

Abstract
Background
Patients prefer oral to intravenous (IV) palliative chemotherapy, provided that oral therapy is not less effective. We compared the efficacy and safety of oral and IV fluoropyrimidines for treatment of colorectal cancer (CRC).
Objectives
To compare the effects of oral and IV fluoropyrimidine chemotherapy in patients treated with curative or palliative intent for CRC.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL; 2016, Issue 5), along with OVID MEDLINE, OVID Embase, and Web of Science databases, in June 2016. We also searched five clinical trials registers, several conference proceedings, and reference lists from study reports and systematic reviews. We contacted pharmaceutical companies to identify additional studies.
Selection criteria
We included randomised controlled trials (RCTs) comparing oral and IV fluoropyrimidine chemotherapy in patients treated with curative or palliative intent for CRC.
Data collection and analysis
Three review authors extracted data and assessed risk of bias independently. We assessed the seven domains in the Cochrane 'Risk of bias' tool and three additional domains: schedules of outcome assessment and/or follow‐up; use of intention‐to‐treat analysis; and baseline comparability of treatment arms.
Main results
We included nine RCTs (total of 10,918 participants) that examined treatment with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy. We included 35 RCTs (total of 12,592 participants) that examined treatment with palliative intent for inoperable advanced or metastatic CRC with chemotherapy (31 first‐line studies, two second‐line studies, and two studies of first‐ or second‐line chemotherapy). All studies included male and female participants, and no studies included participants younger than 18 years of age.
Patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy
• Disease‐free survival (DFS): DFS did not differ between participants treated with oral versus IV fluoropyrimidines (hazard ratio (HR) 0.93, 95% confidence interval (CI) 0.87 to 1.00; seven studies, 8903 participants; moderate‐quality evidence).
• Overall survival (OS): OS did not differ between participants treated with oral versus IV fluoropyrimidines (HR 0.92, 95% CI 0.84 to 1.00; seven studies, 8902 participants analysed; high‐quality evidence).
• Grade ≥ 3 adverse events (AEs): Participants treated with oral fluoropyrimidines experienced less grade ≥ 3 neutropenia/granulocytopenia (odds ratio (OR) 0.14, 95% CI 0.11 to 0.16; seven studies, 8087 participants; moderate‐quality evidence), stomatitis (OR 0.21, 95% CI 0.14 to 0.30; five studies, 4212 participants; low‐quality evidence), and any grade ≥ 3 AEs (OR 0.82, 95% CI 0.74 to 0.90; five studies, 7741 participants; low‐quality evidence). There was more grade ≥ 3 hand foot syndrome (OR 4.59, 95% CI 2.97 to 7.10; five studies, 5731 participants; low‐quality evidence) in patients treated with oral fluoropyrimidines. There were no differences between participants treated with oral versus IV fluoropyrimidines in occurrence of grade ≥ 3 diarrhoea (OR 1.12, 95% CI 0.99 to 1.25; nine studies, 9551 participants; very low‐quality evidence), febrile neutropenia (OR 0.59, 95% CI 0.18 to 1.90; four studies, 2925 participants; low‐quality evidence), vomiting (OR 1.05, 95% CI 0.83 to 1.34; eight studies, 9385 participants; low‐quality evidence), nausea (OR 1.21, 95% CI 0.97 to 1.51; seven studies, 9233 participants; low‐quality evidence), mucositis (OR 0.64, 95% CI 0.25 to 1.62; four studies, 2233 participants; very low‐quality evidence), and hyperbilirubinaemia (OR 1.67, 95% CI 0.52 to 5.38; three studies, 2757 participants; very low‐quality evidence).
Patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy
• Progression‐free survival (PFS): Overall, PFS was inferior in participants treated with oral versus IV fluoropyrimidines (HR 1.06, 95% CI 1.02 to 1.11; 23 studies, 9927 participants; moderate‐quality evidence). Whilst PFS was worse in participants treated with oral compared with IV fluoropyrimidines when UFT/Ftorafur or eniluracil with oral 5‐fluorouracil (5‐FU) was used, PFS did not differ between individuals treated with oral versus IV fluoropyrimidines when capecitabine, doxifluridine, or S‐1 was used.
• OS: Overall, OS did not differ between participants treated with oral versus IV fluoropyrimidines (HR 1.02, 95% CI 0.99 to 1.05; 29 studies, 12,079 participants; high‐quality evidence). OS was inferior in participants treated with oral versus IV fluoropyrimidines when eniluracil with oral 5‐fluorouracil (5‐FU) was used.
• Time to progression (TTP): TTP was inferior in participants treated with oral versus IV fluoropyrimidines (HR 1.07, 95% CI 1.01 to 1.14; six studies, 1970 participants; moderate‐quality evidence).
• Objective response rate (ORR): ORR did not differ between participants treated with oral versus IV fluoropyrimidines (OR 0.98, 95% CI 0.90 to 1.06; 32 studies, 11,115 participants; moderate‐quality evidence).
• Grade ≥ 3 AEs: Participants treated with oral fluoropyrimidines experienced less grade ≥ 3 neutropenia/granulocytopenia (OR 0.17, 95% CI 0.15 to 0.18; 29 studies, 11,794 participants; low‐quality evidence), febrile neutropenia (OR 0.27, 95% CI 0.21 to 0.36; 19 studies, 9407 participants; moderate‐quality evidence), stomatitis (OR 0.26, 95% CI 0.20 to 0.33; 21 studies, 8718 participants; low‐quality evidence), mucositis (OR 0.17, 95% CI 0.12 to 0.24; 12 studies, 4962 participants; low‐quality evidence), and any grade ≥ 3 AEs (OR 0.83, 95% CI 0.74 to 0.94; 14 studies, 5436 participants; low‐quality evidence). There was more grade ≥ 3 diarrhoea (OR 1.66, 95% CI 1.50 to 1.84; 30 studies, 11,997 participants; low‐quality evidence) and hand foot syndrome (OR 3.92, 95% CI 2.84 to 5.43; 18 studies, 6481 participants; moderate‐quality evidence) in the oral fluoropyrimidine arm. There were no differences between oral and IV fluoropyrimidine arms in terms of grade ≥ 3 vomiting (OR 1.18, 95% CI 1.00 to 1.40; 23 studies, 9528 participants; low‐quality evidence), nausea (OR 1.16, 95% CI 0.99 to 1.36; 25 studies, 9796 participants; low‐quality evidence), and hyperbilirubinaemia (OR 1.62, 95% CI 0.99 to 2.64; nine studies, 2699 participants; low‐quality evidence).
Authors' conclusions
Results of this review should provide confidence that treatment for CRC with most of the oral fluoropyrimidines commonly used in current clinical practice is similarly efficacious to treatment with IV fluoropyrimidines. Treatment with eniluracil with oral 5‐FU was associated with inferior PFS and OS among participants treated with palliative intent for CRC, and eniluracil is no longer being developed. Oral and IV fluoropyrimidines have different patterns of side effects; future research may focus on determining the basis for these differences.
Plain language summary
Oral versus intravenous chemotherapy for colorectal cancer
Background
Intravenous (IV) fluoropyrimidines are an essential part of chemotherapy treatment for colorectal cancer (CRC). Patients prefer tablets as long as they work as well and are as safe as IV treatment, because they are easier to take and are more convenient.
Review question
We compared the effects of oral and IV fluoropyrimidine chemotherapy in patients with CRC who were treated with the aim of cure, or who were treated with palliative chemotherapy because the cancer could not be removed by surgery or was metastatic (it had spread from the place where it originated to other places in the body).
Study characteristics
The evidence is current to June 2016. We identified 44 randomised controlled trials involving 23,150 patients which compared oral and IV fluoropyrimidines. All studies included both male and female patients, and no studies included individuals younger than 18 years of age.
Key results
Among patients with CRC who were treated with the aim of cure, disease‐free survival (DFS) and overall survival (OS) did not differ between those who received oral versus IV treatment. In terms of severe side effects, patients who received oral treatment and those who received IV treatment had a similar risk of diarrhoea. Patients who received oral treatment were more likely to develop hand and foot rash but were less likely to have lowered white cell counts (neutropenia) than patients who received IV treatment.
In patients with CRC whose cancer was treated with palliative chemotherapy, overall, those who received oral treatment had worse progression‐free survival (PFS) than those who received IV treatment. Use of two formulations of oral therapy (UFT or Ftorafur, and eniluracil with oral 5‐fluorouracil (5‐FU)) led to worse PFS in patients who received oral compared with IV treatment. Use of three other formulations of oral therapy (capecitabine, S‐1, and doxifluridine) led to similar PFS in patients who received oral compared with IV treatment. OS did not differ between patients treated with oral versus IV fluoropyrimidines. In terms of severe side effects, patients who received oral treatment were more likely to develop diarrhoea and hand and foot rash but were less likely to have lowered white cell counts than those who received IV treatment.
Quality of the evidence
Review authors assessed the quality of evidence for the main outcomes in this review (DFS and PFS) as moderate; the key reason for downgrading quality involved issues with study design. The quality of evidence for OS in patients who were treated with the aim of cure and in patients who were treated with palliative chemotherapy was high. The quality of evidence for side effects ranged from very low to moderate, and was downgraded because of issues with study design, dissimilar results across studies, or not enough data.
Summary of findings
Summary of findings for the main comparison. Oral compared with intravenous fluoropyrimidines for colorectal cancer ‐ Patients treated with curative intent.
| Oral compared with intravenous fluoropyrimidines for colorectal cancer ‐ Patients treated with curative intent | |||||
|
Patient or population: Patients treated with curative intent for colorectal cancer with neoadjuvant and/or adjuvant chemotherapy Setting: Hospital Intervention: Oral fluoropyrimidines Comparison: Intravenous fluoropyrimidines | |||||
| Outcomes | Illustrative comparative risks (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk* | Corresponding risk** | ||||
| Intravenous fluoropyrimidines | Oral fluoropyrimidines | ||||
| Disease‐free survival | 313 per 1000a | 291 per 1000 (272 to 313) |
HR 0.93 (0.87 to 1.00) | 8903 (7 RCTs) | ⊕⊕⊕⊝ MODERATEb |
| Overall survival | 222 per 1000c | 204 per 1000 (186 to 222) |
HR 0.92 (0.84 to 1.00) |
8902 (7 RCTs) |
⊕⊕⊕⊕ HIGH |
| Grade ≥ 3 diarrhoea | 137 per 1000d | 153 per 1000 (135 to 171) |
OR 1.12 (0.99 to 1.25) | 9551 (9 RCTs) | ⊕⊝⊝⊝ VERY LOWb,e,f |
| Grade ≥ 3 hand foot syndrome | 8 per 1000d | 37 per 1000 (24 to 57) |
OR 4.59g (2.97 to 7.10) | 5731 (5 RCTs) | ⊕⊕⊝⊝ LOWb,e |
| Grade ≥ 3 neutropenia/granulocytopenia | 181 per 1000d | 25 per 1000 (20 to 29) |
OR 0.14 (0.11 to 0.16) |
8087 (7 RCTs) |
⊕⊕⊕⊝ MODERATEe |
| *The basis for the assumed risk is provided in footnotes. **The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Pooled estimates from fixed‐effects meta‐analysis are reported in the table CI: Confidence interval; HR: Hazard ratio; RCTs: randomised controlled trials; OR: Odds ratio | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: We are very uncertain about the estimate | |||||
aThe assumed risk for disease‐free survival was based on the 3‐year disease‐free survival rate in the control group from studies in the meta‐analysis (68.7%)
bDowngraded by one level owing to a high risk of bias in included studies.
cThe assumed risk for overall survival was based on the 5‐year overall survival rate in the control group from studies in the meta‐analysis (77.8%)
dThe assumed risk for each grade ≥ 3 AE was the mean risk in the control group from studies in the meta‐analysis
eDowngraded by one level owing to inconsistency of results that was supported by non‐overlapping CIs, high I2 values, and statistically significant heterogeneity of effect estimates
fDowngraded by one level owing to imprecision
gRandom‐effects estimate, OR 2.36 (95% CI 0.52 to 10.74). Pooled effect estimate was sensitive to the meta‐analysis model used
Summary of findings 2. Oral compared with intravenous fluoropyrimidines for colorectal cancer ‐ Patients treated with palliative intent.
| Oral compared with intravenous fluoropyrimidines for colorectal cancer ‐ Patients treated with palliative intent | |||||
|
Patient or population: Patients treated with palliative intent for inoperable advanced or metastatic colorectal cancer with chemotherapy Setting: Hospital Intervention: Oral fluoropyrimidines Comparison: Intravenous fluoropyrimidines | |||||
| Outcomes | Illustrative comparative risks (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk* | Corresponding risk** | ||||
| Intravenous fluoropyrimidines | Oral fluoropyrimidines | ||||
| Progression‐free survival | 398 per 1000a | 422 per 1000 (406 to 442) |
HR 1.06 (1.02 to 1.11) | 9927 (23 RCTs) | ⊕⊕⊕⊝ MODERATEb |
| Overall survival | 336 per 1000c | 343 per 1000 (333 to 353) |
HR 1.02 (0.99 to 1.05) |
12,079 (29 RCTs) |
⊕⊕⊕⊕ HIGH |
| Grade ≥ 3 diarrhoea | 120 per 1000d | 199 per 1000 (180 to 221) |
OR 1.66 (1.50 to 1.84) | 11,997 (30 RCTs) | ⊕⊕⊝⊝ LOWb,e |
| Grade ≥ 3 hand foot syndrome | 13 per 1000d | 51 per 1000 (37 to 71) |
OR 3.92 (2.84 to 5.43) | 6481 (18 RCTs) | ⊕⊕⊕⊝ MODERATEb |
| Grade ≥ 3 neutropenia/granulocytopenia | 331 per 1000d | 56 per 1000 (50 to 60) |
OR 0.17 (0.15 to 0.18) |
11,794 (29 RCTs) |
⊕⊕⊝⊝ LOWb,e |
| *The basis for the assumed risk is provided in footnotes. **The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Pooled estimates from fixed‐effects meta‐analysis are reported in the table CI: Confidence interval; HR: Hazard ratio; RCTs: randomised controlled trials; OR: Odds ratio | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: We are very uncertain about the estimate | |||||
aThe assumed risk for progression‐free survival was based on the 6‐month progression‐free survival rate in the control group from studies in the meta‐analysis (60.2%)
bDowngraded by one level owing to a high risk of bias in included studies
cThe assumed risk for overall survival was based on the 12‐month overall survival rate in the control group from studies in the meta‐analysis (66.4%)
dThe assumed risk for each grade ≥ 3 AE was the mean risk in the control group from the studies in the meta‐analysis
eDowngraded by one level owing to inconsistency of results that was supported by non‐overlapping CIs, high I2 values, and statistically significant heterogeneity of effect estimates
Background
Description of the condition
Worldwide, colorectal carcinoma (CRC) has the third highest incidence rate and the fourth highest mortality rate of all cancers (Ferlay 2013). In 2012, an estimated 1,360,602 new cases and an estimated 693,933 deaths from CRC occurred worldwide (Ferlay 2013). Approximately 20% of patients diagnosed with CRC have distant metastases at diagnosis, and a further 25% to 35% will develop metastases at a later time (Siegel 2017; Van Cutsem 2006; Van der Geest LGM). This contributes to the high mortality rates observed for CRC (Ferlay 2013).
Description of the intervention
Fluoropyrimidines have been an essential part of treatment for CRC for over 40 years.
For patients with colon cancer treated with curative intent, recommendations regarding use of adjuvant chemotherapy following resection of the primary tumour vary, depending on the stage of disease. TNM stage II disease is defined as T3 or T4 but node negative, whilst TNM stage III disease is defined as any T stage and node positive (Edge 2009). Use of adjuvant 5‐fluorouracil (5‐FU)‐based chemotherapy has been demonstrated to improve survival (Francini 1994; IMPACT Investigators 1995; Laurie 1989; Moertel 1990; O'Connell 1997); subsequently, six months' duration of adjuvant 5‐FU/leucovorin (LV) was established as the standard of care for patients with stage III colon cancer (Dencausse 2002; Haller 2005; O'Connell 1998). More recent research has shown that oxaliplatin added to six months of adjuvant 5‐FU/LV chemotherapy leads to further improvement in both five‐year disease‐free survival (DFS) and six‐year overall survival (OS) compared with 5‐FU/LV alone for stage III colon cancer (André 2009).
Survival outcomes for stage II colon cancer are better than for stage III disease, and the survival benefit derived from use of adjuvant chemotherapy is accordingly less in this setting (André 2009; Brenner 2014; Figueredo 2008; Gill 2004; Gray 2007; IMPACT Investigators 1995; Sargent 2009). American Society of Clinical Oncology (ASCO) guidelines state that direct evidence from randomised controlled trials (RCTs) does not support the routine use of adjuvant chemotherapy in stage II disease (Benson 2004). Current National Comprehensive Cancer Network (NCCN) guidelines recommend that for stage II colon cancer, physician and patient discussion should include potential benefits versus risks of adjuvant chemotherapy. This discussion should encompass consideration of high‐risk features (both clinicopathological and molecular), as well as indirect evidence, potential treatment‐related morbidity and patient co‐morbidities, anticipated life expectancy, and patient preferences (NCCN 2016).
The current standard of care for stage II and III rectal carcinoma is curative intent treatment based on a combined‐modality approach. This consists of neoadjuvant chemo‐radiotherapy with 5‐FU, total mesorectal excision (TME), and adjuvant chemotherapy with 5‐FU and oxaliplatin (Weiser 2015).
In patients with inoperable advanced or metastatic CRC, use of palliative intent IV 5‐FU‐based therapy has led to improved survival outcomes (Nordic 1992; Scheithauer 1993). Subsequent advances including optimisation of IV 5‐FU regimens and combination with irinotecan and oxaliplatin chemotherapy have led to further improvements in median OS (Lucas 2011). Over the past decade, anti‐angiogenic therapies have been successfully combined with fluoropyrimidine‐based chemotherapy. A pivotal phase III trial examined bevacizumab (BEV), a humanised monoclonal antibody to vascular endothelial growth factor (VEGF), by randomising participants to irinotecan, fluorouracil, leucovorin (IFL)/placebo (control), and IFL/BEV or 5‐FU/LV/BEV (Hurwitz 2004). Overall, results showed significant improvement in the endpoints of OS, progression‐free survival (PFS), and median duration of response in the IFL/BEV arm. Survival benefits were also reported in a second‐line study which compared oxaliplatin, fluorouracil and leucovorin (FOLFOX4)‐BEV with FOLFOX4 alone (Giantonio 2007) and in the first‐line MAX trial (Tebbutt 2010), which reported that BEV added to the oral fluoropyrimidine capecitabine improved PFS. Subsequently, the benefit of continuing BEV beyond progression in combination with a second‐line fluoropyrimidine‐based chemotherapy was demonstrated in the phase III TML study (Bennouna 2013). Furthermore, the anti‐angiogenic drugs ziv‐aflibercept and ramucirumab, in combination with infusional 5‐FU, leucovorin, and irinotecan (FOLFIRI), were demonstrated to prolong PFS and OS in the second‐line setting (Tabernero 2015; Van Cutsem 2012).
Cetuximab, an epidermal growth factor receptor (EGFR) antibody, added to FOLFIRI in the first‐line setting, was shown to improve efficacy in patients with KRAS wild‐type metastatic CRC (Van Cutsem 2011). Similarly, panitumumab, a fully humanised antibody to EGFR, was shown to be effective for this subset of patients in the first‐ and second‐line setting when combined with fluoropyrimidine chemotherapy (Douillard 2010; Peeters 2010).
How the intervention might work
Intravenous and oral 5‐FU have been used in the treatment of cancer for several decades. Owing to its unpredictable gastrointestinal absorption and marked variation in pharmacokinetics, use of oral 5‐FU alone was abandoned early. Since that time, research has focused on the biomodulation of 5‐FU to improve its therapeutic effectiveness and cytotoxicity. Leucovorin (LV), an intracellular source of reduced folates, acts by stabilising the complex formed by 5‐FU with thymidylate synthase (TS) and 5‐fluoro‐deoxyuridine monophosphate (5‐FdUMP), leading to prolonged TS inhibition and enhanced efficacy. Eniluracil is a potent inactivator of the principal 5‐FU degradation enzyme dihydropyrimidine dehydrogenase (DPD), and co‐administration with oral 5‐FU significantly increased oral bioavailability whilst decreasing 5‐FU pharmacokinetic variability (reviewed in Schilsky 2002b). Development of this combination was discontinued in 2000.
Several other oral fluoropyrimidines have been designed and currently are undergoing clinical trials or are used routinely in the clinic. Doxifluridine (5’‐dFUR) consists of a 5‐FU molecule attached to a pseudo‐pentose, thus it cannot be directly metabolised in deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) synthesis. With exposure to pyrimidine phosphorylases found at higher concentrations in tumours, 5'‐dFUR is preferentially converted to active 5‐FU in malignant tissue (reviewed in Calabresi 1991). Ftorafur (FTO; Tegafur) is a second‐generation fluoropyrimidine prodrug which provides more prolonged and stable release of 5‐FU. UFT, which comprises FTO and uracil in molar proportions of 1:4, is a third‐generation drug designed to improve the therapeutic index of FTO. Uracil, a natural substrate of DPD, is converted preferentially in lieu of FTO owing to its higher molar concentration in this formulation, resulting in a prolonged 5‐FU elimination half‐life. It has been combined with LV under the trade name Orzel. Capecitabine, another third‐generation drug, is the most commonly used oral fluoropyrimidine worldwide. Designed to limit gastrointestinal toxicity, capecitabine resists enzymatic degradation by thymidine phosphorylase (TP) in the intestine and undergoes a three‐stage conversion with eventual transformation to active 5‐FU in the tumour tissue, where TP levels are highest. S‐1 is a combination of FTO and two biomodulators ‐ 5‐chloro‐2,4‐dihydroxypyridine (CDHP) and potassium oxalate (OXO). CDHP is a potent, reversible inhibitor of DPD which is used to achieve prolonged higher concentrations of 5‐FU in the circulation. OXO acts to limit the gastrointestinal toxicity associated with phosphorylation of 5‐FU in the gastrointestinal tract. OXO accumulates in gastrointestinal tissues, where it inhibits phosphorylation of 5‐FU into 5‐fluorouridine‐5′‐monophosphate (5‐FUMP) by orotate phosphoribosyl transferase (OPRT) (reviewed in Hoff 2000 and Malet‐Martino 2002).
More recently, TAS‐102, an oral combination of trifluridine (FTD, a thymidine‐based nucleoside analogue) and tipiracil (a TP inhibitor which improves bioavailability of FTD), was demonstrated to confer an overall survival benefit in the metastatic chemo‐refractory setting (Mayer 2015). At the dosing schedule used in the clinical development of TAS‐102, its clinically relevant mechanism of action consists of incorporation into DNA and subsequent DNA dysfunction, rather than TS inhibition (reviewed in Lenz 2015). We considered its mechanism of action to be distinct from that of the other fluoropyrimidines described here and did not search for studies examining TAS‐102 for inclusion in this review.
Why it is important to do this review
Patients prefer oral over IV administration of palliative chemotherapy for multiple cancers, including CRC, provided that oral therapy is not less effective. Reasons include the convenience of home‐based treatment with a tablet formulation (Borner 2002; Liu 1997; Twelves 2006).
Oral fluoropyrimidine chemotherapy has been compared with IV fluoropyrimidine in patients with CRC who have been treated with curative or palliative intent. However, researchers have reported variable results with respect to efficacy and adverse events (Chau 2009).
Differences in the efficacy and adverse event profiles of IV fluoropyrimidines depend on whether infusional or bolus regimens are used (Meta‐analysis Group in Cancer 1998a; Meta‐analysis Group in Cancer 1998b). Different oral fluoropyrimidines may also have different efficacy and adverse event profiles (Hamaguchi 2015; Hong 2012; Kwakman 2017). For patients treated with palliative intent for inoperable advanced or metastatic CRC, efficacy and adverse event outcomes for oral compared with IV fluoropyrimidines may vary, depending on whether fluoropyrimidines are combined with irinotecan versus oxaliplatin chemotherapy (Chau 2009). Combination cancer therapy can improve efficacy but can also increase toxicity (Braun 2011). Therefore, it is important to assess whether efficacy and adverse event outcomes differ between oral and IV fluoropyrimidines, depending on whether patients with CRC receive chemotherapy alone versus chemo‐radiotherapy (in curative intent studies) or single‐agent versus combination chemotherapy (in palliative intent studies).
We were unable to identify a previous meta‐analysis and systematic review that examined a wide range of oral fluoropyrimidines, nor were we able to find a systematic review that performed subgroup analyses examining chemotherapy versus chemo‐radiotherapy (in curative intent studies) and single‐agent versus combination therapy (in palliative intent studies), infusional versus bolus IV fluoropyrimidine, the oral fluoropyrimidine backbone used, and oxaliplatin‐based versus irinotecan‐based combination therapy.
Objectives
To compare the effects of oral and IV fluoropyrimidine chemotherapy in patients treated with curative or palliative intent for CRC.
Methods
Criteria for considering studies for this review
Types of studies
We included RCTs with treatment arms comparing oral fluoropyrimidine versus IV fluoropyrimidine chemotherapy.
Studies with a cross‐over design from oral to IV fluoropyrimidine, or vice versa, were eligible for inclusion only if the cross‐over design permitted all relevant treatment arms to crossover.
We included studies regardless of publication status and blinding of participants, personnel, and/or outcome assessment. We applied no language restrictions and did not use outcomes as criteria for considering studies for inclusion in this review.
Types of participants
We included patients who were treated with curative intent for CRC and received neoadjuvant (preoperative) and/or adjuvant (postoperative) chemotherapy. For adjuvant chemotherapy, we included patients with stage II or III colon cancer.
We included patients who were treated with palliative intent for inoperable advanced or metastatic CRC and received chemotherapy.
We included only patients for whom a diagnosis of CRC had been confirmed by histopathology or cytology. We did not restrict patients by gender, age, or ethnic group.
If a study included relevant patients as a subgroup and if outcomes related to this subgroup were reported separately, we included the patients who were eligible for this review (e.g. Fuchs 2007).
Types of interventions
Oral fluoropyrimidine treatment included any fluoropyrimidine administered orally (e.g. capecitabine, S‐1, ftorafur, UFT, doxifluridine, 5‐ethynyluracil). IV fluoropyrimidine treatment included agents administered by bolus and by infusion.
For oral and IV fluoropyrimidine treatments, we did not restrict dose, frequency, intensity, and duration of treatment.
We included oral and IV fluoropyrimidine treatments that were administered as a single agent, or in combination with any other cytotoxic agent/s (e.g. irinotecan, oxaliplatin) and targeted therapies (e.g. bevacizumab, cetuximab). In the case of combination therapy, we included only studies in which the same cytotoxic agents and targeted therapies were administered in both the oral and the IV fluoropyrimidine arms.
We also included oral and IV fluoropyrimidine treatments that were administered with radiotherapy (chemo‐radiotherapy). In the case of chemo‐radiotherapy, we included only studies in which radiotherapy was administered in both the oral and the IV fluoropyrimidine arms.
Cross‐over studies were eligible for inclusion only if participants in both the oral and the IV fluoropyrimidine arms received at least three cycles of chemotherapy before crossover.
Types of outcome measures
Primary outcomes
Patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy
Disease‐free survival (DFS), defined as time from randomisation until death from any cause or disease recurrence, whichever occurred first
Patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy
Progression‐free survival (PFS), defined in this review as time from randomisation until death from any cause or disease progression, whichever occurred first
Secondary outcomes
Patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy
Overall survival (OS)
Grade ≥ 3 adverse events (AEs) (diarrhoea, hand foot syndrome (HFS), neutropenia/granulocytopenia, febrile neutropenia, vomiting, nausea, stomatitis, mucositis, hyperbilirubinaemia, any grade ≥ 3 AEs) assessed on the basis of National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) or similar criteria
Patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy
OS
Time to progression (TTP), defined in this review as time from randomisation until disease progression
Objective response rate (ORR), with objective response defined as best response assessed as a complete response (CR) or a partial response (PR) on the basis of Response Evaluation Critieria in Solid Tumours (RECIST) or similar criteria
Incidence of grade ≥ 3 AEs listed above
Search methods for identification of studies
Electronic searches
We searched the following databases with no limitation on publication year or language.
Cochrane Central Register of Controlled Trials (CENTRAL) on 14 June 2016 (2016, Issue 5) in the Cochrane Library (Appendix 1).
MEDLINE (OVID) from 1950 to 14 June 2016 (Appendix 2).
Embase (OVID) from 1974 to 14 June 2016 (Appendix 3).
Web of Science (Web of Knowledge) from 1900 to 16 June 2016 (Appendix 4).
The first three searches were performed by the Cochrane Colorectal Cancer Group Information Specialist.
We searched the following trials registries.
ClinicalTrials.gov (http://clinicaltrials.gov/) on 8 June 2016, with no limitations on the date trial information was received or updated.
World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) (www.who.int/ictrp/en/) on 29 August 2016, with no date restrictions on date of registration.
Current Controlled Trials, using the International Standard Randomised Controlled Trial Number (ISRCTN) Register (International) (www.controlled‐trials.com) on 9 June 2016, with no date limitations.
The Australian New Zealand Clinical Trials Registry (ANZCTR) (www.anzctr.org.au) on 16 June 2016, with no limitations on trial registration or start dates.
European Organisation for Research and Treatment of Cancer (EORTC) clinical trials database (www.eortc.org/clinical‐trials/) on 16 June 2016, with no date limitations.
Searching other resources
We searched for additional trials not identified in the above electronic searches by searching relevant proceedings for oncology meetings and conferences. We searched the following proceedings.
American Society for Clinical Oncology (ASCO), search of the electronic database of meeting abstracts (http://meetinglibrary.asco.org/abstracts) from 2004 to 15 June 2016.
European Society of Medical Oncology (ESMO), handsearched from 2000 to 14 June 2016.
European Cancer Conference, handsearched from 1993 to 14 June 2016.
We searched the reference lists of identified studies and other systematic reviews, and wrote to the following pharmaceutical companies involved in the manufacture of oral fluoropyrimidines: Orzel, Adherex, Roche, Merck Serono, Sanofi Aventis, and Taiho.
Data collection and analysis
Selection of studies
Three review authors (FC and YY or DL) selected trials for inclusion independently, and resolved queries or disagreements with assistance from a fourth review author (NT). We used a standard checklist of inclusion and exclusion criteria to select studies. We listed excluded trials and reasons for their exclusion. We wrote to investigators for clarification when we could not determine eligibility from published report/s for the study.
Data extraction and management
We collected data from the reports for included studies by using Data Extraction Forms that we had piloted successfully. Two or three review authors (FC and YY or DL) performed this independently, and a fourth review author (NT) resolved disagreements.
We collected the following information about the included studies: study design and setting, study eligibility criteria, participant characteristics, intervention(s) given, outcomes assessed, funding sources, and declarations of interest of the primary researchers. We used this information to populate the Characteristics of included studies tables.
When an included study had multiple reports, we used the report with the most recent data for a specific outcome to extract data for that outcome. When applicable and if necessary, we used other study reports to extract additional information required, including study characteristics and information for risk of bias assessments.
We examined retraction statements and errata associated with included studies and, when applicable, updated recorded data accordingly.
If required, we contacted study authors of the included studies for clarification or for additional information, which we then used in analyses of treatment effects and/or risk of bias assessments.
We checked the magnitude and direction of effects reported by studies against the data presented in our review.
Assessment of risk of bias in included studies
Three review authors (FC and YY or DL) independently assessed risk of bias of included studies using the Cochrane 'Risk of bias' tool (Higgins 2011a); NT resolved queries or disagreements. We assessed the following risk of bias domains: random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting, and other sources of bias including the following.
Use of subsequent therapies in treatment arms.
For patients treated with curative intent for CRC who received neoadjuvant chemotherapy, we assessed subsequent treatment with adjuvant chemotherapy.
For patients treated with curative intent for CRC who received adjuvant chemotherapy, we assessed subsequent treatment with chemotherapy following recurrence or new occurrence of CRC.
For patients treated with palliative intent for inoperable advanced or metastatic CRC who received chemotherapy, we assessed subsequent‐line palliative drug therapy following progressive disease.
In factorial trials, assessment of important interactions between effects of different interventions (Higgins 2011b).
We assessed an additional three domains that we judged to be important for risk of bias assessment of included studies.
-
Comparable schedule of assessment and/or follow‐up for outcomes in different treatment arms.
We assessed risk as 'High' if we noted differences in the frequency of outcome assessments between treatment arms, 'Low' if frequency of assessment was the same in the treatment arms, and 'Unclear' if insufficient information was provided to allow assessment.
-
Incomplete outcome data (intention‐to‐treat (ITT) analysis).
We defined ITT analysis as analysis of randomised participants for efficacy and safety outcomes according to allocated treatment, irrespective of whether participants were eligible, received the allocated treatment, received another treatment, or received no treatment.
We assessed risk as 'High' if the efficacy analysis was clearly not an ITT analysis as defined, and/or if ≥ 5% of participants were excluded from the analysis. We assessed risk as 'Unclear' if insufficient information was provided to allow assessment, and we assessed all other studies as 'Low' risk.
-
Comparability of treatment arms at baseline.
This included Eastern Cooperative Oncology Group (ECOG)/Karnofsky/World Health Organization (WHO)/Zubrod performance status (PS); median or mean age; TNM stage and/or stage II vs III for patients treated with curative intent and number of involved organs for patients treated with palliative intent; and difference in the proportion of participants with KRAS‐mutant CRC among those treated with palliative intent using EGFR inhibitors.
We assessed risk as 'High' if differences between treatment arms at baseline were ≥ 15% for PS; ≥ 5 years for age; ≥ 15% for stage or number of involved organs; or ≥ 10% for KRAS mutant status. We assessed risk as 'Unclear' if insufficient information was provided to allow assessment, and we assessed all other studies as 'Low' risk.
We contacted study authors of included studies when we needed clarification or additional information.
Evaluation of risk of bias for outcomes
We assessed risk of bias for all studies that contributed to each of the review outcomes, as follows.
We judged a study contributing to an outcome to be at high risk of bias if we assessed it as having 'High' risk of bias for one or more domains relevant to the outcome.
We judged a study contributing to an outcome to be at low risk of bias if we assessed it as having 'Low' risk of bias for all domains relevant to the outcome.
We judged a study contributing to an outcome to be at unclear risk of bias if we assessed it as having 'Unclear' risk of bias for one or more domains relevant to the outcome, but we did not assess any domain as 'High' risk.
We used risk of bias assessments for each contributing study to summarise risk of bias for each outcome.
Measures of treatment effect
Time‐to‐event data
We expressed effect estimates as hazard ratios (HRs) with 95% confidence intervals (CIs) for the following time‐to‐event outcomes.
-
Patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy.
DFS, OS.
-
Patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy.
PFS, TTP, and OS.
Dichotomous data
We expressed summary statistics as odd ratios (ORs) for the following dichotomous outcomes.
-
Patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy.
Grade ≥ 3 AEs.
-
Patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy.
ORR, grade ≥ 3 AEs.
Statistical methods for data analysis
Specific outcomes
Time‐to‐event outcomes
When possible, we extracted hazard ratios (HRs) and their 95% confidence intervals (CIs) or standard error of the natural logarithm of HR (se(lnHR)) directly from reports of studies or from correspondence with study authors and contacts; if not reported, we estimated these indirectly from the study reports.
A statistician estimated HRs and se(lnHR) indirectly from Kaplan‐Meier survival curves using the method described by Tierney et al. (Tierney 2007). For one study (Douillard 2002), a statistician indirectly estimated the HR and the se(lnHR) for TTP using a ratio of the median TTP to approximate the HR, and the stratified log‐rank P value to approximate the se(lnHR). For studies for which CIs for effect estimates were not reported as 90%, 95%, or 99% CIs for input into Review Manager 5, a statistician used the indirect variance estimation method to determine the se(lnHR) of the reported HR (Tierney 2007).
For patients treated with curative intent for CRC who received neoadjuvant and/or adjuvant chemotherapy, we measured DFS and OS after a minimum of three years' follow‐up.
Dichotomous outcomes
ORR
For ORR, we calculated the OR using the number of participants who achieved an objective response as the number of 'events', and the total number of participants who were assessable or evaluable for response as the 'total'. When the latter information was not specified, we used the number of participants in the ORR population, which was reported for the study as the 'total'. When only the percentage of participants who achieved an objective response in the treatment arms was reported, we used this percentage and the number of participants in the ORR population to calculate the number of 'events'. If this percentage was reported as "less than x%", we used the absolute value of x. For studies that did not specify a separate ORR population, we used the number of participants in the overall analysis population as the 'total'.
For studies that reported ORRs assessed by both Investigator Assessment and an Independent Review Committee (IRC), we used the ORR from the Investigator Assessment, as most studies did not undergo IRC assessment.
Grade ≥ 3 AE outcomes
For grade ≥ 3 AE outcomes, we calculated the OR using the number of participants who experienced grade ≥ 3 AEs as the number of 'events', and the number of participants included in the safety analysis population as the 'total'. When only the percentage of participants who experienced grade ≥ 3 AEs in the treatment arms were reported, we used this percentage and the number of participants in the safety analysis population to calculate the number of 'events'. If this percentage was reported as "less than x %", we used the absolute value of "x". When a separate safety analysis population denominator was not specified, we used the number of participants in the overall analysis population as the 'total'.
We only quantitatively synthesised HFS data that had been assessed as grade ≥ 3 using NCI CTCAE (versions 2.0 to 4.0), as assessments of grade ≥ 3 HFS using other criteria were not considered sufficiently similar.
We quantitatively synthesised hyperbilirubinaemia data that had been assessed as grade ≥ 3 using NCI CTCAE (versions 2.0 to 4.0 and 1981) and WHO (1981 version). Additionally, we considered hyperbilirubinaemia assessed as grade 4 using NCI CTCAE (1994 version), National Cancer Institute of Canada Clinical Trials Group (NCIC‐CTG) Common Toxicity Criteria (CTC) (1991 version), Southwest Oncology Group (SWOG) (1992 version), and Eastern Cooperative Oncology Group (ECOG) CTC to also be sufficiently similar to hyperbilirubinaemia assessed as grade ≥ 3 using NCI CTCAE (versions 2.0 to 4.0 and 1981) and WHO (1981 version), and we included these data in our quantitative synthesis.
Data presented for different populations
When study authors presented efficacy data for both 'per protocol' and ITT populations (as defined in the study report), we used results for the ITT population.
When study authors presented data for both the safety analysis population and those with available safety data, we used data from the safety analysis population.
Non‐inferiority analysis
In our original protocol, we did not hypothesise that one route of fluoropyrimidine administration (oral or IV) was superior to the other. As such, we did not state a priori levels of benefit.
However, in response to a peer reviewer suggestion, we defined non‐inferiority (NI) margins for the primary outcomes DFS and PFS whereby 50%, 70%, 80%, and 90% of the activity of the active control was retained had the original design been one of non‐inferiority, using IV fluoropyrimidines as the historical active control (FDA 2010). We determined these NI margins independent of studies comparing oral versus IV fluoropyrimidines. In response to an editor suggestion, we assessed whether non‐inferiority had been demonstrated if one made the post hoc judgement that retaining at least 80% of the activity of the active control was reasonable to demonstrate this.
Unit of analysis issues
Studies with multiple treatment arms
In the case of studies with multiple treatment arms:
if one or more treatment arms in a study did not contain an oral fluoropyrimidine or IV fluoropyrimidine chemotherapy, we omitted these arms from the analysis;
when two IV fluoropyrimidine treatment arms contained similar regimens with respect to the outcome or subgroup analysis being examined (and it was considered clinically appropriate to pool the arms), we combined these treatment arms to create a single pair‐wise comparison with the oral fluoropyrimidine treatment arm; and
when two IV fluoropyrimidine treatment arms contained regimens that were different with respect to the outcome or subgroup analysis of interest (and it was not considered clinically appropriate to pool the arms), we used these treatment arms in separate comparisons. In such cases, we used half of the sample size of the experimental oral fluoropyrimidine arm for each comparison.
Cross‐over studies
For cross‐over studies, we measured the outcomes DFS, TTP, PFS, ORR, and grade ≥ 3 AEs (not OS) before crossover.
Dealing with missing data
We contacted the study authors to request missing summary data. If study authors provided us with this data, we included these data in the analyses. If this information was not provided to us by study authors, when possible, we extracted and analysed data as described in 'Statistical methods for data analysis'. With respect to missing individual data, we did not use an imputation method for sensitivity analyses of primary (time‐to‐event) outcomes. We identified studies that did not perform an intention‐to‐treat analysis, assessed associated risk of bias (reported in 'Risk of bias' tables), and incorporated this information into our assessments of quality of evidence for all outcomes.
Assessment of heterogeneity
We assessed clinical heterogeneity with focus on included participants, interventions, and measurements of outcomes (Discussion). We assessed statistical heterogeneity using the Chi2 test, with the level of statistical significance set at 5%. We quantified statistical heterogeneity using the I2 statistic, with interpretation of I2 guided by the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011) ‐ 0% to 40%: might not be important; 30% to 60%: may represent moderate heterogeneity; 50% to 90%: may represent substantial heterogeneity; 75% to 100%: considerable heterogeneity.
Assessment of reporting biases
We assessed reporting bias using symmetry of the funnel plot for the co‐primary endpoint PFS, as described in the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2011). As we included only seven studies in the pooled estimate for DFS, we did not examine a funnel plot for this outcome.
Data synthesis
We performed quantitative synthesis of aggregate data using HR and OR effect estimates, and using fixed‐effect model (FEM) meta‐analysis in Review Manager software (RevMan [Computer Program]). We used the generic inverse‐variance method for meta‐analysis of time‐to‐event outcomes, and the Mantel‐Haenszel method for meta‐analysis of dichotomous outcomes (Higgins 2011c).
Multiple included studies reported the outcomes grade ≥ 3 vomiting and nausea and grade ≥ 3 mucositis and stomatitis in combination. We therefore performed quantitative synthesis of these outcomes as follows.
Grade ≥ 3 vomiting included data from studies that reported either grade ≥ 3 vomiting alone, or grade ≥ 3 vomiting or nausea.
Grade ≥ 3 nausea included data from studies that reported either grade ≥ 3 nausea alone, or grade ≥ 3 vomiting or nausea.
Grade ≥ 3 stomatitis included data from studies that reported grade ≥ 3 stomatitis alone, or grade ≥ 3 stomatitis or mucositis.
Grade ≥ 3 mucositis included data from studies that reported either grade ≥ 3 mucositis alone, or grade ≥ 3 stomatitis or mucositis.
Subgroup analysis and investigation of heterogeneity
We used prespecified tests for heterogeneity to compare treatment effects between subgroups (Higgins 2011c), defined by the following intervention characteristics.
Chemotherapy versus chemo‐radiotherapy received (among participants treated with curative intent for CRC)or single‐agent versus combination therapy received (among participants treated with palliative intent for inoperable advanced or metastatic CRC).
Infusional versus bolus IV fluoropyrimidine received.
Type of oral fluoropyrimidine backbone given (e.g. capecitabine vs UFT/Ftorafur vs Eniluracil + oral 5‐FU vs doxifluridine vs S‐1).
Oxaliplatin‐based versus irinotecan‐based therapy received (among participants treated with palliative intent for inoperable advanced or metastatic CRC who received combination chemotherapy).
Bevacizumab (BEV) received versus not received (among participants treated with palliative intent for inoperable advanced or metastatic CRC who received combination chemotherapy) ‐ this was a post hoc analysis for the PFS outcome only.
Sensitivity analysis
We performed the following sensitivity analyses for primary outcomes to evaluate the robustness of meta‐analysis results.
Excluded studies assessed as having 'High' risk of bias (DFS and PFS).
Excluded Seymour 2011, wherein the study population differed from the study populations of most studies (frail and elderly) (PFS).
Excluded studies of second‐line palliative chemotherapy and studies that included a combination of first‐ and second‐line palliative chemotherapy (PFS).
In response to an editor suggestion, for the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, we performed a sensitivity analysis for grade ≥ 3 HFS, which incorporated heterogeneity by using a random‐effects model (REM) for meta‐analysis in Review Manager software (DerSimonian 1986; RevMan [Computer Program]).
'Summary of findings' table
We assessed the quality of evidence for all outcomes using the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) approach (Guyatt 2008a; Guyatt 2008b). We used GRADEpro (GRADEpro [Computer program]) to create 'Summary of findings' tables for the following outcomes, which we assessed as most important.
-
Patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy.
DFS.
OS.
Grade ≥ 3 diarrhoea.
Grade ≥ 3 HFS.
Grade ≥ 3 neutropenia/granulocytopenia.
-
Patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy.
PFS.
OS.
Grade ≥ 3 diarrhoea.
Grade ≥ 3 HFS.
Grade ≥ 3 neutropenia/granulocytopenia.
We classified the quality of evidence into one of four grades.
High quality: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different.
Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect.
Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect.
We downgraded the quality by one (serious concern) or two (very serious concern) levels for the following reasons: risk of bias, inconsistency (unexplained heterogeneity, inconsistency of results), indirectness of evidence (indirect population, intervention, control, outcomes), imprecision of results (wide confidence intervals), and risk of publication bias.
Protocol
The protocol for this review was published on 17 March 2010 (Chionh 2010).
Results
Description of studies
Results of the search
We have presented in Figure 1 the workflow for studies identified and included in the review.
1.
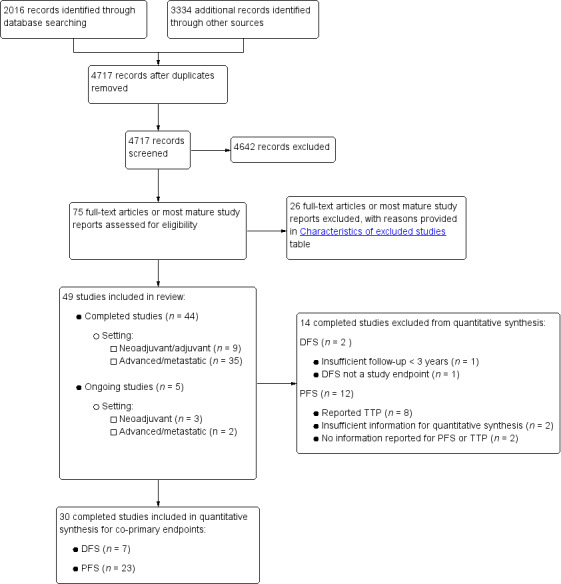
Study flow diagram.
Using the search strategy described, we identified 2016 records from bibliographic databases and 3334 additional records through searches of 'other sources', which included trials registers and conference proceedings. We contacted pharmaceutical companies, and Taiho, Orzel, Adherex, and Roche provided us with lists of potentially eligible studies. After removing duplicates, we screened a total of 4717 records for inclusion.
Of these, we assessed the full‐text reports or the most mature study reports for 75 potentially eligible studies, and we identified 49 studies that met review inclusion criteria. Forty‐four of the included studies were completed studies (Characteristics of included studies), and five were ongoing, with ongoing accrual or follow‐up (Characteristics of ongoing studies). We had two studies translated from Chinese to English (Yu 2005; Mei 2014), and one from Korean to English (Kim 2001a) before we extracted data.
Included studies
Design
We included 44 studies in the review.
The nine studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC included 10,918 randomised participants (Table 3). These included eight phase 3 studies and one study that did not specify the phase of the study. One study of neoadjuvant treatment had a 2 × 2 factorial design (Allegra 2015).
1. Included studies ‐ Patients treated with curative intent for colorectal cancer.
| Treatment setting | Study ID | Phase | Treatment type | Treatment arm/s (oral), n randomised | Treatment arm/s (IV), n randomised | IV arm: bolus vs Infusional |
| Neoadjuvant | Rectal | |||||
| Allegra 2015 | III | Fluoropyrimidine combined with RT | Capecitabine (Grp 2), n = 146 Capecitabine (Grp 5), n = 326 Capecitabine + oxaliplatin (Grp 6), n = 330 |
5‐FU (Grp 1), n = 147 5‐FU (Grp 3), n = 330 5‐FU + oxaliplatin (Grp 4), n = 329 |
Infusional | |
| De la Torre 2008 | III | Fluoropyrimidine combined with RT | UFT (Tegafur/Uracil) + LV with RT, n = 78 | 5‐FU + LV with RT, n = 77 | Bolus | |
| Neoadjuvant/ Adjuvant |
Rectal | |||||
| Hofheinz 2012 | III | Fluoropyrimidine combined with RT | Capecitabine with RT, n = 197 ∙ Adjuvant cohort: n = 116 ∙ Neoadjuvant cohort: n = 81 |
5‐FU with RT, n = 195 ∙ Adjuvant cohort: n = 115 ∙ Neoadjuvant cohort: n = 80 |
Bolus and infusional | |
| Adjuvant | Rectal | |||||
| Kim 2001a | ND | Fluoropyrimidine combined with RT (after completion of 2C of fluoropyrimidine alone) | 5‐dFUR + LV, n = 92 | 5‐FU + LV, n = 74 | Bolus | |
| Colon | ||||||
| De Gramont 2012 | III | Combination chemotherapy ‐ Oxaliplatin + Bevacizumab (BEV) | BEV‐XELOX, n = 952 | BEV‐FOLFOX4, n = 960 | Infusional | |
| Lembersky 2006 | III | Fluoropyrimidine alone | UFT + LV, n = 805 | 5‐FU + LV, n = 803 | Bolus | |
| Shimada 2014 | III | Fluoropyrimidine alone | UFT + LV, n = 551 | 5‐FU + LV, n = 550 | Bolus | |
| Twelves 2012 | III | Fluoropyrimidine alone | Capecitabine, n = 1004 | 5‐FU + LV, n = 983 | Bolus | |
| Colorectal | ||||||
| Pectasides 2015 | III | Combination chemotherapy ‐ fluoropyrimidine + oxaliplatin | CAPOX (capecitabine + oxaliplatin), n = 197 | mFOLFOX6, n = 211 | Infusional | |
IV: intravenous
RT: radiotherapy
5‐FU: 5‐fluorouracil
UFT: tegafur/uracil
LV: leucovorin
ND: no data available
5‐dFUR: doxifluridine
BEV: bevacizumab
The 35 studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC included 12,592 randomised participants (Table 4; Table 5). These included 10 phase 2 and 17 phase 3 studies, along with six studies that did not specify the phase of the study. Study authors described one study as phase 4 in previous abstracts but specified no phase in the journal report (Nogue 2005), and another study as phase 2/3 (Yasui 2015). Three of these studies used a 2 × 2 factorial design (Cassidy 2011a; Kohne 2008; Seymour 2011). Fuchs 2007 used a 3 × 2 factorial design to compare FOLFIRI, irinotecan plus bolus FU/LV (mIFL), and irinotecan plus oral capecitabine (CapeIRI) in period 1 of the trial, which was the only study period of interest for this review.
2. Included studies ‐ Patients treated with palliative intent for inoperable advanced or metastatic colorectal cancer (single‐agent fluoropyrimidines).
| Oral fluoropyrimidine backbone | Study ID | Phase | Treatment line | Treatment arm/s (Oral), n randomised | Treatment arm/s (IV), n randomised | IV arm: Bolus vs Infusional |
| Capecitabine | Hoff 2001 | III | First | Capecitabine, n = 302 | 5‐FU + LV, n = 303 | Bolus |
| Van Cutsem 2001b | III | First | Capecitabine, n = 301 | 5‐FU + LV, n = 301 | Bolus | |
| Doxifluridine (5‐dFUR) | Ahn 2003 | II | First | 5‐dFUR + LV, n = 38 | 5‐FU + LV, n = 39 | Bolus |
| Bajetta 1996 | II | First | 5‐dFUR + LV, n = 67 | 5‐dFUR + LV, n = 63 | Bolus | |
| Eniluracil + oral 5‐FU | ECOG E5296 2012 | III | First | Eniluracil/Oral 5‐FU, n = 61 | 5‐FU, n = 64 | Infusional |
| Schilsky 2002a | III | First | Eniluracil/Oral 5‐FU, n = 488 | 5‐FU + LV, n = 493 | Bolus | |
| Van Cutsem 2001a | III | First | Eniluracil/Oral 5‐FU, n = 268 | 5‐FU + LV, n = 263 | Bolus | |
| Ftorafur/tegafur (FT) | Andersen 1987 | ND | First | Ftorafur, n = 30 | 5‐FU, n = 30 | Bolus |
| Nogue 2005 | Unclear; described as Phase IV in abstracts | First | FT + LV, n = 114 | 5‐FU + LV, n = 123 | Bolus | |
| Ftorafur + uracil (UFT) | Carmichael 2002 | III | First | UFT + LV, n = 190 | 5FU + LV, n = 190 | Bolus |
| Douillard 2002 | III | First | UFT + LV, n = 409 | 5‐FU + LV, n = 407 | Bolus |
IV: intravenous
5‐FU: 5‐fluorouracil
LV: leucovorin
5‐dFUR: doxifluridine
ND: no data available
FT: tegafur
UFT: tegafur + uracil
3. Included studies ‐ Patients treated with palliative intent for inoperable advanced or metastatic colorectal cancer (combination chemotherapy).
| Chemotherapy | Study ID | Phase | Study design ‐ other details | Treatment line | Treatment arm/s (Oral), n randomised | Treatment arm/s (IV), n randomised | IV arm: Bolus vs Infusional |
| Oxaliplatin | Combination with capecitabine | ||||||
| Cassidy 2011a | III | 2 × 2 factorial ‐ following protocol amendment | First | XELOX alone, n = 317 | FOLFOX‐4 alone, n = 317 | Infusional | |
| XELOX + Placebo, n = 350 | FOLFOX‐4 + Placebo, n = 351 | Infusional | |||||
| XELOX + BEV, n = 350 | FOLFOX‐4 + BEV, n = 350 | Infusional | |||||
| Comella 2009 | III | First | OXXEL (Capecitabine + oxaliplatin), n = 158 | OXAFAFU (5‐FU/LV + Oxaliplatin), n = 164 | Bolus | ||
| Diaz‐Rubio 2007 | III | First | XELOX, n = 174 | FUOX (5‐FU + Oxaliplatin), n = 174 | Infusional | ||
| Ducreux 2011 | III | First | XELOX, n = 156 | FOLFOX‐6, n = 150 | Infusional | ||
| Hochster TREE‐1 2008 | ND | First | CapeOx, n = 50 | mFOLFOX6, n = 50 | Infusional | ||
| bFOL, n = 50 | Bolus | ||||||
| Hochster TREE‐2 2008 | ND | First | CapeOx + BEV, n = 74 | mFOLFOX6 + BEV, n = 75 | Infusional | ||
| bFOL + BEV, n = 74 | Bolus | ||||||
| Martoni 2006 | II | First | XELOX, n = 62 | pviFOX, n = 56 | Infusional | ||
| Porschen 2007 | III | First | CAPOX, n = 242 | FUFOX, n = 234 | Infusional | ||
| Rothenberg 2008 | III | Second | XELOX, n = 313 | FOLFOX‐4, n = 314 | Infusional | ||
| Seymour 2011 | ND | 2 × 2 factorial, cross‐over (only from no oxaliplatin to oxaliplatin) | First | Capecitabine or OxCap, n = 229 ∙ Capecitabine, n = 115 ∙ OxCap, n = 114 |
5‐FU or OxFU, n = 230 ∙ 5‐FU, n = 115 ∙ OxFU, n = 115 |
Infusional | |
| Combination with Ftorafur/uracil (UFT) | |||||||
| Douillard 2014 | II | First | UFOX + Cetuximab, n = 152 | FOLFOX4 + Cetuximab, n = 150 | Infusional | ||
| Combination with S‐1 | |||||||
| Mei 2014 | ND | First | SOX, n = 35 | FOLFOX4, n = 35 | Infusional | ||
| Yamada 2013 | III | First | SOX‐BEV, n = 256 | mFOLFOX6‐BEV, n = 256 | Infusional | ||
| Yamazaki 2015 | II | First | SOL (S‐1 + oxaliplatin + oral LV), n = 56 | mFOLFOX6, n = 51 | Infusional | ||
| Irinotecan | Combination with capecitabine | ||||||
| Ducreux 2013 | II | First | XELIRI + BEV, n = 72 | FOLFIRI + BEV, n = 73 | Infusional | ||
| Fuchs 2007 | III | 3 × 2 factorial (Period 1) | First | CapeIRI + Celecoxib/Placebo, n = 145 | FOLFIRI + Celecoxib/Placebo, n = 144 | Infusional | |
| mIFL + Celecoxib/Placebo, n = 141 | Bolus | ||||||
| Kohne 2008 | III | 2 × 2 factorial | First | CAPIRI + Celecoxib/Placebo, n = 44 | FOLFIRI + Celecoxib/Placebo, n = 41 | Infusional | |
| Pectasides 2012 | III | First | XELIRI + BEV, n = 143 | FOLFIRI + BEV, n = 142 | Infusional | ||
| Silvestris 2010 | II | First | XELIRI, n = ND | FOLFIRI, n = ND | Infusional | ||
| Souglakos 2012 | II | First | CAPIRI + BEV, n = 168 | FOLFIRI + BEV, n = 168 | Infusional | ||
| Yu 2005 | ND | First and second | Capecitabine + Irinotecan, n = 27 | 5‐FU + Irinotecan, n = 16 | Infusional | ||
| Combination with Ftorafur/uracil (UFT) | |||||||
| Shigeta 2016 | II | First | TEGAFIRI (UFT, leucovorin, irinotecan) ± BEV, n = 35 | FOLFIRI ± BEV, n = 36 | Infusional | ||
| Combination with S‐1 | |||||||
| Kato 2012 | II | First and second | Sequential IRIS‐BEV, n = 30 | mFOLFIRI‐BEV, n = 30 | Infusional | ||
| Yasui 2015 | II/III | Second | IRIS (Irinotecan + S‐1), n = 213 | FOLFIRI, n = 213 | Infusional | ||
IV: intravenous
BEV: bevacizumab
ND: no data available
UFT: tegafur/uracil
Sample size
Most studies reported a planned sample size with power considerations based on comparisons of efficacy or safety.
Among the studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC, Kim 2001a did not report sample size calculations. Sample size calculations for De Gramont 2012 were based on the DFS hazard rates for BEV‐FOLFOX4 versus FOLFOX4 or BEV‐capecitabine plus oxaliplatin (XELOX) versus FOLFOX4 in patients with stage III disease. However, we compared treatment effects of BEV‐XELOX versus BEV‐FOLFOX4 in this review.
Among the studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC, six studies did not report sample size calculations (Ahn 2003; Andersen 1987; Mei 2014; Silvestris 2010; Van Cutsem 2001b (in abstract form only); Yu 2005), and in three studies, reported sample size calculations did not include power considerations based on comparisons of outcomes between treatment arms (Hochster TREE‐1 2008; Hochster TREE‐2 2008; Martoni 2006). Three other studies used a non‐comparative design (Bajetta 1996; Douillard 2014; Ducreux 2013).
Participants
No studies reported that they included patients younger than 18 years of age (information on youngest age was not provided for Kim 2001a, Lembersky 2006, Van Cutsem 2001a, and Yu 2005). Six out of nine studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC placed an upper limit on the age of eligible patients (Lembersky 2006 (upper limit 60 years); Kim 2001a (upper limit 70 years); Pectasides 2015, Shimada 2014, Twelves 2012 (upper limit 75 years); Bajetta 1996 (upper limit 80 years)). Nine out of 35 studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC placed an upper limit on age of eligible patients (Ahn 2003; Ducreux 2013; Kato 2012; Mei 2014; Shigeta 2016; Yasui 2015; Yu 2005 (upper limit 75 years); Bajetta 1996, Yamada 2013 (upper limit 80 years)).
All studies included both male and female participants.
Treatment type and line of treatment
Among studies of curative intent treatment for CRC, two studies examined neoadjuvant treatment alone for rectal carcinoma (De la Torre 2008; Allegra 2015), and one study explored use of both neoadjuvant and adjuvant treatment for rectal carcinoma (Hofheinz 2012). Six studies examined adjuvant treatment alone, including four studies for colon carcinoma (De Gramont 2012; Lembersky 2006; Shimada 2014; Twelves 2012), one study for rectal carcinoma (Kim 2001a), and one study for carcinoma of the colon or rectum (Pectasides 2015) (Table 3). Among studies that included patients with rectal carcinoma, two studies required the distal border of the tumour to be < 12 cm from the anal verge (Allegra 2015; Kim 2001a), one study required the distal border of the tumour to be < 16 cm from the anal verge (Hofheinz 2012), and two studies did not describe anatomical criteria (De la Torre 2008; Pectasides 2015).
Among studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC, 31 were performed exclusively in the first‐line setting. One study had exclusion criteria that specified “no past history of chemotherapy or chemotherapy ceased for over six months” and included patients in the report who had been given first‐ and second‐line treatment (Yu 2005). Kato 2012 included patients given first‐ or second‐line treatment; if treatment was second‐line, first‐line therapy with FOLFOX was mandated. Two studies were conducted in the second‐line setting ‐ one in combination with oxaliplatin (Rothenberg 2008) and one in combination with irinotecan (Yasui 2015) (Table 4; Table 5).
Location
Among studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC, capecitabine trials were performed in Greece (Pectasides 2015), in Europe (Hofheinz 2012), and in the USA, Europe, Asia, Australia, and other countries (Twelves 2012; De Gramont 2012). The Allegra 2015 study was predominantly performed in North America. UFT studies were conducted at sites in Asia (Shimada 2014), Europe (De la Torre 2008), and North America (Lembersky 2006). The single doxifluridine study was performed in Asia only (Kim 2001a).
Among studies of palliative intent treatment with palliative chemotherapy for inoperable advanced or metastatic CRC, all four S‐1 trials were performed in Asia only (Kato 2012; Yamada 2013; Yamazaki 2015; Yasui 2015). One Asia‐only study used capecitabine (Yu 2005); nine studies were conducted in Europe or included both European and non‐USA sites. Additionally, three European Intergroup studies were carried out ‐ Gruppo Oncologico Aree Metropolitane ‐ GOAM (Martoni 2006); Gruppo Oncologico dell'Italia Meriodionale ‐ GOIM (Silvestris 2010); and European Organisation for Research and Treatment of Cancer ‐ EORTC (Kohne 2008). Two capecitabine studies were conducted in the USA (Hochster TREE‐1 2008; Hochster TREE‐2 2008), and four studies had sites in the USA and in other countries. UFT trials were conducted in Europe and in non‐USA countries (Carmichael 2002; Douillard 2014), and in the USA and in other countries (Douillard 2002). Non‐USA sites in the European UFT trials included Canada, Australia, New Zealand, and Israel (Carmichael 2002); and Asia, South America, Australia, and Israel (Douillard 2014); the Douillard 2002 study also included non‐USA sites in Europe, Canada, and Puerto Rico. One UFT study was based in Japan (Shigeta 2016). Tegafur was used in two European studies (Andersen 1987; Nogue 2005). Eniluracil was used in one USA study (ECOG E5296 2012); one study was performed in the USA and Canada (Schilsky 2002a), and one was an international study (Van Cutsem 2001a). Doxifluridine was used in Europe (Bajetta 1996), and in South Korea (Ahn 2003)(Characteristics of included studies).
Performance status
Although most studies included patients with ECOG PS 2 or less (or the equivalent Karnofsky PS (KPS)), the study population for Seymour 2011 comprised elderly and frail patients who were considered by the treating oncologist to be unsuitable for upfront full‐dose chemotherapy. One study (Andersen 1987) included patients with ECOG PS 3, although the proportion of patients with ECOG PS 3 was not clear (Characteristics of included studies).
Interventions
Among studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC, three studies used fluoropyrimidines alone (not in combination with other chemotherapy or radiotherapy). These studies included the oral fluoropyrimidines UFT (Lembersky 2006; Shimada 2014) and capecitabine (Twelves 2012). Three other studies combined single‐agent fluoropyrimidines with radiotherapy, and included the oral fluoropyrimidines UFT (De la Torre 2008), capecitabine (Hofheinz 2012), and doxifluridine (Kim 2001a). One study of neoadjuvant treatment investigated radiotherapy in combination with oral and intravenous fluoropyrimidines and oxaliplatin (Allegra 2015). Two studies of adjuvant treatment compared combination chemotherapy regimens (De Gramont 2012; Pectasides 2015) (Table 3).
Among studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC, 11 single‐agent studies compared IV fluoropyrimidines with the oral fluoropyrimidines capecitabine, doxifluridine, eniluracil/oral 5‐FU, and Ftorafur (Tegafur) or UFT. All but one study used IV 5‐FU; Bajetta 1996 compared oral and IV doxifluridine. All of the studies that examined IV 5‐FU as a single‐agent used bolus regimens, except ECOG E5296 2012, which used infusional IV 5‐FU (Table 4). All of the 24 studies that included combination chemotherapy used oxaliplatin or irinotecan (Table 5). Of the 14 studies that used oxaliplatin‐based combination chemotherapy, three trials included bolus 5‐FU arms (Comella 2009; Hochster TREE‐1 2008; Hochster TREE‐2 2008). Four studies that used oxaliplatin‐based combination chemotherapy examined combinations with the EGFR‐antibody cetuximab (Douillard 2014) or with BEV (Cassidy 2011a; Hochster TREE‐2 2008; Yamada 2013). Of the ten studies with irinotecan‐based combination chemotherapy, five trials included BEV‐containing arms (Ducreux 2013; Kato 2012; Pectasides 2012; Shigeta 2016; Souglakos 2012). Two further studies with a factorial design randomised participants to CAPIRI versus FOLFIRI plus celecoxib/placebo (Kohne 2008), or CapeIRI versus FOLFIRI versus mIFL plus celecoxib/placebo (Fuchs 2007).
Monitoring of compliance and adherence to oral treatment
Among studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC, only one study (Lembersky 2006) reported monitoring of compliance and adherence to oral treatment.
Among studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC, 10 studies (Ahn 2003; Bajetta 1996; Douillard 2002; Douillard 2014; Ducreux 2011; Martoni 2006; Rothenberg 2008; Schilsky 2002a; Seymour 2011; Shigeta 2016) described oral chemotherapy pill monitoring or use of a patient diary.
Outcomes
Patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy
Of the nine studies, all but one study assessed DFS. Kim 2001a examined rates of local and systemic recurrence but did not report DFS. Although De la Torre 2008 examined DFS, we did not include this study in the DFS meta‐analysis owing to insufficient median follow‐up time (22 months) (Table 3).
All of the studies apart from Kim 2001a reported the OS outcome. We excluded De la Torre 2008 from the meta‐analysis of OS owing to insufficient follow‐up time (Appendix 5).
All of the studies reported outcome data for at least one specific grade ≥ 3 AE of interest for this review, and all provided data that were suitable for meta‐analysis. Included studies reported information for specific grade ≥ 3 AEs: diarrhoea (n = 9), HFS (n = 7), neutropenia/granulocytopenia (n = 7), febrile neutropenia (n = 4), vomiting (n = 8), nausea (n = 7), stomatitis (n = 5), mucositis (n = 4), and hyperbilirubinaemia (n = 4). Two studies of adjuvant treatment (Hofheinz 2012; Kim 2001a) described 'lowered leucocytes' or 'leukopenia' only and were excluded from the meta‐analysis (Appendix 6). One study (Twelves 2012) reported combined data for grade ≥ 3 vomiting and nausea, and one study (De la Torre 2008) reported combined data for grade ≥ 3 stomatitis and mucositis. Table 6 shows the relationships between reported AEs and treatment for the included studies. Included studies used the following AE assessment criteria: ECOG CTC (n = 1), NCI CTCAE version 4.0 (n = 1), NCI CTCAE version 3.0 (n = 1), NCI CTCAE version 2.0 (n = 3), NCIC‐CTG CTC 1991 version (n = 1), NCI CTC 1958 (n = 1), and WHO, version not specified (n = 1).
4. Grade ≥ 3 adverse events ‐ Reported relationships to treatment in different studies.
| Setting | Related | Related and unrelated | Not specified |
| Patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy |
Twelves 2012 | De Gramont 2012 |
Allegra 2015 De la Torre 2008 Hofheinz 2012 Kim 2001a Lembersky 2006 Pectasides 2015 Shimada 2014 |
| Patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy |
Ahn 2003 ECOG E5296 2012 Fuchs 2007 Hoff 2001 Nogue 2005 Schilsky 2002a Seymour 2011 Souglakos 2012 Van Cutsem 2001a Van Cutsem 2001b Yamazaki 2015 |
Cassidy 2011a Douillard 2014 Hochster TREE‐1 2008 Hochster TREE‐2 2008 Kato 2012 Rothenberg 2008 Shigeta 2016 Yamada 2013 Yasui 2015 |
Bajetta 1996 Carmichael 2002 Comella 2009 De la Torre 2008 Diaz‐Rubio 2007 Douillard 2002 Ducreux 2011 Ducreux 2013 Kohne 2008 Martoni 2006 Pectasides 2012 Porschen 2007 Silvestris 2010 Yu 2005 |
CRC: colorectal cancer
Overall, five studies presented data for 'any grade ≥ 3 AEs' (Allegra 2015; De Gramont 2012; Hofheinz 2012; Lembersky 2006; Twelves 2012).
Patients treated with palliative intent for inoperable advanced or metastatic CRC with palliative chemotherapy
Of the 35 studies, all but one study contributed to pooled effect estimates for an efficacy outcome and/or at least one grade ≥ 3 AE outcome (Silvestris 2010).
A total of 25 studies assessed PFS, and eight studies assessed the TTP outcome. Andersen 1987 did not assess either outcome. Hochster TREE‐1 2008, Hochster TREE‐2 2008, Hoff 2001, and Van Cutsem 2001b described TTP as the outcome examined but provided a definition compatible with the definition for PFS provided in this review. Bajetta 1996 stated that time to treatment failure was the examined outcome but provided a definition compatible with the definition for PFS provided in this review. Ahn 2003 described PFS as the examined outcome but provided a definition compatible with the classification for TTP provided in this review. We excluded Hochster TREE‐1 2008 and Hochster TREE‐2 2008 (for the PFS endpoint) and Silvestris 2010 and Yu 2005 (for the TTP endpoint) from our meta‐analyses because we could not estimate the HRs either directly or indirectly from the information provided (Appendix 7). Douillard 2002 presented only median TTP times with a stratified log‐rank P value.
Thirty‐one studies reported the OS outcome. Kato 2012, Martoni 2006, Mei 2014, and Silvestris 2010 did not report the OS outcome, and we excluded Andersen 1987 and Yu 2005 from our quantitative synthesis because we could not estimate the HR either directly or indirectly from the report (Appendix 7).
All 35 studies assessed ORR using the following criteria: WHO 1979 (n = 3), WHO 1981 (n = 4), modified WHO (n = 2), RECIST, version 1.0 (n = 21), RECIST, version not specified (n = 1), ECOG (n = 1), and SWOG (n = 1). Two studies did not specify this information (Van Cutsem 2001a; Yu 2005). We excluded Mei 2014 and Seymour 2011 from meta‐analysis because investigators reported ORR only after two cycles of chemotherapy and at 12 to 14 weeks after the start of treatment, respectively. We excluded Silvestris 2010 because investigators assessed an unclear number of participants for ORR in both arms (Appendix 7). Of the 32 studies included in the meta‐analysis, 22 studies provided information on the number of participants assessable or evaluable for response. One study did not specify a separate ORR analysis population denominator (Van Cutsem 2001a).
All but one included study (Andersen 1987) reported outcome data on grade ≥ 3 AEs of interest for this review. Table 4 shows the relationship between reported AEs and treatment in the included studies. AE assessment criteria included NCI CTCAE, version 3.0 (n = 14); NCI CTCAE, version 2.0 (n = 9); NCI CTCAE, 1994 version (n = 2); NCI CTCAE, 1981 version (n = 1); NCI CTCAE, version not specified (n = 3); WHO, 1981 (n = 1); WHO, version not specified (n = 1); and an adaptation of SWOG, 1992 (n = 1). Two studies did not specify this information. Included studies provided information for specific grade ≥ 3 AEs as follows: diarrhoea (n = 30), HFS (n = 23), neutropenia/granulocytopenia (n = 29), febrile neutropenia (n = 19), vomiting (n = 23), nausea (n = 25), stomatitis (n = 21), mucositis (n = 12), and hyperbilirubinaemia (n = 12). Five trials provided combined grade ≥ 3 stomatitis and mucositis data (Carmichael 2002; Douillard 2002; Shigeta 2016; Yamada 2013; Yasui 2015), and eight studies reported combined grade ≥ 3 nausea and vomiting data (Ahn 2003; Carmichael 2002; Cassidy 2011a; Douillard 2002; Hochster TREE‐1 2008; Hochster TREE‐2 2008; Mei 2014; Nogue 2005). Fourteen studies presented data for 'any grade ≥ 3 AEs'. Three studies did not specify a separate safety analysis denominator (Comella 2009; Porschen 2007; Van Cutsem 2001a). Kato 2012 reported grade ≥ 3 AEs up to 12 weeks.
Four studies had no grade ≥ 3 AE outcomes that were suitable for meta‐analysis. Of these, one study included an unclear number of participants in the safety analysis population and unclear units of analysis (Ahn 2003). Andersen 1987 did not report any grade ≥ 3 AE outcomes. Silvestris 2010 included an unclear number of participants in the safety analysis population for each arm. For Yu 2005, it is unclear whether investigators reported AEs for the entire study population or only for a subset owing to discrepancies between the table title and participant numbers provided in the table (Appendix 8). Two additional included studies reported only 'leukopenia' or lowered 'white blood cells' (Bajetta 1996; Kohne 2008). One study reported grade 2 and 3 HFS (Porschen 2007), and one trial reported toxicities affecting skin/appendages which included but were not confined to HFS (Carmichael 2002). We did not include these studies in the meta‐analysis for neutropenia/granulocytopenia and HFS outcomes, respectively (Appendix 6).
Early stopping
Among studies of curative intent treatment for CRC, one study of neoadjuvant treatment (De la Torre 2008) was stopped early owing to slow accrual after 63% of the number of participants planned for accrual had been randomised. For similar reasons, one study of adjuvant treatment (Pectasides 2015) was prematurely closed after 55% of participants were enrolled.
Among studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC, four studies were stopped early: Nogue 2005 owing to slow accrual, when 85% of the planned number of participants for accrual to the study had been randomised; ECOG E5296 2012 after 125 of the 950 planned participants had been accrued, owing to negative results from two other studies of eniluracil with oral 5‐FU (Schilsky 2002a; Van Cutsem 2001a); Kohne 2008 after enrolment of only 85 participants as a consequence of seven deaths that were assessed as unrelated to disease progression; and Fuchs 2007 after 547 of the 900 participants for Periods 1 and 2 combined had been enrolled. Accrual to this trial had slowed after reports described cardiovascular concerns with celecoxib, although celecoxib/placebo administration was permanently discontinued for patients in January 2005.
Excluded studies
For this review, we classified studies as excluded only when a reader might plausibly expect them to be eligible for inclusion. We have provided reasons for exclusion of 26 such studies in the Characteristics of excluded studies table. We most commonly excluded studies because investigators did not confirm histologically proven colorectal adenocarcinoma as an inclusion criterion, or, in the case of cross‐over studies, because researchers permitted cross‐over in only one arm or treated participants with an insufficient number of chemotherapy cycles before cross‐over.
Risk of bias in included studies
We analysed all nine studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC and 35 studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC for risk of bias using the 10 domains described below (Assessment of risk of bias in included studies; Figure 2; Figure 3).
2.
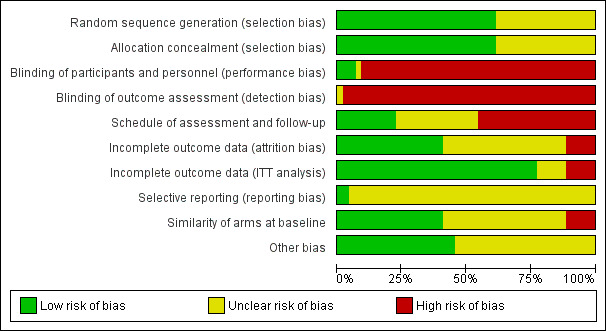
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.1
1 In this graph, the risk of bias for each domain was calculated using the worst assessment documented for that domain in the contributing studies.
3.
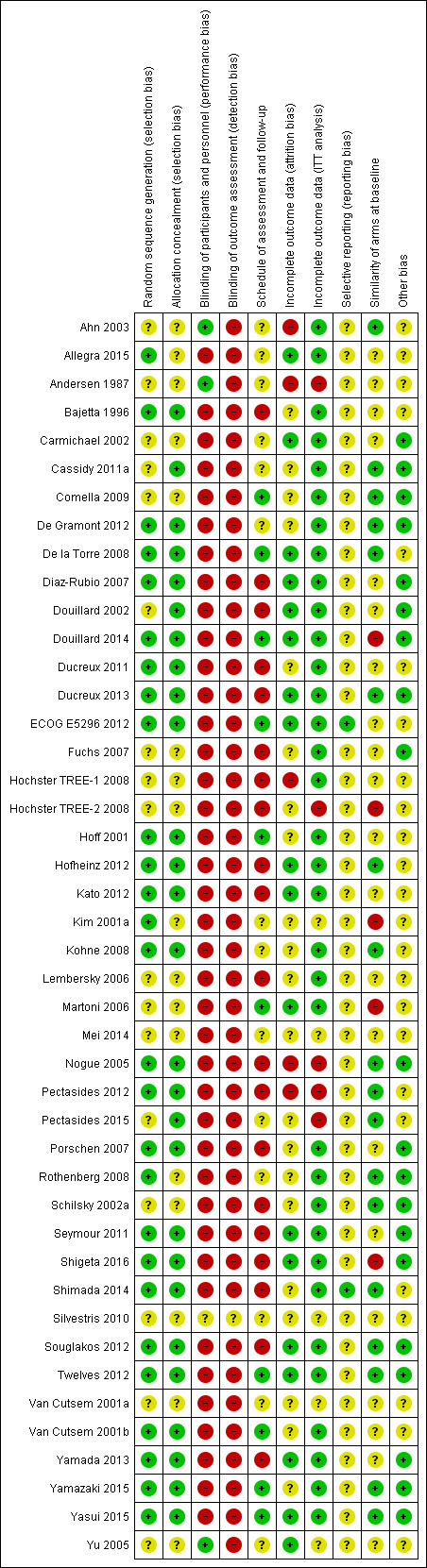
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.1
1 In this summary, the risk of bias for each domain was scored using the worst assessment documented for that domain in the study.
The following section describes risk of bias in the 43 studies that contributed to pooled effect estimates for each outcome (Table 7 and Table 8). We did not include Silvestris 2010 in the pooled effect estimates for any of the outcomes in this review. This study had 'Unclear' risk of bias in all domains.
5. Included studies that contributed to pooled effect estimates for each outcome ‐ Patients treated with curative intent for colorectal cancer.
| Study ID | Outcome | |||||||||||
| Efficacy | Grade ≥ 3 AE | |||||||||||
| DFS | OS | Diarrhoea | HFS |
Neutropenia/ granulocytopenia |
Febrile neutropenia | Vomiting | Nausea | Stomatitis | Mucositis | Hyperbilirubinemia | Any | |
| Allegra 2015 | X | X | X | X | X | X | X | X | X | X | X | |
| De Gramont 2012 | X | X | X | X | X | X | X | X | ||||
| De la Torre 2008 | Oa | Oa | X | Ob | X | X | X | Xc | Xc | |||
| Hofheinz 2012 | X | X | X | X | X | X | X | X | X | X | ||
| Kim 2001a | X | X | ||||||||||
| Lembersky 2006 | X | X | X | X | X | X | X | X | ||||
| Pectasides 2015 | X | X | X | X | X | X | X | X | X | |||
| Shimada 2014 | X | X | X | X | X | X | X | X | X | |||
| Twelves 2012 | X | X | X | Ob | X | Xd | Xd | X | Oe | X | ||
X: Study contributed to the pooled effect estimate for the outcome
O: Study reported the outcome but did not contribute to the pooled effect estimate for the outcome
aInsufficient follow‐up time ‐ median 22 months in each arm (< 3 years)
bAssessed grade ≥ 3 HFS using criteria not considered to be sufficiently similar to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) (versions 2.0 to 4.0)
cReported combined data for grade ≥ 3 stomatitis and mucositis
dReported combined data for grade ≥ 3 vomiting and nausea
eAssessed grade 3 ≥ hyperbilirubinaemia using criteria not considered to be sufficiently similar to NCI CTCAE (versions 2.0 to 4.0 and 1981) and World Health Organisation (WHO) (1981 version)
AE: adverse event
DFS: disease‐free survival
OS: overall survival
HFS: hand foot syndrome
6. Included studies that contributed to pooled effect estimates for each outcome ‐ Patients treated with palliative intent for inoperable advanced or metastatic colorectal cancer.
| Study ID | Outcome | |||||||||||||
| Efficacy | Grade ≥ 3 AE | |||||||||||||
| PFS | TTP | OS | ORR | Diarrhoea | HFS |
Neutropenia/ granulocytopenia |
Febrile neutropenia | Vomiting | Nausea | Stomatitis | Mucositis | Hyperbilirubinemia | Any | |
| Ahn 2003 | X | X | X | Oa | Oa | Oa | Oa | |||||||
| Andersen 1987 | Ob | X | ||||||||||||
| Bajetta 1996 | X | X | X | X | X | |||||||||
| Carmichael 2002 | X | X | X | X | X | X | Xc | Xc | Xd | Xd | Oe | |||
| Cassidy 2011a | X | X | X | X | X | X | X | Xc | Xc | X | X | |||
| Comella 2009 | X | X | X | X | X | X | X | |||||||
| Diaz‐Rubio 2007 | X | X | X | X | X | X | X | X | X | X | X | |||
| Douillard 2002 | X | X | X | X | Of | X | X | Xc | Xc | Xd | Xd | Oe | ||
| Douillard 2014 | X | X | X | X | X | X | X | X | X | X | X | X | X | |
| Ducreux 2011 | X | X | X | X | X | X | X | X | X | X | ||||
| Ducreux 2013 | X | X | X | X | X | X | X | X | X | X | X | |||
| ECOG E5296 2012 | X | X | X | X | X | X | X | X | X | X | X | |||
| Fuchs 2007 | X | X | X | X | X | X | X | X | X | |||||
| Hochster TREE‐1 2008 | Ob | X | X | X | X | X | Xc | Xc | X | |||||
| Hochster TREE‐2 2008 | Ob | X | X | X | X | X | Xc | Xc | X | |||||
| Hoff 2001 | X | X | X | X | Og | X | X | X | X | X | ||||
| Kato 2012 | X | X | X | X | X | X | X | X | X | |||||
| Kohne 2008 | X | X | X | X | X | X | X | X | X | |||||
| Martoni 2006 | X | X | X | X | X | X | X | |||||||
| Mei 2014 | Oh | X | X | Xc | Xc | |||||||||
| Nogue 2005 | X | X | X | X | X | Xc | Xc | X | ||||||
| Pectasides 2012 | X | X | X | X | X | X | X | X | X | X | X | |||
| Porschen 2007 | X | X | X | X | X | X | X | X | ||||||
| Rothenberg 2008 | X | X | X | X | X | X | X | X | X | X | Oi | X | ||
| Schilsky 2002a | X | X | X | X | Og | X | X | X | X | X | ||||
| Seymour 2011 | X | X | Oj | X | X | X | X | X | X | X | ||||
| Shigeta 2016 | X | X | X | X | X | X | X | X | Xd | Xd | X | X | ||
| Silvestris 2010 | Ob | Oa | Oa | Oa | ||||||||||
| Souglakos 2012 | X | X | X | X | X | X | X | X | X | X | ||||
| Van Cutsem 2001a | X | X | X | X | X | X | X | |||||||
| Van Cutsem 2001b | X | X | X | X | Og | X | X | X | X | |||||
| Yamada 2013 | X | X | X | X | X | X | X | X | X | Xd | Xd | X | ||
| Yamazaki 2015 | X | X | X | X | X | X | X | X | X | |||||
| Yasui 2015 | X | X | X | X | X | X | X | Xd | Xd | |||||
| Yu 2005 | Ob | Ob | X | Oa | Oa | Oa | Oa | Oa | ||||||
X: Study contributed to the pooled effect estimate for the outcome
O: Study reported the outcome but did not contribute to the pooled effect estimate for the outcome
aUnclear number of participants assessed for outcomes in both arms
bHazard ratios could not be estimated either directly or indirectly from the provided information
cReported combined data for grade ≥3 vomiting and nausea
dReported combined data for grade ≥3 stomatitis and mucositis
eAssessed grade ≥ 3 hyperbilirubinaemia using Common Toxicity Criteria (CTC), version not specified
fAssessed grade ≥ 3 HFS using CTC, version not specified
gAssessed grade ≥ 3 HFS using criteria not considered to be sufficiently similar to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) (versions 2.0 to 4.0)
hORR reported after 2 cycles of chemotherapy
iAssessed grade ≥3 hyperbilirubinaemia using criteria not considered to be sufficiently similar to NCI CTCAE (versions 2.0 to 4.0 and 1981) and World Health Organisation (WHO) (1981 version)
jORR reported 12 to 14 weeks after start of treatment
AE: adverse event
PFS: progression‐free survival
TTP: time to progression
OS: overall survival
ORR: objective response rate
HFS: hand foot syndrome
Allocation
Random sequence generation
Studies assessed as 'Low' risk used random sequence generation methods including minimisation, varying block size, and computer‐assisted randomisation.
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
We assessed seven curative intent studies as 'Low' risk. Two curative intent studies had 'Unclear' risk of bias owing to unspecified methods of randomisation, and we did not assess any studies as having 'High' risk of bias.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
We assessed 20 palliative intent studies as 'Low' risk. Fifteen palliative intent studies had ‘Unclear’ risk of bias owing to unspecified methods of randomisation, and we did not assess any studies as having 'High' risk of bias.
Allocation concealment
Studies assessed as 'Low' risk used central randomisation by fax, interactive voice response system (IVRS), computer, and central centre.
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
We assessed seven curative intent studies as 'Low' risk. Two curative intent studies had 'Unclear' risk of bias owing to lack of information about allocation concealment, and we did not assess any studies as having 'High' risk of bias.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
We assessed 21 palliative intent studies as 'Low' risk. Fourteen palliative intent studies had 'Unclear' risk of bias owing to lack of information about allocation concealment, and we did not assess any studies as having 'High' risk of bias.
Blinding
Blinding of participants/personnel
One study described a 'double‐blind method' (Yu 2005); however, we judged this to be unclear and unlikely, as investigators did not describe placebo in either the oral or IV treatment arms. No other studies described blinding of participants and/or personnel.
DFS/PFS/TTP/ORR
We judged that lack of blinding of participants and/or personnel would not lead to ‘High’ risk of bias for these outcomes.
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
DFS outcome: We assessed the seven curative intent studies used in the meta‐analysis for this outcome to have 'Low' risk of bias.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
PFS/TTP/ORR outcomes: We assessed the 33 palliative intent studies that contributed to at least one of these outcomes to have ‘Low’ risk of bias.
OS
We judged that lack of blinding of participants and/or personnel would not lead to ‘High’ risk of bias for these outcomes.
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
We assessed the seven curative intent studies used in the meta‐analysis for this outcome to have 'Low' risk of bias.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
We assessed the 29 palliative intent studies used in the meta‐analysis for this outcome to have 'Low' risk of bias.
Grade ≥ 3 AEs
We judged that lack of blinding of participants and personnel would lead to 'High' risk of bias for this outcome.
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
All nine curative intent studies that reported this outcome were open‐label and were deemed at 'High' risk of bias.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
All 31 palliative intent studies that reported this outcome were open‐label and were deemed at 'High' risk of bias.
Blinding of outcome assessment
DFS/PFS/TTP/ORR
We judged that lack of blinding of outcome assessors would lead to 'High' risk of bias for this outcome.
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
DFS outcome: We assessed all seven curative intent studies to have 'High' risk of bias for detection of disease recurrence.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
PFS/TTP/ORR: Of the 33 palliative intent studies that included these outcomes, eight studies had ‘Low’ risk, two had ‘Unclear’ risk, and 23 had 'High’ risk of bias. For all ‘Low’‐risk studies, blinded independent physicians/radiologists or an independent review committee (IRC) assessed response outcomes (Cassidy 2011a; Ducreux 2011; Hoff 2001; Kato 2012; Schilsky 2002a; Souglakos 2012; Van Cutsem 2001b; Yamazaki 2015). The two ‘Unclear’ risk studies used an unspecified method of assessment. In Rothenberg 2008 investigators as well as a blinded IRC assessed tumour response; however it remains unclear whether investigator assessments or IRC assessments were used for the reported PFS outcome. Carmichael 2002 evaluated response data locally, with subsequent central review. However, study authors did not specifically describe the role of the central review in the reported response data. 'High’‐risk studies were other open‐label studies that did not describe using blinded independent radiologists or an IRC to assess response outcomes.
OS
We judged that lack of blinding of outcome assessors would not lead to ‘High’ risk of bias for these outcomes.
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
We assessed seven curative intent studies used in the meta‐analysis for this outcome to have 'Low' risk of bias.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
We assessed 29 palliative intent studies used in the meta‐analysis for this outcome to have 'Low' risk of bias.
Grade ≥ 3 AEs
We judged that lack of blinding of outcome assessors would lead to 'High' risk of bias for this outcome, in particular for assessment of subjective grade ≥ 3 AEs such as HFS, diarrhoea, vomiting, nausea, stomatitis, and mucositis. We did not judge that lack of blinding of outcome assessors would affect assessment of grade ≥ 3 neutropenia/granulocytopenia, febrile neutropenia, or hyperbilirubinaemia, as these rely upon objective laboratory assessments.
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
We assessed all nine curative intent studies used in the meta‐analysis for these outcomes to have 'High' risk of bias.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
We assessed all 31 palliative intent studies used in the meta‐analysis for these outcomes to have 'High' risk of bias.
Incomplete outcome data
Attrition bias
We judged that studies with high percentages (≥ 20%) of non‐evaluable response data in at least one treatment arm had 'High' risk of bias.
ORR in studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
Of the 32 palliative intent studies that contributed to the ORR analysis, 23 studies had 'Low' risk of bias, four had 'Unclear’ risk, and five had ‘High’ risk. For Andersen 1987, ORR data were non‐evaluable for 20% of participants in the IV 5‐FU arm, and for Ahn 2003, ORR data were non‐evaluable for 29% of participants in the 5‐dFUR/LV arm. Twenty‐three per cent of participants in the CapeOx arm had missing confirmed tumour response data in Hochster TREE‐1 2008. Twenty‐four per cent (FT/LV) and 20% (5‐FU/LV) of participants in Nogue 2005 had non‐evaluable data for response owing to protocol deviations in response evaluation methods. In Pectasides 2012, 30.1% of participants in the XELIRI‐BEV arm and 19.7% of those in the FOLFIRI‐BEV arm had non‐evaluable response data owing to treatment discontinuation, early death, missing data, and non‐evaluable disease.
Time‐to‐event outcomes (DFS/PFS/OS/TTP)
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
Of the seven curative intent studies that contributed to DFS or OS (curative intent studies) pooled effect estimates, six studies had ‘Low’ risk of bias, and we judged one study to have ‘Unclear’ risk (Lembersky 2006).
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
Of the 31 palliative intent studies with a time‐to‐event outcome, 23 studies had ‘Low’ risk of bias with no or minimal missing data. Five studies had ‘Unclear’ risk. We judged three studies as having ‘High’ risk (Ahn 2003; Nogue 2005; Pectasides 2012).
Grade ≥ 3 AEs
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
Of the nine curative intent studies included in the meta‐analysis for these outcomes, four had no or minimal missing data, and we assessed these as 'Low' risk. Five studies had an unclear number of participants with missing data and had ‘Unclear’ risk of bias.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
Of the 31 palliative studies that reported this outcome, 14 studies had no or minimal missing data, and we assessed these as 'Low' risk. The other 17 studies had an unclear number of participants with missing data and had ‘Unclear’ risk of bias.
ITT analysis
Efficacy analysis
We judged studies to be at 'Low' risk of bias if an ITT analysis was performed as per the definition in our review, or if < 5% of randomised participants were excluded from the analysis.
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
Six curative intent studies were at 'Low' risk and one study was at 'High' risk of bias (Pectasides 2015).
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
Twenty‐seven palliative intent studies were at 'Low' risk, two studies were at 'Unclear' risk, and four studies were at 'High' risk of bias (Andersen 1987; Hochster TREE‐2 2008; Nogue 2005; Pectasides 2012).
Safety analysis
Most studies performed a safety analysis in the as‐treated population, which included participants who had received at least one dose of chemotherapy.
Selective reporting
ECOG E5296 2012 and Shimada 2014 were the only studies for which a protocol was available. The technical report (ECOG E5296 2012) or the study report (Shimada 2014) included all of the outcomes described in the protocol, and we assessed these studies as ‘Low’ risk. All other studies had ‘Unclear’ risk.
Other potential sources of bias
Schedule of follow‐up and assessment
We judged studies to be at 'High' risk of bias if they used different schedules for assessment of disease recurrence/response, survival, and/or grade ≥ 3 AEs between treatment arms. For example, more frequent AE assessments in a treatment arm compared with the other treatment arm/s may have increased the likelihood of documenting and treating the toxicities of interest earlier. Similarly, more frequent disease recurrence, response, or survival assessments in a treatment arm compared with the other treatment arm/s may have increased the likelihood of documenting recurrence, progression, or death earlier. Variation in assessment schedules occurred because of differences in cycle lengths among treatment arms.
Disease recurrence/response (influences DFS/PFS/TTP/ORR)
This pertains to the detection of disease recurrence, response, and progression events.
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
Of the seven curative intent studies contributing to these outcomes, five had assessments performed at the same time and were at 'Low' risk of bias. Two did not specify the assessment schedule and were at 'Unclear' risk. No studies were at 'High' risk of bias.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
Of the 33 palliative studies contributing to these outcomes, 23 studies had assessments performed at the same time and were at ‘Low’ risk of bias. Four palliative studies did not specify the assessment schedule and were at 'Unclear' risk. Six studies that we assessed as ‘High’ risk had differences in assessment schedules between study arms (Douillard 2002; Ducreux 2011; Nogue 2005; Pectasides 2012; Porschen 2007; Schilsky 2002a).
Survival (influences DFS/PFS/OS)
This pertains to detection of death events.
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
Of seven curative intent studies contributing to these outcomes, five studies had 'Low' risk of bias, and two had 'Unclear' risk.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
Of 29 palliative intent studies contributing to these outcomes, two studies had ‘High’ risk of bias. In Hochster TREE‐1 2008, following treatment discontinuation, investigators collected follow‐up data for participants who consented retrospectively, but provided no information on the number of participants in each arm who consented and were followed up. Shigeta 2016 provided survival follow‐up at the discretion of the treating physician. We assessed a further 19 studies as 'Low' risk. Eight studies were at 'Unclear' risk owing to insufficient information.
Grade ≥ 3 AEs
This pertains to the detection of grade ≥ 3 AEs.
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
Of nine curative studies that contributed to the pooled estimate analysis, three studies (Hofheinz 2012; Lembersky 2006; Shimada 2014) had different AE assessment schedules between arms and were at ‘High’ risk. Two studies were at 'Low' risk, and four studies were at 'Unclear' risk.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
Of the 31 palliative intent studies that contributed to the pooled estimate analysis, we considered 13 to have ‘High’ risk (Bajetta 1996; Diaz‐Rubio 2007; Ducreux 2013; Fuchs 2007; Hochster TREE‐1 2008; Hochster TREE‐2 2008; Kato 2012; Nogue 2005; Pectasides 2012; Schilsky 2002a; Seymour 2011; Souglakos 2012; Yamada 2013). We assessed nine studies as 'Low' risk, and nine studies as having 'Unclear' risk owing to insufficient information.
Baseline similarities
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
Of nine curative intent studies that contributed to meta‐analyses for any of the outcomes in this review, six studies were 'Low' risk, as participants in all treatment arms had similar baseline characteristics with regards to PS, median or mean age, and disease stage. Two studies had 'Unclear' risk, and one study had 'High' risk owing to a difference in mean age of 7.2 years (Kim 2001a).
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
Of the 34 palliative intent studies that contributed to meta‐analyses for any of the outcomes in this review, 12 studies had 'Low' risk of bias, as participants in all treatment arms had similar baseline characteristics with regards to PS, median or mean age, and number of organs involved with metastases, or KRAS mutation status in the case of EGFR inhibitor treatment. Eighteen studies had ’Unclear’ risk of bias. Four studies had ‘High’ risk of bias owing to differences between comparison arms. Hochster TREE‐2 2008 and Shigeta 2016 reported a five‐year difference in median age between oral and IV arms. Martoni 2006 described a 16.5% difference between arms with regards to the percentage with one versus more than one metastatic site at baseline. Douillard 2014 performed a post hoc analysis of participants evaluable for KRAS mutation status and found that a greater proportion of those in the UFOX + cetuximab arm (47/87; 54%) were KRAS mutant than in the FOLFOX4 + cetuximab arm (37/93; 40%). Whilst only 60% of the population was evaluable for KRAS mutation status, we considered that a 14% difference between oral and IV arms would lead to 'High' risk of bias.
Other bias
Studies of curative intent treatment with neoadjuvant and/or adjuvant chemotherapy for CRC
Two curative intent studies provided information about subsequent treatment with adjuvant chemotherapy or chemotherapy following a recurrence or a new occurrence of CRC (Allegra 2015; Twelves 2012); both had 'Low' risk of bias. The remaining seven curative intent studies did not provide this information and had 'Unclear' risk.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
Twenty‐one palliative intent studies reported information about subsequent lines of treatment used for each treatment arm after disease progression. Study authors reported no major differences between treatment arms with regards to the percentage of participants who received subsequent therapy or the type of subsequent therapy used. None were at high risk of bias.
We identified no other reasons for high risk of bias in the included studies.
Risk of bias for outcomes
With respect to efficacy outcomes, we considered DFS in curative intent studies, and PFS, TTP, and ORR in palliative intent studies, to be outcomes at risk of detection bias owing to lack of blinding of outcome assessors. We did not judge OS in both curative intent and palliative intent studies to be at risk of bias owing to lack of blinding of outcome assessors.
With respect to adverse event outcomes, we considered the grade ≥ 3 AEs diarrhoea, HFS, vomiting, nausea, stomatitis, mucositis, and any grade ≥ 3 AEs to be subjective outcomes that were at risk of performance and detection bias if blinding of participants and personnel, and outcome assessors, was lacking, respectively. We considered the grade ≥ 3 AEs neutropenia/granulocytopenia, febrile neutropenia, and hyperbilirubinaemia to be objective outcomes that were not at risk of performance and detection bias from lack of blinding.
Patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy
DFS
We considered all seven curative intent studies that contributed to the pooled effect estimate for the DFS outcome to have high risk of detection bias owing to lack of blinding of outcome assessors (Table 9), and we downgraded this outcome for risk of bias. One study (Pectasides 2015) additionally had high risk of bias owing to lack of an ITT analysis.
7. Risk of bias for studies contributing to the quantitative synthesis for disease‐free survival.
| Risk of bias assessment | ||
| Low | Unclear | High |
| No studies | No studies | Allegra 2015 |
| De Gramont 2012 | ||
| Hofheinz 2012 | ||
| Lembersky 2006 | ||
| Pectasides 2015 | ||
| Shimada 2014 | ||
| Twelves 2012 | ||
OS (curative intent studies)
We did not judge OS (curative intent studies) to be at risk of bias from lack of blinding of outcome assessors. One study (Pectasides 2015) had high risk of bias owing to lack of an ITT analysis. However, this study contributed only 4.2% of the weight for the pooled effect estimate for this outcome, and we did not downgrade this outcome for risk of bias.
Grade ≥ 3 AEs (curative intent studies)
Subjective outcomes
All nine curative intent studies that contributed to the subjective outcomes of grade ≥ 3 AEs diarrhoea, HFS, vomiting, nausea, stomatitis, mucositis, and any grade ≥ 3 AE had high risk of bias owing to lack of blinding; consequently, we downgraded these outcomes for risk of bias.
Four of these nine studies additionally had high risk of bias in other domains. Hofheinz 2012 (which contributed to all subjective grade ≥ 3 AE outcomes), Lembersky 2006 (which contributed to grade ≥ 3 diarrhoea, vomiting, nausea, stomatitis, and any grade ≥ 3 AE outcomes), and Shimada 2014 (which contributed to grade ≥ 3 diarrhoea, HFS, vomiting, and nausea outcomes) had high risk of bias owing to differences in schedules of assessment and/or follow‐up between treatment arms. Kim 2001a (which contributed to grade ≥ 3 diarrhoea and stomatitis) also had high risk of bias owing to a difference in baseline mean age of participants between treatment arms.
Objective outcomes
The grade ≥ 3 AEs neutropenia/granulocytopenia, febrile neutropenia, and hyperbilirubinaemia were objective outcomes and were not at risk of performance and detection bias from lack of blinding.
However, for grade ≥ 3 neutropenia/granulocytopenia (curative intent studies), Lembersky 2006 and Shimada 2014 had high risk of bias owing to differences in schedules of assessment and/or follow‐up between treatment arms. These studies contributed 7.3% of the weight for the pooled effect estimate for this outcome, and we did not downgrade this outcome for risk of bias.
No studies were at high risk of bias for the grade ≥ 3 febrile neutropenia (curative intent studies) outcome, and we did not downgrade this outcome for risk of bias.
For grade ≥ 3 hyperbilirubinaemia (curative intent studies), Hofheinz 2012 and Shimada 2014 were at high risk of bias owing to differences in schedules of assessment and/or follow‐up between treatment arms. These studies contributed 44.3% of the weight for the pooled effect estimate for this outcome, and we downgraded this outcome for risk of bias.
Studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC
PFS
For the PFS outcome, 17 out of 23 studies had high risk of bias (Table 10). These studies contributed 48.5% of the pooled effect estimate for the PFS outcome, and we downgraded the PFS outcome for risk of bias.
8. Risk for bias for studies contributing to the quantitative synthesis for progression‐free survival.
Fifteen of these studies had high risk of detection bias owing to lack of blinding of outcome assessors (Bajetta 1996; Comella 2009; Douillard 2014;Ducreux 2013; ECOG E5296 2012; Fuchs 2007; Kato 2012; Kohne 2008; Pectasides 2012; Porschen 2007; Seymour 2011; Shigeta 2016; Van Cutsem 2001a; Yamada 2013; Yasui 2015). Four of these fifteen studies additionally had high risk of bias in other domains. Douillard 2014 had high risk of bias owing to an imbalance in the proportion of participants with KRAS mutations between oral and IV arms (within the population evaluable for KRAS mutation status) and high risk of detection bias. Pectasides 2012 had high risk of bias owing to detection bias, differences in schedules of assessment and/or follow‐up between arms, lack of an ITT analysis, and attrition bias. Porschen 2007 had high risk of bias owing to differences in schedules of assessment and/or follow‐up between arms. Shigeta 2016 had high risk of bias owing to detection bias, differences in schedules of assessment and/or follow‐up between arms, and a difference in baseline median age of participants between treatment arms.
The remaining two studies (Ducreux 2011; Schilsky 2002a) had low risk of detection bias because tumour responses were reviewed by a blinded independent review panel. However, both studies had high risk of bias owing to differences in schedules of assessment and/or follow‐up between arms.
OS (palliative intent studies)
We did not judge OS (palliative intent studies) to be at risk of bias from lack of blinding of outcome assessors. However, five out of 29 studies had high risk of bias in other domains (Douillard 2014; Hochster TREE‐1 2008; Hochster TREE‐2 2008; Pectasides 2012; Shigeta 2016). These studies contributed only 4.4% to the pooled effect estimate for the OS (palliative intent studies) outcome, and we did not downgrade this outcome for risk of bias.
Douillard 2014 had high risk of bias owing to an imbalance in the proportion of participants with KRAS mutations between oral and IV arms (within the population evaluable for KRAS mutation status). Hochster TREE‐1 2008 and Shigeta 2016 had high risk of bias owing to differences in schedules of assessment and/or follow‐up between arms. Additionally, Shigeta 2016 had high risk of bias owing to a difference in baseline median age of participants between treatment arms. Hochster TREE‐2 2008 had high risk of bias for lack of an ITT analysis and a difference in baseline median age of participants between treatment arms. Pectasides 2012 had high risk of bias owing to lack of an ITT analysis.
TTP
For the TTP outcome, five out of six studies had high risk of bias owing to lack of blinding of outcome assessors (Ahn 2003; Diaz‐Rubio 2007; Douillard 2002; Martoni 2006; Nogue 2005). These studies contributed 93.4% of the pooled effect estimate for the TTP outcome, and we downgraded this outcome for risk of bias.
Three of these five studies had additional judgements of high risk of bias in other domains. Ahn 2003 had high risk of attrition bias, and Douillard 2002 had high risk of bias owing to differences in schedules of assessment and/or follow‐up between arms. Nogue 2005 had high risk of attrition bias and of bias due to differences in schedules of assessment and/or follow‐up between arms, as well as lack of an ITT analysis.
ORR
For the ORR outcome, we considered 25 out of 32 studies to have high risk of bias. These studies contributed 59.3% of the pooled effect estimate for the ORR outcome, and we downgraded this outcome for risk of bias.
Twenty‐three of these studies had high risk of bias owing to lack of blinding of outcome assessors (Ahn 2003; Andersen 1987; Bajetta 1996; Comella 2009; Diaz‐Rubio 2007; Douillard 2002; Douillard 2014; Ducreux 2013; ECOG E5296 2012; Fuchs 2007; Hochster TREE‐1 2008; Hochster TREE‐2 2008; Kato 2012; Kohne 2008; Martoni 2006; Nogue 2005; Pectasides 2012; Porschen 2007; Shigeta 2016; Van Cutsem 2001a; Van Cutsem 2001b; Yasui 2015; Yu 2005). Six of these 23 studies had additional judgements of high risk of bias in other domains. Ahn 2003, Andersen 1987, and Hochster TREE‐1 2008 had high risk of attrition bias; Andersen 1987 additionally had high risk of bias owing to lack of an ITT analysis. Hochster TREE‐2 2008 had high risk of bias owing to lack of an ITT analysis. Nogue 2005 and Pectasides 2012 had high risk of attrition bias owing to differences in schedules of assessment and/or follow‐up between arms, as well as lack of an ITT analysis.
The remaining two studies (Ducreux 2011; Schilsky 2002a) had low risk of detection bias because tumour responses were reviewed by a blinded independent review panel in these studies. However, these two studies had high risk of bias owing to differences in schedules of assessment and/or follow‐up between arms.
Grade ≥ 3 AEs (palliative intent studies)
Subjective outcomes
All 31 palliative intent studies that contributed to the subjective outcomes grade ≥ 3 AEs diarrhoea, HFS, vomiting, nausea, stomatitis, mucositis, and any grade ≥ 3 AE had high risk of bias owing to lack of blinding, and we downgraded these outcomes for risk of bias.
Fourteen of these 31 studies had additional judgements of high risk of bias in other domains (Bajetta 1996; Diaz‐Rubio 2007; Ducreux 2013; Fuchs 2007; Hochster TREE‐1 2008; Hochster TREE‐2 2008; Kato 2012; Nogue 2005; Pectasides 2012; Schilsky 2002a; Seymour 2011; Shigeta 2016; Souglakos 2012; Yamada 2013). With the exception of Shigeta 2016 (high risk of bias caused by a difference in baseline median age of participants between treatment arms), all of these studies had additional high risk of bias owing to differences in schedules of assessment and/or follow‐up between arms. Hochster TREE‐2 2008 also had high risk of bias owing to a difference in baseline median age of participants between treatment arms.
Objective outcomes
The grade ≥ 3 AEs neutropenia/granulocytopenia, febrile neutropenia, and hyperbilirubinaemia were objective outcomes and were not at risk of performance and detection bias from lack of blinding.
However, for the grade ≥ 3 neutropenia/granulocytopenia (palliative intent studies) outcome, 13 out of 29 studies (Diaz‐Rubio 2007; Ducreux 2013; Fuchs 2007; Hochster TREE‐1 2008; Hochster TREE‐2 2008; Kato 2012; Nogue 2005; Pectasides 2012; Schilsky 2002a; Seymour 2011; Shigeta 2016; Souglakos 2012; Yamada 2013) had high risk of bias in domains unrelated to lack of blinding. These studies contributed 29.2% of the pooled effect estimate for the grade ≥ 3 neutropenia/granulocytopenia (palliative intent studies) outcome, and we downgraded this outcome for risk of bias. With the exception of Shigeta 2016 (high risk of bias caused by a difference in baseline median age of participants between treatment arms), all of these studies had additional high risk of bias owing to differences in schedules of assessment and/or follow‐up between arms. Hochster TREE‐2 2008 also had high risk of bias owing to a difference in baseline median age of participants between treatment arms.
No studies were at high risk of bias for grade ≥ 3 febrile neutropenia, and we did not downgrade this outcome for risk of bias.
For the grade ≥ 3 hyperbilirubinaemia (palliative intent studies) outcome, four out of nine studies (Diaz‐Rubio 2007; Kato 2012; Yamada 2013; Shigeta 2016) had high risk of bias in domains unrelated to lack of blinding. These studies contributed 28.5% of the pooled effect estimate for the grade ≥ 3 hyperbilirubinaemia (palliative intent studies) outcome, and we downgraded this outcome for risk of bias. Diaz‐Rubio 2007, Kato 2012, and Yamada 2013 had high risk of bias owing to differences in schedules of assessment and/or follow‐up between arms, and Shigeta 2016 had high risk of bias owing to a difference in baseline median age of participants between treatment arms.
Effects of interventions
We have provided a summary of the results for effects of interventions, shown in Data and analyses. Table 7 (patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy) and Table 8 (patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy) show the studies that contributed to pooled effect estimates for each outcome.
We have also described results of subgroup analyses for the outcomes that we assessed as the most important. For efficacy, these include DFS, PFS, and OS in both curative intent and palliative intent studies. For grade ≥ 3 AEs, these consist of diarrhoea and HFS in both curative intent and palliative intent studies. We have presented results of all other subgroup analyses in Appendix 9, Appendix 10, Appendix 11, and Appendix 12.
We have presented additional information for the outcomes analysed in this review, other than the information used in our quantitative synthesis, in Appendix 5, Appendix 6, Appendix 7, Appendix 8, Appendix 13 , Appendix 14, Appendix 15, Appendix 16, and Appendix 17.
Patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy
Co‐primary outcome
1.1 DFS
For the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, DFS did not differ between participants treated with oral versus IV fluoropyrimidines. The pooled HR from seven studies with 8903 participants was 0.93 (95% CI 0.87 to 1.00) (Analysis 1.1; Table 7). Results show no heterogeneity (Chi² = 5.51, P = 0.48; I² = 0%) among effect estimates for these studies (Figure 4).
1.1. Analysis.
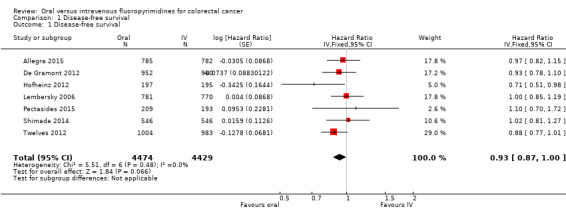
Comparison 1 Disease‐free survival, Outcome 1 Disease‐free survival.
4.
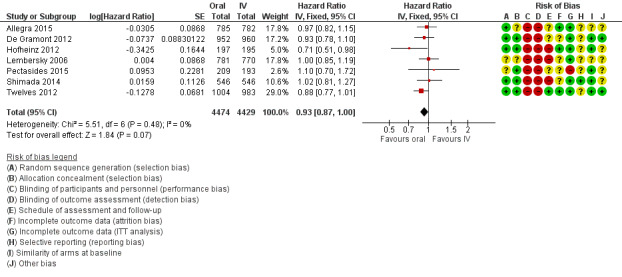
Forest plot of disease‐free survival.
We downgraded the quality of evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We assessed the quality of evidence as moderate (Table 1).
Subgroup analyses:
We observed no subgroup differences in any of the prespecified subgroup analyses (Analysis 1.2; Analysis 1.3; Analysis 1.4; Appendix 9):
1.2. Analysis.
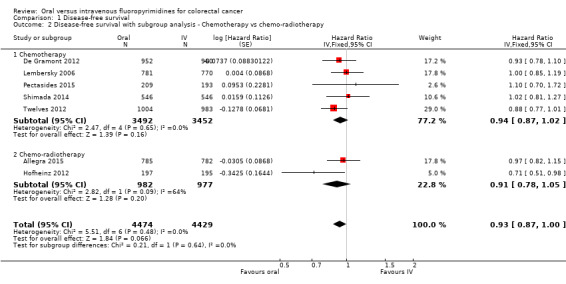
Comparison 1 Disease‐free survival, Outcome 2 Disease‐free survival with subgroup analysis ‐ Chemotherapy vs chemo‐radiotherapy.
1.3. Analysis.
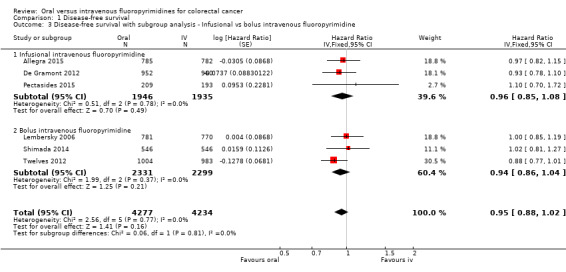
Comparison 1 Disease‐free survival, Outcome 3 Disease‐free survival with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine.
1.4. Analysis.
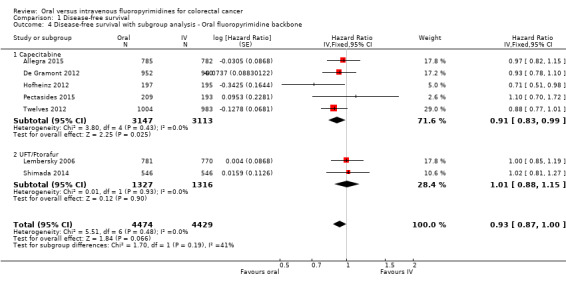
Comparison 1 Disease‐free survival, Outcome 4 Disease‐free survival with subgroup analysis ‐ Oral fluoropyrimidine backbone.
1.2 DFS with subgroup analysis ‐ Treatment type
Chi2 = 0.21, P = 0.64; I2 = 0%.
1.3 DFS with subgroup analysis ‐ Infusional versus bolus intravenous fluoropyrimidine
Chi2 = 0.06, P = 0.81; I2 = 0%.
1.4 DFS with subgroup analysis ‐ Oral fluoropyrimidine backbone
Chi2 = 1.70, P = 0.19; I2 = 41.1%.
Assessment of publication bias for DFS
We did not assess funnel plot asymmetry for the DFS outcome, as we included only seven studies in the meta‐analysis.
Secondary outcomes
2.1 OS (curative intent)
For the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, OS did not differ between participants treated with oral versus IV fluoropyrimidines. The pooled HR from seven studies with 8902 participants was 0.92 (95% CI 0.84 to 1.00) (Analysis 2.1; Table 7). Results show no heterogeneity (Chi² = 4.67, P = 0.59; I² = 0%) among effect estimates for these studies.
2.1. Analysis.
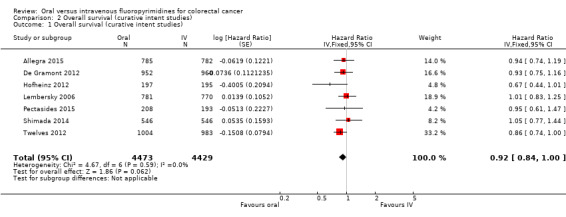
Comparison 2 Overall survival (curative intent studies), Outcome 1 Overall survival (curative intent studies).
We did not identify any factors that reduced the quality of evidence for this outcome, and we assessed the quality of evidence as high (Table 1).
Subgroup analyses
We observed no subgroup differences in any of the prespecified subgroup analyses (Analysis 2.2; Analysis 2.3; Analysis 2.4; Appendix 9):
2.2. Analysis.
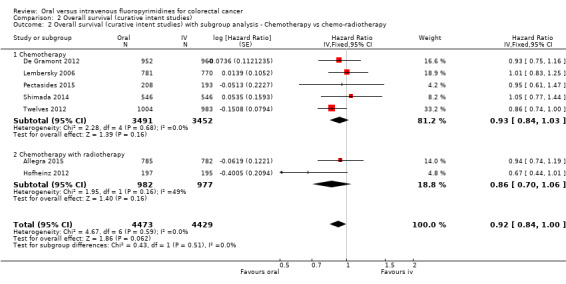
Comparison 2 Overall survival (curative intent studies), Outcome 2 Overall survival (curative intent studies) with subgroup analysis ‐ Chemotherapy vs chemo‐radiotherapy.
2.3. Analysis.
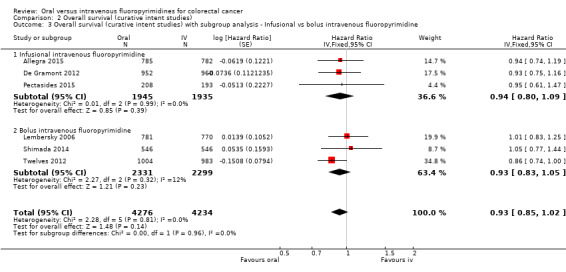
Comparison 2 Overall survival (curative intent studies), Outcome 3 Overall survival (curative intent studies) with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine.
2.4. Analysis.
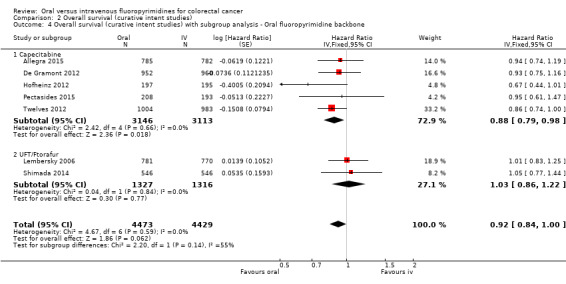
Comparison 2 Overall survival (curative intent studies), Outcome 4 Overall survival (curative intent studies) with subgroup analysis ‐ Oral fluoropyrimidine backbone.
2.2 OS with subgroup analysis ‐ Chemotherapy versus chemo‐radiotherapy
Chi2 = 0.43, P = 0.51; I2 = 0%.
2.3 OS with subgroup analysis ‐ Infusional versus bolus intravenous fluoropyrimidine
Chi2 = 0.00, P = 0.96; I2 = 0%.
2.4 OS with subgroup analysis ‐ Oral fluoropyrimidine backbone
Chi2 = 2.20, P = 0.14; I2 = 54.5%.
Grade ≥ 3 AEs (curative intent studies)
3.1 Grade ≥ 3 diarrhoea (curative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, grade ≥ 3 diarrhoea did not differ between participants treated with oral versus IV fluoropyrimidines. The pooled OR from nine studies with 9551 participants was 1.12 (95% CI 0.99 to 1.25) (Analysis 3.1; Table 7).
3.1. Analysis.
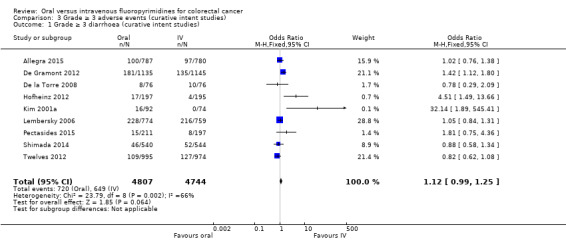
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 1 Grade ≥ 3 diarrhoea (curative intent studies).
We downgraded the quality of evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We further downgraded quality by one level for inconsistency of results, as we noted substantial heterogeneity among included studies (Chi² = 23.79, P = 0.002; I² = 66%), and by one level for imprecision. We assessed the quality of evidence as very low (Table 1).
Subgroup analyses
3.2 Grade ≥ 3 diarrhoea (curative intent studies) ‐ Treatment type
Results show no subgroup differences by treatment type (chemotherapy versus chemo‐radiotherapy): Chi2 = 1.24, P = 0.27; I2 = 19.3% (Analysis 3.2; Appendix 10).
3.2. Analysis.
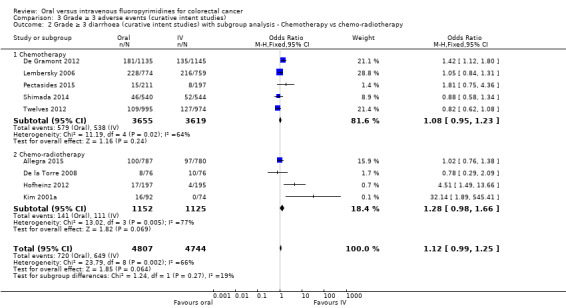
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 2 Grade ≥ 3 diarrhoea (curative intent studies) with subgroup analysis ‐ Chemotherapy vs chemo‐radiotherapy.
3.3 Grade ≥ 3 diarrhoea (curative intent studies) ‐ Infusional versus bolus intravenous fluoropyrimidine
Results show significant subgroup differences between ‘Infusional intravenous fluoropyrimidine’ (pooled OR 1.27, 95% CI 1.06 to 1.53 ‐ indicating more grade ≥ 3 diarrhoea with oral fluoropyrimidines) and ‘Bolus intravenous fluoropyrimidine’ (pooled OR 0.98, 95% CI 0.84 to 1.14 ‐ indicating that grade ≥ 3 diarrhoea did not differ between those treated with oral versus IV fluoropyrimidines) subgroups: Chi2 = 4.52, P = 0.03; I2 = 77.9% (Analysis 3.3; Appendix 10).
3.3. Analysis.
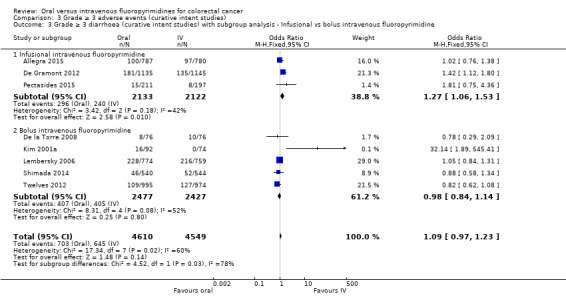
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 3 Grade ≥ 3 diarrhoea (curative intent studies) with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine.
3.4 Grade ≥ 3 diarrhoea (curative intent studies) ‐ Oral fluoropyrimidine backbone
Results show significant differences between subgroups for the different oral fluoropyrimidine backbones.
Pooled effect estimates for the ‘Capecitabine’ and ‘UFT/Ftorafur’ subgroups indicate that grade ≥ 3 diarrhoea did not differ between participants treated with oral versus IV fluoropyrimidines, whilst the OR for the only study in the ‘Doxifluridine’ subgroup (Kim 2001a) indicated that grade ≥ 3 diarrhoea was increased with oral fluoropyrimidines (OR 32.14, 95% CI 1.89 to 545.41). Tests for subgroup differences yielded these results: Chi2 = 6.73, P = 0.03; I2 = 70.3%. Substantial or considerable heterogeneity remained between studies within the ‘Capecitabine’ subgroup (Chi2 = 16.27, P = 0.003; I2 = 75%) (Analysis 3.4; Appendix 10).
3.4. Analysis.
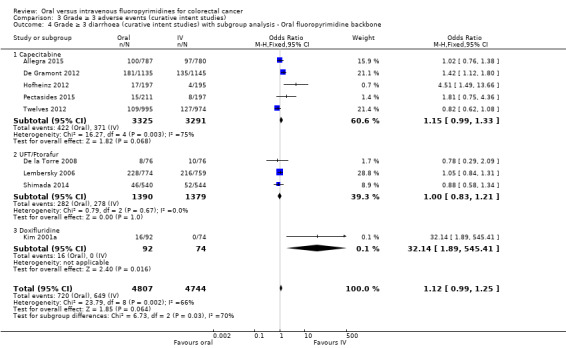
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 4 Grade ≥ 3 diarrhoea (curative intent studies) with subgroup analysis ‐ Oral fluoropyrimidine backbone.
3.5 Grade ≥ 3 hand foot syndrome (curative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, odds of grade ≥ 3 HFS were higher with oral fluoropyrimidine treatment. The pooled OR from five studies with 5731 participants was 4.59 (95% CI 2.97 to 7.10) (Analysis 3.5; Table 7).
3.5. Analysis.
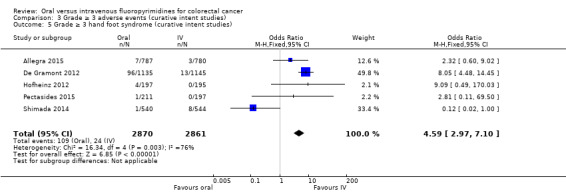
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 5 Grade ≥ 3 hand foot syndrome (curative intent studies).
We downgraded the quality of evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We further downgraded quality by one level for inconsistency of results, as we noted substantial or considerable heterogeneity among included studies (Chi² = 16.34, P = 0.003; I² = 76%). We assessed the quality of evidence as low (Table 1).
In four of the included studies, effect estimates favoured IV fluoropyrimidines, and in three of these, 95% CIs crossed the null value of 1.00 (Allegra 2015; Hofheinz 2012; Pectasides 2015). In one outlier study (Shimada 2014), the effect estimate favoured oral fluoropyrimidines and the upper limit of the 95% CI was 1.00. It is unclear whether this variation in effects was due to clinical diversity (this was the only study for this outcome that utilised UFT and enrolled patients only from Japan) and/or methodological diversity (AE assessments were less frequent in the oral than in the IV treatment arm). In a post hoc sensitivity analysis in which we incorporated heterogeneity into a random‐effects model meta‐analysis, the pooled OR was 2.36 and the 95% confidence interval crossed the null value of 1.00 (95% CI 0.52 to 10.74).
3.6 Grade ≥ 3 neutropenia/granulocytopenia (curative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, the pooled OR for grade ≥ 3 neutropenia/granulocytopenia from seven studies with 8087 participants was 0.14 (95% CI 0.11 to 0.16), favouring oral fluoropyrimidines (Table 7).
We downgraded the quality of evidence by one level for inconsistency of results, as we noted substantial or considerable heterogeneity between the included studies (Chi² = 53.38, P < 0.00001, I² = 89%). We assessed the quality of evidence as moderate (Table 1).
The 95% CIs for effect estimates either favoured the oral fluoropyrimidine group (four studies) or crossed the null value of 1.00 (two studies). Only one outlier study (Allegra 2015) reported that the effect estimate and the 95% CI indicated more grade ≥ 3 neutropenia/granulocytopenia with oral fluoropyrimidine treatment (the only study for this outcome that included combination chemotherapy with radiotherapy) (Analysis 3.6).
3.6. Analysis.
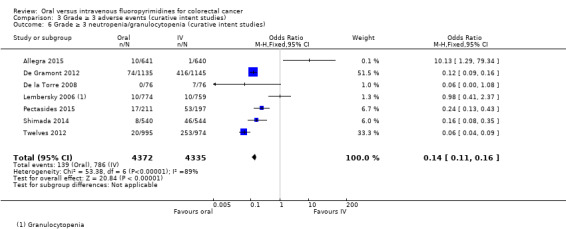
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 6 Grade ≥ 3 neutropenia/granulocytopenia (curative intent studies).
Grade ≥ 3 neutropenia/granulocytopenia (curative intent studies) – study data not suitable for quantitative synthesis
For the Hofheinz 2012 and Kim 2001a studies, wherein neutropenia/granulocytopenia was not specifically reported, the incidence of the grade ≥ 3 AEs ‘lowered leucocytes’ and ‘leukopenia’, respectively, was lower in the oral fluoropyrimidine arms (Appendix 6).
3.7 Grade ≥ 3 febrile neutropenia (curative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, grade ≥ 3 febrile neutropenia events were few in the four studies with 2925 participants that reported this outcome (Analysis 3.7; Table 7). The pooled OR was 0.59 (95% CI 0.18 to 1.90), and we observed no heterogeneity (Chi² = 2.65, P = 0.45; I² = 0%).
3.7. Analysis.
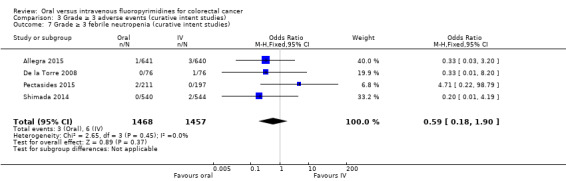
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 7 Grade ≥ 3 febrile neutropenia (curative intent studies).
We downgraded the quality of evidence by two levels for imprecision (small number of events and 95% CI included appreciable benefit and harm) and assessed quality as low.
3.8 Grade ≥ 3 vomiting (curative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, grade ≥ 3 vomiting did not differ between participants treated with oral versus IV fluoropyrimidines. The pooled OR from eight studies with 9385 participants was 1.05 (95% CI 0.83 to 1.34) (Analysis 3.8; Table 7).
3.8. Analysis.
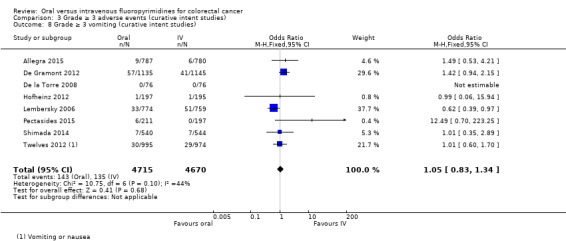
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 8 Grade ≥ 3 vomiting (curative intent studies).
We downgraded the quality of the evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We downgraded quality by one further level for imprecision. The final assessment for quality of evidence was low.
Heterogeneity among effect estimates for these studies was moderate (Chi² = 10.75, P = 0.10; I² = 44%), albeit not statistically significant. However, most of the effect estimates with their 95% CIs crossed the null value of 1.00, with the exception of one outlier study (Lembersky 2006), for which the effect estimate favoured oral fluoropyrimidines.
3.9 Grade ≥ 3 nausea (curative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, grade ≥ 3 nausea did not differ between participants treated with oral versus IV fluoropyrimidines. The pooled OR from seven studies with 9233 participants was 1.21 (95% CI 0.97 to 1.51) (Analysis 3.9; Table 7). Heterogeneity among effect estimates for these studies was minimal (Chi² = 6.40, P = 0.38, I² = 6%).
3.9. Analysis.
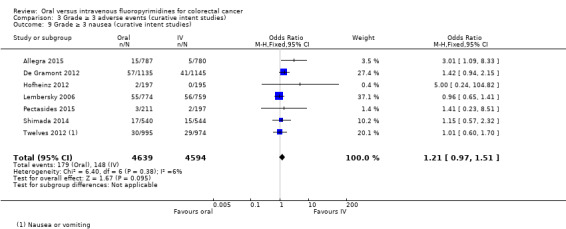
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 9 Grade ≥ 3 nausea (curative intent studies).
We downgraded the quality of the evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We downgraded quality by one further level for imprecision. The final assessment for quality of evidence was low.
3.10 Grade ≥ 3 stomatitis (curative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, the pooled OR for grade ≥ 3 stomatitis from five studies with 4212 participants was 0.21 (95% CI 0.14 to 0.30), favouring oral fluoropyrimidines (Analysis 3.10; Table 7).
3.10. Analysis.
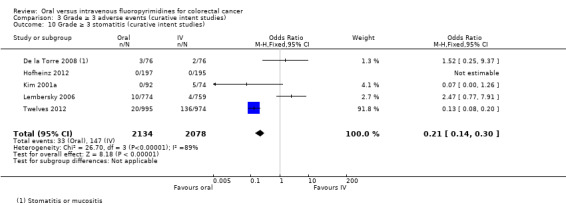
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 10 Grade ≥ 3 stomatitis (curative intent studies).
We downgraded the quality of evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We downgraded quality by one further level for inconsistency of results, as we noted substantial or considerable heterogeneity between the included studies (Chi² = 26.70, P < 0.00001; I² = 89%). We assessed the quality of evidence as low. However, in the included studies, 95% CIs for effect estimates either crossed the null value of 1.00 (three studies) or favoured oral fluoropyrimidines (one study, Twelves 2012).
3.11 Grade ≥ 3 mucositis (curative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, grade ≥ 3 mucositis did not differ between participants treated with oral versus IV fluoropyrimidines. The pooled OR from four studies with 2233 participants was 0.64 (95% CI 0.25 to 1.62) (Analysis 3.11; Table 7). We noted no heterogeneity among effect estimates for these studies (Chi² = 1.56, P = 0.67; I² = 0%).
3.11. Analysis.
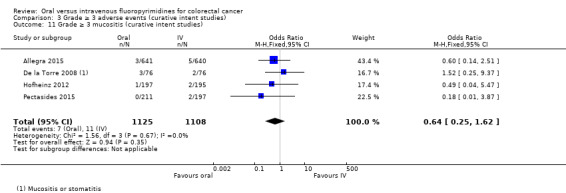
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 11 Grade ≥ 3 mucositis (curative intent studies).
We downgraded the quality of evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We downgraded quality by two further levels for imprecision (small number of events and 95% CI included appreciable benefit and harm). We assessed the quality of evidence as very low.
3.12 Grade ≥ 3 hyperbilirubinaemia (curative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, grade ≥ 3 hyperbilirubinaemia did not differ between participants treated with oral versus IV fluoropyrimidines. The OR of 1.67 (95% CI 0.52 to 5.38) was derived from three studies with 2757 participants (Analysis 3.12; Table 7). However, few events occurred in both arms. Heterogeneity between effect estimates was moderate for these studies (Chi² = 3.45, P = 0.18; I² = 42%).
3.12. Analysis.
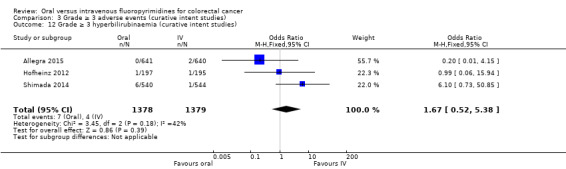
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 12 Grade ≥ 3 hyperbilirubinaemia (curative intent studies).
We downgraded the quality of evidence by one level for risk of bias, as studies at high risk of bias for this outcome contributed 44.3% of the weight for the pooled effect estimate. We downgraded quality by two further levels owing to imprecision (small numbers of events and 95% CIs included appreciable benefit and harm). We assessed the quality of evidence as very low.
3.13 Any grade ≥ 3 AEs (curative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, we found that odds of any grade ≥ 3 AEs were lower with oral fluoropyrimidines, with a pooled OR of 0.82 (95% CI 0.74 to 0.90) from five studies with 7741 participants (Table 7).
We downgraded the quality of evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias. We downgraded quality by one further level for inconsistency of results, as heterogeneity among the included studies was considerable (Chi² = 99.17, P < 0.00001; I² = 96%). We assessed the quality of evidence as low.
The effect estimate for De Gramont 2012 (weight 33.6%) strongly favoured the oral fluoropyrimidine group (HR 0.32, 95% CI 0.26 to 0.39), and in the remaining four studies, 95% CIs for the effect estimates crossed the null value of 1.00 (Analysis 3.13).
3.13. Analysis.
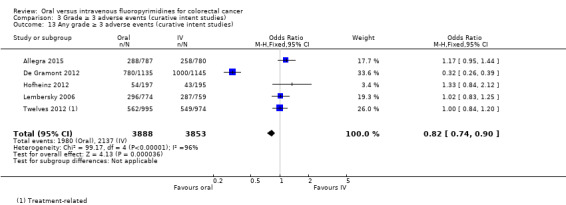
Comparison 3 Grade ≥ 3 adverse events (curative intent studies), Outcome 13 Any grade ≥ 3 adverse events (curative intent studies).
Sensitivity analyses
Excluding studies at 'High' risk of bias
As we assessed all studies contributing to the DFS outcome as having 'High' risk of bias owing to lack of blinding (Table 9), we could not perform a sensitivity analysis that excluded studies at 'High' risk of bias.
Patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy
Co‐primary outcome
4.1 PFS
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, PFS was worse in the oral fluoropyrimidine group. The pooled HR from 23 studies with 9927 participants was 1.06 (95% CI 1.02 to 1.11) (Analysis 4.1; Table 8). Heterogeneity among effect estimates for these studies was minimal (Chi² = 27.08, P = 0.25; I² = 15%).
4.1. Analysis.
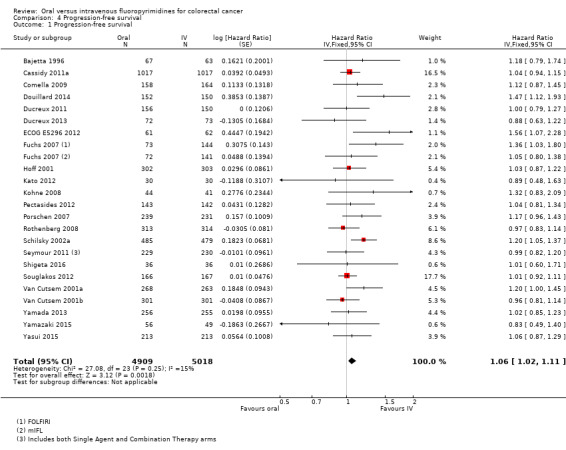
Comparison 4 Progression‐free survival, Outcome 1 Progression‐free survival.
We downgraded the quality of evidence by one level for risk of bias, as studies at high risk of bias for this outcome contributed 48.5% of the weight for the pooled effect estimate. We assessed the quality of evidence as moderate (Table 2).
Subgroup analyses
4.2 PFS with subgroup analysis ‐ Single agent versus combination therapy
We found no evidence of subgroup differences (Chi² = 2.16, P = 0.14; I² = 53.8%) (Analysis 4.2; Appendix 11).
4.2. Analysis.
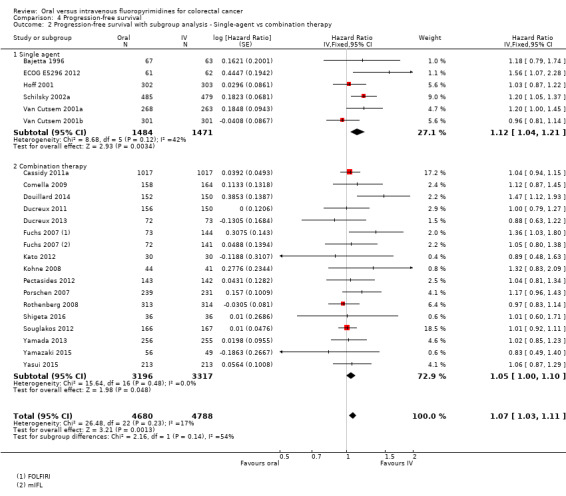
Comparison 4 Progression‐free survival, Outcome 2 Progression‐free survival with subgroup analysis ‐ Single‐agent vs combination therapy.
4.3 PFS with subgroup analysis ‐ Infusional versus bolus intravenous fluoropyrimidine
We found no evidence of subgroup differences (Chi² = 1.33, P = 0.25; I² = 24.7%) (Analysis 4.3; Appendix 11).
4.3. Analysis.
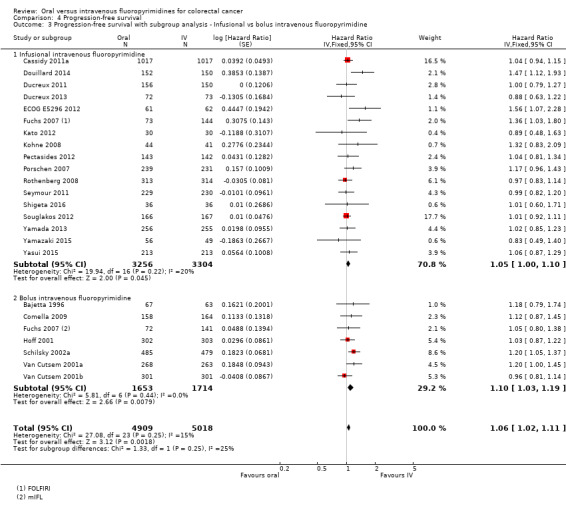
Comparison 4 Progression‐free survival, Outcome 3 Progression‐free survival with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine.
4.4 PFS with subgroup analysis ‐ Oral fluoropyrimidine backbone
Results showed significant subgroup differences by oral fluoropyrimidine backbone (Chi² = 13.46, P = 0.009; I² =70.3%). Pooled effect estimates for the ‘Capecitabine’ and ‘S‐1’ subgroups and the effect estimate for the ‘Doxifluridine’ subgroup (one study, Bajetta 1996) indicated that PFS did not differ between participants treated with oral versus IV fluoropyrimidines. However, pooled effect estimates for the ‘UFT/Ftorafur’ and ‘Eniluracil + oral 5‐FU’ subgroups indicated worse PFS in the oral fluoropyrimidine group (Figure 5; Analysis 4.4; Appendix 11).
5.
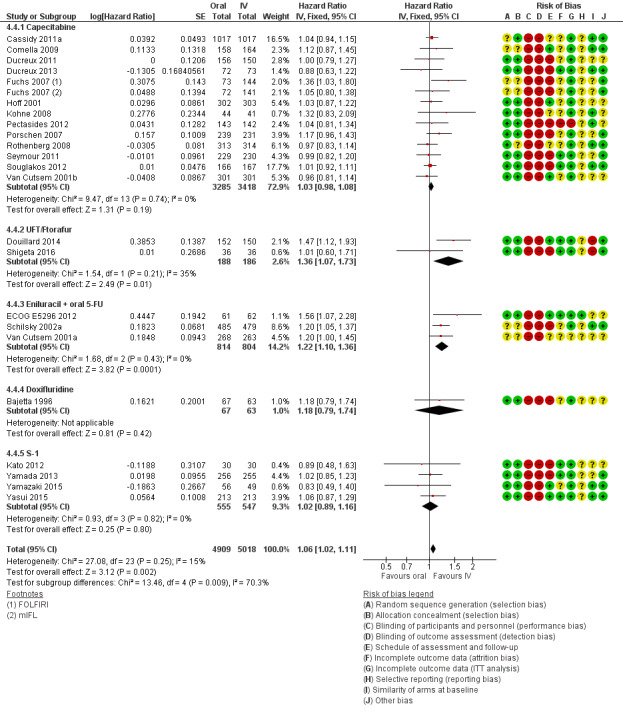
Forest plot of comparison: 4 Progression‐free survival with, outcome: 4.4 Progression‐free survival with subgroup analysis ‐ oral fluoropyrimidine backbone.
4.4. Analysis.
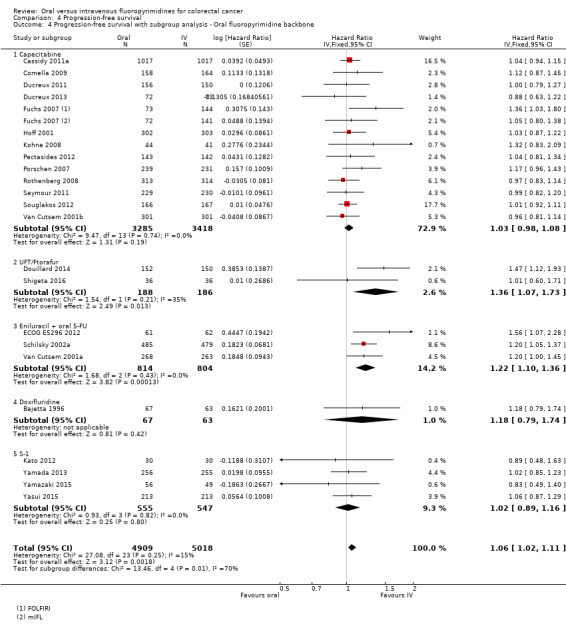
Comparison 4 Progression‐free survival, Outcome 4 Progression‐free survival with subgroup analysis ‐ Oral fluoropyrimidine backbone.
4.5 PFS for combination therapy with subgroup analysis ‐ Oxaliplatin‐based versus irinotecan based
We found no evidence of subgroup differences (Chi² = 0.13, P = 0.72; I² = 0%) (Analysis 4.5; Appendix 11).
4.5. Analysis.
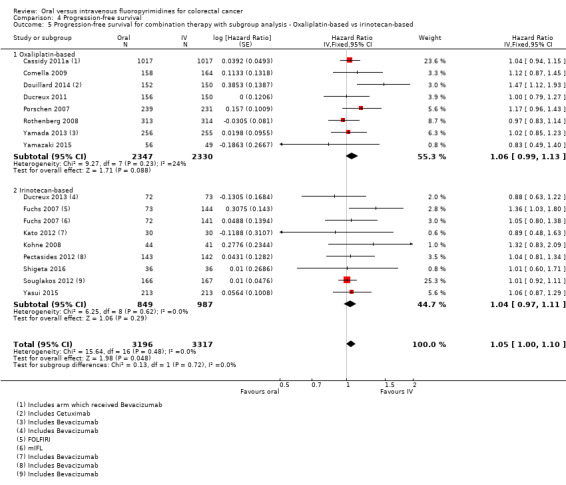
Comparison 4 Progression‐free survival, Outcome 5 Progression‐free survival for combination therapy with subgroup analysis ‐ Oxaliplatin‐based vs irinotecan‐based.
4.6 PFS for combination therapy with subgroup analysis ‐ with bevacizumab versus no bevacizumab
The post hoc subgroup analysis comparing studies of combination chemotherapy that included BEV versus those that did not include BEV found no subgroup differences (Chi² = 1.12, P = 0.29; I² = 11.0%) (Analysis 4.6; Appendix 11).
4.6. Analysis.
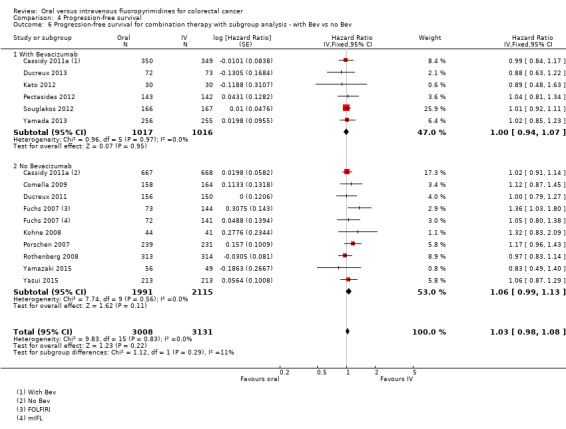
Comparison 4 Progression‐free survival, Outcome 6 Progression‐free survival for combination therapy with subgroup analysis ‐ with Bev vs no Bev.
PFS ‐ study data not suitable for quantitative synthesis
The Hochster TREE‐1 2008 and Hochster TREE‐2 2008 studies reported the median PFS for treatment arms without log‐rank P values. The median PFS for infusional IV fluoropyrimidine arms compared with oral fluoropyrimidine arms (TREE‐1: 8.7 m, 95% CI 6.5 to 9.8 vs 5.9 m, 95% CI 5.1 to 7.4; TREE‐2: 9.9 m, 95% CI 7.9 to 11.7 vs 10.3 m, 95% CI 8.6 to 12.5) and for bolus IV fluoropyrimidine arms compared with oral fluoropyrimidine arms (TREE‐1: 6.9 m, 95% CI 4.2 to 8.0 vs 5.9 m, 95% CI 5.1 to 7.4; TREE‐2: 8.3 m, 95% CI 6.6 to 9.9 vs 10.3 m, 95% CI 8.6 to 12.5) had overlapping 95% CIs for all oral versus IV fluoropyrimidine comparisons (Appendix 7).
Assessment of publication bias for PFS
Visual inspection of a funnel plot of SE(lnHR)s against HRs for the 23 studies quantitatively synthesised for the PFS outcome revealed no asymmetry (Figure 6).
6.
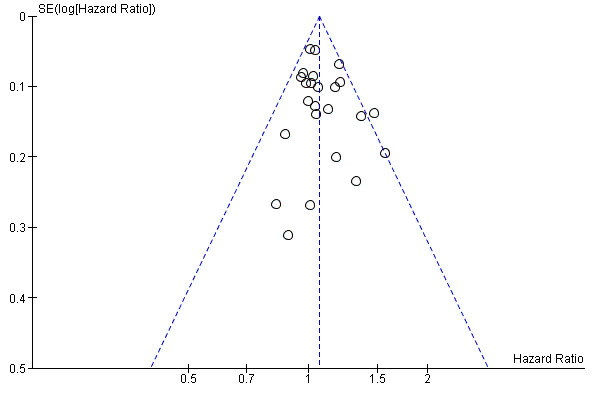
Funnel plot of progression‐free survival.
Secondary outcomes
5.1 OS (palliative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, OS did not differ between participants treated with oral versus IV fluoropyrimidines. The pooled HR from 29 studies with 12,079 participants was 1.02 (95% CI 0.99 to 1.05) (Table 8). Heterogeneity among effect estimates for these studies was minimal (Chi² = 33.69, P = 0.29; I² = 11%) (Analysis 5.1).
5.1. Analysis.
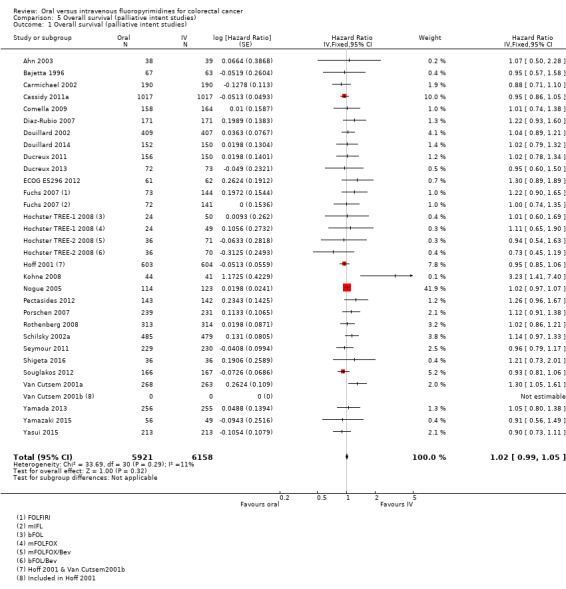
Comparison 5 Overall survival (palliative intent studies), Outcome 1 Overall survival (palliative intent studies).
We did not identify any factors that reduced the quality of evidence for this outcome, and we assessed the quality of evidence as high (Table 2).
Subgroup analyses:
We found no significant subgroup differences for any of the prespecified subgroup analyses (Analysis 5.2; Analysis 5.3; Analysis 5.4; Analysis 5.5; Appendix 11).
5.2. Analysis.
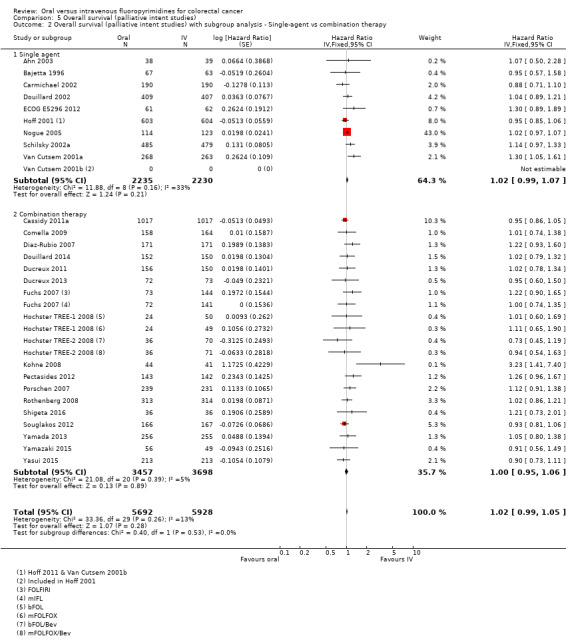
Comparison 5 Overall survival (palliative intent studies), Outcome 2 Overall survival (palliative intent studies) with subgroup analysis ‐ Single‐agent vs combination therapy.
5.3. Analysis.
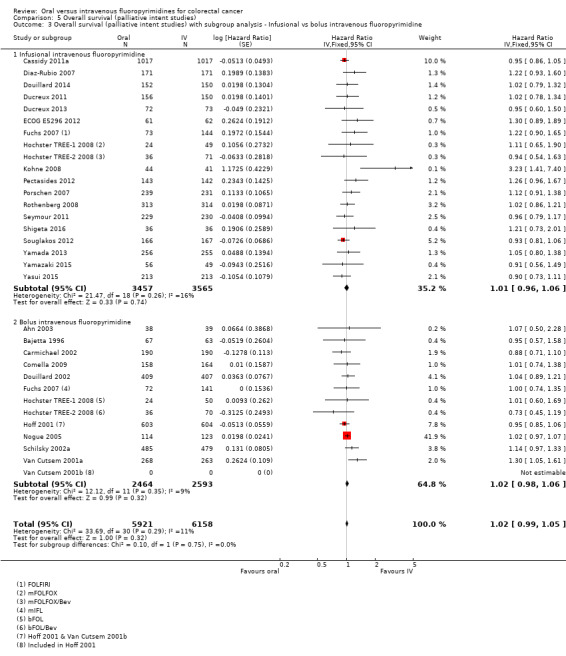
Comparison 5 Overall survival (palliative intent studies), Outcome 3 Overall survival (palliative intent studies) with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine.
5.4. Analysis.
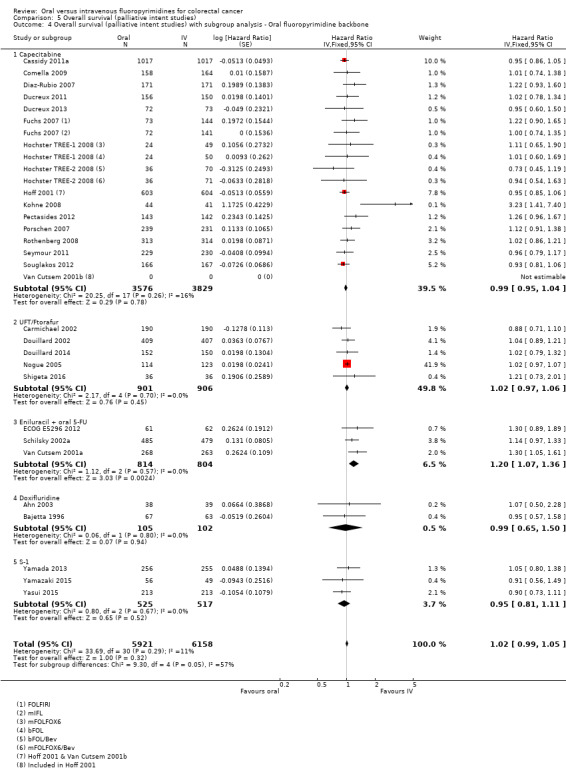
Comparison 5 Overall survival (palliative intent studies), Outcome 4 Overall survival (palliative intent studies) with subgroup analysis ‐ Oral fluoropyrimidine backbone.
5.5. Analysis.
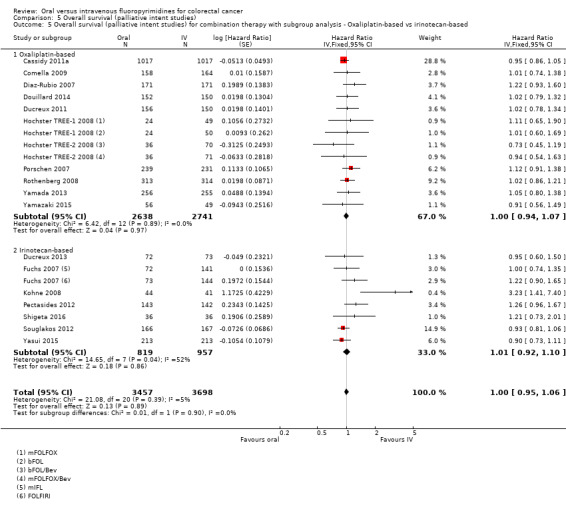
Comparison 5 Overall survival (palliative intent studies), Outcome 5 Overall survival (palliative intent studies) for combination therapy with subgroup analysis ‐ Oxaliplatin‐based vs irinotecan‐based.
5.2 OS with subgroup analysis ‐ Single‐agent versus combination therapy
Chi² = 0.40, P = 0.53; I² = 0%.
5.3 OS with subgroup analysis ‐ Infusional versus bolus intravenous fluoropyrimidine
Chi² = 0.10, P = 0.75; I² = 0%.
5.4 OS with subgroup analysis ‐ Oral fluoropyrimidine backbone
Chi2 = 9.30, P = 0.05; I² = 57.0%.
However, the pooled effect estimate for the 'Capecitabine', 'UFT/Ftorafur', 'Doxifluridine', and 'S‐1' subgroups indicated that OS did not differ between participants treated with oral versus IV fluoropyrimidines, whereas the pooled effect estimate for the 'Eniluracil + oral 5‐FU' subgroup indicated a worse OS in the oral fluoropyrimidine group (Analysis 5.4).
5.5 OS for combination therapy with subgroup analysis‐ Oxaliplatin‐based versus irinotecan‐based
Chi² = 0.01, P = 0.90; I² = 0%.
6.1 TTP
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, TTP was worse in the oral fluoropyrimidine group. The pooled HR from six studies with 1970 participants was 1.07 (95% CI 1.01 to 1.14) (Analysis 6.1; Table 8). We noted no heterogeneity among effect estimates for these studies (Chi² = 4.95, P = 0.42; I² = 0%).
6.1. Analysis.
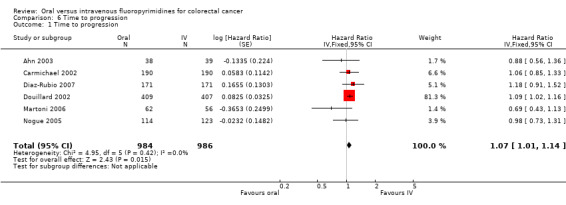
Comparison 6 Time to progression, Outcome 1 Time to progression.
We downgraded the quality of evidence by one level for risk of bias, as studies at high risk of bias for this outcome contributed 93.4% of the weight for the pooled effect estimate. We assessed the quality of evidence as moderate.
7.1 ORR
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, ORR did not differ between participants treated with oral versus IV fluoropyrimidines. The pooled OR from 32 studies with 11,115 participants was 0.98 (95% CI 0.90 to 1.06) (Analysis 7.1; Table 8). Heterogeneity between the included studies was moderate (Chi² = 59.03, P = 0.005; I² = 42%).
7.1. Analysis.
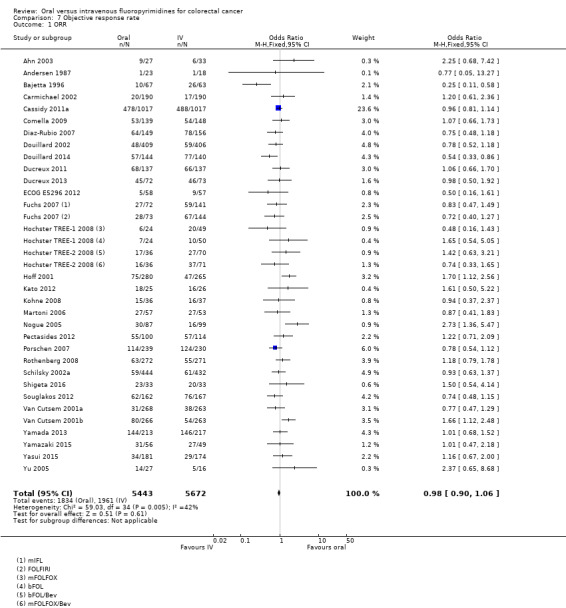
Comparison 7 Objective response rate, Outcome 1 ORR.
We downgraded the quality of evidence by one level for risk of bias, as studies at high risk of bias for this outcome contributed 59.3% of the weight for the pooled effect estimate. We assessed the quality of evidence as moderate.
Grade ≥ 3 AEs (palliative intent studies)
8.1 Grade ≥ 3 diarrhoea (palliative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, odds of grade ≥ 3 diarrhoea were higher in the oral fluoropyrimidine arm. The pooled OR from 30 studies with 11,997 participants was 1.66 (95% CI 1.50 to 1.84) (Analysis 8.1; Table 8).
8.1. Analysis.
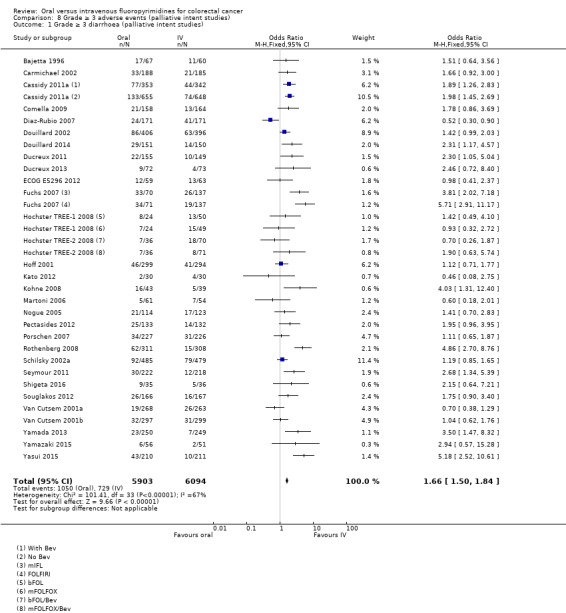
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 1 Grade ≥ 3 diarrhoea (palliative intent studies).
We downgraded the quality of evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We further downgraded quality by one level for inconsistency, as heterogeneity between the included studies was substantial (Chi² = 101.41, P < 0.00001; I² = 67%). The final assessment for quality of evidence was low (Table 2).
We observed that for the included studies, 95% CIs for the effect estimates either crossed the null value of 1.00 or indicated more grade ≥ 3 diarrhoea with oral fluoropyrimidine treatment. One outlier study, which was an exception to this, favoured oral fluoropyrimidines (Diaz‐Rubio 2007). In this study, AE assessments were less frequent in the oral than in the IV treatment arm; however, many other studies included in the meta‐analysis for grade ≥ 3 diarrhoea also had high risk of bias as a result of this methodological issue (Characteristics of included studies).
Subgroup analyses
Results showed subgroup differences for all prespecified subgroup analyses explored. However, substantial or considerable heterogeneity remained between included studies within at least one subgroup (Appendix 12).
8.2 Grade ≥ 3 diarrhoea (palliative intent studies) with subgroup analysis ‐ Single‐agent versus combination therapy
The pooled OR for the ‘Combination therapy’ subgroup favoured IV fluoropyrimidines more than the pooled OR for the ‘Single agent’ subgroup (Chi2 = 21.70, P < 0.00001; I² = 95.4%) (Analysis 8.2).
8.2. Analysis.
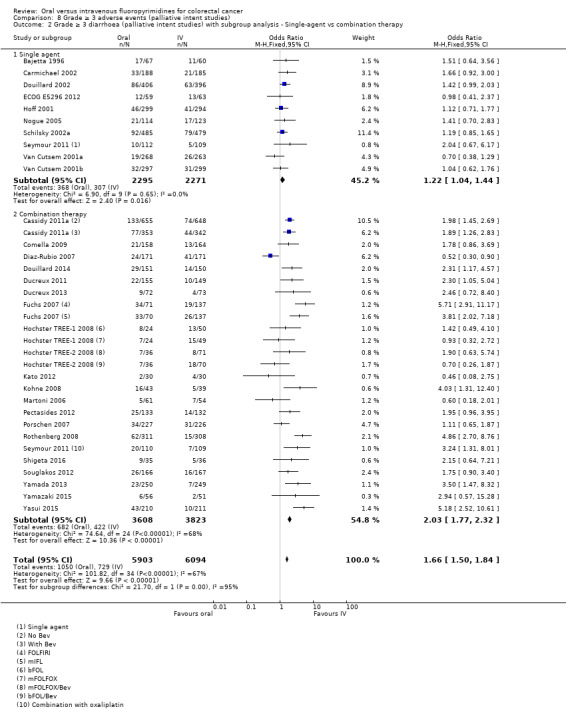
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 2 Grade ≥ 3 diarrhoea (palliative intent studies) with subgroup analysis ‐ Single‐agent vs combination therapy.
8.3 Grade ≥ 3 diarrhoea (palliative intent studies) with subgroup analysis ‐ Infusional versus bolus IV fluoropyrimidine
The pooled OR for the ‘Infusional IV fluoropyrimidine’ subgroup favoured IV fluoropyrimidines more than the pooled effect estimate for the ‘Bolus IV fluoropyrimidine’ subgroup (Chi2 = 15.57, P < 0.0001; I² = 93.6%) (Analysis 8.3).
8.3. Analysis.
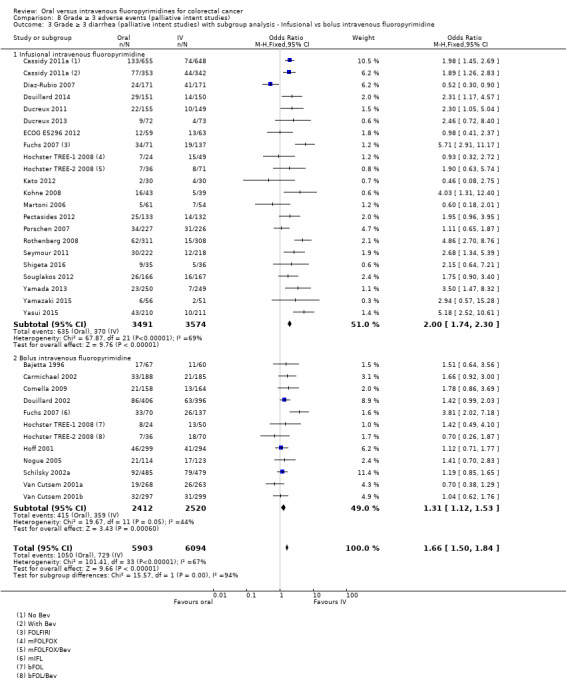
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 3 Grade ≥ 3 diarrhea (palliative intent studies) with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine.
8.4 Grade ≥ 3 diarrhoea (palliative intent studies) with subgroup analysis ‐ Oral fluoropyrimidine backbone
Results showed significant subgroup differences by oral fluoropyrimidine backbone (Chi2 = 21.15, P = 0.0003; I² = 81.1%). The pooled OR for the ‘Capecitabine’, ‘UFT/Ftorafur’ and ‘S‐1’ subgroups indicated worse grade ≥ 3 diarrhoea with oral fluoropyrimidine treatment, and 95% CIs for pooled effect estimates for the ‘Eniluracil + oral 5‐FU’ and ‘Doxifluridine’ (one study, Bajetta 1996) subgroups crossed the null value of 1.00 (Analysis 8.4).
8.4. Analysis.
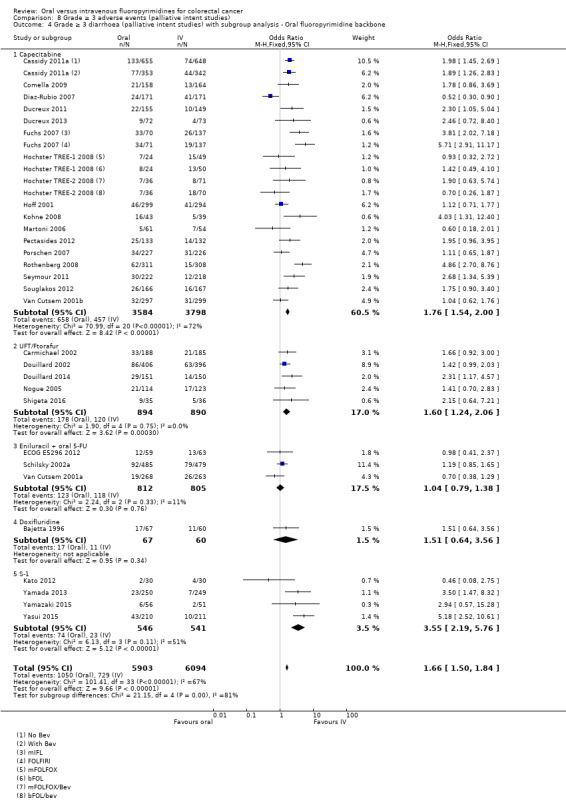
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 4 Grade ≥ 3 diarrhoea (palliative intent studies) with subgroup analysis ‐ Oral fluoropyrimidine backbone.
8.5 Grade ≥ 3 diarrhoea (palliative intent studies) with subgroup analysis for combination therapy ‐ Oxaliplatin‐based versus irinotecan‐based
The pooled OR for the ‘Irinotecan‐based’ subgroup favoured IV fluoropyrimidines more than the pooled effect estimate for the ‘Oxaliplatin‐based’ subgroup (Chi2 = 12.72, P = 0.0004; I² = 92.1% ) (Analysis 8.5).
8.5. Analysis.
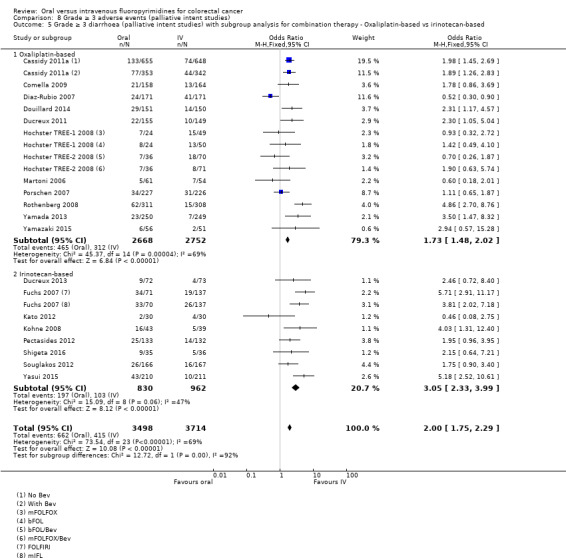
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 5 Grade ≥ 3 diarrhoea (palliative intent studies) with subgroup analysis for combination therapy ‐ Oxaliplatin‐based vs irinotecan‐based.
8.6 Grade ≥ 3 hand foot syndrome (palliative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, results showed greater grade ≥ 3 HFS with oral fluoropyrimidine use. The pooled OR from 18 studies with 6481 participants was 3.92 (95% CI 2.84 to 5.43) (Table 8). Heterogeneity between the included studies was moderate (Chi² = 33.79, P = 0.03; I² = 41%).
We downgraded the quality of evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We assessed the quality of evidence as moderate (Table 2).
We observed that for the included studies, effect estimates with their 95% CIs crossed the null value of 1.00 (10 studies, and one arm of Hochster TREE‐1 2008 and Hochster TREE‐2 2008) or indicated increased grade ≥ 3 HFS with oral fluoropyrimidine treatment (four studies, and one arm of Hochster TREE‐1 2008 and Hochster TREE‐2 2008). One outlier, which was an exception to this, favoured oral fluoropyrimidines (ECOG E5296 2012, the only study for this outcome using Eniluracil + oral 5‐FU). Another study (Shigeta 2016) reported no events in either arm (Analysis 8.6).
8.6. Analysis.
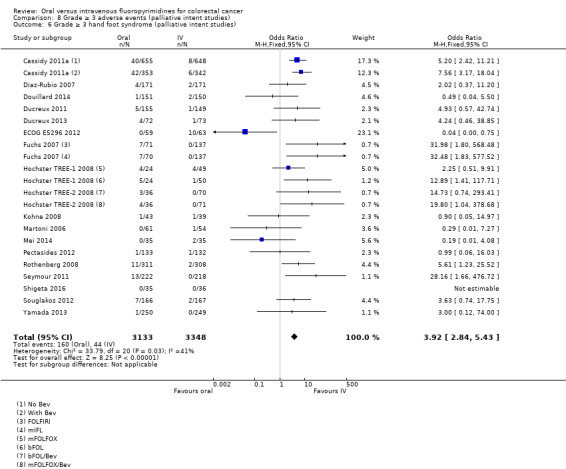
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 6 Grade ≥ 3 hand foot syndrome (palliative intent studies).
Subgroup analyses
8.7 Grade ≥ 3 hand foot syndrome (palliative intent studies) subgroup analysis ‐ Single‐agent versus combination therapy
Results showed subgroup differences in the 'Single‐agent' and 'Combination therapy' subgroups. In the 'Single‐agent' subgroup, grade ≥ 3 HFS did not differ between participants treated with oral versus IV fluoropyrimidines. However, the 'Combination therapy' subgroup showed an increase in grade ≥ 3 HFS with oral fluoropyrimidine treatment (Chi2 = 9.86, P = 0.002; I² = 89.9%). Only two studies were included in the 'Single‐agent' subgroup (one was ECOG E5296 2012, the outlier study), and heterogeneity between these two studies was considerable (Chi2 = 9.56, P = 0.002; I² = 90%) (Analysis 8.7; Appendix 12).
8.7. Analysis.
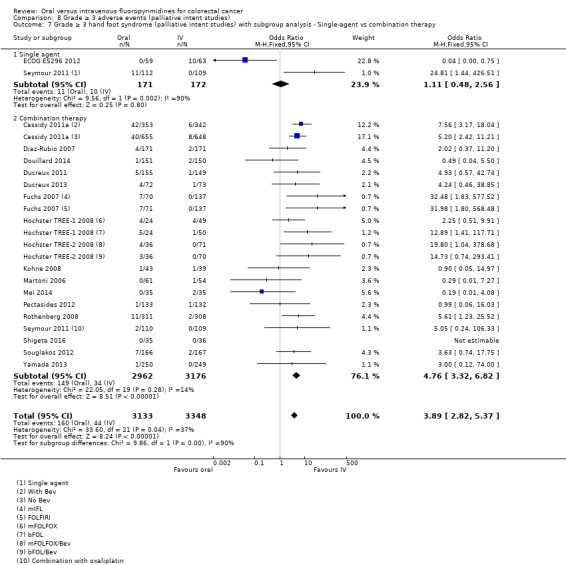
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 7 Grade ≥ 3 hand foot syndrome (palliative intent studies) with subgroup analysis ‐ Single‐agent vs combination therapy.
8.8 Grade ≥ 3 hand foot syndrome (palliative intent studies) subgroup analysis ‐ Infusional versus bolus IV fluoropyrimidine
The pooled OR for the ‘Bolus IV fluoropyrimidine’ subgroup favoured IV fluoropyrimidines more than the pooled effect estimate for the ‘Infusional IV fluoropyrimidine’ subgroup (Chi2 = 4.48, P = 0.03; I² = 77.7%) (Analysis 8.8; Appendix 12). However, heterogeneity between studies within the ‘Infusional IV fluoropyrimidines’ subgroup was moderate (Chi2 = 30.02, P = 0.03; I² = 43%).
8.8. Analysis.
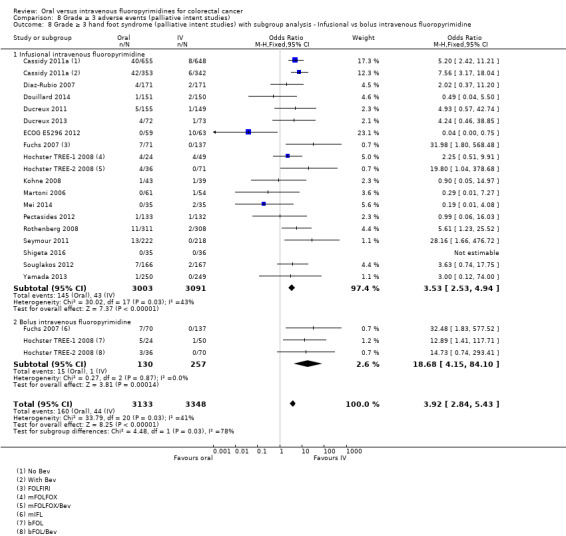
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 8 Grade ≥ 3 hand foot syndrome (palliative intent studies) with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine.
8.9 Grade ≥ 3 hand foot syndrome (palliative intent studies) subgroup analysis ‐ Oral fluoropyrimidine backbone
The effect estimate for the ‘Eniluracil + oral 5‐FU’ subgroup (one study, ECOG E5296 2012) favoured oral fluoropyrimidines, the 95% CI for pooled effect estimates for the ‘UFT/Ftorafur’ and ‘S‐1’ subgroups crossed the null value of 1.00, and the pooled OR for the ‘Capecitabine’ subgroup indicated increased grade ≥ 3 HFS with oral fluoropyrimidine treatment (Chi2 = 19.58, P = 0.0002; I² = 84.7%) (Analysis 8.9; Appendix 12).
8.9. Analysis.
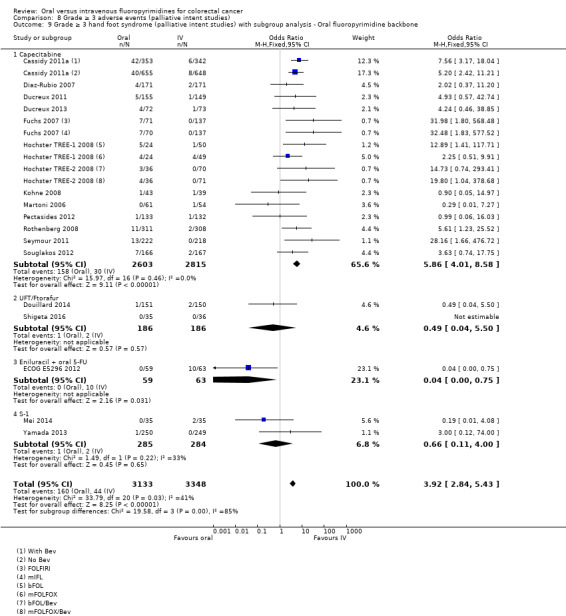
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 9 Grade ≥ 3 hand foot syndrome (palliative intent studies) with subgroup analysis ‐ Oral fluoropyrimidine backbone.
8.10 Grade ≥ 3 hand foot syndrome (palliative intent studies) subgroup analysis for combination therapy ‐ Oxaliplatin‐based versus irinotecan‐based
We found no evidence of subgroup differences (Chi² = 0.32, P = 0.57; I² = 0%) (Analysis 8.10; Appendix 12).
8.10. Analysis.
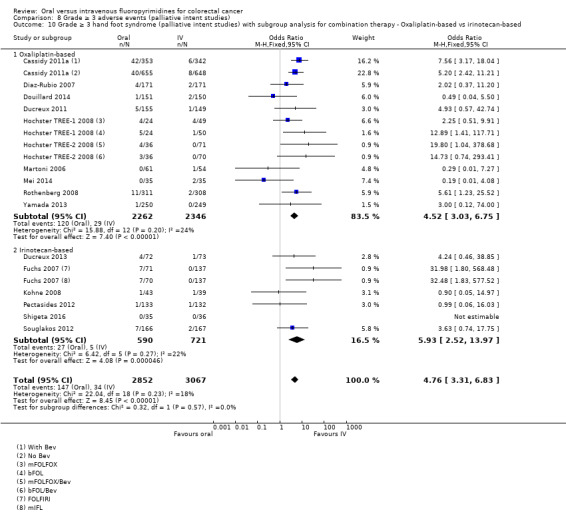
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 10 Grade ≥ 3 hand foot syndrome (palliative intent studies) with subgroup analysis for combination therapy ‐ Oxaliplatin‐based vs irinotecan‐based.
8.11 Grade ≥ 3 neutropenia/granulocytopenia (palliative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, the pooled OR for grade ≥ 3 neutropenia/granulocytopenia from 29 studies with 11,794 participants (Table 8) was 0.17 (95% CI 0.15 to 0.18), favouring oral fluoropyrimidines.
We downgraded the quality of evidence by one level for risk of bias, as studies at high risk of bias for this outcome contributed 29.2% of the weight for the pooled effect estimate. We further downgraded quality by one level for inconsistency of results, as heterogeneity between included studies was substantial to considerable (Chi² = 295.88, P < 0.00001; I² = 90%). We assessed the quality of evidence as low (Table 2).
We observed that for the included studies, effect estimates with their 95% CIs either favoured oral fluoropyrimidines (14 studies and infusional arms of Hochster TREE‐1 2008 and Hochster TREE‐2 2008 studies), or included the null value of 1.00 (13 studies and bolus arms of Hochster TREE‐1 2008 and Hochster TREE‐2 2008 studies) (Analysis 8.11).
8.11. Analysis.
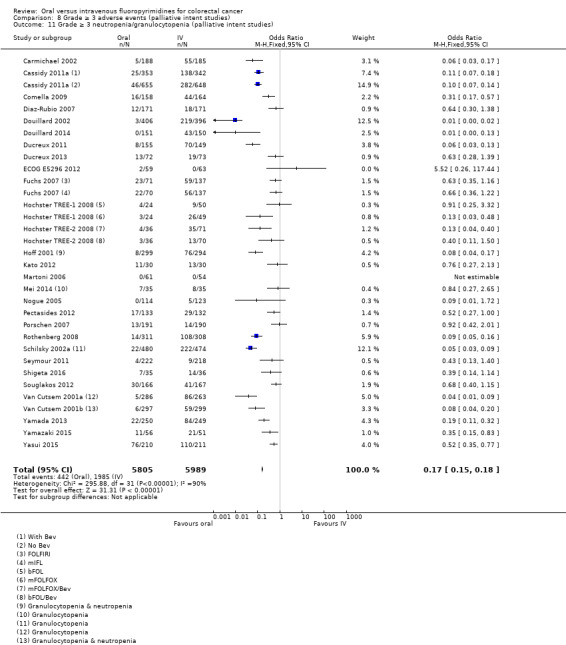
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 11 Grade ≥ 3 neutropenia/granulocytopenia (palliative intent studies).
Grade ≥ 3 neutropenia/granulocytopenia (palliative intent studies) ‐ study data not suitable for quantitative synthesis
For the Kohne 2008 and Silvestris 2010 studies, in which neutropenia/granulocytopenia were not specifically reported, the incidence of the grade ≥ 3 AEs 'white blood cells' and 'leuko/neutropenia', respectively, was similar. For the Bajetta 1996 study, which reported 'leukopenia', the incidence of this grade ≥ 3 AE was lower in the oral fluoropyrimidine arm (Appendix 6).
8.12 Grade ≥ 3 febrile neutropenia (palliative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, the pooled OR for grade ≥ 3 febrile neutropenia from 19 studies with 9407 participants was 0.27 (95% CI 0.21 to 0.36), indicating lower odds of grade ≥ 3 febrile neutropenia in the oral fluoropyrimidine arm (Table 8).
We downgraded the quality of evidence by one level owing to inconsistency of results, with substantial heterogeneity between the included studies (Chi² = 60.67, P < 0.00001; I² = 67%). We assessed the quality of evidence as moderate.
However, we observed that for the included studies, effect estimates with their 95% CIs either crossed the null value of 1.00 (11 studies) or favoured oral fluoropyrimidines (seven studies), with the exception of one outlier study (Yasui 2015) in which participants treated with oral fluoropyrimidines had greater grade ≥ 3 febrile neutropenia (Analysis 8.12).
8.12. Analysis.
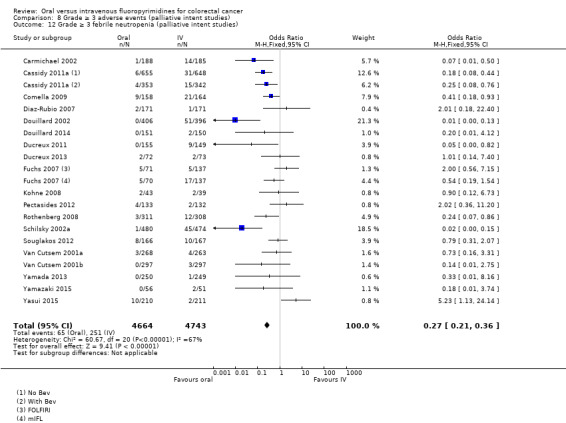
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 12 Grade ≥ 3 febrile neutropenia (palliative intent studies).
8.13 Grade ≥ 3 vomiting (palliative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, the pooled OR of 1.18 (95% CI 1.00 to 1.40) from 23 studies with 9528 participants indicated higher odds of vomiting with oral fluoropyrimidine treatment (Analysis 8.13; Table 8). Heterogeneity among the effect estimates for these studies was minimal (Chi² = 32.08, P = 0.19; I² = 19%). This pooled OR included data from seven studies that combined data for grade ≥ 3 vomiting and nausea (Carmichael 2002; Cassidy 2011a; Douillard 2002; Hochster TREE‐1 2008; Hochster TREE‐2 2008; Nogue 2005; Mei 2014).
8.13. Analysis.
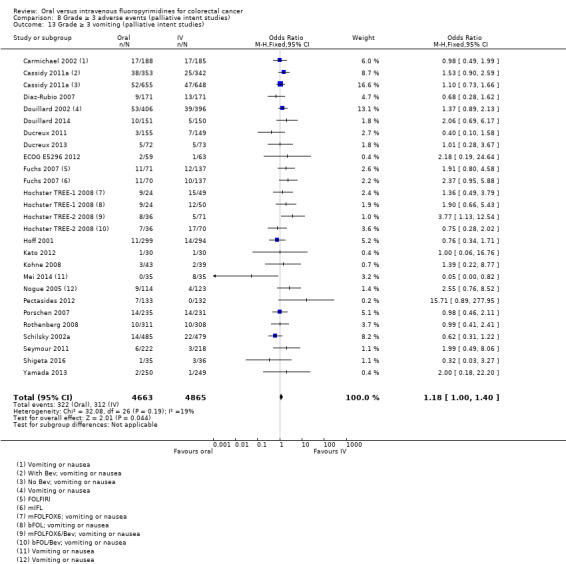
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 13 Grade ≥ 3 vomiting (palliative intent studies).
We downgraded the quality of evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We downgraded quality by one further level as the result of imprecision, and the final assessment for quality of evidence was low.
8.14 Grade ≥ 3 nausea (palliative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, grade ≥ 3 nausea did not differ between participants treated with oral versus IV fluoropyrimidines. The pooled OR from 25 studies with 9796 participants was 1.16 (95% CI 0.99 to 1.36) (Table 8). Heterogeneity between studies was moderate (Chi² = 48.04, P = 0.01; I² = 42%). However, for the included studies, effect estimates with their 95% CIs either crossed the null value of 1.00 or indicated higher odds of grade ≥ 3 nausea with oral fluoropyrimidine treatment, with the exception of two outlier studies (Mei 2014; Schilsky 2002a) (Analysis 8.14).
8.14. Analysis.
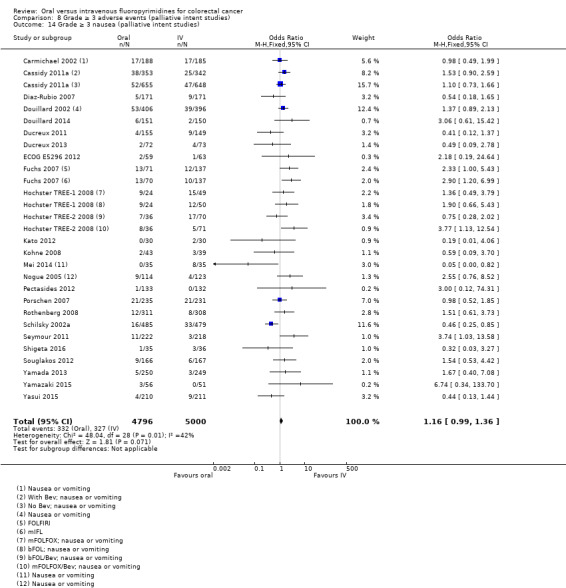
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 14 Grade ≥ 3 nausea (palliative intent studies).
We downgraded the quality of evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We downgraded quality by one further level owing to imprecision. The final assessment for quality of evidence was low.
8.15 Grade ≥ 3 stomatitis (palliative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, the pooled OR for grade ≥ 3 stomatitis from 21 studies with 8718 participants (Table 8) was 0.26 (95% CI 0.20 to 0.33), favouring oral fluoropyrimidines.
We downgraded the quality of evidence by one level owing to risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We downgraded quality by one further level for inconsistency of results, with substantial heterogeneity between the included studies (Chi² = 62.38, P < 0.00001; I² = 66%). We assessed the quality of evidence as low.
We observed that for the included studies, effect estimates with their 95% CIs either crossed the null value of 1.00 (15 studies) or favoured oral fluoropyrimidines (six studies) (Analysis 8.15).
8.15. Analysis.
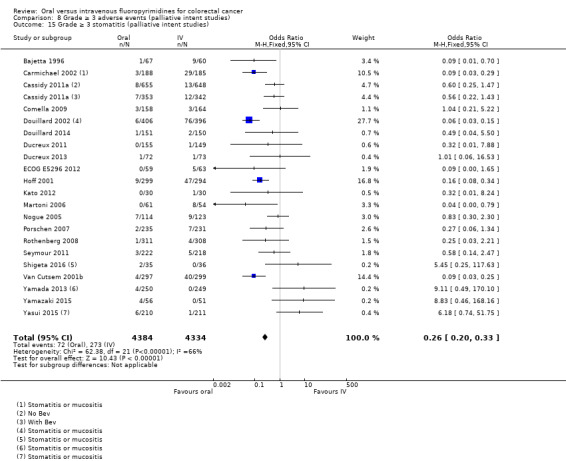
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 15 Grade ≥ 3 stomatitis (palliative intent studies).
8.16 Grade ≥ 3 mucositis (palliative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, the pooled OR for grade ≥ 3 mucositis was 0.17 (95% CI 0.12 to 0.24) from 12 studies with 4962 participants (Table 8), favouring oral fluoropyrimidines.
We downgraded the quality of evidence by one level owing to risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We further downgraded quality by one level owing to inconsistency of results, as heterogeneity between the included studies was substantial or considerable (Chi² = 39.81, P < 0.0001; I² = 75%). We assessed the quality of evidence as low.
We observed that for the included studies, effect estimates with their 95% CIs either crossed the null value of 1.00 (seven studies) or favoured oral fluoropyrimidines (four studies) (Analysis 8.16).
8.16. Analysis.
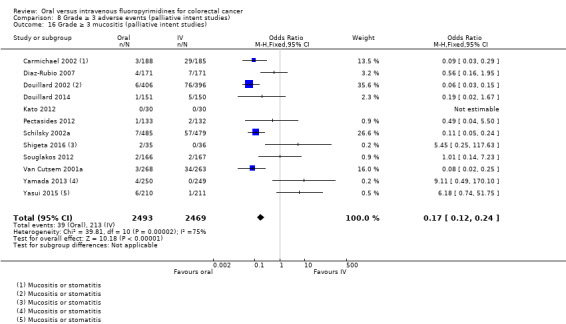
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 16 Grade ≥ 3 mucositis (palliative intent studies).
8.17 Grade ≥ 3 hyperbilirubinaemia (palliative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, grade ≥ 3 hyperbilirubinaemia did not differ between oral and IV fluoropyrimidine arms. The pooled OR from nine studies with 2699 participants was 1.62 (95% CI 0.99 to 2.64). We noted no heterogeneity between the included studies (Chi² = 3.86, P = 0.70; I² = 0%) (Analysis 8.17; Table 8).
8.17. Analysis.
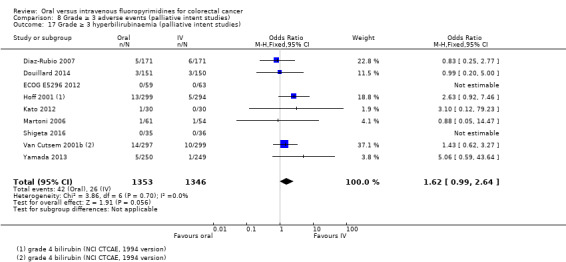
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 17 Grade ≥ 3 hyperbilirubinaemia (palliative intent studies).
We downgraded the quality of evidence by one level owing to risk of bias, as studies at high risk of bias for this outcome contributed 28.5% of the weight for the pooled effect estimate. We downgraded quality by one further level for imprecision, and we assessed the quality of evidence as low.
8.18 Any grade ≥ 3 AEs (palliative intent studies)
For the comparison of oral versus IV fluoropyrimidines in patients treated with palliative intent for CRC with chemotherapy, the pooled OR for any grade ≥ 3 AEs from 14 studies with 5436 participants was 0.83 (95% CI 0.74 to 0.94), favouring oral fluoropyrimidines (Table 8).
We downgraded the quality of evidence by one level for risk of bias, as all studies that contributed to the pooled effect estimate had high risk of bias owing to lack of blinding. We further downgraded quality by one level for inconsistency of results, as heterogeneity between the included studies was substantial or considerable (Chi2 = 69.88, P < 0.00001; I2 = 77%). We assessed the quality of evidence as low.
We observed that for the included studies, 95% CIs for the effect estimates crossed the null value of 1.00 or favoured oral fluoropyrimidines, with the exception of the bolus arm of Hochster TREE‐1 2008, Kohne 2008, and Seymour 2011 (Analysis 8.18).
8.18. Analysis.
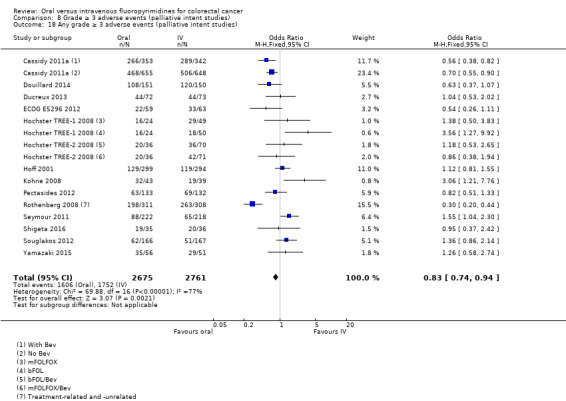
Comparison 8 Grade ≥ 3 adverse events (palliative intent studies), Outcome 18 Any grade ≥ 3 adverse events (palliative intent studies).
Sensitivity analyses
Excluding studies with 'High' risk of bias
When we excluded studies with 'High' risk of bias from the meta‐analysis for the PFS outcome (Table 10), the pooled HR was 1.01 (95% CI 0.96 to 1.07). Whilst results showed no substantial change in the direction or magnitude of the effect estimate compared with the original analysis, which included studies at 'High' risk of bias (HR 1.06, 95% CI 1.02 to 1.11), the 95% CI included the null value of 1.00 (Table 11). We found no heterogeneity (I2 = 0%, P = 0.91).
9. Sensitivity analyses.
| Sensitivity analyses for PFS outcome | Original analysis: (effect estimatea, fixed (95% CI)) | Sensitivity analysis: (effect estimatea, fixed (95% CI)) |
| Excluding studies with 'High' risk of bias | 1.06 (1.02 to 1.11) | 1.01 (95% CI 0.96 to 1.07) |
| Excluding Seymour 2011 study (frail and elderly study population) | 1.06 (1.02 to 1.11) | 1.07 (1.03 to 1.11) |
| Excluding second‐line studies in patients treated with palliative intent for inoperable or metastatic colorectal cancerb | 1.06 (1.02 to 1.11) | 1.07 (1.03 to 1.12) |
aEffect estimates presented as inverse‐variance hazard ratios for time‐to‐event outcomes, and Mantel‐Haenszel odds ratios for adverse events
bAnalysis excluding Kato 2012, Rothenberg 2008, Yasui 2015, and Yu 2005. Kato 2012 and Yu 2005 included patients receiving first‐ or second‐line treatment
PFS: progression‐free survival
CI: confidence interval
Other sensitivity analyses
Results showed no change in direction nor substantial change in magnitude of the pooled effect estimate for PFS when we performed sensitivity analyses excluding the Seymour 2011 study (with a frail and elderly study population) or excluding studies of second‐line chemotherapy (Table 11).
Discussion
Summary of main results
Patients treated with curative intent for colorectal cancer (CRC) with neoadjuvant and/or adjuvant chemotherapy
Efficacy
Our review found that in patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, the co‐primary outcome disease‐free survival (DFS) did not differ between study participants treated with oral versus intravenous (IV) fluoropyrimidines. The pooled hazard ratio (HR) for DFS was 0.93 (95% confidence interval (CI) 0.87 to 1.00). Quantitative synthesis of historical data for the effect of IV fluoropyrimidine‐based therapy in early‐stage CRC demonstrated a 22% reduction in the risk of disease recurrence, with a pooled HR for DFS of 0.78 (95% CI 0.73 to 0.83) (Appendix 18). To retain 50%, 70%, 80%, or 90% of the activity of the active control would lead to non‐inferiority margins of 1.13, 1.08, 1.05, and 1.03, respectively, had the original design been one of non‐inferiority (FDA 2010). If retaining at least 80% of the activity of the active control is required to demonstrate non‐inferiority, the upper bound of the 95% CI for the pooled HR for DFS in our review indicates that this would be met. Overall survival (OS) also did not differ between participants treated with oral versus IV fluoropyrimidines, and pooled HRs for OS and DFS were very similar.
Adverse events
Our review found lower odds of any grade ≥ 3 adverse events (AEs) and grade ≥ 3 neutropenia/granulocytopenia and stomatitis in participants treated with oral fluoropyrimidines. Conversely, odds of grade ≥ 3 hand foot syndrome were higher in the oral fluoropyrimidine group. Grade ≥ 3 diarrhoea, febrile neutropenia, vomiting, nausea, mucositis, and hyperbilirubinaemia did not differ between participants treated with oral versus IV fluoropyrimidines. However, caution in interpreting the results for febrile neutropenia, mucositis, and hyperbilirubinaemia is advised, as the number of events for these outcomes was small, and power to detect a difference between oral and IV fluoropyrimidine groups was low. Heterogeneity was substantial or considerable for grade ≥ 3 diarrhoea, hand foot syndrome (HFS), neutropenia/granulocytopenia, stomatitis, and any grade ≥ 3 AEs. Nevertheless, we observed that for any grade ≥ 3 AEs and for grade ≥ 3 stomatitis, odds ratios (ORs) and associated 95% CIs for the included studies either favoured oral fluoropyrimidines or crossed the null value of 1.00. For grade ≥ 3 neutropenia/granulocytopenia, these either favoured oral fluoropyrimidines or crossed the null value of 1.00, with the exception of one outlier study. For grade ≥ 3 diarrhoea and HFS, these either crossed the null value of 1.00 or indicated worse outcomes with oral fluoropyrimidine treatment.
Factors that potentially contributed to heterogeneity in grade ≥ 3 AEs include the following.
Clinical heterogeneity in study treatment regimens. This included differences in doses and schedules of fluoropyrimidines, and, when relevant, different types, doses, and schedules of additional chemotherapy, biological agents, and/or radiotherapy regimens (Characteristics of included studies).
Variability in the relationship of reported AEs to treatment (Table 6).
Heterogeneity in the toxicity assessment criteria used (Included studies).
Variability in reporting bias by both reporting participants and recording study personnel (Haller 2008; Punt 2008). This may have varied between study populations owing to regional (Haller 2008) or other differences.
Variability in actions taken by participants and treating clinicians in response to AEs (Haller 2008; Punt 2008). In the case of clinicians, this should have been attenuated by the inclusion of guidelines for dose reduction, dose modification, and dose delays in trial protocols.
Differences in the countries and regions of sites participating in the included studies (Included studies). Regional differences in the tolerability profiles of fluoropyrimidines used for curative and palliative intent treatment of CRC have been reported, with greater treatment‐related toxicity observed in the USA than in the rest of the world (Haller 2008). This may be due to differences in patients’ body mass index or body surface area, genetic polymorphisms, cultural and regional differences in medical practice and patient behaviour, and dietary folate intake (Haller 2008; Midgley 2009).
Patients treated with palliative intent for inoperable advanced or metastatic CRC with palliative chemotherapy
Efficacy
Among participants treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy, we found that overall, the co‐primary outcome progression‐free survival (PFS) was worse in those treated with oral fluoropyrimidines. However, results show significant subgroup differences for the PFS outcome by oral fluoropyrimidine backbone. In the ‘Capecitabine’,‘S‐1’, and ‘Doxifluridine’ subgroups, PFS did not differ between individuals treated with oral versus IV fluoropyrimidines, whilst in the ‘UFT/Ftorafur’ and ‘Eniluracil + oral 5‐fluorouracil (FU)’ subgroups, PFS was worse in the oral fluoropyrimidine group. In our review, the pooled HR for PFS was 1.06 (95% CI 1.02 to 1.11). Previous data showed that use of IV fluorouracil‐based palliative chemotherapy for CRC led to a five‐month benefit in PFS compared with primary expectancy (Nordic 1992), with an estimated risk reduction of 62%. To retain 50%, 70%, 80%, or 90% of the activity of the active control would lead to non‐inferiority margins of 1.62, 1.34, 1.21, and 1.10, respectively, had the original design been one of non‐inferiority (FDA 2010). If retaining at least 80% of the activity of the active control is required to demonstrate non‐inferiority, the upper bound of the 95% CI for the pooled HR for PFS in our review indicates that this would be met.
OS did not differ between individuals treated with oral versus IV fluoropyrimidines, and subgroup analyses revealed no significant subgroup differences. However, whilst OS did not differ between individuals treated with oral versus IV fluoropyrimidines when ‘Capecitabine’, ‘UFT/Ftorafur’, ‘S‐1’, and ‘Doxifluridine’ were used, in the ‘Eniluracil + oral 5‐FU’ subgroup, OS was worse in the oral fluoropyrimidine group. The difference in findings for PFS and OS outcomes may be due to variability in utilisation and effects of second‐ or subsequent‐line treatments. We did not have complete information about this for every study included in our review (Risk of bias in included studies). Similar to PFS, time to progression (TTP) was worse in participants treated with oral compared with IV fluoropyrimidines.
Objective response rate (ORR) did not differ between participants treated with oral versus IV fluoropyrimidines. Heterogeneity was moderate between the studies included in this outcome. Factors that potentially contributed to heterogeneity include the following.
Clinical heterogeneity in study treatment regimens. This included differences in doses and schedules of fluoropyrimidines, and, when relevant, different types, doses, and schedules of additional chemotherapy, biological agents, and/or radiotherapy regimens (Characteristics of included studies).
Variability in the reporting of numbers of participants who were assessable or evaluable for response in the included studies. In studies that did not specifically report this number, if in fact some participants were not evaluable or assessable for response, they were treated as non‐responders in the analysis. This may have potentially underestimated the response rate in a given arm. The subsequent magnitude of effect on the pooled effect estimate for ORR would be dependent on the number of participants who were not evaluable or assessable for response in these studies, and the relative distribution of these participants between oral and IV fluoropyrimidine arms.
Variability in the response assessment criteria used across included studies (Included studies).
Adverse events
Our review found lower odds of any grade ≥ 3 AEs, grade ≥ 3 neutropenia/granulocytopenia, febrile neutropenia, stomatitis, and mucositis in participants treated with oral fluoropyrimidines. Conversely, odds of grade ≥ 3 diarrhoea and HFS were higher in the oral fluoropyrimidine group. Grade ≥ 3 vomiting, nausea, and hyperbilirubinaemia did not differ between participants treated with oral versus IV fluoropyrimidines. However, heterogeneity was substantial or considerable for all of the grade ≥ 3 AE outcomes, except HFS, vomiting, nausea, and hyperbilirubinaemia. Nevertheless, we observed that for grade ≥ 3 neutropenia/granulocytopenia, stomatitis, and mucositis, ORs and associated 95% CIs for the included studies either favoured oral fluoropyrimidines or crossed the null value of 1.00. For grade ≥ 3 febrile neutropenia and any grade ≥ 3 AEs, these either favoured oral fluoropyrimidines or crossed the null value of 1.00, with the exception of one and three outlier studies, respectively. For grade ≥ 3 diarrhoea, these either crossed the null value of 1.00 or indicated worse outcomes with oral fluoropyrimidine treatment, with the exception of one outlier study.
Overall completeness and applicability of evidence
The body of evidence that we found was directly relevant and was comprehensive enough to sufficiently address the objectives of this review.
Identified studies included the relevant patient population. Additionally, most of the oral fluoropyrimidines were examined in a wide range of geographical locations. However, the four studies that compared the oral fluoropyrimidine S‐1 versus IV fluoropyrimidines in patients treated with palliative intent for CRC (Yasui 2015; Kato 2012; Yamazaki 2015; Yamada 2013) recruited patients only from Japan. Caucasians receiving S‐1 have been shown to experience more diarrhoea and dehydration, as well as higher rates of toxicity‐related dose reductions, compared with their East Asian counterparts, despite similar 5‐FU exposure (Chuah 2011). Moreover, given the relatively high rates of diarrhoea reported in the oral fluoropyrimidine arm for one of the included studies, which used combination chemotherapy with S‐1 and irinotecan (Yasui 2015), further investigation is required before these results for S‐1 can be applied to other populations (Schmoll 2010). We also identified studies of doxifluridine that had been performed only in Asia and Europe (Ahn 2003; Bajetta 1996; Kim 2001a), but not in other geographical settings (Included studies).
Levels of compliance in clinical trials may not apply to clinical practice outside of trials (Schünemann 2011). In the context of this review, this is a particularly important issue for oral therapy. Lack of patient compliance may have an negative impact on efficacy. Conversely, patients may even demonstrate ‘over‐compliance’, whereby they continue treatment regardless of adverse effects and/or advice and education, and this may impact toxicity (Midgley 2009; Cassidy 2005). These factors may be subject to cultural variation (Haller 2008). Eleven of the 44 completed studies in this review incorporated procedures for monitoring compliance with oral medications. Outside of clinical trials, levels of monitoring in different hospitals and clinics may be subject to wide variability.
The interventions assessed in this review were overall very inclusive. Studies of curative intent treatment for CRC included neoadjuvant treatment alone for rectal carcinoma, neoadjuvant and adjuvant treatment for rectal carcinoma, and adjuvant treatment alone for colon and/or rectal carcinoma. Of note, the addition of oxaliplatin to IV 5‐FU and leucovorin (LV) has been shown to improve DFS and OS in the adjuvant treatment of stage III colon cancer (André 2009). However, we identified only one study that compared oral versus IV fluoropyrimidines in combination with oxaliplatin, without bevacizumab (BEV), for adjuvant treatment of colon cancer (Pectasides 2015), and this study was discontinued prematurely owing to slow accrual. The AVANT (Bevacizumab Plus Oxaliplatin‐Based Chemotherapy as Adjuvant Treatment for Colon Cancer) study (De Gramont 2012) was a large parallel three‐arm study that was designed to show the superiority of adding BEV to oxaliplatin, leucovorin, and 5‐fluorouracil (FOLFOX4) or capecitabine plus oxaliplatin (XELOX), compared with FOLFOX alone. We included in our review the BEV‐XELOX and BEV‐FOLFOX4 treatment arms from this study. However, notably, the addition of BEV was not shown to be of benefit in the AVANT study but was found to be associated with potential detriment for OS.
In studies of individuals treated with palliative intent for CRC, oral versus IV fluoropyrimidines were examined as single agents or in combination with irinotecan or oxaliplatin. Included studies examined bolus as well as infusional IV fluoropyrimidine regimens. In addition, our review identified eight studies that included treatment with BEV and combination chemotherapy (Cassidy 2011a; Ducreux 2013; Hochster TREE‐2 2008; Kato 2012; Pectasides 2012; Shigeta 2016; Souglakos 2012; Yamada 2013). However, we did not identify any studies that examined chemotherapy together with an epidermal growth factor receptor (EGFR) inhibitor in a study population that had been appropriately selected a priori for KRAS wild‐type (wt) status. We also did not identify any studies that included the targeted therapies ziv‐aflibercept, ramucirumab, and panitumumab, which currently are used in clinical practice.
Identified studies addressed the prespecified outcomes for this review. In this review, we compared only efficacy and grade ≥ 3 adverse event outcomes for oral versus IV fluoropyrimidines, as it was not within the scope of the review to examine differences in patient preference, quality of life, and cost‐effectiveness. These factors may influence the decision to use one option over another, and could be included as outcomes in future updates of this review.
The current review aimed to comprehensively assess oral versus IV fluoropyrimidines, regardless of the current state of development of the fluoropyrimidines identified. Of note, development of eniluracil was discontinued in 2000 (Malet‐Martino 2002). The most recent randomised controlled trial (RCT) that examined eniluracil with oral 5‐FU (ECOG E5296 2012) was terminated early on the basis of negative results from two earlier studies of eniluracil with oral 5‐FU (Schilsky 2002a and Van Cutsem 2001a, included in this review). Clinical development of IV doxifluridine for CRC has been abandoned (Saletti 2008).
Capecitabine is currently approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), and is registered by the Therapeutic Goods Administration (TGA) in Australia for treatment of both metastatic CRC and high‐risk stage II/III colon cancer (Pazdur 2016; EMA 2016; TGA 2016). S‐1 is widely used as adjuvant and palliative chemotherapy for CRC in Japan (Miyamoto 2014). Recent guidelines on treatment of Asian patients with mCRC recommended that infusional 5‐FU could be substituted with capecitabine, UFT, or S‐1 (Cheng 2014). These guidelines were developed to reflect current Asian clinical practice, following a consensus meeting in 2012, which included representatives from ten Asian countries (China, Hong Kong, India, Indonesia, Malaysia, the Philippines, Singapore, South Korea, Taiwan, and Thailand) and from two European countries (Germany and Italy).
Quality of the evidence
Efficacy
In patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, we assessed all of the seven studies included in the quantitative synthesis for the primary outcome DFS as having high risk of bias for reasons including lack of blinding of the outcome assessor (detection bias). We did not identify inconsistency in results (P = 0.48; I2 = 0%), indirectness of evidence, or imprecision for this outcome (> 2000 events, 95% CI for the pooled HR excluded appreciable benefit and harm), and the quality of evidence was moderate.
For patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy, we assessed 17 of the 23 studies included in the quantitative synthesis for the primary outcome PFS as having high risk of bias. Fifteen of these studies had high risk of bias for reasons including lack of blinding of outcome assessors, and the remaining two studies had high risk of bias owing to differences in schedules of assessment and/or follow‐up between arms. A sensitivity analysis that excluded the 17 studies at high risk of bias did not lead to substantial changes in direction of the effect estimate nor in its magnitude, although the 95% CI crossed the null value of 1.00. We did not identify inconsistency in results (P = 0.25; I2 = 15%), indirectness of evidence, or imprecision (> 4000 events, and optimum information size was met) for this outcome, and the quality of evidence was moderate.
We assessed all of the other secondary efficacy outcomes in this review as having high or moderate quality of evidence. For OS in both curative intent and palliative intent studies, we did not identify any factors that reduced the quality of evidence, which was assessed as high. In patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy, we assessed the quality of evidence for both TTP and ORR as moderate owing to downgrading by one level for risk of bias (predominantly due to lack of blinding of outcome assessors).
Adverse events
For patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, all of the studies that contributed to the seven subjective AE outcomes grade ≥ 3 diarrhoea, HFS, vomiting, nausea, stomatitis, mucositis, and any grade ≥ 3 AEs had high risk of bias for reasons including lack of blinding of participants, personnel, and outcome assessors. We assessed the quality of evidence as low for five of the seven subjective outcomes ‐ for three outcomes (grade ≥ 3 HFS, stomatitis, and any grade ≥ 3 AEs), we downgraded the quality by one level each for high risk of bias and inconsistency of results, and for two outcomes (grade ≥ 3 vomiting and nausea), we downgraded the quality by one level each for high risk of bias and imprecision. We assessed the quality of evidence as very low for two of the seven subjective outcomes (grade ≥ 3 diarrhoea and mucositis). For grade ≥ 3 diarrhoea, we downgraded the quality by one level each for risk of bias, inconsistency of results, and imprecision; for grade ≥ 3 mucositis, we downgraded the quality by one level for risk of bias and by two levels for imprecision.
With respect to the objective outcomes, we assessed the quality of evidence for grade ≥ 3 neutropenia/granulocytopenia as moderate (downgraded by one level for inconsistency of results), for grade ≥ 3 febrile neutropenia as low (downgraded by two levels for imprecision), and for grade ≥ 3 hyperbilirubinaemia as very low (downgraded by one level for risk of bias, and by two levels for imprecision).
For patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy, all of the studies that contributed to the seven subjective AE outcomes also had high risk of bias for reasons including lack of blinding of participants, personnel, and outcome assessors. We assessed the quality of evidence as moderate for one of the seven subjective outcomes (grade ≥ 3 HFS), and we downgraded quality by one level for risk of bias alone. We assessed the quality of evidence as low for the remaining six subjective outcomes ‐ for four outcomes (grade ≥ 3 diarrhoea, stomatitis, mucositis, and any grade ≥ 3 AEs), we downgraded the quality by one level each for risk of bias and inconsistency of results, and for two outcomes (grade ≥ 3 vomiting and nausea), we downgraded the quality by one level each for risk of bias and imprecision.
With respect to the objective outcomes, we assessed the quality of evidence for grade ≥ 3 febrile neutropenia as moderate (downgraded by one level for inconsistency of results), for grade ≥ 3 neutropenia/granulocytopenia as low (downgraded by one level each for risk of bias and inconsistency of results), and for hyperbilirubinaemia as low (downgraded by one level each for risk of bias and imprecision).
Summary
Overall, the quality of evidence for efficacy outcomes was higher (high or moderate quality) than for adverse event outcomes (very low to moderate quality). Seven of the ten AE outcomes were subjective and were at risk of performance and detection bias from lack of blinding, and all of the studies that contributed to these subjective outcomes were unblinded. Additionally, we further downgraded the quality of evidence for most of these subjective AE outcomes for inconsistency of results and/or imprecision.
Potential biases in the review process
We adhered to having at least two independent review authors select studies, extract data, and conduct risk of bias assessments. These review authors encountered no disagreements that required resolution by a third review author, but a third review author resolved any uncertainties that arose.
In our original protocol, we did not hypothesise that one route of fluoropyrimidine administration (oral or IV) was superior to the other, and we did not state a priori levels of benefit. For the primary outcomes of DFS and PFS, we determined non‐inferiority margins post hoc, whereby 50%, 70%, 80%, and 90% of the activity of the active control (IV fluoropyrimidines) was retained had the original design been one of non‐inferiority. We determined these non‐inferiority margins independent of studies comparing oral versus IV fluoropyrimidine, and we reported all margins. Assessments regarding whether non‐inferiority was demonstrated in this review are potentially at risk of bias, as they are dependent on subjective post hoc judgements about what proportion of the activity of the active control is required to be retained for non‐inferiority to be met.
Agreements and disagreements with other studies or reviews
Reviews including RCTs of multiple oral fluoropyrimidines
A systematic review and meta‐analysis by Sasse et al examined RCTs using capecitabine or UFT/Ftorafur as single agents or in combination therapy (Sasse 2009a). This review used only databases in the systematic search strategy (performed in December 2008) and included no studies using doxifluridine, S‐1, or eniluracil with oral 5‐FU as an oral fluoropyrimidine backbone. Results show some overlap of participants in the list of included studies (Cassidy 2002; Hoff 2001; Van Cutsem 2001b), and this list included a study wherein the co‐intervention was not common to the oral and IV fluoropyrimidine arms (Schmoll 2007). Results presented in the abstract and in the presentation slides show some differences (Sasse 2009a; Sasse 2009b). Quantitative synthesis for the outcomes OS, RR, and PFS included 16 studies, 15 studies, and nine studies respectively (Sasse 2009b). This study combined OS outcome data for patients treated with curative intent and patients treated with palliative intent for CRC. The abstract reported lower ORR and shorter PFS but no significant difference in OS for capecitabine versus infusional IV fluoropyrimidines (cIV); and lower ORR but no difference in PFS or OS for capecitabine versus bolus IV fluoropyrimidines (Sasse 2009a). The abstract reported similar ORR, OS, and PFS for UFT/Ftorafur and bolus 5‐FU (Sasse 2009a). Review authors concluded that "oral fluoropyrimidines are equivalent to bolus 5‐FU in terms of efficacy, but provide less benefit than cIV 5FU." In contrast, our review found no significant subgroup differences between ‘Bolus IV fluoropyrimidine’ and ‘Infusional IV fluoropyrimidine’ subgroups for the PFS outcome.
Reviews of RCTs comparing capecitabine versus IV 5‐FU
A previous systematic review and meta‐analysis pooled results from RCTs comparing capecitabine versus 5‐FU, either alone or in combination therapy for colorectal cancer (Petrelli 2012). Another published individual patient data (IPD) meta‐analysis included six non‐inferiority RCTs from the Roche clinical trials database and included one advanced gastric cancer study (Cassidy 2011b).
Petrelli et al searched databases and American Society of Clinical Oncology (ASCO) conference proceedings and included in their review 17 studies in patients treated with palliative intent for CRC with chemotherapy, including 15 of the studies identified for our review (Cassidy 2011a; Comella 2009; Diaz‐Rubio 2007; Ducreux 2011; Fuchs 2007; Hochster TREE‐1 2008; Hochster TREE‐2 2008; Hoff 2001; Kohne 2008; Martoni 2006; Pectasides 2012; Porschen 2007; Rothenberg 2008; Souglakos 2012; Van Cutsem 2001b). Two studies that we had excluded from our review with reasons were also included (Munoz 2008; Skof 2009). Toxicity outcomes were not restricted to grade ≥ 3 AEs. For efficacy outcomes, review authors also reported significant heterogeneity between the included studies for ORR. Consistent with our findings for the ‘Capecitabine’ subgroup, the pooled HR for both PFS (seven studies) and OS (six studies) in Petrelli 2012 showed no difference between oral and IV fluoropyrimidines.
Reviews of RCTs comparing capecitabine and infusional 5‐FU in combination with irinotecan, and capecitabine and infusional 5‐FU in combination with oxaliplatin
For a systematic review and meta‐analysis published by Montagnani et al, review authors searched databases and conference proceedings for European Society of Medical Oncology (ESMO) and ASCO, and identified only three RCTs comparing capecitabine and infusional 5‐FU in combination with irinotecan for treatment of metastatic CRC (Montagnani 2010). We included two of these studies in our review (Fuchs 2007; Kohne 2008), and we excluded one study (Skof 2009) from our review. The study population for Skof 2009 included selected patients with unresectable liver‐only metastases who had Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 to 1.
Review authors did not report an assessment of heterogeneity for ORR. PFS was worse with oral fluoropyrimidine use (using the pooled HR for PFS reported in the text of the study report). This differed from our findings in the ‘Irinotecan‐based’ subgroup (including any oral fluoropyrimidine), which indicated no difference between oral and IV fluoropyrimidines. For grade ≥ 3 diarrhoea, the findings of Montagnani were consistent with the findings of our analyses for the ‘Irinotecan‐based’ subgroup.
Arkenau et al published a systematic review and meta‐analysis of RCTs comparing capecitabine and infusional 5‐FU in combination with oxaliplatin for treatment of metastatic CRC, with a search strategy including databases, trial registries, and conference proceedings (Arkenau 2008). We included all of the seven RCTs from this study in our review (Cassidy 2011a; Diaz‐Rubio 2007; Ducreux 2011; Hochster TREE‐1 2008; Hochster TREE‐2 2008; Martoni 2006; Porschen 2007).
The HRs for PFS and OS in Arkenau 2008, which showed no evidence of a difference for oral versus IV fluoropyrimidines, were in agreement with results for the ‘Oxaliplatin‐based’ subgroup in our review. Other meta‐analyses of studies that included oxaliplatin‐based combination regimens with a capecitabine arm have reported similar findings for PFS and OS (Cassidy 2008; Cuppone 2008). The pooled estimate for grade ≥ 3 diarrhoea, which indicated worse outcomes with oral fluoropyrimidine treatment, was also similar to that in our 'Oxaliplatin‐based' subgroup.
Schmoll et al published an IPD meta‐analysis of four large RCTs comparing effects of adjuvant treatment with capecitabine or fluorouracil, with or without oxaliplatin, on survival outcomes in resected stage III colon cancer (Schmoll 2014). A total of 8734 participants from two trials that we had included in our review (De Gramont 2012; Twelves 2012), as well as from the NSABP C‐08 and XELOXA (NO16968) trials (Allegra 2011; Haller 2011), were included in a pooled analysis of disease‐free, relapse‐free, and overall survival. The XELOXA study compared capecitabine plus oxaliplatin versus bolus IV FU/folinic acid (FA), and the NSABP C‐08 study compared modified FOLFOX6 (mFOLFOX6) versus mFOLFOX6 with BEV.
In keeping with the findings of our review, the IPD meta‐analysis by Schmoll et al revealed no significant differences in adjusted DFS and OS for capecitabine with or without oxaliplatin compared with IV 5‐FU/LV with or without oxaliplatin.
Authors' conclusions
Implications for practice.
Findings of this review indicate that for patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, moderate‐quality evidence shows that DFS does not differ between patients treated with oral versus IV fluoropyrimidines. For patients treated with palliative intent for CRC with chemotherapy, the HR for PFS for oral versus IV fluoropyrimidine therapy was 1.06 (95% CI 1.02 to 1.11; moderate‐quality evidence). Treatment with UFT/Ftorafur or eniluracil with oral 5‐FU was associated with an inferior PFS compared with IV fluoropyrimidines, but PFS did not differ between individuals treated with oral versus IV fluoropyrimidines when the other oral fluoropyrimidines were used. Overall, OS did not differ between patients treated with oral versus IV fluoropyrimidines. However, treatment with eniluracil with oral 5‐FU versus IV fluoropyrimidines was associated with inferior OS. We also observed differences between grade ≥ 3 adverse event profiles for oral and IV fluoropyrimidines.
The results of this review provide confidence that, for treatment of CRC, most of the oral fluoropyrimidines used commonly in current clinical practice have similar efficacy to IV fluoropyrimidines. For patients treated with palliative intent for CRC, use of eniluracil with oral 5‐FU was associated with an inferior PFS and OS compared with IV fluoropyrimidines, and development of this combination has been ceased. This review did not examine patient preferences, quality of life, and cost‐effectiveness of oral versus IV fluoropyrimidines. In addition to consideration of different adverse effect profiles, these factors may influence the decision to choose one option over the other.
Implications for research.
Future research may focus on understanding the basis for adverse event differences observed with oral versus IV fluoropyrimidines in patients with CRC treated with either curative or palliative intent. For patients treated with palliative intent for CRC, we identified a lack of clinical trials comparing oral and IV fluoropyrimidines used in combination chemotherapy together with EGFR inhibitors, in a study population that has been appropriately selected forKRAS wild‐type status. We also did not identify any studies that included the targeted therapies ziv‐aflibercept, ramucirumab, and panitumumab.
What's new
| Date | Event | Description |
|---|---|---|
| 9 August 2017 | Amended | affiliations amended for two authors in the byline |
Acknowledgements
We would like to acknowledge the following people.
The Cochrane Colorectal Cancer Group (in particular, Dr. Henning Keinke Andersen, Sys Johnsen, Anne Kathrine Helnæs Jensen, Marija Barbateskovic, and Susse Wegeberg) for assistance with the review, including assistance from the Trial Information Specialists with the search strategy for the Cochrane Library, MEDLINE (OVID), and Embase (OVID).
The Australasian Cochrane Centre Training team, for assistance provided.
Diana Zannino, statistician from the Centre for Biostatistics & Clinical Trials; Peter MacCallum Cancer Centre, Australia; for assistance with extracting statistics from manuscripts for the review.
Professor Val Gebski, Professor and Director, Biostatistics and Research Methodology, NHMRC Clinical Trials Centre, Australia, for assistance with non‐inferiority analyses.
Study authors and study contacts (including statisticians, pharmaceutical company liaison staff, and Co‐operative Group staff) who replied to us and assisted with our queries about their studies. In particular, we would like to individually acknowledge the following study authors and study contacts who provided significant assistance with information about their studies: Dr. Abhishek Basu, Dr. Alejandro de la Torre Tomas, Anastasia Eleftheraki and Dr. Angelos Koutras, Mr. Atsuki Shinozaki and Dr. Narikazu Boku, Dr. Bruce Sizer, Dr. Carmen Allegra, Dr. Chikashi Ishioka and Dr. Shunsuke Kato, Dr. Christopher Twelves, Dr. Hirotoshi Hasegawa, Dr. Jean‐Pierre Pignon and Dr. Michel Ducreux, Dr. Jeremey Levin and Dr. Richard Schilsky, Dr. John Souglakos, Mr. Naruhito Takenaka, Dr. Paul Catalano, Dr. Peter Eggleton, Dr. Ralf Hofheinz, Dr. Yuri Barsukov and Dr. Sergey S. Gordeyev, and staff from Taiho who assisted us with our queries.
Dr. V.K. Yeung for translation of two study reports from Chinese to English.
Dr. Hongdo Do for translation of one study report from Korean to English.
Pharmaceutical company liaison staff (from Taiho, Roche, Orzel, and Adherex) who assisted with identifying studies.
Anne McLean, librarian at Austin Hospital, Australia, for assistance with the search strategy and with obtaining full‐text reports or the most mature study reports from the search.
A/Prof Sue‐Anne McLachlan for assistance with the search strategy for the protocol for this review.
Co‐authors of the protocol for this review ‐ Drs. Ainsley Campbell and Shawgi Sukumaran.
Appendices
Appendix 1. Search strategy for CENTRAL, the Cochrane Library
Cochrane Library Issue 5, 20 May 2016 (292 hits in CENTRAL)
1. MeSH descriptor Colorectal Neoplasms explode all trees
2. ((cancer* or carcinoma* or neoplasm* or adenoma* or adenocarcinom* or tumour* or tumor* or polyp* or malignan*) near3 (colorectal* or colon* or rect*))
3. (#1 OR #2)
4. MeSH descriptor Fluorouracil explode all trees
5. MeSH descriptor Antimetabolites, Antineoplastic explode all trees
6. CapeIri or CapeOx or fluoropyrimidine* or $fluorouracil or 5 FU or 5‐FU or 5FU or $uracil or Capecitabine or Xeloda or Tegafur or S1 or S‐1 or Orzel or 776C85 or UFT or Xelox or Xeliri or Capox or Capiri
7. (#4 OR #5 OR #6)
8. (oral* and (intravenous* or infusion*))
9. (#3 AND #7 AND #8)
Appendix 2. Search strategy for MEDLINE (OVID)
MEDLINE (OVID) 1950 to 14 June 2016 (322 hits)
1. exp Colorectal Neoplasms/
2. ((cancer* or carcinoma* or neoplasm* or adenoma* or adenocarcinom* or tumour* or tumor* or polyp* or malignan*) adj3 (colorectal* or colon* or rect*)).mp.
3. 1 or 2
4. exp Fluorouracil/
5. exp Antimetabolites/
6. (CapeIri or CapeOx or fluoropyrimidine* or $fluorouracil or 5 FU or 5‐FU or 5FU or $uracil or Capecitabine or Xeloda or Tegafur or S1 or S‐1 or Orzel or 776C85 or UFT or Xelox or Xeliri or Capox or Capiri).mp.
7. 4 or 5 or 6
8. (oral* and (intravenous* or infusion*)).mp.
9. 3 and 7 and 8
10. randomized controlled trial.pt.
11. controlled clinical trial.pt.
12. randomized.ab.
13. placebo.ab.
14. clinical trials as topic.sh.
15. randomly.ab.
16. trial.ti.
17. 10 or 11 or 12 or 13 or 14 or 15 or 16
18. exp animals/ not humans.sh.
19. 17 not 18
20. 9 and 19
Appendix 3. Search strategy for Embase (OVID)
Embase (OVID) 1974 to 14 June 2016 (498 hits):
1. exp large intestine tumor/
2. ((cancer* or carcinoma* or neoplasm* or adenoma* or adenocarcinom* or tumour* or tumor* or polyp* or malignan*) adj3 (colorectal* or colon* or rect*)).mp.
3. 1 or 2
4. exp fluorouracil/
5. exp antimetabolite/
6. (CapeIri or CapeOx or fluoropyrimidine* or $fluorouracil or 5 FU or 5‐FU or 5FU or $uracil or Capecitabine or Xeloda or Tegafur or S1 or S‐1 or Orzel or 776C85 or UFT or Xelox or Xeliri or Capox or Capiri).mp.
7. 4 or 5 or 6
8. (oral* and (intravenous* or infusion*)).mp.
9. 3 and 7 and 8
10. CROSSOVER PROCEDURE.sh.
11. DOUBLE‐BLIND PROCEDURE.sh.
12. SINGLE‐BLIND PROCEDURE.sh.
13. (crossover* or cross over*).ti,ab.
14. placebo*.ti,ab.
15. (doubl* adj blind*).ti,ab.
16. allocat*.ti,ab.
17. trial.ti.
18. RANDOMIZED CONTROLLED TRIAL.sh.
19. random*.ti,ab.
20. 10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19
21. (exp animal/ or exp invertebrate/ or animal.hw. or nonhuman/) not (exp human/ or human cell/ or (human or humans or man or men or wom?n).ti.)
22. 20 not 21
23. 9 and 22
Appendix 4. Search strategy for Web of Science (Web of Knowledge)
This search was performed on 16 June 2016, with the search dates including 1900 to 2016 (9.6.2016).
| Set | Results | Search Terms |
| # 20 | 904 | #17 AND #9 |
| # 19 | 165,735 | #18 AND #17 |
| # 18 | 3,118,135 | TOPIC: (human) ORTOPIC: (humans) |
| # 17 | 1,641,761 | #16 OR #15 OR #14 OR #13 OR #12 OR #11 OR #10 |
| # 16 | 1,223,762 | TOPIC: (trial) |
| # 15 | 278,002 | TOPIC: (randomly) |
| # 14 | 495,021 | TOPIC: (clinical trial) |
| # 13 | 204,807 | TOPIC: (placebo) |
| # 12 | 650,738 | TOPIC: (randomized) |
| # 11 | 209,393 | TOPIC: (controlled clinical trial) |
| # 10 | 317,506 | TOPIC: (randomized controlled trial) ORTOPIC: (randomised controlled trial) |
| # 9 | 1,131 | #8 AND #7 AND #6 |
| # 8 | 318,494 | #2 OR #1 |
| # 7 | 38,132 | TOPIC: (oral*) ANDTOPIC: (intravenous* or infusion*) |
| # 6 | 286,602 | #5 OR #4 OR #3 |
| # 5 | 286,550 | TOPIC: (fluoropyrimidine* or fluorouracil or 5 FU or 5‐FU or 5FU or uracil or capecitabine or xeloda or tegafur or S1 or S‐1 or orzel or 776C85 or UFT or Xelox or Xeliri or Capox or Capiri or Capeox or Capeiri) |
| # 4 | 40,442 | TOPIC: (fluorouracil) |
| # 3 | 76 | TOPIC: (antimetabolites, antineoplastic) |
| # 2 | 318,494 | TOPIC: (cancer* or carcinoma* or neoplasm* or adenoma* or adenocarcinom* or tumour* or tumor* or polyp* or malignan*) ANDTOPIC: (colorectal* or colon* or rectal or rectum) |
| # 1 | 8,638 | TOPIC: (colorectal neoplasms) |
Appendix 5. Efficacy outcomes in studies not suitable for inclusion in meta‐analysis ‐ Patients treated with curative intent for colorectal cancer
| Study | Chemotherapy arm | 3‐year DFS rate, %; P value | 3‐year OS (curative intent studies) rate, %; P value | HR for OS (curative intent studies) (95% CI) |
| De la Torre 2008 | UFT/LV + RT | 65.6 | 74 | 1.39 (0.66‐2.93) |
| FU/LV + RT | 64.7; P = 0.67 | 87; P = 0.37 | ||
| UFT: tegafur/uracil LV: leucovorin RT: radiotherapy | ||||
Appendix 6. Other information for studies that reported similar adverse event outcomes to "Neutropenia/Granulocytopenia" and "Hand foot syndrome"
| Setting | Study | Outcome in our review | Adverse event reported | Chemotherapy arm | Grade ≥ 3 AE (%) |
| Patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy |
Hofheinz 2012 | Neutropenia/ Granulocytopenia |
Lowered leukocytes | Capecitabine | 1.5 |
| Fluorouracil | 8.2 | ||||
| Kim 2001a | Neutropenia/ Granulocytopenia |
Leukopenia | Doxifluridine + RT | 0 | |
| 5‐FU + RT | 6.8 | ||||
| Patients treated with palliative intent for inoperable advanced or metastatic CRC with chemotherapy |
Bajetta 1996 | Neutropenia/ Granulocytopenia |
Leukopenia | Oral 5‐dFUR | 1.5 |
| IV 5‐dFUR | 15 | ||||
| Silvestris 2010 | Neutropenia/ Granulocytopenia |
Leuko/neutropenia | XELIRI | 17.2 | |
| FOLFIRI | 16.1 | ||||
| Kohne 2008 | Neutropenia/ Granulocytopenia |
White blood cells | CAPIRI‐Celecoxib/Placebo | 14 | |
| FOLFIRI‐Celecoxib/Placebo | 15.4 | ||||
| Carmichael 2002 | HFS | Skin/appendages (including HFS) | UFT/LV | 1 | |
| 5‐FU/LV | 1 | ||||
| Porschen 2007 | HFS | HFS | CAPOX | Only Grade 2/3 HFS included | |
| FUFOX | |||||
| AE: adverse event CRC: colorectal cancer RT: radiotherapy 5‐FU: 5‐fluorouracil 5‐dFUR: doxifluridine IV: intravenous HFS: hand foot syndrome UFT: tegafur/uracil LV: leucovorin | |||||
Appendix 7. Efficacy outcomes in studies not suitable for inclusion in meta‐analysis ‐ Patients treated with palliative intent for inoperable advanced or metastatic colorectal cancer
| Outcome | Study | Line | Chemotherapy arm | Median (95% CI) for PFS, OS, TTP; % for ORR |
| PFS | Hochster TREE‐1 2008a | First | CapeOx | 5.9 m (5.1 to 7.4) |
| mFOLFOX6 | 8.7 m (6.5 to 9.8) | |||
| bFOL | 6.9 m (4.2 to 8.0) | |||
| Hochster TREE‐2 2008a | First | CapeOx + BEV | 10.3 m (8.6 to 12.5) | |
| mFOLFOX6 + BEV | 9.9 m (7.9 to 11.7) | |||
| bFOL + BEV | 8.3 m (6.6 to 9.9) | |||
| OS | Andersen 1987 | First | Ftorafur | 209 d |
| 5‐FU | 211 d | |||
| Yu 2005 | First, Second | Capecitabine + Irinotecan | 17.9 m | |
| 5‐FU + Irinotecan | 14.2 m | |||
| TTP | Silvestris 2010 | First | XELIRI | 8.7 m |
| FOLFIRI | 6.5 m | |||
| Yu 2005 | First, Second | Capecitabine + Irinotecan | 12.5 m | |
| 5‐FU + Irinotecan | 8.4 m | |||
| ORR | Mei 2014 | First | SOX | 51.4 |
| FOLFOX4 | 45.7 | |||
| Seymour 2011 | First | Capecitabine | 14 | |
| OxCap | 32 | |||
| FU | 11 | |||
| OxFU | 38 | |||
| Silvestris 2010 | First | XELIRI | 48.4 | |
| FOLFIRI | 32.2 | |||
|
aOutcome reported as TTP, but treated as PFS in this review based on the definition provided. CI: confidence interval PFS: progression‐free survival OS: overall survival TTP: time to tumour progression ORR: objective response rate BEV: bevacizumab 5‐FU / FU: 5‐fluorouracil | ||||
Appendix 8. Adverse event outcomes in studies not suitable for inclusion in meta‐analysis ‐ Patients treated with palliative intent for inoperable advanced or metastatic colorectal cancer
| Study |
Line of chemotherapy |
Grade ≥3 AE | Chemotherapy arm | Measure of grade ≥ 3 AE |
| Silvestris 2010 | First | Diarrhoea | XELIRI | 12.50% |
| FOLFIRI | 3.20% | |||
| HFS | XELIRI | Grade 3: n = 1 | ||
| FOLFIRI | ||||
| Leuko/neutropenia | XELIRI | 17.2% | ||
| FOLFIRI | 16.1% | |||
| Yu 2005 | First, Second | Diarrhoea | Capecitabine + Irinotecan | 7.40% |
| 5‐FU + Irinotecan | 18.80% | |||
| HFS | Capecitabine + Irinotecan | 0% | ||
| 5‐FU + Irinotecan | 0% | |||
| Neutropenia | Capecitabine + Irinotecan | 0% | ||
| 5‐FU + Irinotecan | 0% | |||
| Nausea/Vomiting | Capecitabine + Irinotecan | 0% | ||
| 5‐FU + Irinotecan | 6.25% | |||
| Stomatitis | Capecitabine + Irinotecan | 0% | ||
| 5‐FU + Irinotecan | 0% | |||
| AE: adverse event HFS: hand foot syndrome 5‐FU: 5‐fluorouracil | ||||
Appendix 9. Summary of subgroup analyses for efficacy outcomes ‐ Patients treated with curative intent for colorectal cancer
| Efficacy outcome | Hazard ratio (fixed, 95% CI) (No. studies/n) for subgroups, test for subgroup differences | ||||||
| Treatment type | Infusional vs bolus IV fluoropyrimidines | Oral fluoropyrimidine backbone | |||||
| Chemotherapy | Chemo‐radiotherapy | Infusional | Bolus | Capecitabine | UFT/Ftorafur | Doxifluridine | |
| DFS | 0.94 (0.87 to 1.02) | 0.91 (0.78 to 1.05) | 0.96 (0.85 to 1.08) | 0.94 (0.86 to 1.04) | 0.91 (0.83 to 0.99) | 1.01 (0.88 to 1.15) | - |
| 5/6944 | 2/1959 | 3/3881 | 3/4630 | 5/6260 | 2/2643 | ||
| Chi² = 0.21, P = 0.64; I2 = 0% | Chi² = 0.06, P = 0.81; I2 = 0% | Chi² = 1.70, P = 0.19; I2 = 41.1% | |||||
| OS | 0.93 (0.84 to 1.03) | 0.86 (0.70 to 1.06) | 0.94 (0.80 to 1.09) | 0.93 (0.83 to 1.05) | 0.88 (0.79 to 0.98) | 1.03 (0.86 to 1.22) | - |
| 5/6943 | 2/1959 | 3/3880 | 3/4630 | 5/6259 | 2/2643 | ||
| Chi² = 0.43, P = 0.51; I2 = 0% | Chi² = 0.00, P = 0.96; I2 = 0% | Chi² = 2.20, P = 0.14; I2 = 54.5% | |||||
| CI: confidence interval IV: intravenous UFT: tegafur/uracil DFS: disease‐free survival OS: overall survival | |||||||
Appendix 10. Summary of subgroup analyses for grade ≥ 3 adverse event outcomes ‐ Patients treated with curative intent for colorectal cancer
| Grade ≥3 AE | Odds ratio (fixed, 95% CI) (No. studies/n) for subgroups, test for subgroup differences | ||||||
| Treatment type | Infusional vs bolus IV fluoropyrimidine | Oral fluoropyrimidine backbone | |||||
| Chemotherapy | Chemo‐radiotherapy | Infusional | Bolus | Capecitabine | UFT/Ftorafur | Doxifluridine | |
| Diarrhoea | 1.08 (0.95 to 1.23) | 1.28 (0.98 to 1.66) | 1.27 (1.06 to 1.53) | 0.98 (0.84 to 1.14) | 1.15 (0.99 to 1.33) | 1.00 (0.83 to 1.21) | 32.14 (1.89 to 545.41) |
| 5/7274 | 4/2277 | 3/4255 | 5/4904 | 5/6616 | 3/2769 | 1/166 | |
| Chi² = 1.24, P = 0.27; I2 = 19.3% | Chi² = 4.52, P = 0.03; I2 = 77.9% | Chi² = 6.73, P = 0.03. I2 = 70.3% | |||||
| Vomiting | 1.03 (0.80 to 1.32) | 1.42 (0.54 to 3.75) | 1.56 (1.07 to 2.26) | 0.78 (0.57 to 1.08) | 1.34 (0.99 to 1.81) | 0.67 (0.44 to 1.01) | - |
| 5/7274 | 3/2111 | 3/4255 | 4/4738 | 5/6616 | 3/2769 | ||
| Chi² = 0.39, P = 0.53; I2 = 0% | Chi² = 7.48, P = 0.006; I2 = 86.6% | Chi² = 7.25, P = 0.007; I2 = 86.2% | |||||
| Nausea | 1.13 (0.90 to 1.42) | 3.19 (1.22 to 8.36) | 1.59 (1.10 to 2.31) | 1.00 (0.76 to 1.33) | 1.40 (1.04 to 1.88) | 1.00 (0.71 to 1.40) | - |
| 5/7274 | 2/1959 | 3/4255 | 3/4586 | 5/6616 | 2/2617 | ||
| Chi² = 4.24, P = 0.04; I2 = 76.4% | Chi² = 3.79, P = 0.05; I2 = 73.6% | Chi² = 2.10, P = 0.15; I2 = 52.3% | |||||
| AE: adverse event CI: confidence interval IV: intravenous UFT: tegafur/uracil | |||||||
Appendix 11. Summary of subgroup analyses for efficacy outcomes ‐ Patients treated with palliative intent for inoperable advanced or metastatic colorectal cancer
| Efficacy outcome | Hazard ratio (fixed, 95% CI) (No. studies/n) for subgroups, test for subgroup differences | ||||||||||
| Single agent vs combination therapy | Infusional vs bolus IV fluoropyrimidine | Oral fluoropyrimidine backbone | Oxaliplatin‐based vs irinotecan‐based | ||||||||
| Single agent | Combination therapy | Infusional | Bolus | Capecitabine | UFT/Ftorafur | Eniluracil + oral 5‐FU | Doxifluridine | S‐1 | Oxaliplatin‐based | Irinotecan‐based | |
| PFS | 1.12 (1.04 to 1.21) | 1.05 (1.00 to 1.10) | 1.05 (1.00 to 1.10) | 1.10 (1.03 to 1.19) | 1.03 (0.98 to 1.08) | 1.36 (1.07 to 1.73) | 1.22 (1.10 to 1.36) | 1.18 (0.79 to 1.74) | 1.02 (0.89 to 1.16) | 1.06 (0.99 to 1.13) | 1.04 (0.97 to 1.11) |
| 6/2955 | 16/6513 | 17/6560 | 7/3367 | 13/6703 | 2/374 | 3/1618 | 1/130 | 4/1102 | 8/4677 | 8/1836 | |
| Chi² = 2.16, P = 0.14; I2 = 53.8% | Chi² = 1.33, P = 0.25; I2 = 24.7% | Chi² = 13.46, P = 0.009; I2 = 70.3% | Chi² = 0.13, P = 0.72; I2 = 0% | ||||||||
| OS | 1.02 (0.99 to 1.07) | 1.00 (0.95 to 1.06) | 1.01 (0.96 to 1.06) | 1.02 (0.98 to 1.06) | 0.99 (0.95 to 1.04) | 1.02 (0.97 to 1.06) | 1.20 (1.07 to 1.36) | 0.99 (0.65 to 1.50) | 0.95 (0.81 to 1.11) | 1.00 (0.94 to 1.07) | 1.01 (0.92 to 1.10) |
| 10/4465 | 18/7155 | 19/7022 | 13/5057 | 16/7405 | 5/1807 | 3/1618 | 2/207 | 3/1042 | 11/5379 | 7/1776 | |
| Chi² = 0.40, P = 0.53; I2 = 0% | Chi² = 0.10, P = 0.75;I2 = 0% | Chi² = 9.30, P = 0.05; I2 = 57.0% | Chi² = 0.01, P = 0.90; I2 = 0% | ||||||||
| TTP | 1.08 (1.01 to 1.14) | 1.05 (0.84 to 1.32) | 1.05 (0.84 to 1.32) | 1.08 (1.01 to 1.14) | 1.05 (0.84 to 1.32) | 1.08 (1.02 to 1.15) | - | 0.88 (0.56 to 1.36) | - | - | |
| 4/1510 | 2/460 | 2/460 | 4/1510 | 2/460 | 3/1433 | 1/77 | |||||
| Chi² = 0.03, P = 0.86; I2 = 0% | Chi² = 0.03, P = 0.86; I2 = 0% | Chi² = 0.89, P = 0.64; I2 = 0% | |||||||||
| ORR | 1.11 (0.94 to 1.31) | 0.93 (0.85 to 1.03) | 0.92 (0.83 to 1.02) | 1.12 (0.96 to 1.29) | 1.01 (0.91 to 1.12) | 0.92 (0.72 to 1.18) | 0.84 (0.62 to 1.13) | 0.52 (0.28 to 0.99) | 1.08 (0.81 to 1.45) | 0.92 (0.83 to 1.03) | 0.97 (0.79 to 1.19) |
| 11/4208 | 21/6907 | 21/6342 | 14/4773 | 17/6690 | 6/1772 | 3/1522 | 2/190 | 4/941 | 12/5201 | 9/1706 | |
| Chi² = 3.14, P = 0.08: I2 = 68.2% | Chi² = 4.69, P = 0.03; I2 = 78.7% | Chi² = 5.81, P = 0.21; I2 = 31.2% | Chi² = 0.15, P = 0.70; I2 = 0% | ||||||||
| CI: confidence interval IV: intravenous UFT: tegafur/uracil 5‐FU: 5‐fluorouracil PFS: progression‐free survival OS: overall survival TTP: time to tumour progression ORR: objective response rate | |||||||||||
Appendix 12. Summary of subgroup analyses for grade ≥ 3 adverse event outcomes ‐ Patients treated with palliative intent for inoperable advanced or metastatic colorectal cancer
| Grade ≥3 AE | Odds ratio (fixed, 95% CI), (No. studies/n) for subgroups, Test for subgroup differences | ||||||||||
| Single‐agent vs combination therapy | Infusional vs bolus IV fluoropyrimidine | Oral fluoropyrimidine backbone | Oxaliplatin‐based vs irinotecan‐based | ||||||||
| Single agent | Combination therapy | Infusional | Bolus | Capecitabine | UFT/Ftorafur | Eniluracil + oral 5‐FU | Doxifluridine | S‐1 | Oxaliplatin‐based | Irinotecan‐based | |
| Diarrhoea | 1.22 (1.04 to 1.44) | 2.03 (1.77 to 2.32) | 2.00 (1.74 to 2.30) | 1.31 (1.12 to 1.53) | 1.76 (1.54 to 2.00) | 1.60 (1.24 to 2.06) | 1.04 (0.79 to 1.38) | 1.51 (0.64 to 3.56) | 3.55 (2.19 to 5.76) | 1.73 (1.48 to 2.02) | 3.05 (2.33 to 3.99) |
| 10/4566 | 21/7431 | 21/7065 | 12/4932 | 17/7382 | 5/1784 | 3/1617 | 1/127 | 4/1087 | 12/5420 | 8/1792 | |
| Chi² = 21.70, P < 0.00001; I2 = 95.4% | Chi² = 15.57, P < 0.0001; I2 = 93.6% | Chi² = 21.15, P = 0.0003; I2 = 81.1% | Chi² = 12.72, P = 0.0004; I2 = 92.1% | ||||||||
| Hand foot syndrome | 1.11 (0.48 to 2.56) | 4.76(3.32 to 6.82) | 3.53 (2.53 to 4.94) | 18.68 (4.15 to 84.10) | 5.86 (4.01 to 8.58) | 0.49 (0.04 to 5.50) | 0.04 (0.00 to 0.75) | - | 0.66 (0.11 to 4.00) | 4.52 (3.03 to 6.75) | 5.93 (2.52 to 13.97) |
| 2/343 | 17/6138 | 18/6094 | 3/387 | 13/5418 | 2/372 | 1/122 | 2/569 | 10/4608 | 6/1311 | ||
| Chi² =9.86 , P = 0.002, I2 = 89.9% | Chi² = 4.48, P = 0.03, I2 = 77.7 % | Chi² = 19.58, P = 0.0002 , I2 = 84.7% | Chi² = 0.32, P = 0.57, I2 = 0% | ||||||||
| Neutropenia/ granulocytopenia | 0.05 (0.04 to 0.07) | 0.24 (0.21 to 0.28) | 0.23 (0.20 to 0.26) | 0.09 (0.07 to 0.11) | 0.21 (0.18 to 0.24) | 0.03 (0.02 to 0.05) | 0.06 (0.04 to 0.09) | - | 0.38 (0.29 to 0.50) | 0.15 (0.13 to 0.18) | 0.59 (0.47 to 0.73) |
| 9/4447 | 21/7347 | 21/6981 | 11/4813 | 16/7228 | 5/1784 | 3/1625 | 5/1157 | 13/5418 | 7/1710 | ||
| Chi² = 102.73, P < 0.00001; I2 = 99.0% | Chi² = 56.29, P < 0.00001; I2 = 98.2% | Chi² = 112.47, P < 0.00001; I2 = 97.3% | Chi² = 91.66, P < 0.00001; I2 = 98.9% | ||||||||
| Febrile neutropenia | 0.05 (0.02 to 0.11) | 0.49 (0.36 to 0.66) | 0.50 (0.35 to 0.71) | 0.13 (0.08 to 0.21) | 0.42 (0.30 to 0.58) | 0.03 (0.01 to 0.11) | 0.08 (0.03 to 0.21) | - | 1.82 (0.67 to 4.92) | 0.26 (0.16 to 0.40) | 1.20 (0.75 to 1.93) |
| 5/3254 | 14/6153 | 13/5624 | 7/3783 | 11/5419 | 3/1476 | 2/1485 | 3/1027 | 8/4492 | 6/1661 | ||
| Chi² = 27.62, P < 0.00001; I2 = 96.4% | Chi² = 20.33, P < 0.00001; I2 = 95.1% | Chi² = 33.08, P < 0.00001; I2 = 90.9% | Chi² = 21.90, P < 0.00001; I2 = 95.4% | ||||||||
| Vomiting | 1.11 (0.83 to 1.47) | 1.22 (1.00 to 1.50) | 1.21 (0.97 to 1.49) | 1.15 (0.89 to 1.49) | 1.25 (1.02 to 1.52) | 1.35 (0.97 to 1.87) | 0.68 (0.36 to 1.31) | - | 0.32 (0.10 to 1.07) | 1.12 (0.90 to 1.40) | 1.85 (1.13 to 3.01) |
| 7/3312 | 17/6216 | 18/6172 | 8/3356 | 13/6029 | 5/1784 | 2/1086 | 3/629 | 10/4959 | 6/1038 | ||
| Chi² = 0.32, P = 0.57; I2 = 0 | Chi² = 0.07, P = 0.79; I2 = 0% | Chi² = 8.08, P = 0.04; I2 = 62.9% | Chi² = 3.35, P = 0.07; I2 = 70.2% | ||||||||
| Nausea | 1.08 (0.81 to 1.44) | 1.20 (0.99 to 1.45) | 1.18 (0.96 to 1.45) | 1.13 (0.87 to 1.46) | 1.31 (1.07 to 1.61) | 1.35 (0.96 to 1.89) | 0.51 (0.28 to 0.91) | - | 0.56 (0.28 to 1.10) | 1.16 (0.93 to 1.44) | 1.29 (0.85 to 1.94) |
| 6/2719 | 20/7077 | 21/7033 | 7/2763 | 13/5769 | 5/1784 | 2/1086 | 5/1157 | 11/5066 | 8/1792 | ||
| Chi² = 0.35, P = 0.55; I2 = 0% | Chi² = 0.08, P = 0.78; I2 = 0% | Chi² = 14.23, P = 0.003; I2 = 78.9% | Chi² = 0.20, P = 0.65; I2 = 0% | ||||||||
| Stomatitis | 0.13 (0.09 to 0.19) | 0.73 (0.49 to 1.07) | 0.65 (0.44 to 0.96) | 0.14 (0.09 to 0.20) | 0.26 (0.18 to 0.37) | 0.15 (0.09 to 0.25) | 0.09 (0.00 to 1.65) | 0.09 (0.01 to 0.70) | 4.45 (1.38 to 14.31) | 0.59 (0.38 to 0.92) | 2.56 (0.80 to 8.24) |
| 8/3071 | 14/5647 | 14/5668 | 7/3050 | 10/5598 | 5/1784 | 1/122 | 1/127 | 4/1087 | 9/4731 | 4/697 | |
| Chi² = 39.19, P < 0.00001; I2 = 97.4% | Chi² = 32.07, P < 0.00001; I2 = 96.9% | Chi² = 28.83, P < 0.00001; I2 = 86.1% | Chi² = 5.27, P = 0.02; I2 =81.0% | ||||||||
| Mucositis | 0.08 (0.05 to 0.13) | 1.17 (0.62 to 2.21) | 1.17 (0.62 to 2.21) | 0.08 (0.05 to 0.13) | 0.63 (0.24 to 1.64) | 0.10 (0.05 to 0.18) | 0.10 (0.05 to 0.19) | - | 7.16 (1.29 to 39.88) | 0.75 (0.32 to 1.77) | 2.12 (0.77 to 5.89) |
| 4/2670 | 8/2292 | 8/2292 | 4/2670 | 3/940 | 4/1547 | 2/1495 | 3/980 | 3/1142 | 5/1150 | ||
| Chi² = 43.07, P < 0.00001; I2 = 97.7% | Chi² = 43.07, P < 0.00001; I2 = 97.7% | Chi² = 31.60, P < 0.00001; I2 = 90.5% | Chi² = 2.33, P = 0.13; I2 = 57.1% | ||||||||
| Hyperbilirubinemia | 1.83 (0.96 to 3.48) | 1.34 (0.62 to 2.90) | 1.34 (0.62 to 2.90) | 1.83 (0.96 to 3.48) | 1.51 (0.87 to 2.61) | 0.99 (0.20 to 5.00) | Not estimable | - | 4.42 (0.74 to 26.40) | 1.26 (0.57 to 2.81) | 3.10 (0.12 to 79.23) |
| 3/1311 | 6/1388 | 7/1510 | 2/1189 | 4/1646 | 2/372 | 1/122 | 2/559 | 4/1257 | 2/131 | ||
| Chi² = 0.37, P = 0.54, I2 = 0% | Chi² = 0.37, P = 0.54, I2 = 0% | Chi² = 1.62, P = 0.45, I2 = 0% | Chi² = 0.28, P = 0.60, I2 = | ||||||||
| Any | 1.09 (0.84 to 1.42) | 0.78 (0.68 to 0.89) | 0.77 (0.68 to 0.88) | 1.24 (0.93 to 1.65) | 0.85 (0.75 to 0.96) | 0.69 (0.44 to 1.10) | 0.54 (0.26 to 1.11) | - | 1.26 (0.58 to 2.74) | 0.65 (0.55 to 0.75) | 1.16 (0.88 to 1.51) |
| 3/936 | 12/4500 | 13/4663 | 3/773 | 10/4835 | 2/372 | 1/122 | 1/107 | 6/3385 | 5/896 | ||
| Chi² = 8.71, P = 0.003; I2 = 88.5% | Chi² = 13.55, P = 0.0002; I2 = 92.6% | Chi² = 3.14, P = 0.37; I2 = 4.4% | Chi² = 13.55, P = 0.0002; I2 = 92.6% | ||||||||
| AE: adverse event CI: confidence interval IV: intravenous UFT: tegafur/uracil 5‐FU: 5‐fluorouracil | |||||||||||
Appendix 13. DFS: Other information for studies where the outcome was suitable for meta‐analysis
| Treatment setting |
Study |
Chemotherapy arm |
Survival rate, % (95% CI) |
|
| 3‐year | 5‐year | |||
| Neoadjuvant | Rectal | |||
| Allegra 2015 | Capecitabine ± oxaliplatin | 66.7 | ||
| 5‐FU ± oxaliplatin | 66.4 | |||
| Neoadjuvant/Adjuvant | Rectal | |||
|
Hofheinz 2012 |
All (Adjuvant/Neoadjuvant) | |||
| Capecitabine | 75 (68 to 81), P = 0.07 | 68 (60 to 74) | ||
| Fluorouracil | 67 (59 to 73) | 54 (45 to 62) | ||
| Adjuvant | ||||
| Capecitabine | 78 (69 to 85) | |||
| Fluorouracil | 69 (59 to 77) | |||
| Neoadjuvant | ||||
| Capecitabine | 71 (60 to 80) | |||
| Fluorouracil | 63 (51 to 73) | |||
| Adjuvant | Colon | |||
|
De Gramont 2012 (Stage III) |
BEV‐XELOX | 75 (72 to 78) | ||
| BEV‐FOLFOX4 | 73 (71 to 76) | |||
|
Lembersky 2006 |
UFT/LV | 74.5 | 67.0 | |
| FU/LV | 74.5 | 68.2 | ||
|
Shimada 2014 |
UFT/LV | 77.8 | 73.6 | |
| 5‐FU/LV | 79.3 | 74.3 | ||
|
Twelves 2012 |
Capecitabine | 64.2 | 60.8 | |
| 5‐FU/FA | 60.6, P = 0.12 | 56.7 | ||
| Colorectal | ||||
| Pectasides 2015 | CAPOX | 79.5 (75.9 to 83.1) | ||
| mFOLFOX6 | 79.8 (76.5 to 83.4), P = 0.784 | |||
| DFS: disease‐free survival CI: confidence interval 5‐FU / FU: 5‐fluorouracil BEV: bevacizumab UFT: tegafur/uracil LV: leucovorin FA: folinic acid | ||||
Appendix 14. OS (curative intent studies): Other information for studies for which the outcome was suitable for meta‐analysis
| Treatment setting | Study | Chemotherapy arm | Survival rate, % (95% CI) |
| Neoadjuvant | Rectal | ||
| Allegra 2015 | Capecitabine ± oxaliplatin | 5 years: 80.8 | |
| 5‐FU ± oxaliplatin | 5 years: 79.9 | ||
| Neoadjuvant/Adjuvant | Rectal | ||
| Hofheinz 2012 | All (Adjuvant/Neoadjuvant) | ||
| Capecitabine | 3 years: 87 (81 to 91) | ||
| Fluorouracil | 3 years: 83 (77 to 88) | ||
| Capecitabine | 5 years: 76 (67 to 82) | ||
| Fluorouracil | 5 years: 67 (58 to 74) | ||
| Capecitabine | 7 years: 71 (60 to 79) | ||
| Fluorouracil | 7 years: 58 (47 to 67) | ||
| Adjuvant | Colon | ||
|
De Gramont 2012 (Stage III) |
BEV‐XELOX | 5 years: 82 (80 to 85) | |
| BEV‐FOLFOX4 | 5 years: 81 (78 to 83) | ||
|
Lembersky 2006 |
UFT/LV | 5 years: 88.4 | |
| FU/LV | 5 years: 87.0 | ||
| UFT/LV | 5 years: 69.6 | ||
| FU/LV | 5 years: 71.5 | ||
| UFT/LV | 5 years: 78.5 | ||
| FU/LV | 5 years: 78.7 | ||
| Shimada 2014 | UFT + LV | 3 years: 93.9 | |
| 5‐FU + l‐LV | 3 years: 94.5 | ||
| UFT + LV | 5 years: 87.5 | ||
| 5‐FU + l‐LV | 5 years: 88.4 | ||
| Twelves 2012 | Capecitabine | 3 years: 81.3 | |
| 5‐FU/FA | 3 years: 77.6, P = 0.05 | ||
| Capecitabine | 5 years: 71.4 | ||
| 5‐FU/FA | 5 years: 68.4 | ||
| Colorectal | |||
| Pectasides 2015 | CAPOX | 3 years: 86.9 (83.4 to 89.9) | |
| mFOLFOX6 | 3 years: 87.2 (84.1 to 91.1), P = 0.844 | ||
| OS: overall survival CI: confidence interval 5‐FU/ FU: 5‐fluorouracil BEV: bevacizumab UFT: tegafur/uracil LV: leucovorin FA: folinic acid | |||
Appendix 15. PFS: Other information for studies for which the outcome was suitable for meta‐analysis
| Study | Line of chemotherapy | Chemotherapy Arm | Median (95% CI) | Survival rate, % (95% CI) |
|
Bajetta 1996 |
First |
Oral 5‐dFUR | 4 m (1 to 23) | |
| IV 5‐dFUR | 7 m (1 to 24) | |||
| Cassidy 2011a | First |
XELOX/XELOX‐placebo/XELOX‐BEV (ITT) | 8.0 m | |
| FOLFOX‐4/FOLFOX‐4‐placebo/FOLFOX‐4‐BEV (ITT) | 8.5 m | |||
| XELOX/XELOX‐placebo/XELOX‐BEV(EPP) | 7.9 m | |||
| FOLFOX‐4/FOLFOX‐4‐placebo/FOLFOX‐4‐BEV (EPP) | 8.5 m | |||
| XELOX‐BEV (ITT) | 9.3 m | |||
| FOLFOX‐4‐BEV (ITT) | 9.4 m | |||
| XELOX (ITT) | 7.3 m | |||
| FOLFOX‐4 (ITT) | 7.7 m | |||
|
Comella 2009 |
First |
OXXEL | 6.6 m (6.0 to 7.0) | |
| OXAFAFU | 6.5 m (5.4 to 7.6) | |||
|
Douillard 2014 |
First |
UFOX + Cetuximab | 6.6 m (5.6 to 7.2) | |
| FOLFOX4 + Cetuximab | 8.2 m (7.5 to 9.2), P (log rank) = 0.0048 |
|||
| Ducreux 2011 | First | XELOX (ITT) | 8.8 m | |
| FOLFOX‐6 (ITT) | 9.3 m | |||
| XELOX (per protocol) | 8.9 m | |||
| FOLFOX‐6 (per protocol) | 9.3 m | |||
|
Ducreux 2013 |
First |
XELIRI + BEV | 9 m (8 to 10) | 6 m: 82 (71 to 90) |
| FOLFIRI + BEV | 9 m (8 to 10) | 6 m: 85 (75 to 92) | ||
| XELIRI+ BEV | 12 m: 25 (16 to 36) | |||
| FOLFIRI + BEV | 12 m: 18 (11 to 28) | |||
|
ECOG E5296 2012 |
First |
Eniluracil/5‐FU | 0.4 y (0.2 to 0.5) | |
| 5‐FU | 0.6 y (0.4 to 0.6), P = 0.021, stratified log‐rank | |||
|
Fuchs 2007 |
First |
CapeIRI + Celecoxib/Placebo | 5.8 m | |
| mIFL + Celecoxib/Placebo | 5.9 m, P = 0.46 (comparison of CapeIRI + Celecoxib/Placebo to mIFL + Celecoxib/Placebo) | |||
| FOLFIRI + Celecoxib/Placebo | 7.6 m, P = 0.015 (comparison of CapeIRI + Celecoxib/Placebo to FOLFIRI + Celecoxib/Placebo) | |||
|
Hoff 2001 |
First |
Capecitabine | 4.3 m (4.1 to 5.1) | |
| 5‐FU/LV | 4.7 m (4.3 to 5.5) | |||
| Kato 2012 | First or second | Sequential IRIS‐BEV | 345 d (312 to 594) | |
| mFOLFIRI‐BEV | 324 d (247 to 475), P = 0.71 | |||
|
Kohne 2008 |
First |
CAPIRI‐Celecoxib/Placebo | 5.9 m (4.4 to 8.9) | 1 y: 22.6 (11.4 to 36.2) |
| FOLFIRI‐Celecoxib/Placebo | 9.6 m (6.9 to 10.9) | 1 y: 29.3 (16.4 to 43.4) | ||
|
Pectasides 2012 |
First |
XELIRI + BEV | 10.2 m (9.0 to 11.5) | |
| FOLFIRI + BEV | 10.8 m (9.7 to 11.8), P = 0.74 | |||
|
Porschen 2007 |
First |
CAPOX | 7.1 m | |
| FUFOX | 8.0 m | |||
|
Rothenberg 2008 |
Second |
XELOX (ITT) | 4.7 m | |
| FOLFOX‐4 (ITT) | 4.8 m | |||
| XELOX (per protocol) | 5.1 m | |||
| FOLFOX‐4 (per protocol) | 5.5 m | |||
|
Schilsky 2002a |
First |
EU/5‐FU | 20 wks (19.1 to 20.9) | |
| 5‐FU/LV | 22.7 wks (18.3 to 24.6), P = 0.0106, log‐rank | |||
|
Seymour 2011 |
First |
Capecitabine | 5.2 m (2.8 to 6.7) | |
| OxCap | 5.8 m (3.3 to 7.4) | |||
| FU | 3.5 m (2.8 to 6.2) | |||
| OxFU | 5.8 m (3.2 to 7.6) | |||
| Shigeta 2016 | First | TEGAFIRI +/‐ BEV | 9.9 m (6.5 to 14.7) | |
| FOLFIRI +/‐ BEV | 10.6 (7.7 to 16.5) | |||
|
Souglakos 2012 |
First |
CAPIRI + BEV | 8.9 m (7.3 to 10.2) | |
| FOLFIRI + BEV | 10.0 m (8.9 to 11.1) | |||
|
Van Cutsem 2001b |
First |
Capecitabine | 5.2 m | |
| 5‐FU/LV | 4.7 m | |||
| Yamada 2013 | First | SOX + BEV | 10.2 m (9.4 to 11.1) | |
| mFOLFOX + BEV | 10.2 m (9.5 to 11.3) | |||
| Yamazaki 2015 | First | SOL | 9.6 m | |
| mFOLFOX6 | 6.9 m | |||
| Yasui 2015 | Second | IRIS | 5.8 m | |
| FOLFIRI | 5.1 m | |||
| PFS: progression‐free survival CI: confidence interval 5‐dFUR: doxifluridine IV: intravenous BEV: bevacizumab ITT: intention‐to‐treat EPP: Expanded Participation Project 5‐FU: 5‐fluorouracil LV: leucovorin | ||||
Appendix 16. TTP: Other information for studies where the outcome was suitable for meta‐analysis
| Study | Line of chemotherapy | Chemotherapy arm | Median (95% CI) | P value |
|
Ahn 2003 |
First |
5‐dFUR + LV | 5.4 m (1 to 18.4) | |
| 5‐FU/LV | 4.7 m (1 to 25.4) | |||
|
Carmichael 2002 |
First |
UFT/LV | 3.4 m (2.6 to 3.8) | |
| 5‐FU/LV | 3.3 m (2.5 to 3.7) | P = 0.591, stratified log‐rank | ||
|
Diaz‐Rubio 2007 |
First |
XELOX | 8.9 m (7.8 to 9.9) | |
| FUOX | 9.5 m (8.1 to 10.8) | |||
|
Douillard 2002 |
First |
UFT/LV | 3.5 m (3.0 to 4.4) | |
| 5‐FU/LV | 3.8 m(3.6 to 5.0) | |||
|
Martoni 2006 |
First |
XELOX | 9 m (8 to 10) | |
| pviFOX | 7 m (5 to 9) | |||
|
Nogue 2005 |
First |
FT/LV | 5.9 m (5.3 to 6.5) | |
| 5‐FU/LV | 6.2 m (5.4 to 6.9) | |||
| TTP: time to progression CI: confidence interval 5‐dFUR: doxifluridine LV: leucovorin 5‐FU: 5‐fluorouracil UFT: tegafur/uracil | ||||
Appendix 17. OS (palliative intent studies): Other information for studies where the outcome was suitable for meta‐analysis
| Study | Line of chemotherapy | Chemotherapy arm | Median (95% CI) | P value | Survival rate, % (95% CI) |
|
Ahn 2003 |
First |
5‐dFUR + LV | 14.9 m (1 to 26.8) | ||
| 5‐FU/LV | 19.5 m (1.9 to 25.4) | ||||
|
Bajetta 1996 |
First |
Oral 5‐dFUR | 10.6 m | ||
| IV 5‐dFUR | 11 m | ||||
|
Carmichael 2002 |
First |
UFT/LV | 12.2 m | ||
| 5‐FU/LV | 10.3 m | P = 0.226, stratified log‐rank | |||
|
Cassidy 2011a |
First |
XELOX/XELOX‐placebo/XELOX‐BEV | 19.8 m | ||
| FOLFOX‐4/FOLFOX‐4‐placebo/FOLFOX‐4‐BEV | 19.5 m | ||||
| XELOX/XELOX‐placebo | 19.0 m | ||||
| FOLFOX‐4/FOLFOX‐4‐placebo | 18.9 m | ||||
| XELOX‐BEV | 21.6 m | ||||
| FOLFOX‐4‐BEV | 21.0 m | ||||
| XELOX | 18.8 m | ||||
| FOLFOX‐4 | 17.7 m | ||||
|
Comella 2009 |
First |
OXXEL | 16.0 m (11.2 to 20.2) | ||
| OXAFAFU | 17.1 m (13.8 to 20.4) | ||||
|
Diaz‐Rubio 2007 |
First |
XELOX | 18.1 m (15.5 to 20.4) | ||
| FUOX | 20.8 m (16.6 to 25.0) | ||||
|
Douillard 2002 |
First |
UFT/LV | 12.4 m (11.2 to 13.6) | ||
| 5‐FU/LV | 13.4 m (11.6 to 15.4) | P = 0.630, stratified log‐rank | |||
| Douillard 2014 | First | UFOX + Cetuximab | 16.8 m (13.9 to 18.5) | ||
| FOLFOX4 + Cetuximab | 18.4 m (15.3 to 20.9) | ||||
| Ducreux 2011 | First | XELOX (ITT) | 19.9 m | ||
| FOLFOX‐6 (ITT) | 20.5 m | ||||
| XELOX (per protocol) | 20.1 m | ||||
| FOLFOX‐6 (per protocol) | 18.9 m | ||||
|
Ducreux 2013 |
First |
XELIRI + BEV | 23 m (21 to 27) | 1 y: 87 (78 to 93) | |
| FOLFIRI + BEV | 23 m (21 to 32) | 1 y: 85 (75 to 91) | |||
| XELIRI + BEV | 2 y: 49 (37 to 60) | ||||
| FOLFIRI + BEV | 2 y: 48 (37 to 59) | ||||
|
ECOG E5296 2012 |
First |
Eniluracil/5‐FU | 1.0 yr (0.6 to 1.3) | ||
| 5‐FU | 1.5 y (0.9 to 1.8) | P = 0.17, stratified log‐rank | |||
|
Fuchs 2007 |
First |
CapeIRI + Celecoxib/Placebo | 18.9 m | ||
| mIFL + Celecoxib/Placebo | 17.6 m | ||||
| FOLFIRI + Celecoxib/Placebo | 23.1 m | ||||
| Douillard 2014 | First |
UFOX + Cetuximab | 16.8 m (13.9 to 18.5) | P = 0.86, log‐rank | |
| FOLFOX4 + Cetuximab | 18.4 m (15.3 to 20.9) | ||||
|
Hochster TREE‐1 2008 |
First |
CapeOx | 17.2 m (12.5 to 22.3) | ||
| bFOL | 17.9 m (11.5 to 24.6) | ||||
| mFOLFOX6 | 19.2 m (14.2 to 24.9) | ||||
|
Hochster TREE‐2 2008 |
First |
CapeOx + BEV | 24.6 m (21.3 to 31.6) | ||
| bFOL + BEV | 20.4 m (18.4 to 25.3) | ||||
| mFOLFOX6 + BEV | 26.1 m (18.0 to NE) | ||||
|
Hoff 2001 |
First |
Capecitabine | 12.5 m (10.5 to 14.2) | ||
| 5‐FU/LV | 13.3 m (12.0‐14.6) | ||||
|
Kohne 2008 |
First |
FOLFIRI‐Celecoxib/Placebo | 19.9 m (18.9, NR) | 1 y: 84.9 (69.4 to 92.9) | |
| CAPIRI‐Celecoxib/Placebo | 14.75 m (10.7 to 18.3) | 1 y: 53.5 (36.0 to 68.2) | |||
|
Nogue 2005 |
First |
FT/LV | 12.4 m (10.3 to 14.5) | ||
| 5‐FU/LV | 12.2 m (8.9 to 15.7) | ||||
|
Pectasides 2012 |
First |
XELIRI + BEV | 20.0 m (15.4 to 24.6) | P = 0.099 | |
| FOLFIRI + BEV | 25.3 m (22.1 to 28.6) | ||||
|
Porschen 2007 |
First |
CAPOX | 16.8 m | ||
| FUFOX | 18.8 m | ||||
|
Rothenberg 2008 |
Second |
XELOX (ITT) | 11.9 m | ||
| FOLFOX‐4 (ITT) | 12.5 m | ||||
| XELOX (EPP) | 12.9 m | ||||
| FOLFOX‐4 (EPP) | 13.2 m | ||||
|
Schilsky 2002a |
First |
EU/5‐FU | 13.3 m (12.0 to 15.1) | ||
| 5‐FU/LV | 14.5 m (12.8 to 16.2) | P = 0.3135, log rank | |||
|
Seymour 2011 |
First |
Capecitabine | 11.0 m (5.4 to 18.0) | ||
| OxCap | 12.4 m (5.8 to 18.0) | ||||
| FU | 10.1 m (5.1 to 17.3) | ||||
| OxFU | 10.7 m (5.7 to 17.2) | ||||
| Shigeta 2016 | First | TEGAFIRI +/‐ BEV | 26.7 m (20.4 to 31.0) | ||
| FOLFIRI +/‐ BEV | 27.7 m (20.0 to 35.0) | ||||
|
Souglakos 2012 |
First |
CAPIRI + BEV | 27.5 m (22.6 to 32.3) | ||
| FOLFIRI + BEV | 25.7 m (23.0 to 28.4) | ||||
|
Van Cutsem 2001a |
First |
Eniluracil/5‐FU | 47.4 wks | ||
| 5‐FU/LV | 63.7 wks | ||||
|
Van Cutsem 2001b |
First |
Capecitabine | 13.2 m | P = 0.33, log‐rank | |
| 5‐FU/LV | 12.1 m | ||||
| Yamada 2013 | First | SOX + BEV | 29.6 m (25.8 to NE) | ||
| mFOLFOX + BEV | 30.9 m (28.6 to 33.1) | ||||
| Yamazaki 2015 | First | SOL | 29.9 m | ||
| mFOLFOX6 | 25.9 m | ||||
| Yasui 2015 | Second | IRIS | 17.8 m | ||
| FOLFIRI | 17.4 m | ||||
| OS: overall survival CI: confidence interval 5‐dFUR: doxifluridine 5‐FU: 5‐fluorouracil LV: leucovorin IV: intravenous UFT: tegafur/uracil BEV: bevacizumab ITT: intention‐to‐treat NE: not estimable NR: not reached EPP: Expanded Participation Project EU: eniluracil | |||||
Appendix 18. Key historical studies used to estimate the activity of the active control for disease‐free survival
NCCTG, ECOG‐NCCTG/INT, SWOG‐INT0035, Siena, NCIC‐CTG, FFCD, and GIVIO studies in Gill 2004
Data and analyses
Comparison 1. Disease‐free survival.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Disease‐free survival | 7 | 8903 | Hazard Ratio (Fixed, 95% CI) | 0.93 [0.87, 1.00] |
| 2 Disease‐free survival with subgroup analysis ‐ Chemotherapy vs chemo‐radiotherapy | 7 | 8903 | Hazard Ratio (Fixed, 95% CI) | 0.93 [0.87, 1.00] |
| 2.1 Chemotherapy | 5 | 6944 | Hazard Ratio (Fixed, 95% CI) | 0.94 [0.87, 1.02] |
| 2.2 Chemo‐radiotherapy | 2 | 1959 | Hazard Ratio (Fixed, 95% CI) | 0.91 [0.78, 1.05] |
| 3 Disease‐free survival with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine | 6 | 8511 | Hazard Ratio (Fixed, 95% CI) | 0.95 [0.88, 1.02] |
| 3.1 Infusional intravenous fluoropyrimidine | 3 | 3881 | Hazard Ratio (Fixed, 95% CI) | 0.96 [0.85, 1.08] |
| 3.2 Bolus intravenous fluoropyrimidine | 3 | 4630 | Hazard Ratio (Fixed, 95% CI) | 0.94 [0.86, 1.04] |
| 4 Disease‐free survival with subgroup analysis ‐ Oral fluoropyrimidine backbone | 7 | 8903 | Hazard Ratio (Fixed, 95% CI) | 0.93 [0.87, 1.00] |
| 4.1 Capecitabine | 5 | 6260 | Hazard Ratio (Fixed, 95% CI) | 0.91 [0.83, 0.99] |
| 4.2 UFT/Ftorafur | 2 | 2643 | Hazard Ratio (Fixed, 95% CI) | 1.01 [0.88, 1.15] |
Comparison 2. Overall survival (curative intent studies).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival (curative intent studies) | 7 | 8902 | Hazard Ratio (Fixed, 95% CI) | 0.92 [0.84, 1.00] |
| 2 Overall survival (curative intent studies) with subgroup analysis ‐ Chemotherapy vs chemo‐radiotherapy | 7 | 8902 | Hazard Ratio (Fixed, 95% CI) | 0.92 [0.84, 1.00] |
| 2.1 Chemotherapy | 5 | 6943 | Hazard Ratio (Fixed, 95% CI) | 0.93 [0.84, 1.03] |
| 2.2 Chemotherapy with radiotherapy | 2 | 1959 | Hazard Ratio (Fixed, 95% CI) | 0.86 [0.70, 1.06] |
| 3 Overall survival (curative intent studies) with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine | 6 | 8510 | Hazard Ratio (Fixed, 95% CI) | 0.93 [0.85, 1.02] |
| 3.1 Infusional intravenous fluoropyrimidine | 3 | 3880 | Hazard Ratio (Fixed, 95% CI) | 0.94 [0.80, 1.09] |
| 3.2 Bolus intravenous fluoropyrimidine | 3 | 4630 | Hazard Ratio (Fixed, 95% CI) | 0.93 [0.83, 1.05] |
| 4 Overall survival (curative intent studies) with subgroup analysis ‐ Oral fluoropyrimidine backbone | 7 | 8902 | Hazard Ratio (Fixed, 95% CI) | 0.92 [0.84, 1.00] |
| 4.1 Capecitabine | 5 | 6259 | Hazard Ratio (Fixed, 95% CI) | 0.88 [0.79, 0.98] |
| 4.2 UFT/Ftorafur | 2 | 2643 | Hazard Ratio (Fixed, 95% CI) | 1.03 [0.86, 1.22] |
Comparison 3. Grade ≥ 3 adverse events (curative intent studies).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Grade ≥ 3 diarrhoea (curative intent studies) | 9 | 9551 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.99, 1.25] |
| 2 Grade ≥ 3 diarrhoea (curative intent studies) with subgroup analysis ‐ Chemotherapy vs chemo‐radiotherapy | 9 | 9551 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.99, 1.25] |
| 2.1 Chemotherapy | 5 | 7274 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.95, 1.23] |
| 2.2 Chemo‐radiotherapy | 4 | 2277 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.28 [0.98, 1.66] |
| 3 Grade ≥ 3 diarrhoea (curative intent studies) with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine | 8 | 9159 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.97, 1.23] |
| 3.1 Infusional intravenous fluoropyrimidine | 3 | 4255 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.27 [1.06, 1.53] |
| 3.2 Bolus intravenous fluoropyrimidine | 5 | 4904 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.84, 1.14] |
| 4 Grade ≥ 3 diarrhoea (curative intent studies) with subgroup analysis ‐ Oral fluoropyrimidine backbone | 9 | 9551 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.99, 1.25] |
| 4.1 Capecitabine | 5 | 6616 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.99, 1.33] |
| 4.2 UFT/Ftorafur | 3 | 2769 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.83, 1.21] |
| 4.3 Doxifluridine | 1 | 166 | Odds Ratio (M‐H, Fixed, 95% CI) | 32.14 [1.89, 545.41] |
| 5 Grade ≥ 3 hand foot syndrome (curative intent studies) | 5 | 5731 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.59 [2.97, 7.10] |
| 6 Grade ≥ 3 neutropenia/granulocytopenia (curative intent studies) | 7 | 8707 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.11, 0.16] |
| 7 Grade ≥ 3 febrile neutropenia (curative intent studies) | 4 | 2925 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.18, 1.90] |
| 8 Grade ≥ 3 vomiting (curative intent studies) | 8 | 9385 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.83, 1.34] |
| 9 Grade ≥ 3 nausea (curative intent studies) | 7 | 9233 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.21 [0.97, 1.51] |
| 10 Grade ≥ 3 stomatitis (curative intent studies) | 5 | 4212 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.14, 0.30] |
| 11 Grade ≥ 3 mucositis (curative intent studies) | 4 | 2233 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.25, 1.62] |
| 12 Grade ≥ 3 hyperbilirubinaemia (curative intent studies) | 3 | 2757 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.52, 5.38] |
| 13 Any grade ≥ 3 adverse events (curative intent studies) | 5 | 7741 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.74, 0.90] |
Comparison 4. Progression‐free survival.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Progression‐free survival | 23 | 9927 | Hazard Ratio (Fixed, 95% CI) | 1.06 [1.02, 1.11] |
| 2 Progression‐free survival with subgroup analysis ‐ Single‐agent vs combination therapy | 22 | 9468 | Hazard Ratio (Fixed, 95% CI) | 1.07 [1.03, 1.11] |
| 2.1 Single agent | 6 | 2955 | Hazard Ratio (Fixed, 95% CI) | 1.12 [1.04, 1.21] |
| 2.2 Combination therapy | 16 | 6513 | Hazard Ratio (Fixed, 95% CI) | 1.05 [1.00, 1.10] |
| 3 Progression‐free survival with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine | 23 | 9927 | Hazard Ratio (Fixed, 95% CI) | 1.06 [1.02, 1.11] |
| 3.1 Infusional intravenous fluoropyrimidine | 17 | 6560 | Hazard Ratio (Fixed, 95% CI) | 1.05 [1.00, 1.10] |
| 3.2 Bolus intravenous fluoropyrimidine | 7 | 3367 | Hazard Ratio (Fixed, 95% CI) | 1.10 [1.03, 1.19] |
| 4 Progression‐free survival with subgroup analysis ‐ Oral fluoropyrimidine backbone | 23 | 9927 | Hazard Ratio (Fixed, 95% CI) | 1.06 [1.02, 1.11] |
| 4.1 Capecitabine | 13 | 6703 | Hazard Ratio (Fixed, 95% CI) | 1.03 [0.98, 1.08] |
| 4.2 UFT/Ftorafur | 2 | 374 | Hazard Ratio (Fixed, 95% CI) | 1.36 [1.07, 1.73] |
| 4.3 Eniluracil + oral 5‐FU | 3 | 1618 | Hazard Ratio (Fixed, 95% CI) | 1.22 [1.10, 1.36] |
| 4.4 Doxifluridine | 1 | 130 | Hazard Ratio (Fixed, 95% CI) | 1.18 [0.79, 1.74] |
| 4.5 S‐1 | 4 | 1102 | Hazard Ratio (Fixed, 95% CI) | 1.02 [0.89, 1.16] |
| 5 Progression‐free survival for combination therapy with subgroup analysis ‐ Oxaliplatin‐based vs irinotecan‐based | 16 | 6513 | Hazard Ratio (Fixed, 95% CI) | 1.05 [1.00, 1.10] |
| 5.1 Oxaliplatin‐based | 8 | 4677 | Hazard Ratio (Fixed, 95% CI) | 1.06 [0.99, 1.13] |
| 5.2 Irinotecan‐based | 8 | 1836 | Hazard Ratio (Fixed, 95% CI) | 1.04 [0.97, 1.11] |
| 6 Progression‐free survival for combination therapy with subgroup analysis ‐ with Bev vs no Bev | 14 | 6139 | Hazard Ratio (Fixed, 95% CI) | 1.03 [0.98, 1.08] |
| 6.1 With Bevacizumab | 6 | 2033 | Hazard Ratio (Fixed, 95% CI) | 1.00 [0.94, 1.07] |
| 6.2 No Bevacizumab | 9 | 4106 | Hazard Ratio (Fixed, 95% CI) | 1.06 [0.99, 1.13] |
Comparison 5. Overall survival (palliative intent studies).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival (palliative intent studies) | 29 | 12079 | Hazard Ratio (Fixed, 95% CI) | 1.02 [0.99, 1.05] |
| 2 Overall survival (palliative intent studies) with subgroup analysis ‐ Single‐agent vs combination therapy | 28 | 11620 | Hazard Ratio (Fixed, 95% CI) | 1.02 [0.99, 1.05] |
| 2.1 Single agent | 10 | 4465 | Hazard Ratio (Fixed, 95% CI) | 1.02 [0.99, 1.07] |
| 2.2 Combination therapy | 18 | 7155 | Hazard Ratio (Fixed, 95% CI) | 1.00 [0.95, 1.06] |
| 3 Overall survival (palliative intent studies) with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine | 29 | 12079 | Hazard Ratio (Fixed, 95% CI) | 1.02 [0.99, 1.05] |
| 3.1 Infusional intravenous fluoropyrimidine | 19 | 7022 | Hazard Ratio (Fixed, 95% CI) | 1.01 [0.96, 1.06] |
| 3.2 Bolus intravenous fluoropyrimidine | 13 | 5057 | Hazard Ratio (Fixed, 95% CI) | 1.02 [0.98, 1.06] |
| 4 Overall survival (palliative intent studies) with subgroup analysis ‐ Oral fluoropyrimidine backbone | 29 | 12079 | Hazard Ratio (Fixed, 95% CI) | 1.02 [0.99, 1.05] |
| 4.1 Capecitabine | 16 | 7405 | Hazard Ratio (Fixed, 95% CI) | 0.99 [0.95, 1.04] |
| 4.2 UFT/Ftorafur | 5 | 1807 | Hazard Ratio (Fixed, 95% CI) | 1.02 [0.97, 1.06] |
| 4.3 Eniluracil + oral 5‐FU | 3 | 1618 | Hazard Ratio (Fixed, 95% CI) | 1.20 [1.07, 1.36] |
| 4.4 Doxifluridine | 2 | 207 | Hazard Ratio (Fixed, 95% CI) | 0.99 [0.65, 1.50] |
| 4.5 S‐1 | 3 | 1042 | Hazard Ratio (Fixed, 95% CI) | 0.95 [0.81, 1.11] |
| 5 Overall survival (palliative intent studies) for combination therapy with subgroup analysis ‐ Oxaliplatin‐based vs irinotecan‐based | 18 | 7155 | Hazard Ratio (Fixed, 95% CI) | 1.00 [0.95, 1.06] |
| 5.1 Oxaliplatin‐based | 11 | 5379 | Hazard Ratio (Fixed, 95% CI) | 1.00 [0.94, 1.07] |
| 5.2 Irinotecan‐based | 7 | 1776 | Hazard Ratio (Fixed, 95% CI) | 1.01 [0.92, 1.10] |
Comparison 6. Time to progression.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Time to progression | 6 | 1970 | Hazard Ratio (Fixed, 95% CI) | 1.07 [1.01, 1.14] |
Comparison 7. Objective response rate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ORR | 32 | 11115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.90, 1.06] |
Comparison 8. Grade ≥ 3 adverse events (palliative intent studies).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Grade ≥ 3 diarrhoea (palliative intent studies) | 30 | 11997 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.66 [1.50, 1.84] |
| 2 Grade ≥ 3 diarrhoea (palliative intent studies) with subgroup analysis ‐ Single‐agent vs combination therapy | 30 | 11997 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.66 [1.50, 1.84] |
| 2.1 Single agent | 10 | 4566 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.22 [1.04, 1.44] |
| 2.2 Combination therapy | 21 | 7431 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.03 [1.77, 2.32] |
| 3 Grade ≥ 3 diarrhea (palliative intent studies) with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine | 30 | 11997 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.66 [1.50, 1.84] |
| 3.1 Infusional intravenous fluoropyrimidine | 21 | 7065 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.00 [1.74, 2.30] |
| 3.2 Bolus intravenous fluoropyrimidine | 12 | 4932 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.31 [1.12, 1.53] |
| 4 Grade ≥ 3 diarrhoea (palliative intent studies) with subgroup analysis ‐ Oral fluoropyrimidine backbone | 30 | 11997 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.66 [1.50, 1.84] |
| 4.1 Capecitabine | 17 | 7382 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.76 [1.54, 2.00] |
| 4.2 UFT/Ftorafur | 5 | 1784 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.60 [1.24, 2.06] |
| 4.3 Eniluracil + oral 5‐FU | 3 | 1617 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.79, 1.38] |
| 4.4 Doxifluridine | 1 | 127 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.51 [0.64, 3.56] |
| 4.5 S‐1 | 4 | 1087 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.55 [2.19, 5.76] |
| 5 Grade ≥ 3 diarrhoea (palliative intent studies) with subgroup analysis for combination therapy ‐ Oxaliplatin‐based vs irinotecan‐based | 20 | 7212 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.00 [1.75, 2.29] |
| 5.1 Oxaliplatin‐based | 12 | 5420 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.73 [1.48, 2.02] |
| 5.2 Irinotecan‐based | 8 | 1792 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.05 [2.33, 3.99] |
| 6 Grade ≥ 3 hand foot syndrome (palliative intent studies) | 18 | 6481 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.92 [2.84, 5.43] |
| 7 Grade ≥ 3 hand foot syndrome (palliative intent studies) with subgroup analysis ‐ Single‐agent vs combination therapy | 18 | 6481 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.89 [2.82, 5.37] |
| 7.1 Single agent | 2 | 343 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.48, 2.56] |
| 7.2 Combination therapy | 17 | 6138 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.76 [3.32, 6.82] |
| 8 Grade ≥ 3 hand foot syndrome (palliative intent studies) with subgroup analysis ‐ Infusional vs bolus intravenous fluoropyrimidine | 18 | 6481 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.92 [2.84, 5.43] |
| 8.1 Infusional intravenous fluoropyrimidine | 18 | 6094 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.53 [2.53, 4.94] |
| 8.2 Bolus intravenous fluoropyrimidine | 3 | 387 | Odds Ratio (M‐H, Fixed, 95% CI) | 18.68 [4.15, 84.10] |
| 9 Grade ≥ 3 hand foot syndrome (palliative intent studies) with subgroup analysis ‐ Oral fluoropyrimidine backbone | 18 | 6481 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.92 [2.84, 5.43] |
| 9.1 Capecitabine | 13 | 5418 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.86 [4.01, 8.58] |
| 9.2 UFT/Ftorafur | 2 | 372 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.04, 5.50] |
| 9.3 Eniluracil + oral 5‐FU | 1 | 122 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.04 [0.00, 0.75] |
| 9.4 S‐1 | 2 | 569 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.11, 4.00] |
| 10 Grade ≥ 3 hand foot syndrome (palliative intent studies) with subgroup analysis for combination therapy ‐ Oxaliplatin‐based vs irinotecan‐based | 16 | 5919 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.76 [3.31, 6.83] |
| 10.1 Oxaliplatin‐based | 10 | 4608 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.52 [3.03, 6.75] |
| 10.2 Irinotecan‐based | 6 | 1311 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.93 [2.52, 13.97] |
| 11 Grade ≥ 3 neutropenia/granulocytopenia (palliative intent studies) | 29 | 11794 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.15, 0.18] |
| 12 Grade ≥ 3 febrile neutropenia (palliative intent studies) | 19 | 9407 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.21, 0.36] |
| 13 Grade ≥ 3 vomiting (palliative intent studies) | 23 | 9528 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.18 [1.00, 1.40] |
| 14 Grade ≥ 3 nausea (palliative intent studies) | 25 | 9796 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.99, 1.36] |
| 15 Grade ≥ 3 stomatitis (palliative intent studies) | 21 | 8718 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.20, 0.33] |
| 16 Grade ≥ 3 mucositis (palliative intent studies) | 12 | 4962 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.12, 0.24] |
| 17 Grade ≥ 3 hyperbilirubinaemia (palliative intent studies) | 9 | 2699 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.62 [0.99, 2.64] |
| 18 Any grade ≥ 3 adverse events (palliative intent studies) | 14 | 5436 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.74, 0.94] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ahn 2003.
| Methods | Randomised controlled trial Phase: II Accrual dates: July 1998 to May 2000 |
|
| Participants | No. randomised: 77 Stage/treatment line: Metastatic, first‐line Countries/sites: Not specified, study authors from a South Korean centre Setting: Hospital Characteristics (Group A/B): Metastatic colorectal adenocarcinoma; age ≤ 75 years (median 58/57 years); male (50/79%); PS ECOG ≤ 2 (PS ECOG 0: 11/3%) |
|
| Interventions | Group A: Oral doxifluridine (5‐dFUR) 333 mg/m2 tds + leucovorin 15 mg bd, D1‐7 and D15‐21 q28d (n randomised = 38) Group B: IV bolus 5‐FU 400 mg/m2/d plus leucovorin 20 mg/m2/d D1‐5, q28d (n randomised = 39) “In the presence of objective response or stable disease, each group of patients was treated for a maximum of 12 cycles. In the case of a complete response, 2 additional cycles were given. The treatment was continued until there was progression, unacceptable toxicity, or a patient’s refusal” (patients and methods, paragraph 2, page 99) |
|
| Outcomes | ORR (WHO criteria, 1981) PFS (treated as TTP in this review, based on the definition provided), OS Grade ≥ 3 AEs (WHO toxicity criteria, version not specified) Median follow‐up: 17.0 m (PFS/OS) |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: None declared Declarations of interest: None declared |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | Low risk | Not specified, blinding unlikely (i) ORR/PFS (treated as TTP): Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: This study was not used for the meta‐analysis for these outcomes |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified (i) ORR/PFS (treated as TTP): High Outcome assessment at risk of bias if there was lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: This study was not used for the meta‐analysis for these outcomes |
| Schedule of assessment and follow‐up | Unclear risk | (i) Response (influences ORR/PFS): Low Quote: "All measurable lesions were assessed every three cycles..." (patients and methods, paragraph 3, page 99) Quote: "Each cycle was repeated every four weeks" (patients and methods, paragraph 2, page 99) Therefore, responses were evaluated at the same frequency in both treatment arms (ii) Survival (influences PFS/OS): Unclear Follow‐up duration and assessment frequency for survival events were not specified (iii) Grade ≥ 3 AEs: Low Quote: "Side effects ... were evaluated at the beginning of each cycle" (patients and methods, paragraph 4, page 99) Therefore, grade ≥ 3 AEs were evaluated at the same frequency in both treatment arms |
| Incomplete outcome data (attrition bias) All outcomes | High risk | (i) ORR/PFS (treated as TTP): High 11/38 (29%) 5‐dFUR/LV and 6/39 (15%) 5‐FU/LV patients were not evaluable. Quote: "... 38 were randomly assigned to group A and 39 to group B" (results, paragraph 1, page 99) Quote: "... the evaluable patients were 27 patients in group A and 33 patients in group B, respectively" (results, paragraph 3, page 99) (ii) OS: Low Although not defined in the Methods, censoring was noted in the KM curves (Fig. 1 and 2, page 100). No evidence suggested bias related to censoring (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "The effects of the treatments were analysed using both the intent‐to‐treat principle and per‐protocol analysis that included only evaluable patients" (patients and methods, paragraph 5, page 99) Safety analysis: Not specified |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) ECOG PS: Low (Table 1, page 99) (ii) Median age: Low (Table 1, page 99) (iii) Number of metastatic organs: Low (Table 1, page 99) |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified |
Allegra 2015.
| Methods | Randomised controlled trial Phase: III Design: Factorial (2 × 2), following protocol amendment in October 2005 Accrual dates: July 2004 to August 2010 |
|
| Participants | No. randomised: 1608 Stage/treatment type: Stage II or III rectal cancer, neoadjuvant Countries/sites: USA, Canada (multiple sites) Setting: Hospital Characteristics (group 1/2/3/4/5/6): Unresected stage II or III rectal adenocarcinoma; age > 18 years (patients ≤ 59 years: 59.2/52.7/56.1/61.4/57.1/61.2%); male (68.0/67.8/67.0/68.1/67.8/67.6%); PS 0/1 |
|
| Interventions | Grp 1 (5‐FU (2 Arm), pre‐amendment): 5‐FU 225 mg/m2/d (n randomised = 147) Grp 2 (CAPE (2 Arm), pre‐amendment): capecitabine 825 mg/m2 oral BD (n randomised = 146) Grp 3 (5‐FU (4 Arm), post‐amendment): 5‐FU 225 mg/m2/d 5 days/wk (n randomised = 330) Grp 4 (5‐FU + OX (4 Arm), post‐amendment): 5‐FU 225 mg/m2/d 5 days/wk, oxaliplatin 50 mg/m2 IV weekly (n randomised = 329) Grp 5 (CAPE (4 Arm), post‐amendment): capecitabine 825 mg/m2 oral BD 5 days/wk (n randomised = 326) Grp 6 (CAPE + OX (4 Arm), post‐amendment): capecitabine 825 mg/m2 oral BD 5 days/wk, oxaliplatin 50 mg/m2 IV weekly (n randomised = 330) All participants received neoadjuvant radiotherapy: 180 cGy per day, 5 doses per week for 25 fractions. Minimal boost: 540 cGy (3 days in 180 cGy fractions) for T3 non‐fixed and distal cancers; 1080 cGy (3 days in 360 cGy fractions) for T4 fixed and/or distal cancers All chemotherapy given for the duration of radiotherapy |
|
| Outcomes | Locoregional control at 3 years OS DFS Time to local‐regional recurrence Grade ≥ 3 AEs (NCI CTCAE, version 4.0) No details on median follow‐up |
|
| Study Details | Journal article and abstract | |
| Funding sources and declarations of interest | Funding sources: US National Cancer Institute at the National Institutes of Health, US Department of Health and Human Services, Public Health Service grants Declarations of interest: None declared |
|
| Notes | Following the October 2005 protocol amendment, oxaliplatin was added to form a 2 × 2 factorial design. Daily dose of fluoropyrimidine chemotherapy was unchanged. However, capecitabine and 5‐FU treatment were given 5 days a week (reduced from 7), coinciding with days of planned radiation therapy | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...randomly assigned to treatment groups using the NSABP biased‐coin minimization algorithm" (methods, paragraph 3, page 2) |
| Allocation concealment (selection bias) | Unclear risk | Unclear ‐ not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Not specified, blinding unlikely (i) DFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified (i) DFS: High Outcome assessment at risk of bias if there was lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | Unclear risk | (i) Locoregional recurrence (influences DFS): Low Quote: "Following surgery, MRI or CT scans were required every 12 months for two years, and proctoscopy or sigmoidoscopy annually for five years" (methods, paragraph 2, page 2) (ii) Survival (influences DFS/OS): Unclear It was unclear whether any follow‐up (e.g. clinical reviews) other than that described above was performed to detect survival events, and whether they were the same in both oral and intravenous arms (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) DFS/OS: Low 19/801 participants from the analysis population on the 5‐FU arm had no follow‐up, whereas 9/794 participants on the capecitabine arm had no follow‐up (correspondence with Dr. Carmen Allegra, received 22 July 2016) (ii) Grade ≥ 3 AEs: Low From the analysis population, 21/801 (3%) and 7/794 (1%) participants in the 5‐FU and CAPE arms, respectively, were missing safety outcome data (Table 2, page 6) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low From the analysis population, 5/806 (0.6%) and 8/802 (1%) participants were excluded from the 5‐FU and CAPE arms, respectively, because they were ineligible (Figure 1, page 4) Safety analysis: Unclear ‐ not specified |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Unclear ‐ information not specified (ii) Median/mean age: Unclear ‐ information not specified (iii) TNM stage: Unclear ‐ information not specified |
| Other bias | Unclear risk | Subsequent therapies: Unclear Quote: "...we do not have complete information concerning the type and use of adjuvant therapy in the study patients" (discussion, paragraph 5, page 7) Risk of bias considerations in a factorial study: Unclear Quote: "... no evidence of oxaliplatin‐treatment‐by‐fluoropyrimidine‐treatment interaction (P = .46)" for 3‐year locoregional recurrence (results, paragraph 2, page 4) No tests for interaction were reported for the other outcomes |
Andersen 1987.
| Methods | Randomised controlled trial Phase: Not specified Accrual dates: Not specified |
|
| Participants | No. randomised: 60 Stage/treatment line: Inoperable/advanced/recurrent colorectal cancer, no prior 5‐FU or Ftorafur and at least 4 weeks elapsed from previous chemotherapy (only those with no prior chemotherapy were analysed) Countries/sites: Authors from Denmark Setting: Hospital Characteristics (Arm A/B): Inoperable/advanced/recurrent colorectal cancer; age (median 55/59 years); male (43/37%); PS (WHO) ≤ 3 |
|
| Interventions | Arm I: Oral Ftorafur 1 gm/m2/d D1‐21, q35d (n randomised = 30) Arm II: IV bolus 5‐FU 500 mg/m2/d D1‐5, q35d (n randomised = 30) Treatment continued until intractable toxicity or PD |
|
| Outcomes | ORR (WHO criteria, 1979) OS Median follow‐up: Not specified |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: None declared Declarations of interest: None declared |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | Low risk | Not specified, blinding unlikely (i) ORR: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: Not outcomes for this study |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified. (i) ORR: High Outcome assessment at risk of bias if there was lack of blinding (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: Not outcomes for this study |
| Schedule of assessment and follow‐up | Unclear risk | (i) Response (influences ORR): Unclear ‐ not specified (ii) Survival (influences OS): This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: Not outcomes for this study |
| Incomplete outcome data (attrition bias) All outcomes | High risk | (i) ORR: High 20% of patients in the 5‐FU arm were not evaluable (Table 1, page 434) (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: Not outcomes for this study |
| Incomplete outcome data (ITT analysis) | High risk | Efficacy analysis: High The efficacy analysis population excluded those who refused treatment, had protocol violations, or were lost to follow‐up (10/60) (Table 1, page 434) Safety analysis: The safety analysis population received at least 1 treatment cycle Quote: "They received at least one treatment cycle with 5‐FU or Ftorafur" (material and methods, paragraph 4, page 434) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | i) PS: Unclear ‐ only median and range reported (Table 1, page 434) ii) Median age: Low (Table 1, page 434) iii) No. of involved organs: Unclear ‐ not specified |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not provided |
Bajetta 1996.
| Methods | Randomised controlled trial Phase: II, non‐comparative Accrual dates: April 1993 to September 1994 |
|
| Participants | No. randomised: 130 Stage/treatment line: Metastatic, first‐line (no previous adjuvant chemotherapy) Countries/sites: Italy, 13 institutions Setting: Hospital Characteristics (Arm A/B): Metastatic colorectal adenocarcinoma; age < 80 years (median 61/61 years); male (52/59%); PS ECOG ≤ 2 |
|
| Interventions | Arm A: Oral doxifluridine (5‐dFUR) 750 mg/m2 bd plus oral levo‐leucovorin 25 mg/dose D1‐4, q12d (n randomised = 67) Arm B: IV 5‐dFUR 3000 mg/m2 plus l‐leucovorin 25 mg D1‐5, q21d (n randomised = 63) "It was initially planned to deliver five cycles to the patients in arm A and three cycles to those in arm B. In the case of a complete response (CR), partial response (PR), or stable disease (SD), the patients were to receive an additional 4 and 2 cycles, respectively. In the case of a subsequent CR or PR following the documentation of SD or PR at the first evaluation, further 4 cycles for arm A and 2 cycles for arm B were administered" (patients and methods, paragraph 4, page 2088) |
|
| Outcomes | ORR (WHO criteria, 1981) Grade ≥ 3 AEs (NCI CTC, 1981) Time to treatment failure (TTF, treated as PFS in this review, based on the definition provided) OS Median follow‐up: 10 m (TTF/OS) |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: None declared Declarations of interest: None declared |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...randomly allocated to receive oral or i.v. 5‐dFUR, balanced in blocks of varying sizes" (patients and methods, paragraph 2, page 2089) |
| Allocation concealment (selection bias) | Low risk | Quote: "... the patients were registered at a central randomization office ..." (patients and methods, paragraph 2, page 2089) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Not specified, blinding unlikely (i) ORR/TTF(treated as PFS): Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified. (i) ORR/TTF(treated as PFS): High Outcome assessment at risk of bias if there was lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/TTF (treated as PFS)): Unclear Quote: "It was initially planned to deliver five cycles to the patients in arm A and three cycles to those in arm B. In the case of a complete response (CR), partial response (PR), or stable disease (SD), the patients were to receive an additional 4 and 2 cycles, respectively. In the case of a subsequent CR or PR following the documentation of SD or PR at the first evaluation, further 4 cycles for arm A and 2 cycles for arm B were administered" (patients and methods, paragraph 4, page 2089) Quote: "Oral 5‐dFUR ... was administered ... for 4 days repeated every 12 days (arm A); intravenous 5‐dFUR was administered ... for 5 consecutive days every 21 days (arm B)" (patients and methods, paragraph 3, page 2089) Therefore, the schedule of assessment for both treatment arms up until the second evaluation was similar However, no information was provided on the response evaluation schedule following the second evaluation (ii) Survival (influences TTF(treated as PFS)/OS): Unclear ‐ not specified (iii) Grade ≥ 3 AEs: High Quote: "Side effects were ... evaluated at the beginning of each cycle" (patients and methods, paragraph 8, page 2089) "Oral 5‐dFUR ... was administered ... for 4 days repeated every 12 days (arm A); intravenous 5‐dFUR was administered ... for 5 consecutive days every 21 days (arm B)" (patients and methods, paragraph 3, page 2089) Therefore, safety evaluations occurred more frequently in arm A |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Low The sum of participants who achieved CR, PR, SD, and “Treatment failure” appears to be the same as the number randomised (Table 2, page 2091) (ii) TTF (treated as PFS)/OS: Low Although censoring was not defined in the Methods, censoring was noted in the KM curves (Figure 1 and Figure 2, page 2091). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote "The activity of the treatments was analyzed using both the intent‐to‐treat principle (including the entire group of patients) and standard analysis (which excludes inadequately treated cases)" (statistical analysis, paragraph 1, page 2089) Safety analysis: Analyses included those who received treatment Quote: "As three of the patients randomized to arm B were censored because no treatment was administered, the safety analysis was based on the results derived from 127 subjects" (results, paragraph 7, page 2090) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Unclear ‐ reported as ECOG PS 0‐1 vs 2 (Table 1, page 2090) (ii) Median age: Low (Table 1, page 2090) (iii) No. of involved organs: Unclear ‐ not specified |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not provided |
Carmichael 2002.
| Methods | Randomised controlled trial Phase: III Accrual dates: May 1996 to July 1997 |
|
| Participants | No. randomised: 380 Stage/treatment line: Metastatic, first‐line Countries/sites: Multi‐nation, 47 sites. Europe (n = 291), Canada (n = 55), Australia (n = 24), New Zealand (n = 5), and Israel (n = 5) Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal adenocarcinoma; age > 18 years (median 61/62 years); male (67/64%); PS ECOG ≤ 2 (ECOG 0: 39/33%) |
|
| Interventions | Arm I: Oral UFT 300 mg/m2/d plus leucovorin (LV) 90 mg/d D1‐28, q35d (n randomised = 190) Arm II: IV bolus 5‐FU 425 mg/m2/d plus LV 20 mg/m2/d D1‐5, q35d (n randomised = 190) |
|
| Outcomes | TTP OS ORR (WHO criteria ‐ modified) Grade ≥ 3 AEs (NCI CTC, version not specified) Median follow‐up ‐ Not specified |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: Taiho Pharmaceutical Company, Bristol‐Myers Squibb Declarations of interest: None declared |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Not specified, blinding unlikely (i) ORR/TTP: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/TTP: Unclear Quote: "Efficacy was evaluated locally with data subsequently centrally reviewed" (patients and methods, paragraph 5, page 3619) However, the role of the central review in the reported response data was not described (ii) OS: Low Not specified. Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Not specified. Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | Unclear risk | (i) Response (influences ORR/TTP): Low Quote: "Tumor reassessment ... was repeated after every two cycles, with an additional computed tomography scan at week 15" (patients and methods, paragraph 5, page 3619) Quote: "On both treatment arms, treatment cycles were to be repeated every 35 days" (patients and methods, paragraph 3, page 3618) Therefore, responses were evaluated at the same frequency in both treatment arms (ii) Survival (influences OS): Unclear The schedule of follow‐up after progression was not specified (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Low 1% (2/190) of patients in both arms were not assessed (Table 2, page 3619) (ii) TTP/OS: Low Although not defined in the Methods, censoring was noted from the KM curves for both outcomes (Fig 1 and 2, pages 3621 and 3622). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Low The greatest percentage of outcome data missing for an AE in any arm was 11%; there were similar percentages missing from each arm (Tables 4, 5 and 6, page 3623) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "All efficacy analyses have been presented by treatment arm as randomized ..." (patients and methods, paragraph 7, page 3619) Safety analysis: Quote: "All 373 patients who received at least one dose of study medication were evaluated for safety and were analyzed according to the treatment arm as treated" (results, paragraph 18, page 3622) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available. |
| Similarity of arms at baseline | Unclear risk | (i) PS: Low (Table 1, page 3619) (ii) Median age: Low (Table 1, page 3619) (iii) No. of involved organs: Unclear ‐ not clearly specified. Quote: "The median number of disease sites was two in both treatment arms" (results, paragraph 4, page 3619) |
| Other bias | Low risk | Subsequent therapies: Low Similar percentages of participants and types of chemotherapy were used in the second‐line setting after progression, in each arm Quote: "Secondary chemotherapy was administered to 41% (78 of 190) of patients receiving UFT/LV, and 39% (75 of 190) of patients receiving 5‐FU/LV" (results, paragraph 16, page 3622) Quote: "The most frequently administered secondary chemotherapy was fluoropyrimidines only, in 49% of the 78 UFT/LV patients and in 47% of the 75 5‐FU/LV patients who took secondary chemotherapy, followed by irinotecan only (28% in each arm). In patients receiving secondary chemotherapy, oxaliplatin alone or in combination with irinotecan was given to 13% (10 of 78) of UFT/LV treated patients and 16% (12 of 75) of 5‐FU/LV treated patients" (results, paragraph 16, page 3622) |
Cassidy 2011a.
| Methods | Randomised controlled trial Phase: III Design: Part One ‐ 2‐arm, 1:1 randomisation to XELOX vs FOLFOX4 Part Two ‐ 2 × 2 factorial (protocol amendment to include randomisation to bevacizumab (BEV) or placebo); 1:1:1:1 randomisation to XELOX + placebo, XELOX + BEV, FOLFOX‐4 + placebo, FOLFOX‐4 + BEV Accrual dates: Part One ‐ July 2003 to May 2004; Part Two ‐ February 2004 to February 2005 |
|
| Participants | No. randomised: 2035 Stage/treatment line: Metastatic, first‐line Countries/sites: Multi‐nation; Europe (n = 1048), Canada (n = 343), Oceania (n = 188), US (n = 178), Central/Eastern Asia (n = 163), South America (n = 65), and South Africa (n = 49) Setting: Hospital Characteristics Part One (Arm I/II): Metastatic colorectal adenocarcinoma; age ≥ 18 years (median 61/62 years); male (61/64%); PS ECOG ≤ 1 (PS ECOG 0: 50/51%) Characteristics Part Two (Arm I/II/III/IV): Metastatic colorectal adenocarcinoma; age ≥ 18 years (median 61/61/60/60 years); male (59/61/53/59%); PS ECOG ≤ 1 (PS ECOG 0: 59/59/60/57%) |
|
| Interventions | Part One: Arm I (XELOX): oxaliplatin 130 mg/m2 D1 plus capecitabine 1000 mg/m2 bd for 14 d, q21d (n randomised = 317) Arm II (FOLFOX4): oxaliplatin 85 mg/m2 D1 plus leucovorin 200 mg/m2/d, IV bolus 5‐FU 400 mg/m2/d and 5‐FU 600 mg/m2/d 22‐hour infusion D1‐2, q14d (n randomised = 317) Part Two: Arm I: XELOX + placebo (n randomised = 350) Arm II: XELOX + BEV 7.5 mg/kg D1, q21d (n randomised = 350) Arm III: FOLFOX‐4 + placebo (n randomised = 351) Arm IV: FOLFOX‐4 + BEV 5 mg/kg D1, q14d (n randomised = 350) Treatment continued until PD or for 48 weeks, whichever came first (study treatment phase). Participants who completed the 48‐week treatment phase without PD were eligible to continue treatment until PD in a post‐study treatment phase |
|
| Outcomes | PFS OS ORR (RECIST, version 1.0) TTF Grade ≥ 3 AEs (NCI CTC, version 3) Median follow‐up: PFS 17.7 m (cut‐off date 31 January 2006), OS cut‐off date 31 July 2008 |
|
| Study Details | Journal articles | |
| Funding sources and declarations of interest | Declarations of interest: Roche, Sanofi‐Aventis Funding sources: Roche |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were assigned to treatment using an interactive voice response system" (patients and methods, paragraph 4, page 2007) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote "... open‐label" (patients and methods, paragraph 1, page 2007) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: Low Quote: "Tumor responses were assessed by investigators and also by an independent response review committee" (patients and methods, paragraph 9, page 2007) Similar ORs were obtained for the response rates, which were "Investigator assessed" and "IRC assessed" (Table 2, page 2010) (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | Unclear risk | (i) Response (influences ORR/PFS): Low Quote: "Tumor assessments (computed tomography scan, magnetic resonance imaging) were ... repeated after every two XELOX cycles and every three FOLFOX‐4 cycles (i.e., every sixth week in both arms and at the end of treatment)" (patients and methods, paragraph 9, page 2007) (ii) Survival (influences PFS/OS): Low Quote: "After completion of study treatment, patients were followed every 3 months until PD and/or death" (patients and methods, paragraph 9, page 2007) (iii) Grade ≥ 3 AEs: Unclear ‐ no schedule was provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Unclear ‐ not specified (ii) PFS/OS: Low Quote: "Patients who were not reported as having died at the time of the analysis were censored using the date they were last known to be alive" (Cassidy et al, British Journal of Cancer, 2011: patients and methods, paragraph 9, page 59). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "The intent‐to‐treat (ITT) patient population included all patients who underwent randomization and signed the informed consent form" (patients and methods, paragraph 11, page 2007) Safety analysis: Quote: "All patients receiving at least one dose of study drug were included in the safety analysis" (patients and methods, paragraph 11, page 2007) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available. |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, 2009) (ii) Median age: Low (Table 1, 2009) (iii) No. of involved organs: Low (Table 1, 2009) |
| Other bias | Low risk | Subsequent therapies: Low Quote: "There were no major imbalances between the treatment groups with respect to the use of second‐line therapy ..." (results, paragraph 9, page 2009) Risk of bias considerations in a factorial study: Low For efficacy analysis ‐ Quote: "Both a clinically relevant and statistically significant (P = .7025) treatment interaction was ruled out" (results, paragraph 4, page 2008) For safety analysis ‐ Quote: "The addition of bevacizumab did not alter the similarities and differences in safety profile between XELOX and FOLFOX‐4" (results, paragraph 14, page 2010) |
Comella 2009.
| Methods | Randomised controlled trial Phase: III Accrual dates: May 2004 to April 2007 |
|
| Participants | No. randomised: 322 Stage/treatment line: Metastatic, first‐line Countries/sites: Italy, 23 Southern Italy Cooperative Oncology group (SICOG) centres Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal adenocarcinoma; age ≥ 18 years (median 65/64 years); male (54/66%); PS ECOG ≤ 2 (PS ECOG 0: 60/61%) |
|
| Interventions | Arm I (OXAFAFU): IV oxaliplatin 85 mg/m2, 6S‐leucovorin 250 mg/m2 D1, IV bolus fluorouracil 850 mg/m2 D2, q14d (n randomised = 164) Arm II (OXXEL): IV oxaliplatin 100 mg/m2 D1 plus capecitabine 1000 mg/m2 bd D1‐D11, q14d (n randomised = 158) Treatment continued until PD, unacceptable toxicity, or participant refusal, or a maximum of 12 cycles |
|
| Outcomes | ORR (WHO criteria, 1981) PFS OS Grade ≥ 3 AEs (WHO criteria, 1981) Median follow‐up: 24 months (PFS/OS) |
|
| Study Details | Journal article and abstracts | |
| Funding sources and declarations of interest | Funding sources: SICOG Declarations of interest: No conflicts of interest |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Not specified, blinding unlikely (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified (i) ORR/PFS: High Outcome assessment at risk of bias if there was lack of blinding (ii)OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | Low risk | (i) Response (influences ORR/PFS): Low Quote: "In both arms, cycles were repeated every 2 weeks" (methods, paragraph 5, page 219) Quote: "CT or MRI scan was repeated after every 4 cycles" (methods, paragraph 4, page 219) Therefore, responses were evaluated at the same frequency in both treatment arms Quote: "Response was ... reassessed 8 weeks after the date of their first documentation; only confirmed responses were computed in the activity analysis" (methods, paragraph 4, page 219) (ii) Survival (influences PFS/OS): Low Quote: "After discontinuation of first‐line treatment, patients were followed every 2 months to assess the disease status and survival" (methods, paragraph 6, page 219) (iii) Grade ≥ 3 AEs: Low Quote: "During treatment, WBC count with differential was performed weekly. Biochemistry, symptoms, body weight, and nonhematological toxicity were checked before each cycle" (methods, paragraph 2, page 218‐9) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Low 11% of patients in the OXXEL arm and 10% of patients in the OXAFAFU arm were not assessed (Comella et al, ASCO Gastrointestinal Cancers Symposium, 2008 ‐ slides associated with abstract 344) (ii) PFS/OS: Low It is likely that censoring has occurred (Figures 1 and 2, pages 222 and 223). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "... 322 eligible patients ... were randomized to the OXXEL (158 patients) or OXAFAFU (164 patients) arm" This was the same as the number of participants at risk at t = 0 in Figure 1 (PFS curve) and Figure 2 (OS curve) (pages 222 and 223), as well as the total number of participants described in the ITT response data (Comella et al, ASCO Gastrointestinal Cancers Symposium, 2008 ‐ slides associated with abstract 344) Safety analysis: Not specified |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, page 220) (ii) Median age: Low (Table 1, page 220) (iii) No. of involved organs: Low (Table 1, page 220) |
| Other bias | Low risk | Subsequent therapies: Low Post‐study treatment was similar in both arms (Comella et al, ASCO Gastrointestinal Cancers Symposium, 2008 ‐ slides associated with abstract 344) |
De Gramont 2012.
| Methods | Randomised controlled trial Phase: III Accrual dates: December 2004 to June 2007 |
|
| Participants | No. randomised: 3451 Stage/treatment type: High‐risk stage II or stage III colon cancer, adjuvant Countries/sites: 330 centres in 34 countries (including USA, Europe, Asia, and Australia) Setting: Hospital Characteristics (Arm A/B/C) for ITT population: Adjuvant colon cancer; age ≥ 18 years (median 58/58/58 years); male (57/51/55%); PS ECOG 0‐1 (PS ECOG 0: 86/85/85%) |
|
| Interventions | Arm A (FOLFOX4): D1 oxaliplatin 85 mg/m2, LV 200 mg/m2, IV bolus fluorouracil 400 mg/m2, followed by IV fluorouracil 600mg/m2 22‐hour continuous infusion on D1 and 2, q14d from W1‐24; W25‐48 observation only (n randomised = 1151) Arm B (Bevacizumab (Bev)‐FOLFOX4): FOLFOX4 + Bev 5 mg/kg D1 q14d from W1‐24, then Bev alone 7.5 mg/kg D1 q21d from W25‐48 (n randomised = 1155) Arm C (Bev‐XELOX): capecitabine 1000 mg/m2 oral bd D1‐14, oxaliplatin 130 mg/m2 D1 + Bev 7.5 mg/kg D1 q21d from W1‐24, then Bev alone 7.5 mg/kg D1 q21d from W25‐48 (n randomised = 1145) Total of 12 courses |
|
| Outcomes | DFS OS Grade ≥ 3 AEs (NCI CTCAE, version 3.0) Median follow‐up: For patients with stage III disease who did not have DFS events at the data cut‐off date ‐ Arm A 48.5 m, Arm B 48.3 m, Arm C 48.3 m Data cut‐off dates: DFS ‐ 30 June 2010, OS ‐ 30 June 2012 |
|
| Study Details | Journal article and abstract | |
| Funding sources and declarations of interest | Funding sources: Genentech, Roche, Chugai Declarations of interest: Honoraria from Roche‐Genentech, Merck‐Serono, Sanofi‐Aventis, Amgen |
|
| Notes | All efficacy results were only reported for stage III participants in all arms | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A block design randomisation procedure (block size of six) was used" (methods, paragraph 3, page 1226) |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomisation was done after surgery using a centralised interactive computerised system" (methods, paragraph 3, page 1226) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "The study had an open‐label design" (methods, paragraph 3, page 1226) (i) DFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) DFS: High Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | Unclear risk | (i) Disease recurrence (influences DFS): Low Quote: "Recurrences or new occurrences were based on investigator tumour assessments, and pre‐scheduled every 6 months after randomisation until year 4, then annually thereafter" (methods, paragraph 7, page 1227) (ii) Survival (influences DFS/OS): Low Quote: "Survival status was assessed every 6 months in the first 4 years after randomisation, then annually thereafter" (methods, paragraph 8, page 1227) (iii) Grade ≥ 3 AEs: Unclear Quote: "Adverse events were monitored until at least 28 days after the last dose of study treatment or end of observation phase" (methods, paragraph 9, page 1227) However, the frequency/schedule of monitoring in both arms was unclear |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) DFS/OS: Low Quote: "Event‐free patients at the clinical cutoff date were censored at the last date at which they were known to be disease‐free" (methods, paragraph 7, page 1227) Quote: "Patients who were still alive at the clinical cutoff date were censored at the date at which they were last confirmed to be alive" (methods, paragraph 8, page 1227) No evidence of bias related to censoring (ii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "Patients who were event‐free at a given time point were censored at that time point" (methods, paragraph 11, page 1228) ITT population included all participants randomised to their allocated treatments (Figure 1, page 1227) Safety analysis: Quote: "The safety population comprised all patients who received at least one dose of study treatment" (methods, paragraph 12, page 1228) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, page 1227) (ii) Median age: Low (Table 1, page 1227) (iii) TNM stage: Low (stage II vs stage III, and stage III N1 vs N2) (Table 1, page 1227) |
| Other bias | Low risk | Subsequent therapies: Low Participants in the Bev‐FOLFOX4 and Bev‐XELOX groups received similar subsequent drug therapy after a recurrence or a new occurrence of colorectal cancer in the stage III ITT population (supplementary appendix, Table 1, page 6) |
De la Torre 2008.
| Methods | Randomised controlled trial Phase: III Accrual dates: January 1999 to September 2004 |
|
| Participants | No. randomised: 155 Stage/treatment type: T3 or T4 rectal adenocarcinoma, with or without nodal metastasis, or any T stage tumours with nodal metastasis; neoadjuvant Countries/sites: Spain, 3 sites Setting: Hospital Characteristics (Arm I/II): Locally advanced rectal adenocarcinoma; age ≤ 80 years (median 65/63 years); male (74/66%); PS WHO 0‐2 (PS ECOG 0: 63/64%) |
|
| Interventions | Arm I (FU+LV): LV 20 mg/m2 followed by IV bolus FU 350 mg/m2 for 5 consecutive days during first and fifth weeks of radiotherapy (n randomised = 77) Arm II (UFT + LV): Single course of oral LV 12.5 mg bd and oral UFT 300 mg/m2/d on D 8‐36 of the radiotherapy course (n randomised = 78) Radiotherapy consisted of a total dose of 45 Gy given in 25 fractions of 1.8 Gy, 5 fractions per week |
|
| Outcomes | DFS OS Grade ≥ 3 AEs (ECOG CTC) Median follow‐up: 22 m, insufficient for DFS outcome |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: None declared Declarations of interest: No conflicts of interest |
|
| Notes | The scientific committee decided to stop the study because of slow accrual after 155 participants (63% of planned accrual) from the 3 participating hospitals had been randomised | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned in blocks of 10 ... " (methods and materials, paragraph 3, page 103) |
| Allocation concealment (selection bias) | Low risk | Quote: "Eligible patients were centrally randomized ..." (methods and materials, paragraph 3, page 103) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "...open‐label clinical trial ..." (methods and materials, paragraph 1, page 103) (i) DFS: This study was not used for the meta‐analysis for this outcome (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) DFS: This study was not used for the meta‐analysis for this outcome (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | Low risk | (i) Disease recurrence (influences DFS) and (ii) Survival (influences DFS/OS) Note: DFS and OS were not included in the meta‐analysis owing to < 3 years of median follow‐up Quote: "Patients were evaluated every 2 months for the first 6 months, every 3 months for the next 6 months, at 6‐month intervals for the next 4 years, and then yearly" (methods and materials, paragraph 10, page 103) (iii) Grade ≥ 3 AEs: Low Quote: "During therapy, patients were monitored weekly for acute toxicity" (methods and materials, paragraph 9, page 103) Quote: "Follow‐up was planned every 3 months following completion of therapy....The same schedule was applied for both arms." (correspondence with Dr. de la Torre, received 13 August 2012) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) Survival (DFS and OS): Low Censoring was noted in the KM curves (Fig. 1, page 106). No evidence of bias related to censoring This study was not used for the meta‐analysis of DFS and OS outcomes (ii) Grade ≥ 3 AEs: Low No missing data from participants in the safety analysis population (correspondence with Dr. de la Torre, received 13 August 2012) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low "One patient randomized to the FU+LV arm and 2 patients randomized to the UFT+LV arm were excluded from all analyses of acute adverse events and outcome" (results, paragraph 3, page 104) Safety analysis: Analyses were also performed in the same population as described above |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, page 104) (ii) Median age: Low (Table 1, page 104) (iii) TNM stage: Low (Table 1, page 104) |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified |
Diaz‐Rubio 2007.
| Methods | Randomised controlled trial Phase: III Accrual dates: April 2002 to August 2004 |
|
| Participants | No. randomised: 348 Stage/treatment line: Metastatic, first‐line Countries/sites: Spain, 29 sites Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal cancer; age ≥ 18 years (median 64/65 years); male (63/58%); KPS ≥ 70% (KPS > 70%: 89/90%) |
|
| Interventions | Arm I (XELOX): capecitabine 1000 mg/m2 bd D1‐14 plus oxaliplatin 130 mg/m2 D1, q21d for 12C (n randomised = 174) Arm II (FUOX): Infusional FU 2250 mg/m2 D1, 8, 15, 22, 29, 36 plus oxaliplatin 85 mg/m2 D1, 15 and 29 q42d for 6C (n randomised = 174) or until PD, intolerable AEs, or participant refusal |
|
| Outcomes | TTP Grade ≥ 3 AEs (NCI CTC, version 2.0); HFS assessed using 3‐grade scale previously described (Blum 1999) ORR (RECIST, version 1.0) OS Median follow‐up: 17.5 m (TTP/OS; cut‐off date 15 June 2006) |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: Treatment of Digestive Tumors (TTD), Madrid, Spain, Roche, Sanofi‐Aventis Declarations of interest: Sanofi‐Aventis, Roche |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned ... using a centrally generated computer randomization code" (patients and methods, paragraph 4, page 4225) |
| Allocation concealment (selection bias) | Low risk | Quote as above |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "...open‐label, phase III trial..." (patients and methods, paragraph 1, page 4225) (i) ORR/TTP: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/TTP: High Quote: "The response was assessed only by the investigators" (patients and methods, paragraph 8, page 4225) Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/TTP): Low Quote: " Imaging studies ... were repeated every 12 weeks during treatment" (patients and methods, paragraph 7, page 4225) (ii) Survival (influences OS): Unclear ‐ not specified (iii) Grade ≥ 3 AEs: High Quote: "Patients were evaluated for adverse events before oxaliplatin administration" (patients and methods, paragraph 5, page 4225) Quote: "XELOX consisted of oral capecitabine ... ... plus oxaliplatin ... on day 1 every 3 weeks. FUOX consisted of FU ... ... plus oxaliplatin ... on days 1, 15, and 29 every 6 weeks" (patients and methods, paragraph 4, page 4225) Therefore, participants were evaluated for AEs more frequently in the FUOX arm than in the XELOX arm |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Low 13% of patients in the XELOX arm and 9% of patients in the FUOX arm were not assessable (Table 2, page 4227) (ii) TTP/OS: Low Although not defined in Methods, censoring was noted in the KM curves (Fig 1 & 2, page 4227). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Low Quote: “Safety was evaluated in all patients who received treatment..." (results, paragraph 5, page 4227) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "...348 patients (intent‐to‐treat population) were ... randomly assigned to treatment: 174 to XELOX and 174 to FUOX. Six patients (three in each treatment arm) did not initiate study treatment, leaving 342 patients who constituted the per‐protocol population" (results, paragraph 1, page 4225) Quote: "The primary statistical analysis of efficacy was ... between groups in the per‐protocol population" (patients and methods, paragraph 9, page 4225) Safety analysis: Quote: “Safety was evaluated in all patients who received treatment (XELOX, n = 171; FUOX, n = 171) ..." (results, paragraph 5, page 4227) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Unclear ‐ presented as KPS ≤ 70% vs > 70% (Table 1, page 4226) (ii) Median age: Low (Table 1, page 4226) (iii) No. of involved organs: Unclear ‐ not specified |
| Other bias | Low risk | Subsequent therapies: Low Quote: "A total of 199 patients (58.2%) received second‐line chemotherapy: 99 patients (57.9%) in the XELOX arm and 100 patients (58.5%) in the FUOX group" (results, paragraph 4, page 4226) Other Quote: "... significantly more patients in the XELOX arm (26%) than in the FUOX arm (16%) had received previous adjuvant chemotherapy (P = .032), which consisted of fluoropyrimidine therapy with or without LV" (results, paragraph 1, page 4225) |
Douillard 2002.
| Methods | Randomised controlled trial Phase: III Accrual dates: June 1995 to August 1997 |
|
| Participants | No. randomised: 816 Stage/treatment line: Metastatic, first‐line Countries/sites: Multi‐nation, 85 sites ‐ USA and Puerto Rico (n = 466), Canada (n = 100), Europe (n = 250) Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal cancer; age > 18 years (median 64/64 years); male (61/60%); PS ECOG 0‐2 (PS ECOG 0: 45/43%) |
|
| Interventions | Arm I (UFT/LV): UFT 300 mg/m2/d and LV 75 mg/d (US) or 90 mg/d (non‐USA countries) D1‐28, q35d (n randomised = 409) Arm II (5‐FU/LV): 5‐FU 425 mg/m2/d plus LV 20 mg/m2/d D1‐5, q28d (n randomised = 407) |
|
| Outcomes | OS TTP ORR (WHO criteria, modified) Grade ≥ 3 AEs (CTC, version not specified) Median follow‐up: Not specified. |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: Taiho Pharmaceutical Company, Bristol‐Myers Squibb Declarations of interest: None declared |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were centrally randomized ..." (patients and methods, paragraph 2, page 3607) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Not specified, blinding unlikely (i) ORR/TTP: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified (i) ORR/TTP: High Outcome assessment at risk of bias if there was lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/TTP): High Quote: "Tumor reassessment, including tumor measurements and a computed tomography scan of the abdomen and pelvis, was repeated after every two courses ..." (patients and methods, paragraph 7, page 3607) Quote: "In the UFT/LV treatment arm ...cycles repeated every 35 days...In the 5‐FU/LV treatment arm ... cycles repeated every 28 days" (patients and methods, paragraph 3, page 3607) Therefore, responses were evaluated more frequently in the 5‐FU/LV arm (ii) Survival (influences OS): Unclear ‐ not specified (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Low 1 patient in the 5‐FU/LV arm (< 1%) and no patients in the UFT/LV arm were not assessable (Table 2, page 3608) (ii) TTP/OS: Low Although not defined in the Methods, censoring was noted in the KM curves (Figure 1, page 3610). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Low Other than data for LFTs, which were not of interest for the review, relevant AEs have < 7% of safety outcome data missing (Table 4, 5, and 6, pages 3611 and 3612) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "All efficacy analyses are presented by treatment arm as randomized" (patients and methods, paragraph 8, page 3608) Safety analysis: Quote: "All 802 patients who received at least one dose of study medication were evaluated for safety and were analyzed based on the treatment arm as treated" (results, paragraph 18, page 3610) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Low (Table 1, page 3608) (ii) Median age: Low (Table 1, page 3608) (iii) No. of involved organs: Unclear ‐ not specified |
| Other bias | Low risk | Subsequent therapies: Low Quote: "Secondary chemotherapy was administered to 52% of patients assigned to UFT/LV and 50% of patients assigned to 5‐FU/LV. Information about the type of drugs included in subsequent chemotherapy was not collected" (results, paragraph 17, page 3610) |
Douillard 2014.
| Methods | Randomised controlled trial Phase: II, non‐comparative Accrual dates: February 2007 to June 2008 |
|
| Participants | No. randomised: 302 Stage/treatment line: Metastatic, first‐line Countries/sites: Multi‐nation; Argentina, Australia, Austria, Belgium, Brazil, France, Germany, Greece, Hong Kong, Israel, Italy, Mexico, Poland, Thailand Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal cancer; age ≥18 years (median 60.0/61.5 years); male (63/63%); KPS ≥ 60 (PS ECOG 0: 79/79%) |
|
| Interventions | Arm I (UFOX + cetuximab): D1 cetuximab (loading dose 400 mg/m2 then 250 mg/m2 weekly, thereafter) followed by oxaliplatin 85 mg/m2 D1 and 15, UFT (tegafur 250 mg/m2/d, uracil 560 mg/m2/d) and folinic acid 90 mg/d D1‐21, q28d (n randomised = 152) Arm II (FOLFOX4 + cetuximab): D1 cetuximab (as per Arm I) followed by oxaliplatin 85 mg/m2 D1 and 15, folinic acid 200 mg/m2, IV bolus 5‐FU 400 mg/m2 and 5‐FU 600 mg/m2 22‐hour infusion D1, 2, 15, and 16, q28d (n randomised = 150) Treatment continued until disease progression, withdrawal of consent, or unacceptable toxicity |
|
| Outcomes | PFS OS ORR (RECIST, version 1.0) Grade ≥ 3 AEs (NCI CTC, version 3.0) Median follow‐up: Clinical cut‐off for PFS ‐ 30 June 2009; clinical cut‐off for OS ‐ 31 August 2011 |
|
| Study Details | Journal article and abstract/poster | |
| Funding sources and declarations of interest | Funding sources: Merck KGaA Declarations of interest: Merck‐Serono, Roche, Amgen |
|
| Notes | Of note, this study was designed before demonstration of KRAS mutation status was a predictive biomarker for cetuximab response. Enrollment was therefore independent of KRAS mutation status. Additionally, recruitment was curtailed after demonstration of KRAS mutation status as a predictive biomarker for cetuximab response | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... patients were randomly assigned ... using a centralized stratified permuted block randomization procedure" (patients and methods, paragraph 4, page 15) |
| Allocation concealment (selection bias) | Low risk | Quote as above |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "...open‐label phase II study" (patients and methods, paragraph 4, page 15) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: High The study was not blinded, and independent review was not performed (correspondence with Dr. Peter Eggleton, received 22 July 2012) Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | Low risk | (i) Response (influences ORR/PFS): Low Quote: "Investigators assessed response to treatment every 8 weeks based on radiological imaging (CT or MRI scans)..." (patients and methods, paragraph 6, page 16) Quote: "After permanent treatment cessation, patients were followed every 3 months to collect data on progression..." (patients and methods, paragraph 6, page 16) Quote: "This particular regimen of UFT and oxaliplatin was chosen to allow a 4‐week dosing cycle, which ensured that all assessments were carried out at the same time in each treatment arm" (patients and methods, paragraph 5, page 15) (ii) Survival (influences PFS/OS): Low Survival assessment was performed 6 weeks after final tumour assessment (correspondence with Dr. Peter Eggleton, received 2 August 2012) Subsequently, quote: "After permanent treatment cessation, patients were followed every 3 months to collect data on ... survival ..." (patients and methods, paragraph 6, page 16) (iii) Grade ≥ 3 AEs: Low Assessments for adverse events followed identical schedules (correspondence with Dr. Peter Eggleton, received 2 August 2012) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Low Similar proportions of participants were not evaluable in both arms (Table 3, page 20) (ii) PFS/OS: Low Although not defined in the Methods, censoring was noted in the KM curves for PFS/OS (Figure 2, page 21). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Low Data related to grade ≥ 3 adverse events were collected until the clinical cut‐off date (30 June 2009). No missing outcome data before that time (correspondence with Dr. Peter Eggleton, received 24 July 2016) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "The primary efficacy analysis of PFS ... was carried out on the intention‐to‐treat (ITT) population, comprising all randomized patients" (patients and methods, paragraph 9, page 16) Safety analysis: Quote: "Safety analyses were performed on the safety population, which comprised all randomized patients who received any dose of any study treatment" (patients and methods, paragraph 10, page 16) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | High risk | (i) PS: Low (Table 1, page 18) (ii) Median age: Low (Table 1, page 18) (iii) No. organs involved: Low (Table 1, page 18) (iv) KRAS mut: High Information on KRAS mut status was available for 93/150 (62%) of the ITT population in the FOLFOX4 + cetuximab arm and 87/152 (57%) of the ITT population in the UFOX + cetuximab arm (Figure 1, page 17) A greater proportion of participants with known KRAS mutant status in the UFOX + cetuximab arm 47/87 (54%) were KRAS mutant than in the FOLFOX4 + cetuximab arm 37/93 (40%) – 14% difference (Table 1, page 18) |
| Other bias | Low risk | Subsequent therapies: Low A similar proportion of participants received different types of subsequent second‐line therapy after disease progression, in each arm (supplementary table 1) |
Ducreux 2011.
| Methods | Randomised controlled trial Phase: III Accrual dates: May 2003 to August 2004 |
|
| Participants | No. randomised: 306 Stage/treatment line: Metastatic, first‐line Countries/sites: France, 33 sites Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal cancer; age ≥ 18 years (median 66/64 years); male (64/60%); PS ECOG ≤ 2 (PS ECOG 0‐1 92/93%) |
|
| Interventions | Arm I (XELOX): oxaliplatin 130 mg/m2 D1 plus oral capecitabine 1000 mg/m2 bd D1‐14, q21d (n randomised = 156) Arm II (FOLFOX): 6 D1 oxaliplatin 100 mg/m2, leucovorin 400 mg/m2, IV bolus 5‐FU 400 mg/m2, and 5‐FU 2400‐3000 mg/m2 46‐hour infusion, q14d (n randomised = 150) Treatment was continued for 24 weeks or until disease progression, whichever came first |
|
| Outcomes | ORR (RECIST, version 1.0) PFS OS Grade ≥ 3 AEs (NCI‐CTC, version 3) Median follow‐up: 18.8 months for ITT population, all outcomes |
|
| Study Details | Journal article and abstract | |
| Funding sources and declarations of interest | Funding sources: Roche Declarations of interest: Roche |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were assigned in a 1:1 ratio to a treatment group by centralised, adaptive randomisation (minimisation method) ..." (materials and methods, paragraph 3, page 683) |
| Allocation concealment (selection bias) | Low risk | Quote as above |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "...open‐label study" (materials and methods, paragraph 1, page 683) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: Low Quote: "Tumour responses were validated in a centralised, blinded review of CT scans by an independent response committee (IRC)" (materials and methods, paragraph 7, page 684) (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/PFS): High Quote: "Tumour assessments (CT scan and MRI) were ... repeated at cycles 3 and 6 in the XELOX group and cycles 4 and 8 in the FOLFOX‐6 group ..." (materials and methods, paragraph 7, page 684) Quote: "XELOX ...every 3 weeks. FOLFOX‐6 ...every 2 weeks" (materials and methods, paragraph 4, page 683) Therefore, the first and second response assessments were performed later in the XELOX arm (weeks 9 and 18) compared with the FOLFOX‐6 arm (weeks 8 and 16) (ii) Survival (influences PFS/OS): Low Quote: "After completion of study treatment, patients were reassessed every 3 months until 18 months after the end of treatment for the last randomised patient" (materials and methods, paragraph 7, page 684) (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Low Quote: "...a small imbalance between the two treatment groups with respect to the percentage of patients not assessable for response (12.2% in XELOX vs. 8.7% in FOLFOX‐6)..." (discussion, paragraph 2, pages 687 and 688) (ii) PFS/OS: Low Although not defined in the Methods, censoring was noted in the KM curves (Figures 2 and 3, pages 687 and 688). No evidence of bias related to censoring. (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "The intent‐to‐treat (ITT) population included all patients who underwent randomisation" (materials and methods, paragraph 10, page 684) Safety analysis: Quote: "All patients receiving at least one dose of study treatment were included in the safety population" (materials and methods, paragraph 10, page 684) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Unclear Reported as ECOG 0‐1 vs 2 (Table 2, page 686) (ii) Median age: Low (Table 2, page 686) (iii) No. of involved organs: Low (Table 2, page 686) |
| Other bias | Unclear risk | Subsequent therapies: Unclear Quote: "Although the protocol did not include monitoring of second‐line therapies following study drug discontinuation, the possibility that second‐line therapies may have influenced the OS results cannot be ruled out" (discussion, paragraph 1, page 687) |
Ducreux 2013.
| Methods | Randomised controlled trial Phase: II, non‐comparative Accrual dates: March 2006 to January 2008 |
|
| Participants | No. randomised: 145 Stage/treatment line: Metastatic, first‐line Countries/sites: 15 centres in France, La Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) study Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal cancer; age 18 to 75 years (median 61/61 years); male (64/48%); PS ECOG 0‐2 (PS ECOG 0: 54/60%) |
|
| Interventions | Arm I (XELIRI + bevacizumab (BEV)): irinotecan 200 mg/m2 D1 and capecitabine 1000 mg/m2 (800 mg/m2 if ≥ 65 years) bd D1‐14 plus BEV 7.5 mg/kg D1, q21d for a maximum of 8 cycles (n randomised = 72) Arm II (FOLFIRI + BEV): Irinotecan 180 mg/m2, leucovorin 400 mg/m2, and IV bolus fluorouracil 400 mg/m2 followed by 2400 mg/m2 46‐hour infusion plus 5 mg/kg BEV D1, q14d for a maximum of 12 cycles (n randomised = 73) For participants whose disease was controlled after 6 months of BEV and chemotherapy, BEV (7.5 mg/kg every 3 weeks) was continued as a single‐agent maintenance therapy in both arms until progressive disease |
|
| Outcomes | PFS ‐ 6 months, PFS Grade ≥ 3 AEs (NCI CTCAE, version 3.0) ORR (RECIST, version 1.0) OS Median follow‐up: 36 months for both Arm I/II; final analysis 15 March 2010 |
|
| Study Details | Journal article, abstract, oral poster presentation, and journal article for translational sub‐study | |
| Funding sources and declarations of interest | Funding sources: Roche, Pfizer, and Chugai Declarations of interest: Roche, Chugai, Pfizer, Amgen, Boeringer, Merck Serono |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed using the minimisation method (using a 10% random factor) (correspondence with Dr. Jean‐Pierre Pignon, received 1 August 2012) |
| Allocation concealment (selection bias) | Low risk | Central randomisation by fax was used (correspondence with Dr. Jean‐Pierre Pignon, received 1 August 2012) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "... open‐label, non‐comparative phase II study" (patients and methods, paragraph 1, page 1237) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: High Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/PFS): Low Quote: "Tumour assessments using abdominal and/or thoracic computed tomography (CT) or magnetic resonance imaging (MRI) were performed at baseline and every 8 weeks until progression" (patients and methods, paragraph 5, page 1238) (ii) Survival (influences PFS/OS): Low Quote: "After disease progression, patients were followed up at least every 2 months until death" (patients and methods, paragraph 4, page 1238) (iii) Grade ≥ 3 AEs: High Quote: "During treatment, physical examination, ECOG performance status, BP, and blood and biochemistry analyses were repeated every cycle" (patients and methods, paragraph 4, page 1237) Quote: "treatment with either ... XELIRI ... every 3 weeks ... or FOLFIRI ... every 2 weeks" (patients and methods, paragraph 3, page 1237) Quote: "During bevacizumab maintenance, clinical examination, BP, blood/urine analysis and ECOG performance status were performed every 3 weeks" (patients and methods, paragraph 4, page 1237 and 1238) Therefore, safety evaluation was performed more frequently in the FOLFIRI + BEV group during combination treatment Safety assessment was 3‐weekly during treatment with BEV alone for both arms |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Low Outcome data were available for all randomised participants, including those who had 'early stopping' (correspondence with Dr. Jean‐Pierre Pignon, received 1 August 2012) (ii) PFS/OS: Low As above (iii) Grade ≥ 3 AEs: Low Quote: "All 145 patients were evaluable for safety..." (results, paragraph 5, page 1239) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Analysis was according to treatment as randomised (Fig. 1, page 1239) Safety analysis: Analysis was according to treatment as randomised (Fig. 1, page 1239) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, page 1239) (ii) Median age: Low (Table 1, page 1239) (iii) No. of involved organs: Low (Table 1, page 1239) |
| Other bias | Low risk | Similar proportions of participants in the XELIRI + BEV and FOLFIRI + BEV groups received different types of subsequent drug therapy following progression on first‐line treatment (supplementary material, supplementary Tables 1 and 2, pages 4‐7) |
ECOG E5296 2012.
| Methods | Randomised controlled trial Phase: III Accrual dates: April 1999 to September 2000 (closed) |
|
| Participants | No. randomised: 125 of planned 950 Stage/treatment line: Metastatic, first‐line Countries/sites: USA, 24 study sites and one Expanded Participation Project (EPP) site Setting: Hospital Characteristics (Arm A/B): Metastatic colorectal cancer; age ≥ 18 years (median 64/65 years); male (64/62%); PS ECOG 0‐2 (PS ECOG 0: 45/43%) |
|
| Interventions | Arm A: Continuous 5‐FU infusion 300 mg/m2/d D1‐28, q35d (n randomised = 64) Arm B: Oral eniluracil 11.5 mg/m2 and oral 5‐FU 1.15 mg/m2 bd D1‐28, q35d (n randomised = 61) |
|
| Outcomes | ORR (ECOG Solid Tumour Response Criteria) PFS OS Grade ≥ 3 AEs (CTC, version 2.0) Median follow‐up: 1.3 years, all outcomes |
|
| Study Details | Technical report and study protocol | |
| Funding sources and declarations of interest | Funding sources: National Cancer Institute, DHHS Declarations of interest: None declared |
|
| Notes | This study was closed early, after accrual of 125 of a planned 950 participants, following results of the Van Cutsem 2001a and Schilsky 2002a studies | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed with permuted blocks within strata algorithm, with override protection for treatment imbalances that could occur within the main institutions of the cooperative group due to the stratified algorithm (correspondence with Dr. Paul Catalano, received 10 July 2012) |
| Allocation concealment (selection bias) | Low risk | Permuted blocks of undisclosed size were generated within strata. Additionally, accruals occurred over time across many treating centres, making it very unlikely that one could decode the randomisation algorithm and predict the next assignment in the sequence (correspondence with Dr. Paul Catalano, received 10 July 2012) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | No blinding occurred in the trial (correspondence with Dr. Paul Catalano, received 10 July 2012) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | No blinding (i) ORR/PFS: High Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | Low risk | (i) Response (influences ORR/PFS): Low Response assessments were performed every 10 weeks. For those with measurable disease, if a CR/PR was achieved, this was confirmed after 4 weeks (E5296 study protocol, page 14) (ii) Survival (influences OS): Low Post‐treatment follow‐up was the same for both arms: every 3 months if < 2 years from study entry, every 6 months if 2 to 5 years from study entry, and every 12 months if > 5 years from study entry (E5296 study protocol, page 20) (iii) Grade ≥ 3 AEs: Low Complete blood count (CBC) was examined weekly and other AE assessments were performed before each treatment cycle (E5296 protocol, page 14). One cycle was every 35 days for both arms (E5296 study protocol, page 6) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Low The sum of those with unevaluable and unknown responses was 5/62 (8%) in the IV 5‐FU arm and 3/61 (5%) in the oral 5‐FU + eniluracil arm (Table 11, page 22) (ii) PFS/OS: Low Survival outcomes were known for all participants who were eligible, were ineligible, and had withdrawn (correspondence with Dr. Paul Catalano, received 10 July 2012) (iii) Grade ≥ 3 AEs: Low Quote: "Toxicity data was submitted for 63 patients randomized to arm A and 59 patients randomized to arm B" (results, paragraph 6, page 9) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Presented results were analysed according to allocated treatment (section 7.1, page 8 and Table 1, page 12). This excluded patients who were found to be ineligible (after randomisation) and who withdrew from the study before treatment (2/125, 1.6%) (results, paragraph 1, page 8 and Table 1, page 12) Safety analysis: Analysis was performed for those who had toxicity data submitted (results, paragraph 6, page 9, and Table 8, pages 20 and 21) |
| Selective reporting (reporting bias) | Low risk | The same outcomes were reported in the study protocol and in the technical report |
| Similarity of arms at baseline | Unclear risk | (i) PS: Low (Table 3, page 13) (ii) Median age: Low (Table 3, page 13) (iii) No. of involved organs: Unclear ‐ not specified |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified |
Fuchs 2007.
| Methods | Randomised controlled trial Phase: III Design: For Period 1 (the only period of interest for this review), 3 × 2 factorial design with randomisation to FOLFIRI vs mIFL vs CapeIRI (open‐label), and randomisation to celecoxib vs placebo (double‐blind) Accrual dates: Period 1 February 2003 to March 2004 |
|
| Participants | No. randomised: 430 Stage/treatment line: Metastatic, first‐line Countries/sites: For Periods 1 and 2, participants were enrolled in USA, Canada, Australia, and New Zealand, at 99 sites Setting: Hospital Characteristics (Arm A/B/C): Metastatic colorectal cancer; age ≥ 18 years (median 61/62/62 years); male (63.9/58.9/54.5%); PS ECOG 0‐1 (PS ECOG 0: 52.1/49.6/48.3%) |
|
| Interventions | Arm A (FOLFIRI + *celecoxib/placebo): irinotecan 180 mg/m2, LV 400 mg/m2, IV bolus FU 400 mg/m2, followed by 5FU 2400 mg/m2 46‐hour infusion, q14d (n randomised = 144) Arm B (mIFL + celecoxib/placebo): irinotecan 125 mg/m2, LV 20 mg/m2, IV bolus FU 500 mg/m2 D1 and 8, q21d (n randomised = 141) Arm C (CapeIRI + celecoxib/placebo): irinotecan 250 mg/m2 D1 and oral capecitabine 1000 mg/m2 bd D1‐14, q21d (n randomised = 145) *Oral celecoxib 400 mg bd or placebo tablets; permanently discontinued on 19 January 2005. Treatment continued until PD, unacceptable toxicity from chemotherapy, or withdrawal of consent |
|
| Outcomes | PFS OS ORR (RECIST, version 1.0) Grade ≥ 3 AEs (NCI CTC, version 2.0) Median follow‐up: 34 months (cut‐off date Nov 17 2006) |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: Pfizer Declarations of interest: Pfizer, Sanofi‐Aventis, Genentech, AstraZeneca, Bristol‐Myers Squibb, Amgen, Imclone Systems Inc, Roche, Boerhinger Ingelheim |
|
| Notes | Study was terminated after enrolment of 547 of a planned 900 participants. Accrual to this trial had slowed after report of cardiovascular concerns with celecoxib, despite celecoxib/placebo administration for all participants was permanently discontinued in January 2005 16 participants (11.1%) in Arm I chose to have bevacizumab after the study amendment (bevacizumab 5 mg/kg IV on D1, repeated every 2 weeks), and 7 participants (5.0%) in Arm II chose to have bevacizumab after the study amendment (bevacizumab 7.5 mg/kg IV on D1, repeated every 3 weeks) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "...randomly assigning patients to one of three open‐label chemotherapy arms ..." (patients and methods, paragraph 2, page 4780) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: High Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/PFS): Low Quote: "During chemotherapy, a follow‐up CT/MRI of the abdomen/pelvis and chest x‐ray or chest CT/MRI were to be performed every 6 weeks. Assessments were performed every 6 weeks until PD or on chemotherapy discontinuation" (patients and methods, paragraph 10, page 4781) (ii) Survival (influences PFS/OS): Low Quote: "After PD, the patient was observed every 3 months for survival" (patients and methods, paragraph 10, page 4781) (iii) Grade ≥ 3 AEs: High Participants were reviewed for safety assessments every week during the first cycle, and then every cycle (communication with Dr. Justin Binko, received 26 July 2012) Quote: "...FOLFIRI ... repeated every 2 weeks ... mIFL ... repeated every 3 weeks. CapeIRI ... repeated every 3 weeks" (patients and methods, paragraph 5, page 4780) Therefore, following cycle 1, participants in the FOLFIRI arm underwent more frequent safety evaluations than those in the mIFL and CapeIRI arms |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Unclear ‐ not specified (ii) PFS/OS: Low PFS‐ Quote: "For patients without documented PD, data were censored on the date of the last tumor assessment with nonprogression status or, for patients who started a second‐line therapy, at the date of the start of new therapy" (patients and methods, paragraph 11, page 4781). No evidence of bias related to censoring OS‐ Quote: "...in the absence of confirmation of death, data were censored at the last date the patient was known to be alive" (patients and methods, paragraph 12, page 4781). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "Efficacy analyses included all patients randomly assigned on an intent‐to‐treat basis" (patients and methods, paragraph 14, page 4781) Safety analysis: Quote: "Safety analyses included all treated patients ..." (patients and methods, paragraph 14, page 4781) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Low (Table 2, page 4783) (ii) Median age: Low (Table 2, page 4783) (iii) No. of involved organs: Unclear ‐ not specified A similar median number of measurable target metastatic lesions for all 3 treatment arms was reported in a published author reply, quote: "The median number of measurable target metastatic lesions was 3.2 for FOLFIRI, 3.1 for mIFL, and 3.1 for CapeIRI" (Fuchs et al, Journal of Clinical Oncology 2008, paragraph 2) |
| Other bias | Low risk | Subsequent therapies: Low Quote: "The rates of utilization of poststudy salvage chemotherapy did not differ significantly between the three first‐line chemotherapy arms in period 1 (77% for FOLFIRI, 75% for mIFL, and 77% for CapeIRI)" (results, paragraph 4, page 4783) Other bias: Some participants in the FOLFIRI and mIFL arms from period 1 received BEV, although this was only a small percentage in each arm Quote: "After activation of this study amendment, patients randomly assigned to FOLFIRI or mIFL during period 1 had the option of adding bevacizumab to their current regimen. Among patients enrolled during period 1, 16 patients on the FOLFIRI arm added bevacizumab to their regimen, and seven patients on the mIFL arm added bevacizumab to their regimen" (patients and methods, paragraph 6, page 4780) Risk of bias considerations in a factorial study: Unclear ‐ tests for interaction not specified |
Hochster TREE‐1 2008.
| Methods | Randomised controlled trial Phase: Not specified Accrual dates: December 2002 to November 2003 |
|
| Participants | No. randomised: 150 Stage/treatment line: Metastatic, first‐line Countries/sites: USA, 33 sites Setting: Hospital Characteristics (Arm I/II/III): Metastatic colorectal cancer; age ≥ 18 years (median 62/62/62.5 years); male (57/62/65%); PS ECOG 0‐1 (PS ECOG 0: 61/58/52%) |
|
| Interventions | Arm I (mFOLFOX6): oxaliplatin 85 mg/m2, LV 350 mg, IV bolus FU 400 mg/m2 and 2400 mg/m2 46‐hour infusion, q14d (n randomised = 50) Arm II (bFOL): oxaliplatin 85 mg/m2 D1 and 15, LV 20 mg/m2, IV bolus FU 500 mg/m2 D1, 8, and 15, q28d (n randomised = 50) Arm III (CapeOx): oxaliplatin 130 mg/m2 D1 and capecitabine 1000 mg/m2 bd D1‐15, q21d (n randomised = 50) Treatment continued until PD, unacceptable toxicity, extended toxicity‐related dose delay or withdrawal of consent |
|
| Outcomes | ORR (RECIST, version 1.0) TTP (treated as PFS in this review, based on the definition provided) Grade ≥ 3 AEs (NCI CTC, version 2.0) OS Median follow‐up: All outcomes ‐ 16.9, 15.1, 15.0 months, for Arms 1 through 3, respectively |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: Sanofi‐Aventis Declarations of interest: Sanofi‐Aventis, Genentech BioOncology, Bristol Myers‐Squibb, Taiho, Samyang Confirma Biotech, Amgen |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "TREE‐1 and TREE‐2 were two sequentially conducted, randomized, open‐label cohorts in this study" (patients and methods, paragraph 1, page 3524) (i) ORR/TTP (treated as PFS): Low Outcome assessment unlikely to be influenced by lack of blinding This study was not used for the meta‐analysis of the TTP (treated as PFS) outcome (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/TTP (treated as PFS): High Outcome assessment at risk of bias from lack of blinding This study was not used for the meta‐analysis of the TTP (treated as PFS) outcome (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/TTP (treated as PFS)): Low Quote: "Tumor assessments were repeated every 12 weeks in TREE‐1 ..." (patients and methods, paragraph 5, page 3524) This study was not used for the meta‐analysis of the TTP (treated as PFS) outcome (ii) Survival (influences TTP (treated as PFS)/OS): High Quote: "After treatment discontinuation, patients in TREE‐2 were followed for survival at 3‐month intervals for at least 2 years and every 6 months thereafter until lost to follow‐up or consent withdrawal; these data were collected for patients in TREE‐1 who consented retrospectively" (patients and methods, paragraph 5, page 3524) No information was provided regarding the relative numbers of participants in each arm of TREE‐1 who consented retrospectively This study was not used for meta‐analysis of the TTP (treated as PFS) outcome (iii) Grade ≥ 3 AEs: High Quote: "Clinical assessments and toxicities were recorded on day 1 of each cycle and at the end of treatment" (patients and methods, paragraph 5, page 3524) Quote: "In TREE‐1, patients received mFOLFOX6 ... every 2 weeks ..., bFOL ... every 4 weeks ... , or CapeOx ... every 3 weeks ..." (patients and methods, paragraph 1, page 3524) Therefore, participants underwent safety evaluations more frequently in the mFOLFOX arm |
| Incomplete outcome data (attrition bias) All outcomes | High risk | (i) ORR: High 23% of participants were missing reported confirmed response data in the CapeOx arm (Table 4, page 3527) (ii) TTP (treated as PFS): Low Quote: "TTP was censored at the last date the patient was known to be progression free for patients who did not have objective tumor progression and who were either still on study at the time of the analysis or who were removed from follow‐up before documentation of objective tumor progression. For patients who received second‐line treatment prior to progression or death, TTP was censored at the time of starting the new therapy" (Table 4, page 3527) No evidence of bias related to censoring This study was not used for the meta‐analysis of TTP (treated as PFS) outcome OS: Unclear ‐ Not specified (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "All analyses are for the as‐treated population, which includes all randomly assigned patients receiving at least one treatment" (patients and methods, paragraph 6, page 3524) Quote: "In TREE‐1, 147 of 150 patients were treated (one was ineligible for prior chemotherapy and two did not start treatment)" (results, paragraph 2, page 3524) Therefore, 2% of randomised participants were excluded from the efficacy analysis population Safety analysis: Same as for efficacy |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available. |
| Similarity of arms at baseline | Unclear risk | (i) PS: Low (Table 1, page 3525) (ii) Median age: Low (Table 1, page 3525) (iii) No. of involved organs: Unclear ‐ not specified |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified for the different arms within TREE‐1 |
Hochster TREE‐2 2008.
| Methods | Randomised controlled trial Phase: Not specified Accrual dates: November 2003 to April 2004 |
|
| Participants | No. randomised: 223 Stage/treatment line: Metastatic, first‐line Countries/sites: USA, 57 sites Setting: Hospital Characteristics (Arm I/II/III): Metastatic colorectal cancer; age ≥ 18 years (median 64/57/62 years); male (61/49/58%); PS ECOG 0‐1 (PS ECOG 0: 61/54/65%) |
|
| Interventions | Arm I (mFOLFOX6 + bevacizumab (BEV)): oxaliplatin 85 mg/m2, LV 350 mg, IV bolus 5FU 400 mg/m2, and 5‐FU 2400 mg/m2 46‐hour infusion, q14d + BEV 5 mg/kg D1, q14d (n randomised = 75) Arm II (bFOL + BEV): oxaliplatin 85 mg/m2 D1 and 15, LV 20 mg/m2, IV bolus FU 500 mg/m2 D1, 8, and 15, q28d + BEV 5 mg/kg D1, q14d (n randomised = 74) Arm III (CapeOx + BEV): oxaliplatin 130 mg/m2 D1 and capecitabine 850 mg/m2 bd D1‐15, q21 plus BEV 7.5 mg/kg D1, q21d (n randomised = 74) Treatment continued until PD, unacceptable toxicity, extended toxicity‐related dose delay, or withdrawal of consent |
|
| Outcomes | ORR (RECIST, version 1.0) TTP (treated as PFS in this review, based on the definition provided) Grade ≥ 3 AEs (NCI CTC, version 2.0) OS Median follow‐up: All outcomes, 17.9, 17.6 and 18.5 months in Arm I‐III, respectively. |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: Sanofi‐Aventis Declarations of interest: Sanofi‐Aventis, Genentech BioOncology, Bristol Myers‐Squibb, Taiho, Samyang Confirma Biotech, Amgen |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "TREE‐1 and TREE‐2 were two sequentially conducted, randomized, open‐label cohorts in this study" (patients and methods, paragraph 1, page 3524) (i) ORR/TTP (treated as PFS): Low Outcome assessment unlikely to be influenced by lack of blinding This study was not used for the meta‐analysis of the TTP (treated as PFS) outcome (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/TTP (treated as PFS): High Outcome assessment at risk of bias from lack of blinding This study was not used for meta‐analysis of the TTP (treated as PFS) outcome (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/TTP (treated as PFS)): Low Quote: "Tumor assessments were repeated ... every 6 weeks in TREE‐2" (patients and methods, paragraph 5, page 3524) This study was not used for meta‐analysis of the TTP (treated as PFS) outcome (ii) Survival (influences TTP (treated as PFS)/OS): Low Quote: "After treatment discontinuation, patients in TREE‐2 were followed for survival at 3‐month intervals for at least 2 years and every 6 months thereafter until lost to follow‐up or consent withdrawal ..." (patients and methods, paragraph 5, page 3524) This study was not used for the meta‐analysis of the TTP (treated as PFS) outcome. (iii) Grade ≥ 3 AEs: High Quote: "Clinical assessments and toxicities were recorded on day 1 of each cycle and at the end of treatment" (patients and methods, paragraph 5, page 3524) Quote: "In TREE‐1, patients received mFOLFOX6 ... every 2 weeks ..., bFOL ... every 4 weeks ... , or CapeOx ... every 3 weeks ... In TREE‐2, patients received one of the same three chemotherapy regimens as in TREE‐1 but with the addition of bevacizumab ..." (patients and methods, paragraph 1, page 3524) Therefore, participants underwent safety evaluations more frequently in the mFOLFOX arm |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Low Confirmed tumour response data were reported for ≥ 85% of participants in all arms (Table 4, page 3527) (ii) TTP (treated as PFS): Low Quote: "TTP was censored at the last date the patient was known to be progression free for patients who did not have objective tumor progression and who were either still on study at the time of the analysis or who were removed from follow‐up before documentation of objective tumor progression. For patients who received second‐line treatment prior to progression or death, TTP was censored at the time of starting the new therapy" (Table 4, page 3527) There was no evidence of bias related to censoring This study was not used for meta‐analysis of the TTP (treated as PFS) outcome OS: Unclear ‐ not specified (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | High risk | Efficacy analysis: High Quote: "All analyses are for the as‐treated population, which includes all randomly assigned patients receiving at least one treatment" (patients and methods, paragraph 6, page 3524). 4/75 (5.3%) , 4/74 (5.4%) and 2/74 (3%) patients were excluded from the mFOLFOX6 + BEV, bFOL + BEV and CapeOx + BEV arms, respectively (from results, paragraph 1, page 3524 and Table 2, page 3525) Safety analysis: Same as for efficacy |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available. |
| Similarity of arms at baseline | High risk | (i) PS: Low (Table 1, page 3525) (ii) Median age: High 5‐year difference between bFOL + BEV and CapeOx + BEV arms (Table 1, page 3525) (iii) No. of involved organs: Unclear ‐ not specified |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified for the different arms within TREE‐2 |
Hoff 2001.
| Methods | Randomised controlled trial Phase: III Accrual dates: September 1996 to February 1998 |
|
| Participants | No. randomised: 605 Stage/treatment line: Metastatic, first‐line Countries/sites: Multi‐nation, 61 sites ‐ USA (n = 48), Canada (n = 9), Brazil (n = 2), and Mexico (n = 2) Setting: Hospital Characteristics (Arm I/II/): Metastatic colorectal cancer; age ≥ 18 years (median 64/63 years); male (59.9/65.0%); KPS ≥ 70% (median 90/90%) |
|
| Interventions | Arm I: capecitabine 1250 mg/m2 bd D1‐14, q21d (n randomised = 302) Arm II: IV bolus 5‐FU 425 mg/m2 plus LV 20 mg/m2 D1‐5, q28d (n randomised = 303) Treatment continued until PD, unacceptable toxicity, or scheduled assessment at 30 weeks. Participants with a tumour response or SD were allowed to enter a continuation phase up to a total of 48 weeks. Treatment continuation beyond 48 weeks (post‐continuation phase) for participants without PD was provided at the discretion of the investigator |
|
| Outcomes | ORR (WHO criteria, 1979) TTP (treated as PFS in this review, based on the definition provided) OS Grade ≥ 3 AEs (NCI CTC, revised December 1994) No details on median follow‐up |
|
| Study Details | Journal articles | |
| Funding sources and declarations of interest | Funding sources: F. Hoffman‐La Roche Declarations of interest: None declared |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... patients were randomly assigned to treatment with capecitabine or 5‐FU/LV according to a computer‐generated randomization code" (patients and methods, paragraph 3, page 2283) |
| Allocation concealment (selection bias) | Low risk | Quote: "The patients were randomized centrally by country in blocks of four patients" (patients and methods, paragraph 3, page 2283) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "... open‐label, randomized, parallel‐group study ..." (patients and methods, paragraph 3, page 2283) (i) ORR/TTP (treated as PFS): Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/TTP (treated as PFS): Low Quote: "Investigator assessments of tumor response were reviewed, solely on the basis of imaging, by an independent review committee (IRC) composed of radiologists who were blinded to the treatment received, the clinical condition of the patient, and the investigator’s evaluation" (patients and methods, paragraph 8, page 2284) The absolute ORR was higher for capecitabine than for 5‐FU/LV for both of these assessments (Table 2, page 2285) (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | Low risk | (i) Response (influences ORR/TTP (treated as PFS)): Low Quote: "Assessments of tumor dimensions and involved sites were performed before the start of treatment and were scheduled after weeks 6, 12, 18, 24, and 30 of therapy. Further assessments were performed after weeks 39 and 48 for patients who received prolonged therapy (up to 48 weeks). Follow‐up assessments for disease progression and survival monitoring were performed every 3 months after the end of treatment" (patients and methods, paragraph 8, page 2284) (ii) Survival (influences TTP (treated as PFS)/OS): Low Quote: "Follow‐up assessments for ... survival monitoring were performed every 3 months after the end of treatment" (patients and methods, paragraph 8, page 2284) (iii) Grade ≥ 3 AEs: Low Quote: "Safety evaluations were conducted at least monthly until 4 weeks after the end of therapy..." (patients and methods, paragraph 9, page 2284) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Low For Investigator assessed responses, missing post‐baseline data for 7.3% of participants in the capecitabine arm and for 12.5% in the 5‐FU/LV arm (Table 2, page 2285) (ii) TTP (treated as PFS)/OS: Unclear ‐ not specified (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "The analyses of efficacy were based on all randomized patients" (patients and methods, paragraph 12, page 2284) Safety analysis: Quote: "The analyses of toxicity were based on the safety population, which included all patients who received at least one dose of study treatment" (patients and methods, paragraph 13, page 2284) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Unclear KPS was reported as a mean/median (Table 1, page 2285) (ii) Median age: Low (Table 1, page 2285) (iii) No. of organs: Unclear‐ not specified |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified |
Hofheinz 2012.
| Methods | Randomised controlled trial Phase: III Accrual dates: March 2002 to December 2007 |
|
| Participants | No. randomised: 401 Stage/treatment type: Rectal adenocarcinoma; adjuvant‐ R0 resection, neoadjuvant‐ cT3‐4 N0 or cTany N+ Countries/sites: Germany, 35 sites Setting: Hospital Characteristics (Arm I/II): Rectal adenocarcinoma; age ≥ 18 years (median 65/64 years); male (65/67%); PS WHO 0‐1 (PS WHO 0: 61/49%) |
|
| Interventions | Adjuvant cohort: Arm I: Two cycles of capecitabine 2500 mg/m2 D1‐14, q21d, followed by chemoradiotherapy 50.4 Gy plus capecitabine 1650 mg/m2 D1‐38, then 3 cycles of capecitabine (n randomised and with post‐randomisation data = 116) Arm II: Two cycles IV bolus fluorouracil 500 mg/m2 D1‐5, repeated D29‐33, followed by chemoradiotherapy 50.4 Gy plus infusional fluorouracil 225 mg/m2 daily, then 2 cycles of bolus fluorouracil (n randomised and with post‐randomisation data = 115) Neoadjuvant cohort: Arm I: Chemoradiotherapy (50.4 Gy plus capecitabine 1650 mg/m2 daily), followed by radical surgery and 5 cycles of capecitabine 2500 mg/m2 per day for 14 days (n randomised and with post‐randomisation data = 81) Arm II: Chemoradiotherapy (50.4 Gy plus infusional fluorouracil 1000 mg/m2 D1‐5 and 29‐33), followed by radical surgery and four cycles of bolus fluorouracil 500 mg/m2 for 5 days (n randomised and with post‐randomisation data = 80) Surgery: TME for tumours of the lower two‐thirds of the rectum and PME for the upper third, assuming a 5 cm distal margin without coning, were mandatory for the adjuvant cohort and recommended for the neoadjuvant cohort. For low‐lying tumours, the decision between low anterior resection and abdominoperineal excision was left to the surgeon’s discretion |
|
| Outcomes | OS (5 years) DFS Grade ≥ 3 AEs (NCI‐CTC, version 2.0) Median follow‐up: 52 months for all outcomes |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: Roche Pharma AG Declarations of interest: Roche Pharma AG, Chugai Pharma, Amgen, Merck KGaA, Ariad, Bristol‐Myers Squibb, Novartis, Merck Sharp & Dohme |
|
| Notes | Study protocol was amended in March 2005, to include patients with locally advanced rectal cancer receiving preoperative chemoradiotherapy (neoadjuvant cohort). Recruitment to the adjuvant cohort was continued | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly allocated ... using permuted blocks with stratification by centre and clinical or pathological tumour stage (T3–4 N0 vs T1–2 Npositive vs T3–4 Npositive)" (methods, paragraph 5, page 580) |
| Allocation concealment (selection bias) | Low risk | Quote: "Local investigators were masked to next assignment in the sequence" (methods, paragraph 5, pages 580 and 581) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "... open‐label, non‐inferiority, phase 3 trial ..." (methods, paragraph 1, page 580) (i) DFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) DFS: High Quote: "The study was open‐label; patients, treating physicians, and data managers and analysts were not masked to group assignment" (methods, paragraph 5, page 581) Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Disease recurrence (influences DFS): Low Quote: "Follow‐up, done for 5 years after the start of therapy..." (methods, paragraph 12, page 582) (ii) Survival (influences DFS/OS): Low Quote: "Follow‐up, done for 5 years after the start of therapy..." (methods, paragraph 12, page 582) (iii) Grade ≥ 3 AEs: High Quote: "Vital signs, haematology, and biochemistry were monitored weekly during chemoradiotherapy and before each chemotherapy cycle" (methods, paragraph 10, page 581) Quote: "Capecitabine was given twice daily ... on days 1–14, and repeated on day 22" (methods, paragraph 8, page 581) Quote: "Fluorouracil bolus was administered on five consecutive days (days 1–5) and repeated on day 29" (methods, paragraph 9, page 581) Therefore, these safety evaluations occurred more frequently in the capecitabine arm |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) DFS: Low Quote: "DFS was analysed using censored failure times ..." (methods, paragraph 15, page 583). No evidence of bias related to censoring (ii) OS: Low OS was also analysed with censoring using the last date of contact or death (correspondence with Dr. Ralf‐Dieter Hofheinz, received 1 August 2012). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Low Data were available for all participants in the analysis population (correspondence with Dr. Ralf‐Dieter Hofheinz, received 1 August 2012) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Participants were analysed according to the treatment arm allocated (Figure 3, page 582) However, quote: "All analyses were based on all patients with post‐randomisation data" (methods, paragraph 13, page 582). Therefore, 9/401 (2.2%) participants were excluded from the analyses (Figure 3, page 582) Safety analysis: Same as for efficacy |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, page 583) (ii) Median age: Low (Table 1, page 583) (iii) TNM stage: Low (Table 1, page 583) |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ similarity of therapy following recurrence or new occurrence of disease not specified |
Kato 2012.
| Methods | Randomised controlled trial Phase: II Accrual dates: November 2007 to February 2010 |
|
| Participants | No. randomised: 60 Stage/treatment line: Unresectable primary or metastases (all enrolled patients had metastases); first‐ or second‐line (if second‐line, first‐line therapy with FOLFOX was mandated), patients who had previous adjuvant chemotherapy had > 6 months elapsed since treatment Countries/sites: Japan, from 12 institutes of theTohoku Clinical Oncology Research and Education Society (T‐CORE) Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal cancer; age 20 to 75 years (median 62.0/62.5 years); male (56.7/60.0%); PS ECOG 0‐1 |
|
| Interventions | Arm I (Sequential IRIS‐bevacizumab (BEV)): D1 irinotecan 150 mg/m2 plus BEV 7.5 mg/kg, followed by S‐1 40‐60 mg* oral bd D3‐16, q21d (n randomised = 30) *S‐1 doses: 80 mg/d if BSA < 1.25 m2; 100 mg/d if BSA 1.25 to 1.5 m2; 120 mg/d if BSA > 1.5 m2 Arm II (mFOLFIRI‐BEV): D1 irinotecan 150 mg/m2, LV 200 mg/m2, IV bolus 5‐FU 400 mg/m2, followed by 5‐FU 2400 mg/m2 46‐hour continuous infusion, plus BEV 5 mg/kg D1, q14d (n randomised = 30) |
|
| Outcomes | PFS ORR (RECIST, version 1.0) Grade ≥ 3 AEs (AE assessments occurred up to 12 weeks) (CTCAE, version 3.0) Median follow‐up: 324 days (range, 41 to 843 days) |
|
| Study Details | Journal article and abstract/poster | |
| Funding sources and declarations of interest | Funding sources: Tohoku Clinical Oncology Research and Education Declarations of interest: Chugai Pharmaceutical Co., Ltd., and Novartis Pharma, Inc. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Dynamic allocation was performed (UMIN‐CTR UMIN000000770) |
| Allocation concealment (selection bias) | Low risk | Central allocation was used (UMIN‐CTR UMIN000000770) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Unblinded (UMIN‐CTR UMIN000000770) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: Low Quote: "Effectiveness was judged comprehensively using blinded tests on the treatment methods by 3 or more physicians not including primary physicians" (patients and methods, paragraph 7, page 103) (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR): Low Imaging was performed at the same time‐points in both arms. Physician assessments for PFS were performed using the same schedule in both arms (correspondence with Dr. Shunsuke Kato, received 15 October 2013) (ii) Survival (influences PFS): Low Physician assessments for PFS were performed using the same schedule in both arms (correspondence with Dr. Shunsuke Kato, received 15 October 2013) (iii) Grade ≥ 3 AEs: High Quote: "With regard to safety data, the patients’ health status was observed and blood samples were tested during weekly medical examinations by the attending physician until 4 weeks after commencing treatment and repeated after the fifth week at the start of each new course of treatment" (patients and methods, paragraph 7, page 103) Treatment cycles were every 3 weeks for the sequential IRIS‐BEV group and every 2 weeks for the mFOLFIRI‐BEV group (patients and methods, paragraph 3, page 103) Therefore, safety evaluations were performed more frequently in the mFOLFIRI‐BEV arm |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Low The reason for 'NE' (non‐evaluable) disease (Table 3, page 105) in 16.7% of the sequential IRIS‐BEV group and 13.3% of the mFOLFIRI‐BEV group was non‐measurable disease in all cases. No missing outcome data (correspondence with Dr. Shunsuke Kato, received 15 October 2013) (ii) PFS: Low Participants lost to follow‐up without progression were censored on the last day if it was confirmed that no progression or death occurred (correspondence with Dr. Shunsuke Kato, received 15 October 2013). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Low The maximum percentage of participants missing data for the grade ≥ 3 AEs of interest was 16.7% (Table 2, page 105). Similar number of participants were missing data in both arms (correspondence with Dr. Shunsuke Kato, received 15 October 2013) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Analysis for efficacy outcomes kept participants in the intervention groups to which they were randomised, regardless of the intervention received (correspondence with Dr. Shunsuke Kato, received 15 October 2013) Safety analysis: Safety analysis population comprised those with available grade ≥ 3 AE data (correspondence with Dr. Shunsuke Kato, received 15 October 2013) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Low (Table 1, page 104) (ii) Median age: Low (Table 1, page 104) (iii) No. of involved organs: Unclear ‐ not specified (Similar number of patients with "Number of metastases ‐ 1/2/3" in both arms) (Table 1, page 104) |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified |
Kim 2001a.
| Methods | Randomised controlled trial Phase: Not specified Accrual dates: October 1997 to February 1999 |
|
| Participants | No. randomised: 166 Stage/treatment type: Stage II/III resected rectal adenocarcinoma, adjuvant Countries/sites: South Korea, single site (Yonsei University College of Medicine) Setting: Hospital Characteristics (Arm I/II): Resected rectal adenocarcinoma; age < 70 years (median 52.3/59.5 years); male (61/64%); PS ECOG ≤ 2 |
|
| Interventions | Arm I: IV bolus 5‐FU 450 mg/m2/daily and leucovorin 20 mg/m2/d D1‐5, q28d for 12 cycles with *radiotherapy (n randomised = 74) Arm II: Oral doxifluridine 700 mg/m2/d with oral leucovorin 20 mg/m2/d D1‐21, q28d for 12 cycles with radiotherapy (n randomised = 92) *Radiotherapy commenced with C3 at a dose of 5400 cGy at 180 Gy/d, 5 days per week for 6 consecutive weeks |
|
| Outcomes | Grade ≥ 3 AEs (WHO criteria, version not specified) Median follow‐up: more than 15 months, range, 6 to 26 months. Less than 3 year follow‐up |
|
| Study Details | Journal article (in Korean) | |
| Funding sources and declarations of interest | Funding sources: None declared Declarations of interest: None declared |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization lists were stratified by a medical statistician, using randomly permuted blocks of varying sizes" (materials and methods, paragraph 1, page 675) |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | (i) DFS: This study was not used for the meta‐analysis for this outcome (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: High Not specified, blinding unlikely. Outcome assessment at risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) DFS: This study was not used for the meta‐analysis for this outcome (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: High Not specified. Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | Unclear risk | (i) Disease recurrence (influences DFS): Unclear ‐ not specified (ii) Survival (influences DFS/OS): This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) DFS: This study was not used for the meta‐analysis for this outcome (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Unclear risk | Efficacy analysis: Unclear Not specified. Note that the number of participants reported in the IV vs oral arm differed substantially‐ 74 vs 92 participants, respectively (Table 1, page 676) Safety analysis: Not specified |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | High risk | (i) PS: Unclear ‐ not specified (ii) Mean age: High 7.2 year difference in mean age between arms (Table 1, page 676) (iii) TNM stage: Low (stage II vs III) Quote: "There was no difference of TNM stage distribution between two groups of patients (P = .454); stage II was 25 in the IV arm and 41 in the oral arm; and stage III was 49 in the IV arm and 51 in the oral arm (results, paragraph 1, page 675) |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ similarity of therapy following recurrence or new occurrence of disease not specified |
Kohne 2008.
| Methods | Randomised controlled trial Phase: III Design: Factorial, 2 × 2 Accrual dates: May 2003 to April 2004 (closed 12 January 2005) |
|
| Participants | No. randomised: 85 Stage/treatment line: Metastatic, first‐line Countries/sites: European Organisation for Research and Treatment of Cancer (EORTC) study Setting: Hospital Characteristics (Arm I/II/III/IV): Metastatic colorectal cancer; age ≥ 18 years (median 66.0/60.5/63.0/65.0 yrs); male (79/64/52/57%); PS WHO 0‐2 (PS WHO 0: 53/64/61/57%) |
|
| Interventions | Arm I (FOLFIRI + celecoxib): irinotecan 180 mg/m2 D1, 15, 22, FA 200 mg/m2 D1, 2, 15, 16, 29, 30, IV bolus 5‐FU 400 mg/m2 followed by 22‐hour continuous infusion 600 mg/m2 D1, 2, 15, 16, 29, 30 + celecoxib 800 mg daily (n randomised = 19) Arm II: FOLFIRI plus placebo (n randomised = 22) Arm III (CAPIRI plus celecoxib): irinotecan 250 mg/m2 D1 and 22 and capecitabine 1000 mg/m2 bd D1‐15 and 22‐36 + celecoxib 800 mg daily (n randomised = 23) Arm IV: CAPIRI plus placebo (n randomised = 21) Treatment continued up to a planned total of 6 cycles or until PD, unacceptable toxicity, or withdrawal of consent. Participants with a response or SD were allowed to continue treatment beyond 6 cycles at the discretion of the investigator |
|
| Outcomes | PFS Grade ≥ 3 AEs (NCI CTC, version 2) ORR (RECIST, version 1.0) OS Median follow‐up: 14.6 months for all outcomes |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: Roche, Pharmacia (currently Pfizer), Aventis (currently Sanofi‐Aventis) Declarations of interest: None declared |
|
| Notes | "...after the enrolment of only 85 patients, recruitment was suspended as a consequence of seven deaths not due to disease progression. One more patient subsequently died following the suspension of recruitment. Six deaths occurred in patients receiving CAPIRI and two in those receiving FOLFIRI ... Five deaths in the CAPIRI arm and both of those in the FOLFIRI arm were deemed to be treatment related. Underlying risk factors could not be identified as a likely explanation for these fatal toxic effects. On the basis of the outcome of this review, the trial was officially closed on 12 January 2005" (results, paragraph 1, page 922) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to receive centrally using a minimization technique" (patients and methods, paragraph 4, page 922) |
| Allocation concealment (selection bias) | Low risk | Quote as above |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Not specified, blinding unlikely (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified. (i) ORR/PFS: High Outcome assessment at risk of bias if there was lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | Unclear risk | (i) Response (influences ORR/PFS): Low Quote: "Evaluation of disease status was carried out every 6 weeks during treatment and every 8 weeks subsequently until the documentation of disease progression" (patients and methods, paragraph 3, page 921) (ii) Survival (influences PFS/OS): Unclear ‐ not specified (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Low Response was not assessable or early death occurred in 8/44(18%) participants in the CAPIRI arm and in 4/41(10%) participants in the FOLFIRI arm (Table 3, page 924) (ii) PFS/OS: Low Quote: "Patients with no evidence of PD at the time of their last visit were censored at that time" (patients and methods, paragraph 3, page 922) (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "... assessed in the intention‐to‐treat (ITT) population" (patients and methods, paragraph 3, page 922) Quote: "...enrollment of ... 85 patients ..." (results, paragraph 1, page 922). This is the same number of participants presented in all of the efficacy analyses. (Table 3 and 4, page 924, and Figure 1, page 925) Safety analysis: Analyses included those who received study drug, quote: "Three patients (4%) did not receive study drugs and are therefore not included in the safety analysis" (results, paragraph 3, page 922) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 2, 923) (ii) Median age: Low (Table 2, 923) (iii) No. of involved organs: Low (Table 2, 923) |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified Risk of bias considerations in a factorial study: Unclear Quote: "...in view of the fact that response rates were lower in both celecoxib arms compared with the corresponding placebo arms for both regimens, it is possible that celecoxib may actually reduce the response to chemotherapy..." (discussion, paragraph 5, page 925) Quote: "Celecoxib did not appear to modulate the toxicity of the chemotherapy; thus it is very unlikely that the toxicity observed with CAPIRI was due to celecoxib" (discussion, paragraph 5, page 925) No tests for interaction were reported |
Lembersky 2006.
| Methods | Randomised controlled trial Phase: III Accrual dates: February 1997 to March 1999 |
|
| Participants | No. randomised: 1608 Stage/treatment type: Stage II/III adenocarcinoma of the colon, adjuvant Countries/sites: National Surgical Adjuvant Breast and Bowel Project (NSABP) study Setting: Hospital Characteristics (Arm I/II): resected Stage II/III colon cancer (age < 60 years: 41.7/41.2%); male (51.6/53.5%); PS ECOG ≤2 |
|
| Interventions | Arm I (FU/LV): LV 500 mg/m2 and IV bolus FU 500 mg/m2 weekly W1 to 6, q56d for 3 cycles (n randomised = 803) Arm II (UFT/LV): UFT 300 mg/m2/d and LV 90 mg/d D1‐28, q35d for 5 cycles (n randomised = 805) |
|
| Outcomes | OS DFS Grade ≥ 3 AEs (NCI toxicity criteria, 1958) Median follow‐up: 62.3 months among surviving patients |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: NSABP Foundation Declarations of interest: Taiho |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Not specified, blinding unlikely. (i) DFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified (i) DFS: High Outcome assessment at risk of bias if there was lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Disease recurrence (influences DFS): Low Quote: "Starting 6 months after the completion of protocol chemotherapy and continuing through 5 years after random assignment, patients were to be re‐evaluated semiannually. Five years after random assignment, a status of disease report was required on a yearly basis" (patients and methods, paragraph 4, page 2060) (ii) Survival (influences DFS/OS): Low Quote as above (iii) Grade ≥ 3 AEs: High Quote: "Before the administration of each cycle of chemotherapy, patients had a physical examination, CBCs, and chemistry profiles including hepatic and renal function studies. During active chemotherapy, patients in both groups underwent weekly CBCs" (patients and methods, paragraph 3, page 2060) Quote: "Patients randomly assigned to the FU+LV group received three 8‐week cycles of intravenous chemotherapy" (patients and methods, paragraph 2, page 2060) Quote: "Patients randomly assigned to the UFT+LV group received five 5‐week cycles ..." (patients and methods, paragraph 2, page 2060) Therefore, participants in the UFT+LV arm had more frequent safety evaluations than those in the FU+LV arm |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i)(ii) DFS/OS: Unclear Quote: "Our primary analyses include all patients who were eligible and had follow‐up information (intent to treat)" (patients and methods, paragraph 8, page 2060) However, no indication whether this follow‐up information was complete for all outcomes (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low ‐ Analyses were performed on what was described as an ’intent to treat’ population but included only those who were eligible and had follow‐up information. Quote: "Fifty patients (3.1%) were deemed ineligible (27 were assigned to the FU+LV arm, and 23 were assigned to the UFT+LV arm)." (results, paragraph 1, page 2060) Safety analysis: Analyses performed on those who were eligible and had follow‐up |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Unclear ‐ not specified (ii) Median age: Unclear ‐ not specified Baseline age was dichotomised into < 60 years vs ≥ 60 years (Table 1, page 2061) (iii) TNM stage: Low (stage II vs stage III and stage III N1 vs N2) N1 vs N2: Low (Table 1, page 2061) |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ similarity of therapy following recurrence or new occurrence of disease not specified |
Martoni 2006.
| Methods | Randomised controlled trial Phase: II Accrual dates: December 2001 to March 2005 |
|
| Participants | No. randomised: 118 Stage/treatment line: Metastatic, first‐line Countries/sites: Gruppo Oncologico Aree Metropolitane (GOAM) study Setting: Hospital Characteristics (Arm A/B): Metastatic colorectal adenocarcinoma; age > 18 years (median 64/67 years); male (50/53.2%); KPS ≥ 70% (Median 90/90%) |
|
| Interventions | Arm A (pviFOX): oxaliplatin 130 mg/m2 D1 and protracted venous infusion 5‐FU 250 mg/m2/d D1‐21, q21d (n randomised = 56) Arm B (XELOX): oxaliplatin 130 mg/m2 D1 and oral capecitabine 1000 mg/m2 bd D1‐14, q21d (n randomised = 62) Treatment continued for 6 cycles or until PD, at the investigators' discretion |
|
| Outcomes | ORR (RECIST, version 1.0) TTP Grade ≥ 3 AEs (CTC criteria, version 2.0) No details on median follow‐up |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: None declared Declarations of interest: No conflicts of interest |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Not specified, blinding unlikely (i) ORR/TTP: Low Outcome unlikely to be influenced by lack of blinding (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified (i) ORR/TTP: High Outcome assessment at risk of bias if there was lack of blinding (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | Low risk | (i) Response (influences ORR/TTP): Low Quote: "Every three cycles, re‐evaluation was scheduled with the recording of the symptoms, weight, KPS, physical examination and chest‐abdominal‐pelvic CT...." (patients and methods, paragraph 5, page 3163) Quote: "In both arms, the treatment was repeated every 21 days ..." (patients and methods, paragraph 3, page 3162) (ii) Survival (influences OS): Not applicable This was not an outcome of interest for this study (iii) Grade ≥ 3 AEs: Low Quote: " ... the recording of the symptoms, side‐effects and physical examination was carried out prior to each cycle, blood count and blood‐chemistry tests for liver and kidney function before and 10 days after each cycle. ..." (patients and methods, paragraph 5, page 3163) Quote: "In both arms, the treatment was repeated every 21 days ..." (patients and methods, paragraph 3, page 3162) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR/TTP: Low Response was not evaluable in 3/56 (5.4%) participants in the pviFOX arm and in 5/62 (8.1%) participants in the XELOX arm (Table 5, page 3166) (ii) OS: N/A Not applicable, as this was not an outcome of interest for this study (iii) Grade ≥ 3 AEs: Low Analyses were performed in those with evaluable data ‐ 54/56 in the pviFOX arm and 61/62 in the XELOX arm (Table 4, page 3165) Although the reasons for participants not having evaluable data were unclear, only a low percentage had non‐evaluable data. |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "Efficacy analyses were based on an intent‐to‐treat analysis" (patients and methods, paragraph 7, page 3164) Quote: "One hundred and twenty‐two patients were enrolled between December 2001 and March 2005. Four patients resulted [sic] ineligible and were excluded from the randomisation" (results, paragraph 1, page 3164) These 118 randomised participants were included in the efficacy analyses (Table 5 and Fig. 1, page 3166) Safety analysis: Analyses were performed in those with evaluable data ‐ 54/56 in the pviFOX arm and 61/62 in the XELOX arm (Table 4, page 3165) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | High risk | (i) PS: Unclear Reported as median KPS only (Table 1, page 3163) (ii) Median age: Low (Table 1, page 3163) (iii) No. of involved organs: High A 16.5% difference between arms with respect to the number of participants with 1 vs more than 1 metastatic site (30/56 (53.6%) in the pviFOX arm and 23/62 (37.1%) in the XELOX arm) (Table 1, page 3163) |
| Other bias | Unclear risk | Subsequent therapies: Quote: "...60 have received a second‐line chemotherapy, 25 and 35 in arms A and B, respectively" (results, paragraph 7, page 3165) Similar proportions in each arm received second‐line therapy, but OS was not an outcome in this study |
Mei 2014.
| Methods | Randomised controlled trial Phase: Not specified Accrual dates: June 2010 to September 2012 |
|
| Participants | No. randomised: 70 Stage/treatment line: Locally advanced or metastatic; first‐line chemotherapy for locally advanced or metastatic CRC (if recurrence occurred after neoadjuvant therapy or adjuvant chemotherapy an interval of at least 6 months from completing therapy was mandated) Countries/sites: Single‐centre study in China (The No. 3 People's Hospital of Zhengzhou, Zhengzhou Tumour Hospital) Setting: Hospital Characteristics (Arm I/II): Locally advanced or metastatic; age 18 to 75 years; PS ECOG 0‐1 (unclear) |
|
| Interventions | Arm I (SOX): L‐OHP 130 mg/m2 D1 IV infusion, S‐1 oral for 14 days. S‐1 dose calculated according to BSA: 80 mg/d for BSA < 1.25 m2; 100 mg/d for BSA ≥ 1.25 m2 but < 1.5 m2; 120 mg/d for BSA ≥ 1.5 m2 but < 1.8 m2; 140 mg/d for BSA > 1.8 m2. Schedule repeated every 3 weeks (n randomised = 35) Arm II (FOLFOX4): L‐OHP 85 mg/m2 IV D1, leucovorin (CF) 200 mg/m2 IV D1‐2, 5‐FU 400 mg/m2 IV bolus D1‐2, infusional 5‐FU 1200 mg/m2 IV continuous over 44 hours (n randomised = 35) Schedule repeated every 2 weeks |
|
| Outcomes | Grade ≥ 3 AEs (NCI‐CTC, version 3.0) ORR (reported only after 2 cycles of chemotherapy) (RECIST, version not specified) No details on median follow‐up |
|
| Study Details | Journal article (in Chinese) | |
| Funding sources and declarations of interest | Funding sources: None declared Declarations of interest: None declared |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Unclear ‐ not specified |
| Allocation concealment (selection bias) | Unclear risk | Unclear ‐ not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Not specified, blinding unlikely (i) ORR: This study was not used for the meta‐analysis for this outcome (ii) OS: Not an outcome for this study (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified. (i) ORR: This study was not used for the meta‐analysis for this outcome (ii) OS: Not an outcome for this study (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | Unclear risk | (i) Response (influences ORR): This study was not used for the meta‐analysis for this outcome (ii) Survival: No survival outcomes in this study (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: This study was not used for the meta‐analysis for this outcome (ii) OS: Not an outcome for this study (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Unclear risk | Efficacy analysis: Unclear, but not an outcome for this study Safety analysis: Quote (translated from Chinese to English): "70 cases are randomly allocated into trial group and control group, each 35 cases" (information and methods, paragraph 1, page 821) Denominator appears to be the randomised population in Table 1, page 821 |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Similarity of arms at baseline | Unclear risk | All Unclear ‐ not specified |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified |
Nogue 2005.
| Methods | Randomised controlled trial Phase: Not specified (journal), abstracts stated phase IV Accrual dates: September 1997 to December 2000 |
|
| Participants | No. randomised: 237 Stage/treatment line: Metastatic/unresectable, first‐line Countries/sites: Spain, 16 sites Setting: Hospital Characteristics (Arm I/II): Metastatic/unresectable colorectal cancer; age ≥ 18 years (median 67/68 years); male (62/68%); KPS ≥ 60% (KPS 100%: 27/29%) |
|
| Interventions | Arm I (FT/LV): Tegafur 750 mg/m2/d D1‐21, q28d with LV 15 mg tds (n randomised = 114) Arm II (5‐FU/LV): IV bolus 5‐FU 425 mg/m2 D1‐5 with LV 20 mg/m2, q28d for 2 cycles, then q35d thereafter (n randomised = 123) Treatment continued until PD, unacceptable toxicity, or withdrawal of consent |
|
| Outcomes | ORR (WHO criteria, 1981) TTP OS Grade ≥ 3 AEs (NCI CTC, version 2.0) No details on median follow‐up |
|
| Study Details | Journal article and abstract | |
| Funding sources and declarations of interest | Funding sources: Prasfarma Almirall‐Prodesfarma, Spain Declarations of interest: No conflicts of interest |
|
| Notes | Owing to slow enrolment, recruitment was suspended after 85% of the expected sample size (246 participants) was randomised. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned in blocks of 4 and stratified by center ..." (patients and methods, paragraph 4, page 2242) |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients ... were centrally randomised to treatment with either oral FT/LV or i.v. 5‐FU/LV" (patients and methods, paragraph 4, page 2242) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "This was a randomised, multicenter, open‐label clinical trial ..." (patients and methods, paragraph 1, page 2242) (i) ORR/TTP: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/TTP: High Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/TTP): High Quote: "Responses were evaluated every 3 cycles" (Salud et al, Journal of Clinical Oncology 2004‐ slides associated with abstract 3547) In the FT/LV arm, treatment was given in 28‐day cycles. In the 5‐FU/LV arm, treatment was given in 28‐day cycles for 2 cycles, and in 35‐day cycles thereafter (patients and methods, paragraph 5, page 2242) Therefore, after cycle 2, response assessments were performed more frequently in the FT/LV treatment arm (ii) Survival (influences OS): Unclear ‐ not specified (iii) Grade ≥ 3 AEs: High Quote: "Before each cycle, adverse events were documented and a physical examination, differential blood count and blood biochemistry test were performed" (patients and methods, paragraph 7, page 2243) In the FT/LV arm, treatment was given in 28‐day cycles. In the 5‐FU/LV arm, treatment was given in 28‐day cycles for 2 cycles, and in 35‐day cycles thereafter (patients and methods, paragraph 5, page 2242) Therefore, after cycle 2, safety assessments were performed more frequently in the FT/LV treatment arm |
| Incomplete outcome data (attrition bias) All outcomes | High risk | (i) ORR/TTP: High 27/114 (24%) participants in the FT/LV arm and 24/123 (20%) in the 5‐FU/LV arm were not evaluable for response owing to “deviations from protocol in the response evaluation methodology” (Fig. 1, page 2244) (ii) OS: Low No participants in the FT/LV arm and 2/123 (1.6%) in the 5‐FU/LV arm were not evaluable for survival (Fig. 1, page 2244) Censoring was noted in the KM curves for OS (Fig. 2, page 2246). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | High risk | Efficacy analysis: High Quote: "All efficacy analyses were conducted on the intent‐to‐treat (ITT) population" (patients and methods, paragraph 10, page 2243) However, 0/144 (0%) and 2/123 (2%) participants randomised to the FT/LV and 5‐FU/LV arms, respectively, were excluded from the survival analysis owing to being unevaluable (Fig. 1, page 2244). The 27/144 (24%) and 24/123 (20%) participants randomised to the FT/LV and 5‐FU/LV arms, respectively, who were not evaluable for response were excluded from the analysis (Fig. 1, page 2244) Safety analysis: Quote: "Toxicity analyses were performed on patients who received at least one dose of study treatment (safety population)" (patients and methods, paragraph 10, page 2243) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, page 2245) (ii) Median age: Low (Table 1, page 2245) (iii) No. of involved organs: Low (Table 1, page 2245) |
| Other bias | Low risk | Subsequent therapies: Low Quote: "Overall, 94 of 237 treated patients continued receiving treatment after the end of the study with second‐line agents. In the FT/LV treatment arm, 48 patients were mainly treated with irinotecan and combination of oxaliplatin + irinotecan in 27% and 20% of cases, respectively. Likewise, in the 5‐FU/LV treatment arm, 46 patients involved in second‐line therapy received irinotecan and combination of oxaliplatin + irinotecan in 30.5% and 28.3% of cases, respectively" (results, paragraph 3, pages 2243 and 2244) |
Pectasides 2012.
| Methods | Randomised controlled trial Phase: III Accrual dates: January 2006 to January 2008 |
|
| Participants | No. randomised: 302 Stage/treatment line: Metastatic, first‐line Countries/sites: Greece, multiple sites Setting: Hospital Characteristics (Arm A/B): Metastatic colorectal cancer; age ≥ 18 years (median 66/66 years); male (55/65%); PS ECOG 0‐2 (PS ECOG 0: 64/66%) |
|
| Interventions | Arm A (XELIRI + bevacizumab (BEV)): irinotecan 240 mg/m2 D1, capecitabine 1000 mg/m2 D1‐14 and BEV 7.5 mg/kg D1, q21d, up to 6 cycles (n randomised and eligible = 143) Arm B (FOLFIRI + BEV): irinotecan 180 mg/m2 D1, leucovorin 200 mg/m2 D1, IV bolus fluorouracil 400 mg/m2 followed by fluorouracil 2400 mg/m2 46‐hour infusion, and BEV 5 mg/kg D1, q14d, up to 12 cycles (n randomised and eligible = 142) Single agent BEV was administered as maintenance until unacceptable toxicity or PD |
|
| Outcomes | PFS ORR (RECIST, version 1.0) OS Grade ≥ 3 AEs (NCI CTC, version 2.0) Median follow‐up: 42 months |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: Hellenic Oncology Research Group (HeCOG) Declarations of interest: Pfizer, Roche Hellas SA, Genesis Pharma SA |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Investigators used stratified blocked randomisation balanced by centre (correspondence with Anastasia Eleftheraki, received 6 July 2012) |
| Allocation concealment (selection bias) | Low risk | Quote: " ... randomization, done centrally at the HeCOG Data Office ..." (methods, paragraph 3, page 2) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: Low This was an open‐label study (correspondence with Anastasia Eleftheraki, received 6 July 2012) Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: High As above, this was an open‐label study (correspondence with Anastasia Eleftheraki, received 6 July 2012) Quote: "No central review of the imaging material was done" (methods, paragraph 5, page 2) Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/PFS): High Quote: "Disease evaluation was carried out after 3 cycles of treatment in group A and after 6 cycles in group B, at the end of treatment, and every 3 months thereafter by chest X‐rays and CT scans of the abdomen and pelvis" (methods, paragraph 5, page 2) Treatments were quote: " ...repeated every 21 days for 6 cycles (group A, XELIRI) or ... repeated every 14 days for 12 cycles (group B, FOLFIRI)" (methods, paragraph 3, page 2) Therefore, disease assessment was performed more frequently in the XELIRI + BEV arm than in the FOLFIRI + BEV arm during treatment (every 9 weeks vs every 12 weeks) (ii) Survival (influences PFS/OS): Low The follow‐up schedule for survival assessment was the same in both arms ‐ approximately every 6 months (correspondence with Anastasia Eleftheraki, received 6 July 2012) (iii) Grade ≥ 3 AEs: High Participants were examined for adverse events every cycle of treatment and 1 month after last treatment administration (both groups) (correspondence with Anastasia Eleftheraki, received 6 July 2012) Treatments were quote: "...repeated every 21 days for 6 cycles (group A, XELIRI) or ... repeated every 14 days for 12 cycles (group B, FOLFIRI)" (methods, paragraph 3, page 2) Therefore, disease assessment was performed more frequently in the FOLFIRI + BEV arm than in the XELIRI + BEV arm during treatment (every 2 weeks vs every 3 weeks) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | (i) ORR/PFS: High Quote: "In group A, 43 patients (30.1%) were not evaluated for response because of treatment discontinuation (24 patients, 16.8%), early death (5, 3.5%), missing data (3, 2.1%), or non‐evaluable disease (11, 7.7%). In group B, 28 patients (19.7%) were not evaluated for response because of treatment discontinuation (5 patients, 3.5%), early death (2, 1.4%), or non‐evaluable disease (21, 14.8%) (results, paragraph 3, page 5) (ii) OS: Low Although not defined in the Methods, censoring was noted in the KM curves for OS (Figure 2, page 5). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Low Outcome data were missing for 5/138 participants in the XELIRI + BEV arm and for 2/132 in the FOLFIRI + BEV arm (results, paragraph 2, page 5 and Table 2, page 6) |
| Incomplete outcome data (ITT analysis) | High risk | Efficacy analysis: High Quote: "PFS, OS and ORR were analyzed on an intent‐to‐treat basis..." (methods, paragraph 10, page 4) However, whilst 302 participants were randomised, 17 of these participants were excluded owing to ineligibility. This left 285 participants who were eligible and included in the analysis for all efficacy outcomes (Figure 1, page 3) Safety analysis: Quote: " ...in the safety analysis ... only the treated population was included" (methods, paragraph 10, page 4) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, page 4) (ii) Median age: Low (Table 1, page 4) (iii) No. of involved organs: Low (Table 1, page 4) |
| Other bias | Unclear risk | Subsequent therapies: Unclear Only information on maintenance BEV was reported Quote: "In total, 64 patients in group A and 46 in group B received Bev as maintenance for a median of 4 cycles (range, 1–35) and 6 cycles (range, 2–48), respectively" (results, paragraph 2, page 5) |
Pectasides 2015.
| Methods | Randomised controlled trial Phase: III Accrual dates: November 2005 to January 2008 |
|
| Participants | No. randomised: 441 Stage/treatment type: High‐risk AJCC stage II or AJCC stage III CRC, adjuvant Countries/sites: Greece, multiple sites Setting: Hospital Characteristics: Characteristics (Arm A/B): High‐risk AJCC stage II CRC (high histological grade, lymphovascular/perineural invasion, mucinous component, T4 stage, extramural vein invasion, symptomatic bowel obstruction or perforation at diagnosis, < 12 lymph nodes removed); or AJCC stage III; age 18 to 75 years (median 62.4/63.7 years); male (56.4/55.5%); PS ECOG 0‐1 (PS ECOG 0 91.7/93.7%) |
|
| Interventions | Arm I (XELOX): capecitabine 1000 mg/m2 bd D1‐14 and oxaliplatin 130 mg/m2 D1, q21d for 8 cycles (n randomised and eligible = 211) Arm II (mFOLFOX6): leucovorin 200 mg/m2, IV bolus 5‐FU 400 mg/m2 and 5‐FU 600 mg/m2 22‐hour infusion D1 and D2 plus oxaliplatin 85 mg/m2 D1, q14d for 12 cycles (n randomised and eligible = 197) |
|
| Outcomes | 3‐year DFS OS Grade ≥ 3 AEs (NCI CTC, version 2.0) Median follow‐up: 74.7 months (range, 0 to 155.5 months) |
|
| Study Details | Journal article and abstract | |
| Funding sources and declarations of interest | Funding sources: Hellenic Oncology Research Groups Declarations of interest: No conflicts of interest |
|
| Notes | The accrual target was 824, but the study was closed prematurely after enrolment of 441 participants owing to slow accrual | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Low risk | Quote: "...randomization, done centrally at the Hellenic Cooperative Oncology Group (HeCOG) data office" (methods, paragraph 2, page 2) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Not specified, blinding unlikely. (i) DFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified (i) DFS: High Outcome assessment at risk of bias if there was lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | Unclear risk | (i) Disease recurrence (influences DFS): Low Quote: "Follow‐up evaluation for disease recurrence was carried out after the completion of treatment in all patients, every 3 months for the first year, every 4 months for the second and third year and every 6 months for the fourth and fifth year ..." (methods, paragraph 4, page 3) (ii) Survival (influences DFS/OS): Unclear ‐ not specified (iii) Safety: Unclear ‐ not specified |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) Recurrence: Unclear ‐ not specified (ii) DFS/OS Censoring was noted in the KM curves (Fig. 2, page 8). There was no evidence of bias related to censoring (ii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | High risk | Efficacy analysis: High Quote: "Analyses of survival parameters and objective response rates were performed in all randomized patients (intention to treat, ITT population) ..." (methods, paragraph 14, page 4) However, this was not an ITT analysis as defined in our review 33 (7.5%) randomised participants were excluded from the analysis owing to ineligibility (Table 1, page 5). Furthermore, whilst the efficacy analysis population was described in the text as all randomised participants, the number of participants at risk at t(0) for PFS and OS in Figure 2 is not consistent with this. 4/197 (2%) and 2/211 (1%) of participants randomised to the mFOLFOX6 and CAPOX arms, respectively, were excluded from the DFS analysis. 4/197 (2%) and 3/211 (1%) of participants randomised to the mFOLFOX6 and CAPOX arms, respectively, were excluded from the OS analysis (Fig. 2, page 8) Safety analysis: Quote: "...analyses of toxicity ... were performed only in patients who did receive treatment (treated patient population)" (methods, paragraph 14, page 4) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Similarity of arms at baseline | Low risk | (i) PS: Low 2% difference in ECOG 1 (Table 1, page 6) (ii) Median age: Low (Table 1, page 6) 1.3 year difference (iii) TNM: Low (stage II vs stage III, stage II T3 vs T4, stage III N1 vs N2) (Table 1, page 6) |
| Other bias | Unclear risk | Subsequent therapies: Unclear regarding subsequent drug therapy after a recurrence or a new occurrence of colorectal cancer |
Porschen 2007.
| Methods | Randomised controlled trial Phase: III Accrual dates: August 2002 to August 2004 |
|
| Participants | No. randomised: 476 Stage/treatment line: Metastatic, first‐line Countries/sites: Germany, 68 sites; Austria, 1 site Setting: Hospital Characteristics (Arm A/B): Metastatic colorectal cancer; age > 18 years (median 64/66 years); male (63/62%); PS ECOG 0‐2 (PS ECOG 0‐1: 93/91%) |
|
| Interventions | Arm A (FUFOX): oxaliplatin 50 mg/m2, leucovorin 500 mg/m2, and 22‐hour infusional FU 2000 mg/m2 D1, 8, 15, and 22, q35d. After cycle 4, oxaliplatin on D1 and 15 of cycle only (n randomised = 234) Arm B (CAPOX): capecitabine 1000 mg/m2 bd D1‐14 and oxaliplatin 70 mg/m2 D1 and 8, q21d. After cycle 6, oxaliplatin on D1 only (n randomised = 242) Treatment continued until PD or severe toxicity |
|
| Outcomes | ORR (RECIST, version 1.0) PFS OS Grade ≥ 3 AEs (NCI CTC, version 2) Median follow‐up: 17.3 months |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: Hoffman La‐Roche, Sanofi‐Aventis Declarations of interest: Roche, Sanofi‐Aventis |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "computer‐based randomization was performed centrally by fax" (patients and methods, paragraph 4, page 4218) |
| Allocation concealment (selection bias) | Low risk | Quote as above |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Not specified, blinding unlikely (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at high risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified (i) ORR/PFS: High Outcome assessment at risk of bias if there was lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/PFS): High Quote: "During therapy, tumor assessments were repeated after three cycles of CAPOX and two cycles of FUFOX ...Follow‐up for disease progression and survival monitoring were performed every 3 months after the end of treatment" (patients and methods, paragraph 5, page 4218) Arm A (FUFOX) was given in 5‐week cycles, and Arm B (CAPOX) was given in 3‐week cycles (Table 1, page 4218) Therefore, tumour assessments were performed more frequently in the CAPOX arm than in the FUFOX arm during treatment (every 9 weeks vs every 10 weeks) (ii) Survival (influences PFS/OS): Low Quote: "Follow‐up for disease progression and survival monitoring were performed every 3 months after the end of treatment" (patients and methods, paragraph 5, page 4218) (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Unclear ‐ not specified (ii) PFS/OS: Low Although not defined in the Methods, censoring was noted in the KM curve for PFS (Fig. 3, page 4220). No evidence of bias related to censoring. (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "The efficacy analysis was based on the intent‐to‐treat population" (patients and methods, paragraph 8, page 4218) 476 participants were randomly assigned, and subsequently, 2 participants were excluded from the allocation population because they did not meet inclusion criteria. Analysis was performed on a population that excluded a further 4 participants owing to withdrawal of consent, and loss to follow‐up after random assignment (excluded 6/476, 1.3%) (Fig. 1, page 4219). Analysis does include participants who were allocated to a treatment arm but did not receive that treatment (total of 239 participants in the CAPOX arm, and 231 in the FUFOX arm) (Fig. 1, page 4219) Safety analysis: Not specified |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Unclear Reported as PS ECOG 0‐1 vs 2 (Table 2, page 4220) (ii) Median age: Low (Table 2, page 4220) (iii) No. of involved organs: Low (Table 2, page 4220) |
| Other bias | Low risk | Subsequent therapies: Low A similar proportion of participants in each arm received the same types of post‐progression treatment Quote: "In both the CAPOX and FUFOX arms, 66% of patients received second‐line treatment. Most of the patients received irinotecan‐based chemotherapy in the second‐line treatment (81% in both CAPOX and FUFOX arms). Additional treatments included reintroduction with oxaliplatin (CAPOX, 13%; FUFOX, 21%), cetuximab (CAPOX, 22%; FUFOX, 21%), or mitomycin (CAPOX, 9%; FUFOX, 9%). On subsequent treatment lines, patients in the CAPOX arm changed to FU (43%) and 29% continued with capecitabine. In the FUFOX arm, 56% continued with FU and 30% received capecitabine. In total, 56% patients of the entire study population received all three drugs: fluoropyrimidine, oxaliplatin, and irinotecan (CAPOX, 57%; FUFOX, 55%)" (results, paragraph 12, pages 4219 and 4220) |
Rothenberg 2008.
| Methods | Randomised controlled trial Phase: III Accrual dates: July 2003 to May 2005 |
|
| Participants | No. randomised: 627 Stage/treatment line: Metastatic, second‐line Countries/sites: 19 countries, 87 centres ‐ Oceania, Central and Eastern Asia, South Africa, Canada, USA, Israel, Mexico, South America, Europe Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal cancer; age ≥ 18 years (median 60.7/59.7 years); male (62/61%); PS ECOG ≤ 2 (PS ECOG 0: 48/46%) |
|
| Interventions | Arm I (XELOX): oxaliplatin 130 mg/m2 D1 plus oral capecitabine 1000 mg/m2 bd D1‐15, q21d, up to 24 weeks of treatment. Could receive treatment beyond week 24 in a post‐study treatment phase until PD (n randomised = 313) Arm II (FOLFOX‐4): oxaliplatin 85 mg/m2 D1 and LV 200 mg/m2/d, IV bolus 5FU 400 mg/m2/d and 22‐hr infusion 600 mg/m2/d D1‐2, q14d. Post study treatment as per Arm I (n randomised = 314) Treatment continued until PD, intolerable AEs, or participant refusal |
|
| Outcomes | PFS OS ORR (RECIST, version 1.0) Grade ≥ 3 AEs (NCI‐CTCAE, version 3) Median follow‐up: 25.7 months |
|
| Study Details | Journal article and abstract | |
| Funding sources and declarations of interest | Funding sources: Roche Declarations of interest: None declared |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Dynamic randomization was used to assign patients to treatment" (patients and methods, paragraph 5, page 1721) |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "The study was open label because of the different routes of administration of the fluoropyrimidine components of these regimens" (patients and methods, paragraph 1, page 1721) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: Quote: "The study was open label because of the different routes of administration of the fluoropyrimidine components of these regimens" (patients and methods, paragraph 1, page 1721) Quote: "Assessments of tumor response were made by investigators and also by an independent response review committee (IRC) that was blinded to treatment assignment" (patients and methods, paragraph 9, page 1721) ‐ Low for ORR Quote: "PFS was the primary end point of the study and was defined as the time from the date of randomization to the first documentation of disease progression by the investigators or death from any cause" (patients and methods, paragraph 12, page 1721) It is unclear if PFS was assessed by a local investigator. If so, this outcome assessment would be at risk of bias from lack of blinding ‐ Unclear for PFS (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | Unclear risk | (i) Response (influences ORR/PFS): Low Quote: "Assessments were then repeated using the same imaging technique approximately every 6 weeks and again within 2 weeks of study completion, withdrawal or treatment discontinuation....Confirmation of response was required after a minimum of 4 weeks" (patients and methods, paragraph 9, page 1721) (ii) Survival (influences PFS/OS): Low Quote: "After completion of study treatment, patients were followed up every 3 months until disease progression or death" (patients and methods, paragraph 9, page 1721) (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Low 13.1% of participants in the XELOX arm and 13.7% in the FOLFOX‐4 arm were missing response data (Rothenberg et al, Journal of Clinical Oncology 2007‐slides associated with abstract 4031) (ii) PFS/OS: Unclear ‐ not specified (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "The intention‐to‐treat (ITT) patient population included all patients who underwent randomization" (patients and methods, paragraph 11, page 1721) The ITT population was used for all efficacy analyses, included PFS (results, paragraph 6, page 1722; Figure 2A, page 1724; Table 2, page 1723), OS (results, paragraph 7, page 1722; Figure 2B, page 1724; Table 2, page 1723) and ORR (results, paragraph 8, page 1723 and Table 2, page 1723) Safety analysis: Quote: "The safety population was defined as all patients receiving at least one dose of study drug" (patients and methods, paragraph 11, page 1721) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, page 1723) (ii) Median age: Low (Table 1, page 1723) (iii) No. of involved organs: Low (Table 1, page 1723) |
| Other bias | Low risk | Subsequent therapies: Low A similar proportion of participants in both arms received further therapy after progression Quote: "A similar proportion of patients in the two treatment groups received further anticancer therapy after discontinuing study treatment (60% with XELOX and 62% with FOLFOX‐4), including drug therapy, surgery and radiotherapy. The most commonly used treatments were 5‐FU (25% in the XELOX group versus 25% in the FOLFOX‐4 group), capecitabine (10% versus 26%), irinotecan (16% versus 21%), cetuximab (15% versus 19%), oxaliplatin (17% versus 14%), radiotherapy (18% versus 14%) and bevacizumab (6% versus 7%)" (results, paragraph 9, page 1723) |
Schilsky 2002a.
| Methods | Randomised controlled trial Phase: III Accrual dates: October 1997 to May 1999 |
|
| Participants | No. randomised: 981 Stage/treatment line: Metastatic, first‐line Countries/sites: USA and Canada, 136 centres Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal cancer; age ≥ 18 years (median 64/64 years); KPS ≥ 70% (KPS 100%: 30/32%) |
|
| Interventions | Arm I (EU/5‐FU): eniluracil 11.5 mg/m2 and 1.15 mg/m2 5‐FU oral bd D1‐28, q35d (n randomised and treated = 485) Arm II (5‐FU/LV): 20 mg/m2 LV and IV 5‐FU 425 mg/m2 D1‐5, q28d (n randomised and treated = 479) Continue treatment until PD, unacceptable toxicity, or withdrawal of consent |
|
| Outcomes | OS ORR (SWOG criteria, 1992 ‐ adapted) PFS Grade ≥ 3 AEs (SWOG criteria, 1992 ‐ adapted) Median follow‐up: N/A |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: None declared Declarations of interest: None declared |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "This was a randomized, multicenter, open‐label, phase III trial..." (patients and methods, paragraph 1, page 1520) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs:High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: Low An independent review panel, blinded to treatment allocation, reviewed all responses (correspondence with Dr. Jeremey Levin, received 23 July, 2012) (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/PFS): High Quote: "Efficacy assessments were performed at baseline and repeated at the beginning of course 3 and every other course thereafter until discontinuation of treatment ... confirmed objective response required two consecutive assessments performed at least 4 weeks apart." (patients and methods, paragraph 6, page 1520) Quote: "Patients randomized to the EU/5‐FU treatment arm received 5‐week courses ... Patients randomized to the 5‐FU/LV treatment arm ... 28‐day cycle" (patients and methods, paragraph 3, page 1520) Therefore, participants in the 5FU/LV arm had more frequent disease assessments (ii) Survival (influences PFS/OS): Low Quote: "At the end of study treatment, all patients were followed quarterly for survival" (patients and methods, paragraph 7, page 1520) (iii) Grade ≥ 3 AEs: High While on treatment, participants were evaluated for safety at the beginning of each cycle, and a final evaluation was performed approximately 28 days after the last dose of study drug in both arms (correspondence with Dr. Jeremey Levin, received 23 July, 2012) Quote: "Patients randomized to the EU/5‐FU treatment arm received 5‐week courses ... Patients randomized to the 5‐FU/LV treatment arm ... 28‐day cycle" (patients and methods, paragraph 3, page 1520) Therefore, participants in the 5FU/LV arm had more frequent safety assessments |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Low Unknown/unable to determine responses in 8% of participants in the EU/5‐FU arm and in 10% in the 5‐FU/LV arm because of loss to follow‐up, withdrawal of consent, or incomplete measurements (Table 4, page 1522) (ii) PFS/OS: Low Quote: "If a patient had not died, duration of survival was censored on the date of last contact" (patients and methods, paragraph 9, page 1520). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Analysis for each outcome kept participants in the intervention groups to which they were randomised, regardless of the intervention received (correspondence with Dr. Jeremey Levin, received 23 July, 2012) However, quote: "Efficacy and safety were summarized for all patients who received at least one dose of study drug" (patients and methods, paragraph 9, page 1520). Of 981 participants randomised, 964 were treated and included in the analyses (1.7% excluded) (results, paragraphs 1 and 6, pages 1521 and 1522) Safety analysis: Analyses as per Efficacy, in all participants who received at least 1 dose of study drug |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, page 1521) (ii) Median age: Low (Table 1, page 1521) (iii) No. of involved organs: Low (Table 1, page 1521) |
| Other bias | Low risk | Subsequent therapies: Low A similar proportion of participants in both arms received further therapy after progression (Table 3, page 1521) |
Seymour 2011.
| Methods | Randomised controlled trial Phase: Not specified Design: Factorial, 2 × 2 Accrual dates: January 2004 to July 2006 |
|
| Participants | No. randomised: 459 Stage/treatment line: Metastatic, first‐line Countries/sites: UK, 61 centres Setting: Hospital Characteristics (Arm A/B/C/D): Metastatic colorectal cancer; “elderly and frail” population considered by the treating oncologist to be unsuitable for upfront full‐dose chemotherapy"; no upper or lower age limit (median 75/75/73/75 years); male (63/60/59/60%); PS WHO ≤ 2 (PS WHO 0: 22/20/20/24%) |
|
| Interventions | Arm A (FU): IV levofolinate 175 mg, IV bolus fluorouracil 320 mg/m2 and fluorouracil 2240 mg/m2 46‐hour infusion, q14d (n randomised = 115) Arm B (OxFU): IV levofolinate 175 mg/m2, oxaliplatin 68 mg/m2, bolus fluorouracil 320 mg/m2, and fluorouracil 1920 mg/m2 46‐hour infusion, q14d (n randomised = 115) Arm C (Cap): capecitabine 1000 mg/m2 bd D1‐15, q21d (n randomised = 115) Arm D (OxCap): oxaliplatin 104 mg/m2 D1 and capecitabine 800 mg/m2 bd D1‐15, q21d (n randomised = 114) Treatment continued until PD or clinical deterioration In Arms A and B, second‐line treatment was considered with OxFU or OxCap, respectively, upon progression |
|
| Outcomes | ORR (reported for after 12‐14 weeks) (RECIST, version 1.0) PFS OS Grade ≥ 3 AEs (CTCAE, version 3.0) No details on median follow‐up |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: Cancer Research UK, National Institute of Health Research. Drugs and infusors supplied by Wyeth and Baxter Declarations of interest: Roche, Sanofi‐Aventis, UK MRC, British Geriatrics Society |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was done by use of the method of minimisation" (methods, paragraph 5, page 1750) |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were randomly assigned in a 1:1:1:1 ratio by telephone with a computerised algorithm developed and maintained centrally at the MRC CTU" (methods, paragraph 5, page 1750) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "Treatment allocation was not masked" (methods, paragraph 5, page 1750) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding This study was not used for the meta‐analysis of the ORR outcome (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: High Outcome assessment at risk of bias from lack of blinding This study was not used for the meta‐analysis of the ORR outcome (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/PFS): Low Quote: "After week 12, radiological response was assessed ..." (methods, paragraph 8, page 1751) This study was not used for the meta‐analysis of the ORR outcome (ii) Survival (influences PFS/OS): Low Quote: "Thereafter, patients without radiological or clinical evidence of deterioration could continue the same regimen, immediately or after a planned break, with reassessment every 12 weeks" (methods, paragraph 9, page 1751) (iii) Grade ≥ 3 AEs: High Quote: "Before each cycle, toxicity was scored ..." (methods, paragraph 7, page 1750) Quote: "The cycle was repeated every 14 days (FU regimen)" (methods, paragraph 6, page 1750) Quote: "The cycle was repeated every 14 days (OxFU regimen)" (methods, paragraph 6, page 1750) Quote: "The cycle was repeated every 21 days (OxCap regimen)" (methods, paragraph 6, page 1750) Quote: "The cycle was repeated every 21 days (Cap regimen)" (methods, paragraph 6, page 1750) Therefore, safety assessments were performed more frequently in the FU and OxFU groups than in the OxCap and Cap arms |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Unclear Quote: "RR and toxic effects are reported as percentage of assessable patients..." (methods, paragraph 13, page 1752). However, the latter was not specified. This study was not used for the meta‐analysis of the ORR outcome (ii) PFS/OS: Low Quote: "For time‐to‐event endpoints, Kaplan‐Meier curves were produced with patients alive and event‐free being censored at the time last seen" (methods, paragraph 13, page 1751). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Low Quote: "440 (96%) patients had complete data for toxic effects" (results, paragraph 7, page 1754) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "PFS was defined as time from randomisation to first progression or death from any cause, assessed by intention to treat" (methods, paragraph 11, page 1751). Furthermore, the same number randomised (Figure 1, page 1751) are included in the analysis population for PFS and OS (Figure 2, page 1753) Safety analysis: Analyses were presented for those with complete data (440/459, 96%) (results, paragraph 7, page 1754) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Low (Table 1, page 1752) (ii) Median age: Low (Table 1, page 1752) (iii) No. of involved organs: Unclear ‐ not specified |
| Other bias | Low risk | Subsequent therapies: Low Quote: "In groups A and C, when progression occurred on the FU or Cap regimens, second‐line treatment was considered with the OxFU or OxCap regimens, respectively. Second‐line therapy in groups B and D, and third‐line therapy in all groups, was at the discretion of the physician" (methods, paragraph 9, page 1751) The salvage therapies described in Figure 1, page 1751, are comparable across the arms Risk of bias considerations in a factorial study: Safety ‐ Low Quote: "Tests for interaction between the two treatment factors showed no evidence of an interaction" (Table 4, page 1755) |
Shigeta 2016.
| Methods | Randomised controlled trial Phase: II Accrual dates: November 2007 to October 2011 |
|
| Participants | No. randomised: 72 Stage/treatment line: Metastatic, first‐line Countries/sites: 1 university hospital and 7 affiliated hospitals in Japan Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal cancer; age 20‐75 years (median 62/67 years); male (58/63%); PS ECOG 0‐1 (PS ECOG 0: 92/94%) |
|
| Interventions | Arm I (FOLFIRI plus bevacizumab (BEV)): BEV 5 mg/kg IV infusion, irinotecan 150 mg/m2, 400 mg/m2 bolus fluorouracil and 2400 mg/m2 infusional fluorouracil (46 hours) (n randomised = 36) Arm II (TEGAFIRI plus BEV): BEV 5 mg/kg IV infusion, irinotecan 150 mg/m2 IV, UFT/LV given oral TDS for 3 weeks, followed by a 7‐day break. LV dose was 75 mg/d for all participants. UFT dose was assigned according to BSA (300 mg/d if BSA < 1.17 m2; 400 mg/d if BSA 1.17 < 1.5 m2; 500 mg/d if BSA 1.5 < 1.83 m2, 600 mg/d if BSA > 1.83 m2) (n randomised = 36) |
|
| Outcomes | PFS OS ORR (RECIST, version 1.0) Grade ≥ 3 AEs (CTCAE, version 3.0) Cut‐off date for PFS ‐ 30 June 2015; median follow‐up 27.1 months (IQR 17.8 to 38.1 months) |
|
| Study Details | Journal article and abstract | |
| Funding sources and declarations of interest | Funding sources: No study funding received Declarations of interest: Merck Serono, Taiho Pharmaceuticals, Yakult Honsha, Chugai Pharmaceuticals, Takeda Pharmaceutical, Otsuka Pharmaceutical, Nippon Kayaku |
|
| Notes | Following the approval of BEV in Japan, the study protocol was amended in 2008. Participants were given the option of receiving BEV, and randomisation was stratified by the addition of BEV. 35% (Arm I) and 40% (Arm II) of participants received BEV | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was done centrally using the minimization method, with stratification by institution, number of metastatic organs (one or more), adhibition of bevacizumab and whether the tumor was unresectable or recurrent" (materials and methods, paragraph 3, page 947) |
| Allocation concealment (selection bias) | Low risk | Quote as above |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "This open‐label randomized phase II trial..." (materials and methods, paragraph 1, page 947) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs:High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: High Outcome assessment at risk of bias due to lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding. |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/PFS): Low Quote: "Lesions were measured every 8 weeks with diagnostic imaging, such as computerized tomography or other methods" (methods, paragraph 6, page 948) (ii) Survival (influences PFS/OS): High Assessment for survival following progressive disease was at the discretion of the attending physicians, in both arms (correspondence with Dr. Hirotoshi Hasegawa, received 21 July 2016) (iii) Grade ≥ 3 AEs: Low The schedule of assessment for safety was the same in both arms (correspondence with Dr. Hirotoshi Hasegawa, received 21 July 2016) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Low A similar proportion of participants was not evaluable in each arm (Table 2, page 951) (ii) PFS/OS: Low One participant in each arm had missing survival data; censoring was performed for both of these participants (correspondence with Dr. Hirotoshi Hasegawa, received 21 July 2016) (iii) Toxicity: Low No incomplete toxicity outcome data (correspondence with Dr. Hirotoshi Hasegawa, received 21 July 2016) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low One participant who was randomised to the TEGAFIRI arm did not receive any chemotherapy and was excluded from the primary analyses. However, for the ITT analyses described in results, paragraph 2, page 5, participants were analysed according to the ITT analysis as defined in our review (i.e. kept in the intervention groups to which they were randomised, regardless of whether any intervention was received or which intervention was actually received) (correspondence with Dr. Hirotoshi Hasegawa, received 21 July 2016) Safety analysis: Safety analysis population included participants who received at least 1 dose of the study drugs (Figure 1, page 947) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Similarity of arms at baseline | High risk | (i) PS: Low 2% difference in ECOG PS 1 (Table 1, page 948) (ii) Median age: High (Table 1, page 948) 5 year difference (iii) No. organs involved: Low 4% difference in number of participants with multiple sites involved (Table 1, page 948) |
| Other bias | Low risk | Subesequent therapies: Low A similar proportion of participants in both arms received second‐line chemotherapy (Table 1, page 948) Other: Use of BEV ‐ A similar proportion of participants in both arms received BEV (Table 1, page 948). Quote: "No significant difference in PFS and OS was detected by addition of bevacizumab between the two groups" (results, paragraph 2, page 950, and shown in Figure 3, pages 950 and 951) |
Shimada 2014.
| Methods | Randomised controlled trial Phase: III Accrual dates: February 2003 to November 2006 |
|
| Participants | No. randomised: 1101 Stage/treatment type: Stage III colon cancer, adjuvant Countries/sites: Japan, 48 sites Setting: Hospital Characteristics (Arm A/B): adjuvant colon cancer; age 20‐75 years (median 61/61 years); male (54/55%); PS ECOG < 0‐1 (PS ECOG 0: 94/95%) |
|
| Interventions | Arm A (5‐FU + leucovorin): 3 courses of 5‐FU 500 mg/m2 and l‐LV 250 mg/m2, D1, 8, 15, 22, 29, 36, q8w (n randomised = 550) Arm B (UFT + leucovorin): 5 courses of UFT 300 mg/m2/d and l‐LV 75 mg/d, D1‐28, q5w (n randomised = 551) |
|
| Outcomes | DFS OS Grade ≥ 3 AEs (NCI CTC, version 2.0) Median follow‐up: 72.0 months |
|
| Study Details | Journal article and abstract, study protocol | |
| Funding sources and declarations of interest | Funding sources: National Cancer Center Research and Development Funds, Ministry
of Health, Labour and Welfare of Japan Declarations of interest: Taiho, Chugai, Yakult, Pfizer, Sanofi, Novartis, Eli Lilly, Bayer, Bristol‐Myers, Merck Serono, Kyowa Kirin, Takeda |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote; "... the patients were randomised ... by the minimisation method of balancing the two arms according to tumour location (i.e. colon versus upper rectum), number of positive lymph node metastases (i.e. ≤ versus >3) and institution" (methods, paragraph 3, page 2233) |
| Allocation concealment (selection bias) | Low risk | Quote: "After confirming the inclusion and exclusion criteria by telephone or fax to the JCOG Data Center, the patients were randomised..." (methods, paragraph 3, page 2233) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "The JCOG0205 is a prospective randomised, open‐label, phase III trial..." (results, paragraph 1, page 2234) (i) DFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) DFS: High Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Disease recurrence (influences DFS) and (ii) Survival (influences DFS/OS): Low Quote: "Total colonoscopy was performed 1 year after surgery. Chest X‐ray, abdominal ultrasonography or computed tomography (CT) scan and pelvic CT scan or magnetic resonance imaging (MRI) were also performed. The serum tumour markers, CEA and CA19‐9, were examined to check for signs of recurrence every 4 months for first 2 years after random assignment and every 6 months for the next 3 years. After 5 years of follow‐up, patients were followed up annually by physical examination and serum tumour markers until November 2011. Optional CT scans were taken when tumour markers were elevated" (methods, paragraph 5, page 2233) Both groups had the same schedule of assessment and follow‐up (iii) Grade ≥ 3 AEs: High Safety evaluation during the treatment period occurred weekly for the first 6 weeks of each 8‐week cycle for the 5‐FU/l‐LV group and in the 1st and 3rd weeks of every 5‐week cycle for the UFT/LV group (study protocol, pages 27‐28) Therefore, safety evaluations occurred more frequently in the 5‐FU/l‐LV group |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) DFS: Low Quote: "DFS ... was censored at the last day when the patient was alive without any evidence of relapse or secondary cancer" (methods, paragraph 7, page 2233). No evidence of bias related to censoring (ii) OS: Low Quote: "OS ... was censored at the last day when patient was alive" (methods, paragraph 7, page 2233). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low 1101 participants were randomised, however 1092 were eligible and were presented in the analyses for DFS and OS Similar proportions of participants from both arms were ineligible and were excluded from the efficacy analyses (4/550, 0.73% in the 5FU/l‐LV arm, and 5/551 (0.91%) in the UFT/LV arm, respectively) (Fig. 1, page 2234) Safety analysis: Safety analysis population included participants who received any study treatment, regardless of eligibility (Fig. 1, page 2234) |
| Selective reporting (reporting bias) | Low risk | All outcomes outlined in the study protocol were reported |
| Similarity of arms at baseline | Low risk | (i) PS: Low 1% difference in ECOG 1 (Table 1, page 2235) (ii) Median age: Low 0 years difference in median age (Table 1, page 2235) (iii) TNM stage: Low (Table 1, page 2235) |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified |
Silvestris 2010.
| Methods | Randomised controlled trial Phase: II Accrual dates: July 2005 to August 2008 |
|
| Participants | No. randomised: Not specified. "A total of 95 consecutive patients were assessable for response" Stage/treatment line: Metastatic, first‐line Countries/sites: Gruppo Oncologico dell'Italia Meriodionale (GOIM) study Setting: Hospital Characteristics: Metastatic colorectal cancer; age > 18 years; PS ECOG < 2 |
|
| Interventions | Arm A (FOLFIRI): irinotecan 180 mg/m2 D1 with LV 100 mg/m2 and IV bolus 5‐FU 400 mg/m2 D1 and D2, and 5‐FU 600 mg/m2 22‐hour infusion, q14d (n randomised not specified) Arm B (XELIRI): irinotecan 250 mg/m2 (200 mg/m2 for participants ≥ 70 years) D1 with capecitabine 1000 mg/m2 bd (750 mg/m2 bd if > 70 years) on D1‐14, q21d (n randomised not specified) |
|
| Outcomes | ORR (RECIST, version 1.0) TTP (median) Grade ≥ 3 AEs (NCI criteria, version not specified) No details on median follow‐up None of these outcomes were suitable for inclusion in meta‐analyses |
|
| Study Details | Update in journal article | |
| Funding sources and declarations of interest | Funding sources: Gruppo Oncologico dell'Italia Meridionale Declarations of interest: No conflicts of interest |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Unclear ‐ not specified |
| Allocation concealment (selection bias) | Unclear risk | Unclear ‐ not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | Unclear risk | (i) ORR/TTP: This study was not used for the meta‐analyses of these outcomes (ii) OS: Not an outcome for this study (iii) Grade ≥ 3 AEs: This study was not used for the meta‐analyses of these outcomes Blinding of participants and personnel was not specified |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | Unclear risk | (i) ORR/TTP: This study was not used for the meta‐analyses of these outcomes (ii) OS: Not an outcome for this study (iii) Grade ≥ 3 AEs: This study was not used for the meta‐analyses of these outcomes Blinding of outcome assessors was not specified |
| Schedule of assessment and follow‐up | Unclear risk | This study was not used for the meta‐analysis of any outcomes Schedule of assessment and follow‐up was not specified |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | This study was not used for the meta‐analysis of any outcomes Information on incomplete outcome data was not specified |
| Incomplete outcome data (ITT analysis) | Unclear risk | This study was not used for the meta‐analysis of any outcomes Information on ITT analysis was not specified |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Similarity of arms at baseline | Unclear risk | This study was not used for the meta‐analysis of any outcomes Information on similarity of arms at baseline was not specified |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified |
Souglakos 2012.
| Methods | Randomised controlled trial Phase: II Accrual dates: June 2005 to June 2008 |
|
| Participants | No. randomised: 336 Stage/treatment line: Metastatic, first‐line Countries/sites: Greece, 23 sites Setting: Hospital Characteristics (Arm A/B): Metastatic colorectal cancer; age ≥ 18 years (median 66/67 years); male (62/66%); PS ECOG 0‐2 (PS ECOG 0: 31/30%) |
|
| Interventions | Arm A (FOLFIRI‐bevacizumab (BEV)): irinotecan 180 mg/m2 D1, FA 200 mg/m2 D1, 2, IV bolus 5‐FU 400 mg/m2 D1 and 600 mg/m2/d 22‐hour infusion D1, 2, plus BEV 5 mg/kg D1, q14d (n randomised = 168) Arm B (CAPIRI‐BEV): capecitabine 2000 mg/m2 D1‐14, irinotecan 250 mg/m2 D1 and BEV 7.5 mg/kg D1, q21d (n randomised = 168) Treatment continued until PD, unacceptable toxicity, or withdrawal of consent |
|
| Outcomes | PFS ORR (RECIST, version 1.0) OS Grade ≥ 3 AEs (NCI CTC, version 3.0) Median follow‐up: 32 months |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: Hellenic Oncology Research Group, University Hospital of Crete Declarations of interest: No conflicts of interest |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Minimisation covariate‐adaptive randomisation was used (correspondence with Dr. John Souglakos, received 31 July 2012) |
| Allocation concealment (selection bias) | Low risk | Centralised Web‐based randomisation was used (correspondence with Dr. John Souglakos, received 31 July 2012) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Not specified, blinding unlikely (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs:High Outcome assessment at risk of bias if there was lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: Low Evaluation of response was performed by independent radiologists (correspondence with Dr. John Souglakos, received 31 July 2012) Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Grade ≥ 3 AEs were reported from the research nurses of participating institutions (correspondence with Dr. John Souglakos, received 31 July 2012) Outcome assessment at risk of bias if there was lack of blinding |
| Schedule of assessment and follow‐up | High risk | (i) Response (influences ORR/PFS): Low Quote: "Response to treatment was evaluated every 8 weeks ..." (patients and methods, paragraph 6, page 454) (ii) Survival (influences PFS/OS): Low Following cessation of treatment or disease progression, participants were followed for survival every 3 months in both arms (correspondence with Dr. John Souglakos, received 31 July 2012) (iii) Grade ≥ 3 AEs: High Quote: "During treatment, a CBC with [sic] was performed weekly. In addition, patients were clinically assessed and blood chemistry was performed before each treatment cycle" (patients and methods, paragraph 6, page 454) Cycles were given every 2 weeks in the FOLFIRI‐BEV arm and every 3 weeks in the CAPIRI‐BEV arm (patients and methods, paragraph 3, page 454) Therefore, safety assessments were performed more frequently in the FOLFIRI‐BEV arm |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Low 4 participants in the CAPIRI‐BEV arm were not evaluable for response owing to clinical deterioration/early death (correspondence with Dr. John Souglakos, received 31 July 2012) (ii) PFS/OS: Low No missing outcome data (correspondence with Dr. John Souglakos, received 31 July 2012) (iii) Grade ≥ 3 AEs: Low No missing outcome data (correspondence with Dr. John Souglakos, received 31 July 2012) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Analyses were described as performed in an ’intent‐to‐treat’ fashion (correspondence with Dr. John Souglakos, received 31 July 2012) 336 participants were randomised, 168 to FOLFIRI‐BEV and 168 to CAPIRI‐BEV. 167 participants allocated to FOLFIRI‐BEV and 166 to CAPIRI‐BEV, who received treatment, were included in the analysis (excluded 3/336, 0.9%) (Figure 1, page 455) Safety analysis: As for efficacy |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, page 455) (ii) Median age: Low (Table 1, page 455) (iii) No. of involved organs: Low (Table 1, page 455) |
| Other bias | Low risk | Subsequent therapies: Low Similar proportions in both arms received second‐line oxaliplatin‐based chemotherapy, irinotecan‐based chemotherapy, cetuximab and BEV beyond progression (Table 4, page 457) |
Twelves 2012.
| Methods | Randomised controlled trial Phase: III Accrual dates: November 1998 to November 2001 |
|
| Participants | No. randomised: 1987 Stage/treatment type: Stage III colon carcinoma, adjuvant Countries/sites: Multination, 164 centres ‐ North and South America, Europe, Asia (Thailand), Israel, and Australia Setting: Hospital Characteristics (Arm I/II): Resected stage III colon carcinoma; age 18 to 75 years (median 62/63 years); male (54/54%); PS ECOG 0‐1 (PS ECOG 0: 85/85%) |
|
| Interventions | Arm I: Eight cycles of oral capecitabine 1250 mg/m2 bd D1‐14, q21d (n randomised = 1004) Arm II: Six cycles of IV bolus leucovorin 20 mg/m2, then fluorouracil 425 mg/m2 D1‐5, q28d (n randomised = 983) |
|
| Outcomes | DFS OS Grade ≥ 3 AEs (NCIC CTC, revised May 1991) Median follow‐up: 6.9 years |
|
| Study Details | Journal articles | |
| Funding sources and declarations of interest | Funding sources: Hoffman La‐Roche Declarations of interest: Sanofi‐Aventis, Hoffman La‐Roche, Merck, Pfizer, AstraZeneca, Baxter |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization, with the use of treatment allocation codes (scratch‐off labels), was stratified by center and performed with a block size of four. The block size was unknown to investigators and monitors" (methods, paragraph 5, page 2698) |
| Allocation concealment (selection bias) | Low risk | Quote as above |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | This was an open‐label study (correspondence with Dr. Chris Twelves, received 22 August 2012) (i) DFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) DFS: High Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | Low risk | (i) Disease recurrence (influences DFS): Low Quote: "Evaluation of efficacy ‐ Patients were assessed every six months for two years after randomization and then yearly" (methods, paragraph 6, page 2698) (ii) Survival (influences DFS/OS): Low Quote as above (iii) Grade ≥ 3 AEs: Low AEs and treatments were recorded throughout chemotherapy and up to 28 days after last study treatment. Safety assessments were performed at weeks 2, 4 (optional), 7, 10, 13, 16, 19, 22, and 25 of treatment. AEs were reported during treatment with trial medication (correspondence with Dr. Chris Twelves, received 22 August 2012) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) DFS: Low 32/1004 participants in the capecitabine arm and 34/983 in the 5FU/LV arm were lost to follow‐up before the 5‐year date after randomisation (correspondence with Dr. Chris Twelves, received 22 August 2012) (ii) OS: Low 93.4% (capecitabine 640/685; 5‐FU/LV 590/632) of participants with an expected 5‐year follow‐up visit had completed 5 years or longer of survival follow‐up at the clinical cut‐off date on 4 June 2007 (correspondence with Dr. Chris Twelves, received 22 August 2012) (ii) Grade ≥ 3 AEs: Low AEs were reported during treatment with trial medication, and all participants were followed for AEs during this time (correspondence with Dr. Chris Twelves, received 22 August 2012) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "The intention‐to‐treat population included all patients who underwent randomization" (methods, paragraph 8, page 2698) Safety analysis: Quote: "The population included in the safety analysis comprised all patients receiving at least one dose of the study drug who were followed up for safety" (methods, paragraph 8, page 2698) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, page 2699) (ii) Median age: Low (Table 1, page 2699) (iii) TNM stage: Low (Table 1, page 2699) |
| Other bias | Low risk | Subsequent therapies: Low Quote: "Overall, 90% (n = 343) and 87% (n = 350) of patients randomized to capecitabine and 5‐FU/FA, respectively, received ≥1 therapeutic intervention at relapse. A similar proportion of patients received systemic treatments at relapse in the two randomization arms (capecitabine versus 5‐FU/FA); these included 5‐FU (57% versus 49%), oxaliplatin (41% versus 35%), irinotecan (36% versus 41%), raltitrexed (6% versus 6%) and cetuximab (4% versus 5%). Capecitabine, however, was later given to more patients in the 5‐FU/FA versus capecitabine arm (24% versus 14%). Locoregional procedures were carried out at relapse in a similar proportion of patients in the capecitabine versus 5‐FU/FA arms, including radiotherapy (18% versus 19%), partial hepatectomy (9% versus 9%) and laparotomy (9% versus 5%)" (Twelves et al, Annals of Oncology 2011 ‐ results, paragraph 2, page 1191) |
Van Cutsem 2001a.
| Methods | Randomised controlled trial Phase: III Accrual dates: Not specified |
|
| Participants | No. randomised: 531 Stage/treatment line: Metastatic, first‐line Countries/sites: International Setting: Hospital Characteristics: Advanced colorectal carcinoma; age not specified; KPS ≥ 70% |
|
| Interventions | Arm I: eniluracil 11.5 mg/m2 and 5‐FU 1.15 mg/m2 oral bd D1‐28, q35d (n randomised and treated = 268) Arm II: IV 5‐FU 425 mg/m2 + LV 20 mg/m2 D1‐5, q28d (n randomised and treated = 263) |
|
| Outcomes | ORR (criteria not specified) PFS OS Grade ≥ 3 AEs (criteria not specified) No details on median follow‐up |
|
| Study Details | Abstract | |
| Funding sources and declarations of interest | Funding sources: None declared Declarations of interest: None declared |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "This multicentre randomised open label phase III study..." (abstract) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: High Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding. |
| Schedule of assessment and follow‐up | Unclear risk | (i) Response (influences ORR/PFS): Unclear ‐ not specified (ii) Survival (influences PFS/OS): Unclear ‐ not specified (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Unclear ‐ not specified (ii) PFS/OS: Unclear ‐ not specified (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Unclear risk | Efficacy analysis: Unclear Not specified Safety analysis: Not specified. |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Unclear ‐ not specified (ii) Median/mean age: Unclear ‐ not specified (iii) No. of involved organs: Unclear ‐ not specified |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified |
Van Cutsem 2001b.
| Methods | Randomised controlled trial Phase: III Accrual dates: October 1996 to February 1998 |
|
| Participants | No. randomised: 602 Stage/treatment line: Metastatic, first‐line Countries/sites: Multi‐nation, 59 centres in Europe, Australia, New Zealand, Taiwan, and Israel Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal carcinoma; age ≥ 18 years (median 64.0/63.5 years); male (57/57%); KPS ≥ 70% (median 90/90%) |
|
| Interventions | Arm I: capecitabine 1250 mg/m2 bd D1‐14, q21d (n randomised = 301) Arm II: LV 20 mg/m2 then IV bolus 5‐FU 425 mg/m2 D1‐5, q28d (n randomised = 301) |
|
| Outcomes | ORR (WHO criteria, 1979) TTP (treated as PFS in this review, based on the definition provided) OS Grade ≥ 3 AEs (NCIC CTC, revised December 1994) No details on median follow‐up |
|
| Study Details | Journal articles | |
| Funding sources and declarations of interest | Funding sources: Hoffman La‐Roche Declarations of interest: None declared |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "After screening to establish eligibility, patients were randomized to treatment with capecitabine or 5‐FU/LV through a computer‐assisted touch‐tone randomization center. The patients were randomized centrally by country, in blocks of six patients, but with Australia, New Zealand, and Taiwan grouped as a single location" (patients and methods, paragraph 2, page 4098) |
| Allocation concealment (selection bias) | Low risk | Quote as above |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "This was an open‐label, randomized, parallel‐group study ..." (patients and methods, paragraph 2, page 4098) (i) ORR/TTP (treated as PFS): Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/TTP (treated as PFS): Low Quote: "Investigator assessments of tumor response were reviewed by an independent review committee (IRC) composed of radiologists. Members of the IRC were blinded to the treatment received, clinical condition of the patient, and to the investigator’s evaluation and measurements. The IRC‐assessed tumor response solely on the basis of x‐ray or other imaging. Oncologists were available for IRC consultation" (patients and methods, paragraph 6, page 4099) There were both Investigator and blinded IRC response assessments. The absolute ORR was higher for capecitabine than 5‐FU/LV for both of these assessments (results, paragraph 4, page 4100) Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | Low risk | (i) Response (influences ORR/TTP (treated as PFS)): Low Quote: "Assessments of tumor dimensions and involved sites were performed before the start of treatment and were scheduled during therapy after weeks 6, 12, 18, 24, and 30. Further assessments were performed after weeks 39 and 48 for patients who received prolonged therapy (48 weeks). Follow‐up assessments for disease progression and survival monitoring were performed every 3 months after the end of treatment" (patients and methods, paragraph 6, page 4099) (ii) Survival (influences TTP(treated as PFS)/OS): Low Follow‐up assessments for disease progression and survival monitoring were performed every 3 months after the end of treatment" (patients and methods, paragraph 6, page 4099) (iii) Safety: Low Quote: "Safety evaluations were conducted at least monthly until 4 weeks after the last administration of therapy ..." (patients and methods, paragraph 7, page 4099) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Low 11.0% of participants in the capecitabine arm and 12.6% in the 5‐FU/LV arm were missing post‐baseline response data (Table 2, page 4100) (ii) TTP (treated as PFS)/OS: Unclear ‐ not specified (iii) Grade ≥ 3 AEs: Unclear ‐ not specified |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy: Low Quote: "All analyses of efficacy are reported for the all‐randomized population ..." (patients and methods, paragraph 10, page 4099) Safety: Quote: "...all analyses of safety are based on the safety population, which included all patients who received at least one dose of study drug" (patients and methods, paragraph 10, page 4099) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Unclear Presented as mean/median only (Table 1, page 4100) (ii) Median age: Low (Table 1, page 4100) (iii) No. of involved organs: Unclear ‐ not specified |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified |
Yamada 2013.
| Methods | Randomised controlled trial Phase: III Accrual dates: February 2009 to March 2011 |
|
| Participants | No. randomised: 512 Stage/treatment line: Inoperable locally advanced or metastatic CRC; first‐line (patients with relapse > 180 days after adjuvant chemotherapy were eligible, but prior treatment with oxaliplatin‐containing adjuvant chemotherapy was not permitted) Countries/sites: Japan, 82 institutions Setting: Hospital Characteristics (Arm I/II): Inoperable locally advanced or metastatic colorectal cancer; age 20 to 80 years (median 63/63 years); male (62.4/66.4%); PS ECOG 0‐1 |
|
| Interventions | Arm I (mFOLFOX6‐bevacizumab (BEV)): D1 Oxaliplatin 85 mg/m2, LV 200 mg/m2, IV bolus 5‐FU 400 mg/m2 followed by 5‐FU 2400 mg/m2 46‐hour continuous infusion, plus BEV 5 mg/kg D1, q14d (n randomised = 256) Arm II (SOX‐BEV): S‐1 40‐60 mg* oral bd D1‐14 and D1 oxaliplatin 130 mg/m2, plus BEV 7.5 mg/kg D1, q21d *S‐1 doses: 80 mg/d if BSA < 1.25 m2; 100 mg/d if 1.25 m2 ≤ BSA <1.5 m2; 120 mg/d if BSA ≥ 1.5 m2 (n randomised = 256) |
|
| Outcomes | PFS OS ORR (RECIST, version 1.0) Grade ≥ 3 AEs (CTCAE, version 3.0) Median follow‐up: PFS 18.4 months (IQR, 13.1 to 24.9 months); OS 23.4 months (IQR, 19.5 to 29.6 months). Data cut‐off date for PFS ‐ June 30, 2012 |
|
| Study Details | Journal article and abstract/poster | |
| Funding sources and declarations of interest | Funding sources: Taiho Declarations of interest: Taiho, Chugai, Pfizer |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was done centrally with the minimisation method,with stratification by institution and whether postoperative adjuvant chemotherapy had been given" (methods, paragraph 5, page 1279) |
| Allocation concealment (selection bias) | Low risk | Quote: "To ensure allocation concealment, a minimisation algorithm with an 80:20 random element was used. The randomisation sequence was generated by a team (EPS Corporation, Tokyo, Japan; independent from the trial sponsor and investigators) who used a validated computer system" (methods, paragraph 5, page 1279) |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "We undertook an open‐label, non‐inferiority, randomised phase 3 trial" (methods, paragraph 1, page 1279) Quote: "Participants, investigators, and data analysts could not be masked to treatment assignment, because we were comparing an oral‐based regimen with an infusional regimen" (methods, paragraph 5, page 1279) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: High Quote: "Progressive disease was assessed solely by the investigator in charge of the patient" (methods, paragraph 8, page 1280) (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | High risk | (ii) Response (influences ORR/PFS): Low Quote: "Lesions were measured every 8 weeks with diagnostic imaging (e.g., CT or MRI)" (methods, paragraph 8, page 1280) Quote: "After initiation of study treatment, target and non‐target lesions were assessed every 8 weeks in the same way as at baseline, with the same imaging conditions (e.g., contrast media and slice thickness). The best overall response was identified" (methods, paragraph 9, page 1280) (ii) Survival (influences PFS/OS): Low Same in both arms (correspondence with Aya Takata, received 14 July 2016) (iii) Grade ≥ 3 AEs: High Quote: "... patients who received SOX plus bevacizumab returned to the hospital once every 3 weeks rather than once every 2 weeks for patients who received mFOLFOX6 plus bevacizumab" (discussion, paragraph 1, page 1284) Therefore, participants in the mFOLFOX6 plus bevacizumab arm would have more frequent opportunities to report AEs |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Low Among participants with measurable disease, only 7% in the mFOLFOX6‐BEV group and 9% in the SOX‐BEV group were non‐evaluable (Takahari et al, Journal of Clinical Oncology 2013 ‐ poster associated with abstract 3519) (ii) PFS/OS: Low Censoring was noted in the KM curves for PFS/OS (Figure 2, page 1282). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Low No missing outcome data for toxicity |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low 0/256 and 1/256 (0.4%) participants randomised to the SOX + BEV and mFOLFOX6 + BEV arms, respectively, were excluded from the analysis post randomisation owing to ineligibility (Figure 1, page 1280) Safety analysis: Quote: "Patients who received at least one dose of the assigned study drugs were included in analyses of ... safety" (methods, paragraph 12, page 1281) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Unclear. All participants were confirmed to be either PS 0 or 1 when enrolled; however, no information was collected about PS status in the CRF (correspondence with Aya Takata, received 14 July 2016) (ii) Median age: Low (Table 1, page 1281) (iii) No. of involved organs: Low (Table 1, page 1281) |
| Other bias | Low risk | Subsequent therapies: Low Quote: "188 patients given mFOLFOX6 plus bevacizumab and 198 given SOX plus bevacizumab received second‐line treatment. Irinotecan was used as second‐line treatment in 122 (65%) of the 188 patients given mFOLFOX6 plus bevacizumab, oxaliplatin in nine (5%), bevacizumab in 70 (37%), cetuximab in ten (5%), and panitumumab in nine (5%). Irinotecan was used in 116 (59%) of the 198 patients given SOX plus bevacizumab, oxaliplatin in 23 (12%), bevacizumab in 67 (34%), cetuximab in 15 (8%), and panitumumab in nine (5%)" (results, paragraph 7, page 1283) |
Yamazaki 2015.
| Methods | Randomised controlled trial Phase: II Accrual dates: July 2008 to July 2009 |
|
| Participants | No. randomised: 107 Stage/treatment line: Metastatic, first‐line Countries/sites: Japan, 22 institutions Setting: Hospital Characteristics (Arm A/B): Metastatic colorectal cancer; age > 20 years (median 60.5/61.0 years); male (58.9/46.9); PS ECOG < 0‐1 (PS ECOG 0: 87.5/81.6%) |
|
| Interventions | Arm A (SOL): S‐1 40‐60 mg bd plus oral LV 25 mg bd D1‐7 and oxaliplatin (L‐OHP) 85 mg/m2 D1, q14d Arm B (mFOLFOX6): D1 oxaliplatin 85 mg/m2, LV 200 mg/m2, IV bolus 5‐FU 400 mg/m2 followed by 5‐FU 2400 mg/m2 46‐hour continuous infusion, q14d |
|
| Outcomes | PFS Grade ≥ 3 AEs (CTCAE, version 3.0) ORR (RECIST, version 3.0) OS PFS cut‐off date 31 March 2010; OS cut‐off date 31 January 2012 (median follow‐up 35 months) |
|
| Study Details | Journal article and abstract/poster | |
| Funding sources and declarations of interest | Funding sources: Taiho, Yakult Declarations of interest: Taiho, Yakult |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to receive SOL or mFOLFOX6 at a central registration center, using a minimization method with stratification according to disease status (unresectable or recurrent disease) and institution" (patients and methods, paragraph 3, page 570‐1) |
| Allocation concealment (selection bias) | Low risk | Quote as above |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "This randomized, open‐label, phase II study... " (patients and methods, paragraph 4, page 571) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: Low Quote: "Response and PFS were evaluated by an independent review committee (IRC)" (patients and methods, paragraph 6, page 571) (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | Low risk | (i) Response (influences ORR/PFS): Low Quote: "Tumors were assessed every 6 weeks until disease progression" (patients and methods, paragraph 6, page 571) (ii) Survival (influences PFS/OS): Low The schedule for assessment of survival was the same in both arms (correspondence with Aya Takata, received 22 July 2016) (iii) Grade ≥ 3 AEs: Low Quote: "Physical examinations and laboratory tests ... were repeated every week during the first four cycles of chemotherapy and every 2 weeks from the fifth cycle onward" (patients and methods, paragraph 6, page 571) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | (i) ORR: Low No missing response data from participants included in the 'full analysis set' (Figure.1, page 572, and Table 2, page 574) (ii) PFS/OS: Low Quote: "Progression‐ free survival (PFS) was defined as the time from randomization to disease progression or death from any cause. Data on patients without documented evidence of progressive disease or death were censored on the date of the last tumor assessment without progression during the protocol treatment" (patients and methods, paragraph 6, page 571). No evidence of bias related to censoring Censoring was also noted on the KM curve for OS (Fig. 2b, page 573). No evidence of bias related to censoring (iii) Grade ≥ 3 AEs: Unclear No information regarding this is available (correspondence with Aya Takata, received 22 July 2016) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low 0/56 (0%) and 2/51 (4%) participants randomised to the SOL and mFOLFOX6 arms, respectively, were excluded from the analysis owing to ineligibility (Fig. 1, page 572) Safety analysis: Low Safety analysis population comprised all participants who were randomised to their respective treatment arms (correspondence with Aya Takata, received 22 July 2016) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Table 1, page 572) (ii) Median age: Low (Table 1, page 572) (iii) No. of involved organs: Low (Table 1, page 572) |
| Other bias | Low risk | Subsequent therapies: Low Quote: "The proportion of patients who received subsequent therapy was slightly higher in the SOL group (100%) than in the mFOLFOX6 group (91.8%). Irinotecan was given to 37 patients (66.1%) in the SOL group and 33 (67.3%) in the mFOLFOX6 group, bevacizumab to 26 patients (46.4%) in the SOL group and 19 (38.8%) in the mFOLFOX6 group, and L‐OHP to 12 (21.4%) in the SOL group and 3 (6.1%) in the mFOLFOX6 group" (results, paragraph 4, page 572) However, of the 12 patients who received post‐treatment containing oxaliplatin in the SOL arm, "7 patients did not discontinue SOL treatment due to disease progression" (Otsuji et al, Journal of Clinical Oncology 2012, poster associated with abstract 586) |
Yasui 2015.
| Methods | Randomised controlled trial Phase: II/III with combined data analysed Accrual dates: January 2006 to January 2008 |
|
| Participants | No. randomised: 426 Stage/treatment line: Metastatic, second‐line (irinotecan‐naive) Countries/sites: Japan, 40 institutions Setting: Hospital Characteristics (Arm I/II): Metastatic colorectal cancer; age 20 to 75 years (median 63.0/61.0 years); male (57.7/56.3%); PS ECOG 0‐1 (PS ECOG 0: 75.1/74.2%) |
|
| Interventions | Arm I (FOLFIRI): folinic acid 200 mg/m2, irinotecan 150 mg/m2, IV bolus fluorouracil 400 mg/m2 D1 and fluorouracil 2400 mg/m2 46h infusion, D1, 15, q28d (n randomised = 213) Arm II (IRIS): irinotecan 125 mg/m2 D1, 15, and S‐1 (40 mg if BSA < 1.25 m2; 50 mg if BSA 1.25 < 1.5 m2; 60 mg if BSA ≥ 1.5 m2) bd D1–14, q28d (n randomised = 213) Treatment continued until PD, unacceptable toxicity, or participant refusal |
|
| Outcomes | PFS OS ORR (RECIST, version 1.0) Grade ≥ 3 AEs (CTCAE, version 3.0) Data collection cut‐off for OS: 29 July 2010, median follow‐up 39.2 months |
|
| Study Details | Journal article and abstract | |
| Funding sources and declarations of interest | Funding sources: Taiho Declarations of interest: Daiichi Sankyo, Taiho, Yakult Honsha |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The patients were centrally randomised to receive either FOLFIRI or IRIS using the minimisation method with stratification by institution, prior therapy (with oxaliplatin vs. without oxaliplatin), and performance status (PS; 0 vs. 1)" (patients and methods, paragraph 2, page 154) Quote: "Assignment of patients was concealed from the investigator" (Muro et al, Lancet Oncology 2010, methods, paragraph 3, page 854) |
| Allocation concealment (selection bias) | Low risk | Quotes as above |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | High risk | Quote: "Treatment assignment was not masked from the investigators or patients" (Muro et al, Lancet Oncology 2010, methods, paragraph 3, page 854) (i) ORR/PFS: Low Outcome assessment unlikely to be influenced by lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | (i) ORR/PFS: High "Treatment assignment was not masked from the investigators or patients" (Muro et al, Lancet Oncology 2010, methods, paragraph 3, page 854) The definition of progression for the primary endpoint of PFS included clinical assessment, quote: "Progression was defined when any of the following three events occurred: (1) PD based on the response evaluation criteria in solid tumours (RECIST) version 1.0; (2) clinical progression judged by the investigator; or (3) death from any cause without progression" (patients and methods, paragraph 5, page 155) Outcome assessment at risk of bias from lack of blinding (ii) OS: Low Outcome assessment unlikely to be influenced by lack of blinding (iii) Grade ≥ 3 AEs: High Outcome assessment at risk of bias from lack of blinding |
| Schedule of assessment and follow‐up | Low risk | (i) Response (influences ORR/PFS): Low Quote: "Tumours were assessed at baseline (within 1 month before enrolment), 2, 3, and 4 months after enrolment, and every 2 months thereafter until progression" (patients and methods, paragraph 5, page 155) (ii) Survival (influences PFS/OS): Low Same in both arms (correspondence with Aya Takata, received 19 July 2016) (iii) Grade ≥ 3 AEs: Low Quote: "Physical examinations and laboratory tests were performed at baseline and repeated at least every 2 weeks during the treatment" (patients and methods, paragraph 5, page 155) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Low 39/213 (18%) in the FOLFIRI arm and 32/213 (15%) in the IRIS arm did not have evaluable response data (Muro et al, Lancet Oncology 2010, results, paragraph 5, page 856) (ii) PFS/OS: Low Quote: "Progression‐free survival was counted from the date of randomisation to the date when the progressive disease was first confirmed by the investigator’s assessment. For patients without documented progressive disease, data was censored on the date of the last tumour assessment with non‐progression status" (Muro et al, Lancet Oncology 2010, methods, paragraph 9, page 855). No evidence of bias related to censoring Quote: "OS was calculated from the date of randomisation to the date of death from any cause. Surviving patients, including those lost to follow‐up, were censored at the date of last confirmation of survival" (patients and methods, paragraph 6, page 155). No evidence of bias related to censoring (iii) Safety: Low No missing safety outcome data (correspondence with Aya Takata, received 19 July 2016) |
| Incomplete outcome data (ITT analysis) | Low risk | Efficacy analysis: Low Quote: "The intent‐to‐treat (ITT) population consisted of all randomised patients..." (patients and methods, paragraph 7, page 155) Safety analysis: Quote: "Safety was assessed in all patients who received at least one dose of the study drug" (Muro et al, Lancet Oncology 2010, methods, paragraph 11, page 855) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Low risk | (i) PS: Low (Muro et al, Lancet Oncology 2010, Table 1, page 855) (ii) Median age: Low (Muro et al, Lancet Oncology 2010, Table 1, page 855) (ii) No. of involved organs: Low (Muro et al, Lancet Oncology 2010, Table 1, page 855) |
| Other bias | Low risk | Subsequent therapies: Low Quote: "Third‐line chemotherapy after failure of the protocol treatment in the second‐line therapy was given to 168 (78.9%) patients in the FOLFIRI group and 153 (71.8%) patients in the IRIS group. In these patients, molecularly targeted agents were concomitantly used in 58 (27.2%) patients (bevacizumab, 45; cetuximab, 17) in the FOLFIRI group and 52 (24.4%) patients (bevacizumab, 38; cetuximab, 16) in the IRIS group, and no marked difference in the use of these agents was evident between the two groups" (results, paragraph 2, page 156) |
Yu 2005.
| Methods | Randomised controlled trial Phase: Not specified Accrual dates: January 2001 to September 2003 |
|
| Participants | No. randomised: 43 Stage/treatment line: Metastatic, first‐line or second‐line if no chemotherapy for longer than 6 months Countries/sites: China Setting: Hospital Characteristics: Metastatic colorectal carcinoma; age ≤ 75 years; KPS 0‐1 (unclear) |
|
| Interventions | Arm I: irinotecan 90‐125 mg/m2 10‐hour infusion, FA 30 mg/m2 + 5‐FU 425 mg/m2/d 48‐hour continuous infusion, q14d for no less than 6 C (n randomised = 16) Arm II: irinotecan 90‐125 mg/m2 10‐hour infusion, q14d and capecitabine 1250 mg/m2/d for 3 months (n randomised = 27) |
|
| Outcomes | ORR (criteria not specified) TTP OS Grade ≥ 3 AEs (criteria not specified) Median follow‐up: 14 months |
|
| Study Details | Journal article | |
| Funding sources and declarations of interest | Funding sources: None declared Declarations of interest: None declared |
|
| Notes | Original article (in Chinese) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) DFS/PFS/TTP/ORR | Low risk | (i) ORR/TTP: Low Quote (translated from Chinese to English): "According to random double‐blind method ..." (materials and methods, paragraph 1, page 558) This is unclear/unlikely, as no placebo was described in the IV or oral arm for different schedules Outcome assessment unlikely to be influenced by lack of blinding This study was not used for the meta‐analysis of the TTP outcome (ii) OS: This study was not used for the meta‐analysis for this outcome (iii) Grade ≥ 3 AEs: This study was not used for the meta‐analyses of these outcomes |
| Blinding of outcome assessment (detection bias) DFS/PFS/TTP/ORR | High risk | Not specified (i) ORR/TTP: High Outcome assessment at risk of bias if there was lack of blinding This study was not used for the meta‐analysis of the TTP outcome (ii) OS: This study was not used for the meta‐analysis of this outcome (iii) Grade ≥ 3 AEs: This study was not used for the meta‐analyses of these outcomes |
| Schedule of assessment and follow‐up | Unclear risk | (i) Response (influences ORR/TTP): Unclear ‐ not specified. This study was not used for the meta‐analysis of the TTP outcome (ii) Survival (influences OS): Unclear ‐ not specified. This study was not used for the meta‐analysis of this outcome (iii) Grade ≥ 3 AEs: Unclear. This study was not used for the meta‐analyses of these outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | (i) ORR: Low The sum of those with CR/PR/SD and PD was the same as the number of participants included in the study for both arms (Table 2, page 558) (ii) TTP/OS: Unclear ‐ not specified. This study was not used for the meta‐analysis of the TTP and OS outcomes (iii) Grade ≥ 3 AEs: Unclear. This study was not used for the meta‐analyses of these outcomes |
| Incomplete outcome data (ITT analysis) | Unclear risk | Efficacy analysis: Unclear ‐ not specified Safety analysis: Not specified. This study was not used for the meta‐analyses of grade ≥ 3 AE outcomes |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available |
| Similarity of arms at baseline | Unclear risk | (i) PS: Unclear ‐ not specified (ii) Median/mean age: Unclear ‐ not specified (iii) No. of involved organs: Unclear ‐ not specified |
| Other bias | Unclear risk | Subsequent therapies: Unclear ‐ not specified |
For the Characteristics of included studies tables, Outcomes listed are those that the study reported that were of interest in this review, regardless of inclusion in quantitative synthesis. Study details refer to the form of report/s used for this review.
PS: performance status
ECOG: Eastern Cooperative Oncology Group
5‐dFUR: doxifluridine
IV: intravenous
5‐FU / FU: 5‐fluorouracil
ORR: objective response rate
WHO: World Health Organisation
PFS: progression‐free survival
TTP: time to tumour progression
AEs: adverse events
OS: overall survival
LV: leucovorin
KM: Kaplan‐Meier
DFS: disease‐free survival
NCI CTCAE: National Cancer Institute Common Terminology Criteria for Adverse Events
NSABP: National Surgical Adjuvant Breast and Bowel Project
CT: computed tomography
MRI: magnetic resonance imaging
WHO: World Health Organisation
CR: complete response
PR: partial response
SD: stable disease
PD: progressive disease
NCI CTC: National Cancer Institute Common Terminology Criteria
TTF: time to treatment failure
UFT: tegafur/uracil
RECIST: Response Evaluation Criteria in Solid Tumours
IRC: independent review committee
ITT: intention‐to‐treat
SICOG: Southern Italy Cooperative Oncology Group
WBC: white blood cell
ASCO: American Society of Clinical Oncology
TTD: Treatment of Digestive Tumors
ECOG CTC: Eastern Cooperative Oncology Group Common Toxicity Criteria
KPS: Karnofsky Performance Scale
LFT: liver function tests
BEV: bevacizumab
FNCLCC: Federation Nationale des Centers de Lutte Contre le Cancer
BP: blood pressure
EPP: Expanded Participation Project
DHHS: Department of Health and Human Services
TME: total mesorectal excision
PME: partial mesorectal excision
T‐CORE: Tohoku Clinical Oncology Research and Education Society
BSA: body surface area
NE: non‐evaluable
EORTC: European Organisation for Research and Treatment of Cancer
FA: folinic acid
GOAM: Gruppo Oncologico Aree Metropolitane
L‐OHP: oxaliplatin
HeCOG: Hellenic Oncology Research Group
CRC: colorectal cancer
AJCC: American Joint Committee on Cancer
SWOG: South‐West Oncology Group
EU: eniluracil
UK MRC: United Kingdom Medical Research Council
MRC CTU: Medical Research Council Clinical Trials Unit
IQR: interquartile range
JCOG: Japan Clinical Oncology Group
CEA: carcinoembryonic antigen
Ca19‐9: cancer antigen 19‐9
GOIM: Gruppo Oncologico dell’Italia Meriodionale
CBC: complete blood count
CRF: case report form
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Ansfield 1977 | RCT, but used a chemotherapy regimen that was not consistent with contemporary practice |
| Bajetta 1997 | This study investigated the role of oral vs IV 5‐dFUR in metastatic CRC, but only in a selected subset of all randomised participants (only those considered to be 5‐FU resistant) |
| Bedikian 1983 | RCT, with cross‐over permitted in only 1 (IV 5‐FU) arm |
| Bjerkeset 1986 | This study included participants with both gastric and colorectal cancer, and results for participants with CRC were inseparable |
| Borner 2002 | RCT with a cross‐over design. Participants in both arms received only 1 cycle of chemotherapy before cross‐over |
| Douglass 1978 | Study did not state that histologically proven colorectal adenocarcinoma was required for inclusion in the trial, other than for hepatomegaly |
| Fan 2005 | This study examined oral capecitabine vs IV calcium folinate/5‐FU in a selected group of randomised participants who had not progressed after 1 cycle of chemotherapy |
| Hahn 1975 | RCT, but used a chemotherapy regimen that was not consistent with contemporary practice |
| Hennig 2008 | RCT with a cross‐over design. Participants received only 6 weeks of IV 5FU/LV or two 3‐week cycles of oral capecitabine before cross‐over to the other arm of treatment |
| Kim 2001b | Histologically proven adenocarcinoma of the rectum was not confirmed as an inclusion criterion for this study. Mean follow‐up period was only 15 months |
| Lima 2005 | RCT with a cross‐over design. Participants in both arms received only 1 cycle of chemotherapy before cross‐over |
| Maetani 1993 | This study did not compare oral and IV fluoropyrimidines. It compared oral UFT with oral ftorafur after surgery |
| Munoz 2008 | Histologically proven colorectal adenocarcinoma was not confirmed as an inclusion criterion for this study |
| NCT00070122 | Study was closed early owing to poor accrual; no publishable results |
| NCT01193452 | Study was ceased early owing to poor accrual, with many elderly patients refusing to have intravenous fluoropyrimidine therapy |
| NCT01196260 | No described comparison between 5‐FU and capecitabine arms |
| NCT01279681 | No clear comparison between oral and IV fluoropyrimidine described |
| NCT01736904 | Histologically proven colorectal adenocarcinoma was not confirmed as an inclusion criterion for this study |
| Pfeiffer 2006 | RCT with a cross‐over design. Participants in the oral capecitabine arm received only 2 cycles of chemotherapy before cross‐over |
| Queißer 1979 | RCT, but participants in both Arms A and B received IV fluoropyrimidine (IV 5‐fluorouracil in Arm A and IV Ftorafur in Arm B) |
| Revazishvili 2008 | It is unclear if this study was a randomised trial |
| Sizer 2006 | Histologically proven colorectal adenocarcinoma was not confirmed as an inclusion criterion for this study |
| Skof 2009 | Histologically proven colorectal adenocarcinoma was not confirmed as an inclusion criterion for this study |
| Tournigand 2012 | Participants were not randomised to oral vs IV fluoropyrimidine for the induction therapy component of the study, according to the most recent efficacy and safety update for this study |
| Twelves 2006 | RCT with a cross‐over design. Participants in the oral capecitabine arm received only 1 cycle of chemotherapy before cross‐over, and participants in the IV 5‐FU/LV arm received 2 cycles of de Gramont IV 5‐FU/LV or 1 cycle of Mayo regimen IV 5‐FU/LV (whichever regimen was used routinely in the individual participating centre) before cross‐over |
RCT: randomised controlled trial
IV: intravenous
5‐dFUR: doxifluridine
CRC: colorectal cancer
5‐FU: 5‐fluorouracil
LV: leucovorin
UFT: tegafur/uracil
Characteristics of ongoing studies [ordered by study ID]
Barsukov 2015.
| Trial name or title | Short‐course radiotherapy with concurrent chemotherapy; a single‐center experience |
| Methods | Prospective randomised trial |
| Participants | Target sample size: 150 Stage/treatment type: Distal rectal cancer, neoadjuvant chemoradiation Countries/sites: Russia, single institution ‐ N.N. Blokhin Russian Research Cancer Center, Colorectal Cancer, Moscow, Russian Federation |
| Interventions | Arm I: 5‐FU 425 mg/m2 IV infusion over 24 hours on D1‐5 of radiotherapy Arm II: capecitabine 2000 mg/m2 oral D1‐14 of radiotherapy Arm III: Tegafur 800 mg/m2 oral D1‐21 of radiotherapy Co‐interventions: short‐course 5 × 5 Gy radiotherapy and surgery 2 to 10 weeks after completion of chemo‐radiotherapy |
| Outcomes | Toxicity, tumour regression |
| Starting date | Start: 2011 |
| Contact information | Professor Y Barsukov. N.N. Blokhin Russian Research Cancer Center, Colorectal Cancer, Moscow, Russian Federation |
| Notes | The last informative response from the contact author indicated that the study was ongoing |
GOIM 2802.
| Trial name or title | Bevacizumab + FOLFOX4 or XELOX2 as first‐line treatment in colorectal cancer. Randomized phase 2 study ‐ GOIM 2802 |
| Methods | Prospective open‐label randomised trial |
| Participants | Target sample size: 120 Stage/treatment line: Metastatic, first‐line Countries/Sites: Italy, multiple sites |
| Interventions | Arm A (FOLFOX4 + Bevacizumab (BEV)): 5‐FU 400 mg/m2 bolus D1, D2, 5‐FU 600 mg/m2 infusion D1‐2, Oxaliplatin 85 mg/m2 D1, BEV 5 mg/kg D1. Repeated every 2 weeks Arm B: Capecitabine 2000 mg/m2 orally BD D1‐7, Oxaliplatin 100 mg/m2 D1, BEV 5 mg/kg D1. Repeated every 2 weeks Participants with an objective response or stable disease after 12 cycles will be randomised to: Arm C: 5‐FU 400 mg/m2 bolus D1, D2, 5‐FU 600 mg/m2 infusion D1‐2, BEV 5 mg/kg D1, every 2 weeks; capecitabine 2000 mg/m2 orally BD D1‐7, BEV 5 mg/kg D1. Repeated every 2 weeks. Participants will receive the same fluoropyrimidine used in Arm A or B Arm D: BEV 5 mg/kg, repeated every 2 weeks |
| Outcomes | Primary: ORR Secondary: AEs, OS, TTP |
| Starting date | 2011 |
| Contact information | Dr Evaristo Maiello, Dipartimento di Oncoematologia. U.O. OncologiaI, IRCCS “Casa Sollievo della Sofferenza”, Viale Cappuccini, 71013 San Giovanni Rotondo (FG), Italy |
| Notes | Study ongoing ‐ EU Clinical Trials register https://www.clinicaltrialsregister.eu/ctr‐search/search?query=2010‐022091‐31 (accessed 7 April 2017) |
Joarder 2012.
| Trial name or title | Neoadjuvant chemoradiation with oral capecitabine versus intravenous 5‐fluorouracil and leucovorin in locally advanced carcinoma rectum – a randomized trial |
| Methods | Open label randomised controlled trial Phase: II |
| Participants | Target sample size: ˜100 participants Stage/treatment type: Locally advanced rectal carcinoma, neoadjuvant chemoradiation Countries/sites: India, single institution – Department of Radiotherapy, R. G. Kar Medical College and Hospital, Kolkata |
| Interventions | Arm A (Study, Capecitabine): External beam radiotherapy (EBRT) 50.4 Gy/28 fractions/5.5 weeks with concomitant capecitabine 825 mg/m2 po BD 5 days per week, for the period of EBRT Arm B (Control, 5‐FU‐LV): EBRT 50.4 Gy/28 fractions/5.5 weeks with concomitant 5‐FU 350 mg/m2/d continuous infusion and LV 20 mg/m2 for 5 days every 4 weeks (D1‐5 and D29‐33) Post‐neoadjuvant chemoradiation, participants undergo definitive surgery after 6 weeks. All participants receive adjuvant chemotherapy for 6 months |
| Outcomes | Primary endpoint: Locoregional response Secondary endpoints: Pathological CR, AEs (CTCAE version 4.0) |
| Starting date | January 2011 |
| Contact information | Dr. Abhishek Basu ‐ drabhishekbasu@yahoo.com |
| Notes | The last informative response from the contact author indicated that the study was ongoing |
Muro 2016.
| Trial name or title | A multinational, randomized, Phase III study of XELIRI with/without Bevacizumab versus FOLFIRI with/without Bevacizumab as second‐line therapy in patients with metastatic colorectal cancer |
| Methods | Randomised controlled trial; open label Phase: III |
| Participants | Target sample size: n = 600 Stage/treatment line: Metastatic, second‐line Countries/sites: Japan, South Korea, and China |
| Interventions | Arm I (FOLFIRI +/‐ bevacizumab): bevacizumab 5 mg/kg IV D1, CPT‐11 180 mg/m2 (150 mg/m2 if homozygous for UGT1A1*6 or UGT1A1*28 OR double heterozygous for UGT1A1*6 and UGT1A1*28), l‐LV 200 mg/m2 or dl‐LV 400 mg/m2 IV D1, Bolus 5‐FU 400 mg/m2 IV bolus D1 and Infusional 5‐FU 2400 mg/m2 IV continuous over 46 hours, in a 2‐week cycle Arm II (XELIRI +/‐ bevacizumab): bevacizumab 7.5 mg/kg IV D1, CPT‐11 200 mg/m2 (150 mg/m2 if homozygous for UGT1A1*6 or UGT1A1*28 OR double heterozygous for UGT1A1*6 and UGT1A1*28) IV D1, capecitabine 800 mg/m2 oral BD D1‐15, in a 3 week cycle |
| Outcomes | OS PFS ORR AEs (CTCAE version 4.0) |
| Starting date | Start: December 2013 Estimated completion date: As of August 2015, n = 650 participants had been enrolled. Estimated study completion date is January 2017 (www.clinicaltrials.gov) |
| Contact information | PI: Dr. Kei Muro ‐ kmuro@aichi‐cc.jp |
| Notes | All patients from South Korea and Japan receive concomitant bevacizumab, and the addition of bevacizumab is a stratification factor |
NCT02280070.
| Trial name or title | Randomised Phase II study of SOX vs mFOLFOX6 as neoadjuvant chemotherapy in patients with resectable rectal cancer |
| Methods | Open‐label randomised controlled trial Phase: II |
| Participants | Target sample size: 110 Stage/treatment type: Resectable rectal cancer, neoadjuvant chemotherapy Countries/sites: Multiple institutions in Japan Setting: Hospital |
| Interventions | Arm I (SOX (S‐1 + L‐OHP)): S‐1 80 mg/m2 oral D 1‐14, L‐OHP 130 mg/m2 D1, in a 3 week cycle until 4 cycles or when discontinuation criteria met Arm II (mFOLFOX6 + L‐OHP): 85 mg/m2 and l‐LV 200 mg/m2 by IV infusion D1, 5‐FU Bolus 400 mg/m2 IV bolus D1 and Infusional 5‐FU 2400 mg/m2 IV continuous over 46 hours, in a 2 week cycle for 6 cycles or when discontinuation criteria met |
| Outcomes | DFS OS AEs (CTCAE v4.0) R0 resection rate Pathological effect |
| Starting date | Start: September 2013 Estimated completion date: August 2020; final data collection date for primary outcome measure (3‐year DFS) is August 2018 (www.clinicaltrials.gov) |
| Contact information | PI: Professor Yoshito Akagi, Kurume University, Japan |
| Notes |
5‐FU: 5‐fluorouracil
IV: intravenous
GOIM: Gruppo Oncologico dell’Italia Meriodionale
BEV: bevacizumab
ORR: objective response rate
AEs: adverse events
OS: overall survival
TTP: time to progression
IRCCS: Institute for Research and Health Care
FG: Foggia
EU: European Union
EBRT: external beam radiotherapy
CR: complete response
CTCAE: Common Terminology Criteria for Adverse Events
CPT‐11: camptothecin‐11
PFS: progression‐free survival
L‐OHP: oxaliplatin
DFS: disease‐free survival
Differences between protocol and review
Terminology
We changed the term "intravenous 5‐FU chemotherapy" to "intravenous fluoropyrimidines" to correctly reflect the intended comparison of oral versus IV fluoropyrimidines in this review.
We changed the term "randomised trials" to “randomised controlled trials”.
We changed the term "Grade 3 or 4 toxicity" to "Grade ≥ 3 AEs".
Eligibility criteria
We clarified eligibility criteria regarding inclusion of studies with a cross‐over design to exclude studies in which participants in one or more of the treatment arms received fewer than three cycles of chemotherapy before cross‐over. We judged that reasonable comparisons with respect to efficacy and adverse event outcomes for this type of study design could not be performed in such studies.
Outcomes
We included TTP in participants with inoperable advanced or metastatic CRC who were treated with oral versus IV fluoropyrimidine chemotherapy as a secondary outcome. Multiple studies reported TTP rather than PFS as an efficacy outcome.
We provided further detail regarding the criteria used for grade ≥ 3 AE and ORR assessments.
Search methods
Trial Information Specialists from the Cochrane Colorectal Cancer Group updated search strategies for the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE (OVID), and Embase (OVID).
We formulated the search strategy for the Web of Science (Web of Knowledge) databases with the assistance of Anne McLean (librarian at Austin Hospital, Australia).
We included search strategies for the databases listed above in the 'Appendices' section of the review.
We additionally searched the World Health Organization International Clinical Trials Registry Platform (WHO ICTRP), in keeping with the Methodological Expectations of Cochrane Intevention Reviews (MECIR) guidelines (Chandler 2013).
We did not search CancerLit separately, as we searched the broader MEDLINE database. We did not search Current Contents and Science Citation Index separately. Current Contents is integrated with Web of Science, and Science Citation Index Expanded was already included in the Web of Science search. After discussion with the hospital Drug Information Pharmacist, we did not search the International Pharmaceutical Abstracts database.
We modified the years of searching the proceedings for meeting and conferences.
Data collection and analysis
We changed the independent review authors for study selection, data extraction and management, and assessment of risk of bias to FC, YY, and DL, owing to attrition of AC and SS as authors of this review.
For studies that included patients treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, we clarified that these were included only for DFS and OS outcomes if median follow‐up time was three years or longer; however, they could be included for grade ≥ 3 AEs outcome if median follow‐up time was less than three years.
We provided additional detail regarding assessment of risk of bias.
We provided additional detail regarding analysis of studies with multiple treatment arms.
In response to a peer reviewer suggestion, we performed analyses to assess whether oral fluoropyrimidines are non‐inferior to IV fluoropyrimidines, had the original design been one of non‐inferiority. In response to an editor suggestion, we further assessed whether non‐inferiority had been demonstrated if one made the post hoc judgement that retaining at least 80% of the activity of the active control was reasonable to demonstrate this.
We described pre‐specified subgroup analyses more clearly, in particular to reflect the comparison of any oral fluoropyrimidines versus any IV fluoropyrimidine. We included additional (but still pre‐specified) subgroup analyses to assess important subgroup analyses defined by the following intervention characteristics: received chemotherapy versus received chemo‐radiotherapy (among participants treated with curative intent for CRC)or received single‐agent versus combination therapy (among participants treated with palliative intent for inoperable advanced or metastatic CRC); received infusional versus bolus IV fluoropyrimidine; type of oral fluoropyrimidine backbone given (e.g. capecitabine vs UFT/Ftorafur vs Eniluracil + oral 5‐FU vs doxifluridine vs S‐1); and oxaliplatin‐based versus irinotecan‐based therapy (among participants treated with palliative intent for inoperable advanced or metastatic CRC who received combination chemotherapy).
After identifying the included studies, we performed a post hoc subgroup analysis to compare combination chemotherapy regimens with and without bevacizumab for the co‐primary endpoint of PFS in studies of palliative intent treatment with chemotherapy for inoperable advanced or metastatic CRC.
We provided additional detail in the review regarding the sensitivity analyses performed.
In response to an editor suggestion, we performed a sensitivity analysis for grade ≥ 3 HFS among participants treated with curative intent for CRC with neoadjuvant and/or adjuvant chemotherapy, and used a random‐effects model for meta‐analysis.
We assessed quality of the evidence using the GRADE approach, and reported this for key outcomes using 'Summary of findings' tables, in keeping with MECIR guidelines (Chandler 2013; Tovey 2012).
Contributions of authors
Fiona Chionh: protocol development, with contribution from all protocol co‐authors; co‐ordinating the review; study selection; data extraction; risk of bias assessment; correspondence with study authors and study contacts; data analysis; interpretation of data; writing the review.
Yvonne Yeung: study selection; data extraction; risk of bias assessment; writing the review.
David Lau: study selection; data extraction; risk of bias assessment; interpretation of data; writing the review.
Timothy Price: contribution to protocol development; clinical advice; writing the review.
Niall Tebbutt: contribution to protocol development; study selection; risk of bias assessment; interpretation of data; clinical advice; writing the review.
Declarations of interest
FC has been provided with honorarium from Roche to speak at a non‐promotional educational meeting. DL has received an educational grant from Roche. TP is on advisory boards for Roche and Amgen. NT is on advisory boards for Roche, Amgen and Bayer.
Edited (no change to conclusions)
References
References to studies included in this review
Ahn 2003 {published data only (unpublished sought but not used)}
- Ahn J‐H, Kim T‐W, Lee J‐H, Min Y‐J, Kim J‐G, Kim JC, et al. Oral doxifluridine plus leucovorin in metastatic colorectal cancer: randomized phase II trial with intravenous 5‐fluorouracil plus leucovorin. American Journal of Clinical Oncology 2003;26(1):98‐102. [DOI] [PubMed] [Google Scholar]
Allegra 2015 {published and unpublished data}
- Allegra CJ, Yothers G, O'Connell MJ, Beart RW, Wozniak TF, Pitot HC, et al. Neoadjuvant 5‐FU or capecitabine plus radiation with or without oxaliplatin in rectal cancer patients: a phase III randomized clinical trial. Journal of the National Cancer Institute 2015;107(11):pii: djv248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegra CJ, Yothers G, O'Connell MJ, Roh MS, Beart RW, Petrelli NJ, et al. Neoadjuvant therapy for rectal cancer: mature results from NSABP protocol R‐04. Journal of Clinical Oncology 2014;32(Suppl 3):abstr 390. [Google Scholar]
- Allegra CJ, Yothers G, O'Connell MJ, Roh MS, Lopa SH, Petrelli NJ, et al. Final results from NSABP protocol R‐04: neoadjuvant chemoradiation (RT) comparing continuous infusion (CIV) 5‐FU with capecitabine (Cape) with or without oxaliplatin (Ox) in patients with stage II and III rectal cancer. Journal of Clinical Oncology 2014;32(5s):abstr 3603. [Google Scholar]
- O'Connell MJ, Colangelo LH, Beart RW, Petrelli NJ, Allegra CJ, Sharif S, et al. Capecitabine and oxaliplatin in the preoperative multimodality treatment of rectal cancer: surgical end points from National Surgical Adjuvant Breast and Bowel Project trial R‐04. Journal of Clinical Oncology 2014;32(18):1927‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh MS, Yothers GA, O'Connell MJ, Beart RW, Pitot HC, Shields AF, et al. The impact of capecitabine and oxaliplatin in the preoperative multimodality treatment in patients with carcinoma of the rectum: NSABP R‐04. Journal of Clinical Oncology 2011;29(Suppl):abstr 3503. [Google Scholar]
Andersen 1987 {published data only (unpublished sought but not used)}
- Andersen E, Pedersen H. Oral ftorafur versus intravenous 5‐fluorouracil. A comparative study in patients with colorectal cancer. Acta Oncologica 1987;26(6):433‐6. [DOI] [PubMed] [Google Scholar]
Bajetta 1996 {published data only (unpublished sought but not used)}
- Bajetta E, DiBartolomeo M, Somma L, Moreschi M, Comella G, Turci D, et al. Randomized phase II noncomparative trial of oral and intravenous doxifluridine plus levo‐leucovorin in untreated patients with advanced colorectal carcinoma. Cancer 1996;78(10):2087‐93. [DOI] [PubMed] [Google Scholar]
Carmichael 2002 {published data only (unpublished sought but not used)}
- Carmichael J, Popiela T, Radstone D, Falk S, Borner M, Oza A, et al. Randomized comparative study of tegafur/uracil and oral leucovorin versus parenteral fluorouracil and leucovorin in patients with previously untreated metastatic colorectal cancer. Journal of Clinical Oncology 2002;20(17):3617‐27. [DOI] [PubMed] [Google Scholar]
Cassidy 2011a {published data only (unpublished sought but not used)}
- Arkenau HT, Arnold D, Cassidy J, Diaz‐Rubio E, Douillard JY, Hochster H, et al. Efficacy of oxaliplatin plus capecitabine or infusional fluorouracil/leucovorin in patients with metastatic colorectal cancer: a pooled analysis of randomized trials. Journal of Clinical Oncology 2008;26(36):5910‐7. [DOI] [PubMed] [Google Scholar]
- Cassidy J, Clarke S, Diaz‐Rubio E, Scheithauer W, Figer A, Wong R, et al. Randomized phase III study of capecitabine plus oxaliplatin compared with fluorouracil/folinic acid plus oxaliplatin as first‐line therapy for metastatic colorectal cancer. Journal of Clinical Oncology 2008;26(12):2006‐12. [DOI] [PubMed] [Google Scholar]
- Cassidy J, Clarke S, Diaz‐Rubio E, Scheithauer W, Figer A, Wong R, et al. XELOX vs FOLFOX‐4 as first‐line therapy for metastatic colorectal cancer: NO16966 updated results. British Journal of Cancer 2011;105(1):58‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Comella 2009 {published data only (unpublished sought but not used)}
- Comella P, Massidda B, Filippelli G, Farris A, Natale D, Barberis G, et al. Randomised trial comparing biweekly oxaliplatin plus oral capecitabine versus oxaliplatin plus i.v. bolus fluorouracil/leucovorin in metastatic colorectal cancer patients: results of the Southern Italy Cooperative Oncology study 0401. Journal of Cancer Research and Clinical Oncology 2009;135(2):217‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comella P, Massidda B, Filippelli G, Farris A, Natale D, Barberis G, et al. Randomized comparison of capecitabine versus fluorouracil/leucovorin in combination with oxaliplatin in metastatic colorectal cancer patients: Southern Italy Cooperative Oncology Group trial 0401. ASCO 2008 Gastrointestinal Cancers Symposium. 2008:abstr 344.
- Comella P, Massidda B, Filippelli G, Farris A, Natale D, Maiorino L, et al. Capecitabine or folinic acid/fluorouracil i.v. bolus plus Eloxatin evaluation (COFFE trial) in metastatic colorectal carcinoma (MCRC): final results of the Southern Italy Cooperative Oncology Group (SICOG) phase III trial 0401. ECCO 14 The European Cancer Conference Barcelona 23‐27 September; 2007. 2007:248, abstr 3044.
De Gramont 2012 {published data only (unpublished sought but not used)}
- Gramont A, Cutsem E, Schmoll H‐J, Tabernero J, Clarke S, Moore MJ, et al. Bevacizumab plus oxaliplatin‐based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncology 2012;13:1225‐33. [DOI] [PubMed] [Google Scholar]
- Gramont A, Cutsem E, Tabernero J, Moore M, Cunningham D, Rivera F, et al. AVANT: results from a randomized, three‐arm multinational phase III study to investigate bevacizumab with either XELOX or FOLFOX4 versus FOLFOX4 alone as adjuvant treatment for colon cancer. Journal of Clinical Oncology 2011;29(Suppl 4):abstr 362. [Google Scholar]
De la Torre 2008 {published and unpublished data}
- Torre A, Garcia‐Berrocal MI, Arias F, Marino A, Valcarcel F, Magallon R, et al. Preoperative chemoradiotherapy for rectal cancer: randomized trial comparing oral uracil and tegafur and oral leucovorin vs. intravenous 5‐fluorouracil and leucovorin. International Journal of Radiation Oncology Biology Physics 2008;70(1):102‐10. [DOI] [PubMed] [Google Scholar]
Diaz‐Rubio 2007 {published data only (unpublished sought but not used)}
- Diaz‐Rubio E, Tabernero J, Gomez‐Espana A, Massuti B, Sastre J, Chaves M, et al. Phase III study of capecitabine plus oxaliplatin compared with continuous‐infusion fluorouracil plus oxaliplatin as first‐line therapy in metastatic colorectal cancer: final report of the Spanish Cooperative Group for the Treatment of Digestive Tumors Trial. Journal of Clinical Oncology 2007;25(27):4224‐30. [DOI] [PubMed] [Google Scholar]
Douillard 2002 {published data only (unpublished sought but not used)}
- Douillard JY, Hoff PM, Skillings JR, Eisenberg P, Davidson N, Harper P, et al. Multicenter phase III study of uracil/tegafur and oral leucovorin versus fluorouracil and leucovorin in patients with previously untreated metastatic colorectal cancer. Journal of Clinical Oncology 2002;20(17):3605‐16. [DOI] [PubMed] [Google Scholar]
Douillard 2014 {published and unpublished data}
- Douillard J‐Y, Zemelka T, Fountzilas G, Barone C, Schlichting M, Eggleton S, et al. Randomized phase II study evaluating UFOX plus cetuximab versus FOLFOX4 plus cetuximab as first‐line therapy in metastatic colorectal cancer: FUTURE. Annals of Oncology 2012;23(Suppl 4):iv5‐iv18, abstr 0‐0.0017. [DOI] [PubMed] [Google Scholar]
- Douillard JY, Zemelka T, Fountzilas G, Barone C, Schlichting M, Heighway J, et al. FOLFOX4 With Cetuximab vs. UFOX With Cetuximab as First‐Line Therapy in Metastatic Colorectal Cancer: The Randomized Phase II FUTURE Study. Clinical Colorectal Cancer 2014;13(1):14‐26. [DOI] [PubMed] [Google Scholar]
Ducreux 2011 {published data only (unpublished sought but not used)}
- Ducreux M, Bennouna J, Hebbar M, Ychou M, Lledo G, Conroy T, et al. Capecitabine plus oxaliplatin (XELOX) versus 5‐fluorouracil/leucovorin plus oxaliplatin (FOLFOX‐6) as first‐line treatment for metastatic colorectal cancer. Journal international du cancer [International Journal of Cancer] 2011;128:682‐90. [DOI] [PubMed] [Google Scholar]
- Ducreux M, Bennouna J, Hebbar M, Ychou M, Lledo G, Conroy T, et al. Efficacy and safety findings from a randomized phase III study of capecitabine (X) + oxaliplatin (O) (XELOX) vs. infusional 5‐FU/LV + O (FOLFOX‐6) for metastatic colorectal cancer (MCRC). Journal of Clinical Oncology 2007;25(18S):abstr 4029. [Google Scholar]
Ducreux 2013 {published and unpublished data}
- Ducreux M, Adenis A, Mendiboure J, Francois E, Boucher E, Chauffert M, et al. Efficacy and safety of bevacizumab (BEV)‐ based combination regimens in patients with metastatic colorectal cancer (mCRC): Randomized phase II study of BEV + FOLFIRI versus BEV + XELIRI (FNCLCC ACCORD 13/0503 study). Journal of Clinical Oncology 2009;27(15S):abstr 4086. [Google Scholar]
- Ducreux M, Adenis A, Pignon J‐P, François E, Chauffert B, Ichanté JL, et al. Efficacy and safety of bevacizumab‐based combination regimens in patients with previously untreated metastatic colorectal cancer: final results from a randomised phase II study of bevacizumab plus 5‐fluorouracil, leucovorin plus irinotecan versus bevacizumab plus capecitabine plus irinotecan (FNCLCC ACCORD13/0503 study). European Journal of Cancer 2013;49:1236‐45. [DOI] [PubMed] [Google Scholar]
- Malka D, Boige V, Jacques N, Vimond N, Adenis A, Boucher E, et al. Clinical value of circulating endothelial cell levels in metastatic colorectal cancer patients treated with first‐line chemotherapy and bevacizumab. Annals of Oncology 2012;23(4):919‐27. [DOI] [PubMed] [Google Scholar]
ECOG E5296 2012 {unpublished data only}
- Levy D, Leshne G, DiGiuseppe S, Bucher T. Analysis of E5296: phase III trial comparing a 28 day schedule of daily oral 5‐FU plus eniluracil to protracted intravenous infusion in previously untreated patients with advanced colorectal cancer. Boston (MA): Dana‐Farber Cancer Institute 2005 (Revised July 2, 2012):Technical Report No.: 1080E.
Fuchs 2007 {published and unpublished data}
- Fuchs CS, Marshall J, Barrueco. Capecitabine and irinotecan as first‐line treatment of advanced colorectal cancer‐ author reply. Journal of Clinical Oncology 2008;26(11):1908‐9. [DOI] [PubMed] [Google Scholar]
- Fuchs CS, Marshall J, Mitchell E, Wierzbicki R, Ganju V, Jeffery M, et al. Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first‐line treatment of metastatic colorectal cancer: results from the BICC‐C study. Journal of Clinical Oncology 2007;25(30):4779‐86. [DOI] [PubMed] [Google Scholar]
Hochster TREE‐1 2008 {published data only (unpublished sought but not used)}
- Hochster HS, Hart LL, Ramanathan RK, Childs BH, Hainsworth JD, Cohn AL, et al. Safety and efficacy of oxaliplatin and fluoropyrimidine regimens with or without bevacizumab as first‐line treatment of metastatic colorectal cancer: results of the TREE Study. Journal of Clinical Oncology 2008;26(21):3523‐9. [DOI] [PubMed] [Google Scholar]
Hochster TREE‐2 2008 {published data only (unpublished sought but not used)}
- Hochster HS, Hart LL, Ramanathan RK, Childs BH, Hainsworth JD, Cohn AL, et al. Safety and efficacy of oxaliplatin and fluoropyrimidine regimens with or without bevacizumab as first‐line treatment of metastatic colorectal cancer: results of the TREE Study. Journal of Clinical Oncology 2008;26(21):3523‐9. [DOI] [PubMed] [Google Scholar]
Hoff 2001 {published data only (unpublished sought but not used)}
- Hoff PM, Ansari R, Batist G, Cox J, Kocha W, Kuperminc M, et al. Comparison of oral capecitabine versus intravenous fluorouracil plus leucovorin as first‐line treatment in 605 patients with metastatic colorectal cancer: results of a randomized phase III study. Journal of Clinical Oncology 2001;19(8):2282‐92. [DOI] [PubMed] [Google Scholar]
- Cutsem E, Hoff PM, Harper P, Bukowski RM, Cunningham D, Dufour P, et al. Oral capecitabine vs intravenous 5‐fluorouracil and leucovorin: integrated efficacy data and novel analyses from two large, randomised, phase III trials. British Journal of Cancer 2004;90(6):1190‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hofheinz 2012 {published and unpublished data}
- Hofheinz RD, Wenz F, Post S, Matzdorff A, Laechelt S, Hartmann JT, et al. Chemoradiotherapy with capecitabine versus fluorouracil for locally advanced rectal cancer: a randomised, multicentre, non‐inferiority, phase 3 trial. Lancet Oncology 2012;13(6):579‐88. [DOI] [PubMed] [Google Scholar]
Kato 2012 {published and unpublished data}
- Gamoh M, Andoh H, Yamaguchi T, Maeda S, Sasaki Y, Suzuki T, et al. A randomized pilot study comparing safety and efficacy of irinotecan plus S‐1 plus bevacizumab (IRIS1BV) and modified FOLFIRI plus BV (mFOLFIRI+BV) in patients (pts) with metastatic colorectal cancer (MCRC): The result of T‐core0702. Annals of Oncology 2011;22(Suppl 9):ix49‐ix50, abstr 109. [Google Scholar]
- Kato S, Andoh H, Gamoh M, Yamaguchi T, Murakawa Y, Sasaki Y, et al. A randomized pilot study comparing safety and efficacy of irinotecan plus S‐1 (IRIS) plus bevacizumab (BV) and modified (m) FOLFIRI plus BV in patients (pts) with metastatic colorectal cancer (mCRC): first report of T‐CORE0702. Journal of Clinical Oncology 2011;4(Suppl):abstr 496. [Google Scholar]
- Kato S, Andoh H, Gamoh M, Yamaguchi T, Murakawa Y, Shimodaira H, et al. Safety verification trials of mFOLFIRI and sequential IRIS + bevacizumab as first‐ or second‐line therapies for metastatic colorectal cancer in Japanese patients. Oncology 2012;83:101‐7. [DOI] [PubMed] [Google Scholar]
Kim 2001a {published data only (unpublished sought but not used)}
- Kim NK, Lee KY, Park JK, Yun SH, Roh JK, Min JS. Prospective Randomized Trials Comparing Intravenous 5‐Fluorouracil and Oral Doxifluridine as a Postoperative Adjuvant Treatment for Advanced Rectal Cancer [Korean]. Journal of the Korean Surgical Society 2001;60(2):195‐9. [DOI] [PubMed] [Google Scholar]
- Min JS, Kim NK, Park JK, Yun SH, Noh JK. A prospective randomized trial comparing intravenous 5‐fluorouracil and oral doxifluridine as postoperative adjuvant treatment for advanced rectal cancer. Annals of Surgical Oncology 2000;7(9):674‐9. [DOI] [PubMed] [Google Scholar]
Kohne 2008 {published data only (unpublished sought but not used)}
- Kohne CH, Greve J, Hartmann JT, Lang I, Vergauwe P, Becker K, et al. Irinotecan combined with infusional 5‐fluorouracil/folinic acid or capecitabine plus celecoxib or placebo in the first‐line treatment of patients with metastatic colorectal cancer. EORTC study 40015. Annals of Oncology 2008;19(5):920‐6. [DOI] [PubMed] [Google Scholar]
Lembersky 2006 {published data only (unpublished sought but not used)}
- Lembersky BC, Wieand HS, Petrelli NJ, O'Connell MJ, Colangelo LH, Smith RE, et al. Oral uracil and tegafur plus leucovorin compared with intravenous fluorouracil and leucovorin in stage II and III carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project protocol C‐06. Journal of Clinical Oncology 2006;24(13):2059‐64. [DOI] [PubMed] [Google Scholar]
Martoni 2006 {published data only (unpublished sought but not used)}
- Martoni AA, Pinto C, Fabio F, Lelli G, Llimpe FLR, Gentile AL, et al. Capecitabine plus oxaliplatin (xelox) versus protracted 5‐fluorouracil venous infusion plus oxaliplatin (pvifox) as first‐line treatment in advanced colorectal cancer: a GOAM phase II randomised study (FOCA trial). European Journal of Cancer 2006;42(18):3161‐8. [DOI] [PubMed] [Google Scholar]
Mei 2014 {published data only}
- Mei Z, Yanhua Z, Weiyan G, Hongxia H, Lige Y, Tiandong K. Clinical observation of S‐1 plus oxaliplatin in the treatment of locally advanced or metastatic colorectal cancer [Chinese]. Cancer Research and Clinic 2014;26(12):820‐2. [Google Scholar]
Nogue 2005 {published data only (unpublished sought but not used)}
- Nogue M, Salud A, Batiste‐Alentorn E, Saigi E, Losa F, Cirera L, et al. Randomised study of tegafur and oral leucovorin versus intravenous 5‐fluorouracil and leucovorin in patients with advanced colorectal cancer. European Journal of Cancer 2005;41(15):2241‐9. [DOI] [PubMed] [Google Scholar]
- Salud A, Saigi E, Batiste‐Alentorn E, Losa F, Cirera L, Mendez M, et al. Randomized phase IV trial of oral tegafur and low dose leucovorin versus intravenous 5‐fluorouracil and leucovorin in the treatment of advanced colorectal cancer (ACC): final results. Journal of Clinical Oncology 2004;22(14S):abstr 3547. [Google Scholar]
Pectasides 2012 {published and unpublished data}
- Pectasides D, Papaxoinis G, Kalogeras K, Eleftheraki A, Xanthakis I, Makatsoris T, et al. XELIRI‐‐bevacizumab versus FOLFIRI‐bevacizumab as first‐line treatment in patients with metastatic colorectal cancer: a Hellenic Cooperative Oncology Group phase III trial with collateral biomarker analysis. BMC Cancer 2012;12(1):271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pectasides 2015 {published data only (unpublished sought but not used)}
- Maniadakis N, Fragoulakis V, Pectasides D, Fountzilas G. XELOX versus FOLFOX6 as an adjuvant treatment in colorectal cancer: an economic analysis. Current Medical Research and Opinion 2009;25(3):797‐805. [DOI] [PubMed] [Google Scholar]
- Pectasides D, Karavasilis V, Papaxoinis G, Gourgioti G, Makatsoris T, Raptou G, et al. Randomized phase III clinical trial comparing the combination of capecitabine and oxaliplatin (CAPOX) with the combination of 5‐fluorouracil, leucovorin and oxaliplatin (modified FOLFOX6) as adjuvant therapy in patients with operated high‐risk stage II or stage III colorectal cancer. BMC Cancer 2015;15:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pectasides DG, Papaxoinis G, Xanthakis I, Kouvatseas G, Kotoula V, Xiros N, et al. Randomized phase III trial of FOLFOX versus XELOX as adjuvant chemotherapy in patients with early‐stage colorectal adenocarcinoma. Journal of Clinical Oncology 2014;32(5s):abstr 3617. [Google Scholar]
Porschen 2007 {published data only (unpublished sought but not used)}
- Arkenau HT, Arnold D, Cassidy J, Diaz‐Rubio E, Douillard JY, Hochster H, et al. Efficacy of oxaliplatin plus capecitabine or infusional fluorouracil/leucovorin in patients with metastatic colorectal cancer: a pooled analysis of randomized trials. Journal of Clinical Oncology 2008;26(36):5910‐7. [DOI] [PubMed] [Google Scholar]
- Porschen R, Arkenau HT, Kubicka S, Greil R, Seufferlein T, Freier W, et al. Phase III study of capecitabine plus oxaliplatin compared with fluorouracil and leucovorin plus oxaliplatin in metastatic colorectal cancer: a final report of the AIO Colorectal Study Group. Journal of Clinical Oncology 2007;25(27):4217‐23. [DOI] [PubMed] [Google Scholar]
Rothenberg 2008 {published data only (unpublished sought but not used)}
- Rothenberg M, Navarro M, Butts C, Bang Y, Cox J, Goel R, et al. Phase III trial of capecitabine + oxaliplatin (XELOX) vs. 5‐fluorouracil (5‐FU), leucovorin (LV), and oxaliplatin (FOLFOX4) as 2nd‐line treatment for patients with metastatic colorectal cancer (MCRC). Journal of Clinical Oncology 2007;25(18S):abstr 4031. [Google Scholar]
- Rothenberg ML, Cox JV, Butts C, Navarro M, Bang YJ, Goel R, et al. Capecitabine plus oxaliplatin (XELOX) versus 5‐fluorouracil/folinic acid plus oxaliplatin (FOLFOX‐4) as second‐line therapy in metastatic colorectal cancer: a randomized phase III noninferiority study. Annals of Oncology 2008;19(10):1720‐6. [DOI] [PubMed] [Google Scholar]
Schilsky 2002a {published and unpublished data}
- Schilsky RL, Levin J, West WH, Wong A, Colwell B, Thirlwell MP, et al. Randomized, open‐label, phase III study of a 28‐day oral regimen of eniluracil plus fluorouracil versus intravenous fluorouracil plus leucovorin as first‐line therapy in patients with metastatic/advanced colorectal cancer. Journal of Clinical Oncology 2002;20(6):1519‐26. [DOI] [PubMed] [Google Scholar]
Seymour 2011 {published data only (unpublished sought but not used)}
- Seymour MT, Thompson LC, Wasan HS, Middleton G, Brewster AE, Shepherd SF, et al. Chemotherapy options in elderly and frail patients with metastatic colorectal cancer (MRC FOCUS2): an open‐label, randomised factorial trial. Lancet 2011;377(9779):1749‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shigeta 2016 {published and unpublished data}
- Hasegawa H, Endo T, Ochiai D, Uchida H, Okabayashi T, Imai S, et al. A randomized Phase II study of tegafur/uracil (UFT), oral leucovorin and irinotecan, combination therapy (TEGAFIRI)+bevacizumab versus FOLFIRI+bevacizumab for the first‐line treatment of patients with metastatic colorectal cancer. European Journal of Cancer 2013;49 (Suppl 2):S557, abstr 2390. [Google Scholar]
- Shigeta K, Hasegawa H, Okabayashi K, Tsuruta M, Ishii Y, Endo T, et al. Randomized phase II trial of TEGAFIRI (tegafur/uracil, oral leucovorin, irinotecan) compared with FOLFIRI (folinic acid,5‐fluorouracil, irinotecan) in patients with unresectable/recurrent colorectal cancer. International Journal of Cancer 2016;139(4):946‐54. [DOI] [PubMed] [Google Scholar]
Shimada 2014 {published data only (unpublished sought but not used)}
- Shimada Y, Hamaguchi T, Mizusawa J, Saito N, Kanemitsu Y, Takiguchi N, et al. Randomised phase III trial of adjuvant chemotherapy with oral uracil and tegafur plus leucovorin versus intravenous fluorouracil and levofolinate in patients with stage III colorectal cancer who have undergone Japanese D2/D3 lymph node dissection: Final results of JCOG0205. European Journal of Cancer 2014;50(13):2231‐2240. [DOI] [PubMed] [Google Scholar]
- Shimada Y, Hamaguchi T, Moriya Y, Saito N, Kanemitsu Y, Takiguchi N, et al. Randomized phase III study of adjuvant chemotherapy with oral uracil and tegafur plus leucovorin versus intravenous fluorouracil and levofolinate in patients (pts) with stage III colon cancer (CC): final results of Japan Clinical Oncology Group study (JCOG0205). Journal of Clinical Oncology 2012;30(Suppl):abstr 3524. [Google Scholar]
Silvestris 2010 {published data only (unpublished sought but not used)}
- Silvestris N, Maiello E, Vita F, Cinieri S, Santini D, Russo A, et al. Update on capecitabine alone and in combination regimens in colorectal cancer patients. Cancer Treatment Reviews 2010;36(Suppl 3):S46‐55. [DOI] [PubMed] [Google Scholar]
Souglakos 2012 {published and unpublished data}
- Souglakos J, Ziras N, Kakolyris S, Boukovinas I, Kentepozidis N, Makrantonakis P, et al. Randomised phase‐II trial of CAPIRI (capecitabine, irinotecan) plus bevacizumab vs FOLFIRI (folinic acid, 5‐fluorouracil, irinotecan) plus bevacizumab as first‐line treatment of patients with unresectable/metastatic colorectal cancer (mCRC). British Journal of Cancer 2012;106(3):453‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Twelves 2012 {published and unpublished data}
- Scheithauer W, McKendrick J, Begbie S, Borner M, Burns WI, Burris HA, et al. Oral capecitabine as an alternative to i.v. 5‐fluorouracil‐based adjuvant therapy for colon cancer: safety results of a randomized, phase III trial. Annals of Oncology 2003;14(12):1735‐43. [DOI] [PubMed] [Google Scholar]
- Twelves C, Scheithauer W, McKendrick J, Seitz JF, Hazel G, Wong A, et al. Capecitabine versus 5‐fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results from the X‐ACT trial with analysis by age and preliminary evidence of a pharmacodynamic marker of efficacy. Annals of Oncology 2012;23(5):1190‐7. [DOI] [PubMed] [Google Scholar]
- Twelves C, Wong A, Nowacki MP, Abt M, Burris H, 3rd, Carrato A, et al. Capecitabine as adjuvant treatment for stage III colon cancer. New England Journal of Medicine 2005;352(26):2696‐704. [DOI] [PubMed] [Google Scholar]
Van Cutsem 2001a {published and unpublished data}
- Cutsem E, Sorensen J, Cassidy J, Daniel F, Harper P, Bailey N, et al. International phase III study of oral eniluracil (EU) plus 5‐fluorouracil (5‐FU) versus intravenous (IV) 5‐FU plus leucovorin (LV) in the treatment of advanced colorectal cancer (ACC). Journal of Clinical Oncology, Proceedings of American Society of Clinical Oncology 2001;20:abstr 522. [Google Scholar]
Van Cutsem 2001b {published data only (unpublished sought but not used)}
- Cutsem E, Hoff PM, Harper P, Bukowski RM, Cunningham D, Dufour P, et al. Oral capecitabine vs intravenous 5‐fluorouracil and leucovorin: integrated efficacy data and novel analyses from two large, randomised, phase III trials. British Journal of Cancer 2004;90(6):1190‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutsem E, Twelves C, Cassidy J, Allman D, Bajetta E, Boyer M, et al. Oral capecitabine compared with intravenous fluorouracil plus leucovorin in patients with metastatic colorectal cancer: results of a large phase III study. Journal of Clinical Oncology 2001;19(21):4097‐106. [DOI] [PubMed] [Google Scholar]
Yamada 2013 {published and unpublished data}
- Matsumoto H, Yamada Y, Takahari D, Baba H, Yoshida K, Komatsu Y, et al. The SOFT study: A randomized phase III trial of S‐1/oxaliplatin (SOX)plus bevacizumab versus 5‐FU/l‐LV/oxaliplatin (mFOLFOX6) plus bevacizumab in patients with metastatic colorectal cancer [SOFT Study Group]. European Journal of Cancer 2013;49(Suppl 2):S486, abstr 2167. [Google Scholar]
- Takahari D, Yamada Y, Matsumoto H, Baba H, Yoshida K, Nakamura M, et al. A randomized phase III trial of S‐1/oxaliplatin (SOX) plus bevacizumab versus 5‐FU/l‐LV/oxaliplatin (mFOLFOX6) plus bevacizumab in patients with metastatic colorectal cancer: the SOFT study. Journal of Clinical Oncology 2013;15(suppl):abstr 3519. [Google Scholar]
- Yamada Y, Takahari D, Matsumoto H, Baba H, Nakamura M, Yoshida K, et al. Leucovorin, fluorouracil, and oxaliplatin plus bevacizumab versus S‐1 and oxaliplatin plus bevacizumab in patients with metastatic colorectal cancer (SOFT): an open‐label, non‐inferiority, randomised phase 3 trial. Lancet Oncology 2013;14:1278‐86. [DOI] [PubMed] [Google Scholar]
Yamazaki 2015 {published and unpublished data}
- Ojima H, Yamakazi K, Kuwano H, Otsuji T, Kato T, Shimada K, et al. Randomized Phase II study of S‐1, oral leucovorin, and oxaliplatin combination therapy (SOL) versus mFOLFOX6 in patients with untreated metastatic colorectal cancer (mCRC). European Journal of Cancer 2011;47(Suppl 1):427, abstr 6118. [Google Scholar]
- Otsuji T, Yamazaki K, Ojima H, Kuwano H, Kato T, Shimada K, et al. Updated survival results of the randomized phase II study of S‐1, oral leucovorin, and oxaliplatin combination therapy (SOL) versus mFOLFOX6 in patients with untreated metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2012;30(Suppl 4):abstr 586. [Google Scholar]
- Yamazaki K, Kuwano H, Ojima H, Otsuji T, Kato T, Shimada K, et al. A randomized phase II study of combination therapy with S‐1, oral leucovorin, and oxaliplatin (SOL) and mFOLFOX6 in patients with previously untreated metastatic colorectal cancer. Cancer Chemotherapy and Pharmacology 2015;75(3):569‐77. [DOI] [PubMed] [Google Scholar]
Yasui 2015 {published and unpublished data}
- Baba H, Muro K, Yasui H, Shimada Y, Tsuji A, Sameshima S, et al. Updated results of the FIRIS study: A phase II/III trial of 5‐FU/l‐leucovorin/irinotecan (FOLFIRI) versus irinotecan/S‐1 (IRIS) as second‐line chemotherapy for metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2011;29(Suppl):abstr 3562. [Google Scholar]
- Muro K, Boku N, Shimada Y, Tsuji A, Sameshima S, Baba H, et al. Irinotecan plus S‐1 (IRIS) versus fluorouracil and folinic acid plus irinotecan (FOLFIRI) as second‐line chemotherapy for metastatic colorectal cancer: a randomised phase 2/3 non‐inferiority study (FIRIS study). Lancet Oncology 2010;11(9):853‐60. [DOI] [PubMed] [Google Scholar]
- Yasui H, Muro K, Shimada Y, Tsuji A, Sameshima S, Baba H, et al. A phase 3 non‑inferiority study of 5‑FU/l‑leucovorin/irinotecan(FOLFIRI) versus irinotecan/S‑1 (IRIS) as second‑line chemotherapy for metastatic colorectal cancer: updated results of the FIRIS study. Journal of Cancer Research and Clinical Oncology 2015;141:153‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yu 2005 {published data only (unpublished sought but not used)}
- Yu B‐M, Wu W‐Q. Irinotecan combined with fluoropyrimidine in treatment for advanced/metastatic colorectal carcinoma. Chung‐Hua Wai Ko Tsa Chih [Chinese Journal of Surgery] 2005;43(9):557‐60. [PubMed] [Google Scholar]
References to studies excluded from this review
Ansfield 1977 {published data only}
- Ansfield F, Klotz J, Nealon T, Ramirez G, Minton J, Hill G, et al. A phase III study comparing the clinical utility of four regimens of 5‐fluorouracil: a preliminary report. Cancer 1977;39(1):34‐40. [DOI] [PubMed] [Google Scholar]
Bajetta 1997 {published data only}
- Bajetta E, Bartolomeo M, Somma L, Vecchio M, Artale S, Zunino F, et al. Doxifluridine in colorectal cancer patients resistant to 5‐fluorouracil (5‐FU) containing regimens. European Journal of Cancer Part A 1997;33(4):687‐90. [DOI] [PubMed] [Google Scholar]
Bedikian 1983 {published data only}
- Bedikian AY, Stroehlein J, Korinek J, Karlin D, Bodey GP. A comparative study of oral tegafur and intravenous 5‐fluorouracil in patients with metastatic colorectal cancer. American Journal of Clinical Oncology 1983;6(2):181‐6. [DOI] [PubMed] [Google Scholar]
Bjerkeset 1986 {published data only}
- Bjerkeset T, Fjosne HE. Comparison of oral ftorafur and intravenous 5‐fluorouracil in patients with advanced cancer of the stomach, colon or rectum. Oncology 1986;43(4):212‐5. [DOI] [PubMed] [Google Scholar]
Borner 2002 {published data only}
- Borner MM, Schoffski P, Wit R, Caponigro F, Comella G, Sulkes A, et al. Patient preference and pharmacokinetics of oral modulated UFT versus intravenous fluorouracil and leucovorin: a randomised crossover trial in advanced colorectal cancer. European Journal of Cancer 2002;38(3):349‐58. [DOI] [PubMed] [Google Scholar]
Douglass 1978 {published data only}
- Douglass HO, Jr, Lavin PT, Woll J, Conroy JF, Carbone P. Chemotherapy of advanced measurable colon and rectal carcinoma with oral 5‐fluorouracil, alone or in combination with cyclophosphamide or 6‐thioguanine, with intravenous 5‐fluorouracil or beta‐2'‐deoxythioguanosine or with oral 3(4‐methyl‐cyclohexyl)‐1(2‐chlorethyl)‐1‐nitrosourea: a Phase II‐III study of the Eastern Cooperative Oncology Group (EST 4273). Cancer 1978;42(6):2538‐45. [DOI] [PubMed] [Google Scholar]
Fan 2005 {published data only}
- Fan L, Liu W‐C, Zhang Y‐J, Ren J, Pan B‐R, Liu D‐H, et al. Oral xeloda plus bi‐platinu two‐way combined chemotherapy in treatment of advanced gastrointestinal malignancies. World Journal of Gastroenterology 2005;11(28):4300‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hahn 1975 {published data only}
- Hahn RG, Moertel CG, Schutt AJ, Bruckner HW. A double‐blind comparison of intensive course 5‐flourouracil by oral vs. intravenous route in the treatment of colorectal carcinoma. Cancer 1975;35(4):1031‐5. [DOI] [PubMed] [Google Scholar]
Hennig 2008 {published data only}
- Hennig IM, Naik JD, Brown S, Szubert A, Anthoney DA, Jackson DP, et al. Severe sequence‐specific toxicity when capecitabine is given after Fluorouracil and leucovorin. Journal of Clinical Oncology 2008;26(20):3411‐7. [DOI] [PubMed] [Google Scholar]
Kim 2001b {published data only (unpublished sought but not used)}
- Kim NK, Min JS, Park JK, Yun SH, Sung JS, Jung HC, et al. Intravenous 5‐fluorouracil versus oral doxifluridine as preoperative concurrent chemoradiation for locally advanced rectal cancer: prospective randomized trials. Japanese Journal of Clinical Oncology 2001;31(1):25‐9. [DOI] [PubMed] [Google Scholar]
Lima 2005 {published data only}
- Lima APR, Giglio A. Randomized crossover trial of intravenous 5‐FU versus oral UFT both modulated by leucovorin: a one‐centre experience. European Journal of Cancer Care 2005;14(2):151‐4. [DOI] [PubMed] [Google Scholar]
Maetani 1993 {published data only}
- Maetani S. Multicenter clinical trial of UFT with FT for patients with colorectal cancer. Therapeutic Research 1993;14(6):226‐32. [Google Scholar]
Munoz 2008 {published data only (unpublished sought but not used)}
- Munoz Llarena A, Salud Salvia A, García Girón C, Murias Rosales A, Burillo M, Arizcun Sánchez‐Morate A, et al. Randomized phase III trial, with irinotecan and capecitabine (XELIRI) versus irinotecan, 5FU and folinic acid (Saltz regimen) in first‐line treatment on metastatic colorectal cancer patients (CRC). ASCO 2008 Gastrointestinal Cancers Symposium. 2008:abstr 379.
NCT00070122 {published and unpublished data}
- NCT00070122. Combination chemotherapy and bevacizumab in treating patients with locally recurrent colorectal cancer. http://clinicaltrials.gov/ct2/show/NCT00070122 (accessed 8 June 2016).
NCT01193452 {published and unpublished data}
- NCT01193452. S‐1/leucovorin (SL) versus sLV5FU2 as the first‐line treatment for elderly patients with colorectal cancer. http://clinicaltrials.gov/ct2/show/NCT01193452 (accessed 8 June 2016).
NCT01196260 {published data only}
- NCT01196260. Combination chemotherapy treatments in patients with colorectal cancer stage II and III. http://clinicaltrials.gov/ct2/show/NCT01196260 (accessed 8 June 2016).
NCT01279681 {published data only}
- NCT01279681. Randomized phase III trial of mFOLFOX7 or XELOX plus bevacizumab versus 5‐fluorouracil/leucovorin or capecitabine plus bevacizumab as first‐line treatment in elderly patients with metastatic colorectal cancer. http://clinicaltrials.gov/ct2/show/NCT01279681 (accessed 8 June 2016).
NCT01736904 {published data only (unpublished sought but not used)}
- NCT01736904. wXELIRI versus FOLFIRI regimen in the treatment of advanced colorectal cancer patients. http://clinicaltrials.gov/ct2/show/NCT01736904 (accessed 8 June 2016).
Pfeiffer 2006 {published data only}
- Pfeiffer P, Mortensen JP, Bjerregaard B, Eckhoff L, Schonnemann K, Sandberg E, et al. Patient preference for oral or intravenous chemotherapy: A randomised cross‐over trial comparing capecitabine and Nordic fluorouracil/leucovorin in patients with colorectal cancer. European Journal of Cancer 2006;42(16):2738‐43. [DOI] [PubMed] [Google Scholar]
Queißer 1979 {published data only}
- Queißer W, Schaefer J, Arnold H, Drings P, Geldmacher J, Hartwich G, et al. A prospective multi‐centre study of the response of metastatic gastrointestinal tumours [Prospektive kontrollierte studie bei metastasierten gastrointestinalen tumoren: vergleich zwischen 5‐fluorouracil ‐ carmustin und ftorafur ‐ carmustin]. Deutsche Medizinische Wochenschrift 1979;104(35):1231‐6. [DOI] [PubMed] [Google Scholar]
Revazishvili 2008 {published data only (unpublished sought but not used)}
- Revazishvili P, Shavdia M, Chvamichva R, Shavidia N. Oral ftorafur in treatment of advanced colorectal cancer. Annals of Oncology 2008;19(Suppl 8):viii147, abstr 437. [Google Scholar]
Sizer 2006 {published data only (unpublished sought but not used)}
- Sizer B, Makris A, Barone C, Mainwaring P, Eggleton P. QoL and resource use analysis of tegafur‐uracil/LV or 5‐FU/LV in first‐line metastatic colorectal cancer (mCRC): final results of a multicenter phase II study. Journal of Clinical Oncology, 2006 ASCO Annual Meeting Proceedings Part I 2006;24(18S):abstr 3631. [Google Scholar]
Skof 2009 {published data only (unpublished sought but not used)}
- Skof E, Rebersek M, Hlebanja Z, Ocvirk J. Capecitabine plus Irinotecan (XELIRI regimen) compared to 5‐FU/LV plus Irinotecan (FOLFIRI regimen) as neoadjuvant treatment for patients with unresectable liver‐only metastases of metastatic colorectal cancer: a randomised prospective phase II trial. BMC Cancer 2009;9:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tournigand 2012 {published data only}
- Tournigand C, Samson B, Scheithauer W, Lledo G, Viret F, Andre T, et al. Bevacizumab (Bev) with or without erlotinib as maintenance therapy, following induction first‐line chemotherapy plus Bev, in patients (pts) with metastatic colorectal cancer (mCRC): Efficacy and safety results of the International GERCOR DREAM phase III trial. Abstract No: LBA3500, Presenter: Christophe Tournigand, MD, PhD. 2012 ASCO Annual Meeting, Gastrointestinal (Colorectal) Cancer Track‐ Oral Abstract Session. Available from: http://www.asco.org/ASCOv2/MultiMedia/Virtual+Meeting?&vmview=vm_session_presentations_view&confID=114&sessionID=4814 (accessed 26 September 2012).
- Tournigand C, Samson B, Scheithauer W, Lledo G, Viret F, Andre T, et al. Bevacizumab (Bev) with or without erlotinib as maintenance therapy, following induction first‐line chemotherapy plus Bev, in patients (pts) with metastatic colorectal cancer (mCRC): Efficacy and safety results of the International GERCOR DREAM phase III trial. Journal of Clinical Oncology 2012;30(Suppl):abstr LBA3500. [Google Scholar]
Twelves 2006 {published data only}
- Twelves C, Gollins S, Grieve R, Samuel L. A randomised cross‐over trial comparing patient preference for oral capecitabine and 5‐fluorouracil/leucovorin regimens in patients with advanced colorectal cancer. Annals of Oncology 2006;17(2):239‐45. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
Barsukov 2015 {published data only}
- Barsukov Y, Perevoshikov A, Kuzmichev D, Mammadli Z, Polynovskiy A. Combined treatment of resectable distal rectal cancer with different fluoropyrimidines. European Journal of Surgical Oncology 2012;38(9):832, abstr 324. [Google Scholar]
GOIM 2802 {unpublished data only}
- GOIM 2802. Bevacizumab + FOLFOX4 or XELOX2 as first‐line treatment in colorectal cancer. Randomized phase II study. https://www.clinicaltrialsregister.eu/ctr‐search/trial/2010‐022091‐31/IT (accessed 29 August 2016).
Joarder 2012 {published data only}
- Joarder R, Basu A, Choudhury KB, Dasgupta C, Dasgupta P, Ghosh K. Neoadjuvant chemoradiation with oral capecitabine versus intravenous 5‐fluorouracil and leucovorin in locally advanced carcinoma rectum – a randomized trial: an interim report. Journal of Cancer Research and Therapeutics 2012;8(Suppl 3):S159. [Google Scholar]
Muro 2016 {published data only}
- Muro K, Kim TW, Park YS, Uetake H, Nishina T, Nozawa H, et al. A multinational, randomized, phase III trial of XELIRI (+bevacizumab) versus FOLFIRI (+bevacizumab) as the second‐line chemotherapy for metastatic colorectal cancer: Asian XELIRI project (AXEPT). Journal of Clinical Oncology 2016;34(Suppl 4S):abstr TPS780. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT02280070 {published data only}
- NCT02280070. RP II study of SOX vs mFOLFOX6 in patients with resectable rectal cancer (KSCC1301). https://clinicaltrials.gov/ct2/show/record/NCT02280070 (accessed 27 June 2016).
Additional references
Allegra 2011
- Allegra CJ, Yothers G, O'Connell MJ, Sharif S, Petrelli NJ, Colangelo LH, et al. Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: results of NSABP protocol C‐08. Journal of Clinical Oncology 2011;29(1):11‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
André 2009
- André T, Boni C, Navarro M, Tabernero J, Hickish T, Topham C, et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. Journal of Clinical Oncology 2009;27(19):3109‐16. [DOI] [PubMed] [Google Scholar]
Arkenau 2008
- Arkenau HT, Arnold D, Cassidy J, Diaz‐Rubio E, Douillard JY, Hochster H, et al. Efficacy of oxaliplatin plus capecitabine or infusional fluorouracil/leucovorin in patients with metastatic colorectal cancer: a pooled analysis of randomized trials. Journal of Clinical Oncology 2008;26(36):5910‐7. [DOI] [PubMed] [Google Scholar]
Bennouna 2013
- Bennouna J, Sastre J, Arnold D, Österlund P, Greil R, Cutsem E, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncology 2013;14(1):29‐37. [DOI] [PubMed] [Google Scholar]
Benson 2004
- Benson AB 3rd, Schrag D, Somerfield MR, Cohen AM, Figueredo AT, Flynn PJ, et al. American Society of Clinical Oncology recommendations on adjuvant chemotherapy for stage II colon cancer. Journal of Clinical Oncology 2004;22(16):3408‐19. [DOI] [PubMed] [Google Scholar]
Blum 1999
- Blum JL, Jones SE, Buzdar AU, LoRusso PM, Kuter I, Vogel C, et al. Multicenter Phase II study of capecitabine in paclitaxel‐refractory metastatic breast cancer. Journal of Clinical Oncology 1999;17(2):485‐93. [DOI] [PubMed] [Google Scholar]
Bosset 2006
- Bosset JF, Collette L, Calais G, Mineur L, Maingon P, Radosevic‐Jelic L, et al. Chemotherapy with preoperative radiotherapy in rectal cancer. New England Journal of Medicine 2006;355(11):1114‐23. [DOI] [PubMed] [Google Scholar]
Braun 2011
- Braun MS, Seymour MT. Balancing the efficacy and toxicity of chemotherapy in colorectal cancer. Therapeutic Advances in Medical Oncology 2011;3(1):43‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brenner 2014
- Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet 2014;383(9927):1490‐502. [DOI] [PubMed] [Google Scholar]
Bujko 2006
- Bujko K, Nowacki MP, Nasierowska‐Guttmejer A, Michalski W, Bebenek M, Kryj M. Long‐term results of a randomized trial comparing preoperative short‐course radiotherapy with preoperative conventionally fractionated chemoradiation for rectal cancer. British Journal of Surgery 2006;93(10):1215‐23. [DOI] [PubMed] [Google Scholar]
Cafiero 2003
- Cafiero F, Gipponi M, Lionetto R, P.A.R. Cooperative Study Group. Randomised clinical trial of adjuvant postoperative RT vs. sequential postoperative RT plus 5‐FU and levamisole in patients with stage II‐III resectable rectal cancer: a final report. Journal of Surgical Oncology 2003;83(3):140‐6. [DOI] [PubMed] [Google Scholar]
Calabresi 1991
- Calabresi F, Repetto M. Doxifluridine in advanced colorectal cancer. Journal of Surgical Oncology Supplement 1991;2:124‐8. [DOI] [PubMed] [Google Scholar]
Cassidy 2002
- Cassidy J, Twelves C, Cutsem E, Hoff P, Bajetta E, Boyer M, et al. First‐line oral capecitabine therapy in metastatic colorectal cancer: a favorable safety profile compared with intravenous 5‐fluorouracil/leucovorin. Annals of Oncology 2002;13(4):566‐75. [DOI] [PubMed] [Google Scholar]
Cassidy 2005
- Cassidy J. Benefits and drawbacks of the use of oral fluoropyrimidines as single‐agent therapy in advanced colorectal cancer. Clinical Colorectal Cancer 2005;5(Suppl 1):S47‐50. [DOI] [PubMed] [Google Scholar]
Cassidy 2008
- Cassidy J, Clarke S, Diaz‐Rubio E, Scheithauer W, Figer A, Wong R, et al. XELOX vs. FOLFOX4: Update of efficacy results from XELOX‐1/NO16966, a randomized phase III trial of first‐line treatment for patients (pts) with metastatic colorectal cancer (MCRC). ASCO 2008 Gastrointestinal Cancer Symposium. 2008:abstr 341.
Cassidy 2011b
- Cassidy J, Saltz L, Twelves C, Cutsem E, Hoff P, Kang Y, et al. Efficacy of capecitabine versus 5‐fluorouracil in colorectal and gastric cancers: a meta‐analysis of individual data from 6171 patients. Annals of Oncology 22;12:2604‐9. [DOI] [PubMed] [Google Scholar]
Chandler 2013
- Chandler J, Churchill R, Higgins J, Lasserson T, Tovey D. Methodological standards for the conduct of new Cochrane Intervention Reviews. Version 2.3. 02 December 2013. Available from: http://editorial‐unit.cochrane.org/mecir.
Chau 2009
- Chau I, Cunningham D. Treatment in advanced colorectal cancer: what, when and how?. British Journal of Cancer 2009;100(11):1704‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cheng 2014
- Cheng AL, Li J, Vaid AK, Ma BB, Teh C, Ahn JB, et al. Adaptation of international guidelines for metastatic colorectal cancer: an asian consensus. Clinical Colorectal Cancer 2014;13(3):145‐55. [DOI] [PubMed] [Google Scholar]
Chionh 2010
- Chionh F, Campbell A, Sukumaran S, Price T, Tebbutt N. Oral versus intravenous fluoropyrimidines for colorectal cancer. Cochrane Database of Systematic Reviews 2010, Issue 3. [DOI: 10.1002/14651858.CD008398] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chuah 2011
- Chuah B, Goh BC, Lee SC, Soong R, Lau F, Mulay M, et al. Comparison of the pharmacokinetics and pharmacodynamics of S‐1 between Caucasian and East Asian patients. Cancer Science 2011;102(2):478‐83. [DOI] [PubMed] [Google Scholar]
Cuppone 2008
- Cuppone F, Bria E, Maio M, Carlini P, Sperduti I, Milella M, et al. Meta‐analysis of randomized clinical trials (RCTS) exploring capecitabine (C) versus 5‐fluorouracil (FU) in combination with oxaliplatin (OX) as 1st‐line chemotherapy for metastatic colorectal cancer (MCRC). Annals of Oncology 2008;19(Suppl 8):viii131, abstr 380P. [Google Scholar]
Deeks 2011
- Deeks J, Higgins J, Altman D, on behalf of the Cochrane Statistical Methods Group (Editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins J, Green S (Editors).Cochrane Handbook of Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Dencausse 2002
- Dencausse Y, Hartung G, Sturm J, Kopp‐Schneider A, Hagmüller E, Wojatschek C, et al. Adjuvant chemotherapy in stage III colon cancer with 5‐fluorouracil and levamisole versus 5‐fluorouracil and leucovorin. Onkologie 2002;25(5):426‐30. [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled clinical trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
Douillard 2010
- Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first‐line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. Journal of Clinical Oncology 2010;28(31):4697‐705. [DOI] [PubMed] [Google Scholar]
Edge 2009
- Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A 3rd (Editors). Chapter 14: Colon and Rectum. AJCC Cancer staging manual. 7th Edition. Springer, 2009:173‐206. [Google Scholar]
EMA 2016
- Xeloda (capecitabine) . European Medicines Agency 2016, updated 26 July 2016. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000316/human_med_001157.jsp&mid=WC0b01ac058001d124 (accessed 12 September 2016).
FDA 2010
- U.S. Department of Health and Human Services, Food, Drug Administration. Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for Industry‐ Non‐inferiority Clinical Trials. FDA 2010.
Ferlay 2013
- Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. Globocan 2012 v1.0, Cancer Incidence and Mortality Worldwide, IARC CancerBase No. 11 [Internet]. International Agency for Research on Cancer; 2013. Available from: http://globocan.iarc.fr (accessed on 10 March 2016).
Figueredo 2008
- Figueredo A, Coombes ME, Mukherjee S. Adjuvant Therapy for completely resected Stage II Colon Cancer. Cochrane Database of Systematic Reviews 2008, Issue 3. [DOI: 10.1002/14651858.CD005390.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Francini 1994
- Francini G, Petrioli R, Lorenzini L, Mancini S, Armenio S, Tanzini G, et al. Folinic acid and 5‐fluorouracil as adjuvant chemotherapy in colon cancer. Gastroenterology 1994;106(4):899‐906. [DOI] [PubMed] [Google Scholar]
Gerard 2006
- Gérard JP, Conroy T, Bonnetain F, Bouché O, Chapet O, Closon‐Dejardin MT, et al. Preoperative radiotherapy with or without concurrent fluorouracil and leucovorin in T3‐4 rectal cancers: results of FFCD 9203. Journal of Clinical Oncology 2006;24(28):4620‐5. [DOI] [PubMed] [Google Scholar]
Giantonio 2007
- Giantonio BJ, Catalano PJ, Meropol NJ, O'Dwyer PJ, Mitchell EP, Alberts SR, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. Journal of Clinical Oncology 2007;25(12):1539‐44. [DOI] [PubMed] [Google Scholar]
Gill 2004
- Gill S, Loprinzi CL, Sargent DJ, Thomé SD, Alberts SR, Haller DG, et al. Pooled analysis of fluorouracil‐based adjuvant therapy for stage II and III colon cancer: who benefits and by how much?. Journal of Clincal Oncology 2004;22(10):1797‐806. [DOI] [PubMed] [Google Scholar]
GRADEpro [Computer program] [Computer program]
- Hamilton (ON): GRADE Working Group, McMaster University. GRADEpro GDT. Hamilton (ON): GRADE Working Group, McMaster University, 2015.
Grage 1981
- Grage TB, Moss SE. Adjuvant chemotherapy in cancer of the colon and rectum: demonstration of effectiveness of prolonged 5‐FU chemotherapy in a prospectively controlled, randomized trial. The Surgical clinics of North America 1981;61(6):1321‐9. [DOI] [PubMed] [Google Scholar]
Gray 2007
- Quasar Collaborative Group, Gray R, Barnwell J, McConkey C, Hills RK, Williams NS, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. The Lancet 2007;370(9604):2020‐9. [DOI] [PubMed] [Google Scholar]
Guyatt 2008a
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ (Clinical research ed.) 2008;336(7650):924‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guyatt 2008b
- Guyatt GH, Oxman AD, Kunz R, Vist GE, Falck‐Ytter Y, Schünemann HJ, et al. What is "quality of evidence" and why is it important to clinicians?. BMJ (Clinical research ed.) 2008;336(7651):995‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Haller 2005
- Haller DG, Catalano PJ, Macdonald JS, O'Rourke MA, Frontiera MS, Jackson DV, et al. Phase III study of fluorouracil, leucovorin and levamisole in high‐risk stage II and III colon cancer: final report of Intergroup 0089. Journal of Clinical Oncology 2005;23(34):8671‐8. [DOI] [PubMed] [Google Scholar]
Haller 2008
- Haller DG, Cassidy J, Clarke SJ, Cunningham D, Cutsem E, Hoff PM, et al. Potential regional differences for the tolerability profiles of fluoropyrimidines. Journal of Clinical Oncology 2008;26(13):2118‐23. [DOI] [PubMed] [Google Scholar]
Haller 2011
- Haller DG, Tabernero J, Maroun J, Braud F, Price T, Cutsem E, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. Journal of Clinical Oncology 2011;29(11):1465‐71. [DOI] [PubMed] [Google Scholar]
Hamaguchi 2015
- Hamaguchi T, Shimada Y, Mizusawa J. Kinugasa Y, Kanemitsu Y, Ohue M, et al. Randomized phase III study of adjuvant chemotherapy with S‐1 versus capecitabine (cape) in patients with stage III colon cancer (CC): Results of Japan Clinical Oncology Group study (JCOG0910). Journal of Clinical Oncology 2015;33(Suppl):abstr 3512. [Google Scholar]
Higgins 2011a
- Higgins J, Altman DG, Sterne J on behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group. Chapter 8: Assessing risk of bias in included studies. In: Higgins J, Green S (editors). Cochrane Handbook of Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins J, Deeks J, Altman DG on behalf of the Cochrane Statistical Methods Group. Chapter 16: Special topics in statistics. Higgins J, Green S (editors). Cochrane Handbook of Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration. Available from www.cochrane‐handbook.org, 2011. [Google Scholar]
Higgins 2011c
- Deeks J, Higgins J and Altman DJ on behalf of the Cochrane Statistical Methods Group. Chapter 9: Analysing data and undertaking meta‐analyses. Higgins J, Green S (editors). Cochrane Handbook of Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. Available from www.cochrane‐handbook.org: The Cochrane Collaboration, 2011, The Cochrane Collaboration, 2011. [Google Scholar]
Hoff 2000
- Hoff PM. The tegafur‐based dihydropyrimidine dehydrogenase inhibitory fluoropyrimidines, UFT/leucovorin (ORZEL) and S‐1: a review of their clinical development and therapeutic potential. Investigational New Drugs 2000;18(4):331‐42. [DOI] [PubMed] [Google Scholar]
Hong 2012
- Hong YS, Park YS, Lim HY, Lee J, Kim TW, Kim K‐P, et al. S‐1 plus oxaliplatin versus capecitabine plus oxaliplatin for first‐line treatment of patients with metastatic colorectal cancer: a randomised, non‐inferiority phase 3 trial. Lancet Oncology 2012;13:1125‐32. [DOI] [PubMed] [Google Scholar]
Hurwitz 2004
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. New England Journal of Medicine 2004;350(23):2335‐42. [DOI] [PubMed] [Google Scholar]
IMPACT Investigators 1995
- International Multicentre Pooled Analysis of Colon Cancer Trials (IMPACT) investigators. Efficacy of adjuvant fluorouracil and folinic acid in colon cancer. The Lancet 1995;345(8955):939‐44. [PubMed] [Google Scholar]
Kwakman 2017
- Kwakman JJM, Simkens LHJ, Rooijen JM, Wouw AJ, Loosveld OJL, Creemers GJM, et al. Randomised phase 3 study of S‐1 versus capecitabine, with bevacizumab optional in both arms, in the first‐line treatment of metastatic colorectal cancer (mCRC), the SALTO study of the Dutch Colorectal Cancer Group. European Journal of Cancer 2017;71 (Supplement 1):S5. [Google Scholar]
Laurie 1989
- Laurie JA, Moertel CG, Fleming TR, Wieand HS, Leigh JE, Rubin J, et al. Surgical adjuvant therapy of large‐bowel carcinoma: an evaluation of levamisole and the combination of levamisole and fluorouracil. The North Central Cancer Treatment Group and the Mayo Clinic. Journal of Clinical Oncology 1989;7(10):1447‐56. [DOI] [PubMed] [Google Scholar]
Lenz 2015
- Lenz HJ, Stintzing S, Loupakis F. TAS‐102, a novel antitumor agent: a review of the mechanism of action. Cancer Treatment Reviews 2015;41(9):7777‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 1997
- Liu G, Franssen E, Fitch MI, Warner E. Patient preferences for oral versus intravenous palliative chemotherapy. Journal of Clinical Oncology 1997;15(1):110‐5. [DOI] [PubMed] [Google Scholar]
Lucas 2011
- Lucas AS, O'Neil BH, Goldberg RM. A decade of advances in cytotoxic chemotherapy for metastatic colorectal cancer. Clinical Colorectal Cancer 2011;10(4):238‐44. [DOI] [PubMed] [Google Scholar]
Malet‐Martino 2002
- Malet‐Martino M, Martino R. Clinical studies of three oral prodrugs of 5‐fluorouracil (capecitabine, UFT, S‐1): a review. Oncologist 2002;7(4):288‐323. [DOI] [PubMed] [Google Scholar]
Mayer 2015
- Mayer RJ, Cutsem E, Falcone A, Yoshino T, Garcia‐Carbonero R, Mizunuma N, et al. Randomized trial of TAS‐102 for refractory metastatic colorectal cancer. The New England Journal of Medicine 2015;372(20):1909‐19. [DOI] [PubMed] [Google Scholar]
Meta‐analysis Group in Cancer 1998a
- Meta‐analysis Group in Cancer. Efficacy of intravenous continuous infusion of fluorouracil compared with bolus administration in advanced colorectal cancer. Journal of Clinical Oncology 1998;16:301‐8. [DOI] [PubMed] [Google Scholar]
Meta‐analysis Group in Cancer 1998b
- Meta‐analysis Group in Cancer. Toxicity of fluorouracil in patients with advanced colorectal cancer: Effect of administration schedule and prognostic factors. Journal of Clinical Oncology 1998;16:3537‐41. [DOI] [PubMed] [Google Scholar]
Midgley 2009
- Midgley R, Kerr DJ. Capecitabine: have we got the dose right?. Nature Clinical Practice Oncology 2009;6(1):17‐24. [DOI] [PubMed] [Google Scholar]
Miyamoto 2014
- Miyamoto Y, Sakamoto Y, Yoshida N, Baba H. Efficacy of S‐1 in colorectal cancer. Expert Opinion on Pharmacotherapy 2014;15(12):1761‐70. [DOI] [PubMed] [Google Scholar]
Moertel 1990
- Moertel CG, Fleming TR, Macdonald JS, Haller DG, Laurie JA, Goodman PJ, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon cancer. New England Journal of Medicine 1990;322(6):352‐8. [DOI] [PubMed] [Google Scholar]
Montagnani 2010
- Montagnani F, Chiriatti A, Licitra S, Aliberti C, Fiorentini G. Differences in efficacy and safety between capecitabine and infusional 5‐fluorouracil when combined with irinotecan for the treatment of metastatic colorectal cancer. Clinical Colorectal Cancer 2010;9(4):243‐7. [DOI] [PubMed] [Google Scholar]
NCCN 2016
- National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology. Colon Cancer (Version 2.2016). https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf (accessed 14 September 2016).
Nordic 1992
- Nordic Gastrointestinal Tumor Adjuvant Therapy Group. Expectancy or primary chemotherapy in patients with advanced asymptomatic colorectal cancer: a randomized trial. Journal of Clinical Oncology 1992;10(6):904‐11. [DOI] [PubMed] [Google Scholar]
O'Connell 1997
- O'Connell MJ, Mailliard JA, Kahn MJ, Macdonald JS, Haller DG, Mayer RJ, et al. Controlled trial of fluorouracil and low‐dose leucovorin given for 6 months as postoperative adjuvant therapy for colon cancer. Journal of Clinical Oncology 1997;15(1):246‐50. [DOI] [PubMed] [Google Scholar]
O'Connell 1998
- O'Connell MJ, Laurie JA, Kahn M, Fitzgibbons RJ Jr, Erlichman C, Shepherd L, et al. Prospectively randomized trial of postoperative adjuvant chemotherapy in patients with high‐risk colon cancer. Journal of Clinical Oncology 1998;16(1):295‐300. [DOI] [PubMed] [Google Scholar]
Pazdur 2016
- Pazdur R. Cancer drug information: FDA approval for capecitabine. National Cancer Institute at the National Institutes of Health 2012, updated 2 July 2013. Available from: http://www.cancer.gov/cancertopics/druginfo/fda‐capecitabine (accessed 13 September 2016).
Peeters 2010
- Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second‐line treatment in patients with metastatic colorectal cancer. Journal of Clinical Oncology : official journal of the American Society of Clinical Oncology 2010;28(31):4706‐13. [DOI] [PubMed] [Google Scholar]
Petrelli 2012
- Petrelli F, Cabiddu M, Barni S. 5‐Fluorouracil or capecitabine in the treatment of advanced colorectal cancer: a pooled‐analysis of randomized trials. Medical Oncology 2012;29(2):1020‐9. [DOI] [PubMed] [Google Scholar]
Punt 2008
- Punt CJ, Koopman M. Capecitabine and irinotecan as first‐line treatment of advanced colorectal cancer. Journal of Clinical Oncology 2008;26(11):1907‐8; author reply 8‐9. [DOI] [PubMed] [Google Scholar]
Quasar 2007
- Quasar Collaborative Group, Gray R, Barnwell J, McConkey C, Hills RK, WilliamsNS, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020‐9. [DOI] [PubMed] [Google Scholar]
RevMan [Computer Program] [Computer program]
- Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Saletti 2008
- Saletti P, Sessa C, Cavalli F. Commentary 2008: 25 or 50 years later?. Journal of Clinical Oncology 26;30:4854‐5. [DOI] [PubMed] [Google Scholar]
Sargent 2009
- Sargent D, Sobrero A, Grothey A, O'Connell MJ, Buyse M, Andre T, et al. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. Journal of Clinical Oncology 2009;27(6):872‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sasse 2009a
- Sasse AD, Sasse EC, dos Santos LV, Lima JS, Nascente CM, Saito HP, et al. Oral fluoropyrimidines versus 5‐fluorouracil for colorectal cancer: Results of a systematic review and meta‐analysis. Journal of Clinical Oncology 2009;27(15s):abstr4111. [Google Scholar]
Sasse 2009b
- Sasse AD, Sasse EC, dos Santos LV, Lima JS, Nascente CM, Saito HP, et al. Oral fluoropyrimidines versus 5‐fluorouracil for colorectal cancer: Results of a systematic review and meta‐analysis. Abstract No: 4111, Andre D Sasse, MD, PhD. 2009 ASCO Annual Meeting, Gastrointestinal (Colorectal) Cancer Track‐ General Poster Session. http://www.asco.org/ASCOv2/MultiMedia/Virtual+Meeting?&vmview=vm_session_presentations_view&confID=65&sessionID=3128 (accessed 26 September 2012).
Scheithauer 1993
- Scheithauer W, Rosen H, Kornek GV, Sebesta C, Depisch D. Randomised comparison of combination chemotherapy plus supportive care with supportive care alone in patients with metastatic colorectal cancer. British Medical Journal 1993;306(6880):752‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schilsky 2002b
- Schilsky RL. Inhibiting 5‐fluorouracil breakdown: a broken down approach to 5‐fluorouracil modulation. Clinical Colorectal Cancer 2002;2(1):51‐2. [DOI] [PubMed] [Google Scholar]
Schmoll 2007
- Schmoll HJ, Cartwright T, Tabernero J, Nowacki MP, Figer A, Maroun J, et al. Phase III trial of capecitabine plus oxaliplatin as adjuvant therapy for stage III colon cancer: a planned safety analysis in 1,864 patients. Journal of Clinical Oncology 2007;25(1):102‐9. [DOI] [PubMed] [Google Scholar]
Schmoll 2010
- Schmoll HJ. S‐1 for advanced colorectal cancer: do we need another oral fluorouracil prodrug?. Lancet Oncology 2010;11(9):808‐9. [DOI] [PubMed] [Google Scholar]
Schmoll 2014
- Schmoll HJ, Twelves C, Sun W, O'Connell MJ, Cartwright T, McKenna E, et al. Effect of adjuvant capecitabine or fluorouracil, with or without oxaliplatin, on survival outcomes in stage III colon cancer and the effect of oxaliplatin on post‐relapse survival: a pooled analysis of individual patient data from four randomised controlled trials. Lancet Oncology 2014;15(13):1481‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann H, Oxman A, Vist G, Higgins J, Deeks J, Glasziou P, et al on behalf of the Cochrane Applicability and Recommendations Methods Group. Chapter 12: Interpreting results and drawing conclusions. In: Higgins J, Green S (editors). Cochrane Handbook of Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. www.cochrane‐handbook.org.
Siegel 2017
- Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal cancer statistics, 2017. Ca: A Cancer Journal for Clinicians, 2017 Mar 1;doi: 10.3322/caac.21395:[Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne J, Egger M, Moher D, on behalf of the Cochrane Bias Methods Group (Editors). Chapter 10: Addressing reporting bias. In: Higgins J, Green S (Editors). Cochrane Handbook of Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Taal 2001
- Taal BG, Tinteren H, Zoetmulder FA, NACCP group. Adjuvant 5FU plus levamisole in colonic or rectal cancer: improved survival in stage II and III. British Journal of Cancer 2001;85(10):1437‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tabernero 2015
- Tabernero J, Yoshino T, Cohn AL, Obermannova R, Bodoky G, Garcia‐Carbonero R, et al. Ramucirumab versus placebo in combination with second‐line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first‐line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double‐blind, multicentre, phase 3 study. The Lancet Oncology 2015;16(5):499‐508. [DOI] [PubMed] [Google Scholar]
Tebbutt 2010
- Tebbutt NC, Wilson K, Gebski VJ, Cummins MM, Zannino D, Hazel GA, et al. Capecitabine, bevacizumab, and mitomycin in first‐line treatment of metastatic colorectal cancer: results of the Australasian Gastrointestinal Trials Group Randomized Phase III MAX Study. Journal of Clinical Oncology 2010;28(19):3191‐8. [DOI] [PubMed] [Google Scholar]
TGA 2016
- Capecitabine colorectal. [Australian Government Department of Health. Therapeutic Goods Administration]. Available from: https://tga‐search.clients.funnelback.com/s/search.html?query=capecitabine%20colorectal%20&collection=tga‐artg (accessed 12 September 2016).
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time‐to‐event data into meta‐analysis. Trials 2007;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tovey 2012
- Tovey D (Editor). Standards for the reporting of new Cochrane Intervention Reviews. Version 1.1. December 2012. Available from: http://editorial‐unit.cochrane.org/mecir.
Van Cutsem 2006
- Cutsem E, Nordlinger B, Adam R, Köhne C‐H, Pozzo C, Poston G, et al. Towards a pan‐European consensus on the treatment of patients with colorectal liver metastases. European Journal of Cancer 2006;42(14):2212‐21. [DOI] [PubMed] [Google Scholar]
Van Cutsem 2011
- Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first‐line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. Journal of Clinical Oncology 2011;29(15):2011‐9. [DOI] [PubMed] [Google Scholar]
Van Cutsem 2012
- Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausova J, Macarulla T, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin‐based regimen. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30(28):3499‐506. [DOI] [PubMed] [Google Scholar]
Van der Geest LGM
- Geest LGM, Lam‐Boer J, Koopman M, Verhoef C, Elferink MAG, Wilt JHW. Nationwide trends in incidence, treatment and survival of colorectal cancer patients with synchronous metastases. Clinical & Experimental Metastasis 2015;32:457‐65. [DOI] [PubMed] [Google Scholar]
Weiser 2015
- Weiser MR, Zhang Z, Schrag D. Locally advanced rectal cancer: time for precision therapeutics. American Society of Clinical Oncology Education Book 2015;e192‐6:doi: 10.14694/EdBook_AM.2015.35.e192.. [DOI] [PubMed] [Google Scholar]