 1/2 and Akt activation, in vitro. J Biochem Mol Biol. 2007;40:899–910. [DOI] [PubMed] [Google Scholar]
- 65. Delesque‐Touchard N, Pendaries C, Volle‐Challier C, Millet L, Salel V, Herve C, Pflieger AM, Berthou‐Soulie L, Prades C, Sorg T, Herbert JM, Savi P, Bono F. Regulator of G‐protein signaling 18 controls both platelet generation and function. PLoS One. 2014;9:e113215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang Y, Neo SY, Han J, Yaw LP, Lin SC. RGS16 attenuates galphaq‐dependent p38 mitogen‐activated protein kinase activation by platelet‐activating factor. J Biol Chem. 1999;274:2851–2857. [DOI] [PubMed] [Google Scholar]
- 67. Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. RGS family members: Gtpase‐activating proteins for heterotrimeric G‐protein alpha‐subunits. Nature. 1996;383:172–175. [DOI] [PubMed] [Google Scholar]
- 68. Chen CK, Wieland T, Simon MI. RGS‐r, a retinal specific RGS protein, binds an intermediate conformation of transducin and enhances recycling. Proc Natl Acad Sci USA. 1996;93:12885–12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Druey KM, Blumer KJ, Kang VH, Kehrl JH. Inhibition of G‐protein‐mediated map kinase activation by a new mammalian gene family. Nature. 1996;379:742–746. [DOI] [PubMed] [Google Scholar]
- 70. Neill JD, Duck LW, Sellers JC, Musgrove LC, Scheschonka A, Druey KM, Kehrl JH. Potential role for a regulator of G protein signaling (RGS3) in gonadotropin‐releasing hormone (GNRH) stimulated desensitization. Endocrinology. 1997;138:843–846. [DOI] [PubMed] [Google Scholar]
- 71. Nagata Y, Oda M, Nakata H, Shozaki Y, Kozasa T, Todokoro K. A novel regulator of G‐protein signaling bearing GAP activity for Galphai and Galphaq in megakaryocytes. Blood. 2001;97:3051–3060. [DOI] [PubMed] [Google Scholar]
- 72. Yowe D, Weich N, Prabhudas M, Poisson L, Errada P, Kapeller R, Yu K, Faron L, Shen M, Cleary J, Wilkie TM, Gutierrez‐Ramos C, Hodge MR. RGS18 is a myeloerythroid lineage‐specific regulator of G‐protein‐signalling molecule highly expressed in megakaryocytes. Biochem J. 2001;359:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lopez‐Ilasaca M. Signaling from G‐protein‐coupled receptors to mitogen‐activated protein (MAP)‐kinase cascades. Biochem Pharmacol. 1998;56:269–277. [DOI] [PubMed] [Google Scholar]