

SUMMARY
Throughout the developing nervous system, considerable synaptic re-organization takes place as postsynaptic neurons extend dendrites and incoming axons refine their synapses, strengthening some and eliminating others. It is well accepted that these processes rely on synaptic activity; however, the mechanisms that lead to this developmental reorganization are not fully understood. Here, we explore the regulation of cap-dependent translation, a mechanism known to play a role in synaptic growth and plasticity. Using sympathetic ganglia in α3 nicotinic acetylcholine receptor (nAChR)-knockout (KO) mice, we establish that electrophysiologically silent synapses between preganglionic axons and postsynaptic sympathetic neurons do not refine, and the growth of dendrites and the targeting of synapses on postsynaptic neurons are impaired. Remarkably, genetically removing 4E-BP, a suppressor of cap-dependent translation, from these α3 nAChR-KO mice largely restores these features. We conclude that synaptic connections can re-organize and refine without postsynaptic activity during post-natal development when 4E-BP-regulated cap-dependent translation is enhanced.
Graphical Abstract
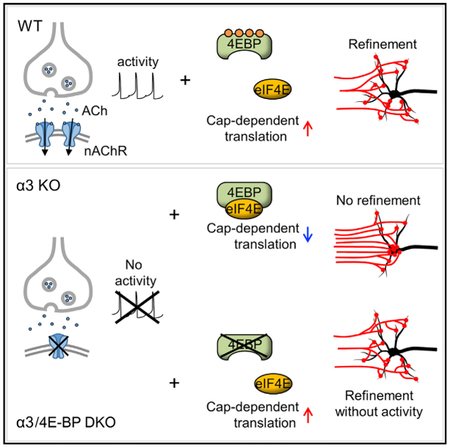
In Brief
Synaptic activity is required for synaptic refinement and reorganization during post-natal development. Chong et al. find that silent synapses in superior cervical ganglia (SCG) refine when 4E-BP is genetically removed, suggesting that enhanced cap-dependent translation promotes synaptic refinement in the absence of postsynaptic activity.
INTRODUCTION
During early post-natal development, as circuits begin to form, the emergent synaptic activity remodels axons and dendrites to refine synaptic connections and improve circuit performance. This activity-dependent refinement occurs widely in the developing nervous system (Purves and Lichtman, 1980; Sanes and Lichtman, 1999; Katz and Crowley, 2002; Bleckert and Wong, 2011; Hong and Chen, 2011; Buffelli et al., 2002; Zou et al., 2004; Schlaggar et al., 1993; Takeuchi et al., 2014; Yasuda et al., 2011), suggesting that this refinement is regulated by common mechanisms; however, in spite of its widespread occurrence, the underlying mechanisms are not fully understood. It is generally accepted that synaptic refinement takes place through an activity-dependent competitive process(es) that eliminates some inputs and strengthens others (Cohen-Cory, 2002; Lichtman and Colman, 2000; Katz and Shatz, 1996; Zhang and Poo, 2001; Kano and Hashimoto, 2009). And, interfering with pre- or postsynaptic activity in developing circuits or manipulating postsynaptic calcium influx prevents inputs from refining (Fitzsimonds and Poo, 1998; Balice-Gordon and Lichtman, 1994; Hata and Stryker, 1994; Hashimoto et al., 2011), indicating clearly that the processes that lead to synaptic refinement require activity. Given recent evidence that activity-dependent mechanisms promote cap-dependent mRNA translation in developing dendrites and axons and play essential roles in forming neural circuits (Bramham and Wells, 2007; Lin et al., 2016; Jung et al., 2012; Wang et al., 2010), we asked whether cap-dependent translation has any role in re-organizing neural connections during post-natal development.
Briefly, a critical step in regulating cap-dependent translation is binding of the eukaryotic translation initiation factor 4E (eIF4E) to the 5′ cap structure of mRNAs (Gingras et al., 1999; Sonenberg and Hinnebusch, 2009). The availability of eIF4E is controlled by 4E-binding proteins (4E-BPs), important regulators of cap-dependent mRNA translation and major downstream targets of the mechanistic target of rapamycin complex 1 (mTORC1), a serine-threonine kinase (Richter and Sonenberg, 2005; Thoreen et al., 2012). Hypophosphorylated 4E-BP represses the initiation of cap-dependent translation by sequestering eIF4E, whereas hyperphosphorylated 4E-BP releases eIF4E and allows eIF4E to bind to eIF4G, a molecular interaction that is required to initiate cap-dependent translation and de novo protein synthesis (Gingras et al., 1999; Sonenberg and Hinnebusch, 2009).
To determine whether cap-dependent translational mechanisms have a role in synaptic refinement during post-natal development, we investigated synapses in the superior cervical ganglion (SCG), a well-established model that is known to undergo considerable synaptic reorganization and refinement during the first post-natal month as circuits are being established (Purves and Lichtman, 1984). Briefly, all SCG neurons are innervated exclusively by a homogeneous group of excitatory cholinergic preganglionic axons that are anatomically well defined and readily accessible for stimulation and labeling. Moreover, SCG neurons receive no other excitatory innervation, nor do they receive inhibitory innervation; therefore, refinement of preganglionic axons is not confounded by other inputs to the postsynaptic neurons. Furthermore, fast synaptic transmission between preganglionic axons and sympathetic neurons in the SCG is mediated by only 1 class of postsynaptic receptors: α3-containing nicotinic acetylcholine receptors (nAChRs) (Xu et al., 1999; Rassadi et al., 2005). Therefore, deleting the α3 nAChR subunit gene from mice (referred to here as α3 knockout [KO]) completely abolishes synaptic transmission in the SCG (Rassadi et al., 2005; Krishnaswamy and Cooper, 2009), even though the presynaptic terminals establish synapses that appear ultrastructurally normal (Krishnaswamy and Cooper, 2009). In addition, one can rapidly restore function to these synapses by infecting postsynaptic neurons with α3-expressing adenoviruses (Krishnaswamy and Cooper, 2009).
Using α3-KO mice of different post-natal ages, we show that SCG neurons remain innervated by multiple preganglionic axons and we find defects in the growth of dendrites on α3-KO sympathetic neurons and in the targeting of synapses by preganglionic axon terminals. Un-silencing these synapses by virally introducing α3 normalized synaptic targeting to dendrites, and enabled the preganglionic axons to refine. In α3-KO sympathetic neurons, we found lower levels of phosphorylated 4E-BP1 compared to that in wild-type (WT) controls. Additionally, in proteomic experiments, we observed changes in levels for a large number of proteins in the SCG of α3-KO mice. Interestingly, genetically removing 4E-BP from α3-KO mice largely restored these protein levels toward those in WT, even though the SCG had no synaptic transmission. Furthermore, we show that removing 4E-BP from α3-KO mice reversed the defects both in dendritic growth on SCG neurons and in targeting of synapses, and, remarkably, the preganglionic inputs refined in the absence of postsynaptic activity. Our findings highlight a role for cap-dependent translation in re-organizing connections, particularly in circuits where synapses have been silenced.
RESULTS
The Absence of Preganglionic Refinement in the SCG of α3-KO Mice
To estimate the number of preganglionic synaptic inputs onto sympathetic neurons in WT SCG, we measured the discrete jumps in the evoked excitatory postsynaptic potentials (EPSPs) while gradually increasing the stimulus to the preganglionic nerve to recruit innervating axons (Figure 1). In post-natal day 1–3 (P1–P3) mice, approximately 7–8 preganglionic axons converged onto postsynaptic sympathetic neurons, each axon evoking a small EPSP of approximately equal strength (Figure 1A). Over the first post-natal month, as neonatal pups start to regulate internal organs to maintain homeostasis, activity in the autonomic nervous system increases dramatically and some preganglionic axons innervating sympathetic neurons are gradually eliminated. By P8–P9, sympathetic neurons were innervated by approximately 5–6 axons, and, by P28–P30, only 2–3 axons innervated SCG neurons (Figures 1B and 1F). In addition, over this time period, the strength of the inputs had increased markedly. At P56–P64, the neurons remained innervated by 2–3 axons (Figure 1F).
Figure 1. Preganglionic Axons Innervating Neurons in α3-KO SCG Do Not Refine.

(A–D) Left: representative compound EPSPs in an SCG neuron from (A) P1–P3 WT, (B) P28 WT, (C) P1–P3 α3-KO, and (D) P28 α3-KO mice evoked by increasing stimuli to the preganglionic nerve. Middle: the EPSPs evoked by individual preganglionic axons are shown. DI, disparity index. Right: distribution of SCG neurons innervated by the number of inputs; each distribution was fit with a Gaussian function. The distribution for P28 α3 KO is not significantly different from P1–P3 WT or α3 KO (p > 0.2), but it is significantly different from P28 WT (p < 0.001). Each distribution in (A)–(D) contains data from at least 4 mice. n, number of neurons.
(E) The average difference in strength between the strongest and second strongest inputs, expressed as a percentage of the maximum compound EPSP.
(F) The average number of axons innervating an SCG neuron in WT (solid line) and α3-KO (dotted line) mice at P1, P4, P28, and P60.
For (E) and (F), error bars represent ± SEM.
***p < 0.001.
We quantified the disparity in strength of the EPSPs on each neuron in two ways. In one, we calculated a disparity index, defined as the ratio of the SD of the EPSPs evoked by each axon divided by the mean (SD/M) (Hashimoto and Kano, 2003; see the Experimental Procedures and Supplemental Information). From P1–P3 to P28–P30, we observed a significant increase in the mean disparity index (DI) (Figures 1A and 1B), reflecting the increase in strength of 1 or more inputs. In the second method, we computed the difference in strength between the strongest and second strongest inputs, expressed as a percentage of the maximum compound EPSP. At P1–P3, the difference between the strongest and the next strongest input was <5%, whereas at P28–P30 the difference was ~35% (Figure 1E). These results establish that, over the first post-natal month, preganglionic inputs innervating sympathetic neurons refine their connections by eliminating some axons and strengthening others.
To test whether refinement is dependent on synaptic activity, as it is elsewhere in the nervous system (Lichtman and Colman, 2000; Katz and Shatz, 1996; Zhang and Poo, 2001), we examined preganglionic innervation in the SCG of postnatal α3-KO mice. Synapses are electrophysiologically silent in the SCG when α3-containing nAChRs are absent, although synapses are morphologically intact (Krishnaswamy and Cooper, 2009). We first infected these mice for 1–2 days with α3-encoding adenoviral vectors (Ad-α3) so that we could measure the convergence of preganglionic axons. At P1–P3, approximately 7–8 preganglionic axons converged onto post-synaptic sympathetic neurons, with each axon evoking a small EPSP of approximately equal strength (Figure 1C), comparable to that of WT at P1–P3. In contrast to WT, preganglionic axons innervating α3-KO SCG neurons showed no refinement, and the neurons continued to be innervated by 7–8 axons over the next 2 months of post-natal life (Figures 1D and 1F). Moreover, EPSPs measured from α3-KO neurons showed little disparity during the same period of time (Figures 1D and 1E). These results suggest that postsynaptic activity during the first post-natal month is necessary both for the elimination of some preganglionic axons and the strengthening of those inputs that persist.
Preganglionic Axons Refine when Synaptic Activity Is Restored in α3-KO SCG
To examine whether preganglionic axons in α3-KO mice are capable of refining when postsynaptic activity is restored, we infected mice with Ad-α3 at P28–P30, and we examined functional innervation of SCG neurons 1 month later (at P56–P64). We found that rescued α3-KO SCG neurons were innervated by only 2–3 preganglionic axons (Figure 2A), similar to that of WT neurons at P60 (Figure 2C), whereas α3-KO SCG neurons (not rescued) at P60 continued to be innervated by 7–8 axons (Figure 2B). In addition to the shift in convergence, the EPSPs evoked by 1–2 preganglionic axons on rescued neurons strengthened considerably, increasing the disparity among EPSPs to levels in WT SCG (Figures 2A, 2C, and 2D). From these data, we conclude that, to refine and strengthen, synapses made by preganglionic axons require retrograde signals downstream of postsynaptic activity. Furthermore, our findings demonstrate that presynaptic inputs onto postsynaptic sympathetic neurons maintain their ability to refine in an activity-dependent manner, well beyond the post-natal period.
Figure 2. Refinement of Preganglionic Axons Requires Postsynaptic Activity.

(A–C) Left: representative compound EPSPs evoked in an SCG neuron from (A) α3-KO mice 1 month after rescue (see inset), (B) P60 α3-KO mice, and (C) P60 WT mice. Middle: the EPSPs evoked by individual preganglionic axons are shown. DI, disparity index. Right: distribution of the SCG neurons innervated by the number of inputs; each distribution is fit with a Gaussian function. The distribution for α3-KO rescue neurons is not significantly different from P60 WT (p > 0.2), but it is significantly different from P60 α3 KO (p < 0.001). Each distribution in (A)–(C) contains data from at least 4 mice. n, number of neurons.
(D) The average difference in strength between the strongest and second strongest inputs, expressed as a percentage of the maximum compound EPSP. Error bars represent ± SEM. ***p < 0.001.
The Extension of Dendrites and Targeting of Synapses Are Defective in the Absence of Postsynaptic Activity
In addition to the convergence of preganglionic axons, we asked whether the absence of postsynaptic activity modifies the organization of pre- and postsynaptic structures, including the growth of dendrites, and/or the manner in which preganglionic axons target their synapses onto SCG neurons.
To quantify dendritic growth, we sparsely labeled SCG neurons with the lipophilic dye 3,3′-Dioctadecyloxacarbo-cyanine perchlorate (DiO). At P1, total dendritic outgrowth (TDO) on WT and α3-KO neurons was similar (Figures 3A–3C); however, TDO on WT neurons was significantly greater than that on α3-KO neurons by as early as P4, and, by P28, TDO on WT neurons was twice that of α3-KO neurons (Figures 3A–3C). These results are consistent with a role for activity in promoting dendritic growth (Haas et al., 2006; Niell et al., 2004; Wong and Ghosh, 2002; Cline, 2001). The main defect in dendritic growth on sympathetic neurons developing without excitatory synaptic transmission was that they could not maintain primary branches (Figure 3D): the number of primary dendrites on α3-KO neurons decreased from five at P1–P4 to three at P28. On the other hand, we found no significant difference in the length of the dendritic branches on α3-KO neurons compared to neurons in WT SCG, nor did we find differences in the number of secondary or higher-order branches, once normalized for primary branch number (Figure 3E; Figure S1). These results suggest that, apart from maintaining primary dendrites, most aspects of dendritic growth on sympathetic neurons do not depend on synaptic activity.
Figure 3. Primary Dendrites Are Not Maintained without Postsynaptic Activity, and Preganglionic Axons Target Silent Synapses to the Soma.

(A and B) Maximum-intensity projections of DiO-labeled SCG neurons from (A) WT and (B) α3-KO mice labeled at P1 (top) and at P28 (bottom). Axons are marked by an asterisk. Scale bar, 20 μm. In each panel, neurons are from different ganglia and have been tiled for comparison.
(C) Average total dendritic outgrowth (TDO) per neuron at P1, P4, and P28; filled circles represent WT and open circles represent α3 KO in this panel and (D).
(D) Average number of primary dendrites per neuron at P1, P4, and P28.
(E) Average length of primary dendrites, secondary branches, and tertiary branches at P28; filled columns represent WT and open columns represent α3 KO.
For (C)–(E), error bars represent ± SEM. *p < 0.05 and ***p < 0.001. WT: for P1, n = 23 neurons (10 mice); for P4, n = 28 neurons (10 mice); and for P28, n = 34 neurons (12 mice). α3 KO: for P1, n = 20 neurons (10 mice); for P4, n = 24 neurons (10 mice); and for P28, n = 36 neurons (14 mice).
(F and G) Maximum-intensity projections of DiO-labeled P28 SCG neurons (green) innervated by Dil-labeled preganglionic axons (red) in (F) WT and (G) α3-KO SCG. Scale bar, 20 μm. Inset: the boxed region at higher magnification shows preganglionic axons (red) with varicosities along a segment of dendrite (green). Scale bar, 2 μm.
(H) Percentage of neuronal cell body covered by preganglionic axons for P28 SCG neurons of different sizes. (WT, filled circles, n = 50 neurons (16 mice), α3 KO, open circles, n = 38 neurons (13 mice)).
(I) DiO-labeled neuron (green) from a P28 WT SCG immunostained for VAChT (red). Axon is marked by an asterisk. VAChT puncta not touching the neurons were removed for clarity.
(J–N) Skeletonized reconstructions showing dendritic arbors (black), axon (gray, marked by an asterisk), and preganglionic axon varicosities (red), determined by VAChT staining, as in (A). (J) WT neuron from (I), (K) α3-KO neuron at P28, (L) WT neuron at P1, (M) α3-KO neuron at P1, and (N) α3-KO SCG neuron 1 month after rescue with Ad-α3 are shown. Neurons in (I)–(K) and (N) are shown at the same scale, and neurons in (L) and (M) are magnified for clarity.
(O) Average distribution of varicosities on the cell body (open) and dendrites (filled) in WT and α3-KO SCG at P1, P4, and P28 and on α3-KO neurons 1 month after rescue with Ad-α3. Error bars represent ± SEM. ***p < 0.001. WT: for P1, n = 6 neurons (3 mice); for P4, n = 9 neurons (5 mice); and for P28, n = 10 neurons (4 mice). α3 KO: for P1, n = 6 neurons (5 mice); for P4, n = 10 neurons (4 mice); and for P28, n = 11 neurons (4 mice). For α3-KO rescue, n = 12 neurons (8 mice).
Preganglionic axons mainly target their synapses to the dendrites of sympathetic neurons (Forehand, 1985). Given the decrease in primary dendrites when synaptic transmission is absent, we asked whether preganglionic axons targeted their synapses differently on α3-KO and WT SCG neurons. To address this, we sparsely labeled preganglionic axons in WT and α3-KO SCG at P28 with 1,1′-Dioctadecyl-3,3,3′,3′-tetrame-thylindocarbocyanine perchlorate (DiI) while simultaneously labeling sympathetic neurons with DiO, and we found a striking difference in how presynaptic axons converged onto SCG neurons (Figures 3F and 3G). In α3-KO SCG, the preganglionic axons grew extensively over the somas of sympathetic neurons (Figures 3G and 3H); the proportion of the neuron’s soma covered by preganglionic axons in α3-KO SCG was at least 50% on average, likely an underestimation because the preganglionic axons were sparsely labeled in these experiments. In contrast, in WT SCG, the proportion of the soma covered by preganglionic axons was less than 5% (Figures 3F and 3H). Our ultrastructural studies support these results: comparing random sections from α3-KO and WT SCG, we detected 6 times as many varicosities bordering neuronal cell bodies in α3-KO SCG (Figures S2A and S2B). These results indicate that, in the absence of synaptic activity, preganglionic axons more readily contact the cell bodies of sympathetic neurons.
This prominent growth of preganglionic axons over SCG neuronal cell bodies in 1-month-old α3-KO mice suggests that, without synaptic activity, the axons target silent synapses onto the soma instead of preferentially onto dendrites. To determine whether this mis-targeting of synapses in α3-KO SCG is already present at birth or whether it evolves during the first post-natal month, as preganglionic innervation increases without activity, we stained varicosities of Dillabeled axons at P1, P4, and P28 for vesicular acetylcholine transporter protein (VAChT). VAChT is highly localized in cholinergic presynaptic terminals, and over 90% of these VAChT-positive varicosities co-localized with postsynaptic density-93 (PSD-93) puncta (Krishnaswamy and Cooper, 2009; Figures S2C and S2D), suggesting that these are sites of synaptic contact.
Over the first post-natal month, the number of synapses on WT SCG neurons increased ~2-fold (Figure S2E, inset). In α3-KO mice, synapses continued to be established on SCG neurons during the first post-natal month, even when synaptic transmission was absent and synapses were electrophysiologically silent. When normalized to the size of the dendritic tree, the number of synapses per α3-KO neuron at P28 was ~40% greater than the number for age-matched controls (Figure S2E).
Preganglionic axons target their synapses mainly to dendrites of sympathetic neurons in WT SCG. We found that over 90% of the VAChT-positive varicosities on P1 and P4 SCG neurons were located on dendrites, and, by P28, this proportion increased to 95% (Figures 3I, 3J, 3L, and 3O). As well, in α3-KO SCG at P1–P4, synapses were mainly distributed on dendrites and not statistically different from those on age-matched WT neurons (Figures 3M and 3O). In contrast, at P28, when the preganglionic axons converged on the soma in α3-KO neurons, over 50% of the VAChT-positive varicosities were located on the cell body (Figures 3K and 3O). Moreover, rescuing synaptic activity in the SCG of α3-KO mice at P28 (and assessed at P56) resulted in a shift in the distribution of presynaptic varicosities from the soma to the dendrites (Figures 3N and 3O), without any effect on the size of the dendritic tree. These findings indicate that synaptic activity reorganizes preganglionic synapses and targets them to dendrites of sympathetic neurons.
Levels of Phosphorylated 4E-BP Are Decreased in α3-KO SCG
Our findings demonstrate clearly that postsynaptic activity during early post-natal development is required for appropriate refinement and remodelling of synaptic connections in the SCG. Inactivity of sympathetic neurons in the SCG of α3-KO mice presumably exerts its effects by altering the synthesis of various proteins (Wells et al., 2000; Steward and Schuman, 2001; Alvarez-Castelao and Schuman, 2015). Since a growing body of evidence points to a link among regulated mRNA translation, neuronal development, and synaptic plasticity (Bramham and Wells, 2007; Jung et al., 2012), we asked whether the effects of synaptic activity on developing neural circuits in sympathetic ganglia might be mediated, at least in part, through mechanisms that regulate mRNA translation. For example, work in Drosophila, as well as in hippocampal neuronal primary cultures, has suggested that postsynaptic translational mechanisms can respond to changes in synaptic activity and participate in retrograde regulation of synaptic plasticity (Henry et al., 2012; Penney et al., 2012, 2016). We focused our attention on the eIF4E 4E-BPs, proteins whose phosphorylation status regulates the initiation of translation (Ma and Blenis, 2009; Sonenberg and Hinnebusch, 2007; Gingras et al., 1999; Figure 4A).
Figure 4. Levels of Phosphorylated 4E-BP Are Higher in WT SCG Than in α3-KO SCG.

(A) Illustration showing cap-dependent translation, governed by the availability of eIF4E, which is regulated by 4E-BP, a major downstream target of the mTORC1 complex. Hypophosphorylated 4E-BP represses the initiation of cap-dependent translation by sequestering eIF4E. When hyperphosphorylated, 4E-BP releases eIF4E, which interacts with eIF4G and other initiation factors to initiate cap-dependent translation.
(B) Confocal images showing immunostaining for P-4E-BP1 (green) and MAP-1A (white) in WT and α3-KO SCG at P28. Scale bar, 20 μm.
(C) P-4E-BP1 mean fluorescence intensity per neuron normalized to MAP-1A in WT and α3-KO neurons at P1, P4, and P28. Filled columns represent WT and open columns represent α3 KO.
(D) Confocal images showing immunostaining for total 4E-BP1 (green) and MAP-1A (white) in WT and α3-KO SCG at P28. Scale bar, 20 μm.
(E) Total 4E-BP1 mean fluorescence intensity per neuron normalized to MAP-1A in WT and α3-KO neurons at P1, P4, and P28. Filled columns represent WT and open columns represent α3 KO. For (C) and (E), error bars represent ± SEM. ***p < 0.001. For WT in (C) and (E), n = 90 neurons (4 mice) for P1, n = 90 neurons (4 mice) for P4, n = 300 neurons (4 mice) for P28; and for α3 KO, n = 90 neurons (4 mice) for P1, n = 90 neurons (4 mice) for P4, and n = 300 neurons (4 mice) for P28.
(F) Distribution of SCG neurons innervated by the number of inputs in WT (gray) and 4E-BP-KO (red) mice at P1–P3, P8–P9, and P28. Each distribution is fit with a Gaussian function. The distributions at P1–P3 and at P28 are not significantly different (p > 0.2), whereas at P8–P9, p < 0.05. Each distribution contains data from at least 4 mice. n, number of neurons; DI, disparity index. Data for WT P1–P3 and P28 are from Figures 1A and 1B.
(G) Left: extracellular recordings from the SCG postganglionic nerve at P28 in response to supramaximal stimulation of the preganglionic axons. Compound action potentials were evoked in 4E-BP-KO SCG, but not in α3/4E-BP-DKO SCG. Right: intracellular recordings from an SCG neuron in response to stimulation of the preganglionic axons. EPSPs and action potentials were recorded in SCG neurons from P28 4E-BP-KO mice, but not detected in SCG neurons from α3/4E-BP-DKO mice; inset shows that SCG neurons are capable of firing action potentials. Scale bars represent 1 mV and 5 ms for the CAP and 20 mV and 10 ms for the EPSPs on 4E-BP-KO neurons and 2 mV and 20 ms on α3/4E-BP DKO. The inset is 20 mV and 10 ms.
(H) Graph shows percentage of SCG neurons that had EPSPs in response to supramaximal stimulation of the preganglionic nerve. WT (8 mice); 4E-BP KO (6 mice); α3 KO (4 mice); and α3/4E-BP DKO (8 mice). n, number of neurons. These results demonstrate that there is no synaptic transmission in α3-KO or α3/4E-BP-DKO SCG.
At birth, there was no significant difference in the levels of phosphorylated 4E-BP1 in α3-KO SCG neurons when compared to age-matched WT neurons (Figure 4C); however, at 1 month, phosphorylated 4E-BP1 in α3-KO SCG was ~25%–30% less than in WT SCG (Figures 4B and 4C), while total 4E-BP1 was at comparable levels (Figures 4D and 4E; Figures S3D–S3F). This finding raised the intriguing possibility that the effects of synaptic activity on developing SCG neurons are mediated, in part, by the downstream phosphorylation of 4E-BPs and the regulation of translation.
Since 4E-BP functions as a repressor of translation, one might expect that the genetic removal of 4E-BPs would enhance cap-dependent translation, independent of synaptic transmission. To test this idea, we examined SCG neurons in mice with a deletion in 4E-BP genes. SCG neurons express both 4E-BP1 and 4E-BP2 at comparable levels (Figures S3B and S3C) (in contrast to CNS neurons, which primarily express 4E-BP2); therefore, we used mice with a deletion in both genes (4E-BP½1/2−/−; referred to simply as 4E-BP KO).
In the SCG from 4E-BP-KO mice, we observed no significant difference in dendritic growth on SCG neurons during the first post-natal month compared to that on age-matched SCG neurons in WT mice (Figure S1). We also did not detect any significant difference in the targeting of preganglionic axons compared to that in the SCG of age-matched WT mice (Figure S4A). Moreover, at P1–P3, sympathetic neurons in the SCG of 4E-BP-KO mice were innervated by 7–8 axons, similar to WT SCG neurons (Figure 4F). By P8–P9, there was a mild acceleration in the elimination of preganglionic axons (average ~5 axons in WT versus average ~4 axons in 4E-BP KO); however, at P28, the preganglionic innervation of the SCG neurons in 4E-BP mutant mice was indistinguishable from that of WT SCG neurons (Figure 4F).
SCG Proteomic Profile Is Significantly Changed in the Absence of Synaptic Activity and Largely Restored by the Removal of 4E-BP
Next, we conducted a proteomic profiling of SCG from 1-month-old WT, α3-KO, and 4E-BP-KO mice. In addition, we investigated whether removing 4E-BP influences the profile of SCG when synaptic transmission is absent. Therefore, we crossed 4E-BP KO with α3 KO to generate α3/4E-BP-double-knockout (DKO) mutant mice; the SCG in these mice had no 4E-BP or synaptic transmission (Figures 4G and 4H).
Using a data-independent acquisition SWATH workflow (Gillet et al., 2012), we quantified over 2,100 proteins expressed in the SCG, including representatives from all the major molecular function and protein classes (Figure S5; see the Experimental Procedures). Between α3-KO and WT SCG, at least 83 proteins were expressed at significantly different levels (greater than ±1.25-fold change at a q-value < 0.05); several of these are involved in mitochondrial function. Figure 5A shows a heatmap for these 83 proteins (Z score ratios), and it indicates that loss of synaptic activity led to significant changes in the SCG proteome. To determine the impact of removing 4E-BP on translation in the SCG when synaptic activity is absent, we examined the proteome of α3/4E-BP-DKO SCG. Of the 83 proteins whose levels were altered in α3-KO SCG, over 60% (51/83) were reversed and closer to levels in WT SCG (Figure 5A; Table S3). On the other hand, a similar comparison between WT and 4E-BP-KO SCG revealed insignificant differences for >99% of all proteins between the two groups, suggesting that, when fast synaptic transmission was intact, loss of 4E-BP had little effect on the proteome in the SCG.
Figure 5. Genetic Removal of 4E-BP Bypasses the Need for Synaptic Activity and Restores Dendritic Morphology, Synaptic Targeting, and Refinement of Inputs in α3/4E-BP-DKO Mice.

(A) Heatmap showing the log2 Z score ratios of protein levels for 83 proteins whose levels were significantly different (±1.25x, q-value < 0.05) between α3-KO and WT SCG. Many of the differences were restored in α3/4E-BP DKO. All mice were P28. See Figure S5 and Table S3.
(B) Maximum-intensity projections of DiO-labeled P28 SCG neurons from α3/4E-BP-DKO mice. All neurons are shown at the same scale; axons are marked by an asterisk. Scale bar, 20 μm. Neurons are from different ganglia and have been tiled for comparison.
(C) Average total dendritic outgrowth (TDO) per neuron at P28 in α3-KO (from Figure 3C) and α3/4E-BP-DKO SCG.
(D) Average number of primary dendrites per neuron at P28 in α3-KO (from Figure 3D) and α3/4E-BP-DKO SCG.
(E) Skeletonized reconstruction of a P28 neuron in α3/4E-BP-DKO SCG showing dendritic arbors (black), axon (gray, marked by an asterisk), and the locations of preganglionic axon varicosities (red), determined by VAChT staining. Scale bar, 20 μm.
(F) Average distribution of varicosities on the cell body (open) and dendrites (filled) in α3-KO (from Figure 3O) and α3/4E-BP-DKO SCG at P1, P4, and P28.
For (C), (D), and (F), error bars represent ± SEM. ***p < 0.001. For (C) and (D), α3/4E-BP DKO: n = 24 neurons (10 mice). For (F), α3/4E-BP DKO: at P1 n = 6 neurons (3 mice); at P4, n = 7 neurons (4 mice); and at P28, n = 10 neurons (4 mice).
(G) Left: EPSPs evoked on a P28 SCG neuron from α3/4E-BP-DKO mice. Middle: EPSPs evoked by individual preganglionic axons. DI, disparity index. Right: distribution of P28 SCG neurons in α3/4E-BP-DKO (red) and α3-KO (gray; from Figure 1D) mice innervated by the number of inputs. The distribution is fit with a Gaussian function.
(H) The average difference in strength between the strongest and second strongest inputs, expressed as a percentage of the maximum compound EPSP in WT, α3-KO, and α3/4E-BP-DKO SCG. Error bars represent ± SEM. ***p < 0.001.
Loss of 4E-BP Restores Synaptic Refinement and Remodelling in the Absence of Activity
Since removing 4E-BP from α3-KO SCG restores the majority of proteins toward their WT levels, we examined whether it also restores the synaptic organization in the SCG of α3/4E-BP-DKO mice. At P28, total dendritic outgrowth on SCG neurons in α3/4E-BP-DKO mice was approximately twice that of age-matched α3-KO neurons and about 90% of WT neurons (Figures 5B and 5C). Also, α3/4E-BP-DKO SCG neurons at 1 month maintained 5 primary dendrites, in contrast to 3 primary dendrites maintained on age-matched neurons in the SCG in α3-KO mice (Figure 5D). These results show that the genetic removal of 4E-BP largely restores normal dendritic growth, even though the SCG had no synaptic transmission.
In addition, the removal of 4E-BP from α3-KO mice influenced the targeting of synapses by preganglionic axons. At the end of the first post-natal month, the distribution of synapses on sympathetic neurons in the SCG of α3/4E-BP-DKO mice was similar to that on WT neurons, and it was significantly different from that on α3-KO SCG. Over 95% of synapses on the SCG neurons in α3/4E-BP-DKO mice were located on dendrites, in contrast to that on α3-KO SCG, where over 50% were located to the soma (Figures 5E and 5F; Figure S4A). These results show that genetic removal of 4E-BP largely restores dendritic targeting of presynaptic innervation on SCG neurons that are synaptically silent during development.
Since removing 4E-BP from α3-KO neurons restored the targeting of synapses to dendrites and increased dendritic growth, we wondered whether synaptic refinement would be restored in double-mutant mice even in the absence of postsynaptic activity. To address this possibility, we compared the number of preganglionic inputs that converged onto SCG neurons in α3/4E-BP-DKO mice at P28 to the number that converged onto SCG neurons in α3-KO mice.
Remarkably, SCG neurons in 1-month-old α3/4E-BP-DKO mice were innervated by only ~3 preganglionic axons, significantly fewer than the 7–8 on age-matched α3-KO neurons (Figure 5G), but not statistically different from the 2–3 axons innervating SCG neurons in 4E-BP-KO or WT mice. These results indicate that removal of 4E-BP from α3-KO mice restores the ability of preganglionic axons to refine, even though synaptic transmission is absent and synapses remain silent.
The persistent preganglionic inputs to SCG neurons in α3/4E-BP-DKO mice increased in strength, but unlike WT SCG, the disparity among these inputs was small, but greater than that in α3 KO; this is reflected both by the disparity index (Figure 5G) and by the difference in strength between the strongest input and second strongest input (Figure 5H). These results suggest that activity-dependent mechanisms controlling axon elimination are distinct from those that control the differential strengthening of refined connections.
DISCUSSION
During the first post-natal month, connections between preganglionic inputs and postsynaptic sympathetic neurons in the mouse SCG undergo a robust refinement and dendritic remodeling. In this study, we demonstrate that this developmental reorganization is mediated by synaptic activity and that when synaptic activity is absent, regulating cap-dependent translation influences the re-organization of synaptic connections. Our work shows that in mice lacking fast nicotinic transmission in SCG because of a deletion in the α3 nAChR subunit gene (α3 KO), the reorganization of synaptic connections is severely disrupted: the targeting of synapses on postsynaptic neurons is impaired, and preganglionic axons fail to refine. Virally re-introducing the missing α3 and re-establishing synaptic transmission restores these features to those in WT SCG. These results clearly establish that synaptic activity is both necessary and sufficient for the reorganization and refinement of synaptic connections in the mouse SCG. A key finding of our study is that enhancing cap-dependent translation by genetic removal of the translational repressor 4E-BP leads to a remarkable recovery in the reorganization of connections, and synapse elimination occurred even though fast synaptic transmission was absent; this demonstrates a remarkable degree of plasticity by silent synapses. Sympathetic neurons in mice homozygous for mutations in both α3 and 4E-BP (α3/4E-BP-DKO) closely resemble those in WT type mice in terms of the number of presynaptic inputs, dendritic growth, and synaptic distribution. Our results indicate that when postsynaptic activity is absent, considerable reorganization of synaptic connections can still take place by enhancing cap-dependent translation.
Synaptic refinement is a competitive process in which converging axons of similar type compete with one another to innervate a target. As development proceeds, the synaptic strength of 1 or more axons increases; it is thought that this differential in synaptic strength among competing axons gives the stronger inputs an advantage for consolidation, while weaker inputs are eliminated (Lichtman and Colman, 2000; Busetto et al., 2000; Bosman et al., 2008; Turney and Lichtman, 2012; Lee et al., 2014; but see Wang et al. 2011). In the SCG of α3/4E-BP-DKO mice, there is less differential strengthening among inputs; nonetheless, synapse elimination of preganglionic axons occurs in the absence of synaptic transmission. Therefore, in sympathetic ganglia, a differential in synaptic strength does not determine which axons are retained and/or which are eliminated, challenging the notion that a differential in synaptic strength determines which synapses are stabilized and which are eliminated (Katz and Shatz, 1996; Piochon et al., 2016).
The Effects of Activity and Removing 4E-BP on Protein Levels
Our proteomic experiments demonstrate that a significant number of proteins are altered when postsynaptic activity is absent and the loss of 4E-BP in α3-KO mutant mice largely restores the proteome. In its basal state, hypo-phosphorylated 4E-BP suppresses cap-dependent translation by sequestering eIF4E and preventing the formation of the 5′ cap. Several signals initiate cap-dependent translation, directly or indirectly, by hyperphosphorylating 4E-BP and releasing eIF4E. We show that, in SCG neurons, fast excitatory synaptic transmission is one such signal that leads to increased 4E-BP phosphorylation and, by extension, cap-dependent translation. Similarly, deletion of 4E-BP removes this sequestration of eIF4E, leading to enhanced cap-dependent translation and altered protein levels. How organisms tolerate genetic alteration of major regulators of cap-dependent translation, such as loss of 4E-BP or heterozygocity for eIF4E, is the subject of intense experimental investigations. Recent discoveries suggest that specific mRNAs and, therefore, specific pathways rather than overall maintenance of proteins are affected as a result of these manipulations (Truitt et al., 2015; Gkogkas et al. 2013).
Consistent with this notion, we found that loss of 4E-BP had little effect on the development and innervation of SCG neurons or on the global proteome profile of SCG; of the 2,100 specific proteins identified at the SCG, only a few showed differential expression as a result of the loss of 4E-BP. By contrast, the absence of excitatory synaptic transmission had a significant effect on the global proteome profile: we found that the levels of at least 83 proteins were significantly altered in α3-KO SCG, indicating that these proteins are regulated by pathways downstream of postsynaptic activity and possibly involved in the refinement of presynaptic inputs, the stability of primary dendritic branches, and the targeting of synapses by preganglionic axons. Relevantly, deleting 4E-BP from α3-KO mice changed the proteomic profile of SCG and shifted the levels of more than half of these proteins toward values found in WT SCG.
Interestingly, the levels of a large number of proteins were not statistically different between WT and α3-KO SCG, suggesting that these proteins are not directly regulated by postsynaptic activity. Of particular interest are proteins implicated in synapse elimination elsewhere in the nervous system. For example, the major histocompatibility complex class 1 molecules H2-Db and H2-Kb are involved in refinement of retinal ganglion cell axons innervating the lateral geniculate nucleus (LGN) through a mechanism that regulates the expression of postsynaptic AMPA receptors (Lee et al., 2014). We found no difference in the levels of major histocompatibility complex class 1 molecule H2-Db between α3-KO and WT SCG, and we did not detect H2-Kb. Moreover, we detected no statistical difference in the level of the complement proteins C1q and C3, molecules required for the elimination of retinal ganglion cell synapses in the LGN (Stevens et al., 2007; Stephan et al., 2012). These results suggest that other molecules and pathways are involved in synapse elimination in the SCG.
Our proteomic analysis provides a comprehensive profile of activity-dependent protein expression in mouse SCG, not only revealing that postsynaptic activity during the first post-natal month has a profound effect on the proteomic landscape in the SCG but also confirming that the removal of 4E-BP can largely normalize the proteomic landscape in the absence of activity. These experiments pave the way for future work aimed at identifying a mechanistic link between individual or a network of multiple proteins and the developmental program of refinement at the SCG.
Distribution of Silent Synapses
As axons are being eliminated in WT SCG during the first post-natal month, those that persist strengthen their inputs by increasing the number of synaptic contacts on sympathetic neurons, and, by the first post-natal month, the overall number of synapses on SCG neurons is ~2-fold greater than at birth. Although the increase in synapses overlaps in time with axon elimination, our results from α3-KO SCG suggest that these two processes are regulated by different mechanisms. Synaptic contacts in α3-KO SCG continue to increase over the first postnatal month in an activity-independent manner, whereas synapse elimination depends critically on activity. Moreover, in α3-KO adult SCG, the density of synaptic contacts on sympathetic neurons is approximately 40% greater than that on neurons in WT SCG, even though axon elimination does not occur. This indicates that the increase in synapses and the elimination of axons are likely regulated by different mechanisms.
Postsynaptic activity plays a crucial role in determining how preganglionic axons target their synapses onto sympathetic neurons during post-natal development. At birth, most synapses are targeted to dendrites in both WT and α3-KO SCG. Yet, over the first post-natal month, as synapses are continually formed, the preganglionic axons in WT SCG continue to target their synapses to dendrites, while most synapses in α3-KO SCG at 1 month are targeted to and hyperinnervate the soma of these synaptically inactive neurons, as if, in the absence of activity, sympathetic neurons actively seek innervation. Furthermore, restoring postsynaptic activity to α3-KO SCG neurons results in a shift in distribution of synapses from the soma to the dendrites, indicating that the targeting of synapses to the dendrites of sympathetic neurons remains plastic and the distribution of synapses is directed by mechanisms downstream of postsynaptic activity. Notably, when 4E-BP is deleted from α3-KO mice, electrophysiologically silent synaptic contacts continue to increase in the first post-natal month at a density similar to WT. These results suggest that, upon the removal of 4E-BP, the preganglionic axons target their synapses to dendrites, bypassing the normal requirement for postsynaptic activity.
Dendritic Growth
Dendrites on neurons in α3-KO SCG grow poorly over the first post-natal month. This is consistent with several studies demonstrating that disrupting synaptic activity influences the growth of dendrites (Wong and Ghosh, 2002; Cline and Haas, 2008; Lefebvre et al., 2015). However, our findings are at odds with previous work showing that cutting the preganglionic nerve at birth had no effect on the growth of dendrites on denervated neurons over the first post-natal month (Voyvodic, 1987). It is not clear why inactivity produced by the deletion of α3 has a different effect on dendritic growth by SCG neurons than inactivity produced by cutting the preganglionic nerve. It seems unlikely that α3 has some unanticipated role in dendritic growth and stabilization, apart from its role in forming functional nAChRs and mediating fast synaptic transmission, because, when placed in culture, α3-KO and WT SCG neurons extend dendrites that are statistically similar (Figures S1J and S1K).
Neurons in the SCG receive cholinergic-nicotinic synaptic transmission before birth (Rubin, 1985), raising the possibility that a reduction in synaptic activity in α3-KO SCG in utero might have influenced the growth of dendrites. However, this seems unlikely because: (1) the activity is low in the sympathetic nervous system prenatally, as the mother maintains the pups’ homeostasis in utero; and (2) at birth, dendrites on α3-KO SCG are not statistically different from control WT neurons.
A clear distinction between α3-KO neurons and denervated neurons is that α3-KO neurons receive morphological synapses. Conceivably, the difference in dendritic growth between α3-KO SCG neurons and denervated WT neurons is that α3-KO SCG neurons have electrophysiologically silent synapses. If the silent synapses signal inappropriately to molecules downstream of the postsynaptic complex to perturb dendritic growth (Quach et al., 2013), it might account for the difference between dendritic growth on inactive α3-KO SCG neurons and those on denervated neurons. Whatever the molecules involved, we show that 4E-BP-regulated translation can act on such pathways to control dendritic growth.
In summary, our findings demonstrate that 4E-BP-regulated mechanisms are involved in the growth of dendrites, the targeting of synapses, and the refinement of preganglionic axons when postsynaptic activity is absent. Our findings suggest that, in the absence of synaptic activity, where synaptic refinement is curtailed, genetic removal of 4E-BP activates a molecular program that can compensate for the lack of activity-dependent mechanisms that normally direct synaptic reorganization and refinement of presynaptic inputs.
EXPERIMENTAL PROCEDURES
Experimental Model and Subject Details
Mice
Mice with a deletion in the alpha 3 nicotinic subunit gene (Xu et al., 1999; referred to as α3 KO) were maintained on an outcrossed background (Krishnaswamy and Cooper, 2009). To generate 4EBP1/2−/−; α3−/− mice (referred to as α3/4E-BP DKO) a series of crosses were performed between 4EBP1/2−/− mice (Tsukiyama-Kohara et al., 2001; Banko et al., 2005) and −3 KO mice. Genotypes for all three genes were determined by PCR (Figure S3A). Our results include data from both male and female mice. All procedures for animal handling were carried out according to the guidelines of the Canadian Council on Animal Care.
Method Details
Electrophysiological Recordings
For intracellular recordings, we used 80–120 mΩ glass microelectrodes attached to a high inertial precision microdrive. The recording electrode was filled with 1M KAc and connected to the head stage of an Axoclamp 2A amplifier used in current-clamp mode. On-line stimulation and data acquisition were done with N-Clamp and offline data analysis was performed using Igor Pro software. To measure dendrites on Ad-α3 infected neurons, electrodes were filled with 10mM Alexa Fluor 488 hydrazide in 200mM KCl.
To measure the convergence of preganglionic axons innervating a sympathetic neuron, the preganglionic nerve was stimulated with voltages of increasing strength while holding the neuron at ~-90 mV to prevent EPSPs from triggering action potentials. In some experiments, we also included QX314 in the recording electrode to prevent action potentials. Increasing the strength of the stimulus to the preganglionic nerve activates axons of different thresholds which results in discrete jumps in the amplitude of the EPSPs. We used these discrete jumps as a measure of the number of axons innervating the neuron. To isolate the EPSP evoked by individual axons, we averaged at least 10 traces for each discrete jump and calculated the difference between the average EPSP evoked by that axon and all axons of lower threshold and the average EPSP evoked only by axons of lower threshold. To calculate the disparity index, DI, for each neuron, we divided the standard deviation, SD, of the EPSPs by the mean EPSP (Hashimoto and Kano, 2003).
Adenoviruses
Full-length α3 neuronal nAChR subunit cDNA was ligated with either the synapsin or human ubiquitin C promoter into pAdTrack (Ad-α3/Syn or Ad-α3/Ubi) and infected mice with either Ad-α3/Syn or Ad-α3/Ubi adenovirus at a concentration of ~107 pfu/mL.
Lipophilic Tracer Labeling
We used lipophilic tracers DiI and DiO to sparsely label a random subset of preganglionic axons and postsynaptic neurons in the SCG. After labeling, ganglia were kept in the dark in 1X PBS for 5 - 6 days to allow for tracers to diffuse along the axons. Ganglia were sliced into 100 μm sections.
Immunohistochemistry
Details are described in Supplemental Experimental Procedures.
Image Acquisition and Anaiysis
Images were acquired on an upright confocal microscope (BX-61W, Olympus) with a 60X, NA 1.42 PlanApo N oil-immersion objective. Laser lines were activated sequentially. All analysis was performed with ImageJ (NIH, Bethesda, MD). Dendrite analysis. Only neurons with complete dendritic arbors and an identifiable axon were analyzed. For images of isolated neurons, we removed all DiO-labeled neurites that were not connected to the neuron of interest, as determined from 3D reconstructions. To quantify the length and number of dendritic branches, we reconstructed neurons in 3D.
Synaptic targeting.
For synaptic targeting, we identified VAChT puncta located on a neuron of interest on each plane of a z stack. VAChT puncta that co-localized with the DiO membrane label, were at least 0.5 μm in diameter, and spanned at least two optical slices were counted as putative synapses.
Preganglionic axonal targeting.
To estimate the percentage of the cell body surface covered by an axon, on each plane of the cell body, we measured the circumference of the cell body (DiO labeled from postganglionic nerve), and the proportion of the cell body circumference occupied by an axon (DiI labeled from preganglionic nerve) on that plane.
P-4E-BP1, 4E-BP1 and MAP-1A fluorescence intensity.
For intensity analysis, average fluorescence intensity for each ROI was measured from the MAP-1A channel, and ROI were transferred to the P-4E-BP1 or 4E-BP1 channel to measure the corresponding fluorescence intensity.
Proteomics
Full details for the proteomic experiments are in the Supplemental Experimental Procedures.
Quantification and Statistical Analysis
Values of n and p values are reported in the Figures and corresponding figure legends. In all figures, error bars represent ± SEM, *p < 0.05, ***p < 0.001. To test for statistical differences between two samples, we used unpaired two tailed t tests assuming equal variance; for three or more samples, we used a one-way ANOVA to determine if one or more samples were significantly different. If the p value calculated from the F-statistic was less than 0.05, we used a post hoc Tukey HSD test to identify which pairs of samples were significantly different from each other.
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the raw proteomics reported in this paper are ProteomeXchange: PXD007141 and MassIVE: MSV000081386, and can also be accessed at ftp://massive.ucsd.edu/MSV000081386.
Supplementary Material
Highlights.
Postsynaptic activity regulates synaptic refinement at the SCG
Postsynaptic activity influences 4E-BP phosphorylation in the SCG
Loss of postsynaptic activity alters the proteomic profile of the SCG
In the absence of postsynaptic activity, removal of 4E-BP restores synaptic refinement
ACKNOWLEDGMENTS
This work was supported by Canadian Institute for Health Research operating grants to A.P.H., N. Sonenberg, and E.C. We thank Professor L. Cooper for comments on the manuscript. The proteomics analysis was partially funded by an NIH shared instrumentation grant for the TripleTOF system at the Buck Institute (1S10 OD016281). N.B. received a fellowship from the Glenn Foundation for Medical Research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, five figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.040.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alvarez-Castelao B, and Schuman EM (2015). The Regulation of Synaptic Protein Turnover. J. Biol. Chem 290, 28623–28630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balice-Gordon RJ, and Lichtman JW (1994). Long-term synapse loss induced by focal blockade of postsynaptic receptors. Nature 372, 519–524. [DOI] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, and Klann E (2005). The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J. Neurosci. 25, 9581–9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleckert A, and Wong ROL (2011). Identifying roles for neurotransmission in circuit assembly: insights gained from multiple model systems and experimental approaches. BioEssays 33, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman LW, Takechi H, Hartmann J, Eilers J, and Konnerth A (2008). Homosynaptic long-term synaptic potentiation of the “winner” climbing fiber synapse in developing Purkinje cells. J. Neurosci 28, 798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, and Wells DG (2007). Dendritic mRNA: transport, translation and function. Nat. Rev. Neurosci 8, 776–789. [DOI] [PubMed] [Google Scholar]
- Buffelli M, Busetto G, Cangiano L, and Cangiano A (2002). Perinatal switch from synchronous to asynchronous activity of motoneurons: link with synapse elimination. Proc. Natl. Acad. Sci. USA 99, 13200–13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busetto G, Buffelli M, Tognana E, Bellico F, and Cangiano A (2000). Hebbian mechanisms revealed by electrical stimulation at developing rat neuromuscular junctions. J. Neurosci 20, 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline HT (2001). Dendritic arbor development and synaptogenesis. Curr. Opin. Neurobiol 11, 118–126. [DOI] [PubMed] [Google Scholar]
- Cline H, and Haas K (2008). The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: a review of the synaptotrophic hypothesis. J. Physiol. 586, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Cory S (2002). The developing synapse: construction and modulation of synaptic structures and circuits. Science 298, 770–776. [DOI] [PubMed] [Google Scholar]
- Fitzsimonds RM, and Poo MM (1998). Retrograde signaling in the development and modification of synapses. Physiol. Rev. 78, 143–170. [DOI] [PubMed] [Google Scholar]
- Forehand CJ (1985). Density of somatic innervation on mammalian autonomic ganglion cells is inversely related to dendritic complexity and preganglionic convergence. J. Neurosci. 5, 3403–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, and Aebersold R (2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell Proteomics. 11, O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A-C, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, and Sonenberg N (1999). Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 13, 1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, et al. (2013). Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas K, Li J, and Cline HT (2006). AMPA receptors regulate experience-dependent dendritic arbor growth in vivo. Proc. Natl. Acad. Sci. USA 103, 12127–12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, and Kano M (2003). Functional differentiation of multiple climbing fiber inputs during synapse elimination in the developing cerebellum. Neuron 38, 785–796. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Tsujita M, Miyazaki T, Kitamura K, Yamazaki M, Shin H-S, Watanabe M, Sakimura K, and Kano M (2011). Postsynaptic P/Q-type Ca2+ channel in Purkinje cell mediates synaptic competition and elimination in developing cerebellum. Proc. Natl. Acad. Sci. USA 108, 9987–9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata Y, and Stryker MP (1994). Control of thalamocortical afferent rear-rangement by postsynaptic activity in developing visual cortex. Science 265, 1732–1735. [DOI] [PubMed] [Google Scholar]
- Henry FE, McCartney AJ, Neely R, Perez AS, Carruthers CJ, Stuenkel EL, Inoki K, and Sutton MA (2012). Retrograde changes in presynaptic function driven by dendritic mTORC1. J. Neurosci. 32, 17128–17142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong YK, and Chen C (2011). Wiring and rewiring of the retinogeniculate synapse. Curr. Opin. Neurobiol 21, 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Yoon BC, and Holt CE (2012). Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat. Rev. Neurosci 13, 308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, and Hashimoto K (2009). Synapse elimination in the central nervous system. Curr. Opin. Neurobiol 19, 154–161. [DOI] [PubMed] [Google Scholar]
- Katz LC, and Crowley JC (2002). Development of cortical circuits: lessons from ocular dominance columns. Nat. Rev. Neurosci 3, 34–42. [DOI] [PubMed] [Google Scholar]
- Katz LC, and Shatz CJ (1996). Synaptic activity and the construction of cortical circuits. Science 274, 1133–1138. [DOI] [PubMed] [Google Scholar]
- Krishnaswamy A, and Cooper E (2009). An activity-dependent retrograde signal induces the expression of the high-affinity choline transporter in cholinergic neurons. Neuron 61, 272–286. [DOI] [PubMed] [Google Scholar]
- Lee H, Brott BK, Kirkby LA, Adelson JD, Cheng S, Feller MB, Datwani A, and Shatz CJ (2014). Synapse elimination and learning rules co-regulated by MHC class I H2-Db. Nature 509, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre JL, Sanes JR, and Kay JN (2015). Development of dendritic form and function. Annu. Rev. Cell Dev. Biol 31, 741–777. [DOI] [PubMed] [Google Scholar]
- Lichtman JW, and Colman H (2000). Synapse elimination and indelible memory. Neuron 25, 269–278. [DOI] [PubMed] [Google Scholar]
- Lin TV, Hsieh L, Kimura T, Malone TJ, and Bordey A (2016). Normalizing translation through 4E-BP prevents mTOR-driven cortical mislamination and ameliorates aberrant neuron integration. Proc. Natl. Acad. Sci. USA 113, 11330–11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, and Blenis J (2009). Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol 10, 307–318. [DOI] [PubMed] [Google Scholar]
- Niell CM, Meyer MP, and Smith SJ (2004). In vivo imaging of synapse formation on a growing dendritic arbor. Nat. Neurosci 7, 254–260. [DOI] [PubMed] [Google Scholar]
- Penney J, Tsurudome K, Liao EH, Elazzouzi F, Livingstone M, Gonzalez M, Sonenberg N, and Haghighi AP (2012). TOR is required for the retrograde regulation of synaptic homeostasis at the Drosophila neuromuscular junction. Neuron 74, 166–178. [DOI] [PubMed] [Google Scholar]
- Penney J, Tsurudome K, Liao EH, Kauwe G, Gray L, Yanagiya A, R Calderon M, Sonenberg N, and Haghighi AP (2016). LRRK2 regulates retrograde synaptic compensation at the Drosophila neuromuscular junction. Nat. Commun. 7, 12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piochon C, Kano M, and Hansel C (2016). LTD-like molecular pathways in developmental synaptic pruning. Nat. Neurosci 19, 1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves D, and Lichtman JW (1980). Elimination of synapses in the developing nervous system. Science 210, 153–157. [DOI] [PubMed] [Google Scholar]
- Purves D, and Lichtman J (1984). Principles of Neural Development (Massachusetts: Sinauer Associates Inc.). [Google Scholar]
- Quach DH, Oliveira-Fernandes M, Gruner KA, and Tourtellotte WG (2013). A sympathetic neuron autonomous role for Egr3-mediated gene regulation in dendrite morphogenesis and target tissue innervation. J. Neurosci. 33, 4570–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassadi S, Krishnaswamy A, Pié B, McConnell R, Jacob MH, and Cooper E (2005). A null mutation for the alpha3 nicotinic acetylcholine (ACh) receptor gene abolishes fast synaptic activity in sympathetic ganglia and reveals that ACh output from developing preganglionic terminals is regulated in an activity-dependent retrograde manner. J. Neurosci. 25, 8555–8566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, and Sonenberg N (2005). Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433, 477–480. [DOI] [PubMed] [Google Scholar]
- Rubin E (1985). Development of the rat superior cervical ganglion: initial stages of synapse formation. J. Neurosci. 5, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, and Lichtman JW (1999). Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 22, 389–442. [DOI] [PubMed] [Google Scholar]
- Schlaggar BL, Fox K, and O’Leary DD (1993). Postsynaptic control of plasticity in developing somatosensory cortex. Nature 364, 623–626. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, and Hinnebusch AG (2007). New modes of translational control in development, behavior, and disease. Mol. Cell 28, 721–729. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, and Hinnebusch AG (2009). Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Barres BA, and Stevens B (2012). The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 35, 369–389. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- Steward O, and Schuman EM (2001). Protein synthesis at synaptic sites on dendrites. Annu. Rev. Neurosci 24, 299–325. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Asano H, Katayama Y, Muragaki Y, Imoto K, and Miyata M (2014). Large-scale somatotopic refinement via functional synapse elimination in the sensory thalamus of developing mice. J. Neurosci. 34, 1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, and Sabatini DM (2012). A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, Seo Y, Barna M, and Ruggero D (2015). Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell 162, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama-Kohara K, Poulin F, Kohara M, DeMaria CT, Cheng A, Wu Z, Gingras A-C, Katsume A, Elchebly M, Spiegelman BM, et al. (2001). Adipose tissue reduction in mice lacking the translational inhibitor 4E-BP1. Nat. Med 7, 1128–1132. [DOI] [PubMed] [Google Scholar]
- Turney SG, and Lichtman JW (2012). Reversing the outcome of synapse elimination at developing neuromuscular junctions in vivo: evidence for synaptic competition and its mechanism. PLoS Biol. 10, e1001352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voyvodic JT (1987). Development and regulation of dendrites in the rat superior cervical ganglion. J. Neurosci. 7, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DO, Martin KC, and Zukin RS (2010). Spatially restricting gene expression by local translation at synapses. Trends Neurosci. 33, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu H, and Zhang ZW (2011). Elimination of redundant synaptic inputs in the absence of synaptic strengthening. J. Neurosci. 31, 16675–16684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells DG, Richter JD, and Fallon JR (2000). Molecular mechanisms for activity-regulated protein synthesis in the synapto-dendritic compartment. Curr. Opin. Neurobiol 10, 132–137. [DOI] [PubMed] [Google Scholar]
- Wong ROL, and Ghosh A (2002). Activity-dependent regulation of dendritic growth and patterning. Nat. Rev. Neurosci 3, 803–812. [DOI] [PubMed] [Google Scholar]
- Xu W, Gelber S, Orr-Urtreger A, Armstrong D, Lewis RA, Ou C-N, Patrick J, Role L, De Biasi M, and Beaudet AL (1999). Megacystis, mydriasis, and ion channel defect in mice lacking the α3 neuronal nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA 96, 5746–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M, Johnson-Venkatesh EM, Zhang H, Parent JM, Sutton MA, and Umemori H (2011). Multiple forms of activity-dependent competition refine hippocampal circuits in vivo. Neuron 70, 1128–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LI, and Poo MM (2001). Electrical activity and development of neural circuits. Nat. Neurosci 4 (Suppl), 1207–1214. [DOI] [PubMed] [Google Scholar]
- Zou D-J, Feinstein P, Rivers AL, Mathews GA, Kim A, Greer CA, Mombaerts P, and Firestein S (2004). Postnatal refinement of peripheral olfactory projections. Science 304, 1976–1979. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the raw proteomics reported in this paper are ProteomeXchange: PXD007141 and MassIVE: MSV000081386, and can also be accessed at ftp://massive.ucsd.edu/MSV000081386.