

Abstract
Background
Epilepsy is a common neurological condition with a worldwide prevalence of around 1%. Approximately 60% to 70% of people with epilepsy will achieve a longer‐term remission from seizures, and most achieve that remission shortly after starting antiepileptic drug treatment. Most people with epilepsy are treated with a single antiepileptic drug (monotherapy) and current guidelines from the National Institute for Health and Care Excellence (NICE) in the United Kingdom for adults and children recommend carbamazepine or lamotrigine as first‐line treatment for partial onset seizures and sodium valproate for generalised onset seizures; however a range of other antiepileptic drug (AED) treatments are available, and evidence is needed regarding their comparative effectiveness in order to inform treatment choices.
Objectives
To compare the time to withdrawal of allocated treatment, remission and first seizure of 10 AEDs (carbamazepine, phenytoin, sodium valproate, phenobarbitone, oxcarbazepine, lamotrigine, gabapentin, topiramate, levetiracetam, zonisamide) currently used as monotherapy in children and adults with partial onset seizures (simple partial, complex partial or secondary generalised) or generalised tonic‐clonic seizures with or without other generalised seizure types (absence, myoclonus).
Search methods
We searched the following databases: Cochrane Epilepsy's Specialised Register, CENTRAL, MEDLINE and SCOPUS, and two clinical trials registers. We handsearched relevant journals and contacted pharmaceutical companies, original trial investigators, and experts in the field. The date of the most recent search was 27 July 2016.
Selection criteria
We included randomised controlled trials of a monotherapy design in adults or children with partial onset seizures or generalised onset tonic‐clonic seizures (with or without other generalised seizure types).
Data collection and analysis
This was an individual participant data (IPD) review and network meta‐analysis. Our primary outcome was 'time to withdrawal of allocated treatment', and our secondary outcomes were 'time to achieve 12‐month remission', 'time to achieve six‐month remission', 'time to first seizure post‐randomisation', and 'occurrence of adverse events'. We presented all time‐to‐event outcomes as Cox proportional hazard ratios (HRs) with 95% confidence intervals (CIs). We performed pairwise meta‐analysis of head‐to‐head comparisons between drugs within trials to obtain 'direct' treatment effect estimates and we performed frequentist network meta‐analysis to combine direct evidence with indirect evidence across the treatment network of 10 drugs. We investigated inconsistency between direct estimates and network meta‐analysis via node splitting. Due to variability in methods and detail of reporting adverse events, we have not performed an analysis. We have provided a narrative summary of the most commonly reported adverse events.
Main results
IPD was provided for at least one outcome of this review for 12,391 out of a total of 17,961 eligible participants (69% of total data) from 36 out of the 77 eligible trials (47% of total trials). We could not include IPD from the remaining 41 trials in analysis for a variety of reasons, such as being unable to contact an author or sponsor to request data, data being lost or no longer available, cost and resources required to prepare data being prohibitive, or local authority or country‐specific restrictions.
We were able to calculate direct treatment effect estimates for between half and two thirds of comparisons across the outcomes of the review, however for many of the comparisons, data were contributed by only a single trial or by a small number of participants, so confidence intervals of estimates were wide.
Network meta‐analysis showed that for the primary outcome ‘Time to withdrawal of allocated treatment,’ for individuals with partial seizures; levetiracetam performed (statistically) significantly better than both current first‐line treatments carbamazepine and lamotrigine; lamotrigine performed better than all other treatments (aside from levetiracetam), and carbamazepine performed significantly better than gabapentin and phenobarbitone (high‐quality evidence). For individuals with generalised onset seizures, first‐line treatment sodium valproate performed significantly better than carbamazepine, topiramate and phenobarbitone (moderate‐ to high‐quality evidence). Furthermore, for both partial and generalised onset seizures, the earliest licenced treatment, phenobarbitone seems to perform worse than all other treatments (moderate‐ to high‐quality evidence).
Network meta‐analysis also showed that for secondary outcomes ‘Time to 12‐month remission of seizures’ and ‘Time to six‐month remission of seizures,’ few notable differences were shown for either partial or generalised seizure types (moderate‐ to high‐quality evidence). For secondary outcome ‘Time to first seizure,’ for individuals with partial seizures; phenobarbitone performed significantly better than both current first‐line treatments carbamazepine and lamotrigine; carbamazepine performed significantly better than sodium valproate, gabapentin and lamotrigine. Phenytoin also performed significantly better than lamotrigine (high‐quality evidence). In general, the earliest licenced treatments (phenytoin and phenobarbitone) performed better than the other treatments for both seizure types (moderate‐ to high‐quality evidence).
Generally, direct evidence and network meta‐analysis estimates (direct plus indirect evidence) were numerically similar and consistent with confidence intervals of effect sizes overlapping.
The most commonly reported adverse events across all drugs were drowsiness/fatigue, headache or migraine, gastrointestinal disturbances, dizziness/faintness and rash or skin disorders.
Authors' conclusions
Overall, the high‐quality evidence provided by this review supports current guidance (e.g. NICE) that carbamazepine and lamotrigine are suitable first‐line treatments for individuals with partial onset seizures and also demonstrates that levetiracetam may be a suitable alternative. High‐quality evidence from this review also supports the use of sodium valproate as the first‐line treatment for individuals with generalised tonic‐clonic seizures (with or without other generalised seizure types) and also demonstrates that lamotrigine and levetiracetam would be suitable alternatives to either of these first‐line treatments, particularly for those of childbearing potential, for whom sodium valproate may not be an appropriate treatment option due to teratogenicity.
Antiepileptic drug monotherapy (single drug treatment) for epilepsy
Background
Epilepsy is a common neurological disorder in which abnormal electrical discharges from the brain cause recurrent seizures. We studied two types of epileptic seizures in this review: partial seizures that start in one area of the brain, and generalised onset tonic‐clonic seizures that start in both cerebral hemispheres simultaneously.
For around 70% of people with epilepsy seizures can be controlled, and for the majority, seizures are controlled with a single antiepileptic drug. Currently in the UK, National Institute for Health and Care Excellence (NICE) guidelines for adults and children recommend carbamazepine or lamotrigine as the first treatment options to try for individuals with newly diagnosed partial seizures and sodium valproate for individuals with newly diagnosed generalised tonic‐clonic seizures; however a range of other antiepileptic drug treatments are available.
The choice of the first antiepileptic drug for an individual with newly diagnosed seizures is of great importance and should be made taking into account high‐quality evidence of how effective the drugs are at controlling seizures and whether they are associated with side effects. It is also important that drugs appropriate for different seizure types are compared to each other.
Review methods
The antiepileptic drugs of interest to this review were carbamazepine, phenytoin, sodium valproate, phenobarbitone, oxcarbazepine, lamotrigine, gabapentin, topiramate, levetiracetam, zonisamide. In this review, we evaluated the evidence from 77 randomised controlled clinical trials comparing two or more of the drugs of interest based on how effective the drugs were at controlling seizures (i.e. whether people had recurrence of seizures or had long periods of freedom from seizures (remission)) and how tolerable any related side effects of the drugs were. We were able to combine data for 12,391 people from 36 of the 77 trials; for the remaining 5570 people from 41 trials, data were not available to use in this review.
We performed two types of analysis in this review; firstly we combined data available where pairs of drugs had been compared directly in clinical trials and secondly we performed an analysis to combine all information from the clinical trials across the 'network' of 10 drugs. This analysis allowed us to compare drugs in the network that had not previously been compared to each other in clinical trials.
Key results
Out of the 45 possible pairwise comparisons of the 10 drugs of interest in the review, data from clinical trials were available for just over half of these comparisons but for many only a single trial had made a comparison of the two drugs and the comparison did not include many people.
Our 'network' analysis showed that the oldest drugs in the network (phenobarbitone and phenytoin) were better options in terms of seizure control than the other drugs but that these older drugs were the worst in terms of long‐term retention (withdrawing from the treatment) compared to the newer drugs such as lamotrigine and levetiracetam.
The most commonly reported side effects across all drugs were drowsiness or fatigue, headache or migraine, gastrointestinal disturbances (stomach upsets), dizziness or faintness and rash or skin disorders.
Quality of the evidence
This review provides high‐quality evidence for individuals with partial seizures and moderate‐ to high‐quality evidence for individuals with generalised tonic‐clonic seizures, as less information is available for some of the drugs of interest for people with this seizure type.
Conclusions
The results of this review support the NICE guidelines that carbamazepine and lamotrigine are suitable first treatment options for individuals with partial onset seizures and also show that levetiracetam would also be a suitable treatment. Results of this review also support the use of sodium valproate as the first‐line treatment for individuals with generalised tonic‐clonic seizures and also show that lamotrigine and levetiracetam would be suitable alternatives, particularly for those who are pregnant or considering becoming pregnant, for whom sodium valproate may not be an appropriate treatment option.
How up‐to‐date is this review?
The review authors searched for studies that had been published up to 27 July 2016.
Summary of findings
Summary of findings for the main comparison.
Summary of findings ‐ Time to withdrawal of allocated treatment for individuals with partial seizures
| Antiepileptic drug monotherapy for epilepsy: time to withdrawal of allocated treatment for individuals with partial seizures | ||||||
|
Patient or population: adults and children with partial seizures Settings: outpatients Intervention: phenobarbitone, phenytoin, sodium valproate, lamotrigine, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: carbamazepine | ||||||
|
Intervention (experimental treatment)a,b |
Comparison (reference treatment) |
No of participants (studies) with direct evidence |
Relative effect HR (95% CI) Direct evidence (pairwise meta‐analysis)c Heterogeneity: I2 |
Relative effect HR (95% CI) Direct plus indirect evidence (network meta‐analysis)c |
Proportion of direct evidence (%)d | Quality of the evidence (GRADE) |
| Phenobarbitone | Carbamazepine | 520 (4 studies) |
1.57 (1.16 to 2.13) I2 = 0% |
1.55 (1.18 to 2.04) | 52.5% | ⊕⊕⊕⊕ highe,f |
| Phenytoin | Carbamazepine | 428 (3 studies) |
1.03 (0.74 to 1.42) I2 = 63.6% |
1.13 (0.92 to 1.38) | 12.8% | ⊕⊕⊕⊕ highe,f,g |
| Sodium Valproate | Carbamazepine | 814 (5 studies) |
0.94 (0.73 to 1.19) I2 = 0% |
1.04 (0.86 to 1.25) | 40.1% | ⊕⊕⊕⊕ highe,f |
| Lamotrigine | Carbamazepine | 2268 (9 studies) |
0.76 (0.61 to 0.95) I2 = 39.3% |
0.75 (0.65 to 0.86) | 28.9% | ⊕⊕⊕⊕ highe,f |
| Oxcarbazepine | Carbamazepine | 562 (2 studies) |
4.62 (0.95 to 22.4) I2 = 0% |
1.09 (0.84 to 1.42) | 5.7% | ⊕⊕⊕⊕ highe,f |
| Topiramate | Carbamazepine | 937 (2 studies) |
1.04 (0.52 to 2.07) I2 = 0% |
1.18 (0.98 to 1.43) | 7.4% | ⊕⊕⊕⊕ highe,f |
| Gabapentin | Carbamazepine | 954 (2 studies) |
1.14 (0.84 to 1.55) I2 = 0% |
1.20 (1.00 to 1.43) | 87.1% | ⊕⊕⊕⊕ highe,f |
| Levetiracetam | Carbamazepine | 1567 (3 studies) |
0.70 (0.52 to 0.94) I2 = 0% |
0.82 (0.69 to 0.97) | 37.9% | ⊕⊕⊕⊕ highe,f |
| Zonisamide | Carbamazepine | 583 (1 study) |
1.08 (0.81 to 1.44) I2 = NA) |
1.08 (0.79 to 1.48) | 100% | ⊕⊕⊕⊕ highe,f |
| Abbreviations: CI: confidence interval; HR: hazard ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHR < 1 indicates an advantage to the experimental treatment cHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis). dProportion of the estimate contributed by direct evidence. eSeveral trials contributing direct evidence or contributing to the network meta‐analysis were at high risk of bias for at least one domain (see Risk of bias in included studies); we performed numerous sensitivity analyses in the case of particular sources of bias or inconsistencies within individual participant data provided to us (see Sensitivity analysis for full details). Results of sensitivity analyses showed similar numerical results and no changes to conclusions, therefore we judged that any risks of bias within the trials included in these analyses have not influenced the overall results (no downgrade of quality of evidence). fNo indication of inconsistency between direct evidence and network meta‐analysis results (no downgrade of quality of evidence). gLarge amount of heterogeneity present in pairwise meta‐analysis; no change to conclusions when analysis was repeated with random‐effects, and heterogeneity likely due to difference in trial designs (e.g. age of participants). Despite heterogeneity, numerical results from direct evidence and from network results are similar and conclusions the same (no downgrade of quality of evidence).
Summary of findings 2.
Summary of findings ‐ Time to withdrawal of allocated treatment for individuals with partial seizures
| Antiepileptic drug monotherapy for epilepsy: time to withdrawal of allocated treatment for individuals with partial seizures | ||||||
|
Patient or population: adults and children with partial seizures Settings: outpatients Intervention: carbamazepine, phenobarbitone, phenytoin, sodium valproate, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: lamotrigine | ||||||
|
Intervention (experimental treatment)a,b |
Comparison (reference treatment) |
No of participants (studies) with direct evidence |
Relative effect HR (95% CI) Direct evidence (pairwise meta‐analysis)c Heterogeneity: I2 |
Relative effect HR (95% CI) Direct plus indirect evidence (network meta‐analysis)3 |
Proportion of direct evidence (%)d | Quality of the evidence (GRADE) |
| Carbamazepine | Lamotrigine | 2268 (9 studies) |
1.31 (1.05 to 1.64) I2 = 39.3% |
1.34 (1.17 to 1.53) | 28.9% | ⊕⊕⊕⊕ highe,f |
| Phenobarbitone | Lamotrigine | No direct evidence | No direct evidence I2: NA |
2.08 (1.52 to 2.86) | 0% | ⊕⊕⊕⊕ highe,f |
| Phenytoin | Lamotrigine | 90 (1 study) |
0.91 (0.47 to 1.76) I2: NA |
1.52 (1.18 to 1.92) | 11.6% | ⊕⊕⊕⊕ highe,f |
| Sodium Valproate | Lamotrigine | 221 (3 studies) |
0.71 (0.51 to 1.00) I2 = 45.1% |
1.39 (1.11 to 1.72) | 5.1% | ⊕⊕⊕⊝ moderatee,g |
| Oxcarbazepine | Lamotrigine | 506 (1 study) |
0.69 (0.12 to 4.14) I2: NA |
1.46 (1.11 to 1.92) | 4.4% | ⊕⊕⊕⊕ highe,f |
| Topiramate | Lamotrigine | 648 (1 study) |
1.18 (0.86 to 1.62) I2: NA |
1.59 (1.29 to 1.95) | 20.9% | ⊕⊕⊕⊕ highe,f |
| Gabapentin | Lamotrigine | 659 (1 study) |
0.62 (0.06 to 6.01) I2: NA |
1.60 (1.31 to 1.96) | 1% | ⊕⊕⊕⊕ highe,f |
| Levetiracetam | Lamotrigine | 240 (1 study) |
0.86 (0.58 to 1.28) I2: NA |
1.10 (0.89 to 1.35) | 23.7% | ⊕⊕⊕⊕ highe,f |
| Zonisamide | Lamotrigine | No direct evidence | No direct evidence I2: NA |
1.45 (1.03 to 2.04) | 0% | ⊕⊕⊕⊕ highe,f |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHR < 1 indicates an advantage to the experimental treatment. cHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis). dProportion of the estimate contributed by direct evidence. eSeveral trials contributing direct evidence or contributing to the network meta‐analysis were at high risk of bias for at least one domain (see Risk of bias in included studies); we performed numerous sensitivity analyses in the case of particular sources of bias or inconsistencies within individual participant data provided to us (see Sensitivity analysis for full details). Results of sensitivity analyses showed similar numerical results and no changes to conclusions, therefore we judged that any risks of bias within the trials included in these analyses have not influenced the overall results (no downgrade of quality of evidence). fNo indication of inconsistency between direct evidence and network meta‐analysis results (no downgrade of quality of evidence). gConfidence intervals of estimate from direct evidence and from network meta‐analysis do not overlap indicating potential inconsistency (quality of the evidence downgraded once due this potential inconsistency, see Effects of interventions for further discussion).
Summary of findings 3.
Summary of findings ‐ Time to withdrawal of allocated treatment for individuals with generalised seizures
| Antiepileptic drug monotherapy for epilepsy: time to withdrawal of allocated treatment for individuals with generalised seizures | ||||||
|
Patient or population: adults and children with generalised seizures* Settings: outpatients Intervention: carbamazepine, phenobarbitone, phenytoin, lamotrigine, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide. Comparison: sodium valproate | ||||||
|
Intervention (experimental treatment)a,b |
Comparison (reference treatment) |
No of participants (studies) with direct evidence |
Relative effect HR (95% CI) Direct evidence (pairwise meta‐analysis)c Heterogeneity: I2 |
Relative effect HR (95% CI) Direct plus indirect evidence (network meta‐analysis)c |
Proportion of direct evidence (%)d | Quality of the evidence (GRADE) |
| Carbamazepine | Sodium Valproate | 405 (4 studies) |
0.79 (0.45 to 1.37) I2 = 6.6% |
1.42 (1.09 to 1.85) | 27.3% | ⊕⊕⊕⊕ highe,f |
| Phenobarbitone | Sodium Valproate | 94 (2 studies) |
1.79 (0.65 to 5.00) I2 = 0% |
2.09 (1.17 to 3.75) | 19.4% | ⊕⊕⊕⊝ moderatee,f,g |
| Phenytoin | Sodium Valproate | 326 (3 studies) |
1.52 (0.68 to 3.33) I2 = 22.6% |
1.30 (0.79 to 2.15) | 19.3% | ⊕⊕⊕⊕ highe,f |
| Lamotrigine | Sodium Valproate | 387 (3 studies) |
0.46 (0.22 to 0.97) I2 = 0% |
0.90 (0.60 to 1.35) | 14.8% | ⊕⊕⊕⊕ highe,f |
| Oxcarbazepine | Sodium Valproate | No direct evidence | No direct evidence I2: NA |
1.42 (0.29 to 6.92) | 0% | ⊕⊕⊕⊝ moderatee,f,g |
| Topiramate | Sodium Valproate | 443 (2 studies) |
1.04 (0.52 to 2.07) I2 = 48.5% |
1.76 (1.22 to 2.53) | 22.4% | ⊕⊕⊕⊝ moderatee,f,h |
| Gabapentin | Sodium Valproate | No direct evidence | No direct evidence I2: NA |
1.28 (0.16 to 10.5) | 0% | ⊕⊕⊕⊝ moderatee,f,g |
| Levetiracetam | Sodium Valproate | 512 (1 study) |
0.68 (0.30 to 1.59) I2: NA) |
1.05 (0.58 to 1.90) | 18.6% | ⊕⊕⊕⊕ highe,f |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
*Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHR < 1 indicates an advantage to the experimental treatment. cHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis). dProportion of the estimate contributed by direct evidence. eSeveral trials contributing direct evidence or contributing to the network meta‐analysis were at high risk of bias for at least one domain (see Risk of bias in included studies); we performed numerous sensitivity analyses in the case of particular sources of bias or inconsistencies within individual participant data provided to us (see Sensitivity analysis for full details). Results of sensitivity analyses showed similar numerical results and no changes to conclusions, therefore we judged that any risks of bias within the trials included in these analyses have not influenced the overall results (no downgrade of quality of evidence). fNo indication of inconsistency between direct evidence and network meta‐analysis results (no downgrade of quality of evidence). gWide or very wide confidence intervals on the network meta‐analysis estimate (downgraded once for imprecision). hConfidence intervals of estimate from direct evidence and from network meta‐analysis do not overlap indicating potential inconsistency (quality of the evidence downgraded once due this potential inconsistency, see Effects of interventions for further discussion).
Summary of findings 4.
Summary of findings ‐ Time to 12‐month remission for individuals with partial seizures
| Antiepileptic drug monotherapy for epilepsy: time to 12‐month remission for individuals with partial seizures | ||||||
|
Patient or population: adults and children with partial seizures Settings: outpatients Intervention: phenobarbitone, phenytoin, sodium valproate, lamotrigine, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: carbamazepine | ||||||
|
Intervention (experimental treatment)a,b |
Comparison (reference treatment) |
No of participants (studies) with direct evidence |
Relative effect HR (95% CI) Direct evidence (pairwise meta‐analysis)c Heterogeneity: I2 |
Relative effect HR (95% CI) Direct plus indirect evidence (network meta‐analysis)c |
Proportion of direct evidence (%)d | Quality of the evidence (GRADE) |
| Phenobarbitone | Carbamazepine | 525 (4 studies) |
1.41 (1.04 to 1.91) I2 = 0% |
1.02 (0.76 to 1.35) | 56.1% | ⊕⊕⊕⊕ highe,f |
| Phenytoin | Carbamazepine | 430 (3 studies) |
1.00 (0.76 to 1.32) I2 = 54.8% |
1.03 (0.85 to 1.25) | 18.6% | ⊕⊕⊕⊕ highe,f,g |
| Sodium Valproate | Carbamazepine | 816 (5 studies) |
1.03 (0.85 to 1.25) I2 = 46.4% |
1.05 (0.89 to 1.25) | 27.6% | ⊕⊕⊕⊕ highe,f |
| Lamotrigine | Carbamazepine | 891 (2 studies) |
1.02 (0.69 to 1.50) I2 = 0% |
1.16 (0.98 to 1.37) | 17.5% | ⊕⊕⊕⊕ highe,f |
| Oxcarbazepine | Carbamazepine | 555 (2 studies) |
1.13 (0.62 to 2.05) I2 = 0% |
0.98 (0.78 to 1.25) | 21% | ⊕⊕⊕⊕ highe,f |
| Topiramate | Carbamazepine | 925 (2 studies) |
0.94 (0.48 to 1.83) I2 = 0% |
1.08 (0.92 to 1.27) | 7.2% | ⊕⊕⊕⊕ highe,f |
| Gabapentin | Carbamazepine | 651 (1 study) |
0.61 (0.06 to 5.82) I2: NA |
1.20 (0.99 to 1.47) | 10.5% | ⊕⊕⊕⊕ highe,f |
| Levetiracetam | Carbamazepine | 1567 (3 studies) |
1.08 (0.81 to 1.42) I2 = 60.8% |
1.35 (1.09 to 1.69) | 14.2% | ⊕⊕⊕⊕ highe,f,g |
| Zonisamide | Carbamazepine | 582 (1 study) |
1.05 (0.85 to 1.30) I2: NA |
1.05 (0.81 to 1.35) | 100% | ⊕⊕⊕⊕ highe,f |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHR < 1 indicates an advantage to the experimental treatment. cHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis). dProportion of the estimate contributed by direct evidence. eSeveral trials contributing direct evidence or contributing to the network meta‐analysis were at high risk of bias for at least one domain (see Risk of bias in included studies); we performed numerous sensitivity analyses in the case of particular sources of bias or inconsistencies within individual participant data provided to us (see Sensitivity analysis for full details). Results of sensitivity analyses showed similar numerical results and no changes to conclusions, therefore we judged that any risks of bias within the trials included in these analyses have not influenced the overall results (no downgrade of quality of evidence). fNo indication of inconsistency between direct evidence and network meta‐analysis results (no downgrade of quality of evidence). gLarge amount of heterogeneity present in pairwise meta‐analysis; no change to conclusions when analysis was repeated with random‐effects and heterogeneity likely due to difference in trial designs (e.g. age of participants). Despite heterogeneity, numerical results from direct evidence and from network results are similar and conclusions the same (no downgrade of quality of evidence).
Summary of findings 5.
Summary of findings ‐ Time to 12‐month remission for individuals with partial seizures
| Antiepileptic drug monotherapy for epilepsy: time to 12‐month remission for individuals with partial seizures | ||||||
|
Patient or population: adults and children with partial seizures Settings: outpatients Intervention: carbamazepine, phenobarbitone, phenytoin, sodium valproate, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: lamotrigine | ||||||
|
Intervention (experimental treatment)a,b |
Comparison (reference treatment) |
No of participants (studies) with direct evidence |
Relative effect HR (95% CI) Direct evidence (pairwise meta‐analysis)c Heterogeneity: I2 |
Relative effect HR (95% CI) Direct plus indirect evidence (network meta‐analysis)c |
Proportion of direct evidence (%)d | Quality of the evidence (GRADE) |
| Carbamazepine | Lamotrigine | 891 (2 studies) |
0.98 (0.67 to 1.45) I2 = 0% |
0.86 (0.72 to 1.02) | 17.5% | ⊕⊕⊕⊕ highe,f |
| Phenobarbitone | Lamotrigine | No direct evidence | No direct evidence I2: NA |
0.88 (0.62 to 1.22) | 0% | ⊕⊕⊕⊕ highe,f |
| Phenytoin | Lamotrigine | No direct evidence | No direct evidence I2: NA |
0.89 (0.68 to 1.13) | 0% | ⊕⊕⊕⊕ highe,f |
| Sodium Valproate | Lamotrigine | 221 (3 studies) |
0.72 (0.56 to 0.93) I2 = 0% |
0.91 (0.73 to 1.33) | 39.9% | ⊕⊕⊕⊕ highe,f |
| Oxcarbazepine | Lamotrigine | 499 (1 study) |
1.49 (0.33 to 6.67) I2: NA |
0.85 (0.66 to 1.09) | 2.8% | ⊕⊕⊕⊕ highe,f |
| Topiramate | Lamotrigine | 636 (1 study) |
0.98 (0.29 to 3.25) I2: NA |
0.93 (0.75 to 1.15) | 2.5% | ⊕⊕⊕⊕ highe,f |
| Gabapentin | Lamotrigine | 647 (1 study) |
0.74 (0.08 to 6.58) I2: NA |
1.04 (0.84 to 1.30) | 10.1% | ⊕⊕⊕⊕ highe,f |
| Levetiracetam | Lamotrigine | 240 (1 study) |
1.02 (0.70 to 1.49) I2: NA |
1.16 (0.93 to 1.47) | 26.6% | ⊕⊕⊕⊕ highe,f |
| Zonisamide | Lamotrigine | No direct evidence | No direct evidence I2: NA |
0.91 (0.67 to 1.22) | 0% | ⊕⊕⊕⊕ highe,f |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHR < 1 indicates an advantage to the experimental treatment. cHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis). dProportion of the estimate contributed by direct evidence. eSeveral trials contributing direct evidence or contributing to the network meta‐analysis were at high risk of bias for at least one domain (see Risk of bias in included studies); we performed numerous sensitivity analyses in the case of particular sources of bias or inconsistencies within individual participant data provided to us (see Sensitivity analysis for full details). Results of sensitivity analyses showed similar numerical results and no changes to conclusions, therefore we judged that any risks of bias within the trials included in these analyses have not influenced the overall results (no downgrade of quality of evidence). fNo indication of inconsistency between direct evidence and network meta‐analysis results (no downgrade of quality of evidence).
Summary of findings 6.
Summary of findings ‐ Time to 12‐month remission for individuals with generalised seizures
| Antiepileptic drug monotherapy for epilepsy: time to withdrawal of allocated treatment for individuals with generalised seizures | ||||||
|
Patient or population: adults and children with generalised seizures* Settings: outpatients Intervention: carbamazepine, phenobarbitone, phenytoin, lamotrigine, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: sodium valproate | ||||||
|
Intervention (experimental treatment)a,b |
Comparison (reference treatment) |
No of participants (studies) with direct evidence |
Relative effect HR (95% CI) Direct evidence (pairwise meta‐analysis)c |
Relative effect HR (95% CI) Direct plus indirect evidence (network meta‐analysis)c |
Proportion of direct evidence (%)d | Quality of the evidence (GRADE) |
| Carbamazepine | Sodium Valproate | 412 (4 studies) |
0.99 (0.69 to 1.39) I2 = 0% |
1.06 (0.88 to 1.27) | 51.1% | ⊕⊕⊕⊕ highe,f |
| Phenobarbitone | Sodium Valproate | 98 (2 studies) |
0.86 (0.40 to 1.89) I2 = 42.3% |
1.33 (0.87 to 2.04) | 13% | ⊕⊕⊕⊕ highe,f |
| Phenytoin | Sodium Valproate | 269 (4 studies) |
1.15 (0.71 to 1.82) I2 = 0% |
0.91 (0.67 to 1.25) | 44.9% | ⊕⊕⊕⊕ highe,f |
| Lamotrigine | Sodium Valproate | 387 (3 studies) |
0.77 (0.38 to 1.56) I2 = 0% |
1.35 (0.57 to 3.13) | 35.7% | ⊕⊕⊕⊕ highe,f |
| Oxcarbazepine | Sodium Valproate | No direct evidence | No direct evidence I2: NA |
1.82 (0.50 to 6.67) | 0% | ⊕⊕⊕⊝ moderatee,f,g |
| Topiramate | Sodium Valproate | 441 (2 studies) |
0.52 (0.26 to 1.04) I2 = 58.5% |
1.12 (0.83 to 1.52) | 10.6% | ⊕⊕⊕⊕ highe,f,h |
| Gabapentin | Sodium Valproate | No direct evidence | No direct evidence I2: NA |
0.79 (0.10 to 6.25) | 0% | ⊕⊕⊕⊝ moderatee,f,g |
| Levetiracetam | Sodium Valproate | 512 (1 study) |
0.91 (0.49 to 1.70) I2: NA |
1.41 (0.83 to 2.44) | 55.2% | ⊕⊕⊕⊕ highe,f |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
*Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHR < 1 indicates an advantage to the experimental treatment. cHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis). dProportion of the estimate contributed by direct evidence. eSeveral trials contributing direct evidence or contributing to the network meta‐analysis were at high risk of bias for at least one domain (see Risk of bias in included studies); we performed numerous sensitivity analyses in the case of particular sources of bias or inconsistencies within individual participant data provided to us (see Sensitivity analysis for full details). Results of sensitivity analyses showed similar numerical results and no changes to conclusions, therefore we judged that any risks of bias within the trials included in these analyses have not influenced the overall results (no downgrade of quality of evidence). fNo indication of inconsistency between direct evidence and network meta‐analysis results (no downgrade of quality of evidence). gWide or very wide confidence intervals on the network meta‐analysis estimate (downgraded once for imprecision). hLarge amount of heterogeneity present in pairwise meta‐analysis; no change to conclusions when analysis was repeated with random‐effects and heterogeneity likely due to difference in trial designs (e.g. age of participants). Despite heterogeneity, numerical results from direct evidence and from network results are similar and conclusions the same (no downgrade of quality of evidence).
Background
Description of the condition
Epilepsy is a common neurological condition in which recurrent, unprovoked seizures occur due to abnormal electrical discharges in the brain, with an estimated incidence of 33 to 57 per 100,000 person‐years worldwide (Annegers 1999; Hirtz 2007; MacDonald 2000; Olafsson 2005; Sander 1996), accounting for approximately 1% of the global burden of disease (WHO 1994). The lifetime risk of epilepsy onset is estimated to be 1300 to 4000 per 100,000 person years (Hauser 1993; Juul‐Jenson 1983), and the lifetime prevalence could be as large as 70 million people world‐wide (Ngugi 2010). It is believed that with effective drug treatment, up to 70% of individuals with active epilepsy have the potential to become seizure‐free and go into long‐term remission shortly after starting drug therapy (Cockerell 1995; Hauser 1993; Sander 2004), and that around 70% of individuals can achieve seizure freedom using a single AED (AED) in monotherapy (Cockerell 1995). The remaining 30% of individuals experience refractory or drug‐resistant seizures, which often require treatment with combinations of AEDs or alternative treatments such as epilepsy surgery (Kwan 2000).
Epilepsy is not a single condition, but is in fact a heterogeneous group of conditions ranging from those with a purely genetic cause to those that are symptomatic of a brain injury (e.g. stroke) or other abnormality (e.g. tumour). We also recognise a range of differing seizure types, and epilepsy syndromes that have been classified by the International League Against Epilespy (ILAE), a classification that continues to be revised as our understanding of the genetics and basic biology of epilepsy improves (Berg 2010; Commission 1981; Commission 1989)
The simplest dichotomy in epilepsy is between partial onset (or focal) and generalised onset seizures. Partial onset seizures originate in one part of the brain and include simple partial, complex partial and secondary generalised seizures (Berg 2010). Generalised seizures originate in both cerebral hemispheres simultaneously and include generalised tonic‐clonic seizures, absence seizures and myoclonic seizures. In this review we focus on this dichotomy rather than specific epilepsy syndromes.
Description of the intervention
For the treatment of partial and generalised onset seizures we included in our evidence base the following 10 AEDs, which at the time of publication of the protocol of this review (December 2014) were licensed and used in clinical practice for use as monotherapy in at least one country (eMC 2014; FDA 2014):
carbamazepine;
phenobarbitone;
phenytoin;
sodium valproate;
oxcarbazepine;
lamotrigine;
gabapentin;
topiramate;
levetiracetam;
zonisamide.
Carbamazepine, sodium valproate, phenytoin and phenobarbitone are among the earliest drugs licensed for treating epileptic seizures. Carbamazepine and sodium valproate have been commonly used as monotherapy for partial onset and generalised onset seizures for over 30 years (Shakir 1980), while phenytoin and phenobarbitone have been used in monotherapy for over 50 years (Gruber 1962).
These traditionally used drugs have all been recommended as first‐line treatments due to their effects across a range of seizure types, however they are also associated with a number of adverse effects. Phenytoin and phenobarbitone are no longer considered as first‐line agents in the USA and much of Europe due to worries over adverse events (Wallace 1997; Wilder 1995). Both drugs have been shown to be teratogenic (associated with malformations of an embryo or fetus) and are associated with low folic acid levels and megaloblastic anaemia (a blood disorder marked by the appearance of very large red blood cells (Carl 1992; Gladstone 1992; Meador 2008; Morrow 2006; Nulman 1997)). Phenytoin is particularly associated with fetal hydantoin syndrome, the name given to a group of birth defects associated with exposure to phenytoin (Scheinfeld 2003), and phenobarbitone has been associated with behavioural disturbances, particularly in children (de Silva 1996; Trimble 1988). These agents are however still used as first‐line drugs in low‐ to middle‐income countries (Ogunrin 2005; Pal 1998).
Carbamazepine and sodium valproate are also associated with congenital abnormalities (Canger 1999; Gladstone 1992; Morrow 2006; Nulman 1997; Tomson 2011). Systematic reviews have shown sodium valproate to have the highest incidence of congenital malformations of traditional first‐line AEDs (Meador 2008; Weston 2016), particularly spina bifida, as well as cardiac, craniofacial, skeletal and limb defects known as 'valproate syndrome' (Ornoy 2009). A recent study has shown an increased prevalence of neurodevelopmental disorders following prenatal sodium valproate exposure (Bromley 2013). A recently published Cochrane Review found that levetiracetam and lamotrigine exposure carried the lowest risk of overall congenital malformation, however information regarding specific malformations was lacking (Weston 2016).
In the last 20 years, a second‐generation of AEDs including oxcarbazepine, lamotrigine, gabapentin, topiramate and, most recently, levetiracetam and zonisamide, have been licensed as monotherapy following demonstrations of efficacy, or non‐inferiority within the European Union, compared to the traditional AEDs (for example, Baulac 2012; Bill 1997; Brodie 1995a; Brodie 1999; Brodie 2007; Chadwick 1998; Christe 1997; Dam 1989; Guerreiro 1997; SANAD A 2007, SANAD B 2007; Privitera 2003; Reunanen 1996; Rowan 2005; Steiner 1999; Trinka 2013). Comparative studies have also shown the newer AEDs to be generally well tolerated as monotherapy in both adults and children and related to fewer adverse events, fewer serious adverse events, fewer teratogenic effects and fewer drug interactions with concomitant AEDs and other concomitant medications than the traditional first‐line AEDs (French 2004; French 2007).
Current guidelines from the National Institute for Health and Care Excellence (NICE) for adults and children recommend carbamazepine or lamotrigine as first‐line treatment for partial onset seizures and sodium valproate for generalised onset seizures, on the condition that women and girls of childbearing age are made aware of the potential teratogenic effects of the drug (NICE 2012).
How the intervention might work
AEDs suppress seizures by reducing neuronal excitability, hence reducing the probability that a seizure will occur. Different AEDs have different mechanisms of action; therefore certain AEDs are more effective at treating different seizure types. For example, there are reports of efficacy for sodium valproate in generalised epilepsy syndromes such as juvenile myoclonic epilepsy and absence epilepsy (Bourgeois 1987; Delgado‐Escueta 1984; Grünewald 1993; Jeavons 1977; Penry 1989), while carbamazepine, on the other hand, is reported to exacerbate some generalised seizure types such as myoclonic and absence seizures (Liporace 1994; Shields 1983; Snead 1985).
The majority of traditional AEDs are thought to have multiple mechanisms of action such as blocking ion channels, binding with neurotransmitter receptors or inhibiting the metabolism or reuptake of neurotransmitters. However the precise mechanism of action is not known for all AEDs, particularly sodium valproate. It is thought that one of the mechanisms of action of phenytoin, sodium valproate, carbamazepine, oxcarbazepine and lamotrigine is via blocking of sodium channels (Brodie 1996; Faigle 1990; Granger 1995; Grant 1992; Lees 1993; McLean 1986; Pinder 1977; Ragsdale 1991; Willow 1985), while phenobarbitone binds with gamma‐aminobutyric acid (GABA) A receptors (Rho 1996).
Zonisamide is thought to have multiple mechanisms of action (Endoh 1994; Kawai 1994; Okada 1998; Sackellares 2004; Schauf 1987; Suzuki 1992; Zhu 1999), while the mechanism of actions of gabapentin and topiramate are not fully understood (Brodie 1996; Coulter 1993; Hill 1993; McClean 1995; McLean 1999; White 1997). Levetiracatam has a novel mode of action which is different from that of other AEDs (Cho 2011); it is thought to exhibit its antiepileptic effect by binding to synaptic vesicle protein 2A (encoded within the SV2A gene), influencing excitatory neurotransmitter release (Gillard 2006; Lynch 2004).
Why it is important to do this review
Given that up to 70% of individuals with a new epilepsy diagnosis enter a long‐term remission of seizures shortly after starting drug therapy (Cockerell 1995; Hauser 1993; Sander 2004), the correct choice of first‐line antiepileptic therapy for individuals with newly diagnosed seizures is of great importance. There are currently over 50 AEDs available worldwide for the treatment of all epilepsy syndromes (Epilepsy Foundation of America 2013), and therefore it is important that the choice of first AEDs is based on the highest‐quality evidence regarding potential benefits and harms of various treatments.
We have published a series of Cochrane systematic Reviews investigating pairwise monotherapy comparisons using individual participant data (Marson 2000; Nevitt 2016; Nolan 2013b; Nolan 2013c; Nolan 2015; Nolan 2016a; Nolan 2016b; Nolan 2016d). Each Cochrane Review and meta‐analysis provides high‐quality evidence for each pair of drugs but does not inform a choice among the range of drugs available. Furthermore, direct evidence from randomised controlled trials (RCTs) is not available for some drug comparisons such as between oxcarbazepine and phenobarbitone; therefore it is not possible to make pairwise comparisons of treatment effects between all 10 drugs included in this review. Also, pairwise comparisons between certain drugs are unlikely to be made in the future, such as comparisons with phenobarbitone, which is no longer considered to be a first‐line treatment, so it is unlikely that a RCT will be designed in the future to compare oxcarbazepine with phenobarbitone (Tudur Smith 2007). However, it is possible to estimate an indirect treatment effect size between oxcarbazepine and phenobarbitone using existing evidence comparing oxcarbazepine with phenytoin and phenytoin with phenobarbitone (Nolan 2013b; Nolan 2016d). By similar methodology, an indirect pairwise comparison is possible for all 10 drugs in our treatment network. Indirect comparisons are also valuable in the case that a limited amount of data are available to inform a direct comparison or in the case that evidence informing a direct comparison is of poor methodological quality. The power and precision of a treatment effect estimate can be increased by 'borrowing strength' from the indirect evidence in the network of treatments (Bucher 1997). Eight of the AEDs included in this review have been included in an IPD network meta‐analysis of epilepsy monotherapy drugs (Tudur Smith 2007). We wish to update the information in this network meta‐analysis with new evidence from trials published since 2007 and including evidence for two drugs, which were licensed for use as monotherapy after 2007.
As noted in the series of Cochrane Reviews investigating pairwise monotherapy comparisons, the important efficacy outcomes in epilepsy monotherapy trials often require analysis of time‐to‐event data (for example, time to first seizure after randomisation or time to withdrawal of allocated treatment). Although methods have been developed to synthesise time‐to‐event data using summary information (Parmar 1998; Williamson 2002), the appropriate statistics are not commonly reported in published epilepsy trials (Altman 1995; Nolan 2013a).
Furthermore, although seizure data have been collected in most epilepsy monotherapy trials, we have seen little uniformity in the definition and reporting of outcomes. For example, trials may report time to 12‐month remission but not time to first seizure or vice versa, or some trials may define time to first seizure from the date of randomisation but others use date of achieving maintenance dose. Trial investigators have also adopted differing approaches to the analysis, particularly with respect to the censoring of time‐to‐event data. For these reasons, we performed the pairwise meta‐analyses using IPD, which helps to overcome these problems and is considered to be the 'gold standard' approach to synthesis of censored data (Parmar 1998). We therefore also performed the network meta‐analysis of epilepsy monotherapy drugs as an IPD analysis.
Objectives
To compare the time to withdrawal of allocated treatment, remission and first seizure of 10 AEDs (carbamazepine, phenytoin, sodium valproate, phenobarbitone, oxcarbazepine, lamotrigine, gabapentin, topiramate, levetiracetam, zonisamide) currently used as monotherapy in children and adults with partial onset seizures (simple partial, complex partial or secondary generalised) or generalised tonic‐clonic seizures with or without other generalised seizure types (absence, myoclonus).
Methods
Criteria for considering studies for this review
Types of studies
We included RCTs using either:
an adequate method of allocation concealment (e.g. sealed, opaque envelopes);
a quasi method of randomisation (e.g. allocation by date of birth).
Trials may be double‐blind, single‐blind or unblinded. We included only trials of a monotherapy design; in other words, all participants are randomised to treatment with a single drug. We excluded trials with an add‐on (polytherapy), or withdrawal to monotherapy designs.
We included trials of parallel designs. We excluded trials of a cross‐over design, as this design is not appropriate for assessing treatment decisions at the time of epilepsy diagnosis and the cross‐over design is also inappropriate for measuring our primary time‐to‐event outcome 'time to withdrawal of allocated treatment', as a withdrawal of allocated treatment in the first treatment period would mean than the participant could not cross into the second treatment period, potentially leading to a large amount of incomplete outcome data and therefore a reduction in statistical power. Furthermore, the use of cross‐over designs is no longer recommended in epilepsy due to concerns over trial duration, large proportions of dropouts, unblinding of masked treatments as participants cross into the second period, and potential carryover effects; a particular concern in trials of a monotherapy design that aim to assess the effect of a single treatment (Engel 2008; Wyllie 2006).
Types of participants
Children or adults with partial onset seizures (simple partial, complex partial, or secondarily generalised tonic‐clonic seizures) or generalised onset tonic‐clonic seizures (with or without other generalised seizure types). We did not include participants with other generalised seizure types alone (for example absence seizures alone without generalised tonic‐clonic seizures) as guidelines for the first‐line treatment of other generalised seizure types are different from the guidelines for generalised tonic‐clonic seizures (NICE 2012), and due to documented evidence that certain drugs of interest in our review may exacerbate some generalised seizure types (How the interventions might work). We also considered individuals with a new diagnosis of epilepsy, or who had had a relapse following antiepileptic monotherapy withdrawal.
We excluded trials that considered AEDs as treatment for conditions other than epilepsy.
Types of interventions
We included the 10 AEDs currently licensed and commonly used as monotherapy in our network of treatments: carbamazepine, phenytoin, sodium valproate, phenobarbitone, oxcarbazepine, lamotrigine, gabapentin, topiramate, levetiracetam, zonisamide.
Included trials had to make at least one pairwise comparison between at least two of the 10 AEDs included in our network. For trials with three treatment arms or more, we included treatment arms only of the 10 AEDs included in our network; treatment arms of drugs not included in our network were excluded from analysis. We did not make pairwise comparisons (direct or indirect) between any AEDs not specified above. We made pairwise comparisons (based on direct or indirect evidence, or both) between all 10 drugs (Data synthesis).
We included trials with multiple arms of the same drug as long as at least one arm of another drug from our network was included (e.g. multiple doses of gabapentin compared to carbamazepine in Chadwick 1998). We pooled multiple dose arms of the same drug in our analysis; dose comparisons are outside the scope of this review.
Types of outcome measures
We investigated the following outcomes in this review (Primary outcomes; Secondary outcomes). Reporting of these outcomes in the original trial report was not an eligibility requirement for inclusion in this review.
Primary outcomes
Time to withdrawal of allocated treatment (retention time). This is a combined outcome reflecting both efficacy and tolerability, as treatment may be withdrawn due to continued seizures, adverse effects or a combination of both. This is an outcome to which the participant makes a contribution, and is the primary effectiveness outcome measure recommended by the Commission on Antiepileptic Drugs of the International League Against Epilepsy (Glauser 2006; ILAE 1998).
Secondary outcomes
Time to achieve 12‐month seizure‐free period (remission) after randomisation
Time to achieve six‐month seizure‐free period (remission) after randomisation
Time to first seizure post randomisation
Occurrence of adverse events (to be reported narratively) (Data synthesis)
Search methods for identification of studies
We searched the following databases with no language restrictions:
the Cochrane Epilepsy Specialised Register (26 July 2016) using the search strategy outlined in Appendix 1;
the Cochrane Central Register of Controlled Trials (CENTRAL; 2016, issue 7) via the Cochrane Register of Studies Online (CRSO, 26 July 2016) using the search strategy outlined in Appendix 2;
MEDLINE (Ovid, 1946 to 26 July 2016) using the search strategy outlined in Appendix 3;
SCOPUS (1823 to 09 September 2014) using the search strategy outlined in Appendix 4;
ClinicalTrials.gov searched on 26 July 2016) using the search strategy outlined in Appendix 5;
World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) search portal searched on 26 July 2016), using the search strategy outlined in Appendix 6.
We had originally searched SCOPUS as an alternative to Embase, but this is no longer necessary, because randomised and quasi‐randomised controlled trials in Embase are now included in CENTRAL. We have not, therefore, updated the SCOPUS search.
We also reviewed reference lists of retrieved trials to search for additional reports of relevant trials, reviewed relevant conference proceedings and contacted experts in the field for details of any ongoing or unpublished trials.
Data collection and analysis
Selection of studies
One author (SJN) screened all titles and abstracts of all records identified by the electronic searches as described in Search methods for identification of reviews, according to the inclusion criteria specified above (Types of studies; Types of participants; Types of interventions). Subsequently, two authors (SJN and AGM) independently assessed full‐text publications according to the same inclusion criteria specified above. We resolved disagreements by discussion or by consulting a third author (CT) where necessary. We recorded the reasons for exclusion of trials at both stages of screening. We contacted trial authors for clarification if the eligibility of a trial was unclear from the published information.
Data extraction and management
Requesting individual participant data
For all trials meeting our inclusion criteria, two authors (SJN and AGM) sent a data‐request form to the first or corresponding author, or both, of the trial or to the trial sponsor where appropriate (referred to as data providers in this review).
Our data‐request form asked data providers if the following information was available (tick yes or no).
-
Trial methods:
method of generation of random list;
method of concealment of randomisation;
stratification factors;
blinding methods.
-
Participant covariates:
sex;
age;
seizure types;
epilepsy status (newly diagnosed/relapsed seizures following drug withdrawal);
time between first seizure and randomisation;
number of seizures prior to randomisation (with dates);
presence of neurological signs;
electroencephalography (EEG) results;
computed tomography (CT) or magnetic resonance imaging (MRI) results;
aetiology of seizures (if known).
-
Follow‐up data:
treatment allocation;
date of randomisation;
dates of follow‐up;
dates of seizures post randomisation or seizure frequency data between follow‐up visits;
dates of treatment withdrawal and reason(s) for treatment withdrawal;
starting dose of treatment;
dates of dose changes;
adverse events reported.
We also requested any available, related documents such as case report forms, trial protocols, clinical summaries etc. from data providers.
In the event of no response to our IPD request, we sent a follow‐up email to the original data provider contacted. If we still received no response for a particular trial, we attempted to contact another trial author or sponsor where appropriate. If a data provider was unable to make IPD available to us, we recorded the quoted reason why IPD could not be made available and we requested any aggregate data related to our outcome not reported in the publication.
If data could not be obtained (no response to any requests or IPD was not available), two independent authors (SJN and MS) assessed whether any relevant and appropriate aggregate level data was reported in the trial publication or could be indirectly estimated via the methods described in Parmar 1998 and Williamson 2002. We resolved any disagreements on extracted aggregate data by discussion or by consulting a third author (CT) if necessary.
Management of individual participant data
We stored all obtained data on a secure, dedicated network drive accessible only to the statisticians performing analysis (SJN, MS, CT). We checked all provided data for consistency and prepared them for analysis according to a pre‐specified procedure prepared by one author (SJN) (available on request) and piloted by two authors (SJN and MS). For each trial where IPD were supplied, we reproduced results from trial findings where possible and we performed the following consistency checks:
trial details cross‐checked against any published report of the trial; original trial authors to be contacted if missing data, errors or inconsistencies were found;
review of the chronological randomisation sequence by checking the balance of prognostic factors, taking account of factors stratified for in randomisation procedure.
We discussed any inconsistencies in the provided data with the corresponding data providers. If large or major inconsistencies were present, which could not be resolved by data providers, we did not include the data in any analyses. If minor inconsistencies were present, we analysed the data and conducted sensitivity analyses to test the robustness of results (Sensitivity analysis).
Following consistency checking and data cleaning, we prepared datasets for analysis and calculated outcomes for this review according to the methodology summarised below. We followed a 'standard operating procedure' for the data cleaning and preparation of data for analysis for all datasets to ensure a standardised and consistent approach to analysis throughout this review. Further details of this procedure can be obtained from the corresponding author on request.
Preparation of individual participant data for analysis
For the analysis of time to withdrawal of allocated treatment as a time‐to‐event outcome, we defined an 'event' as either the withdrawal of the allocated treatment due to poor seizure control or adverse events, or both. We also classed non‐compliance with the treatment regimen or the addition of another AED as 'events'. We censored the outcome if treatment was withdrawn because the individual achieved a period of remission, if a participant withdrew from allocated treatment for reasons not related to the treatment (such as loss to follow‐up) or if the individual was still on allocated treatment at the end of follow‐up. Two authors (SJN and AG) independently reviewed reasons for treatment withdrawal for classification as events or censored observations, and we resolved any disagreements by mutual discussion or by involving a third author (CT).
If seizure data were provided or recorded in terms of the number of seizures recorded between clinic visits rather than specific dates of seizures, to enable the calculation of time‐to‐event outcomes, we applied linear interpolation to estimate dates of seizures between follow‐up visits. For example, if the trial recorded four seizures between two visits that occurred on 1 March 2010 and 1 May 2010 (interval of 61 days), then the date of the first seizure would be approximately 13 March 2010. This allowed the computation of an estimate of the time to six‐month remission, 12‐month remission, and first seizure.
We calculated time to six‐month and 12‐month remission from the date of randomisation to the date (or estimated date) the individual had first been free of seizures for six or 12 months respectively. If the person had one or more seizures in the titration period, a six‐month or 12‐month seizure‐free period could also occur between the estimated date of the last seizure in the titration period and the estimated date of the first seizure in the maintenance period
We calculated time to first seizure from the date of randomisation to the date that their first seizure was estimated to have occurred. If seizure data were missing for a particular visit, these outcomes were censored at the previous visit. These outcomes were also censored if the individual died or if follow‐up ceased prior to the occurrence of the event of interest.
Two trials were designed in strata based on whether recommended treatment would be carbamazepine or sodium valproate (Privitera 2003; Trinka 2013). Within the two strata, participants were randomised to topiramate (Privitera 2003) or levetiracetam (Trinka 2013) compared to the recommended treatment of carbamazepine or sodium valproate depending on the strata. To ensure that randomised comparisons were made, we analysed data for these two trials according to the separate strata in this review (i.e. treated as two trials Privitera 2003 carbamazepine branch and Privitera 2003 sodium valproate branch).
Assessment of risk of bias in included studies
Two authors (SJN, JW) independently assessed risk of bias in all included trials using the Cochrane tool for assessing risk of bias (Higgins 2011). The following methodological criteria are assessed according to this tool:
selection bias (sequence generation and allocation concealment);
performance bias (blinding of participants and personnel);
detection bias (blinding of outcome assessment);
attrition bias (incomplete outcome data);
reporting bias (selective outcome reporting).
We resolved any disagreements by discussion. In theory, a review using IPD should overcome issues of reporting biases as unpublished data can be provided and unpublished outcomes calculated. Any selective reporting bias detected could be assessed with the Outcome Reporting Bias in Trials (ORBIT) classification system (Kirkham 2010). As specified in Data extraction and management, we asked the data providers to provide trial methods such as randomisation and blinding methods, and we discussed any missing data and or inconsistencies, or both with them.
Measures of treatment effect
We summarised all time‐to‐event outcomes using the hazard ratio (HR) as the measure of treatment effect. We calculated outcomes from IPD provided where possible or extracted summary statistics from published trials. We did not attempt to analyse or synthesise adverse event data; a large range of different adverse events are thought to be associated with the 10 different drugs and such data were collected and presented in different ways across trials. For these reasons, we believe a synthesis of adverse event data would present only selective, and potentially misleading information, while a narrative description of adverse event data from IPD or extracted from published trials would be the most informative way of presenting these data.
Unit of analysis issues
We did not encounter any unit of analysis issues. For inclusion in the review, the unit of allocation had to be the individual. Trials of a repeated‐measures (longitudinal) nature or of a cross‐over design were not eligible for inclusion.
Dealing with missing data
For all included trials, we conducted an assessment of the proportion of missing outcome, demographic and covariate data and made a judgement regarding the extent and nature of missing data (e.g. missing at random, missing not at random). We attempted to contact all trial authors in order to request relevant data; we included any information regarding missing data in such requests (Data extraction and management). If further information regarding missing data could not be provided and we judged that an important proportion of data (particularly outcome data) were missing, we conducted sensitivity analyses to investigate the potential impact of the missing data (for example, best case scenario or worst case scenario analyses, assuming those with missing outcome data all had a favourable or unfavourable outcome, respectively).
Assessment of heterogeneity
We used a fixed‐effect model for all pairwise and network meta‐analyses in the first instance as we anticipated that our specific inclusion criteria would result in eligible studies of a similar design and populations and our use of IPD to standardise definitions of outcomes. Also, our previous reviews of this topic have not showed any important heterogeneity (Marson 2000; Nevitt 2016; Nolan 2013b; Nolan 2013c; Nolan 2015; Nolan 2016a; Nolan 2016b; Nolan 2016d); see Data synthesis for further details of pairwise and network meta‐analysis.
For each pairwise comparison, we assessed the presence of heterogeneity statistically using the Q test (P value less than 0.10 for significance) and the I2 statistic with the following interpretation (Higgins 2003):
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity.
We also assessed the presence of heterogeneity by visually inspecting forest plots, particularly in terms of the magnitude and direction of effects. If substantial or considerable heterogeneity (i.e. I2 of 50% or over) was found to be present, which we were not able to explain by differences in characteristics of the trials and participants, we planned to perform network meta‐analysis with a random‐effects model.
It was not possible to directly calculate an I2 statistic for the network meta‐analysis due to the between‐study covariance structure required for the network meta‐analysis model (see Data synthesis). However, for this model, we were able to estimate an R statistic, which compares the impact of heterogeneity in the fixed‐effect and random‐effects models (Jackson 2012) and it has been previously shown that R can be used to calculate I2 as follows: I2 = (R2 ‐ 1)/R2 (Higgins 2002)
Therefore we estimated an I2 statistic for the whole treatment network for each analysis and interpreted as above. We also presented an estimate of Tau2 (an estimate of the between‐study variance in random‐effects meta‐analysis) for each analysis and we have taken both statistics into account when interpreting the presence of any important heterogeneity in the treatment network.
Assessment of reporting biases
Two authors (SJN and JW) undertook a full 'Risk of bias' assessment for each eligible trial, including risk of reporting biases. In theory, a review using IPD should overcome issues of reporting biases, as unpublished data can be provided and unpublished outcomes calculated. As specified in Data extraction and management, we asked the data providers for trial methods, such as randomisation and blinding methods, and we discussed any missing data and inconsistencies with them.
If we suspected selective reporting bias in the review, we intended to assess the magnitude and impact of this selective reporting bias using the ORBIT classification system (Kirkham 2010), however we did not have any major concerns about selective reporting bias in this review. The approach to this review (re‐analysis of IPD) helps to overcome issues of reporting bias, as unpublished data can be provided and unpublished outcomes calculated.
Data synthesis
Figure 1 and Figure 2 visually present the network of 45 pairwise comparisons from the 10 antiepileptic treatments of interest to this review.
Figure 1.

Network plot of pairwise comparisons in all included studies, studies providing individual participant data (IPD) and studies without IPD
Note that the size of the node indicates the number of studies the drug is included in and the thickness of the edges corresponds to the number of participants contributing to the comparison (i.e. larger node = more studies, thicker edge = more participants).
CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Figure 2.
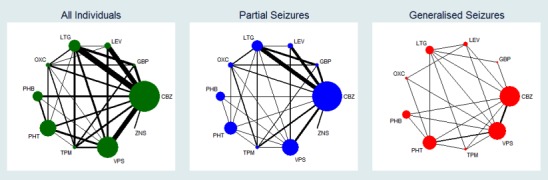
Network plot of pairwise comparisons for all included participants (total 17,961 participants), participants with partial seizures and participants with generalised tonic‐clonic seizures with or without other seizure types (shortened to 'generalised seizures' for brevity).
11978 participants were classified as experiencing partial seizures (66.7% of total), 4407 participants were classified as experiencing generalised seizures (24.5% of total) and 1576 had an unclassified or missing seizure type (8.8% of total).
Note that the size of the node indicates the number of studies the drug is included in and the thickness of the edges corresponds to the number of participants contributing to the comparison (i.e. larger node = more studies, thicker edge = more participants).
CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Pairwise and Network meta‐analysis
We used the statistical software package SAS (version 9.3) (SAS 2011) to perform all data cleaning, consistency checking and data preparation (see Data extraction and management) and Stata version 14 (StataCorp 2015) to perform all synthesis of direct and indirect evidence .
We requested data for one trial, Biton 2001, via data sharing portal ClinicalStudyDataRequest.com and the data were provided to us via a remote secure data access system that allowed analysis in SAS‐based statistical software and export of analysis results. We were unable to combine this dataset with the other datasets to perform the analyses described below in Stata version 14, therefore we treated the results exported from the data access system as aggregate data in sensitivity analysis (see Sensitivity analysis).
We took an intention‐to‐treat approach (as far as possible) to analysis; in other words, we analysed participants in the group to which they had been randomised in an individual trial, irrespective of which treatment they had actually received. Therefore, for time‐to‐event outcomes, 'time to six‐month remission', 'time to 12‐month remission' and 'time to first seizure post randomisation', participants were not censored if treatment was withdrawn. For the primary outcome, time to withdrawal of allocated treatment, we considered withdrawals due to lack of efficacy (i.e. recurrent seizures), poor tolerability (i.e. adverse events) or a combination of both poor efficacy and tolerability. Other withdrawals such as losses to follow‐up, non treatment‐related deaths, administrative trial reasons etc. were censored at the time of withdrawal.
For all time‐to‐event outcomes, we investigated the relationship between the time to the event and treatment effect of the AEDs. We fitted a Cox proportional hazards regression model, stratified by trial to preserve the within‐trial randomisation, to the entire individual participant dataset. We fitted this model via the 'mvmeta_make' command in Stata version 14 to produce a dataset in the correct format to perform network meta‐analysis with the 'mvmeta' command (White 2009); in other words, a dataset with trial‐specific estimates of treatment effect (log HR), the associated variance of the treatment effect and covariances where applicable (i.e. correlation between treatment effects for trials with more than two treatment arms).
The Cox proportional hazards model assumes that ratio of hazards (risks) between the two treatment groups is constant over time. To assess the validity of this assumption, we tested the statistical significance of time‐varying covariates for all covariates in the primary model. If we had reason to believe that the proportional hazards assumption had been violated in the primary model, in sensitivity analysis we fitted a parametric, accelerated failure‐time model, stratified by trial, to the entire individual participant dataset via the 'mvmeta_make' command and compared these results to those of the primary analysis (White 2009). An accelerated failure‐time model assumes that treatment effect accelerates or decelerates over time, rather than remains constant as assumed by the Cox proportional hazards model.
We calculated direct pairwise treatment effect estimates (where possible) using the 'metan' command (Palmer 2016) in Stata version 14 to pool trial‐specific log hazard ratios from the Cox proportional hazards model as described above.
We performed network meta‐analysis via the 'mvmeta' command in Stata version 14 assuming equal heterogeneity for all comparisons (i.e. a between‐study covariance structure (variance‐covariance matrix) proportional to unknown parameter Tau2) (White 2009). It was necessary to make an assumption regarding the between‐study covariance structure for a network without pairwise comparisons between all treatments of interest. However, due to this assumption regarding heterogeneity, we could not calculate an I2 statistic directly from the model and had to estimated it (see Assessment of heterogeneity). Network meta‐analysis provided treatment effect estimates combining direct and indirect evidence.
We performed pairwise and network meta‐analyses with a treatment by epilepsy type interaction (see Subgroup analysis and investigation of heterogeneity for further details).
For clinical interest and relevance, we have presented HR estimates from the network model (direct and indirect evidence combined) for each AED in the network compared to the current recommended first‐line treatments (carbamazepine or lamotrigine for partial onset seizures and sodium valproate for generalised onset seizures) and for all comparisons by epilepsy type in the main results of this review via forest plots.
Often rankings of treatments (i.e. the probability that each treatment in the network is the best) are presented for network meta‐analysis; however due to the treatment by epilepsy type interaction in this model, we could not calculate rankings by epilepsy type. Instead, we informally 'ranked' treatments by ordering according to their treatment effect sizes compared to the reference treatment (e.g. better or worse than carbamazepine) on the forest plots presented.
Investigation of consistency in network meta‐analysis
A key assumption made in network meta‐analysis is that treatment effect is 'exchangeable' across all included trials; in other words, the indirect comparison made between two treatments is a feasible comparison to make (known as the transitivity assumption) and that the indirect evidence is consistent with the direct evidence where a comparison exists (known as the consistency assumption).
Transitivity requires that all treatments are "jointly randomisable"; in other words, all 10 AEDs could feasibly be randomised in the same trial and those that are not treatment arms in any given trial are "missing at random" (Lu 2006). This assumption cannot be formally tested statistically; transitivity must be judged by careful consideration of trial settings and characteristics, treatment mechanisms and participant demographics to investigate if any differences would be expected to modify relative treatment effects. Given that all of the 10 drugs within this network are licenced as monotherapy treatments for individuals with newly diagnosed partial onset seizures or generalised onset tonic‐clonic seizures (with or without other generalised seizure types) and have all been used within trials of similar designs, we have no concerns over this transitivity assumption in this network.
The consistency assumption can be evaluated statistically comparing the difference between the direct treatment effect estimate and the indirect estimate for each loop of evidence. Given the complexity of the network model fitted (with treatment by epilepsy type interaction) and the number of multi‐arm trials included in analysis, we performed node splitting in Stata version 14 via the command 'network sidesplit' (Dias 2010; White 2015) to formally estimate differences between direct and indirect evidence for each comparison. In order to examine any clinical inconsistency (i.e. important differences in numerical results between direct, indirect and network results), we have presented HR estimates for direct evidence, indirect evidence (from the node splitting model) and direct plus indirect evidence from the network models for each pairwise comparison via forest plots and discuss the potential origins and implications of any apparent inconsistency. Secondly, we fitted a ‘design‐by‐treatment’ inconsistency model in Stata version 14 via mvmeta (White 2009); this method evaluates both loop and design inconsistencies, particularly within multi‐arm trials (Higgins 2012).
Adverse events
Due to the wide range of events reported in the trials and the different methods of recording and reporting of adverse events, we have not analysed adverse event data in meta‐analysis but have provided a narrative report according to the definition of the events within the data provided to us or in the published paper.
Subgroup analysis and investigation of heterogeneity
There are strong clinical beliefs that certain AEDs are more effective in certain seizure types than others, for example carbamazepine is more effective in partial onset seizures and sodium valproate is more effective in generalised onset seizures (Marson 2000), suggesting that there is a treatment‐by‐seizure type (partial or generalised) interaction. Without taking account of this potential interaction in our analysis, we believe that the key assumption of an exchangeable treatment effect across all included trials would be violated.
To account for this, we conducted all analyses separately by epilepsy type (partial onset or generalised onset) according to the classification of main seizure type at baseline and performed all network meta‐analysis with a treatment‐by‐epilepsy‐type interaction. We classified partial seizures (simple or complex) and partial secondarily generalised seizures as partial epilepsy. We classified primarily generalised seizures as generalised epilepsy. We then judged exchangeability of treatment effect separately by analyses of seizure type.
We also performed an analysis adjusted for age at entry into the trial (an interaction between treatment and age (centred) added to initial Cox proportional hazards model described in Data synthesis) and we compared results to primary analysis with adjustment only for seizure type.
We would have liked to explore other participant covariates specified in Data extraction and management as potential modifiers of treatment effect and as potential sources of heterogeneity or inconsistency, or both, such as seizure frequency before randomisation (time since first ever seizure and/or number of seizures before randomisation) and aetiology of seizures (if known according to pre‐treatment investigations such as EEG, CT and/or MRI scan); however, due to large proportions of missing data for most of these covariates and variability in the definitions of data provided to us for these covariates (see Included studies), an additional adjusted analysis was not appropriate. We will consider other options to explore these covariates for an update of this review.
Sensitivity analysis
As described in Data synthesis, we applied a fixed‐effect model principally to pairwise and network meta‐analysis, and fitted a random‐effects model to both pairwise and network meta‐analysis models in sensitivity analysis, and compared the results.
Also as described in Data synthesis, we applied a Cox proportional hazards model principally to pairwise and network meta‐analysis. We fitted an accelerated failure‐time model, which does not make the assumption of constant treatment effect over time, to both pairwise and network meta‐analysis models in sensitivity analysis and compared the results.
As specified in Data extraction and management, we discussed any inconsistencies in the provided data with the corresponding data providers and performed sensitivity analyses to investigate the impact of any missing data (see Dealing with missing data). If large or major inconsistencies were present, which could not be resolved by the data providers, we would not include the data in any analyses. If minor inconsistencies were present, we included the data in analyses and pursued sensitivity analyses to test the robustness of results included in these data. We performed the following sensitivity analyses due to inconsistencies in IPD provided and compared the results of sensitivity analyses to those of the primary analysis:
In Stephen 2007 there were minor inconsistencies between rates of seizure recurrence and reasons for withdrawal between the data provided and the published paper, which the trial authors could not resolve. Therefore we performed sensitivity analysis excluding Stephen 2007 from all analyses.
In Reunanen 1996, participants were considered to have completed the trial and hence treatment was withdrawn if they experienced a seizure after week six. This does not correspond with the treatment withdrawal definition used in this review (see Primary outcomes and Data extraction and management). Therefore, we performed sensitivity analysis excluding Reunanen 1996 for the analysis of 'Time to withdrawal of allocated treatment.'
In Banu 2007, there were minor inconsistencies between rates of seizure recurrence between the data provided and the published paper, which the authors could not resolve. Therefore we performed sensitivity analysis excluding Banu 2007 from analysis of 'Time to first seizure.' (Data for first seizure recurrence only were available, so this trial did not contribute to outcomes of time to six‐month remission and time to 12‐month remission).
Nieto‐Barrera 2001 did not include seizures that occurred during the first four weeks of the trial in efficacy analyses, and dates of seizures before week four were not supplied to us. Therefore, we calculated seizure outcomes as the time to first seizure and time to six‐month remission after week four rather than after randomisation. We performed sensitivity analysis excluding seizure data for Nieto‐Barrera 2001 from analysis of 'Time to first seizure' (this trial was 24 weeks' duration so did not contribute to outcomes of time to six‐month remission and time to 12‐month remission).
In Placencia 1993, there were minor inconsistencies between reasons for withdrawal between the data provided and the published paper. We compared reasons for withdrawal in the data provided with reasons reported in the publication and performed a sensitivity analysis for the analysis of 'Time to withdrawal of allocated treatment', with withdrawals reclassified according to definitions from the published paper (this sensitivity analysis was also performed in a previously published Cochrane Review, see Nolan 2016b for further details).
Given that misclassification of seizure type is a recognised problem in epilepsy (whereby some individuals with generalised seizures have been mistakenly classed as having partial onset seizures and vice versa) and such misclassification did impact upon the results of a review in our series of pairwise reviews for monotherapy in epilepsy comparing phenytoin and sodium valproate in which nearly 50% of participants analysed may have had their seizure type misclassified (Nolan 2016d), we investigated the potential impact of misclassification on results in a sensitivity analysis. Given clinical evidence that individuals with generalised onset seizures are unlikely to have an 'age of onset' greater than 25 to 30 years (Malafosse 1994), we examined the distribution of age at onset for individuals with generalised seizures. We identified 1164 participants classified as experiencing generalised seizures and estimated age of onset as greater than 30 years (age of first seizure provided directly in IPD or estimated to be within one year of age of entry into trial for newly diagnosed participants). We performed two sensitivity analyses to investigate misclassification:
re‐classification of all individuals with generalised seizures and age of onset greater than 30 years as having partial onset seizures. We then repeated network meta‐analysis with the interaction term of treatment by seizure type with the reclassified seizure type.
re‐classification of all individuals with generalised seizure types and age at onset greater than 30 years and those with missing seizure type into an 'unclassified seizure type' group. We then repeated network meta‐analysis with the interaction term of treatment by seizure type, where seizure type is partial epilepsy compared to generalised or unclassified epilepsy.
We were unable to perform network meta‐analysis with a 'three‐way' interaction (i.e. partial epilepsy compared to generalised epilepsy compared to unclassified epilepsy) due to small numbers of participants with unclassified epilepsy on some of the treatments.
Where possible, if IPD were not available for analysis, we attempted to extract aggregate data. Where aggregate hazard ratios and standard errors or confidence intervals could be extracted or estimated from trial publications by seizure type for our outcomes of interest, we incorporated these estimates into network meta‐analysis and compared the results of these sensitivity analyses to those of the primary analysis. As described in Data synthesis, we were provided with IPD for one trial (Biton 2001), in a remote data access system therefore we could not combine this dataset with the other datasets to perform IPD analysis. We also treated our exported results for this trial as aggregate data in sensitivity analysis.
'Summary of findings' table and quality of the evidence
We have presented six 'Summary of findings' tables for our primary outcome and first secondary outcome by epilepsy type and by reference treatment (see Data synthesis for further information);
Time to withdrawal of allocated treatment for individuals with partial seizures (reference treatment carbamazepine) (see Table 1)
Time to withdrawal of allocated treatment for individuals with partial seizures (reference treatment lamotrigine) (see Table 2)
Time to withdrawal of allocated treatment for individuals with generalised seizures (reference treatment sodium valproate) (see Table 3)
Time to 12‐month remission for individuals with partial seizures (reference treatment carbamazepine) (see Table 4)
Time to 12‐month remission for individuals with partial seizures (reference treatment lamotrigine) (see Table 5)
Time to 12‐month remission for individuals with generalised seizures (reference treatment sodium valproate) (see Table 6)
We have presented the tables following the approach of Salanti 2014 as far as possible ‐ for pairwise comparisons, we have presented the relative effect from direct evidence from pairwise meta‐analysis, number of studies and participants contributing to direct evidence, the relative effect from direct plus indirect evidence from network meta‐analysis, proportion of direct evidence, and quality of the evidence.
We determined quality of the evidence using the GRADE approach (GRADE 2008), whereby we downgraded evidence in the presence of high risk of bias, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results or high probability of publication bias. We downgraded evidence by one level if we considered the limitation to be serious and two levels if we considered it to be very serious. In this context of network meta‐analysis we also considered the proportion of direct evidence and inconsistency of direct and indirect evidence when determining quality of the evidence.
Results
Description of studies
Results of the search
We identified 6762 records from the databases and search strategies outlined in Electronic searches. We found three further records by handsearching and checking reference lists of included studies. We removed 3032 duplicate records and screened 3733 records (title and abstract) for inclusion in the review. We excluded 3591 records based on title and abstract and assessed 142 full‐text articles for inclusion in the review. We excluded 31 studies (described in 32 full‐text articles) from the review (see Excluded studies below) and included 77 trials in the review, which were reported in 95 full‐text articles (see Included studies below). We identified seven studies as ongoing (ACTRN12615000556549; ACTRN12615000639527; ACTRN12615000640505; ACTRN12615000641594; ACTRN12615000643572; NCT01891890; NCT02201251) and seven studies (described in eight records) as awaiting classification (translation: Chen 2013; Korean Zonisamide Study 1999; Park 2001; Rysz 1994; Xu 2012) or further information: IRCT201202068943N1; NCT00154076). See Figure 3 for PRISMA study flow diagram (Moher 2009).
Figure 3.
Study flow diagram
Included studies
We included 77 trials in the review (Aikia 1992; Banu 2007; Baulac 2012; Bidabadi 2009; Bill 1997; Biton 2001; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2002; Brodie 2007; Callaghan 1985; Capone 2008; Castriota 2008; Chadwick 1998; Chen 1996; Cho 2011; Christe 1997; Consoli 2012; Cossu 1984; Craig 1994; Czapinski 1997; Dam 1989; de Silva 1996; Dizdarer 2000; Donati 2007; Eun 2012; Feksi 1991; Forsythe 1991; Fritz 2006; Gilad 2007; Guerreiro 1997; Heller 1995; Jung 2015; Kalviainen 2002; Kopp 2007; Korean Lamotrigine Study Group 2008; Kwan 2009; Lee 2011; Lukic 2005; Mattson 1985; Mattson 1992; Mitchell 1987; Miura 1990; Motamedi 2013; NCT01498822; NCT01954121; Nieto‐Barrera 2001; Ogunrin 2005; Pal 1998; Placencia 1993; Privitera 2003; Pulliainen 1994; Ramsey 1983; Ramsey 1992; Ramsey 2007; Ramsey 2010; Rastogi 1991; Ravi Sudhir 1995; Resendiz 2004; Reunanen 1996; Richens 1994; Rowan 2005; Saetre 2007; SANAD A 2007; SANAD B 2007; Shakir 1981; So 1992; Steiner 1999; Steinhoff 2005; Stephen 2007; Suresh 2015; Thilothammal 1996; Trinka 2013; Turnbull 1985; Verity 1995; Werhahn 2015).
Seven trials were available in abstract form only (Bidabadi 2009; Czapinski 1997; Fritz 2006; Kalviainen 2002; Kopp 2007; Lukic 2005; Ramsey 2007), one was available in English only as a clinical trial summary report (Korean Lamotrigine Study Group 2008) and two trials were available only as an online summary (NCT01498822; NCT01954121). Three trials were published in Italian (Capone 2008; Castriota 2008; Cossu 1984) and one in Spanish (Resendiz 2004) and were translated into English. One of the published reports contained results on two separate RCTs run on very similar protocols; although the two trials were reported within the same publication we treated them as separate trials within this systematic review (Brodie 1995a; Brodie 1995b).
Characteristics of included trials
Twenty‐five trials were designed to recruit individuals with partial seizures only (Baulac 2012; Bidabadi 2009; Castriota 2008; Chadwick 1998; Cho 2011; Cossu 1984; Czapinski 1997; Dizdarer 2000; Donati 2007; Eun 2012; Gilad 2007; Jung 2015; Lee 2011; Mattson 1985; Mattson 1992; Mitchell 1987; NCT01498822; NCT01954121; Nieto‐Barrera 2001; Ramsey 2007; Resendiz 2004; SANAD A 2007; So 1992; Suresh 2015; Werhahn 2015). Three trials were designed to recruit individuals with generalised tonic‐clonic seizures with or without other generalised seizure types or unclassified seizure types only (Ramsey 1992; SANAD B 2007; Thilothammal 1996). The remaining 49 trials recruited individuals with partial or generalised tonic‐clonic seizures with or without other generalised seizure types (Aikia 1992; Banu 2007; Bill 1997; Biton 2001; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2002; Brodie 2007; Callaghan 1985; Capone 2008; Chen 1996; Christe 1997; Consoli 2012; Craig 1994; Dam 1989; de Silva 1996; Feksi 1991; Forsythe 1991; Fritz 2006; Guerreiro 1997; Heller 1995; Kalviainen 2002; Kopp 2007; Korean Lamotrigine Study Group 2008; Kwan 2009; Lukic 2005; Miura 1990; Motamedi 2013; Ogunrin 2005; Pal 1998; Placencia 1993; Privitera 2003; Pulliainen 1994; Ramsey 1983; Ramsey 2010; Rastogi 1991; Ravi Sudhir 1995; Reunanen 1996; Richens 1994; Rowan 2005; Saetre 2007; Shakir 1981; Steiner 1999; Steinhoff 2005; Stephen 2007; Trinka 2013; Turnbull 1985; Verity 1995). However five trials did not describe the number of participants with each seizure type recruited (Capone 2008; Dam 1989; Forsythe 1991; Fritz 2006; Saetre 2007).
Forty‐seven trials recruited only individuals with new onset seizures and no previous AED treatment (Aikia 1992; Baulac 2012; Bill 1997; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2007; Castriota 2008; Chen 1996; Cho 2011; Christe 1997; Cossu 1984; Craig 1994; Czapinski 1997; Dam 1989; de Silva 1996; Donati 2007; Eun 2012; Forsythe 1991; Guerreiro 1997; Heller 1995; Jung 2015; Kalviainen 2002; Kopp 2007; Lukic 2005; Mitchell 1987; Miura 1990; Motamedi 2013; NCT01498822; NCT01954121; Ogunrin 2005; Pal 1998; Placencia 1993; Privitera 2003; Pulliainen 1994; Ramsey 1983; Ramsey 1992; Ravi Sudhir 1995; Resendiz 2004; Saetre 2007; Steiner 1999; Steinhoff 2005; Stephen 2007; Suresh 2015; Thilothammal 1996; Turnbull 1985; Werhahn 2015). Three trials recruited individuals with new onset post‐stroke seizures (Consoli 2012; Capone 2008; Gilad 2007), seven trials recruited individuals with new onset or long‐term untreated seizures (Banu 2007; Callaghan 1985; Feksi 1991; Lee 2011; Korean Lamotrigine Study Group 2008; Nieto‐Barrera 2001; Trinka 2013), six trials recruited individuals with new onset, untreated or under‐treated seizures (Mattson 1985; Mattson 1992; Ramsey 2007; Ramsey 2010; Rowan 2005; So 1992), five trials recruited individuals with new onset or relapsed seizures following a period of remission (Chadwick 1998; Kwan 2009; Reunanen 1996; Richens 1994; Verity 1995), three trials recruited individuals with new onset, relapsed seizures following a period of remission or individuals whose previous treatment with an AED had failed (SANAD A 2007; SANAD B 2007; Shakir 1981) and six trials did not state if individuals had received previous AED treatment (Biton 2001; Brodie 2002; Bidabadi 2009; Dizdarer 2000; Fritz 2006; Rastogi 1991).
Twenty‐eight trials were single‐centre and conducted in Bangladesh (Banu 2007) Iran (Bidabadi 2009; Motamedi 2013), Ireland (Callaghan 1985), Italy (Capone 2008; Castriota 2008; Cossu 1984), Taiwan (Chen 1996), Republic of Korea (Cho 2011), the UK (Craig 1994; Forsythe 1991; Stephen 2007; Turnbull 1985), Turkey (Dizdarer 2000), Kenya (Feksi 1991), Israel (Gilad 2007), Germany (Kopp 2007), Serbia and Montenegro (Lukic 2005), the USA (Mitchell 1987), Japan (Miura 1990), Nigeria (Ogunrin 2005), India (Pal 1998; Rastogi 1991; Ravi Sudhir 1995; Suresh 2015; Thilothammal 1996), Ecuador (Placencia 1993) and Finland (Pulliainen 1994).
Forty‐five trials were multicentre, conducted in centres across the USA (Biton 2001; Mattson 1985; Mattson 1992; Ramsey 1983; Ramsey 1992; Ramsey 2007; Ramsey 2010; Rowan 2005), the UK (Brodie 1995a; Brodie 1995b; Brodie 1999; de Silva 1996; Heller 1995; Richens 1994; SANAD A 2007; SANAD B 2007; Steiner 1999; Verity 1995), the UK and New Zealand (Shakir 1981), Europe (Consoli 2012; Dam 1989; Donati 2007; Kalviainen 2002; Saetre 2007; Steinhoff 2005; Werhahn 2015), Europe and Australia (Brodie 2002; Reunanen 1996; Trinka 2013), Europe and South Africa (Brodie 2007), Europe and Mexico (Nieto‐Barrera 2001), Europe, South America and South Africa (Christe 1997), South America and South Africa (Bill 1997; Guerreiro 1997), Republic of Korea (Eun 2012; Jung 2015; Korean Lamotrigine Study Group 2008; Lee 2011; NCT01498822), China (NCT01954121), Hong Kong (Kwan 2009), Mexico (Resendiz 2004), Asia, Australia and Europe (Baulac 2012), Europe, Australia, Canada and South Africa (Chadwick 1998), the USA, Canada, Europe and South America (Privitera 2003).
Four trials did not state whether they were single‐ or multicentre; these trials were conducted in Finland (Aikia 1992), Poland (Czapinski 1997), Germany (Fritz 2006) and the USA (So 1992).
Twenty trials recruited adults and children (Biton 2001; Brodie 1995a; Brodie 1995b; Callaghan 1985; Chadwick 1998; Cho 2011; Feksi 1991; Korean Lamotrigine Study Group 2008; Nieto‐Barrera 2001; Placencia 1993; Privitera 2003; Ramsey 1992; Ramsey 2010; Rastogi 1991; Reunanen 1996; SANAD A 2007; SANAD B 2007; Shakir 1981; Steinhoff 2005; Stephen 2007).
Fifteen trials recruited children; four trials recruited children under the age of 12 years (Bidabadi 2009; Eun 2012; Mitchell 1987; Thilothammal 1996), one trial recruited children under 14 years (Forsythe 1991), three trials recruited children under 15 years (Banu 2007; Chen 1996; Dizdarer 2000), three trials recruited children under 16 years (de Silva 1996; Jung 2015; Verity 1995), one trial recruited children under 17 years (Donati 2007) and three trials recruited children under 18 years (Guerreiro 1997; Pal 1998; Resendiz 2004).
Thirty‐nine trials recruited adults; two trials defined adults as over the age of 13 years (Heller 1995; So 1992), four trials defined adults as over the age of 14 years (Ogunrin 2005; Ravi Sudhir 1995; Steiner 1999; Turnbull 1985); four trials defined adults as over the age of 15 years (Cossu 1984; Dam 1989; Fritz 2006; Pulliainen 1994), nine trials defined adults as over the age of 16 years (Bill 1997; Brodie 2002; Brodie 2007; Christe 1997; Lee 2011; NCT01498822; NCT01954121; Richens 1994; Trinka 2013), nine trials defined adults as over the age of 18 (Baulac 2012; Consoli 2012; Czapinski 1997; Kwan 2009; Lukic 2005; Mattson 1985; Mattson 1992; Ramsey 1983; Suresh 2015) and four trials did not state the minimum age of an ‘adult’ in the trial (Aikia 1992; Capone 2008; Castriota 2008; Gilad 2007). Seven trials recruited elderly participants; two trials recruited participants over the age of 65 years (Brodie 1999; Saetre 2007) and five trials recruited individuals over the age of 60 years (Craig 1994; Motamedi 2013; Ramsey 2007; Rowan 2005; Werhahn 2015).
Three trials did not state the age ranges of eligible participants (Kalviainen 2002; Kopp 2007; Miura 1990).
Table 7 shows the number of participants randomised to each of the 10 drugs, split according to the trials for which individual participant data were available and not available:
Table 1.
Number of participants randomised to each drug
| Trial\Drug | CBZ | PHB | PHT | VPS | LTG | OXC | LEV | TPM | GBP | ZNS | Total |
Total randomiseda |
| Trials providing individual participant data | ||||||||||||
| Banu 2007 | 54 | 54 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 108 | 108 |
| Baulac 2012 | 301 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 282 | 583 | 583 |
| Bill 1997 | 0 | 0 | 144 | 0 | 0 | 143 | 0 | 0 | 0 | 0 | 287 | 287 |
| Biton 2001 | 0 | 0 | 0 | 69 | 66 | 0 | 0 | 0 | 0 | 0 | 135 | 136 |
| Brodie 1995a | 66 | 0 | 0 | 0 | 70 | 0 | 0 | 0 | 0 | 0 | 136 | 136 |
| Brodie 1995b | 63 | 0 | 0 | 0 | 61 | 0 | 0 | 0 | 0 | 0 | 124 | 124 |
| Brodie 1999 | 48 | 0 | 0 | 0 | 102 | 0 | 0 | 0 | 0 | 0 | 150 | 150 |
| Brodie 2007 | 291 | 0 | 0 | 0 | 0 | 0 | 288 | 0 | 0 | 0 | 579 | 579 |
| Chadwick 1998 | 74 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 218 | 0 | 292 | 292 |
| Craig 1994 | 0 | 0 | 81 | 85 | 0 | 0 | 0 | 0 | 0 | 0 | 166 | 166 |
| de Silva 1996 | 54 | 10 | 54 | 49 | 0 | 0 | 0 | 0 | 0 | 0 | 167 | 173 |
| Dizdarer 2000 | 26 | 0 | 0 | 0 | 0 | 26 | 0 | 0 | 0 | 0 | 52 | 52 |
| Eun 2012 | 41 | 0 | 0 | 0 | 43 | 0 | 0 | 0 | 0 | 0 | 84 | 84 |
| Guerreiro 1997 | 0 | 0 | 94 | 0 | 0 | 99 | 0 | 0 | 0 | 0 | 193 | 193 |
| Heller 1995 | 61 | 58 | 63 | 61 | 0 | 0 | 0 | 0 | 0 | 0 | 243 | 243 |
| Kwan 2009 | 0 | 0 | 0 | 44 | 37 | 0 | 0 | 0 | 0 | 0 | 81 | 81 |
| Lee 2011 | 53 | 0 | 0 | 0 | 57 | 0 | 0 | 0 | 0 | 0 | 110 | 110 |
| Mattson 1985 | 155 | 155 | 165 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 475 | 475 |
| Mattson 1992 | 236 | 0 | 0 | 244 | 0 | 0 | 0 | 0 | 0 | 0 | 480 | 480 |
| Nieto‐Barrera 2001 | 202 | 0 | 0 | 0 | 420 | 0 | 0 | 0 | 0 | 0 | 622 | 622 |
| Ogunrin 2005 | 19 | 18 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 55 | 55 |
| Pal 1998 | 0 | 47 | 47 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 94 | 94 |
| Placencia 1993 | 95 | 97 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 192 | 192 |
| Privitera 2003 (CBZ branch)b | 129 | 0 | 0 | 0 | 0 | 0 | 0 | 266 | 0 | 0 | 395 | 395 |
| Privitera 2003 (VPS branch)b | 0 | 0 | 0 | 78 | 0 | 0 | 0 | 147 | 0 | 0 | 225 | 225 |
| Ramsey 1992 | 0 | 0 | 50 | 86 | 0 | 0 | 0 | 0 | 0 | 0 | 136 | 136 |
| Ramsey 2010 | 0 | 0 | 128 | 0 | 0 | 0 | 0 | 133 | 0 | 0 | 261 | 261 |
| Reunanen 1996 | 121 | 0 | 0 | 0 | 230 | 0 | 0 | 0 | 0 | 0 | 351 | 351 |
| Richens 1994 | 151 | 0 | 0 | 149 | 0 | 0 | 0 | 0 | 0 | 0 | 300 | 300 |
| SANAD A 2007 | 378 | 0 | 0 | 0 | 378 | 210 | 0 | 378 | 377 | 0 | 1721 | 1721 |
| SANAD B 2007 | 0 | 0 | 0 | 238 | 239 | 0 | 0 | 239 | 0 | 0 | 716 | 716 |
| Steiner 1999 | 0 | 0 | 95 | 0 | 86 | 0 | 0 | 0 | 0 | 0 | 181 | 181 |
| Stephen 2007 | 0 | 0 | 0 | 109 | 117 | 0 | 0 | 0 | 0 | 0 | 226 | 227 |
| Trinka 2013 (CBZ branch)b | 503 | 0 | 0 | 0 | 0 | 0 | 493 | 0 | 0 | 0 | 996 | 999 |
| Trinka 2013 (VPS branch)b | 0 | 0 | 0 | 353 | 0 | 0 | 350 | 0 | 0 | 0 | 703 | 703 |
| Turnbull 1985 | 0 | 0 | 70 | 70 | 0 | 0 | 0 | 0 | 0 | 0 | 140 | 140 |
| Verity 1995 | 130 | 0 | 0 | 130 | 0 | 0 | 0 | 0 | 0 | 0 | 260 | 260 |
| Werhahn 2015 | 121 | 0 | 0 | 0 | 118 | 0 | 122 | 0 | 0 | 0 | 361 | 361 |
| Total | 3372 | 439 | 1009 | 1765 | 2024 | 478 | 1253 | 1163 | 595 | 282 | 12,380 | 12,391 |
| Trials not providing individual participant data | ||||||||||||
| Trial\Drug | CBZ | PHB | PHT | VPS | LTG | OXC | LEV | TPM | GBP | ZNS | Total |
Total randomiseda |
| Aikia 1992 | 0 | 0 | 18 | 0 | 0 | 19 | 0 | 0 | 0 | 0 | 37 | 37 |
| Bidabadi 2009 | 36 | 35 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 71 | 71 |
| Brodie 2002 | 0 | 0 | 0 | 0 | 151 | 0 | 0 | 0 | 158 | 0 | 309 | 309 |
| Callaghan 1985 | 59 | 0 | 58 | 64 | 0 | 0 | 0 | 0 | 0 | 0 | 181 | 181 |
| Capone 2008 | 17 | 0 | 0 | 0 | 0 | 0 | 18 | 0 | 0 | 0 | 35 | 35 |
| Castriota 2008 | 14 | 0 | 0 | 0 | 0 | 0 | 13 | 0 | 0 | 0 | 27 | 27 |
| Chen 1996 | 26 | 25 | 0 | 25 | 0 | 0 | 0 | 0 | 0 | 0 | 76 | 76 |
| Cho 2011 | 15 | 0 | 0 | 0 | 0 | 0 | 16 | 0 | 0 | 0 | 31 | 31 |
| Christe 1997 | 0 | 0 | 0 | 121 | 0 | 128 | 0 | 0 | 0 | 0 | 249 | 249 |
| Consoli 2012 | 66 | 0 | 0 | 0 | 0 | 0 | 62 | 0 | 0 | 0 | 128 | 128 |
| Cossu 1984 | 6 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 12 | 12 |
| Czapinski 1997 | 30 | 30 | 30 | 30 | 0 | 0 | 0 | 0 | 0 | 0 | 120 | 120 |
| Dam 1989 | 100 | 0 | 0 | 0 | 0 | 94 | 0 | 0 | 0 | 0 | 194 | 194 |
| Donati 2007 | 28 | 0 | 0 | 29 | 0 | 55 | 0 | 0 | 0 | 0 | 112 | 112 |
| Feksi 1991 | 152 | 150 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 302 | 302 |
| Forsythe 1991 | 23 | 0 | 20 | 21 | 0 | 0 | 0 | 0 | 0 | 0 | 64 | 64 |
| Fritz 2006 | 0 | 0 | 0 | 0 | 21 | 27 | 0 | 0 | 0 | 0 | 48 | 48 |
| Gilad 2007 | 32 | 0 | 0 | 0 | 32 | 0 | 0 | 0 | 0 | 0 | 64 | 64 |
| Jung 2015 | 64 | 0 | 0 | 0 | 0 | 0 | 57 | 0 | 0 | 0 | 121 | 121 |
| Kalviainen 2002 | 70 | 0 | 0 | 0 | 73 | 0 | 0 | 0 | 0 | 0 | 143 | 143 |
| Kopp 2007 | 6 | 0 | 0 | 3 | 0 | 0 | 6 | 0 | 0 | 0 | 15 | 15 |
| Korean Lamotrigine Study Group 2008 | 129 | 0 | 0 | 0 | 264 | 0 | 0 | 0 | 0 | 0 | 393 | 393 |
| Lukic 2005 | 0 | 0 | 0 | 38 | 35 | 0 | 0 | 0 | 0 | 0 | 73 | 73 |
| Mitchell 1987 | 15 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 33 | 33 |
| Miura 1990 | 66 | 0 | 51 | 46 | 0 | 0 | 0 | 0 | 0 | 0 | 163 | 163 |
| Motamedi 2013 | 0 | 0 | 0 | 0 | 50 | 0 | 50 | 0 | 0 | 0 | 100 | 100 |
| NCT01498822 | 0 | 0 | 0 | 0 | 0 | 178 | 175 | 0 | 0 | 0 | 353 | 353 |
| NCT01954121 | 215 | 0 | 0 | 0 | 0 | 0 | 218 | 0 | 0 | 0 | 433 | 433 |
| Pulliainen 1994 | 23 | 0 | 20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 43 | 43 |
| Ramsey 1983 | 42 | 0 | 45 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 87 | 87 |
| Ramsey 2007c | ? | 0 | 0 | 0 | 0 | 0 | ? | 0 | 0 | 0 | 37 | 37 |
| Rastogi 1991 | 0 | 0 | 45 | 49 | 0 | 0 | 0 | 0 | 0 | 0 | 94 | 94 |
| Ravi Sudhir 1995 | 20 | 0 | 20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 40 | 40 |
| Resendiz 2004 | 42 | 0 | 0 | 0 | 0 | 0 | 0 | 46 | 0 | 0 | 88 | 88 |
| Rowan 2005 | 198 | 0 | 0 | 0 | 200 | 0 | 0 | 0 | 195 | 0 | 593 | 593 |
| Saetre 2007 | 92 | 0 | 0 | 0 | 93 | 0 | 0 | 0 | 0 | 0 | 185 | 185 |
| Shakir 1981 | 0 | 0 | 15 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 33 | 33 |
| So 1992 | 17 | 0 | 0 | 16 | 0 | 0 | 0 | 0 | 0 | 0 | 33 | 33 |
| Suresh 2015 | 30 | 0 | 0 | 0 | 0 | 0 | 30 | 0 | 0 | 0 | 60 | 60 |
| Steinhoff 2005 | 88 | 0 | 0 | 30 | 121 | 0 | 0 | 0 | 0 | 0 | 239 | 239 |
| Thilothammal 1996 | 0 | 51 | 52 | 48 | 0 | 0 | 0 | 0 | 0 | 0 | 151 | 151 |
| Totalc | 1721 | 315 | 374 | 538 | 1040 | 501 | 645 | 46 | 353 | 0 | 5570 | 5570 |
| Grand totalc | 5093 | 754 | 1383 | 2303 | 3064 | 979 | 1898 | 1209 | 948 | 282 | 17,950 | 17,961 |
CBZ: carbamazepine; GBP: gabapentin; IPD: individual participant data; ITT: intention to treat; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aDrug allocated missing for 11 participants in the IPD provided. bTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review. cOne trial provided the total number randomised but not the numbers randomised to each group. The 37 participants randomised are counted in the overall totals.
5093 participants were randomised to carbamazepine and we were provided with 66% of IPD
3064 participants were randomised to lamotrigine and we were provided with 66% of IPD
2303 participants were randomised to sodium valproate and we were provided with 77% of IPD
1898 participants were randomised to levetiracetam and we were provided with 66% of IPD
1383 participants were randomised to phenytoin and we were provided with 73% of IPD
1209 participants were randomised to topiramate and we were provided with 96% of IPD
979 participants were randomised to oxcarbazepine and we were provided with 49% of IPD
948 participants were randomised to gabapentin and we were provided with 63% of IPD
754 participants were randomised to phenobarbitone and we were provided with 58% of IPD
282 participants were randomised to zonisamide and we were provided with 100% of IPD
One trial with 37 participants (Ramsey 2010, IPD not provided) randomised individuals to carbamazepine or levetiracetam but did not state how many individuals were randomised to each drug and for 11 individuals the randomised drug was missing from the IPD
In total, we were provided with data for 12,391 out of a total of 17,961 eligible participants (69% of total data) from 36 out of the 77 eligible trials (47%).
Trials with individual participant data
Individual participant data were available for 36 trials recruiting 12,391 participants (Banu 2007; Baulac 2012; Bill 1997; Biton 2001; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2007; Chadwick 1998; Craig 1994; de Silva 1996; Dizdarer 2000; Eun 2012; Guerreiro 1997; Heller 1995; Kwan 2009; Lee 2011; Mattson 1985; Mattson 1992; Nieto‐Barrera 2001; Ogunrin 2005; Pal 1998; Placencia 1993; Privitera 2003; Ramsey 1992; Ramsey 2010; Reunanen 1996; Richens 1994; SANAD A 2007; SANAD B 2007; Steiner 1999; Stephen 2007; Trinka 2013; Turnbull 1985; Verity 1995; Werhahn 2015).
Table 8; Table 9 and Table 10 show the participant characteristics from the trials providing IPD. Data were available for the following participant characteristics (percentage of 12,391 participants with data available): sex (99.5%, data missing for 75 participants), seizure type (96%, data missing for 555 participants), drug randomised (99.9%, data missing for 11 participants), age at randomisation (99%, data missing for 98 participants), number of seizures in six months prior to randomisation (83%, data missing for 2135 participants), and time since first seizure to randomisation (37%, data missing for 7820 participants).
Table 2.
Characteristics of participants providing individual participant data (categorical variables)
| Trial | Gender | Epilepsy type | Epilepsy type reclassifiedc | ||||||
| Male | Female | Missing | Genb | Partial | Missing | Genb | Partial | Unclassifiedd | |
| Banu 2007 | 61 (56%) | 47 (44%) | 0 (0%) | 49 (45%) | 59 (55%) | 0 (0%) | 49 (45%) | 59 (55%) | 0 (0%) |
| Baulac 2012 | 347 (60%) | 236 (40%) | 0 (0%) | 0 (0%) | 583 (100%) | 0 (0%) | 0 (0%) | 583 (100%) | 0 (0%) |
| Bill 1997 | 174 (61%) | 113 (39%) | 0 (0%) | 105 (37%) | 182 (63%) | 0 (0%) | 75 (26%) | 182 (63%) | 30 (10%) |
| Biton 2001 | 60 (44%) | 75 (55%) | 1 (1%) | 46 (34%) | 82 (60%) | 8 (6%) | 33 (24%) | 82 (60%) | 21 (15%) |
| Brodie 1995a | 56 (41%) | 80 (59%) | 0 (0%) | 54 (40%) | 82 (60%) | 0 (0%) | 34 (25%) | 82 (60%) | 20 (15%) |
| Brodie 1995b | 56 (45%) | 68 (55%) | 0 (0%) | 62 (50%) | 62 (50%) | 0 (0%) | 39 (31%) | 62 (50%) | 23 (19%) |
| Brodie 1999 | 83 (55%) | 67 (45%) | 0 (0%) | 45 (30%) | 105 (70%) | 0 (0%) | 0 (0%) | 105 (70%) | 45 (30%) |
| Brodie 2007 | 319 (55%) | 260 (45%) | 0 (0%) | 113 (20%) | 466 (80%) | 0 (0%) | 50 (9%) | 466 (80%) | 63 (11%) |
| Chadwick 1998 | 157 (54%) | 135 (46%) | 0 (0%) | 0 (0%) | 292 (100%) | 0 (0%) | 0 (0%) | 292 (100%) | 0 (0%) |
| Craig 1994 | 71 (43%) | 92 (55%) | 3 (2%) | 86 (52%) | 80 (48%) | 0 (0%) | 2 (1%) | 80 (48%) | 84 (51%) |
| de Silva 1996 | 86 (50%) | 81 (47%) | 6 (3%) | 84 (49%) | 89 (51%) | 0 (0%) | 84 (49%) | 89 (51%) | 0 (0%) |
| Dizdarer 2000 | 21 (40%) | 31 (60%) | 0 (0%) | 0 (0%) | 52 (100%) | 0 (0%) | 0 (0%) | 52 (100%) | 0 (0%) |
| Eun 2012 | 48 (57%) | 36 (43%) | 0 (0%) | 0 (0%) | 84 (100%) | 0 (0%) | 0 (0%) | 84 (100%) | 0 (0%) |
| Guerreiro 1997 | 100 (52%) | 93 (48%) | 0 (0%) | 50 (26%) | 143 (74%) | 0 (0%) | 45 (23%) | 143 (74%) | 5 (3%) |
| Heller 1995 | 117 (48%) | 126 (52%) | 0 (0%) | 141 (58%) | 102 (42%) | 0 (0%) | 82 (34%) | 102 (42%) | 59 (24%) |
| Kwan 2009 | 40 (49%) | 41 (51%) | 0 (0%) | 48 (59%) | 29 (36%) | 4 (5%) | 25 (31%) | 29 (36%) | 27 (33%) |
| Lee 2011 | 57 (52%) | 53 (48%) | 0 (0%) | 15 (14%) | 95 (86%) | 0 (0%) | 6 (5%) | 95 (86%) | 9 (8%) |
| Mattson 1985 | 413 (87%) | 58 (12%) | 4 (1%) | 1 (0%) | 474 (100%) | 0 (0%) | 1 (0%) | 474 (100%) | 0 (0%) |
| Mattson 1992 | 445 (93%) | 35 (7%) | 0 (0%) | 0 (0%) | 480 (100%) | 0 (0%) | 0 (0%) | 480 (100%) | 0 (0%) |
| Nieto‐Barrera 2001 | 329 (53%) | 293 (47%) | 0 (0%) | 3 (1%) | 619 (99%) | 0 (0%) | 1 (0%) | 619 (100%) | 2 (0%) |
| Ogunrin 2005 | 34 (62%) | 21 (38%) | 0 (0%) | 45 (82%) | 10 (18%) | 0 (0%) | 26 (47%) | 10 (18%) | 19 (35%) |
| Pal 1998 | 47 (50%) | 45 (48%) | 2 (2%) | 34 (36%) | 60 (64%) | 0 (0%) | 34 (36%) | 60 (64%) | 0 (0%) |
| Placencia 1993 | 67 (35%) | 125 (65%) | 0 (0%) | 59 (31%) | 133 (69%) | 0 (0%) | 35 (18%) | 133 (69%) | 24 (13%) |
|
Privitera 2003 (CBZ branch)a |
215 (54%) | 180 (46%) | 0 (0%) | 88 (22%) | 285 (72%) | 22 (6%) | 51 (13%) | 285 (72%) | 59 (15%) |
|
Privitera 2003 (VPS branch)a |
112 (50%) | 113 (50%) | 0 (0%) | 131 (58%) | 78 (35%) | 16 (7%) | 86 (38%) | 78 (35%) | 61 (27%) |
| Ramsey 1992 | 73 (54%) | 63 (46%) | 0 (0%) | 136 (100%) | 0 (0%) | 0 (0%) | 110 (81%) | 0 (0%) | 26 (19%) |
| Ramsey 2010 | 126 (48%) | 135 (52%) | 0 (0%) | 150 (57%) | 53 (20%) | 58 (22%) | 80 (31%) | 53 (20%) | 128 (49%) |
| Reunanen 1996 | 188 (54%) | 163 (46%) | 0 (0%) | 114 (32%) | 237 (68%) | 0 (0%) | 71 (20%) | 237 (68%) | 43 (12%) |
| Richens 1994 | 153 (51%) | 147 (49%) | 0 (0%) | 154 (51%) | 146 (49%) | 0 (0%) | 87 (29%) | 146 (49%) | 67 (22%) |
| SANAD A 2007 | 922 (54%) | 755 (44%) | 44 (3%) | 25 (1%) | 1491 (87%) | 205 (12%) | 16 (1%) | 1491 (87%) | 214 (12%) |
| SANAD B 2007 | 420 (59%) | 282 (39%) | 14 (2%) | 463 (65%) | 52 (7%) | 201 (28%) | 397 (55%) | 52 (7%) | 267 (37%) |
| Steiner 1999 | 101 (56%) | 80 (44%) | 0 (0%) | 91 (50%) | 90 (50%) | 0 (0%) | 55 (30%) | 90 (50%) | 36 (20%) |
| Stephen 2007 | 114 (50%) | 112 (49%) | 1 (0%) | 32 (14%) | 154 (68%) | 41 (18%) | 29 (13%) | 154 (68%) | 44 (19%) |
|
Trinka 2013 (CBZ branch)a |
551 (55%) | 448 (45%) | 0 (0%) | 141 (14%) | 858 (86%) | 0 (0%) | 48 (5%) | 858 (86%) | 93 (9%) |
|
Trinka 2013 (VPS branch)a |
398 (57%) | 305 (43%) | 0 (0%) | 513 (73%) | 190 (27%) | 0 (0%) | 285 (41%) | 190 (27%) | 228 (32%) |
| Turnbull 1985 | 73 (52%) | 67 (48%) | 0 (0%) | 77 (55%) | 63 (45%) | 0 (0%) | 42 (30%) | 63 (45%) | 35 (25%) |
| Verity 1995 | 122 (47%) | 138 (53%) | 0 (0%) | 152 (58%) | 108 (42%) | 0 (0%) | 152 (58%) | 108 (42%) | 0 (0%) |
| Werhahn 2015 | 215 (60%) | 146 (40%) | 0 (0%) | 0 (0%) | 361 (100%) | 0 (0%) | 0 (0%) | 361 (100%) | 0 (0%) |
| Total | 6971(56%) | 5345 (43%) | 75 (1%) | 3307 (27%) | 8529 (69%) | 555 (4%) | 2130 (17%) | 8529 (69%) | 1732 (14%) |
aTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review. bGen: Generalised tonic‐clonic seizures with or without other seizure types cSee Sensitivity analysis for further details of misclassification of epilepsy type dUnclassified seizures defined as missing seizure type or generalised onset seizures and age of onset of seizures over the age of 30 years (see Sensitivity analysis for further details)
Table 3.
Characteristics of participants providing individual participant data (continuous variables)
| Trial | Age (years) | Epilepsy duration (years) |
Number of seizures in the last 6 months |
|||||||
| Mean | SD | Range | Missing | Median | Range | Missing | Median | Range | Missing | |
| Banu 2007 | 5.7 | 3.5 | 1 to 15 | 0 | 1.2 | 0 to 11.5 | 0 | 24 | 1 to 7200 | 5 |
| Baulac 2012 | 36.4 | 15.9 | 18 to 75 | 0 | 0.2 | 0 to 17.7 | 30 | 2 | 1 to 30 | 1 |
| Bill 1997 | 26.8 | 10.7 | 15 to 91 | 1 | 0.4 | 0 to 25 | 0 | 3 | 0 to 252 | 0 |
| Biton 2001 | 32 | 14.5 | 12 to 76 | 0 | 1 | 0 to 53 | 27 | 2 | 0 to 100 | 2 |
| Brodie 1995a | 34 | 15.8 | 14 to 71 | 0 | 1 | 0 to 18 | 0 | 4 | 1 to 960 | 0 |
| Brodie 1995b | 30 | 14.1 | 14 to 81 | 0 | 0.5 | 0 to 19.4 | 0 | 3 | 1 to 1020 | 0 |
| Brodie 1999 | 76.9 | 6 | 65 to 94 | 0 | NA | NA | 150 | 3 | 0 to 163 | 0 |
| Brodie 2007 | 39 | 16.2 | 15 to 82 | 0 | NA | NA | 579 | 3 | 1 to 1410 | 4 |
| Chadwick 1998 | 35 | 16.6 | 12 to 86 | 0 | 0.5 | 0 to 7.7 | 5 | 4 | 1 to 146 | 6 |
| Craig 1994 | 78.2 | 7.1 | 61 to 95 | 3 | NA | NA | 166 | 3 | 0 to 99 | 3 |
| de Silva 1996 | 9.9 | 3.6 | 3 to 16 | 6 | 0.5 | 0 to 13.7 | 6 | 3 | 1 to 900 | 6 |
| Dizdarer 2000 | 10.8 | 2.3 | 4 to 15 | 0 | NA | NA | 52 | 3 | 1 to 60 | 0 |
| Eun 2012 | 8.8 | 2.1 | 5 to 13 | 0 | 0.4 | 0 to 4.5 | 0 | 3 | 2 to 11 | 0 |
| Guerreiro 1997 | 18.6 | 9.7 | 5 to 53 | 1 | 0.4 | 0 to 20 | 0 | 2 | 0 to 157 | 0 |
| Heller 1995 | 32.3 | 14.8 | 13 to 77 | 3 | 1 | 0 to 40 | 4 | 2 | 1 to 579 | 3 |
| Kwan 2009 | 33.9 | 10.9 | 16 to 56 | 0 | NA | NA | 81 | 1 | 0 to 540 | 0 |
| Lee 2011 | 35.8 | 12.2 | 16 to 60 | 0 | NA | NA | 110 | 2 | 0 to 200 | 0 |
| Mattson 1985 | 41 | 15.5 | 18 to 82 | 4 | 2 | 0.5 to 59 | 5 | 1 | 1 to 100 | 7 |
| Mattson 1992 | 47.1 | 16.1 | 18 to 83 | 0 | 3 | 1 to 68 | 19 | 12 | 1 to 2248 | 38 |
| Nieto‐Barrera 2001 | 27.2 | 21.4 | 2 to 84 | 1 | NA | NA | 622 | 3 | 1 to 9000 | 0 |
| Ogunrin 2005 | 27.5 | 8.5 | 14 to 55 | 0 | 7 | 3 to 11.5 | 18 | 12 | 6 to 42 | 0 |
| Pal 1998 | 11.4 | 5 | 2 to 18 | 0 | 2.5 | 0.5 to 17 | 2 | NA | NA | 94 |
| Placencia 1993 | 29 | 17.6 | 2 to 68 | 0 | 5 | 0.5 to 44 | 0 | 2 | 0 to 100 | 0 |
|
Privitera 2003 (CBZ branch)a |
34.4 | 18.4 | 6 to 80 | 0 | NA | NA | 395 | 4 | 0 to 2400 | 0 |
|
Privitera 2003 (VPS branch)a |
32.8 | 19.4 | 6 to 84 | 0 | NA | NA | 225 | 4 | 0 to 20000 | 0 |
| Ramsey 1992 | 20.9 | 14.2 | 3 to 64 | 0 | 0 | 0 to 3 | 0 | NA | NA | 136 |
| Ramsey 2010 | 34.1 | 14.8 | 12 to 78 | 0 | NA | NA | 261 | 4 | 0 to 570 | 0 |
| Reunanen 1996 | 32.1 | 14.2 | 12 to 72 | 2 | 0.7 | 0 to 27 | 3 | 3 | 1 to 145 | 1 |
| Richens 1994 | 33 | 14.9 | 16 to 79 | 2 | NA | NA | 300 | 4 | 2 to 101 | 5 |
| SANAD A 2007 | 38.4 | 18.3 | 5 to 86 | 44 | NA | NA | 1721 | 4 | 0 to 1185 | 49 |
| SANAD B 2007 | 22.5 | 14.1 | 5 to 77 | 14 | NA | NA | 716 | 3 | 0 to 2813 | 17 |
| Steiner 1999 | 34.1 | 16.7 | 13 to 75 | 1 | 1.3 | 0 to 28.5 | 1 | 3 | 1 to 600 | 0 |
| Stephen 2007 | 36 | 16.9 | 13 to 80 | 2 | NA | NA | 227 | 18 | 6 to 1080 | 37 |
|
Trinka 2013 (CBZ branch)1 |
42.8 | 17.2 | 16 to 89 | 0 | NA | NA | 999 | NA | NA | 999 |
|
Trinka 2013 (VPS branch)1 |
36.5 | 17.8 | 16 to 85 | 1 | NA | NA | 703 | NA | NA | 703 |
| Turnbull 1985 | 35.2 | 16.1 | 14 to 70 | 0 | 0.75 | 0.1 to 30 | 0 | 2 | 0 to 60 | 0 |
| Verity 1995 | 10.1 | 2.9 | 5 to 16 | 13 | 0.3 | 0 to 5.9 | 32 | 3 | 1 to 104 | 12 |
| Werhahn 2015 | 71.5 | 7.2 | 60 to 95 | 0 | NA | NA | 361 | 2 | 1 to 96 | 7 |
| Total (missing) | 98 | 7820 | 2135 | |||||||
Abbreviations: SD: Standard deviation
aTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review.
Table 4.
Characteristics of participants providing individual participant data (baseline investigations)
| Trial | Electroencephalographic (EEG) |
Computerised Tomography (CT) /Magnetic Resonance Imaging (MRI) |
Neurological exams | ||||||
| Normal | Abnormal | Missing | Normal | Abnormal | Missing | Normal | Abnormal | Missing | |
| Banu 2007 | 49 (45%) | 54 (50%) | 5 (5%) | 21 (19%) | 5 (5%) | 82 (76%) | 0 (0%) | 0 (0%) | 108 (100%) |
| Baulac 2012 | 0 (0%) | 0 (0%) | 583 (100%) | 0 (0%) | 0 (0%) | 583 (100%) | 478 (82%) | 103 (18%) | 2 (0%) |
| Bill 1997 | 126 (44%) | 152 (53%) | 9 (3%) | 173 (60%) | 69 (24%) | 45 (16%) | 0 (0%) | 0 (0%) | 287 (100%) |
| Biton 2001 | 0 (0%) | 0 (0%) | 136 (100%) | 0 (0%) | 0 (0%) | 136 (100%) | 89 (65%) | 46 (34%) | 1 (1%) |
| Brodie 1995a | 62 (46%) | 72 (53%) | 2 (1%) | 82 (60%) | 12 (9%) | 42 (31%) | 123 (90%) | 13 (10%) | 0 (0%) |
| Brodie 1995b | 76 (61%) | 42 (34%) | 6 (5%) | 72 (58%) | 20 (16%) | 32 (26%) | 108 (87%) | 16 (13%) | 0 (0%) |
| Brodie 1999 | 0 (0%) | 0 (0%) | 150 (100%) | 62 (41%) | 87 (58%) | 1 (1%) | 90 (60%) | 60 (40%) | 0 (0%) |
| Brodie 2007 | 0 (0%) | 0 (0%) | 579 (100%) | 0 (0%) | 0 (0%) | 579 (100%) | 493 (85%) | 86 (15%) | 0 (0%) |
| Chadwick 1998 | 107 (37%) | 179 (61%) | 6 (2%) | 0 (0%) | 0 (0%) | 292 (100%) | 0 (0%) | 0 (0%) | 292 (100%) |
| Craig 1994 | 28 (17%) | 74 (45%) | 64 (39%) | 0 (0%) | 0 (0%) | 166 (100%) | 0 (0%) | 0 (0%) | 166 (100%) |
| de Silva 1996 | 0 (0%) | 0 (0%) | 173 (100%) | 0 (0%) | 0 (0%) | 173 (100%) | 152 (88%) | 15 (9%) | 6 (3%) |
| Dizdarer 2000 | 18 (35%) | 34 (65%) | 0 (0%) | 50 (96%) | 2 (4%) | 0 (0%) | 0 (0%) | 0 (0%) | 52 (100%) |
| Eun 2012 | 6 (7%) | 78 (93%) | 0 (0%) | 75 (89%) | 9 (11%) | 0 (0%) | 83 (99%) | 1 (1%) | 0 (0%) |
| Guerreiro 1997 | 92 (48%) | 99 (51%) | 2 (1%) | 126 (65%) | 12 (6%) | 55 (28%) | 0 (0%) | 0 (0%) | 193 (100%) |
| Heller 1995 | 0 (0%) | 0 (0%) | 243 (100%) | 0 (0%) | 0 (0%) | 243 (100%) | 222 (91%) | 19 (8%) | 2 (1%) |
| Kwan 2009 | 0 (0%) | 0 (0%) | 81 (100%) | 0 (0%) | 0 (0%) | 81 (100%) | 0 (0%) | 0 (0%) | 81 (100%) |
| Lee 2011 | 58 (53%) | 52 (47%) | 0 (0%) | 74 (67%) | 36 (33%) | 0 (0%) | 110 (100%) | 0 (0%) | 0 (0%) |
| Mattson 1985 | 126 (27%) | 343 (72%) | 6 (1%) | 308 (65%) | 119 (25%) | 48 (10%) | 0 (0%) | 0 (0%) | 475 (100%) |
| Mattson 1992 | 0 (0%) | 0 (0%) | 480 (100%) | 0 (0%) | 0 (0%) | 480 (100%) | 0 (0%) | 0 (0%) | 480 (100%) |
| Nieto‐Barrera 2001 | 0 (0%) | 0 (0%) | 622 (100%) | 0 (0%) | 0 (0%) | 622 (100%) | 0 (0%) | 0 (0%) | 622 (100%) |
| Ogunrin 2005 | 0 (0%) | 0 (0%) | 55 (100%) | 37 (67%) | 0 (0%) | 18 (33%) | 55 (100%) | 0 (0%) | 0 (0%) |
| Pal 1998 | 0 (0%) | 0 (0%) | 94 (100%) | 0 (0%) | 0 (0%) | 94 (100%) | 24 (26%) | 70 (74%) | 0 (0%) |
| Placencia 1993 | 180 (94%) | 12 (6%) | 0 (0%) | 0 (0%) | 0 (0%) | 192 (100%) | 0 (0%) | 0 (0%) | 192 (100%) |
|
Privitera 2003 (CBZ branch)a |
0 (0%) | 0 (0%) | 395 (100%) | 0 (0%) | 0 (0%) | 395 (100%) | 0 (0%) | 0 (0%) | 395 (100%) |
|
Privitera 2003 (VPS branch)a |
0 (0%) | 0 (0%) | 225 (100%) | 0 (0%) | 0 (0%) | 225 (100%) | 0 (0%) | 0 (0%) | 225 (100%) |
| Ramsey 1992 | 0 (0%) | 0 (0%) | 136 (100%) | 0 (0%) | 0 (0%) | 136 (100%) | 0 (0%) | 0 (0%) | 136 (100%) |
| Ramsey 2010 | 0 (0%) | 0 (0%) | 261 (100%) | 0 (0%) | 0 (0%) | 261 (100%) | 0 (0%) | 0 (0%) | 261 (100%) |
| Reunanen 1996 | 13 (4%) | 13 (4%) | 325 (93%) | 16 (5%) | 5 (1%) | 330 (94%) | 305 (87%) | 46 (13%) | 0 (0%) |
| Richens 1994 | 0 (0%) | 0 (0%) | 300 (100%) | 0 (0%) | 0 (0%) | 300 (100%) | 0 (0%) | 0 (0%) | 300 (100%) |
| SANAD A 2007 | 0 (0%) | 0 (0%) | 1721 (100%) | 0 (0%) | 0 (0%) | 1721 (100%) | 1267 (74%) | 410 (24%) | 44 (3%) |
| SANAD B 2007 | 0 (0%) | 0 (0%) | 716 (100%) | 0 (0%) | 0 (0%) | 716 (100%) | 595 (83%) | 107 (15%) | 14 (2%) |
| Steiner 1999 | 103 (57%) | 71 (39%) | 7 (4%) | 111 (61%) | 33 (18%) | 37 (20%) | 165 (91%) | 16 (9%) | 0 (0%) |
| Stephen 2007 | 51 (22%) | 121 (53%) | 55 (24%) | 0 (0%) | 0 (0%) | 227 (100%) | 0 (0%) | 0 (0%) | 227 (100%) |
|
Trinka 2013 (CBZ branch)1 |
0 (0%) | 0 (0%) | 999 (100%) | 0 (0%) | 0 (0%) | 999 (100%) | 0 (0%) | 0 (0%) | 999 (100%) |
|
Trinka 2013 (VPS branch)1 |
0 (0%) | 0 (0%) | 703 (100%) | 0 (0%) | 0 (0%) | 703 (100%) | 0 (0%) | 0 (0%) | 703 (100%) |
| Turnbull 1985 | 70 (50%) | 70 (50%) | 0 (0%) | 17 (12%) | 10 (7%) | 113 (81%) | 0 (0%) | 0 (0%) | 140 (100%) |
| Verity 1995 | 0 (0%) | 0 (0%) | 260 (100%) | 0 (0%) | 0 (0%) | 260 (100%) | 0 (0%) | 0 (0%) | 260 (100%) |
| Werhahn 2015 | 117 (32%) | 242 (67%) | 2 (1%) | 78 (22%) | 282 (78%) | 1 (0%) | 0 (0%) | 0 (0%) | 361 (100%) |
| Total | 1282 (10%) | 1708 (14%) | 9401 (75%) | 1302 (11%) | 701 (6%) | 10,388 (83%) | 4359 (36%) | 1008 (8%) | 7024 (56%) |
aTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review.
Thirteen trials (Baulac 2012; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2007; de Silva 1996; Eun 2012; Heller 1995; Lee 2011; Ogunrin 2005; Pal 1998; Reunanen 1996; Steiner 1999) provided the results of neurological examinations for 5367 participants (43%). Seventeen trials (Banu 2007; Bill 1997; Brodie 1995a; Brodie 1995b; Chadwick 1998; Craig 1994; Dizdarer 2000; Eun 2012; Guerreiro 1997; Lee 2011; Mattson 1985; Placencia 1993; Reunanen 1996; Steiner 1999; Stephen 2007; Turnbull 1985; Werhahn 2015) provided electroencephalographic (EEG) results for 2990 participants (24%). Fifteen trials (Banu 2007; Bill 1997; Brodie 1995a; Brodie 1995b; Brodie 1999; Dizdarer 2000; Eun 2012; Guerreiro 1997; Lee 2011; Mattson 1985; Ogunrin 2005; Reunanen 1996; Steiner 1999; Turnbull 1985; Werhahn 2015) provided computerised tomography/magnetic resonance imaging (CT/MRI) results for 2083 participants (16%).
Trials without individual participant data
The remaining 41 trials recruiting 5570 participants did not provide IPD for the review (Aikia 1992; Bidabadi 2009; Brodie 2002; Callaghan 1985; Capone 2008; Castriota 2008; Chen 1996; Cho 2011; Christe 1997; Consoli 2012; Cossu 1984; Czapinski 1997; Dam 1989; Donati 2007; Feksi 1991; Forsythe 1991; Fritz 2006; Gilad 2007; Jung 2015; Kalviainen 2002; Kopp 2007; Korean Lamotrigine Study Group 2008; Lukic 2005; Mitchell 1987; Miura 1990; Motamedi 2013; NCT01498822; NCT01954121; Pulliainen 1994; Ramsey 1983; Ramsey 2007; Rastogi 1991; Ravi Sudhir 1995; Resendiz 2004; Rowan 2005; Saetre 2007; Shakir 1981; So 1992; Steinhoff 2005; Suresh 2015; Thilothammal 1996).
In response to our direct requests for IPD, trial authors or government sponsors of nine trials confirmed that data were no longer available (Callaghan 1985; Capone 2008; Consoli 2012; Forsythe 1991; Pulliainen 1994; Ramsey 1983; Shakir 1981; So 1992; Thilothammal 1996).
Data could not be provided for three pharmaceutical trials where data were requested via ClinicalStudyDataRequest.Com, due to the cost and resource of locating and preparing data (Kalviainen 2002; Saetre 2007) and due to country‐specific restrictions regarding anonymisation of data (Steinhoff 2005). For three further pharmaceutical company‐sponsored trials, data were not available could not be provided due to time elapsed since the trial was completed (Brodie 2002; Christe 1997; Donati 2007).
The authors of three trials confirmed that the data we required had not been collected (Chen 1996; Lukic 2005; Mitchell 1987) and the authors of two trials stated that data could not be provided due to local authority/ethical restrictions (Cho 2011; Jung 2015).
We were unable to make contact with the authors or sponsors of 14 trials to request data (Aikia 1992; Bidabadi 2009; Castriota 2008; Cossu 1984; Dam 1989; Fritz 2006; Kopp 2007; Miura 1990; Motamedi 2013; Ramsey 2007; Rastogi 1991; Ravi Sudhir 1995; Resendiz 2004; Suresh 2015).
We received an initially positive response from the authors or government sponsors of three trials but no data were provided (Czapinski 1997; Gilad 2007; Rowan 2005) and for two pharmaceutical trials, data could not be made available until a final manuscript had been published for the trials (NCT01498822; NCT01954121). Our IPD request to the sponsor of one trial is still ongoing (Korean Lamotrigine Study Group 2008); if data are provided at a later date for these trials, they will be included in an update of this review.
An author of Feksi 1991 provided access to an IPD dataset, but this was not the final dataset used for the analysis published by the original trial authors. The pharmaceutical company that sponsored the trial, Ciba‐Geigy, who at that time held the product licence for carbamazepine, held the final dataset. Since the trial was undertaken, there have been a number of mergers and restructures within the industry, and the current owners of the data are Novartis. Unfortunately, Novartis were unable to locate the data for this trial. The dataset that we had for this trial contained a number of problems and inconsistencies, and we therefore decided not to include this trial in the meta‐analysis. This was the only trial with major inconsistencies that prevented the inclusion of this IPD in analysis; for details of minor inconsistencies between IPD and published results, see Sensitivity analysis and Other potential sources of bias.
Two trials (Forsythe 1991; Shakir 1981) presented times at which the allocated drug was withdrawn and the reason for withdrawal in the trial publication for each individual. However only Shakir 1981 provided this information according to seizure type, so only results for Shakir 1981 could be incorporated into the analysis of 'Time to withdrawal of allocated treatment' (see Sensitivity analysis). Shakir 1981 presented 'Time on trial drug' in months for each participant, therefore to calculate 'Time to withdrawal of allocated treatment', we assumed that if 'Time spent on trial drug' was five months, the individual spent five full months (152 full days) on the trial drug before withdrawal.
Three trials presented sufficient detail to extract individual withdrawal (Gilad 2007; Steinhoff 2005) or seizure times (Gilad 2007; Consoli 2012) from survival curves, however this information was not separated by seizure type for Consoli 2012 so we could not include the results in analysis for this trial.
A further four trials reported summary statistics or graphical data for one of more outcomes of interest of the review; however none of these trials presented information by seizure type so we could not include the results in analysis (Brodie 2002; Christe 1997; Rowan 2005; Saetre 2007).
The remaining 31 trials did not report any published results relevant to this review (Aikia 1992; Bidabadi 2009; Callaghan 1985; Capone 2008; Castriota 2008; Chen 1996; Cossu 1984; Czapinski 1997; Dam 1989; Donati 2007; Feksi 1991; Fritz 2006; Jung 2015; Kalviainen 2002; Kopp 2007; Korean Lamotrigine Study Group 2008; Lukic 2005; Mitchell 1987; Miura 1990; Motamedi 2013; NCT01498822; NCT01954121; Pulliainen 1994; Ramsey 1983; Ramsey 2007; Rastogi 1991; Ravi Sudhir 1995; Resendiz 2004; So 1992; Suresh 2015; Thilothammal 1996). Details of outcomes considered and a summary of results of each trial for which IPD were not available to us can be found in Table 11.
Table 5.
Summary of results of trials without individual participant data
| Trial | Summary of resultsb |
| Aikia 1992 | 1. MANOVA revealed no significant interaction effect of group and time 2. MANOVA revealed no significant interaction effect of group and time 3. MANOVA revealed no significant interaction effect of group and time 4. MANOVA revealed no significant interaction effect of group and time |
| Bidabadi 2009 | 1. CBZ: 64%, PHB: 63% 2. No statistically significant difference between groups 3. No statistically significant difference between groups 4. Mean seizure frequency: CBZ: 0.66, PHB: 0.8 5. Mean duration (seconds): CBZ: 12.63; PHB: 15 |
| Brodie 2002 | 1. Median time to exit: GBP: 69 days, LTG: 48 days HR: 1.043 (95% confidence interval 0.602 to 1.809) 2. Proportion of evaluable population completing the study – GBP: 71.6%, LTG: 67.1% No difference between groups for time to withdrawal for any reason 3. No difference between groups for time to first seizure 4. GBP: 76.1%, LTG: 76.8% (ITT population) 5. Withdrawals during titration: GBP: 7, LTG: 10 Withdrawals after titration: GBP: 10, LTG: 13 |
| Callaghan 1985 | 1a. PHT: 67%; CBZ: 37%; VPS: 53% 1b. PHT: 12%; CBZ: 37%; VPS: 25% 1c. PHT: 21%; CBZ: 25%; VPS: 22% 2. PHT: 10%; CBZ: 8%; VPS:11% |
| Capone 2008 | 1. CBZ: 76%, LEV: 76% 2. Proportion with AEs: CBZ: 65%, LEV: 50% 3. CBZ: 2 discontinuations due to failure to control seizures and interactions with other medications LEV: 3 discontinuations – 1 death from stroke and 2 due to AEs |
| Castriota 2008 | 1. No significant difference between groups 2. No significant difference between groups |
| Chen 1996 | 1. No significant difference between groups 2. No significant difference between groups 3. 2 children from PHB group, 1 child from CBZ group and no children from VPS group withdrew from the study because of allergic reactions 4. No significant difference between groups |
| Cho 2011 | 1. Overall effect on sleep parameters was comparable between groups. LEV group PSG significant increase post treatment compared to baseline in sleep efficiency (P = 0.039) and in decrease of wake time after sleep onset (P = 0.047), no significant change in other sleep parameters. CBZ group post treatment compared to baseline significant increases in the percentage of slow wave sleep (P = 0.038), no significant change in other sleep parameters 2. No significant difference between baseline and post‐treatment between the 2 groups |
| Christe 1997 | 1. OXC 56.6% ; VPS 53.8% 2. No significant difference between groups 3. OXC 40.6% ; VPS 33.9% 4. Efficacy no significant difference between groups Tolerability no significant difference between groups Therapeutic effect no significant difference between groups 5. Proportion of participants experiencing at least 1 AE regardless of relationship to trial drug OXC 89.8%; VPS 87.6% 6. Seizure frequency per week OXC (n = 106) mean 0.17 median 0, VPS (n = 106) mean 0.40, median 0 |
| Consoli 2012 | 1. No significant difference between groups 2. Completed study LEV 52/62, CBZ 54/66, withdrawals: 8 poor compliance (LEV 4, CBZ 4); 7 severe adverse effect (LEV 3, CBZ 4); 7 unknown cause (LEV 3, CBZ 4) 3. Attention deficit on digital span end of follow up greater in CBZ group than LEV (P = 0.03) Stroop test worse in CBZ than LEV (P = 0.02) No significant difference between groups for other scales. Impairment of activities of daily living greater CBZ than LEV (P = 0.05) 4. 4 participants (LEV 2, CBZ 2) had abnormal EEG at baseline, normal at end of treatment. Drug dose reduction (LEV 4, CBZ 2). Remaining participants unmodified versus baseline 5. No significant difference between groups |
| Cossu 1984 | 1. Significant decrease in visual‐verbal memory for CBZ and acoustic memory for PHB. No significant differences for other tests |
| Czapinski 1997 | 1. PHB: 60%, PHT: 59%; CBZ: 62%; VPS: 64% 2. PHB: 33%, PHT: 23%; CBZ: 30%; VPS: 23% |
| Dam 1989 | 1. Baseline OXC mean 2.9 (SD 7.0), median 1, range 0‐60 CBZ mean 5.8 (SD 14.7) median 1, range 0‐99 Maintenance phases OXC mean 0.4 (SD 3.0) median 0, range 0‐27 CBZ mean 0.3 (SD 1.4) median 0, range 0‐12 2. Severe side effects CBZ 25, OXC 13, statistically significant difference favouring OXC (P = 0.04) Participants without any side effects CBZ 25, OXC 29 no significant difference between groups (P = 0.22) 3. Global efficacy no significant difference between groups (P = 0.77); global tolerability (P = 0.11) Participants very good/good CBZ 69 (73%), OXC 76 (84%) Participants poor/very poor CBZ 26 (27%), OXC 15 (16%) 4. Nature of side effects same between groups, included tiredness, headache, dizziness, ataxia. Participants withdrawn due to severe side effects CBZ 16, OXC 9 5. Clinically relevant changes observed in 2 participants only, both CBZ group, both stopped treatment |
| Donati 2007 | 1. Comparison of cognitive results no significant difference between treatment groups (P = 0.195) No significant difference between treatment groups for secondary variables (psychomotor speed, alertness, memory and learning, attention, intelligence scores) 2. OXC 58%; CBZ 46%; VPS 54% 3. Most common (> 10% reported) side effects OXC fatigue and headache; CBZ fatigue and rash VPS headache, increased appetite, alopecia 4. Good/very good: OXC investigators 84%, participants 82%, parents/carers 86%; Combined CBZ/VPS investigators 77%, participants 73%, parents/carers 80% |
| Feksi 1991 | 1. Minor adverse effects reported in PHB: 58 participants (39%) reported 86 AEs, CBZ: 46 participants (30%) reported 68 AEs 2. All withdrawals: PHB: 18%, CBZ: 17% Withdrawals due to side‐effects: PHB: 5%, CBZ: 3% 3. Seizure‐free: PHB: 54%, CBZ: 52% > 50% reduction of seizures: PHB: 23%, CBZ: 29% 50% reduction‐50% increase in seizures: PHB: 15%, CBZ: 13% > 50% increase in seizures: PHB: 8%, CBZ: 6% |
| Forsythe 1991 | 1. Significant difference favouring VPS test of speed of information processing No significant differences between treatment groups for any other cognitive tests 2. PHT: 30%; CBZ: 39%; VPS:33% |
| Fritz 2006 | 1. Seizure freedom: LTG: 38%, OXC: 44% < 50% seizure reduction: LTG: 48%, OXC: 55% 2. Both groups showed improvement in verbal learning and in 1/4 measures of attention. In addition, participants under OXC improved in word fluency. Improved mood was reported with OXC only. |
| Gilad 2007 | 1. Number of participants experiencing early seizures as first event: LTG 2/32, CBZ 3/32 Number of participants remaining seizure‐free in the follow‐up period: LTG 23/32 (72%), CBZ 14/32 (44%) P = 0.05 2. Incidence of side effects: LTG 2/32 (6.25%), CBZ 12/32 (37.5%) P = 0.05 3. Withdrawals from study due to side effects LTG 1/32 (3%), CBZ 10/32 (31%), P = 0.02 |
| Jung 2015 | 1. No difference between groups in terms of social competence; school competence; internalising behaviour problems; externalising behaviour problems; total behaviour problems and anxiety. Significant decrease in depression in LEV group compared to CBZ group (P = 0.027) 2. LEV 95.7% , CBZ 97.1% , P = 0.686 3. LEV 66.7%, CBZ 57.8% , P = 0.317 4. LEV 33.3%, CBZ 46.9%. Number of AEs not significantly different between groups |
| Kalviainen 2002 | 1.CBZ: 53% LTG: 56% 2. No significant difference between groups in overall cognitive score. In terms of individual assessments, only Stroop test B showed a statistically significant advantage for LTG. |
| Kopp 2007 | 1. No significant difference between groups 2. No significant difference between groups |
| Korean Lamotrigine Study Group 2008 | 1. LTG: 65% CBZ: 70% 2. Total seizure‐free rate LTG: 62% CBZ: 63% Time to first seizure: mean (SD): weeks LTG: 10 (5.09), CBZ: 10.82 (6.44) |
| Lukic 2005 | 1. LTG: 54%, VPS: 55 %, no difference by seizure type 2. LTG: 69%, VPS:68 % |
| Mitchell 1987 | 1. No significant differences between treatment groups 2. Compliance: trend towards better compliance in CBZ group (not significant) Randomised participants only: trend towards higher rate withdrawal from treatment in PHB group (not significant). More mild systemic side‐effects in CBZ group (significant). 3 children switched from CBZ to PHB and 1 from PHB to CB following adverse reactions 3. 6 months: excellent/good: PHB = 15, CBZ = 13 12 months: excellent/good: PHB = 13, CBZ = 9 |
| Miura 1990 | 1. Partial seizures ‐ PHT: 32%; CBZ: 40%; VPS : 41% Generalised seizures ‐ PHT :35%; CBZ: 15%; VPS: 7% 1. Partial seizures ‐ PHT: 24%; CBZ: 24%; VPS : 25% Generalised seizures ‐ PHT :13%; CBZ: 0%; VPS: 0% |
| Motamedi 2013 | 1. Seizure recurrence at 2 weeks ‐ LTG: 43% LEV: 35%, p=0.42 Seizure recurrence at 4 weeks ‐ LTG: 39% LEV: 33%, p=0.53 Seizure recurrence at 8 weeks ‐ LTG: 35% LEV: 28%, p=0.50 Seizure recurrence at 12 weeks ‐ LTG: 33% LEV: 24%, p=0.35 Seizure recurrence at 20 weeks ‐ LTG: 31% LEV: 13%, p=0.03 2. No significant difference between groups 3. Proportion with AEs ‐ LTG: 53%, LEV: 67% |
| NCT01498822 | 1. LEV: 12.7%, OXC: 23.4% 2. Median months: LEV: 7.6, OXC: NA (fewer than 50% of participants in the OXC group had seizure recurrence) 3. LEV: 53.8%, OXC: 58.5% 4. LEV: 34.7%, OXC: 40.9% |
| NCT01954121 | 1. LEV: 47.3%, CBZ: 68.4% 2. LEV: 48.4%, CBZ: 70.2% 3. Number of events: LEV: 88, CBZ: 45 4. Number of events: LEV: 87, CBZ: 39 5. Number of events: LEV: 97, CBZ: 57 |
| Pulliainen 1994 | 1. Compared to CBZ, participants on PHT became slower (motor speed of the hand) and their visual memory decreased. There was an equal decrease in negative mood (helplessness, irritability, depression) on PHT and CBZ 2. 3 participants taking PHT complained of tiredness, and 1 participant taking CBZ complained of facial skin problems, another tiredness and memory problems |
| Ramsey 1983 | 1. Incidence of major side effects (proportion of analysed participants): PHT 23%; CBZ: 23% Minor side effects: cognitive impairment and sedation twice as likely on CBZ compared to PHT. Other minor side effects similar between groups. 2. Treatment failures among analysed participants: PHT 4/35 (11%); CBZ: 5/35 (14%) Seizure control (among analysed participants with no major side effects): PHT: 86%; CBZ: 82% 3. Significantly lower mean LDH level at 24 weeks in CBZ participants than PHT participants. Other laboratory results similar across treatment groups |
| Ramsey 2007c | 1. 8 discontinuations; due to generalised rash (n = 1), excessive tiredness (n = 1), withdrew consent (n = 2), renal transplant (n = 1), lost to follow‐up (n = 2), died (n = 1) 2. 6 participants reported treatment‐emergent side effects. 3. No participants withdrew due to lack of seizure control |
| Rastogi 1991 | 1(a). PHT: 51%, VPS: 49% 1(b). PHT : 24%, VPS: 35% 1(c). PHT: 18%, VPS: 10% 1(d). PHT: 2%, VPS: 6% 2. All reported AEs were minor and similar rates between groups |
| Ravi Sudhir 1995 | 1. No significant differences between any tests of cognitive function taken before treatment and after 10‐12 weeks for both treatment groups |
| Resendiz 2004 | 1. Six months of seizure freedom: CBZ: 81%, TPM: 91% 50% reduction of seizures: CBZ: 84% TPM: 97% The average number of seizures was significantly less in the TPM group compared to the CBZ group at 6 and 9 months 2. AEs were mild and similar between groups 3. No significant differences between groups |
| Rowan 2005 | 1. Significant difference between 3 treatment groups (P = 0.00022) CBZ more early terminators than GBP (P = 0.008) or LTG (P < 0.0001) 2. LTG 51.4%, GBP 47.4%, CBZ 64.3% no significant difference between groups P = 0.09 3. No difference between groups for time to first, second, fifth and tenth seizure (P values = 0.18, 0.13, 0.74, 0.95 respectively) 4. More systemic toxicities on GBP than CBZ or LTG No significant differences in neuro‐toxicities between treatment groups over 12 months 5. Mean serum levels: 6 weeks GBP 8.67 ± 4.83; µg/mL, CBZ 6.79 ± 2.92 µg/mL and LTG 2.87 ± 1.60 µg/mL 52 weeks GBP 8.54 ± 5.57 µg/mL, CBZ 6.48 ± 3.72 µg/mL and LTG 3.46 ± 1.68 µg/mL Overall medical compliance 89% without significant group differences 6. 3 months LTG 49.7%, GBP 43.3%, CBZ 36.0% significant difference between groups P = 0.02 6 months LTG 37.2%, GBP 33.0%, CBZ 28.9% no significant difference between groups P = 0.22 12 months LTG 28.6%, GBP 23.2%, CBZ 22.8% no significant difference between groups P = 0.33 |
| Saetre 2007 | 1. LTG 68 (73%), CBZ 61 (67%), no significant difference between groups 2. LTG 59 (63%), CBZ 69 (76%), not significant difference P = 0.068 ITT analysis 3. LTG 71 (76%), CBZ 81 (89%), significant difference, P = 0.0234 ITT analysis 4.Hazard ratio (lamotrigine/carbamazepine) 1.50, 95% CI 0.94–2.40, p value 0.092 5. During treatment period LTG 82 (88%) reported 378 AEs, CBZ 79 (86%) reported 310 AEs. No significant differences between groups for any AEs except for immune system Withdrew due to AE LTG 13 (14%), CBZ 23 (25%), P = 0.078 6. No difference between groups even when changes over time corrected for age, gender and baseline score |
| Shakir 1981 | 1. PHT: 33%; VPS: 39% 2. All reported AEs were minor and similar rates between groups |
| So 1992 | 1. VPS 7/11 (64%), CBZ 9/14 (64%) 2. At least one AE reported VPS 15/16 (94%), CBZ 16/17 (94%) |
| Steinhoff 2005 | 1. FE CBZ group 83/88 (94.3%), LTG group 78/88 (88.6%) no significant difference between groups GE VPS group 25/30 (83.3%) LTG group 20/33 (60.6%) no significant difference between groups 2. FE CBZ group 81%, LTG group 91%, not a significant difference between groups GE VPS group 97%, LTG group 88%, not stated as significant or non‐significant difference 3. At least 1 AE FE CBZ 81 participants (91%), LTG 68 participants (77.3%) GE VPS 25 participants (83.3%), LTG 24 participants (72.7%) Serious AEs FE CBZ 8 participants (9%), LTG 6 participants (7%) GE VPS 1 participant (3%), LTG 5 participants (15%) AEs considered related to study drug FE CBZ 65 participants (74%), LTG 38 participants (43%) GE VPS 16 participants (53%), LTG 15 participants (45.5%) |
| Suresh 2015 | 1. Mean quality‐of‐life score at baseline CBZ group 31.14 ± 1.83, LEV group 29.76 ± 1.71 (P value = 0.5861) Mean quality of life score after 26 weeks of treatment CBZ group 58.41 ± 1.89, LEV 64.58 ± 2.02 (P value = 0.0302) 2.28 participants in CBZ group, 28 in LEV group Seizure freedom 4 weeks CBZ group 85.72%, LEV group 85.72% (P value = 1); 12 weeks CBZ group 89.29%, LEV group 93.34% (P value = 0.4595); 26 weeks CBZ group 96.43%, LEV group 100% (P value = 0.1212); 6 months CBZ group 71.42% (20 participants), LEV group 78.57% (22 participants) (P value = 0.2529) 3. Participants experiencing at least 1 AE, CBZ group 36.66%, LEV group 40% (P value = 0.77) |
| Thilothammal 1996 | 1. PHB: 31%, PHT: 27%, VPS: 21% 2. PHB: 33%, PHT: 63%, VPS: 31% |
AE: adverse event; CBZ: carbamazepine; EEG: electroencephalogram; FE: focal epilepsies; GBP: gabapentin; GE: generalised epilepsies; ITT: intention to treat; LDH: lactic acid dehydrogenase; LEV: levetiracetam; LTG: lamotrigine; MANOVA: repeated measures analysis of variance; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; SD: standard deviation; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aFor further details of adverse events see Table 22 and Table 23. bSee Table 7 for details of treatment arms in each trial and number of participants randomised to each arm. cResults not split by treatment arm for Ramsey 2007.
Excluded studies
We excluded 31 studies from the review; three were cross‐over trials (Cereghino 1974; Gruber 1962; Loiseau 1984), three studies were terminated early with no results available (EUCTR2004‐004053‐26‐SE; EUCTR2010‐018284‐42‐NL; ISRCTN73223855), two were not fully randomised (Baxter 1998; Kaminow 2003), one did not recruit participants with epilepsy (Taragano 2003) and the other 22 did not have a monotherapy design (Albani 2006; Alsaadi 2002; Alsaadi 2005; ; Ben‐Menachem 2003; Beydoun 1997; Beydoun 1998; Beydoun 2000; Bittencourt 1993; Canadian Group 1999; Chung 2012; DeToledo 2000; Fakhoury 2004; French 2012; Gilliam 1998; Hakami 2012; Kerr 1999; Kerr 2001; Reinikainen 1984; Reinikainen 1987; Rosenow 2012; Simonsen 1975a; Simonsen 1975b). See Characteristics of excluded studies for further information.
Risk of bias in included studies
For further details, see the Characteristics of included studies and Figure 4.
Figure 4.
Risk of bias summary: review authors' judgements about each risk of bias item for each included trial
Allocation
Trials for which we received IPD (information reported in published papers or provided with IPD)
One trial used alternate allocation (quasi randomisation) which we judged to be at high risk of selection bias (Dizdarer 2000). One trial described an adequate method of randomisation, use of a random number list, but reported that allocation was concealed by sealed, opaque envelopes, although this method was not used for all participants in the trial (Placencia 1993), so we also judged this trial to be at high risk of selection bias.
Twenty trials described adequate methods of generation of random sequence and allocation concealment and we judged them to be at low risk of bias. One trial used a random number list and central allocation (Ogunrin 2005). Four trials used block randomisation, of which three concealed allocation with sealed, opaque envelopes (de Silva 1996; Heller 1995; Mattson 1992) and one used central pharmacy allocation (Chadwick 1998). Ten trials used a computer‐generated random sequence. Of these, seven concealed allocation with sealed, opaque envelopes (Bill 1997; Brodie 1995a; Brodie 1995b; Brodie 1999; Guerreiro 1997; Nieto‐Barrera 2001; Reunanen 1996), two used a telephone interactive voice‐response system (Baulac 2012; Brodie 2007) and one used central pharmacy allocation (Werhahn 2015). Five trials used a computer‐generated minimisation programme: four used central telephone allocation (Richens 1994; SANAD A 2007; SANAD B 2007; Verity 1995) and one used central pharmacy allocation (Craig 1994).
Two trials were described as randomised but gave no information about the generation of the random list (unclear risk of bias for generation of random sequence). One of these trials concealed allocation with sealed, opaque envelopes (Banu 2007) and one used a telephone interactive voice‐response system (Trinka 2013) (both low risk of bias for allocation concealment). Five trials gave no information about allocation concealment (unclear risk of bias). Of these, three used a computer‐generated random sequence (Biton 2001; Eun 2012; Privitera 2003) and two used random number tables (Pal 1998; Ramsey 1992) (all low risk of bias for generation of random sequence).
The remaining seven trials were described as randomised but gave no details of methods of generation of random sequence and allocation concealment and we judged them to be at unclear risk of bias (Kwan 2009; Lee 2011; Mattson 1985; Ramsey 2010; Steiner 1999; Stephen 2007; Turnbull 1985).
Trials for which no IPD were available (information reported in published papers only)
We judged two trials to be at high risk of selection bias: one trial reported a method of quota allocation and did not report how allocation was concealed (Forsythe 1991) and the other reported a method of randomisation and allocation concealment based on two Latin squares which seemed to take into account the drug preference of participants (the “drug of first preference” was selected from the randomisation list on a sequential basis) (Callaghan 1985).
Five trials described adequate methods of generation of random sequence and allocation concealment and we judged them to be at low risk of bias. Of these, one trial used a random number list and sealed, opaque envelopes (Feksi 1991) and four trials used a computer‐generated random sequence, including three trials that used central telephone randomisation (Donati 2007; Rowan 2005; Shakir 1981) and one trial that used central pharmacy allocation (Jung 2015).
Six trials gave no information about allocation concealment (unclear risk of bias).Of these, two used block randomisation (Brodie 2002; Chen 1996), one used random number tables (Resendiz 2004) and three used a computer‐generated random sequence (Consoli 2012; Motamedi 2013; Thilothammal 1996) (all low risk of bias for generation of random sequence).
The remaining 28 trials were described as randomised but gave no details of methods of generation of random sequence and allocation concealment and we judged them to be at unclear risk of bias: six were published as abstracts only (Bidabadi 2009; Czapinski 1997; Fritz 2006; Kalviainen 2002; Kopp 2007; Lukic 2005); three were published only as only summary results (Korean Lamotrigine Study Group 2008; NCT01498822; NCT01954121); and nineteen were published as full‐text articles (Aikia 1992; Capone 2008; Castriota 2008; Cho 2011, Christe 1997; Cossu 1984; Dam 1989; Gilad 2007; Mitchell 1987; Miura 1990; Pulliainen 1994; Ramsey 1983; Ramsey 2007; Rastogi 1991; Ravi Sudhir 1995; Saetre 2007; So 1992; Steinhoff 2005; Suresh 2015).
Blinding
Trials for which we received IPD (information reported in published papers or provided with IPD)
Five trials reported that participants, personnel and outcome assessors were blinded via the use of matching placebo tablets (Baulac 2012; Biton 2001; Ogunrin 2005; Ramsey 2010; Steiner 1999). Eleven trials reported that participants and personnel were double‐blinded but gave no information about blinding of outcome assessors (Banu 2007; Bill 1997; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2007; Guerreiro 1997; Mattson 1985; Mattson 1992; Privitera 2003; Werhahn 2015). We judged all of these trials to be at low risk of performance bias but unclear risk of detection bias.
Two trials reported that outcome assessors were blinded but that participants and personnel were not blinded (Craig 1994; Pal 1998) and two trials gave no information about blinding so we judged them to be at unclear risk of performance and detection bias (Placencia 1993; Turnbull 1985).
Fifteen trials were of an open‐label design and judged to be at high risk of performance and detection bias (de Silva 1996; Dizdarer 2000; Eun 2012; Heller 1995; Kwan 2009; Lee 2011; Nieto‐Barrera 2001; Ramsey 1992; Reunanen 1996; Richens 1994; SANAD A 2007; SANAD B 2007; Stephen 2007; Trinka 2013; Verity 1995) and one trial could not blind participants and personnel by design but did not state whether outcome assessors were blinded (Chadwick 1998).
Trials for which no IPD were available (information reported in published papers only)
Five trials reported that outcome assessors were blinded. Of these, three did not state whether participants and personnel were blinded (Chen 1996; Cho 2011; Pulliainen 1994) and in the other two trials participants and personnel were not blinded (Forsythe 1991; Jung 2015). Eleven trials reported that participants and personnel were double‐blinded but gave no information about blinding of outcome assessors (Aikia 1992; Brodie 2002; Christe 1997; Cossu 1984; Dam 1989; Motamedi 2013; Ramsey 1983; Ramsey 2007; Rowan 2005; Saetre 2007; So 1992). We judged all of these trials to be at low risk of performance bias but unclear risk of detection bias.
Twelve trials were of an open‐label design and we judged them to be at high risk of performance and detection bias (Castriota 2008; Consoli 2012; Donati 2007; Gilad 2007; Korean Lamotrigine Study Group 2008; Lukic 2005; Mitchell 1987; NCT01498822; NCT01954121; Resendiz 2004; Steinhoff 2005, Suresh 2015)
Thirteen trials gave no information about blinding so we judged them to be at unclear risk of performance and detection bias. Of these, five were published as abstracts only (Bidabadi 2009; Czapinski 1997; Fritz 2006; Kalviainen 2002; Kopp 2007) and eight were published as full‐text articles (Callaghan 1985; Capone 2008; Feksi 1991; Miura 1990; Rastogi 1991; Ravi Sudhir 1995; Shakir 1981; Thilothammal 1996).
Incomplete outcome data
Trials for which we received individual participant data (information reported in published papers or provided with IPD)
In theory, a review using IPD should overcome issues of attrition bias, as unpublished data can be provided, unpublished outcomes calculated, and all randomised participants can be analysed by an intention‐to‐treat approach. All 36 trials (Banu 2007; Baulac 2012; Bill 1997; Biton 2001; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2007; Chadwick 1998; Craig 1994; de Silva 1996; Dizdarer 2000; Eun 2012; Guerreiro 1997; Heller 1995; Kwan 2009; Lee 2011; Mattson 1985; Mattson 1992; Nieto‐Barrera 2001; Ogunrin 2005; Pal 1998; Placencia 1993; Privitera 2003; Ramsey 1992; Ramsey 2010; Reunanen 1996; Richens 1994; SANAD A 2007; SANAD B 2007; Steiner 1999; Stephen 2007; Trinka 2013; Turnbull 1985; Verity 1995; Werhahn 2015) provided individual participant data for all randomised individuals and reported the extent of follow‐up for each individual. We queried any missing data with the original trial authors. From the information provided by the trial authors, we deemed the small amount of missing data present (see Included studies) to be missing at random and not affecting our analysis so we judged them to be at low risk of bias.
Trials for which no IPD were available (information reported in published papers only)
Seven trials, which were published as abstracts only, did not give enough information to assess selective reporting so we judged them to have unclear risk of bias (Bidabadi 2009; Czapinski 1997; Fritz 2006; Kalviainen 2002; Kopp 2007; Lukic 2005; Ramsey 2007). Three trials excluded the small proportion of participants who withdrew from the trial from analysis but it is unclear whether this would have influenced analysis (Castriota 2008; Chen 1996; Suresh 2015) and two trials did not clearly report whether participants had withdrawn from the trial (Cho 2011; Rastogi 1991) so we also judged these trials to be at unclear risk of bias.
Twelve trials reported attrition rates and used an intention‐to‐treat approach to analysis so we judged them to be at low risk of attrition bias (Brodie 2002; Callaghan 1985; Capone 2008; Cossu 1984; Forsythe 1991; Gilad 2007; Mitchell 1987; Miura 1990; Rowan 2005; Saetre 2007; Shakir 1981; Thilothammal 1996). The remaining 17 trials excluded participants from analysis and did not use an intention‐to‐treat approach to analysis and we judged them to be at high risk of attrition bias (Aikia 1992; Christe 1997; Consoli 2012; Dam 1989; Donati 2007; Feksi 1991; Jung 2015; Korean Lamotrigine Study Group 2008; Motamedi 2013; NCT01498822; NCT01954121; Pulliainen 1994; Ramsey 1983; Ravi Sudhir 1995; Resendiz 2004; So 1992; Steinhoff 2005).
Selective reporting
Trials for which we received IPD (information reported in published papers or provided with IPD)
We requested trial protocols in all IPD requests and protocols were provided for 20 out of the 36 trials providing IPD (Baulac 2012; Bill 1997; Biton 2001; Brodie 1995a; Brodie 1995b; Brodie 1999; de Silva 1996; Guerreiro 1997; Heller 1995; Mattson 1985; Mattson 1992; Nieto‐Barrera 2001; Ogunrin 2005; Reunanen 1996; Richens 1994; SANAD A 2007; SANAD B 2007; Steiner 1999; Verity 1995; Werhahn 2015).
In theory, a review using IPD should overcome issues of reporting biases, as unpublished data can be provided and unpublished outcomes calculated, so we judged all trials providing IPD to be at low risk of bias. We received sufficient IPD to calculate the four outcomes ('Time to withdrawal of allocated treatment', 'Time to six‐month remission, 'Time to 12‐month remission', and 'Time to first seizure') for 20 of the 36 trials (Baulac 2012; Bill 1997; Brodie 2007; de Silva 1996; Dizdarer 2000; Guerreiro 1997; Heller 1995; Kwan 2009; Mattson 1985; Mattson 1992; Placencia 1993; Privitera 2003; Richens 1994; SANAD A 2007; SANAD B 2007,Stephen 2007; Trinka 2013; Turnbull 1985; Verity 1995; Werhahn 2015)
We could not calculate 'Time to 12‐month remission' for nine trials as the duration of the trial was less than 12 months (Biton 2001; Brodie 1995a; Brodie 1995b; Chadwick 1998; Eun 2012; Lee 2011; Ramsey 1992; Reunanen 1996; Steiner 1999) and we could not calculate 'Time to 12‐month remission' or 'Time to six‐month remission' for three trials as the duration of the trial was less than six months (Brodie 1999; Nieto‐Barrera 2001; Ramsey 2010).
Withdrawal information was not available for two trials so we could not calculate 'Time to withdrawal of allocated treatment' (Craig 1994; Pal 1998). For two trials we could only calculate 'Time to first seizure': the trial duration of Ogunrin 2005 was 12 weeks, and all randomised participants completed the trial without withdrawing; and Banu 2007 did not record the dates of all seizures after randomisation and dates of withdrawal for allocated treatment for all participants.
Trials for which no IPD were available (information reported in published papers only)
Protocols were not available for any of the 41 trials without IPD available, so we made a judgement of the risk of bias based on the information included in the publications or from the IPD we received (see the Characteristics of included studies tables for more information).
We judged two trials to be at high risk of reporting bias; one trial reported results for outcomes that were not defined in the methods section (Suresh 2015) and one trial did not provide online results for all listed outcomes (NCT01954121).
In 25 trials, expected efficacy and tolerability outcomes were well reported in the methods and results therefore we judged these trials to be at low risk of selective reporting bias (Aikia 1992; Brodie 2002; Callaghan 1985; Chen 1996; Cho 2011, Christe 1997; Consoli 2012; Dam 1989; Donati 2007; Feksi 1991; Gilad 2007; Jung 2015; Korean Lamotrigine Study Group 2008; Mitchell 1987; Motamedi 2013; NCT01498822; Ramsey 1983; Rastogi 1991; Resendiz 2004; Rowan 2005; Saetre 2007; Shakir 1981; So 1992; Steinhoff 2005; Thilothammal 1996).
Seven trials that were published as abstracts only (Bidabadi 2009; Czapinski 1997; Fritz 2006; Kalviainen 2002; Kopp 2007; Lukic 2005; Ramsey 2007) and one trial with a very brief description of methods (Capone 2008) did not give enough information to assess selective reporting so we judged them to have unclear risk of bias. Six trials reported only cognitive outcomes rather than expected efficacy or tolerability outcomes and it was unclear if such outcomes were planned a priori, therefore we also judged these trials to have unclear risk of bias (Castriota 2008; Cossu 1984; Forsythe 1991; Miura 1990; Pulliainen 1994; Ravi Sudhir 1995).
Other potential sources of bias
We detected another source of bias in eight trials.
Following consistency checks of IPD for Placencia 1993; Stephen 2007 and Banu 2007, we found some inconsistencies between the data provided and the results in the publications in terms of withdrawal and seizure recurrences, respectively, which the trial authors could not resolve. We performed sensitivity analysis to investigate the impact of the inconsistent data on our outcomes (see Sensitivity analysis). Furthermore, we received IPD for another trial (Feksi 1991), but too many inconsistencies were present for this data to be usable (see Included studies for further details).
We included one trial with very small participant numbers (six participants randomised to each drug) and very short‐term follow‐up (three weeks) (Cossu 1984), and one trial that terminated early with only 20% of target sample size recruited (Consoli 2012). It is unlikely that either of these trials were adequately powered and of sufficient duration to detect differences. Another trial had several other potential sources of bias (Mitchell 1987); the trial was likely underpowered to detect differences between the treatments, one of the tools for outcome assessment was not fully validated, and non‐randomised children from a related pilot study were included in analysis for some of the outcomes. In one trial, it was unclear if all participants were receiving AED monotherapy treatment (‘total number of AEDs’ described in Table 1 of the publication), so we judged this trial to be at unclear risk of bias (Gilad 2007).
No other sources of bias were identified in the remaining 69 trials.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6
For brevity throughout the results section, we refer to participants with generalised tonic‐clonic seizures with or without other generalised seizure types as 'participants with generalised seizures.'
Figure 1 and Figure 2 visually present the network of 45 pairwise comparisons from the 10 antiepileptic treatments. Figure 1 also demonstrates the network of the trials with and without IPD provided for analysis and Figure 2 also presents the network of evidence for participants with partial seizures and with generalised seizures. We note that zonisamide has only been used in a single trial recruiting individuals with partial onset seizures only (Baulac 2012), therefore zonisamide does not feature in the network of evidence for generalised seizures and there are 36 pairwise comparisons in this network.
Table 12 shows the total number of participants contributing to each analysis (Table 13 shows the reported reasons for withdrawal from treatment across all studies) and Table 14; Table 15; Table 16; Table 17; Table 18; Table 19; Table 20; Table 21 and Figure 5; Figure 6; Figure 7; Figure 8; Figure 9; Figure 10; and Figure 11 show the results for each of the outcomes below. Results highlighted in bold in the tables indicate statistically significant results and HR less than 1 indicates an advantage to the second drug in the comparison. All results presented were calculated with a fixed‐effect analysis.
Table 6.
Number of participants contributing individual participant data to analyses
| Trial | Time to withdrawal from allocated treatmentc | Time to first seizure | Time to 12‐month remissiond | Time to six‐month remissiond | ||||||||||||
| Cens | Event | Total | Missing | Cens | Event | Total | Missing | Cens | Event | Total | Missing | Cens | Event | Total | Missing | |
| Banu 2007a | 0 | 0 | 0 | 108 | 39 | 69 | 108 | 0 | 0 | 0 | 0 | 108 | 0 | 0 | 0 | 108 |
| Baulac 2012 | 392 | 191 | 583 | 0 | 388 | 186 | 574 | 9 | 251 | 323 | 574 | 9 | 194 | 380 | 574 | 9 |
| Bill 1997 | 232 | 55 | 287 | 0 | 137 | 145 | 282 | 5 | 190 | 92 | 282 | 5 | 136 | 146 | 282 | 5 |
| Biton 2001 | 99 | 36 | 135 | 1 | 64 | 71 | 135 | 1 | 0 | 0 | 0 | 136 | 90 | 45 | 135 | 1 |
| Brodie 1995a | 78 | 58 | 136 | 0 | 69 | 67 | 136 | 0 | 0 | 0 | 0 | 136 | 122 | 14 | 136 | 0 |
| Brodie 1995b | 79 | 45 | 124 | 0 | 52 | 72 | 124 | 0 | 0 | 0 | 0 | 124 | 96 | 28 | 124 | 0 |
| Brodie 1999 | 95 | 55 | 150 | 0 | 70 | 80 | 150 | 0 | 0 | 0 | 0 | 150 | 106 | 44 | 150 | 0 |
| Brodie 2007 | 323 | 256 | 579 | 0 | 350 | 229 | 579 | 0 | 260 | 319 | 579 | 0 | 177 | 402 | 579 | 0 |
| Chadwick 1998 | 69 | 223 | 292 | 0 | 102 | 190 | 292 | 0 | 0 | 0 | 0 | 292 | 193 | 99 | 292 | 0 |
| Craig 1994 | 0 | 0 | 0 | 166 | 68 | 81 | 149 | 17 | 117 | 30 | 147 | 19 | 58 | 89 | 147 | 19 |
| de Silva 1996 | 100 | 67 | 167 | 6 | 18 | 149 | 167 | 6 | 22 | 145 | 167 | 6 | 19 | 148 | 167 | 6 |
| Dizdarer 2000 | 44 | 8 | 52 | 0 | 40 | 12 | 52 | 0 | 11 | 41 | 52 | 0 | 8 | 44 | 52 | 0 |
| Eun 2012 | 75 | 9 | 84 | 0 | 52 | 32 | 84 | 0 | 0 | 0 | 0 | 84 | 35 | 49 | 84 | 0 |
| Guerreiro 1997 | 151 | 42 | 193 | 0 | 106 | 84 | 190 | 3 | 112 | 78 | 190 | 3 | 84 | 106 | 190 | 3 |
| Heller 1995 | 166 | 77 | 243 | 0 | 66 | 177 | 243 | 0 | 78 | 165 | 243 | 0 | 49 | 194 | 243 | 0 |
| Kwan 2009 | 60 | 21 | 81 | 0 | 38 | 29 | 67 | 14 | 68 | 13 | 81 | 0 | 30 | 50 | 80 | 1 |
| Lee 2011 | 73 | 37 | 110 | 0 | 79 | 31 | 110 | 0 | 0 | 0 | 0 | 110 | 39 | 71 | 110 | 0 |
| Mattson 1985 | 267 | 208 | 475 | 0 | 226 | 238 | 464 | 11 | 325 | 149 | 474 | 1 | 281 | 193 | 474 | 1 |
| Mattson 1992 | 308 | 172 | 480 | 0 | 165 | 303 | 468 | 12 | 334 | 133 | 467 | 13 | 242 | 225 | 467 | 13 |
| Nieto‐Barrera 2001 | 511 | 111 | 622 | 0 | 310 | 312 | 622 | 0 | 0 | 0 | 0 | 622 | 431 | 191 | 622 | 0 |
| Ogunrin 2005a | 0 | 0 | 0 | 55 | 29 | 26 | 55 | 0 | 0 | 0 | 0 | 55 | 0 | 0 | 0 | 55 |
| Pal 1998 | 0 | 0 | 0 | 94 | 41 | 49 | 90 | 4 | 82 | 8 | 90 | 4 | 63 | 27 | 90 | 4 |
| Placencia 1993 | 158 | 32 | 190 | 2 | 121 | 71 | 192 | 0 | 132 | 60 | 192 | 0 | 69 | 123 | 192 | 0 |
|
Privitera 2003 (CBZ branch)b |
221 | 174 | 395 | 0 | 208 | 187 | 395 | 0 | 316 | 79 | 395 | 0 | 194 | 201 | 395 | 0 |
|
Privitera 2003 (VPS branch)b |
111 | 114 | 225 | 0 | 119 | 106 | 225 | 0 | 180 | 45 | 225 | 0 | 106 | 119 | 225 | 0 |
| Ramsey 1992 | 113 | 23 | 136 | 0 | 81 | 44 | 125 | 11 | 0 | 0 | 0 | 136 | 78 | 47 | 125 | 11 |
| Ramsey 2010 | 192 | 69 | 261 | 0 | 197 | 64 | 261 | 0 | 0 | 0 | 0 | 261 | 0 | 0 | 0 | 261 |
| Reunanen 1996 | 288 | 63 | 351 | 0 | 216 | 135 | 351 | 0 | 0 | 0 | 0 | 351 | 328 | 23 | 351 | 0 |
| Richens 1994 | 210 | 76 | 286 | 14 | 91 | 199 | 290 | 10 | 92 | 198 | 290 | 10 | 77 | 213 | 290 | 10 |
| SANAD A 2007 | 857 | 815 | 1672 | 49 | 383 | 1261 | 1644 | 77 | 577 | 1067 | 1644 | 77 | 355 | 1326 | 1681 | 40 |
| SANAD B 2007 | 400 | 299 | 699 | 17 | 182 | 511 | 693 | 23 | 167 | 526 | 693 | 23 | 96 | 610 | 706 | 10 |
| Steiner 1999 | 108 | 73 | 181 | 0 | 100 | 81 | 181 | 0 | 0 | 0 | 0 | 181 | 157 | 24 | 181 | 0 |
| Stephen 2007 | 160 | 67 | 227 | 0 | 81 | 140 | 221 | 6 | 172 | 55 | 227 | 0 | 137 | 90 | 227 | 0 |
|
Trinka 2013 (CBZ branch)b |
760 | 239 | 999 | 0 | 572 | 427 | 999 | 0 | 780 | 219 | 999 | 0 | 336 | 663 | 999 | 0 |
|
Trinka 2013 (VPS branch)b |
583 | 120 | 703 | 0 | 456 | 247 | 703 | 0 | 484 | 219 | 703 | 0 | 191 | 512 | 703 | 0 |
| Turnbull 1985 | 91 | 49 | 140 | 0 | 75 | 65 | 140 | 0 | 47 | 93 | 140 | 0 | 36 | 104 | 140 | 0 |
| Verity 1995 | 187 | 59 | 246 | 14 | 59 | 187 | 246 | 14 | 84 | 162 | 246 | 14 | 19 | 227 | 246 | 14 |
| Werhahn 2015 | 195 | 166 | 361 | 0 | 249 | 96 | 345 | 16 | 211 | 150 | 361 | 0 | 178 | 183 | 361 | 0 |
| Total | 7756 | 4109 | 11,865 | 526 | 5699 | 6453 | 12,152 | 239 | 5092 | 4369 | 9461 | 2930 | 4810 | 7010 | 11,820 | 571 |
Abbreviation: cens = censored
aFor two studies we could only calculate 'Time to first seizure'; the study duration of Ogunrin 2005 was 12 weeks, and all randomised participants completed the study without withdrawing; and Banu 2007 did not record the dates of all seizures after randomisation and dates of withdrawal for allocated treatment for all participants. bTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review. cWithdrawal information was not available for two trials so we could not calculate 'Time to withdrawal of allocated treatment' (Craig 1994; Pal 1998). dWe could not calculate 'Time to 12‐month remission' for nine trials as the duration of the study was less than 12 months (Biton 2001; Brodie 1995a; Brodie 1995b; Chadwick 1998; Eun 2012; Lee 2011; Ramsey 1992; Reunanen 1996; Steiner 1999) and we could not calculate 'Time to 12‐month remission' or 'Time to six‐month remission' for three trials as the duration of the study was less than six months (Brodie 1999; Nieto‐Barrera 2001; Ramsey 2010).
Table 7.
Reasons for withdrawal from allocated treatment
| Reason for withdrawal |
Classification for analysis |
Randomised drug4b | ||||||||||
| CBZ | PHB | PHT | VPS | LTG | OXC | TPM | GBP | LEV | ZNS | Total | ||
| Adverse events | Event | 505 (45%) | 24 (20%) | 93 (35%) | 132 (28%) | 235 (41%) | 56 (41%) | 259 (48%) | 73 (20%) | 134 (39%) | 31 (32%) | 1542 (38%) |
| Inadequate response | Event | 232 (20%) | 20 (16%) | 46 (17%) | 140 (29%) | 144 (26%) | 36 (26%) | 148 (27%) | 223 (62%) | 89 (26%) | 23 (24%) | 1101 (27%) |
| Both adverse events and inadequate response |
Event | 148 (13%) | 51 (41%) | 54 (20%) | 107 (22%) | 32 (6%) | 11 (8%) | 46 (8%) | 32 (9%) | 0 (0%) | 0 (0%) | 481 (12%) |
| Protocol violation/non compliance | Event | 102 (9%) | 15 (12%) | 41 (15%) | 11 (2%) | 68 (12%) | 27 (20%) | 0 (0%) | 21 (6%) | 21 (6%) | 3 (3%) | 309 (8%) |
| Withdrew consent | Event | 121 (11%) | 13 (11%) | 25 (9%) | 64 (13%) | 65 (11%) | 2 (1%) | 55 (10%) | 4 (1%) | 68 (20%) | 35 (36%) | 452 (11%) |
| Othera | Event | 29 (3%) | 0 (0%) | 7 (3%) | 24 (5%) | 26 (5%) | 5 (4%) | 37 (7%) | 9 (2%) | 32 (9%) | 4 (4%) | 173 (4%) |
| Total eventsb | 1137 (35%) | 123 (38%) | 266 (31%) | 478 (28%) | 570 (29%) | 137 (29%) | 545 (47%) | 362 (61%) | 344 (27%) | 96 (34%) | 4058 (34%) | |
| Illness or death | Censored | 34 (2%) | 10 (5%) | 17 (3%) | 7 (1%) | 20 (1%) | 1 (0%) | 10 (2%) | 9 (4%) | 0 (0%) | 0 (0%) | 108 (1%) |
| Remission of seizures | Censored | 49 (2%) | 4 (2%) | 38 (6%) | 75 (6%) | 40 (3%) | 12 (4%) | 44 (7%) | 21 (9%) | 0 (0%) | 0 (0%) | 283 (4%) |
| Lost to follow‐up | Censored | 81 (4%) | 31 (16%) | 51 (9%) | 63 (5%) | 33 (3%) | 24 (7%) | 18 (3%) | 0 (0%) | 41 (5%) | 0 (0%) | 342 (4%) |
| Otherc | Censored | 104 (5%) | 6 (3%) | 22 (4%) | 82 (7%) | 31 (2%) | 5 (2%) | 26 (4%) | 26 (12%) | 0 (0%) | 25 (13%) | 327 (4%) |
| Completed study | Censored | 1829 (87%) | 139 (73%) | 468 (79%) | 949 (81%) | 1272 (91%) | 291 (87%) | 501 (84%) | 166 (75%) | 868 (95%) | 161 (87%) | 6644 (86%) |
| Total censoredb | 2097 (65%) | 190 (62%) | 596 (69%) | 1176 (72%) | 1396 (71%) | 333 (71%) | 599 (53%) | 222 (39%) | 909 (73%) | 186 (66%) | 7704 (66%) | |
| Missingd | 24 | 7 | 1 | 26 | 12 | 8 | 14 | 11 | 0 | 0 | 103 | |
| Totale | 3258 | 320 | 863 | 1680 | 1978 | 478 | 1158 | 595 | 1253 | 282 | 11,865 | |
CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aOther treatment‐related reasons included: physician's decision, drug‐related death, pregnancy or perceived remission, or non specific (drug‐related) reason. bProportions for specific reasons indicate proportion of total events or total censored. Proportion for total events and total censored indicate the proportion of total participants. cOther non treatment‐related reasons included: epilepsy diagnosis changed, participants developed other medical disorders including neurological and psychiatric disorders or non specific (non drug‐related) reason. dWe treated those with missing reasons for withdrawal as censored in analysis and performed a sensitivity analysis treating these individuals as having withdrawal 'events.' Results of sensitivity analysis were practically identical and conclusions unchanged so we have presented the results treating these individuals as censored. eFour studies did not contribute to analysis of time to withdrawal of allocated treatment (Banu 2007; Craig 1994; Ogunrin 2005; Pal 1998).
Table 8.
Pairwise and network meta‐analysis results ‐ Time to withdrawal of allocated treatment for individuals with partial seizures
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence (network meta‐analysis) | ||||
| Number of studies | Number of participants | HR (95% CI)b,c | I² statisticd | Direct evidence (%)e | HR (95% CI)b,c | |
| CBZ vs PHB | 4 | 520 | 1.57 (1.16 to 2.13) | 0% | 52.5% | 1.55 (1.18 to 2.04) |
| CBZ vs PHT | 3 | 428 | 1.03 (0.74 to 1.42) | 63.6% | 12.8% | 1.13 (0.92 to 1.38) |
| CBZ vs VPS | 5 | 814 | 0.94 (0.73 to 1.19) | 0% | 40.1% | 1.04 (0.86 to 1.25) |
| CBZ vs LTG | 9 | 2268 | 0.76 (0.61 to 0.95) | 39.3% | 28.9% | 0.75 (0.65 to 0.86) |
| CBZ vs OXC | 2 | 562 | 4.62 (0.95 to 22.4) | 0% | 5.7% | 1.09 (0.84 to 1.42) |
| CBZ vs TPM | 2 | 937 | 1.04 (0.52 to 2.07) | 0% | 7.4% | 1.18 (0.98 to 1.43) |
| CBZ vs GBP | 2 | 954 | 1.14 (0.84 to 1.55) | 0% | 87.1% | 1.20 (1.00 to 1.43) |
| CBZ vs LEV | 3 | 1567 | 0.70 (0.52 to 0.94) | 0% | 37.9% | 0.82 (0.69 to 0.97) |
| CBZ vs ZNS | 1 | 583 | 1.08 (0.81 to 1.44) | NA | 100% | 1.08 (0.79 to 1.48) |
| PHB vs PHT | 3 | 404 | 0.67 (0.50 to 0.91) | 65% | 15.2% | 0.73 (0.55 to 0.96) |
| PHB vs VPS | 2 | 75 | 0.68 (0.34 to 1.36) | 23% | 8.8% | 0.67 (0.48 to 0.92) |
| PHB vs LTG | No direct evidence | 0% | 0.48 (0.35 to 0.66) | |||
| PHB vs OXC | No direct evidence | 0% | 0.70 (0.48 to 1.03) | |||
| PHB vs TPM | No direct evidence | 0% | 0.76 (0.55 to 1.06) | |||
| PHB vs GBP | No direct evidence | 0% | 0.77 (0.55 to 1.07) | |||
| PHB vs LEV | No direct evidence | 0% | 0.53 (0.38 to 0.73) | |||
| PHB vs ZNS | No direct evidence | 0% | 0.70 (0.46 to 1.06) | |||
| PHT vs VPS | 4 | 168 | 1.00 (0.60 to 1.64) | 58.5% | 9% | 0.92 (0.70 to 1.21) |
| PHT vs LTG | 1 | 90 | 1.10 (0.57 to 2.14) | NA | 11.6% | 0.66 (0.52 to 0.85) |
| PHT vs OXC | 2 | 325 | 0.65 (0.32 to 1.32) | 0% | 40.4% | 0.97 (0.69 to 1.35) |
| PHT vs TPM | 1 | 53 | 0.77 (0.38 to 1.57) | NA | 10.9% | 1.05 (0.80 to 1.39) |
| PHT vs GBP | No direct evidence | 0% | 1.06 (0.81 to 1.40) | |||
| PHT vs LEV | No direct evidence | 0% | 0.73 (0.56 to 0.95) | |||
| PHT vs ZNS | No direct evidence | 0% | 0.96 (0.66 to 1.39) | |||
| VPS vs LTG* | 3 | 221 | 1.40 (1.00 to 1.96) | 45.1% | 5.1% | 0.72 (0.58 to 0.90) |
| VPS vs OXC | No direct evidence | 0% | 1.05 (0.76 to 1.44) | |||
| VPS vs TPM | 2 | 111 | 1.66 (1.24 to 2.23) | 48.1% | 33.7% | 1.14 (0.88 to 1.48) |
| VPS vs GBP | No direct evidence | 0% | 1.15 (0.89 to 1.49) | |||
| VPS vs LEV | 1 | 190 | 1.14 (0.73 to 1.75) | NA | 17.2% | 0.79 (0.61 to 1.03) |
| VPS vs ZNS | No direct evidence | 0% | 1.04 (0.73 to 1.50) | |||
| LTG vs OXC | 1 | 506 | 0.69 (0.12 to 4.14) | NA | 4.4% | 1.46 (1.11 to 1.92) |
| LTG vs TPM | 1 | 648 | 1.18 (0.86 to 1.62) | NA | 20.9% | 1.59 (1.29 to 1.95) |
| LTG vs GBP | 1 | 659 | 0.62 (0.06 to 6.01) | NA | 1% | 1.60 (1.31 to 1.96) |
| LTG vs LEV | 1 | 240 | 0.86 (0.58 to 1.28) | NA | 23.7% | 1.10 (0.89 to 1.35) |
| LTG vs ZNS | No direct evidence | 0% | 1.45 (1.03 to 2.04) | |||
| OXC vs TPM | 1 | 496 | 0.87 (0.16 to 4.73) | NA | 4.9% | 1.09 (0.82 to 1.44) |
| OXC vs GBP | 1 | 507 | 0.90 (0.08 to 9.96) | NA | 2.3% | 1.10 (0.83 to 1.45) |
| OXC vs LEV | No direct evidence | 0% | 0.75 (0.55 to 1.03) | |||
| OXC vs ZNS | No direct evidence | 0% | 0.99 (0.66 to 1.49) | |||
| TPM vs GBP | 1 | 649 | 1.04 (0.12 to 9.33) | NA | 1.1% | 1.01 (0.82 to 1.25) |
| TPM vs LEV | No direct evidence | 0% | 0.69 (0.54 to 0.89) | |||
| TPM vs ZNS | No direct evidence | 0% | 0.91 (0.64 to 1.31) | |||
| GBP vs LEV | No direct evidence | 0% | 0.69 (0.54 to 0.88) | |||
| GBP vs ZNS | No direct evidence | 0% | 0.90 (0.63 to 1.30) | |||
| LEV vs ZNS | No direct evidence | 0% | 1.32 (0.93 to 1.88) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details. cNote that HR < 1 indicates an advantage to the second drug in the comparison; results highlighted in bold are statistically significant. dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence. eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
For comparisons marked with a *, confidence intervals of direct evidence and network meta‐analysis do not overlap indicating that inconsistency may be present in the results.
Table 9.
Pairwise and network meta‐analysis results ‐ Time to withdrawal of allocated treatment for individuals with generalised seizures
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence (network meta‐analysis) | ||||
| Number of studies | Number of participants | HR (95% CI)b,c | I² statisticd | Direct evidence (%)5 | HR (95% CI)b,c | |
| CBZ vs PHB | 3 | 156 | 1.21 (0.51 to 2.86) | 11.8% | 27.3% | 1.47 (0.83 to 2.61) |
| CBZ vs PHT | 2 | 118 | 2.68 (0.95 to 7.57) | 0% | 11.3% | 0.92 (0.59 to 1.42) |
| CBZ vs VPS | 4 | 405 | 1.26 (0.73 to 2.20) | 6.6% | 27.3% | 0.70 (0.54 to 0.92) |
| CBZ vs LTG | 7 | 302 | 1.23 (0.72 to 2.10) | 0% | 39.2% | 0.63 (0.45 to 0.89) |
| CBZ vs OXC | 1 | 9 | 0.39 (0.03 to 4.35) | NA | 3.9% | 1.00 (0.21 to 4.81) |
| CBZ vs TPM | 2 | 101 | 1.10 (0.51 to 2.36) | 0% | 23.2% | 1.24 (0.90 to 1.71) |
| CBZ vs GBP | 1 | 6 | 0.49 (0.03 to 7.90) | NA | 8.5% | 0.90 (0.11 to 7.29) |
| CBZ vs LEV | 2 | 251 | 1.22 (0.74 to 2.02) | 0% | 57% | 0.74 (0.44 to 1.23) |
| PHB vs PHT | 2 | 95 | 1.56 (0.49 to 4.99) | 0% | 16.1% | 0.62 (0.32 to 1.24) |
| PHB vs VPS | 2 | 94 | 0.56 (0.20 to 1.54) | 0% | 19.4% | 0.48 (0.27 to 0.86) |
| PHB vs LTG | No direct evidence | 0% | 0.43 (0.22 to 0.83) | |||
| PHB vs OXC | No direct evidence | 0% | 0.68 (0.13 to 3.60) | |||
| PHB vs TPM | No direct evidence | 0% | 0.84 (0.44 to 1.60) | |||
| PHB vs GBP | No direct evidence | 0% | 0.61 (0.07 to 5.34) | |||
| PHB vs LEV | No direct evidence | 0% | 0.50 (0.23 to 1.09) | |||
| PHT vs VPS | 3 | 326 | 0.66 (0.30 to 1.45) | 22.6% | 19.3% | 0.77 (0.46 to 1.27) |
| PHT vs LTG | 1 | 91 | 1.11 (0.42 to 2.94) | NA | 14.9% | 0.69 (0.39 to 1.20) |
| PHT vs OXC | 2 | 155 | 1.05 (0.44 to 2.52) | 0% | 37.9% | 1.09 (0.21 to 5.56) |
| PHT vs TPM | 1 | 150 | 1.68 (0.49 to 5.69) | NA | 11.2% | 1.35 (0.79 to 2.30) |
| PHT vs GBP | No direct evidence | 0% | 0.98 (0.12 to 8.30) | |||
| PHT vs LEV | No direct evidence | 0% | 0.80 (0.42 to 1.55) | |||
| VPS vs LTG | 3 | 387 | 0.46 (0.22 to 0.97) | 0% | 14.8% | 0.90 (0.60 to 1.35) |
| VPS vs OXC | No direct evidence | 0% | 1.42 (0.29 to 6.92) | |||
| VPS vs TPM* | 2 | 443 | 0.53 (0.27 to 1.07) | 48.5% | 22.4% | 1.76 (1.22 to 2.53) |
| VPS vs GBP | No direct evidence | 0% | 1.28 (0.16 to 10.5) | |||
| VPS vs LEV | 1 | 512 | 0.68 (0.30 to 1.59) | NA | 18.6% | 1.05 (0.58 to 1.90) |
| LTG vs OXC | 1 | 10 | 2.09 (0.34 to 12.8) | NA | 7.6% | 1.58 (0.33 to 7.67) |
| LTG vs TPM | 1 | 14 | 1.10 (0.42 to 2.89) | NA | 7.3% | 1.96 (1.25 to 3.08) |
| LTG vs GBP | 1 | 7 | 2.63 (0.27 to 25.7) | NA | 13.8% | 1.42 (0.17 to 11.6) |
| LTG vs LEV | No direct evidence | 0% | 1.17 (0.63 to 2.19) | |||
| OXC vs TPM | 1 | 14 | 1.31 (0.24 to 7.32) | NA | 9% | 1.24 (0.26 to 5.94) |
| OXC vs GBP | 1 | 7 | 1.26 (0.11 to 14.1) | NA | 12.7% | 0.90 (0.08 to 9.96) |
| OXC vs LEV | No direct evidence | 0% | 0.74 (0.14 to 3.86) | |||
| TPM vs GBP | 1 | 11 | 0.96 (0.11 to 8.67) | NA | 14.6% | 0.73 (0.09 to 5.89) |
| TPM vs LEV | No direct evidence | 0% | 0.60 (0.33 to 1.09) | |||
| GBP vs LEV | No direct evidence | 0% | 0.82 (0.10 to 7.10) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details cNote that HR < 1 indicates an advantage to the second drug in the comparison; results highlighted in bold are statistically significant. dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence. eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
For comparisons marked with a *, confidence intervals of direct evidence and network meta‐analysis do not overlap indicating that inconsistency may be present in the results
Table 10.
Pairwise and network meta‐analysis results ‐ Time to 12‐month remission of seizures for individuals with partial seizures
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence (network meta‐analysis) | ||||
| Number of studies | Number of participants | HR (95% CI)b,c | I² statisticd | Direct evidence (%)e | HR (95% CI)b,c | |
| CBZ vs PHB | 4 | 525 | 1.41 (1.04 to 1.91) | 0% | 56.1% | 1.02 (0.76 to 1.35) |
| CBZ vs PHT | 3 | 430 | 1.00 (0.76 to 1.32) | 54.8% | 18.6% | 1.03 (0.85 to 1.25) |
| CBZ vs VPS | 5 | 816 | 1.03 (0.85 to 1.25) | 46.4% | 27.6% | 1.05 (0.89 to 1.25) |
| CBZ vs LTG | 2 | 891 | 1.02 (0.69 to 1.50) | 0% | 17.5% | 1.16 (0.98 to 1.37) |
| CBZ vs OXC | 2 | 555 | 1.13 (0.62 to 2.05) | 0% | 21% | 0.98 (0.78 to 1.25) |
| CBZ vs TPM | 2 | 925 | 0.94 (0.48 to 1.83) | 0% | 7.2% | 1.08 (0.92 to 1.27) |
| CBZ vs GBP | 1 | 651 | 0.61 (0.06 to 5.82) | NA | 10.5% | 1.20 (0.99 to 1.47) |
| CBZ vs LEV | 3 | 1567 | 1.08 (0.81 to 1.42) | 60.8% | 14.2% | 1.35 (1.09 to 1.69) |
| CBZ vs ZNS | 1 | 582 | 1.05 (0.85 to 1.30) | NA | 100% | 1.05 (0.81 to 1.35) |
| PHB vs PHT | 4 | 465 | 0.80 (0.59 to 1.10) | 0% | 0.2% | 1.01 (0.75 to 1.37) |
| PHB vs VPS | 2 | 80 | 0.85 (0.51 to 1.40) | 4.4% | 15.6% | 1.04 (0.75 to 1.43) |
| PHB vs LTG | No direct evidence | 0% | 1.14 (0.82 to 1.59) | |||
| PHB vs OXC | No direct evidence | 0% | 0.96 (0.67 to 1.41) | |||
| PHB vs TPM | No direct evidence | 0% | 1.06 (0.76 to 1.47) | |||
| PHB vs GBP | No direct evidence | 0% | 1.19 (0.83 to 1.69) | |||
| PHB vs LEV | No direct evidence | 0% | 1.33 (0.93 to 1.92) | |||
| PHB vs ZNS | No direct evidence | 0% | 1.03 (0.70 to 1.52) | |||
| PHT vs VPS | 4 | 245 | 1.04 (0.78 to 1.40) | 0% | 41.6% | 1.03 (0.80 to 1.32) |
| PHT vs LTG | No direct evidence | 0% | 1.12 (0.88 to 1.45) | |||
| PHT vs OXC | 2 | 318 | 1.21 (0.73 to 2.03) | 0% | 29.9% | 0.95 (0.70 to 1.30) |
| PHT vs TPM | No direct evidence | 0% | 1.05 (0.81 to 1.35) | |||
| PHT vs GBP | No direct evidence | 0% | 1.18 (0.88 to 1.56) | |||
| PHT vs LEV | No direct evidence | 0% | 1.32 (0.98 to 1.75) | |||
| PHT vs ZNS | No direct evidence | 0% | 1.02 (0.74 to 1.41) | |||
| VPS vs LTG | 3 | 221 | 1.37 (1.07 to 1.77) | 0% | 39.9% | 1.10 (0.88 to 1.37) |
| VPS vs OXC | No direct evidence | 0% | 0.93 (0.70 to 1.23) | |||
| VPS vs TPM | 2 | 111 | 1.11 (0.87 to 1.40) | 0% | 67.8% | 1.02 (0.80 to 1.30) |
| VPS vs GBP | No direct evidence | 0% | 1.14 (0.88 to 1.47) | |||
| VPS vs LEV | 1 | 190 | 1.14 (0.84 to 1.55) | NA | 34.7% | 1.28 (0.97 to 1.67) |
| VPS vs ZNS | No direct evidence | 0% | 0.99 (0.74 to 1.35) | |||
| LTG vs OXC | 1 | 499 | 1.49 (0.33 to 6.67) | NA | 2.8% | 0.85 (0.66 to 1.09) |
| LTG vs TPM | 1 | 636 | 0.98 (0.29 to 3.25) | NA | 2.5% | 0.93 (0.75 to 1.15) |
| LTG vs GBP | 1 | 647 | 0.74 (0.08 to 6.58) | NA | 10.1% | 1.04 (0.84 to 1.30) |
| LTG vs LEV | 1 | 240 | 1.02 (0.70 to 1.49) | NA | 26.6% | 1.16 (0.93 to 1.47) |
| LTG vs ZNS | No direct evidence | 0% | 0.91 (0.67 to 1.22) | |||
| OXC vs TPM | 1 | 487 | 0.66 (0.17 to 2.47) | NA | 3.7% | 1.10 (0.83 to 1.45) |
| OXC vs GBP | 1 | 498 | 0.49 (0.05 to 4.74) | NA | 9.8% | 1.23 (0.95 to 1.59) |
| OXC vs LEV | No direct evidence | 0% | 1.37 (1.05 to 1.79) | |||
| OXC vs ZNS | No direct evidence | 0% | 1.06 (0.76 to 1.52) | |||
| TPM vs GBP | 1 | 635 | 0.75 (0.09 to 6.00) | NA | 11.2% | 1.12 (0.87 to 1.45) |
| TPM vs LEV | No direct evidence | 0% | 1.25 (0.96 to 1.64) | |||
| TPM vs ZNS | No direct evidence | 0% | 0.97 (0.72 to 1.32) | |||
| GBP vs LEV | No direct evidence | 0% | 1.12 (0.88 to 1.43) | |||
| GBP vs ZNS | No direct evidence | 0% | 0.87 (0.63 to 1.20) | |||
| LEV vs ZNS | No direct evidence | 0% | 0.78 (0.56 to 1.09) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details. cNote that HR < 1 indicates an advantage to the second drug in the comparison; results highlighted in bold are statistically significant. dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence. eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
Table 11.
Pairwise and network meta‐analysis results ‐ Time to 12‐month remission of seizures for individuals with generalised seizures
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence (network meta‐analysis) | ||||
| Number of studies | Number of participants | HR (95% CI)b,c | I² statisticd | Direct evidence (%)5 | HR (95% CI)b,c | |
| CBZ vs PHB | 3 | 158 | 0.53 (0.28 to 1.00) | 0% | 42.6% | 1.25 (0.83 to 1.89) |
| CBZ vs PHT | 2 | 121 | 1.11 (0.61 to 2.02) | 64.5% | 9.3% | 0.86 (0.65 to 1.16) |
| CBZ vs VPS | 4 | 412 | 1.01 (0.72 to 1.43) | 0% | 51.1% | 0.94 (0.79 to 1.14) |
| CBZ vs LTG | 1 | 9 | 1.33 (0.29 to 6.03) | NA | 7% | 1.28 (0.54 to 3.03) |
| CBZ vs OXC | 1 | 9 | 0.77 (0.15 to 3.89) | NA | 5.6% | 1.72 (0.47 to 6.25) |
| CBZ vs TPM | 2 | 101 | 1.15 (0.52 to 2.53) | 0% | 27.2% | 1.06 (0.78 to 1.45) |
| CBZ vs GBP | 1 | 6 | 2.19 (0.23 to 21.2) | NA | 10.9% | 0.75 (0.10 to 5.88) |
| CBZ vs LEV | 2 | 251 | 1.02 (0.65 to 1.59) | 77.4% | 16.6% | 1.33 (0.81 to 2.22) |
| PHB vs PHT | 3 | 130 | 1.30 (0.61 to 2.78) | 53% | 10.9% | 0.68 (0.44 to 1.08) |
| PHB vs VPS | 2 | 98 | 1.15 (0.53 to 2.49) | 42.3% | 13% | 0.75 (0.49 to 1.15) |
| PHB vs LTG | No direct evidence | 0% | 1.01 (0.40 to 2.63) | |||
| PHB vs OXC | No direct evidence | 0% | 1.37 (0.35 to 5.26) | |||
| PHB vs TPM | No direct evidence | 0% | 0.85 (0.51 to 1.41) | |||
| PHB vs GBP | No direct evidence | 0% | 0.60 (0.07 to 5.00) | |||
| PHB vs LEV | No direct evidence | 0% | 1.06 (0.56 to 2.04) | |||
| PHT vs VPS | 4 | 269 | 0.87 (0.55 to 1.40) | 0% | 44.9% | 1.10 (0.80 to 1.49) |
| PHT vs LTG | No direct evidence | 0% | 1.47 (0.60 to 3.57) | |||
| PHT vs OXC | 2 | 154 | 0.77 (0.41 to 1.47) | 0% | 41.2% | 2.00 (0.53 to 7.69) |
| PHT vs TPM | No direct evidence | 0% | 1.23 (0.81 to 1.85) | |||
| PHT vs GBP | No direct evidence | 0% | 0.87 (0.11 to 7.14) | |||
| PHT vs LEV | No direct evidence | 0% | 1.56 (0.87 to 2.78) | |||
| VPS vs LTG | 3 | 387 | 0.77 (0.38 to 1.56) | 0% | 35.7% | 1.35 (0.57 to 3.13) |
| VPS vs OXC | No direct evidence | 0% | 1.82 (0.50 to 6.67) | |||
| VPS vs TPM | 2 | 441 | 0.52 (0.26 to 1.04) | 58.5% | 10.6% | 1.12 (0.83 to 1.52) |
| VPS vs GBP | No direct evidence | 0% | 0.79 (0.10 to 6.25) | |||
| VPS vs LEV | 1 | 512 | 0.91 (0.49 to 1.70) | NA | 55.2% | 1.41 (0.83 to 2.44) |
| LTG vs OXC | 1 | 10 | 0.58 (0.13 to 2.64) | NA | 9.2% | 1.35 (0.33 to 5.56) |
| LTG vs TPM | 1 | 14 | 1.13 (0.33 to 3.82) | NA | 15.1% | 0.83 (0.35 to 2.00) |
| LTG vs GBP | 1 | 7 | 1.64 (0.18 to 14.8) | NA | 12.5% | 0.59 (0.07 to 5.00) |
| LTG vs LEV | No direct evidence | 0% | 1.05 (0.40 to 2.78) | |||
| OXC vs TPM | 1 | 14 | 1.95 (0.51 to 7.50) | NA | 11.4% | 0.62 (0.17 to 2.27) |
| OXC vs GBP | 1 | 7 | 2.83 (0.29 to 27.6) | NA | 10.9% | 0.44 (0.04 to 4.35) |
| OXC vs LEV | No direct evidence | 0% | 0.78 (0.20 to 3.13) | |||
| TPM vs GBP | 1 | 11 | 1.45 (0.18 to 11.7) | NA | 15.9% | 0.71 (0.09 to 5.56) |
| TPM vs LEV | No direct evidence | 0% | 1.27 (0.68 to 2.33) | |||
| GBP vs LEV | No direct evidence | 0% | 1.79 (0.21 to 14.3) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details. cNote that HR < 1 indicates an advantage to the second drug in the comparison; results in highlighted in bold are statistically significant. dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence. eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
Table 12.
Pairwise and network meta‐analysis results ‐ Time to six‐month remission of seizures for individuals with partial seizures
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence (network meta‐analysis) | ||||
| Number of studies | Number of participants | HR (95% CI)b,c | I² statisticd | Direct evidence (%)5 | HR (95% CI)b,c | |
| CBZ vs PHB | 4 | 525 | 1.24 (0.95 to 1.61) | 0% | 31.3% | 0.95 (0.76 to 1.19) |
| CBZ vs PHT | 3 | 430 | 0.85 (0.66 to 1.09) | 4.2% | 23.3% | 1.03 (0.88 to 1.20) |
| CBZ vs VPS | 5 | 816 | 1.06 (0.90 to 1.25) | 56.5% | 16.6% | 1.10 (0.96 to 1.25) |
| CBZ vs LTG | 7 | 1535 | 1.15 (0.89 to 1.48) | 0% | 26.4% | 1.11 (0.98 to 1.27) |
| CBZ vs OXC | 2 | 555 | 1.15 (0.65 to 2.04) | 0% | 16.6% | 0.98 (0.82 to 1.18) |
| CBZ vs TPM | 2 | 925 | 1.05 (0.64 to 1.72) | 0% | 8.8% | 1.11 (0.96 to 1.28) |
| CBZ vs GBP | 2 | 943 | 0.81 (0.52 to 1.27) | 0% | 73.7% | 1.16 (0.99 to 1.35) |
| CBZ vs LEV | 3 | 1567 | 1.06 (0.84 to 1.33) | 37.9% | 20.4% | 1.04 (0.93 to 1.16) |
| CBZ vs ZNS | 1 | 582 | 1.00 (0.82 to 1.20) | NA | 100% | 1.00 (0.83 to 1.20) |
| PHB vs PHT | 4 | 465 | 0.79 (0.60 to 1.05) | 0% | 31.1% | 1.08 (0.85 to 1.37) |
| PHB vs VPS | 2 | 80 | 0.67 (0.42 to 1.08) | 0% | 9.1% | 1.15 (0.89 to 1.49) |
| PHB vs LTG | No direct evidence | 0% | 1.16 (0.90 to 1.52) | |||
| PHB vs OXC | No direct evidence | 0% | 1.03 (0.77 to 1.39) | |||
| PHB vs TPM | No direct evidence | 0% | 1.16 (0.89 to 1.54) | |||
| PHB vs GBP | No direct evidence | 0% | 1.22 (0.93 to 1.59) | |||
| PHB vs LEV | No direct evidence | 0% | 1.10 (0.85 to 1.41) | |||
| PHB vs ZNS | No direct evidence | 0% | 1.04 (0.78 to 1.41) | |||
| PHT vs VPS | 5 | 245 | 0.90 (0.70 to 1.15) | 0% | 26.5% | 1.06 (0.88 to 1.30) |
| PHT vs LTG | 1 | 90 | 0.88 (0.25 to 3.03) | NA | 1.20% | 1.09 (0.88 to 1.32) |
| PHT vs OXC | 2 | 318 | 1.21 (0.79 to 1.87) | 0% | 33.2% | 0.95 (0.75 to 1.22) |
| PHT vs TPM | No direct evidence | 0% | 1.09 (0.88 to 1.33) | |||
| PHT vs GBP | No direct evidence | 0% | 1.12 (0.91 to 1.39) | |||
| PHT vs LEV | No direct evidence | 0% | 1.02 (0.84 to 1.22) | |||
| PHT vs ZNS | No direct evidence | 0% | 0.97 (0.76 to 1.23) | |||
| VPS vs LTG | 3 | 221 | 1.22 (0.97 to 1.52) | 0% | 32.1% | 1.02 (0.85 to 1.22) |
| VPS vs OXC | No direct evidence | 0% | 0.90 (0.72 to 1.12) | |||
| VPS vs TPM | 2 | 111 | 1.08 (0.87 to 1.34) | 0% | 61.7% | 1.02 (0.83 to 1.23) |
| VPS vs GBP | No direct evidence | 0% | 1.05 (0.87 to 1.28) | |||
| VPS vs LEV | 1 | 190 | 1.09 (0.88 to 1.33) | NA | 40.5% | 0.95 (0.79 to 1.14) |
| VPS vs ZNS | No direct evidence | 0% | 0.91 (0.72 to 1.14) | |||
| LTG vs OXC | 1 | 499 | 1.08 (0.27 to 4.32) | NA | 2.4% | 0.88 (0.73 to 1.08) |
| LTG vs TPM | 1 | 636 | 0.89 (0.70 to 1.13) | NA | 1.7% | 1.00 (0.85 to 1.18) |
| LTG vs GBP | 1 | 647 | 1.46 (0.16 to 13.0) | NA | 1.6% | 1.04 (0.88 to 1.22) |
| LTG vs LEV | 1 | 240 | 0.83 (0.59 to 1.17) | NA | 17.8% | 0.93 (0.80 to 1.10) |
| LTG vs ZNS | No direct evidence | 0% | 0.89 (0.71 to 1.12) | |||
| OXC vs TPM | 1 | 487 | 0.86 (0.26 to 2.86) | NA | 3.3% | 1.14 (0.93 to 1.37) |
| OXC vs GBP | 1 | 498 | 1.35 (0.15 to 12.1) | NA | 2.1% | 1.18 (0.96 to 1.43) |
| OXC vs LEV | No direct evidence | 0% | 1.06 (0.86 to 1.32) | |||
| OXC vs ZNS | No direct evidence | 0% | 1.01 (0.78 to 1.32) | |||
| TPM vs GBP | 1 | 635 | 1.56 (0.2 to 12.5) | NA | 1.6% | 1.04 (0.88 to 1.23) |
| TPM vs LEV | No direct evidence | 0% | 0.93 (0.79 to 1.12) | |||
| TPM vs ZNS | No direct evidence | 0% | 0.89 (0.70 to 1.14) | |||
| GBP vs LEV | No direct evidence | 0% | 0.90 (0.75 to 1.09) | |||
| GBP vs ZNS | No direct evidence | 0% | 0.86 (0.68 to 1.10) | |||
| LEV vs ZNS | No direct evidence | 0% | 0.95 (0.77 to 1.19) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details. cNote that HR < 1 indicates an advantage to the second drug in the comparison; results in highlighted in bold are statistically significant. dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence. eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
Table 13.
Pairwise and network meta‐analysis results ‐ Time to six‐month remission of seizures for individuals with generalised seizures
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence (network meta‐analysis) | ||||
| Number of studies | Number of participants | HR (95% CI)b,c | I² statisticd | Direct evidence (%)e | HR (95% CI)b,c | |
| CBZ vs PHB | 3 | 158 | 0.56 (0.33 to 0.96) | 13.2% | 28.2% | 1.28 (0.92 to 1.79) |
| CBZ vs PHT | 2 | 121 | 1.44 (0.82 to 2.55) | 31.4% | 13% | 0.87 (0.68 to 1.10) |
| CBZ vs VPS | 4 | 412 | 1.11 (0.81 to 1.53) | 29.9% | 30.7% | 0.95 (0.84 to 1.09) |
| CBZ vs LTG | 5 | 254 | 0.58 (0.25 to 1.32) | 0% | 0.1% | 1.20 (0.99 to 1.49) |
| CBZ vs OXC | 1 | 9 | 0.79 (0.17 to 3.56) | NA | 4.6% | 1.30 (0.42 to 4.00) |
| CBZ vs TPM | 2 | 101 | 1.00 (0.55 to 1.79) | 0% | 32.8% | 1.11 (0.78 to 1.59) |
| CBZ vs GBP | 1 | 6 | 0.71 (0.07 to 6.90) | NA | 10% | 1.75 (0.23 to 12.5) |
| CBZ vs LEV | 2 | 251 | 1.00 (0.72 to 1.37) | 57.9% | 26.7% | 1.14 (0.85 to 1.52) |
| PHB vs PHT | 3 | 130 | 1.31 (0.67 to 2.53) | 0% | 22.7% | 0.68 (0.47 to 0.98) |
| PHB vs VPS | 2 | 98 | 1.50 (0.72 to 3.11) | 7.5% | 15.3% | 0.75 (0.53 to 1.05) |
| PHB vs LTG | No direct evidence | 0% | 0.94 (0.64 to 1.39) | |||
| PHB vs OXC | No direct evidence | 0% | 1.01 (0.31 to 3.23) | |||
| PHB vs TPM | No direct evidence | 0% | 0.87 (0.53 to 1.41) | |||
| PHB vs GBP | No direct evidence | 0% | 1.37 (0.17 to 11.1) | |||
| PHB vs LEV | No direct evidence | 0% | 0.88 (0.57 to 1.37) | |||
| PHT vs VPS | 4 | 394 | 1.03 (0.68 to 1.54) | 0% | 36.8% | 1.10 (0.85 to 1.43) |
| PHT vs LTG | 1 | 91 | 1.96 (0.37 to 10.2) | NA | 4.4% | 1.39 (1.03 to 1.89) |
| PHT vs OXC | 2 | 154 | 0.71 (0.42 to 1.21) | 0% | 45.1% | 1.49 (0.48 to 4.76) |
| PHT vs TPM | No direct evidence | 0% | 1.28 (0.84 to 1.96) | |||
| PHT vs GBP | No direct evidence | 0% | 2.00 (0.26 to 16.7) | |||
| PHT vs LEV | No direct evidence | 0% | 1.32 (0.89 to 1.92) | |||
| VPS vs LTG | 3 | 387 | 0.84 (0.48 to 1.47) | 0% | 43.5% | 1.27 (1.03 to 1.56) |
| VPS vs OXC | No direct evidence | 0% | 1.35 (0.44 to 4.17) | |||
| VPS vs TPM | 2 | 441 | 0.67 (0.38 to 1.19) | 58.7% | 12.9% | 1.16 (0.81 to 1.67) |
| VPS vs GBP | No direct evidence | 0% | 1.82 (0.24 to 14.3) | |||
| VPS vs LEV | 1 | 512 | 0.88 (0.60 to 1.30) | NA | 48.6% | 1.19 (0.86 to 1.64) |
| LTG vs OXC | 1 | 10 | 0.80 (0.20 to 3.26) | NA | 7.6% | 1.08 (0.35 to 3.33) |
| LTG vs TPM | 1 | 14 | 0.59 (0.30 to 1.16) | NA | 10% | 0.92 (0.62 to 1.37) |
| LTG vs GBP | 1 | 7 | 0.73 (0.08 to 6.57) | NA | 11% | 1.45 (0.19 to 11.1) |
| LTG vs LEV | No direct evidence | 0% | 0.93 (0.65 to 1.33) | |||
| OXC vs TPM | 1 | 14 | 1.34 (0.40 to 4.54) | NA | 9.4% | 0.86 (0.28 to 2.63) |
| OXC vs GBP | 1 | 7 | 0.91 (0.10 to 8.20) | NA | 10.7% | 1.35 (0.15 to 12.5) |
| OXC vs LEV | No direct evidence | 0% | 0.88 (0.27 to 2.78) | |||
| TPM vs GBP | 1 | 11 | 0.68 (0.08 to 5.45) | NA | 13.9% | 1.56 (0.21 to 12.5) |
| TPM vs LEV | No direct evidence | 0% | 1.02 (0.65 to 1.61) | |||
| GBP vs LEV | No direct evidence | 0% | 0.65 (0.08 to 5.00) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details. cNote that HR < 1 indicates an advantage to the second drug in the comparison; results highlighted in bold are statistically significant. dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence. eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
Table 14.
Pairwise and network meta‐analysis results ‐ Time to first seizure for individuals with partial seizures
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence (network meta‐analysis) | ||||
| Number of studies | Number of participants | HR (95% CI)b,c | I² statisticd | Direct evidence (%)e | HR (95% CI)b,c | |
| CBZ vs PHB | 6 | 581 | 0.99 (0.78 to 1.26) | 54.3% | 21% | 0.79 (0.64 to 0.97) |
| CBZ vs PHT | 4 | 432 | 0.91 (0.72 to 1.16) | 16.1% | 27.1% | 0.98 (0.85 to 1.13) |
| CBZ vs VPS | 5 | 813 | 1.01 (0.86 to 1.19) | 32% | 34.6% | 1.20 (1.06 to 1.37) |
| CBZ vs LTG | 9 | 2252 | 0.98 (0.75 to 1.27) | 0% | 40.7% | 1.29 (1.17 to 1.42) |
| CBZ vs OXC | 2 | 555 | 1.47 (0.57 to 3.81) | 57.3% | 4.8% | 1.09 (0.89 to 1.32) |
| CBZ vs TPM | 2 | 925 | 1.03 (0.51 to 2.08) | 69.3% | 1.5% | 1.12 (0.97 to 1.29) |
| CBZ vs GBP | 2 | 943 | 1.64 (1.14 to 2.36) | 17.7% | 49% | 1.44 (1.25 to 1.66) |
| CBZ vs LEV | 3 | 1552 | 1.18 (0.85 to 1.65) | 0% | 26.2% | 1.14 (0.99 to 1.30) |
| CBZ vs ZNS | 1 | 581 | 1.30 (0.97 to 1.73) | NA | 100% | 1.30 (0.97 to 1.73) |
| PHB vs PHT | 5 | 463 | 1.07 (0.83 to 1.37) | 27.7% | 33.6% | 1.24 (0.99 to 1.56) |
| PHB vs VPS* | 2 | 80 | 0.71 (0.43 to 1.17) | 9.1% | 12.8% | 1.53 (1.20 to 1.94) |
| PHB vs LTG | No direct evidence | 0% | 1.63 (1.30 to 2.06) | |||
| PHB vs OXC | No direct evidence | 0% | 1.38 (1.04 to 1.83) | |||
| PHB vs TPM | No direct evidence | 0% | 1.42 (1.11 to 1.83) | |||
| PHB vs GBP | No direct evidence | 0% | 1.83 (1.42 to 2.35) | |||
| PHB vs LEV | No direct evidence | 0% | 1.44 (1.12 to 1.85) | |||
| PHB vs ZNS | No direct evidence | 0% | 1.64 (1.15 to 2.35) | |||
| PHT vs VPS | 5 | 245 | 0.96 (0.72 to 1.29) | 0% | 25.4% | 1.23 (1.02 to 1.48) |
| PHT vs LTG | 1 | 90 | 0.77 (0.38 to 1.54) | NA | 6% | 1.31 (1.10 to 1.57) |
| PHT vs OXC | 2 | 318 | 1.46 (0.88 to 2.44) | 23.9% | 36.1% | 1.11 (0.87 to 1.41) |
| PHT vs TPM | 1 | 53 | 2.32 (0.95 to 5.70) | NA | 4% | 1.14 (0.93 to 1.40) |
| PHT vs GBP | No direct evidence | 0% | 1.47 (1.20 to 1.80) | |||
| PHT vs LEV | No direct evidence | 0% | 1.16 (0.95 to 1.41) | |||
| PHT vs ZNS | No direct evidence | 0% | 1.32 (0.96 to 1.82) | |||
| VPS vs LTG | 3 | 215 | 1.57 (1.23 to 2.00) | 39.4% | 10% | 1.07 (0.92 to 1.24) |
| VPS vs OXC | No direct evidence | 0% | 0.90 (0.72 to 1.14) | |||
| VPS vs TPM | 2 | 111 | 1.18 (0.93 to 1.50) | 0% | 70.2% | 0.93 (0.77 to 1.13) |
| VPS vs GBP | No direct evidence | 0% | 1.20 (0.99 to 1.44) | |||
| VPS vs LEV | 1 | 190 | 1.27 (0.94 to 1.72) | NA | 31% | 0.94 (0.77 to 1.15) |
| VPS vs ZNS | No direct evidence | 0% | 1.08 (0.78 to 1.48) | |||
| LTG vs OXC | 1 | 499 | 0.87 (0.23 to 3.25) | NA | 5.5% | 0.84 (0.69 to 1.03) |
| LTG vs TPM | 1 | 636 | 0.73 (0.57 to 0.93) | NA | 2.3% | 0.87 (0.75 to 1.01) |
| LTG vs GBP | 1 | 647 | 0.63 (0.07 to 5.42) | NA | 4.4% | 1.12 (0.96 to 1.30) |
| LTG vs LEV | 1 | 229 | 0.84 (0.53 to 1.35) | NA | 15.9% | 0.88 (0.75 to 1.04) |
| LTG vs ZNS | No direct evidence | 0% | 1.01 (0.74 to 1.36) | |||
| OXC vs TPM | 1 | 487 | 0.55 (0.15 to 2.06) | NA | 5.4% | 1.03 (0.84 to 1.27) |
| OXC vs GBP | 1 | 498 | 0.73 (0.08 to 6.49) | NA | 4.6% | 1.32 (1.08 to 1.63) |
| OXC vs LEV | No direct evidence | 0% | 1.05 (0.83 to 1.32) | |||
| OXC vs ZNS | No direct evidence | 0% | 1.19 (0.84 to 1.69) | |||
| TPM vs GBP | 1 | 635 | 1.31 (0.15 to 11.2) | NA | 3.5% | 1.28 (1.09 to 1.51) |
| TPM vs LEV | No direct evidence | 0% | 1.01 (0.83 to 1.23) | |||
| TPM vs ZNS | No direct evidence | 0% | 1.15 (0.84 to 1.59) | |||
| GBP vs LEV | No direct evidence | 0% | 0.79 (0.65 to 0.96) | |||
| GBP vs ZNS | No direct evidence | 0% | 0.90 (0.65 to 1.24) | |||
| LEV vs ZNS | No direct evidence | 0% | 1.14 (0.83 to 1.57) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; zNS: Zonisamide
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details. cNote that HR < 1 indicates an advantage to the second drug in the comparison; results highlighted in bold are statistically significant dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence. eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
For comparisons marked with a *, confidence intervals of direct evidence and network meta‐analysis do not overlap indicating that inconsistency may be present in the results.
Table 15.
Pairwise and network meta‐analysis results ‐ Time to first seizure for individuals with generalised seizures
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence (network meta‐analysis) | ||||
| Number of studies | Number of participants | HR (95% CI)2,3 | I² statistic4 | Direct evidence(%)5 | HR (95% CI)2,3 | |
| CBZ vs PHB | 5 | 237 | 0.55 (0.33 to 0.92) | 50.4% | 35.5% | 1.10 (0.80 to 1.51) |
| CBZ vs PHT | 3 | 150 | 0.88 (0.51 to 1.54) | 0% | 26.6% | 0.76 (0.59 to 0.98) |
| CBZ vs VPS | 4 | 411 | 1.37 (0.98 to 1.92) | 84.1% | 10.4% | 0.88 (0.76 to 1.03) |
| CBZ vs LTG | 7 | 302 | 1.49 (0.94 to 2.35) | 0% | 0.3% | 0.98 (0.70 to 1.37) |
| CBZ vs OXC | 1 | 9 | 1.55 (0.38 to 6.31) | NA | 9% | 1.09 (0.36 to 3.36) |
| CBZ vs TPM | 2 | 101 | 1.19 (0.56 to 2.50) | 62% | 9% | 1.15 (0.89 to 1.48) |
| CBZ vs GBP | 1 | 6 | 2.83 (0.31 to 25.5) | NA | 10.7% | 0.79 (0.10 to 6.08) |
| CBZ vs LEV | 2 | 251 | 1.04 (0.65 to 1.64) | 0% | 44.9% | 1.19 (0.78 to 1.83) |
| PHB vs PHT | 4 | 161 | 1.41 (0.76 to 2.62) | 46.9% | 20.3% | 0.69 (0.48 to 1.00) |
| PHB vs VPS | 2 | 98 | 1.87 (0.87 to 4.00) | 69.8% | 6.5% | 0.80 (0.57 to 1.12) |
| PHB vs LTG | No direct evidence | 0% | 0.89 (0.56 to 1.42) | |||
| PHB vs OXC | No direct evidence | 0% | 1.00 (0.31 to 3.20) | |||
| PHB vs TPM | No direct evidence | 0% | 1.05 (0.70 to 1.56) | |||
| PHB vs GBP | No direct evidence | 0% | 0.72 (0.09 to 5.68) | |||
| PHB vs LEV | No direct evidence | 0% | 1.09 (0.64 to 1.85) | |||
| PHT vs VPS | 4 | 394 | 1.11 (0.71 to 1.74) | 0% | 36.4% | 1.16 (0.88 to 1.53) |
| PHT vs LTG | 1 | 91 | 1.00 (0.40 to 2.46) | NA | 16.2% | 1.29 (0.85 to 1.97) |
| PHT vs OXC | 2 | 154 | 0.60 (0.33 to 1.10) | 49.7% | 25.2% | 1.44 (0.46 to 4.56) |
| PHT vs TPM | 1 | 150 | 0.63 (0.18 to 2.26) | NA | 9.8% | 1.51 (1.06 to 2.15) |
| PHT vs GBP | No direct evidence | 0% | 1.05 (0.13 to 8.14) | |||
| PHT vs LEV | No direct evidence | 0% | 1.57 (0.96 to 2.58) | |||
| VPS vs LTG | 3 | 377 | 0.64 (0.37 to 1.11) | 23.2% | 31.3% | 1.11 (0.77 to 1.60) |
| VPS vs OXC | No direct evidence | 0% | 1.24 (0.40 to 3.84) | |||
| VPS vs TPM* | 2 | 441 | 0.42 (0.23 to 0.80) | 46.4% | 21% | 1.30 (1.01 to 1.68) |
| VPS vs GBP | No direct evidence | 0% | 0.90 (0.12 to 6.92) | |||
| VPS vs LEV | 1 | 512 | 0.82 (0.48 to 1.40) | NA | 34% | 1.35 (0.86 to 2.13) |
| LTG vs OXC | 1 | 10 | 0.94 (0.25 to 3.57) | NA | 12.2% | 1.12 (0.36 to 3.48) |
| LTG vs TPM | 1 | 14 | 0.61 (0.28 to 1.30) | NA | 13.1% | 1.17 (0.78 to 1.77) |
| LTG vs GBP | 1 | 7 | 1.72 (0.20 to 14.9) | NA | 11.9% | 0.81 (0.11 to 6.25) |
| LTG vs LEV | No direct evidence | 0% | 1.22 (0.71 to 2.10) | |||
| OXC vs TPM | 1 | 14 | 1.90 (0.50 to 7.19) | NA | 13.6% | 1.05 (0.34 to 3.24) |
| OXC vs GBP | 1 | 7 | 1.83 (0.20 to 16.5) | NA | 13.3% | 0.73 (0.08 to 6.49) |
| OXC vs LEV | No direct evidence | 0% | 1.09 (0.33 to 3.62) | |||
| TPM vs GBP | 1 | 11 | 0.96 (0.11 to 8.29) | NA | 13.2% | 0.69 (0.09 to 5.32) |
| TPM vs LEV | No direct evidence | 0% | 1.04 (0.63 to 1.71) | |||
| GBP vs LEV | No direct evidence | 0% | 1.50 (0.19 to 12.0) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details. cNote that HR < 1 indicates an advantage to the second drug in the comparison; results in highlighted in bold are statistically significant. dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence. eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
For comparisons marked with a *, confidence intervals of direct evidence and network meta‐analysis do not overlap indicating that inconsistency may be present in the results
Figure 5.
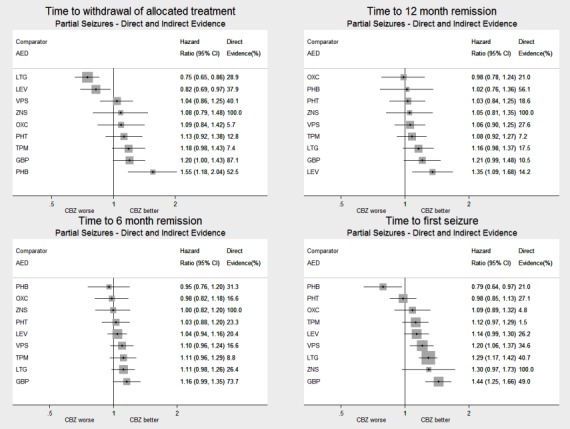
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with partial seizures, all drugs compared to carbamazepine (CBZ)
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Figure 6.
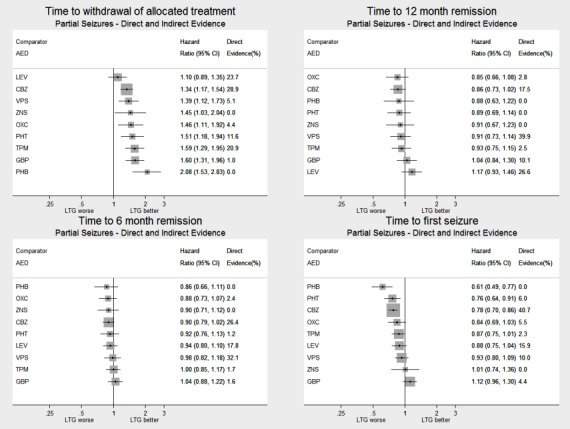
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with partial seizures, all drugs compared to lamotrigine (LTG)
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Figure 7.
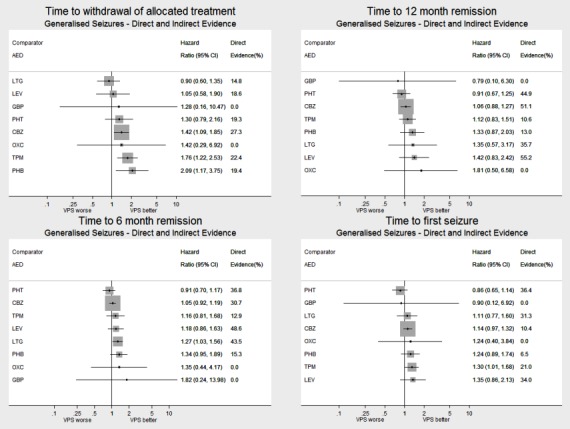
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with generalised seizures, all drugs compared to sodium valproate (VPS)
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Figure 8.
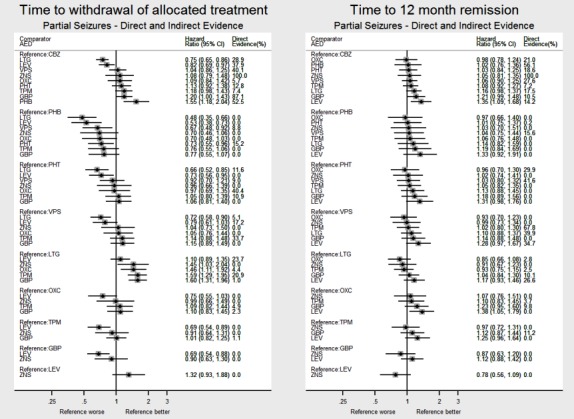
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with partial seizures, all pairwise comparisons for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Figure 9.
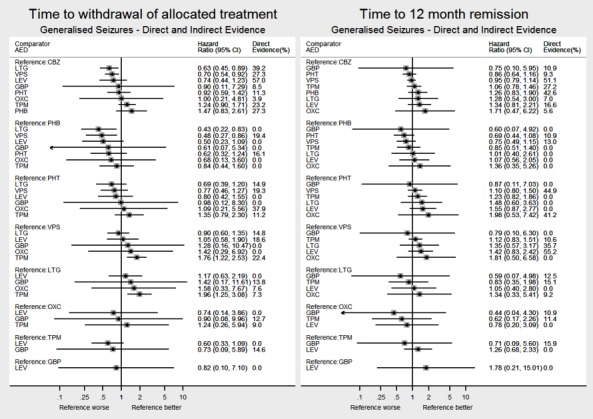
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with generalised seizures, all pairwise comparisons for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Figure 10.
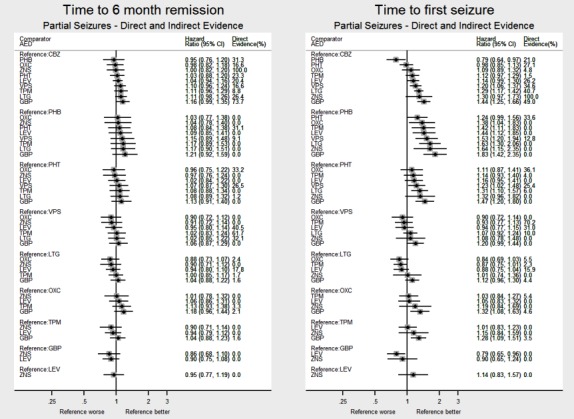
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with partial seizures, all pairwise comparisons for time to six‐month remission and time to first seizure.
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Figure 11.
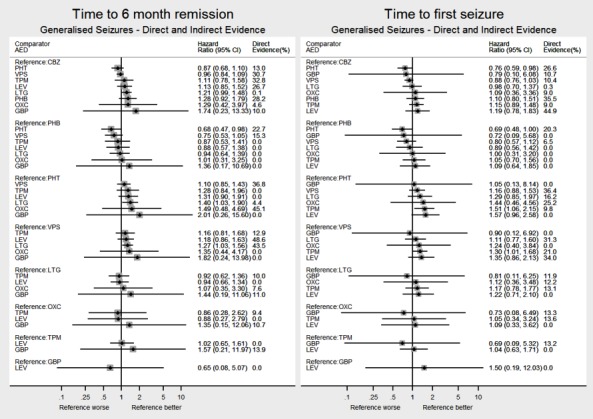
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with generalised seizures, all pairwise comparisons for time to six‐month remission and time to first seizure.
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
All tables and figures of results indicate the proportion of the treatment effect estimate that is contributed by direct evidence (ranging from 0% where no direct comparison exists to 100% for the carbamazepine vs zonisamide comparison, which is disconnected from the rest of the network ‐ see Figure 1). We note that due to the limited amount of evidence for individuals with generalised seizures for some comparisons in the network; some confidence intervals of treatment effect sizes are very wide.
We investigated inconsistency of the direct and network meta‐analysis estimates via node splitting (Dias 2010) and via ‘design‐by treatment’ inconsistency models (Higgins 2012) – see Data synthesis for further detail. Figure 12; Figure 13; Figure 14; Figure 15; Figure 16 and Figure 17 display investigations of inconsistency graphically. Figures show direct evidence, indirect evidence and network meta‐analysis results (direct plus indirect evidence) for all treatments compared to first‐line treatments carbamazepine and lamotrigine for individuals with partial seizures and sodium valproate for individuals with generalised seizures. Numerical results from investigations of inconsistency for all pairwise comparisons are available from the corresponding author on request.
Figure 12.
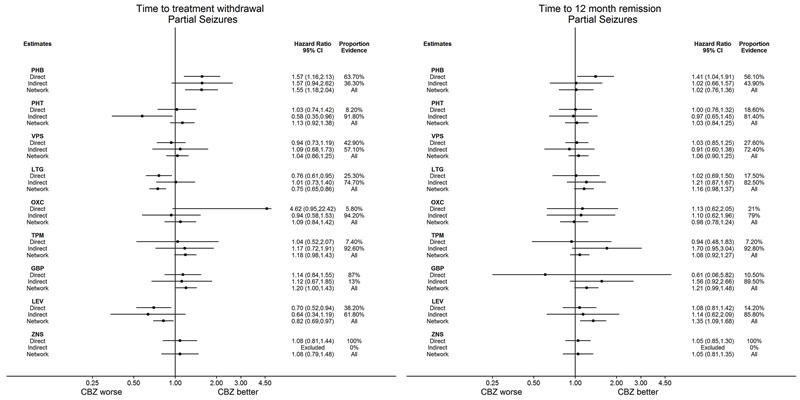
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with partial seizures compared to carbamazepine (CBZ) for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Figure 13.
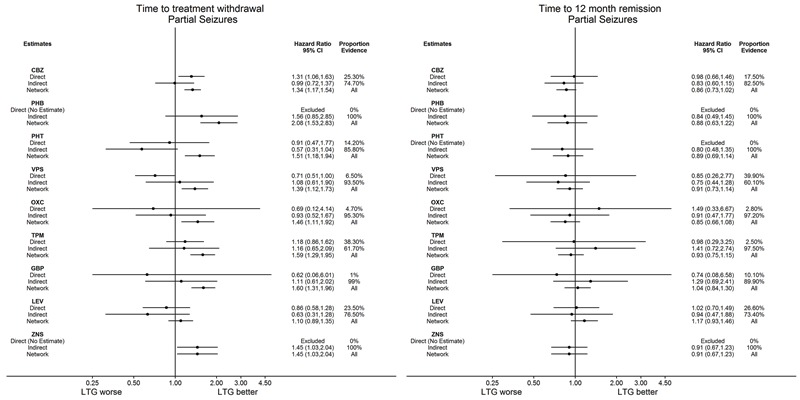
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with partial seizures compared to lamotrigine (LTG) for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Figure 14.
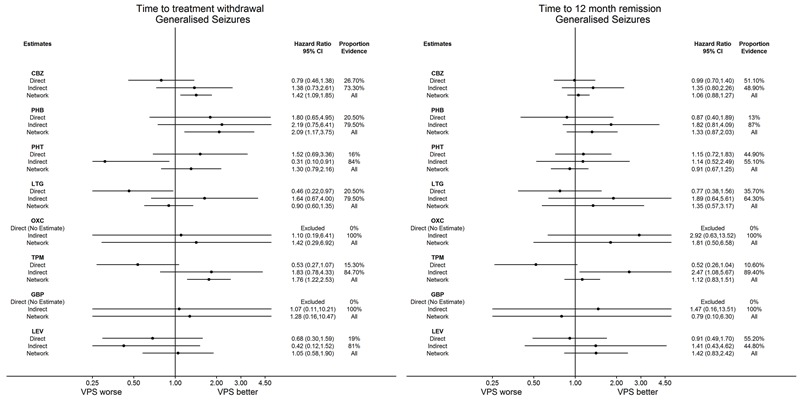
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: Direct, Indirect and Network estimates for individuals with generalised seizures compared to sodium valproate (VPS) for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Figure 15.
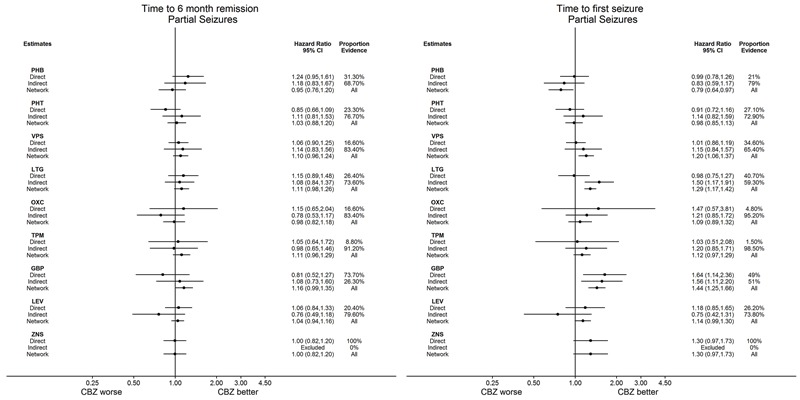
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with partial seizures compared to carbamazepine (CBZ) for time to six‐month remission and time to first seizure.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Figure 16.
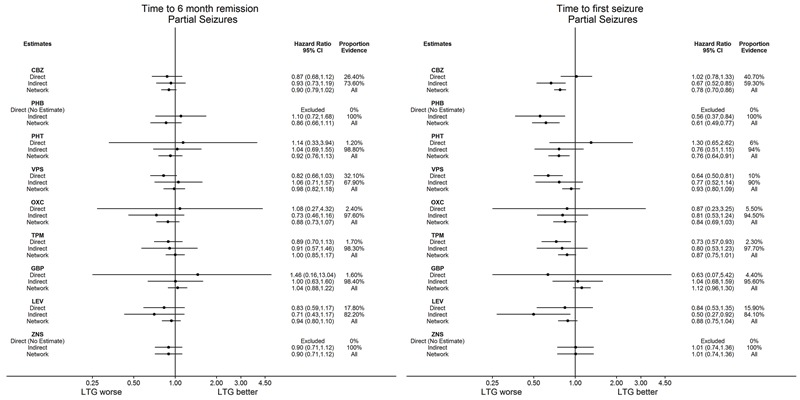
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with partial seizures compared to lamotrigine (LTG) for time to six‐month remission and time to first seizure.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Figure 17.
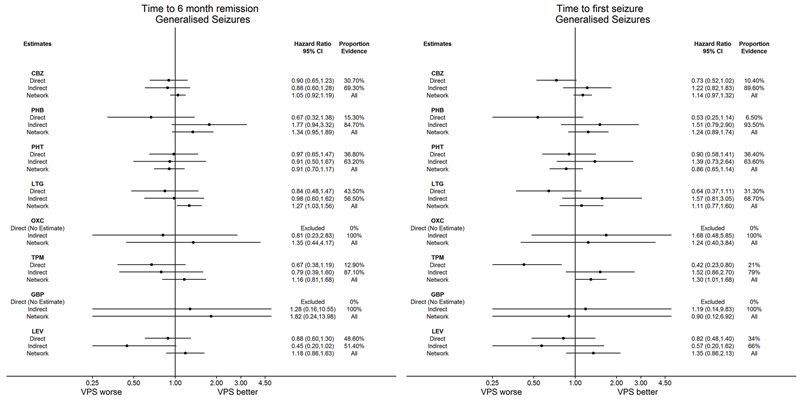
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with generalised seizures compared to sodium valproate (VPS) for time to six‐month remission and time to first seizure.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
We note for the interpretation of these plots that direct evidence comes from the trials that compared the drugs head‐to‐head, indirect evidence comes from the trials that did not compare the drugs head‐to‐head, and direct plus indirect evidence comes from the whole network (head‐to‐head comparisons and indirect comparisons for all drugs).
We examined the numerical results, particularly overlap of confidence intervals of the direct evidence, indirect evidence and network meta‐analysis results. We anticipate that numerical results for the network meta‐analysis will be the most precise. We note potentially important clinical inconsistency to be present where confidence intervals of results from direct evidence and direct plus indirect evidence do not overlap and we consider possible reasons and origins of this inconsistency. Our main concern is statistically significant differences between direct evidence and network meta‐analysis results; however we also note where confidence intervals of results from indirect evidence do not overlap with the confidence intervals of the other estimates.
We conducted a number of sensitivity analyses for each outcome (see Sensitivity analysis for further information). For brevity, we only summarise the conclusions of the sensitivity analyses below rather than presenting full numerical results but these can be made available on request from the corresponding review author.
Time to withdrawal of allocated treatment
The number of participants that contributed to analysis of our primary outcome was 11,865 out of 12,391 participants (96%).
Table 13 shows the reported reasons for withdrawal from treatment across all studies and how we treated each of these reasons in analysis. We note that in some trials, participants many have withdrawn from treatment for a combination of reasons; for the purpose of analysis we have made a judgement regarding the primary reason for withdrawal.
Out of the 11,865 participants who contributed data, 4058 (34%) of individuals prematurely withdrew; fewest participants withdrew from levetiracetam (27%) and sodium valproate (28%) and the most participants withdrew from gabapentin (47%) and phenobarbitone (38%).
The most commonly reported reason for withdrawal from treatment was due to adverse events (38% of all withdrawal 'events'); fewest participants withdrew from gabapentin (20%) and phenobarbitone (20%) due to adverse events and the most participants withdrew from carbamazepine (45%) and topiramate (48%) due to adverse events. Inadequate response (i.e. lack of seizure control) was reported as the reason for withdrawal for 27% of participants ranging from 16% of participants on phenobarbitone to 62% of participants on gabapentin.
We censored 7704 participants out of 11,865 (66%) in the analysis. The majority of censored participants were still taking their allocated treatment at last follow‐up; ranging by drug from 73% (phenobarbitone) to 95% (levetiracetam) of censored participants. Very few participants were lost to follow‐up in the trials (ranging from 0% (gabapentin and zonisamide) to 16% (phenobarbitone)).
For 103 participants, reason for withdrawal was missing (ranging by drug from 0 participants (levetiracetam and zonisamide) to 26 participants (sodium valproate)). We treated those with missing reason for withdrawal as censored in analysis and performed a sensitivity analysis treating these individuals as having withdrawal 'events.' Results of sensitivity analysis were practically identical and conclusions unchanged so we present the results treating these individuals as censored.
We also note that information reported in Table 13 does not take account of randomisation within trials and should be interpreted as exploratory.
Direct evidence
Table 14 (individuals with partial seizures) and Table 15 (individuals with generalised seizures) show the number of trials and participants contributing direct evidence for each of the pairwise comparisons in the network. Twenty out of 45 comparisons had no direct evidence for individuals with partial seizures. Thirteen out of 36 comparisons had no direct evidence for individuals with generalised seizures and eight comparisons for individuals with generalised seizures had fewer than 20 individuals contributing direct evidence resulting in wide confidence intervals around the treatment effect estimate for these comparisons.
The comparisons with the most participants contributing to analysis were carbamazepine vs lamotrigine and carbamazepine vs levetiracetam for individuals with partial seizures and sodium valproate vs levetiracetam and sodium valproate vs topiramate for individuals with generalised seizures.
Table 14 and Table 15 also show estimates for heterogeneity in the direct treatment effects. No substantial heterogeneity was present (I2 greater than 50%) for any comparison for individuals with generalised seizures.
For three comparisons for individuals with partial seizures, substantial heterogeneity was present (I2 greater than 50%). The heterogeneity in these comparisons seemed to originate from difference in trial designs contributing to the pooled result; that is, pooling of trials recruiting children only, adults only or elderly participants only and pooling of double‐blind and open‐label trials (see Nolan 2016b for further discussion of the importance of blinding to the outcome of time‐to‐treatment withdrawal). Repeating analysis with random‐effects did not change conclusions for two of the comparisons (carbamazepine vs phenytoin and phenytoin vs sodium valproate); but for one comparison (phenobarbitone vs phenytoin), when repeating analysis with random‐effects there was no longer a statistically significant advantage to phenytoin: HR 0.42 (0.16 to 1.06)
Network meta‐analysis results (direct plus indirect evidence)
Figure 5 shows how each treatment performed compared to first‐line treatment carbamazepine for individuals with partial seizures (ordered by treatment effect estimate); lamotrigine and levetiracetam are significantly better than carbamazepine, and carbamazepine is significantly better than gabapentin and phenobarbitone.
Figure 6 shows how each treatment performs compared to first‐line treatment lamotrigine for individuals with partial seizures (ordered by treatment effect estimate); lamotrigine is significantly better than all treatments except for levetiracetam.
Figure 7 shows how each treatment performs compared to first‐line treatment sodium valproate for individuals with generalised seizures (ordered by treatment effect estimate); sodium valproate is significantly better than carbamazepine, topiramate and phenobarbitone.
Table 14 and Figure 8 (individuals with partial seizures) and Table 15 and Figure 9 (individuals with generalised seizures) show treatment effect estimates for all pairwise comparisons in the network combining direct with indirect evidence.
In addition to the results described above; for individuals with partial seizures, levetiracetam seems to perform better than most other drugs and for individuals with generalised seizures, lamotrigine seems to perform better than most other drugs. For both individuals with partial seizures and individuals with generalised seizures, phenobarbitone seems to perform worse than most other drugs.
As described further in Assessment of heterogeneity, we could not directly calculate an I2 statistic for the network meta‐analysis but the estimated I2 statistic was 11.7%. When repeating network meta‐analysis with random‐effects the Tau2 statistic was 0.0037, numerical results for treatment effects were very similar (the same to one or two decimal places) and conclusions remained unchanged.
Investigation of inconsistency (node‐splitting)
We fitted the ‘design‐by‐treatment’ inconsistency model to 17 variables and regressed it on 23 designs, five of which were multi‐arm trials (up to five treatment arms). Accounting for the multi‐arm trials, this resulted in an overall test for inconsistency with 36 degrees of freedom, which was not significant (Chi2 statistic (36) = 45.6, P value = 0.1312, heterogeneity (Tau) = 5.65 x 10‐10). Furthermore, there was no significant evidence of inconsistency within any of the 23 designs.
Table 14 (individuals with partial seizures) and Table 15 (individuals with generalised seizures) show treatment effect estimates from direct evidence and from direct plus indirect evidence, Figure 12 and Figure 13 show treatment effect estimates for direct, indirect, and direct plus indirect evidence for individuals with partial seizures compared to carbamazepine and lamotrigine respectively and Figure 14 for individuals with generalised seizures compared to sodium valproate.
We note that for most pairwise comparisons, numerical results of direct evidence and network meta‐analysis are similar, mostly in the same direction and confidence intervals of estimates overlap. For all pairwise comparisons, results from network meta‐analysis are more precise than results from direct evidence (in some cases much more precise where limited direct evidence exists, for example see carbamazepine compared to oxcarbazepine, Figure 12).
For the following comparisons, conclusions drawn from direct evidence and from network meta‐analysis are different (see Table 14 and Table 15).
Direct evidence shows a significant advantage to one of the drugs and the network meta‐analysis results show no significant difference between the drugs: sodium valproate vs topiramate (partial seizures).
Direct evidence shows no significant difference between the drugs and network meta‐analysis shows a significant advantage for one of the drugs: carbamazepine vs gabapentin, lamotrigine vs oxcarbazepine, lamotrigine vs topiramate, lamotrigine vs gabapentin (all partial seizures); carbamazepine vs sodium valproate, carbamazepine vs lamotrigine, phenobarbitone vs sodium valproate, sodium valproate vs topiramate, lamotrigine vs topiramate (all generalised seizures).
No direct evidence exists between the drugs while network meta‐analysis shows a significant advantage for one of the drugs: phenobarbitone vs lamotrigine, phenobarbitone vs levetiracetam, lamotrigine vs zonisamide, topiramate vs levetiracetam, gabapentin vs levetiracetam (all partial seizures); phenobarbitone vs lamotrigine (generalised seizures).
For the following comparisons, confidence intervals for the results from indirect evidence do not overlap with:
direct evidence: carbamazepine vs phenytoin (generalised seizures), phenobarbitone vs phenytoin (generalised seizures);
network meta‐analysis results: lamotrigine vs phenytoin (partial seizures), carbamazepine vs phenytoin (generalised seizures), lamotrigine vs phenytoin (generalised seizures).
For the following comparisons, confidence intervals for the results from direct evidence and from network meta‐analysis do not overlap which indicates potential inconsistency is present (see Table 13, Table 14, Figure 12; Figure 13 and Figure 14): sodium valproate vs lamotrigine (partial seizures), sodium valproate vs topiramate (generalised seizures).
For the comparison of sodium valproate vs lamotrigine for individuals with partial seizures, from direct evidence only, there is a statistically significant advantage to sodium valproate (HR 1.40 (1.00 to 1.96), however from the network meta‐analysis results, the direction of effect changes to a statistically significant advantage to lamotrigine (HR 0.72 (0.58 to 0.90)). However, for this comparison, only 5.1% of the network estimate is contributed from direct evidence and a moderate amount of heterogeneity is present in this estimate (I2 = 45%), likely due to variability in the trial design of the three trials contributing to this estimate (for example, one trial (SANAD B 2007) was designed to only recruit individuals with generalised or unclassified seizures but did recruit a small number of individuals with partial seizures who contribute to this outcome).
For the comparison of sodium valproate vs topiramate for individuals with generalised seizures, from direct evidence, there is no significant difference between the drugs (HR 0.53 (0.27 to 1.07)), however from the network meta‐analysis results, a statistically significant advantage is shown for sodium valproate (HR 1.76 (1.22 to 2.53)). As above, for this comparison, only 22.4% of the network estimate is contributed from direct evidence and a moderate amount of heterogeneity is present in this estimate (I2 = 48.5%). Again, this heterogeneity is likely due to difference in trial design of the two trials contributing direct evidence (see characteristics of Privitera 2003 for details of stratification).
Furthermore, the 'design‐by treatment' inconsistency model does not show any significant evidence of inconsistency within the network. Therefore, we are not concerned about any impact of this observed inconsistency of numerical results on the conclusions of the review.
Subgroup and sensitivity analysis
See Sensitivity analysis for full details and rationale of all sensitivity analyses conducted.
We performed an additional analysis adjusted for age (as well as epilepsy type ‐ see Subgroup analysis and investigation of heterogeneity). Numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We were able to incorporate aggregate or extracted individual‐level data for 471 participants for four additional trials (Biton 2001; Gilad 2007; Steinhoff 2005; Shakir 1981). Numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We performed two sensitivity analyses to investigate the possibility of generalised seizures being misclassified; in the first analysis we reclassified those with generalised seizures and age of onset greater than 30 years as having partial onset seizures and in the second analysis we reclassified generalised seizure types and age at onset greater than 30 years and those with missing seizure type into an 'unclassified seizure type' group.
For the first analysis; numerical results for individuals with generalised seizures were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions. However, for individuals with partial seizures, most numerical results were similar but the most notable change was that phenytoin was now significantly better than all other treatments.
There was a large amount of heterogeneity present in this sensitivity analysis; the estimated I2 statistic was 98% and when repeating network meta‐analysis with random‐effects, Tau2 was 7.074 and confidence intervals of all treatment effect estimates were very wide so that no significant differences were present between any effect sizes. We are unsure why this sensitivity analysis has introduced a large amount of heterogeneity into analysis for this outcome but not for the other outcomes (as described below). Due to this uncertainty, we do not encourage interpretation of this sensitivity analysis.
For the second analysis of seizure type classification, numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We assessed the validity of the proportional hazards assumption of the Cox model used in the network meta‐analysis (see Data synthesis for further details); numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
We excluded one trial (Stephen 2007) from all analyses due to inconsistencies in provided data. Numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
Another trial (Reunanen 1996) was excluded from analysis due to the definition of withdrawal from allocated treatment. Again, numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
For one trial (Placencia 1993), we performed an additional analysis with different definitions of withdrawal from allocated treatment. Again, numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
Time to achieve 12‐month seizure‐free period (remission) after randomisation
The number of participants that contributed to analysis of our secondary outcome, 'Time to achieve 12‐month seizure‐free period' was 9461 out of 12,391 participants (76%).
Direct evidence
Table 16 (individuals with partial seizures) and Table 17 (individuals with generalised seizures) show the number of trials and participants contributing direct evidence for each of the pairwise comparisons in the network. Twenty‐two out of 45 comparisons had no direct evidence for individuals with partial seizures. Fifteen out of 36 comparisons had no direct evidence for individuals with generalised seizures and nine comparisons for individuals with generalised seizures had fewer than 20 individuals contributing direct evidence resulting in wide confidence intervals around the treatment effect estimate for these comparisons.
The comparisons with the most participants contributing to analysis were carbamazepine vs levetiracetam and carbamazepine vs topiramate for individuals with partial seizures and sodium valproate vs levetiracetam and sodium valproate vs topiramate for individuals with generalised seizures.
Table 16 and Table 17 also show estimates of heterogeneity in the direct treatment effects. For three comparisons for individuals with partial seizures and for four comparisons for individuals with generalised seizures, substantial heterogeneity was present (I2 greater than 50%).
The heterogeneity in these comparisons seemed to originate from differences in trial designs contributing to the pooled result; that is, pooling of trials recruiting children only, adults only or elderly participants only and pooling trials with or without treatment strata (see Data extraction and management for further details). None of the treatment effects with substantial heterogeneity present were statistically significant so conclusions would not change for these treatment effects if random‐effects were applied.
Network meta‐analysis results (direct plus indirect evidence)
Figure 5 shows how each treatment performs compared to first‐line treatment carbamazepine for individuals with partial seizures (ordered by treatment effect estimate); carbamazepine is significantly better than levetiracetam.
Figure 6 shows how each treatment performs compared to first‐line treatment lamotrigine for individuals with partial seizures (ordered by treatment effect estimate); there is no significant difference between lamotrigine and the other treatments.
Figure 7 shows how each treatment performs compared to first‐line treatment sodium valproate for individuals with generalised seizures (ordered by treatment effect estimate); there is no significant difference between sodium valproate and the other treatments.
Table 16 and Figure 8 (individuals with partial seizures) and Table 17 and Figure 9 (individuals with generalised seizures) show treatment effect estimates for all pairwise comparisons in the network combining direct with indirect evidence. In addition to the results described above; there are few notable differences between any of the treatments for either individuals with partial seizures or individuals with generalised seizures.
As described further in Assessment of heterogeneity, we could not directly calculate an I2 statistic for the network meta‐analysis but the estimated I2 statistic was 17.3%. When repeating network meta‐analysis with random‐effects, the Tau2 statistic was 0.005, numerical results for treatment effects were very similar (the same to one or two decimal places) and conclusions remained unchanged.
Investigation of inconsistency (node‐splitting)
We fitted the ‘design‐by‐treatment’ inconsistency model was fitted to 17 variables and regressed it on 18 designs, five of which were multi‐arm trials (up to five treatment arms). Accounting for the multi‐arm trials, this resulted in an overall test for inconsistency with 29 degrees of freedom, which was not significant (Chi2 statistic (29) = 14.3, P value = 0.990, heterogeneity (Tau) = 0.154). Furthermore, there was no significant evidence of inconsistency within any of the 18 designs.
Table 16 (individuals with partial seizures) and Table 17 (individuals with generalised seizures) show treatment effect estimates from direct evidence, and from direct plus indirect evidence, Figure 12 and Figure 13 show treatment effect estimates for direct, indirect, and direct plus indirect evidence for individuals with partial seizures compared to carbamazepine and lamotrigine respectively and Figure 14 for individuals with generalised seizures compared to sodium valproate.
We note that for most pairwise comparisons, numerical results of direct evidence and network meta‐analysis are similar, mostly in the same direction and confidence intervals of estimates overlap. For all pairwise comparisons, results from network meta‐analysis are more precise than results from direct evidence (in some cases much more precise where limited direct evidence exists, for example see carbamazepine compared to gabapentin, Figure 12).
For the following comparisons, conclusions drawn from direct evidence and from network meta‐analysis are different (see Table 16 and Table 17).
Direct evidence shows a significant advantage to one of the drugs and the network meta‐analysis results show no significant difference between the drugs: carbamazepine vs phenobarbitone (for both partial seizures and generalised seizures).
Direct evidence shows no significant difference between the drugs and network meta‐analysis shows a significant advantage for one of the drugs: carbamazepine vs levetiracetam, sodium valproate vs lamotrigine (all partial seizures).
No direct evidence exists between the drugs while network meta‐analysis shows a significant advantage for one of the drugs: oxcarbazepine vs levetiracetam (partial seizures).
For the following comparisons, confidence intervals for the results from indirect evidence do not overlap with:
direct evidence: sodium valproate vs topiramate (generalised seizures);
network meta‐analysis results: none.
Confidence intervals overlap for the results from direct evidence and from network meta‐analysis for all comparisons, therefore there is no indication that inconsistency is present in the results (see Table 16, Table 17, Figure 12; Figure 13 and Figure 14).
Subgroup and sensitivity analysis
See Sensitivity analysis for full details and rationale of all sensitivity analyses conducted.
We performed an additional analysis adjusted for age (as well as epilepsy type ‐ see Subgroup analysis and investigation of heterogeneity). Numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
No trials reported aggregate or summary data for this outcome, therefore we did not perform any sensitivity analysis incorporating aggregate data.
We performed two sensitivity analyses to investigate the possibility of generalised seizures being misclassified; in the first analysis we reclassified those with generalised seizures and age of onset greater than 30 years as having partial onset seizures and in the second analysis we reclassified those with generalised seizure types and age at onset greater than 30 years, and those with missing seizure type into an 'unclassified seizure type' group. For both analyses, numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We assessed the validity of the proportional hazards assumption of the Cox model used in the network meta‐analysis (see Data synthesis for further details); there was no evidence the assumption was violated for any of the covariates in the network meta‐analysis, so we did not perform any sensitivity analysis.
We excluded one trial (Stephen 2007) from all analyses due to inconsistencies in provided data. Numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
Time to achieve six‐month seizure‐free period (remission) after randomisation
The number of participants that contributed to analysis of our secondary outcome, 'Time to achieve six‐month seizure‐free period' was 11,820 out of 12,391 participants (95%).
Direct evidence
Table 18 (individuals with partial seizures) and Table 19 (individuals with generalised seizures) show the number of trials and participants contributing direct evidence for each of the pairwise comparisons in the network. Twenty‐one out of 45 comparisons had no direct evidence for individuals with partial seizures. Fourteen out of 36 comparisons had no direct evidence for individuals with generalised seizures and eight comparisons for individuals with generalised seizures had fewer than 20 individuals contributing direct evidence resulting in wide confidence intervals around the treatment effect estimate for these comparisons.
The comparisons with the most participants contributing to analysis were carbamazepine vs levetiracetam and carbamazepine vs topiramate for individuals with partial seizures and sodium valproate vs levetiracetam and sodium valproate vs topiramate for individuals with generalised seizures.
Table 18 and Table 19 also show estimates of heterogeneity in the direct treatment effects. For one comparison for individuals with partial seizures and for two comparisons for individuals with generalised seizures, substantial heterogeneity was present (I2 greater than 50%).
The heterogeneity in these comparisons seemed to originate from differences in trial designs contributing to the pooled result; that is, pooling of trials recruiting children only, adults only or elderly participants only and pooling trials with or without treatment strata (see Data extraction and management for further details). None of the treatment effects with substantial heterogeneity present were statistically significant so conclusions would not change for these treatment effects if random‐effects were applied.
Network meta‐analysis results (direct plus indirect evidence)
Figure 5 shows how each treatment performs compared to first‐line treatment carbamazepine for individuals with partial seizures (ordered by treatment effect estimate); there is no significant difference between carbamazepine and the other treatments.
Figure 6 shows how each treatment performs compared to first‐line treatment lamotrigine for individuals with partial seizures (ordered by treatment effect estimate); there is no significant difference between lamotrigine and the other treatments.
Figure 7 shows how each treatment performs compared to first‐line treatment sodium valproate for individuals with generalised seizures (ordered by treatment effect estimate); sodium valproate is significantly better than lamotrigine.
Table 18 and Figure 10 (individuals with partial seizures) and Table 19 and Figure 11 (individuals with generalised seizures) show treatment effect estimates for all pairwise comparisons in the network combining direct with indirect evidence. In addition to the results described above; there are few notable differences between any of the treatments for either individuals with partial seizures or individuals with generalised seizures.
As described further in Assessment of heterogeneity, we could not directly calculate an I2 statistic for the network meta‐analysis but the estimated I2 statistic was 0%. When repeating network meta‐analysis with random‐effects, Tau2 was 7 x 10‐22. As no heterogeneity was present and Tau2 was negligible, numerical results for treatment effects and conclusions were identical.
Investigation of inconsistency (node‐splitting)
We fitted the ‘design‐by‐treatment’ inconsistency model to 17 variables and regressed it on 23 designs, five of which were multi‐arm trials (up to five treatment arms). Accounting for the multi‐arm trials, this resulted in an overall test for inconsistency with 37 degrees of freedom which was not significant (Chi2 statistic (37) = 36.2, P value = 0.508, heterogeneity (Tau) = 8.09 x 10‐12). Furthermore, there was no significant evidence of inconsistency within any of the 23 designs.
Table 18 (individuals with partial seizures) and Table 19 (individuals with generalised seizures) show treatment effect estimates from direct evidence, and from direct plus indirect evidence, Figure 15 and Figure 16 show treatment effect estimates for direct, indirect, and direct plus indirect evidence for individuals with partial seizures compared to carbamazepine and lamotrigine respectively and Figure 17 for individuals with generalised seizures compared to sodium valproate.
We note that for most pairwise comparisons, numerical results of direct evidence and network meta‐analysis are similar, mostly in the same direction and confidence intervals of estimates overlap. For all pairwise comparisons, results from network meta‐analysis are more precise than results from direct evidence (in some cases much more precise where limited direct evidence exists, for example see lamotrigine compared to gabapentin, Figure 16).
For the following comparisons, conclusions drawn from direct evidence and from network meta‐analysis are different (see Table 18 and Table 19).
Direct evidence shows a significant advantage to one of the drugs and the network meta‐analysis results show no significant difference between the drugs: carbamazepine vs phenobarbitone (generalised seizures).
Direct evidence shows no significant difference between the drugs and network meta‐analysis shows a significant advantage for one of the drugs: sodium valproate vs lamotrigine (generalised seizures).
No direct evidence exists between the drugs while network meta‐analysis shows a significant advantage for one of the drugs: none.
For the following comparisons, confidence intervals for the results from indirect evidence do not overlap with:
direct evidence: carbamazepine vs phenobarbitone (generalised seizures);
network meta‐analysis results: none.
Confidence intervals overlap for the results from direct evidence and from network meta‐analysis for all comparisons, therefore there is no indication that inconsistency is present in the results (see Table 18, Table 19, Figure 15; Figure 16 and Figure 17).
Subgroup and sensitivity analysis
See Sensitivity analysis for full details and rationale of all sensitivity analyses conducted.
We performed an additional analysis adjusted for age (as well as epilepsy type ‐ see Subgroup analysis and investigation of heterogeneity). Numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We were able to incorporate aggregate or extracted individual‐level data for 135 participants for one additional trial (Biton 2001). Numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
We performed two sensitivity analyses to investigate the possibility of generalised seizures being misclassified; in the first analysis we reclassified those with generalised seizures and age of onset greater than 30 years as having partial onset seizures and in the second analysis we reclassified generalised seizure types and age at onset greater than 30 years, and those with missing seizure type into an 'unclassified seizure type' group. For both analyses, numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We assessed the validity of the proportional hazards assumption of the Cox model used in the network meta‐analysis (see Data synthesis for further details); there was no evidence the assumption was violated for any of the covariates in the network meta‐analysis so we did not perform any sensitivity analysis.
We excluded one trial (Stephen 2007) from all analyses due to inconsistencies in provided data. Numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
Time to first seizure post randomisation
The number of participants that contributed to analysis of our secondary outcome, 'Time to first seizure post randomisation' was 12,152 out of 12,391 participants (98%).
Direct evidence
Table 20 (individuals with partial seizures) and Table 21 (individuals with generalised seizures) show the number of trials and participants contributing direct evidence for each of the pairwise comparisons in the network. Twenty out of 45 comparisons had no direct evidence for individuals with partial seizures. Thirteen out of 36 comparisons had no direct evidence for individuals with generalised seizures and eight comparisons for individuals with generalised seizures had fewer than 20 individuals contributing direct evidence resulting in wide confidence intervals around the treatment effect estimate for these comparisons.
The comparisons with the most participants contributing to analysis were carbamazepine vs lamotrigine and carbamazepine vs levetiracetam for individuals with partial seizures and sodium valproate vs levetiracetam and sodium valproate vs topiramate for individuals with generalised seizures.
Table 20 and Table 21 also show estimates of heterogeneity in the direct treatment effects. For three comparisons for individuals with partial seizures and for four comparisons for individuals with generalised seizures, substantial heterogeneity was present (I2 greater than 50%). The heterogeneity in these comparisons seemed to originate from differences in trial designs contributing to the pooled result; that is, pooling of trials recruiting children only, adults only or elderly participants only and pooling trials with or without treatment strata (see Data extraction and management for further details). For the comparisons for individuals with partial seizures, none of the treatment effects with substantial heterogeneity present were statistically significant so conclusions would not change for these treatment effects if random‐effects were applied. For the comparisons for individuals with generalised seizures, repeating analysis with random‐effects did not change conclusions for two of the comparisons (carbamazepine vs sodium valproate and phenytoin vs sodium valproate); but for one comparison (carbamazepine vs phenobarbitone), when repeating analysis with random‐effects there was no longer a statistically significant advantage to phenobarbitone: HR 0.59 (0.27 to 1.26)
Network meta‐analysis results (direct plus indirect evidence)
Figure 5 shows how each treatment performs compared to first‐line treatment carbamazepine for individuals with partial seizures (ordered by treatment effect estimate); phenobarbitone is significantly better than carbamazepine and carbamazepine is significantly better than sodium valproate, lamotrigine and gabapentin.
Figure 6 shows how each treatment performs compared to first‐line treatment lamotrigine for individuals with partial seizures (ordered by treatment effect estimate); phenobarbitone, phenytoin and carbamazepine are significantly better than lamotrigine.
Figure 7 shows how each treatment performs compared to first‐line treatment sodium valproate for individuals with generalised seizures (ordered by treatment effect estimate); sodium valproate is significantly better than topiramate.
Table 20 and Figure 10 (individuals with partial seizures) and Table 21 and Figure 11 (individuals with generalised seizures) show treatment effect estimates for all pairwise comparisons in the network combining direct with indirect evidence. In addition to the results described above; for individuals with partial seizures, phenobarbitone and phenytoin seems to perform better than most other drugs and for individuals with generalised seizures, phenytoin seems to perform better than most other drugs. There were few notable differences between the newer drugs (oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide) for either individuals with partial seizures or individuals with generalised seizures.
As described further in Assessment of heterogeneity, we could not directly calculate an I2 statistic for the network meta‐analysis the estimated I2 statistic was 0%. When repeating network meta‐analysis with random‐effects, Tau2 was 9 x 10‐21. As no heterogeneity was present and Tau2 was negligible, numerical results for treatment effects and conclusions were identical.
Investigation of inconsistency (node‐splitting)
We fitted the ‘design‐by‐treatment’ inconsistency model to 17 variables and regressed it on 23 designs, seven of which were multi‐arm trials (up to five treatment arms). Accounting for the multi‐arm trials, this resulted in an overall test for inconsistency with 43 degrees of freedom, which was not significant (Chi2 statistic (43) = 38.2, P value = 0.680, heterogeneity (Tau) = 0.094). Furthermore, there was no significant evidence of inconsistency within any of the 23 designs.
Table 20 (individuals with partial seizures) and Table 21 (individuals with generalised seizures) show treatment effect estimates from direct evidence, and from direct plus indirect evidence, Figure 15 and Figure 16 show treatment effect estimates for direct, indirect, and direct plus indirect evidence for individuals with partial seizures compared to carbamazepine and lamotrigine respectively and Figure 17 for individuals with generalised seizures compared to sodium valproate.
We note that for most pairwise comparisons, numerical results of direct evidence and network meta‐analysis are similar, mostly in the same direction and confidence intervals of estimates overlap. For all pairwise comparisons, results from network meta‐analysis are more precise than results from direct evidence (in some cases much more precise where limited direct evidence exists, for example see lamotrigine compared to gabapentin, Figure 15).
For the following comparisons; conclusions drawn from direct evidence and from network meta‐analysis are different (see Table 20 and Table 21).
Direct evidence shows a significant advantage to one of the drugs and the network meta‐analysis results show no significant difference between the drugs: sodium valproate vs lamotrigine (partial seizures); carbamazepine vs phenobarbitone (generalised seizures).
Direct evidence shows no significant difference between the drugs and network meta‐analysis shows a significant advantage for one of the drugs: carbamazepine vs phenobarbitone, carbamazepine vs sodium valproate, carbamazepine vs lamotrigine, phenobarbitone vs sodium valproate, phenytoin vs sodium valproate, phenytoin vs lamotrigine, oxcarbazepine vs gabapentin (all partial seizures), carbamazepine vs phenytoin, phenobarbitone vs phenytoin (generalised seizures).
No direct evidence exists between the drugs while network meta‐analysis shows a significant advantage for one of the drugs: phenobarbitone vs lamotrigine, phenobarbitone vs oxcarbazepine, phenobarbitone vs topiramate, phenobarbitone vs gabapentin, phenobarbitone vs levetiracetam, phenobarbitone vs zonisamide, phenytoin vs gabapentin, gabapentin vs levetiracetam (all partial seizures).
Confidence intervals for the results from indirect evidence overlapped with the confidence intervals from direct evidence and from network meta‐analysis for all comparisons.
For the following comparisons, confidence intervals for the results from direct evidence and from network meta‐analysis do not overlap, which indicates potential inconsistency is present (see Table 20, Table 21, Figure 15; Figure 16 and Figure 17): phenobarbitone vs sodium valproate (partial seizures), sodium valproate vs topiramate (generalised seizures).
For the comparison of phenobarbitone vs sodium valproate for individuals with partial seizures, from direct evidence, there is no significant difference between the drugs (HR 0.71 (0.43 to 1.17)), however from the network meta‐analysis results, a statistically significant advantage is shown for phenobarbitone (HR 1.53 (1.20 to 1.94)). For this comparison, only 12.8% of the network estimate is contributed from direct evidence and only 80 individuals contribute to this estimate. This small sample size and imprecision for the direct evidence is likely because sodium valproate is not considered to be a first‐line treatment for partial seizures and although phenobarbitone is a broad spectrum agent for the treatment of many seizure types, it is no longer used as a first‐line treatment (see NICE 2012 and Description of the intervention).
For the comparison of sodium valproate vs topiramate for individuals with generalised seizures, from direct evidence only, there is a statistically significant advantage to topiramate (HR 0.42 (0.23 to 0.80)), however from the network meta‐analysis results, the direction of effect changes to a statistically significant advantage to sodium valproate (HR 1.30 (1.01 to 1.68)). Furthermore, for this comparison, only 21% of the network estimate is contributed from direct evidence and a moderate amount of heterogeneity is present in this estimate (I2 = 46%). The same two trials contribute evidence to this outcome as ‘Time to withdrawal of allocated treatment;’ see above for discussion of the differences in design of these trials.
Furthermore, the 'design‐by treatment' inconsistency model does not show any significant evidence of inconsistency within the network. Therefore, we are not concerned about any impact of this observed inconsistency of numerical results on the conclusions of the review.
Subgroup and sensitivity analysis
See Sensitivity analysis for full details and rationale of all sensitivity analyses conducted.
We performed an additional analysis adjusted for age (as well as epilepsy type ‐ see Subgroup analysis and investigation of heterogeneity). Numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We were able to incorporate aggregate or extracted individual‐level data for 199 participants from two additional trials (Biton 2001; Gilad 2007). Numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
We performed two sensitivity analyses to investigate the possibility of generalised seizures being misclassified; in the first analysis we reclassified those with generalised seizures and age of onset greater than 30 years as having partial onset seizures, and in the second analysis we reclassified generalised seizure types and age at onset greater than 30 years, and those with missing seizure type into an 'unclassified seizure type' group. For both analyses, numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We assessed the validity of the proportional hazards assumption of the Cox model used in the network meta‐analysis (see Data synthesis for further details); most numerical results of this sensitivity analysis were similar, however there were a few changes in conclusions from those above, most notably that lamotrigine became significantly better than gabapentin, and that sodium valproate was no longer significantly better than topiramate (or any other treatment).
We excluded one trial (Stephen 2007) from all analyses due to inconsistencies in provided data. Numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with partial seizures) and conclusions remained unchanged.
We excluded another trial (Banu 2007) from analysis due to inconsistencies in provided data. Again, numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with partial seizures) and conclusions remained unchanged.
We excluded one trial (Nieto‐Barrera 2001) from analysis as we were not provided with seizure dates in the first four weeks of the trial. Again, numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with partial seizures) and conclusions remained unchanged.
Occurence of adverse events
We were provided with individual participant data for adverse events experienced during the trial for 23 trials (Banu 2007; Baulac 2012; Biton 2001; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2007; Chadwick 1998; Dizdarer 2000; Eun 2012; Kwan 2009; Lee 2011; Nieto‐Barrera 2001; Ogunrin 2005; Privitera 2003; Ramsey 2010; Reunanen 1996; SANAD A 2007; SANAD B 2007; Steiner 1999; Stephen 2007; Trinka 2013; Werhahn 2015). The remaining 13 trials providing IPD, did not provide detailed IPD for adverse events, so we extracted information regarding adverse events from the trial publications (Bill 1997; Craig 1994; de Silva 1996; Guerreiro 1997; Heller 1995; Mattson 1985; Mattson 1992; Pal 1998; Placencia 1993; Ramsey 1992; Richens 1994; Turnbull 1985; Verity 1995). No adverse events data was reported in three of these publications (de Silva 1996; Heller 1995; Turnbull 1985).
We were also able to extract a summary of adverse event data from 26 trials not providing IPD ((Brodie 2002; Callaghan 1985; Capone 2008; Christe 1997; Consoli 2012; Dam 1989; Donati 2007; Feksi 1991; Gilad 2007; Jung 2015; Kalviainen 2002; Korean Lamotrigine Study Group 2008; Motamedi 2013; NCT01498822; NCT01954121; Pulliainen 1994; Ramsey 1983; Rastogi 1991; Resendiz 2004; Rowan 2005; Saetre 2007; Shakir 1981; So 1992; Steinhoff 2005; Suresh 2015; Thilothammal 1996).
No adverse event data was reported in 15 publications (Aikia 1992; Bidabadi 2009; Castriota 2008; Chen 1996; Cho 2011; Cossu 1984; Czapinski 1997; Forsythe 1991; Fritz 2006; Kopp 2007; Lukic 2005; Mitchell 1987; Miura 1990; Ramsey 2007; Ravi Sudhir 1995)
Due to the wide range of events reported in the trials and the different methods of recording and reporting of adverse events, we have not analysed adverse event data in meta‐analysis and provide a narrative report. We took the following approach to the negative synthesis of adverse events. One review author (SJN) grouped verbatim or reported terms extracted from publications or provided in IPD under higher level definitions and discussed any uncertainties in definition with the senior clinical author (AGM). We took the definitions used in this review from a previous review in our series of IPD monotherapy reviews (Nolan 2016a), with further definitions added as appropriate when reviewing the reported terms.
Table 22 describes the number of adverse events and the number of participants experiencing adverse events respectively by drug. Table 23 describes the frequency of some of the most commonly associated side effects of AEDs by drug.
Table 16.
Adverse events ‐ number of participants and number of events
| Drug | Number of participants randomised | Number of participants reporting adverse eventsa,b | Number of events reporteda,b |
| CBZ | 5134 | 3023 | 9769 |
| PHB | 754 | 271 | 181 |
| PHT | 1384 | 614 | 1513 |
| VPS | 2303 | 1294 | 3599 |
| LTG | 3107 | 1608 | 6296 |
| OXC | 978 | 623 | 1000 |
| TPM | 1898 | 920 | 6316 |
| GBP | 1209 | 506 | 2580 |
| LEV | 948 | 1441 | 4258 |
| ZNS | 282 | 182 | 606 |
| Total | 18,045 | 10,482 | 36,118 |
CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aAdverse event data were provided as detailed individual participant data for 23 trials and we extracted summary adverse event information from 36 trial publications. No adverse event data were reported in 18 trial publications. bSome trial publications reported only on the “most common” adverse events, the totals and frequencies are likely to be an underestimation of the true number of events and number of individuals experiencing events. Furthermore, detailed information was provided in the more recent trial publications and individual participant data requests of more recent trials, often involving newer antiepileptic drugs, such as LTG, LEV and TPM; which may indicate that these newer drugs are associated with more adverse events than older drugs such as PHB and PHT, for which less detailed information was available.
Table 17.
Adverse events ‐ frequency of most commonly reported events
| Event (general description)a,b,c | CBZ | PHB | PHT | VPS | LTG | OXC | TPM | GBP | LEV | ZNS | Total |
| Accidental injury | 100 | 0 | 100 | 28 | 110 | 5 | 95 | 36 | 58 | 8 | 540 |
| Anorexia or weight loss | 126 | 0 | 126 | 24 | 116 | 6 | 394 | 58 | 62 | 25 | 937 |
| Anxiety/depression | 203 | 0 | 203 | 59 | 171 | 32 | 309 | 82 | 163 | 16 | 1238 |
| Aphasia | 59 | 7 | 66 | 11 | 26 | 4 | 106 | 22 | 11 | 2 | 314 |
| Asthenia | 59 | 1 | 60 | 26 | 41 | 1 | 31 | 33 | 37 | 10 | 299 |
| Ataxia | 172 | 37 | 209 | 32 | 55 | 17 | 61 | 40 | 32 | 8 | 663 |
| Cognitive (memory, concentration, confusion etc.) | 321 | 41 | 362 | 100 | 204 | 44 | 439 | 127 | 73 | 19 | 1730 |
| Dental problems | 93 | 0 | 93 | 28 | 62 | 5 | 61 | 24 | 70 | 7 | 443 |
| Dizziness/faintness | 617 | 0 | 617 | 171 | 348 | 140 | 269 | 160 | 394 | 23 | 2739 |
| Drowsiness/fatigue | 1270 | 1 | 1271 | 422 | 539 | 233 | 628 | 326 | 477 | 33 | 5200 |
| Fever or viral infection | 379 | 0 | 379 | 68 | 172 | 24 | 84 | 58 | 338 | 37 | 1539 |
| Gastrointestinal disturbances | 683 | 20 | 703 | 246 | 394 | 33 | 236 | 142 | 284 | 42 | 2783 |
| Hair loss | 47 | 0 | 47 | 130 | 22 | 15 | 39 | 8 | 16 | 3 | 327 |
| Headache or migraine | 843 | 0 | 843 | 264 | 556 | 137 | 315 | 171 | 596 | 47 | 3772 |
| Impotence | 90 | 24 | 114 | 13 | 17 | 0 | 27 | 32 | 11 | 3 | 331 |
| Increased/worsened seizures | 151 | 0 | 151 | 31 | 164 | 6 | 58 | 48 | 140 | 6 | 755 |
| Infection | 121 | 0 | 121 | 19 | 90 | 4 | 56 | 27 | 63 | 5 | 506 |
| Laboratory results abnormal | 367 | 0 | 367 | 103 | 117 | 8 | 47 | 19 | 90 | 32 | 1150 |
| Menstrual problems | 110 | 0 | 110 | 28 | 31 | 1 | 22 | 18 | 39 | 4 | 363 |
| Mood or behavioural change | 279 | 41 | 320 | 128 | 163 | 25 | 415 | 121 | 121 | 15 | 1628 |
| Nausea/vomiting | 413 | 1 | 414 | 167 | 233 | 53 | 132 | 92 | 142 | 20 | 1667 |
| Pain | 345 | 1 | 346 | 65 | 250 | 6 | 154 | 48 | 251 | 25 | 1491 |
| Paraesthesia or tingling | 56 | 0 | 56 | 22 | 33 | 2 | 708 | 34 | 28 | 7 | 946 |
| Problems sleeping/nightmares | 108 | 1 | 109 | 46 | 197 | 16 | 147 | 31 | 101 | 14 | 770 |
| Rash or skin disorder | 701 | 17 | 718 | 46 | 420 | 73 | 163 | 113 | 125 | 31 | 2407 |
| Renal/urinary disorder | 152 | 0 | 152 | 27 | 78 | 2 | 92 | 57 | 93 | 21 | 674 |
| Respiratory disorder | 233 | 0 | 233 | 53 | 124 | 4 | 190 | 23 | 131 | 17 | 1008 |
| Tremor or twitch | 171 | 1 | 172 | 258 | 219 | 19 | 56 | 23 | 51 | 2 | 972 |
| Visual disturbance/nystagmus | 199 | 0 | 199 | 53 | 96 | 33 | 86 | 59 | 33 | 8 | 766 |
| Weight gain | 259 | 0 | 259 | 347 | 167 | 22 | 71 | 258 | 70 | 1 | 1454 |
CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aVerbatim or reported terms extracted from publications or provided in individual participant data were grouped under the definitions by one review author (SJN) and any uncertainties in definition were discussed with the senior clinical author (AGM).
bAdverse event data were provided as detailed individual participant data for 23 trials and we extracted summary adverse event information from 36 trial publications. No adverse event data were reported in 18 trial publications.
cSome trial publications reported only on the “most common” adverse events, the totals and frequencies are likely to be an underestimation of the true number of events and number of individuals experiencing events. Furthermore, detailed information was provided in the more recent trial publications and individual participant data requests of more recent trials, often involving newer antiepileptic drugs, such as LTG, LEV and TPM; which may indicate that these newer drugs are associated with more adverse events than older drugs such as PHB and PHT ,for which less detailed information was available.
The most commonly occurring adverse events across all drugs were drowsiness/fatigue, headache or migraine, gastrointestinal disturbances, dizziness/faintness and rash or skin disorders.
Drowsiness/fatigue was the most commonly reported adverse event of carbamazepine, phenytoin, sodium valproate, oxcarbazepine and gabapentin. Headache or migraine was the most commonly reported adverse event of lamotrigine, levetiracetam and zonisamide, Paraesthesia (tingling or 'pins and needles') was the most commonly reported adverse event of topiramate and cognitive disorders (memory or concentration difficulties, confusion etc.) and mood or behaviour changes (including aggression) were the most commonly reported adverse event of phenobarbitone.
We note that as some trial publications reported only on the “most common” adverse events, the totals and frequencies are likely to be an underestimation of the true number of events and number of individuals experiencing events. Furthermore in general, more detailed information was provided in the more recent trial publications and IPD requests of more recent trials often involving newer AEDs such as lamotrigine, levetiracetam and topiramate; which may indicate that these newer drugs are associated with more adverse events than older drugs such as phenobarbitone and phenytoin, for which less detailed information was available.
Such limitations must be taken into account when interpreting Table 22 and Table 23 as well as the definitions of adverse events in the review, which were defined by the review authors rather than according to dictionary terminology (such as MedDRA®); we encourage only general comparison of the relative frequencies of different adverse events experienced by participants on different drugs and we do not encourage direct interpretation of numerical frequencies of adverse events.
Discussion
Summary of main results
For brevity throughout the results section, we refer to participants with generalised tonic‐clonic seizures with or without other generalised seizure types as 'participants with generalised seizures.'
Individual participant data were provided for at least one outcome of this review for 12,391 participants with partial seizures or generalised seizures randomised to carbamazepine, phenytoin, sodium valproate, phenobarbitone, oxcarbazepine (oxcarbazepine), lamotrigine, gabapentin, topiramate, levetiracetam or zonisamide (zonisamide) in 36 trials.
We calculated ‘direct estimates’ via meta‐analysis of the head‐to‐head comparisons of the drugs within the trials and performed network meta‐analysis to combine this direct evidence with indirect evidence across the network of 10treatments. Network meta‐analysis provided a total of 45 pairwise comparisons for individuals with partial seizures and 36 pairwise comparisons for individuals with generalised seizures (no participants with generalised onset seizures were randomised to zonisamide).
Direct estimates could be calculated for between half and two thirds of comparisons across the outcomes of the review, however for many of the comparisons, data were contributed by only a single trial or by a small number of participants, or both. Where pooling of head‐to‐head data was possible, direct evidence was generally quite consistent, and where substantial heterogeneity was present between trials (I2 greater than 50%), it is likely that the heterogeneity originated from variability in design of the pooled trials such as pooling of trials recruiting different age groups, pooling of double‐blind and open‐label trials and pooling of trials with and without treatment stratification.
Network meta‐analysis showed that for our primary outcome, ‘Time to withdrawal of allocated treatment,’ for individuals with partial seizures; lamotrigine and levetiracetam were significantly better than first‐line treatment carbamazepine, which was significantly better than gabapentin and phenobarbitone. Lamotrigine was significantly better than all treatments except levetiracetam. For individuals with generalised onset seizures, first‐line treatment sodium valproate performed significantly better than carbamazepine, topiramate and phenobarbitone.
For ‘Time to 12‐month remission of seizures’ and ‘Time to six‐month remission of seizures,’ few notable differences were shown for either seizure type; only that carbamazepine was significantly better than levetiracetam for individuals with partial seizures (12‐month remission) and sodium valproate was significantly better than lamotrigine for individuals with generalised seizures (six‐month remission). Network meta‐analysis also showed that for ‘Time to first seizure,’ for individuals with partial seizures; phenobarbitone was significantly better than both first‐line treatments carbamazepine and lamotrigine; first‐line treatment carbamazepine performed significantly better than sodium valproate, gabapentin and first‐line treatment lamotrigine and phenytoin also performed significantly better than lamotrigine. In general, the earliest licenced treatments (phenytoin and phenobarbitone) performed better than the other treatments for both seizure types.
Results from network meta‐analysis were more precise than results from head‐to‐head comparisons, often much more precise for comparisons where there was limited direct evidence, reflecting the added precision of network meta‐analysis over pairwise meta‐analysis. Across outcomes for the majority of pairwise comparisons, numerical results of direct evidence and network meta‐analysis were similar, mostly in the same direction, confidence intervals of estimates overlapped and there was little indication of inconsistency between direct and network meta‐analysis results. For the few pairwise comparisons where confidence intervals of direct estimates and network meta‐analysis estimates did not overlap, generally direct evidence was limited and contributed only a small proportion of evidence to the network meta‐analysis estimates.
Adverse event data were recorded and reported variably in individual participant datasets and trial publications, therefore we have not attempted to analyse these data and have provided only a narrative report of commonly reported adverse events. The most commonly reported adverse events across all drugs were drowsiness/fatigue, headache or migraine, gastrointestinal disturbances, dizziness/faintness and rash or skin disorders, with some drug‐specific variations (e.g. paraesthesia (tingling or 'pins and needles') was the most commonly reported adverse event of topiramate, and cognitive disorders (memory or concentration difficulties, confusion etc.) and mood or behaviour changes (including aggression) were the most commonly reported adverse event of phenobarbitone).
Overall completeness and applicability of evidence
We have gratefully received IPD for 12,391 out of a total of 17,961 eligible participants (69% of total data) from 36 out of the 77 eligible trials (47%) randomising participants to one of 10 AEDs. We received between 49% and 100% of participant data across the 10 drugs.
Data from the remaining 41 trials could not be provided for a variety of reasons reported by trial authors or sponsors, including data lost or no longer available, cost and resources required to prepare data was prohibitive, local authority‐ or country‐specific restrictions. Furthermore for 15 of these trials, at the time of writing, we have been unable to make contact with an author or sponsor to request data and two trials are currently available only as ClinicalTrials.gov summaries. If data can be made available for any of these additional trials at a later date, they will be included in an update of this review.
Figure 1 shows network plots of pairwise comparisons in all included trials, trials providing IPD and trials without IPD. Visually, the plot of the trials providing IPD is very similar to the plot of all included trials; therefore it is likely that the 69% of participant data we received is a representative sample of all eligible participants and that the 31% of missing participant data can generally be treated as ‘missing at random.’
Specifically, we were provided with IPD for all direct pairwise comparisons in the total network except for oxcarbazepine compared to sodium valproate and oxcarbazepine compared to levetiracetam. In fact, out of all drugs included in the network, we received the lowest proportion of data for oxcarbazepine (49%). The lack of data for these comparisons may have contributed to imprecision of some effect sizes relating to oxcarbazepine (see Figure 9 and Figure 11), therefore we encourage caution when interpreting results relating to oxcarbazepine from this review. We note that the 51% of IPD missing for oxcarbazepine mostly comes from trials for which we could not establish contact with an author or sponsor to request IPD. If additional data can be included in an update for oxcarbazepine, we expect the precision of these estimates to improve.
Figure 2 shows network plots of pairwise comparisons in all eligible participants, from participants with partial seizures and from participants with generalised seizures. The majority of participants recruited into the trials were classified as experiencing partial seizures (66.7% of participants in all trials and 67.5% of participants with IPD provided); this majority is demonstrated in the visual similarity of the network plot for individuals with partial seizures compared to the plot of all participants and reflected in the relative precision of the results of this review for partial seizures compared to generalised seizures.
While a majority of partial seizures compared to generalised seizures is reflective of clinical practice (around 60% of individuals with epilepsy experience partial seizures, NINDS 2015), the proportion of individuals with partial seizures recruited to the trials in this review is even greater. This likely in part reflects the challenges of undertaking trials in children, highlights the need for more large and high‐quality trials.
The remaining participants were classified as experiencing generalised seizures (24.5% of participants in all trials and 26.5% of participants with IPD provided) or unclassified/missing seizure type (8.8% of participants in all trials and 6% of participants with IPD provided). Misclassification of seizure type is a recognised problem in epilepsy (whereby some individuals with generalised seizures have been mistakenly classed as having partial onset seizures and vice versa). The potential impact of this misclassification on results has been shown in our series of Cochrane IPD reviews of monotherapy for epilepsy (Nolan 2016d) whereby up to 50% of individuals classified as experiencing generalised seizures may have had their seizure type misclassified, as an age of seizure onset of over 30 years is unlikely for generalised seizures (Malafosse 1994). Investigation of misclassification within this review (reclassification of 1164 participants with generalised seizures and age of onset of over 30 years, 36% of individuals originally classified at experiencing generalised seizures) did not show any important changes to treatment effect sizes and no changes to conclusions.
This does not, however, indicate that misclassification of seizure type has not occurred in these trials; rather that the primary analysis results are robust to any misclassification. Trials included in this review were published between 1981 and 2015 and a proportion of trials classified generalised and partial onset seizures according to the International League Against Epilepsy (ILAE) classification of 1981 (Commission 1981), rather than the revised ILAE classification in 1989 (Commission 1989) or recently revised terminology (Berg 2010), which may have led to misclassification. Furthermore, several trials were conducted in low‐income countries in Africa, Asia and Central or South America, without access to the same facilities such as EEGs or MRI scanners as trials conducted in the USA and Europe. Within these trials, it is likely that seizure type would have been classified clinically, which may have further contributed to misclassification in these trials
In reality, it is likely that fewer than 20% of participants recruited into all of these trials (17% of participants included in IPD analysis were classified as having generalised seizures following reclassification in sensitivity analysis) experienced generalised seizures which is a lower proportion than would be expected in clinical practice (NINDS 2015). For this reason, treatment effect sizes for generalised seizures, particularly those that are imprecise, should be treated as less applicable than the treatment effect sizes for partial seizures.
In order to provide more precise evidence, applicable to individuals with generalised seizures, it is important both to ensure accurate seizure classification (as far as possible) and to increase the proportion of individuals with generalised seizures recruited into trials of AEDs to better reflect the ‘real world’ ratio of partial to generalised seizures. Increased recruitment of participants may not be straightforward, particularly as those with new onset generalised seizures are expected to be children and adolescents, and recruitment of children into clinical trials comes with difficulties (Joseph 2015); however, if targeted recruitment strategies could be implemented and the evidence base for individuals with generalised seizures increased, this may better inform treatment decisions for this population, particularly for those of childbearing potential, for whom first‐line treatment sodium valproate may not be appropriate (NICE 2012).
Quality of the evidence
This review provides mostly high‐quality evidence for the relative effectiveness of 10 commonly used anti‐epileptic drugs for the treatment of partial seizures and generalised tonic‐clonic seizures. Where limited data were available for a comparison and confidence intervals around treatment effect size results were wide (mostly for individuals with generalised seizures) or where potential inconsistency existed between direct estimates and network meta‐analysis estimates, we judged the quality of the evidence to be moderate and additional data from future trials may impact on these treatment effect estimates (see Table 1; Table 2; Table 3; Table 4; Table 5; Table 6).
Direct estimates and network meta‐analysis estimates were generally consistent and despite some methodological concerns in several trials contributing to analyses, which may have introduced bias into analyses, or inconsistencies present within individual participant data, (see Risk of bias in included studies); numerous sensitivity analyses were performed to test the robustness of the results in the presence of these biases (see Sensitivity analysis for full details); results of sensitivity analyses were numerically similar and did not lead to any changes to conclusions, therefore it is unlikely that any methodological inadequacies of individual trials has influenced the overall pooled network meta‐analysis results
Potential biases in the review process
The search strategies for this review were extensive and we are confident that we have identified all relevant evidence for this review including ongoing trials.
We have taken an IPD approach to analysis, which has many advantages, such as allowing the standardisation of definitions of outcomes across trials, and reducing attrition and reporting biases, as we can perform additional analyses and calculate additional outcomes from unpublished data. For the outcomes we used in this review that are of a time‐to‐event nature, an IPD approach is considered to be the 'gold standard' approach to analysis (Parmar 1998). Furthermore, the use of IPD in this analysis has allowed us to consider the relationship between treatment effect and seizure type via an interaction term in the network meta‐analysis model and present results separately according to seizure type in the context of the recommended first‐line treatment of the seizure type; an approach which would not have been possible without the use of IPD.
Despite the advantages of an IPD approach, for reasons out of our control, we were not able to obtain IPD for 5570 participants from 41 eligible trials, and for the majority of these trials, no aggregate data were available for our outcomes of interest in trial publications. It is inevitable that the exclusion of 31% of eligible participants may be a source of bias in our analyses, however as discussed in more detail above in Overall completeness and applicability of evidence, we believe that the 69% of participants we were able to include in IPD analyses were a representative sample of the total participants included in all eligible trials and that the benefits of an IPD approach outweigh the limitations.
The majority of IPD requested were provided to us directly but for one trial (Biton 2001) we requested data via data sharing portal ClinicalStudyDataRequest.com and data were provided to us via a remote secure data access system, which allowed analysis in SAS‐based statistical software and export of analysis results. We were unable to combine this dataset with the other datasets to perform the analyses described in Data synthesis, therefore we treated the results exported from the data access system as aggregate data in sensitivity analysis (see Sensitivity analysis). As described above, numerical results were similar and conclusions unchanged following the addition of aggregate data to the IPD analyses, therefore the restricted access format of this single trial does not seem to have impacted on the results of the review; however, we are concerned for updates of this review in particular and for future meta‐analyses of IPD in general, that the provision of data in different formats and the increased use of remote access systems may restrict the analyses that it is possible to perform across all eligible datasets and subsequently impact on meta‐analytic results and the scope of clinical questions that are able to be addressed.
Agreements and disagreements with other studies or reviews
In 2007 our group published a network meta‐analysis (NMA) including IPD for over 6418 participants from 20 trials (also included in the current review) comparing direct and indirect evidence from carbamazepine, phenobarbitone, phenytoin, sodium valproate, lamotrigine, oxcarbazepine, topiramate and gabapentin (Tudur Smith 2007). Results of this NMA showed for partial onset seizures that lamotrigine performed better than all other drugs in terms of treatment withdrawal but may not perform better than carbamazepine in terms of seizure control. Phenobarbitone performed better than other drugs in terms of seizure control but at the expense of increased treatment failure. Overall for individuals with partial seizures, lamotrigine, carbamazepine and oxcarbazepine seemed to provide the best balance of seizure control and treatment failure. As in the current review, data for individuals with generalised seizures were limited and results suggested that sodium valproate or phenytoin may provide the best combination of seizure control and treatment failure.
The present review was designed to update the information in the previous NMA with new evidence from trials published since 2007 and including evidence for two drugs, which were licensed for use as monotherapy after 2007 (levetiracetam and zonisamide).
The results of this review generally agree with the results of the NMA, in addition to providing evidence of the comparative effectiveness of the two new drugs within the spectrum of commonly used anti‐epileptic drugs, and further highlight that nearly 10 years on, data for individuals with generalised seizures are still limited.
Authors' conclusions
Current guidelines from the National Institute for Health and Care Excellence (NICE) in the UK for adults and children recommend carbamazepine or lamotrigine as first‐line treatment for partial onset seizures, and sodium valproate for generalised onset seizures (NICE 2012); however given the range of treatment options available to individuals with new onset seizures, including many recently licenced 'second generation' anti‐epileptic drugs (AEDs), the choice of first‐line treatment for an individual must be made based on the highest‐quality evidence of the relative effectiveness and tolerability of AEDs compared to one another.
Results of this review demonstrate that generally the earliest licenced AEDs such as phenytoin and phenobarbitone provide increased seizure control, in terms of delaying recurrence of first seizure and earlier remission, compared to newer AEDs. However, this comes at the expense of earlier treatment failure and it is newer AEDs such as lamotrigine and levetiracetam that perform the best in terms of treatment retention. Considering the optimum balance of efficacy (seizure control) and tolerability (treatment retention), for individuals with partial seizures, carbamazepine, lamotrigine and levetiracetam seem to be the best treatment options whereas for individuals with generalised tonic‐clonic seizures (with or without other seizure types), sodium valproate, lamotrigine and levetiracetam seem to be the best treatment options. Zonisamide, the most recently licenced AED for monotherapy treatment, may be an effective treatment option for individuals with partial onset seizures; however further evidence from randomised controlled trials is needed and the effectiveness of this drug has yet to be evaluated in a published clinical trial for individuals with generalised seizures.
Overall, the high‐quality evidence provided by this review is in line with NICE guidelines that carbamazepine and lamotrigine are suitable first‐line treatments for individuals with partial onset seizures and also demonstrates that levetiracetam may be a suitable alternative. High‐quality evidence from this review is also in line with the use of sodium valproate as the first‐line treatment for individuals with generalised tonic‐clonic seizures (with or without other seizure types) and also demonstrates that lamotrigine and levetiracetam would be suitable alternative first‐line treatments, particularly for those of child bearing potential, for whom sodium valproate may not be an appropriate treatment option. Evidence for the relative effectiveness of other AEDs for individuals with generalised seizures is limited and of moderate quality; further evidence from randomised controlled trials is needed.
This review highlights the need for the design of future AED monotherapy trials that are well powered to detect a difference between particular AEDs while recruiting a sample of individuals representative of the wider population in terms of age and seizure type. An approach to best reflect and inform clinical practice, as well as being statistically powerful, would be to recruit heterogeneous populations for whom epilepsy syndromes have been adequately defined, with testing for interaction between treatment and epilepsy syndrome. In view of potential problems of misclassification, syndromes will have to be well defined, with adequate checking mechanisms to ensure that classifications are accurate and a system to recognise uncertainty surrounding epilepsy syndromes in individuals within trials.
The choice of outcomes at the design stage of a trial and the presentation of the results of outcomes, particularly of a time‐to‐event nature, require very careful consideration. While the majority of trials of a monotherapy design do record and report outcomes measuring efficacy and tolerability (adverse events), there is little uniformity between the definition of the outcomes and the reporting of the summary statistics related to the outcomes (Nolan 2013b), making an aggregate data approach to meta‐analysis in reviews of monotherapy trials impossible. Where trial authors cannot or will not make individual participant data (IPD) available for analysis, review authors are left with no choice but to exclude a proportion of relevant evidence from their review, which will inevitably have some impact upon the interpretation of results of the review and applicability of the evidence and conclusions. The International League Against Epilepsy recommends that trials of a monotherapy design should adopt a primary effectiveness outcome of 'time to withdrawal of allocated treatment (retention time)' and should be of a duration of at least 48 weeks to allow for assessment of longer‐term outcomes, such as remission (ILAE 1998). If trials followed these recommendations, an aggregate data approach to meta‐analysis may be feasible, reducing the resources and time required from an IPD approach.
The provision of accessible, standardised and high‐quality data (whether provided at the aggregate or IPD level) is essential to allow updates of this review and future reviews of AED therapy as further information becomes available, particularly for recently licenced and future treatment options.
Acknowledgements
We are very grateful to Graham Chan, Cochrane Epilepsy Information Specialist for developing electronic search strategies for this review.
We are grateful to the translators of the Spanish, Italian, Persian and Chinese trials.
We are greatly indebted to the original trial investigators and sponsors for their time and efforts in making individual participant data available for this review.
Appendices
Appendix 1. Search strategy for Cochrane Epilepsy's Specialized Register
#1 MeSH DESCRIPTOR Carbamazepine Explode All
#2 Carbamezepine OR CBZ OR SPD417 OR Apo‐Carbamazepine OR Atretol OR Biston OR Calepsin OR Carbagen OR Carbamazepen OR Carbatrol OR Carbazepine OR Carbelan OR Epitol OR Equetro OR Finlepsin OR Karbamazepin OR Lexin OR Neurotol OR Novo‐Carbamaz OR Nu‐Carbamazepine OR Sirtal OR Stazepin OR Stazepine OR Taro‐Carbamazepine OR Tegretal OR Tegretol OR Telesmin OR Teril OR Timonil
#3 #1 OR #2
#4 MeSH DESCRIPTOR Phenytoin Explode All
#5 Dihydantoin OR Diphenylhydantoin OR Diphenylhydantoine OR Diphenylhydatanoin OR Fenitoina OR Phenytoine OR Phenytoinum OR Aleviatin OR Antisacer OR Auranile OR Causoin OR Citrullamon OR Citrulliamon OR Comital OR Comitoina OR Convul OR Danten OR Dantinal OR Dantoinal OR Dantoine OR Denyl OR Di‐Hydan OR Di‐Lan OR Di‐Phetine OR Didan OR Difenilhidantoina OR Difenin OR Difetoin OR Difhydan OR Dihycon OR Dilabid OR Dilantin OR Dilantine OR Dillantin OR Dintoin OR Dintoina OR Diphantoin OR Diphedal OR Diphedan OR Diphenat OR Diphenin OR Diphenine OR Dipheninum OR Diphentoin OR Diphentyn OR Diphenylan OR Ditoinate OR Ekko OR Elepsindon OR Enkelfel OR Epamin OR Epanutin OR Epasmir OR Epdantoin OR Epdantoine OR Epelin OR Epifenyl OR Epihydan OR Epilan OR Epilantin OR Epinat OR Epised OR Eptal OR Eptoin OR Fenantoin OR Fenidantoin OR Fentoin OR Fenylepsin OR Fenytoin OR Fenytoine OR Gerot‐epilan‐D OR Hidan OR Hidantal OR Hidantilo OR Hidantina OR Hidantomin OR Hindatal OR Hydantal OR Hydantin OR Hydantoin OR Hydantoinal OR Hydantol OR Ictalis OR Idantoil OR Idantoin OR Iphenylhydantoin OR Kessodanten OR Labopal OR Lehydan OR Lepitoin OR Lepsin OR Mesantoin OR Minetoin OR Neos‐Hidantoina OR Neosidantoina OR Novantoina OR Novophenytoin OR Om‐hidantoina OR Om‐Hydantoine OR Oxylan OR Phanantin OR Phanatine OR Phenatine OR Phenatoine OR Phenhydan OR Phenhydanin OR Phenitoin OR Phentoin OR Phentytoin OR Phenytek OR Phenytex OR Ritmenal OR Saceril OR Sanepil OR Silantin OR Sinergina OR Sodanthon OR Sodantoin OR Sodanton OR Solantin OR Solantoin OR Solantyl OR Sylantoic OR Tacosal OR Thilophenyl OR TOIN OR Zentronal OR Zentropil OR PHT
#6 #4 OR #5
#7 MeSH DESCRIPTOR Valproic Acid Explode All
#8 Avugane OR Baceca OR Convulex OR Delepsine OR Depacon OR Depakene OR Depakine OR Depakote OR Deproic OR Epiject OR Epilex OR Epilim OR Episenta OR Epival OR Ergenyl OR Mylproin OR Orfiril OR Orlept OR Selenica OR Stavzor OR Valcote OR Valparin OR Valpro OR Valproate OR Valproic OR VPA
#9 #7 OR #8
#10 MeSH DESCRIPTOR Phenobarbital Explode All
#11 Fenobarbital OR Phenobarbitol OR Phenobarbitone OR "Phenobarbituric Acid" OR Phenylethylbarbiturate OR "Phenylethylbarbituric Acid" OR Phenylethylmalonylurea OR Adonal OR Aephenal OR Agrypnal OR Amylofene OR Aphenylbarbit OR Aphenyletten OR Barbenyl OR Barbinal OR Barbiphen OR Barbiphenyl OR Barbipil OR Barbita OR Barbivis OR Barbonal OR Barbophen OR Bardorm OR Bartol OR Bialminal OR Blu‐Phen OR Cabronal OR Calmetten OR Calminal OR Cardenal OR Chinoin OR Codibarbita OR Coronaletta OR Cratecil OR Damoral OR Dezibarbitur OR Dormina OR Dormiral OR Dormital OR Doscalun OR Duneryl OR Ensobarb OR Ensodorm OR Epanal OR Epidorm OR Epilol OR Episedal OR Epsylone OR Eskabarb OR Etilfen OR Euneryl OR Fenbital OR Fenemal OR Fenosed OR Fenylettae OR Gardenal OR Gardepanyl OR Glysoletten OR Haplopan OR Haplos OR Helional OR Hennoletten OR Henotal OR Hypnaletten OR Hypnette OR Hypno‐Tablinetten OR Hypnogen OR Hypnolone OR Hypnoltol OR Hysteps OR Lefebar OR Leonal OR Lephebar OR Lepinal OR Lepinaletten OR Linasen OR Liquital OR Lixophen OR Lubergal OR Lubrokal OR Lumen OR Lumesettes OR Lumesyn OR Luminal OR Lumofridetten OR Luphenil OR Luramin OR Molinal OR Neurobarb OR Nirvonal OR Noptil OR Nova‐Pheno OR Nunol OR Parkotal OR Pharmetten OR Phen‐Bar OR Phenaemal OR Phenemal OR Phenemalum OR Phenobal OR Phenobarbyl OR Phenoluric OR Phenolurio OR Phenomet OR Phenonyl OR Phenoturic OR Phenyletten OR Phenyral OR Phob OR Polcominal OR Prominal OR Promptonal OR Seda‐Tablinen OR Sedabar OR Sedicat OR Sedizorin OR Sedlyn OR Sedofen OR Sedonal OR Sedonettes OR Sevenal OR Sinoratox OR Solfoton OR Solu‐Barb OR Sombutol OR Somnolens OR Somnoletten OR Somnosan OR Somonal OR Spasepilin OR Starifen OR Starilettae OR Stental OR Talpheno OR Teolaxin OR Teoloxin OR Thenobarbital OR Theoloxin OR Triabarb OR Tridezibarbitur OR Triphenatol OR Versomnal OR Zadoletten OR Zadonal OR PB
#12 #10 OR #11
#13 Oxcarbazepine
#14 "GP 47680" OR OCBZ OR Oxcarbamazepine OR Actinium OR Barzepin OR Carbox OR Deprectal OR Lonazet OR Oxalepsy OR Oxetol OR Oxpin OR Oxrate OR Oxtellar OR Oxypine OR Pharozepine OR Prolepsi OR Timox OR Trexapin OR Trileptal OR Trileptin OR OXC
#15 #13 OR #14
#16 Lamotrigine
#17 "GW 273293" OR Lamotrigina OR Lamotriginum OR Lamictal OR Lamotrine OR Lamitrin OR Lamictin OR Lamogine OR Lamitor OR LTG
#18 #16 OR #17
#19 Gabapentin
#20 Gabapentine OR Gabapentino OR Gabapentinum OR Gabapetin OR Aclonium OR Fanatrex OR Gabarone OR Neogab OR Gralise OR Neurontin OR Novo‐Gabapentin OR Nupentin OR GBP
#21 #19 OR #20
#22 Topiramate
#23 Tipiramate OR Topiramatum OR "Topiramic acid" OR Topamax OR TPM
#24 #22 OR #23
#25 Levetiracetam
#26 Levetiracetamum OR Levitiracetam OR Keppra OR LEV
#27 #25 OR #26
#28 Zonisamide
#29 Zonisamida OR Zonisamidum OR Zonegran OR Exceglan OR Excegram OR Excegran OR ZNS
#30 #28 OR #29
#31 #3 OR #6 OR #9 OR #12 OR #15 OR #18 OR #21 OR #24 OR #27 OR #30
#32 ((adjunct* or "add‐on" or "add on" or adjuvant* or combination* or polytherap*) not (monotherap* or alone or singl*)):TI
#33 #31 NOT #32
#34 #33 AND INREGISTER
Appendix 2. CENTRAL via CRSO search strategy
#1 MESH DESCRIPTOR Carbamazepine EXPLODE ALL TREES
#2 biston OR carbamazepin OR carbamazepina OR carbamazepine OR carbamazepinee OR carbamazepines OR carbamazepinesr OR carbamazepinetreated OR carbatrol OR cbz OR epitol OR equetro OR neurotop OR tegretol OR teril OR timonil
#3 #1 OR #2
#4 MESH DESCRIPTOR Phenytoin EXPLODE ALL TREES
#5 dilantin OR epanutin OR eptoin OR fenitoina OR phenytek OR phenytoin OR phenytoine OR phenytoinum
#6 #4 OR #5
#7 MESH DESCRIPTOR Valproic Acid EXPLODE ALL TREES
#8 convulex OR depacon OR depakene OR depakine OR depakote OR dpa OR epilim OR epival OR stavzor OR valproate OR valproic OR vpa
#9 #7 OR #8
#10 MESH DESCRIPTOR Phenobarbital EXPLODE ALL TREES
#11 luminal OR phenobarbital OR phenobarbitalprophylaxe OR phenobarbitals OR phenobarbitol OR phenobarbitone
#12 #10 OR #11
#13 ocbz OR oxcarbazepina OR oxcarbazepine OR trileptal
#14 epilepax OR lamictal OR lamotrigina OR lamotrigine OR lamotriginer OR lamotrigines
#15 gabapentin OR gabapentin1000 OR gabapentina OR gabapentine OR gabapentinin OR gabapentinoid OR gabapentinoids OR gabapentinului OR neurontin
#16 qudexy OR topamax OR topiramate OR topiramate50mg OR topiramateshowed OR topiramatewere OR topiramato OR tpm
#17 keppra OR levetiracetam OR levetiracetame OR levitiracetam
#18 zonegran OR zonisamide OR zonisamidemay OR zonisamidetreated
#19 #3 OR #6 OR #9 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18
#20 (epilep* OR seizure* OR convuls*):TI,AB,KY
#21 MESH DESCRIPTOR Epilepsy EXPLODE ALL TREES
#22 MESH DESCRIPTOR Seizures EXPLODE ALL TREES
#23 #20 OR #21 OR #22
#24 #19 AND #23
#25 ((adjunct* OR "add‐on" OR "add on" OR adjuvant* OR combination* OR polytherap*) NOT (monotherap* or alone or singl*)):TI
#26 #24 NOT #25
#27 ("Conference Abstract"):PT AND INEMBASE
#28 #26 NOT #27
Appendix 3. MEDLINE search strategy
This strategy is based on the Cochrane Highly Sensitive Search Strategy for identifying randomized trials (Lefebvre 2011).
1. exp Carbamazepine/
2. (Carbamezepine or CBZ or SPD417 or Apo‐Carbamazepine or Atretol or Biston or Calepsin or Carbagen or Carbamazepen or Carbatrol or Carbazepine or Carbelan or Epitol or Equetro or Finlepsin or Karbamazepin or Lexin or Neurotol or Novo‐Carbamaz or Nu‐Carbamazepine or Sirtal or Stazepin or Stazepine or Taro‐Carbamazepine or Tegretal or Tegretol or Telesmin or Teril or Timonil).mp.
3. 1 or 2
4. exp Phenytoin/
5. (Dihydantoin or Diphenylhydantoin or Diphenylhydantoine or Diphenylhydatanoin or Fenitoina or Phenytoine or Phenytoinum or Aleviatin or Antisacer or Auranile or Causoin or Citrullamon or Citrulliamon or Comital or Comitoina or Convul or Danten or Dantinal or Dantoinal or Dantoine or Denyl or Di‐Hydan or Di‐Lan or Di‐Phetine or Didan or Difenilhidantoina or Difenin or Difetoin or Difhydan or Dihycon or Dilabid or Dilantin or Dilantine or Dillantin or Dintoin or Dintoina or Diphantoin or Diphedal or Diphedan or Diphenat or Diphenin or Diphenine or Dipheninum or Diphentoin or Diphentyn or Diphenylan or Ditoinate or Ekko or Elepsindon or Enkelfel or Epamin or Epanutin or Epasmir or Epdantoin or Epdantoine or Epelin or Epifenyl or Epihydan or Epilan or Epilantin or Epinat or Epised or Eptal or Eptoin or Fenantoin or Fenidantoin or Fentoin or Fenylepsin or Fenytoin or Fenytoine or Gerot‐epilan‐D or Hidan or Hidantal or Hidantilo or Hidantina or Hidantomin or Hindatal or Hydantal or Hydantin or Hydantoin or Hydantoinal or Hydantol or Ictalis or Idantoil or Idantoin or Iphenylhydantoin or Kessodanten or Labopal or Lehydan or Lepitoin or Lepsin or Mesantoin or Minetoin or Neos‐Hidantoina or Neosidantoina or Novantoina or Novophenytoin or Om‐hidantoina or Om‐Hydantoine or Oxylan or Phanantin or Phanatine or Phenatine or Phenatoine or Phenhydan or Phenhydanin or Phenitoin or Phentoin or Phentytoin or Phenytek or Phenytex or Ritmenal or Saceril or Sanepil or Silantin or Sinergina or Sodanthon or Sodantoin or Sodanton or Solantin or Solantoin or Solantyl or Sylantoic or Tacosal or Thilophenyl or TOIN or Zentronal or Zentropil or PHT).mp.
6. 4 or 5
7. exp Valproic Acid/
8. (Avugane or Baceca or Convulex or Delepsine or Depacon or Depakene or Depakine or Depakote or Deproic or Epiject or Epilex or Epilim or Episenta or Epival or Ergenyl or Mylproin or Orfiril or Orlept or Selenica or Stavzor or Valcote or Valparin or Valpro or Valproate or Valproic or VPA).mp.
9. 7 or 8
10. exp Phenobarbital/
11. (Fenobarbital or Phenobarbitol or Phenobarbitone or "Phenobarbituric Acid" or Phenylethylbarbiturate or "Phenylethylbarbituric Acid" or Phenylethylmalonylurea or Adonal or Aephenal or Agrypnal or Amylofene or Aphenylbarbit or Aphenyletten or Barbenyl or Barbinal or Barbiphen or Barbiphenyl or Barbipil or Barbita or Barbivis or Barbonal or Barbophen or Bardorm or Bartol or Bialminal or Blu‐Phen or Cabronal or Calmetten or Calminal or Cardenal or Chinoin or Codibarbita or Coronaletta or Cratecil or Damoral or Dezibarbitur or Dormina or Dormiral or Dormital or Doscalun or Duneryl or Ensobarb or Ensodorm or Epanal or Epidorm or Epilol or Episedal or Epsylone or Eskabarb or Etilfen or Euneryl or Fenbital or Fenemal or Fenosed or Fenylettae or Gardenal or Gardepanyl or Glysoletten or Haplopan or Haplos or Helional or Hennoletten or Henotal or Hypnaletten or Hypnette or Hypno‐Tablinetten or Hypnogen or Hypnolone or Hypnoltol or Hysteps or Lefebar or Leonal or Lephebar or Lepinal or Lepinaletten or Linasen or Liquital or Lixophen or Lubergal or Lubrokal or Lumen or Lumesettes or Lumesyn or Luminal or Lumofridetten or Luphenil or Luramin or Molinal or Neurobarb or Nirvonal or Noptil or Nova‐Pheno or Nunol or Parkotal or Pharmetten or Phen‐Bar or Phenaemal or Phenemal or Phenemalum or Phenobal or Phenobarbyl or Phenoluric or Phenolurio or Phenomet or Phenonyl or Phenoturic or Phenyletten or Phenyral or Phob or Polcominal or Prominal or Promptonal or Seda‐Tablinen or Sedabar or Sedicat or Sedizorin or Sedlyn or Sedofen or Sedonal or Sedonettes or Sevenal or Sinoratox or Solfoton or Solu‐Barb or Sombutol or Somnolens or Somnoletten or Somnosan or Somonal or Spasepilin or Starifen or Starilettae or Stental or Talpheno or Teolaxin or Teoloxin or Thenobarbital or Theoloxin or Triabarb or Tridezibarbitur or Triphenatol or Versomnal or Zadoletten or Zadonal or PB).mp.
12. 10 or 11
13. (Oxcarbazepine or "GP 47680" or OCBZ or Oxcarbamazepine or Actinium or Barzepin or Carbox or Deprectal or Lonazet or Oxalepsy or Oxetol or Oxpin or Oxrate or Oxtellar or Oxypine or Pharozepine or Prolepsi or Timox or Trexapin or Trileptal or Trileptin or OXC).mp.
14. (Lamotrigine or "GW 273293" or Lamotrigina or Lamotriginum or Lamictal or Lamotrine or Lamitrin or Lamictin or Lamogine or Lamitor or LTG).mp.
15. (Gabapentin or Gabapentine or Gabapentino or Gabapentinum or Gabapetin or Aclonium or Fanatrex or Gabarone or Neogab or Gralise or Neurontin or Novo‐Gabapentin or Nupentin or GBP).mp.
16. (Topiramate or Tipiramate or Topiramatum or "Topiramic acid" or Topamax or TPM).mp.
17. (Levetiracetam or Levetiracetamum or Levitiracetam or Keppra or LEV).mp.
18. (Zonisamide or Zonisamida or Zonisamidum or Zonegran or Exceglan or Excegram or Excegran or ZNS).mp.
19. 3 or 6 or 9 or 12 or 13 or 14 or 15 or 16 or 17 or 18
20. ((adjunct$ or "add‐on" or "add on" or adjuvant$ or combination$ or polytherap$) not (monotherap$ or alone or singl$)).ti.
21. 19 not 20
22. exp Epilepsy/
23. exp Seizures/
24. (epilep$ or seizure$ or convuls$).tw.
25. 22 or 23 or 24
26. exp Pre‐Eclampsia/ or exp Eclampsia/
27. 25 not 26
28. (randomized controlled trial or controlled clinical trial or pragmatic clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
29. clinical trials as topic.sh.
30. trial.ti.
31. 28 or 29 or 30
32. exp animals/ not humans.sh.
33. 31 not 32
34. 21 and 27 and 33
35. remove duplicates from 34
Appendix 4. SCOPUS search strategy
(((TITLE (carbamazepine OR carbamezepine OR cbz OR spd417 OR apo‐carbamazepine OR atretol OR biston OR calepsin OR carbagen OR carbamazepen OR carbatrol OR carbazepine OR carbelan OR epitol OR equetro OR finlepsin OR karbamazepin OR lexin OR neurotol OR novo‐carbamaz OR nu‐carbamazepine OR sirtal OR stazepin OR stazepine OR taro‐carbamazepine OR tegretal OR tegretol OR telesmin OR teril OR timonil OR phenytoin OR dihydantoin OR diphenylhydantoin OR diphenylhydantoine OR diphenylhydatanoin OR fenitoina OR phenytoine OR phenytoinum OR aleviatin OR antisacer OR auranile OR causoin OR citrullamon OR citrulliamon OR comital OR comitoina OR convul OR danten OR dantinal OR dantoinal OR dantoine OR denyl OR di‐hydan OR di‐lan OR di‐phetine OR didan OR difenilhidantoina OR difenin OR difetoin OR difhydan OR dihycon OR dilabid OR dilantin OR dilantine OR dillantin OR dintoin OR dintoina OR diphantoin OR diphedal OR diphedan OR diphenat OR diphenin OR diphenine OR dipheninum OR diphentoin OR diphentyn OR diphenylan OR ditoinate OR ekko OR elepsindon OR enkelfel OR epamin OR epanutin OR epasmir OR epdantoin OR epdantoine OR epelin OR epifenyl OR epihydan OR epilan OR epilantin OR epinat OR epised OR eptal OR eptoin OR fenantoin OR fenidantoin OR fentoin OR fenylepsin OR fenytoin OR fenytoine OR gerot‐epilan‐d OR hidan OR hidantal OR hidantilo OR hidantina OR hidantomin OR hindatal OR hydantal OR hydantin OR hydantoin OR hydantoinal OR hydantol OR ictalis OR idantoil OR idantoin OR iphenylhydantoin OR kessodanten OR labopal OR lehydan OR lepitoin OR lepsin OR mesantoin OR minetoin OR neos‐hidantoina OR neosidantoina OR novantoina OR novophenytoin OR om‐hidantoina OR om‐hydantoine OR oxylan OR phanantin OR phanatine OR phenatine OR phenatoine OR phenhydan OR phenhydanin OR phenitoin OR phentoin OR phentytoin OR phenytek OR phenytex OR ritmenal OR saceril OR sanepil OR silantin OR sinergina OR sodanthon OR sodantoin OR sodanton OR solantin OR solantoin OR solantyl OR sylantoic OR tacosal OR thilophenyl OR toin OR zentronal OR zentropil OR pht OR "Valproic Acid" OR avugane OR baceca OR convulex OR delepsine OR depacon OR depakene OR depakine OR depakote OR deproic OR epiject OR epilex OR epilim OR episenta OR epival OR ergenyl OR mylproin OR orfiril OR orlept OR selenica OR stavzor OR valcote OR valparin OR valpro OR valproate OR valproic OR vpa OR phenobarbital OR fenobarbital OR phenobarbitol OR phenobarbitone OR "Phenobarbituric Acid" OR phenylethylbarbiturate OR "Phenylethylbarbituric Acid" OR phenylethylmalonylurea OR adonal OR aephenal OR agrypnal OR amylofene OR aphenylbarbit OR aphenyletten OR barbenyl OR barbinal OR barbiphen OR barbiphenyl OR barbipil OR barbita OR barbivis OR barbonal OR barbophen OR bardorm OR bartol OR bialminal OR blu‐phen OR cabronal OR calmetten OR calminal OR cardenal OR chinoin OR codibarbita OR coronaletta OR cratecil OR damoral OR dezibarbitur OR dormina OR dormiral OR dormital OR doscalun OR duneryl OR ensobarb OR ensodorm OR epanal OR epidorm OR epilol OR episedal OR epsylone OR eskabarb OR etilfen OR euneryl OR fenbital OR fenemal OR fenosed OR fenylettae OR gardenal OR gardepanyl OR glysoletten OR haplopan OR haplos OR helional OR hennoletten OR henotal OR hypnaletten OR hypnette OR hypno‐tablinetten OR hypnogen OR hypnolone OR hypnoltol OR hysteps OR lefebar OR leonal OR lephebar OR lepinal OR lepinaletten OR linasen OR liquital OR lixophen OR lubergal OR lubrokal OR lumen OR lumesettes OR lumesyn OR luminal OR lumofridetten OR luphenil OR luramin OR molinal OR neurobarb OR nirvonal OR noptil OR nova‐pheno OR nunol OR parkotal OR pharmetten OR phen‐bar OR phenaemal OR phenemal OR phenemalum OR phenobal OR phenobarbyl OR phenoluric OR phenolurio OR phenomet OR phenonyl OR phenoturic OR phenyletten OR phenyral OR phob OR polcominal OR prominal OR promptonal OR seda‐tablinen OR sedabar OR sedicat OR sedizorin OR sedlyn OR sedofen OR sedonal OR sedonettes OR sevenal OR sinoratox OR solfoton OR solu‐barb OR sombutol OR somnolens OR somnoletten OR somnosan OR somonal OR spasepilin OR starifen OR starilettae OR stental OR talpheno OR teolaxin OR teoloxin OR thenobarbital OR theoloxin OR triabarb OR tridezibarbitur OR triphenatol OR versomnal OR zadoletten OR zadonal OR pb OR oxcarbazepine OR "GP 47680" OR ocbz OR oxcarbamazepine OR actinium OR barzepin OR carbox OR deprectal OR lonazet OR oxalepsy OR oxetol OR oxpin OR oxrate OR oxtellar OR oxypine OR pharozepine OR prolepsi OR timox OR trexapin OR trileptal OR trileptin OR oxc OR lamotrigine OR "GW 273293" OR lamotrigina OR lamotriginum OR lamictal OR lamotrine OR lamitrin OR lamictin OR lamogine OR lamitor OR ltg OR gabapentin OR gabapentine OR gabapentino OR gabapentinum OR gabapetin OR aclonium OR fanatrex OR gabarone OR neogab OR gralise OR neurontin OR novo‐gabapentin OR nupentin OR gbp OR topiramate OR tipiramate OR topiramatum OR "Topiramic acid" OR topamax OR tpm OR levetiracetam OR levetiracetamum OR levitiracetam OR keppra OR lev OR zonisamide OR zonisamida OR zonisamidum OR zonegran OR exceglan OR excegram OR excegran OR zns)) OR (ABS(carbamazepine OR carbamezepine OR cbz OR spd417 OR apo‐carbamazepine OR atretol OR biston OR calepsin OR carbagen OR carbamazepen OR carbatrol OR carbazepine OR carbelan OR epitol OR equetro OR finlepsin OR karbamazepin OR lexin OR neurotol OR novo‐carbamaz OR nu‐carbamazepine OR sirtal OR stazepin OR stazepine OR taro‐carbamazepine OR tegretal OR tegretol OR telesmin OR teril OR timonil OR phenytoin OR dihydantoin OR diphenylhydantoin OR diphenylhydantoine OR diphenylhydatanoin OR fenitoina OR phenytoine OR phenytoinum OR aleviatin OR antisacer OR auranile OR causoin OR citrullamon OR citrulliamon OR comital OR comitoina OR convul OR danten OR dantinal OR dantoinal OR dantoine OR denyl OR di‐hydan OR di‐lan OR di‐phetine OR didan OR difenilhidantoina OR difenin OR difetoin OR difhydan OR dihycon OR dilabid OR dilantin OR dilantine OR dillantin OR dintoin OR dintoina OR diphantoin OR diphedal OR diphedan OR diphenat OR diphenin OR diphenine OR dipheninum OR diphentoin OR diphentyn OR diphenylan OR ditoinate OR ekko OR elepsindon OR enkelfel OR epamin OR epanutin OR epasmir OR epdantoin OR epdantoine OR epelin OR epifenyl OR epihydan OR epilan OR epilantin OR epinat OR epised OR eptal OR eptoin OR fenantoin OR fenidantoin OR fentoin OR fenylepsin OR fenytoin OR fenytoine OR gerot‐epilan‐d OR hidan OR hidantal OR hidantilo OR hidantina OR hidantomin OR hindatal OR hydantal OR hydantin OR hydantoin OR hydantoinal OR hydantol OR ictalis OR idantoil OR idantoin OR iphenylhydantoin OR kessodanten OR labopal OR lehydan OR lepitoin OR lepsin OR mesantoin OR minetoin OR neos‐hidantoina OR neosidantoina OR novantoina OR novophenytoin OR om‐hidantoina OR om‐hydantoine OR oxylan OR phanantin OR phanatine OR phenatine OR phenatoine OR phenhydan OR phenhydanin OR phenitoin OR phentoin OR phentytoin OR phenytek OR phenytex OR ritmenal OR saceril OR sanepil OR silantin OR sinergina OR sodanthon OR sodantoin OR sodanton OR solantin OR solantoin OR solantyl OR sylantoic OR tacosal OR thilophenyl OR toin OR zentronal OR zentropil OR pht OR "Valproic Acid" OR avugane OR baceca OR convulex OR delepsine OR depacon OR depakene OR depakine OR depakote OR deproic OR epiject OR epilex OR epilim OR episenta OR epival OR ergenyl OR mylproin OR orfiril OR orlept OR selenica OR stavzor OR valcote OR valparin OR valpro OR valproate OR valproic OR vpa OR phenobarbital OR fenobarbital OR phenobarbitol OR phenobarbitone OR "Phenobarbituric Acid" OR phenylethylbarbiturate OR "Phenylethylbarbituric Acid" OR phenylethylmalonylurea OR adonal OR aephenal OR agrypnal OR amylofene OR aphenylbarbit OR aphenyletten OR barbenyl OR barbinal OR barbiphen OR barbiphenyl OR barbipil OR barbita OR barbivis OR barbonal OR barbophen OR bardorm OR bartol OR bialminal OR blu‐phen OR cabronal OR calmetten OR calminal OR cardenal OR chinoin OR codibarbita OR coronaletta OR cratecil OR damoral OR dezibarbitur OR dormina OR dormiral OR dormital OR doscalun OR duneryl OR ensobarb OR ensodorm OR epanal OR epidorm OR epilol OR episedal OR epsylone OR eskabarb OR etilfen OR euneryl OR fenbital OR fenemal OR fenosed OR fenylettae OR gardenal OR gardepanyl OR glysoletten OR haplopan OR haplos OR helional OR hennoletten OR henotal OR hypnaletten OR hypnette OR hypno‐tablinetten OR hypnogen OR hypnolone OR hypnoltol OR hysteps OR lefebar OR leonal OR lephebar OR lepinal OR lepinaletten OR linasen OR liquital OR lixophen OR lubergal OR lubrokal OR lumen OR lumesettes OR lumesyn OR luminal OR lumofridetten OR luphenil OR luramin OR molinal OR neurobarb OR nirvonal OR noptil OR nova‐pheno OR nunol OR parkotal OR pharmetten OR phen‐bar OR phenaemal OR phenemal OR phenemalum OR phenobal OR phenobarbyl OR phenoluric OR phenolurio OR phenomet OR phenonyl OR phenoturic OR phenyletten OR phenyral OR phob OR polcominal OR prominal OR promptonal OR seda‐tablinen OR sedabar OR sedicat OR sedizorin OR sedlyn OR sedofen OR sedonal OR sedonettes OR sevenal OR sinoratox OR solfoton OR solu‐barb OR sombutol OR somnolens OR somnoletten OR somnosan OR somonal OR spasepilin OR starifen OR starilettae OR stental OR talpheno OR teolaxin OR teoloxin OR thenobarbital OR theoloxin OR triabarb OR tridezibarbitur OR triphenatol OR versomnal OR zadoletten OR zadonal OR pb OR oxcarbazepine OR "GP 47680" OR ocbz OR oxcarbamazepine OR actinium OR barzepin OR carbox OR deprectal OR lonazet OR oxalepsy OR oxetol OR oxpin OR oxrate OR oxtellar OR oxypine OR pharozepine OR prolepsi OR timox OR trexapin OR trileptal OR trileptin OR oxc OR lamotrigine OR "GW 273293" OR lamotrigina OR lamotriginum OR lamictal OR lamotrine OR lamitrin OR lamictin OR lamogine OR lamitor OR ltg OR gabapentin OR gabapentine OR gabapentino OR gabapentinum OR gabapetin OR aclonium OR fanatrex OR gabarone OR neogab OR gralise OR neurontin OR novo‐gabapentin OR nupentin OR gbp OR topiramate OR tipiramate OR topiramatum OR "Topiramic acid" OR topamax OR tpm OR levetiracetam OR levetiracetamum OR levitiracetam OR keppra OR lev OR zonisamide OR zonisamida OR zonisamidum OR zonegran OR exceglan OR excegram OR excegran OR zns))) AND ((TITLE‐ABS‐KEY(epilep* OR "infantile spasm" OR "ring chromosome 20" OR "R20" OR "myoclonic encephalopathy" OR "pyridoxine dependency") OR (TITLE‐ABS‐KEY(syndrome) W/2 (aicardi OR angelman OR doose OR dravet OR janz OR jeavons OR "landau kleffner" OR "lennox gastaut" OR ohtahara OR panayiotopoulos OR rasmussen OR rett OR "sturge weber" OR tassinari OR "unverricht lundborg" OR west)) OR TITLE(seizure OR convuls*) OR (TITLE‐ABS‐KEY(lafora*) W/4 (disease OR epilep*) AND NOT (TITLE(dog OR canine) OR INDEXTERMS(dog OR canine)))) AND NOT (TITLE(*eclampsia) OR INDEXTERMS(*eclampsia)) AND NOT INDEX(medl)) AND (TITLE(randomiz* OR randomis* OR controlled OR placebo OR blind* OR unblind* OR "parallel group" OR crossover OR "cross over" OR cluster OR "head to head") OR ABS(randomiz* OR randomis* OR controlled OR placebo OR blind* OR unblind* OR "parallel group" OR crossover OR "cross over" OR cluster OR "head to head") PRE/2 (trial OR method OR procedure OR study) AND NOT INDEX(medl))) AND NOT (TITLE((adjunct* OR "add‐on" OR "add on" OR adjuvant* OR combination* OR polytherap*) AND NOT (monotherap* OR alone OR singl*)))
Appendix 5. ClinicalTrials.gov search strategy
Intervention: Carbamazepine OR Phenytoin OR Valproic Acid OR Phenobarbital OR Oxcarbazepine OR Lamotrigine OR Gabapentin OR Topiramate OR Levetiracetam OR Zonisamide
Condition: epilepsy
Appendix 6. ICTRP search strategy
Intervention: Carbamazepine OR Phenytoin OR Valproic Acid OR Phenobarbital OR Oxcarbazepine OR Lamotrigine OR Gabapentin OR Topiramate OR Levetiracetam OR Zonisamide
Condition: epilepsy
Recruitment status: All
Data and analyses
This review has no analyses.
Differences between protocol and review
Review structure
The title was changed in December 2014 to specify that the review uses individual participant data.
Additional headings were added to the Data extraction and management and Data synthesis and text was re‐ordered for easier reading.
Synthesis
We intended to test the proportional hazards assumption of the Cox regression model for each outcome of each trial by testing the statistical significance of a time‐varying covariate in the model for each trial and perform sensitivity analyses via interval censored (piecewise) Cox models. However, on reflection, we are unsure of the relevance and importance of the violation of this assumption for a single trial within the whole network. Therefore, instead, we tested the statistical significance of time‐varying covariates for all covariates in the primary model (stratified by trial) and if the proportional hazards assumption appeared to be violated, we performed an alternative, more flexible sensitivity analysis fitting parametric accelerated failure time model to the IPD dataset in preparation for network meta‐analysis and compared these results to the results of the primary analysis.
We stated in the protocol that we would "investigate inconsistency via the Bucher Method (Bucher 1997), which applies a z‐test to the difference between the direct treatment effect estimate and the indirect estimate for each loop of evidence. Given the simplicity of this test, the influence of the precision of the treatment effect estimate on the result of this test and the complexity introduced by multi‐arm trials and therefore association between treatment effects estimated from arms of the same trial, we used a conservative significance threshold of 10% (P value < 0.1) to judge the presence of heterogeneity. " Given the complexity of the network model fitted (with treatment by epilepsy type interaction) and the number of multi‐arm trials included in analysis, we felt that a more formal and less conservative method was needed, therefore we performed node splitting (Dias 2010) to formally estimate differences between direct and indirect evidence for each comparison and we fitted a ‘design‐by‐treatment’ inconsistency model, a method which evaluates both loop and design inconsistencies, particularly within multi‐arm trials (Higgins 2012).
Details of how adverse events will be presented in the review has been added (a narrative report rather than formal analysis).
Sensitivity analysis
Protocol‐defined sensitivity analyses were vague in detail as it was unknown exactly what kind of sensitivity analyses may be required. Specific details of required sensitivity analyses are now given.
We stated in the protocol that we intended to perform sensitivity analyses by "excluding any trial judged to be at high risk of bias for any methodological aspect." We performed several sensitivity analyses relating to inconsistencies between data provided to us and published results (mainly described in Other potential sources of bias) and the only other sources of bias (according to the Cochrane 'Risk of bias' tool) in the trials providing IPD was the open‐label design. Given the long‐term and pragmatic nature of these trials, we do not necessarily consider an open‐label design to induce bias (as further discussed in Overall completeness and applicability of evidence), therefore we did not feel such a sensitivity analysis was appropriate.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Randomised, double‐blinded, parallel‐group trial conducted in Finland 2 treatment arms: OXC and PHT |
|
| Participants | Adult participants with newly diagnosed epilepsy and "normal intellectual capacity" with a minimum of 2 seizures in the last 2 years or 1 seizure and an epileptiform EEG Number randomised: OXC = 19, PHT = 18 Number completed and included in analysis: OXC = 14, PHT = 15 (see Notes) 11 male participants (38%) out of 29 included participants 21 participants with partial epilepsy (72%) out of 29 included participants Mean age of included participants (SD): OXC = 33.6 (14) years, PHT = 32.7 (12.5) years |
|
| Interventions | Monotherapy with OXC or PHT 4‐ to 8‐week titration period followed by a maintenance phase of 12 months. Doses achieved not stated Range of follow‐up not stated |
|
| Outcomes | Neuropsychological assessment and cognitive functioning in 3 major areas at baseline, 6 months' and 12 months' follow‐up: Verbal learning and memory Sustained attention Simple psychomotor speed |
|
| Notes | Participants experiencing inadequate seizure control, adverse events or those who were non‐compliant were withdrawn from the trial and excluded from analysis (5 from OXC group and 3 from PHT group). Results presented only for 29 participants (OXC = 14 and PHT = 15) completing the trial Outcomes chosen for this review were not reported; contact could not be made with trial author to provide IPD |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were "randomly assigned" to treatment; no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The study followed a double blind design" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "The study followed a double blind design"; no further information provided about whether outcome assessor was blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ITT approach not taken: results reported only for 29 participants (OXC = 14 and PHT = 15) who completed 12‐month follow‐up. 8 participants experiencing inadequate seizure control, adverse events or those who were non‐compliant (OXC = 5 and PHT = 3) were excluded from analysis and results |
| Selective reporting (reporting bias) | Low risk | No protocol available and outcomes chosen for this review not reported. Neuropsychological and cognitive outcomes well reported and treatment withdrawal rates reported |
| Other bias | Low risk | None identified |
| Methods | Single‐centre, double‐blind RCT of participants recruited from clinical referral to a multidisciplinary child development centre at a children's hospital in Dhaka, Bangladesh 2 treatment arms: CBZ and PHB |
|
| Participants | 108 children aged 2‐15 years with 2 or more generalised tonic‐clonic, partial, or secondarily generalised seizures in the previous year Number randomised: CBZ = 54, PHB = 54 61 boys (56%) 59 participants with partial epilepsy (55%) 26 participants had previous AED treatment (24%) Mean age (range): 6 years (1‐15 years) |
|
| Interventions | Monotherapy with CBZ (immediate release) or PHB Starting daily dose: CBZ = 1.5 mg/kg/d, PHB = 5 mg/kg/d, maximum daily dose: CBZ = 4 mg/kg/d, PHB = 16 mg/kg/d Trial duration: 12 months, range of follow‐up: 0‐22 months |
|
| Outcomes | Seizure control: seizure freedom during the last quarter of the 12‐month follow‐up Time to first seizure after randomisation Time to treatment withdrawal due to adverse events Change in behaviour from baseline according to age‐appropriate questionnaire Incidence of behavioural side‐effects |
|
| Notes | We received IPD for all randomised participants from the trial author. We received reasons for withdrawal of allocated treatment as well as the date of the last follow‐up visit, but withdrawal of allocated treatment did not always coincide with the date of the last follow‐up visit (i.e. several participants had the allocated treatment substituted for the other trial drug and continued to be followed up). Dates of withdrawal of allocated treatment could not be provided; therefore, we could not calculate 'Time to withdrawal of allocated treatment'. We received the date of first seizure after randomisation, but dates of other seizures in the follow‐up time could not be provided; therefore, we calculated 'Time to first seizure' for all participants, but we could not calculate the time to 6‐ and 12‐month remission | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were "randomly assigned to treatment"; the method of randomisation was not stated and not provided by the trial authors |
| Allocation concealment (selection bias) | Low risk | Allocation was concealed by sealed envelopes prepared on a different site to the site of recruitment of participants |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants, a psychologist, and a therapist were blinded throughout the trial. The treating physician was unblinded for practical and ethical reasons |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | A researcher performing outcome assessment was blinded throughout the trial but unblinded for analysis. It was unclear if this could have influenced the results |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates were reported. We analysed all randomised participants from the IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | We calculated 1 outcome for this review from the IPD provided (see footnote 2). We could not calculate other outcomes for this review as the appropriate data were not recorded/not available. All cognitive outcomes from the trial were well reported |
| Other bias | High risk | There were inconsistencies between rates of seizure recurrence between the data provided and the published paper, which the trial authors could not resolve |
| Methods | Randomised, double‐blind, parallel‐group, non‐inferiority trial, conducted in 120 centres in Asia, Australia, and Europe 2 treatment arms: CBZ and ZNS |
|
| Participants | Participants aged 18‐75 years with newly diagnosed epilepsy, at least 2 partial seizures (with or without secondary generalisation) or generalised tonic‐clonic seizures without clear focal origin in the previous 12 months and at least 1 seizure in the previous 3 months, and had not previously received AEDs or had been treated with 1 AED for no more than 2 weeks. Number randomised: CBZ = 301, ZNS = 282 347 (60%) male participants 100% partial epilepsy Mean age (range): 36 (18‐75 years) |
|
| Interventions | Monotherapy with CBZ or ZNS Titration over 4 weeks to a target dose of CBZ = 600 mg/d and ZNS = 300 mg/d Range of follow‐up: 0‐29 months |
|
| Outcomes | Proportion of participants who achieved seizure freedom for 26 weeks or more (maintenance period) in the per‐protocol population Incidence of treatment‐emergent results Time to 26‐week (6‐month) remission Time to 52‐week (12‐month) remission Proportion of participants with no seizures for at least 52 weeks Time to withdrawal because of absence of efficacy or adverse events |
|
| Notes | IPD provided for all outcomes of this review by trial sponsor Eisai | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomisation scheme was generated centrally by computer programme, which produced a randomisation list with a pseudo‐random number generator |
| Allocation concealment (selection bias) | Low risk | Allocation concealment was achieved by the use of a telephone interactive voice‐response system to dispense the allocated treatment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants, investigators, and sponsor personnel administering medication, assessing outcomes, and analysing data were masked to the allocation. Masking was maintained by use of matching placebo tablets. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants, investigators, and sponsor personnel administering medication, assessing outcomes, and analysing data were masked to the allocation. Masking was maintained by use of matching placebo tablets. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Six‐month, systematic, simple randomised trial of children referred to a child neurology clinic (the author was from Guilan University of Medical Sciences, Iran, so it was likely that the trial was also conducted there) 2 treatment arms: CBZ and PHB |
|
| Participants | Children aged 2‐12 years with partial seizures with secondary generalisation Number randomised: CBZ = 36, PHB = 35 36 boys (53%) 100% of participants with partial epilepsy Percentage newly diagnosed was not stated Age range: 2‐12 years |
|
| Interventions | Monotherapy with CBZ or PHB Doses started or achieved not stated Trial duration: 6 months, range of follow‐up not stated |
|
| Outcomes | Proportion seizure‐free Response rate and rate of side‐effects Seizure frequency and seizure duration |
|
| Notes | The trial was reported in abstract form only with very limited information. Outcomes chosen for this review were not reported; IPD were not available, trial author could not be contacted | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial was described as a 'systematic simple randomised study'; no further information was provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No attrition rates were reported; it was unclear if all participants were analysed |
| Selective reporting (reporting bias) | Unclear risk | There was no protocol available; the trial was available in abstract format only. Outcomes for this review were not available |
| Other bias | Low risk | None identified |
| Methods | Multicentre, double‐blind, parallel‐group trial conducted in centres in Argentina, Brazil, Mexico, South Africa 2 treatment arms: OXC and PHT |
|
| Participants | Participants aged 16‐65 years with newly diagnosed epilepsy with partial or generalised tonic clonic seizures A minimum of 2 seizures, separated by at least 48 h, within 6 months preceding trial entry No previous AED, except emergency treatment of seizures for a maximum of 3 weeks prior to trial entry Number randomised: total = 287, OXC = 143, PHT = 144 174 male participants (61%); 182 participants with partial epilepsy (63%) Mean age (range) = 26 (15‐91) years |
|
| Interventions | Monotherapy with OXC or PHT 8‐week titration period started with 300 mg OXC or 100 mg PHT, increased bi‐weekly, based on clinical response After 8 weeks participants were to be on a three‐times‐a‐day regimen with daily doses of 450 mg‐2400 mg OXC or 150 mg‐800 mg PHT Continued during 48‐week maintenance with adjustment according to clinical response A third long‐term, open‐label extension phase followed the maintenance period. Double‐blind results only were reported Range of follow‐up = 0‐19 months |
|
| Outcomes | The proportion of seizure‐free participants who had at least one seizure during the maintenance period Time to premature discontinuation due to adverse experiences Rate of premature discontinuations for any reason Overall assessments of efficacy and tolerability and therapeutic effect Individual adverse experiences Laboratory values Seizure frequency during maintenance |
|
| Notes | IPD provided for all outcomes of this review from trial sponsor Novartis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Treatment groups randomised in 1:1 ratio across centres via computer‐generated randomisation numbers over balanced blocks of size 6 |
| Allocation concealment (selection bias) | Low risk | Allocation concealment was achieved with sequentially‐numbered packages that were identical and contained identical tablets (information provided by trial statistician) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Trial conducted in 2 phases: 56‐week, double‐blind phase followed by long‐term, open‐label extension. Double‐blind phase results reported only. Blind achieved with divisible OXC and PHT tablets identical in appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported in both treatment phases, participants withdrawing from treatment were no longer followed up so seizure outcomes had to be censored at time of withdrawal and therefore analyses for remission and seizure outcomes could not adopt an ITT approach |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, double‐blind, parallel group, multicentre trial conducted in the USA. 2 treatment arms: LTG and VPS |
|
| Participants | Participants > 12 years with newly diagnosed or previously diagnosed epilepsy of any seizure type, not currently using an AED Number randomised: LTG = 66, VPS = 69, ITT population: LTG = 65, VPS = 68 (2 participants withdrew before drug escalation phase) 60 male participants (44%) 82 participants with partial epilepsy (60%) Proportion newly diagnosed not stated Mean age (range): 32 (12‐76) years |
|
| Interventions | Monotherapy with LTG or VPS Dose‐escalation phase of 8 weeks to target doses of LTG = 200 mg/d and VPS = 20 mg/kg/d Trial duration: 32 weeks |
|
| Outcomes | Weight change The proportion of participants seizure‐free during the entire trial Incidence of the most common drug‐related adverse events Time to withdrawal from the trial |
|
| Notes | IPD provided for remote analysis by trial sponsor Glaxo Smith Kline for time to treatment withdrawal, time to first seizure and time to six‐month remission. IPD had to be treated as aggregate data in network meta‐analysis due to remote access to data. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐ generated randomisation scheme was used |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and personnel double‐ blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Results presented to investigator in a " blinded " fashion |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, double‐blind, parallel‐group trial conducted in 8 centres in the UK. 2 treatment arms: LTG and CBZ |
|
| Participants | Adults and children > 13 years with newly diagnosed epilepsy. None had received previous AED treatment. Number randomised: LTG = 70, CBZ = 66 56 male participants (41%) 82 with partial epilepsy (60%); Mean age (range): 34 (14‐71) years |
|
| Interventions | Monotherapy with LTG or CBZ 4‐week escalation phase leading to LTG = 150 mg/d, CBZ = 600 mg/d Range of follow‐up = 0‐14 months |
|
| Outcomes | Time to first seizure after 6 weeks of treatment Time to withdrawal Proportion of randomised participants remaining seizure‐free during the last 40 and 24 weeks of trial Percentages of participants who reported adverse events |
|
| Notes | IPD provided by trial sponsor Glaxo Smith Kline for time to treatment withdrawal, time to first seizure and time to six‐month remission | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence (information provided by drug manufacturer). Stratification by seizure type |
| Allocation concealment (selection bias) | Low risk | Allocation concealed by individual sealed, opaque envelopes (information provided by drug manufacturer) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind achieved using LTG tablets formulated to be identical in appearance to CBZ tablets |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Trial investigator blinded, not stated if other outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, double‐blind, parallel‐group trial conducted in 8 centres in the UK 2 treatment arms: LTG and CBZ |
|
| Participants | Adults and children > 13 years with newly diagnosed epilepsy. None had received previous AED treatment. Number randomised: LTG = 61, CBZ = 63 56 male participants (45%) 62 participants with partial epilepsy (50%) Mean age (range): 30 (14‐81) years |
|
| Interventions | Monotherapy with LTG or CBZ 4‐week escalation phase leading to LTG = 150 mg/d, CBZ = 600 mg/d Range of follow‐up = 0‐13 months |
|
| Outcomes | Time to first seizure after 6 weeks of treatment Time to withdrawal Proportion of randomised participants remaining seizure‐free during the last 40 and 24 weeks of trial Percentages of participants who reported adverse events |
|
| Notes | IPD provided by trial sponsor Glaxo Smith Kline for time to treatment withdrawal, time to first seizure and time to six‐month remission | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence (information provided by drug manufacturer). Stratification by seizure type |
| Allocation concealment (selection bias) | Low risk | Allocation concealed by individual sealed, opaque envelopes (information provided by drug manufacturer) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind achieved using LTG tablets formulated to be identical in appearance to CBZ tablets |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Trial investigator blinded, not stated if other outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, multicentre, double‐blind, parallel‐group trial conducted in the UK 2 treatment arms: LTG and CBZ randomised in a 2:1 ratio |
|
| Participants | Adults > 65 years with newly diagnosed epilepsy with ≥ 2 seizures in the previous year with at least 1 seizure in the last 6 months. None had received previous AED treatment. Number randomised: LTG = 102, CBZ = 48 83 male participants (55%) 105 participants with partial epilepsy (70%) Mean age (range): 77 (65‐94) years |
|
| Interventions | Monotherapy with LTG or CBZ 4‐week escalation phase leading to LTG = 100 mg/d, CBZ = 400 mg/d Range of follow‐up = 0‐13.5 months |
|
| Outcomes | Time to first seizure after 6 weeks of treatment Time to withdrawal Percentage of participants reporting an adverse event Proportion of participants who were both seizure‐free in the last 16 weeks of the trial and did not discontinue treatment |
|
| Notes | IPD provided by trial sponsor Glaxo Smith Kline for time to treatment withdrawal and time to first seizure (plus seizure‐freedom rates at 24 weeks) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence (information provided by drug manufacturer). Participants randomised in a 2:1 ratio (LTG:CBZ) |
| Allocation concealment (selection bias) | Low risk | Allocation concealed by individual sealed, opaque envelopes (information provided by drug manufacturer) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind achieved using LTG tablets formulated to be identical in appearance to CBZ tablets |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Trial investigator blinded, not stated if other outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, multicentre, double‐blind trial conducted in 41 centres in Europe and Australia 2 treatment arms: GBP and LTG |
|
| Participants | Participants > 16 years with at least 2 partial seizures with or without secondary generalisation or primary generalised tonic clonic seizures in the last 12 months. All participants were untreated in the previous 6 months or AED naive Number randomised: GBP = 158, LTG = 151. Evaluable population (exclusions due to protocol violations): GBP = 148, LTG = 143 152 male participants (52%) out of evaluable population 233 participants with partial epilepsy (80%) out of evaluable population Mean age of evaluable population (SD, range): GBP: 35.8 years (16.4, 13‐78), LTG: 37.9 (16.7, 16‐78) |
|
| Interventions | Monotherapy with GBP or LTG Titration of 2 weeks for GBP to a dose range of 1200 mg/d‐3600 mg/d and titration of 6 weeks for LTG to a dose range of 100 mg/d‐300 mg/d Titration period followed by 24‐week maintenance period. Range of follow‐up not stated |
|
| Outcomes | Time to exit Percentage of completers/time to withdrawal for any reason Time to first seizure Percentage who remained seizure‐free during the final 12 weeks of the 30‐week evaluation period Withdrawal rate due to adverse events |
|
| Notes | IPD requested from trial sponsor Pfizer but data could not be provided due to time elapsed since the trial was completed. Additional information provided in a clinical study report. Aggregate data extracted for time to exit from the trial and time to first seizure extracted from the publication | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed with permuted blocks, stratified within each centre by seizure type. |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Masking was achieved by double‐dummy dosing. A dose range was permitted within the trial to maintain the blind of two drugs with different titration rates (2 weeks for GBP and 6 weeks for LTG) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants included in an ITT analysis (even though demographics presented for 'evaluable population' only) |
| Selective reporting (reporting bias) | Low risk | All efficacy and tolerability outcomes specified in the methods sections were reported well in the results section. No protocol was available. |
| Other bias | Low risk | None identified |
| Methods | Randomised, double‐blind, parallel‐group trial conducted at 85 centres in 12 European countries and in South Africa 2 treatment arms: CBZ (controlled release) and LEV |
|
| Participants | Adults (> 16 years) with 2 partial or generalised tonic–clonic seizures separated by at least 48 h in the previous year with at least one seizure in the last 3 months Number randomised: CBZ = 291, LEV = 288 319 male participants (55%) 466 participants with partial epilepsy (80%) Mean age (range): 39 (15‐82 years) |
|
| Interventions | Monotherapy with CBZ or LEV Titration for 2 weeks to target dose of CBZ = 400 mg/d, LEV = 1000 mg/d Range of follow‐up = 0‐28 months |
|
| Outcomes | Proportion of per‐protocol (PP) participants achieving at least 6 months of seizure freedom at the last evaluated dose One year seizure‐freedom rate 6‐month and 1‐year seizure‐freedom rate by dose level Time to trial withdrawal Incidence of adverse events |
|
| Notes | IPD provided for all outcomes of this review by trial sponsor UCB | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised following a central 1:1 randomisation scheme with a statistical block size of 2 and stratified by seizure category |
| Allocation concealment (selection bias) | Low risk | Participants were randomised using an interactive voice‐response system |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | To ensure blinding, LEV and CBZ‐CR tablets were identically encapsulated. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Single‐centre, randomised, parallel‐group trial of participants referred for assessment at Cork Regional Hospital, Ireland 3 treatment arms: CBZ, PHT, VPS |
|
| Participants | Adults and children with a minimum of 2 untreated generalised or partial seizures in the 6 months preceding the trial Number randomised: PHT = 58, CBZ = 59, VPS = 64 95 male participants (52%) 79 participants (44%) with partial epilepsy Age range: 4‐75 years |
|
| Interventions | Monotherapy with CBZ, PHT or VPS Mean daily dose achieved: PHT = 5.4 mg/kg, CBZ = 10.9 mg/kg, VPS = 15.6 mg/kg Duration of treatment (range in months): 14‐24 months |
|
| Outcomes | Seizure control:
Side effects |
|
| Notes | Outcomes chosen for this review were not reported. IPD not available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation based on two Latin squares without stratification. The first, second and third preference of drug for the participant appears to have been taken into account in the process. Unclear if assignment was completely random |
| Allocation concealment (selection bias) | High risk | An independent person (department secretary) selected the “drug of first preference” from randomisation list on a sequential basis. Allocation not adequately concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attirition rates reported. ITT approach taken, all randomised participants analysed |
| Selective reporting (reporting bias) | Low risk | Primary outcomes (seizure control) and secondary outcomes (side effects) reported sufficiently |
| Other bias | Low risk | None identified |
| Methods | Randomised trial of participants with epileptic seizures following stroke conducted in Italy 2 treatment arms: CBZ (controlled release) and LEV |
|
| Participants | Participants with "vascular epilepsy", newly onset following stroke. Not stated if participants had been previously treated with AEDs Number randomised: CBZ = 17, LEV = 18 17 male participants (49%) Proportion of participants with partial epilepsy not stated Mean age: 70 (43‐90) years |
|
| Interventions | Monotherapy with CBZ or LEV Dose achieved: CBZ: 400 mg/d‐1200 mg/d, LEV = 1000 mg/d‐3000 mg/d Trial duration and range of follow‐up not stated |
|
| Outcomes | Seizure freedom Adverse events during the trial Discontinuations of the trial drug |
|
| Notes | The trial was published in Italian; the characteristics and outcomes were translated. Outcomes chosen for this review were not reported; IPD were not available (author confirmed that the data had been lost) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial was described as randomised ('randomizzazione' in Italian); no further information was available |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, no formal statistical analysis performed so withdrawals did not influence results |
| Selective reporting (reporting bias) | Unclear risk | Methods brief, efficacy and tolerability reported in the results. Outcomes chosen for this review not reported. No protocol available so unclear which outcomes were planned a priori |
| Other bias | Low risk | None identified |
| Methods | Randomised, open‐label trial to evaluate event‐related potentials on the effect of CBZ and LEV cognitive functions, conducted in Italy 2 treatment arms, CBZ (controlled release) and LEV |
|
| Participants | Participants with newly diagnosed partial epilepsy Number randomised: CBZ = 14, LEV = 13 14 male participants (52%) 100% of participants had partial epilepsy Mean age (years): CBZ = 38, LEV = 42, range not stated |
|
| Interventions | Monotherapy with CBZ or LEV Fifteen‐day titration to CBZ = 800 mg/d and LEV = 100 mg/d Trial duration: 24 weeks (assessments at baseline and 12 weeks), range of follow‐up not stated |
|
| Outcomes | Event‐related potential recordings Neuropsychological assessments |
|
| Notes | The trial was published in Italian; the characteristics and outcomes were translated. Outcomes chosen for this review were not reported; IPD were not available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial was described as randomised ('randomizzazione' in Italian); no further information was available |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Attrition rates reported (3 dropouts from the CBZ group, 11% of total participants). These participants are excluded from analysis; this is not an ITT approach |
| Selective reporting (reporting bias) | Unclear risk | Cognitive outcomes described in methods section well reported in results section. No seizure outcomes or adverse events reported and outcomes chosen for this review not reported. No protocol available so unclear if seizure outcomes were planned a priori |
| Other bias | Low risk | None identified |
| Methods | Randomised (partially), double‐blind, multicenter trial conducted at 25 sites in Europe, Australia, South Africa and Canada. 4 treatment arms: GBP (3 arms, 300 mg/d, 900 mg/d and 1800 mg/d) and CBZ. Dose of GBP was masked within the treatment arm but CBZ was given open‐label due to difficulties of blinding tablets and capsules and differing titration periods for the two drugs. |
|
| Participants | Participants with newly diagnosed partial epilepsy, with at least 2 unprovoked partial or generalised tonic clonic seizures in the 6 months prior to trial entry, who were AED naive or had received fewer than 2 weeks of AED therapy, which had to be discontinued before trial entry. Participants with a seizure recurrence after at least 2 years of remission were also eligible. Number randomised: CBZ = 74, GBP = 218 157 male participants (54%) 100% participants with partial epilepsy Mean age (range): 35 (12‐86 years) |
|
| Interventions | Monotherapy with GBP or CBZ Titration period of 7 d for GBP to target doses 300 mg/d, 900 mg/d or 1800 mg/d. Titration period of 21 d for CBZ to target dose 600 mg/d. Titration period followed by an evaluation period of 24 weeks and an optional open‐label period Range of follow‐up: 0‐77 months |
|
| Outcomes | Time to exit Time to exit event plus withdrawals because of adverse events Completion rate (percentage of participants attending end‐of‐phase visit) Exit event rate (percentage of participants who experienced an exit event during the evaluation phase) Adverse event withdrawal rate (percentage of participants who withdrew because of adverse events during either titration or evaluation phases) Exit plus adverse event withdrawal rate (the sum of the exit rate plus the adverse event withdrawal rate) Incidence of adverse events |
|
| Notes | IPD provided for all outcomes of this review by trial sponsor Pfizer. In primary analysis, three arms of GBP are pooled and compared to CBZ (see Data extraction and management) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A randomisation schedule was prepared separately for each trial centre in blocks of four and eight |
| Allocation concealment (selection bias) | Low risk | Trial medication was distributed centrally via a pharmacy |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The trial was partially double‐blinded (the dose of GBP was blinded but GBP was not blinded compared to CBZ). Given that the main comparison made in this review is GBP compared to CBZ rather than comparisons between the doses of GBP, this trial is be treated as an open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specifically stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, parallel‐group trial conducted in Taiwan 3 treatment arms: CBZ, PHB, VPS |
|
| Participants | Children with 2 or more previously untreated unprovoked epileptic seizures Number randomised: CBZ = 26, PHB = 25, VPS = 25; number analysed: CBZ = 25, PHB = 23, VPS = 25 (see notes) 38 boys (52%) 38 participants with partial epilepsy (52%) Mean age (range) for participants analysed: CBZ = 10.8 (7‐15 years), PHB = 9.9 (7‐15 years), VPS = 9.9 (7‐15 years) |
|
| Interventions | Monotherapy with CBZ, PHB or VPS Dose started or achieved not stated Trial duration: 12 months, range of follow‐up: not stated |
|
| Outcomes | Cognitive/psychometric outcomes: IQ (WISC‐R scale) and developmental delay (Bender‐Gestalt test) Auditory event‐related potentials (neurophysiological outcome) Incidence of allergic reactions Seizure control |
|
| Notes | 2 children from the PHB group and 1 child from the CBZ group withdrew from the trial because of allergic reactions. Published results were presented for children who completed the trial only. Outcomes chosen for this review were not reported; IPD were not available |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were allocated with "simple randomisation of block size 3" |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The cognitive assessor was "single blinded", implying that participants and personnel were unblinded, but no further information was provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The cognitive assessor was "single blinded" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Withdrawal rates were reported; results were presented only for those who completed the trial (3/73 (4%) excluded from analysis). An ITT approach was not taken but unclear whether the exclusion of this small proportion of participants would influence results |
| Selective reporting (reporting bias) | Low risk | All cognitive, efficacy, and tolerability outcomes specified in the methods sections were reported well in the results section. No protocol was available. Outcomes chosen for this review were not reported |
| Other bias | Low risk | None identified |
| Methods | Randomised trial conducted in Republic of Korea 2 treatment arms: CBZ (controlled release) and LEV |
|
| Participants | Participants with newly diagnosed partial epilepsy who had their first seizure between 6 and 1 month prior to entry into the trial and had not taken any AEDs previously. Number completing the trial: CBZ = 15, LEV = 16 (number randomised not stated) 22 male participants (71%) 100% of participants had partial epilepsy Mean age (SD, range): CBZ = 29.8 (9.31, 15‐49), LEV = 31.4 (15.3, 15‐66) years |
|
| Interventions | Monotherapy with CBZ or LEV Treatment regimens were CBZ = 400 mg/d and LEV = 1000 mg/d Trial duration 4‐6 weeks, range of follow‐up not stated |
|
| Outcomes | Change in overnight PSG scores (sleep latency, REM sleep latency, total sleep time, sleep efficiency, percentage of each sleep stage, arousal index, and Wake time After Sleep Onset) from baseline after 4‐6 weeks of treatment Change in sleep questionnaires (sleep diaries, the Pittsburg Sleep Quality Index, the Korean version of the Epworth Sleepiness Scale, Beck’s depression inventory‐2 and the Hospital Anxiety Scale) and National Hospital Seizure Severity Scale (NHS3) from baseline after 4‐6 weeks of treatment |
|
| Notes | IPD could not be provided for the trial due to concerns over institutional review board approval (information provided by corresponding author). Outcomes chosen for this review were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial described as randomised, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | PSG scores were interpreted by a certified physician who was blinded to treatment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number randomised not stated, results provided only for those who completed the trial |
| Selective reporting (reporting bias) | Low risk | All sleep, efficacy, and tolerability outcomes specified in the methods sections were reported well in the results section. No protocol was available. Outcomes chosen for this review were not reported |
| Other bias | Low risk | None identified |
| Methods | Multicentre, double‐blind, parallel‐group trial conducted in centres in Europe, Brazil and South Africa 2 treatment arms: OXC and VPS |
|
| Participants | Participants aged 16‐65 years with newly diagnosed epilepsy with partial or generalised tonic clonic seizures A minimum of 2 seizures, separated by at least 48 h, within 6 months preceding trial entry No previous AED, except emergency treatment of seizures for a maximum of 3 weeks prior to trial entry Number randomised: OXC = 128, VPS = 121 127 male participants (51%) 154 participants with partial epilepsy (62%) Mean age (range): OXC: 32.45 (15‐65), VPS: 32.47 (15‐64) |
|
| Interventions | Monotherapy with OXC or VPS Titration period of 8 weeks to target doses of 900 mg/d‐2400 mg/d of OXC or VPS Titration period followed by 48‐week maintenance period and the possibility of a long‐term open‐label extension of 1 year |
|
| Outcomes | The proportion of seizure‐free participants who had at least 1 seizure during the maintenance period Time to premature discontinuation due to adverse experiences Rate of premature discontinuations for any reason Overall assessments of efficacy and tolerability and therapeutic effect Individual adverse experiences Seizure frequency during maintenance |
|
| Notes | IPD requested from trial sponsor Novartis but data could not be provided due to time elapsed since the trial was completed. Aggregate data extracted from graph of time to premature discontinuation in the publication | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomised in a 1:1 ratio, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The trial treatment with OXC or VPS was administered as non‐divisible film‐coated tablets of identical appearance containing 300 mg of active substance. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rates reported, only those who reached the maintenance period were included in efficacy analyses. This is not an ITT approach |
| Selective reporting (reporting bias) | Low risk | All efficacy and tolerability outcomes specified in the methods sections were reported well in the results section. No protocol was available. |
| Other bias | Low risk | None identified |
| Methods | Multicentre, open‐label randomised trial conducted in two centres in Italy 2 treatment arms: CBZ and LEV |
|
| Participants | Participants > 18 years with late post‐stroke seizures (2 weeks to 3 years after stroke) seen in the Cerebrovascular Unit between September 2008 and March 2009. No previous AED treatments were allowed except for emergency treatments Number randomised: CBZ = 66, LEV = 62. Number completing the trial: CBZ = 54, LEV = 52 58 male participants (55%) of those completing the trial 74 participants with partial epilepsy (74%) of those completing the trial Mean age of those completing trial (SD): CBZ = 69.7 (13.2), LEV = 74.1 (11.3) |
|
| Interventions | Monotherapy with CBZ or LEV 2‐week titration period to CBZ: 600 mg/d or LEV: 1000 mg/d Titration period followed by 52‐week maintenance period. Range of follow‐up not stated |
|
| Outcomes | Frequency of seizures during the treatment period Rentention of treatment from the first intake Changes in cognitive measures and quality‐of‐life measures at the end of the treatment period:
Changes in EEG assessments at the end of the treatment period Tolerability of treatment |
|
| Notes | Contact made with trial author who provided additional information for one of the trial centres but full IPD dataset unavailable. Aggregate data extracted from graph of time to seizure recurrence in the publication. Trial was terminated early due to financial reasons when 128 out of a target 630 participants had been recruited |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation numbers were sequentially assigned across centres, and a computer‐generated randomisation scheme was used to provide balanced blocks of participants for each treatment group within each centre |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open label trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rates reported, only those who completed the trial were included in efficacy analyses. This is not an ITT approach |
| Selective reporting (reporting bias) | Low risk | All efficacy, cognitive and tolerability outcomes specified in the methods sections were reported well in the results section. No protocol was available. |
| Other bias | High risk | Likely that trial is underpowered from the early termination with 20% of target sample size recruited |
| Methods | Randomised, double‐blind trial to assess short‐term therapy of CBZ and PHB on cognitive and memory function conducted in Italy Three treatment arms: CBZ, PHB, and placebo |
|
| Participants | Participants with newly diagnosed and untreated temporal lobe epilepsy with no seizures in the previous month Number randomised: CBZ = 6, PHB = 6 1 man and 5 women in each group 100% partial (temporal lobe epilepsy), 100% newly diagnosed Mean age (SD): CBZ = 26.33 (9.73) years, PHB = 18.5 (2.56) years. Age range: 15‐45 years |
|
| Interventions | Monotherapy with CBZ or PHB Dose started and achieved not stated Trial duration: 3 weeks; all participants completed in 3 weeks |
|
| Outcomes | Changes in memory function from baseline after 3 weeks of treatment (verbal, visual, (visual‐verbal and visual‐non‐verbal), acoustic, tactile, and spatial). | |
| Notes | The trial was published in Italian; the characteristics and outcomes were translated. Outcomes chosen for this review were not reported; contact could not be made with trial author to provide IPD | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial was described as randomised ('randomizzazione' in Italian); no further information was available |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Trial was described as double blind ('condizioni di doppia cecità' in Italian), we assume this refers to participants and personnel |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed this short trial and contributed to analysis |
| Selective reporting (reporting bias) | Unclear risk | Cognitive and memory outcomes described in methods section well reported in results section. No seizure outcomes or adverse events reported and outcomes chosen for this review not reported. No protocol available so unclear if seizure outcomes were planned a priori |
| Other bias | High risk | Very small participant numbers and very short‐term follow‐up. Unclear if this trial was adequately powered and of sufficient duration to detect differences |
| Methods | Parallel design, RCT conducted in the UK 2 treatment arms: PHT and VPS |
|
| Participants | Participants > 60 years with newly onset seizures (1 or more generalised tonic‐clonic seizures or 2 or more partial seizures) Number randomised: PHT = 81, VPS = 85 71 male participants (43%) 80 participants with partial epilepsy (48%) Mean age (range): 78 (61‐95 years) |
|
| Interventions | Monotherapy with PHT or VPS Starting doses: PHT: 200 mg/d, VPS: 400 mg/d Median daily dose achieved: PHT 247 mg (range 175‐275); VPS: 688 mg (range 400‐1000) Range of follow‐up: 0‐22 months |
|
| Outcomes | Psychological tests (cognitive function, anxiety and depression) Adverse event frequency Seizure control |
|
| Notes | Trial paper reports on a subset of 38 participants. Full individual participant dataset provided by trial authors and used for this review includes all 166 participants randomised in the trial. IPD provided for 3/4 outcomes of this review ('withdrawal from allocated treatment' not available). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computerised stratified minimisation programme, stratified for age group, gender and seizure type |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled allocation, prescription disclosed to general practitioner and consultant |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and personnel unblinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The main investigator performing cognitive testing was blinded to allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported. ITT analysis undertaken with all randomised participants from IPD (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcome measures reported in published report or provided in IPD (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | 36‐month randomised, comparative trial conducted in Poland 4 treatment arms: CBZ, PHB, PHT, VPS |
|
| Participants | Adults with newly diagnosed epilepsy Number randomised: CBZ = 30, PHT = 30, PHB = 30, VPS = 30 100% of participants had partial epilepsy Age range: 18‐40 years Percentage male and range of follow‐up not mentioned (outcome recorded at 3 years) |
|
| Interventions | Monotherapy with CBZ, PHT, PHB or VPS Starting doses CBZ = 400 mg/d, PHT = 200 mg/d, PHB = 100 mg/d, VPS: 600 mg/d Dose achieved not stated |
|
| Outcomes | Proportion achieving 24‐month remission at 3 years and exclusions after randomisation due to adverse effects or no efficacy | |
| Notes | Abstract only. Outcomes chosen for this review were not reported, contact made with trial authors but IPD not available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial randomised but no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | "Exclusion rates" reported for all treatment groups, no further information provided |
| Selective reporting (reporting bias) | Unclear risk | No protocol available, trial available in abstract format only. Outcomes for this review not available |
| Other bias | Low risk | None identified |
| Methods | Randomised, multicentre, double‐blind trial conducted in 20 centres across four European countries 2 treatment arms: CBZ and OXC |
|
| Participants | Participants aged 15‐65 years with newly diagnosed and previously untreated epilepsy Number randomised: total of 235 but 41 excluded for protocol violations (number randomised by treatment group not stated). Number analysed: CBZ = 100, OXC = 94 96 male participants (49%) out of those analysed Proportion with partial epilepsy not stated Median age (range): 33 (14‐63) |
|
| Interventions | Monotherapy with CBZ or OXC Starting daily dose CBZ: 200 mg OXC: 300 mg. Mean daily dose (range) achieved CBZ: 684 (300 mg‐1400 mg), OXC: 1040 (300 mg‐1800 mg) Titration period of 4‐8 weeks followed by a maintenance period of 48 weeks Mean (range) duration of follow‐up (maintenance period): 336 (10‐390) days |
|
| Outcomes | Changes in seizure frequency between baseline and the end of each maintenance period Changes in EEG tracings between baseline and the end of each maintenance period Global evaluation of therapeutic efficacy and tolerability by the investigator at the end of each maintenance period Side effects observed by participants and investigators each visit Laboratory tests (while blood cell counts and liver function tests, blood pressure and pulse, drug trough serum levels) |
|
| Notes | Trial authors could not be contacted to request IPD. Outcomes chosen for this review were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial described as randomised, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Trial was of double‐blind design |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rate reported, up to 30% of randomised participants who did not complete the trial were excluded from analyses; this is not an ITT approach |
| Selective reporting (reporting bias) | Low risk | Efficacy, and tolerability outcomes specified in the methods sections were reported well in the results section. No protocol was available. Outcomes chosen for this review were not reported |
| Other bias | Low risk | None identified |
| Methods | Randomised, parallel‐group, open‐label paediatric trial conducted in 2 centres in the UK 4 treatment arms: CBZ, PHB, PHT, VPS |
|
| Participants | Children with newly diagnosed epilepsy (2 or more untreated partial or generalised tonic‐clonic seizures in the 12 months preceding the trial) Number randomised: CBZ = 54, PHB = 10, PHT = 54, VPS = 49 86 boys (50%) 89 children with partial epilepsy (51%) Mean age (range): 10 (3‐16) years |
|
| Interventions | Monotherapy with CBZ, PHT, PHB or VPS Median daily dose achieved: CBZ = 400 mg/d, PHT = 175 mg/d, PHB = not stated (see notes), VPS= 600 mg/d Range of follow‐up (months): 10‐164 |
|
| Outcomes | Time to first seizure recurrence after start of therapy Time to 12‐month remission from all seizures Adverse effects and withdrawals due to adverse events |
|
| Notes | IPD provided for all randomised participants. All outcomes in this review calculated from IPD. 6 of the first 10 children assigned to PHB had unacceptable adverse effects, so no further children were assigned to PHB. The 10 children randomised to PHB were retained in analysis. IPD provided for all outcomes of this review by the Medical Research Council |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation list generated using permuted blocks of size 8 or 16 with stratification for centre, seizure type and presence of neurological signs |
| Allocation concealment (selection bias) | Low risk | Allocation concealed via 4 batches of concealed, opaque envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Unblinded, authors state masking of treatment would not be “practicable or ethical” and would “undermine compliance.” Lack of masking could have led to early withdrawal of the PHB arm from the trial, which was likely to have influenced the overall results |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Unblinded, authors state masking of treatment would not be “practicable or ethical” and would “undermine compliance.” Lack of masking could have led to early withdrawal of the PHB arm from the trial, which was likely to have influenced the overall results |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Prospective quasi‐randomised, open‐label trial conducted at a single hospital in Turkey 2 treatment arms: CBZ and OXC |
|
| Participants | Children with partial epilepsy (not stated how many were newly diagnosed) Number randomised: CBZ = 26, OXC = 26 21 boys (40%) 100% of participants had partial epilepsy Mean age (range): 11 (4‐15 years) |
|
| Interventions | Monotherapy with CBZ or OXC CBZ prescribed at 20‐25 mg/kg/d and OXC at 30‐50 mg/kg/d Range of follow‐up: 3.5 to 26 months |
|
| Outcomes | Seizure recurrence Most common side effects Number of participants switching treatment |
|
| Notes | IPD provided for all outcomes of this review by trial author. Trial publication available as abstract only, additional data provided by trial authors | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quasi‐randomisation by alternately allocating participants to CBZ or OXC (information provided by trial authors) |
| Allocation concealment (selection bias) | High risk | Allocation was not concealed (alternate allocation) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐ label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐ label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Multicentre, randomised, open‐label trial conducted at 21 sites in seven European countries between December 2001 and December 2003 3 treatment arms: CBZ, OXC, VPS (randomised in a 1:2:1 ratio) |
|
| Participants | Children and adolescents (aged 6‐17) with newly diagnosed partial seizures. Participants must have had at least 2 unprovoked partial seizures (simple and complex partial and partial evolving into secondarily generalised seizures) in the 3 months prior to study entry Number randomised: CBZ = 28, OXC = 55, VPS = 29 51 male participants (46%) 100% of participants had partial epilepsy Median age (range): 10 (6‐16) |
|
| Interventions | Monotherapy with CBZ, OXC or VPS Dose achieved (mean (SD)): CBZ =14.4 (3.6) mg/kg/d, VPS = 20.7 (7.5) mg/kg/d Study duration: 6 months, Range of follow‐up not stated |
|
| Outcomes | Cognitive testing: Computerized Visual Searching Task, assessing mental information processing speed and attention. Rey Auditory Verbal Learning Test and Raven’s Standard Progressive matrices for children: psychomotor speed, alertness, memory and learning, and non‐verbal intelligence. Percentage of participants remaining seizure‐free throughout treatment Most common adverse events Treatment satisfaction on a 4‐point scale from poor to very good |
|
| Notes | IPD requested from trial sponsor Novartis but data could not be provided due to time elapsed since the trial was completed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An interactive voice‐response system was used to automate the randomisation of participants to treatment groups within age strata |
| Allocation concealment (selection bias) | Low risk | An interactive voice‐response system was used to automate the randomisation of participants to treatment groups within age strata |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label study (justified as primary and secondary cognitive outcomes were objective) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label study (justified as primary and secondary cognitive outcomes were objective) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rate reported. Most results reported only for the per‐protocol population who completed the study |
| Selective reporting (reporting bias) | Low risk | All cognitive, efficacy, and tolerability outcomes specified in the methods sections were reported well in the results section. No protocol was available. Outcomes chosen for this review were not reported |
| Other bias | Low risk | None detected |
| Methods | Randomised, multicentre, open‐label, parallel‐group trial conducted in 7 hospitals in Republic of Korea 2 treatment arms: LTG and CBZ |
|
| Participants | Children aged 6‐12 years with a new diagnosis of partial epilepsy and at least 2 seizures in the last 6 months. Number randomised: LTG = 43, CBZ = 41 48 male participants (57%) 100% of participants had partial epilepsy Not stated if any participants had received previous AED treatment Mean age (range): 9 (5‐13) years |
|
| Interventions | Monotherapy with LTG or CBZ 8‐week escalation phase leading to LTG = 3‐6 mg/kg/d, CBZ = 10‐20 mg/kg/d Range of follow‐up: 0.5‐28 months |
|
| Outcomes | Seizure‐free rate over 6 months (maintenance period) by treatment group Change in cognition (neuropsychological), behaviour and quality of life from screening to the end of the maintenance phase by treatment group Incidence of adverse events |
|
| Notes | IPD provided by trial author for time to treatment withdrawal, time to first seizure and time to 6‐month remission | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Each centre received a separate and independent computer‐ generated random code list |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, parallel‐group trial conducted among residents of the Nakuru district, a semi‐urban population of rural Kenya 2 treatment arms: CBZ and PHB |
|
| Participants | Participants had a history of generalised tonic‐clonic seizures and at least 2 generalised tonic‐clonic seizures within the preceding year (with or without other seizure types) and untreated in the 3 months prior to the trial. 79 (26%) participants had been treated in the past with AEDs Number randomised: PHB = 150, CBZ = 152 173 male participants (57%) 115 of participants with partial epilepsy (38%) Mean age (range): 21 (6‐65 years) |
|
| Interventions | Monotherapy with CBZ or PHB Starting doses: PHB: 6‐10 years: 30 mg/d, 11‐15 years: 45 mg/d, > 16 years: 60 mg/d CBZ: 6‐10 years of age: 400 mg/d, 11‐15 years of age: 500 mg/d, > 16 years of age: 600 mg/d Dose achieved not stated Range of follow‐up: participants followed up for up to 1 year |
|
| Outcomes | Adverse effects Withdrawals from allocated treatment Seizure frequency (during second 6 months of trial) |
|
| Notes | IPD were made available but not used because of inconsistencies and problems with the data provided (see Included studies for further details) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants randomised with random number list |
| Allocation concealment (selection bias) | Low risk | Allocation concealed via sealed, opaque envelopes (information provided by trial author) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rates reported, results presented only for participants completing 12 months' follow‐up (results not presented for 53 (17.5%) participants out of 302 who withdrew from treatment), approach is not ITT |
| Selective reporting (reporting bias) | Low risk | No protocol available, outcomes chosen for this review not reported. Seizure outcomes and adverse events well reported |
| Other bias | High risk | Inconsistencies with IPD and published results so IPD could not be used (see Included studies for further details) |
| Methods | Single‐centre, randomised, parallel‐group trial conducted in the UK 3 treatment arms: CBZ, PHT, VPS |
|
| Participants | Children with at least 3 newly diagnosed generalised or partial seizures within a period of 6 months Number randomised: CBZ = 23, PHT = 20, VPS = 21 No information on epilepsy type or sex Age range: 5‐14 years |
|
| Interventions | Monotherapy with CBZ, PHT or VPS Mean dose: CBZ = 17.9 mg/d, PHT = 6.1 mg/d, VPS: 25.3 mg/d Trial duration: 12 months, range of follow‐up not stated |
|
| Outcomes | Cognitive assessments Summary of withdrawals from randomised drug |
|
| Notes | Outcomes chosen for this review were not reported IPD not available, but could be constructed from the publication for the outcome 'time to withdrawal of allocated drug' |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quota allocation by sex, age, seizure type and current treatment is an inadequate randomisation method |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Personnel and participants (and parents) unblinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors single‐blinded for cognitive testing |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, results reported and analysed for all participants randomised and all who completed various stages of follow‐up |
| Selective reporting (reporting bias) | Unclear risk | One of four outcomes for this review reported. Cognitive outcomes described in methods section well reported in results section. Adverse effects reported, no seizure outcomes reported and outcomes chosen for this review not reported. No protocol available so unclear if seizure outcomes were planned a priori |
| Other bias | Low risk | None identified |
| Methods | Prospective, open‐label, randomised trial conducted in Germany 2 treatment arms: LTG and OXC |
|
| Participants | Participants with untreated epilepsy, number newly diagnosed not stated Number randomised: LTG = 21, OXC = 27 26 male participants (54%) Proportion of participants with partial epilepsy not stated Age range: 15‐61 |
|
| Interventions | Monotherapy with LTG or OXC Doses started or achieved not stated Range of follow‐up and trial duration not stated |
|
| Outcomes | Seizure reduction Cognition, mood and health‐related quality of life |
|
| Notes | Abstract only. Trial authors could not be contacted to request IPD Results refer to reduction of seizures to only "simple seizures" remaining so we assume that this population of participants has the eligible seizure type for this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Treatments were "randomly assigned", no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Abstract only, attrition rate not stated. Insufficient information to make a judgement |
| Selective reporting (reporting bias) | Unclear risk | Abstract only, insufficient information to make a judgement |
| Other bias | Low risk | None identified |
| Methods | Randomised, single‐centre, open‐label, parallel‐group trial conducted at Tel Aviv University and Medical Centre, Israel 2 treatment arms: LTG and CBZ |
|
| Participants | Adults admitted to the neurological department with a first seizure event after an ischaemic stroke Number randomised: LTG = 32, CBZ = 32 46 male participants (72%) 100% of participants had partial epilepsy Unclear if any participants had received previous AED treatment Mean age (range): 67.5 (38‐90) years |
|
| Interventions | Monotherapy with LTG or CBZ for 12 months Dose escalation phase (length not stated) leading to LTG 100 mg/d, CBZ 300 mg/d Range of follow‐up: not stated |
|
| Outcomes | The appearance of a second seizure under treatment or by finishing the 12‐month follow‐up without seizures Tolerability: incidence of adverse events Withdrawals due to adverse events |
|
| Notes | Contact made with trial author who was willing to provide IPD but data never received. Aggregate data extracted from graphs in the publication. Stated in the title of the paper that LTG and CBZ were monotherapy treatments but Table 1 of the paper refers to Total no. AED, unclear if all participants were receiving monotherapy treatment | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised in a 1:1 ratio, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rate reported, all randomised participants included in analysis |
| Selective reporting (reporting bias) | Low risk | No protocol available. Seizure outcomes and adverse events well reported |
| Other bias | Unclear risk | Unclear if all participants were receiving monotherapy treatment |
| Methods | Multicentre, double‐blind, parallel‐group trial conducted in centres in Argentina and Brazil 2 treatment arms: OXC and PHT |
|
| Participants | Participants aged > 5 years with newly diagnosed epilepsy with partial seizures or generalised tonic‐clonic seizures A minimum of 2 seizures, separated by at least 48 h, within 6 months preceding trial entry No previous AED, except emergency treatment of seizures for a maximum of 3 weeks prior to trial entry Number randomised: OXC = 997, PHT = 94 100 male participants (52%); 143 of participants had partial epilepsy (74%) Mean age (range): 18.5 (5‐53) years |
|
| Interventions | Monotherapy with OXC or PHT 8‐week titration period started with 150 mg OXC or 50 mg PHT, increased bi‐weekly, based on clinical response to a regimen with daily doses of 450 mg‐2400 mg OXC or 150 mg‐800 mg PHT Continued during 48‐week maintenance with adjustment according to clinical response A third long‐term, open‐label extension phase followed the maintenance period. Double‐blind results only were reported Range of follow‐up: 1‐28 months |
|
| Outcomes | The proportion of seizure‐free participants who had at least 1 seizure during the maintenance period Time to premature discontinuation due to adverse experiences Rate of premature discontinuations for any reason Overall assessments of efficacy and tolerability and therapeutic effect Individual adverse experiences Laboratory values Seizure frequency during maintenance |
|
| Notes | IPD provided for all outcomes of this review by trial sponsor Novartis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Treatment groups randomised in 1:1 ratio across centres via computer‐generated randomisation numbers over balanced blocks of size 6 |
| Allocation concealment (selection bias) | Low risk | Allocation concealment was achieved with sequentially‐numbered packages which were identical and contained identical tablets (information provided by trial statistician) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Trial conducted in 2 phases: 56‐week, double‐blind phase followed by long‐term, open‐label extension. Double‐blind phase results reported only Blind achieved with divisible OXC and PHT tablets identical in appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated whether outcome assessor was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported in both treatment phases, participants withdrawing from treatment were no longer followed up so seizure outcomes had to be censored at time of withdrawal and therefore analyses for remission and seizure outcomes could not adopt an ITT approach |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, parallel‐group, open‐label trial conducted in 2 centres in the UK 4 treatment arms: CBZ, PHB, PHT, VPS |
|
| Participants | Adults with newly diagnosed epilepsy (2 or more untreated partial or generalised tonic‐clonic seizures in the 12 months preceding the trial) Number randomised: CBZ = 61, PHB = 58, PHT = 63, VPS = 61 117 male participants (48%) 102 participants with partial epilepsy (42%) Mean age (range): 32 (13‐77) years |
|
| Interventions | Monotherapy with CBZ, PHB, PHT or VPS Median daily dose achieved: CBZ = 600 mg/d, PHB = 105 mg/d, PHT = 300 mg/d, VPS = 800 mg/d Range of follow‐up: 0‐166 months |
|
| Outcomes | Time to first seizure recurrence after start of therapy Time to 12‐month remission from all seizures Adverse effects and withdrawals due to adverse events |
|
| Notes | IPD provided for all outcomes of this review by the Medical Research Council | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation list generated using permuted blocks of size 8 or 16 with stratification for centre, seizure type and presence of neurological signs |
| Allocation concealment (selection bias) | Low risk | Allocation concealed via 4 batches of concealed, opaque envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Unblinded, authors state masking of treatment would not be “practical” and would have “introduced bias due to a very large drop‐out rate.” Lack of blinding may have influenced the withdrawal rate. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Unblinded, authors state masking of treatment would not be “practical” and would have “introduced bias due to a very large drop‐out rate.” Lack of blinding may have influenced the withdrawal rate. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Multicenter, randomised, open‐label, non‐inferiority trial conducted across 7 centres in Republic of Korea 2 treatment arms: CBZ and LEV |
|
| Participants | Children aged 4‐16 years with newly diagnosed focal epilepsy, no previous anti‐epileptic therapy and "above borderline" intelligence Number randomised: CBZ = 64, LEV = 57 (ITT population) 69 male participants (57%) 100% of participants with partial epilepsy Mean age (SD): CBZ = 8.05 (3.02), LEV = 9.28 (3.37) years |
|
| Interventions | Monotherapy with CBZ or LEV 4‐week dose titration period to a minimal target dose of CBZ = 20/mg/kg/d or LEV = 40/mg/kg/d Trial duration: 52 weeks, range of follow‐up not stated |
|
| Outcomes | Neuropsychological outcomes; change from baseline to 52 weeks in neurocognitive (Korean‐WISC‐III or Korean‐Wechsler Preschool and Primary Scale of Intelligence‐III), behavioural (Korean‐CBCL), and emotional (Children's Depression Inventory and Revised Children's Manifest Anxiety Scale) function assessments Mean percentage change in seizure frequency from baseline Seizure‐freedom rates Incidence of adverse events |
|
| Notes | IPD could not be provided for the trial due to restrictions on data sharing from the Korean Food and Drug Administration (information provided by corresponding author). Outcomes chosen for this review were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised independently at each centre using a computerised random code assignment based on stratified permuted block randomisation that were designed separately and independently for each participating centre |
| Allocation concealment (selection bias) | Low risk | At each centre, allocation concealment was carried out by the pharmacy in order to blind those assessing outcomes from the trial medication |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial for participants and personnel |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Those assessing outcomes were blinded to trial medication (pharmacy allocation) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 7 randomised participants did not take any trial medication so were not included in ITT population. Results for neuropsychological outcomes recorded only for those who completed the trial ‐ 81/121 participants (67%) |
| Selective reporting (reporting bias) | Low risk | All neuropsychological, efficacy, and tolerability outcomes specified in the methods sections were reported well in the results section. No protocol was available. Outcomes chosen for this review were not reported |
| Other bias | Low risk | None identified |
| Methods | Open‐label, multicentre, randomised trial. Authors based in Denmark and Finland 2 treatment arms: CBZ (slow release) and LTG |
|
| Participants | Participants with newly onset partial and/or generalised tonic clonic seizures Number randomised: CBZ = 70, LTG = 73 No information provided about age and gender or previous AED use |
|
| Interventions | Monotherapy with CBZ or LTG for 52 weeks Mean dosage during maintenance period: CBZ = 549 mg/d, LTG = 146 mg/d Range of follow‐up not stated |
|
| Outcomes | Seizure freedom Cognitive assessments |
|
| Notes | IPD requested from trial sponsor Glaxo Smith Kline but data could not be located. Abstract publication only available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Treatments were "randomly assigned", no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Abstract only, attrition rate not stated. Insufficient information to make a judgement |
| Selective reporting (reporting bias) | Unclear risk | Abstract only, insufficient information to make a judgement |
| Other bias | Low risk | None identified |
| Methods | Randomised trial of outpatients of a hospital in Berlin, Germany 3 treatment arms: CBZ, LEV, VPS |
|
| Participants | Newly diagnosed ("de novo") participants Number randomised: CBZ = 6, LEV = 6, VPS = 3 12 (80%) partial epilepsy No information on age or gender |
|
| Interventions | Monotherapy with CBZ, LEV or VPS Doses started or achieved not stated Assessments performed at 6 and 12 weeks |
|
| Outcomes | Cognitive performance Neuropsychological assessment |
|
| Notes | Abstract only. Trial authors could not be contacted to request IPD | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Treatments were "randomly assigned", no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Abstract only, attrition rate not stated. Insufficient information to make a judgement |
| Selective reporting (reporting bias) | Unclear risk | Abstract only, insufficient information to make a judgement |
| Other bias | Low risk | None identified |
| Methods | Phase IV, open‐label, randomised, multicentre trial conducted in 21 centres in Republic of Korea 2 treatment arms: CBZ and LTG |
|
| Participants | Participants were untreated epileptics who had at least 2 unprovoked seizures (partial or generalised tonic clonic) during the last 24 weeks before the study start, more than 24 h apart Number randomised: CBZ = 129, LTG = 264 (ITT population) 154 male participants (39%) 288 participants (73%) with partial epilepsy Mean age (SD): CBZ = 37.6 (15.8), LTG = 34.2 (16.3) years |
|
| Interventions | Monotherapy with CBZ or LTG Permitted doses LTG: 100 mg/d–500 mg/d for LTG , CBZ: 400 mg/d–1200mg/d |
|
| Outcomes | Retention rate at study end Terminal 24‐week seizure‐free rate and time interval from the end of dose titration phase to the first seizure |
|
| Notes | Full text of the trial published in Korean. Abstract and clinical trial summary available in English. IPD request for this trial ongoing. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial described as randomised, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rate reported, not all participants included in analysis, which is not an ITT approach |
| Selective reporting (reporting bias) | Low risk | Results for all outcomes summarised for all listed outcomes |
| Other bias | Low risk | None identified |
| Methods | Randomised, open‐label trial conducted in 2 hospitals in Hong Kong 2 treatment arms: LTG and VPS |
|
| Participants | Chinese patients with newly diagnosed, untreated epilepsy or a recurrence of seizures after a period of remission with AED therapy completely withdrawn for at least a year, aged 18‐55 years and not receiving AED therapy were recruited from the Prince of Wales Hospital and United Christian Hospital in Hong Kong. Number randomised: LTG = 37, VPS = 44 40 male participants (49%) 29 participants with partial epilepsy (36%) Mean age (range): 34 (16‐56 years) |
|
| Interventions | Monotherapy with LTG or VPS Titration of 4 weeks to target dose of LTG = 100 mg/d and VPS = 800 mg/d Range of follow‐up: 0‐15 months |
|
| Outcomes | Difference in mean fasting serum insulin concentration at 12 months between the 2 treatment groups Difference in mean changes from baseline at various time points in metabolic and endocrine measurements and BMI between the 2 treatment groups and by gender Frequency of common adverse events experienced by at least 10% of participants by treatment group |
|
| Notes | IPD provided for all outcomes of this review by trial author | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised stratified for sex and hospital, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, multicentre, open‐label, parallel‐group trial conducted in the Korea 2 treatment arms: LTG and CBZ |
|
| Participants | Adults over the age of 16 with newly diagnosed partial epilepsy or untreated partial epilepsy for at least one year Number randomised: LTG = 57, CBZ = 53 57 male participants (52%) 95 participants with partial epilepsy (86%) Not stated how many participants had received previous AED treatment Mean age (range): 36 (16‐60) years |
|
| Interventions | Monotherapy with LTG or CBZ 8‐week escalation phase leading to LTG = 200 mg/d, CBZ = 600 mg/d Range of follow‐up: 0‐16.5 months |
|
| Outcomes | Change of neuropsychological and cognitive scores from baseline: general intellectual ability, learning and memory, attention and executive function (group‐by‐time interaction) Frequency of psychological and health‐related quality of life symptoms Proportion with seizure freedom during the maintenance period |
|
| Notes | IPD provided by trial author for time to treatment withdrawal, time to first seizure and time to six‐month remission | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block randomisation (block size four) via a computer randomisation programme (information provided by trial author) |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Prospective, open‐label randomised trial conducted in Serbia and Montenegro 2 treatment arms: LTG and VPS |
|
| Participants | Participants with newly diagnosed, previously untreated epilepsy Number randomised: LTG = 35, VPS = 38 51 (70%) with partial epilepsy Median age (range): 34 (18‐76) years No information on gender |
|
| Interventions | Monotherapy with LTG or VPS All participants to be followed up for at least 6 months |
|
| Outcomes | Seizure freedom Retention on treatment |
|
| Notes | Abstract of interim results only available. Contact was made with trial author who was unable to provide IPD | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial described as randomised, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Interim report, proportion of participants completing the trial period presented. Unclear if an ITT approach was taken to analysis |
| Selective reporting (reporting bias) | Unclear risk | Abstract only, insufficient information to make a judgement |
| Other bias | Low risk | None identified |
| Methods | Multicentre, randomised, parallel‐group, double‐blinded trial over 10 centres in the USA with separate randomisation schemes used for each seizure type. 4 treatment arms: CBZ, PHB, PHT and primidone |
|
| Participants | Adults with previously untreated or under‐treated simple or complex partial or secondary generalised tonic‐clonic seizures Number randomised: PHB: 155, PHT = 165, CBZ = 155 413 male participants (87%) 99.8% of participants with partial epilepsy Mean age (range): 41 (18‐82) years |
|
| Interventions | Monotherapy with PHT or CBZ Median daily dose achieved: CBZ = 800 mg/d, PHB = 160 mg/d, PHT = 400 mg/d Range of follow‐up: 0‐78 months |
|
| Outcomes | Participant retention/time to drug failure (length of time participant continued to take randomised drug) Composite scores of seizure frequency (seizure rates and total seizure control) and toxicity Incidence of side effects |
|
| Notes | IPD provided for all outcomes of this review by the Department of Veterans Affairs | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants randomised with stratification for seizure type. Method of randomisation not stated and not provided by authors |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind (participants and personnel) achieved using an additional blank tablet |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Double‐blind, multicentre trial across 13 Veteran’s Affairs medical centres (USA) 2 treatment arms: CBZ and VPS |
|
| Participants | Adults (18‐70 years) with previously untreated or under‐treated complex partial seizures, secondarily generalised tonic‐clonic seizures Number randomised: CBZ = 236, VPS = 244 445 male participants (93%) 100% of participants had partial epilepsy. Mean age (range): 47 (18‐83) years |
|
| Interventions | Monotherapy with CBZ or VPS Mean daily dose achieved by month 12 CBZ = 722+‐ 230 mg/d, VPS = 2099 +‐824 mg/d Range of follow‐up: 0‐73 months |
|
| Outcomes | Total number of seizures (of each type) during 12 months Number of seizures per month Percentage of participants with seizures completely controlled Time to first seizure Seizure rating score (severity of seizures) at 12 and 24 months Composite score (combined score for the control of seizures and incidence of adverse events) Incidence of systemic and neurologic adverse events (and severity) Time to treatment failure |
|
| Notes | IPD provided for all outcomes of this review by the Department of Veterans Affairs | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised to treatment using random permuted blocks with a different randomisation scheme for two seizure groups (complex partial and secondarily generalised tonic clonic) |
| Allocation concealment (selection bias) | Low risk | Treatment allocation was concealed via sealed, opaque envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blind (participants and personnel) achieved with additional matching placebo tablets |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear if outcome assessment was blinded, no information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, double‐blind, single‐centre, parallel paediatric trial conducted in Los Angeles, USA 2 treatment arms: CBZ and PHB |
|
| Participants | Children with newly diagnosed epilepsy Number randomised: PHB = 18, CBZ = 15 20 boys (61%) 100% of participants had partial epilepsy Mean age (range): PHB = 7.89 (2‐12 years), CBZ = 6.07 (2‐12 years) |
|
| Interventions | Monotherapy with PHB or CBZ Doses started and achieved not stated Trial duration: 12 months Range of follow‐up: not reported |
|
| Outcomes | Change in cognitive, intelligence (IQ), behavioural, and psychometric scores between baseline, 6 months, and 12 months Compliance, drug changes, and withdrawal rates Seizure control at 6 and 12 months (excellent/good/fair/poor) |
|
| Notes | 33 participants were randomised to PHB (18) and CBZ (15) in this trial; 6 children were enrolled into a 6‐month pilot trial (PHB (4) CBZ (2)) prior to the randomised trial. The 6 children were included in 6‐month follow‐up psychometric data Outcomes for this review were not reported; IPD were not available |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | 33 children were "randomised using a scheme that balanced drug distribution by age and sex"; no further details were provided on the randomisation scheme. 6 non‐randomised children were also used in some analyses |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The trial blinded participants (and parents); clinicians were unblinded for clinical follow‐up |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The trial blinded participants (and parents); clinicians were unblinded for clinical follow‐up |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates were reported; results were reported for all children who completed each stage of follow‐up |
| Selective reporting (reporting bias) | Low risk | Cognitive/behavioural outcomes, seizure control outcomes, and adverse events were all well reported. No protocol was available; outcomes for this review were not reported |
| Other bias | High risk | There was evidence that the trial may have been underpowered to detect differences (e.g. 55% power to find a 5‐point difference in IQ score). The behavioural questionnaire was not fully validated. Non‐randomised children from a pilot trial were included in the results for psychometric outcomes and medical outcomes |
| Methods | Prospective, randomised trial of participants newly referred to the pediatric clinic of Kitasato University School of Medicine, Japan 3 treatment arms: CBZ, PHT and VPS |
|
| Participants | Children aged 1‐14 with previously untreated partial seizures and/or generalised tonic‐clonic seizures Number randomised: CBZ = 66, PHT = 51, VPS = 46 116 participants with partial epilepsy (71%) No information on age and gender |
|
| Interventions | Monotherapy with PHT or CBZ Initial daily dose: CBZ = 13.0 +/‐ 1.6 mg/kg/d, PHT = 7.2 +/‐ 1.4 mg/kg/d, VPS = 22.9 +/‐ 4.9 mg/kg/d Range of follow‐up: 6‐66 months, mean follow‐up: 34 months in CBZ group, 37 in PHT group and 40 in VPS group |
|
| Outcomes | Proportion of all randomised participants with seizure recurrence (by seizure type) Proportion of participants with optimum plasma levels with seizure recurrence (by seizure type) |
|
| Notes | Very limited information available, the trial was reported in a summary publication of 3 different studies (other 2 studies are not monotherapy designs). Outcomes chosen for this review were not reported, IPD not available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial was described as "randomised" but no further details were provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Ranges of follow‐up given for both treatment groups. Results reported "at the end of follow up," no withdrawals or exclusions mentioned, all participants included in analysis |
| Selective reporting (reporting bias) | Unclear risk | Seizure recurrence outcomes described well reported. No adverse events reported; no protocol available so unclear if adverse events were planned a priori. Outcomes for this review not available |
| Other bias | Low risk | None identified |
| Methods | Double‐blind randomised trial performed in a single centre in Tehran, Iran 2 treatment arms: LEV and LTG |
|
| Participants | Participants > 60 years who were referred to the neurologic clinic at Sina University Hospital, Iran in 2012. Participants must have had a diagnosis of epilepsy for at least 1 year and experienced a minimum of 1 unprovoked partial or generalised epileptic seizure over the last 6 months Number randomised: LEV = 50, LTG = 50 55 male participants (58%) out of 95 participants who completed the trial 67 participants with partial epilepsy (71%) out of 95 participants who completed the trial Mean age (SD, range): 72.4 (5.87, 63‐85) years for all randomised participants |
|
| Interventions | Monotherapy with LEV or LTG LEV was initiated with 250 mg twice daily and was increased to 500 mg twice daily, LTG was initiated with 25 mg daily and was increased up to a maximum dose of 100 mg twice daily Trial duration: 20 weeks, range of follow‐up not stated |
|
| Outcomes | Seizure recurrence Abnormal laboratory values Adverse events |
|
| Notes | The trial was published in Persian; the characteristics and outcomes were translated. Outcomes chosen for this review were not reported; contact could not be made with trial author to provide IPD | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐based table was generated by balanced block randomisation |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The participant received a drug with a specific code and did not know the name of the drug. The physician in charge of the participant follow‐up was unaware of the drug provided for the participant |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only those who completed the trial were included in analyses, five participants excluded |
| Selective reporting (reporting bias) | Low risk | Seizure recurrence outcomes and adverse events were all well reported. No protocol was available; outcomes for this review were not reported |
| Other bias | Low risk | None identified |
| Methods | Phase 4, randomised, parallel‐design, open‐label trial in Republic of Korea 2 treatment arms: LEV and OXC |
|
| Participants | Participants aged16‐80 years with newly diagnosed partial epilepsy Partcipants must have had at least 2 seizures separated by a minimum of 48 h and 1 in the 6 months prior to screening and no AEDs in the previous 6 months Number enrolled: LEV = 175, OXC = 178 190 male participants (54%) 100% of participants had partial epilepsy Mean age (SD): LEV = 39.5 (16.7), OXC = 42.7 (17.3) |
|
| Interventions | Monotherapy with LEV or OXC Titration for 2 weeks up to a maximum of LEV = 1000 mg/d‐3000 mg/d, OXC = 900 mg/d‐24,000 mg/d Trial duration: 50 weeks, range of follow‐up not stated |
|
| Outcomes | Percentage of participants with a treatment failure after 50 weeks Time to the first seizure defined as the time from the first dose of medication to the occurrence of the first seizure during the 48 weeks' treatment period Percentage of subjects who achieved seizure freedom for 24 consecutive weeks during the 48 weeks' treatment period at any time Percentage of subjects who achieved seizure freedom during the 48 weeks' treatment period |
|
| Notes | Trial registered as NCT001498822 on ClincalTrials.gov and listed as completed and trial results published online but no published manuscript was available. Trial sponsored by UCB Korea, inquiries regarding this trial made to the sponsor. Data cannot be made available until a manuscript has been published; if IPD is provided at a future date, this trial will be included in analyses | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial described as randomised, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rate reported, not all participants included in analysis which is not an ITT approach |
| Selective reporting (reporting bias) | Low risk | Results for all outcomes reported online for listed outcomes |
| Other bias | Low risk | None identified |
| Methods | Phase 3, randomised, open‐label, parallel‐group trial conducted in China 2 treatment arms: CBZ and LEV |
|
| Participants | Chinese participants > 16 years, recent onset partial seizures, at least 2 unprovoked seizures in the year preceding randomisation, of which at least 1 unprovoked seizure occurred in the 3 months preceding randomisation Number enrolled: CBZ = 215, LEV = 218 233 male participants (54%) 100% of participants had partial epilepsy Mean age (SD): CBZ = 33.3 (14.3), LEV = 37.8 (16.2) |
|
| Interventions | Monotherapy with CBZ or LEV Titration of 3 weeks to CBZ = 400 mg/d, LEV = 1000 mg/d |
|
| Outcomes | Proportion of subjects remaining seizure‐free during the 6‐month evaluation period Proportion of subjects retained in the trial for the duration of the period covering the up‐titration period, stabilization period, and evaluation period Time to first seizure or discontinuation due to an adverse event (AE)/lack of efficacy (LOE) during the evaluation period Time to first seizure during the evaluation period Time to first seizure during the period covering the up‐titration period, stabilisation period, and evaluation period from the first dose of trial drug |
|
| Notes | Trial registered as NCT01954121 on ClincalTrials.gov and listed as completed and trial results published online but no published manuscript was available. Trial sponsored by UCB SA, inquiries regarding this trial made to the sponsor. Data cannot be made available until a manuscript has been published; if IPD is provided at a future date, this trial will be included in analyses | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial described as randomised, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rate reported, not all participants included in analysis which is not an ITT approach |
| Selective reporting (reporting bias) | High risk | Results reported online for only some of the outcomes, no statistical analysis reported for the Time to First Seizure outcomes. |
| Other bias | Low risk | None identified |
| Methods | Randomised, multicentre, open‐label, parallel‐group trial conducted in Europe and Mexico 2 treatment arms: LTG and CBZ randomised in a 2:1 ratio |
|
| Participants | Adults and children over the age of 2 years with newly diagnosed or currently untreated partial epilepsy with ≥ two seizures in the previous 6 months and with at least 1 seizure in the last 3 months Number randomised: LTG = 420, CBZ = 202 329 male participants (53%) 619 participants with partial epilepsy (99.5%) Not stated how many participants had received previous AED treatment Mean age (range): 27 (2‐84) years |
|
| Interventions | Monotherapy with LTG or CBZ 6‐week escalation phase leading to minimum of LTG 2 mg/kg/d age range 2‐12 years, 200 mg/d age range 13‐64 years and 100 mg/d age > 65 years. CBZ aged 2‐12 years 5 mg/kg‐40 mg/kg, age > 12 years 100 mg/d‐1500 mg/d Range of follow‐up: 0‐245 days |
|
| Outcomes | Proportion of participants seizure‐free during the last 16 weeks of treatment Efficacy success: proportion of participants who did not withdraw before the end of week 18 and were seizure‐free in the last 16 weeks of the trial Time to withdrawal from the trial (proportion of participants completing the trial) Proportion of participants experiencing adverse events Withdrawals due to adverse events |
|
| Notes | IPD provided by trial sponsor Glaxo Smith Kline for time to treatment withdrawal and time to first seizure (plus seizure‐freedom rates at 24 weeks) Dates of seizures during the first 4 weeks not provided with IPD |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence. Participants randomised in a 2:1 ratio (LTG:CBZ), stratified by age group and country |
| Allocation concealment (selection bias) | Low risk | Allocation concealed by individual sealed, opaque envelopes (information provided by drug manufacturer) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Protocol provided. Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Double‐blinded, parallel‐group, randomised trial conducted in a single centre in Nigeria. 3 treatment arms: CBZ, PHB, PHT |
|
| Participants | Consecutive newly diagnosed participants aged ≥ 14 years presenting at the outpatient neurology clinic of the University Teaching Hopsital, Benin City, Nigeria with recurrent, untreated afebrile seizures Number randomised: PHT = 18, PHB = 18, CBZ = 19 34 male participants (62%) 10 participants with partial epilepsy (18%) Mean age (range): 27.5 years (14‐55 years) |
|
| Interventions | Monotherapy with PHT or CBZ Median daily dose (range): CBZ = 600 mg (400 mg‐1200 mg), PHT = 200 mg (100 mg‐300 mg), PHB = 120 mg (60 mg‐180 mg) All participants followed up for 12 weeks |
|
| Outcomes | Cognitive measures (reaction times, mental speed, memory, attention) | |
| Notes | IPD provided for all randomised participants by the trial author. Trial duration was 12 weeks; all participants completed the trial without withdrawing, therefore outcomes, time to withdrawal of allocated drug, time to six‐month remission and time to 12‐month remission could not be calculated. Time to first seizure calculated from IPD provided | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Trial randomised using simple randomisation. Each participant was asked to pick one from a table of numbers (1‐60), numbers corresponded to allocation of 1 of 3 drugs (information provided by trial author) |
| Allocation concealment (selection bias) | Low risk | Recruitment/randomisation of participants and allocations of treatments took place on different sites (information provided by trial author) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants single‐blinded. Research assistant recruiting participants and counselling on medication adherence was not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators performing cognitive assessments were single‐blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants completed the trial. All randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. One outcome for this review calculated from IPD provided (see footnote 2). Other outcomes for this review not available due to short trial length. All cognitive outcomes from the trial well reported |
| Other bias | Low risk | None identified |
| Methods | Randomised, parallel‐group trial conducted in a rural district of West Bengal, India 2 treatment arms: PHB and PHT |
|
| Participants | Children from a rural district of a developing country (India) who had experienced 2 or more unprovoked seizures within the 12 months preceding the trial and had been untreated in the 3 months preceding the trial Number randomised: PHB = 47 ; PHT = 47 47 boys (50%) 60 children had partial epilepsy (64%) Mean age (range): 11 (2‐18) years |
|
| Interventions | Monotherapy with PHB or PHT Maintenance doses: PHT = 5 mg/kg/d, PHB = 3 mg/kg/d. Daily dose achieved not stated Range of follow‐up: 0.5‐13 months |
|
| Outcomes | Time to first seizure Proportion seizure‐free in each trial quarter Proportion of adverse events including behavioural side effects |
|
| Notes | IPD provided for remission and seizure outcomes of this review by the trial author. Withdrawal information not available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | First 10 participants randomised from a pre‐prepared balanced random number list, following participants randomised by minimisation with stratification by age group and presence of cerebral impairment |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants, parents and treating physicians unblinded for “practical and ethical reasons.” Withdrawal information from treatments not available, however lack of blinding may have influenced withdrawal rates |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors single‐blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, parallel‐group trial conducted in the context of existing community health care in a rural highland area of a developing country (Ecuador) 2 treatment arms: CBZ and PHB |
|
| Participants | Participants with a history of at least 2 afebrile seizures and no previous AED treatment in the 4 weeks preceding the trial were eligible Number randomised: PHB = 97, CBZ = 95 67 male participants (35%) 133 participants with partial epilepsy (69%) Mean age (range): 29 (2‐68) years |
|
| Interventions | Monotherapy with PHB or CBZ Minimum maintenance doses by age groups: 2‐5 years: PHB: 15 mg/d, CBZ: 150 mg/d; 6‐0 years: PHB: 30 mg/d, CBZ: 300 mg/d; 11‐15 years: PHB: 45 mg/d, CBZ: 500 mg/d; > 16 years: PHB: 60 mg/d, CBZ: 600 mg/d. Doses gradually increased Doses achieved not stated |
|
| Outcomes | Proportion seizure‐free at 3‐, 6‐, and 12‐month follow‐ups Proportion seizure‐free, with more than 50% seizure reduction and no change in seizure frequency in 6‐ to 12‐month follow‐up period Incidence of adverse effects Trial duration: 12 months Range of follow‐up: 3.5‐23 months |
|
| Notes | We received IPD for all outcomes used in this review from the trial author. Results in the published paper were given for 139 participants who completed 6 months' follow‐up, but we received IPD for all 192 participants randomised | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants randomised with random number list, no information provided on method of generating random list |
| Allocation concealment (selection bias) | High risk | Allocation concealed using sealed, opaque envelopes but method not used for all participants (information provided by trial author) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported or calculated with the IPD provided (see footnote 2) |
| Other bias | High risk | Inconsistencies between number and reasons of withdrawals between the data and the published paper, which could not be resolved by the author |
| Methods | Multinational, randomised, double‐blind trial was conducted at 115 centres across the USA, Canada, Europe and South America Four treatments: CBZ, VPS and TPM (2 arms, 100 mg/d and 200 mg/d) ‐ see Notes |
|
| Participants | Participants > 6 years and > 30 kg in weight, with a diagnosis of epilepsy within the 3 months before trial entry and no previous AED treatment except emergency treatment Number randomised (ITT population): CBZ = 126, TPM = 266 (CBZ branch), VPS = 78, TPM = 147 (VPS branch) 327 male participants (53%) 363 participants with partial epilepsy (59%) Mean age (range): 34 (6‐84 years) |
|
| Interventions | Monotherapy with CBZ, VPS or TPM Starting doses: CBZ = 200 mg/d, VPS = 250 mg/d, TPM = 25 mg/d Target doses (after 4‐week titration): CBZ = 600 mg/d, VPS = 1000 mg/d, TPM = 100 or 200 mg/d (see Notes) Range of follow‐up: 0‐29 months |
|
| Outcomes | Time to exit Time to first seizure Proportion of seizure‐free participants during the last 6 months of double‐blind treatment Safety assessment: most commonly occurring adverse events |
|
| Notes | IPD provided for all outcomes of this review by trial sponsor Johnson & Johnson. Trial designed in 2 strata based on whether recommended treatment would be CBZ or VPS. Within the 2 strata, participants were randomised to 10 mg/d TPM, 200 mg/d TPM or CBZ/VPS depending on the strata. Data analysed according to the separate strata in this review with the 2 TPM doses analysed together (see Data extraction and management) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was balanced using permuted blocks of size three and stratified by trial centre, according to a computer‐generated randomisation schedule prepared by the trial sponsor |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Trial was double‐blinded for the first 6 months, followed by an open‐label phase |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants from the ITT population analysed from IPD provided (see footnote 2). Eight participants with no follow‐up data were excluded from ITT population |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Single‐centre, randomised, parallel‐group trial of participants, referrals to the outpatient department of neurology of the Central Hospital of Paijat‐Hame, Finland. 2 treatment arms: CBZ and PHT |
|
| Participants | Adults (eligible age range 15‐57) with newly diagnosed epilepsy Number randomised: PHT = 20, CBZ = 23 20 male participants (47%) 10 participants with partial epilepsy (23%) Mean age (SD) years: PHT = 31.5 (11.3), CBZ = 26.8 (13.2) |
|
| Interventions | Monotherapy with PHT or CBZ Dose information not reported Trial duration: 6 months, range of follow‐up not stated |
|
| Outcomes | Cognitive assessments (visual motor speed, co‐ordination, attention and concentration, verbal and visuospatial learning, visual and recognition memory, reasoning, mood, handedness) Harmful side effects |
|
| Notes | 59 participants were randomised but 16 were subsequently excluded. Results were presented only for the 43 participants who completed the entire trial. Outcomes chosen for this review were not reported. IPD not available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment groups, method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Cognitive outcome assessor was blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 16/59 (27%) of participants excluded from analysis. Results presented only for participants who completed the trial |
| Selective reporting (reporting bias) | Unclear risk | Cognitive outcomes described in methods section well reported in results section. Adverse effects reported, no seizure outcomes reported and outcomes chosen for this review not reported. No protocol available so unclear if seizure outcomes were planned a priori |
| Other bias | Low risk | None identified |
| Methods | Randomised, 'two compartment' parallel trial, conducted in the USA 2 treatment arms: CBZ and PHT |
|
| Participants | Adults, previously untreated, with at least 2 seizures or at least 1 seizure and an EEG with paroxysmal features Number randomised: PHT = 45, CBZ = 42 60 male participants (69%) 55 participants with partial epilepsy (63%) Mean age (range) 37.4 (18‐77) years |
|
| Interventions | Monotherapy with PHT or CBZ Mean daily dose achieved (for the 54 participants with no major side effects): PHT = 5.35 mg/kg/d, CBZ = 9.32 mg/kg/d Trial duration: 2 years. Range of follow‐up not reported |
|
| Outcomes | Laboratory measures Side effects (major and minor) Seizure control/treatment failure |
|
| Notes | 7 participants on CBZ and 10 participants on PHT were “dropped for non‐compliance” and excluded from analysis Outcomes chosen for this review were not reported. IPD not available |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants randomly assigned to treatment groups, method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind (participants and personnel) achieved with additional blank tablet |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear if outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 17/87 (19.5%) of participants excluded from analysis for "non‐compliance". Results presented only for participants who completed the trial |
| Selective reporting (reporting bias) | Low risk | All efficacy and tolerability outcomes specified in the methods sections reported well in the results section. No protocol available. Outcomes chosen for this review were not reported |
| Other bias | Low risk | None identified |
| Methods | Open‐label, parallel‐design, multicentre RCT conducted at 16 centres in the USA 2 treatment arms: PHT and VPS randomised in a 2:1 ratio |
|
| Participants | Participants with at least 2 newly‐diagnosed and previously untreated primary generalised tonic clonic seizures within 14 days of starting the trial Number randomised: PHT = 50, VPS = 86 73 male participants (54%) 0% participants with partial epilepsy (all generalised epilepsy) Mean age (range): 21 (3‐64 years) |
|
| Interventions | Monotherapy with PHT or VPS Starting doses PHT: 3 mg/kg/d‐ 5 mg/kg/d, VPS: 10 mg/kd/d‐15 mg/kg/d, doses gradually increased. Doses achieved not stated Range of follow‐up: 0‐11 months |
|
| Outcomes | Time to first generalised tonic clonic seizure 6‐month seizure recurrence rates Adverse events |
|
| Notes | IPD provided for 3/4 outcomes of this review by the Department of Veteran's Affairs (maximum follow‐up 6 months, therefore trial could not contribute to outcome, 'Time to 12‐month remission') | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants randomised on a 2:1 ratio VPS:PHT using randomisation tables in each centre (information provided by trial author) |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial; trial authors state that differences in adverse events of PHT and VPS would "quickly unblind" the trial anyway |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial; trial authors state that differences in adverse events of PHT and VPS would "quickly unblind" the trial anyway |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Double‐blind, multi‐centre, RCT conducted in the USA 2 treatment arms: CBZ and LEV |
|
| Participants | Adults > 60 years with new onset partial seizures (previously untreated or under treated) Interim results: 37 participants recruited (numbers recruited to each arm not stated) 28 male participants (76%) 100% of participants had partial epilepsy Age: 20 participants aged 60‐69 years and 17 participants > 70 years |
|
| Interventions | Monotherapy with CBZ or LEV Intitial doses: CBZ = 100 mg/d, LEV = 250 mg/d. Target doses: CBZ = 400 mg/d, LEV = 1000 mg/d Interim results, range of follow‐up not stated |
|
| Outcomes | Discontinuations from the trial Treatment‐emergent side effects Seizure control |
|
| Notes | Trial available as abstract only. Attempts to contact the principal investigator and trial sponsor for further information were unsuccessful | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial described as randomised, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind trial ‐ trial drugs were over encapsulated and all participants received similar appearing active medication |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Interim report, 7/37 participants recruited had discontinued treatment. Unclear if an ITT approach would be taken to analysis |
| Selective reporting (reporting bias) | Unclear risk | Abstract only, insufficient information to make a judgement |
| Other bias | Low risk | None identified |
| Methods | Randomised, multicentre, double‐blind trial conducted in the USA 2 treatment arms: PHT and TPM |
|
| Participants | Participants 12–65 years (inclusive), weighed at least 50 kg and experienced 1–20 unprovoked, complex partial or primary/secondarily generalised tonic–clonic seizures within the past 3 months, either as newly diagnosed epilepsy or as epilepsy relapse from remission Number randomised: PHT = 128, TPM = 133 126 male participants (48%) 53 participants with partial epilepsy (20%) Mean age (range): 34 (12‐78 years) |
|
| Interventions | Monotherapy with PHT or TPM Short titration (1 day) to target dose of PHT = 300 mg/d and TPM = 100 mg/d Range of follow‐up: 0‐2.5 months |
|
| Outcomes | Time to first complex partial seizure or generalised tonic clonic seizure Participant retention (time to discontinuation of treatment) Incidence and summary of adverse events |
|
| Notes | IPD provided by trial sponsor Johnson & Johnson for time to withdrawal and time to first seizure, trial duration insufficient to measure remission outcomes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial described as randomised, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and personnel double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinded assessment of results and serum AED level |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Parallel‐design RCT conducted in Meerut, India 2 treatment arms: PHT and VPS |
|
| Participants | Participants with at least 2 partial or generalised tonic‐clonic seizures per month Unclear if participants were newly diagnosed Number randomised: PHT = 45; VPS = 49 70 male participants (74%) 27 participants with partial epilepsy (29%) Age range: PHT: 12‐42 years; VPS: 8‐52 years |
|
| Interventions | Monotherapy with PHT or VPS Average daily dose achieved: PHT: 5.6 mg/kg/d, VPS: 18.8 mg/kg/d Participants were evaluated after 4, 12 and 24 weeks of treatment No information on range of follow‐up |
|
| Outcomes | Reduction in frequency of seizures:
Adverse effects Seizure control |
|
| Notes | Outcomes chosen for this review were not reported. IPD not available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants "randomly allocated irrespective of seizure type," no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Frequency of seizures reported for all randomised participants, no information provided on withdrawal rates/attrition rates etc |
| Selective reporting (reporting bias) | Low risk | Frequency of seizures during treatment well reported, most common adverse events reported No protocol available to compare with a priori analysis plan, outcomes for this review not reported |
| Other bias | Low risk | None identified |
| Methods | Single‐centre, randomised, parallel‐group trial of participants referred to the Neurology Clinic of Nehru Hospital, Chandigarh, India. 2 treatment arms: CBZ and PHT |
|
| Participants | Newly diagnosed and drug‐naive adult participants > 14 attending the Neurology Clinic of Nehru Hospital, Chandigarh, India Number randomised: PHT = 20, CBZ = 20 28 male participants (70%) 11 participants with partial epilepsy (27.5%) Mean age (range): PHT group 23.4 (14‐44 years), CBZ 24.4 (14‐45 years) |
|
| Interventions | Monotherapy with PHT or CBZ Initial daily dose: PHT = 5 mg/kg/d, CBZ = 10 mg/kg/d Trial duration 10‐12 weeks. Range of follow‐up not reported |
|
| Outcomes | Cognitive measures before and after treatments (verbal, performance, memory, visuomotor, perceptomotor organisation, visual organisation, dysfunction) | |
| Notes | 6 participants on CBZ and 8 participants on PHT were excluded from final analysis of cognitive assessments who were lost to follow‐up or who had uncontrolled seizures Outcomes chosen for this review were not reported. IPD not available |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The subjects were randomised to one of the two trial groups," no further information given on methods of randomisation |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 14/40 (35%) of participants excluded from analysis who were lost to follow‐up or experienced uncontrolled seizures. Results presented only for participants who completed the trial |
| Selective reporting (reporting bias) | Unclear risk | Cognitive outcomes described in methods section well reported in results section. No seizure outcomes or adverse events reported and outcomes chosen for this review not reported. No protocol available, so unclear if seizure outcomes were planned a priori |
| Other bias | Low risk | None identified |
| Methods | Randomised, open‐label trial conducted in several hospitals in Mexico 2 treatment arms: CBZ and TPM |
|
| Participants | Participants aged 2‐18 years with newly diagnosed partial epilepsy with or without secondary generalisation with at least two unprovoked seizures > 24 h apart and at least 1 seizure in the last 6 months. Participants must have no established treatment and have received no antiepileptic treatment within the past 30 days Number randomised: CBZ = 42, TPM = 46. Number included in analysis CBZ = 32, TPM = 33 100% partial epilepsy 33 male participants (60%) included in analysis Mean age (range): CBZ = 10 (5‐17) years, TPM = 8 (2‐16) years for participants included in analysis |
|
| Interventions | Monotherapy with CBZ or TPM Treatments titrated to a maximum of CBZ = 20 mg/kg/d‐25 mg/kg/d, TPM = 9 mg/kg/d Follow‐up assessments at 6 and 9 months, range of follow‐up not stated |
|
| Outcomes | Seizure freedom and frequency of seizures during the trial Adverse events during the trial Laboratory results |
|
| Notes | The trial was published in Spanish; the characteristics and outcomes were translated. Outcomes chosen for this review were not reported; contact could not be made with trial author to provide IPD Results presented only for those who completed the trial. Those with less than 35% reduction of seizures were excluded from analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Random number tables used to assign participants to treatment groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Open‐label trial |
| Selective reporting (reporting bias) | Low risk | Attrition rates reported (23 drops outs, 10 for CBZ and 13 for TPM). Only those who completed the trial were included in analysis (non responders to treatment excluded), this is not an ITT approach |
| Other bias | Low risk | No protocol available. Seizure outcomes and adverse events well reported |
| Methods | Randomised, double‐blind, parallel‐group trial conducted in 56 centres in Europe and Australia. 3 treatment arms: LTG (200 mg/d), LTG (100 mg/d) and CBZ |
|
| Participants | Adults and children > 12 years with newly diagnosed, currently untreated or recurrent epilepsy with ≥ two seizures in the previous 6 months and with at least 1 seizure in the last 3 months. Participants must not have taken AEDs in the previous 6 months Number randomised: LTG (200 mg) = 115, LTG (100 mg) = 115, CBZ = 121 188 male participants (54%) 237 participants with partial epilepsy (68%) Not stated how many participants had received previous AED treatment Mean age (range): 32 (12‐72) years |
|
| Interventions | Monotherapy with LTG or CBZ for 30 weeks 4‐week escalation phase leading to LTG = 100 mg/d, LTG = 200 mg/d, CBZ = 600 mg/d Range of follow‐up: 0‐378 days |
|
| Outcomes | Proportion completing seizure‐free after the first 6 weeks of treatment Time to first seizure Time to withdrawal Frequency of adverse events with at least 5% incidence in any treatment group |
|
| Notes | IPD provided by trial sponsor Glaxo Smith Kline for time to treatment withdrawal, time to first seizure and time to six‐month remission. Participants considered to complete the trial if they experienced a seizure after the first 6 weeks. In primary analysis, two arms of LTG pooled and compared to CBZ and separate doses of LTG compared to CBZ in sensitivity analysis (see Data extraction and management) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐ generated random sequence (information provided by drug manufacturer) |
| Allocation concealment (selection bias) | Low risk | Allocation concealed by individual, sealed, opaque envelopes (information provided by drug manufacturer) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐ label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐ label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Open‐label, multicentre trial across 22 centres in the UK 2 treatment arms: CBZ and VPS |
|
| Participants | Adults with newly onset primary generalised epilepsy or partial epilepsy with/without generalisation or with a recurrence of seizures following withdrawal of AED treatment were eligible given that no anticonvulsants had been received in the previous 6 months Number randomised: CBZ = 151, VPS = 149 153 (51%) male participants (51%) 147 participants with partial epilepsy (49%) Mean age (range): 33 (16‐79) years |
|
| Interventions | Monotherapy with CBZ or VPS Mean daily dose achieved by month 24: CBZ = 516 mg/d, VPS = 924 mg/d Range of follow‐up: 0.5‐90 months |
|
| Outcomes | Remission analysis (time to 6‐, 12‐ and 24‐month remission) Retention analysis (time to treatment failure) Adverse event incidence Incidence of treatment failures due to poor seizure control and adverse events |
|
| Notes | IPD provided for all outcomes of this review by trial sponsor Sanofi. Participants with other generalised seizure types (e.g. myoclonic/absence) were included in the trial, but efficacy analyses were based solely on generalised tonic clonic seizures. Results in the published paper were given for 181 participants out of 300 analysed by ITT (participants randomised and with data for at least 1 follow‐up visit). IPD is provided for all 300 participants randomised and used for analyses in this review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised to treatment using a computerised minimisation programme with stratification for age, sex, seizure type and centre |
| Allocation concealment (selection bias) | Low risk | Treatment allocation was concealed via central telephone allocation from the Trial Office at Sanofi Winthrop LTD |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, double‐blind, parallel‐group trial conducted in 18 Veterans Affairs Medical Centres in the USA 3 treatment arms: LTG, CBZ and GBP |
|
| Participants | Adults > 60 years with newly diagnosed seizures, untreated or treated with sub‐therapeutic AED levels, with at least 1 seizure in the previous 3 months. Number randomised: CBZ = 198, GBP = 195, LTG = 200 570 male participants (96%) 446 participants with partial epilepsy (75%) Not stated how many participants had received previous AED treatment Mean age (years): CBZ = 71.9, GBP = 72.9, LTG = 71.9. Range not stated |
|
| Interventions | Monotherapy with CBZ, GBP, LTG 6‐week escalation phase leading to CBZ = 600 mg/d, GBP = 1500 mg/d, LTG = 150 mg/d Trial duration: 12 months. Range of follow‐up: not stated |
|
| Outcomes | Retention in the trial for 12 months Seizure freedom at 12 months Time to first, second, fifth and tenth seizure (time to seizures) Drug toxicity (incidence of systemic and neurologic toxicities) Serum drug levels and compliance Seizure‐free retention rates |
|
| Notes | IPD requested from trial sponsor, the Department of Veterans Affairs, USA. At the time of review, IPD has not been received. Aggregate data extracted from graphs in the publication | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block randomisation (varying sizes) performed by site via a computer‐generated list |
| Allocation concealment (selection bias) | Low risk | Telephone randomisation used and pharmacy dispensed a prescription of the allocated drug (part of a blinded drug kit) to participants |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blind achieved with double dummy tablets, doses of both increased and decreased simultaneously |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported. Most of the randomised participants included in analysis, 3 excluded due to site closure (not related to treatment) |
| Selective reporting (reporting bias) | Low risk | No protocol available but case report forms of data collected provided by the sponsor. Seizure outcomes and adverse events well reported |
| Other bias | Low risk | None identified |
| Methods | Randomised, double‐blind, parallel‐group trial conducted in 29 centres across Croatia, Finland, France, Finland and Norway 2 treatment arms: LTG and CBZ |
|
| Participants | Adults > 65 years with newly diagnosed seizures, with a history of at least 2 seizures and at least 1 seizure in the previous 6 months. Participants must not have taken AEDs for more than 2 weeks in the previous 6 months and never taken CBZ or LTG Number randomised: LTG = 93, CBZ = 92 102 male participants (54%) Proportion with partial epilepsy not stated Not stated how many participants had received previous AED treatment Mean age: 74 (65‐91) years |
|
| Interventions | Monotherapy with LTG or CBZ 4‐week escalation phase leading to LTG = 100 mg/d, CBZ = 400 mg/d Trial duration 40 weeks. Range of follow‐up: not stated |
|
| Outcomes | Retention in the trial (time to treatment withdrawal for any cause) Seizure freedom after week 4 Seizure freedom after week 20 Time to first seizure Adverse event reports Tolerability according to the Liverpool Adverse Event profile (AEP) |
|
| Notes | IPD requested from trial sponsor Glaxo Smith Kline but data could not be located. Aggregate summary data extracted from the publication |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised, no other information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blind achieved with double dummy tablets, packaged together |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all participants who received trial treatment were included in an ITT analysis |
| Selective reporting (reporting bias) | Low risk | No protocol available but clinical trial summary provided by the sponsor. Seizure outcomes and adverse events well reported |
| Other bias | Low risk | None identified |
| Methods | Randomised, multicentre, open‐label, parallel‐group trial conducted in the UK 5 treatment arms: LTG, CBZ, GBP, TPM and OXC |
|
| Participants | Adults and children > 4 years with newly diagnosed partial epilepsy, relapsed partial epilepsy or failed treatment with a previous drug not used in this trial. Number randomised: CBZ = 378, LTG = 378, OXC = 210, TPM = 378, GBP = 377 922 male participants (54%) 1491 partial epilepsy (87%) 309 had received previous AED treatment (18%) Mean age(range): 38 (5‐86) years |
|
| Interventions | Monotherapy for LTG, CBZ, GBP, TPM or OXC Titration doses and maintenance doses decided by treating clinician Range of follow‐up: 0‐86 months |
|
| Outcomes | Time to treatment failure Time to 1‐year (12 month) remission Time to 2‐year remission Time to first seizure Health‐related quality of life via the NEWQOL (Newly Diagnosed Epilepsy Quality of Life Battery) Health economic assessment and cost effectiveness of the drugs (cost per QALY gained and cost per seizure avoided) Frequency of clinically important adverse events |
|
| Notes | IPD provided for time to treatment withdrawal, time to first seizure, time to six‐month, time to 12‐month and time to 24‐month remission (trial conducted at our site) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer minimisation programme stratified by centre, sex and treatment history |
| Allocation concealment (selection bias) | Low risk | Telephone randomisation to a central randomisation allocation service |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, multicentre, open‐label, parallel‐group trial conducted in the UK 3 treatment arms: LTG, GBP, TPM |
|
| Participants | Adults and children > 4 years with newly diagnosed or relapsed generalised or unclassified epilepsy, or failed treatment with a previous drug not used in this trial. Number randomised: LTG = 239, VPS = 238; TPM = 239 420 male participants (59%) 52 partial epilepsy (7%) 108 had received previous AED treatment (15%) Mean age (range): 22.5 (5‐77) years |
|
| Interventions | Monotherapy for LTG, GBP or TPM Titration doses and maintenance doses decided by treating clinician Range of follow‐up: 0‐83.5 months |
|
| Outcomes | Time to treatment failure Time to 1‐year (12‐month) remission Time to 2‐year remission Time to first seizure Health‐related quality of life via the NEWQOL (Newly Diagnosed Epilepsy Quality of Life Battery) Health economic assessment and cost effectiveness of the drugs (cost per QALY gained and cost per seizure avoided) Frequency of clinically important adverse events |
|
| Notes | IPD provided for time to treatment withdrawal, time to first seizure, time to six‐month, time to 12‐month and time to 24‐month remission (trial conducted at our site) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer minimisation programme stratified by centre, sex and treatment history |
| Allocation concealment (selection bias) | Low risk | Telephone randomisation to a central randomisation allocation service |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Parallel‐design RCT conducted at 2 centres (Glasgow, Scotland and Wellington, New Zealand) 2 treatment arms: PHT and VPS |
|
| Participants | 21 (64%) of participants previously untreated, 12 (36%) of participants continued to have seizures on previous drug therapies. Original treatments gradually withdrawn before PHT or VPS treatment introduced. Number randomised: PHT = 15, VPS = 18 12 male participants (36%) 19 participants with partial epilepsy (58%) Mean age (range): 23 (7‐55 years) |
|
| Interventions | Monotherapy with PHT or VPS Starting doses: PHT: < 12 years 150 mg/d, older participants: 300 mg/d, VPS: < 12 years 300‐400 mg/d, older participants: 800‐1200 mg/d. Doses achieved not stated. Mean follow‐up (range): 30 (9‐48 months) |
|
| Outcomes | Seizures during treatment Adverse events |
|
| Notes | Outcomes chosen for this review were not reported IPD not available but could be constructed from the publication for the outcome 'Time to treatment withdrawal.' |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants "randomly divided", using telephone randomisation (information provided by trial author) |
| Allocation concealment (selection bias) | Low risk | Centralised telephone randomisation used (information provided by trial author) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Results reported for all randomised participants, time on treatment reported for all randomised participants. No losses to follow‐up reported |
| Selective reporting (reporting bias) | Low risk | No protocol available, outcomes chosen for this review not reported, Seizure and adverse event outcomes well reported |
| Other bias | Low risk | None identified |
| Methods | Randomised double‐blind study conducted in the USA 2 treatment arms: CBZ and VPS |
|
| Participants | Participants between the ages of 10 and 70 who had experienced at least two complex partial seizures who were previously untreated or insufficiently treated Number randomised: CBZ = 17, VPS = 16 15 male participants (45%) 100% of participants with partial epilepsy Mean age (range): CBZ = 32.5 (13‐65), VPS = 31.3 (17‐57) |
|
| Interventions | Monotherapy with CBZ or VPS Doses started or achieved not stated 4‐week titration period followed by a 24‐week maintenance period. Range of follow‐up not stated |
|
| Outcomes | Proportion of participants free of complex partial seizures during the maintenance period Proportion of participants reporting specific adverse events |
|
| Notes | Outcomes for this review were not reported; IPD were not available due to time elapsed since the trial was conducted | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomised in a 1:1 ratio, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blind study |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rates reported. Only those who entered the maintenance period were included in analysis; this is not an ITT analysis |
| Selective reporting (reporting bias) | Low risk | Efficacy, and tolerability outcomes specified in the methods sections were reported well in the results section. No protocol was available. Outcomes chosen for this review were not reported |
| Other bias | Low risk | None identified |
| Methods | Randomised, double‐blind, multicentre trial conducted in the UK 2 treatment arms: LTG and PHT |
|
| Participants | Participants aged 14‐75 years with two or more partial, secondarily generalised, or primary generalised tonic‐clonic seizures Number randomised: PHT = 95, LTG = 86 101 male participants (56%) 90 participants with partial epilepsy (50%) Mean age (range): 34 (13‐75 years) |
|
| Interventions | Monotherapy with LTG or PHT Titrated for 2 weeks to a target dose of LTG = 150 mg/d, PHT = 300 mg/d Range of follow‐up: 0‐15 months |
|
| Outcomes | Percentage of participants remaining on treatment Percentage of participants remaining seizure free in the last 24 and last 16 weeks of treatment Number of seizures (percentage change from baseline) in the last 24 weeks and 16 weeks of treatment Time to first seizure after the first 6 weeks of treatment (dose‐titration period) Time to discontinuation Incidence of adverse events and adverse events leading to discontinuation Quality of Life according to the Side Effects and Life Satisfaction (SEALs) inventory |
|
| Notes | IPD provided by trial sponsor Glaxo Smith Kline for time to treatment withdrawal, time to first seizure and time to six‐month remission | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation was stratified according to seizure type, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All participants, personnel and outcome assessors involved in the trial were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All participants, personnel and outcome assessors involved in the trial were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, open‐label, parallel‐group trial conducted in 24 centres across Germany. 4 treatment arms: LTG (two arms), CBZ and VPS Participants with partial and generalised epilepsy randomised separately to LTG or CBZ and LTG or VPS respectively |
|
| Participants | Adults and children > 12 years with newly diagnosed epilepsy; at least 1 seizure and EEG imaging suggesting epilepsy Number randomised not stated, number included in analysis: LTG = 88, CBZ = 88 (partial); LTG = 33, VPS = 30 (generalised) 106 male participants (64%) in partial epilepsy group, 27 male participants (43%) in the generalised epilepsy group 166 out of 239 total included in analysis have partial epilepsy (69%) Not stated how many participants had received previous AED treatment Mean age (years): LTG (partial) = 46.6, CBZ = 43.1, LTG (generalised) = 22.3, VPS = 23.3 Range not stated |
|
| Interventions | Monotherapy with LTG, CBZ or VPS 4‐week escalation phase leading to LTG = 100 mg/d‐200 mg/d, CBZ = 600 mg/d‐1200 mg/d in adults and 600 mg/d‐1000 mg/d in children aged 11‐15, VPS = 600 mg/d‐1200 mg/d for children aged 6‐14, 600 mg/d‐1500 mg/d for adolescents over 14 years and 1200 mg/d‐2100 mg/d for adults Trial duration: 26 weeks, range of follow‐up: not stated |
|
| Outcomes | Number of seizure‐free patients during trial weeks 17‐24 "Leaving the study" (retention rates) Adverse event rates |
|
| Notes | IPD requested from trial sponsor Glaxo Smith Kline but data could not be provided due to restrictions over the de‐identification of datasets from trials conducted in Germany. Aggregate data extracted from graphs in the publication. Data from participants with partial epilepsy is the randomised comparison of LTG and CBZ and data from participants with generalised epilepsy is the randomised comparison of LTG and VPS (see Data extraction and management) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised, no other information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number of participants randomised to each group not reported (254 randomised and 239 analysed in the four arms of the trial). Reasons for exclusion stated but not which drug these participants were randomised to |
| Selective reporting (reporting bias) | Low risk | No protocol available but clinical trial summary provided by the sponsor. Seizure outcomes and adverse events well reported |
| Other bias | Low risk | None identified |
| Methods | Randomised, single‐centre, open‐label trial conducted in Scotland, UK 2 treatment arms LTG and VPS |
|
| Participants | Participants of at least 13 years with a minimum of 2 newly onset unprovoked seizures of any type and no previous exposure to LTG or VPS Number randomised: LTG = 117, VPS =109 114 male participants (50%) 154 participants with partial epilepsy (68%) Mean age (range): 36 (13 ‐ 80 years) |
|
| Interventions | Monotherapy with LTG or VPS Titration of 5‐10 weeks to target doses of LTG = 200 mg/d and VPS = 1000 mg/d Range of follow‐up: 0‐51 months |
|
| Outcomes | Percentage of randomised participants achieving a minimum period of 12 months' seizure freedom Percentage of randomised participants withdrawing due to adverse events Percentage of randomised participants with lack of efficacy at maximum tolerated dose Changes in levels of androgenic hormone levels (testosterone, androstenedione and sex hormone‐binding globulin levels) Changes in weight and BMI from baseline |
|
| Notes | IPD provided for all outcomes of this review by trial author | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial described as randomised, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants from the ITT population analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | High risk | There were inconsistencies between rates of seizure recurrence and reasons for withdrawal between the data provided and the published paper, which the authors could not resolve |
| Methods | Randomised, single‐centre, open‐label trial conducted in Bengaluru, India. 2 treatment arms: CBZ and LEV |
|
| Participants | Participants aged 18‐60 years diagnosed newly with focal or partial seizures with or without secondary generalisation referred to the Department of Neurology at Vydehi Institute of Medical Sciences and Research Center Number randomised CBZ = 30, LEV = 30 30 male participants (50%) 100% participants with partial epilepsy Mean age (range): not provided for all randomised participants |
|
| Interventions | Monotherapy with CBZ or LEV Starting dose of CBZ = 200 mg/d, LEV = 500 mg/d titrated to a maximum dose of CBZ 1200 mg/d, LEV 300 mg/d Trial duration: 1 year, range of follow‐up: not stated |
|
| Outcomes | Quality of Life by the QOLIE‐10 questionnaire before and after 26 weeks of therapy Treatment efficacy (seizure freedom at 4 weeks, 12 weeks, 26 weeks and 6 months) Treatment safety (proportion of participants experiencing at least 1 adverse event) |
|
| Notes | Outcomes chosen for this review were not reported; contact could not be made with trial author to provide IPD | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial described as randomised, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Attrition rates reported, two participants lost to follow‐up in each group not included in analysis. This is not an ITT approach but unlikely that this small amount of missing data would influence the overall results |
| Selective reporting (reporting bias) | High risk | Only one outcome is predefined in the methods section (Quality of Life), other results reported were not predefined |
| Other bias | Low risk | None identified |
| Methods | Parallel‐design RCT conducted in Madras (Chennai), India Three treatment arms: PHB, PHT, VPS |
|
| Participants | Children with more than 1 previously untreated generalised tonic clonic (afebrile) seizure Number randomised: PHB group = 51, PHT = 52, VPS = 48 81 boys (54%) 0% partial epilepsy (all had generalised epilepsy) Age range: 4‐12 years |
|
| Interventions | Monotherapy with PHT or VPS Starting doses: PHB: 3 mg/kg/d‐ 5 mg/kg/d PHT: 5 mg/kg/d‐ 8 mg/kg/d, VPS: 15 mg/kg/d‐ 50 mg/kg/d Dose achieved not stated Range of follow‐up (months): 22‐36 |
|
| Outcomes | Proportion with recurrence of seizures Adverse events |
|
| Notes | Outcomes chosen for this review were not reported. IPD not available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants randomised via a computer‐generated list of random numbers |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double–blinded using additional placebo tablets, unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double–blinded using additional placebo tablets, unclear who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed |
| Selective reporting (reporting bias) | Low risk | No protocol available, outcomes chosen for this review not reported, Seizure and adverse event outcomes well reported |
| Other bias | Low risk | None identified |
| Methods | Multi‐centre, open label, randomised, two parallel group stratified trial carried out in a community setting between February 2005 and October 2007 in 269 centres across 23 European countries and Australia. Four treatment arms: CBZ (controlled release), LEV (two arms) and VPS (extended release) ‐ see notes |
|
| Participants | Patients aged ≥16 years were included if they had two or more unprovoked seizures in the previous 2 years with at least one during the previous 6 months. Participants must not have received one of the trial drugs previously or treated for epilepsy with any other AED in the previous 6 months Number randomised (ITT population): CBZ = 503, LEV = 492 (CBZ branch), LEV = 349, VPS = 353 (VPS branch). 949 male participants (56%) 1048 participants with partial epilepsy (62%) Mean age (range): 40 (16 ‐ 89 years). |
|
| Interventions | Monotherapy with CBZ, LEV or VPS Titration over two weeks to target doses CBZ‐CR=600 mg/day, LEV=1000 mg/day, VPS‐ER=1000 mg/day, Range of follow up: 0 to 28.5 months |
|
| Outcomes | Time to withdrawal from trial medication (treatment withdrawal) after randomisation Time to first seizure after randomisation Treatment withdrawal rates at 6 and 12 months Seizure‐freedom rates at 6 and 12 months Change of baseline in quality of life measures (QOLIE‐31‐P and EQ‐5D) Treatment emergent adverse events (intensity and seriousness) |
|
| Notes | IPD provided for all outcomes of this review by trial sponsor UCB. Trial designed in 2 strata based on whether recommended treatment would be CBZ or VPS. Data analysed according to the separate strata in this review (see Data extraction and management) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation was stratified, no further information provided |
| Allocation concealment (selection bias) | Low risk | Treatment allocation was concealed by use of an interactive voice‐response system via telephone to manage the randomisation process |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants from the ITT population analysed from IPD provided (see footnote 2). 8 randomised participants excluded from ITT population due to no informed consent or lack of compliance with good clinical practice. |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Single‐centre, parallel‐design RCT conducted in Newcastle, UK 2 treatment arms: PHT and VPS |
|
| Participants | Participants with ≥ 2 partial or generalised tonic‐clonic seizures in the past 3 years. Participants were previously untreated but started on AED treatment within 3 months of their most recent seizure Number randomised: PHT = 70, VPS = 70 73 male participants (52%) 63 participants with partial epilepsy (45%) Mean age (range): 35 (14‐70 years) |
|
| Interventions | Monotherapy with PHT or VPS Starting doses: PHT 300 mg/d, VPS 600 mg/d. Dose achieved not stated Range of follow‐up: 3.5‐52 months |
|
| Outcomes | Time to 2‐year remission Time to first seizure Adverse events |
|
| Notes | IPD provided for all outcomes included in this review by trial author | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants randomised with stratification for age group, gender and seizure type. Method of randomisation not stated or provided by author |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Open‐label, multicentre trial across 63 centres in UK and Ireland 2 treatment arms: CBZ and VPS |
|
| Participants | Children with newly onset primary generalised epilepsy or partial epilepsy with/without generalisation or with a recurrence of seizures following withdrawal of AED treatment were eligible given that no anticonvulsants had been received in the previous 6 months Number randomised: CBZ = 130, VPS = 130 122 boys (47%) 108 participants with partial epilepsy (42%) Mean age (range): 10 (5‐16) years |
|
| Interventions | Monotherapy with CBZ or VPS Mean daily dose achieved by month 24 CBZ = 450 mg/d, VPS = 700 mg/d Range of follow‐up: 2‐59 months |
|
| Outcomes | Remission analysis (time to 6‐, 12‐ and 24‐month remission) Retention analysis (time to treatment failure) Adverse event incidence Incidence of treatment failures due to poor seizure control and adverse events |
|
| Notes | IPD provided for all outcomes of this review by trial sponsor Sanofi. Results in the published paper are given for 244 children out of 260 analysed by "intention to treat" (children randomised and with data for at least one follow‐up visit). IPD is provided for all 260 children randomised and will be used for analyses in this review | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised to treatment using a computerised minimisation programme with stratification for age, sex, seizure type and centre |
| Allocation concealment (selection bias) | Low risk | Treatment allocation was concealed via central telephone allocation from the Trial Office at Sanofi Winthrop LTD |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, ITT approach, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
| Methods | Randomised, double‐blind, parallel‐group trial conducted in 47 centres across Germany, Austria and Switzerland. 3 treatment arms: LTG, CBZ and LEV |
|
| Participants | Adults > 60 years with newly diagnosed partial seizures, with a history of at least 2 seizures and at least 1 seizure in the previous 6 months. Participants must not have taken AEDs for more than 4 weeks Number randomised: LTG = 118, CBZ = 121, LEV = 122 215 male participants (60%) 100% of participants with partial epilepsy Not stated how many participants had received previous AED treatment Mean age(range): 71.5 (60‐95) years |
|
| Interventions | Monotherapy with LEV, LTG or CBZ for 58 weeks 6‐week escalation phase leading to CBZ = 400 mg/d. LEV+ 1000 mg/d, LTG = 100 mg/d Range of follow‐up: 0‐54 months |
|
| Outcomes | Retention rate at week 58 Time to discontinuation from randomisation Seizure‐freedom rates at week 30 and week 58 Time to first seizure from randomisation Time to first drug‐related adverse event Adverse events (by severity) |
|
| Notes | IPD provided by trial author for time to treatment withdrawal, time to first seizure, time to six‐month and time to 12‐month remission | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A randomisation list for each centre (random permuted blocks) was prepared by the Interdisciplinary Centre for Clinical Trials (IZKS), Mainz, Germany |
| Allocation concealment (selection bias) | Low risk | The pharmacy of the University Hospital Mainz encapsulated the trial drugs and labelled the blinded medication including the randomisation number. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and trial investigator blinded by the use of matching capsules |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Trial investigator blinded, not stated if other outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported, all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
1. Abbreviations: ADL: activities of daily living; AED: antiepileptic drug; BMI: body mass index; CBCL: child behavior checklist; CBZ: carbamazepine; EEG: electroencephalography; GBP: gabapentin; IPD: Iindividual participant data; ITT: intention to treat; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; PSB: polysomnography; RCT: randomised controlled trial; REM: rapid eye movement; TPM: topiramate; VPS: sodium valproate; WISC: Wechsler Intelligence Scale for Children; ZNS: zonisamide
2. Attrition bias and reporting bias are reduced in trials for which IPD were provided, as attrition rates and unpublished outcome data were requested
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Albani 2006 | Conversion to monotherapy design, monotherapy comparison not possible |
| Alsaadi 2002 | Conversion to monotherapy design, monotherapy comparison not possible |
| Alsaadi 2005 | Conversion to monotherapy design, monotherapy comparison not possible |
| Baxter 1998 | Participants randomised to LTG and physician's choice of CBZ or VPS. No fully randomised comparison between the drugs |
| Ben‐Menachem 2003 | Conversion to monotherapy design, monotherapy comparison not possible |
| Beydoun 1997 | Conversion to monotherapy design, monotherapy comparison not possible |
| Beydoun 1998 | Conversion to monotherapy design, monotherapy comparison not possible |
| Beydoun 2000 | Conversion to monotherapy design, monotherapy comparison not possible |
| Bittencourt 1993 | Conversion to monotherapy design, monotherapy comparison not possible |
| Canadian Group 1999 | Conversion to monotherapy design, monotherapy comparison not possible |
| Cereghino 1974 | Cross‐over design is not appropriate for measuring long‐term outcomes |
| Chung 2012 | Conversion to monotherapy design, monotherapy comparison not possible |
| DeToledo 2000 | Conversion to monotherapy design, monotherapy comparison not possible |
| EUCTR2004‐004053‐26‐SE | Trial terminated early, no results available |
| EUCTR2010‐018284‐42‐NL | Trial terminated early, no results available |
| Fakhoury 2004 | Conversion to monotherapy design, monotherapy comparison not possible |
| French 2012 | Conversion to monotherapy design, monotherapy comparison not possible |
| Gilliam 1998 | Conversion to monotherapy design, monotherapy comparison not possible |
| Gruber 1962 | Cross‐over design is not appropriate for measuring long‐term outcomes |
| Hakami 2012 | Conversion to monotherapy design, monotherapy comparison not possible |
| ISRCTN73223855 | Trial terminated early, no results available |
| Kaminow 2003 | Participants randomised to LTG and physician's choice of CBZ PHT or VPS. No fully randomised comparison between the drugs |
| Kerr 1999 | Conversion to monotherapy design, monotherapy comparison not possible |
| Kerr 2001 | Conversion to monotherapy design, monotherapy comparison not possible |
| Loiseau 1984 | Cross‐over design is not appropriate for measuring long‐term outcomes |
| Reinikainen 1984 | Conversion to monotherapy design, monotherapy comparison not possible |
| Reinikainen 1987 | Conversion to monotherapy design, monotherapy comparison not possible |
| Rosenow 2012 | Conversion to monotherapy design, monotherapy comparison not possible |
| Simonsen 1975a | Conversion to monotherapy design, monotherapy comparison not possible |
| Simonsen 1975b | Conversion to monotherapy design, monotherapy comparison not possible |
| Taragano 2003 | Included participants primarily had dementia, only a subset had epilepsy |
CBZ: carbamazepine; LTG: lamotrigine; PHT: phenytoin; VPS: sodium valproate
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | Randomised trial conducted in China 2 treatment arms: CBZ and OXC |
| Participants | Children aged 2‐14 years, who were newly diagnosed with focal epilepsy between October 2009 and December 2011 Number randomised: CBZ = 60, OXC = 58 |
| Interventions | Monotherapy with CBZ or OXC Doses started and achieved not stated |
| Outcomes | Response rates Seizure‐free rates Adverse event rates |
| Notes | 2 publications of the trial available only in Chinese (English abstract). Awaiting translation of the full text |
| Methods | Randomised, double‐blind trial conducted at Neurology clinic of Ahvaz Golestan Hospital, Iran 2 treatment arms: OXC or PHT |
| Participants | Participants > 65 years with partial and secondary generalised epilepsy |
| Interventions | Monotherapy with PHT or OXC for 6 months Maximum dose: PHT = 600 mg/d, OXC = 600 mg/d |
| Outcomes | Seizure symptoms Adverse events |
| Notes | Trial registered as IRCT201202068943N1 on the Iranian Registry of Clinical Trials. We have attempted to contact the trial authors for more information |
| Methods | Randomised, double‐blind, parallel‐design trial 2 treatment arms: CBZ and ZNS |
| Participants | People newly diagnosed with epilepsy Number randomised: 171 (not stated by treatment group) Number entering dose escalation phase: CBZ = 82, ZNS = 73 |
| Interventions | Monotherapy with CBZ or ZNS 4 weeks titration to maximum dose of CBZ: 600 mg/d, ZNS: 300 mg/d |
| Outcomes | Terminal remission rate at week 24 Time interval to first seizure recurrence Adverse events |
| Notes | Full text of the trial available only in Korean (English Abstract). Awaiting translation of the full text |
| Methods | Phase 4, randomised, parallel‐design, open‐label safety trial 2 treatment arms: TPM and ZNS |
| Participants | Participants > 13 years with at least 2 seizures and 1 in the 3 months prior to screening and no AEDs in the previous 4 months Estimated number enrolled = 140 |
| Interventions | Monotherapy with TPM or ZNS Initial doses: TPM = 25 mg/d, ZNS = 100 mg/d. Maximum doses: TPM = 400 mg/d, ZNS = 600 mg/d |
| Outcomes | Cognitive function (change from baseline at 24 weeks) |
| Notes | Trial registered as NCT00154076 on ClincalTrials.gov and listed as completed but no results published. Trial sponsored by Eisai Korea, inquiries regarding this trial made to the sponsor but no data could be provided. If more information on this trial can be found, this trial will be included in future updates of the review. |
| Methods | Randomised, open‐label, parallel‐group trial conducted in Republic of Korea 2 treatment arms: OXC and TPM |
| Participants | Children with newly diagnosed epilepsy Number randomised: OXC = 20, TPM = 25 |
| Interventions | Monotherapy OXC or TPM Doses started and achieved not stated Trial duration: 16 weeks |
| Outcomes | Seizure freedom Seizure frequency Adverse events |
| Notes | Full text of the trial available only in Korean (English abstract). Awaiting translation of the full text |
| Methods | 2‐arm trial of CBZ and PHT. Unclear from information provided in the English abstract if the trial is randomised |
| Participants | 64 participants with untreated partial (n = 9), partial complex (n = 27), partial secondary generalised (n = 22), or primary generalised seizures (n = 6) |
| Interventions | Monotherapy with CBZ or PHT. Unclear how many participants were allocated to each drug |
| Outcomes | Somatosensoric evoked potentials (mean wave amplitude, mean proximal conduction time, mean central conduction time) |
| Notes | Full‐text available only in Polish, abstract available in English. Full‐text is awaiting translation before eligibility can be judged |
| Methods | Randomised trial conducted in China 4 treatment arms: LEV, LTG, OXC and TPM |
| Participants | Participants with newly diagnosed, complex partial epilepsy/complex partial secondary generalized seizures Number randomised: LEV = 68, LTG = 70, OXC = 57, TPM = 58 |
| Interventions | Monotherapy with LEV, LTG, OXC or TPM Doses started and achieved not stated |
| Outcomes | "Effective rate" (assumed to be efficacy/seizure freedom) One‐year retention rate Cause of drug withdrawal |
| Notes | Full text of the trial available only in Chinese (English abstract). Awaiting translation of the full text |
Abbreviations: AED: antiepileptic drug; CBZ: carbamazepine; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHT: phenytoin; TPM: topiramate; ZNS: zonisamide
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | EpiNet‐First Trial 2: Comparison of efficacy of levetiracetam and sodium valproate in people with previously untreated epilepsy who have generalised seizures |
| Methods | Phase 4 randomised, open‐label, pragmatic trial conducted across sites in New Zealand and Europe 2 treatment arms: LEV and VPS |
| Participants | Individuals > 5 years with ≥ 2 spontaneous generalised seizures that require AED treatment (provided all seizures have not been absence seizures) Target sample size = 506 |
| Interventions | Monotherapy with LEV or VPS Target doses LEV: 250 mg‐4000 mg, VPS: 250 mg‐400 mg |
| Outcomes | Time to 12‐month remission from seizures Proportion of participants who achieve a seizure‐free 12‐month remission by 18 months AND who have not changed to a different AED Time to treatment failure due to either inadequate seizure control, or due to unacceptable adverse events Time to treatment failure due to inadequate seizure control. Time to treatment failure due to unacceptable adverse events. Time to first seizure Time to 24‐month remission. Serious adverse events attributed to the trial medication or other AED. Quality of life as assessed by the QOLIE31 and QOLIE48 questionnaires |
| Starting date | May 2015 |
| Contact information | Dr Peter Bergin (pbergin@adhb.govt.nz) |
| Notes | Trial is registered as ACTRN12615000556549 on the Australian New Zealand Clinical Trials Registry and is listed as currently recruiting participants (correct to August 2016) Estimated finish date is May 2018 |
| Trial name or title | EpiNet‐First Trial 3: Comparison of efficacy of levetiracetam and lamotrigine in people with previously untreated epilepsy who have generalised seizures, and for whom sodium valproate is not deemed an acceptable anti‐epileptic drug |
| Methods | Phase 4 randomised, open‐label, pragmatic trial conducted across sites in New Zealand and Europe 2 treatment arms: LEV and LTG |
| Participants | Individuals > 5 years with ≥ 2 spontaneous generalised seizures that require AED treatment (provided all seizures have not been absence seizures) Target sample size = 664 |
| Interventions | Monotherapy with LEV or LTG Target doses LEV: 250 mg‐4000 mg, LTG: 250 mg‐400 mg |
| Outcomes | Time to 12‐month remission from seizures Proportion of participants who achieve a seizure‐free 12‐month remission by 18 months AND who have not changed to a different AED Time to treatment failure due to either inadequate seizure control, or due to unacceptable adverse events Time to treatment failure due to inadequate seizure control. Time to treatment failure due to unacceptable adverse events. Time to first seizure Time to 24‐month remission Serious adverse events attributed to the trial medication or other antiepileptic medication Quality of life as assessed by the QOLIE31 and QOLIE48 questionnaires |
| Starting date | May 2015 |
| Contact information | Dr Peter Bergin (pbergin@adhb.govt.nz) |
| Notes | Trial is registered as ACTRN12615000639527 on the Australian New Zealand Clinical Trials Registry and is listed as currently recruiting participants (correct to August 2016) Estimated finish date is May 2018 |
| Trial name or title | EpiNet‐First Trial 4: Comparison of efficacy of levetiracetam, lamotrigine and sodium valproate in people with previously untreated epilepsy who have unclassified seizures |
| Methods | Phase 4 randomised, open‐label, pragmatic trial conducted across sites in New Zealand and Europe Three treatment arms: LEV, LTG and VPS |
| Participants | Individuals > 5 years with ≥ 2 spontaneous generalised seizures that require AED treatment (provided all seizures have not been absence seizures) Target sample size = 1176 |
| Interventions | Monotherapy with LEV, LTG or VPS Target doses LEV: 250 mg‐4000 mg, LTG 250 mg‐400 mg, VPS: 250 mg‐400 mg |
| Outcomes | Time to 12‐month remission from seizures Proportion of participants who achieve a seizure‐free 12‐month remission by 18 months AND who have not changed to a different AED Time to treatment failure due to either inadequate seizure control, or due to unacceptable adverse events Time to treatment failure due to inadequate seizure control. Time to treatment failure due to unacceptable adverse events. Time to first seizure Time to 24‐month remission Serious adverse events attributed to the trial medication or other AED Quality of life as assessed by the QOLIE31 and QOLIE48 questionnaires |
| Starting date | May 2015 |
| Contact information | Dr Peter Bergin (pbergin@adhb.govt.nz) |
| Notes | Trial is registered as ACTRN12615000640505 on the Australian New Zealand Clinical Trials Registry and is listed as currently recruiting participants (correct to August 2016) Estimated finish date is May 2018 |
| Trial name or title | EpiNet‐First Trial 5: Comparison of efficacy of levetiracetam and lamotrigine in people with previously untreated epilepsy who have unclassified seizures, and for whom sodium valproate is not deemed an acceptable AED |
| Methods | Phase 4 randomised, open‐label, pragmatic trial conducted across sites in New Zealand and Europe 2 treatment arms: LEV and LTG |
| Participants | Individuals > 5 years with ≥ 2 spontaneous generalised seizures that require AED treatment (provided all seizures have not been absence seizures) Target sample size = 664 |
| Interventions | Monotherapy with LEV or LTG Target doses LEV: 250 mg‐4000 mg, LTG: 250 mg‐400 mg |
| Outcomes | Time to 12‐month remission from seizures Proportion of participants who achieve a seizure‐free 12‐month remission by 18 months AND who have not changed to a different AED Time to treatment failure due to either inadequate seizure control, or due to unacceptable adverse events Time to treatment failure due to inadequate seizure control. Time to treatment failure due to unacceptable adverse events. Time to first seizure Time to 24‐month remission Serious adverse events attributed to the trial medication or other antiepileptic medication Quality of life as assessed by the QOLIE31 and QOLIE48 questionnaires |
| Starting date | May 2015 |
| Contact information | Dr Peter Bergin (pbergin@adhb.govt.nz) |
| Notes | Trial is registered as ACTRN12615000641594 on the Australian New Zealand Clinical Trials Registry and is listed as currently recruiting participants (correct to August 2016) . Estimated finish date is May 2018 |
| Trial name or title | EpiNet‐First Trial 1: Comparison of efficacy of levetiracetam, lamotrigine and carbamazepine in people with previously untreated epilepsy who have focal seizures |
| Methods | Phase 4 randomised, open‐label, pragmatic trial conducted across sites in New Zealand and Europe 3 treatment arms: CBZ, LEV and LTG |
| Participants | Individuals > 5 years with ≥ 2 spontaneous generalised seizures that require AED treatment (provided all seizures have not been absence seizures) Target sample size = 1467 |
| Interventions | Monotherapy with CBZ, LEV or LTG Target doses CBZ: 250 mg‐4000 mg, LEV: 250 mg‐4000 mg, LTG: 250 mg‐400 mg |
| Outcomes | Time to 12‐month remission from seizures Proportion of participants who achieve a seizure‐free 12‐month remission by 18 months AND who have not changed to a different AED Time to treatment failure due to either inadequate seizure control, or due to unacceptable adverse events Time to treatment failure due to inadequate seizure control. Time to treatment failure due to unacceptable adverse events. Time to first seizure Time to 24‐month remission Serious adverse events attributed to the trial medication or other antiepileptic medication Quality of life as assessed by the QOLIE31 and QOLIE48 questionnaires |
| Starting date | May 2015 |
| Contact information | Dr Peter Bergin (pbergin@adhb.govt.nz) |
| Notes | Trial is registered as ACTRN12615000643572 on the Australian New Zealand Clinical Trials Registry and is listed as currently recruiting participants (correct to August 2016) Estimated finish date is May 2018 |
| Trial name or title | Cognitive AED outcomes in pediatric localization related epilepsy (COPE) |
| Methods | Phase 3 randomised, single‐blinded (outcome assessor) trial conducted at 12 sites in the USA 3 treatment arms: LEV, LTG and OXC |
| Participants | Children aged 5‐16 at the time of enrolment with localisation‐related partial epilepsy with or without secondary generalised Participants must be AED naive or have had less than a weeks' exposure to AEDs Estimated enrolment = 300 |
| Interventions | Monotherapy with LEV, LTG or OXC Assessments made at 3 and 6 months |
| Outcomes | Conners' Continuous Performance Test (CPT) Confidence Interval Child Behavior Checklist Weschler Intelligence Scale for Children‐IV Processing Speed Story Memory Symbol Digit Modalities Test Grooved Pegboard Columbia Suicidality Severity Rating Scale Youth Self Report Affective Reactivity Scale Pediatric Neuro‐QOL Parenting Stress Index Pediatric Inventory for Parents |
| Starting date | August 2013 |
| Contact information | Emory University |
| Notes | Trial registered as NCT01891890 on ClincalTrials.gov and listed as ongoing but not recruiting participants (correct to August 2016). Estimated finishing date is April 2017. |
| Trial name or title | A study to investigate the safety of the drugs topiramate and levetiracetam in treating children recently diagnosed with epilepsy |
| Methods | Phase 3, randomised, open‐label, parallel‐group trial conducted in multiple centres in the USA, South America, Asia and Europe 2 treatment arms: LEV and TPM |
| Participants | Participants with a clinical diagnosis of new‐onset or recent‐onset epilepsy characterised by partial‐onset seizures (with or without secondary generalisation) or primary generalised tonic‐clonic seizures with no previous treatment for epilepsy (except emergency treatment) Estimated enrolment = 282 |
| Interventions | Monotherapy with LEV or TPM Maximum recommended doses: LEV = 3000 mg/d, TPM = 400 mg/d |
| Outcomes | Percentage of participants with kidney stones Change from baseline in weight Z‐score at month 12 Change from baseline in height at month 12 Change from baseline in bone mineral density (BMD) at Month 12 (other measures of weight, height and bone density specified on trial registration page) |
| Starting date | October 2014 |
| Contact information | Janssen Research & Development |
| Notes | Trial registered as NCT012201251 on ClincalTrials.gov and listed as currently recruiting participants (correct to August 2016). Estimated finishing date is March 2018. |
Abbreviations: AED: antiepileptic drug; CBZ: carbamazepine; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; VPS: sodium valproate; TPM: topiramate
Contributions of authors
SJN wrote the protocol under the supervision of AGM and CT. MS and JW commented on drafts of the protocol.
SJN and AGM screened all studies for inclusion in the review. SJN and JW performed independent risk of bias assessments on all included trials.
SJN, CTS and AGM requested all individual participant data
SJN and MS prepared individual participant data for analysis, SJN conducted analyses of the review and interpreted results under the supervision of CTS (statistical interpretation) and AGM (clinical interpretation).
SJN wrote the text of the review with the input of MS, JW, CTS and AGM.
Sources of support
Internal sources
-
National Institute of Health Research, UK.
This review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Epilepsy. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
External sources
No sources of support supplied
Declarations of interest
SJN was funded between 2011 and 2014 as part of a three‐year research programme, ‘Clinical and cost effectiveness of interventions for epilepsy in the National Health Service (NHS)’, which receives financial support from the National Institute of Health Research (NIHR).
JW was funded between 2011 and 2014 as part of a three‐year research programme, ‘Clinical and cost effectiveness of interventions for epilepsy in the National Health Service (NHS)’, which receives financial support from the National Institute of Health Research (NIHR).
AGM: a consortium of pharmaceutical companies (GSK, EISAI, UCB Pharma) funded the National Audit of Seizure Management in Hospitals (NASH) through grants paid to the University of Liverpool. Professor Tony Marson is Theme Leader for Managing Complex Needs at NIHR CLAHRC NWC.
Notes
Sarah J Nolan (author of the protocol) is now Sarah J Nevitt
New
References
References to studies included in this review
- Aikia M, Kalviainen R, Sivenius J, Halonen T, Riekkinen PJ. Cognitive effects of oxcarbazepine and phenytoin monotherapy in newly diagnosed epilepsy: one year follow‐up. Epilepsy Research 1992;11(3):199‐203. [DOI] [PubMed] [Google Scholar]
- Banu SH, Jahan M, Koli UK, Ferdousi S, Khan NZ, Neville B. Side effects of phenobarbital and carbamazepine in childhood epilepsy: randomised controlled trial. BMJ 2007;334(7605):1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulac M, Brodie MJ, Patten A, Segieth J, Giorgi L. Efficacy and tolerability of zonisamide versus controlled‐release carbamazepine for newly diagnosed partial epilepsy: a phase 3, randomised, double‐blind, non‐inferiority trial. Lancet Neurology 2012;11(7):579‐88. [DOI] [PubMed] [Google Scholar]
- Bidabadi E. Comparison of the effects of phenobarbital versus carbamazepine as single drug therapy in partial seizure with secondary generalization in children. Epilepsia 2009;50(Suppl 10):167. Abstract no: p772 28th International Epilepsy Congress; Budapest 2009. [Google Scholar]
- Bill PA, Vigonius U, Pohlmann H, Guerreiro CA, Kochen S, Saffer D, et al. A double‐blind controlled clinical trial of oxcarbazepine versus phenytoin in adults with previously untreated epilepsy. Epilepsy Research 1997;27(3):195‐204. [DOI] [PubMed] [Google Scholar]
- Biton V, Levisohn P, Hoyler S, Vuong A, Hammer AE. Lamotrigine versus valproate monotherapy‐associated weight change in adolescents with epilepsy: results from a post hoc analysis of a randomized, double‐blind clinical trial. Journal of Child Neurology 2003;18(2):133‐9. [DOI] [PubMed] [Google Scholar]; Biton V, Mirza W, Montouris G, Vuong A, Hammer AE, Barrett PS. Weight change associated with valproate and lamotrigine monotherapy in patients with epilepsy. Neurology 2001;56(2):172‐7. [DOI] [PubMed] [Google Scholar]; Edwards KR, Sackellares JC, Vuong A, Hammer AE, Barrett PS. Lamotrigine monotherapy improves depressive symptoms in epilepsy: a double‐blind comparison with valproate. Epilepsy & Behavior 2001;2(1):28‐36. [DOI] [PubMed] [Google Scholar]; Sackellares JC, Kwong WJ, Vuong A, Hammer AE, Barrett PS. Lamotrigine monotherapy improves health‐related quality of life in epilepsy: a double‐blind comparison with valproate. Epilepsy & Behavior 2002;3(4):376‐82. [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Richens A, Yuen AW. Double‐blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. UK Lamotrigine/Carbamazepine Monotherapy Trial Group. Lancet 1995;345(8948):476‐9. [DOI] [PubMed] [Google Scholar]; Gillham R, Kane K, Bryant‐Comstock L, Brodie MJ. A double‐blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy with health‐related quality of life as an outcome measure. Seizure 2000;9(6):375‐9. [DOI] [PubMed] [Google Scholar]; Severi S, Muscas GC, Bianchi A, Zolo P. Efficacy and safety of lamotrigine monotherapy in partial epilepsy. Bollettino ‐ Lega Italiana Contro L'Epilessia 1994;86‐7:149‐51. [Google Scholar]
- Brodie MJ, Richens A, Yuen AW. Double‐blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. UK Lamotrigine/Carbamazepine Monotherapy Trial Group. Lancet 1995;345(8948):476‐9. [DOI] [PubMed] [Google Scholar]; Gillham R, Kane K, Bryant‐Comstock L, Brodie MJ. A double‐blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy with health‐related quality of life as an outcome measure. Seizure 2000;9(6):375‐9. [DOI] [PubMed] [Google Scholar]; Severi S, Muscas GC, Bianchi A, Zolo P. Efficacy and safety of Lamotrigine monotherapy in partial epilepsy. Bollettino ‐ Lega Italiana Contro L'Epilessia 1994;86‐7:149‐51. [Google Scholar]
- Brodie MJ, Overstall PW, Giorgi L. Multicentre, double‐blind, randomised comparison between lamotrigine and carbamazepine in elderly patients with newly diagnosed epilepsy: The UK Lamotrigine Elderly Study Group. Epilepsy Research 1999;37(1):81‐7. [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Chadwick DW, Anhut H, Otte A, Messmer S‐L, Maton S, et al. Gabapentin Study Group. Gabapentin versus lamotrigine monotherapy: a double‐blind comparison in newly diagnosed epilepsy. Epilepsia 2002;43(9):993‐1000. [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Perucca E, Ryvlin P, Ben‐Menachem E, Meencke HJ, Levetiracetam Monotherapy Study Group. Comparison of levetiracetam and controlled‐release carbamazepine in newly diagnosed epilepsy. Neurology 2007;68(6):402‐8. [DOI] [PubMed] [Google Scholar]
- Callaghan N, Kenny RA, O'Neill B, Crowley M, Goggin T. A prospective study between carbamazepine, phenytoin and sodium valproate as monotherapy in previously untreated and recently diagnosed patients with epilepsy. Journal of Neurology, Neurosurgery & Psychiatry 1985;48(7):639‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]; Goggin T, Casey C, Callaghan N. Serum levels of sodium valproate, phenytoin and carbamazepine and seizure control in epilepsy. Irish Medical Journal 1986;79(6):150‐6. [PubMed] [Google Scholar]
- Capone L, Carrieri L, Centrone G, Olivieri G, Ragno G, Roca M, et al. Randomised open trial of 35 subjects with epileptic seizures after stroke, treated with levetiracetam or carbamazepine CR. Bollettino ‐ Lega Italiana Contro L'Epilessia 2008;138:203‐6. [Google Scholar]
- Castriota O, Guido M, Goffredo R, Claudio T, Specchio LM. Event‐related potentials (ERPS) in the evaluation of the effect of levetiracetam and carbamazepine on cognitive functions in adult newly diagnosed epileptic patients. Preliminary results of an opened randomised trial. Bollettino ‐ Lega Italiana Contro L'Epilessia 2008;138:235‐7. [Google Scholar]
- Chadwick DW, Anhut H, Greiner MJ, Alexander J, Murray GH, Garofalo EA, et al. A double‐blind trial of gabapentin monotherapy for newly diagnosed partial seizures. International Gabapentin Monotherapy Study Group 945‐77. Neurology 1998;51(5):1282‐8. [DOI] [PubMed] [Google Scholar]; Sasanelli F, Amodeo M, Colombo A, Molini GE. Gabapentin versus carbamazepine: monotherapy in newly‐diagnosed patients with partial epilepsy: preliminary results. Bollettino ‐ Lega Italiana Contro L'Epilessia 1996;95‐96:185‐6. [Google Scholar]
- Chen YJ, Kang WM, So WC. Comparison of antiepileptic drugs on cognitive function in newly diagnosed epileptic children: a psychometric and neurophysiological study. Epilepsia 1996;37(1):81‐6. [DOI] [PubMed] [Google Scholar]
- Cho YW, Kim DH, Motamedi GK. The effect of levetiracetam monotherapy on subjective sleep quality and objective sleep parameters in patients with epilepsy: compared with the effect of carbamazepine‐CR monotherapy. Seizure 2011;20(4):336‐9. [DOI] [PubMed] [Google Scholar]
- Christe W, Kramer G, Vigonius U, Pohlmann H, Steinhoff BJ, Brodie MJ, et al. A double‐blind controlled clinical trial: oxcarbazepine versus sodium valproate in adults with newly diagnosed epilepsy. Epilepsy Research 1997;26(3):451‐60. [DOI] [PubMed] [Google Scholar]
- Consoli D, Bosco D, Postorino P, Galati F, Plastino M, Perticoni GF, et al. EpIC Study. Levetiracetam versus carbamazepine in patients with late poststroke seizures: a multicenter prospective randomized open‐label study (EpIC Project).. Cerebrovascular Diseases 2012;34(4):282‐9. [DOI] [PubMed] [Google Scholar]
- Cossu G, Monaco F, Piras MR, Grossi E. Short‐term therapy with carbamazepine and phenobarbital: effects on cognitive functioning in temporal lobe epilepsy. Bollettino Lega Italiana contro l'Epilessia 1984;45/46:377‐9. [Google Scholar]
- Craig I, Tallis R. Impact of valproate and phenytoin on cognitive function in elderly patients: results of a single‐blind randomized comparative study. Epilepsia 1994;35(2):381‐90. [DOI] [PubMed] [Google Scholar]; Tallis R, Easter D. Multicenter comparative trial of valproate and phenytoin. Epilepsia 1994;35(Suppl 7):62. [DOI] [PubMed] [Google Scholar]
- Czapinski P, Terczynski A. Open randomized comparative trial of sodium valproate and carbamazepine in adult onset epilepsy. Neurologia i Neurochirurgia Polska 1996;30(3):419‐26. [PubMed] [Google Scholar]; Czapinski P, Terczynski A, Czapinska E. Randomised 36‐month comparative study of valproic acid, phenytoin, phenobarbital and carbamazepine efficacy in patients with newly diagnosed epilepsy with partial complex seizures. Epilepsia 1997;38(Suppl(3)):42. [Google Scholar]
- Dam M, Ekberg R, Loyning Y, Waltimo O, Jakobsen K. A double‐blind study comparing oxcarbazepine and carbamazepine in patients with newly diagnosed, previously untreated epilepsy. Epilepsy Research 1989;3(1):70‐6. [DOI] [PubMed] [Google Scholar]
- Silva M, MacArdle B, McGowan M, Hughes E, Stewart J, Neville BG, et al. Randomised comparative monotherapy trial of phenobarbitone, phenytoin, carbamazepine, or sodium valproate for newly diagnosed childhood epilepsy. Lancet 1996;347(9003):709‐13. [DOI] [PubMed] [Google Scholar]
- Dizdarer G, Kangin M, Sutcuoglu S, Ozmen Y, Yaprak I. A comparison of carbamazepine and oxcarbazepine in partial epilepsies. Epilepsia 2000;41(Suppl Florence):67‐8. [Google Scholar]
- Donati F, Gobbi G, Campistol J, Rapatz G, Daehler M, Sturm Y, et al. Oxcarbazepine Cognitive Study Group. The cognitive effects of oxcarbazepine versus carbamazepine or valproate in newly diagnosed children with partial seizures. Seizure 2007;16(8):670‐9. [DOI] [PubMed] [Google Scholar]
- Eun S‐H, Eun B‐L, Lee JS, Hwang YS, Kim KJ, Lee Y‐M, et al. Effects of lamotrigine on cognition and behavior compared to carbamazepine as monotherapy for children with partial epilepsy. Brain & Development 2012;34(10):818‐23. [DOI] [PubMed] [Google Scholar]
- Feksi AT, Kaamugisha J, Sander JW, Gatiti S, Shorvon SD. Comprehensive primary health care antiepileptic drug treatment programme in rural and semi‐urban Kenya. ICBERG (International Community‐based Epilepsy Research Group). Lancet 1991;337(8738):406‐9. [DOI] [PubMed] [Google Scholar]
- Berg I, Butler A, Ellis M, Foster J. Psychiatric aspects of epilepsy in childhood treated with carbamazepine, phenytoin or sodium valproate: a random trial. Developmental Medicine & Child Neurology 1993;35(2):149‐57. [DOI] [PubMed] [Google Scholar]; Forsythe I, Butler R, Berg I, McGuire R. Cognitive impairment in new cases of epilepsy randomly assigned to carbamazepine, phenytoin and sodium valproate. Developmental Medicine & Child Neurology 1991;33(6):524‐34. [DOI] [PubMed] [Google Scholar]
- Fritz N, Elger C, Helmstaedter C. Effects of lamotrigine and oxcarbazepine on seizures, cognition, mood and health‐related quality of life in patients with untreated epilepsy. Epilepsia 2006;47(Suppl 3):157. [Google Scholar]
- Gilad R, Sadeh M, Rapoport A, Dabby R, Boaz M, Lampl Y. Monotherapy of lamotrigine versus carbamazepine in patients with poststroke seizure. Clinical Neuropharmacology 2007;30(4):189‐95. [DOI] [PubMed] [Google Scholar]
- Guerreiro MM, Vigonius U, Pohlmann H, Manreza ML, Fejerman N, Antoniuk SA, et al. A double‐blind controlled clinical trial of oxcarbazepine versus phenytoin in children and adolescents with epilepsy. Epilepsy Research 1997;27(3):205‐13. [DOI] [PubMed] [Google Scholar]
- Heller AJ, Chesterman P, Elwes RD, Crawford P, Chadwick D, Johnson AL, et al. Phenobarbitone, phenytoin, carbamazepine, or sodium valproate for newly diagnosed adult epilepsy: a randomised comparative monotherapy trial. Journal of Neurology, Neurosurgery & Psychiatry 1995;58(1):44‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung DE, Yu R, Yoon JR, Eun BL, Kwon SH, Lee YJ, et al. Neuropsychological effects of levetiracetam and carbamazepine in children with focal epilepsy. Neurology 2015;84(23):2312‐9. [DOI] [PubMed] [Google Scholar]
- Kalviainen R, Tigaran S, Keranen T, Peltola J, Aikia M. Open, multicenter, randomised comparison of cognitive effects of lamotrigine and slow‐release carbamazepine monotherapy in newly diagnosed patients with epilepsy. Epilepsia 2002;43(Suppl 8):47. 12121295 [Google Scholar]
- Kopp UA, Gaus V, Wandschneider B, Schmitz B. Cognitive side effects of levetiracetam in monotherapy: comparison with carbamazepine and valproate. Epilepsia 2007;48(Suppl 3):39‐40, Abstract No: P178. [Google Scholar]
- Korean Lamotrigine Study Group. An open, randomized, multicenter comparative clinical trial of lamotrigine and carbamazepine as initial monotherapy in previously untreated epilepsies. Journal of Korean Epilepsy Society 2008;12(1):27‐34. [Google Scholar]
- Kwan P, Yip FP, Hui ACF, Leung H, Ng PW, Hui KF, et al. Effects of valproate or lamotrigine monotherapy on the reproductive endocrine and insulin‐related metabolic profile in Chinese adults with epilepsy: a prospective randomized study. Epilepsy & Behavior 2009;14(4):610‐6. [DOI] [PubMed] [Google Scholar]
- Ju KM, Lee SA, Lee HW, Heo K, Shin DJ, Song HK, et al. Effect of seizures on cognition, behavior, and quality of life during carbamazepine or lamotrigine monotherapy in patients with newly diagnosed partial epilepsy. Epilepsy currents / American Epilepsy Society 2011;11(Suppl 1):399, Abstract no: 3.090. [Google Scholar]; Lee S‐A, Lee H‐W, Heo K, Shin D‐J, Song H‐K, Kim O‐J, et al. Cognitive and behavioral effects of lamotrigine and carbamazepine monotherapy in patients with newly diagnosed or untreated partial epilepsy. Seizure 2011;20(1):49‐54. [DOI] [PubMed] [Google Scholar]; Lee SA, Kim MJ, Lee HW, Heo K, Shin DJ, Song HK, et al. The effect of recurrent seizures on cognitive, behavioral, and quality‐of‐life outcomes after 12 months of monotherapy in adults with newly diagnosed or previously untreated partial epilepsy. Epilepsy & Behavior 2015;53:202‐8. [DOI] [PubMed] [Google Scholar]
- Lukic SR, Spasic MJ, Lukic NR. Comparison of valproate and lamotrigine for the treatment of newly diagnosed epilepsy: interim report. Epilepsia 2005;46(Suppl 6):278. [Google Scholar]
- Mattson RH, Cramer JA, Collins JF, Smith DB, Delgado‐Escueta AV, Browne TR, et al. Comparison of carbamazepine, phenobarbital, phenytoin, and primidone in partial and secondarily generalized tonic‐clonic seizures. New England Journal of Medicine 1985;313(3):145‐51. [DOI] [PubMed] [Google Scholar]
- Mattson RH, Cramer JA, Collins JF. A comparison of valproate with carbamazepine for the treatment of complex partial seizures and secondarily generalized tonic‐clonic seizures in adults. The Department of Veterans Affairs Epilepsy Cooperative Study No. 264 Group. New England Journal of Medicine 1992;327(11):765‐71. [DOI] [PubMed] [Google Scholar]
- Mitchell WG, Chavez JM. Carbamazepine versus phenobarbital for partial onset seizures in children. Epilepsia 1987;28(1):56‐60. [DOI] [PubMed] [Google Scholar]
- Miura H. Aspects of antiepileptic drug therapy in children. No to Hattatsu [Brain & Development] 1990;22(2):154‐9. [PubMed] [Google Scholar]
- Motamedi M, Ghini MR, Etemadi P, Ramim T. Levetiracetam and lamotrigine in old aged onset of epilepsy: a randomized double‐blind clinical trial. Tehran University Medical Journal 2013;71(9):568‐76. [Google Scholar]
- NCT01498822. Levetiracetam versus oxcarbazepine as monotherapy to evaluate efficacy and safety in subjects with partial epilepsy. ClinicalTrials.gov (accessed 23 August 2016).
- NCT01954121. Open‐label, randomized, active‐controlled study of LEV used as monotherapy in patients with partial‐onset seizures. ClinicalTrials.gov (accessed 23 August 2016).
- Nieto‐Barrera M, Brozmanova M, Capovilla G, Christe W, Pedersen B, Kane K, et al. Lamictal vs. Carbamazepine Study Group. A comparison of monotherapy with lamotrigine or carbamazepine in patients with newly diagnosed partial epilepsy. Epilepsy Research 2001;46(2):145‐55. [DOI] [PubMed] [Google Scholar]
- Ogunrin O, Adamolekun B, Ogunniyi A. Cognitive effects of anti‐epileptic drugs in Nigerians with epilepsy. African Journal of Neurological Sciences 2005;24(1):18‐24. [Google Scholar]
- Pal DK, Das T, Chaudhury G, Johnson AL, Neville BG. Randomised controlled trial to assess acceptability of phenobarbital for childhood epilepsy in rural India. Lancet 1998;351(9095):19‐23. [DOI] [PubMed] [Google Scholar]
- Placencia M, Sander JW, Shorvon SD, Roman M, Alarcon F, Bimos C, et al. Antiepileptic drug treatment in a community health care setting in northern Ecuador: a prospective 12‐month assessment. Epilepsy Research 1993;14(3):237‐44. [DOI] [PubMed] [Google Scholar]
- Privitera MD, Brodie MJ, Mattson RH, Chadwick DW, Neto W, Wang S, EPMN study group. Topiramate, carbamazepine and valproate monotherapy: double‐blind comparison in newly diagnosed epilepsy. Acta Neurologica Scandinavica 2003;107(3):165‐75. [DOI] [PubMed] [Google Scholar]; Wheless JW, Neto W, Wang S, EPMN study group. Topiramate, carbamazepine, and valproate monotherapy: double‐blind comparison in children with newly diagnosed epilepsy. Journal of Child Neurology 2004;19(2):135‐41. [DOI] [PubMed] [Google Scholar]
- Pulliainen V, Jokelainen M. Effects of phenytoin and carbamazepine on cognitive functions in newly diagnosed epileptic patients. Acta Neurologica Scandinavica 1994;89(2):81‐6. [DOI] [PubMed] [Google Scholar]
- Ramsay RE, Wilder BJ, Berger JR, Bruni J. A double‐blind study comparing carbamazepine with phenytoin as initial seizure therapy in adults. Neurology 1983;33(7):904‐10. [DOI] [PubMed] [Google Scholar]
- Ramsay RE, Wilder BJ, Murphy JV, Holmes GL, Uthman B, Slater J, et al. Efficacy and safety of valproic acid versus phenytoin as sole therapy for newly diagnosed primary generalized tonic‐clonic seizures. Journal of Epilepsy 1992;5(1):55‐60. [Google Scholar]; Wilder BJ, Ramsay RE, Murphy JV, Karas BJ, Marquardt K, Hammond EJ. Comparison of valproic acid and phenytoin in newly diagnosed tonic‐clonic seizures. Neurology 1983;33(11):1474‐6. [DOI] [PubMed] [Google Scholar]
- Ramsay T, Bainbridge J, Fredericks T, Slater J, Nemire R, Ramsay RE. Results of a randomized double‐blind comparison of levetiracetam and carbamazepine in new onset seizures in a geriatric population. Epilepsia 2007;48(Suppl 6):36‐7. 18047600 [Google Scholar]
- Ramsay E, Faught E, Krumholz A, Naritoku D, Privitera M, Schwarzman L, et al. Capss Study Group. Efficacy, tolerability, and safety of rapid initiation of topiramate versus phenytoin in patients with new‐onset epilepsy: a randomized double‐blind clinical trial. Epilepsia 2010;51(10):1970‐7. [DOI] [PubMed] [Google Scholar]
- Rastogi P, Mehrotra TN, Agarwala RK, Singh VS. Comparison of sodium valproate and phenytoin as single drug treatment in generalised and partial epilepsy. Journal of the Association of Physicians of India 1991;39(8):606‐8. [PubMed] [Google Scholar]
- Ravi Sudhir RV, Sawhney IMS, Prabhakar S, Pershad D, Nain CK. Comparative cognitive effects of phenytoin and carbamazepine in adult epileptics. Neurology India 1995;43(4):186‐92. [PubMed] [Google Scholar]
- Resendiz‐Aparicio JC, Rodriguez‐Rodriguez E, Contreras‐Bernal J, Ceja‐Moreno H, Barragan‐Perez E, Garza‐Morales S, et al. A randomised open trial comparing monotherapy with topiramate versus carbamazepine in the treatment of paediatric patients with recently diagnosed epilepsy. Revista de Neurologia 2004;39(3):201‐4. [PubMed] [Google Scholar]
- Reunanen M, Dam M, Yuen AW. A randomised open multicentre comparative trial of lamotrigine and carbamazepine as monotherapy in patients with newly diagnosed or recurrent epilepsy. Epilepsy Research 1996;23(2):149‐55. [DOI] [PubMed] [Google Scholar]
- Richens A, Davidson DL, Cartlidge NE, Easter DJ. A multicentre comparative trial of sodium valproate and carbamazepine in adult onset epilepsy. Adult EPITEG Collaborative Group. Journal of Neurology, Neurosurgery & Psychiatry 1994;57(6):682‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan AJ, Ramsay RE, Collins JF, Pryor F, Boardman KD, Uthman BM, et al. Cooperative Study 428 Group. New onset geriatric epilepsy: a randomized study of gabapentin, lamotrigine, and carbamazepine. Neurology 2005;64(11):1868‐73. [DOI] [PubMed] [Google Scholar]
- Saetre E, Perucca E, Isojarvi J, Gjerstad L, Group LAMS. An international multicenter randomized double‐blind controlled trial of lamotrigine and sustained‐release carbamazepine in the treatment of newly diagnosed epilepsy in the elderly. Epilepsia 2007;48(7):1292‐302. [DOI] [PubMed] [Google Scholar]
- Marson AG, Al‐Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW, et al. SANAD Study Group. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet 2007;369(9566):1000‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson AG, Al‐Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW, et al. SANAD Study Group. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy: an unblinded randomised controlled trial. Lancet 2007;369(9566):1016‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakir RA, Johnson RH, Lambie DG, Melville ID, Nanda RN. Comparison of sodium valproate and phenytoin as single drug treatment in epilepsy. Epilepsia 1981;22(1):27‐33. [DOI] [PubMed] [Google Scholar]
- So EL, Lai CW, Pellock J, Mashman J, Brugger A. Safety and efficacy of valproate and carbamazepine in the treatment of complex partial seizures. Journal of Epilepsy 1992;5(3):149‐52. [Google Scholar]
- Steiner TJ, Dellaportas CI, Findley LJ, Gross M, Gibberd FB, Perkin GD, et al. Lamotrigine monotherapy in newly diagnosed untreated epilepsy: a double‐blind comparison with phenytoin. Epilepsia 1999;40(5):601‐7. [DOI] [PubMed] [Google Scholar]
- Steinhoff BJ, Ueberall MA, Siemes H, Kurlemann G, Schmitz B, Bergmann L, LAM Safe Study Group. The LAM‐SAFE Study: lamotrigine versus carbamazepine or valproic acid in newly diagnosed focal and generalised epilepsies in adolescents and adults. Seizure 2005;14(8):597‐605. [DOI] [PubMed] [Google Scholar]
- Stephen LJ, Sills GJ, Leach JP, Butler E, Parker P, Hitiris N, et al. Sodium valproate versus lamotrigine: a randomised comparison of efficacy, tolerability and effects on circulating androgenic hormones in newly diagnosed epilepsy. Epilepsy Research 2007;75(2‐3):122‐9. [DOI] [PubMed] [Google Scholar]
- Suresh SH, Chakraborty A, Virupakshaiah A, Kumar N. Efficacy and safety of levetiracetam and carbamazepine as monotherapy in partial seizures. Epilepsy research and treatment 2015;2015:Article ID 415082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thilothammal N, Banu K, Ratnam RS. Comparison of phenobarbitone, phenytoin with sodium valproate: randomized, double‐blind study. Indian Pediatrics 1996;33(7):549‐55. [PubMed] [Google Scholar]
- Trinka E, Marson AG, Paesschen W, Kälviäinen R, Marovac J, Duncan B, et al. KOMET: an unblinded, randomised, two parallel‐group, stratified trial comparing the effectiveness of levetiracetam with controlled‐release carbamazepine and extended‐release sodium valproate as monotherapy in patients with newly diagnosed epilepsy. Journal of Neurology, Neurosurgery and Psychiatry 2013;84(10):1138‐47. [DOI] [PubMed] [Google Scholar]
- Turnbull DM, Howel D, Rawlins MD, Chadwick DW. Which drug for the adult epileptic patient: phenytoin or valproate?. British Medical Journal 1985;290(6471):815‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]; Turnbull DM, Rawlins MD, Weightman D, Chadwick DW. A comparison of phenytoin and valproate in previously untreated adult epileptic patients. Journal of Neurology, Neurosurgery & Psychiatry 1982;45(1):55‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easter D, O'Bryan‐Tear CG, Verity C. Weight gain with valproate or carbamazepine ‐ a reappraisal. Seizure 1997;6(2):121‐5. [DOI] [PubMed] [Google Scholar]; Verity CM, Hosking G, Easter DJ. A multicentre comparative trial of sodium valproate and carbamazepine in paediatric epilepsy. The Paediatric EPITEG Collaborative Group. Developmental Medicine & Child Neurology 1995;37(2):97‐108. [DOI] [PubMed] [Google Scholar]
- Werhahn KJ, Trinka E, Dobesberger J, Ruckes C, Krämer G. Levetiracetam is superior to carbamazepine‐SR in newly diagnosed epilepsy in the elderly: results of the step‐one trial. Epilepsia 2012;53 Suppl. 5:49, Abstract no: p164. [Google Scholar]; Werhahn KJ, Trinka E, Dobesberger J, Unterberger I, Baum P, Deckert‐Schmitz M, et al. A randomized, double‐blind comparison of antiepileptic drug treatment in the elderly with new‐onset focal epilepsy. Epilepsia 2015;56(3):450‐9. [DOI] [PubMed] [Google Scholar]; Wild I, Noack‐Rink M, Ramirez F, Tofighy A, Werhahn K. Comparative effectiveness of the antiepileptic drugs (AEDs) levetiracetam, valproate and carbamazepine among patients aged 60 years and over with newly diagnosed epilepsy. Journal of Neurology 2014;261(Suppl 1):S159‐S60, Abstract no: EP2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
- Albani F, Baruzzi A, Primo Study Group. Oxcarbazepine long‐term treatment retention in patients switched over from carbamazepine. Neurological Sciences 2006;27(3):173‐5. [DOI] [PubMed] [Google Scholar]
- Alsaadi TM, Thieman C, Zusman EE. Levetiracetam monotherapy for adults with localization‐related epilepsy. Epilepsy and Behavior 2002;3(5):471‐4. [DOI] [PubMed] [Google Scholar]
- Alsaadi TM, Shatzel A, Marquez AV, Jorgensen J, Farias S. Clinical experience of levetiracetam monotherapy for adults with epilepsy: 1‐year follow‐up study. Seizure 2005;14(2):139‐42. [DOI] [PubMed] [Google Scholar]
- Baxter L, Cheesbrough A. An open randomised comparison of Lamictal (lamotrigine) with physicians preferred choice of either valproate or carbamazepine as monotherapy in patients over 12 years of age with newly diagnosed epilepsy. Clinical summary report1998.
- Ben‐Menachem E. Preliminary efficacy of levetiracetam in monotherapy. Epileptic Disorders 2003;5(Suppl 1):S51‐5. [PubMed] [Google Scholar]
- Beydoun A, Fischer J, Labar DR, Harden C, Cantrell D, Uthman BM, et al. Gabapentin monotherapy: II. A 26‐week, double‐blind, dose‐controlled, multicenter study of conversion from polytherapy in outpatients with refractory complex partial or secondarily generalized seizures. The US Gabapentin Study Group 82/83. Neurology 1997;49(3):746‐52. [DOI] [PubMed] [Google Scholar]
- Beydoun A, Fakhoury T, Nasreddine W, Abou‐Khalil B. Conversion to high dose gabapentin monotherapy in patients with medically refractory partial epilepsy. Epilepsia 1998;39(2):188‐93. [DOI] [PubMed] [Google Scholar]
- Beydoun A, Sachdeo RC, Rosenfeld WE, Krauss GL, Sessler N, Mesenbrink P, et al. Oxcarbazepine monotherapy for partial‐onset seizures: a multicenter, double‐blind, clinical trial. Neurology 2000;54(12):2245‐51. [DOI] [PubMed] [Google Scholar]
- Bittencourt PR, Antoniuk SA, Bigarella MM, Costa JC, Doro MP, Ferreira AS, et al. Carbamazepine and phenytoin in epilepsies refractory to barbiturates: efficacy, toxicity and mental function. Epilepsy Research 1993;16(2):147‐55. [DOI] [PubMed] [Google Scholar]
- Bawden HN, Camfield CS, Camfield PR, Cunningham C, Darwish H, Dooley JM, et al. The cognitive and behavioural effects of clobazam and standard monotherapy are comparable. Canadian Study Group for Childhood Epilepsy. Epilepsy Research 1999;33(2‐3):133‐43. [DOI] [PubMed] [Google Scholar]
- Cereghino JJ, Brock JT, Meter JC, Penry JK, Smith LD, White BG. Carbamazepine for epilepsy. A controlled prospective evaluation. Neurology 1974;24(5):401‐10. [DOI] [PubMed] [Google Scholar]
- Chung S, Ceja H, Gawlowicz J, Avakyan G, McShea C, Schiemann J, et al. Levetiracetam extended release conversion to monotherapy for the treatment of patients with partial‐onset seizures: a double‐blind, randomised, multicentre, historical control study. Epilepsy Research 2012;101(1‐2):92‐102. [DOI] [PubMed] [Google Scholar]
- DeToledo JC, Ramsay RE, Lowe MR, Greiner M, Garofalo EA. Increased seizures after discontinuing carbamazepine: results from the gabapentin monotherapy trial. Therapeutic Drug Monitoring 2000;22(6):753‐6. [DOI] [PubMed] [Google Scholar]
- EUCTR2004‐004053‐26‐SE. An explorative use open‐label, multi‐center, randomized trial studying the safety and efficacy of levetiracetam (500 mg/day to 3000 mg/day) and valproate (600 mg/day to 3000 mg/day) as monotherapy in newly diagnosed patients over the age of 65 years ‐ Scandinavian elderly study. www.clinicaltrialsregister.eu (accessed 23 August 2016).
- EUCTR2010‐018284‐42‐NL. A trial comparing efficacy, safety and tolerance between levetiracetam and valproic acid in children with epilepsy. www.clinicaltrialsregister.eu (accessed 11 September 2015).
- Fakhoury TA, Hammer AE, Vuong A, Messenheimer JA. Efficacy and tolerability of conversion to monotherapy with lamotrigine compared with valproate and carbamazepine in patients with epilepsy. Epilepsy & Behavior 2004;5(4):532‐8. [DOI] [PubMed] [Google Scholar]
- French JA, Temkin NR, Shneker BF, Hammer AE, Caldwell PT, Messenheimer JA. Lamotrigine XR conversion to monotherapy: first study using a historical control group. Neurotherapy 2012;9(1):176‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliam F, Vazquez B, Sackellares JC, Chang GY, Messenheimer J, Nyberg J, et al. An active‐control trial of lamotrigine monotherapy for partial seizures. Neurology 1998;51(4):1018‐25. [DOI] [PubMed] [Google Scholar]
- Gruber CM, Brock JT, Dyken MD. Comparison of the effectiveness of phenobarbital, mephobarbital, primidone, dipheylhydantoin, ethotoin, metharbital, and methylphenylhydantion in motor seizures. Clinical Pharmacology & Therapeutics 1962;3:23‐8. [DOI] [PubMed] [Google Scholar]
- Hakami T, O'Brien TJ, Petty SJ, Sakellarides M, Christie J, Kantor S, et al. Monotherapy with levetiracetam versus older AEDs: a randomized comparative trial of effects on bone health. Calcified Tissue International 2016;98(6):556‐65. [DOI] [PubMed] [Google Scholar]; Hakami T, Todaro M, Petrovski S, MacGregor L, Velakoulis D, Tan M, et al. Substitution monotherapy with levetiracetam vs older antiepileptic drugs: a randomized comparative trial. Archives of Neurology 2012;69(12):1563‐71. [DOI] [PubMed] [Google Scholar]
- ISRCTN73223855. A randomised controlled trial of phenobarbitone and phenytoin for newly diagnosed epilepsy in adults. www.isrctn.com (accessed 11 September 2015).
- Kaminow L, Schimschock JR, Hammer AE, Vuong A. Lamotrigine monotherapy compared with carbamazepine, phenytoin, or valproate monotherapy in patients with epilepsy. Epilepsy & Behavior 2003;4(6):659‐66. [DOI] [PubMed] [Google Scholar]
- Kerr M, Kane K, Moorat A. Switching patients to monotherapy: efficacy and tolerability for Lamictal(r) compared to valproate. European Journal of Neurology 1999;6(Suppl 3):17. [Google Scholar]
- Kerr M, Moorat A, Curtis P. Lamotrigine compared to valproate: a randomised open label switch to monotherapy study in patients with uncontrolled epilepsy. Journal of the Neurological Sciences 2001;187(Suppl 1):S291‐2. [Google Scholar]
- Loiseau P, Cohadon S, Jogeix M, Legroux M, Dartigues JF. Efficacy of sodium valproate in partial epilepsy. Crossed study of valproate and carbamazepine. Revue Neurologique 1984;140(6‐7):434‐7. [PubMed] [Google Scholar]
- Reinikainen K, Keranen T, Hallikainen E, Riekkinen PJ. Substitution of diphenylhydantoin by oxcarbazepine or carbamazepine: double‐blind study. Acta Neurologica Scandinavica 1984;69(Suppl 98):89‐90. [Google Scholar]
- Reinikainen KJ, Keranen T, Halonen T, Komulainen H, Riekkinen PJ. Comparison of oxcarbazepine and carbamazepine: a double‐blind study. Epilepsy Research 1987;1(5):284‐9. [DOI] [PubMed] [Google Scholar]
- Rosenow F, Schade‐Brittinger C, Burchardi N, Bauer S, Klein KM, Weber Y, et al. LaLiMo Study Group. The LaLiMo Trial: lamotrigine compared with levetiracetam in the initial 26 weeks of monotherapy for focal and generalised epilepsy‐‐an open‐label, prospective, randomised controlled multicenter study. Journal of Neurology, Neurosurgery & Psychiatry 2012;83(11):1093‐8. [DOI] [PubMed] [Google Scholar]
- Simonsen N, Olsen PZ, Kuhl V. Carbamazepine versus diphenylhydantoin in psychomotor epilepsy. A double blind study of antiepileptic effect and side effects. Ugeskrift for Laeger 1975;137(41):2392‐6. [PubMed] [Google Scholar]
- Simonsen N, Zander Olsen P, Kuhl V. A double blind study of carbamazepine and diphenylhydantoin in temporal lobe epilepsy. Acta Neurologica Scandinavica 1975;52(Suppl 60):39‐42. [DOI] [PubMed] [Google Scholar]
- Taragano FE, Loñ L, Sarasola D, Serrano C. Is oxcarbazepine better than phenytoin for the treatment of dementia patients complicated with secondary epilepsy? A comparative study. Revista Neurologica Argentina 2003;28(4):209‐16. [Google Scholar]
References to studies awaiting assessment
- Chen YB, Hao YP, Hao XS, Liang D. Clinical efficacy of oxcarbazepine suspension in children with focal epilepsy. Zhongguo Dang Dai Er Ke Za Zhi 2013;15(5):340‐2. [PubMed] [Google Scholar]; Chen YB, Wang JT, Wang LJ, Liang D. Clinical observation of oxcarbazepine suspension monotherapy for 2 to 4‐year‐old patients newly diagnosed as partial epilepsy. Chinese Journal of Neurology 2012;45(10):730‐3. [Google Scholar]
- IRCT201202068943N1. Efficacy of oxcarbazepine and phenytoin in control of epilepsy in the elderly. www.irct.ir/searchresult.php?id=8943&number=1 (accessed 23 August 2016).
- Korean Zonisamide Study Group. Double‐blind, randomized, comparative clinical trial of zonisamide and carbamazepine as initial monotherapy in newly diagnosed epilepsy. Journal of Korean Epilepsy Society 1999;3(1):50‐7. [Google Scholar]
- NCT00154076. A multicenter comparative trial of zonisamide and topiramate as initial monotherapy in untreated epilepsies. ClinicalTrials.gov (accessed 23 August 2016).
- Park WS, Kim CW, Park SP, Kwon SH. Safety and efficacy of topiramate monotherapy in children with recent‐onset seizures. Journal of Korean Epilepsy Society 2001;5(1):65‐9. [Google Scholar]
- Rysz A. Effect of monotherapy with phenytoin or carbamazepine on somatosensory evoked potentials in patients with newly diagnosed epilepsy. Polski Tygodnik Lekarski 1994;49(4‐5):79‐81. [PubMed] [Google Scholar]
- Xu F, Sun HB. Comparative study for retention rate of new antiepileptic drugs as an initial monotherapy in complex partial epilepsy. Chinese Journal of New Drugs 2012;21(11):1265‐8. [Google Scholar]
References to ongoing studies
- ACTRN12615000556549. EpiNet‐First Trial 2: comparison of efficacy of levetiracetam and sodium valproate in people with previously untreated epilepsy who have generalised seizures. In: Auckland District Health B, editor. 2015 (accessed 23 August 2016).
- ACTRN12615000639527. EpiNet‐First Trial 3: comparison of efficacy of levetiracetam and lamotrigine in people with previously untreated epilepsy who have generalised seizures, and for whom sodium valproate is not deemed an acceptable anti‐epileptic drug. In: Auckland District Health B, editor. 2015 (accessed 23 August 2016).
- ACTRN12615000640505. EpiNet‐First Trial 4: comparison of efficacy of levetiracetam, lamotrigine and sodium valproate in people with previously untreated epilepsy who have unclassified seizures. In: Auckland District Health B, editor. 2015 Vol. (accessed 23 August 2016).
- ACTRN12615000641594. EpiNet‐First Trial 5: comparison of efficacy of levetiracetam and lamotrigine in people with previously untreated epilepsy who have unclassified seizures, and for whom sodium valproate is not deemed an acceptable anti‐epileptic drug. In: Auckland District Health B, editor. 2015 (accessed 23 Agust 2016).
- ACTRN12615000643572. EpiNet‐First Trial 1: comparison of efficacy of levetiracetam, lamotrigine and carbamazepine in people with previously untreated epilepsy who have focal seizures. In: Auckland District Health B, editor. 2015 (accessed 23 August 2016).
- NCT01891890. Cognitive AED outcomes in pediatric localization related epilepsy (COPE). ClinicalTrials.gov (accessed 23 August 2016).
- NCT02201251. A study to investigate the safety of the drugs topiramate and levetiracetam in treating children recently diagnosed with epilepsy. ClinicalTrials.gov (accessed 23 August 2016).
Additional references
- Altman DG, Stavola BL, Love SB, Stepniewska KA. Review of survival analyses published in cancer journals. British Journal of Cancer 1995;72:511‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annegers JF, Dubinsky S, Coan SP, Newmark ME, Roht L. The incidence of epilepsy and unprovoked seizures in multiethnic, urban health maintenance organizations. Epilepsia 1999;40(4):502‐6. [DOI] [PubMed] [Google Scholar]
- Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005‐2009. Epilepsia 2010;51:676‐85. [DOI] [PubMed] [Google Scholar]
- Bourgeois B, Beaumanoir A, Blajev B, Cruz N, Despland PA, Egli M, et al. Monotherapy with valproate in primary generalized epilepsies. Epilepsia 1987;28(Suppl 2):S8‐11. [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Dichter MA. Antiepileptic drugs. New England Journal of Medicine 1996;334(3):168‐75. [DOI] [PubMed] [Google Scholar]
- Bromley RL, Mawer GE, Briggs M, Cheyne C, Clayton‐Smith J, García‐Fiñana M, et al. The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. Journal of Neurology, Neurosurgery and Psychiatry 2013;84(6):637‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta‐analysis of randomized controlled trials. Journal of Clinical Epidemiology 1997;50(6):683‐91. [DOI] [PubMed] [Google Scholar]
- Canger R, Battino D, Canevini MP, Fumarola C, Guidolin L, Vignoli A, et al. Malformations in offspring of women with epilepsy: a prospective study. Epilepsia 1999;40(9):1231‐6. [DOI] [PubMed] [Google Scholar]
- Carl GF, Smith ML. Phenytoin‐folate interactions: differing effects of the sodium salt and the free acid of phenytoin. Epilepsia 1992;33(2):372‐5. [DOI] [PubMed] [Google Scholar]
- Cockerell OC, Johnson AL, Sander JW, Hart YM, Shorvon SD. Remission of epilepsy: results from the National General Practice Study of Epilepsy. Lancet 1995;346(8968):140‐4. [DOI] [PubMed] [Google Scholar]
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 1981;22(4):489‐501. [DOI] [PubMed] [Google Scholar]
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30(4):389‐99. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Sombati S, DeLorenzo RJ. Selective effects of topiramate on sustained repetitive firing and spontaneous bursting in cultured hippocampal neurons. Epilepsia 1993;34(Suppl 2):123. [DOI] [PubMed] [Google Scholar]
- Delgado‐Escueta AV, Enrile‐Bacsal F. Juvenile myoclonic epilepsy of Janz. Neurology 1984;34(3):285‐94. [DOI] [PubMed] [Google Scholar]
- Dias S, Welton NJ, Caldwell DM, Ades AE. Checking consistency in mixed treatment comparison meta‐analysis. Statistics in Medicine 2010;29((7‐8)):932‐44. [DOI] [PubMed] [Google Scholar]
- Electronic Medicines Compendium (eMC). www.medicines.org.uk/emc/ (accessed 11 October 2016).
- Endoh A, Kinno I, Kawai M, Hiramatsu M, Mori A. Effect of zonisamide on neurotransmitter in mouse brain. Neurosciences 1994;20(suppl):P173‐6. [Google Scholar]
- Engel J, Pedley TA, Aicardi J. Antiepileptic Drugs. Epilepsy: A Comprehensive Textbook. Vol. 3, Lippincott Williams & Wilkins, 2008:1488‐9. [Google Scholar]
- Epilepsy Foundation of America, Inc. Seizure and Epilepsy Medicines. www.epilepsy.com/learn/treating‐seizures‐and‐epilepsy/seizure‐and‐epilepsy‐medicines?gclid=COarmvHni7kCFXLJtAodYREA1w (accessed 20 August 2013).
- Faigle JW, Menge GP. Metabolic characteristics of oxcarbazepine (Trileptal) and their beneficial implications for enzyme induction and drug interactions. Behaviour Neurology 1990;3(1):21‐30. [DOI] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration. www.fda.gov/ (accessed 11 October 2016).
- French JA, Kanner AM, Bautista J, Abou‐Khalil B, Browne T, Harden CL, et al. Efficacy and tolerability of the new antiepileptic drugs I: treatment of new onset epilepsy: report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2004;62(8):1252‐60. [DOI] [PubMed] [Google Scholar]
- French JA, Kanner AM, Bautista J, Abou‐Khalil B, Browne T, Harden CL, et al. Appendix C: Efficacy and tolerability of the new antiepileptic drugs I: Treatment of new onset epilepsy: Report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. CONTINUUM Lifelong Learning in Neurology 2007;13:203‐11. [DOI] [PubMed] [Google Scholar]
- Gillard M, Chatelain P, Fuks B. Binding characteristics of levetiracetam to synaptic vesicle protein 2A (SV2A) in human brain and in CHO cells expressing the human recombinant protein. European Journal of Pharmacology 2006;536(1‐2):102‐8. [DOI] [PubMed] [Google Scholar]
- Gladstone DJ, Bologa M, Maguire C, Pastuszak A, Koren G. Course of pregnancy and fetal outcome following maternal exposure to carbamazepine and phenytoin: a prospective study. Reproductive Toxicology 1992;6(3):257‐61. [DOI] [PubMed] [Google Scholar]
- Glauser T, Ben‐Menachem E, Bourgeois B, Cnaan A, Chadwick D, Guerreiro C, et al. ILAE treatment guidelines: evidence‐based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 2006;47(7):1094‐120. [DOI] [PubMed] [Google Scholar]
- GRADE Working Group. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336:924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger P, Biton B, Faure C, Vige X, Depoortere H, Graham D, et al. Modulation of the gamma‐aminobutyric acid type A receptor by the antiepileptic drugs carbamazepine and phenytoin. Molecular Pharmacology 1995;47:1189‐96. [PubMed] [Google Scholar]
- Grant SM, Faulds D. Oxcarbazepine. A review of its pharmacology and therapeutic potential in epilepsy, trigeminal neuralgia and affective disorders. Drugs 1992;43:873‐88. [DOI] [PubMed] [Google Scholar]
- Grünewald R, Panayiotopoulos CP. Juvenile myoclonic epilepsy: a review. Archives of Neurology 1993;50:594‐8. [DOI] [PubMed] [Google Scholar]
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota 1935 ‐ 1984. Epilepsia 1993;34:453‐68. [DOI] [PubMed] [Google Scholar]
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21:1539‐58. [DOI] [PubMed] [Google Scholar]
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Higgins JPT, Jackson D, Barrett JK, Lu G, Ades, White IR. Consistency and inconsistency in network meta‐analysis: concepts and models for multi‐arm studies. Research Synthesis Methods 2012;3:98‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DR, Suman‐Chauhan N, Woodruff GN. Localization of [3H] gabapentin to a novel site in rat brain: autoradiographic studies. European Journal of Pharmacology 1993;244:303‐9. [DOI] [PubMed] [Google Scholar]
- Hirtz D, Thurman DJ, Gwinn‐Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. How common are the "common" neurologic disorders?. Neurology 2007;68:326‐37. [DOI] [PubMed] [Google Scholar]
- ILAE Commission on Antiepileptic Drugs. Considerations on designing clinical trials to evaluate the place of new antiepileptic drugs in the treatment of newly diagnosed and chronic patients with epilepsy. Epilepsia 1998;39(7):799‐803. [DOI] [PubMed] [Google Scholar]
- Jackson D, White IR, Riley R. Quantifying the impact of between‐study heterogeneity in multivariate meta‐analyses. Statistics in Medicine 2012;31:3805‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeavons PM, Clark JE, Maheshwan MC. Treatment of generalized epilepsies of childhood and adolescence with sodium valproate. Developmental Medicine and Child Neurology 1977;19:9‐25. [DOI] [PubMed] [Google Scholar]
- Joseph PD, Craig JC, Caldwell PH. Clinical trials in children. British Journal of Clinical Pharmacology 2015 Mar;79(3):357‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juul‐Jenson P, Foldspang A. Natural history of epileptic seizures. Epilepsia 1983;24:297‐312. [DOI] [PubMed] [Google Scholar]
- Kawai M, Hiramatsu M, Endo A, Kinno I, Endo Y, Suh M, et al. Effect of zonisamide on release of aspartic acid and gamma‐aminobutyric acid from the hippocampal slices of E1 mice. Neurosciences 1994;20:115‐9. [Google Scholar]
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:c365. [DOI] [PubMed] [Google Scholar]
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. New England Journal of Medicine 2000;342:314‐9. [DOI] [PubMed] [Google Scholar]
- Lees G, Leach MJ. Studies on the mechanism of action of the novel anti‐convulsant lamotrigine (Lamictal) using primary neuroglial cultures from rat cortex. Brain Research 1993;612:190‐99. [DOI] [PubMed] [Google Scholar]
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Liporace JD, Sperling MR, Dichter MA. Absence seizures and carbamazepine in adults. Epilepsia 1994;35(5):1026‐8. [DOI] [PubMed] [Google Scholar]
- Lu G, Ades A. Assessing evidence inconsistency in mixed treatment comparisons. Journal of the American Statistical Association 2006;101(474):447‐59. [Google Scholar]
- Lynch BA, Lambeng N, Nocka K, Kensel‐Hammes P, Bajjalieh SM, Matagne A, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. National Academy of Sciences of the United States of America. 2004; Vol. 101(26):9861‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BK, Johnson AL, Goodridge DM, Cockerell OC, Sander JWA, Shorvon SD. Factors predicting prognosis of epilepsy after presentation with seizures. Annals of Neurology 2000;48:833‐41. [PubMed] [Google Scholar]
- Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R. Idiopathic Generalised Epilepsies: Clinical, Experimental and Genetic. Eastleigh: John Libbey and Company, 1994. [Google Scholar]
- Marson AG, Williamson PR, Hutton JL, Clough HE, Chadwick DW. Carbamazepine versus valproate monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2000, Issue 3. [DOI: 10.1002/14651858.CD001030] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClean MJ. Gabapentin. Epilepsia 1995;36(Suppl 2):72‐86. [DOI] [PubMed] [Google Scholar]
- McLean MJ, MacDonald RL. Sodium valproate, but not ethosuximide, produces use‐ and voltage‐dependent limitation of high frequency repetitive firing of action potentials of mouse central neurons in cell culture. Journal of Pharmacology and Experimental Therapeutics 1986;237:1001‐11. [PubMed] [Google Scholar]
- McLean MJ. Gabapentin in the management of convulsive disorders. Epilepsia 1999;40(Suppl 6):S39‐50; discussion S73‐4. [DOI] [PubMed] [Google Scholar]
- Meador K, Reynolds M, Crean S, Fahrbach K, Probst C. Pregnancy outcomes in women with epilepsy: a systematic review and meta‐analysis of published pregnancy registries and cohorts. Epilepsy Research 2008;81:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group (2009). Preferred Reporting Items for Systematic Reviews and Meta‐Analyses: The PRISMA Statement. PLoS Medicine 2009;6(7):e1000097. [DOI: 10.1371/journal.pmed1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow J, Russel A, Guthrie E, Parsons L, Robertson I, Waddell R, et al. Malformation risks of antiepileptic drugs in pregnancy: a prospective study from the UK Epilepsy and Pregnancy Register. Journal of Neurology, Neurosurgery and Neuropsychiatry 2006;77(2):193‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevitt SJ, Sudell M, Tudur Smith C, Marson A. Topiramate versus carbamazepine monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2016, Issue 12. [DOI: 10.1002/14651858.CD012065.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngugi AK, Bottomley C, Kleinschmidt I, Sander JW, Newton CR. Estimation of the burden of active and life‐time epilepsy: a meta‐analytic approach. Epilepsia 2010;51:883‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institute for Health and Care Excellence. The epilepsies: the diagnosis and management of the epilepsies in adults and children and primary and secondary care. NICE guidelines [CG137] Published date: January 2012. Available from www.nice.org.uk/guidance/cg137.
- National Institute of Neurological Disorders and Stroke (NINDS). The epilepsies and seizures: hope through research. www.ninds.nih.gov/disorders/epilepsy/curing_the_epilepsies_brochure.pdf (accessed 11 October 2016).
- Nolan SJ, Sutton L, Marson A, Tudur Smith C. Consistency of outcome and statistical reporting of time‐to‐event data: the impact on Cochrane Reviews and meta‐analyses in epilepsy. 21st Cochrane Colloquium: Better Knowledge for Better Health. Quebec City, 2013:114‐5. [Google Scholar]
- Nolan SJ, Tudur Smith C, Pulman J, Marson AG. Phenobarbitone versus phenytoin monotherapy for partial onset seizures and generalised onset tonic‐clonic seizures. Cochrane Database of Systematic Reviews 2013, Issue 1. [DOI: 10.1002/14651858.CD002217.pub2] [DOI] [PubMed] [Google Scholar]
- Nolan SJ, Muller M, Tudur Smith C, Marson AG. Oxcarbazepine versus phenytoin monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2013, Issue 5. [DOI: 10.1002/14651858.CD003615.pub3] [DOI] [PubMed] [Google Scholar]
- Nolan SJ, Marson AG, Weston J, Tudur Smith C. Carbamazepine versus phenytoin monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2015, Issue 8. [DOI: 10.1002/14651858.CD001911.pub2] [DOI] [PubMed] [Google Scholar]
- Nolan SJ, Tudur Smith C, Weston J, Marson AG. Lamotrigine versus carbamazepine monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2016, Issue 11. [DOI: 10.1002/14651858.CD001031.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan SJ, Marson AG, Weston J, Tudur Smith C. Carbamazepine versus phenobarbitone monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2016, Issue 12. [DOI: 10.1002/14651858.CD001904.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan SJ, Marson AG, Pulman J, Tudur‐Smith C. Phenytoin versus valproate monotherapy for partial onset seizures and generalised onset tonic‐clonic seizures: an individual participant data review. Cochrane Database of Systematic Reviews 2016, Issue 4. [DOI: 10.1002/14651858.CD001769.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nulman I, Scolnik D, Chitayat D, Farkas LD, Koren G. Findings in children exposed in utero to phenytoin and carbamazepine monotherapy: independent effects of epilepsy and medications. American Journal of Medical Genetics 1997;68(1):18‐24. [PubMed] [Google Scholar]
- Okada M, Kawata Y, Mizuno K, Wada K, Kondo T, Kaneko S. Interaction between Ca2+, K+, carbamazepine and zonisamide on hippocampal extracellular glutamate monitored with a microdialysis electrode. British Journal of Pharmacology 1998;124:1277‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olafsson E, Ludvigsson P, Gudmundsson G, Hesdorfer D, Kjartansson O, Hauser WA. Incidence of unprovoked seizures and epilepsy in Iceland and assessment of the epilepsy syndrome classification: a prospective study. Lancet Neurology 2005;4:627‐34. [DOI] [PubMed] [Google Scholar]
- Ornoy A. Valproic acid in pregnancy: how much are we endangering the embryo and fetus?. Reproductive Toxicology 2009;28(1):1‐10. [DOI] [PubMed] [Google Scholar]
- Palmer TM, Sterne JAC. Meta‐Analysis in Stata: An Updated Collection from the Stata Journal, Second Edition. Stata Press, 2016. [Google Scholar]
- Parmar MKB, Torri V, Stewart L. Extracting summary statistics to perform meta‐analysis of the published literature for survival endpoints. Statistics in Medicine 1998;17:2815‐34. [DOI] [PubMed] [Google Scholar]
- Penry JK, Dean JC, Riela AR. Juvenile myoclonic epilepsy: long term response to therapy. Epilepsia 1989;30:19‐23. [DOI] [PubMed] [Google Scholar]
- Pinder RM, Brogden RN, Speight TM, Avery GS. Sodium valproate: a review of its pharmacological properties and therapeutic efficacy in epilepsy. Drugs 1977;13:81‐123. [DOI] [PubMed] [Google Scholar]
- Ragsdale DS, Scheuer T, Catterall WA. Frequency and voltage‐dependent inhibition of type IIA Na+ channels, expressed in a mammalian cell line, by local anesthetic, antiarrhythmic, and anticonvulsant drugs. Molecular Pharmacology 1991;40(5):756‐65. [PubMed] [Google Scholar]
- Rho JM, Donevan, SD, Rogawski MA. Direct activation of GABAA receptors by barbiturates in cultured rat hippocampal neurons. Journal of Physiology 1996;497(2):509‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackellares JC, Ramsay RE, Wilder BJ, Browne TR 3rd, Shellenberger MK. Randomized, controlled clinical trial of zonisamide as adjunctive treatment for refractory partial seizures. Epilepsia 2004;45(6):610‐7. [DOI] [PubMed] [Google Scholar]
- Salanti G, Giovane C, Chaimani A, Caldwell DM, Higgins JP. Evaluating the quality of evidence from a network meta‐analysis. PloS one 2014 Jul 3;9(7):e99682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JW, Shorvon SD. Epidemiology of the epilepsies. Journal of Neurology, Neurosurgery, and Psychiatry 1996;61(5):433‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JW. The use of anti‐epileptic drugs ‐ principles and practice. Epilepsia 2004;45(6):28‐34. [DOI] [PubMed] [Google Scholar]
- SAS Institute Inc. Base SAS. Version 9.3. Cary, NC, USA: SAS Institute Inc, 2011.
- Schauf CL. Zonisamide enhances slow sodium inactivation in Myxicola. Brain Research 1987;413:185‐8. [DOI] [PubMed] [Google Scholar]
- Scheinfeld N. Phenytoin in cutaneous medicine: its uses, mechanisms and side effects. Dermatology Online Journal 2003;9(3):6. [PubMed] [Google Scholar]
- Shakir RA. Sodium valproate, phenytoin and carbamazepine as sole anticonvulsants. The place of sodium valproate in the treatment of epilepsy. London: Academic Press Inc (London) Ltd and the Royal Society of Medicine, 1980:7‐16. [Google Scholar]
- Shields WD, Saslow E. Myoclonic, atonic, and absence seizures following institution of carbamazepine therapy in children. Neurology 1983;33:1487‐9. [DOI] [PubMed] [Google Scholar]
- Snead OC, Hosey LC. Exacerbation of seizures in children by carbamazepine. New England Journal of Medicine 1985;313(15):916‐21. [DOI] [PubMed] [Google Scholar]
- StataCorp LP. Stata Statistical Software: Release 14. College Station, TX: StataCorp LP, 2015.
- Suzuki S, Kawakami K, Nishimura S, Watanabe Y, Yagi K, Seino M, et al. Zonisamide blocks T‐type calcium channel in cultured neurons of rat cerebral cortex. Epilepsy Research 1992;12:21‐7. [DOI] [PubMed] [Google Scholar]
- Tomson T, Battino D, Bonizzoni E, Craig J, Lindhout D, Sabers A, et al. Dose‐dependent risk of malformation with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurology 2011;10(7):609‐17. [DOI] [PubMed] [Google Scholar]
- Trimble MR, Cull C. Children of school age: the influence of antiepileptic drugs on behavior and intellect. Epilepsia 1988;29(Suppl 3):S15‐19. [DOI] [PubMed] [Google Scholar]
- Tudur Smith C, Marson AG, Chadwick DW, Williamson PR. Multiple treatment comparisons in epilepsy monotherapy trials. Trials 2007;5(8):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace H, Shorvon SD, Hopkins A, O'Donoghue M. National Society of Epilepsy Guidelines. London: Royal College of Physicians, 1997. [Google Scholar]
- Weston J, Bromley R, Jackson CF, Adab N, Clayton‐Smith J, Greenhalgh J, et al. Monotherapy treatment of epilepsy in pregnancy: congenital malformation outcomes in the child. Cochrane Database of Systematic Reviews 2016, Issue 11. [DOI: 10.1002/14651858.CD010224.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
- White HS, Brown SD, Woodhead JH, Skeen GA, Wolf HH. Topiramate enhances GABA‐mediated chloride flux and GABA‐evoked chloride currents in murine brain neurons and increases seizure threshold. Epilepsy Research 1997;28:167‐79. [DOI] [PubMed] [Google Scholar]
- White IR. Multivariate random‐effects meta‐analysis. Stata Journal2009; Vol. 9, issue 1:40‐56.
- White IR. Network meta‐analysis. Stata Journal 2015;15(4):951‐85. [Google Scholar]
- World Health Organization. Global comparative assessments in the health sector: disease burden, expenditures and intervention packages. Collected reprints from the Bulletin of the World Health Organization. 1994. Available from apps.who.int/iris/handle/10665/41177 (accessed 13 June 2017).
- Wilder BJ. Phenytoin: clinical use. Antiepileptic drugs. New York: Raven Press, 1995:339‐44. [Google Scholar]
- Williamson PR, Tudur Smith C, Hutton JL, Marson AG. Aggregate data meta‐analysis with time‐to‐event outcomes. Statistics in Medicine 2002;21(11):3337‐51. [DOI] [PubMed] [Google Scholar]
- Willow M, Gonoi T, Catterall WA. Voltage clamp analysis of the inhibitory actions of diphenyihydantoin and carbamazepine on voltage sensitive sodium channels in neuroblastoma cells. Molecular Pharamcology 1985;27:549‐58. [PubMed] [Google Scholar]
- Wyllie E, Gupta A, Lachhwani DK. Antiepileptic Medications. The Treatment of Epilepsy: Principles & Practice. 4th Edition. Lippincott Williams & Wilkins, 2006:650. [Google Scholar]
- Zhu W, Rogawski A. Zonisamide depresses excitatory synaptic transmission by a presynaptic action. Epilepsia 1999;40(suppl 7):245. [Google Scholar]
References to other published versions of this review
- Nolan SJ, Sudell M, Weston J, Tudur Smith C, Marson AG. Antiepileptic drug monotherapy for epilepsy: a network meta‐analysis. Cochrane Database of Systematic Reviews 2014, Issue 12. [DOI: 10.1002/14651858.CD011412] [DOI] [PMC free article] [PubMed] [Google Scholar]