

Abstract
Deep sequencing genomic analysis is becoming increasingly common in clinical research and practice, enabling accurate identification of diagnostic, prognostic, and predictive determinants. Variant calling, distinguishing between true mutations and experimental errors, is a central task of genomic analysis and often requires sophisticated statistical, computational, and/or heuristic techniques. Although variant callers seek to overcome noise inherent in biological experiments, variant calling can be significantly affected by outside factors including those used to prepare, store, and analyze samples. The goal of this review is to discuss known experimental features, such as sample preparation, library preparation, and sequencing, alongside diverse biological and clinical variables, and evaluate their effect on variant caller selection and optimization.
Keywords: Variant calling, Genomics, Clinical oncology, Bioinformatics, Computational biology
Graphical Abstract
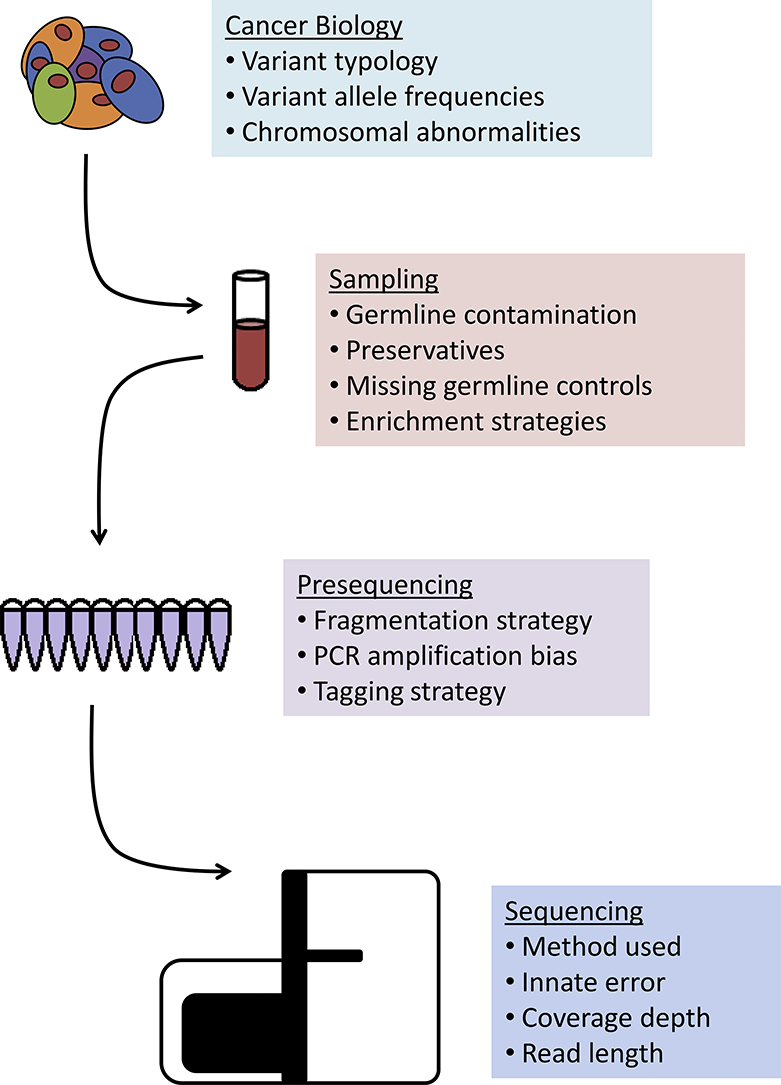
1. Introduction
Genomic sequencing is a multi-step process that converts clinical samples to actionable mutational knowledge. In a typical clinical genomics workflow, clinical investigators identify patients for whom genomic sequencing is appropriate, clinical and experimental staff are responsible for sample collection and storage, a sequencing group prepares the sample for sequencing and then runs it through a high-throughput sequencer, and the raw outputs of the sequencing are then given to computing specialists who are responsible for processing the data, which is then returned to the clinical investigators and experimentalists for interpretation and clinical decision-making. The accuracy of computational data analysis and its final interpretation can be significantly influenced by biological, clinical, and laboratory confounders, such as a sample preparation, storage, or the sequencing technology used [[1], [2], [3], [4], [5]]. Here, we will discuss common experimental and biological challenges faced in clinical genomic sequencing and how they affect a choice of computing tools used for their analysis.
Clinical genomic studies are generally focused on the identification of genetic variants from DNA sequencing data, where variants are defined as single nucleotide variants (SNVs), small insertions and deletions (indels), and structural variants (SVs). The primary computational challenge in DNA sequencing data analysis is identifying and differentiating “true variants” from “noise” for a given sample, a task referred to as variant calling. Variant callers are vastly diverse in terms of their core mathematical algorithms and acceptable inputs (Fig. 1), and we refer our readers to Xu [6] for his careful mathematical overview.
Fig. 1.

Comparison of common variant caller features in selected variant callers. A comparative analysis of variant callers with variant type identified, sequencing type, and major capabilities listed. Color codes: blue, feature present; orange, feature absent; yellow, possible with parameter tuning; white, insufficient data [111], [112], [113], [114], [115], [116], [117], [118], [119], [120], [121], [122], [123], [124], [125], [126], [128], [130].
The oldest group of variant callers used in clinical genomics includes those specifically designed for germline variant identification (i.e., hereditary variants naturally occurring in the human population), including HaplotypeCaller from the Genome Analysis Tool Kit (GATK) [7], MAQ [8], and SAMtools mpileup [9], which are still maintained and have shown reliable performance. The newer group of variant callers is primarily designed for somatic variant calling (i.e., detecting non-hereditary mutations in somatic cells, especially as they relate to carcinogenesis) that either use paired tumor-normal samples to distinguish between germline polymorphisms, somatic variants, and sequencing errors (examples include MuTect2 [10], VarDict [11], and VarScan2 [12]) or somatic callers that allow for unpaired sample use, important in the case of archival samples where matched normal samples are not available, an example of which is LoFreqStar [[13], [14], [15]]. More recently, sequencing methods have begun to include unique molecular identifiers (UMIs) or barcodes and require specialized callers such as DeepSNVMiner [16] and smCounter2 [17,18], with enhanced performance at low variant allele frequencies (VAFs, the percentage of reads containing a variant in a given sample) [[17], [18], [19], [20]].
Due to the diversity of variant callers and their outputs, some groups have generated holistic surveys of the variant caller landscape using simulated data sets [5] or standardized variant profiles, such as those generated by the Genome In A Bottle consortium [[21], [22], [23]]. Significant effort has also been devoted to a variety of crowdsourced competition-based benchmarking efforts [24,25]. Recently, there has been an effort to develop standardized benchmarking measures for variant calling, but these projects are still in their early phases [26]. The results of these efforts have been positive, but practical guides for how to translate them into clinical decision-making have yet to be developed.
As genomic analysis becomes more common in clinical practice, it is critical for the scientific community to understand not only how to accurately model mutations and experimental error but also how to choose an appropriate variant caller and how various aspects of clinical and laboratory workflow can affect the performance of the algorithms they use [27]. The goal of this review is to discuss known experimental features, such as sample preparation, library preparation, and sequencing alongside diverse biological and clinical features and evaluate their effect on variant caller selection and optimization.
2. Experimental Features: From Sample Preparation to Sequencing
The performance of variant callers can be affected by experimental confounders, such as sample preparation, library preparation, and sequencing technology (see Graphical Abstract), with several variant callers explicitly designed to address one or more of these experimental confounders.
2.1. Sample Acquisition and Storage
Sample acquisition, preparation, and storage protocols can play a major role in variant calling performance. In modern clinical genomics studies, pairing optimal sample preparation protocols with appropriate variant callers is critical to accurate variant identification.
2.1.1. Sampling and Contamination
For traditional clinical assays, such as cell counts or microscopic pathology, a small amount of cellular contamination associated with sample acquisition (e.g., epithelial cells from a needle puncture) has a negligible effect. Conversely, the presence of surrounding tissues in a genomic sample can affect the statistical error estimations used to quantify VAF and can adversely affect lower-bound variant detection limits (see Fig. 2 for a visual representation of the consequences of this effect) [2]. For example, healthy germline tissue could dilute the genetic material of cancer clones, thus artificially reducing the VAF of any somatic mutations. Significant germline contamination and low VAF may lead to false negative calls, or most often a failure to call true variants due to a reduction of VAF below cutoff values. Such contamination can be addressed by including paired germline samples [28,29], which allow variant callers such as JointSNVMix [30], Strelka [31,32], MuTect2 [10] or SNVSniffer [33] to account for both background error rates and germline variants, with the latter eliminated from somatic variant calls [2].
Fig. 2.

Effect of variant caller sensitivity and VAF on variant detection. Simulated traces of wild-type and variant frequencies in a genomic region. (A) Simulated germline sample showing most variant frequencies clustered around 0, 0.5, and 1. This allows variant calling algorithms to exclude VAFs outside of narrow ranges as experimental error. (B) Simulated cancer somatic sample showing wider ranges of variant frequencies, including very low frequencies. Optimal variant caller tuning can increase the sensitivity of variant callers to detect rare variants.
For more accurate estimation of background error rates, pooled unpaired normal samples can be coupled with paired germline samples and utilized in EBCall [34]. However, care must be taken to ensure any normal sample data are generated using the same sequencing workflow as that of the experimental samples because variations in library preparation method and sequencing platform can significantly alter background error rates (see Sections 2.2 and 2.3).
In cases when no paired germline samples are available, LoFreqStar [13] provides reliable outputs for tumor-only samples [2], yet still inferior accuracy when compared to the average performance when paired germline samples are used [2]. Furthermore, LumosVar [35] provides a creative solution to overcome the absence of germline paired samples by integrating multiple data sources, including known germline SNPs from the dbSNP database [36], pooled unpaired normal samples (when available), and an innovative expectation maximization approach for more accurate allelic copy number and clonal sample fraction estimation [35].
2.1.2. Preservation and Storage
Almost all clinical samples, whether fresh or from long-term storage, include preservative or stabilizer. The most common preservation method of archival tissue samples is formalin fixation and paraffin embedding (FFPE) [37,38]. FFPE samples are readily available and easy to store, but formalin fixation can have profound effects on variant calling because it introduces artificial base alterations and DNA fragmentation [4]. Similarly, the storage duration of an archival sample can negatively affect DNA extraction yield [39]. These alterations can affect genome alignment and increase estimated background error rates, making it more difficult to accurately detect rare variants [4]. Because of these aligner effects, it is important to select a variant caller that performs local realignments and/or performs well with large amounts of clipped reads (i.e., reads that have had some part of their sequence ignored or deleted during alignment), with VarDict [11] accomplishing both of these goals with a novel approach to address reads clipped during alignment (Fig. 1) as well as Platypus [40] and DeepSNVMiner [16,40].
2.2. Library Preparation
Before a sample can be sequenced, it must be processed to convert whole genomic DNA molecules into a collection of DNA fragments that are appropriate for accurate genomic sequencing and analysis. This process is referred to as library preparation. Library preparation workflows can vary depending on experimental goals, and some of the most important steps that might affect variant caller performance include DNA amplification, isolation of specific genomic regions of interest (e.g., in the case of exome or targeted sequencing), and ligation of index sequences and/or UMIs (Fig. 3) [41].
Fig. 3.

Detailed molecular steps involved in a typical library preparation protocol. Factors affecting variant caller performance at each stage of the library preparation workflow (whole genome sequencing is used as an example).
2.2.1. DNA Amplification
Amplification of DNA fragments during library preparation (i.e., DNA amplification) is the largest contributor to background error in many library preparation protocols [42]. Because most modern amplification strategies are based on polymerase chain reactions (PCR), they are subject to all of the biases of PCR, but these biases become exponentially more influential for low variant frequencies or relatively low-coverage regions of interest [43]. Common features of PCR-induced bias include overrepresentation of repetitive sequences and under-representation of high-GC regions [42], in addition to mutations that can be introduced by polymerase errors (Fig. 3) [43,44]. It is not currently fully understood how amplification-related biases affect the output of variant callers. However, UMI-aware variant callers, such as DeepSNVMiner [16], MAGERI [45], and smCounter2 [17,18] can use UMI sequences known beforehand to more accurately model sources of error in sequencing experiments, especially those generated by PCR amplification (see Section 2.2.3 for more details on UMI-based sequencing).
2.2.2. Isolation of Regions of Interest
Library preparation protocols can vary substantially depending on the sequencing modalities, which are utilized to enrich for specific regions of interest (i.e., exons, targeted genes, etc.). These sequencing modalities include whole genome sequencing (WGS, sequencing of both exons and introns of the genome), whole exome sequencing (WES, sequencing of exons of the genome) and targeted sequencing panels (sequencing of smaller numbers of genes of interest). For example, in WGS, library preparation includes DNA isolation and fragmentation (usually through sonication), with subsequent ligation and possible amplification. Library preparation for WES and targeted sequencing, when compared to WGS, often includes several extra steps related to capturing their specific fragments of interest. In particular, in WES, biotinylated probes hybridize to (or “capture”) DNA fragments associated with known exons, which are then isolated using streptavidin beads [46], while in targeted sequencing specific primers linked to sequencing tags enrich for the genes of interest, and this enrichment is an essential step of the protocol [47]. More detailed descriptions of these and other sequencing modalities can be found in the Illumina Sequencing Method Explorer [48].
The nature of different sequencing modalities and their subsequent library preparation result in variable capture efficiency, which can be especially problematic at borders between sequenced and sequenced regions [49] and in the presence of repetitive sequences and other regions that are traditionally difficult to align and call [50]. Furthermore, these result in substantial differences at a later sequencing step, such as different amounts of coverage (i.e., the percentage of the genome that has sequencing reads that align to it), sequencing depth (i.e., the mean number of reads that align to any given point in the genomic region of interest), and sequencing uniformity (i.e., the consistency of sequencing depth across the region of interest). By definition, WGS has higher coverage than WES or targeted sequencing, which have generally had higher depth at a cost of lower uniformity [51].
Thus it is important to select variant callers that have been benchmarked for use with specific sequencing modalities [51]. For example, variant callers such as MuTect2 [10] and Strelka2 [32] show better performance in sequencing modalities with higher depth and lower coverage, such as WES. In modalities that may have lower uniformity, such as targeted sequencing, it is important to select variant callers that perform local realignment, including LoFreqStar [13] and VarDict [11] (also see Fig. 1 and Table 1).
Table 1.
Variant callers and laboratory protocols to address selected sequencing scenarios.
| Features | Laboratory intervention | Recommended variant callers |
|---|---|---|
| Germline contamination | Germline control | JointSNVMix [30], Strelka2 [32], MuTect2 [10], SNVSniffer [33] |
| Highly admixed sample | Germline sample + normal controls | VarDict [11], Platypus [40], EBCall [34] |
| Archival FFPE sample | VarDict [11], Platypus [40], DeepSNVMiner [16] | |
| Cell-free DNA | iDES [20], SiNVICT [82] | |
| Unique molecular identifiers (UMIs) | DeepSNVMiner [16], iDES [20], MAGERI [45], smCounter2 [17,18] | |
| Exome sequencing | Dual-indexed sequencing | MuTect2 [10], Strelka2 [32], EBCall [34] |
| Targeted sequencing | LoFreqStar [13], VarDict [11] | |
| Bead-based sequencing | MuTect2 [10], VarDict [11], HapMuC [66] | |
| Single-molecule real-time (SMRT) | GenomicConsensus [70] | |
| Nanopore | PoreSeq [127] | |
| Important low-frequency variants | UMI-based sequencing | DeepSNVMiner [16], smCounter2 [18], MuTect2 [10], VarDict [11], Platypus [40] |
| Chromosomal instability | VarDict [11], Seurat [98], Platypus [40] | |
| Pediatric patient | MuTect2 [10], Platypus [40], SomVarIUS [129], Pisces [109] | |
| Hereditary disease | Germline control | VarDict [11], Platypus [40], Pisces [109], VarScan2 [12], HaplotypeCaller [7] |
2.2.3. Sequencing Identifiers
Nearly all modern library preparation approaches involve adding an identifier sequence (also known as “index sequences”) to the input DNA fragments, which is typically a unique 8 base pair-long sequence added to the 3′ end of the fragment. In most cases, the primary motivation for this molecular indexing is to identify/mark individual samples in a multiplexed sequencing run (i.e., a sequencing run containing multiple samples). However, the use of these index sequences can also result in errors known as index swaps, which occur during sequencing and library preparation [52]. Index swaps represent events in which a sample identifier sequence in a multiplexed sequencing run is swapped to that of another sample in the sample pool, effectively registering a DNA fragment as originating from the wrong sample. This swapping is believed to occur when a piece of DNA containing an index sequence, whether free in solution or attached to a fragment in its native sample, erroneously anneals to a fragment from a nearby sample during amplification [52].
Index swapping usually occurs at a low rate, yet there are situations that can lead to much higher rates of index swapping having a significant effect on variant calling [52]. For example, sequencers that use patterned flow cells with Exclusion Amplification (ExAmp) chemistry, such as Illumina HiSeqX, HiSeq4000, and NovaSeq platforms [52,53] can lead to more common index swaps, attributed to the excess of sequencing primers in the presence of specific ExAmp reagent mixes [53]. Exome sequencing per se is also associated with higher rates of index swapping due to PCR amplification during the exome capture process, when fragments from multiple samples in close spatial proximity may be bound by the same bead [52]. Such index swapping events occur in 3% of reads on average, yet can rise to up to 6% [52], which is sufficient to alter measured VAFs for affected samples in ways that may lead to both false positive and false negative calls.
Currently, the most reliable approaches to address index swapping are implemented at the library preparation phase of a sequencing experiment and include modifications to the sequencing chemistry to generate dual-indexed libraries, which for example is available in kit form from Illumina [53]. Computationally, the index swapping challenge has currently been extensively addressed for single-cell RNA sequencing applications [54,55], with high potential to be extended to other sequencing modalities in the future.
In addition to sample membership, sequencing identifiers have recently been successfully utilized in targeted sequencing for rare variants [56] and in such cases have been referred to as molecular barcodes or unique molecular identifiers (UMIs). UMIs are often longer than typical sequencing identifiers ranging from a minimum of 8 bases to over 28 bases, allowing variant callers to better distinguish between rare variants and errors resulting from library preparation or sequencing [57,58]. Fully utilizing data generated using UMI-based techniques usually requires specialized variant callers such as DeepSNVMiner [16], MAGERI [45], or smCounter2 [17,18]. Unfortunately, large amounts of genomic data lack these UMIs, although UMI-based sequencing may become standard practice in the future.
2.3. Sequencing Methods
Different sequencing methods have very different chemical and signal acquisition approaches, including slide-based and bead-based sequencing by synthesis, single molecule real-time (SMRT) sequencing, and nanopore sequencing, which can lead to fundamental differences in their sequence outputs [59]. Most commercial sequencing methods are characterized by high robustness with low error rates yet can result in systemic biases such as those introduced by amplification, similar to the effects of amplification during library preparation [60]. In fact, an early polymerase error during library preparation would result in a homogeneous distribution of aberrant sequences potentially mixed with non-aberrant, but an early polymerase error during slide-based sequencing by synthesis will result in a single spatially localized read. Many sequencing services and equipment providers continuously update their methods and kits to minimize known sources of error, but in some cases, these updates can involve new sequencing chemistry or detection methods that necessitate specialized variant calling tools or optimizations [6,57,58]. For example, Illumina now offers dual-indexed sequencing kits [53] to address the index swapping issue discussed in Section 2.2.3. Use of these kits improves variant calling confidence for rare variants by decreasing false negative results [52]. Molecular barcoding and its associated variant callers are another well recognized example of this type of specialization (Section 2.2.3) [16,17,20,45].
2.3.1. Slide-Based Sequencing by Synthesis (Illumina)
One of the most popular high-throughput sequencing methods in use today is slide- or plate-based sequencing by synthesis, which operates by generating a signal trace each time a new nucleotide is added to newly synthesized strands of DNA that are bound to the slide or plate in tight cluster [59,61,62]. There are a variety of different methods to generate these signal traces, but the most common is optical excitation and quenching [63]. Because sequencing by synthesis uses bridge PCR or a similar process to generate signals, it is vulnerable to amplification errors (see Section 2.2.1), especially early in the sequencing run, when a polymerase error may be propagated to all of the daughter strands of a given reaction cluster [64], thus altering the cluster's sequencing “trace” (i.e., the series of signals that correspond to each nucleotide in the sequencing fragment). However, these errors are generally biased to specific nucleotides and occur at known rates, so there is some potential for variant callers to model this error. Another related type of error that is especially prevalent in this sequencing method is index swapping, which is discussed in Section 2.2.3. Because slide-based sequencing machines are by far the most common, the majority of available genomic data has been generated with this method, and thus, the majority of variant callers, including Mutect2 [10], LoFreqStar [13], and JointSNVMix [30], have been primarily benchmarked and tested using these data or simulated data informed by these data.
2.3.2. Bead-Based Sequencing by Synthesis (Life Technologies/Ion Torrent)
Conceptually, bead-based sequencing methods are very similar to slide-based sequencing by synthesis, but the use of beads allows for additional flexibility when reading signals from the sequencing reaction because beads are not bound to the single plane of a slide. Notably, although some bead-based approaches use pyrosequencing, they can also use direct electronic excitation [61]. This difference in signaling detection can affect base quality scores and even result in sequence truncation [65], both of which can affect aligner and variant caller performance. Variant callers with options to set read quality cutoffs (such as MuTect2 [10]) may show better or more reliable performance for bead-based approaches. Furthermore, it can be beneficial to select variant callers that execute local realignment as a part of their method, such as VarDict or HapMuC [11,66].
2.3.3. Single-Molecule Real-Time Sequencing (Pacific Biosciences)
SMRT sequencing represents a significant conceptual departure from typical sequencing-by-synthesis methods. As its name suggests, this approach is capable of sequencing single long molecules of DNA, which it accomplishes using zero-mode waveguide chambers seeded with a single DNA polymerase [67]. This allows the technology to accurately detect the fluorescent output of a single nucleotide incorporation reaction. SMRT technology is capable of sequencing very large DNA molecules, often in the size of kilobases, which can have significant advantages in applications that involve highly repetitive sequences or other features not amenable to more conventional short-read alignments. However, SMRT sequencing's error profile is very different than that of typical sequencing-by-synthesis reactions [68]. For example, the error rate of SMRT sequencing is substantially higher, with a high propensity for indels, but each DNA molecule is analyzed in isolation without modification (rather than relying on clusters of synthesized sequences), so multiple passes over the same molecule can generate a more accurate consensus sequence [69]. These error profiles and long read lengths generally require specialized variant calling algorithms, such as GenomicConsensus [70] distributed by PacBio, and accurately aligning and calling variants from these data are currently an area of active research [71,72].
2.3.4. Nanopore Sequencing (Oxford)
As its name suggests, Oxford nanopore technology uses molecular motors to drive DNA molecules through nanopores, and the changes in electrical current induced as each base on the DNA molecule passes through the pore are measured and used to determine the sequence of the DNA molecule. Like other single-molecule methods (such as SMRT discussed in Section 2.3.3), this technology has relatively high error rates and produces long reads, so it requires specialized alignment and variant calling algorithms, including PoreSeq [71,72]. Although most long-read alignment and variant calling utilities support both SMRT and nanopore input data, their performance between the two technologies can differ significantly [69,73]. However, variant calling for long-read sequencing technologies is currently an active area of research, and disparities in variant calling performance between nanopore and SMRT sequencing studies are largely unknown [73].
3. Biological Features
In addition to experimental confounders discussed above, many of fundamental aspects of tissue and tumor biology are important for variant caller selection, with the two most important features for variant calling being the presence of rare variants and chromosomal instability.
3.1. Low Variant Allele Frequencies
Many treatment methods, including radiation and chemotherapy, can act as selective forces that drive clonal evolution in the tumor [74], characterized by the outgrowth of rare cell populations that are resistant to the outside factors. For instance, in treatment-resistant or relapsed liquid tumors, this is represented by a so-called minimal residual disease (MRD) [75,76], which often results in false negative variant calls as the variants may exhibit VAFs below the cutoff used by a typical variant caller. In these situations, sequencing techniques that utilize barcoding followed by variant callers designed to detect low VAFs, such as DeepSNVMiner [16] and smCounter2 [17,18], are advised, and if possible, longitudinal samples should be taken to monitor disease dynamics [77].
The accuracy of VAFs, as representative of the true variant frequencies in the tumor, can also be dependent on the sampling method used. For example, a core needle biopsy from a solid tumor might only sample a small area of the total tumor mass, and a rare variant in the biopsy may be a dominant variant in another region of the tumor. In this case, the reliability of VAFs as a diagnostic or prognostic criterion will be decreased. In such cases, paired tumor and germline control samples are recommended to help distinguish between germline and rare somatic variants. These paired samples can be used as inputs for any number of paired-sample variant callers (Fig. 1), including VarDict [11] or Platypus [40], which use germline control samples to model background error.
Another example in which detection of low VAFs is critical is variant calling from cell-free DNA (cfDNA) samples. The most recent highly promising use for cell-free DNA sequencing in oncology is identification and characterization of circulating tumor DNA [78], which allows for early diagnostic, prognostic, and predictive capabilities. Cell-free sequencing typically involves very small amounts of input DNA and relies on numerous rounds of PCR amplification to generate sufficient material for a sequencing run, thus resulting in the accumulation of amplification errors at a rate beyond that seen in more typical applications [79]. UMI-based sequencing methods and their associated variant callers (discussed in Section 2.2.3), such as DeepSNVMiner [16], MAGERI [45], and smCounter2 [17,18], have shown acceptable performance [80,81], however they might be insufficient due to higher than usual rates of amplification errors [18]. To overcome this limitation, a variety of cfDNA-specific software packages have been developed and/or extended such as SiNVICT [82] and iDES [20], the latter of which couples the advantages of UMI-based variant calling with “background polishing” to control for known sequencing errors. Other tools, such as PEC [79] are specifically designed to be incorporated into the existing variant calling pipelines and seek to specifically address PCR-related errors as the pre-processing step.
3.2. Chromosomal Instability
In addition to small insertions or deletions (i.e., indels) or single nucleotide mutations, many cancers are characterized by larger structural variants (SVs), which can be markers of disease aggressiveness and treatment outcomes [[83], [84], [85], [86]]. For most cancers, variants at every scale, from the loss of whole chromosomes or chromosome arms [87] to single nucleotide polymorphisms [1], can have varying degrees of clinical relevance, and there is evidence that these different scales of variants can have synergistic or novel effects when present in the same tumor [3], with potential interfaces between small-scale mutations and cytogenetic features [[88], [89], [90]]. A canonical example of this type of interaction between different variant types is Philadelphia chromosome and its role in chronic myelogenous leukemia. In particular, the Philadelphia chromosome is an SV generated by reciprocal translocation between chromosomes 9 and 22 that produces the BCR-ABL1 fusion protein. This fusion protein is a primary driver of chronic myelogenous leukemia pathogenesis and can be successfully targeted using a variety of therapies (e.g., imatinib) [91]. However, secondary point mutations in the kinase domain of the BCR-ABL1 fusion protein can confer resistance to treatment, which drastically affects patient outcome [92].
Many variant callers are focused on the identification of relatively small variants [11,30,[93], [94], [95], [96]] and do not explicitly take into account larger structural chromosomal alterations, which can be far more difficult to identify without specialized SV identification algorithms. In current clinical practice, most malignancies in which SVs are important, such as hematologic malignancies, are experimentally analyzed using dedicated cytogenetic methods such as fluorescence in situ hybridization (FISH) [83,84,97]. A few currently available variant callers do attempt to identify SVs, including VarDict [11], Seurat [98], and Platypus [40], but their accuracy and effectiveness has yet to exceed that of preferred cytogenetic laboratory methods.
4. Clinical Features
Clinical features, including but not limited to patient age and presence of heritable mutations, are also important considerations in variant caller selection.
4.1. Patient Age
Patient age is an important clinical factor in variant caller selection, especially for pediatric malignancies. Many pediatric cancers have genomic profiles that are significantly different from their adult counterparts [[99], [100], [101]]. Their somatic mutation frequency is generally lower, and they often have a smaller set of potentially relevant mutations [100]. Thus, variant callers with high sensitivity for identifying rare variants, such as MuTect2 [2,10], would be recommended. Furthermore, because SVs, especially translocations, also play key causative roles in many pediatric cancers [102,103], it may be important to select variant callers that can also accurately call SVs such as Platypus [40], but more reliable SV data may be generated by non-sequencing methods (see Section 3.2 for more information on SV calling).
4.2. Heritability
Several malignancies are associated with a variety of inherited variants that affect their diagnosis, prognosis, and clinical outcomes [[104], [105], [106]]. Some of the most common examples of these inherited cancer syndromes include Lynch syndrome, which represents a collection of polymorphisms in DNA mismatch repair genes that causes colon cancer [107], and hereditary breast and ovarian cancer (HBOC) syndrome, which represents polymorphisms in the BRCA1 or BRCA2 genes [108]. As inherited variants associated with cancer predisposition are germline rather than somatic, it is important to select a variant caller that distinguishes between germline and somatic mutations, such as VarDict [11], Platypus [40], Pisces [109], and VarScan2 [12]. An alternative is to specifically call variants from germline samples, in which case GATK's HaplotypeCaller [7] is routinely recommended. The accurate identification of these inherited variants in parents will not only help with diagnosis, prognosis, and disease monitoring, but also guide preventative measures, such as more aggressive prophylactic monitoring in offspring [110].
5. Conclusions
Variant calling is one of the canonical challenges in computational genomics. Variant callers are both numerous and diverse in terms of algorithmic designs as well as intended uses. Some variant callers are designed for broad use with a wide variety of sample types and sequencing workflows, whereas others are optimized for a single class of sample types or a single sequencing method. However, regardless of design goals, all variant callers must solve the fundamental problem of distinguishing between true mutations and experimental noise. In many cases, the frequency of rare variants can drift below the levels of experimental noise in a system, which makes the accurate variant calling for such cases extremely challenging. This challenge in distinguishing true variants from noise can be partially addressed by selecting variant callers with the best performance for a given data set. Optimizing variant calling in complex clinical environments requires detailed understanding of both the clinical and laboratory workflows, including sample acquisition and sequencing approach, as well as disease features. Knowledge of these factors allows selection of optimal variant calling pipeline for each genomic experiment, enabling most biologically relevant data interpretation.
Declarations of Interest
None.
Financial Support
Antonina Mitrofanova is supported by Rutgers School of Health Professions Start-up funds.
Contributor Information
Zachary S. Bohannan, Email: zach.bohannan@shp.rutgers.edu.
Antonina Mitrofanova, Email: antonina.mitrofanova@rutgers.edu.
References
- 1.Deng N., Zhou H., Fan H., Yuan Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget. 2017;8:110635–110649. doi: 10.18632/oncotarget.22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bohnert R., Vivas S., Jansen G. Comprehensive benchmarking of SNV callers for highly admixed tumor data. PLoS One. 2017;12 doi: 10.1371/journal.pone.0186175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan S.H., Ngeow J. Germline mutation contribution to chromosomal instability. Endocr Relat Cancer. 2017;24:T33–T46. doi: 10.1530/ERC-17-0062. [DOI] [PubMed] [Google Scholar]
- 4.Oh E., Choi Y.L., Kwon M.J., Kim R.N., Kim Y.J. Comparison of accuracy of whole-exome sequencing with formalin-fixed paraffin-embedded and fresh frozen tissue samples. PLoS One. 2015;10 doi: 10.1371/journal.pone.0144162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laehnemann D., Borkhardt A., McHardy A.C. Denoising DNA deep sequencing data—high-throughput sequencing errors and their correction. Brief Bioinform. 2016;17:154–179. doi: 10.1093/bib/bbv029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu C. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput Struct Biotechnol J. 2018;16:15–24. doi: 10.1016/j.csbj.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lai Z., Markovets A., Ahdesmaki M., Chapman B., Hofmann O. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016;44:e108. doi: 10.1093/nar/gkw227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilm A., Aw P.P., Bertrand D., Yeo G.H., Ong S.H. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012;40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raymond V.M., Gray S.W., Roychowdhury S., Joffe S., Chinnaiyan A.M. Germline findings in tumor-only sequencing: points to consider for clinicians and laboratories. J Natl Cancer Inst. 2016;108 doi: 10.1093/jnci/djv351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones S., Anagnostou V., Lytle K., Parpart-Li S., Nesselbush M. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med. 2015;7 doi: 10.1126/scitranslmed.aaa7161. (283ra53) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andrews T.D., Jeelall Y., Talaulikar D., Goodnow C.C., Field M.A. DeepSNVMiner: a sequence analysis tool to detect emergent, rare mutations in subsets of cell populations. PeerJ. 2016;4 doi: 10.7717/peerj.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu C., Nezami Ranjbar M.R., Wu Z., DiCarlo J., Wang Y. Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller. BMC Genomics. 2017;18:5. doi: 10.1186/s12864-016-3425-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu C., Gu X., Padmanabhan R., Wu Z., Peng Q. smCounter2: an accurate low-frequency variant caller for targeted sequencing data with unique molecular identifiers. Bioinformatics. 2018 doi: 10.1093/bioinformatics/bty790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng Q., Vijaya Satya R., Lewis M., Randad P., Wang Y. Reducing amplification artifacts in high multiplex amplicon sequencing by using molecular barcodes. BMC Genomics. 2015;16:589. doi: 10.1186/s12864-015-1806-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newman A.M., Lovejoy A.F., Klass D.M., Kurtz D.M., Chabon J.J. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547–555. doi: 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zook J.M., Chapman B., Wang J., Mittelman D., Hofmann O. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol. 2014;32:246–251. doi: 10.1038/nbt.2835. [DOI] [PubMed] [Google Scholar]
- 22.Hwang S., Kim E., Lee I., Marcotte E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci Rep. 2015;5:17875. doi: 10.1038/srep17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cornish A., Guda C. A comparison of variant calling pipelines using genome in a bottle as a reference. Biomed Res Int. 2015;2015:456479. doi: 10.1155/2015/456479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boutros P.C., Margolin A.A., Stuart J.M., Califano A., Stolovitzky G. Toward better benchmarking: challenge-based methods assessment in cancer genomics. Genome Biol. 2014;15:462. doi: 10.1186/s13059-014-0462-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boutros P.C., Ewing A.D., Ellrott K., Norman T.C., Dang K.K. Global optimization of somatic variant identification in cancer genomes with a global community challenge. Nat Genet. 2014;46:318–319. doi: 10.1038/ng.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krusche P., Trigg L., Boutros P.C., Mason C.E., De La Vega F.M. Best practices for benchmarking germline small variant calls in human genomes. Nat Biotechnol. 2019 doi: 10.1038/s41587-019-0054-x. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buckley A.R., Standish K.A., Bhutani K., Ideker T., Lasken R.S. Pan-cancer analysis reveals technical artifacts in TCGA germline variant calls. BMC Genomics. 2017;18:458. doi: 10.1186/s12864-017-3770-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robison K. Application of second-generation sequencing to cancer genomics. Brief Bioinform. 2010;11:524–534. doi: 10.1093/bib/bbq013. [DOI] [PubMed] [Google Scholar]
- 30.Roth A., Ding J., Morin R., Crisan A., Ha G. JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics. 2012;28:907–913. doi: 10.1093/bioinformatics/bts053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saunders C.T., Wong W.S., Swamy S., Becq J., Murray L.J. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 32.Kim S., Scheffler K., Halpern A.L., Bekritsky M.A., Noh E. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods. 2018;15:591–594. doi: 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y., Loewer M., Aluru S., Schmidt B. SNVSniffer: an integrated caller for germline and somatic single-nucleotide and indel mutations. BMC Syst Biol. 2016;10(Suppl. 2):47. doi: 10.1186/s12918-016-0300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shiraishi Y., Sato Y., Chiba K., Okuno Y., Nagata Y. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013;41 doi: 10.1093/nar/gkt126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halperin R.F., Carpten J.D., Manojlovic Z., Aldrich J., Keats J. A method to reduce ancestry related germline false positives in tumor only somatic variant calling. BMC Med Genomics. 2017;10:61. doi: 10.1186/s12920-017-0296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paskal W., Paskal A.M., Debski T., Gryziak M., Jaworowski J. Aspects of modern biobank activity - comprehensive review. Pathol Oncol Res. 2018;24(4):771–785. doi: 10.1007/s12253-018-0418-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donczo B., Guttman A. Biomedical analysis of formalin-fixed, paraffin-embedded tissue samples: the holy grail for molecular diagnostics. J Pharm Biomed Anal. 2018;155:125–134. doi: 10.1016/j.jpba.2018.03.065. [DOI] [PubMed] [Google Scholar]
- 39.Schroder C., Steimer W. gDNA extraction yield and methylation status of blood samples are affected by long-term storage conditions. PLoS One. 2018;13 doi: 10.1371/journal.pone.0192414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rimmer A., Phan H., Mathieson I., Iqbal Z., Twigg S.R.F. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46:912–918. doi: 10.1038/ng.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J.R., Quach B., Furey T.S. Correcting nucleotide-specific biases in high-throughput sequencing data. BMC Bioinforma. 2017;18:357. doi: 10.1186/s12859-017-1766-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aird D., Ross M.G., Chen W.-S., Danielsson M., Fennell T. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011;12:R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sabina J., Leamon J.H. Bias in whole genome amplification: causes and considerations. Methods Mol Biol. 2015;1347:15–41. doi: 10.1007/978-1-4939-2990-0_2. [DOI] [PubMed] [Google Scholar]
- 44.Chu W.K., Edge P., Lee H.S., Bansal V., Bafna V. Ultraaccurate genome sequencing and haplotyping of single human cells. Proc Natl Acad Sci U S A. 2017;114:12512–12517. doi: 10.1073/pnas.1707609114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shugay M., Zaretsky A.R., Shagin D.A., Shagina I.A., Volchenkov I.A. MAGERI: computational pipeline for molecular-barcoded targeted resequencing. PLoS Comput Biol. 2017;13 doi: 10.1371/journal.pcbi.1005480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Warr A., Robert C., Hume D., Archibald A., Deeb N. Vol. 5. 2015. Exome Sequencing: Current and Future Perspectives. G3 (Bethesda) (1543-50) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kou R., Lam H., Duan H., Ye L., Jongkam N. Benefits and challenges with applying unique molecular identifiers in next generation sequencing to detect low frequency mutations. PLoS One. 2016;11:e0146638. doi: 10.1371/journal.pone.0146638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Illumina . 2019. Sequencing method explorer. [Google Scholar]
- 49.Meienberg J., Zerjavic K., Keller I., Okoniewski M., Patrignani A. New insights into the performance of human whole-exome capture platforms. Nucleic Acids Res. 2015;43 doi: 10.1093/nar/gkv216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dolgalev I., Sedlazeck F., Busby B. 2017. DangerTrack: A scoring system to detect difficult-to-assess regions; p. 443. F1000Res 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meynert A.M., Ansari M., FitzPatrick D.R., Taylor M.S. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinforma. 2014;15:247. doi: 10.1186/1471-2105-15-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Costello M., Fleharty M., Abreu J., Farjoun Y., Ferriera S. Characterization and remediation of sample index swaps by non-redundant dual indexing on massively parallel sequencing platforms. BMC Genomics. 2018;19:332. doi: 10.1186/s12864-018-4703-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Illumina . 2017. Effects of index misalignment on multiplexing and downstream analysis. [Google Scholar]
- 54.Griffiths J.A., Richard A.C., Bach K., Lun A.T.L., Marioni J.C. Detection and removal of barcode swapping in single-cell RNA-seq data. Nat Commun. 2018;9:2667. doi: 10.1038/s41467-018-05083-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Larsson A.J.M., Stanley G., Sinha R., Weissman I.L., Sandberg R. Computational correction of index switching in multiplexed sequencing libraries. Nat Methods. 2018;15:305–307. doi: 10.1038/nmeth.4666. [DOI] [PubMed] [Google Scholar]
- 56.Kim M.J., Kim S.C., Kim Y.J. A universal analysis pipeline of hybrid capture-based targeted sequencing data with unique molecular indexes (UMIs) Genomics Inform. 2018;16 doi: 10.5808/GI.2018.16.4.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jabara C.B., Jones C.D., Roach J., Anderson J.A., Swanstrom R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a primer ID. Proc Natl Acad Sci U S A. 2011;108:20166–20171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmitt M.W., Kennedy S.R., Salk J.J., Fox E.J., Hiatt J.B. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A. 2012;109:14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reuter J.A., Spacek D.V., Snyder M.P. High-throughput sequencing technologies. Mol Cell. 2015;58:586–597. doi: 10.1016/j.molcel.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ross M.G., Russ C., Costello M., Hollinger A., Lennon N.J. Characterizing and measuring bias in sequence data. Genome Biol. 2013;14:R51. doi: 10.1186/gb-2013-14-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goodwin S., McPherson J.D., McCombie W.R. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yohe S., Thyagarajan B. Review of clinical next-generation sequencing. Arch Pathol Lab Med. 2017;141:1544–1557. doi: 10.5858/arpa.2016-0501-RA. [DOI] [PubMed] [Google Scholar]
- 63.Bentley D.R., Balasubramanian S., Swerdlow H.P., Smith G.P., Milton J. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Paz A.M., Cybulski T.R., Marblestone A.H., Zamft B.M., Church G.M. High-resolution mapping of DNA polymerase fidelity using nucleotide imbalances and next-generation sequencing. Nucleic Acids Res. 2018;46(13):e78. doi: 10.1093/nar/gky296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salipante S.J., Kawashima T., Rosenthal C., Hoogestraat D.R., Cummings L.A. Performance comparison of Illumina and ion torrent next-generation sequencing platforms for 16S rRNA-based bacterial community profiling. Appl Environ Microbiol. 2014;80:7583–7591. doi: 10.1128/AEM.02206-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Usuyama N., Shiraishi Y., Sato Y., Kume H., Homma Y. HapMuC: somatic mutation calling using heterozygous germ line variants near candidate mutations. Bioinformatics. 2014;30:3302–3309. doi: 10.1093/bioinformatics/btu537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eid J., Fehr A., Gray J., Luong K., Lyle J. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 68.Yang Y., Botton M.R., Scott E.R., Scott S.A. Sequencing the CYP2D6 gene: from variant allele discovery to clinical pharmacogenetic testing. Pharmacogenomics. 2017;18:673–685. doi: 10.2217/pgs-2017-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chu J., Mohamadi H., Warren R.L., Yang C., Birol I. Innovations and challenges in detecting long read overlaps: an evaluation of the state-of-the-art. Bioinformatics. 2017;33:1261–1270. doi: 10.1093/bioinformatics/btw811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.PacBio . 2019. GenomicConsensus. [Google Scholar]
- 71.Berlin K., Koren S., Chin C.S., Drake J.P., Landolin J.M. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol. 2015;33:623–630. doi: 10.1038/nbt.3238. [DOI] [PubMed] [Google Scholar]
- 72.Li H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics. 2016;32:2103–2110. doi: 10.1093/bioinformatics/btw152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Magi A., Semeraro R., Mingrino A., Giusti B., D'Aurizio R. Nanopore sequencing data analysis: state of the art, applications and challenges. Brief Bioinform. 2017;19(6):1256–1272. doi: 10.1093/bib/bbx062. [DOI] [PubMed] [Google Scholar]
- 74.Zhao B., Hemann M.T., Lauffenburger D.A. Modeling tumor clonal evolution for drug combinations design. Trends Cancer. 2016;2:144–158. doi: 10.1016/j.trecan.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Malmberg E.D., Rehammar A., Pereira M.B., Abrahamsson J., Samuelsson T. Accurate and sensitive analysis of minimal residual disease in acute myeloid leukemia using deep sequencing of single nucleotide variations. J Mol Diagn. 2018;21(1):149–162. doi: 10.1016/j.jmoldx.2018.08.004. [DOI] [PubMed] [Google Scholar]
- 76.Perrot A., Lauwers-Cances V., Corre J., Robillard N., Hulin C. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood. 2018;132(23):2456–2464. doi: 10.1182/blood-2018-06-858613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dogliotti I., Drandi D., Genuardi E., Ferrero S. New molecular technologies for minimal residual disease evaluation in B-cell lymphoid malignancies. J Clin Med. 2018;7 doi: 10.3390/jcm7090288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yi X., Ma J., Guan Y., Chen R., Yang L. The feasibility of using mutation detection in ctDNA to assess tumor dynamics. Int J Cancer. 2017;140:2642–2647. doi: 10.1002/ijc.30620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim C.S., Mohan S., Ayub M., Rothwell D.G., Dive C. In silico error correction improves cfDNA mutation calling. Bioinformatics. 2018 doi: 10.1093/bioinformatics/bty1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang N., Li Y., Liu Z., Qin H., Du D. The characteristics of ctDNA reveal the high complexity in matching the corresponding tumor tissues. BMC Cancer. 2018;18:319. doi: 10.1186/s12885-018-4199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kukita Y., Matoba R., Uchida J., Hamakawa T., Doki Y. High-fidelity target sequencing of individual molecules identified using barcode sequences: de novo detection and absolute quantitation of mutations in plasma cell-free DNA from cancer patients. DNA Res. 2015;22:269–277. doi: 10.1093/dnares/dsv010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kockan C., Hach F., Sarrafi I., Bell R.H., McConeghy B. SiNVICT: ultra-sensitive detection of single nucleotide variants and indels in circulating tumour DNA. Bioinformatics. 2017;33:26–34. doi: 10.1093/bioinformatics/btw536. [DOI] [PubMed] [Google Scholar]
- 83.Wan T.S. Cancer cytogenetics: an introduction. Methods Mol Biol. 2017;1541:1–10. doi: 10.1007/978-1-4939-6703-2_1. [DOI] [PubMed] [Google Scholar]
- 84.Palumbo E., Russo A. Chromosome imbalances in cancer: molecular cytogenetics meets genomics. Cytogenet Genome Res. 2016;150:176–184. doi: 10.1159/000455804. [DOI] [PubMed] [Google Scholar]
- 85.Grade M., Difilippantonio M.J., Camps J. Patterns of chromosomal aberrations in solid tumors. Recent Results Cancer Res. 2015;200:115–142. doi: 10.1007/978-3-319-20291-4_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mrozek K., Harper D.P., Aplan P.D. Cytogenetics and molecular genetics of acute lymphoblastic leukemia. Hematol Oncol Clin North Am. 2009;23:991–1010. doi: 10.1016/j.hoc.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sansregret L., Vanhaesebroeck B., Swanton C. Determinants and clinical implications of chromosomal instability in cancer. Nat Rev Clin Oncol. 2018;15(3):139–150. doi: 10.1038/nrclinonc.2017.198. [DOI] [PubMed] [Google Scholar]
- 88.Moorman A.V., Enshaei A., Schwab C., Wade R., Chilton L. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood. 2014;124:1434–1444. doi: 10.1182/blood-2014-03-562918. [DOI] [PubMed] [Google Scholar]
- 89.Idossa D., Lasho T.L., Finke C.M., Ketterling R.P., Patnaik M.M. Mutations and karyotype predict treatment response in myelodysplastic syndromes. Am J Hematol. 2018;93(11):1420–1426. doi: 10.1002/ajh.25267. [DOI] [PubMed] [Google Scholar]
- 90.Brown A., Geiger H. Chromosome integrity checkpoints in stem and progenitor cells: transitions upon differentiation, pathogenesis, and aging. Cell Mol Life Sci. 2018;75:3771–3779. doi: 10.1007/s00018-018-2891-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Druker B.J. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112:4808–4817. doi: 10.1182/blood-2008-07-077958. [DOI] [PubMed] [Google Scholar]
- 92.O'Hare T., Eide C.A., Deininger M.W. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110:2242–2249. doi: 10.1182/blood-2007-03-066936. [DOI] [PubMed] [Google Scholar]
- 93.Koboldt D.C., Chen K., Wylie T., Larson D.E., McLellan M.D. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Larson D.E., Harris C.C., Chen K., Koboldt D.C., Abbott T.E. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fang L.T., Afshar P.T., Chhibber A., Mohiyuddin M., Fan Y. An ensemble approach to accurately detect somatic mutations using SomaticSeq. Genome Biol. 2015;16:197. doi: 10.1186/s13059-015-0758-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goya R., Sun M.G., Morin R.D., Leung G., Ha G. SNVMix: predicting single nucleotide variants from next-generation sequencing of tumors. Bioinformatics. 2010;26:730–736. doi: 10.1093/bioinformatics/btq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rack K.A., van den Berg E., Haferlach C., Beverloo H.B., Costa D. European recommendations and quality assurance for cytogenomic analysis of haematological neoplasms. Leukemia. 2019 doi: 10.1038/s41375-019-0378-z. https://www.nature.com/articles/s41375-019-0378-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Christoforides A., Carpten J.D., Weiss G.J., Demeure M.J., Von Hoff D.D. Identification of somatic mutations in cancer through Bayesian-based analysis of sequenced genome pairs. BMC Genomics. 2013;14:302. doi: 10.1186/1471-2164-14-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sturm D., Bender S., Jones D.T., Lichter P., Grill J. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer. 2014;14:92–107. doi: 10.1038/nrc3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tarlock K., Meshinchi S. Pediatric acute myeloid leukemia: biology and therapeutic implications of genomic variants. Pediatr Clin North Am. 2015;62:75–93. doi: 10.1016/j.pcl.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 102.Northcott P.A., Lee C., Zichner T., Stutz A.M., Erkek S. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511:428–434. doi: 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen X., Bahrami A., Pappo A., Easton J., Dalton J. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7:104–112. doi: 10.1016/j.celrep.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ohmoto A., Yachida S., Morizane C. Genomic features and clinical management of patients with hereditary pancreatic cancer syndromes and familial pancreatic cancer. Int J Mol Sci. 2019;20 doi: 10.3390/ijms20030561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Alldredge J., Randall L. Germline and somatic tumor testing in gynecologic cancer care. Obstet Gynecol Clin North Am. 2019;46:37–53. doi: 10.1016/j.ogc.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Couch F.J., Shimelis H., Hu C., Hart S.N., Polley E.C. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017;3:1190–1196. doi: 10.1001/jamaoncol.2017.0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carethers J.M., Stoffel E.M. Lynch syndrome and lynch syndrome mimics: the growing complex landscape of hereditary colon cancer. World J Gastroenterol. 2015;21:9253–9261. doi: 10.3748/wjg.v21.i31.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hoang L.N., Gilks B.C. Hereditary breast and ovarian cancer syndrome: moving beyond BRCA1 and BRCA2. Adv Anat Pathol. 2018;25:85–95. doi: 10.1097/PAP.0000000000000177. [DOI] [PubMed] [Google Scholar]
- 109.Dunn T., Berry G., Emig-Agius D., Jiang Y., Lei S. Pisces: an accurate and versatile variant caller for somatic and germline next-generation sequencing data. Bioinformatics. 2018 doi: 10.1093/bioinformatics/bty849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Green R.F., Ari M., Kolor K., Dotson W.D., Bowen S. Evaluating the role of public health in implementation of genomics-related recommendations: a case study of hereditary cancers using the CDC science impact framework. Genet Med. 2019;21:28–37. doi: 10.1038/s41436-018-0028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cantarel B.L., Weaver D., McNeill N., Zhang J., Mackey A.J. BAYSIC: a Bayesian method for combining sets of genome variants with improved specificity and sensitivity. BMC Bioinforma. 2014;15:104. doi: 10.1186/1471-2105-15-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.CaVEMan . 2014. Genome Research Ltd. [Google Scholar]
- 113.Gerstung M., Beisel C., Rechsteiner M., Wild P., Schraml P. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun. 2012;3:811. doi: 10.1038/ncomms1814. [DOI] [PubMed] [Google Scholar]
- 114.Wang W., Wang P., Xu F., Luo R., Wong M.P. FaSD-somatic: a fast and accurate somatic SNV detection algorithm for cancer genome sequencing data. Bioinformatics. 2014;30:2498–2500. doi: 10.1093/bioinformatics/btu338. [DOI] [PubMed] [Google Scholar]
- 115.Garrison E., Marth G. 2012. Haplotype-based variant detection from short-read sequencing. [arXiv:1207.3907 [q-bio]] [Google Scholar]
- 116.Sengupta S., Gulukota K., Zhu Y., Ober C., Naughton K. Ultra-fast local-haplotype variant calling using paired-end DNA-sequencing data reveals somatic mosaicism in tumor and normal blood samples. Nucleic Acids Res. 2016;44 doi: 10.1093/nar/gkv953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Carrot-Zhang J., Majewski J. LoLoPicker: detecting low allelic-fraction variants from low-quality cancer samples. Oncotarget. 2017;8:37032–37040. doi: 10.18632/oncotarget.16144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ding J., Bashashati A., Roth A., Oloumi A., Tse K. Feature-based classifiers for somatic mutation detection in tumour-normal paired sequencing data. Bioinformatics. 2012;28:167–175. doi: 10.1093/bioinformatics/btr629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fan Y., Xi L., Hughes D.S., Zhang J., Zhang J. MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 2016;17:178. doi: 10.1186/s13059-016-1029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kassahn K.S., Holmes O., Nones K., Patch A.M., Miller D.K. Somatic point mutation calling in low cellularity tumors. PLoS One. 2013;8 doi: 10.1371/journal.pone.0074380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Radenbaugh A.J., Ma S., Ewing A., Stuart J.M., Collisson E.A. RADIA: RNA and DNA integrated analysis for somatic mutation detection. PLoS One. 2014;9 doi: 10.1371/journal.pone.0111516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hansen N.F., Gartner J.J., Mei L., Samuels Y., Mullikin J.C. Shimmer: detection of genetic alterations in tumors using next-generation sequence data. Bioinformatics. 2013;29:1498–1503. doi: 10.1093/bioinformatics/btt183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Spinella J.F., Mehanna P., Vidal R., Saillour V., Cassart P. SNooPer: a machine learning-based method for somatic variant identification from low-pass next-generation sequencing. BMC Genomics. 2016;17:912. doi: 10.1186/s12864-016-3281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim S., Jeong K., Bhutani K., Lee J., Patel A. Virmid: accurate detection of somatic mutations with sample impurity inference. Genome Biol. 2013;14:R90. doi: 10.1186/gb-2013-14-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kalatskaya I., Trinh Q.M., Spears M., McPherson J.D., Bartlett J.M.S. ISOWN: accurate somatic mutation identification in the absence of normal tissue controls. Genome Med. 2017;9:59. doi: 10.1186/s13073-017-0446-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Muller E., Goardon N., Brault B., Rousselin A., Paimparay G. OutLyzer: software for extracting low-allele-frequency tumor mutations from sequencing background noise in clinical practice. Oncotarget. 2016;7:79485–79493. doi: 10.18632/oncotarget.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Szalay T., Golovchenko J.A. De novo sequencing and variant calling with nanopores using PoreSeq. Nat Biotechnol. 2015;33:1087–1091. doi: 10.1038/nbt.3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wei Z., Wang W., Hu P., Lyon G.J., Hakonarson H. SNVer: a statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic Acids Res. 2011;39:e132. doi: 10.1093/nar/gkr599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Smith K.S., Yadav V.K., Pei S., Pollyea D.A., Jordan C.T. SomVarIUS: somatic variant identification from unpaired tissue samples. Bioinformatics. 2016;32:808–813. doi: 10.1093/bioinformatics/btv685. [DOI] [PubMed] [Google Scholar]
- 130.Vallania F.L., Druley T.E., Ramos E., Wang J., Borecki I. High-throughput discovery of rare insertions and deletions in large cohorts. Genome Res. 2010;20:1711–1718. doi: 10.1101/gr.109157.110. [DOI] [PMC free article] [PubMed] [Google Scholar]