

Abstract
The aims of this work were to characterize ipatasertib exposure–response (E‐R) relationships in a phase II study and to quantitatively assess benefit‐risk using a clinical utility index approach to support ipatasertib phase III dose selection in patients with metastatic castration‐resistant prostate cancer. Logistic regression and Cox proportional‐hazards models characterized E‐R relationships for safety and efficacy endpoints, respectively. Exposure metrics with and without considering dose interruptions/reductions (modifications) were tested in the E‐R models. Despite a steeper E‐R relationship when accounting for dose modifications, similar dose‐response projections were generated. The clinical utility index analysis assessed important attributes, weights, and clinically meaningful cutoff/tradeoff values based on predefined minimal, target, and optimistic product profiles. Ipatasertib 400 mg daily, showing the highest probability of achieving the minimal product profiles and better benefit‐risk balance than other doses (200–500 mg daily), was selected for further development in metastatic castration‐resistant prostate cancer.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑The 200 and 400 mg daily ipatasertib doses in combination with abiraterone were evaluated in a phase II trial in metastatic castration‐resistant prostate cancer, where limited ipatasertib dose modifications were observed. When compared with 200 mg, the 400 mg daily dose provided better efficacy with manageable safety profiles.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑Is ipatasertib 400 mg daily the optimal dose to balance efficacy and safety in metastatic castration‐resistant prostate cancer based on available information?
☑What are the differences in exposure–response (E‐R) trends and dose‐response projections from E‐R models using exposures with and without considering dose modifications?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑Based on the E‐R–based product‐profile–driven clinical utility index analysis, ipatasertib 400 mg daily dose showed optimal benefit‐risk balance when combined with abiraterone in metastatic castration‐resistant prostate cancer. Generally, the E‐R trend is steeper when modeled considering dose interruptions/reductions, but both models generate similar dose–response projections.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑This clinical utility index analysis framework can be used for dose optimization during clinical drug development. The selection of an exposure metric and E‐R modeling approaches may depend on the analysis objective.
The benefit‐risk assessment of a therapeutic agent can support decision making in both drug development and regulatory evaluation. The traditional way of assessing benefit‐risk involves characterizing dose/exposure–response (E‐R) relationships on primary efficacy and safety endpoints separately and then qualitatively comparing those relationships to support the decision making of dose/formulation selection, go/no go in clinical development, and so on. However, with this implicit approach, it is difficult to balance benefit with risk when there are multiple important attributes (e.g., efficacy, safety, compliance). Therefore, the clinical utility index (CUI; also called multiattribute utility), a more structured quantitative approach that brings all of the attributes onto the same scale and reduces them to a single measure, allows more transparent and efficient benefit‐risk assessment and decision making.1, 2, 3, 4
There are typically the following four steps in the E‐R–based CUI development: (i) characterize the dose/E‐R relationships of efficacy and safety endpoints, (ii) select important attributes and assign a weight to each attribute, (iii) determine a utility function for each attribute using cutoff/tradeoff values as criteria to define clinically meaningful changes, and (iv) obtain the overall CUI with uncertainty and conduct sensitivity analysis to test the robustness of assumptions.3 In practice, determination of important attributes, weights, and clinically meaningful cutoff/tradeoff values requires intensive development team discussion. Predefined product profiles (PPs) can be a good anchor point for the discussion and help the team reach agreement. Several CUI analyses in the literature have supported decision making in early clinical development, including dose regimen selection for combination therapy,4, 5 go/no go decision for a backup molecule,6 dosing algorithm optimization,7 and others.8, 9, 10 In addition, a series of articles by Raju et al.11, 12, 13 built on the US Food and Drug Administration qualitative benefit‐risk framework14 by combining benefits and risks in terms of a common metric (e.g., gain in adjusted years of remaining life vs. placebo), which is equivalent to a CUI. These articles reviewed the Food and Drug Administration decisions on drugs for multiple myeloma,11 non‐small cell lung cancer,12 multiple sclerosis, and tuberculosis.13
This article summarizes the development of E‐R modeling and simulation and application of PP‐driven CUI to assess benefit‐risk and support phase III dose selection of ipatasertib in combination with abiraterone in metastatic castration‐resistant prostate cancer (mCRPC). Ipatasertib is a potent, selective, adenosine triphosphate‐competitive, small‐molecule inhibitor of the activated form of Akt that disrupts oncogenic phosphoinositide 3‐kinase/Akt signaling. Based on preclinical data suggesting cooperativity between phosphoinositide 3‐kinase/Akt and hormonal blockade, ipatasertib was evaluated in combination with abiraterone (one of the current standard of care in mCRPC) in a phase Ib/II study (A.MARTIN; NCT01485861) in mCRPC.15, 16 In the A.MARTIN phase II study, the patients received placebo or ipatasertib 200 mg or 400 mg daily in combination with abiraterone.16 The primary endpoint of the study was radiographic progression‐free survival (rPFS). Adverse events (AEs) more common with ipatasertib included diarrhea, hyperglycemia, rash, nausea, and vomiting, which are generally consistent with the phosphoinositide 3‐kinase/Akt pathway inhibitor class. To capture the effect of dose modifications, an exposure metric based on the actual dose (steady‐state area under the curve (AUC) based on the average dose up to the event (AUCss,event); details in Methods) was used in the E‐R modeling for both efficacy and safety endpoints. To convert the nominal dose to actual dose to enable the dose–response projections, a dose‐intensity (DI) model was developed to characterize the relationship between the nominal dose and DI of that efficacy or safety event. The E‐R models were then coupled with their corresponding DI models to project the dose‐response relationships over the range of 0–500 mg once daily ipatasertib with abiraterone. The CUI analysis was then performed to help determine which dose was most likely to provide an optimal balance between efficacy and safety. Key components of CUI, including important attributes, weights, and clinically meaningful cutoff/tradeoff values, were determined based on the predefined ipatasertib minimal, target, and optimistic PPs. The overall utility and probability of achieving the PPs were calculated for ipatasertib 400 mg and the doses around it (200 mg, 300 mg, and 500 mg) to determine the dose with optimal benefit‐risk balance. In addition, E‐R analyses and doseresponse projections with and without considering dose modifications were compared. The results from the comparisons may help justify the exposure metric selection in future E‐R analyses in trials with sizable dose modifications.
Methods
Study design
The data used in the analyses were from a double‐blinded, randomized phase II part of a phase Ib/II clinical trial (A.MARTIN; NCT01485861), which evaluated ipatasertib and abiraterone combination therapy vs. abiraterone monotherapy in patients with mCRPC who received prior docetaxel chemotherapy.16 The patients were randomized 1:1:1 to one of the following three arms, all in combination with abiraterone 1,000 mg and prednisone 10 mg daily orally: ipatasertib 400 mg, ipatasertib 200 mg, or placebo. Randomization was stratified by prior enzalutamide treatment (yes or no), number of chemotherapies (one vs. more), and progression by prostate‐specific antigen only vs. other. The patients received study treatment until disease progression, intolerable toxicity, elective withdrawal from the study, study completion, or termination. The date of radiographic progression was defined by the first event in either bone scan or soft tissue disease that meets the definition of progression adapted from the recommendations of the Prostate Cancer Clinical Trials Working Group.17 Full details of the study design and results have been published.16 The clinical trial was conducted in accordance with the Declaration of Helsinki and in compliance with good clinical practice guidelines and quality assurance procedures.
Response and exposure data
Efficacy of the E‐R analysis was characterized by rPFS hazard ratio (HR). The E‐R analysis of safety included diarrhea, hyperglycemia, and rash, which were common AEs with ipatasertib. Because most hyperglycemia events were asymptomatic and quickly reversible after clinical management, diarrhea and rash were expected to have a larger impact on patients’ tolerability to the study treatment and were included in the PPs. Therefore, only diarrhea and rash E‐R results are shown.
A population pharmacokinetics (PK) model was built with data from five phase I and phase II ipatasertib clinical studies,16, 18, 19, 20, 21 including the A.MARTIN phase II study. Ipatasertib PK over the clinical dose range was adequately described by a two‐compartment model with first‐order absorption, first‐order elimination, and dose‐dependent bioavailability (Figure S1). The relative bioavailability increased with ipatasertib dose.
To capture the dose modification effect, AUCss,event, the predicted ipatasertib average steady‐state AUC up to event based on actual dose, was employed as the exposure metric in the E‐R analyses. The AUCss,event was calculated as the average dose up to and including the time of event (e.g., tumor progression or occurrence of AEs) divided by the population model derived individual apparent clearance. For patients without AEs or tumor progression, the AUCss,event was set to be the average steady‐state AUC for the entire treatment duration, calculated as the average dose during the treatment duration divided by the model‐predicted individual apparent clearance. Given that AEs and progression event onset times were generally more than 1 week after the first dose and the effective half‐life of ipatasertib is relatively short (approximately 24 hours), the AUCss,event was considered to be an appropriate exposure metric in the E‐R analysis.
E‐R analysis
For exposure–rPFS, a Cox proportional‐hazards model relating ipatasertib AUCss,event to the hazard of radiographic progression was developed using R 3.2.3.22 The model was evaluated by comparing the model‐predicted cumulative probability of rPFS vs. time, with the corresponding distribution determined by nonparametric Kaplan–Meier analysis. For exposure–safety, logistic regression models relating AUCss,event to the probability of grade ≥2 or grade ≥3 AEs were utilized. Odds ratios for an event were calculated, and the exposure metric was incorporated into the model via the logit function. Logistic regression models were fitted in R 3.2.3 using the glm function for binomial likelihood and a logit link function.22 The models were then evaluated by examining the agreement between the observed probability of AEs by dose and the associated model‐predicted median and 95% confidence interval.
DI modeling
Given that the exposure metric based on actual dose (AUCss,event) was used in the E‐R modeling, to project the expected dose‐response relationship based on the nominal dose, a DI model characterizing the relationship between the nominal dose and DI was developed for each response endpoint (rPFS, diarrhea, and rash) using R 3.2.3.22 Of note, unlike the conventional DI, the DI in this study is end point/event specific. The DI for each end point was calculated as the total dose up to and including the time of event divided by the treatment duration and the nominal dose. The DI is typically a left‐skewed variable, but because of accidental dosing errors, a few patients in each response end point had a DI > 1.
The DI model was developed in the following two steps: (i) the percentage of patients who had a DI of 1 or greater modeled using logistic regression and (ii) the logit‐transformed DI in patients with DI < 1 modeled using linear regression. The two steps can be summarized by the following equations:
| (1) |
| (2) |
where Intercept1 and Slope1 are the coefficients describing the probability that DI is 1 or greater, Intercept2 and Slope2 are the coefficients describing the central tendency of the DI as a function of the nominal dose, given that DI is less than 1, and ε is a normally distributed random variable with variance σ2 describing the distribution of dose intensities in the range 0–1.
Dose‐response projections
Simulations were conducted to estimate the safety and efficacy responses at multiple ipatasertib daily doses (0, 200, 300, 400, and 500 mg). To convert nominal dose to actual dose, the first step in the simulation was to simulate the DI of each patient. For each simulated patient, Eq. (1) was simulated using the binomial distribution to identify whether that patient had a DI of 1 or greater. If so, the patient was assigned a DI of 1. If not, Eq. (2) was simulated to identify the particular value of DI between 0 and 1 from the distribution. The simulated DI for each patient was then applied to the corresponding E‐R model to project the response at different nominal dose levels. For each response end point, a total of 1,000 replicates of 1,000 patients per replicate were simulated for each dose using R 3.2.3.22
Additional exploratory E‐R analysis
To explore the difference between using an exposure metric with and without considering dose modifications, E‐R analyses were also conducted using an AUC based on nominal dose (AUCss). The E‐R trends and dose‐projection results were compared with those from the E‐R analyses using AUCss,event.
Benefit‐risk analysis
The CUI approach was applied in the benefit‐risk analysis, and the CUI was calculated as a weighted sum of individual utility functions for multiple selected attributes:
where w i are positive weights summing to 1, U i(x i) are individual attribute utility functions, and x i is modeled with a probability distribution from the dose‐response projection, yielding a probabilistic CUI. To minimize input requirements, the U i(x i) were assumed to be linear, therefore fully specified by two points, and taken from the minimal and optimistic PP values. For convenience, the individual utility functions, and hence the CUI, are assumed to be 0 at the minimal PP and 1 at the optimistic PP:
where x i0 is such that U i(x i0) = 0, and b i is the slope (1 − 0)/(x i1 − x i0) where x i1 satisfies U i(x i1) = 1. Of note, U i(x i) was defined as a continuous variable; therefore, the CUI was allowed to fall below 0 and above 1 without constraint.
Dose‐response projections from the E‐R modeling provided uncertainty ranges around each mean prediction in the form of a 2.5–97.5% range. The attribute distribution at each dose was fitted by a log‐normal distribution using the ratio of the 95th and 5th percentiles to determine the log‐normal σ parameter (as ln(x .95/x .05)/(1.645 × 2)) and then the mean of the distribution to determine the log‐normal μ parameter (to satisfy mean = exp(μ + σ2/2)). These distributions were translated by the CUI model into uncertainty around the CUI.
The probability distribution for CUI at each dose was calculated from the probability distributions for the attributes. The attribute distributions were assumed log‐normal; however, the CUI, a weighted sum of linear transformations of lognormal variables, is no longer log‐normal and was calculated numerically using all combinations of 39 evenly spaced percentiles from 2.5 to 97.5% for each attribute (39 × 39 combinations for two attributes), treated as equally likely. The attributes were transformed to utility by the linear utility functions, weighted, and added to form the CUI. The final CUI distributions in each case were summarized with the mean and the 10th to 90th percentiles and were plotted vs. dose, as were the probabilities of reaching the CUIs corresponding to the minimal, target, and optimistic PPs (0, about 0.5, and 1). The CUI analyses were conducted using Microsoft Excel 2010 (Microsoft Corp., Redmond, WA) (Data S1 and Data S2).
Results
A total of 253 patients enrolled in the A.MARTIN phase II study provided evaluable PK, rPFS, and safety data for the analysis. A limited number of patients across treatment arms had dose modifications mainly as a result of AEs: 14.6% (12 of 82) in the placebo group, 14.9% (13 of 87) in the 200 mg group, and 39.3% (33 of 84) in the 400 mg group.
The E‐R analysis of the rPFS HR did not demonstrate a statistically significant (P < 0.05) association between ipatasertib exposure (AUCss,event) and rPFS HR, with a slight trend of higher exposure leading to a lower rPFS HR (Table S1). DI modeling results for rPFS described the observed DI data reasonably well as shown in Figure S2a. Coupled with the DI model, the dose‐response predictions based on the Cox proportional‐hazards model showed generally good agreement between the model‐predicted rPFS HR and the corresponding observed data (Figure 1). There was an underestimation of the uncertainty for rPFS HR in the 200 mg arm, which is likely because of the underlying assumption of the Cox proportional‐hazards model that rPFS HR is 1 without uncertainty at a dose of 0.
Figure 1.
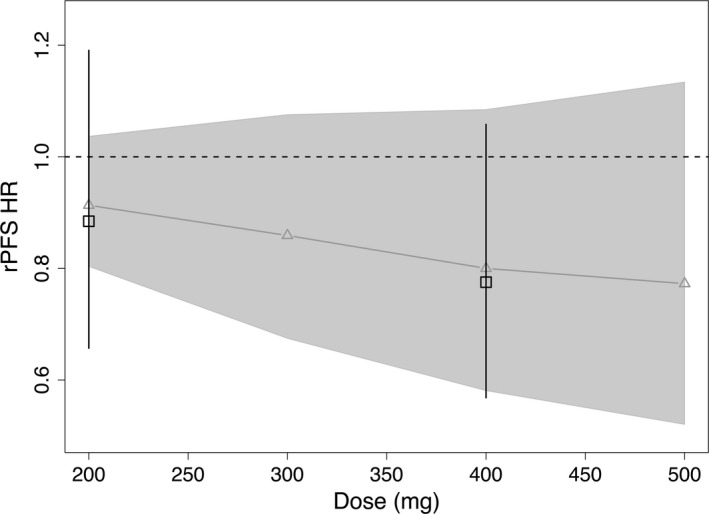
Dose‐response projections of rPFS HR based on exposure–rPFS model and rPFS dose‐intensity model. Black squares and error bars are the observed rPFS HR and 90% confidence interval. Gray curves (and triangles) and shaded area are the model‐predicted rPFS HR and 90% confidence interval. HR, hazard ratio; rPFS, radiographic progression‐free survival.
The E‐R analyses of AEs indicated a statistically significant (P < 0.05) association between ipatasertib exposure (AUCss,event) and both diarrhea and rash (Table S2). The probability of experiencing a grade ≥2 or grade ≥3 diarrhea or rash event increased with increasing ipatasertib exposure (Figure S3). DI modeling results for diarrhea and rash are shown in Figure S2b–e. Coupled with the corresponding DI models, the dose‐response projections based on the logistic regression E‐R models showed good agreement between the model‐predicted and observed probability of grade ≥2 and grade ≥3 diarrhea and rash (Figure 2).
Figure 2.
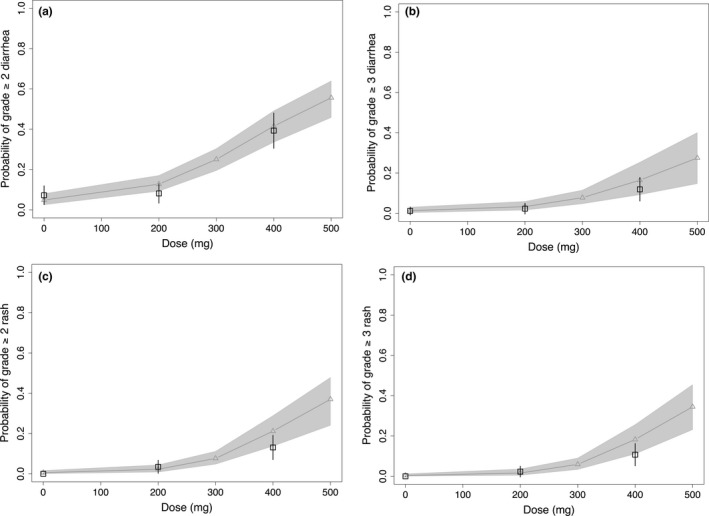
Dose‐response projections for grade ≥2 diarrhea (a), grade ≥3 diarrhea (b), grade ≥2 rash (c), and grade ≥3 rash (d) based on exposure–adverse event models and the corresponding dose‐intensity models. Black squares and error bars are the observed median and 90% confidence interval. Gray curves (and triangles) and shaded area are the model‐predicted median and 90% confidence interval.
The comparison between the E‐R relationships of diarrhea or rash from models with AUCss,event and with steady‐state AUCss indicated that the E‐R trends were generally flatter in the latter (grade ≥ 2 diarrhea in Figure 3 a,b and other AEs in Figure S4a–c). For numerical comparisons, slope (β) estimates from the Cox proportional‐hazards and logistic regression models were summarized in Tables S1 and S2. Despite the different E‐R trends, the dose‐response projections based on these two models were similar (grade ≥ 2 diarrhea in Figure 3 c,d, and other AEs and rPFS HR in Figure S4d–g) with slightly larger variability in the predictions with the AUCss model, which is because the AUCss model cannot explain some of the variability from the dose modifications.
Figure 3.
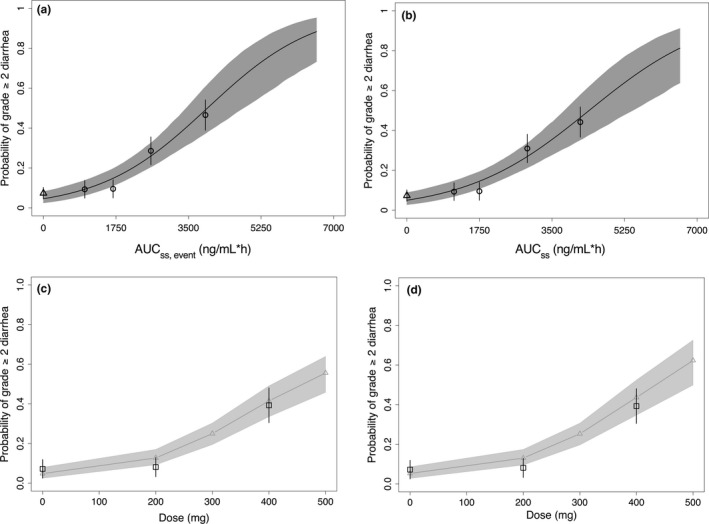
Comparison between exposure–response models with (AUC ss,event as exposure metric, left panels) and without (AUC ss as exposure metric, right panels) considering dose modifications using grade ≥2 diarrhea as an example. Comparison plots for all other response endpoints are shown in Figure S4. (a) and (b) are the logistic regression modeling results, and (c) and (d) are the dose–response projection results. Projections in (c) were from the exposure–response model (AUC ss,event as exposure metric) coupled with the dose‐intensity model of grade ≥2 diarrhea, whereas projections in (d) were from the exposure–response model (AUC ss as exposure metric) alone. In (a) and (b), symbols and error bars represent mean observations and associated 95% confidence intervals of the mean, respectively, for placebo patients (triangle) and for treated patients stratified by quartiles of ipatasertib exposure AUC ss,event (circles). The curves and associated shaded area represent the mean model predictions and associated 95% confidence intervals of the mean prediction, respectively. In (c) and (d), black squares and error bars are the observed medians and 90% confidence intervals. Gray curves and shaded area are the model‐predicted median and 90% confidence interval. AUC ss,event, area under the curve based on the average dose up to the event; AUC ss, area under the curve based on nominal dose.
Based on the PPs, rPFS HR (efficacy) and diarrhea and rash (safety) were selected as key attributes with the cutoff/tradeoff values summarized in Table 1. Given that the AEs are generally manageable and reversible, a clinical team discussion led to the choice of a slightly higher weight for efficacy (w 1 = 0.6) than for AEs (with total weight summing to 1). Within AEs, the relative weight of diarrhea was set at 0.75 (w 2 = 0.3) with the remaining weight on rash (w 3 = 0.1). Sensitivity analyses were conducted to test (i) excluding rash with w 1:w 2:w 3 = 0.6:0.4:0 and (ii) grade ≥2 vs. grade ≥3 AEs. Therefore, a total of four scenarios were tested in the CUI analyses (Table 1), and the results of these four scenarios all supported the 400 mg daily as the optimal dose. Figure 4 shows the results of scenario 2 (rPFS HR, grade ≥3 diarrhea and rash as key attributes) as an example. The expected utility of efficacy (rPFS HR) increased with dose, whereas the expected utility of combined grade ≥3 diarrhea and rash decreased with dose, faster at higher doses (Figure 4 a). Weighted and combined into a CUI, these three measures produced almost constant mean (expected) CUI across 300–400 mg, with substantially lower CUIs at 200 and 500 mg (Figure 4 a). However, the probability of reaching minimal or target PPs peaked at 400 mg (Figure 4 b), likely because the wider uncertainty at 400 mg when compared with 300 mg gave a higher chance of reaching the criteria of PPs.
Table 1.
Summary of scenarios evaluated in the clinical utility index analyses
| Scenario | Key attributes | Minimal PP (utility = 0) | Target PP | Optimistic PP (utility = 1) | Weight |
|---|---|---|---|---|---|
| 1 | rPFS HR | 0.73 | 0.70 | 0.65 | 0.6 |
| Diarrhea grade ≥ 3 | 20% | 15% | 10% | 0.4 | |
| 2 | rPFS HR | 0.73 | 0.70 | 0.65 | 0.6 |
| Diarrhea grade ≥ 3 | 20% | 15% | 10% | 0.3 | |
| Rash grade ≥ 3 | 15% | 10% | 5% | 0.1 | |
| 3 | rPFS HR | 0.73 | 0.70 | 0.65 | 0.6 |
| Diarrhea grade ≥ 2 | 45% | 35% | 25% | 0.4 | |
| 4 | rPFS HR | 0.73 | 0.70 | 0.65 | 0.6 |
| Diarrhea grade ≥ 2 | 45% | 35% | 25% | 0.3 | |
| Rash grade ≥ 2 | 18% | 12% | 6% | 0.1 |
HR, hazard ratio; PP, product profile; rPFS, radiographic progression‐free survival.
Figure 4.
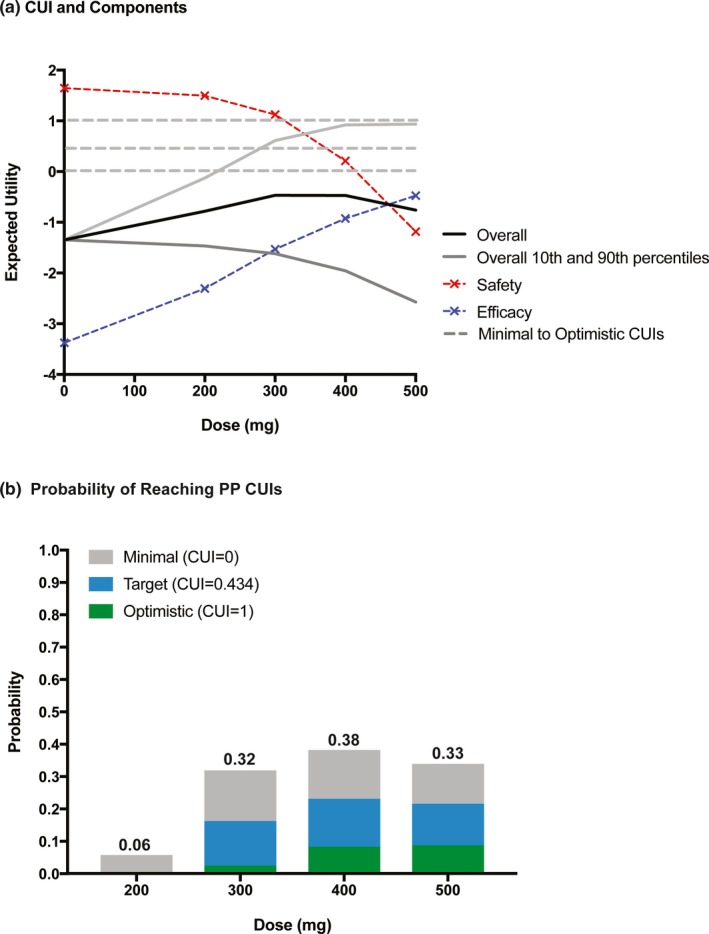
CUI distribution (a) and probabilities of reaching PPs (b) vs. dose using scenario 2 (details in Table 1) as an example. CUI, clinical utility index; PP, product profiles.
Sensitivity analyses indicated that less weight on efficacy favored lower doses and vice versa. When the efficacy weight (w 1) was lowered from 0.60 to 0.50, the peak of the mean CUI curve fell to 300 mg, but the highest chance of reaching the target PP CUI remained at 400 mg. The range of w 1 over which the mean CUI peaks at 400 mg is 0.60–0.75; the range over which the chance of reaching the target PP peaks at 400 mg is 0.48–0.63 (and for the minimal PP, 0.53–0.67). The results from the other three scenarios were generally similar, and they all supported the 400 mg daily ipatasertib as the optimal dose (Figure S5).
Discussion
As demonstrated in this study, with multiple key attributes and a large overall uncertainty on the exposure–rPFS relationship, E‐R understanding and probabilistic benefit‐risk assessment were crucial for the dose optimization of ipatasertib in mCRPC. A good E‐R understanding depends on good clinical trial design, including the number of doses tested. The A.MARTIN phase II study tested two dose levels: 400 mg continuous daily dose, which is comparable to the cumulative dose obtained with the maximum tolerated dose determined in the phase I study (600 mg 21 days on and 7 days off in a 28‐day cycle), and 200 mg continuous daily dose, which achieved robust pathway inhibition in tissue and surrogate markers in a phase I study with the potential for better tolerability during a longer treatment interval.18 This two‐dose, phase II study design provided a relatively large range of exposure levels, which helped the characterization of the E‐R relationships. Literature suggests that CUI analysis has been more common in nononcology indications,3, 4, 6, 7, 8, 9 likely because nononcology phase II trials in patients normally include multiple treatment arms with different doses and/or regimens to cover a large range of exposure, which enables more informative E‐R analyses. In oncology, CUI analysis has been conducted with dose‐escalation cohort data in trials focusing on a specific indication (typically in phase Ib), and the results have been used to guide dose selection for subsequent expansion cohort.5, 10
Benefit‐risk assessment via CUI requires determination of the important attributes, weights, and clinically meaningful cutoff/tradeoff values, all of which call for intensive discussions with the clinical development team. Because of its predefined and comprehensive nature, PPs can be a good basis to initiate the discussion. The minimal and optimistic PPs can provide clinically meaningful cutoff/tradeoff values for the key attributes and help bring them onto the same scale in the utility calculation. In addition, utility levels derived from minimal, target, and optimistic PPs can be used as benchmarks to compare with the overall expected utility curve and to calculate the probability of reaching these PP utility levels based on the uncertainty in the overall utility. Weight assignment may require difficult tradeoff judgments, but sensitivity analysis can help show its impact on the overall utility.
In contrast to the traditional qualitative approach, the CUI, as a more structured quantitative approach, excels when multiple (more than two) important attributes (e.g., efficacy, safety, compliance) need to be taken into account in the benefit‐risk assessment. In this study, the CUI analysis did allow more transparent and efficient decision making for the phase III dose selection. Beyond dose selection, the CUI can also be applied to support other key decision making in drug development 4, 5, 6, 7, 8, 9, 10 and regulatory evaluation.11, 12, 13, 14
In this study, the E‐R relationship of rPFS was not statistically significant (P > 0.05). This is not unexpected given that the A.MARTIN phase II trial was for hypothesis generation and did not have adequate power to detect clinically meaningful differences between treatment and placebo arms at an ɑ (type 1 error) level of 5%.16 Accordingly, the uncertainty band was wide for the rPFS HR dose‐response projection (Figure 1), which was then carried forward to the utility calculations (Figures 4 and S5). In addition, to compare with responses at a higher dose, we cautiously extrapolated the response projections to 500 mg given the expected large overlap in predicted exposures between 400 and 500 mg. Because of the lack of observed data at 500 mg, relatively large uncertainty was shown in the response projections and CUI calculations.
In oncology clinical trials with advanced cancer patients, dose modifications, especially interruptions and reductions mainly because of AEs, are common. In such a case, using nominal dose‐derived exposure as a baseline predictor of response will not represent the underlying E‐R relationship. In the E‐R analyses here, we used exposure metrics based on both the nominal dose and actual dose (considering dose modifications). As shown in Figures 3 a,b and S4a–c, the E‐R trends in the models with AUCss of nominal dose are generally flatter than those in the models with AUCss,event. One possible reason is illustrated in Figure 5. In the case of dose interruptions/reductions, the true exposures (or doses) will decrease from the nominal doses (from solid red circles to open red circles). If nominal dose‐based exposures (green solid squares) are used to determine the E‐R relationship (green curve), the curve will be shifted clockwise from the true E‐R curve (red curve). Therefore, the E‐R trend without considering dose interruptions/reductions will generally be flatter than the true E‐R relationship. The distance between the true E‐R curve and the E‐R curve without considering dose modifications is determined by the DI level, and the DI level depends on the event (e.g., AEs, rPFS) type and its onset time. If the event is the cause of dose modifications (e.g., certain AEs), the DI level can be high and the distance between those two E‐R curves can be short because dose modifications happen after that event. If the event has a late onset and happens after most of the dose modifications, then the DI level can be low, and the two E‐R curves can be far apart.
Figure 5.
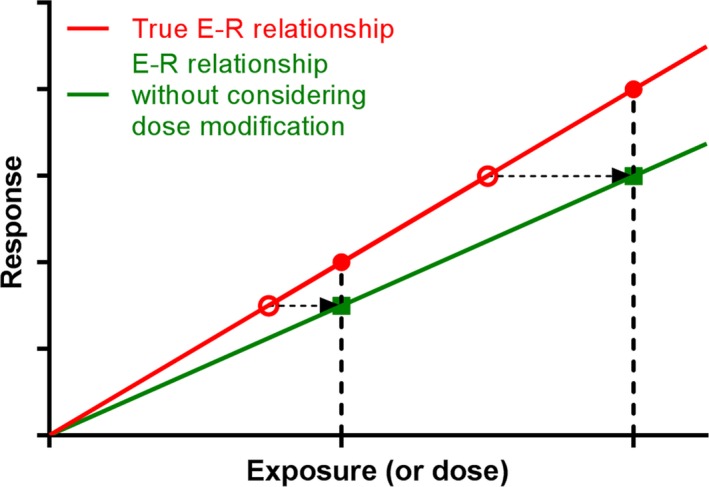
E‐R relationship with and without considering dose modifications (using interruptions/reductions as an example). Detailed descriptions are in the Discussion section. E‐R, exposure–response.
Despite the difference between E‐R trends (Figures 3 a,b and S4a–c), the dose‐response projections from these two models were similar (Figures 3 c,d and S4d–g). However, the E‐R model with exposure considering dose modifications has the flexibility of coupling with an alternative DI model to predict the response of a new trial (e.g., a trial with better management of AEs, leading to less dose modifications). Of note, using the exposure based on the actual dose may also be bias prone in this type of E‐R analysis. Therefore, we recommend using nominal dose‐based exposure for this type of E‐R analysis. If there is a need to capture the dose‐modification effect, a longitudinal PK‐pharmacodynamic modeling or a time‐to‐event modeling approach depending on the data type may be more comprehensive and appropriate.
In summary, this work characterized the ipatasertib E‐R relationships for both efficacy (rPFS) and safety (diarrhea and rash) in mCRPC. Based on the dose‐response relationships projected using coupled E‐R and DI models, PP‐driven CUI analysis balanced efficacy and safety to support dose selection for phase III. Similar analyses using this framework may be useful to support dose selection and other key decision making in clinical drug development. In addition, exposure metrics with and without considering dose modifications were found to produce different E‐R relationships but similar dose‐response projections. These results can be used to guide exposure metric selection in E‐R analyses in trials with sizable dose modifications.
Funding
This study was supported by Genentech, South San Francisco, CA.
Conflict of Interest
R.Z., Q.L., L.M., D.M., E.C., W.Y., H.M., J.Y.J., and N.B.: Genentech employees and Roche shareholders. B.P. and R.W.: Certara employees.
Author Contributions
R.Z., B.P., R.W., Q.L., L.M., D.M., E.C., W.Y., H.M., J.Y.J., and N.B. wrote the manuscript. N.B., R.Z., L.M., D.M., W.Y., and H.M. designed the research. R.Z., L.M., D.M., E.C., W.Y., and H.M. performed the research. R.Z., B.P., R.W., and Q.L. analyzed the data.
Supporting information
Figure S1. Visual predictive check of A.MARTIN phase II study ipatasertib concentration by doses.Figure S2. Dose‐intensity modeling results for radiographic progression‐free survival (a), grade ≥2 diarrhea (b), grade ≥3 diarrhea (c), grade ≥2 rash (d), and grade ≥3 rash (e).Figure S3. Exposure–adverse event logistic regression modeling results for grade ≥2 diarrhea (a), grade ≥3 diarrhea (b), grade ≥2 rash (c), and grade ≥3 rash (d).Figure S4. Comparison between exposure–response modeling with (area under the curve based on the average dose up to the event (AUCss,event) as exposure metric, left panels) and without (area under the curve based on nominal dose (AUCss) as exposure metric, right panels) considering dose modification.Figure S5. Clinical utility index distribution and probabilities of reaching product profiles vs. dose for scenario 1 (a), scenario 3 (b), and scenario 4 (c).
Table S1. Exposure–radiographic progression‐free survival hazard ratio Cox proportional‐hazards model parameter estimates.Table S2. Exposure–adverse event logistic regression model parameter estimates.
Data S1. Clinical utility index code for scenarios 3 and 4.
Data S2. Clinical utility index code for scenarios 1 and 2.
Acknowledgment
The authors thank Ms Sheila Dayog for providing data flow management support.
The results of the study were previously presented in part at the American Society of Clinical Pharmacology and Therapeutics meeting in March 2017 and Population Approach Group in Europe in June 2018.
References
- 1. Khan, A.A. , Perlstein, I. & Krishna, R. The use of clinical utility assessments in early clinical development. AAPS J 11, 33–38 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poland, B. et al The clinical utility index as a practical multiattribute approach to drug development decisions. Clin. Pharmacol. Ther. 86, 105–108 (2009). [DOI] [PubMed] [Google Scholar]
- 3. Ouellet, D. Benefit‐risk assessment: the use of clinical utility index. Expert. Opin. Drug Saf. 9, 289–300 (2010). [DOI] [PubMed] [Google Scholar]
- 4. de Greef‐van der Sandt, I. et al A quantitative benefit‐risk assessment approach to improve decision making in drug development: application of a multicriteria decision analysis model in the development of combination therapy for overactive bladder. Clin. Pharmacol. Ther. 99, 442–451 (2016). [DOI] [PubMed] [Google Scholar]
- 5. Freise, K.J. , Jones, A.K. , Verdugo, M.E. , Menon, R.M. , Maciag, P.C. & Salem, A.H. Moving beyond maximum tolerated dose for targeted oncology drugs: use of clinical utility index to optimize venetoclax dosage in multiple myeloma patients. Clin. Pharmacol. Ther. 102, 970–976 (2017). [DOI] [PubMed] [Google Scholar]
- 6. Ouellet, D. , Werth, J. , Parekh, N. , Feltner, D. , McCarthy, B. & Lalonde, R.L. The use of a clinical utility index to compare insomnia compounds: a quantitative basis for benefit‐risk assessment. Clin. Pharmacol. Ther. 85, 277–282 (2009). [DOI] [PubMed] [Google Scholar]
- 7. Manner, D.H. , Luo, J. , Qu, Y. , Berry, S. , Gaydos, B.L. & Jacober, S.J. A clinical utility index for selecting an optimal insulin dosing algorithm for LY2605541 in patients with type 2 diabetes pretreated with basal insulin. Diabetes Technol. Ther. 16, 499–505 (2014). [DOI] [PubMed] [Google Scholar]
- 8. Korsan, B. , Dykstra, K. & Pullman, W. Transparent tradeoffs: a clinical utility index openly evaluates a product's attributes and chance of success. In: Pharmaceutical Executive (2005).
- 9. Bettinger, T.L. , Shuler, G. , Jones, D.R. & Wilson, J.P. Schizophrenia: multi‐attribute utility theory approach to selection of atypical antipsychotics. Ann. Pharmacother. 41, 201–207 (2007). [DOI] [PubMed] [Google Scholar]
- 10. Dai, G. et al Determination of optimal dose and dosing regimen for phase II with clinical utility index: application to an anti‐tumor agent. Clin. Pharmacol. Ther., 81 (suppl.). Abstract PI‐23 (2007). [Google Scholar]
- 11. Raju, G.K. et al A benefit‐risk analysis approach to capture regulatory decision‐making: multiple myeloma. Clin. Pharmacol. Ther. 103, 67–76 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Raju, G.K. et al A benefit‐risk analysis approach to capture regulatory decision‐making: non‐small cell lung cancer. Clin. Pharmacol. Ther. 100, 672–684 (2016). [DOI] [PubMed] [Google Scholar]
- 13. Raju, G.K. et al Benefit‐risk analysis for decision‐making: an approach. Clin. Pharmacol. Ther. 100, 654–671 (2016). [DOI] [PubMed] [Google Scholar]
- 14. US Food and Drug Administration . Benefit‐risk assessment in drug regulatory decision‐making. Draft PDUFA VI Implementation Plan (FY 2018‐2022) <https://www.fda.gov/ForIndustry/UserFees/PrescriptionDrugUserFee/ucm326192.htm>. Accessed March 30, 2018.
- 15. Manning, B.D. & Cantley, L.C. AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Bono, J.S. et al Randomized phase II study of Akt blockade with or without ipatasertib in abiraterone‐treated patients with metastatic prostate cancer with and without PTEN loss. Clin. Cancer Res. 25, 928–936 (2019). [DOI] [PubMed] [Google Scholar]
- 17. Scher, H.I. et al Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J. Clin. Oncol. 26, 1148–1159 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saura, C. et al A first‐in‐human phase I study of the ATP‐competitive AKT inhibitor ipatasertib demonstrates robust and safe targeting of AKT in patients with solid tumors. Cancer Discov. 7, 102–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. US National Institutes of Health . Safety and clinical pharmacology of GDC‐0068 in combination with docetaxel, fluoropyrimidine plus oxaliplatin, paclitaxel, or enzalutamide in participants with advanced solid tumors <https://clinicaltrials.gov/show/NCT01362374> (2011). Accessed January 31, 2018.
- 20. US National Institutes of Health . <https://clinicaltrials.gov/show/NCT01562275> (2011). Accessed January 31, 2018.
- 21. Bang, Y.J. et al JAGUAR: a randomized phase II study of the AKT inhibitor ipatasertib (GDC‐0068) versus placebo in combination with mFOLFOX6 chemotherapy in patients (pts) with locally advanced or metastatic HER2‐negative gastric (G) or gastroesophageal junction (GEJ) adenocarcinoma. J. Clin. Oncol. 32 (5 suppl.). Abstract TPS4147 (2014). [Google Scholar]
- 22. R Foundation for Statistical Computing . R: a language and environment for statistical computing. <http://www.R-project.org/> (2014). Accessed January 31, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Visual predictive check of A.MARTIN phase II study ipatasertib concentration by doses.Figure S2. Dose‐intensity modeling results for radiographic progression‐free survival (a), grade ≥2 diarrhea (b), grade ≥3 diarrhea (c), grade ≥2 rash (d), and grade ≥3 rash (e).Figure S3. Exposure–adverse event logistic regression modeling results for grade ≥2 diarrhea (a), grade ≥3 diarrhea (b), grade ≥2 rash (c), and grade ≥3 rash (d).Figure S4. Comparison between exposure–response modeling with (area under the curve based on the average dose up to the event (AUCss,event) as exposure metric, left panels) and without (area under the curve based on nominal dose (AUCss) as exposure metric, right panels) considering dose modification.Figure S5. Clinical utility index distribution and probabilities of reaching product profiles vs. dose for scenario 1 (a), scenario 3 (b), and scenario 4 (c).
Table S1. Exposure–radiographic progression‐free survival hazard ratio Cox proportional‐hazards model parameter estimates.Table S2. Exposure–adverse event logistic regression model parameter estimates.
Data S1. Clinical utility index code for scenarios 3 and 4.
Data S2. Clinical utility index code for scenarios 1 and 2.