

Abstract
Background
Neuropathic pain, which is caused by a lesion or disease affecting the somatosensory system, may be central or peripheral in origin. Neuropathic pain often includes symptoms such as burning or shooting sensations, abnormal sensitivity to normally painless stimuli, or an increased sensitivity to normally painful stimuli. Neuropathic pain is a common symptom in many diseases of the nervous system. Opioid drugs, including morphine, are commonly used to treat neuropathic pain. Most reviews have examined all opioids together. This review sought evidence specifically for morphine; other opioids are considered in separate reviews.
Objectives
To assess the analgesic efficacy and adverse events of morphine for chronic neuropathic pain in adults.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, and Embase for randomised controlled trials from inception to February 2017. We also searched the reference lists of retrieved studies and reviews, and online clinical trial registries.
Selection criteria
We included randomised, double‐blind trials of two weeks' duration or longer, comparing morphine (any route of administration) with placebo or another active treatment for neuropathic pain, with participant‐reported pain assessment.
Data collection and analysis
Two review authors independently extracted data and assessed trial quality and potential bias. Primary outcomes were participants with substantial pain relief (at least 50% pain relief over baseline or very much improved on Patient Global Impression of Change scale (PGIC)), or moderate pain relief (at least 30% pain relief over baseline or much or very much improved on PGIC). Where pooled analysis was possible, we used dichotomous data to calculate risk ratio (RR) and number needed to treat for an additional beneficial outcome (NNT) or harmful outcome (NNH). We assessed the quality of the evidence using GRADE and created 'Summary of findings' tables.
Main results
We identified five randomised, double‐blind, cross‐over studies with treatment periods of four to seven weeks, involving 236 participants in suitably characterised neuropathic pain; 152 (64%) participants completed all treatment periods. Oral morphine was titrated to maximum daily doses of 90 mg to 180 mg or the maximum tolerated dose, and then maintained for the remainder of the study. Participants had experienced moderate or severe neuropathic pain for at least three months. Included studies involved people with painful diabetic neuropathy, chemotherapy‐induced peripheral neuropathy, postherpetic neuralgia criteria, phantom limb or postamputation pain, and lumbar radiculopathy. Exclusions were typically people with other significant comorbidity or pain from other causes.
Overall, we judged the studies to be at low risk of bias, but there were concerns over small study size and the imputation method used for participants who withdrew from the studies, both of which could lead to overestimation of treatment benefits and underestimation of harm.
There was insufficient or no evidence for the primary outcomes of interest for efficacy or harm. Four studies reported an approximation of moderate pain improvement (any pain‐related outcome indicating some improvement) comparing morphine with placebo in different types of neuropathic pain. We pooled these data in an exploratory analysis. Moderate improvement was experienced by 63% (87/138) of participants with morphine and 36% (45/125) with placebo; the risk difference (RD) was 0.27 (95% confidence interval (CI) 0.16 to 0.38, fixed‐effects analysis) and the NNT 3.7 (2.6 to 6.5). We assessed the quality of the evidence as very low because of the small number of events; available information did not provide a reliable indication of the likely effect, and the likelihood that the effect will be substantially different was very high. A similar exploratory analysis for substantial pain relief on three studies (177 participants) showed no difference between morphine and placebo.
All‐cause withdrawals in four studies occurred in 16% (24/152) of participants with morphine and 12% (16/137) with placebo. The RD was 0.04 (‐0.04 to 0.12, random‐effects analysis). Adverse events were inconsistently reported, more common with morphine than with placebo, and typical of opioids. There were two serious adverse events, one with morphine, and one with a combination of morphine and nortriptyline. No deaths were reported. These outcomes were assessed as very low quality because of the limited number of participants and events.
Authors' conclusions
There was insufficient evidence to support or refute the suggestion that morphine has any efficacy in any neuropathic pain condition.
Plain language summary
Morphine for neuropathic pain in adults
Bottom line
There is very low quality evidence that morphine taken by mouth has any important effect on pain in people with moderate or severe neuropathic pain.
Background
Neuropathic pain comes from damaged nerves. It is different from pain messages that are carried along healthy nerves from damaged tissue (a fall or cut, or arthritic knee). Neuropathic pain is often treated by different medicines (drugs) to those used for pain from damaged tissue, which we often think of as painkillers. Medicines that are sometimes used to treat depression or epilepsy can be effective in some people with neuropathic pain. Opioid painkillers are sometimes used to treat neuropathic pain.
Opioid painkillers are drugs like morphine. Morphine is derived from plants or synthesised by chemists. Morphine is widely available for use as a painkiller, usually given by mouth.
Our definition of a good result was someone with a high level of pain relief and able to keep taking the medicine without side effects making them want to stop.
Study characteristics
In February 2017, we searched for clinical trials in which morphine was used to treat neuropathic pain in adults. Five studies satisfied the inclusion criteria, randomising 236 participants to treatment with morphine, placebo, or other drugs. Studies lasted four to seven weeks. Few studies reported beneficial outcomes that would be regarded as clinically relevant.
Key results
Four small studies reported that pain was reduced by between a quarter and a third in some people. This level of pain reduction was experienced by 6 in 10 participants with morphine and 4 in 10 with placebo. Between 1 and 2 in 10 participants withdrew from treatment with both morphine and placebo, but the reasons were not given. Side effects were poorly reported, but were more common with morphine than with placebo, and included drowsiness, dizziness, constipation, feeling sick, dry mouth, and decreased appetite.
Quality of the evidence
The evidence was of very low quality. This means that the research did not provide a reliable indication of the likely effect, and the likelihood that the effect will be substantially different is very high. Small studies like those in this review tend to overestimate results of treatment compared to the effects found in larger, better designed studies. There were other problems that might lead to over‐optimistic results. The very low quality evidence and the lack of any important benefit mean that we need new, longer‐lasting, large trials before we will know if morphine is useful for the treatment of neuropathic pain.
Summary of findings
Summary of findings for the main comparison. Morphine compared with placebo for neuropathic pain.
| Morphine compared with placebo for neuropathic pain | ||||||
|
Patient or population: adults with neuropathic pain (any origin) Settings: community Intervention: oral morphine (any dose) Comparison: placebo | ||||||
| Outcomes (at trial end) | Probable outcome with morphine | Probable outcome with placebo | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments |
| Reported outcomes approximating to 30% reduction in pain | 39/62 | 21/55 | Not analysed | 62 participants 2 cross‐over studies |
Very low quality | Downgraded 3 levels due to small number of studies, participants, and events, and several source of potential bias. |
| At least 50% reduction in pain | 28/62 | 14/55 | Not analysed | 62 participants 2 cross‐over studies |
Very low quality | Downgraded 3 levels due to small number of studies, participants, and events, and several source of potential bias. |
| PGIC much or very much improved | 13/32 | 11/28 | Not analysed | 32 participants 1 study |
Very low quality | Downgraded 3 levels due to small number of studies, participants, and events, and several source of potential bias. |
| Any pain‐related outcome (moderate pain improvement) | 630 per 1000 | 360 per 1000 | RD 0.27 (0.16 to 0.38) NNT 3.7 (2.6 to 6.5) |
263 participants 4 studies |
Very low quality | Downgraded 3 levels due to small number of studies, participants, and events, and several source of potential bias. |
| Serious adverse events | None reported | None reported | Not analysed | 289 participants 4 studies |
Very low quality | Downgraded 3 levels due to small number of studies, participants, and events, and several source of potential bias. |
| Death | None reported | None reported | Not analysed | 289 participants 4 studies |
Very low quality | Downgraded 3 levels due to small number of studies, participants, and events, and several source of potential bias. |
| Any adverse event | Inadequate information | Inadequate information | Not analysed | 289 participants 4 studies |
Very low quality | Downgraded 3 levels due to small number of studies, participants, and events, and several source of potential bias. |
| All‐cause withdrawal | 160 per 1000 | 120 per 1000 | RD 0.04 (‐0.04 to 0.12 | 289 participants 4 studies |
Very low quality | Downgraded 3 levels due to small number of studies, participants, and events, and several source of potential bias. |
| CI: confidence interval; NNT: number needed to treat for an additional beneficial outcome; PGIC: Patient Global Impression of Change; RD: risk difference. | ||||||
| Descriptors for levels of evidence (EPOC 2015):
High quality: This research provides a very good indication of the likely effect. The likelihood that the effect will be substantially differenta is low.
Moderate quality: This research provides a good indication of the likely effect. The likelihood that the effect will be substantially differenta is moderate.
Low quality: This research provides some indication of the likely effect. However, the likelihood that it will be substantially differenta is high.
Very low quality: This research does not provide a reliable indication of the likely effect. The likelihood that the effect will be substantially differenta is very high. a Substantially different: a large enough difference that it might affect a decision. | ||||||
Background
This review is based on a template for reviews of drugs used to relieve neuropathic pain. The aim is for all reviews to use the same methods, based on new criteria for what constitutes reliable evidence in chronic pain (Moore 2010a; Appendix 1).
Description of the condition
Neuropathic pain is a consequence of a pathological maladaptive response of the nervous system to 'damage' from a wide variety of potential causes (Colloca 2017). It is characterised by pain in the absence of a noxious stimulus, or where minor or moderate nociceptive stimuli evoke exaggerated levels of pain. Neuropathic pain may be spontaneous (continuous or paroxysmal) in its temporal characteristics or be evoked by sensory stimuli (dynamic mechanical allodynia where pain is evoked by light touch of the skin).
Neuropathic pain is heterogeneous in aetiology, pathophysiology, and clinical appearance. The 2011 International Association for the Study of Pain definition of neuropathic pain is "pain caused by a lesion or disease of the somatosensory system" (Jensen 2011), and is based on a definition agreed at an earlier consensus meeting (Treede 2008). Neuropathic pain is associated with a variety of sensory loss (numbness) and sensory gain (allodynia) clinical phenomena, the exact pattern of which varies between people and disease, perhaps reflecting different pain mechanisms operating in an individual person and, therefore, potentially predictive of response to treatment (Demant 2014; Helfert 2015; von Hehn 2012). A new approach of subgrouping people with peripheral neuropathic pain of different aetiologies according to intrinsic sensory profiles has generated three profiles that may be related to pathophysiological mechanisms and may be useful in clinical trial design to enrich the study population for treatment responders (Baron 2017).
Preclinical research hypothesises a bewildering array of possible pain mechanisms that may operate in people with neuropathic pain, which largely reflect pathophysiological responses in both the central and peripheral nervous systems, including neuronal interactions with immune cells (Baron 2012; Calvo 2012; von Hehn 2012). Overall, the benefits of drugs for neuropathic pain treatments, even the most effective, are modest (Finnerup 2015; Moore 2013a), and a robust classification of neuropathic pain is not yet available (Finnerup 2013).
Neuropathic pain is usually divided according to the cause of nerve injury. There may be many causes, but some common causes of neuropathic pain include diabetes (painful diabetic neuropathy (PDN)), shingles (postherpetic neuralgia (PHN)), amputation (stump and phantom limb pain), neuropathic pain after surgery or trauma, stroke or spinal cord injury, trigeminal neuralgia, and HIV infection. Sometimes the cause is unknown.
Many people with neuropathic pain conditions are significantly disabled with moderate or severe pain for many years. Chronic pain conditions comprised five of the 11 top‐ranking conditions for years lived with disability in 2010 (Vos 2012), and are responsible for considerable loss of quality of life and employment, and increased healthcare costs (Moore 2014a). One US study found the healthcare costs were three‐fold higher for people with neuropathic pain than matched controls (Berger 2004). A UK study and a German study showed a two‐ to three‐fold higher level of use of healthcare services in people with neuropathic pain than people without (Berger 2009; Berger 2012). For PHN, for example, studies demonstrated large loss of quality of life and substantial costs (Scott 2006; van Hoek 2009).
In systematic reviews, the overall prevalence of neuropathic pain in the general population is reported to be between 7% and 10% (van Hecke 2014), and about 7% in a systematic review of studies published since 2000 (Moore 2014a). In individual countries, prevalence rates have been reported as 3.3% in Austria (Gustorff 2008), 6.9% in France (Bouhassira 2008), and up to 8% in the UK (Torrance 2006). In a community study of recent joint pain, features of neuropathic pain were common and were present in over half of those people reporting pain of at least moderate severity (Soni 2013). Some forms of neuropathic pain, such as PDN and postsurgical chronic pain (which is often neuropathic in origin), are increasing (Hall 2008). The prevalence of PHN is likely to fall if vaccination against the herpes virus becomes widespread.
Estimates of incidence vary between individual studies for particular origins of neuropathic pain, often because of small numbers of cases. In primary care in the UK, between 2002 and 2005, the incidences (per 100,000 person‐years' observation) were 28 (95% confidence interval (CI) 27 to 30) for PHN, 27 (95% CI 26 to 29) for trigeminal neuralgia, 0.8 (95% CI 0.6 to 1.1) for phantom limb pain, and 21 (95% CI 20 to 22) for PDN (Hall 2008). Other studies have estimated an incidence of 4 in 100,000 per year for trigeminal neuralgia (Katusic 1991; Rappaport 1994), and 12.6 per 100,000 person‐years for trigeminal neuralgia and 3.9 per 100,000 person‐years for PHN in a study of facial pain in the Netherlands (Koopman 2009). One systematic review of chronic pain demonstrated that some neuropathic pain conditions, such as PDN, can be more common than other neuropathic pain conditions, with prevalence rates up to 400 per 100,000 person‐years (McQuay 2007).
Neuropathic pain is difficult to treat effectively, with only a minority of people experiencing a clinically relevant benefit from any one intervention (Kalso 2013; Moore 2013a). A multidisciplinary approach is now advocated, combining pharmacological interventions with physical or cognitive (or both) interventions. The evidence of benefit from interventional management is very weak, or non‐existent (Dworkin 2013). Conventional analgesics such as paracetamol and nonsteroidal anti‐inflammatory drugs (NSAID) are not thought not to be effective, but without evidence to support or refute that view (Moore 2015a; Wiffen 2016). Some people may derive some benefit from a topical lidocaine patch or low‐concentration topical capsaicin, although evidence about benefits is uncertain (Derry 2012; Derry 2014). High‐concentration topical capsaicin may benefit some people with PHN (Derry 2017). Treatment is often by so‐called 'adjuvant analgesics' (pain modulators) such as antidepressants (duloxetine and amitriptyline; Lunn 2014; Moore 2014b; Moore 2015b; Sultan 2008), or antiepileptic drugs (gabapentin or pregabalin; Moore 2009; Moore 2014c; Wiffen 2013). Evidence for efficacy of opioids is inconclusive (Derry 2016; Gaskell 2016; Stannard 2016; Wiffen 2015).
The proportion of people who achieve worthwhile pain relief (typically at least 50% pain intensity reduction; Moore 2013b) is small, generally only 10% to 25% more than with placebo, with numbers needed to treat for an additional beneficial outcome (NNT) usually between 4 and 10 (Kalso 2013; Moore 2013a). Neuropathic pain is not particularly different from other chronic pain conditions in that only a small proportion of trial participants have a good response to treatment (Moore 2013a).
The current National Institute for Health and Care Excellence (NICE) guidance for the pharmacological management of neuropathic pain suggests offering a choice of amitriptyline, duloxetine, gabapentin, or pregabalin as initial treatment for neuropathic pain (with the exception of trigeminal neuralgia), with switching if the first, second, or third drugs tried are not effective or not tolerated (NICE 2013). This concurs with other guidance (Finnerup 2015).
Description of the intervention
Morphine in one form or another has been available for millennia, and appeared in Pliny's Historia Naturalis (AD 77) as opium, the resin derived from poppy sap. Morphine was extracted from opium in 1803 and named as such by Sertürner, a German pharmacist, in 1817 (Rey 1993). Oral morphine was first recommended in England for the treatment of cancer pain in the 1950s. This was often in the form of the so‐called 'Brompton cocktail' containing cocaine and alcohol in addition to morphine or diamorphine. Treatment moved towards oral morphine alone as morphine demonstrated effective pain relief without the adverse effects linked to the 'cocktail'.
Following the publication of World Health Organization (WHO) guidelines in the mid‐1980s, the oral administration of aqueous morphine solution every four hours by the clock became commonplace for moderate to severe cancer pain (WHO 1986). Morphine in a modified‐release tablet was first marketed around the same time, allowing the dosage interval to be extended to 12 hours.
Morphine, usually as the sulphate or hydrochloride salt, is available in four oral formulations: an elixir or solution of morphine in various concentrations; an immediate‐release tablet; a number of different preparations of modified‐release tablets or capsules; and modified‐release suspensions. A table showing the brand names of morphine preparations is provided in Appendix 2. Modified‐release tablets are available in both 12‐hour and 24‐hour release patterns and should be swallowed whole. They have lower maximum plasma concentrations and longer time to maximum concentration than equal doses of immediate‐release formulations (Collins 1998).
The wide range of formulations and dosages allows great flexibility (Grahame‐Smith 2002). Potent opioid analgesics are indicated for the relief of pain in malignant disease and often have the additional very useful actions of relieving anxiety, producing drowsiness, and allowing sleep (Grahame‐Smith 2002). However, all opioid analgesics have the potential to produce adverse effects including respiratory depression, nausea and vomiting, constipation, urinary retention, cognitive impairment, and itching. During chronic opioid therapy, progressively higher doses may be required to sustain the analgesic effect (tolerance) and people can be at risk of opioid withdrawal syndrome upon sudden cessation of the opioid or administration of an antagonist (physiological dependence).
Morphine is also available for intravenous, intramuscular, or subcutaneous injection, and is used for injection around the spinal cord or within the brain in some circumstances. Sublingual, buccal, and rectal routes of administration of morphine are sometimes used. None of these ways of administering morphine is likely to be used for neuropathic pain, but we did not exclude them from this review.
How the intervention might work
Opioids such as morphine bind to specific opioid receptors in the nervous system and other tissues; there are three principal classes of receptors (mu, kappa, and delta) although others have been suggested, and subtypes of receptors are considered to exist. Binding of opioid agonists such as morphine to receptors brings about complex cellular changes that, for the most part, acutely inhibit injury‐induced activation of nociceptive pathways. The outcomes include decreased perception of pain, decreased reaction to pain, and increased pain tolerance. As a result, people treated with morphine report decreased pain and decreased reaction to tissue injury. However, paradoxical excitatory effects of morphine administration have been observed in some nociceptive neurons and in the brain, particularly after prolonged administration.
Why it is important to do this review
One UK survey found that weak and strong opioids were used frequently for treating neuropathic pain (Hall 2013). Morphine (a strong opioid) has long been considered as the preferred choice of opioid. It is widely, though still not universally, available across the world, is comparatively inexpensive, and is effective orally. It is listed in the WHO Essential Medicines List (WHO 2011). Since the early‐2000s, a marked increase in prescribing of opioids for non‐cancer pain in general, despite a relatively modest evidence base, has in some countries led to widespread diversion with consequent abuse, misuse, and mortality. Concurrently, suspicion has arisen that opioid‐induced hyperalgesia, together with tolerance to the analgesic effects of opioids, may in reality result in a lesser degree of benefit for opioids in neuropathic pain than previously assumed.
The standards used to assess evidence in chronic pain trials have evolved substantially in recent years, with particular attention being paid to trial duration, withdrawals, and statistical imputation following withdrawal, all of which can substantially alter estimates of efficacy. The most important change is the move from using mean pain scores, or mean change in pain scores, to the number of people who have a large decrease in pain (by at least 50%) and who continue in treatment, ideally in trials of 8 to 12 weeks' duration or longer. Pain intensity reduction of 50% or more correlates with improvements in comorbid symptoms, function, and quality of life. These standards are set out in the PaPaS Author and Referee Guidance for pain studies of the Cochrane Pain, Palliative and Supportive Care Group (PaPaS 2012).
This Cochrane Review assesses the evidence using methods that make both statistical and clinical sense, and uses developing criteria for what constitutes reliable evidence in chronic pain (Moore 2010a). Trials included and analysed meet a minimum of reporting quality (blinding, randomisation), validity (duration, dose and timing, diagnosis, outcomes, etc.), and size (ideally at least 500 participants in a comparison in which the number needed to treat is 4 or above; Moore 1998). This approach sets high standards for the demonstration of efficacy and marks a departure from how reviews were conducted previously.
Taking this newer, more rigorous approach is particularly important for opioids in chronic non‐cancer pain. Opioids in clinical trials in non‐cancer pain are associated with very high withdrawal rates of up to 60% over about 12 weeks (Moore 2010b). Many withdrawals occur within the first few weeks, when patients experience pain relief but cannot tolerate the drug. The common practice of using the last observed results carried forward to the end of the trial many weeks later (last observation carried forward (LOCF)) can, therefore, produce results based largely on people no longer in the trial, and who in clinical practice could not achieve pain relief because they could not take the tablets. The newer standards, outlined in Appendix 1, would not allow this LOCF effect and may produce very different results. For example, one large analysis of pooled data from trials in osteoarthritis and chronic low back pain conducted over about 12 weeks judged oxycodone effective, but an analysis of the same data using the new clinically meaningful standards showed it to be significantly worse than placebo (Lange 2010).
One previous Cochrane Review demonstrated the limitations of our knowledge about opioids in neuropathic pain, except in short duration studies of 24 hours or less (McNicol 2013). These limitations were confirmed by more recent reviews specific to particular opioids (Derry 2016; Gaskell 2016; Stannard 2016; Wiffen 2015). A review specific to morphine is timely, given its wide availability and use, and distinctive patterns of metabolism to an array of byproducts some of which may worsen, and others improve, pain (Ball 1985; Faura 1996; Hand 1987).
Objectives
To assess the analgesic efficacy and adverse events of morphine for chronic neuropathic pain in adults.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) with double‐blind assessment of participant outcomes following two weeks or more of treatment, although the emphasis of the review was on studies with a duration of eight weeks or longer. We required full journal publication, with the exception of online clinical trial results summaries of otherwise unpublished clinical trials, and abstracts with sufficient data for analysis. We did not include short abstracts (usually meeting reports). We excluded studies that were non‐randomised, studies of experimental pain, case reports, and clinical observations.
Types of participants
Studies included adults aged 18 years and above with one or more chronic neuropathic pain condition including (but not limited to):
cancer‐related neuropathy;
central neuropathic pain;
complex regional pain syndrome (CRPS) Type II;
HIV neuropathy;
painful diabetic neuropathy (PDN);
phantom limb pain;
postherpetic neuralgia (PHN);
postoperative or traumatic neuropathic pain;
spinal cord injury;
trigeminal neuralgia.
Where we included studies with more than one type of neuropathic pain, we analysed results according to the primary condition if identifiable.
Types of interventions
Morphine at any dose, by any route, administered for the relief of neuropathic pain and compared with placebo or any active comparator.
Types of outcome measures
We anticipated that studies would use a variety of outcome measures, with most studies using standard subjective scales (numerical rating scale (NRS) or visual analogue scale (VAS)) for pain intensity or pain relief, or both. We were particularly interested in Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) definitions for moderate and substantial benefit in chronic pain studies (Dworkin 2008). These were defined as:
at least 30% pain relief over baseline (moderate);
at least 50% pain relief over baseline (substantial);
much or very much improved on Patient Global Impression of Change scale (PGIC; moderate);
very much improved on PGIC (substantial).
These outcomes are different from those used in most earlier reviews, concentrating as they do on dichotomous outcomes where pain responses do not follow a normal (Gaussian) distribution. People with chronic pain desire high levels of pain relief, ideally more than 50% pain intensity reduction, and ideally resulting in no worse than mild pain (Moore 2013b; O'Brien 2010).
Primary outcomes
Participant‐reported pain relief of 30% or greater.
Participant‐reported pain relief of 50% or greater.
PGIC much or very much improved.
PGIC very much improved.
Secondary outcomes
Any pain‐related outcome indicating some improvement, such as better function.
Withdrawals due to lack of efficacy, adverse events, and for any cause.
Participants experiencing any adverse event.
Participants experiencing any serious adverse event. Serious adverse events typically include any untoward medical occurrence or effect that at any dose results in death, is life‐threatening, requires hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, is a congenital anomaly or birth defect in the person's offspring, is an 'important medical event' that may jeopardise the person, or may require an intervention to prevent one of the above characteristics or consequences.
Specific adverse events, particularly somnolence and dizziness.
Search methods for identification of studies
Electronic searches
We searched the following databases, without language restrictions:
Cochrane Central Register of Controlled Trials (CENTRAL, via CRSO) (15 February 2017);
MEDLINE (via Ovid) (1946 to 15 February 2017);
Embase (via Ovid) (1974 to 15 February 2017).
Search strategies are available in Appendix 3 (CENTRAL), Appendix 4 (MEDLINE), and Appendix 5 (Embase).
Searching other resources
We reviewed the bibliographies of any RCTs identified and review articles, and searched clinical trial databases (ClinicalTrials.gov (ClinicalTrials.gov) and the WHO International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/)) to identify additional published or unpublished data. We did not contact investigators or study sponsors.
Data collection and analysis
We planned to perform separate analyses for efficacy outcomes according to particular neuropathic pain conditions, but there were insufficient data. We combined different neuropathic pain conditions in analyses of efficacy for exploratory purposes only.
We pooled information from different pain conditions for adverse events and withdrawals, where possible.
Selection of studies
We determined eligibility by reading the abstract of each study identified by the search. We eliminated studies that clearly did not satisfy the inclusion criteria, and obtained full copies of the remaining studies. Two review authors made the decisions. Two review authors read these studies independently and reached agreement by discussion. We did not anonymise the studies before assessment. We created a PRISMA flow chart of the process (Figure 1).
1.
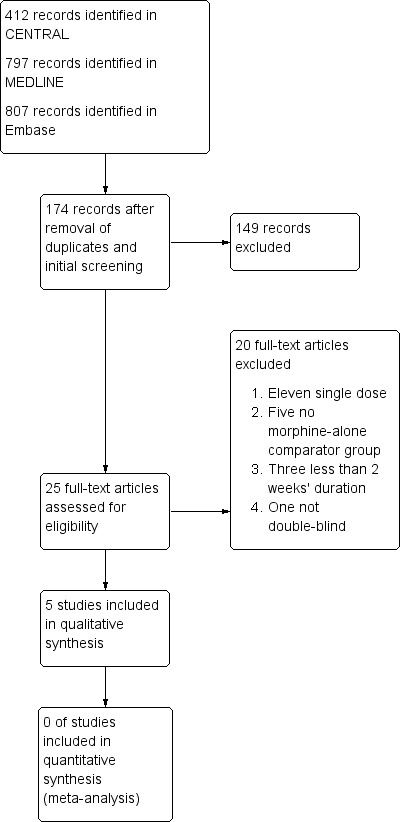
Study flow diagram.
Data extraction and management
Two review authors extracted the data independently using a standard form and checked for agreement before entry into Review Manager 5 (RevMan 2014), or any other analysis tool. We included information about the pain condition and number of participants treated, drug and dosing regimen, study design (placebo or active control), study duration and follow‐up, analgesic outcome measures and results, withdrawals, and adverse events (participants experiencing any adverse event or a serious adverse event).
Assessment of risk of bias in included studies
We used the Oxford Quality Score as the basis for inclusion (Jadad 1996), limiting inclusion to studies that were randomised and double‐blind as a minimum.
Two review authors independently assessed risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Chapter 8, Higgins 2011), and adapted from those used by the Cochrane Pregnancy and Childbirth Group, with any disagreements resolved by discussion. We assessed the following for each study.
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (any truly random process, e.g. random number table; computer random number generator); unclear risk of bias (when the method used to generate the sequence was not clearly stated). We excluded studies at a high risk of bias that used a non‐random process (e.g. odd or even date of birth; hospital or clinic record number).
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions prior to assignment determines whether intervention allocation could have been foreseen in advance of, or during, recruitment, or changed after assignment. We assessed the methods as: low risk of bias (e.g. telephone or central randomisation; consecutively numbered, sealed, opaque envelopes); unclear risk of bias (when the method was not clearly stated). We excluded studies that did not conceal allocation and were, therefore, at a high risk of bias (e.g. open list).
Blinding of participants and personnel (checking for possible performance bias), and blinding of outcome assessment (checking for possible detection bias). We assessed the methods used to blind study personnel and participants (all outcomes were self‐assessed) from knowledge of which intervention a participant received. We assessed the methods as: low risk of bias (study stated that it was blinded and described the method used to achieve blinding, e.g. identical tablets, matched in appearance and smell); unclear risk of bias (study stated that it was blinded but did not provide an adequate description of how it was achieved). We excluded studies at a high risk of bias that were not double‐blind.
Incomplete outcome data (checking for possible attrition bias due to the amount, nature, and handling of incomplete outcome data). We assessed the methods used to deal with incomplete data as: low risk of bias (fewer than 10% of participants did not complete the study or used 'baseline observation carried forward' analysis, or both); unclear risk of bias (used LOCF analysis); or high risk of bias (used 'completer' analysis).
Selective reporting (checking for possible reporting bias). We checked if an a priori study protocol was available and if all outcomes of the study protocol were reported in the publication of the study. We assigned a low risk of reporting bias if the study protocol was available and all the study's prespecified (primary and secondary) outcomes that were of interest in the review had been reported in the prespecified way, or if the study protocol was not available but it was clear that the published reports contained all expected outcomes, including those that were prespecified (convincing text of this nature may be uncommon). We assigned a high risk of reporting bias if not all the study's prespecified primary outcomes were reported; one or more primary outcomes was reported using measurements, analysis methods, or subsets of the data (subscales) that were not prespecified; one or more reported primary outcomes were not prespecified (unless clear justification for their reporting was provided, such as an unexpected adverse event); one or more outcomes of interest in the review was reported incompletely so that it could not be entered in a meta‐analysis; the study report did not include results for a key outcome that would be expected to have been reported for such a study.
Size of study (checking for possible biases confounded by small size (Dechartres 2013; Dechartres 2014; Moore 1998; Nüesch 2010; Thorlund 2011)). We assessed studies as being at low risk of bias (200 participants or more per treatment arm); unclear risk of bias (50 to 199 participants per treatment arm); or high risk of bias (fewer than 50 participants per treatment arm).
Measures of treatment effect
We calculated NNTs as the reciprocal of the absolute risk reduction (McQuay 1998). For unwanted effects, the NNT becomes the number needed to treat for an additional harmful outcome (NNH) and is calculated in the same manner. We used dichotomous data to calculate risk difference (RD) with 95% CI using a fixed‐effect model unless we found significant statistical heterogeneity (see below). We did not use continuous data in analyses.
Unit of analysis issues
We planned to split the control treatment arm between active treatment arms in a single study if the active treatment arms were not combined for analysis.
Dealing with missing data
We used intention‐to‐treat (ITT) analysis where the ITT population consisted of participants who were randomised, took at least one dose of the assigned study medication, and provided at least one postbaseline assessment. We assigned zero improvement to missing participants wherever possible.
Assessment of heterogeneity
We planned to deal with clinical heterogeneity by combining studies that examined similar conditions, and to assess statistical heterogeneity visually (L'Abbé 1987), and with the use of the I2 statistic. When the I2 value was greater than 50%, we considered possible reasons for this.
Assessment of reporting biases
The aim of this review was to use dichotomous outcomes of known utility and of value to people with pain (Hoffman 2010; Moore 2010c; Moore 2010d; Moore 2010e; Moore 2013b). The review did not depend on what the authors of the original studies chose to report or not, though clearly difficulties arose in studies that did not report any dichotomous results. We extracted and used continuous data, which probably will reflect efficacy and utility poorly, only when useful for illustrative purposes.
We assessed publication bias using a method designed to detect the amount of unpublished data with a null effect required to make any result clinically irrelevant (usually taken to mean an NNT of 10 or higher; Moore 2008).
Data synthesis
We planned to use a fixed‐effect model for meta‐analysis, but would use a random‐effects model for meta‐analysis if there was significant clinical heterogeneity and it was considered appropriate to combine studies.
Quality of the evidence
We used the GRADE system to assess the quality of the evidence related to the key outcomes listed in Types of outcome measures, as appropriate (Appendix 6). Two review authors (TC, RAM) independently rated the quality of each outcome.
We paid particular attention to inconsistency, where point estimates varied widely across studies or CIs of studies showed minimal or no overlap (Guyatt 2011), and potential for publication bias, based on the amount of unpublished data required to make the result clinically irrelevant (Moore 2008).
In addition, there may be circumstances where the overall rating for a particular outcome needs to be adjusted as recommended by GRADE guidelines (Guyatt 2013a). For example, where there were so few data that the results were highly susceptible to the random play of chance, or if a study used LOCF imputation in circumstances where there were substantial differences in adverse event withdrawals, one would have no confidence in the result, and would need to downgrade the quality of the evidence by three levels, to very low quality. In circumstances where there were no data reported for an outcome, we reported the level of evidence as very low quality (Guyatt 2013b).
In addition, we are aware that many Cochrane Reviews are based largely or wholly on small underpowered studies, and of the danger of making conclusive assessments of evidence based on inadequate information (AlBalawi 2013; Brok 2009; Roberts 2015; Turner 2013).
'Summary of findings' table
We have included a 'Summary of findings' table as set out in the PaPaS author guide (PaPaS 2012), and recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Chapter 11, Higgins 2011). The table includes, where possible, outcomes equivalent to moderate or substantial benefit of at least 30% and at least 50% pain intensity reduction, PGIC (possibly at least substantial improvement and at least moderate improvement) (Dworkin 2008), any pain‐related outcome, serious adverse events, any adverse events, withdrawals due to lack of efficacy, withdrawals due to adverse events, and death (a particular serious adverse event). We were aware that there would be limited information, and in the event, we chose to concentrate on those outcomes where some information was available.
For the 'Summary of findings' table we used the following descriptors for levels of evidence (EPOC 2015):
high: this research provides a very good indication of the likely effect. The likelihood that the effect will be substantially differenta is low;
moderate: this research provides a good indication of the likely effect. The likelihood that the effect will be substantially differenta is moderate;
low: this research provides some indication of the likely effect. However, the likelihood that it will be substantially differenta is high;
very low: this research does not provide a reliable indication of the likely effect. The likelihood that the effect will be substantially differenta is very high.
a Substantially different: a large enough difference that it might affect a decision.
Subgroup analysis and investigation of heterogeneity
We planned all analyses to be according to individual neuropathic pain conditions, because placebo response rates for the same outcome can vary between conditions, as can the drug‐specific effects (Moore 2009).
We did not plan subgroup analyses since experience of previous reviews indicates that there would be too few data for any meaningful subgroup analysis (Derry 2016; Gaskell 2016; McNicol 2013; Stannard 2016; Wiffen 2015).
Sensitivity analysis
We planned no sensitivity analysis because the evidence base was known to be too small to allow reliable analysis; we did not pool results from neuropathic pain of different origins in the primary analyses. We would have examined details of dose‐escalation schedules to see if this could provide some basis for a sensitivity analysis, but there were too few data.
Results
Description of studies
Results of the search
A flow diagram of the search results is shown in Figure 1.
The database search identified 2016 records. After removing duplicates and initial screening there were 174 records. We excluded 149 irrelevant records. Our searches identified 25 studies using oral morphine (target doses 90 mg/day to 180 mg/day) to treat chronic neuropathic pain. We excluded 20 full‐text articles at this stage and documented the reasons (see Characteristics of excluded studies table). Five studies fulfilled the criteria using the appropriate morphine interventions and were included (Gilron 2005; Gilron 2015; Huse 2001; Khoromi 2007; Wu 2008) (see Characteristics of included studies table).
Included studies
The five included studies all had a cross‐over design with two to four treatment periods. Details are to be found in the Characteristics of included studies table. The five studies randomised 236 participants to treatment, and 152 (64%) participants completed all treatment periods. Included studies involved people with PDN, chemotherapy‐induced peripheral neuropathy, PHN, phantom limb or postamputation pain, and lumbar radiculopathy. Participants typically had to have at least moderate pain (score of 3/10 or greater) for three months to enter the studies, and average baseline pain intensities ranged between 4.7/10 and 7/10.
Gilron 2005 investigated 57 participants in a single‐centre, randomised, double‐blind, placebo and active comparator controlled, four‐arm, four‐period cross‐over study. Participants had a diagnosis of PDN and PHN with moderate pain scores for three months or more at randomisation. The mean age was 64 years (range 40 to 81), and 56% of participants were men. Participants received active daily placebo (lorazepam 1.6 mg), morphine (target 120 mg), gabapentin (target 2300 mg), or gabapentin plus morphine (target gabapentin 2400 mg plus morphine 60 mg) orally, titrated to maximum tolerated doses over three weeks, maintained at the maximum dose for the fourth week, then tapered in the fifth week to include a three‐day washout. Participants then received the next intervention in the cross‐over sequence. The total duration of the study was 20 weeks (4 × 5 weeks). Analyses included participants who withdrew during the period, but imputation method was not mentioned. Of the 57 participants randomised, 41 completed all treatment periods.
Gilron 2015 investigated 52 participants in a single‐centre, randomised, double‐blind, active comparator controlled, three‐arm, three‐period cross‐over study. Participants had diagnoses of PDN, chemotherapy‐induced peripheral neuropathy, or PHN, with pain scores of at least 3/10 for six months or more at randomisation. The mean age was 66 years (range 49 to 80), and 73% were men. Participants received morphine (target 100 mg), nortriptyline (target 100 mg), or morphine plus nortriptyline (target morphine 100 mg plus nortriptyline 100 mg) orally, titrated to maximum tolerated doses over 24 days, maintained at the maximum dose for one week, then tapered over one week, followed by a four‐day washout. Participants then received the next intervention in the cross‐over sequence. The total duration of the study was 18 weeks (3 × 6 weeks). Analyses included participants who withdrew during the period, but imputation method was not mentioned. Of the 52 participants randomised, 36 completed all treatment periods.
Huse 2001 investigated 12 participants in a single‐centre, randomised, double‐blind, placebo‐controlled, two‐arm, two‐period cross‐over study. Participants were leg amputees with phantom limb pain, with minimum VAS pain scores of 3/10 at randomisation. The mean age was 51 years (range 30 to 71), and 83% were men. Participants started treatment with a test infusion of morphine or placebo, then received the same intervention orally for four weeks, titrated to the maximum tolerated dose (70 mg to 300 mg). There was a washout period of one to two weeks before treatment started with the other intervention. There were no withdrawals.
Khoromi 2007 investigated 55 participants in a single‐centre, randomised, double‐blind, placebo‐ and active‐comparator controlled, four‐arm, four‐period cross‐over study. Participants had a diagnosis of lumbar radiculopathy with average leg pain scores of at least 4/10 at randomisation. The median age was 53 years (range 19 to 65), and 55% were men. Participants received daily oral active placebo (benztropine 0.5 mg to 1 mg), morphine (15 mg to 90 mg), nortriptyline (25 mg to 100 mg), or morphine plus nortriptyline (morphine 15 mg to 90 mg plus nortriptyline 25 mg to 100 mg), titrated to maximum tolerated dose for five weeks, then maintained for two weeks before a further two weeks of dose tapering before switching to the next intervention in the cross‐over sequence. The total duration of the study was 36 weeks (4 × 9 weeks). The efficacy analysis included only participants who completed two or more treatment periods; those who dropped out during the first or second treatment periods were considered to be missing. Of the 55 participants randomised, 28 completed all treatment periods.
Wu 2008 investigated 60 participants in a single‐centre, randomised, double‐blind, placebo‐ and active‐comparator controlled, three‐arm, three‐period cross‐over study. Participants had a diagnosis of persistent postamputation pain with minimum pain scores of 3/10 for six months or longer at randomisation. The mean age was 63 years (standard deviation 16), and 78% were men. Participants received daily oral placebo, morphine (target 180 mg), or the sodium channel blocker mexiletine (target 1200 mg), titrated to the maximum tolerated dose over four weeks, followed by two weeks at the maximum dose, then a two‐week drug taper and one‐week washout before switching to the next intervention in the cross‐over sequence. The total duration of the study was 27 weeks (3 × 9 weeks). It is unclear whether results were analysed using LOCF imputation for withdrawals, though the authors reported that a separate analysis for completers of all three periods gave similar results to the whole population. Of the 60 participants randomised, 35 completed all treatment periods.
Studies generally excluded participants who had any contraindication to the study drugs, any significant comorbidity or other pain condition that might interfere with the study, or a history of alcohol or substance abuse.
Two studies allowed continued use of stable dose of non‐opioid analgesics other than any used in the study (Gilron 2005; Gilron 2015), while Huse 2001 reported that all analgesic and psychotropic medication use was noted in the patient diary. Khoromi 2007 required that participants tapered off any opioids before the start of the study and took no opioids, selective serotonin reuptake inhibitors (SSRIs) or tricyclic medications outside of the protocol during the study, and made no changes to any other stable analgesic medication regimen, but anti‐inflammatory medications and paracetamol (acetaminophen) were allowed as rescue medications. Wu 2008 required that all pain and psychotropic medication was stopped at least two weeks before the study started, but allowed paracetamol and NSAIDs throughout the study.
Excluded studies
See Characteristics of excluded studies tables.
We excluded 20 studies after reading the full articles. All studies were randomised.
Eleven studies were single dose trials (Domzal 1986; Eide 1994; Jadad 1992; Kalman 2002; Persson 1998; Rowbotham 1991; Serinken 2016; Siddall 2000; Wasan 2006; Watt 1996; Wu 2002).
Five studies did not have morphine treatment alone to compare with placebo or an active comparator (Galer 2005; Nalamachu 2013; Patarica‐Huber 2011; Raja 2002; Rocha 2014).
Three studies had treatment periods of less than two weeks (Attal 2002; Dellemijn 1994; Harke 2001).
One study was not double‐blind (Xiong 2008).
Risk of bias in included studies
A 'Risk of bias' graph is available in Figure 2, and 'Risk of bias' summary in Figure 3. Incomplete reporting in the cross‐over studies and small size contributed to high risk of bias in nearly all studies, but other risks of bias were generally low. All studies scored 5/5 on the Oxford Quality Score.
2.
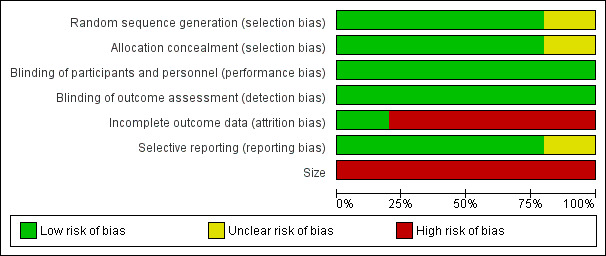
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
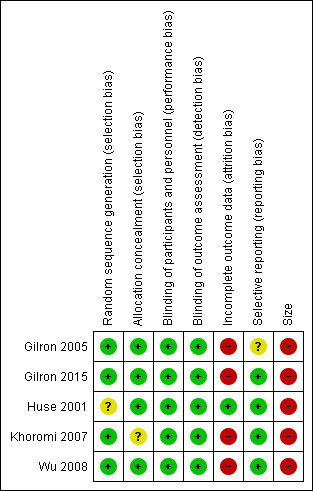
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
We judged the studies at low risk of bias for random sequence generation and allocation concealment. All studies clearly described their methods of random allocation of participants and sequence generation to the treatment arms, with the except of: Huse 2001, who did not clearly state their exact method of random sequence generation, even though they stated it was randomised in the methods; in addition, Khoromi 2007 did not provide details of methods for concealing allocation. We judged both studies at unclear risk of bias. No studies were at high risk of selection bias.
Blinding
We judged the studies to be at low risk of performance and detection bias. All five studies were double‐blind, and clearly described their methods of blinding. We excluded open‐label and single‐blind trials (see Characteristics of excluded studies).
Incomplete outcome data
We judged most studies to be at high risk of bias for attrition bias, with 20% or more withdrawals before treatment ended in all but one study (Huse 2001). Not all participants were accounted for neither were all imputation methods described. In the five studies, only 64% of participants randomised completed all the treatment phases and provided data for all treatments.
Selective reporting
We judged reporting bias to be low risk overall. We judged Gilron 2005 at unclear risk of selective reporting because in the methods, the authors specified Global Pain Relief at the end of treatment as an outcome, but reported no results. They stated they would only carry out analyses if the global result was significant. We judged the other studies at low risk of bias.
Other potential sources of bias
Four studies began with between 50 and 60 participants, but all of them lost participants and ended with numbers below 50. Therefore, we judged the risk of bias to be high. One study had only 12 participants (Huse 2001).
Effects of interventions
See: Table 1
Table 1 includes the following outcomes: reported outcomes approximating to 30% reduction, at least 50% reduction in pain, PGIC much or very much improved, any pain‐related outcome (moderate pain improvement), serious adverse events, participants experiencing any adverse event, and all‐cause withdrawal. These were not entirely what was intended, but were dictated largely by their importance and the available evidence.
Morphine versus placebo
One of the four studies did not include a placebo, so provided no data for these analyses (Gilron 2015).
Participants with at least 30% pain relief
Two studies reported outcomes closely approximating to at least 30% pain relief.
Huse 2001 reported on participants who achieved more than 25% pain intensity reduction (separately as 25% to 50%, and greater than 50%); 6/12 participants with morphine and 2/12 participants with placebo achieved more than 25% pain intensity reduction.
Wu 2008 reported that 33/50 participants had a 33% decrease in pain intensity with morphine and 19/43 participants with placebo.
Participants with at least 50% pain relief
Two studies provided information on participants achieving at least 50% pain relief.
Huse 2001 reported that 5/12 participants had more than 50% pain intensity reduction with morphine and 1/12 participants with placebo.
Wu 2008 reported that 23/50 participants had a 50% decrease in pain intensity with morphine and 13/43 participants with placebo.
Patient Global Impression of Change much or very much improved
Two studies reported the number of participants with at least moderate relief, using a 6‐point global scale (pain worse, no relief, slight relief, moderate relief, a lot of relief, or complete relief), which we judged equivalent to PGIC much or very much improved.
Gilron 2005 reported that 35/44 participants achieved at least moderate pain relief with morphine and 13/42 participants with placebo.
Khoromi 2007 reported that 13/32 participants achieved at least moderate pain with morphine and 11/28 participants with placebo.
Patient Global Impression of Change very much improved
Khoromi 2007 reported that 8/32 participants had 'a lot or complete relief' with morphine and 5/28 participants with placebo, using a 6‐point scale (pain worse, no relief, slight relief, moderate relief, a lot of relief, or complete relief), which we judged equivalent to PGIC very much improved.
Any pain‐related outcome indicating some improvement
All studies reported group mean data for various measures of pain intensity or pain relief, with modest reductions from baseline in all treatment arms, which were slightly more for active treatment than placebo. Details are in Appendix 7.
Moderate pain improvement
Four studies (263 participants) provided data relating to improvement that would approximate IMMPACT) definitions of moderate improvement (Dworkin 2008). Gilron 2005 reported at least moderate pain relief over four weeks in PDN and PHN, Huse 2001 reported more than 25% pain intensity reduction over four weeks in phantom limb pain, Khoromi 2007 reported at least moderate pain relief over seven weeks in lumbar radiculopathy, and Wu 2008 reported at least 33% pain reduction over six weeks in postamputation pain. We did not plan to pool data from different pain conditions, but in the absence of more data, we decided to pool these studies in an exploratory analysis, using a random‐effects method.
The proportion of participants experiencing moderate pain improvement with morphine was 63% (87/138, range 41% to 80%).
The proportion of participants experiencing moderate pain improvement with placebo was 36% (45/125, range 17% to 44%).
The RD for morphine compared with placebo was 0.27 (95% CI 0.16 to 0.38) (Analysis 1.1; Figure 4); the NNT was 3.7 (95% CI, 2.6 to 6.5).
1.1. Analysis.

Comparison 1 Morphine versus placebo, Outcome 1 Moderate improvement.
4.
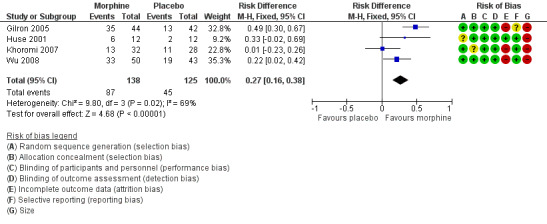
Forest plot of comparison: 1 Morphine versus placebo, outcome: 1.1 Moderate improvement.
We downgraded the evidence for moderate pain improvement to very low quality because of the small size of studies, the different pain conditions studied, there were only 132 actual events, and the studies did not report an ITT analysis or adequately report how they handled withdrawals. As expected from small studies with both clinical and methodological heterogeneity, the I2 statistic was moderate at 69%. Approximately 450 participants would be needed in studies of null effect to increase the NNT to 10.
Substantial pain improvement
Three studies (177 participants) provided data relating to improvement that would approximate substantial improvement (Dworkin 2008). Huse 2001 reported greater than 50% pain reduction over four weeks in phantom limb pain, Khoromi 2007 reported a global impression of a lot of improvement over seven weeks in lumbar radiculopathy, and Wu 2008 reported at least 50% pain reduction over six weeks in postamputation pain. Although the total numbers were below our limit of 200, we performed an analysis for hypothesis‐testing purposes.
The proportion of participants experiencing substantial pain improvement with morphine was 38% (36/94, range 25% to 46%).
The proportion of participants experiencing substantial pain improvement with placebo was 23% (19/83, range 8% to 30%).
The RD for morphine compared with placebo was 0.15 (95% CI ‐0.02 to 0.32) (Analysis 1.2); the NNT was not calculated.
1.2. Analysis.
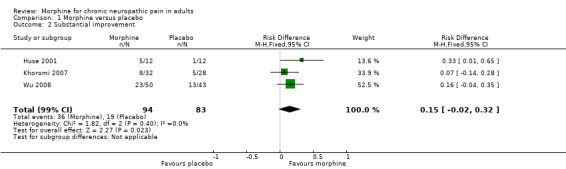
Comparison 1 Morphine versus placebo, Outcome 2 Substantial improvement.
We downgraded the evidence for substantial pain improvement to very low quality because of the small size of studies, the different pain conditions studied, there were only 55 actual events, the studies did not report an ITT analysis or adequately report how they handed withdrawals, and because there was no significant effect.
Withdrawals
Four studies provided information on all‐cause withdrawals (Gilron 2005; Huse 2001; Khoromi 2007; Wu 2008).
The proportion of participants who withdrew for any reason with morphine was 16% (24/152, range 0% to 22%).
The proportion of participants who withdrew for any reason with placebo was 12% (16/137, range 0% to 23%).
The RD for morphine compared with placebo was 0.04 (95% CI ‐0.04, 0.12) (Analysis 1.3); the NNH was not calculated.
1.3. Analysis.
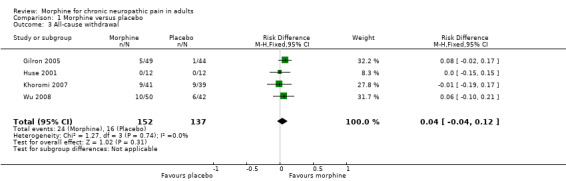
Comparison 1 Morphine versus placebo, Outcome 3 All‐cause withdrawal.
We downgraded the evidence for this outcome to very low quality because of the small size of studies and pooled data set, and because there were only 40 actual events.
There was insufficient information regarding adverse event or lack of efficacy withdrawals.
Adverse events
Adverse events were inconsistently reported and there was insufficient information for any meaningful analysis. Events reported were typical of opioids and included drowsiness, dizziness, constipation, dry mouth, nausea, and decreased appetite. There were two serious adverse, one with morphine alone (pneumonia) and one with morphine combined with nortriptyline (leg oedema) in Gilron 2015. There were no reported deaths.
Morphine versus active comparators
Four studies reported active comparisons with morphine. These were:
nortriptyline and nortriptyline plus morphine in two studies (Gilron 2015; Khoromi 2007);
gabapentin and gabapentin plus morphine in one study (Gilron 2005);
mexiletine in one study (Wu 2008).
None of the studies provided sufficient information for analysis for any outcome. Combination pharmacotherapy for neuropathic pain is considered in another Cochrane Review (Chaparro 2012).
One study reported serious adverse events without a placebo comparator (Gilron 2015). One serious adverse event occurred in participants treated with morphine alone and another with morphine plus nortriptyline.
Discussion
We identified five included studies with 236 participants. The cross‐over design involved substantial withdrawals, so that 152 (64%) participants completed all treatment periods. These studies involved people with PDN, chemotherapy‐induced peripheral neuropathy, PHN, phantom limb or postamputation pain, and lumbar radiculopathy. Participants had at least moderate initial pain (scoring 3/10 or greater), with mean baseline pain intensity scores ranging between 4.7/10 and 7/10.
Summary of main results
The primary pain outcomes of this review were 'substantial' pain relief, ideally a reduction in pain intensity by 50% or more, and 'moderate' pain relief, a reduction by 30% or more, and sustained over the duration of the trial, ideally three months. These outcomes are judged as desirable by people with pain (Moore 2013a). Few studies reported pain outcomes of interest to people with neuropathic pain, and we could not perform analyses according to different types of neuropathic pain. This is important because different types of neuropathic pain can respond differently to the same treatment when studies are otherwise identical (Moore 2009). Studies did not report any predefined useful outcomes relating to harm.
The evidence that morphine is beneficial for pain or any other outcome in neuropathic pain is severely limited. Therefore, the conclusion of this review is different from that of a review of all opioids that included short duration studies, conducted in 2005 (McNicol 2013).
Overall completeness and applicability of evidence
Study participants were typical of people with neuropathic pain who are eligible to take part in clinical trials. As is usual, exclusion criteria included other significant comorbidities or pain from other causes, contraindications to morphine or other opioids, and a history of addiction or drug or alcohol abuse.
This review faces the same issue as many Cochrane Reviews that are based largely or wholly on small underpowered studies, with the consequent danger of making conclusive assessments of evidence based on inadequate information (AlBalawi 2013; Brok 2009; Overall 1969; Roberts 2015; Turner 2013). This is exacerbated by the problems of having too few events to overcome random chance effects (Moore 1998; Thorlund 2011), as well there being a risk of bias in small studies (Dechartres 2013; Dechartres 2014; Nüesch 2010). Other potential problems derive from the included studies all being cross‐over studies (Elbourne 2002), the fact that important efficacy and harm outcomes were not reported, and the relative short duration of the studies of four to seven weeks of treatment not allowing for longer term issues of tolerance. What constitutes an adequate study duration is a matter of debate (Tayeb 2016).
All of these issues make the evidence less than complete and not easily applicable to many people with neuropathic pain. While problems of bias surrounding the studies might be expected to produce a large treatment effect, the was no such large treatment effect for the primary outcome of at least 50% pain intensity reduction.
This paucity of evidence contrasts with one UK survey that found that weak and strong opioids were used frequently for treating neuropathic pain, either alone or in combination with other drugs (Hall 2013). The lack of high quality evidence for long term benefit with morphine reflects a similar result with oxycodone, buprenorphine, and other opioids (Derry 2016; Gaskell 2014; Haroutiunian 2012; McNicol 2013; Stannard 2016; Wiffen 2015). The lack of evidence of efficacy combined with substantial evidence of harm has led to calls for referral to a pain management specialist (ideally with expertise in opioid use) if daily dosing exceeds 80 mg to 100 mg morphine equivalents, particularly if pain and function are not substantially improved (Franklin 2014). An overview review is currently looking for studies of higher doses of opioids (Els 2016); it is unlikely to find much evidence.
This review did not address some important issues for morphine. Intercurrent medical conditions, dehydration, or simple effects of ageing upon hepatic or renal function can reduce the clearance of morphine and lead to accumulation of high blood levels. The same is true for morphine's bioactive metabolites, particularly the potent active metabolite morphine‐6‐glucuronide (Ball 1985; Faura 1996; Faura 1998; Hand 1987; Klimas 2014; Sear 1989).
Quality of the evidence
All five included studies had at least one major risk of bias. Poor reporting of useful pain outcomes rendered the evidence quality low to very low. We were unable to carry out planned pooled analyses due to lack of data, and exploratory pooled analyses were typically on only about 200 participants, where chance effects are possible (Moore 1998; Thorlund 2011). In view of the small sample sizes, as well as uncertainties for other possible risks of bias, we chose to downgrade the quality of the evidence three levels to very low quality. Very low quality means that this research does not provide a reliable indication of the likely effect. The likelihood that the effect will be substantially different is very high.
Potential biases in the review process
We know of no potential biases in the review process.
Agreements and disagreements with other studies or reviews
This review agrees with previous reviews and Cochrane Reviews that there appears to be no body of good clinical studies assessing the efficacy of morphine for neuropathic pain, beyond short term and single‐dose experimental studies (McNicol 2013).
Our results are more cautious than those of Finnerup 2015, who assessed the evidence for strong opioids, including morphine, to have moderate quality, but made a weak recommendation for their use. Other groups have also made weak recommendations for using strong opioids, including morphine, in the short term (Sommer 2015). The reasons for their disagreement with the present review lie in different attitudes to the importance of sources of potential weakness in designs of cross‐over studies, short duration, imputation methods or completer analyses, and small size. Some reviews share our concerns, at least in part, and can agree that on the basis of inadequate evidence, NNT values of about 4 can be obtained as long as concerns of outcome, duration, and bias are ignored (Colloca 2017; Edelsberg 2011).
Authors' conclusions
Implications for practice.
For people with neuropathic pain
There is no convincing evidence that oral morphine at doses up to 180 mg/day is effective in relieving neuropathic pain in the longer term.
For clinicians
There is no convincing evidence that morphine at doses up to 180 mg/day is effective in relieving neuropathic pain in the longer term. There is no good evidence demonstrating high levels of analgesia (at least 50% pain reduction, for example) in good quality studies lasting at least 12 weeks. That does not exclude the possibility that an as‐yet undefined subgroup of people may get a good response with morphine.
For policy makers
There is no convincing evidence that morphine at doses up to 180 mg/day is effective in relieving neuropathic pain in the longer term. There is no good evidence demonstrating high levels of analgesia (at least 50% pain reduction, for example) in good quality studies lasting at least 12 weeks. That does not exclude the possibility that an as‐yet undefined subgroup of people may get a good response with morphine.
For funders
There is no strongly convincing evidence to support the suggestion that morphine has any efficacy in relieving neuropathic pain, but we cannot exclude the possibility that an as‐yet undefined subgroup of people may get a good response with morphine. In the absence of any additional supporting evidence, morphine should probably be available only at the discretion of a pain specialist with particular expertise in neuropathic pain.
Implications for research.
General
The design of studies in neuropathic pain, and the outcomes, are well understood, but as the number of people experiencing good pain relief with morphine over the longer term (12 weeks) is likely to be small, an enriched‐enrolment randomised‐withdrawal (EERW) design might provide the highest sensitivity to detect a signal (Moore 2015c). Since combination therapy for neuropathic pain has been reported to be more effective than monotherapy with any drug (Chaparro 2012), and combination therapy is common clinical practice, studies examining morphine in combination with a gabapentinoid or antidepressant could be of interest.
Design
Reporting of clinically relevant outcomes using appropriate imputation for withdrawal would improve the relevance of the findings for clinical practice. The use of EERW designs for comparison with classic trial designs indicates that good quality EERW designs of long duration may be appropriate for neuropathic pain.
Stratification by phenotype might be an interesting possibility for future studies (Baron 2017), as well as the possibility of measuring pain scores with activity (including dynamic tactile allodynia) versus at rest or on average/worst/best over prior 24 hours.
While pain is important, other outcomes relating to function, sleep, fatigue, and quality of life are important, and are probably closely linked (Hoffman 2010).
Measurement (endpoints)
Assessment of neuropathic pain and other symptoms should be based on dichotomous participant‐reported outcomes of confirmed clinical utility.
Comparison between active treatments
Without knowing whether morphine is effective, there seems little point in comparing it with other treatments.
What's new
| Date | Event | Description |
|---|---|---|
| 28 May 2019 | Amended | Contact details updated. |
| 11 October 2017 | Review declared as stable | See Published notes. |
History
Protocol first published: Issue 4, 2015 Review first published: Issue 5, 2017
| Date | Event | Description |
|---|---|---|
| 22 May 2017 | Amended | Amended Date next stage expected. |
Notes
No new studies likely to change the conclusions are expected. Therefore, this review has now been stabilised following discussion with the authors and editors. If appropriate, we will update the review if new evidence likely to change the conclusions is published, or if standards change substantially which necessitate major revisions.
Acknowledgements
Institutional support was provided by the Oxford Pain Relief Trust.
Cochrane Review Group funding acknowledgement: this project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Pain, Palliative and Supportive Care Review Group (PaPaS). The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Methodological considerations for chronic pain
There have been several changes in how the efficacy of conventional and unconventional treatments is assessed in chronic painful conditions. The outcomes are now better defined, particularly with new criteria for what constitutes moderate or substantial benefit (Dworkin 2008); older trials may only report participants with 'any improvement'. Newer trials tend to be larger, avoiding problems from the random play of chance. Newer trials also tend to be of longer duration, up to 12 weeks, and longer trials provide a more rigorous and valid assessment of efficacy in chronic conditions. New standards have evolved for assessing efficacy in neuropathic pain, and we are now applying stricter criteria for the inclusion of trials and assessment of outcomes, and are more aware of problems that may affect our overall assessment. We summarised some of the recent insights that must be considered in this new review below.
Pain results tend to have a U‐shaped distribution rather than a bell‐shaped distribution. This is true in acute pain (Moore 2011a; Moore 2011b), back pain (Moore 2010e), arthritis (Moore 2010d), and fibromyalgia (Straube 2010); in all cases, average results usually describe the experience of almost no‐one in the trial. Data expressed as averages are potentially misleading, unless they can be proven to be suitable.
As a consequence, we have to depend on dichotomous results (the individual either has or does not have the outcome) usually from pain changes or patient global assessments. The Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) group has helped with their definitions of minimal, moderate, and substantial improvement (Dworkin 2008). In arthritis, trials of less than 12 weeks' duration, and especially those shorter than eight weeks, overestimate the effect of treatment (Moore 2010d); the effect is particularly strong for less effective analgesics, and this may also be relevant in neuropathic‐type pain.
The proportion of people with at least moderate benefit can be small, even with an effective medicine, falling from 60% with an effective medicine in arthritis to 30% in fibromyalgia (Moore 2009; Moore 2010d; Moore 2013a; Moore 2014b; Straube 2008; Sultan 2008). One Cochrane Review of pregabalin in neuropathic pain and fibromyalgia demonstrated different response rates for different types of chronic pain (higher in painful diabetic neuropathy and postherpetic neuralgia and lower in central pain and fibromyalgia) (Moore 2009). This indicates that different neuropathic pain conditions should be treated separately from one another, and that pooling should not be done unless there are good reasons for doing so.
Individual patient analyses indicate that people who get good pain relief (moderate or better) have major benefits in many other outcomes, affecting quality of life in a significant way (Moore 2010c; Moore 2014a).
Imputation methods such as last observation carried forward (LOCF), used when participants withdraw from clinical trials, can overstate drug efficacy especially when adverse event withdrawals with drug are greater than those with placebo (Moore 2012).
Appendix 2. Brand names for morphine products worldwide
| A: | Actiskenan, Algedol, Amidiaz, Analmorph, Anafil, Analfin, Anamorph, Astramorph, Avinza |
| C: | Capros, Cloruro Morfico Braun, Compensan, Continue, Contalgin |
| D: | DepoDur, Depolan, Dimorf, Dolcontin, Dolo Moff, Doloral, DOLQ, Doltard, Dulcontin, Duralgin, Duralmor, Duramorph |
| E: | Embeda, Epidural, Epimorph |
| F: | Filnarine |
| G: | GNO, Graten, G‐Morphine |
| I: | Infumorph (morphine for intrathecal microinfusion by implanted device) |
| K: | Kadian, Kapabloc, Kapanol |
| L: | LA Morph, Loceptin, Longphine |
| M: | M‐Beta, M‐Dolor, M‐Eslon, M‐Long, M‐Retard, M‐Stada, Malfin, Maxidon, MCR, Meconium, Meslon, Micro‐Morphine, MIR, Mogetic, Morapid, Moraxen, Morcap, Moretal, Morfenil, Morficontin, Morfin, Morfin Meda, Morfina, Morin, Morph, Morphanton, Morphex, Morphgesic, Morphin, Morphini, Morphinum, Morphiphar, Morphitec, Morphium, Morstel, MOS, Moscontin, Morstel, Mortificontin, MS Contin, MS Direct, MS Long, MS Mono, MS/L, MS/S, MSI, MSIR, MSP, MSR, MST Continus, MST Unicontinus, Mundidol, MXL |
| N: | Neocalmans, Noceptin, Novo‐Morphine |
| O: | Oblioser, Oglos, OMS Concentrate, Onkomorphin, Opitard, Opsalvina, Oramorph, Ordine |
| P: | Painbreak, PMS‐Morphine |
| Q: | Q‐Med Morphine |
| R: | RA Morph, Ratio‐Morphine, Relimal, Relipain, Repriadol, Rescudose, RMS, Roxanol |
| S: | S‐Morphine, Sevre‐Long, Sevredol, Skenan, Slo‐Morph, Slovalgin, SRM‐Rhotard, Statex, Stellaphine, Stellorphinad, Stellorphine, Substitol |
| U: | Uni Mist |
| V: | Vendal |
| Z: | Zomorph |
Appendix 3. Search strategy for CENTRAL via CRSO
MESH DESCRIPTOR Neuralgia EXPLODE ALL TREES (718)
MESH DESCRIPTOR Peripheral Nervous System Diseases EXPLODE ALL TREES (2963)
MESH DESCRIPTOR Somatosensory Disorders EXPLODE ALL TREES (796)
((pain* or discomfort*) adj10 (central or complex or nerv* or neuralg* or neuropath*)):TI,AB,KY (3931)
((neur* or nerv*) adj6 (compress* or damag*)):TI,AB,KY (732)
#1 or #2 or #3 or #4 or #5 (7377)
MESH DESCRIPTOR Morphine EXPLODE ALL TREES (3659)
(Morphine or Dulcontin or Kapanol or Morcap or (MST Continus) or Oramorph or Roxanol or Sevredol or Slo‐Morph or Zomorph):TI,AB,KY (8571)
#7 or #8 (8571)
#6 and #9 (412)
Appendix 4. Search strategy for MEDLINE via Ovid
exp NEURALGIA/ (16807)
exp PERIPHERAL NERVOUS SYSTEM DISEASES/ (131463)
exp SOMATOSENSORY DISORDERS/ (18817)
((pain* or discomfort*) adj10 (central or complex or nerv* or neuralg* or neuropath*)).mp. (46313)
((neur* or nerv*) adj6 (compress* or damag*)).mp. (53946)
1 or 2 or 3 or 4 or 5 (210458)
Morphine/ (36140)
(Morphine or Dulcontin or Kapanol or Morcap or (MST Continus) or Oramorph or Roxanol or Sevredol or Slo‐Morph or Zomorph).mp. (50379)
7 or 8 (50379)
randomized controlled trial.pt. (447705)
randomized.ab. (340542)
placebo.ab. (168289)
drug therapy.fs. (1934054)
randomly.ab. (234814)
trial.ab. (356016)
groups.ab. (1462628)
10 or 11 or 12 or 13 or 14 or 15 or 16 (3658507)
6 and 9 and 17 (1985)
limit 18 to "all adult (19 plus years)" (797)
Appendix 5. Search strategy for Embase via Ovid
exp NEURALGIA/ (93428)
exp PERIPHERAL NERVOUS SYSTEM DISEASES/ (62857)
exp SOMATOSENSORY DISORDERS/ (84384)
((pain* or discomfort*) adj10 (central or complex or nerv* or neuralg* or neuropath*)).mp. (95312)
((neur* or nerv*) adj6 (compress* or damag*)).mp. (81538)
1 or 2 or 3 or 4 or 5 (332505)
morphine/ (96373)
(Morphine or Dulcontin or Kapanol or Morcap or MST Continus or Oramorph or Roxanol or Sevredol or Slo‐Morph or Zomorph).mp. (109025)
7 or 8 (109025)
random*.ti,ab. (1179013)
factorial*.ti,ab. (29726)
(crossover* or cross over* or cross‐over*).ti,ab. (87323)
placebo*.ti,ab. (253712)
(doubl* adj blind*).ti,ab. (177963)
assign*.ti,ab. (308446)
allocat*.ti,ab. (113656)
RANDOMIZED CONTROLLED TRIAL/ (479705)
DOUBLE‐BLIND PROCEDURE/ (141056)
CROSSOVER PROCEDURE/ (55223)
10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19 (1656149)
6 and 9 and 20 (1684)
limit 21 to (adult <18 to 64 years> or aged <65+ years>) (807)
Appendix 6. GRADE: criteria for assigning grade of evidence
The GRADE system uses the following criteria for assigning a quality level to a body of evidence (Chapter 12, Higgins 2011).
High: randomised trials; or double‐upgraded observational studies.
Moderate: downgraded randomised trials; or upgraded observational studies.
Low: double‐downgraded randomised trials; or observational studies.
Very low: triple‐downgraded randomised trials; or downgraded observational studies; or case series/case reports.
Factors that may decrease the quality level of a body of evidence are:
limitations in the design and implementation of available studies suggesting high likelihood of bias;
indirectness of evidence (indirect population, intervention, control, outcomes);
unexplained heterogeneity or inconsistency of results (including problems with subgroup analyses);
imprecision of results (wide confidence intervals);
high probability of publication bias.
Factors that may increase the quality level of a body of evidence are:
large magnitude of effect;
all plausible confounding would reduce a demonstrated effect or suggest a spurious effect when results show no effect;
dose‐response gradient.
Appendix 7. Summary of efficacy in individual studies
| Study | Treatment | Pain outcome | Other efficacy outcome |
| Gilron 2005 | Morphine: target 120 mg daily Morphine + gabapentin: target 60 mg + 2400 mg daily Gabapentin: target 3200 mg daily Placebo: lorazepam 1.6 mg daily n = 57 (cross‐over) |
Participants who completed a treatment period with at least moderate pain relief on maximum tolerated dose Morphine: 35/44 (80%) Gabapentin: 27/44 (61%) Placebo (lorazepam): 13/42 (31%) Question: global pain relief assessed in response to questions from the research nurse on the following scale: pain worse, no relief, slight relief, moderate relief, a lot of relief, or complete relief. |
On maximum tolerated dose, mean ± SE: Mean PI (0‐10 NRS) [Baseline: 5.72 ± 0.23] Morphine: 3.70 ± 0.34 Gabapentin: 4.15 ± 0.33 Placebo: 4.49 ± 0.34 McGill Pain Questionnaire PI (10‐cm VAS) [Baseline: 5.0 ± 0.4] Morphine: 3.3 ± 0.4 Gabapentin: 3.5 ± 0.4 Placebo: 3.9 ± 0.4 McGill Pain Questionnaire Present PI (0‐3) [Baseline: 2.40 ± 0.16] Morphine: 1.57 ± 0.16 Gabapentin: 1.64 ± 0.16 Placebo: 2.07 ± 0.16 SF‐36 Bodily Pain (0‐100) [Baseline: 52.1 ± 2.7] Morphine: 64.4 ± 2.9 Gabapentin: 65.6 ± 2.9 Placebo: 56.0 ± 3.0 |
| Gilron 2015 | Morphine: target 100 mg daily Nortriptyline: target 100 mg daily Morphine + nortriptyline: target 100 mg + 100 mg daily n = 51 (received treatment) (cross‐over) |
Participant‐reported pain relief ≥ 30%: Morphine: 24/47 (51.3%) Nortriptyline: 30/45 (65.8%) Participant‐reported pain relief ≥ 50%: Morphine: 10/47 (25.6%) Nortriptyline: 17/45 (36.8%) PGIC much or very much improved: Participants with at least moderate relief on maximum tolerated dose Morphine: 74.4% Nortriptyline: 70.7% Reports "Overall effect of treatment on global pain relief scores was not significant (P = 0.17)." PGIC very much improved: Not reported |
Average daily pain throughout trial ± SE (0‐10 NRS) Morphine: 3.4 ± 0.4 Nortriptyline: 3.1 ± 0.4 Global pain relief (pain worse, no relief, slight relief, moderate relief, a lot of relief, or complete relief) Worst pain (0‐10 NRS) Morphine: 4.4 ± 0.4 Nortriptyline: 4.1 ± 0.4 Nocturnal pain (0‐10 NRS) Morphine: 2.2 ± 0.4 Nortriptyline: 2.0 ± 0.4 |
| Huse 2001 | Morphine: target 70‐300 mg daily Placebo n = 12 (cross‐over) |
> 25% and ≤ 50% PI reduction Morphine: 1/12 (8%) Placebo: 1/12 (8%) > 50% PI reduction Morphine: 5/12 (42%) Placebo: 1/12 (8%) |
At end of treatment phase: Mean PI ± SD (10‐cm VAS) [Baseline: 4.65 ± 1.06] Morphine: 3.26 ± 1.59 Placebo: 3.99 ± 1.23 Data also for sensory pain and affective pain components, not required. |
| Khoromi 2007 | Morphine: target dose 90 mg daily Nortriptyline: target dose 100 mg daily Morphine + nortriptyline: target dose 90 mg + 100 mg daily Placebo (benztropine) 0.25‐1 mg daily n = 55 (cross‐over) |
Global pain relief (moderate + a lot + complete relief): Morphine: 13/32 (41%) Nortriptyline: 12/31 (39%) Placebo: 11/28 (39%) Global pain relief (a lot + complete relief): Morphine: 8/32 (25%) Nortriptyline: 10/31 (32%) Placebo: 5/28 (18%) |
Average leg pain (scale 0‐10): [Baseline 4.9 ± 2.4] Morphine: 3.4 ± 2.8 (7% reduction) Nortriptyline: 3.0 ± 2.7 (14% reduction) Placebo: 3.7 ± 2.7 |
| Wu 2008 | Morphine SR: target dose 180 mg daily Mexiletine: target dose 1200 mg daily Placebo n = 60 (cross‐over) |
33% decrease in PI: Morphine: 33/50 (66%) Mexiletine: 16/42 (38%) Placebo: 19/43 (44%) 50% decrease in PI: Morphine: 23/50 (46%) Mexiletine: 11/42 (26%) Placebo: 13/43 (30%) |
Any pain‐related outcome: Average reduction in PI from baseline (0‐10): Morphine: 2.8 (95% CI 2.3 to 3.4) Mexiletine: 1.5 (95% CI 0.9 to 2.2) Placebo: 1.4 (95% CI 0.6 to 2.2) No difference between groups for overall functional activity and pain‐related interference with daily activities. |
CI: confidence interval; n: number of participants in study; NRS: numerical rating scale; PI: pain intensity; PGIC: Patient Global Impression of Change; SD: standard deviation; SE: standard error of the mean; SF‐36: 36‐item Short Form quality of life instrument; SR: sustained release; VAS: visual analogue scale.
Appendix 8. Summary of adverse events and withdrawals in individual studies
| Study | Treatment | Adverse events | Withdrawals |
| Gilron 2005 | Morphine: target 120 mg daily Morphine + gabapentin: target 60 mg + 2400 mg daily Gabapentin: target 3200 mg daily Placebo: lorazepam 1.6 mg daily n = 57 (cross‐over) |
Any AE: Did not report number of participants with any AE Reported only moderate to severe AEs occurring in > 5% for any treatment SAEs: not reported Specific AEs: morphine (%); gabapentin (%); placebo (%) Constipation: 38.6; 2.1; 4.7 Sedation: 15.9; 8.3; 14.0 Dry mouth: 4.6; 6.3; 0 Insomnia: 2.3; 8.3; 7.0 Vomiting: 0; 0; 0 Pruritus: 6.8; 2.1; 0 Cognitive dysfunction: 2.3; 2.1; 2.3 Dizziness: 0; 2.1; 0 Nausea: 4.6; 0; 0 Ataxia: 4.6; 0; 0 Oedema: 4.6; 0; 2.3 Anxiety: 2.3; 8.3; 0 Blurry vision: 4.6; 0; 2.3 Diarrhoea: 0; 2.1; 2.3 Restless legs: 0; 0; 0 Headache: 2.3; 0; 2.3 Decreased appetite: 4.6; 0; 0 |
Randomised: 57 Total withdrawals: 16 Withdrew at end of first treatment period: 13 Withdrew at end of second period: 3 (AEs ‐ included in efficacy analysis) Did not report treatment at/before withdrawal. |
| Gilron 2015 | Morphine: target 100 mg daily Nortriptyline: target 100 mg daily Morphine + nortriptyline: target 100 mg + 100 mg daily n = 51 (received treatment) (cross‐over) |
Any AEs: Did not report number of participants with any AE. SAEs: Morphine: 1/47 Morphine + nortriptyline: 1/45 Specific AEs: Morphine (%); nortriptyline (%) Dry mouth: 12.8; 48.8 Constipation: 46.2; 4.7 Somnolence: 18.0; 7.0 Nausea: 2.6; 0 Dizziness: 7.7; 2.3 Insomnia: 5.1; 2.3 Fatigue: 7.7; 2.3 Anorexia: 5.1; 0 Erectile dysfunction: 0; 7.0 Pruritus: 5.1; 0 Blurry vision: 2.6; 2.3 Weight gain: 0; 0 Urinary retention: 0; 0 Inability to concentrate: 2.6; 0 Diarrhoea: 0; 0 Headache: 0; 0 Vomiting: 0; 0 Excessive sweating: 0; 0 Reflux: 0; 0 Weakness: 0; 0 |
Withdrawals due to AEs: Morphine: 8/47 (1 SAE) Nortriptyline: 3/45 Morphine + nortriptyline: 5/44 (1 SAE) Withdrawals due to other reasons: Morphine: 1/47 Nortriptyline: 1/45 Morphine + nortriptyline: 1/44 Withdrawals LoE: None reported |
| Huse 2001 | Morphine: target 70‐300 mg daily Placebo n = 12 (cross‐over) |
9 specific AEs reported, but number of participants experiencing any AE or specific AEs not reported. Data given for intensity of AEs. No significant difference between treatments except for constipation. |
Withdrawals (any cause): Morphine: 0/12 Placebo: 0/12 |
| Khoromi 2007 | Morphine: target dose 90 mg daily Nortriptyline: target dose 100 mg daily Morphine + nortriptyline: target dose 90 mg + 100 mg daily Placebo (benztropine): 0.25‐1 mg daily n = 55 (cross‐over) |
Any AE: Reported for 28 study completers only, and AEs with incidence > 5% for any treatment. Morphine: 26/28 Nortriptyline: 19/28 Placebo: 14/28 SAE: None reported. Specific AEs: In completers (n = 28) Drowsiness Morphine: 7/28 Nortriptyline: 2/28 Placebo: 1/28 Dizziness Morphine: 4/28 Nortriptyline: 2/28 Placebo: 1/28 Decreased appetite Morphine: 2/28 Nortriptyline: 0/28 Placebo: 0/28 |
All‐cause withdrawals: Morphine: 9/41 Nortriptyline: 3/34 Placebo: 9/39 AE withdrawals: Morphine: 5/41 Nortriptyline: 2/34 Placebo: 1/39 LoE withdrawals: Morphine: 0/41 Nortriptyline: 0/34 Placebo: 3/39 Other withdrawals: Morphine: 4/41 Nortriptyline: 1/34 Placebo: 5/39 (1 non‐compliance ‐ took opioid) |
| Wu 2008 | Morphine SR: target dose 180 mg daily Mexiletine: target dose 1200 mg daily Placebo n = 60 (cross‐over) |
Any AE: Morphine: 27/50 Mexiletine: 7/42 Placebo: 7/43 SAE: None reported. Specific AEs: Constipation Morphine: 17/50 (14%) Mexiletine: 2/42 (5%) Placebo: 2/43 (5%) Nausea Morphine: 4/50 (8%) Mexiletine: 0/42 (0%) Placebo: 1/43 (2%) Drowsiness Morphine: 9/50 (18%) Mexiletine: 4/42 (10%) Placebo: 3/43 (7%) Dizziness Morphine: 2/50 (4%) Mexiletine: 2/42 (5%) Placebo: 2/43 (5%) |
All‐cause withdrawals: Morphine: 10/50 Mexiletine: 6/42 Placebo: 5/43 35 participants completed all treatment phases. |
AE: adverse event; LoE: lack of efficacy; n: number of participants in study; SAE: serious adverse event.
Data and analyses
Comparison 1. Morphine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Moderate improvement | 4 | 263 | Risk Difference (M‐H, Fixed, 95% CI) | 0.27 [0.16, 0.38] |
| 2 Substantial improvement | 3 | 177 | Risk Difference (M‐H, Fixed, 95% CI) | 0.15 [‐0.02, 0.32] |
| 3 All‐cause withdrawal | 4 | 289 | Risk Difference (M‐H, Fixed, 95% CI) | 0.04 [‐0.04, 0.12] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Gilron 2005.
| Methods |
Allocation: randomised. Control: placebo and active comparator. Blinding: double‐blind. Arm: 4‐arm cross‐over. Centre: single. Study dates: recruitment February 2001 to November 2003. Setting and location: Kingston General Hospital, Ontario, Canada, |
|
| Participants |
Inclusion criteria: people with painful diabetic neuropathy or postherpetic neuralgia; moderate pain for ≥ 3 months; aged 18‐89 years. Exclusion criteria: allergy to study medications, another painful condition, recent myocardial infarction, unstable angina, congestive heart failure, any central neurological disorder, seizures, mood disorder, drug or alcohol abuse, pregnancy, lactation. Baseline characteristics n: 57. Gender: F 25, M 32. Age: mean 64 years (range 40‐81). Number randomised: 57. Number completed: 41. Baseline pain intensity: 5.7/10 (SD 1.7). |
|
| Interventions | Morphine: target dose 120 mg daily. Morphine + gabapentin: target dose 60 mg + 2400 mg daily. Gabapentin: target dose 3200 mg daily. Placebo: lorazepam 1.6 mg. Duration: 4 × 5 weeks. Titration to maximum tolerated or ceiling dose over first 3 weeks. Lower doses with slower titration for older/smaller people. 4th week on maximum tolerated dose. 5th week was 4‐day dose tapering + 3‐day complete washout. Additional analgesics: non‐opioid drugs other than gabapentin were permitted at a steady dose throughout the trial. |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Oxford Quality Score: R2, DB2, W1. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "With the use of a balanced Latin‐square crossover design, 23 patients were allocated, in a double‐blind, randomized fashion, to one of four treatment sequences." |
| Allocation concealment (selection bias) | Low risk | Comment: medication prepared remotely according to random schedule and assigned consecutive numbers. Allocation in sequence of enrolment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Medications were placed in blue and gray gelatin capsules by the investigational pharmacist in order to maintain double‐blind conditions. Patients received identical‐appearing blue and gray capsules during each treatment regimen in accord with a double‐dummy design." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: participant‐reported outcomes, participant blinded to intervention. |
| Incomplete outcome data (attrition bias) All outcomes | High risk |
Quote: "57 underwent randomization, and a total of 16 withdrew during the treatment periods ‐ 13 before completing the second treatment period, and 3 because of adverse effects but after completing at least two treatment periods (these 3 were therefore included in the efficacy analysis). Forty‐one patients completed the trial." Comment: 28% of participants withdrew. |
| Selective reporting (reporting bias) | Unclear risk | Comment: in the methods, the authors planned to record and report global pain relief. No results reported, but global results probably used to test for difference (0.05 level) before pair‐wise comparisons made. All other outcomes reported. |
| Size | High risk | Comment: 57 participants began, but number completing < 50. |
Gilron 2015.
| Methods |
Allocation: randomised. Control: active comparator. Blinding: double‐blind. Arm: 3‐arm cross‐over. Centre: single. Study dates: January 2010 to May 2014. Setting and location: Kingston General Hospital, Ontario, Canada. |
|
| Participants |
Inclusion criteria: participants with peripheral neuropathic pain (score ≥ 4/10); postherpetic neuralgia, post‐traumatic neuropathy, or chemotherapy‐induced peripheral neuropathy for > 6 months; daily pain (score 3/10 for at least 6 months); aged 18‐89 years. Exclusion criteria: neuropathy attributable to hypothyroidism, vitamin B12 deficiency, connective tissue disease, and amyloidosis; major organ system disease; cardiovascular autonomic neuropathy; baseline postural hypotension (20 mmHg); ongoing sedation or ataxia; men with ongoing urinary retention; hypersensitivity to any of the study medications, and painful conditions as severe as their neuropathic pain; psychiatric or substance abuse disorder. Baseline characteristics n: 52 (1 withdrew before treatment). Gender: F 14, M 38. Age: mean 66 years (range 49‐80). Number randomised: 52. Number completed: 36. Baseline pain intensity: 5.3/10 (SD 1.4). |
|
| Interventions | Morphine: target dose 100 mg daily. Nortriptyline: target dose 100 mg daily. Morphine + nortriptyline: target dose 100 mg + 100 mg daily. Duration: 3 × 6 weeks. Titration to maximum tolerated dose over 24 days, maintained at the maximum dose for 1 week, then tapered over 1 week, followed by a 4‐day washout. Additional analgesics: participants allowed to continue gabapentin, pregabalin, NSAIDs, or paracetamol (acetaminophen) at a steady dose. |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Stated "all enrolled patients included in the efficacy analyses", but did not mention imputation. Oxford Quality Score: R2, DB2, W1. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "as per a balanced Latin square crossover design, patients were allocated, in a double‐blind randomized fashion, to 1 of 3 possible sequences." |
| Allocation concealment (selection bias) | Low risk | Quote: "A trial pharmacist prepared a concealed allocation schedule by computer randomizing these 3 sequences, in blocks of 3, to a consecutive number series. Patients were assigned the next consecutive number, and the corresponding sequence of study medications was dispensed." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Two sets of medications were encapsulated in yellow (nortriptyline) and white (sustained‐release morphine) capsules. Active drug and placebo capsules for each drug were identical in appearance across treatments." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: participants blinded and participant‐reported outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: > 30% attrition, almost all due to adverse events. Imputation method unclear. |
| Selective reporting (reporting bias) | Low risk | Comment: all planned outcomes in the methods were reported in the results. |
| Size | High risk | Comment: 52 participants began (51 took medication), but number completing < 50. |
Huse 2001.
| Methods |
Allocation: randomised. Control: placebo. Blinding: double‐blind. Arm: 2‐arm cross‐over. Centre: single. Setting and location: Pain Clinic, University Hospital, Tubingen, Germany. |
|
| Participants |
Inclusion criteria: leg amputees with phantom limb pain, pain intensity score ≥ 3/10 VAS, aged 18‐75 years. Exclusion criteria: neurological or psychiatric disorders, severe illness, pregnancy, breastfeeding, morphine‐specific sensitivities. Baseline characteristics n: 12. Gender: F 2, M 10. Age: mean 51 years (range 30‐71). Number randomised: 12. Number completed: 12. Baseline pain intensity: 4.7/10 (SD 1.1). |
|
| Interventions | Morphine: target 70‐300 mg. Placebo: oral glucose tablet. Duration: 2 × 4‐week treatment periods, preceded by 4‐week treatment‐free baseline period. Each treatment phase initiated by IV test infusion (same intervention as treatment phase + metoclopramide 5 mg), and treatments separated by 1‐ to 2‐week taper and washout. Starting dose of morphine not reported, possibly calculated from IV infusion. Titration allowed. 11 participants took 70‐160 mg daily; 1 took 300 mg daily. Additional analgesics: use of all analgesic and psychotropic medication was noted in the pain diary. |
|
| Outcomes |
Primary outcomes
At the end of each treatment period: Pain‐related Self‐assessment Scale and Self‐rating Depression Scale. Secondary outcomes
|
|
| Notes | Oxford Quality Score: R2, DB2, W1. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: states "randomized", but method used to generate random sequence not described. |
| Allocation concealment (selection bias) | Low risk | Quote: "a physician who had no contact with the patients was responsible for the randomized distribution of the patients to the experimental conditions and kept the code." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Tablets were put into exactly identical pills by the pharmacy of the hospital." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: participant‐reported outcomes, participant blinded to intervention. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: all followed up and 0 lost during treatment phase. |
| Selective reporting (reporting bias) | Low risk | Comment: all planned outcomes in the methods were reported in the results. |
| Size | High risk | Comment: 12 participants (n < 50). |
Khoromi 2007.
| Methods |
Allocation: randomised. Control: placebo and active comparator. Blinding: double‐blind. Arm: 4‐arm, 4‐period cross‐over. Centre: single. Study dates: February 2001 to March 2004. Setting and location: NIH Clinical Center, Bethesda, Maryland, USA. |
|
| Participants |
Inclusion criteria: lumbar radiculopathy (diagnostic criteria described in paper), average leg pain score ≥ 4/10, willingness to stop other medications, aged 18‐65 years. Exclusion criteria: serious medical illness; prostatic disease; pregnancy or lactation; history of narcotic or alcohol abuse; narrow angle glaucoma; seizures; fibromyalgia; severe pain other than back or legs; polyneuropathy and peripheral vascular disease associated with symptoms of numbness or burning; allergy to morphine, nortriptyline, or benztropine; multisomatoform disorder; unwilling to taper off current opioids. Baseline characteristics n: 55. Gender: F 25, M 30. Age: median 53 years (range 19‐65). Number randomised: 55 Number completed: 28. Baseline pain intensity: 5.0/10 (SD 2.3) (average overall). |
|
| Interventions | Morphine: target dose 90 mg daily. Nortriptyline: target dose 100 mg daily. Morphine + nortriptyline: target dose 90 mg + 100 mg. Placebo (benztropine) 0.25 mg to 1 mg daily. Duration: 4 × 9 weeks; 5 weeks of dose escalation, 2 weeks at maximum tolerated dose, 2 weeks of dose tapering. Additional analgesics: no opioids, SSRIs and tricyclic medications outside of protocol during study; no changes to other stable analgesic medication regimen; anti‐inflammatory medications and paracetamol (acetaminophen) were allowed as rescue medications. |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Efficacy analyses included only participants who had completed ≥ 2 treatment periods (34 participants; 28 participants completed whole study)
No information on how missing data were handled (withdrawals or missing points). Oxford Quality Score: R2, DB2, W1. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was performed by the NIH Pharmaceutical Development Service. Patients were assigned by random numbers within blocks of four to one of four treatment sequences specified by a Latin square." |
| Allocation concealment (selection bias) | Unclear risk | Comment: method used to conceal allocation not described. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk |
Quote: "Patients and staff were blinded to the randomisation order." Comment: double‐dummy method used, with 'active' placebo to mimic adverse events. Tablets were colour coded by pharmacists for the active treatments and active placebos. Note: switches from calling them 'pills' to 'capsules' mid‐way through. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: participant‐reported outcomes, participant blinded to intervention. |
| Incomplete outcome data (attrition bias) All outcomes | High risk |
Comment: analysis for study completers (n = 28) and those completing ≥ 2 treatment periods (n = 34). Comment: no information for how missing data points handled. |
| Selective reporting (reporting bias) | Low risk | Comment: all planned outcomes in the methods were reported in the results. |
| Size | High risk | Comment: 55 participants began, but number completing < 50. |
Wu 2008.
| Methods |
Allocation: randomised. Control: active comparator and placebo. Blinding: double‐blind. Arm: 3‐arm, 3‐period cross‐over. Centre: single. Study Dates: 1999‐2003. Setting and location: university hospital, USA. |
|
| Participants |
Inclusion criteria: adults with persistent postamputation pain ≥ 3/10 for ≥ 6 months. Exclusion criteria: allergy to morphine or mexiletine, cardiac conduction defects, myocardial infraction last 3 months, pulmonary disease, alcohol or substance abuse, seizures, dementia, pregnancy, lactation, hepatic disease, haematological diseases. Baseline characteristics n: 60. Gender: F 13, M 47. Age: mean 63 years (SD 16) Number randomised: 60. Number completed: 35. Baseline pain intensity: about 7/10. |
|
| Interventions | Morphine SR: target dose 180 mg daily. Mexiletine: target dose 1200 mg daily. Placebo. Duration: 3 × 9 weeks. Each treatment period included 4‐week titration (dose increased every 3‐4 days), 2‐week maintenance, 2‐week drug taper and 1‐week washout. Additional analgesics: all opioids, antiepileptic drugs, benzodiazepines, mexiletine, baclofen, other neuroleptic pain medicine discontinued ≥ 2 weeks before start of study. Paracetamol and NSAIDs allowed throughout. |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | No mention of imputation for missing data. Separate analysis for completers of all 3 periods gave similar results. Oxford Quality Score: R2, DB2, W1. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The randomization sequence was computer generated by a biostatistician ..." |
| Allocation concealment (selection bias) | Low risk | Quote: "the sequence of drug and placebo treatment periods for each subject was provided in sealed enveloped to the investigational pharmacy and the monitoring committee." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk |
Quote: "Patients and investigators were unaware of the contents of the study drug and randomisation scheme. The investigational pharmacy manufactured the study drug as gelatin capsules, which are readily dissolved in the upper gastrointestinal tract within 15 min." Quote: "sustained release morphine tablets were placed in a sealed capsules along with an inert powder (lactose) to prevent the subject from recognizing the medication. Mexiletine and placebo were similarly packaged in sealed capsules that were identical in appearance." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: participant‐reported outcomes, participant blinded to intervention. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: 20% attrition ‐ reasons per group not given. |
| Selective reporting (reporting bias) | Low risk | Comment: all planned outcomes in the methods were reported in the results. |
| Size | High risk | Comment: 60 participants began, but number completing < 50. |
DB: double blind (Oxford Quality Score); F: female; IV: intravenous; M: male; n: number of participants in study; NRS: numerical rating scale; NSAID: nonsteroidal anti‐inflammatory drug; R: randomised (Oxford Quality Score); SD: standard deviation; SR: sustained release; SSRI: selective serotonin reuptake inhibitor; VAS: visual analogue scale; W: withdrawals (Oxford Quality Score).
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Attal 2002 | Open‐label intravenous morphine to determine maximum tolerated dose. Double‐blind phase using predetermined maximum dose or placebo for single infusion. |
| Dellemijn 1994 | Treatment duration only 1 week for each intervention. |
| Domzal 1986 | Single dose, effect approximately 20 hours. |
| Eide 1994 | Single doses, 8 participants. |
| Galer 2005 | Report of 3 trials in non‐neuropathic pain, morphine combination used. |
| Harke 2001 | Neuropathic responsive to spinal cord stimulation, morphine vs placebo, 8 days, inadequate dose of morphine. |
| Jadad 1992 | Single doses. |
| Kalman 2002 | Single‐blind, single doses. |
| Nalamachu 2013 | No morphine‐only treatment arm. |
| Patarica‐Huber 2011 | No morphine‐only treatment arm. |
| Persson 1998 | Single dose/infusion. |
| Raja 2002 | 26 (41%) participants switched from morphine to methadone. |
| Rocha 2014 | Morphine only as 'acute pain medication,' presumably for breakthrough pain. |
| Rowbotham 1991 | Single doses. |
| Serinken 2016 | Single doses. |
| Siddall 2000 | 15 participants, cross‐over, single doses, investigator‐assessed pain intensity. |
| Wasan 2006 | Single doses, 3‐hour observations. |
| Watt 1996 | Single‐blind. |
| Wu 2002 | Single doses, observations over 70 minutes. |
| Xiong 2008 | Not indexed as blinded in Embase. |
Differences between protocol and review
We made changes to the search strategy and reran the searches in September 2016 and February 2017 to be more specific, to ensure that searches were up‐to‐date, and to check with other recently published reviews.
We removed CRPS Type I from the list of possible neuropathic pain conditions because there is some controversy over whether this fulfils new diagnostic criteria. In the event, we found no studies in CRPS.
Tiers of evidence is not now used in favour of an updated version of GRADE that performs essentially the same function. To continue with both methods seemed redundant.
We added 'chronic' to the title, and included selective reporting bias in risk of bias assessment. We had omitted assessment of blinding for performance bias in the protocol (although all studies had to be double‐blind); this has been corrected.
We report risk difference rather than risk ratio.
Contributions of authors
SD and RAM wrote the protocol.
SD, TC, and JC searched for and selected studies for inclusion and carried out data extraction.
RAM, SD, TC, and PW generated the initial draft of the review
All review authors were involved in the analysis and in writing the full review.
Sources of support
Internal sources
-
Oxford Pain Relief Trust, UK.
General institutional support
External sources
-
The National Institute for Health Research (NIHR), UK.
NIHR Cochrane Programme Grant: 13/89/29 ‐ Addressing the unmet need of chronic pain: providing the evidence for treatments of pain
Declarations of interest
TC: none known.
JC: none known.
PW: none known.
SD: none known.
DBC: none known; DBC is a specialised pain physician and has managed patients with chronic pain.
DA: is a specialist pain physician and manages patients with neuropathic pain. He has received lecture fees from Grünenthal (2014, 2015) and Pfizer (2016).
PC: none known; PC is a specialist pain physician and manages patients with neuropathic pain. PC received support from Boston Scientific (2014) for travel and accommodation at a scientific meeting; Boston Scientific does not market drugs.
RAM: has received grant support from Grünenthal relating to individual patient level analyses of trial data regarding tapentadol in osteoarthritis and back pain (2015). He has received honoraria for attending boards with Menarini concerning methods of analgesic trial design (2014), with Novartis (2014) about the design of network meta‐analyses, and RB on understanding pharmacokinetics of drug uptake (2015). He has received honoraria from Omega Pharma (2016) and Futura Pharma (2016) for providing advice on trial and data analysis methods.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Gilron 2005 {published data only}
- Gilron I, Bailey JM, Dongsheng T, Holden RR, Weaver DF, Houlden RL. Morphine, gabapentin, or their combination for neuropathic pain. New England Journal of Medicine 2005;352(13):1324‐34. [DOI: 10.1056/NEJMoa042580] [DOI] [PubMed] [Google Scholar]
Gilron 2015 {published data only}
- Gilron I, Tu D, Holden RR, Jackson AC, DuMerton‐Shore D. Combination of morphine with nortriptyline for neuropathic pain. Pain 2015;156(8):1440‐8. [DOI: 10.1097/j.pain.0000000000000149] [DOI] [PubMed] [Google Scholar]
Huse 2001 {published data only}
- Huse E, Larbig W, Flor H, Birbaumer N. The effect of opioids on phantom limb pain and cortical reorganization. Pain 2001;90(1‐2):47‐55. [DOI: 10.1016/S0304-3959(00)00385-7] [DOI] [PubMed] [Google Scholar]
Khoromi 2007 {published data only}
- Khoromi S, Cui L, Nackers L, Max MB. Morphine, nortriptyline and their combination vs. placebo in patients with chronic lumbar root pain. Pain 2007;130(1‐2):66‐75. [DOI: 10.1016/j.pain.2006.10.029] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 2008 {published data only}
- Wu CL, Agarwal S, Tella PK, Klick B, Clark MR, Haythornthwaite JA, et al. Morphine versus mexiletine for treatment of postamputation pain. Anesthesiology 2008;109(2):289‐96. [DOI: 10.1097/ALN.0b013e31817f4523] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Attal 2002 {published data only}
- Attal N, Guiriman F, Brasseur L, Gaude V, Chauvin M, Bouhassira D. Effects of IV morphine in central pain: a randomized placebo‐controlled study. Neurology 2002;58(4):554‐63. [PUBMED: 11865132] [DOI] [PubMed] [Google Scholar]
Dellemijn 1994 {published data only}
- Dellemijn PL, Verbiest HB, Vliet JJ, Roos PJ, Vecht CJ. Medical therapy of malignant nerve pain. A randomised double‐blind explanatory trial with naproxen versus slow‐release morphine. European Journal of Cancer 1994;30A(9):1244‐50. [PUBMED: 7999406] [DOI] [PubMed] [Google Scholar]
Domzal 1986 {published data only}
- Domzal T, Szymanski H, Zaleska B. Morphine epidural block in lumbosacral pain [Morfinowe blokady nadtwardowkowe w bolu ledzwiowo‐krzyzowym]. Neurologia i Neurochirurgia Polska 1986;20(3):214‐7. [PUBMED: 2946972] [PubMed] [Google Scholar]
Eide 1994 {published data only}
- Eide PK, Jorum E, Stubhaug A, Bremnes J, Breivik H. Relief of post‐herpetic neuralgia with the N‐methyl‐D‐aspartic acid receptor antagonist ketamine: a double‐blind, cross‐over comparison with morphine and placebo. Pain 1994;58(3):347‐54. [DOI: 10.1016/0304-3959(94)90129-5] [DOI] [PubMed] [Google Scholar]
Galer 2005 {published data only}
- Galer BS, Lee D, Ma T, Nagle B, Schlagheck TG. MorphiDex (morphine sulfate/dextromethorphan hydrobromide combination) in the treatment of chronic pain: three multicenter, randomized, double‐blind, controlled clinical trials fail to demonstrate enhanced opioid analgesia or reduction in tolerance. Pain 2005;115(3):284‐95. [DOI: 10.1016/j.pain.2005.03.004] [DOI] [PubMed] [Google Scholar]
Harke 2001 {published data only}
- Harke H, Gretenkort P, Ladleif HU, Rahman S, Harke O. The response of neuropathic pain and pain in complex regional pain syndrome I to carbamazepine and sustained‐release morphine in patients pretreated with spinal cord stimulation: a double‐blinded randomized study. Anesthesia and Analgesia 2001;92(2):488‐95. [DOI: 10.1213/00000539-200102000-00039] [DOI] [PubMed] [Google Scholar]
Jadad 1992 {published data only}
- Jadad AR, Carroll D, Glynn CJ, Moore RA, McQuay HJ. Morphine responsiveness of chronic pain: double‐blind randomised crossover study with patient‐controlled analgesia. Lancet 1992;339(8806):1367‐71. [DOI: 10.1016/0140-6736(92)91194-D] [DOI] [PubMed] [Google Scholar]
Kalman 2002 {published data only}
- Kalman S, Osterber A, Sorensen J, Boivie J, Bertler A. Morphine responsiveness in a group of well‐defined multiple sclerosis patients: a study with i.v. morphine. European Journal of Pain 2002;6(1):69‐80. [DOI: 10.1053/eujp.2001.0307] [DOI] [PubMed] [Google Scholar]
Nalamachu 2013 {published data only}
- Nalamachu S, Ruck D, Nalamasu R, Fasbinder S, Bansal R. Open‐label study to evaluate the efficacy and safety of extended‐release hydromorphone in patients with chronic neuropathic pain. Journal of Opioid Management 2013;9(1):43‐9. [DOI: 10.5055/jom.2013.0416] [DOI] [PubMed] [Google Scholar]
Patarica‐Huber 2011 {published data only}
- Patarica‐Huber E, Boskov N, Pjevic M. Multimodal approach to therapy‐related neuropathic pain in breast cancer. Journal of B.U.ON. 2011;16(1):40‐5. [PUBMED: 21674848] [PubMed] [Google Scholar]
Persson 1998 {published data only}
- Persson J, Hasselstrom J, Wiklund B, Heller A, Svensson JO, Gustafsson LL. The analgesic effect of racemic ketamine in patients with chronic ischemic pain due to lower extremity arteriosclerosis obliterans. Acta Anaesthesiologica Scandinavia 1998;42(7):750‐8. [DOI: 10.1111/j.1399-6576.1998.tb05317.x] [DOI] [PubMed] [Google Scholar]
Raja 2002 {published data only}
- Raja SN, Haythornthwaite JA, Pappagallo M, Clark MR, Travison TG, Sabeen S, et al. Opioids versus antidepressants in postherpetic neuralgia. Neurology 2002;59(7):1015‐21. [DOI: 10.1212/WNL.59.7.1015] [DOI] [PubMed] [Google Scholar]
Rocha 2014 {published data only}
- Rocha Rde O, Teixeria MJ, Yeng LT, Cantara MG, Faria VG, Liggieri V, et al. Thoracic sympathetic block for the treatment of complex regional pain syndrome type I: a double‐blind randomized controlled study. Pain 2014;155(11):2274‐81. [DOI: 10.1016/j.pain.2014.08.015] [DOI] [PubMed] [Google Scholar]
Rowbotham 1991 {published data only}
- Rowbotham MC, Reisner‐Keller LA, Fields HL. Both intravenous lidocaine and morphine reduce the pain of postherpetic neuralgia. Neurology 1991;41(7):1024‐8. [PUBMED: 1712433] [DOI] [PubMed] [Google Scholar]
Serinken 2016 {published data only}
- Serinken M, Eken C, Gungor F, Emet M, Al B. Comparison of intravenous morphine versus paracetamol in sciatica: a randomized placebo controlled trial. Academic Emergency Medicine 2016;23(6):674‐8. [DOI: 10.1111/acem.12956] [DOI] [PubMed] [Google Scholar]
Siddall 2000 {published data only}
- Siddall PJ, Molloy AR, Walker S, Mather LE, Ruthowski SB, Cousins MJ. The efficacy of intrathecal morphine and clonidine in the treatment of pain after spinal cord injury. Anesthesia and Analgesia 2000;91(6):1493‐8. [DOI: 10.1097/00000539-200012000-00037] [DOI] [PubMed] [Google Scholar]
Wasan 2006 {published data only}
- Wasan AD, Kaptchuk TJ, Davar G, Jamison RN. The association between psychopathology and placebo analgesia in patients with discogenic low back pain. Pain Medicine 2006;7(3):217‐28. [DOI: 10.1111/j.1526-4637.2006.00154.x] [DOI] [PubMed] [Google Scholar]
Watt 1996 {published data only}
- Watt JW, Wiles JR, Bowsher DR. Epidural morphine for postherpetic neuralgia. Anaesthesia 1996;51(7):647‐51. [DOI: 10.1111/j.1365-2044.1996.tb07846.x] [DOI] [PubMed] [Google Scholar]
Wu 2002 {published data only}
- Wu CL, Tella P, Staats PS, Vaslav R, Kazim DA, Wesselmann U, et al. Analgesic effects of intravenous lidocaine and morphine on postamputation pain. Anesthesiology 2002;96(2):841‐8. [DOI: 10.1097/00000542-200204000-00010] [DOI] [PubMed] [Google Scholar]
Xiong 2008 {published data only}
- Xiong J, Liang C, Wu X, Zhao YX, Fu L, Zhou YF. Efficacy and safety of oxycodone‐acetaminophen tablet in 142 patients with different types of moderate and advanced cancer pain. Chinese Journal of New Drugs 2008;17(21):1877‐9. [EMBASE: 2008602294] [Google Scholar]
Additional references
AlBalawi 2013
- AlBalawi Z, McAlister FA, Thorlund K, Wong M, Wetterslev J. Random error in cardiovascular meta‐analyses: how common are false positive and false negative results?. International Journal of Cardiology 2013;168(2):1102‐7. [DOI: 10.1016/j.ijcard.2012.11.048] [DOI] [PubMed] [Google Scholar]
Ball 1985
- Ball M, McQuay HJ, Moore RA, Allen MC, Fisher A, Sear J. Renal failure and the use of morphine in intensive care. Lancet 1985;1(8432):784‐6. [DOI: 10.1016/S0140-6736(85)91448-5] [DOI] [PubMed] [Google Scholar]
Baron 2012
- Baron R, Wasner G, Binder A. Chronic pain: genes, plasticity, and phenotypes. Lancet Neurology 2012;11(1):19‐21. [DOI: 10.1016/S1474-4422(11)70281-2] [DOI] [PubMed] [Google Scholar]
Baron 2017
- Baron R, Maier C, Attal N, Binder A, Bouhassira D, Cruccu G, et al. Peripheral neuropathic pain: a mechanism‐related organizing principle based on sensory profiles. Pain 2017;158(2):261‐72. [DOI: 10.1097/j.pain.0000000000000753] [DOI] [PMC free article] [PubMed] [Google Scholar]
Berger 2004
- Berger A, Dukes EM, Oster G. Clinical characteristics and economic costs of patients with painful neuropathic disorders. Journal of Pain 2004;5(3):143‐9. [DOI: 10.1016/j.jpain.2003.12.004] [DOI] [PubMed] [Google Scholar]
Berger 2009
- Berger A, Toelle T, Sadosky A, Dukes E, Edelsberg J, Oster G. Clinical and economic characteristics of patients with painful neuropathic disorders in Germany. Pain Practice 2009;9(1):8‐17. [DOI: 10.1111/j.1533-2500.2008.00244.x] [DOI] [PubMed] [Google Scholar]
Berger 2012
- Berger A, Sadosky A, Dukes E, Edelsberg J, Oster G. Clinical characteristics and patterns of healthcare utilization in patients with painful neuropathic disorders in UK general practice: a retrospective cohort study. BMC Neurology 2012;12:8. [DOI: 10.1186/1471-2377-12-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bouhassira 2008
- Bouhassira D, Lantéri‐Minet M, Attal N, Laurent B, Touboul C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 2008;136(3):380‐7. [DOI: 10.1016/j.pain.2007.08.013] [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta‐analyses may be inconclusive ‐ Trial Sequential Analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta‐analyses. International Journal of Epidemiology 2009;38(1):287‐98. [DOI: 10.1093/ije/dyn188] [DOI] [PubMed] [Google Scholar]
Calvo 2012
- Calvo M, Dawes JM, Bennett DL. The role of the immune system in the generation of neuropathic pain. Lancet Neurology 2012;11(7):629‐42. [DOI: 10.1016/S1474-4422(12)70134-5] [DOI] [PubMed] [Google Scholar]
Chaparro 2012
- Chaparro LE, Wiffen PJ, Moore RA, Gilron I. Combination pharmacotherapy for the treatment of neuropathic pain in adults. Cochrane Database of Systematic Reviews 2012, Issue 7. [DOI: 10.1002/14651858.CD008943.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Collins 1998
- Collins SL, Faura CC, Moore RA, McQuay HJ. Peak plasma concentrations after oral morphine: a systematic review. Journal of Pain and Symptom Management 1998;16(6):388‐402. [DOI: 10.1016/S0885-3924(98)00094-3] [DOI] [PubMed] [Google Scholar]
Colloca 2017
- Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, et al. Neuropathic pain. Nature Reviews Disease Primers 2017;3:17002. [DOI: 10.1038/nrdp.2017.2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dechartres 2013
- Dechartres A, Trinquart L, Boutron I, Ravaud P. Influence of trial sample size on treatment effect estimates: meta‐epidemiological study. BMJ 2013;346:f2304. [DOI: 10.1136/bmj.f2304] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dechartres 2014
- Dechartres A, Altman DG, Trinquart L, Boutron I, Ravaud P. Association between analytic strategy and estimates of treatment outcomes in meta‐analyses. JAMA 2014;312:623‐30. [DOI: 10.1001/jama.2014.8166] [DOI] [PubMed] [Google Scholar]
Demant 2014
- Demant DT, Lund K, Vollert J, Maier C, Segerdahl M, Finnerup NB, et al. The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double‐blind, placebo‐controlled phenotype‐stratified study. Pain 2014;155(11):2263‐73. [DOI: 10.1016/j.pain.2014.08.014] [DOI] [PubMed] [Google Scholar]
Derry 2012
- Derry S, Moore RA. Topical capsaicin (low concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2012, Issue 9. [DOI: 10.1002/14651858.CD010111] [DOI] [PMC free article] [PubMed] [Google Scholar]
Derry 2014
- Derry S, Wiffen PJ, Moore RA, Quinlan J. Topical lidocaine for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2014, Issue 7. [DOI: 10.1002/14651858.CD010958.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Derry 2016
- Derry S, Stannard C, Cole P, Wiffen PJ, Knaggs R, Aldington D, et al. Fentanyl for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2016, Issue 10. [DOI: 10.1002/14651858.CD011605.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Derry 2017
- Derry S, Rice ASC, Cole P, Tan T, Moore RA. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2017, Issue 1. [DOI: 10.1002/14651858.CD007393.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dworkin 2008
- Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. Journal of Pain 2008;9(2):105‐21. [DOI: 10.1016/j.jpain.2007.09.005] [DOI] [PubMed] [Google Scholar]
Dworkin 2013
- Dworkin RH, O'Connor AB, Kent J, Mackey SC, Raja SN, Stacey BR, et al. Interventional management of neuropathic pain: NeuPSIG recommendations. Pain 2013;154(11):2249‐61. [DOI: 10.1016/j.pain.2013.06.004] [DOI] [PMC free article] [PubMed] [Google Scholar]
Edelsberg 2011
- Edelsberg JS, Lord C, Oster G. Systematic review and meta‐analysis of efficacy, safety, and tolerability data from randomized controlled trials of drugs used to treat postherpetic neuralgia. Annals of Pharmacotherapy 2011;45(12):1483‐90. [DOI: 10.1345/aph.1P777] [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI: 10.1093/ije/31.1.140] [DOI] [PubMed] [Google Scholar]
Els 2016
- Els C, Hagtvedt R, Kunyk D, Sonnenberg B, Lappi VG, Straube S. High‐dose opioids for chronic non‐cancer pain: an overview of Cochrane reviews. Cochrane Database of Systematic Reviews 2016, Issue 7. [DOI: 10.1002/14651858.CD012299] [DOI] [Google Scholar]
EPOC 2015
- Effective Practice, Organisation of Care (EPOC). 23. Worksheets for preparing a Summary of Findings using GRADE. Resources for review authors, 2015. Oslo: Norwegian Knowledge Centre for the Health Services. Available at: epoc.cochrane.org/epoc‐specific‐resources‐review‐authors (accessed 13 February 2017).
Faura 1996
- Faura CC, Moore RA, Horga JF, Hand CW, McQuay HJ. Morphine and morphine‐6‐glucuronide plasma concentrations and effect in cancer pain. Journal of Pain and Symptom Management 1996;11(2):95‐102. [DOI: 10.1016/0885-3924(95)00148-4] [DOI] [PubMed] [Google Scholar]
Faura 1998
- Faura CC, Collins SL, Moore RA, McQuay HJ. Systematic review of factors affecting the ratios of morphine and its major metabolites. Pain 1998;74(1):43‐53. [DOI: 10.1016/S0304-3959(97)00142-5] [DOI] [PubMed] [Google Scholar]
Finnerup 2013
- Finnerup NB, Scholz J, Attal N, Baron R, Haanpää M, Hansson P, et al. Neuropathic pain needs systematic classification. European Journal of Pain 2013;17(7):953‐6. [DOI: 10.1002/j.1532-2149.2012.00282.x] [DOI] [PubMed] [Google Scholar]
Finnerup 2015
- Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta‐analysis. Lancet Neurology 2015;14(2):162‐73. [DOI: 10.1016/S1474-4422(14)70251-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
Franklin 2014
- Franklin GM, American Academy of Neurology. Opioids for chronic noncancer pain: a position paper of the American Academy of Neurology. Neurology 2014;83(14):1277‐84. [DOI: 10.1212/WNL.0000000000000839] [DOI] [PubMed] [Google Scholar]
Gaskell 2014
- Gaskell H, Moore RA, Derry, S, Stannard C. Oxycodone for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2014, Issue 6. [DOI: 10.1002/14651858.CD010692.pub2] [DOI] [PubMed] [Google Scholar]
Gaskell 2016
- Gaskell H, Derry S, Stannard C, Moore RA. Oxycodone for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2016, Issue 7. [DOI: 10.1002/14651858.CD010692.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Grahame‐Smith 2002
- Grahame‐Smith DG, Aronson JK. Oxford Textbook of Clinical Pharmacology and Drug Therapy. 3rd Edition. Oxford (UK): Oxford University Press, 2002. [Google Scholar]
Gustorff 2008
- Gustorff B, Dorner T, Likar R, Grisold W, Lawrence K, Schwarz F, et al. Prevalence of self‐reported neuropathic pain and impact on quality of life: a prospective representative survey. Acta Anaesthesiologica Scandinavica 2008;52(1):132‐6. [DOI: 10.1111/j.1399-6576.2007.01486.x] [DOI] [PubMed] [Google Scholar]
Guyatt 2011
- Guyatt GH, Oxman AD, Kunz R, Woodcock J, Brozek J, Helfand M, et al. GRADE guidelines: 7. Rating the quality of evidence ‐ inconsistency. Journal of Clinical Epidemiology 2011;64(12):1294‐302. [DOI: 10.1016/j.jclinepi.2011.03.017] [DOI] [PubMed] [Google Scholar]
Guyatt 2013a
- Guyatt G, Oxman AD, Sultan S, Brozek J, Glasziou P, Alonso‐Coello P, et al. GRADE guidelines: 11. Making an overall rating of confidence in effect estimates for a single outcome and for all outcomes. Journal of Clinical Epidemiology 2013;66:151‐7. [DOI: 10.1016/j.jclinepi.2012.01.006] [DOI] [PubMed] [Google Scholar]
Guyatt 2013b
- Guyatt GH, Oxman AD, Santesso N, Helfand M, Vist G, Kunz R, et al. GRADE guidelines: 12. Preparing summary of findings tables ‐ binary outcomes. Journal of Clinical Epidemiology 2013;66:158‐72. [DOI: 10.1016/j.jclinepi.2012.01.012] [DOI] [PubMed] [Google Scholar]
Hall 2008
- Hall GC, Carroll D, McQuay HJ. Primary care incidence and treatment of four neuropathic pain conditions: a descriptive study, 2002‐2005. BMC Family Practice 2008;9:26. [DOI: 10.1186/1471-2296-9-26] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hall 2013
- Hall GC, Morant SV, Carroll D, Gabriel ZL, McQuay HJ. An observational descriptive study of the epidemiology and treatment of neuropathic pain in a UK general population. BMC Family Practice 2013;14:28. [DOI: 10.1186/1471-2296-14-28] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hand 1987
- Hand CW, Blunnie WP, Claffey LP, McShane AJ, McQuay HJ, Moore RA. Potential analgesic contribution from morphine‐6‐glucuronide in CSF. Lancet 1987;2(8569):1207‐8. [DOI] [PubMed] [Google Scholar]
Haroutiunian 2012
- Haroutiunian S, McNicol ED, Lipman AG. Methadone for chronic non‐cancer pain in adults. Cochrane Database of Systematic Reviews 2012, Issue 11. [DOI: 10.1002/14651858.CD008025.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Helfert 2015
- Helfert SM, Reimer M, Höper J, Baron R. Individualized pharmacological treatment of neuropathic pain. Clinical Pharmacology and Therapeutics 2015;97(2):135‐42. [DOI: 10.1002/cpt.19] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hoffman 2010
- Hoffman DL, Sadosky A, Dukes EM, Alvir J. How do changes in pain severity levels correspond to changes in health status and function in patients with painful diabetic peripheral neuropathy?. Pain 2010;149(2):194‐201. [DOI: 10.1016/j.pain.2009.09.017] [DOI] [PubMed] [Google Scholar]
Jadad 1996
- Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJ, Gavaghan DJ, et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Controlled Clinical Trials 1996;17(1):1‐12. [DOI: 10.1016/0197-2456(95)00134-4] [DOI] [PubMed] [Google Scholar]
Jensen 2011
Kalso 2013
- Kalso E, Aldington DJ, Moore RA. Drugs for neuropathic pain. BMJ 2013;347:f7339. [DOI: 10.1136/bmj.f7339] [DOI] [PubMed] [Google Scholar]
Katusic 1991
- Katusic S, Williams DB, Beard CM, Bergstralh EJ, Kurland LT. Epidemiology and clinical features of idiopathic trigeminal neuralgia and glossopharyngeal neuralgia: similarities and differences, Rochester, Minnesota, 1945‐1984. Neuroepidemiology 1991;10:276‐81. [DOI: 10.1159/000110284] [DOI] [PubMed] [Google Scholar]
Klimas 2014
- Klimas R, Mikus G. Morphine‐6‐glucuronide is responsible for the analgesic effect after morphine administration: a quantitative review of morphine, morphine‐6‐glucuronide, and morphine‐3‐glucuronide. British Journal of Anaesthesia 2014;113(6):935‐44. [DOI: 10.1093/bja/aeu186] [DOI] [PubMed] [Google Scholar]
Koopman 2009
- Koopman JS, Dieleman JP, Huygen FJ, Mos M, Martin CG, Sturkenboom MC. Incidence of facial pain in the general population. Pain 2009;147(1‐3):122‐7. [DOI: 10.1016/j.pain.2009.08.023] [DOI] [PubMed] [Google Scholar]
L'Abbé 1987
- L'Abbé KA, Detsky AS, O'Rourke K. Meta‐analysis in clinical research. Annals of Internal Medicine 1987;107:224‐33. [DOI: 10.7326/0003-4819-107-2-224] [DOI] [PubMed] [Google Scholar]
Lange 2010
- Lange B, Kuperwasser B, Okamoto A, Steup A, Häufel T, Ashworth J, et al. Efficacy and safety of tapentadol prolonged release for chronic osteoarthritis pain and low back pain. Advances in Therapy 2010;27(6):381‐99. [DOI: 10.1007/s12325-010-0036-3] [DOI] [PubMed] [Google Scholar]
Lunn 2014
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD007115.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
McNicol 2013
- McNicol ED, Midbari A, Eisenberg E. Opioids for neuropathic pain. Cochrane Database of Systematic Reviews 2013, Issue 8. [DOI: 10.1002/14651858.CD006146.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
McQuay 1998
- McQuay H, Moore R. An Evidence‐Based Resource for Pain Relief. Oxford (UK): Oxford University Press, 1998. [ISBN: 0‐19‐263048‐2] [Google Scholar]
McQuay 2007
- McQuay HJ, Smith LA, Moore RA. Chronic pain. In: Stevens A, Raftery J, Mant J, Simpson S editor(s). Health Care Needs Assessment, 3rd Series. Oxford (UK): Radcliffe Publishing, 2007. [ISBN: 978‐1‐84619‐063‐6] [Google Scholar]
Moore 1998
- Moore RA, Gavaghan D, Tramèr MR, Collins SL, McQuay HJ. Size is everything ‐ large amounts of information are needed to overcome random effects in estimating direction and magnitude of treatment effects. Pain 1998;78(3):209‐16. [DOI: 10.1016/S0304-3959(98)00140-7] [DOI] [PubMed] [Google Scholar]
Moore 2008
- Moore RA, Barden J, Derry S, McQuay HJ. Managing potential publication bias. In: McQuay HJ, Kalso E, Moore RA editor(s). Systematic Reviews in Pain Research: Methodology Refined. Seattle (WA): IASP Press, 2008:15‐24. [ISBN: 978‐0‐931092‐69‐5] [Google Scholar]
Moore 2009
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007076.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010a
- Moore RA, Eccleston C, Derry S, Wiffen P, Bell RF, Straube S, et al. "Evidence" in chronic pain ‐ establishing best practice in the reporting of systematic reviews. Pain 2010;150(3):386‐9. [DOI: 10.1016/j.pain.2010.05.011] [DOI] [PubMed] [Google Scholar]
Moore 2010b
- Moore RA, Straube S, Derry S, McQuay HJ. Chronic low back pain analgesic studies ‐ a methodological minefield. Pain 2010;149(3):431‐4. [DOI: 10.1016/j.pain.2010.02.032] [DOI] [PubMed] [Google Scholar]
Moore 2010c
- Moore RA, Straube S, Paine J, Phillips CJ, Derry S, McQuay HJ. Fibromyalgia: moderate and substantial pain intensity reduction predicts improvement in other outcomes and substantial quality of life gain. Pain 2010;149(2):360‐4. [DOI: 10.1016/j.pain.2010.02.039] [DOI] [PubMed] [Google Scholar]
Moore 2010d
- Moore RA, Smugar SS, Wang H, Peloso PM, Gammaitoni A. Numbers‐needed‐to‐treat analyses ‐ do timing, dropouts, and outcome matter? Pooled analysis of two randomized, placebo‐controlled chronic low back pain trials. Pain 2010;151(3):592‐7. [DOI: 10.1016/j.pain.2010.07.013] [DOI] [PubMed] [Google Scholar]
Moore 2010e
- Moore RA, Moore OA, Derry S, Peloso PM, Gammaitoni AR, Wang H. Responder analysis for pain relief and numbers needed to treat in a meta‐analysis of etoricoxib osteoarthritis trials: bridging a gap between clinical trials and clinical practice. Annals of the Rheumatic Diseases 2010;69(2):374‐9. [DOI: 10.1136/ard.2009.107805] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2011a
- Moore RA, Straube S, Paine J, Derry S, McQuay HJ. Minimum efficacy criteria for comparisons between treatments using individual patient meta‐analysis of acute pain trials: examples of etoricoxib, paracetamol, ibuprofen, and ibuprofen/paracetamol combinations after third molar extraction. Pain 2011;152(5):982‐9. [DOI] [PubMed] [Google Scholar]
Moore 2011b
- Moore RA, Mhuircheartaigh RJ, Derry S, McQuay HJ. Mean analgesic consumption is inappropriate for testing analgesic efficacy in post‐operative pain: analysis and alternative suggestion. European Journal of Anaesthesiology 2011;28(6):427‐32. [DOI: 10.1097/EJA.0b013e328343c569] [DOI] [PubMed] [Google Scholar]
Moore 2012
- Moore RA, Straube S, Eccleston C, Derry S, Aldington D, Wiffen P, et al. Estimate at your peril: imputation methods for patient withdrawal can bias efficacy outcomes in chronic pain trials using responder analyses. Pain 2012;153(2):265‐8. [DOI: 10.1016/j.pain.2011.10.004] [DOI] [PubMed] [Google Scholar]
Moore 2013a
- Moore A, Derry S, Eccleston C, Kalso E. Expect analgesic failure; pursue analgesic success. BMJ 2013;346:f2690. [DOI: 10.1136/bmj.f2690] [DOI] [PubMed] [Google Scholar]
Moore 2013b
- Moore RA, Straube S, Aldington D. Pain measures and cut‐offs ‐ 'no worse than mild pain' as a simple, universal outcome. Anaesthesia 2013;68(4):400‐12. [DOI: 10.1111/anae.12148] [DOI] [PubMed] [Google Scholar]
Moore 2014a
- Moore RA, Derry S, Taylor RS, Straube S, Phillips CJ. The costs and consequences of adequately managed chronic non‐cancer pain and chronic neuropathic pain. Pain Practice 2014;14(1):79‐94. [DOI: 10.1111/papr.12050] [DOI] [PubMed] [Google Scholar]
Moore 2014b
- Moore RA, Cai N, Skljarevski V, Tölle TR. Duloxetine use in chronic painful conditions ‐ individual patient data responder analysis. European Journal of Pain 2014;18(1):67‐75. [DOI: 10.1002/j.1532-2149.2013.00341.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2014c
- Moore RA, Wiffen PJ, Derry S, McQuay HJ. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2014, Issue 4. [DOI: 10.1002/14651858.CD007938.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2015a
- Moore RA, Chi CC, Wiffen PJ, Derry S, Rice ASC. Oral nonsteroidal anti‐inflammatory drugs for neuropathic pain. Cochrane Database of Systematic Reviews 2015, Issue 10. [DOI: 10.1002/14651858.CD010902.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2015b
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2015, Issue 7. [DOI: 10.1002/14651858.CD008242.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2015c
- Moore RA, Wiffen PJ, Eccleston C, Derry S, Baron R, Bell RF, et al. Systematic review of enriched enrolment, randomised withdrawal trial designs in chronic pain: a new framework for design and reporting. Pain 2015;156(8):1382‐95. [DOI: 10.1097/j.pain.0000000000000088] [DOI] [PubMed] [Google Scholar]
NICE 2013
- National Institute for Health and Care Excellence (NICE). Neuropathic pain ‐ pharmacological management: the pharmacological management of neuropathic pain in adults in non‐specialist settings, 2013 (updated 2017). nice.org.uk/guidance/cg173 (accessed 23 February 2017).
Nüesch 2010
- Nüesch E, Trelle S, Reichenbach S, Rutjes AW, Tschannen B, Altman DG, et al. Small study effects in meta‐analyses of osteoarthritis trials: meta‐epidemiological study. BMJ 2010;341:c3515. [DOI: 10.1136/bmj.c3515] [DOI] [PMC free article] [PubMed] [Google Scholar]
O'Brien 2010
- O'Brien EM, Staud RM, Hassinger AD, McCulloch RC, Craggs JG, Atchison JW, et al. Patient‐centered perspective on treatment outcomes in chronic pain. Pain Medicine 2010;11(1):6‐15. [DOI: 10.1111/j.1526-4637.2009.00685] [DOI] [PubMed] [Google Scholar]
Overall 1969
- Overall JE. Classical statistical hypothesis testing within the context of Bayesian theory. Psychological Bulletin 1969;71(4):285‐92. [DOI: 10.1037/h0026860] [DOI] [Google Scholar]
PaPaS 2012
- Cochrane Pain, Palliative and Supportive Care Group (PaPaS) author and referee guidance. papas.cochrane.org/papas‐documents (accessed 23 February 2017).
Rappaport 1994
- Rappaport ZH, Devor M. Trigeminal neuralgia: the role of self‐sustaining discharge in the trigeminal ganglion. Pain 1994;56(2):127‐38. [DOI: 10.1016/0304-3959(94)90086-8] [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rey 1993
- Rey R. History of Pain. Paris (France): Editions La Decouverte, 1993. [ISBN: 2‐7071‐2256‐4] [Google Scholar]
Roberts 2015
- Roberts I, Ker K, Edwards P, Beecher D, Manno D, Sydenham E. The knowledge system underpinning healthcare is not fit for purpose and must change. BMJ 2015;350:h2463. [DOI: 10.1136/bmj.h2463] [DOI] [PubMed] [Google Scholar]
Scott 2006
- Scott FT, Johnson RW, Leedham‐Green M, Davies E, Edmunds WJ, Breuer J. The burden of herpes zoster: a prospective population based study. Vaccine 2006;24(9):1308‐14. [DOI: 10.1016/j.vaccine.2005.09.026] [DOI] [PubMed] [Google Scholar]
Sear 1989
- Sear JW, Hand CW, Moore RA, McQuay HJ. Studies on morphine disposition: influence of renal failure on the kinetics of morphine and its metabolites. British Journal of Anaesthesia 1989;62(1):28‐32. [DOI: 10.1093/bja/62.1.28] [DOI] [PubMed] [Google Scholar]
Sommer 2015
- Sommer C, Welsch P, Klose P, Schaefert R, Petzke F, Häuser W. Opioids in chronic neuropathic pain. A systematic review and meta‐analysis of efficacy, tolerability and safety in randomized placebo‐controlled studies of at least 4 weeks duration [Opioide bei chronischem neuropathischem Schmerz]. Schmerz (Berlin, Germany) 2015;29(1):35‐46. [DOI: 10.1007/s00482-014-1455-x] [DOI] [PubMed] [Google Scholar]
Soni 2013
- Soni A, Batra R, Gwilym S, Spector T, Hart D, Arden N, et al. Neuropathic features of joint pain: a community‐based study. Arthritis and Rheumatism 2013;65(7):1942‐9. [DOI: 10.1002/art.37962] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stannard 2016
- Stannard C, Gaskell H, Derry S, Aldington D, Cole P, Cooper TE, et al. Hydromorphone for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2016, Issue 5. [DOI: 10.1002/14651858.CD011604.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2008
- Straube S, Derry S, McQuay HJ, Moore RA. Enriched enrolment: definition and effects of enrichment and dose in trials of pregabalin and gabapentin in neuropathic pain. A systematic review. British Journal of Clinical Pharmacology 2008;66(2):266‐75. [DOI: 10.1111/j.1365-2125.2008.03200] [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2010
- Straube S, Derry S, Moore RA, Paine J, McQuay HJ. Pregabalin in fibromyalgia ‐ responder analysis from individual patient data. BMC Musculoskeletal Disorders 2010;11:150. [DOI: 10.1186/1471-2474-11-150] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sultan 2008
- Sultan A, Gaskell H, Derry S, Moore RA. Duloxetine for painful diabetic neuropathy and fibromyalgia pain: systematic review of randomised trials. BMC Neurology 2008;8:29. [DOI: 10.1186/1471-2377-8-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tayeb 2016
- Tayeb BO, Barreiro AE, Bradshaw YS, Chui KK, Carr DB. Durations of opioid, nonopioid drug, and behavioral clinical trials for chronic pain: adequate or inadequate?. Pain Medicine 2016;17(11):2036‐46. [DOI: 10.1093/pm/pnw245] [DOI] [PubMed] [Google Scholar]
Thorlund 2011
- Thorlund K, Imberger G, Walsh M, Chu R, Gluud C, Wetterslev J, et al. The number of patients and events required to limit the risk of overestimation of intervention effects in meta‐analysis ‐ a simulation study. PloS One 2011;6(10):e25491. [DOI: 10.1371/journal.pone.0025491] [DOI] [PMC free article] [PubMed] [Google Scholar]
Torrance 2006
- Torrance N, Smith BH, Bennett MI, Lee AJ. The epidemiology of chronic pain of predominantly neuropathic origin. Results from a general population survey. Journal of Pain 2006;7(4):281‐9. [DOI: 10.1016/j.jpain.2005.11.008] [DOI] [PubMed] [Google Scholar]
Treede 2008
- Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 2008;70(18):1630‐5. [DOI: 10.1212/01.wnl.0000282763.29778.59] [DOI] [PubMed] [Google Scholar]
Turner 2013
- Turner RM, Bird SM, Higgins JP. The impact of study size on meta‐analyses: examination of underpowered studies in Cochrane reviews. PloS One 2013;8(3):e59202. [DOI: 10.1371/journal.pone.0059202] [DOI] [PMC free article] [PubMed] [Google Scholar]
van Hecke 2014
- Hecke O, Austin SK, Khan RA, Smith BH, Torrance N. Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 2014;155(4):654‐62. [DOI: 10.1016/j.pain.2013.11.013] [DOI] [PubMed] [Google Scholar]
van Hoek 2009
- Hoek AJ, Gay N, Melegaro A, Opstelten W, Edmunds WJ. Estimating the cost‐effectiveness of vaccination against herpes zoster in England and Wales. Vaccine 2009;27(9):1454‐67. [DOI: 10.1016/j.vaccine.2008.12.024] [DOI] [PubMed] [Google Scholar]
von Hehn 2012
- Hehn CA, Baron R, Woolf CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 2012;73(4):638‐52. [DOI: 10.1016/j.neuron.2012.02.008] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vos 2012
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990‐2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380(9859):2163‐96. [DOI: 10.1016/S0140-6736(12)61729-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 1986
- World Health Organization. Cancer Pain Relief, 1986. who.int/iris/handle/10665/43944. Geneva: WHO, (accessed 23 February 2017).
WHO 2011
- World Health Organization. WHO Essential Medicines List 17th edition, 2011. whqlibdoc.who.int/hq/2011/a95053_eng.pdf (accessed 23 February 2017).
Wiffen 2013
- Wiffen PJ, Derry S, Moore RA, Aldington D, Cole P, Rice ASC, et al. Antiepileptic drugs for neuropathic pain and fibromyalgia ‐ an overview of Cochrane reviews. Cochrane Database of Systematic Reviews 2013, Issue 11. [DOI: 10.1002/14651858.CD010567.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiffen 2015
- Wiffen PJ, Derry S, Moore RA, Stannard C, Aldington D, Cole P, et al. Buprenorphine for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2015, Issue 9. [DOI: 10.1002/14651858.CD011603.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiffen 2016
- Wiffen PJ, Knaggs R, Derry S, Cole P, Phillips T, Moore RA. Paracetamol (acetaminophen) with or without codeine or dihydrocodeine for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2016, Issue 12. [DOI: 10.1002/14651858.CD012227.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]