

Abstract
Chronic neurological impairments can manifest from repetitive traumatic brain injury (rTBI), particularly when subsequent injuries occur before the initial injury completely heals. Herein, we apply post-traumatic sleep as a physiological biomarker of vulnerability, hypothesizing that a second TBI during post-traumatic sleep worsens neurological and histological outcomes compared to one TBI or a second TBI after post-traumatic sleep subsides. Mice received sham or diffuse TBI by midline fluid percussion injury; brain-injured mice received one TBI or rTBIs at 3- or 9-h intervals. Over 40 h post-injury, injured mice slept more than shams. Functional assessments indicated lower latencies on rotarod and increased Neurological Severity Scores for mice with rTBIs within 3 h. Anxiety-like behaviors in the open field task were increased for mice with rTBIs at 3 h. Based on pixel density of silver accumulation, neuropathology was greater at 28 days post-injury (DPI) in rTBI groups than sham and single TBI. Cortical microglia morphology was quantified and mice receiving rTBI were de-ramified at 14 DPI compared to shams and mice receiving a single TBI, suggesting robust microglial response in rTBI groups. Orexin-A–positive cells were sustained in the lateral hypothalamus with no loss detected, indicating that loss of wake-promoting neurons did not contribute to post-traumatic sleep. Thus, duration of post-traumatic sleep is a period of vulnerability that results in exacerbated injury from rTBI. Monitoring individual post-traumatic sleep is a potential clinical tool for personalized TBI management, where regular sleep patterns may inform rehabilitative strategies and return-to-activity guidelines.
Keywords: concussion, inflammation, pathology, repetitive TBI, sleep
Introduction
The enduring consequences of repetitive traumatic brain injury (rTBI) have garnered substantial scientific and public attention in response to clinical reports of progressive neurological dysfunction after injury.1 As a result, translational research continues to focus on the cognitive and neurological consequences of rTBI and multiple concussions.2 rTBI can alter mood and behavior while exacerbating neurological dysfunction.1,3,4 Further, given the frequency of TBIs in athletes and military personnel, the societal and policy implications of such consequences are substantial.2 After a concussion, the first priority is a quick, effective, and sustained return to activity, including work, active duty responsibilities, school, and extracurricular activities.5,6 Although guidelines are useful tools for return-to-activity prognosis, post-concussion management needs a personalized approach that is based on patient assessment, symptoms, and response to care.7–9
TBI is characterized by two pathological phases: 1) cellular injury resulting from primary impact and 2) the ensuing secondary injury mediated by pathological processes.10 The primary injury begins with the application of a mechanical force to the brain, which initiates a pathological cascade of secondary injury processes, including inflammation, that contribute to the clinical morbidities associated with TBI.11 Brain injury can lead to both short- and long-term impairments, including cognitive,12 neurological, and behavioral deficits,13 as well as increased risk for developing neurodegenerative disease,14 post-traumatic headaches,15 and/or psychiatric disorders.16 After TBI, acute post-traumatic neurological deficits exist, defined as a transient consequence of TBI-related pathology that impairs brain activity and behavioral function, which can naturally recover, in part, over time post-injury.17 Post-traumatic morbidities, or long-term consequences of injury-related pathological processes, can emerge in a delayed manner because of impaired brain circuit function and activation, including problems with cognition, sensory processing, and affective/emotional function.18,19
TBI is a highly heterogeneous disease, and individual patients exhibit combinations of different clinical symptoms to varying degrees. Thus, the availability of objective and quantifiable TBI biomarkers can allow for monitoring physiological processes and aid diagnosis, prognosis, and return-to-activity decisions after TBI and rTBI.20–22 Further, the identification of such biomarkers could elucidate TBI pathophysiology and aid the development of targeted therapies.21 We previously showed that post-traumatic sleep, an increase in acute sleep after diffuse TBI, may be indicative of underlying injury-induced inflammation.23,24 Early sleep disruptions have previously been used as a predictor of late-onset morbidities after TBI.4,25 In the clinic, sleep disturbances experienced immediately after trauma (<3 months) predicted neuropsychiatric symptoms 1 year after injury.25 As such, sleep becomes a plausible physiological biomarker. Herein, we identify sleep as a physiological biomarker, or marker of physio- and pathological processes involved in the diagnosis and prognosis of TBI and rTBI.
In the current study, post-traumatic sleep was tested as an objective biomarker of vulnerability to further injury after a second TBI (Fig. 1A). Previous data from our laboratory indicated that post-traumatic sleep occurs for up to 6 h after diffuse brain injury in the mouse regardless of injury severity or time of day that injury occurs.23 Further, we found that the temporal profile of secondary injury cascades may be driving the increase in post-traumatic sleep,23,26 and although post-traumatic sleep did not appear to be a tractable therapeutic target (by sleep disruption), it may still have value as a biomarker.27 These findings support post-traumatic sleep as a possible physiological biomarker that could track brain vulnerability to a subsequent TBI. Thus, we hypothesized that a second TBI occurring during the period of post-traumatic sleep worsens neurological and histological outcomes compared to one TBI or a second TBI after post-traumatic sleep resolves. We found that the delivery of a second TBI during the 6-h post-traumatic window resulted in functional deficits, anxiety-like behaviors, and significant neuropathology. We conclude that monitoring acute individual post-traumatic sleep has potential clinical relevance as a tool for personalized TBI management, where return of characteristic sleep patterns after a single TBI may inform rehabilitative strategies and return-to-activity guidelines.
FIG. 1.

(A) Experimental study design. Baseline sleep was recorded after acclimation to sleep cages. Rotarod training and baseline scores were obtained pre-injury. Sleep was monitored for 40 h uninterrupted. At 2, 5, and 7 days post-injury (DPI), mice were tested on the rotarod and modified Neurological Severity Score (NSS). At 7, 14, and 28 DPI, mice underwent functional testing (open field, elevated plus maze [EPM], forced swim task [FST], and novel object recognition [NOR]) and brains were collected from subsets of each group. (B) To control for anesthetic used during the induction of injury, uninjured shams were split in two groups receiving a second exposure to isoflurane 3 or 9 h after the initial exposure. The single-TBI group also received a second exposure to isoflurane 3 or 9 h before injury. (C) TBI suppressed righting reflex times regardless of the interval length between the second injuries. A two-way ANOVA comparing the righting reflex times between rTBI groups revealed no significant differences between the first and second hit regardless of the time interval between injuries (F(1,29) = 0.1757; p = 0.6782; mean ± SEM; TBI, n = 13; rTBI 3 h, n = 13; rTBI 9 h, n = 18). ANOVA, analysis of variance; N/A, not applicable; rTBI, repetitive traumatic brain injury; SEM, standard error of the mean.
Methods
All animal studies were conducted in accordance with the guidelines established by the internal Institutional Animal Care and Use Committee and the National Institutes of Health (NIH) guidelines for the care and use of laboratory animals. Studies are reported following the Animal Research: Reporting In Vivo Experiments guidelines. Randomization of animals was achieved by assigning individuals to treatment groups before initiation of the study to ensure equal distribution among groups. A power analysis was performed to identify group sizes that enable statistically robust detection of injury-induced deficits while minimizing the number of animals needed; this was based on preliminary data and previously published work from our group.23 Data collection was stopped at pre-determined final endpoints based on days post-injury (DPI) for each animal. Animals were excluded from the study if post-operative weight decreased by ≥15% of pre-surgical weight or baseline rotarod score was not met. All animal behavior was scored by investigators blinded to the treatment groups.
Animals
Male C57BL/6 mice (Harlan Laboratories, Inc., Indianapolis, IN) were used for all experiments (n = 53). Mice were housed in a 14-h light/10-h dark cycle at a constant temperature (23 ± 2°C), with food and water available ad libitum according to the Association for Assessment and Accreditation of Laboratory Animal Care International. All mice used in this study were singly housed as necessitated by the sleep recording system. Following shipment, mice were acclimated to their environment for at least 3 days before any experiments. After surgery, mice were evaluated daily during post-operative care by physical examination and documentation of each animal's condition. Animal care was approved by the Institutional Animal Care and Use Committee at the University of Arizona (Phoenix, AZ).
Midline fluid percussion injury
Mice (20–24 g) were subjected to midline fluid percussion injury (mFPI) consistent with methods previously described.23,27–30 Group sizes are indicated in the Results section and figure legends for individual studies. Mice were anesthetized using 5% isoflurane in 100% oxygen for 5 min, and the head of the mouse was placed in a stereotaxic frame with continuously delivered isoflurane at 2.5% by nosecone. While anesthetized, body temperature was maintained using a Deltaphase® isothermal heating pad (Braintree Scientific Inc., Braintree, MA). A midline incision was made exposing bregma and lambda, and fascia was removed from the surface of the skull. A trephine (3-mm outer diameter) was used for the craniotomy, centered on the sagittal suture between bregma and lambda without disruption of the dura. An injury cap prepared from the female portion of a Luer-Loc needle hub was fixed over the craniotomy using cyanoacrylate gel and methyl-methacrylate (Hygenic Corp., Akron, OH). The incision was sutured at the anterior and posterior edges, and topical Lidocaine ointment was applied. The injury hub was closed using a Luer-Loc cap, and mice were placed in a heated recovery cage and monitored until they were ambulatory before being returned to their respective sleep cages.
For injury induction 24 h post-surgery, mice were reanesthetized with 5% isoflurane delivered for 5 min. The cap was removed from the injury-hub assembly, and the dura was visually inspected through the hub to ensure it was intact with no debris. The hub was then filled with normal saline and attached to an extension tube connected to the male end of the fluid percussion device (Custom Design and Fabrication; Virginia Commonwealth University, Richmond, VA). An injury of moderate severity for our injury model (1.4 atm) was administered by releasing the pendulum onto the fluid-filled cylinder after the return of a toe-pinch response. Sham-injured mice underwent the same procedure, except that the pendulum was not released. Injured mice were monitored for presence of a forearm fencing response, and righting reflex times were recorded as indicators of injury severity.31 The righting reflex time is the total time from the initial impact until the mouse spontaneously rights itself from a supine position. The fencing response is a tonic posturing characterized by extension and flexion of opposite arms that has been validated as an overt indicator of injury severity.31 The injury hub was removed, and the brain was inspected for uniform herniation and integrity of the dura. The dura was intact in all mice, so none were excluded as technical failures. The incision was cleaned with saline and closed using sutures. Moderately brain-injured mice had righting reflex recovery times greater than 6 min and a positive fencing response.
Mice subjected to a moderate mFPI regained gross neurological function with no intervention, and therefore these injuries were most consistent with mild-moderate TBI, with a Glasgow Coma Score of 9–13, in which patients are generally responsive, but likely disoriented.17 Sham injured mice recovered a righting reflex immediately when removed from the injury device (within 20 sec). To control for anesthetic used during the induction of injury, uninjured shams were split into two groups receiving a second exposure to isoflurane 3 or 9 h after the initial exposure (Fig. 1B). The single-TBI group also received a second exposure to isoflurane 3 or 9 h before injury. After spontaneously righting, mice were placed in a heated recovery cage and monitored until they were ambulatory (approximately 5–15 min) before being returned to their piezoelectric sleep cage (see below). Adequate measures were taken to minimize pain or discomfort.
Sleep recordings
The non-invasive sleep cage system (Signal Solutions, Lexington, KY) used in this study consisted of 16 separate units that simultaneously monitored sleep and wake states, as previously published.27,32,33 Each cage unit housed a single mouse inside 18 × 18 cm walled compartments with attached food and water structures.32 The cages had open bottoms resting on polyvinylidine difluoride (PVDF) sensors that served as the cage floor.32 The non-invasive high-throughput PVDF sensors were coupled to an input differential amplifier that classified motions consistent with either activity related to wake or inactivity and regular breathing movements associated with sleep.27,32 Briefly, sleep was primarily characterized by periodic (3 Hz) and regular amplitude signals recorded from the PVDF sensors, typical of respiration from a sleeping mouse. In contrast, signals characteristic of wake were both the absence of characteristic sleep signals and higher-amplitude, irregular signals associated with volitional movements, even during quiet wake. The piezoelectric signals in 2-sec epochs were classified by a linear discriminant classifier algorithm based on multiple signal variables to produce a binary label of “sleep” or “wake.”32 Mice sleep in a polycyclic manner (often more than 40 sleep episodes per hour34); therefore, mouse sleep was quantified as the minutes spent sleeping per hour, presented as a percentage for each hour. In the current study, brain-injured mice slept more during the dark cycles, but less during the light cycles, compared to control shams. To accurately measure this change in sleep, each mouse's percent sleep was converted to cumulative minutes slept and the absolute change in cumulative minutes was compared to control sham sleep.
Rotarod
Sensorimotor function was assessed using the Economex Rotarod system from Columbus Instruments (Columbus, OH). Mice were acclimated 3 days before surgery/injury. Mice were placed on the stationary rod and allowed to explore for 30 sec. After exploration, mice were placed on the rod at a constant speed of 5 revolutions per minute (rpm). If the mouse fell off the rod, it was placed back on the rod and the timer was restarted (until the mice could walk 15 sec at 5 rpm). Next, mice were placed on the rod with an initial rotation speed of 5 rpm and an acceleration of 0.2 rpm/sec. The trial ended when the mouse fell off the rod; the acclimation period ended after two trials. After acclimation, mice were trained over 3 consecutive days before surgery/injury and the last training session was recorded as baseline. Testing occurred at 2, 5, and 7 DPI. For the training and testing phase, mice were placed on the stationary rod and the motor was started at 5 rpm with an acceleration of 0.2 rpm/sec. Two trials were run back to back, and mice were returned to cages thereafter. After 10 min, mice preformed a third trial. Time spent on the rotarod from the best two trials were averaged to generate a time score for each mouse.
Neurological Severity Score
Post-traumatic neurological impairments were assessed at 2, 5, and 7 DPI using an 8-point Neurological Severity Score (NSS) paradigm adapted from those previously used in experimental models of TBI.35–38 One point was given for failure on an individual task, whereas no points were given if a mouse completed a task successfully. Mice were observed for hindlimb flexion, startle reflex, and seeking behavior (presence of these behaviors was considered successful task completion). Mice traversed in sequence, 3-, 2-, and 1-cm beams. The beams were elevated, and mice were given 1 min to travel 30 cm along the beams. The task was scored as a success if the mouse traveled 30 cm with normal fore- and hindlimb position (fore-/hindlimb did not hang from the beam). Mice were also required to balance on a 0.5-cm beam and a 0.5-cm round rod for 3 sec in a stationary position with front paws between hind paws. The resulting non-parametric data are presented as a composite score ranging from 0 to 8, representing performance on all tasks combined. High final NSS scores were indicative of task failure and interpreted as neurological impairment.
Open field
The open field task was used to assess locomotor activity and anxiety-like behavior as previously described.39 At 7, 14, and 28 DPI, mice were placed in an empty arena (45 × 45 × 30 cm, W × L × H) and allowed to explore freely for 5 min. Mouse movement was tracked by an overhead camera, and the distance traveled, time spent in the center of the arena (29.5 × 29.5 cm), and number of entries into the center were calculated using EthoVision XT 10 software (Noldus Information Technology, Leesburg, VA).
Forced swim task
Depressive-like behavior was assessed using the forced swim task (FST), as previously described.40,41 For 6 min, mice were placed into glass cylinders (15 cm diameter × 24.5 cm H) filled with water (25°C). The first minute was excluded from analysis as an acclimation phase. Mice were visually recorded using an overhead camera. Videos were analyzed using EthoVision XT 10 software (Noldus Information Technology) for time spent actively swimming and time spent immobile.
Novel object recognition
Cognitive impairment was tested using the novel object recognition (NOR) test as previously published.7,24 The test consisted of three phases: habituation, training, and testing. On the day of the test, mice were placed in an open field (42 × 21 × 21 cm) for 1 h of habituation. Mice were removed, and two identical objects were placed in opposing quadrants of the field for the training phase. Mice were then placed in the center of the open field and given 5 min to explore the objects. After training, mice were returned to their home cages. Testing began 4 h after training. One familiar object was placed in an original location, and one novel object was placed in the opposing quadrant of the open field. Mice were placed into the center and given 5 min to explore. For testing, times spent actively investigating the novel and familiar object were quantified. Investigation of an object included the mice sniffing, touching, or climbing onto an object while facing the object. If an animal climbed onto an object and sniffed into the air, this time was not calculated into the exploration of the novel object. Testing data are displayed as the percentage of total investigation time spent with each object and as a discrimination index (DI) in which 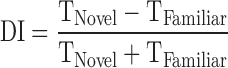 .
.
Tissue preparation
At pre-determined time points (7, 14, and 28 days) post-injury or sham operation, mice were given an overdose of sodium pentobarbital and transcardially perfused with 4% paraformaldehyde after a phosphate-buffered saline flush. Brains were transferred to fresh fixative solution and sent to NeuroScience Associates Inc. (Knoxville, TN) to be processed for histological and immunohistochemical staining. Brains were embedded into a gelatin matrix where they could be frozen and sectioned from one solid block (MultiBrain® Technology; NeuroScience Associates). Sections of 40-μm thickness were taken in the coronal plane and wet-mounted on 2% gelatin-subbed slides. Three series of tissue sections were labeled for neuropathology, neuroinflammation, and Orexin-A (see below). Both stained and unstained brain sections were returned to our laboratory.
Aminocupric silver technique
Neuropathology, indicated by argyrophilic reaction product, was examined using the de Olmos amino cupric silver (Ag) technique according to proprietary protocols.42–45 Brain sections were counterstained with Neutral Red to stain normal cell bodies. Stained sections were analyzed in our laboratory. Primary quantification of Ag-stained sections was carried out for hemispheres. Immunostained slides were imaged using a Zeiss Imager A2 microscope with AxioCam MRc5 digital camera (Carl Zeiss, Jena, Germany)). Digital images were taken of the right hemisphere of all brains using fixed exposure settings to ensure consistency, though mFPI is expected to exert equal forces bilaterally. Images were converted to grayscale, followed by digitally thresholding the pixel distribution to separate positively stained pixels from unstained pixels (ImageJ software; NIH, Bethesda, MD; https://imagej.nih.gov/ij/). Thresholded images were each then segmented into white and black pixels, indicating positive and negative staining, respectively. Finally, the percentage of positive pixels was calculated within the hemisphere.
Immunohistochemistry
One series of brain sections was labeled for ionized calcium binding protein-1 (Iba-1), and one series was labeled for Orexin-A by NeuroScience Associates using their proprietary protocols. Digital photomicrographs of Orexin-A tissue were captured from the lateral hypothalamus at 200 × total magnification. Digital photomicrographs of Iba-1–immunostained tissue were captured from the cortex at 400 × total magnification. Care was taken to ensure that illumination and contrast settings were identical between treatment groups at the time of image acquisition.
Microglial identification and morphology analysis
Stained sections (n = 4 per group) were analyzed after Iba-1 staining to determine the average ramification of microglia post-injury. The area of interest, the primary somatosensory barrel fields (S1BF), was chosen based on previous work that demonstrated a multi-focal concentration of neuropathology and microglial activation in the S1BF after mFPI in the rodent.24,46–48 Sections were screened using a Zeiss (AXIO imager A2) microscope with attached digital camera (AxioCam MRc5). Images were captured with proprietary Zen software (Carl Zeiss) at 200 × magnification. The area of interest was examined in both hemispheres and in two different coronal planes: an anterior section (approximately bregma −1.555mm) and a posterior section (approximately bregma −2.255mm). A total of four photomicrographs per brain were analyzed in ImageJ (NIH). All photomicrographs were converted to binary and skeletonized images.49 In addition to creating skeletonized images, cell somas were manually counted for each photomicrograph. The Analyze Skeleton Plugin was then applied to the skeleton image, which tags skeletal features relevant to microglia ramification. For each photomicrograph, there was an original image, binary image, and skeleton image used to convert an entire original photomicrograph to a plugin-tagged image. We summarized the number of process endpoints and length from the Analyze Skeleton Plugin data output and normalized all data by the number of microglia cell somas per image to calculate the number of microglia endpoints per cell and microglia process length per cell.
Statistical analyses
For each analysis, shams receiving isoflurane 3 h apart and shams receiving isoflurane 9 h apart were statistically compared. No differences were detected between sham groups, so shams were combined from each treatment. Percent sleep, mean bout length, bout episode duration, and differences in rotarod performance were analyzed using a repeated-measures two-way analysis of variance (ANOVA) followed by Tukey's multiple comparisons test. Cumulative sleep (min), open field (sec), elevated plus maze (EPM; sec), FST (sec), NOR (ratio/sec), hemispheric silver stain area (percent), and Orexin-A–positive cells (number) were analyzed using a one-way ANOVA followed by Tukey's multiple comparisons test. Non-parametric NSS data were analyzed using a Kruskal-Wallis test, followed by Dunn's multiple comparisons test. Differences in microglia cell counts/field, endpoints per cell, summed process length, and a spatial-temporal interaction between region and time post-injury were analyzed using a two-way ANOVA followed by a Tukey's multiple comparisons test. For all continuous variables, the assumption that variables were normally distributed was verified using a combination of density and q-q plots and Shapiro-Wilk tests to ensure validity of the analytical approaches used. Resulting critical values are included in the figure legends. All normally distributed data were screened using the Grubb's outlier test for statistical outliers; outliers are reported in the Results section. All of the aforementioned analyses were conducted, and associated figures were constructed, using GraphPad Prism (version 6; GraphPad Software Inc., La Jolla, CA); results are shown as mean ± standard error of the mean (SEM), with statistical significance assigned at the 95% confidence level (α < 0.05).
To investigate whether the NSS or rotarod behavioral tests or cumulative sleep were predictive of silver stain pathology, microglia cell count, number of microglia process endpoints per cell, or summed microglia process lengths, we fit multiple linear regression models by maximum likelihood implemented using the lm function in R statistical software (R Foundation for Statistical Computing, Vienna, Austria).50 We used silver stain pathology, microglia cell count, endpoints per cell, and summed process lengths as outcome variables in four separate regression models (i.e., one model for each outcome), with NSS scores, rotarod scores, and cumulative sleep over the first 6 h post-injury (CumSleep) as predictor variables. We included the TBI group as a categorical variable in all models to control for confounding; this variable had four levels (sham, TBI, rTBI 3 h, and rTBI 9 h) with sham as the reference level. Shapiro-Wilk tests were used to assess normality of continuous variables; results indicated that all continuous variables were approximately normally distributed (p = 0.08–0.46). Multi-variate imputation by chained equations implemented in the R package MICE was used to replace missing data points with values estimated directly from the data by predictive mean matching, which assumed values were missing at random.51 Five imputations were conducted using 50 Markov chain Monte Carlo iterations for each imputation, with a random number generator seed of 500. Convergence of iterations for each of the five imputations was assessed using trace plots, and plausibility of the imputed values was assessed by plotting imputed against observed values. We pooled fitted regression models for each outcome variable to produce combined coefficient estimates (β). Statistical significance was assigned to β estimates if 95% confidence intervals (CIs) excluded zero.
Results
Righting reflex times were not different regardless of interval between injuries
We previously reported on the suppression of the righting reflex response in mice after mFPI27 as an injury-induced deficit and indicator of injury severity. Diffuse brain injury resulted in a suppression of the righting reflex in brain-injured mice. A second brain injury resulted in a second suppression of the righting reflex that was not significantly different between injury groups that received two hits (Fig. 1C). Results from a two-way ANOVA comparing only groups that received two brain injuries (rTBI 3 h, rTBI 9 h) showed no significant difference of righting reflex times, indicating that the second TBI was of similar severity across all groups. All sham animals righted immediately and were not used in this analysis.
Both diffuse traumatic brain injury and repetitive traumatic brain injury increased percent sleep and decreased mean bout length of sleep
We previously reported that a single diffuse TBI increases post-traumatic sleep immediately after injury.23 We measured percent sleep and mean bout length in mice over the first 40 h post-injury (Fig. 2A,B). Over the full 40-h time course, there was a significant time effect on percent sleep, and significant time and group effect on mean bout length. To justify the study design and confirm our previous findings that TBI results in an increase in immediate sleep over the first 6 h which is resolved by 9 h post-injury, percent sleep was analyzed over the first 9 h. A two-way ANOVA followed by a Tukey's multiple comparison test indicated that all brain-injured mice slept significantly more over 6 h compared to uninjured shams, rTBI 9 h mice slept more than uninjured shams in hour 7 post-injury, and all brain-injured mice slept comparable to shams in hours 8 and 9 post-injury (group effect, F(3,44) = 7.203; p = 0.0005). As expected, the first 3 h post-injury (in the light cycle) resulted in the rTBI 3 h, rTBI 9 h, and the single-TBI groups exhibiting significantly increased sleep compared to uninjured shams at 5:00 pm, and the rTBI 3 h group exhibiting significantly increased sleep compared to uninjured shams at 7:00 pm (Fig. 2C). There was no group effect or time effect over this 3-h light period on mean bout length of sleep (Fig. 2D). In the subsequent 10 h, or the first dark cycle post-injury, the brain-injured mice slept more than shams, but this increase failed to reach significance (Fig. 2E); however, all brain-injured mice had significantly shorter bouts compared to uninjured shams (Fig. 2F). In the subsequent 14-h light cycle, rTBI 3 h and rTBI 9 h mice slept significantly less than uninjured shams at 6:00 am, and TBI mice slept less than shams at 12 noon (Fig. 2G); all brain-injured mice had significantly shorter mean bout length compared to uninjured shams (Fig. 2H). In the second 10-h dark cycle, the rTBI 3 h and rTBI 9 h mice slept significantly more than uninjured shams (Fig. 2I), and there was a significant time and group effect on mean bout length, with injured mice having shorter bouts compared to shams (Fig. 2J). Brain-injured mice slept significantly more, but had shorter mean bout lengths, thereby warranting analysis of sleep fragmentation as previously published.33 Frequency of individual sleep bouts with different episode durations was analyzed and all brain-injured mice had significantly more bouts of 8–63 seconds in length compared to uninjured shams (Supplementary Fig. 1) (see online supplementary material at http://www.liebertpub.com). Further, the rTBI 3 h group had more 8- to 15-sec bouts than the TBI and rTBI 9 h group. One mouse was excluded from the TBI group from all sleep analyses because it was a significant outlier (Grubb's outlier test).
FIG. 2.

(A) A single diffuse TBI and rTBI modulated post-traumatic sleep. There was a significant time effect (F(39, 1716) = 9.084; p < 0.0001) and interaction (F(117,1716) = 2.202; p < 0.0001), but no significant group effect (F(3,45) = 1.108; p = 0.3558) on the percent sleep of mice over 40 h post-injury. (B) There was a significant time effect (F(39, 1716) = 5.095; p < 0.0001), group effect (F(3,44) = 13.88; p < 0.0001), and interaction (F(117,1716) = 1.799; p < 0.0001) on the mean bout length of sleep over 40 h post-injury. (C) There was a significant time effect (F(2, 88) = 8.270; p = 0.0005), group effect (F(3,44) = 7.159; p = 0.0005), and interaction (F(6,88) = 0.9913; p = 0.4362) on the percent sleep of mice over the first 3 h post-injury during the light cycle. Tukey's post-hoc analysis indicated that all brain-injured mice slept significantly more than uninjured sham at 5:00 pm, and rTBI 3 h mice slept more than uninjured shams at 7:00 pm. (D) There was a no significant time effect (F(2,88) = 0.08350; p = 0.9200), group effect (F(3,44) = 1.532; p = 0.2196), or interaction (F(6,88) = 0.7826; p = 0.5858) on mean bout length of sleep over the first 3 h post-injury. (E) Over the first 10-h dark cycle, there was a significant time (F(9,396) = 1.904; p = 0.0499) and interaction effect (F(27,396) = 2.868; p < 0.0001), but no group effect (F(3,44) = 2.551; p = 0.0678), on percent sleep. (F) Over the first 10-h dark cycle, there was no time effect (F(9,396) = 0.7521; p = 0.6610), but there was a significant group effect (F(3,44) = 14.24; p < 0.001) and interaction (F(27,396) = 2.312; p = 0.0003) on mean bout length. Tukey's post-hoc analysis indicated that all brain-injured mice slept significantly shorter bouts at 11:00 pm and 1:00, 2:00, 3:00, 4:00, and 5:00 am compared to uninjured shams. At 12 midnight, the rTBI 3 h and rTBI 9 h mice slept shorter bouts than shams. (G) Across the first full 14-h light cycle, there was no time effect (F(13,572) = 0.8581; p = 0.5980) or interaction (F(39,572) = 1.026; p = 0.4292), but a significant group effect (F(3,44) = 3.591; p = 0.0209), on percent sleep, with rTBI 3 h and rTBI 9 h mice sleeping significantly less than uninjured shams at 6:00 am and TBI mice sleeping less than shams at 12 noon. (H) Across the first full 14-h light cycle, there was no time effect (F(13,572) = 1.019; p = 0.4311) or interaction (F(39,572) = 1.214; p = 0.1786), but a significant group effect (F(3,44) = 17.50; p < 0.0001), on mean bout length, with all brain-injured mice having significantly shorter bouts than uninjured shams from 6:00 am to 4:00 pm and rTBI 3 h mice having shorter bouts than shams at 5:00 and 6:00 pm. (I) During the second full 10-h dark cycle, there was a significant time (F(9,396) = 3.899; p < 0.0001) and group effect (F(3,44) = 3.577; p = 0.0212) on percent sleep, but no interaction (F(27,396) = 1.236; p = 0.1961). Tukey's post-hoc test indicated that rTBI 9 h mice slept significantly more than shams at 11:00 pm and 12 midnight, and rTBI 3 h mice slept significantly more than uninjured shams at 1:00 am. (J) During the second full 10-h dark cycle, there was a significant time (F(9,396) = 2.364; p = 0.0131) and group effect (F(3,44) = 3.773; p = 0.0171) on mean bout length, but no interaction (F(27,396) = 0.9791; p = 0.4971). *Indicates significance from sham (mean ± SEM; sham, n = 15; TBI, n = 12; rTBI 3 h, n = 13; rTBI 9 h, n = 8). rTBI, repetitive traumatic brain injury; SEM, standard error of the mean.
Diffuse traumatic brain injury and repetitive traumatic brain injury resulted in changes in cumulative time spent sleeping
Brain-injured mice slept less in the light cycle, but more in the dark cycle, so each mouse's percent sleep was converted to cumulative minutes slept and the absolute change in cumulative minutes was compared to control sham sleep. All brain-injured mice had a change in cumulative minutes spent sleeping compared to uninjured shams (Fig. 3A–C). The rTBI 3 h group had a significantly higher absolute change in cumulative sleep compared to the single-TBI and the rTBI 9 h group (Fig. 3D).
FIG. 3.

Brain-injured mice slept less during the light cycle, but more during the dark cycle, so each mouse's percent sleep was converted to cumulative minutes slept and the absolute change in cumulative minutes was compared to control sham sleep. A single diffuse TBI and rTBI resulted in a change in the absolute value of cumulative sleep measured in minutes compared to uninjured shams. Mice subjected to a (A) single diffuse TBI, (B) rTBI 3 h, or (C) rTBI 9 h resulted primarily in increased cumulative sleep during the dark cycles and decreased sleep during the light cycles. We further analyzed this sleep as the absolute change in cumulative sleep compared to shams, and results indicated that (D) rTBI 3 h mice had a significantly greater cumulative change in sleep compared to both shams and the rTBI 9 h mice (F(2,30) = 0.8654; p = 0.0154). #Indicates significance from TBI; +indicates significance from rTBI 9 h (mean ± SEM; sham, n = 15; TBI, n = 12; rTBI 3 h, n = 13; rTBI 9 h, n = 8). rTBI, repetitive traumatic brain injury; SEM, standard error of the mean.
Both diffuse traumatic brain injury and repetitive traumatic brain injury resulted in neurological function deficits
To assess motor function, the rotarod was used as previously published.24,27,30 Motor function was tested as the latency to stay on the rotarod over 7 DPI, with significant effects of both time post-injury and between groups. Tukey's post-hoc analysis indicated that compared to uninjured shams, all brain-injured mice had significantly shorter latencies to fall from the rod (Fig. 4A). There were no significant differences in latencies to fall between mice receiving one TBI and mice receiving rTBI 3 h or rTBI 9 h. Similarly, all brain-injured groups demonstrated gross neurological dysfunction, as evaluated by the modified NSS, as previously published.27 A nonparametric Kruskal-Wallis test with a post-hoc Dunn test and Bonferroni correction for multiple comparisons supported an overall time and group effect, and all brain-injured mice had significantly higher NSS scores than shams (Fig. 4B).
FIG. 4.

(A) Diffuse TBI induced motor deficits measured by the rotarod task. There was a significant effect of time after injury on motor performance (F(2,98) = 21.97; p < 0.0001). There was also a significant group effect on latency to stay on the rod (F(3, 49) = 22.92; p < 0.0001). Tukey's post-hoc test indicated that compared to uninjured shams, all brain-injured groups demonstrated significantly shorter latencies to fall from the rotarod. (B) Diffuse TBI induced neurological function deficits measured by a modified Neurological Severity Score (NSS). A nonparametric Kruskal-Wallis rank-sum test indicated significant time (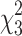 = 89.39; p < 0.001) and group (
= 89.39; p < 0.001) and group (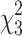 = 14.45; p = 0.002) effects, with the TBI, rTBI 3 h, and rTBI 9 h groups having significantly higher NSS scores compared to uninjured shams. (C) Anxiety-like behavior was assessed at 7 days post-injury (DPI) using the open field task, and a one-way ANOVA indicated no group effect on time spent in the center (F(3,49) = 1.124; p = 0.3485). (D) There was an overall effect on distance traveled (F(3,49) = 4.245; p = 0.0096), and Tukey's post-hoc analysis indicated that the rTBI 3 h group traveled shorter distances compared to uninjured shams and the single-TBI group. (E) There was no difference between groups in the time to the first center entry (F(3,49) = 0.3361; p = 0.777) (F) or the total number of entries into the center (F(3,49) = 1.285; p = 0.2898) in the open field task. (G) A one-way ANOVA indicated no overall group effect on the time spent in the open arms of an elevated plus maze (EPM) at 7 DPI (F(3,48) = 0.4126; p = 0.7447). (H) A one-way ANOVA indicated no overall group effect on time spent immobile during a forced swim task (FST; F(3,40) = 1.166; p = 0.3347). (I) A one-way ANOVA indicated no overall group effect on discrimination index measured by the novel object recognition (NOR) task (F(3,47) = 1.340; p = 0.2728) at 7 DPI. (J) A one-way ANOVA indicated an overall group effect on total investigation time (F(3,47 = 2.940; p = 0.0427), and Tukey's multiple comparisons test indicated that the rTBI 9 h group spent significantly less time investigating objects compared to the single-TBI group. *Indicates significance from sham; #indicates significance from TBI (mean ± SEM; sham, n = 16; TBI, n = 15; rTBI 3 h, n = 9; rTBI 9 h, n = 13). ANOVA, analysis of variance; rTBI, repetitive traumatic brain injury; SEM, standard error of the mean.
= 14.45; p = 0.002) effects, with the TBI, rTBI 3 h, and rTBI 9 h groups having significantly higher NSS scores compared to uninjured shams. (C) Anxiety-like behavior was assessed at 7 days post-injury (DPI) using the open field task, and a one-way ANOVA indicated no group effect on time spent in the center (F(3,49) = 1.124; p = 0.3485). (D) There was an overall effect on distance traveled (F(3,49) = 4.245; p = 0.0096), and Tukey's post-hoc analysis indicated that the rTBI 3 h group traveled shorter distances compared to uninjured shams and the single-TBI group. (E) There was no difference between groups in the time to the first center entry (F(3,49) = 0.3361; p = 0.777) (F) or the total number of entries into the center (F(3,49) = 1.285; p = 0.2898) in the open field task. (G) A one-way ANOVA indicated no overall group effect on the time spent in the open arms of an elevated plus maze (EPM) at 7 DPI (F(3,48) = 0.4126; p = 0.7447). (H) A one-way ANOVA indicated no overall group effect on time spent immobile during a forced swim task (FST; F(3,40) = 1.166; p = 0.3347). (I) A one-way ANOVA indicated no overall group effect on discrimination index measured by the novel object recognition (NOR) task (F(3,47) = 1.340; p = 0.2728) at 7 DPI. (J) A one-way ANOVA indicated an overall group effect on total investigation time (F(3,47 = 2.940; p = 0.0427), and Tukey's multiple comparisons test indicated that the rTBI 9 h group spent significantly less time investigating objects compared to the single-TBI group. *Indicates significance from sham; #indicates significance from TBI (mean ± SEM; sham, n = 16; TBI, n = 15; rTBI 3 h, n = 9; rTBI 9 h, n = 13). ANOVA, analysis of variance; rTBI, repetitive traumatic brain injury; SEM, standard error of the mean.
Repetitive traumatic brain injury 3 h resulted in persisting anxiety-like behavior, but a single diffuse traumatic brain injury or repetitive traumatic brain injury resulted in no depressive-like behavior
Repetitive brain injury with a 3-h interinjury interval resulted in anxiety-like behavior assessed by the open field task as previously published.52 There was no overall effect on time spent in the center of the arena, but at 7 DPI, results from Tukey's multiple comparisons test suggested that the rTBI 3 h group traveled significantly shorter distances compared to both uninjured shams and mice that received a single TBI (Fig. 4C,D). These deficits persisted to 28 DPI, at which results from Tukey's post-hoc test suggested that the rTBI 3 h group traveled significantly shorter distances compared to uninjured shams (Supplementary Fig. 2) (see online supplementary material at http://www.liebertpub.com). There was no change in the time to the first center entry (Fig. 4E) or total number of center entries (Fig. 4G) between groups at 7 or at 14 and 28 DPI (Supplementary Fig. 2) (see online supplementary material at http://www.liebertpub.com). At 7 DPI, there was no overall group effect on time spent in the open arms of an EPM (Fig. 4G). At 14 and 28 DPI, there was no overall group effect on time spent in the open arms (Supplementary Fig. 3) (see online supplementary material at http://www.liebertpub.com). One mouse in the TBI group was excluded from the EPM analyses as a significant outlier (Grubb's outlier test). Depressive-like behavior was assessed at 7 DPI using the FST as previously published.52 There was no overall effect on the amount of time spent immobile during the task (Fig. 4H).
A single traumatic brain injury and repetitive traumatic brain injury resulted in significantly less exploration during novel object task
An NOR task was used to assess cognitive behavior as previously published.27 At 7 DPI, there was no overall group effect on DI (Fig. 4I), but there was an overall effect on the investigation time (Fig. 4J). Results from Tukey's multiple comparisons test suggested that the rTBI 9 h group spent significantly less time investigating objects during the task compared to mice that received a single TBI (Fig. 4J). At 7 DPI, 2 mice were excluded from NOR, 1 in the TBI group and 1 in the rTBI 9 h group because they were significant outliers (Grubb's outlier test). There was no injury effect on DI across groups at 14 (Supplementary Fig. 4A) (see online supplementary material at http://www.liebertpub.com) or 28 DPI (Supplementary Fig. 4C) (see online supplementary material at http://www.liebertpub.com). There was an overall group effect on total time spent exploring objects at 14 (Supplementary Fig. 4B) (see online supplementary material at http://www.liebertpub.com) and 28 DPI (Supplementary Fig. 4D) (see online supplementary material at http://www.liebertpub.com).
A single traumatic brain injury or repetitive traumatic brain injury did not result in loss of orexigenic neurons
The lateral hypothalamus was assessed for total number of Orexin-A–positive stained neurons (Fig. 5A). There were no significant differences in total number of Orexin-A–positive neurons at 7, 14, or 28 DPI compared to uninjured sham (Fig. 5B–D).
FIG. 5.

Diffuse TBI or rTBI did not result in the loss of orexigenic neurons in the lateral hypothalamus. (A) Cells positive for Orexin-A were counted in the lateral hypothalamus. There was no significant change in number of Orexin-A–positive cells across all groups at (B) 7 DPI (F(3,13) = 0.8792; p = 0.4772), (C) 14 DPI (F(3,12) = 0.5323; p = 0.6687), (D) or 28 DPI (F(3,13) = 1.167; p = 0.3598; mean ± SEM; sham, n = 15; TBI, n = 11; rTBI 3 h, n = 10; rTBI 9 h, n = 11). rTBI, repetitive traumatic brain injury; SEM, standard error of the mean.
Repetitive traumatic brain injury, but not a single traumatic brain injury, resulted in sustained neuropathology
As we have previously published, de Olmos amino cupric Ag stain was used as an indicator of neuropathology.48 There was an overall effect on the percent area positive for Ag stain accumulation at 7, 14, and 28 DPI (Fig. 6). Results from Tukey's multiple comparisons test indicated that the rTBI 3 h group had significantly more hemispheric staining compared to uninjured shams at 7, 14, and 28 DPI (Fig. 6A–C). The rTBI 3 h group also had significantly more hemispheric staining at 28 DPI compared to mice receiving a single TBI (Fig. 6C). Similarly, results from Tukey's multiple comparisons test suggested that the rTBI 9 h group had significantly more hemispheric staining compared to uninjured shams at 7 and 28 DPI, and significantly more staining at 7 DPI compared to mice receiving a single TBI (Fig. 6A,C).
FIG. 6.

Repetitive TBI, but not a single TBI, resulted in sustained neuropathology measured by an increase in the percent hemisphere area stained with silver stain. Representative images show silver staining of entire hemispheres collected at 7, 14, and 28 DPI. (A) A one-way ANOVA indicated a significant injury effect in hemispheric silver staining at 7 DPI (F(3,11) = 7.672; p = 0.0048), and a Tukey's post-hoc test indicated a significant increase in the rTBI 3 h mice compared to uninjured shams, and the rTBI 9 h group compared to both uninjured shams and the single-TBI group. (B) At 14 DPI, a one-way ANOVA indicated a significant injury effect (F(3,11) = 4.188; p = 0.0332), and post-hoc analysis indicated that the rTBI 3 h group had a significant increase in silver staining compared to uninjured shams. (C) At 28 DPI, a one-way ANOVA indicated a significant injury effect (F(3,12) = 8.231; p = 0.0030) and a post-hoc analysis indicated a sustained increase in silver staining in the rTBI 3 h group compared to both uninjured sham and the single-TBI group as well as a significant increase in staining in the rTBI 9 h group compared to uninjured sham. *Indicates significance from sham; #indicates significance from TBI (mean ± SEM; sham, n = 15; TBI, n = 10; rTBI 3 h, n = 9; rTBI 9 h, n = 12). ANOVA, analysis of variance; rTBI, repetitive traumatic brain injury; SEM, standard error of the mean.
Microglia were de-ramified in the cortex as a result of repetitive traumatic brain injury, but not a single traumatic brain injury
Microglia were quantified for ramification using a computer-aided skeleton analysis as we have previously published.53 Data were collected in duplicate (two coronal sections) for each mouse subjected to a single TBI, rTBI 3 h, rTBI 9 h, or control sham surgery; two photomicrographs were acquired from the cortex near the injury site (primary motor cortex [M1]) of each mouse and averaged. Our data summarize microglia morphology for the averaged images for each mouse assigned to each time point. A photomicrograph of microglia at 400 × total magnification in the cortex, cropped and zoomed in at 630 × total magnification, is shown (Fig. 7A). A representative workflow including the binary image, skeleton, overlay, and tagged skeleton images are shown (Fig. 7A). There was an increase in total microglia cell count in rTBI 9 h mice compared to uninjured shams at 7 and 14 DPI (Fig. 7B,C). There was no overall effect on number of microglia process endpoints per cell or the summed microglia process length per cell at 7 or 28 DPI (Fig. 7B,D). Overall effects on both microglia process endpoints per cell and the summed microglia process length per cell were supported at 14 DPI (Fig. 7C). Results from Tukey's multiple comparisons test supported a significant decrease in number of microglia process endpoints per cell in rTBI 9 h mice compared to uninjured shams (Fig. 7C). At 14 DPI, results from Tukey's multiple comparisons test suggested a significant decrease in both number of process endpoints per cell and summed microglia process length per cell in both the rTBI 3 h and rTBI 9 h groups compared to both uninjured shams and a single TBI (Fig. 7C).
FIG. 7.

Microglia were de-ramified in the cortex as a result of rTBI, but not a single TBI. (A) For each photomicrograph, an original image was converted to a binary image, skeleton, overlay, and tagged image for analysis using a computer-aided skeleton analysis to analyze changes in ramified morphology of Iba-1–stained microglia in the cortex. (B) At 7 DPI, there was an overall effect on the total number of microglia (F(3,10) = 4.403; p = 0.0322), with rTBI 9 h mice having an increase in total microglia compared to uninjured shams. There was no change in number of microglia process endpoints per cell (F(3,10) = 1.669; p = 0.2359) or the summed microglia process length per cell (F(3,10) = 2.498; p = 0.1192) at 7 DPI. (C) At 14 DPI, there was a significant injury effect on total number of microglia (F(3,11) = 5.280; p = 0.0169) and number of microglia process endpoints per cell (F(3,11) = 4.133; p = 0.0344), and a Tukey's post-hoc test indicated an increase in total microglia cell count, but a decrease in number of microglia process endpoints per cell measured in the rTBI 9 h group compared to uninjured shams. There was also a significant injury effect on summed microglia process length per cell (F(3,11) = 9.267; p = 0.0024), and the post-hoc analysis indicated a decrease in both the rTBI 9 h and rTBI 3 h group compared to both uninjured sham and the single-TBI group. (D) The increase in total microglia and the de-ramification was resolved by 28 DPI, and there was no overall effect on total number of microglia (F(3,13) = 1.429; p = 0.2794), number of process endpoints per cell (F(3,13) = 1.687; p = 0.2186), or summed microglia process length per cell (F(3,13) = 0.8631; p = 0.4848). *Indicates significance from sham; #indicates significance from TBI. DPI, days post-injury; Iba-1, ionized calcium binding protein-1; rTBI, repetitive traumatic brain injury.
Cumulative post-injury sleep over the first 6 h predicted de-ramification of microglia
Results from regression analysis indicated that microglia process endpoints per cell declined by −0.38 (95% CI, −0.57, −0.20) for every minute increase in cumulative sleep, and that the rTBI 3 h group had microglia process endpoints that were lower across all time points by −25.54 endpoints per cell (95% CI, −50.20, −1.88), on average, irrespective of cumulative minutes slept (Fig. 8A; Supplementary Table 1) (see online supplementary material at http://www.liebertpub.com). Although summed microglia process lengths for all injured groups were predicted to decline by an average of −0.15 (95% CI, −0.29, −0.03) for every minute increase in cumulative sleep, the TBI and rTBI 9 h groups had 29.33 (95% CI, 12.42, 46.24) and 18.82 μm (95% CI, 1.29, 36.36) longer microglia process lengths, on average, than sham or rTBI 3 h mice (Fig. 8B; Supplementary Table 2) (see online supplementary material at http://www.liebertpub.com). Scores on rotarod and NSS behavioral tests were not significant predictors of silver stain pathology (p > 0.05); however, the rTBI 3 h and rTBI 9 h groups were predicted to have 5.34 (95% CI, 2.79, 7.88) and 3.97 (95% CI, 2.04, 5.89) higher silver stain pathologies defined as percent area of silver stain, on average, than shams or single-TBI mice (Supplementary Table 3) (see online supplementary material at http://www.liebertpub.com). No statistically significant relationships existed between microglia cell count and any of the considered predictors (p > 0.05; Supplementary Table 4) (see online supplementary material at http://www.liebertpub.com). Estimated precision of the aforementioned significant β estimates, determined by the coefficient of variation (CV = 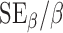 ), was moderate but generally poorer than desirable (i.e., CV ≤0.20), ranging from 0.23 to 0.38.
), was moderate but generally poorer than desirable (i.e., CV ≤0.20), ranging from 0.23 to 0.38.
FIG. 8.

Regression lines from fitted multiple linear regression models for each of two measures of neuroinflammation, microglia process endpoints per cell (A) and summed microglia process lengths (B), with cumulative sleep during the first 6 h post-injury as the predictor variable, controlling for the TBI group. Gray shaded areas for each group-specific regression line represent 95% prediction intervals, not confidence intervals. rTBI, repetitive traumatic brain injury.
Discussion
Sports- and military-related concussions have received increased attention in both young adult, professional athletes, and military personnel. Concussions are TBIs initiated by a pathological event and evolve into a disease with substantial morbidity. Clinical data suggest that two back-to-back concussions or repetitive mild concussions may have particularly severe consequences.54 As a research community, identification of prognostic indicators that can determine the severity of symptoms and length of recovery from a sports- or military-related concussion is an overarching goal.55–57 During the acute phase of concussion, neurological symptoms can manifest in four domains: cognitive, somatic, sleep disturbances, and emotional, which together represent the full impact of disease.55,56,58,59 In the current study, we investigated those four domains after diffuse brain injury in the mouse, identifying post-traumatic sleep as a physiological biomarker of brain injury that defined a period of brain vulnerability to a second TBI.
We previously reported that acute sleep increases over the first 6 h after experimental diffuse TBI.23 The current study showed an increase in sleep across all brain-injured groups compared to uninjured shams over the first 3 h post-injury during the light cycle, which confirms our previous finding. Over the first 40 h post-injury, we analyzed the absolute change in cumulative time spent sleeping compared to sham control, where the rTBI 3 h mice had a significant increase compared to rTBI 9 h. The rTBI 3 h mice slept significantly more than shams over the first and second dark cycle post-injury and significantly less than shams with shorter bout lengths during the second light cycle. Our results also indicated that rTBI 9 h mice slept more than shams during the second dark cycle. Increased sleep during the dark cycle, when mice typically exhibit prolonged periods of wakefulness, suggests hypersomnia, or excessive daytime sleepiness, which parallels clinical data in TBI survivors.60,61 Similarly, mice subjected to focal TBI induced by controlled cortical impact demonstrated less wakefulness during the dark cycle compared to sham mice,62 and mice subjected to lateral FPI spent less time awake over both the light and dark cycle compared to sham mice,63 based on electroencepalography/electromyography analyses. Decreased bout lengths suggest that rTBI 3 h mice may have fragmented sleep, which, based on clinical reports, frequently follows TBI and has been reported to worsen neuropsychiatrical, behavioral, and physical symptoms (see Lucke-Wold and colleagues64 for review). To investigate measures of sleep quality in the current study, sleep fragmentation was calculated using relative frequency histograms for episode durations as previously published.33 Data indicated that TBI and rTBI led to fragmented sleep, defined as an increase in frequency of shorter bouts, compared to uninjured shams, where the rTBI 3 h mice had more 8- to 15-sec bouts than all other groups. Mice subjected to a single TBI or repeat injuries using a closed-head acceleration-deceleration model spend significantly more time awake over a 24-h period measured 1 month post-injury compared to uninjured shams.65 Given that wake is inversely proportional to sleep, the possibility exists for acute post-traumatic sleep to evolve into sleep issues in the more chronic post-injury period.
TBI results in functional, cognitive, emotional, and behavioral disorders16,66 that can be exacerbated by disruptions in sleep.64 In the current study, we show that both a single diffuse TBI and rTBI resulted in functional deficits. All brain-injured mice had significantly decreased latencies to stay on the rotarod compared to shams, with rTBI 3 h spending the least amount of time on the rotarod at all time points. Similarly, all brain-injured mice had significant neurological deficits across all time points compared to sham on the NSS, with the rTBI 3 h mice having the highest scores. These collective findings were expected and support that experimental TBI induces acute sensorimotor deficits, as previously published.24,26,27 Importantly, we note that the rTBI 3 h mice had the poorest performance on both tasks, whereas the rTBI 9 h mice performed similar to mice with a single TBI, suggesting a vulnerability to exacerbated injury from a subsequent TBI while post-traumatic sleep lingers. Our results reinforce published reports suggesting mice that receive repetitive closed TBIs or a single closed TBI exhibit decreased rotarod performance over 7 DPI.67 Our findings also indicate that at 7 DPI, rTBI 3 h mice moved significantly shorter distances in the arena compared to mice with a single TBI and shams, which resolved at 14 DPI but returned at 28 DPI.
These results support the hypothesis that a second TBI occurring during the period of post-traumatic sleep worsens functional outcome compared to mice receiving one TBI or a second TBI after post-traumatic sleep subsides. A single TBI or rTBI did not result in anxiety-like behavior, defined as less time spent in the center of an open field, longer duration before entering the center of an arena, or less entries to the center of an arena. Although there was an injury effect on exploration time during the novel object task, changes in the discrimination index across groups were insignificant, suggesting no measureable cognitive deficits for these groups. There was an injury-induced decrease in total investigation time at 7 DPI, however, with the rTBI 9 h mice exploring less than uninjured shams, which extended through 14 and 28 DPI for the rTBI 9 h and TBI groups. A more complex cognitive task, such as the Barnes maze, might have shown cognitive deficits, given that other research groups have reported deficits on the Barnes maze after repetitive closed TBI in the mouse.67
Orexin-A is a neuropeptide expressed exclusively by lateral hypothalamic neurons with axonal projections throughout the brain.68 The orexin system, made up of orexin-producing neurons, plays a vital role in the regulation of sleep/wakefulness,68 and the intracerebroventricular administration of orexin during the light period may increase wake time and decrease sleep time in the rodent.69,70 Further, sleep fragmentation has been observed in both orexin knockout mice71 and orexin receptor knockout mice.72 Clinically, patients with severe TBI reportedly had a significantly reduced number of orexin neurons,73 and orexin-A levels measured in cerebral spinal fluid after brain injury were lower in 95% of patients with moderate or severe TBI compared to control.74 To further elucidate the role of orexin in TBI-induced sleep disturbances, a recent study subjected hypocretin (orexin) knockout mice to controlled cortical impact brain injury and found that sleep was not altered by experimental TBI in hypocretin knockout mice.75 These findings collectively suggest that deficiencies in orexin-A may be linked to the development of sleep disorders in TBI survivors. We previously reported increased sleep after diffuse brain injury in the mouse over the first 6 h post-injury, and we hypothesized that this increase is caused by injury-induced inflammation23 as opposed to acute loss of orexin neurons.
In the current study, we investigated orexin-A neurons and found that neither diffuse TBI nor rTBI reduced neuron numbers in the lateral hypothalamus. These findings were similar to published studies that found no acute loss of orexin neurons after controlled cortical impact in the mouse62 and no orexin cell loss at 4 weeks post-injury in mice subjected to lateral FPI. However, after lateral fluid percussion in the rat, reduced number of orexin-A–positive neurons has been reported at 30 DPI.76 This TBI-induced loss of hypocretin (orexin) neurons has also been reported 32 days after controlled cortical impact in the ipsilateral hypothalamus of the mouse.75 Although we do not report a brain injury-induced loss of orexin neurons, it is possible that neuronal function, receptor expression, or circulating orexin levels contributed to sleep disturbances in our mice.
In the current study, we quantitatively analyzed neuropathology identified with silver stain and microglia de-ramification by skeletal analysis of Iba-1–stained tissue. Regardless of the interinjury interval, rTBI resulted in significant neuropathology at all time points and significant de-ramification of microglia at 14 DPI. These findings are consistent with previously published rTBI studies in the rodent that indicated that repetitive closed TBI increases microglia activity in both the mouse77 and rat,78 and repetitive controlled cortical impact increases microglia activation in the rat.79 We previously reported increased silver stain after a single diffuse TBI in the rat,48 as well as de-ramification of microglia assessed by skeletal analysis.49 To translate past findings in the rat to present findings in the mouse, we conducted a post-hoc analysis to investigate whether a single diffuse TBI increased neuropathology and de-ramification of microglia compared to uninjured shams only. Results from unpaired t-tests supported significantly increased hemispheric silver stain in mice subjected to a single TBI compared to uninjured shams at 14 and 28 DPI (Supplementary Table 5) (see online supplementary material at http://www.liebertpub.com), which is in line with our previously reported findings in the rat.48 However, we did not observe significant de-ramification measured by the skeletal analysis between mice subjected to a single diffuse TBI and uninjured shams. The current study analyzed microglia in the S1BF based on previously published data from our group in the rat.49,80 Based on the silver stain pathology presented here, vulnerable cortical regions might differ between the rat and mouse, such that skeletal analysis of the primary motor cortex (M1 and M2) may indicate de-ramification after a single TBI.
Shitaka and colleagues previously summarized animal models of repeated mild TBI and concluded that pathological features of the injury were subtle in comparison to the extent of behavioral deficits, hypothesizing that previously utilized histological assessments may not be sufficiently sensitive to detect structural damage responsible for the impairments.81 In the current study, we utilized multiple linear regression to investigate whether pathological damage (silver stain) or neuroinflammation could be predicted by behavioral tests or post-injury sleep monitoring. For pathology, regression results supported the findings of our more simplistic statistical tests with improved resolution (e.g., hemispheric silver stain significantly increased in the rTBI 3 h and rTBI 9 h mice), but behavioral tests were not supported as significant predictors of silver stain pathology. However, we found that neuroinflammation and the de-ramification of microglia, measured as microglia process length, worsened in all injured groups with increased sleep during the first 6 h post-injury, whereas this neuroinflammation slightly decreased in shams with increased sleep (Fig. 8), although injured animals had shorter process lengths, on average, than did shams. Additionally, based on microglia endpoints per cell, the rTBI 3 h group had sustained neuroinflammation regardless of the duration of post-injury sleep, which supports findings of previous studies that posited acute post-injury sleep may be indicative of underlying pathological processes, including inflammation, and that receiving multiple TBIs within a short time frame may be detrimental to outcome. Our inflammation versus sleep regression models importantly have considerable predictive power that might be useful for a personalized medicine approach to treatment of TBIs in clinical settings.
We conducted post-hoc power analyses using our data and regression results by the R package Webpower,82 which suggested that, given our group-specific sample sizes and observed small effect sizes (0.0–0.11) for regression models that evaluated relationships with behavioral tests, power was 0.10–0.49 at the 95% confidence level. Even at a more relaxed 90% confidence level, the power to detect relationships with behavioral tests remained poor (0. 20–0.62). Assuming similar effect sizes, we would have needed ≥65 total animals within each TBI group, or at least 260 total animals, to achieve a minimally acceptable power of ≥0.80 at the 90% and 95% confidence levels. In contrast, power to detect the significant relationships that we found between two inflammation metrics and sleep was >0.92 at the 95% confidence level, given the moderate observed effect sizes (0.26–0.34). The power to detect these latter relationships specific to each TBI group, however, was poor (0.26–0.52); we would have needed ≥26 animals within each TBI group or a total of 104 animals, to achieve power of ≥0.80 at the 90% and 95% confidence levels, assuming similar effect sizes. Thus, our findings do not support that no relationships exist between the behavioral tests and pathology or between the TBI group-specific neuroinflammation and acute post-traumatic sleep, but instead point to an overarching issue of small sample sizes generally accepted in the field when evaluating a single outcome measure.
In the current study, we sought to identify a relationship between acute behavioral outcomes and enduring pathology. Currently, no framework exists linking brain injury to functional deficits that predict recovery,83 wherein such a relationship could enhance personalized medicine for TBI survivors. Further, accurate prediction of outcome could drive the design of successful clinical trials. Data from the current study suggest that sleep may be a personalized predictor for outcome after TBI. In patients, sleep can be measured through actigraphy assessments, which are inexpensive, non-invasive, and objective. Sleep after TBI is currently being measured in clinical settings using actigraphy wrist bands.84 This technology could be implemented to replace subjective self-reporting of sleep disturbances and provide a reliable personalized predictor for return-to-activity decisions. Discordant opinions suggest that individuals should not be allowed to sleep or should be frequently awakened after mild TBI or concussion, whereas prevailing hypotheses suggest sleep is restorative and neuroprotective.85,86 For the individuals suffering from concussion, promotion of sleep coupled with actigraphy monitoring may be a plausible physiological biomarker for treatment. Further, assessing sleep using actigraphy monitoring would provide a personalized outcome for individuals, wherein a return to baseline sleep could inform coaches, team physicians, and military personnel when an individual could return to activity.
Limitations of this study identified by the data and results require further support for 1) acute post-traumatic sleep as a predictor of vulnerability to a second brain injury; 2) the bidirectional relationship between the physiological feature of sleep induced by injury and the pathological feature of inflammation; and 3) statistical models that use functional/behavior assessments to predict pathology. In the current study, rTBI 3 h mice slept significantly more during the dark cycles but significantly less during the light cycles. We have previously hypothesized that the increase in sleep after diffuse TBI may be, in part, caused by an increase in inflammatory cytokines that have dual roles as sleep regulatory cytokines. In the current study, sleep is fragmented and decreased in the light cycle, in the absence of loss of orexin neurons, suggesting that other mechanisms need to be explored, including the investigation of orexin neuron function and contribution of neuroinflammation. Further, the non-invasive sleep cages utilized in this study limit the discrimination of stages of sleep (i.e., non-REM [rapid eye movement], REM); thus, future studies should focus on more detailed analyses of TBI-induced sleep. Larger total sample sizes would improve power and resolution, which would allow for identification of salient predictive relationships in studies with multiple longitudinal endpoints, specifically regarding behavior-predicted pathology and multiple injury groups. Last, the current study was conducted using adult male mice, but findings from a recent study support sexual dimorphism in the inflammatory response to focal TBI in the mouse.87 Similarly, sex differences in rats subjected to a focal TBI have been reported in response to minocycline administration as a treatment of neuroinflammation.88 Additional studies are therefore necessary to investigate the current hypothesis in female mice, as well as juveniles.
In conclusion, results are consistent with TBI disrupting sleep in the rodent.23,24,62,76,89,90 In concordance with previous reports, our results show that repetitive TBI results in sensorimotor, neurological, and affective deficits (Supplementary Table 6) (see online supplementary material at http://www.liebertpub.com). There is a critical need for objective, personalized assessment criteria for TBI patients and this need is intensified by the increasing concern over the consequences of repetitive TBIs, especially if a second injury occurs before complete recovery from the initial injury.77 In the current study, we show that if mice receive a second TBI 3 h after the initial TBI, but during ongoing post-traumatic sleep, then the consequence is more severe functional and histopathological outcomes than mice that receive a second TBI after sleep from the initial impact subsides. Based on these observations, the previously identified period of acute sleep induced by TBI (6 h) represents a window of vulnerability to exacerbated damage from a second injury. Thus, post-traumatic sleep may serve as a personalized physiological biomarker defining a period of time when the brain is more vulnerable to a second TBI. Further, our results raise the consideration that post-traumatic sleep is a potential clinical tool for personalized TBI management, where regular sleep patterns may inform rehabilitative strategies and return-to-activity guidelines.
Supplementary Material
Acknowledgments
Research reported in this article was supported, in part, by National Institute of Neurological Disorders and Stroke of the NIH under award numbers R21 NS072611 and RO1 NS-065052 and PCH Mission Support Funds. The generosity of the Diane and Bruce Halle Foundation supported J.L.H. during the conduct of these studies, as well as NIH F31 NS-090921. R.K.R. was supported by a Science Foundation Arizona Bisgrove Scholarship during the conduct of these studies. The authors thank Anthony Romero for his contribution to behavior scoring. The authors also thank Cody Zurhellen for optimizing the Orexin-A staining. J.L. is a member of the University of Arizona Bio5 Institute.
Author Disclosure Statement
No competing financial interests exist.
References
- 1. Bailes J.E., Dashnaw M.L., Petraglia A.L., and Turner R.C. (2014). Cumulative effects of repetitive mild traumatic brain injury. Prog. Neurol. Surg. 28, 50–62 [DOI] [PubMed] [Google Scholar]
- 2. Rivara F.P., and Graham R. (2014). Sports-related concussions in youth: report from the Institute of Medicine and National Research Council. JAMA 311, 239–240 [DOI] [PubMed] [Google Scholar]
- 3. Merritt V.C., Clark A.L., Crocker L.D., Sorg S.F., Werhane M.L., Bondi M.W., Schiehser D.M., and Delano-Wood L. (2018). Repetitive mild traumatic brain injury in military veterans is associated with increased neuropsychological intra-individual variability. Neuropsychologia 119, 340–348 [DOI] [PubMed] [Google Scholar]
- 4. Bryan C.J. (2013). Repetitive traumatic brain injury (or concussion) increases severity of sleep disturbance among deployed military personnel. Sleep 36, 941–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Iverson G.L., and Gioia G.A. (2016). Returning to School Following Sport-Related Concussion. Phys. Med. Rehabil. Clin. North Am. 27, 429–436 [DOI] [PubMed] [Google Scholar]
- 6. McKee A.C., and Robinson M.E. (2014). Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement. 10, 3 Supppl., S242-S253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. List M., Vukonich M., Nord W., and Huntington M. (2014). Sports concussion management: a review of the evidence. S. D. Med. 67, 370–373 [PubMed] [Google Scholar]
- 8. Collins M.W., Kontos A.P., Reynolds E., Murawski C.D., and Fu F.H. (2014). A comprehensive, targeted approach to the clinical care of athletes following sport-related concussion. Knee Surg. Sports Traumatol. Arthrosc. 22, 235–246 [DOI] [PubMed] [Google Scholar]
- 9. Gregory E., West T.A., Cole W.R., Bailie J.M., McCulloch K.L., Ettenhofer M.L., Cecchini A., and Qashu F.M. (2017). Use of a multi-level mixed methods approach to study the effectiveness of a primary care progressive return to activity protocol after acute mild traumatic brain injury/concussion in the military. Contemp. Clin. Trials 52, 95–100 [DOI] [PubMed] [Google Scholar]
- 10. Werner C., and Engelhard K. (2007). Pathophysiology of traumatic brain injury. Br. J. Anaesth. 99, 4–9 [DOI] [PubMed] [Google Scholar]
- 11. Ziebell J.M., and Morganti-Kossmann M.C. (2010). Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 7, 22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Albensi B.C., and Janigro D. (2003). Traumatic brain injury and its effects on synaptic plasticity. Brain Inj. 17, 653–663 [DOI] [PubMed] [Google Scholar]
- 13. Yeates K.O., Taylor H.G., Wade S.L., Drotar D., Stancin T., and Minich N. (2002). A prospective study of short- and long-term neuropsychological outcomes after traumatic brain injury in children. Neuropsychology 16, 514–523 [DOI] [PubMed] [Google Scholar]
- 14. Masel B.E., and DeWitt D.S. (2010). Traumatic brain injury: a disease process, not an event. J. Neurotrauma 27, 1529–1540 [DOI] [PubMed] [Google Scholar]
- 15. Theeler B., Lucas S., Riechers R.G., II, and Ruff R.L. (2013). Post-traumatic headaches in civilians and military personnel: a comparative, clinical review. Headache 53, 881–900 [DOI] [PubMed] [Google Scholar]
- 16. Arciniegas D.B., Topkoff J., and Silver J.M. (2000). Neuropsychiatric aspects of traumatic brain injury. Curr. Treat. Options Neurol. 2, 169–186 [DOI] [PubMed] [Google Scholar]
- 17. Lifshitz J., Rowe R.K., Griffiths D.R., Evilsizor M.N., Thomas T.C., Adelson P.D., and McIntosh T.K. (2016). Clinical relevance of midline fluid percussion brain injury: acute deficits, chronic morbidities and the utility of biomarkers. Brain Inj. 30, 1293–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen A.J., and D'Esposito M. (2010). Traumatic brain injury: from bench to bedside to society. Neuron 66, 11–14 [DOI] [PubMed] [Google Scholar]
- 19. McAllister T.W. (1992). Neuropsychiatric sequelae of head injuries. Psychiatr. Clin. North Am. 15, 395–413 [PubMed] [Google Scholar]
- 20. Pasinetti G.M., Fivecoat H., and Ho L. (2010). Personalized medicine in traumatic brain injury. Psychiatr. Clin. North Am. 33, 905–913 [DOI] [PubMed] [Google Scholar]
- 21. Toman E., Harrisson S., and Belli T. (2016). Biomarkers in traumatic brain injury: a review. J. R. Army Med. Corps. 162, 103–108 [DOI] [PubMed] [Google Scholar]
- 22. Kulbe J.R., and Geddes J.W. (2016). Current status of fluid biomarkers in mild traumatic brain injury. Exp. Neurol. 275, Pt. 3, 334–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rowe R.K., Striz M., Bachstetter A.D., Van Eldik L.J., Donohue K.D., O'Hara B.F., and Lifshitz J. (2014). Diffuse brain injury induces acute post-traumatic sleep. PLoS One 9, e82507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harrison J.L., Rowe R.K., Ellis T.W., Yee N.S., O'Hara B.F., Adelson P.D., and Lifshitz J. (2015). Resolvins AT-D1 and E1 differentially impact functional outcome, post-traumatic sleep, and microglial activation following diffuse brain injury in the mouse. Brain Behav. Immun. 47, 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rao V., McCann U., Han D., Bergey A., and Smith M.T. (2014). Does acute TBI-related sleep disturbance predict subsequent neuropsychiatric disturbances? Brain Inj. 28, 20–26 [DOI] [PubMed] [Google Scholar]
- 26. Rowe R.K., Harrison J.L., Zhang H., Bachstetter A.D., Hesson D.P., O'Hara B.F., Greene M.I., and Lifshitz J. (2018). Novel TNF receptor-1 inhibitors identified as potential therapeutic candidates for traumatic brain injury. J. Neuroinflammation 15, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rowe R.K., Harrison J.L., O'Hara B.F., and Lifshitz J. (2014). Recovery of neurological function despite immediate sleep disruption following diffuse brain injury in the mouse: clinical relevance to medically untreated concussion. Sleep 37, 743–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lifshitz J. (2008). Fluid percussion injury, in: Animal Models of Acute Neurological Injuries. Chen J., Zu X.M., Xu Z.C., and Zhang J.H. (eds). Humana: Totowa, NJ [Google Scholar]
- 29. Rowe R.K., Harrison J.L., O'Hara B.F., and Lifshitz J. (2014). Diffuse brain injury does not affect chronic sleep patterns in the mouse. Brain Inj. 28, 504–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harrison J.L., Rowe R.K., O'Hara B.F., Adelson P.D., and Lifshitz J. (2014). Acute over-the-counter pharmacological intervention does not adversely affect behavioral outcome following diffuse traumatic brain injury in the mouse. Exp. Brain Res. 232, 2709–2719 [DOI] [PubMed] [Google Scholar]
- 31. Hosseini A.H., and Lifshitz J. (2009). Brain injury forces of moderate magnitude elicit the fencing response. Med. Sci. Sports Exerc. 41, 1687–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Donohue K.D., Medonza D.C., Crane E.R., and O'Hara B.F. (2008). Assessment of a non-invasive high-throughput classifier for behaviours associated with sleep and wake in mice. Biomed. Eng. Online 7, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mang G.M., Nicod J., Emmenegger Y., Donohue K.D., O'Hara B.F., and Franken P. (2014). Evaluation of a piezoelectric system as an alternative to electroencephalogram/ electromyogram recordings in mouse sleep studies. Sleep 37, 1383–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McShane B.B., Galante R.J., Jensen S.T., Naidoo N., Pack A.I., and Wyner A. (2010). Characterization of the bout durations of sleep and wakefulness. J. Neurosci. Methods 193, 321–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ziebell J.M., Bye N., Semple B.D., Kossmann T., and Morganti-Kossmann M.C. (2011). Attenuated neurological deficit, cell death and lesion volume in Fas-mutant mice is associated with altered neuroinflammation following traumatic brain injury. Brain Res. 1414, 94–105 [DOI] [PubMed] [Google Scholar]
- 36. Semple B.D., Bye N., Rancan M., Ziebell J.M., and Morganti-Kossmann M.C. (2010). Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2–/– mice. J. Cereb. Blood Flow Metab. 30, 769–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen Y., Constantini S., Trembovler V., Weinstock M., and Shohami E. (1996). An experimental model of closed head injury in mice: pathophysiology, histopathology, and cognitive deficits. J. Neurotrauma 13, 557–568 [DOI] [PubMed] [Google Scholar]
- 38. Pleasant J.M., Carlson S.W., Mao H., Scheff S.W., Yang K.H., and Saatman K.E. (2011). Rate of neurodegeneration in the mouse controlled cortical impact model is influenced by impactor tip shape: implications for mechanistic and therapeutic studies. J. Neurotrauma 28, 2245–2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nagai T., Yu J., Kitahara Y., Nabeshima T., and Yamada K. (2012). D-Serine ameliorates neonatal PolyI:C treatment-induced emotional and cognitive impairments in adult mice. J. Pharmacol. Sci. 120, 213–227 [DOI] [PubMed] [Google Scholar]
- 40. Porsolt R.D., Anton G., Blavet N., and Jalfre M. (1978). Behavioural despair in rats: a new model sensitive to antidepressant treatments. Eur. J. Pharmacol. 47, 379–391 [DOI] [PubMed] [Google Scholar]
- 41. Porsolt R.D. (1979). Animal model of depression. Biomedicine 30, 139–140 [PubMed] [Google Scholar]
- 42. DeOlmos J.S., and Ingram W.R. (1971). An improved cupric-silver method for impregnation of axonal and terminal degeneration. Brain Res. 33, 523–529 [DOI] [PubMed] [Google Scholar]
- 43. de Olmos J.S., Beltramino C.A., and de Olmos de Lorenzo S. (1994). Use of an amino-cupric-silver technique for the detection of early and semiacute neuronal degeneration caused by neurotoxicants, hypoxia, and physical trauma. Neurotoxicol. Teratol. 16, 545–561 [DOI] [PubMed] [Google Scholar]
- 44. Switzer R.C., III (2000). Application of silver degeneration stains for neurotoxicity testing. Toxicol. Pathol. 28, 70–83 [DOI] [PubMed] [Google Scholar]
- 45. Mikics E., Baranyi J., and Haller J. (2008). Rats exposed to traumatic stress bury unfamiliar objects—a novel measure of hyper-vigilance in PTSD models? Physiol. Behav. 94, 341–348 [DOI] [PubMed] [Google Scholar]
- 46. Cao T., Thomas T.C., Ziebell J.M., Pauly J.R., and Lifshitz J. (2012). Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience 225, 65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ziebell J.M., Taylor S.E., Cao T., Harrison J.L., and Lifshitz J. (2012). Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J. Neuroinflammation 9, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lifshitz J., and Lisembee A.M. (2012). Neurodegeneration in the somatosensory cortex after experimental diffuse brain injury. Brain Struct. Funct. 217, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morrison H., Young K., Qureshi M., Rowe R.K., and Lifshitz J. (2017). Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci. Rep. 7, 13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. R Core Team. (2018). R: A Language and Environment for Stastical Computing. R Foundation for Statistical Computing: Vienna, Austria [Google Scholar]
- 51. van Buuren S., and Groothuis-Oudshoorn K. (2011). mice: Multivariate Imputation by Chained Equations in R. J. Stat. Softw. 45, 1–67 [Google Scholar]
- 52. Rowe R.K., Ziebell J.M., Harrison J.L., Law L.M., Adelson P.D., and Lifshitz J. (2016). Aging with traumatic brain injury: effects of age at injury on behavioral outcome following diffuse brain injury in rats. Dev. Neurosci. 38, 195–205 [DOI] [PubMed] [Google Scholar]
- 53. Morrison H.W., and Filosa J.A. (2013). A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J. Neuroinflammation 10, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McLendon L.A., Kralik S.F., Grayson P.A., and Golomb M.R. (2016). The controversial second impact syndrome: a review of the literature. Pediatr. Neurol. 62, 9–17 [DOI] [PubMed] [Google Scholar]
- 55. Kostyun R.O., Milewski M.D., and Hafeez I. (2015). Sleep disturbance and neurocognitive function during the recovery from a sport-related concussion in adolescents. Am. J. Sports Med. 43, 633–640 [DOI] [PubMed] [Google Scholar]
- 56. Lau B.C., Collins M.W., and Lovell M.R. (2012). Cutoff scores in neurocognitive testing and symptom clusters that predict protracted recovery from concussions in high school athletes. Neurosurgery 70, 371–379; discussion, 379. [DOI] [PubMed] [Google Scholar]
- 57. Murdaugh D.L., Ono K.E., Reisner A., and Burns T.G. (2018). Assessment of sleep quantity and sleep disturbances during recovery from sports-related concussion in youth athletes. Arch. Phys. Med. Rehabil. 99, 960–966 [DOI] [PubMed] [Google Scholar]
- 58. Lau B.C., Kontos A.P., Collins M.W., Mucha A., and Lovell M.R. (2011). Which on-field signs/symptoms predict protracted recovery from sport-related concussion among high school football players? Am. J. Sports Med. 39, 2311–2318 [DOI] [PubMed] [Google Scholar]
- 59. Lau B., Lovell M.R., Collins M.W., and Pardini J. (2009). Neurocognitive and symptom predictors of recovery in high school athletes. Clin. J. Sport Med. 19, 216–221 [DOI] [PubMed] [Google Scholar]
- 60. Billiard M., and Podesta C. (2013). Recurrent hypersomnia following traumatic brain injury. Sleep Med. 14, 462–465 [DOI] [PubMed] [Google Scholar]
- 61. Watson N.F., Dikmen S., Machamer J., Doherty M., and Temkin N. (2007). Hypersomnia following traumatic brain injury. J. Clin. Sleep Med. 3, 363–368 [PMC free article] [PubMed] [Google Scholar]
- 62. Willie J.T., Lim M.M., Bennett R.E., Azarion A.A., Schwetye K.E., and Brody D.L. (2012). Controlled cortical impact traumatic brain injury acutely disrupts wakefulness and extracellular orexin dynamics as determined by intracerebral microdialysis in mice. J. Neurotrauma 29, 1908–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lim M.M., Elkind J., Xiong G., Galante R., Zhu J., Zhang L., Lian J., Rodin J., Kuzma N.N., Pack A.I., and Cohen A.S. (2013). Dietary therapy mitigates persistent wake deficits caused by mild traumatic brain injury. Sci. Transl. Med. 5, 215ra173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lucke-Wold B.P., Smith K.E., Nguyen L., Turner R.C., Logsdon A.F., Jackson G.J., Huber J.D., Rosen C.L., and Miller D.B. (2015). Sleep disruption and the sequelae associated with traumatic brain injury. Neurosci. Biobehav. Rev. 55, 68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Petraglia A.L., Plog B.A., Dayawansa S., Chen M., Dashnaw M.L., Czerniecka K., Walker C.T., Viterise T., Hyrien O., Iliff J.J., Deane R., Nedergaard M., and Huang J.H. (2014). The spectrum of neurobehavioral sequelae after repetitive mild traumatic brain injury: a novel mouse model of chronic traumatic encephalopathy. J. Neurotrauma 31, 1211–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rao V., and Lyketsos C. (2000). Neuropsychiatric sequelae of traumatic brain injury. Psychosomatics 41, 95–103 [DOI] [PubMed] [Google Scholar]
- 67. Mouzon B., Chaytow H., Crynen G., Bachmeier C., Stewart J., Mullan M., Stewart W., and Crawford F. (2012). Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J. neurotrauma 29, 2761–2773 [DOI] [PubMed] [Google Scholar]
- 68. Inutsuka A., and Yamanaka A. (2013). The physiological role of orexin/hypocretin neurons in the regulation of sleep/wakefulness and neuroendocrine functions. Front. Endocrinol. (Lausanne) 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Piper D.C., Upton N., Smith M.I., and Hunter A.J. (2000). The novel brain neuropeptide, orexin-A, modulates the sleep-wake cycle of rats. Eur. J. Neurosci. 12, 726–730 [DOI] [PubMed] [Google Scholar]
- 70. Hagan J.J., Leslie R.A., Patel S., Evans M.L., Wattam T.A., Holmes S., Benham C.D., Taylor S.G., Routledge C., Hemmati P., Munton R.P., Ashmeade T.E., Shah A.S., Hatcher J.P., Hatcher P.D., Jones D.N., Smith M.I., Piper D.C., Hunter A.J., Porter R.A., and Upton N. (1999). Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc. Natl. Acad. Sci. U. S. A. 96, 10911–10916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chemelli R.M., Willie J.T., Sinton C.M., Elmquist J.K., Scammell T., Lee C., Richardson J.A., Williams S.C., Xiong Y., Kisanuki Y., Fitch T.E., Nakazato M., Hammer R.E., Saper C.B., and Yanagisawa M. (1999). Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98, 437–451 [DOI] [PubMed] [Google Scholar]
- 72. Willie J.T., Chemelli R.M., Sinton C.M., Tokita S., Williams S.C., Kisanuki Y.Y., Marcus J.N., Lee C., Elmquist J.K., Kohlmeier K.A., Leonard C.S., Richardson J.A., Hammer R.E., and Yanagisawa M. (2003). Distinct narcolepsy syndromes in Orexin receptor-2 and Orexin null mice: molecular genetic dissection of non-REM and REM sleep regulatory processes. Neuron 38, 715–730 [DOI] [PubMed] [Google Scholar]
- 73. Baumann C.R., Bassetti C.L., Valko P.O., Haybaeck J., Keller M., Clark E., Stocker R., Tolnay M., and Scammell T.E. (2009). Loss of hypocretin (orexin) neurons with traumatic brain injury. Ann. Neurol. 66, 555–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Baumann C.R., Stocker R., Imhof H.G., Trentz O., Hersberger M., Mignot E., and Bassetti C.L. (2005). Hypocretin-1 (orexin A) deficiency in acute traumatic brain injury. Neurology 65, 147–149 [DOI] [PubMed] [Google Scholar]
- 75. Thomasy H.K., and Opp M.R. (2018). Hypocretin mediates sleep and wake disturbances in a mouse model of traumatic brain injury. J. Neurotrauma. October 3. doi: 10.1089/neu.2018.5810. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Skopin M.D., Kabadi S.V., Viechweg S.S., Mong J.A., and Faden A.I. (2015). Chronic decrease in wakefulness and disruption of sleep-wake behavior after experimental traumatic brain injury. J. Neurotrauma 32, 289–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Robinson S., Berglass J.B., Denson J.L., Berkner J., Anstine C.V., Winer J.L., Maxwell J.R., Qiu J., Yang Y., Sillerud L.O., Meehan W.P. Mannix R., III, and Jantzie L.L. (2017). Microstructural and microglial changes after repetitive mild traumatic brain injury in mice. J. Neurosci. Res. 95, 1025–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tyburski A.L., Cheng L., Assari S., Darvish K., and Elliott M.B. (2017). Frequent mild head injury promotes trigeminal sensitivity concomitant with microglial proliferation, astrocytosis, and increased neuropeptide levels in the trigeminal pain system. J. Headache Pain 18, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bai R., Gao H., Han Z., Huang S., Ge X., Chen F., and Lei P. (2017). Flow cytometric characterization of T cell subsets and microglia after repetitive mild traumatic brain injury in rats. Neurochem. Res. 42, 2892–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ziebell J.M., Taylor S.E., Cao T., Harrison J.L., and Lifshitz J. (2012). Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J. Neuroinflammation 9, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shitaka Y., Tran H.T., Bennett R.E., Sanchez L., Levy M.A., Dikranian K., and Brody D.L. (2011). Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J. Neuropathol. Exp. Neurol. 70, 551–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang Z. (2018). Practical Statistical Power Analysis using Webpower and R. ISDSA: Granger, IN [Google Scholar]
- 83. Falcon M.I., Jirsa V., and Solodkin A. (2016). A new neuroinformatics approach to personalized medicine in neurology: The Virtual Brain. Curr. Opin. Neurol. 29, 429–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Imbach L.L., Valko P.O., Li T., Maric A., Symeonidou E.R., Stover J.F., Bassetti C.L., Mica L., Werth E., and Baumann C.R. (2015). Increased sleep need and daytime sleepiness 6 months after traumatic brain injury: a prospective controlled clinical trial. Brain 138, 726–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Xie L., Kang H., Xu Q., Chen M.J., Liao Y., Thiyagarajan M., O'Donnell J., Christensen D.J., Nicholson C., Iliff J.J., Takano T., Deane R., and Nedergaard M. (2013). Sleep drives metabolite clearance from the adult brain. Science 342, 373–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Eugene A.R., and Masiak J. (2015). The neuroprotective aspects of sleep. MEDtube Sci. 3, 35–40 [PMC free article] [PubMed] [Google Scholar]
- 87. Villapol S., Loane D.J., and Burns M.P. (2017). Sexual dimorphism in the inflammatory response to traumatic brain injury. Glia 65, 1423–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Taylor A.N., Tio D.L., Paydar A., and Sutton R.L. (2018). Sex differences in thermal, stress, and inflammatory responses to minocycline administration in rats with traumatic brain injury. J. Neurotrauma 35, 630–638 [DOI] [PubMed] [Google Scholar]
- 89. Elliott M.B., Oshinsky M.L., Amenta P.S., Awe O.O., and Jallo J.I. (2012). Nociceptive neuropeptide increases and periorbital allodynia in a model of traumatic brain injury. Headache 52, 966–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Borniger J.C., Ungerleider K., Zhang N., Karelina K., Magalang U.J., and Weil Z.M. (2018). Repetitive brain injury of juvenile mice impairs environmental enrichment-induced modulation of REM sleep in adulthood. Neuroscience 375, 74–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.