

Abstract
Background
There is increasing focus on providing high quality care for people at the end of life, irrespective of disease or cause, and in all settings. In the last ten years the use of care pathways to aid those treating patients at the end of life has become common worldwide. The use of the Liverpool Care Pathway (LCP) in the UK has been criticised. In England the LCP was the subject of an independent review, commissioned by a Health Minister. The resulting Neuberger Review acknowledged that the LCP was based on the sound ethical principles that provide the basis of good quality care for patients and families when implemented properly. It also found that the LCP often was not implemented properly, and had instead become a barrier to good care; it made over 40 recommendations, including education and training, research and development, access to specialist palliative care services, and the need to ensure care and compassion for all dying patients. In July 2013, the Department of Health released a statement that stated the use of the LCP should be "phased out over the next 6‐12 months and replaced with an individual approach to end of life care for each patient".
The impact of opioids was a particular concern because of their potential influence on consciousness, appetite and thirst in people near the end of life. There was concern that impaired patient consciousness may lead to an earlier death, and that effects of opioids on appetite and thirst may result in unnecessary suffering. This rapid review, commissioned by the National Institute for Health Research, used standard Cochrane methodology to examine adverse effects of morphine, fentanyl, oxycodone, and codeine in cancer pain studies as a close approximation to possible effects in the dying patient.
Objectives
To determine the impact of opioid treatment on patient consciousness, appetite and thirst in randomised controlled trials of morphine, fentanyl, oxycodone or codeine for treating cancer pain.
Search methods
We assessed adverse event data reported in studies included in current Cochrane reviews of opioids for cancer pain: specifically morphine, fentanyl, oxycodone, and codeine.
Selection criteria
We included randomised studies using multiple doses of four opioid drugs (morphine, fentanyl, oxycodone, and codeine) in cancer pain. These were taken from four existing or ongoing Cochrane reviews. Participants were adults aged 18 and over. We included only full journal publication articles.
Data collection and analysis
Two review authors independently extracted adverse event data, and examined issues of study quality. The primary outcomes sought were numbers of participants experiencing adverse events of reduced consciousness, appetite, and thirst. Secondary outcomes were possible surrogate measures of the primary outcomes: delirium, dizziness, hallucinations, mood change and somnolence relating to patient consciousness, and nausea, vomiting, constipation, diarrhoea, dyspepsia, dysphagia, anorexia, asthenia, dehydration, or dry mouth relating to appetite or thirst.
Comparative measures of harm were known to be unlikely, and we therefore calculated the proportion of participants experiencing each of the adverse events of interest with each opioid, and for all four opioid drugs combined.
Main results
We included 77 studies with 5619 randomised participants. There was potential bias in most studies, with small size being the most common; individual treatment groups had fewer than 50 participants in 60 studies. Participants were relatively young, with mean age in the studies typically between 50 and 70 years. Multiple major problems with adverse event reporting were found, including failing to report adverse events in all participants who received medication, all adverse events experienced, how adverse events were collected, and not defining adverse event terminology or whether a reporting system was used.
Direct measures of patient consciousness, patient appetite, or thirst were not apparent. For opioids used to treat cancer pain adverse event incidence rates were 25% for constipation, 23% for somnolence, 21% for nausea, 17% for dry mouth, and 13% for vomiting, anorexia, and dizziness. Asthenia, diarrhoea, insomnia, mood change, hallucinations and dehydration occurred at incidence rates of 5% and below.
Authors' conclusions
We found no direct evidence that opioids affected patient consciousness, appetite or thirst when used to treat cancer pain. However, somnolence, dry mouth, and anorexia were common adverse events in people with cancer pain treated with morphine, fentanyl, oxycodone, or codeine.
We are aware that there is an important literature concerning the problems that exist with adverse event measurement, reporting, and attribution. Together with the known complications concerning concomitant medication, data collection and reporting, and nomenclature, this means that these adverse events cannot always be attributed unequivocally to the use of opioids, and so they provide only a broad picture of adverse events with opioids in cancer pain. The research agenda includes developing definitions for adverse events that have a spectrum of severity or importance, and the development of appropriate measurement tools for recording such events to aid clinical practice and clinical research.
Plain language summary
Impact of morphine, fentanyl, oxycodone or codeine on patient consciousness, appetite and thirst when used to treat cancer pain
Description of the problem
Care pathways are packages of care designed to ensure that patients have appropriate and effective care in particular situations. Such pathways are commonly used, and often produce good results, but they can also be used as a tick box solution that acts as a barrier to good care. Care pathways have been used to ensure appropriate care for people who are dying in hospice settings.
The Liverpool Care Pathway was devised for use in hospices, and has been used in general hospital settings to care for dying patients. Its use has been criticised. A government review of the use of end‐of‐life care pathways in the NHS in the UK recommended they should not be used because they were being misused.
A concern, mainly raised by relatives, was that opioids were over‐prescribed, used to hasten death, to reduce consciousness, and diminish the patient's desire or ability to accept food or drink.
The purpose of this review
This Cochrane review was commissioned to look at harms (adverse events) associated with the use of opioids to treat cancer pain particularly relating to patient consciousness, appetite or thirst.
How the information was gathered
Ideally, when writing this review we would have looked at medical trials of opioid use in older people receiving end‐of‐life care, but there are no trials in this area. So, we looked at trials of people being treated with opioids for cancer pain, as the information these trials provide is likely to be the closest that is available to opioid use in end‐of‐life care ‐ although people treated for cancer pain are not usually at the end of their lives.
What we found
This review identified 77 studies with over 5,000 people who received various treatments. The population in these trials was mainly aged between 50 and 70 years. Trial quality was generally poor; particular problems included small study size, and not reporting adverse events in all patients, or all recorded adverse events. Known problems with adverse event measurement, recording, and reporting made assessment even more difficult.
For all four opioids together, 1 in 4 people experienced constipation and somnolence (sleepiness, drowsiness), 1 in 5 experienced nausea and dry mouth, and 1 in 8 experienced vomiting, loss of appetite, and dizziness. Weakness, diarrhoea, insomnia (difficulty in sleeping), mood change, hallucinations and dehydration occurred at rates of 1 in 20 people and below. These results may contribute to understanding the effects of opioids on consciousness, appetite, and thirst in end‐of‐life care in all patients deemed to be people who are dying.
Background
This review assesses the impact of four opioid drugs (morphine, fentanyl, oxycodone, and codeine) on patient consciousness, appetite, and thirst in randomised controlled trials (RCT) in cancer‐related pain. It has been commissioned by the National Institute for Health Research (NIHR) in the UK as a rapid review. The specific research question concerned the effects of opioids on level of consciousness, and inability of patients to eat and drink. It is recognised that participants with pain from cancer in these clinical trials are not the same as the (mostly older) people considered to be within a short time of dying, but it is the closest available randomised trial data set.
Description of the condition
Pain is often the first symptom causing someone to seek medical advice that eventually leads to a diagnosis of cancer; 30% to 50% of all people with cancer will experience moderate to severe pain (Portenoy 1999). For those with advanced cancer 75% to 90% will experience pain severe enough to have a major impact on daily living.
Description of the intervention
The four opioids chosen are those most commonly used to treat cancer‐related pain, and for which Cochrane reviews have either been published and updated or are near publication. As with all treatments, benefit and harm needs to be considered in making choices about the care of patients. Recently concern has been expressed about the adverse events associated with opioids, especially in terms of the impact on patient consciousness, appetite and thirst in people near the end of life. There is fear that impaired patient consciousness may lead to an earlier death, and that effects of opioids on appetite and thirst may result in unnecessary suffering (DH 2013).
Why it is important to do this review
There is increasing focus on providing high quality care for people at the end of life, irrespective of disease or cause, and in all settings. In the last ten years the use of care pathways to aid those treating patients at the end of life has become common worldwide (Bailey 2005; Bookbinder 2005; Ellershaw 2003), despite a lack of evidence from randomised trials (Chan 2013). A randomised comparison of the Liverpool Care Pathway (LCP) and usual care in cancer in hospital wards showed no difference in quality of care or survival times between them, though most outcomes were numerically superior in the LCP arm (Costantini 2014).
The pathways are intended to improve the care and dignity of those facing the last days of life, but questions around their use have been raised. In the UK, for example, criticisms emerged that the LCP was in fact leading to poor care, as reported in the media and confirmed in part by a review panel. A number of commentaries provide different perspectives on the background and future of care of the dying (Knights 2013; George 2014; Regnard 2014).
In response, the Minister of State for Care Services in England set up a panel with a wide‐ranging set of complementary interests and expertise in end‐of‐life care to review the use and experience of the LCP. This group, chaired by Baroness Julia Neuberger, published its report in July 2013 (DH 2013), recommending that the LCP be phased out and replaced by individualised end‐of‐life care plans. One key issue reported to the review body was that junior doctors felt that training was on how to fill in documents, rather than the principles of terminal care: how to care for the patient was becoming secondary to form filling.
Comments in the report (DH 2013) included:
‘The Review heard that, if a patient became more agitated or in greater pain as they died, they often became peaceful because the right drugs were given to them at the right time and in the right dose. But there were complaints that opiate pain killers and tranquillisers were being used inappropriately as soon as the LCP was initiated.’
‘At the end of life, a person may become overhydrated, and there is no moral or legal obligation to continue to administer and clinically assisted hydration or nutrition if they are having no beneficial effect. But there can be no clinical justification for denying a drink to a dying patient who wants one, unless doing so would cause them distress. In hospitals in particular, there appear to have been many instances demonstrating an inadequate understanding of the LCP’s direction on oral hydration. Refusing food and drink is a decision for the patient, not clinical staff, to make.’
Opioids are known both to have sedative properties and to have an impact on the gastrointestinal system. This review was commissioned by the NIHR to help answer one question raised by the review of the LCP, namely whether opioids given for relief of cancer pain have an adverse effect on patient consciousness, appetite, and thirst. The review will seek to quantify how often these symptoms or effects are reported in RCTs of four commonly‐used opioid drugs when used to treat cancer pain. This information may be directly relevant to patients in the last days or hours of life, or may highlight deficiencies in the available evidence and serve to direct further studies, or both.
Although the motivation for this review is firmly UK‐based, the LCPs are used worldwide, and the findings of the review are likely to be of value, or at least interest, outside the UK. In addition, while the Neuberger review raised over 40 different points relating to clinical practice and clinical research, the purpose of this review of opioid adverse events in treating cancer pain related only to one of those points.
Review limitations
It is important to acknowledge ab initio a major limitation of the review, namely whether an evaluation of RCTs is able to answer the question. RCTs typically exclude patients at the end of life in the last few weeks or days of life, and the review is predicated on the assumption that an evaluation of adverse events during a clinical trial in a selected, sometimes healthier patient sample, can provide relevant information about the nature of these side effects in dying patients. Any assumption that cancer studies may be extrapolated to all populations receiving end‐of‐life care is fundamentally speculative because the studies reviewed will not provide data from populations that possess the same type of physiological and functional disturbances, comorbidities, and concurrent treatments as the populations of dying people that are the reference group for the review.
The point of the review is to examine whether there is any available evidence on adverse events recorded in cancer pain studies that provides information relevant to end‐of‐life care, and for future research. The specific research question concerned the effects of opioids on level of consciousness, and inability of patients to eat and drink.
Objectives
To determine the impact of opioid treatment on patient consciousness, appetite and thirst in randomised controlled trials of morphine, fentanyl, oxycodone or codeine for treating cancer pain.
Methods
Criteria for considering studies for this review
Types of studies
We assessed adverse event data reported in studies included in Cochrane reviews of opioids for cancer pain: morphine (Wiffen 2013), fentanyl (Hadley 2012), oxycodone (Schmidt‐Hansen 2010 together with the authors of an ongoing update, supplemented by additional searches), and a completed but unpublished review of codeine (Schremmer 2013 ‐ full review in press).
For inclusion in the individual reviews, studies were: RCTs using single or multiple doses, with parallel or cross‐over design, and of any duration. Studies that did not state that they were randomised were excluded, as were quasi‐randomised studies, studies with fewer than 10 participants (Moore 1998), and studies that did not deal with cancer‐related pain, or did not assess pain as an outcome measure. Studies reported only as short abstracts (usually from meetings) were not included because they contain insufficient information to assess methodological quality and risk of bias. For this review, single dose studies were excluded as they are less likely to give useful data on adverse events.
Types of participants
Our original intention was to include any relevant data from adults and children with cancer pain requiring treatment with opioids. However, as none of the Cochrane reviews identified any relevant studies in children (the oxycodone review looked only for studies in adults), this review was limited to adults.
Types of interventions
Morphine, fentanyl, oxycodone, or codeine preparations compared with either placebo, an alternative formulation of morphine or an active control. Any route of administration was permitted, though morphine, codeine and oxycodone were likely to be used by the oral route of administration, while fentanyl was used in the form of transdermal patches.
Types of outcome measures
The primary outcomes are numbers of participants experiencing adverse events of level of consciousness or inability to eat or drink.
Secondary surrogate outcomes included numbers of participants reporting:
delirium, dizziness, hallucinations, mood change, asthenia, and somnolence that may relate to patient consciousness;
nausea, vomiting, constipation, diarrhoea, dyspepsia, dysphagia, anorexia, dehydration, or dry mouth that may relate to appetite or thirst.
Where studies report these adverse effects, we looked to see if concomitant medication could also have contributed.
Search methods for identification of studies
Studies already identified and included in the four Cochrane reviews were considered. The codeine review (Schremmer 2013) was available as a protocol and the authors kindly provided access to their completed but unpublished review.The oxycodone review (Schmidt‐Hansen 2010) was in the process of updating, and again authors provided information about any additional studies. This was supplemented by a brief search of PubMed for any other additional studies of oxycodone as this review was not yet completed, and two additional studies were identified.
Data collection and analysis
Selection of studies
Two review authors (PW, SD) independently screened and assessed papers retrieved from the four reviews. Disagreements were resolved by discussion with all authors.
Data extraction and management
Existing characteristics of included studies tables were imported and any further information on relevant outcomes was added.
Assessment of risk of bias in included studies
We imported the risk of bias assessments for individual studies from the four included reviews and checked that they were correct and conformed to the most recent standards (AUREF 2012). We used the following standard parameters:
A 'Risk of bias' table was completed for each included study, using methods adapted from those described by the Cochrane Pregnancy and Childbirth Group. Two authors independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) with any disagreements resolved by discussion. The following were assessed for each study:
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (any truly random process, e.g. random number table; computer random number generator); unclear risk of bias: the trial may or may not be free of bias. Studies with high risk of bias (any non‐random process, e.g. odd or even date of birth; hospital or clinic record number) were excluded.
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions before assignment assessed whether intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment. The methods were assessed as: low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes); unclear risk of bias (methods not clearly stated). Studies with high risk of bias (open random allocation; unsealed or non‐opaque envelopes, alternation; date of birth) were excluded.
Blinding of outcome assessment (checking for possible detection bias). The methods used to blind study participants and outcome assessors from knowledge of which intervention a participant received were assessed. Studies were considered at low risk of bias if they stated that they were blinded and described the method used to achieve blinding (e.g. identical tablets, matched in appearance and smell); at unknown risk if they state that they were blinded, but do not provide an adequate description of how it was achieved, and at high risk if they were single blind or open‐label studies.
Incomplete adverse event outcome data ‐ patient level. Studies were considered at low risk of bias if all participants who took the study medication were included. Where more than 10% of participants were not included in AE reports then these studies were considered to be high risk. Any thing else was considered to be unclear.
Selective reporting bias for adverse events. Studies were considered at low risk of bias if all adverse events were reported. Where there was clear evidence of partial reporting (e.g. most common or more than a given rate) then these studies were considered to be high risk. Anything else was considered to be unclear.
Size (checking for possible biases confounded by small size). Small studies have been shown to overestimate treatment effects, probably due to methodological weaknesses (Moore 2010, Nüesch 2010). Studies were considered at low risk of bias if they had 200 or more participants, at unclear risk if they had between 50 and 200 participants, and at high risk if they had fewer than 50 participants.
We also assessed studies using the Oxford Quality Score (Jadad 1996).
Measures of treatment effect
Where possible we planned to use dichotomous data (patients experiencing an adverse event) to calculate risk ratio (RR) with 95% confidence intervals (CIs) using a fixed‐effect model unless significant statistical heterogeneity was found. Where that was possible, and where there was statistical significance, we would calculate numbers needed to harm (NNH) as the reciprocal of the absolute risk increase (McQuay 1997).
Dealing with missing data
The completeness of reporting of adverse event data in clinical trials is known to be a significant problem (Derry 2008; Edwards 1999; Ioannidis 2001; Loke 2001). Issues include how adverse events are recorded (diaries versus spontaneous reporting, for example), and whether all adverse events are reported in publications, where often only those with 3%, 5%, or even 10% incidence are recorded. For none of these issues is there a suitable mechanism for dealing with it, nor is it known which if any of the methods used for recording adverse events provides the 'best', or 'truest' result. For these reasons data as reported in the studies was taken as reported, with no method used to deal with the potential for missing data.
One other possible problem is nomenclature. For example, consciousness has a spectrum from fully alert on the one side to unconscious on the other. Words used to describe states of consciousness include sleepiness, drowsiness, or somnolence (Tassi 2001). We combined slightly different reporting nomenclature from different trials, so somnolence, for instance, included both drowsiness and sleepiness.
Assessment of heterogeneity
Assessment of statistical heterogeneity would use the I2 statistic if appropriate, however we did not carry out any meta‐analysis.
Data synthesis
We planned to undertake head to head comparisons of these drugs. If data were available, we planned to use Review Manager 5.2 (RevMan 2012) for statistical meta‐analysis. Where results were statistically significant, we would have calculated the numbers needed to treat for harm (NNTH) for adverse events (Cook 1995).
Due to the absence of direct head to head comparisons we have chosen to calculate the proportion of participants experiencing each of the adverse events of interest with each opioid to allow a simple comparison of rates, and for all opioid drugs combined.
Subgroup analysis and investigation of heterogeneity
The evidence base was expected to be small, so subgroup analyses were conducted only for the individual drugs. Any subgroup analysis required at least two studies with at least 200 participants.
Sensitivity analysis
We did not plan any sensitivity analyses.
Results
Description of studies
Results of the search
The only studies considered were those included in Cochrane reviews of morphine (Wiffen 2013), fentanyl (Hadley 2012) oxycodone (Schmidt‐Hansen 2010 together with the authors of an ongoing update, supplemented by additional searches), and a completed but unpublished review of codeine (Schremmer 2013 ‐ full review in press).
Included studies
We included 77 studies (Figure 1).
1.
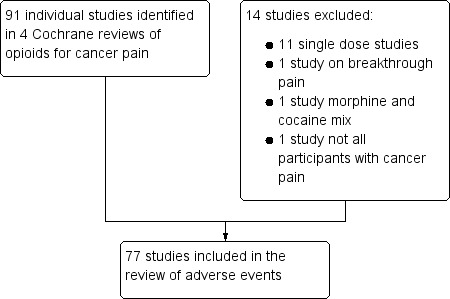
Flow diagram of studies in the review
Morphine in various oral formulations was the sole opioid in 43 studies with 2160 participants (Arkinstall 1989; Babul 1998; Boureau 1992; Broomhead 1997; Cundiff 1989; Currow 2007; Dale 2009; De Conno 1995; Dellemijn 1994; Deschamps 1992; Ferrell 1989; Finn 1993; Flöter 1997; Gillette 1997; Gourlay 1997; Guo‐Zhu 1997; Hagen 2005; Hanks 1987; Hanks 1995; Harris 2003; Homsi 2010; Hoskin 1989; Kerr 2000; Klepstad 2003; Knudsen 1985; Kossman 1983; Melzack 1979; Mignault 1995; Mizuguchi 1990; O'Brien 1997; Panich 1993; Portenoy 1989; Ridgway 2010; Rodriguez 1994; Smith 1991; Thirlwell 1989; Todd 2002; Vainio 1988; Ventafridda 1989; Vielvoye‐Kerkmeer 2002; Walsh 1985; Walsh 1992; Wilkinson 1992).
Morphine in various oral formulations was compared with another opioid in 18 studies with 1382 participants. The other opioids were transdermal fentanyl (Ahmedzai 1997; Mercadante 2008; Oztürk 2008; van Seventer 2003; Wong 1997), oral oxycodone (Bruera 1998; Heiskanen 1997; Kalso 1990; Lauretti 2003; Mercadante 2010; Mucci LoRusso 1998), methadone (Bruera 2004; Ventafridda 1986), hydromorphone (Hanna 2008; Moriarty 1999), tramadol (Leppart 2001; Wilder‐Smith1994), and dextropropoxyphene (Mercadante 1998).
Fentanyl in various transdermal formulations was the sole opioid in four studies with 801 participants (Kongsgaard 1998; Kress 2008; Mystakidou 2005; Pistevou‐Gompaki 2004).
Oxycodone in various oral forms was the sole opioid in six studies with 574 participants (Ahmedzai 2012; Gabrail 2004; Kaplan 1998; Parris 1998; Salzman 1999; Stambaugh 2001).
Oxycodone in various oral formulations was compared with another opioid in two studies with 371 participants. The other opioids were hydromorphone (Hagen 1997) and tapentadol (Imanaka 2013).
Codeine in various oral forms was the sole opioid in two studies with 110 participants (Carlson 1990; Dhaliwal 1995).
Codeine was compared with another opioid in two studies with 221 participants. The other opioids were tramadol (Rico 2000) and tramadol or hydrocodone (Rodriguez 2007).
Participants in the studies were usually equally men and women, with a mean age between 50 and 70 years, and an age range typically between 30 and 87 years. In some of the larger trials with a slightly higher mean age, over half the participants were aged over 65 years (Imanaka 2013).
While all studies were of patients with cancer‐related pain, not all specified the type of cancer. Where reported most studies were of mixed cancer types.
Excluded studies
We excluded 14 studies after reading the full reports. Reasons for exclusion of individual studies are in the Characteristics of excluded studies table. The most common reason for exclusion was that studies only investigated a single dose of opioid.
Risk of bias in included studies
Figure 2 and Figure 3 illustrate the 'Risk of bias' assessments by category for each included study. The Oxford Quality Scores were 1/5 for seven studies, 2/5 for 18 studies, 3/5 for 13 studies, 4/5 for 26 studies, and 5/5 for 13 studies.
2.
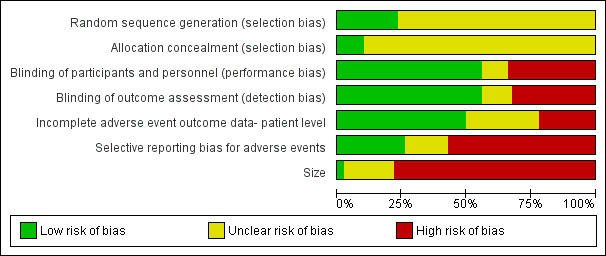
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
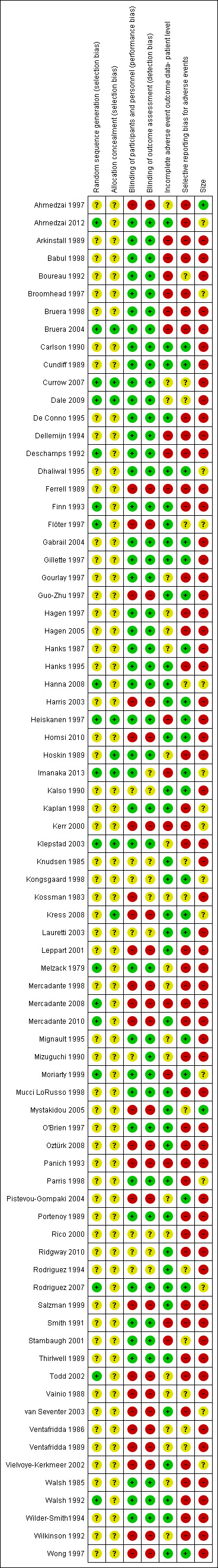
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All studies were randomised. Random sequence generation and allocation concealment were unclear in most studies (Figure 2).
Blinding
A number of the studies were open, and for these risk of bias was high (Figure 2).
Incomplete outcome data
Over half of the studies failed to include all participants randomised and given at least one dose of opioid when reporting adverse events (Figure 2). These were judged to be at high risk of bias.
Selective reporting
A number of studies reported only the most common adverse events, or those occurring at more than a given incidence (5%, for example); these were judged to be at high risk of bias (Figure 2).
Other potential sources of bias
Small size was a major potential source of bias. Studies were typically small: 20 of the 77 studies randomised 100 or more participants, and these involved 60% of the total participant numbers; 45 of the 77 studies involved fewer than 50 participants, with only 25% of total participants. Individual treatment groups included fewer than 50 participants in 60 studies, between 50 and 199 participants in 15 studies, and 200 or more in only two studies (Figure 3).
Effects of interventions
It was not possible to perform any pooled comparative analysis because of the varied nature of the comparisons made in the different studies, We therefore provide a narrative report on the primary outcomes, and a pooled analysis of adverse event incidence rates for secondary outcomes, by individual opioid, and by all four opioids combined.
Primary outcomes
There were few direct mentions of events approximating to our primary outcome of patient consciousness, patient appetite, or thirst. Only one study (Imanaka 2013) reported classification methods used for adverse events.
For morphine, one study reported stupor in 9/98 participants, without defining what was meant by stupor, or what the cause might be. We judged that this was possibly a translation problem (the study originated in Thailand; Panich 1993), and we included this under somnolence in our analysis of secondary outcomes.
For oxycodone, Kalso reported sedation in most participants, but also reported somnolence in 4/19 (Kalso 1990); it is difficult to interpret both of these events. On the other hand Gabrail (Gabrail 2004) mentioned sedation in 13/41 participants on oxycodone, but did not mention somnolence as a separate adverse event; we interpreted this as a different definition of somnolence or drowsiness.
Appetite was reported specifically in only one study (Imanaka 2013), who reported that 24/172 participants had decreased appetite without commenting further. Studies reporting the outcome of anorexia did not provide further details (Lauretti 2003).
Secondary outcomes
Results of surrogate adverse events for individual opioids and for all four opioids combined is in Summary of results A. For a number of adverse events a large number of participants reported on their presence or absence, over 2,000 for nausea, vomiting, constipation, and somnolence, and over 1,000 for dizziness. Other adverse events were reported less frequently; the reasons are unknown, but include low incidence adverse events often not being reported by studies.
There was a general consistency in event rates between different opioids, with constipation, somnolence, and nausea all reported by over 20% of participants.
Summary of results A: pooled adverse event incidence rates for individual opioids and all four combined
| Morphine ‐ oral | Fentanyl ‐ TD | Oxycodone ‐ oral | Codeine ‐ oral | All opioids | ||||||
| Adverse event | Events/total | Percent | Events/total | Percent | Events/total | Percent | Events/total | Percent | Events/total | Percent |
| Nausea | 267/1205 | 22 | 93/664 | 14 | 201/885 | 23 | 61/171 | 36 | 622/2925 | 21 |
| Vomiting | 115/869 | 13 | 43/587 | 7 | 119/866 | 14 | 40/171 | 23 | 317/2493 | 13 |
| Constipation | 355/1189 | 30 | 105/632 | 17 | 196/885 | 22 | 52/171 | 30 | 708/2877 | 25 |
| Diarrhoea | 18/416 | 4 | No data | 29/383 | 8 | 0/99 | 0 | 47/898 | 5 | |
| Dyspepsia | No data | No data | No data | No data | No data | |||||
| Decreased appetite/Anorexia | 38/354 | 11 | 9/20 | 45 | 38/221 | 17 | 2/99 | 2 | 87/694 | 13 |
| Dry mouth | 104/222 | 47 | 3/117 | 3 | 37/469 | 8 | 18/134 | 13 | 162/942 | 17 |
| Dysphagia | No data | No data | No data | No data | No data | |||||
| Dehydration | No data | 2/117 | 2 | No data | No data | 2/117 | 2 | |||
| Somnolence | 290/1205 | 24 | 26/204 | 13 | 172/715 | 24 | 29/134 | 22 | 517/2258 | 23 |
| Delirium | 1/54 | 2 | No data | 6/172 | 3 | No data | 7/226 | 3 | ||
| Dizziness | 89/592 | 15 | 4/117 | 3 | 61/488 | 13 | 21/134 | 16 | 175/1331 | 13 |
| Insomnia | 1/20 | 5 | 5/137 | 4 | 21/435 | 5 | 0/99 | 0 | 27/691 | 4 |
| Asthenia | No data | 2/117 | 2 | 14/208 | 7 | 5/94 | 5 | 21/419 | 5 | |
| Hallucinations | 6/305 | 2 | 2/117 | 2 | 0/60 | 0 | 4/59 | 7 | 12/541 | 2 |
| Mood change | 20/451 | 4 | No data | No data | No data | 20/451 | 4 | |||
The most appropriate surrogate measure of consciousness was probably somnolence, into which we pooled several definitions including drowsiness, sleepiness, and stupor. It was reported by 517/2258 participants (23%); severity was not usually mentioned. Delirium was reported in 7/226 participants.
The most appropriate surrogate measure of patient appetite was probably anorexia. Together with the report of decreased appetite it was reported by 87/898 participants (13%); neither severity nor consequences were mentioned.
There was no obvious appropriate surrogate measure of thirst, but dry mouth occurred in 162/942 participants (17%), with no indication of severity.
Many participants received concomitant medication. Where clearly identified, these are listed in the Characteristics of included studies table. In many cases they included medications that could produce the adverse events of interest. It would have been helpful if these medications were clearly specified in every study.
Discussion
The title of this rapid review was registered on 7 March 2014, the protocol submitted on 11 March and published on 31 March. The full review was submitted on 2 May 2014, revisions after peer review completed by 15 May, and the expected date of publication is early June 2014. The process will have been completed in about 13 weeks. While the review has been rapid it has not compromised on methodological quality. Rapidity was achieved by a combination of using studies identified by previous and ongoing Cochrane reviews, an experienced review team, thoughtful peer reviewers, and by a prepared and proactive editorial base.
Summary of main results
Direct measures of patient consciousness, patient appetite, or thirst were not apparent. The results showed that several adverse events that are likely to impact on the quality of life were common with opioids used to treat cancer pain, with incidence rates of 25% for constipation, 23% for somnolence, 21% for nausea, 17% for dry mouth, and 13% for vomiting, anorexia, and dizziness. Asthenia, diarrhoea, insomnia, mood change, hallucinations and dehydration occurred at incidence rates of 5% and below. For a variety of reasons discussed below, none of these can be attributed unequivocally to the use of opioid.
None of the included studies was undertaken in a ‘dying patient’ population.
Overall completeness and applicability of evidence
The assessment of adverse events is fraught with problems. Firstly, even young, fit people taking no medicines and with no known medical problems report high rates of adverse events over periods as short as three days, with fatigue reported by 40% (Meyer 1996; Reidenberg 1968). High levels of adverse event reports can be found in patients given placebo in clinical trials of statins and in adults not in a clinical trial over a short time period (Rief 2006). These can include what might be regarded as very important events, like chest pain, as well as events like diarrhoea or nausea. In addition, adverse event incidence depends on the assessment method, whether reporting is spontaneous or elicited in any way, with elicited events give much higher overall adverse event rates than those reported spontaneously (Edwards 1999; Olsen 1999; Rief 2009). There is some evidence that expectations from investigators and participants can influence adverse event profiles (Rief 2009). Moreover, for some adverse events patients accommodate quickly when doses of opioids are stabilised, and event rates may be higher when dose titration is occurring. This may be the case in some of these relatively short duration studies, but few of them were carried out in opioid‐naive patients as many were switching participants from one opioid formulation to another.
Added to this is the well‐known problem that trial reports often failed to provide details on how adverse drug reactions were defined or recorded (Loke 2001; Nuovo 2007). In this review, for instance, some adverse events were only reported if they were considered to be drug related, or new, or unexpected. And where people are being treated for cancer, the use of concomitant medications or comorbid conditions or both may confound results.
This background limits the confidence we can have in adverse event reports, except in the broadest sense.
In this review there were major problems with completeness of the evidence. One reason was the tendency for studies to report only the most commonly occurring adverse events, above 5%, for instance. Another tendency was to report adverse events on subsections of the whole population randomised and receiving at least one dose of opioid, participants completing all phases of a cross‐over study, for instance. Together these mean that, particularly for less common adverse events, we have reports on only a proportion of the total population exposed.
Severity of adverse events was not usually reported. Severity is intertwined with definitions and meaning of outcomes, and particularly the interpretation of any adverse event incidence rate. For example, consciousness has a spectrum between fully alert and unconscious, with drowsiness, sleepiness, somnolence, and stupor being points of severity along the spectrum, some of which may have particular clinical and human relevance in different circumstances. There is little literature to help, as most studies of consciousness are concerned with the difference between consciousness and unconsciousness, and with no good definitions of different states of consciousness (Tassi 2001). Much the same might be said about decreased appetite or anorexia.
The applicability of the evidence on adverse events to the population of patients with cancer pain studied is high. These tended, however, to be relatively young, with mean ages in individual studies mostly between 50 and 70 years. The situation is probably quite different in the older, more frail population likely to be representative of people being treated at the end of life, who are often being given drugs other than opioids, and in whom the incidence of adverse drug reactions is known to be high (Avorn 2008). Using lessons learned from cancer for end‐of‐life care is acknowledged to be difficult (Murray 2008). We think it unlikely that the type of cancer will influence the response to analgesia in these studies of moderate to severe pain at baseline.
Quality of the evidence
Most of the studies were small, and others had problems of incomplete and selective reporting of adverse event outcome data, as well as an absence of clear definitions of what some adverse events actually meant.
Potential biases in the review process
We were unaware of any potential biases in the review process other than taking only studies included in four cancer pain Cochrane reviews. This is unlikely to be a major concern, as there are relatively few RCTs for other opioids, but the four included reviews excluded studies not using pain as an outcome measure. It is not impossible that meaningful studies of opioids in cancer reported only adverse event data, and we are unaware of any large body of evidence that may have been overlooked. To the best of our knowledge there is no literature on adverse events of opioids more relevant for end‐of‐life care.
Agreements and disagreements with other studies or reviews
Only one other systematic review has reported on adverse events of opioids, in that case in chronic non‐cancer pain (Moore 2005). Figure 4 compares adverse event incidence rates found in cancer and chronic non‐cancer pain. They are very similar, though it should be noted that only common adverse events were assessed in chronic non‐cancer pain; non appearance of an adverse event in the figure does not mean that it was not present.
4.
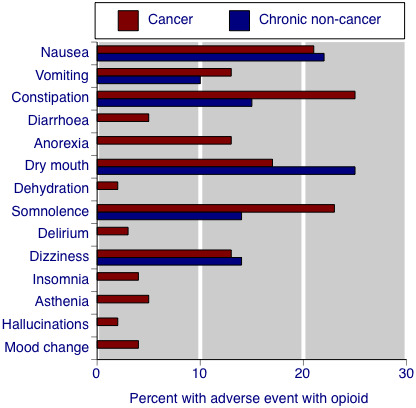
Comparison of adverse event rates in randomised studies of opioids in cancer pain and chronic non‐cancer pain
Authors' conclusions
Implications for practice.
We found no evidence that opioids were associated with patient consciousness, appetite or thirst when used to treat cancer pain. However, somnolence, dry mouth, and anorexia were common adverse events in people with cancer pain treated with morphine, fentanyl, oxycodone, or codeine. Rates were similar to those in chronic non‐cancer pain. Both these populations entered into randomised trials were likely to be considerably younger and much less frail than people treated with opioids at the end of life. It is likely that opioids used for end‐of‐life care will to some degree affect patient consciousness, appetite, and thirst, but it is not possible to quantify the effect or to identify circumstances where problems may be greater or lesser.
Implications for research.
In order to address the issues raised by the Neuberger Review, research into the effect of opioids on levels of consciousness, and effects on appetite and thirst in dying patients should be commissioned. This is no easy task, however, and there are many, possibly major, issues that would need to be overcome in defining what that research may comprise.
There are two immediate implications for research, and we limit comments to these two.
Definitions
Perhaps the most frustrating aspect is the issue of how adverse events are recorded, and the issues of seriousness, severity, and definition. There are a number of systems for recording diagnoses and adverse events, including the International Statistical Classification of Diseases and Related Health Problems (ICD), and MedDRA (the Medical Dictionary for Regulatory Activities Terminology), a controlled vocabulary widely used as a medical coding scheme for adverse events. These are often used, but because of their broad, generic, nature, often fail to pick up important nuances in specific circumstances.
Adverse events often display a spectrum of seriousness. For example, the induction agent propofol exhibits cardiac events that include bradycardia (1 in 9), asystole (1 in 660), and bradycardia‐related death (1 in 100,000) (Tramèr 1997), and a spectrum of gastrointestinal harm exists with NSAIDs, encompassing dyspepsia, endoscopically detected ulcers and erosions, hospital admission for bleeding ulcer, and death from bleeding ulcer (Tramèr 2000).
One of the issues with spectrums of harm is that the most serious events are rare and difficult to capture, and often more common, surrogate, measures are used in their place. All of which is fine as long as the spectrum can be well established, and there are well‐established definitions that can be followed. Even then, establishing the value of a surrogate measure can be difficult despite very considerable evidence (Moore 2009; Moore 2013).
It is likely that there are spectrums of outcome for consciousness (from fully alert, to drowsy, sleepy, somnolent, sedated, and then unconscious). But this all depends on how words are used. For example, in discussing results from one RCT done more than 20 years ago with an author, it was clear that the trial report of sedation actually meant fatigue, or tiredness. For eating and drinking it is likely that similar principles apply.
Therefore one clear implication for research is for:
a set of clear definitions of the various sections of each spectrum that is of clinical interest;
the development of measurement tools or aids;
testing the tools;
investigating whether a spectrum of response can be determined.
Data recording and reporting
Some studies produced good quality adverse event data in tables, but most studies did not do this. There are groups working on adverse event reporting standards, and the CONSORT group has provided useful guidance on adverse event reporting (Ioannidis 2004). A Cochrane Adverse Effects Methods Group also exists.
The problem, though, is that while such groups do excellent work on the generic problems of adverse event recording and reporting, this may still fail to be useful to a specific set of harms in specific circumstances. A key need, therefore, is to develop a set of recommendations on adverse events (and perhaps beneficial events) that are specific to end‐of‐life care, in order that they may be tested and used in clinical practice and clinical trials.
What's new
| Date | Event | Description |
|---|---|---|
| 28 May 2019 | Amended | Contact details updated. |
| 11 October 2017 | Review declared as stable | No new studies likely to change the conclusions are expected. |
History
Protocol first published: Issue 4, 2014 Review first published: Issue 5, 2014
| Date | Event | Description |
|---|---|---|
| 25 July 2017 | Review declared as stable | See Published notes. |
| 13 January 2015 | Amended | Minor corrections. |
| 2 October 2014 | Amended | Minor typo corrected. |
| 16 June 2014 | Amended | Minor change to wording to remove possible ambiguity in 'Implications for practice'. |
Notes
A restricted search in July 2017 did not identify any potentially relevant studies likely to change the conclusions. Therefore, this review has now been stabilised following discussion with the authors and editors. If appropriate, we will update the review if new evidence likely to change the conclusions is published, or if standards change substantially which necessitate major revisions.
Acknowledgements
We wish to thank Dr Bee Wee for useful discussions that led to the initiation of this review, Mia Schmidt‐Hansen and Carmen Schremmer for allowing us access to their ongoing Cochrane reviews, and for several thoughtful peer reviewers of protocol and final review who provided useful additional guidance and insight.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ahmedzai 1997.
| Methods | Design: multicentre, randomised, open label, two‐period cross‐over study. Initial opioid dose calculated using manufacturers recommendations, with dose titration at start of each period to achieve pain control. Assessed at baseline and 8, 16, 23, 31 days, and by daily patient diary Duration: 2 x 15 days, no washout between periods + titration Setting: Palliative care centres, UK |
|
| Participants | Adult cancer patients requiring strong opioid analgesia and receiving stable dose of morphine for at least 48 hours Life expectancy > 1 month N = 202 M 112, F 90 Mean age 62 years (range 18 to 89) |
|
| Interventions |
MIR was used freely to titrate pain at the start of study and at cross‐over Where possible other medication remained unchanged, but other analgesics allowed: e.g. NSAIDs, permitted radiotherapy, nerve blocks |
|
| Outcomes | Sleep, rescue medication, drowsiness: VAS, daily diary Pain and mood: Memorial Pain Assessment Card, twice daily QoL (self‐rated): EORTC (European Organisation for Research and Treatment of Cancer) QLQ‐C30 Performance status (clinician rated): WHO scale Treatment preference Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Presented as numbers of AEs but not clear whether this is events or participants. Denominator unclear. |
| Selective reporting bias for adverse events | High risk | Only commonest events reported |
| Size | Low risk | > 200 participants per treatment arm |
Ahmedzai 2012.
| Methods | Design: randomised double blind, active controlled, double dummy, parallel group study. Pre‐study opioid and laxative stopped prior to randomisation Duration: 4 weeks Setting: Probably community‐ not clearly stated |
|
| Participants | Cancer pain‐ moderate or severe requiring round the clock opioid therapy equivalent to Oxycodone 20‐80mg/day. Participants who had chemotherapy in previous 2 weeks excluded or radiotherapy that could influence bowel function or pain N = 184 M 94, F 90 Mean age 63 years (range 36 ‐ 84) |
|
| Interventions | 1. Oxycodone prolonged release 2. Oxycodone prolonged release with naloxone |
|
| Outcomes | Pain control using BPI‐SF Bowel function Use of rescue medication Use of laxatives QoL Adverse events |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | 'pseudo random number generator in a computer programme' |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | 'treatments were masked in a double dummy fashion' |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | 'treatments were masked in a double dummy fashion' |
| Incomplete adverse event outcome data‐ patient level | Low risk | > 90% of participants included |
| Selective reporting bias for adverse events | High risk | Incomplete breakdown of AEs |
| Size | Unclear risk | 185 participants |
Arkinstall 1989.
| Methods | Design: randomised, double blind (double dummy), two‐period cross‐over study. Prestudy stabilisation period to achieve adequate control of pain, no change in dose for ≥ 3 days, and mean daily rescue medication ≤ 50% of titrated daily dose Duration: 2 x 10 days, no washout, + dose stabilisation phase Setting: Hospital/acute /surgery/community |
|
| Participants | Cancer pain N = 29 Mean age 63 years Mean weight 61.1 kg |
|
| Interventions |
Rescue medication: MIR |
|
| Outcomes | PI: VAS PPI: McGill‐Melzack Pain Questionnaire (6‐point categorical scale) Use of rescue medication Treatment preference Plasma morphine concentrations in last 3 days of both phases Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "by means of random allocation". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "matching placebos were used to maintain blinding" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "matching placebos were used to maintain blinding" |
| Incomplete adverse event outcome data‐ patient level | High risk | AEs reported on fewer than 90% participants |
| Selective reporting bias for adverse events | High risk | Only frequent AEs reported‐mean data only |
| Size | High risk | < 50 participants per treatment arm |
Babul 1998.
| Methods | Design: Randomised, double blind (double dummy), two‐period cross‐over study. Dose stabilisation using morphine; non‐morphine participants transferred to morphine Duration: 2 x 7 days + dose stabilisation phase Setting: not specified |
|
| Participants | Cancer pain N = 27 (22 completed and evaluated) M 13, F 9 Mean age 55 years |
|
| Interventions |
Dose ratio oral:rectal = 1:1 Non‐opioid analgesics continued Rescue medication: MIR |
|
| Outcomes | PI: VAS x 4 daily PPI (6‐point categorised scale: no pain 0, mild pain 1, discomforting pain 2, distressing pain 3, horrible pain 4, excruciating pain 5) Nausea, sedation: 100 mm VAS ‐ spontaneous + investigator‐reported |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Double blind conditions maintained by use of matching placebos" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Double blind conditions maintained by use of matching placebos" |
| Incomplete adverse event outcome data‐ patient level | High risk | AEs reported on fewer than 90% participants |
| Selective reporting bias for adverse events | High risk | 'AEs consistent with use of opioid analgesics in patients with advanced cancer' |
| Size | High risk | < 50 participants per treatment arm |
Boureau 1992.
| Methods | Design: Multicentre, randomised, double blind (double dummy), two‐period cross‐over study Duration: 2 x 7 days, with no washout Setting: not stated |
|
| Participants | Cancer pain. Participants on stable dose morphine for previous 48 h with adequate pain relief. Participants all taking < 400 mg morphine/24 h N = 52 (44 analysed) M 34, F 18 Age 62 years (SD 11) |
|
| Interventions | Previous daily dose of morphine given in 2 doses (12‐hourly)
Morphine dose: 108 mg/day (SD 57; range 40 to 260) |
|
| Outcomes | PI: VAS x 3 daily Verbal rating scale (5‐point) Rescue medication Participant preferences QoL indices (activity mood sleep) by participant and investigator |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "four patient block randomisation method". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double dummy design with placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double dummy design with placebo |
| Incomplete adverse event outcome data‐ patient level | High risk | AEs reported on fewer than 90% participants |
| Selective reporting bias for adverse events | Unclear risk | Unsure if full list of AEs reported |
| Size | High risk | < 50 participants per treatment arm |
Broomhead 1997.
| Methods | Design: multicentre, randomised, double blind (double dummy), parallel group study. Participants titrated with MIR during 3 to 14 day run‐in period to achieve adequate control of pain, no change in dose for 3 consecutive days, and ≤ 2 doses of rescue medication/day Duration of treatment: 7 days ± 1 day + titration phase Setting: outpatients |
|
| Participants | Cancer pain of moderate to severe intensity N = Phase 1: 19, Phase 2 169 received treatment and were randomised (152 completers) Mean age 61 years |
|
| Interventions | Phase 1:
Phase 2 (main study): As phase1 but no placebo Other non‐opioid medication was allowed Rescue medication: MIR for all groups |
|
| Outcomes | Elapsed time to re‐medication and total amount of rescue medication Pain intensity (VAS) daily Verbal PI (four point) Verbal PR (four point) Sleep quality Global assessment over 7 days Adverse events (5‐point) |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double dummy design with placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double dummy design with placebo |
| Incomplete adverse event outcome data‐ patient level | High risk | AEs reported on fewer than 90% participants |
| Selective reporting bias for adverse events | High risk | Not all AEs reported |
| Size | Unclear risk | 50 to 199 participants per treatment arm |
Bruera 1998.
| Methods | Design: Randomised, double blind (double dummy), two‐period cross‐over study. Stable analgesic requirements for ≥ 3 days with rescue medication ≤ 20% daily dose Duration: 2 x 7 days, with no washout Setting: Palliative care programme |
|
| Participants | Cancer pain N = 32 (23 completed and assessed) M 9, F 23 |
|
| Interventions |
Dose adjustment allowed if greater than 3 rescue doses in previous 24 hours Rescue medication: IR Oxycodone or MIR; no other opioids or analgesics allowed |
|
| Outcomes | PI: VAS x 4 daily and 5‐point categorical scale Participant preferences Nausea and sedation scale Adverse event checklist Rescue analgesia |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "blinding was maintained by double dummy technique using matching placebos" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "blinding was maintained by double dummy technique using matching placebos" |
| Incomplete adverse event outcome data‐ patient level | High risk | AEs reported on fewer than 90% participants |
| Selective reporting bias for adverse events | High risk | Reported as no difference between groups |
| Size | High risk | < 50 participants per treatment arm |
Bruera 2004.
| Methods | Design: Randomised, double blind, parallel group study. Dose titrated over first 8 days Duration: 4 weeks Setting: Palliative care groups |
|
| Participants | Advanced cancer and pain requiring the initiation of strong opioids N = 103 M 37, F 66 Median age 60 years (range 26 to 87) |
|
| Interventions |
Dose adjustments allowed Non‐opioid analgesics discontinued Rescue medication: 5 mg methadone or MIR every 4 h as needed |
|
| Outcomes | PI: VAS Sedation, confusion, nausea, constipation: VAS Edmonton staging system for cancer pain: daily assessments for 8 d then weekly assessment Global impression of change |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "the random allocation sequence was generated centrally by computer generated numbers" |
| Allocation concealment (selection bias) | Low risk | "allocation code was kept in a sealed envelope" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "capsules containing the drug were identical" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "capsules containing the drug were identical" |
| Incomplete adverse event outcome data‐ patient level | High risk | AEs reported on fewer than 90% participants |
| Selective reporting bias for adverse events | High risk | Only AEs leading to withdrawal and mean data for some AEs was reported |
| Size | High risk | < 50 participants in each treatment arm |
Carlson 1990.
| Methods | Design: randomised, double blind, parallel group, first‐dose 6‐hour observation period in which patients were randomised to ketorolac, paracetamol plus codeine, or placebo. Thereafter participants receiving placebo were reassigned to one of the two active treatments and observed for 7 days with drugs taken x 4 daily Setting: not stated |
|
| Participants | Moderate to severe cancer pain; histologically confirmed diagnosis of cancer (most common types: genitourinary, lung, breast, gastrointestinal) N = 75 M 43, F 32 Mean age 62 years |
|
| Interventions | First‐dose 6‐hour observation period:
|
|
| Outcomes | PI: four‐point scale (0‐3)
PR:five‐point scale (0‐4) Time to remedication Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not clearly stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "identical‐appearing capsules" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "identical‐appearing capsules" |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | Low risk | All AEs reported |
| Size | High risk | < 50 participants per treatment arm |
Cundiff 1989.
| Methods | Design: randomised, double blind (double dummy), two‐period cross‐over. Morphine titrated upwards until not more than 20% total daily morphine given as rescue over a 2‐day period Duration: 4 ‐ 7 days (time to reach steady state). Cross‐over to start at ⅓ pre‐study equivalent then titrate up Setting: in‐ and outpatients |
|
| Participants | Cancer pain N = 23 (14 evaluable) M 9, F 5 Mean age 45 years (range 31 to 72) |
|
| Interventions |
Rescue medication: 15 mg MIR tablets |
|
| Outcomes | Dose and frequency of rescue medication Nurse assessed PI Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "random assignment". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double dummy technique.... placebo physically indistinguishable from the alternative therapy" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "double dummy technique.... placebo physically indistinguishable from the alternative therapy" |
| Incomplete adverse event outcome data‐ patient level | Low risk | All participants reported |
| Selective reporting bias for adverse events | Low risk | All AEs reported |
| Size | High risk | < 50 participants per treatment arm |
Currow 2007.
| Methods | Design: Randomised, double blind, two‐period cross‐over study Duration: 2 x 7 days + 1 day cross‐over on day 8 Setting: community and hospital |
|
| Participants | Cancer pain N = 42 M 28, F 14 Mean age 64 years (36 ‐ 82) |
|
| Interventions |
Co‐analgesics allowed at stable doses |
|
| Outcomes | PI: VAS, every 4 h while awake PR: categorical scale, daily Adverse events Sleep, nausea and vomiting, constipation, confusion, somnolence: categorical scale |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomisation was allocated from a central computer generated random number sequence" |
| Allocation concealment (selection bias) | Low risk | "the process was blinded at all times to participants and treating clinicians" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "identical placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "identical placebo" |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Data not reported |
| Selective reporting bias for adverse events | Unclear risk | Only mean data with no denominator |
| Size | High risk | < 50 participants per treatment arm |
Dale 2009.
| Methods | Design: Randomised, double blind, two‐period cross‐over study. After titration of dose, participants randomised to receive either a single dose of MIR at bedtime followed by another dose 4 h later, or a double dose of MIR with a placebo dose 4 h later Duration: 2 x 1 night on each treatment Setting: hospital inpatients |
|
| Participants | Cancer pain N = 22 (19 completed) M 11, F 8 Mean age 57 years (45 ‐ 74) |
|
| Interventions |
|
|
| Outcomes | PI: 11‐point NRS Participant preference BPI, Edmonton symptom assessment scale |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "concealed procedure performed by hospital pharmacist using restricted randomisation table" |
| Allocation concealment (selection bias) | Low risk | "concealed procedure performed by hospital pharmacist using restricted randomisation table" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "placebo tablets identical in appearance and taste" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "placebo tablets identical in appearance and taste" |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Adverse events reported with patient numbers but not clear if everything was reported |
| Selective reporting bias for adverse events | Unclear risk | Only mean data with no denominator |
| Size | High risk | < 50 participants per treatment arm |
De Conno 1995.
| Methods | Design: randomised, double blind (double dummy), two‐period cross‐over study. Assessments at 10, 20, 30, 40, 60, 90, 120, 180 and 240 mins daily Duration: 2 x 2 days Setting: outpatients |
|
| Participants | Advanced or metastatic cancer with PI > 30/100 mm at baseline, opioid‐naive
N = 34 M 23, F 11 Mean age 59 (SD 8.8; range 38 to 70) |
|
| Interventions |
Single dose administered on each of two days then crossover to other treatment. Use of NSAIDs allowed for first day |
|
| Outcomes | PI: VAS Nausea and sedation: VAS Number of vomiting episodes Time to pain relief |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 0. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly allocated ... according to a predetermined allocation sequence". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double blind double dummy technique" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "double blind double dummy technique" |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | High risk | Only nausea, vomiting & sedation reported |
| Size | High risk | < 50 participants per treatment arm |
Dellemijn 1994.
| Methods | Design: randomised, double blind (double dummy), two‐period cross‐over study Duration 2 x 1 week, 6 h washout period Setting: not stated |
|
| Participants | Malignant nerve pain due to cancer (severe) N = 20 (16 evaluable) M 10, F 10 Age 42 ‐ 81 years |
|
| Interventions |
Rescue medication: paracetamol and domperidone |
|
| Outcomes | PI: 101‐point numerical rating scale after 7 days PR: 6‐point categorical after 7 days Participant preference Rescue medication Adverse events: 4‐point scale |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | 'randomised'. Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | 'double blind, dummy technique' |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | 'double blind, dummy technique' |
| Incomplete adverse event outcome data‐ patient level | High risk | AEs reported on fewer than 90% participants |
| Selective reporting bias for adverse events | High risk | mean data only |
| Size | High risk | < 50 participants per treatment arm |
Deschamps 1992.
| Methods | Design: randomised, double blind (double dummy), two‐period cross‐over study with titration phase Duration 2 x 7 days, no washout Setting: outpatients |
|
| Participants | Metastatic cancer with pain requiring opioids N = 20 Mean age 57 years (range 40 to 72) |
|
| Interventions |
No dose adjustment allowed after titration MIR (solution) for breakthrough; no other opioids/analgesics allowed |
|
| Outcomes | PI: 100 mm VAS Adverse events: verbal (6‐point) severity scale Participant preference |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomised by Pharmaceutical company...using randomisation table" |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "titration and trial phases conducted under double blind conditions with double dummy technique" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "titration and trial phases conducted under double blind conditions with double dummy technique" |
| Incomplete adverse event outcome data‐ patient level | High risk | AEs reported on fewer than 90% participants |
| Selective reporting bias for adverse events | High risk | Common AEs only reported as mean scores |
| Size | High risk | < 50 participants per treatment arm |
Dhaliwal 1995.
| Methods | Design: Randomised, double blind, two‐period cross‐over study Duration: 2 x 7 days |
|
| Participants | Chronic cancer pain N = 35 participants (30 completers: 13 women, 17 men) Mean age 64 years |
|
| Interventions |
Rescue medication: paracetamol 300 mg plus codeine 30 mg once or twice every 4 hours |
|
| Outcomes | PI: 100 mm VAS and five‐point NRS (0‐4) Doses of rescue medication per day Pain disability index Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not clearly stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "matching placebos" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "matching placebos" |
| Incomplete adverse event outcome data‐ patient level | Low risk | > 90% of participants included |
| Selective reporting bias for adverse events | Low risk | All AEs included |
| Size | Unclear risk | < 50 participants per treatment arm |
Ferrell 1989.
| Methods | Design: Randomised, open label, parallel group study. Participants remained on current short acting analgesics (oxycodone, hydromorphone, codeine or short‐acting morphine) or switched to MS Contin. Historical control of patients receiving MS Contin ≥ 2 weeks, who remained on the treatment Duration: 6 weeks Setting: Oncology units in 2 US hospitals |
|
| Participants | Chronic cancer pain, receiving short‐acting oxycodone, hydromorphone, codeine, morphine N = 83 M 36, F 47 Mean age 60 years (range 21 ‐ 87) |
|
| Interventions |
Doses not stated |
|
| Outcomes | Pain Experience Measure Tool PPI: McGill City of Hope QoL |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 0. Total = 1/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | High risk | QoL study, AEs not reported |
| Selective reporting bias for adverse events | High risk | QoL study, AEs not reported |
| Size | High risk | < 50 participants per treatment arm |
Finn 1993.
| Methods | Design: Randomised, double blind (double dummy), two‐period cross‐over study Duration of study: 6 days (day 1: usual MIR; days 2 and 3 either Mm/r or MIR (with matched placebo); days 4 and 5 cross‐over) Setting: outpatients |
|
| Participants | Cancer pain requiring > 60 mg MIR/daily N = 37 (34 completed) Mean age 59 years |
|
| Interventions |
Dose adjustment allowed Non‐opioid medications continued Rescue medication: paracetamol, MIR or sub‐cut/IM morphine |
|
| Outcomes | PI: VAS x 3 daily, and 4‐point categorical (Karnofsky) Adverse events Use of rescue medication Participant preference |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomisation by using randomisation schedule provided to the responsible pharmacist" |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "blinded drug supplies packaged daily by the responsible pharmacist" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "blinded drug supplies packaged daily by the responsible pharmacist" |
| Incomplete adverse event outcome data‐ patient level | Low risk | > 90% of participants included |
| Selective reporting bias for adverse events | High risk | Only selected AEs reported |
| Size | High risk | < 50 participants per treatment group |
Flöter 1997.
| Methods | Design: randomised, open label, parallel group study. Initial 7 ‐ 14 day titration with Kapanol or Mm/r Duration: 14 days + titration phase Setting: in‐ and outpatient |
|
| Participants | Mixed pain: 27/91 Kapenol and 26/74 MST had cancer pain N = 165 M 98, F 67 Mean age 55 years Weight 69 kg |
|
| Interventions |
Paracetamol, NSAIDs, antidepressants allowed; advised not to alter. Other opioids not permitted Rescue medication: MIR 10 mg |
|
| Outcomes | PI: VAS (physician assessment of pain control)
Quality of sleep
Rescue medication
Well being etc (patient diary) Adverse events |
|
| Notes | Oxford Quality Score: R = 2, DB = 0, W = 1. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomisation performed using a random number generator" |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open |
| Incomplete adverse event outcome data‐ patient level | Low risk | > 90% of participants included |
| Selective reporting bias for adverse events | Unclear risk | No details of specific AEs |
| Size | Unclear risk | 50 ‐ 200 participants per treatment arm |
Gabrail 2004.
| Methods | Design: Randomised, double‐blind, two‐period cross‐over study. Prestudy open label stabilisation phase to establish fixed dosage that provided adequate analgesia for at least 2 consecutive days, required no more than 2 doses of rescue medication/day, and produced tolerable AEs ‐ used to calculate equianalgesic dose for study Duration: 2 x 7 ‐ 10 days + 3 ‐ 10‐day stabilisation phase Setting: outpatient |
|
| Participants | Moderate to severe pain secondary to cancer, requiring long‐term outpatient treatment with an opioid analgesic N = 44 (37 analysed for efficacy) M 21, F 23 Mean age 59 years (range 26 ‐ 81) |
|
| Interventions |
Dose adjustment allowed in first 3 days only Rescue medication: 15 mg oral morphine sulphate (IR) every 4‐6 hours as needed Other permitted medication not reported |
|
| Outcomes | PI: 11‐point NRS and BPI QoL: BPI (pain interference with 7 domains of quality of life: general activity, mood, walking ability, normal work, relations with other people, sleep, and enjoyment of life) Global assessment of treatment: participant and physician Adverse events Karnovsky performance status |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study medication was "over encapsulated" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study medication was "over encapsulated" |
| Incomplete adverse event outcome data‐ patient level | Low risk | > 90% of participants included |
| Selective reporting bias for adverse events | Low risk | All treatment related AEs reported |
| Size | High risk | < 50 participants per treatment arm |
Gillette 1997.
| Methods | Design: randomised, double blind (double dummy), two‐period cross‐over study. Prestudy dose stabilisation (≤ 5 days) using MIR syrup 5 mg/ml to achieve adequate pain control and determine dosage used on study Duration: 2 x 6 days, no washout Setting: hospital |
|
| Participants | Advanced cancer and severe pain N = 27 Mean age 61.3 years, weight 60 kg |
|
| Interventions |
No dose adjustment allowed Rescue medication: drugs other than morphine (not required) |
|
| Outcomes | PI: VAS x 4 daily Adverse events: 5‐point verbal scale Sleep quality on days 6 and 12 Morphine concentrations on days 6 and 12 |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double blind, dummy technique" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "double blind, dummy technique" |
| Incomplete adverse event outcome data‐ patient level | Low risk | > 90% of participants included |
| Selective reporting bias for adverse events | Low risk | All AEs reported |
| Size | High risk | < 50 participants per treatment arm |
Gourlay 1997.
| Methods | Design: randomised, double blind (double dummy), two‐period cross‐over study. Prestudy dose stabilisation phase using MIR to achieve adequate pain relief and constant dose ≥ 2 consecutive days with ≤ 2 doses of rescue medication Duration: 2 x 7 days (no washout) Setting: not stated |
|
| Participants | Cancer patients requiring at least 40 mg morphine/24 hours
N = 29 M 15, F 9 (completers) |
|
| Interventions |
Rescue medication: dextromoramide |
|
| Outcomes | PI: VAS and categorical scale PR: categorical scale Sleep Participant global assessment Adverse events Plasma morphine concentrations |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "matching placebo opaque capsules" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "matching placebo opaque capsules" |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Numbers not reported |
| Selective reporting bias for adverse events | High risk | Selective reporting of mean data |
| Size | High risk | < 50 participants per treatment arm |
Guo‐Zhu 1997.
| Methods | Design: randomised, open label, parallel group study comparing 2 doses of Mm/r capsules with 2 doses of Mm/r tablets (4 groups in total). No dose titration; lower dose given to those who had not used, or rarely used, opiates previously Duration of treatment: 7 days Setting: hospital |
|
| Participants | Terminal cancer and moderate to severe pain
N = 120 M 72, F 48 Mean age approximately 55 years |
|
| Interventions |
Rescue medication not mentioned Use of antidepressants, non opioid drugs, acupuncture and TCM prohibited |
|
| Outcomes | PI: 10‐point NRS PR: 5‐point categorical scale Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 0. Total = 1/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly divided". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | Low risk | > 90% of participants included |
| Selective reporting bias for adverse events | Low risk | All AEs appear to be included |
| Size | High risk | < 50 participants per treatment arm |
Hagen 1997.
| Methods | Design: Randomised, double‐blind, two‐phase cross‐over study Duration: 2 x 7 days, no washout Setting: not stated |
|
| Participants | Chronic cancer pain and stable opioid analgesic requirements (≤ 2 rescue doses of opioid analgesic per 24‐hour period, calculated over ≥ 3 days) N = 44 (31 completed) M 13, F 18 Mean age 56 years (SD 3) |
|
| Interventions |
Dose changes were permitted Rescue medication: Oxycodone IR at approximately 10% of the daily scheduled dose No other opioids were permitted. Stable non opioid analgesics (e.g. corticosteroids, antidepressants, anticonvulsants, bisphosphonates and psychostimulants) continued at the same dose level throughout the study |
|
| Outcomes | PI: VAS x 4 daily, and 5‐point categorical scale Nausea and sedation: VAS, x 4 daily Adverse events at end of each period Treatment preference: participant and investigator |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 0. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blind crossover using matching placebos |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double blind double dummy crossover using matching placebos |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | No denominator given |
| Selective reporting bias for adverse events | High risk | Limited reporting of AEs |
| Size | High risk | < 50 participants per treatment arm |
Hagen 2005.
| Methods | Design: randomised, double blind (double dummy), multicentre, two‐period cross‐over study. Prestudy dose titration if required Duration: 2 x 1 week Setting: not stated |
|
| Participants | Chronic cancer pain with stable analgesic requirements N = 29 M 12, F 13 (completers) Age 53 years (± 10) |
|
| Interventions |
No dose adjustments permitted, but MIR allowed for breakthrough pain No other opioids allowed, but NSAIDs, antidepressants, anticonvulsants etc allowed to continue at pre‐trial doses |
|
| Outcomes | PI: VAS and categorical (least, worse and average pain, 12‐hourly) Nausea and sedation: VAS Participant preference Blood levels of morphine |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "blinding maintained using the double placebo technique" (double dummy) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "blinding maintained using the double placebo technique" (double dummy) |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Not clear if all included‐no denominator |
| Selective reporting bias for adverse events | High risk | Only most common reported |
| Size | High risk | < 50 participants per treatment arm |
Hanks 1987.
| Methods | Design: randomised, double blind (double dummy), two‐period cross‐over study Duration: 2 x 2 days Setting: continuing care unit |
|
| Participants | Cancer pain N = 27 (18 completed) M 7, F 11 Mean age M 72 years (range 59 to78), F 68 years (53 to 82) |
|
| Interventions |
|
|
| Outcomes | PI: VAS and 5‐point categorical scale Alertness, nausea, mood, sleep assessment and appetite: VAS Global rating Participant preference |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double dummy technique" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "double dummy technique" |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Denominator not stated |
| Selective reporting bias for adverse events | Low risk | ' no specific adverse events were encountered apart from drowsiness in one patient' |
| Size | High risk | < 50 participants per treatment arm |
Hanks 1995.
| Methods | Design: randomised, double blind (double dummy), two phase cross‐over study. Assessments x 4 on last day of each treatment phase Duration: 2 x 3 days Setting: |
|
| Participants | Advanced malignant disease with pain requiring at least 400 mg morphine/day N = 25 (19 completed) M 11, F 14 Mean age 56 years (range 35 ‐ 69) |
|
| Interventions |
Exact dose made up with standard CR morphine tablets (30 ‐ 100 mg), but dose remained constant throughout study Rescue medication: morphine elixir ≤ 1/6 total daily dose |
|
| Outcomes | PI: VAS Symptom score: categorical 4‐point. Scores taken x 4 on days 3 and 6 Use of rescue medication Morphine plasma concentrations in 4 participants |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "patients were randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double blind double dummy crossover ...." "identical tablets" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "double blind double dummy crossover ...." "identical tablets" |
| Incomplete adverse event outcome data‐ patient level | Low risk | > 90% of participants included |
| Selective reporting bias for adverse events | High risk | Selective reporting of AEs |
| Size | High risk | < 50 participants per treatment arm |
Hanna 2008.
| Methods | Design: randomised, double blind (double dummy), parallel group study. Dose titration with hydromorphone IR or MIR from days 2 to 9 to achieve adequate pain control for ≥ 2 consecutive days and ≤ 3 doses of rescue medication, then stable on hydromorphone‐OROS or Mm/r at same dose for days 10 to 15 Duration: up to 24 days Setting: Inpatients and outpatients |
|
| Participants | Chronic cancer pain requiring 60 mg to 540 mg oral morphine/day N = 200 (163 completed IR phase, 133 CR phase) M 97, F 103 Mean age 60 years |
|
| Interventions |
Dose adjustment allowed Rescue medication: hydromorphone IR or MIR at ≤ 1/6 total daily dose Concomitant chemotherapy or radiotherapy and non‐opioid analgesics allowed, but not other opioids |
|
| Outcomes | Worst pain in previous 24 hours PI: BPI, VAS PR: VAS Participant judgement of PR (v good, excellent) |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomised 1:1 with central computer generated randomisation list" |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "matching placebo capsules and tablets were used" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "matching placebo capsules and tablets were used" |
| Incomplete adverse event outcome data‐ patient level | Low risk | > 90% of participants included |
| Selective reporting bias for adverse events | Unclear risk | Only reported AEs in at least 5% of patients. Serious AEs reported |
| Size | Unclear risk | 50 ‐ 199 participants per treatment arm |
Harris 2003.
| Methods | Design: randomised, open label, parallel study. Assessed every hour for 12 hours, then every day for 2 days, then weekly Duration: unclear Setting: outpatients |
|
| Participants | Patients with end stage cancer and severe pain, some opioid‐naive N = 62 M 47, F 15 Most participants aged 30 ‐ 80 years |
|
| Interventions |
Equivalent dose of rescue allowed at intervals ≥ 1 hour All participants also received paracetamol or diclofenac, and metoclopramide. No other analgesics |
|
| Outcomes | PR: 3‐point scale (1 = total, 2 = satisfactory, 3 = unsatisfactory) Use of rescue medication Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised was achieved by sampling with replacement" Not clear what this means. |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | Low risk | > 90% of participants included |
| Selective reporting bias for adverse events | Low risk | All AEs reported |
| Size | High risk | < 50 participants per treatment arm |
Heiskanen 1997.
| Methods | Design: Randomised, double‐blind (double dummy), two‐phase cross‐over study. Prestudy open label titration phase (maximum 21 days) to achieve effective pain relief with acceptable adverse effects for ≥ 48 hours Duration: 2 x 3 ‐ 6 days + titration phase up to 21 days Setting: Not stated |
|
| Participants | Chronic cancer pain requiring opioid analgesics N = 45 (27 analysed) M 16, F 11 Mean age 60 years (range 39 ‐ 76) |
|
| Interventions |
Dose adjustment allowed Rescue medication: Oxycodone IR or MIR in a dose of approximately 1/6 to 1/8 of the daily dose of controlled‐release formulation Stable NSAIDs continued at the same dose |
|
| Outcomes | PI: 4‐point VRS (none, slight, moderate, severe) x 4 daily Acceptability of therapy: 5‐point VRS (very poor, poor, fair, good, excellent) x 2 daily Adverse events Modified Specific Drug Effect Questionnaire: participants and investigator, at end of study |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer generated randomisation for the open‐label titration phase and again for the double‐blind phase was performed by the Purdue Frederick Company and a list of randomisation codes was kept by the hospital pharmacist" |
| Allocation concealment (selection bias) | Low risk | "list of randomisation codes was kept by the hospital pharmacy". Probably adequate |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double dummy method, "matched placebo tablets" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double dummy method, "matched placebo tablets" |
| Incomplete adverse event outcome data‐ patient level | High risk | AEs reported on fewer than 90% participants |
| Selective reporting bias for adverse events | Low risk | Appears to be a complete list |
| Size | High risk | < 50 participants per treatment arm |
Homsi 2010.
| Methods | Design: randomised, open label, parallel group study comparing two brands of Mm/r. Dose stabilised before randomisation Duration: 5 days Setting: inpatients and outpatients |
|
| Participants | Chronic cancer pain requiring ≥ 30 mg oral morphine/day N = 37 (32 evaluated) M 18, F 14 Median age 64 years (27 ‐ 79) |
|
| Interventions |
Rescue medication: MIR Adjuvant analgesics permitted if morphine dose stable |
|
| Outcomes | PR: categorical 4‐point scale Rescue medication: dose and frequency Participant preference (how is this possible? not a cross‐over) Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "patients were randomised based on a number list" |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open label study |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | Low risk | All AEs reported |
| Size | High risk | < 50 participants per treatment arm |
Hoskin 1989.
| Methods | Design: randomised, double blind, single dose study to test effect of loading dose of MIR together with first dose of Mm/r Duration: 12 hours Setting: Inpatients |
|
| Participants | Advanced cancer with pain requiring regular oral morphine ≤ 800 mg/day N = 19 Age 51 ‐ 84 years |
|
| Interventions |
|
|
| Outcomes | Plasma morphine levels PI: VAS and 4‐point categorical scale PR: VAS Adverse events: categorical + nurse assessment |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "prospectively randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Low risk | "randomisation code was kept in the hospital pharmacy" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "preparations were prepared so as to have an identical taste and physical appearance" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "preparations were prepared so as to have an identical taste and physical appearance" |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | >90% participants included |
| Selective reporting bias for adverse events | High risk | Mean scores only |
| Size | High risk | < 50 participants per treatment arm |
Imanaka 2013.
| Methods | Design: randomised, double blind, parallel‐group comparison of oral tapentadol and oral oxycodone. One week titration period to establish efficacy, and then those with PI ≤3 /10 carried on into 4‐week maintenance phase with stable dose. Those not meeting this criterion continued into maintenance phase with dose adjustment Adverse events coded using the Medical Dictionary for Regulatory Activities (MedDRA), version 15.0 |
|
| Participants | Japanese and Korean patients with moderate or severe cancer‐related pain (PI ≥ 4/10) N = 340 (randomised and received study drug), 236 completed M 190, F 150 Mean age 66 years, with 54% over 65 years |
|
| Interventions |
Rescue medication: MIR Non‐study opioids, neuroleptics, SNRIs, and some antidepressants not permitted. SSRIs, TCAs, anti‐anxiety agents hypnotics, and anticonvulsants could continue unchanged if stable before start of study |
|
| Outcomes | PI: 11‐point NRS, and numbers with 30% and 50% reduction in PI during last 3 days of drug administration PGIC: 7‐point scale AE monitored using MedDRA v 15, and reasons for discontinuation also monitored |
|
| Notes | Oxford Quality Score: R = 2, DB = 1, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer generated" |
| Allocation concealment (selection bias) | Low risk | "Interactive Voice Response System assigned unique treatment code" "blind not broken until all patients completed study" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Interactive Voice Response System" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Method for double blinding not given |
| Incomplete adverse event outcome data‐ patient level | High risk | Only AE with ≥5% incidence reported |
| Selective reporting bias for adverse events | Low risk | All patients randomised used for AE reporting |
| Size | Unclear risk | <200 participants per treatment roup |
Kalso 1990.
| Methods | Design: randomised, double blind, two‐phase cross‐over study. PCA titration with allocated drug until pain‐free. After 48 hours, conversion to oral every 4 hours. Dose adjustment allowed. After 96 hours, cross‐over with PCA titration followed by oral Duration: 2 x 2 days with pre‐ and post‐phase, 8 days in total Setting: not stated |
|
| Participants | Metastasized cancer and severe pain, requiring a change from weaker narcotic analgesic agents N = 20 M 11, F 9 Age 20 ‐ 75 years |
|
| Interventions |
Any pre‐existing treatment with NSAIDs was continued, but opioids stopped |
|
| Outcomes | PI: VAS (0 ‐ 10) every 4 hours from 8am to 8 pm Adverse events (moderate = 1, severe = 2) on the second day of each study period Quality of sleep Participant preference |
|
| Notes | Oxford Quality Score: R = 1, DB = 1, W = 0. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "in a randomised double blind crossover study". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "in a randomised double blind crossover study" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "in a randomised double blind crossover study" |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | Low risk | All AEs reported |
| Size | High risk | < 50 participants per treatment arm |
Kaplan 1998.
| Methods | Design: randomised, double‐blind, parallel‐group study. Original protocol did not permit titration or use of rescue medication, but later amended to include open label titration using MIR to determine adequate dose before randomisation and MIR for breakthrough pain Duration: 6 days + titration phase where appropriate Setting: in‐ and outpatients |
|
| Participants | Cancer pain requiring strong single‐entity opioid or ≥ 10 tablets/day of a fixed‐dose opioid/non opioid analgesic. Stable opioid dose N = 164 (156 in efficacy analysis, 160 in safety analysis) M 93, F 67 Mean age 59 years (SD 1) |
|
| Interventions |
Rescue medication: oxycodone IR 5 mg ≤ 1/6 total daily dose (after protocol amendment) Prestudy opioid analgesics stopped ≤ 4 hours before start of study |
|
| Outcomes | PI: 4‐point VRS (0 = none, 1 = slight, 2 = moderate, 3 = severe) x 4 daily Acceptability of treatment: 5‐point VRS (1 = very poor, 2 = poor, 3 = fair, 4 = good, 5 = excellent) x 2 daily Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "all doses were encapsulated in green size #00, lactose‐filled capsules" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "all doses were encapsulated in green size #00, lactose‐filled capsules" |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | High risk | Selective reporting and clinician assessed |
| Size | Unclear risk | <200 participants per treatment arm |
Kerr 2000.
| Methods | Design: multicentre, randomised, open label, two‐phase cross‐over study. Dose stabilisation for 3 ‐ 14 days using MIR, once stable then randomised to different formulations of Mm/r Duration: 2 x 10 days (± 1 d) No washout Setting: not stated |
|
| Participants | Chronic moderate or severe cancer pain requiring opioid analgesics N = 134 (114 analysed for efficacy) M 66, F 48 Age 61 years (range 36 ‐ 84) |
|
| Interventions |
No dose adjustment allowed Rescue medication: MIR, typically 1/8 ‐ 1/6 total daily dose, every 2 ‐ 4 hours Antiemetics given as needed |
|
| Outcomes | PI: VAS, average pain, least pain and worst pain in 24 hours Interference with daily activities Participant preference Average daily dose of MIR Investigator global assessment QoL |
|
| Notes | Oxford Quality Score: R = 2, DB = 0, W = 1. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "balanced randomisation". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "open label" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "open label" |
| Incomplete adverse event outcome data‐ patient level | High risk | Gives numbers of withdrawals due to AEs and numbers of AEs |
| Selective reporting bias for adverse events | High risk | Selective reporting |
| Size | Unclear risk | <200 participants per treatment arm |
Klepstad 2003.
| Methods | Design: randomised, double blind (double dummy), parallel group study. Initial dose 60 mg morphine per day, then titrated to pain relief; study stopped 2 days after achieving stable analgesic dose (≤3 on 7‐point pain VRS and ≤ 2 doses of rescue medication) Setting: hospital |
|
| Participants | Malignant disease with pain despite treatment with weak opioids for mild to moderate pain N = 40 (36 started the titration phase) Age 57 to 71 |
|
| Interventions |
No other opioids allowed, but NSAIDs continued Rescue medication: ketobemidone |
|
| Outcomes | PI: VAS Participant satisfaction: 5‐point VRS Use of rescue medication Adverse events: nausea, loss of sleep, tiredness, loss of appetite, constipation, vertigo: 4‐point categorical scale QoL: EORTC QLQ‐C30 |
|
| Notes | NB Data in section 3.4 of paper are incorrect (typo); Table 4 correct ‐ confirmed with author Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "hospital pharmacy performed a computerised randomisation" |
| Allocation concealment (selection bias) | Low risk | "none of the pharmacists arranging the study drugs were involved in other parts of the study" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double blind, double dummy" "placebo tablets identical in appearance and taste" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "double blind, double dummy" "placebo tablets identical in appearance and taste" |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Cannot tell what the denominator is |
| Selective reporting bias for adverse events | High risk | mean intensity of 6 AEs reported |
| Size | High risk | < 50 participants per treatment arm |
Knudsen 1985.
| Methods | Design: randomised, double blind, two‐phase cross‐over study Duration: 2 x 7 days Setting: home and hospital |
|
| Participants | Chronic pain due to advanced cancer N = 18 Age 38 ‐ 66 years |
|
| Interventions |
Dose of morphine was the same as was used in 24 hours before study No details of rescue medication or concomitant medication |
|
| Outcomes | PI: VAS Sedation: VAS |
|
| Notes | Oxford Quality Score: R = 1, DB = 1, W = 0. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "consecutively randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double blind" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double blind" |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | Unclear risk | Cannot tell id all AEs were reported |
| Size | High risk | < 50 participants per treatment arm |
Kongsgaard 1998.
| Methods | Design: multicentre, enriched enrolment, randomised withdrawal study Duration: 7 day stabilisation phase, 15 day open label titration phase, and 9 day double blind, placebo controlled, parallel group phase Assessment by daily patient diary and clinical visits at trial entry, beginning and end of double blind period, and 3 month intervals for follow‐up Setting: hospital‐based |
|
| Participants | Adult cancer patients with cancer pain recurring after potentially curative therapy, not currently amenable to curative therapy. Requiring equivalent of 60 to 300 mg oral morphine daily, with acceptable toxicity and pain relief. Karnofsky performance > 50 Titration phase: N = 138 (131 enrolled after stabilisation, 7 directly) M 85, F 53 Mean age 59 years (range 24 to 83) |
|
| Interventions | Stabilisation phase: oral morphine (≥ 60 mg to ≤ 300 mg daily) titrated to provide adequate pain control with acceptable adverse effects 15 day dose‐titration period: fixed conversion table used to convert morphine to fentanyl and titration to maintain adequate pain control with acceptable adverse effects. New fentanyl patch applied every 72 hours 9 day double blind period: fentanyl patch or placebo at same dose as at end of titration period (median dose 50 μg/h) Medication for concurrent illness continued Rescue medication (rapid release morphine) available |
|
| Outcomes | Withdrawals due to inadequate analgesia PI: VAS x 2 daily Rescue medication, daily Well‐being: VAS x 2 daily Investigator assessment of pain intensity using 7‐point scale (no pain‐intolerable pain) Investigator global assessment of trial medication (excellent, good, moderate, poor) Adverse events |
|
| Notes | Oxford Quality Score: R1, DB1, W1. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Method not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Method not described |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | Low risk | All AEs reported |
| Size | Unclear risk | 50 ‐ 199 participants per treatment group (double blind phase) |
Kossman 1983.
| Methods | Design: randomised, parallel group study Duration: 7 days Setting: probably inpatient |
|
| Participants | Cancer pain N = 20 No further details |
|
| Interventions |
No details of dose, rescue medication, or concomitant medication |
|
| Outcomes | PI: VAS Pain duration Quality of sleep: VAS |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 0. Total = 1/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Data not reported |
| Selective reporting bias for adverse events | Unclear risk | Data not reported |
| Size | High risk | < 50 participants per treatment arm |
Kress 2008.
| Methods | Design: multicentre, randomised, open label, parallel group study Duration: 30 days of treatment plus 7 days follow‐up Assessment by daily patient diary and weekly clinic visits. Aim to determine non‐inferiority and compare safety of new formulation patch (FIT) with standard formulation patch and oral morphine. Participants switched to FIT using standardised conversion ratio, based on previous 24 hour intake or 12.5 μg/h if opioid naïve; previous analgesics phased out Dose adjustment allowed throughout study to meet needs of individual participants Setting: in‐ or outpatients |
|
| Participants | Adult cancer patients with chronic cancer‐related pain requiring long term (> 30 days) strong (WHO Step 3) opioid treatment, either step up or rotation. Karnofsky score >50/100 at baseline N = 220 Mean age 63 (±11) years M 132, F 88 |
|
| Interventions | Fentanyl Improved Transdermal (FIT) patch, n = 117 Standard opioid treatment, n = 103 (65 transdermal fentanyl (Durogesic patch), 38 Oramorph) New patches applied every 72 hours, Oramorph given every 12 hours Dose adjustment permitted if breakthrough pain became regular (upward) or if significant adverse events were experienced alongside adequate pain control and no rescue medication (downward) Other treatment continued, including radiotherapy and chemotherapy, and both pharmacological and non‐pharmacological pain‐modulating interventions. Rescue medication: morphine, administered as preferred by participant or investigator |
|
| Outcomes | PI: NRS (0 to 10), daily Tolerability (constipation, nausea sleep disturbance, daytime drowsiness), using 4‐point ordinal scale (absent, mild, moderate, severe) Rescue medication Adverse events, serious adverse events Primary endpoint was relative area under the curve of PI expressed as a % maximum possible area under the curve |
|
| Notes | Oxford Quality Score: R1, DB0, W1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Low risk | Interactive voice response system |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | Low risk | All AEs reported |
| Size | Unclear risk | 50 ‐ 199 participants per treatment arm |
Lauretti 2003.
| Methods | Design: randomised, double blind, two‐period cross‐over study. Pre‐study 7‐day open label titration with MIR to determine suitable morphine dose Duration: 2 x 14 days, no washout Setting: not stated |
|
| Participants | Cancer pain not adequately controlled with tramadol/NSAID combination N = 26 (22 evaluated) M 15, F 7 Mean age 59 ± 19 years |
|
| Interventions |
Doses assigned by pharmacist Rescue medication: MIR 10 mg All participants taking oral amitriptyline 25 mg at bedtime |
|
| Outcomes | PI: VAS Participant satisfaction Adverse events Use of rescue medication Nausea and vomiting: VAS |
|
| Notes | Oxford Quality Score: R = 1, DB = 1, W = 1. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Method not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Method not described |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | Low risk | All AEs reported |
| Size | High risk | < 50 participants per treatment arm |
Leppart 2001.
| Methods | Design: randomised, open label prospective study. Dose stabilisation for 7 days with IR formulations (starting at tramadol 25 mg ‐ 50 mg and MIR 5 mg), then converted to SR formulations Duration: 7 day dose stabilisation, 28 days maintenance (35 days in total) Setting: outpatients |
|
| Participants | Cancer pain of at least moderate intensity (≥ 45/100). Participants opioid naive N = 40 |
|
| Interventions |
All participants given metoclopramide for first 3 days, then as needed. No dose during night, but last evening dose increased by 50% |
|
| Outcomes | PI: VAS and 5‐point VRS QoL: EORTC C30 |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "open randomised prospective study". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open label |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | High risk | Only most common and most rare AEs reported |
| Size | High risk | < 50 participants per treatment arm |
Melzack 1979.
| Methods | Design: randomised, double blind, two‐phase cross‐over study. 20 participants completed cross‐over in same environment; 7 completed cross‐over in different environments; 17 too ill to complete cross‐over Duration: 2 x 14 days Setting: in‐ and outpatients |
|
| Participants | Advanced malignant disease with pain requiring narcotics N = 44 (30 completed both phases) |
|
| Interventions |
Prochlorperazine 5 ‐ 10 mg given for nausea |
|
| Outcomes | PPI: 6‐point VRS scale Confusion, nausea, drowsiness: 4‐point VRS rated by participants, nurse, and relative |
|
| Notes | 17 participants too ill to cross over Oxford Quality Score: R = 2, DB = 2, W = 0. Total = 4/5 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Gellerman randomised table was used" |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "final solutions were identical in colour and consistency, and could not be distinguished" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "final solutions were identical in colour and consistency, and could not be distinguished" |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Unable to determine |
| Selective reporting bias for adverse events | High risk | Selective reporting of AEs |
| Size | High risk | < 50 per treatment arm |
Mercadante 1998.
| Methods | Design: randomised, open‐label study Records made in first 10 days of therapy and last 4 weeks of life Duration: long‐term; average length in study 38 days Setting: home |
|
| Participants | Advanced cancer patients with pain not responding to non‐opioids N = 32 |
|
| Interventions |
Participants allowed to switch from dextropropoxyphene to Mm/r Non‐opioid drugs were continued, as were other palliative treatments |
|
| Outcomes | Performance status Mean opioid dose Days on dextropropoxyphene in group1 Days on morphine in each group PI: VAS Adverse events: 4‐point categorical scale |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open label |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Unable to determine |
| Selective reporting bias for adverse events | High risk | Selective reporting and mean data |
| Size | High risk | < 50 participants per treatment arm |
Mercadante 2008.
| Methods | Design: multicentre, randomised, open, parallel group study. Assessment at baseline and weekly intervals Duration: 4 weeks Setting: outpatients |
|
| Participants | Adult cancer patients requiring strong opioids who had received opioids for mild to moderate pain, including tramadol and codeine at doses of at least 300 mg and 180 mg respectively without adequate analgesia. Expected survival > 3 months Breast cancer was the most frequent diagnosis (16 patients), and mixed nociceptive‐neuropathic syndromes (18 patients) the most dominant pain type N = 108 M 36, F 34 (completers) Mean age 59 years (range 18‐78) (completers) |
|
| Interventions | Fixed starting dose of study medication, adjusted to balance analgesia and adverse effects
Rescue medication: oral morphine at 1/6 daily 24 hour oral equivalent requirement Use of other medication permitted, including those for palliation of symptoms |
|
| Outcomes | Nausea, drowsiness, confusion: 4‐point scale (not at all, slight, a lot, severe) Constipation: 4‐point scale (0 = 1 passage every 1 to 2 days, 1 = one passage every 3 to 4 days, 2 = one passage > 4 days, 3 = rectal measures) Distress score calculated from sum of symptom intensities PI: NRS (0 to 10) Time to achieve dose stabilisation Number of daily dose changes Opioid escalation index QoL: Spitzer QoL index |
|
| Notes | Oxford Quality Score: R2, DB0, W1. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "computer generated" |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | High risk | AEs reported on fewer than 90% participants |
| Selective reporting bias for adverse events | High risk | mean intensity by drug only |
| Size | High risk | < 50 participants per treatment arm |
Mercadante 2010.
| Methods | Design: randomised, open label, parallel group study for control of breakthrough pain Duration: 4 weeks with extension up to 8 weeks Setting: outpatient or home care |
|
| Participants | Pancreatic cancer pain with PI ≥ 4/10, requiring opioids N = 60 randomised, but only 46 (M 19, F 27) completed baseline observations, 39 completed 4 weeks, 17 completed 8 weeks Mean age 63 years |
|
| Interventions |
Dose escalated according to clinical need (if PI > 4/10, or > 3 breakthrough pain medications per day) Adjuvant and symptomatic drugs prescribed as indicated Rescue medication: MIR at 1/6 total daily dose |
|
| Outcomes | Average PI in last 24 hours: NRS (0 ‐ 10) Opioid‐related symptoms: Nausea and vomiting, drowsiness and confusion (0 ‐ 3) Constipation rating scale (0 ‐ 3) |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomised by computer system" |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open label |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | High risk | Selective reporting. Mean symptom scores |
| Size | High risk | < 50 participants per treatment arm |
Mignault 1995.
| Methods | Design: randomised, double blind, two‐phase cross‐over 5‐day study. Prestudy titration period to establish total daily requirement Duration: 2 x 5 days + titration period Setting: not stated |
|
| Participants | Moderate to severe cancer pain N = 27 (19 included in analysis) Mean age 57 years (range 38 ‐ 69) Weight 65 (47 ‐ 104) kg |
|
| Interventions |
Rescue medication: MIR |
|
| Outcomes | PI: VAS x 4 daily Adverse events: 4‐point categorical scale Participant global rating: 4‐point scale Participant preference: 4‐point scale |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "blinding maintained by administration of active and placebo tablets each day" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "blinding maintained by administration of active and placebo tablets each day" |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Unable to determine |
| Selective reporting bias for adverse events | Low risk | Incidence of 9 different AEs |
| Size | High risk | < 50 participants per treatment arm |
Mizuguchi 1990.
| Methods | Design: multicentre, randomised, single blind (double dummy), cross‐over study Duration: 2 x 3 days. No washout Setting: not clear, probably inpatient |
|
| Participants | Cancer pain N = 46 Mean age 58 years (±12) |
|
| Interventions |
Previoius medication continued |
|
| Outcomes | PI: 4‐point categorical scale PR: 6‐point categorical scale Sleep Adverse events Global assessment |
|
| Notes | Oxford Quality Score: R = 2, DB = 0, W = 1. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomisation by code". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "matching placebos were prepared" but states single blind |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "matching placebos were prepared" |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Unable to determine |
| Selective reporting bias for adverse events | High risk | Selective reporting |
| Size | High risk | < 50 participants per treatment arm |
Moriarty 1999.
| Methods | Design: multicentre, randomised, double blind (double dummy), two‐phase cross‐over study Participants stabilised on Mm/r during 1 ‐ 3 day run‐in to confirm stability of pain control Duration: 2 x 3 days, plus pre‐study stabilisation. No washout Setting: not stated |
|
| Participants | Cancer pain adequately controlled with Mm/r N = 100 M 53, F 47 Age > 18 years |
|
| Interventions |
No other opioids allowed, antiemetics permitted Range of escape medication: MIR solution, diamorphine solution, diamorphine tablets and dextromoramide |
|
| Outcomes | PI: VAS and 6‐point categorical scale
Nausea: 4‐point scale Participant preference |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomisation schedule prepared by clinical supplies dept" |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "matching placebos" "double dummy technique" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "matching placebos" "double dummy technique" |
| Incomplete adverse event outcome data‐ patient level | High risk | Not reported at patient level |
| Selective reporting bias for adverse events | Low risk | All events reported |
| Size | Unclear risk | 50 ‐ 199 participants per treatment group |
Mucci LoRusso 1998.
| Methods | Design: multicentre, randomised, double blind, parallel group study Duration: 12 days Setting: General cancer patients |
|
| Participants | Chronic cancer‐related pain, requiring 30 ‐ 340 mg oxycodone, or equivalent, daily N = 100 55% male Mean age 59 years (range 30‐83) |
|
| Interventions |
Immediate release oxycodone 5 mg and MIR15 mg for breakthrough No other opioids permitted during study, but non‐opioid analgesics and adjuvant medications were allowed provided they had been given on a regular basis (not as needed) before the study |
|
| Outcomes | PI: 4‐point categorical scale, before each dose
Participant global rating of therapy: 5‐point categorical scale Participant preference QoL: FACT ‐ G 28 item questionnaire Adverse events: Specific Drug Effect Questionnaire, VAS |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "block randomisation was used" Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double dummy technique" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "double dummy technique" |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | High risk | Only reported when experienced by 10% or more of patients |
| Size | High risk | < 50 participants per treatment arm |
Mystakidou 2005.
| Methods | Design: randomised, open label, parallel group study All participants underwent palliative radiotherapy before randomisation. Fixed starting dose of study medication, adjusted to patient requirements Assessment at baseline, 3, 7, 14, 28 days, and 2 months Duration: 2 months Setting: Possibly outpatients‐ not clearly stated |
|
| Participants | Adult cancer patients with painful bony metastasis and moderate/severe chronic cancer pain requiring strong opioids Primary cancer location: lung, kidney/bladder, gastrointestinal, breast, unknown, other) Site of bony metastasis: thoracic spine, lumbar spine, cervical spine, thoracic and lumbar spine, pelvis, femur, scapula Other metastases: brain, gastrointestinal, lung, adrenal N = 460 (422 eligible) Mean age 61 (25 to 88) years (eligible) M 219, F 203 (eligible) |
|
| Interventions |
Fentanyl dose was increased when treatment satisfaction ≤ 2 and pain score ≥ 3 Rescue medication: fentanyl‐treated participants could receive paracetamol and codeine twice in first 12 hours after patch application |
|
| Outcomes | PI: Greek brief pain inventory (G‐BPI), 0 to 10 Overall treatment satisfaction: VRS 4‐point scale (not at all satisfied, fairly satisfied, satisfied, completely satisfied) QoL: VAS, 0 to 10 European Collaborative Oncology Group status Adverse events |
|
| Notes | Oxford Quality Score: R1, DB0, W1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | Unclear risk | Probably selective reporting as only data on 5 different AEs reported |
| Size | Low risk | > 200 participants per treatment arm |
O'Brien 1997.
| Methods | Design: Multicentre, randomised, double blind (double dummy), cross‐over study Duration: 2 x 7 days Setting: GP practices, hospitals or hospices |
|
| Participants | Cancer pain, fully titrated to pain control with twice‐daily Mm/r, with dose stable for ≥ 3 days N = 85 Mean age 64 years |
|
| Interventions |
NSAIDs, steroids, other long‐term therapy for any chronic non related condition continued unchanged MIR tablets for breakthrough pain |
|
| Outcomes | PI: BS‐11 scale Use of rescue medication Sleep Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised" Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double blind" and "double dummy" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "double blind" and "double dummy" |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | High risk | Highly selective ‐ most frequent AEs |
| Size | High risk | < 50 participants per treatment arm |
Oztürk 2008.
| Methods | Design: randomised, open label, parallel group study Duration: 15 days Setting: hospital and home |
|
| Participants | Lung cancer requiring WHO step 3 opioids for pain; 18 of fentanyl patients were treated in hospital, and 16 of morphine patients were treated in hospital, others were visited by doctors at home N = 50 M/F not reported Mean age 55 years (completers, range not stated) |
|
| Interventions |
Starting level: Participants requiring 200 to 400 mg tramadol used 25 μg/h TDF patches Participants requiring 500 to 600 mg oral tramadol used 50 μg/h TDF patches 120 mg slow release morphine Dose increased if inadequate response to maximum 100 mg/h TDF or 180 mg Mmr(41% and 23% changed, two participants in each group increased dose twice) Rescue medication: both groups given subcutaneous morphine if pain ‘unbearable’ (NRS > 3) |
|
| Outcomes | Pain: NRS (0‐10) Activities of daily living: Eastern Cooperative Oncology Group Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | High risk | Selective reporting |
| Size | High risk | < 50 participants per treatment arm |
Panich 1993.
| Methods | Design: randomised, single blind, two‐phase cross‐over study Duration 2 x 7 days, no washout Setting: Pain clinic in Thailand |
|
| Participants | Severe cancer pain
N = 73 (49 reported) Mean age 53 years (± 10) Weight 46.5 kg (± 10.6) |
|
| Interventions |
Paracetamol or narcotic injection for breakthrough pain |
|
| Outcomes | Nurse assessment of pain: VAS Nurse assessment of sleep duration: 4‐point categorical scale Participant preference Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised into 2 groups" Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Single blind ‐ nurse blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Single blind ‐ nurse blinded |
| Incomplete adverse event outcome data‐ patient level | High risk | <90% participants included |
| Selective reporting bias for adverse events | High risk | Selective reporting |
| Size | High risk | < 50 participants in each treatment arm (for analysis reported) |
Parris 1998.
| Methods | Design: multicentre, randomised, double‐blind (double dummy), parallel group study Duration: 5 days Setting: Not stated |
|
| Participants | Cancer‐related pain requiring 6 to 12 tablets or capsules per day of fixed‐combination analgesics for acceptable control. Most common cancer diagnoses were breast, gastrointestinal, lung, and gynaecological N = 103 50% female Mean age 57 years (range 31‐80) |
|
| Interventions |
Stable doses of non opioid analgesics or analgesic adjuvants allowed after protocol amendment Rescue medication: participants needing titration of analgesic or supplemental medication were required to discontinue from the study |
|
| Outcomes | PI: 4‐point categorical scale (0 ‐ 3) x 4 daily Acceptability of therapy: 5‐point categorical scale (1 ‐ 5), x 2 daily Adverse events, daily |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double dummy method |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double dummy method |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | High risk | Selective reporting of >5% of patients with an AE |
| Size | Unclear risk | 50 ‐ 199 participants per treatment arm |
Pistevou‐Gompaki 2004.
| Methods | Design: multicentre, randomised, open label, parallel group study All participants received palliative radiotherapy (unclear whether before or during medication). Assessed at baseline (before radiotherapy) and at 2‐weekly intervals during and after radiotherapy Duration: 3 months Setting: outpatients |
|
| Participants | Adult cancer patients with painful bony metastasis. Moderate/severe pain refractory to common analgesics, no previous strong opioids Primary cancer location (lung, prostate, breast, stomach/gallbladder, kidney, multiple myeloma, unknown); site of bony metastasis (thoracic spine, lumbar spine, cervical spine, thoracic and lumbar spine, pelvis, limbs, scapula); other metastases (brain, lymph, lung, liver) N = 26 (24 eligible) M 19, F 7 Age range 54 to 72 years |
|
| Interventions |
All participants received radiotherapy 3 fentanyl and 2 paracetamol plus codeine participants also received iv bisphosphonates |
|
| Outcomes | PI: VAS (0 to 10) QoL: Greek Brief Pain Inventory (G‐BPI) 0 to 10 |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total =2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | >90% participants included |
| Selective reporting bias for adverse events | Low risk | Complete |
| Size | High risk | < 50 participants per treatment arm |
Portenoy 1989.
| Methods | Design: randomised, double blind, parallel group comparison of 2 strengths of Mm/r. Prestudy stabilisation period (1 ‐ 2 days) of MIR 30 mg every 4 h with 15 mg every 2 h for breakthrough Duration: 3 days Setting: not stated |
|
| Participants | Severe cancer pain, requiring approximately 200 mg morphine /24h N = 51 (49 evaluated) M 29, F 22 Mean age 52 years Weight 66.3 kg |
|
| Interventions | Dosing regimen:
Mm/r 1 x 100 mg 12‐hourly, n = 25 Mm/r 3 x 30 mg 12‐hourly, n = 26 Rescue medication: 15 mg morphine available every 2 hours as required |
|
| Outcomes | PI: 5‐point categorical scale, 3 x daily Adverse events Bowel function |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "the blind condition was maintained by dispensing . . . in opaque capsules" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "the blind condition was maintained by dispensing . . . in opaque capsules" |
| Incomplete adverse event outcome data‐ patient level | Low risk | >90% participants included |
| Selective reporting bias for adverse events | High risk | Selective reporting‐ mean intensity data |
| Size | High risk | < 50 participants per treatment arm |
Rico 2000.
| Methods | Design: randomised, double blind, two‐way cross‐over Duration: 2 x 7 days with 3‐day washout Setting: not stated |
|
| Participants | Oncologic pain N = 44 30 women, 14 men Mean age 55 years Baseline pain > 6/10 |
|
| Interventions | (1) codeine 120 mg daily to max 320 mg daily (average max dose 49 ± 15 mg x 4 daily) (2) tramadol 160 mg daily to max 400 mg daily (average max dose 68 ± 24 mg x 4 daily) All participants also received paracetamol 500 mg x 4 daily |
|
| Outcomes | PI: 10 cm VAS Patient preference Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 1, W = 1. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not clearly stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Method not clearly stated, used the same number of drops and both had bitter taste |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Method not clearly stated, used the same number of drops and both had bitter taste |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Denominator not clear |
| Selective reporting bias for adverse events | High risk | Selective reporting of a few AEs |
| Size | High risk | < 50 participants per treatment arm |
Ridgway 2010.
| Methods | Design: multicentre, randomised, double blind, two‐way cross‐over study of once a day Mm/r formulation vs twice a day Mm/r formulation. Prestudy stabilisation period ≥ 3 days, to achieve adequate pain control with ≤ 4 doses of rescue medication/day, followed by fixed dose Duration: 2 x 2 weeks, plus pre study stabilisation. No washout Setting: not stated but 8 sites in Lithuania and Poland |
|
| Participants | Cancer pain requiring 30 mg ‐ 240 mg/day morphine equivalent (stable for ≥ 2 weeks) N = 38 M 24, F 14 Mean age 58 years (range 42 ‐ 81) |
|
| Interventions |
No further dose adjustment allowed MIR allowed for breakthrough at approximately 10% of the daily dose |
|
| Outcomes | Use of rescue medication: average daily doses over last 7 days PI: NRS (0‐10), current, least, worst Patient impression of treatment: 5‐point VRS Patient preference |
|
| Notes | Oxford Quality Score: R = 1, DB = 1, W = 1. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised to a treatment sequence". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "blinded by over encapsulating with gelatin capsules" but does not say identical |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "blinded by over encapsulating with gelatin capsules" but does not say identical |
| Incomplete adverse event outcome data‐ patient level | Low risk | <90% participants included |
| Selective reporting bias for adverse events | High risk | Most common treatment related |
| Size | High risk | < 50 participants per treatment arm |
Rodriguez 1994.
| Methods | Design: multicentre, randomised, double blind, parallel group study Duration: 7 days Setting: Oncology departments in Spain |
|
| Participants | Cancer pain, with pre study intensity > 70/100 mm N = 149 eligible, 121 participated 70% men Mean age 61 years Baseline VAS PI > 70/100 |
|
| Interventions |
No other medication allowed Rescue medication: paracetamol 300 mg and codeine 15 mg |
|
| Outcomes | PI: VAS, daily Adverse events: check list ‐ severity judged by investigators | |
| Notes | Oxford Quality Score: R = 1, DB = 1, W = 1. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double blind". Method not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double blind". Method not described |
| Incomplete adverse event outcome data‐ patient level | Low risk | <90% participants included |
| Selective reporting bias for adverse events | Unclear risk | All AEs by events appear to be included |
| Size | High risk | < 50 participants per treatment arm |
Rodriguez 2007.
| Methods | Design: randomised, double blind, parallel group study Duration: 2‐day run in, then 21‐day treatment period Setting: not stated |
|
| Participants | Persistent moderate or severe cancer pain (primarily gastric, breast, prostate, lung) N = 177 M 88, F 89 Mean age 60 years |
|
| Interventions |
If no PR (VAS ≥ 4/10) dose could be doubled. If this caused intolerable adverse events it could be reduced by 25% Rescue medication: |
|
| Outcomes | PI: 10 cm VAS PR: five‐point scale (0‐4) Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "computer‐generated schedule" |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "study drugs had similar characteristics such as color, shape, and dimensions and were packaged in identical containers" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "study drugs had similar characteristics such as color, shape, and dimensions and were packaged in identical containers" |
| Incomplete adverse event outcome data‐ patient level | Low risk | <90% participants included |
| Selective reporting bias for adverse events | Low risk | Comprehensive list |
| Size | Unclear risk | 50 ‐ 199 participants per treatment arm |
Salzman 1999.
| Methods | Design: multicentre, randomised, open label, parallel group study. For participants receiving non opioid therapy, dosing regimen stabilised ≥ 1 week before initiation of study medication and remained stable for the duration of the study Duration: up to 21 days Setting: outpatient |
|
| Participants | Stable cancer pain not adequately controlled by previous analgesic therapy with or without opioids N = 48, 35 completed titration period M 21, F 27 Mean age 61 years (range 25 ‐ 91) |
|
| Interventions |
Starting dose for opioid‐naive patients = 20 mg/day, and for non‐opioid‐naive patients the starting dose was based on the prior 3 days of analgesic therapy. Titrated to maximum 400 mg daily to achieve PI ≤ 'slight' (1.5) for 48 h with ≤ 2 doses of rescue medication Stable non opioid medication continued, no other opioids allowed Rescue medication: oxycodone IR 5 mg, 10 mg, or 1/6 total dose, depending on daily dose, and taken no more than once every 4 hours |
|
| Outcomes | PI: 4‐point categorical scale, daily Adverse events: 4‐point categorical scale, daily Time to stable pain control: time to achieve PI ≤ 'slight' (1.5) for 48 h with ≤ 2 doses of rescue medication |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open label |
| Incomplete adverse event outcome data‐ patient level | Low risk | <90% participants included |
| Selective reporting bias for adverse events | High risk | Selective reporting. Treatment related AEs in >10% patients |
| Size | High risk | < 50 participants per treatment arm |
Smith 1991.
| Methods | Design: multicentre, randomised double blind (double dummy), two‐way cross‐over study. Prestudy titration to satisfactory pain control with Mm/r, then fixed dose Duration: 2 x 3 or 4 days Setting: not stated |
|
| Participants | Cancer pain N = 25 (20 completed) M 8, F 12 Age 35 ‐ 69 years |
|
| Interventions |
Rescue medication: aqueous morphine, dextromoramide, solpadeine (paracetamol codeine, caffeine) |
|
| Outcomes | Outcome measures:
PI: VAS x 3 ‐ 4 daily Morphine levels |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 0. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly allocated". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "patients received either MSC 100mg or 200mg with appropriate alternative placebo tablets". Double dummy method |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "patients received either MSC 100mg or 200mg with appropriate alternative placebo tablets". Double dummy method |
| Incomplete adverse event outcome data‐ patient level | High risk | No data for AEs |
| Selective reporting bias for adverse events | High risk | No data for AEs |
| Size | High risk | < 50 participants per treatment arm |
Stambaugh 2001.
| Methods | Design: Randomised, double‐blind, two‐way cross‐over study. Initial open label titration to determine dose required to achieve < moderate pain with ≤ 2 doses of rescue medication daily Duration: up to 35 days, consisting of a titration period of 2‐21 days, followed by two double‐blind cross‐over periods each lasting 3‐7 days, with no washout Setting: home, outpatient |
|
| Participants | Moderate or severe cancer‐related pain, not requiring > 240 mg/day oral oxycodone equivalent for pain relief. Primary site mostly bone N = 40, 30 completed both phases M 10, F 20 (completers) Mean age 60 years (range 34 ‐ 83) |
|
| Interventions | Open label titration with immediate‐release oxycodone, using starting dose calculated from past 3 days of analgesia therapy Oxycodone CR 12‐hourly + placebo to maintain blinding Oxycodone IR 6‐hourly Rescue medication: Oxycodone IR 5 mg No other opioids permitted, but concurrent, stable therapy with acetaminophen, NSAIDs, or analgesic adjuvants and co‐analgesics was allowed |
|
| Outcomes | PI: NRS (0 ‐ 10) PR: 11‐point categorical scale Acceptability of treatment: 5‐point categorical scale (1 ‐ 5) Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "three tablets identical in appearance" "identical medication blister packs" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "three tablets identical in appearance" "identical medication blister packs" |
| Incomplete adverse event outcome data‐ patient level | High risk | Reported only 30 or 31 completers of 40 initial starters reported adverse events |
| Selective reporting bias for adverse events | Unclear risk | Drug related adverse events |
| Size | High risk | < 50 participants per treatment arm |
Thirlwell 1989.
| Methods | Design: randomised, double blind (double dummy), two‐phase cross‐over study Duration: 2 x ≥ 5 days, no washout Setting: not stated |
|
| Participants | Cancer pain requiring oral opioid therapy
N = 28 (23 analysed) M 13, F 10 Age 58 years (± 12) |
|
| Interventions |
Dose determined from pre study use. Same dose used for each phase No non‐study opioids allowed. Non‐opioids were allowed Rescue medication: MIR |
|
| Outcomes | PI: 4‐point categorical scale x 4 daily Use of rescue medication Plasma morphine concentrations Adverse events: severity graded on 4‐point scale |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned allocation technique". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double dummy technique" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "double dummy technique" |
| Incomplete adverse event outcome data‐ patient level | Low risk | <90% participants included |
| Selective reporting bias for adverse events | High risk | Some patients were withdrawn during phase 1 (steady state) |
| Size | High risk | < 50 participants per treatment arm |
Todd 2002.
| Methods | Design: multicenter, randomised, open label, two‐phase cross‐over study of two dosing regimens of MIR Duration: 2 x 2 days, no washout Setting: inpatients |
|
| Participants | Cancer‐related pain adequately treated with MIR, stable for ≥ 2 days, with ≤ 2 doses of rescue medication N = 24 (20 completed) Median age 62 years (range 40 ‐ 89) M 10, F 14 |
|
| Interventions |
Rescue medication: MIR |
|
| Outcomes | Use of rescue medication during night PI for overnight and morning pain: NRS (0 ‐ 10) Adverse events: 4‐point VRS |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated randomised list |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Reported events not patients |
| Selective reporting bias for adverse events | High risk | Selective AEs reported |
| Size | High risk | < 50 participants per treatment arm |
Vainio 1988.
| Methods | Design: randomised, open label, parallel group study Duration: 14 days Setting: hospital and home |
|
| Participants | Cancer patients with tumour compression or infiltration of brachial or lumbar plexus. Mean baseline PI VAS > 8/10 N = 30 Mean age 53 years (range 23 ‐ 86) |
|
| Interventions |
Doses adjusted to patient need |
|
| Outcomes | PI: 10 cm VAS PID calculation after 24 hours and 2 weeks Karnofsky performance Adverse event profile |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 0. Total = 1/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open label |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Not clear what is reported |
| Selective reporting bias for adverse events | Unclear risk | Not clear what is reported |
| Size | High risk | < 50 participants per treatment arm |
van Seventer 2003.
| Methods | Design: multicentre, randomised, open label, parallel group study. Assessements by investigator and participant at baseline, 7 and 28 days. Participants also kept a daily diary Duration: 4 weeks Setting: Community |
|
| Participants | Moderate‐severe cancer‐related pain requiring opioid treatment, with life expectancy ≥ 3 months. Participants could be opioid naïve or using opioids for mild‐to‐moderate pain before entry. Participants using opioids for moderate‐to‐severe pain in 30 days preceding study entry were excluded N = 131 M 85, F 46 Mean age 65 (±12) years |
|
| Interventions |
Rescue medication: 10 mg severedol every 2 ‐ 4 hours, as required Concomitant medication recorded |
|
| Outcomes | Pain control: Shortened Wisconsin brief pain inventory: 11‐point scale (0 = no, 10 = extreme), daily Global assessment of pain relief, sleep, interruption of daily activities and caregiver's activities, troublesome side effects: 4‐point scale (1 = not at all, 4 = very much) at start and 28 days Overall assessment: 11‐point scale (0 = very poor, 10 = very good) Constipation: questionnaire (bowel function normal, constipated, diarrhoeal) at start, 7 and 28 days Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not adequately described but states "centrally randomised" |
| Allocation concealment (selection bias) | Unclear risk | Method not adequately described but states "centrally randomised" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | Low risk | <90% participants included |
| Selective reporting bias for adverse events | High risk | Selective reporting of most serious or most frequent |
| Size | Unclear risk | 50 ‐ 200 participants per treatment arm |
Ventafridda 1986.
| Methods | Design: randomised, open label, parallel group study. Initial dose based on pain level and previous treatment, then titrated to adequate control Duration: 14 days Setting: home |
|
| Participants | Cancer pain, severe, uncontrolled N = 66 randomised, 54 included M 31, F 23 Mean age 55 years |
|
| Interventions |
All participants received diclofenac 150 mg daily and haloperidol 20 mg/day by injection |
|
| Outcomes | PI: 5‐point categorical scale (Integrated pain score) Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 0. Total = 1/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "not blinded" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "not blinded" |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | No denominator for data |
| Selective reporting bias for adverse events | Unclear risk | Presented data for days with AEs |
| Size | High risk | < 50 participants per treatment arm |
Ventafridda 1989.
| Methods | Design: randomised, open label, parallel group study. Initial dose based on pain level and previous treatment, then titrated to adequate control Duration: 14 days Setting: home |
|
| Participants | Cancer pain; no previous strong opioids N = 70 M 39, F 31 Mean age 57 years (range 28 ‐ 88) |
|
| Interventions |
Participants also received diclofenac 75 mg 3 x daily; haloperidol 20 mg in 2 doses daily |
|
| Outcomes | Outcome measures:
Integrated score PI scale 0 ‐ 240 Pain intensity assessed using 5 key words: slight 1, troublesome 2.5, exhausting 5, terrible 7.5, killing 10. Adverse events were recorded |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 0. Total = 1/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open label |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | No denominator for data |
| Selective reporting bias for adverse events | Unclear risk | Present data for days with AEs |
| Size | High risk | < 50 participants per treatment arm |
Vielvoye‐Kerkmeer 2002.
| Methods | Design: randomised, open label (presumed), parallel study. Participants stabilised on morphine (Mm/r, x 2 daily) over maximum of 14 days, then once stable for 3 days (pain controlled and ≤ 2 doses of rescue medication/day), randomised to different dosing regimens Duration: 6 ‐ 7 days + stabilisation Setting: outpatient |
|
| Participants | Moderate to severe chronic cancer pain N = 153 enrolled, 110 entered treatment phase No demographic details provided |
|
| Interventions | Mm/r x 1 daily, n = 52 Mm/r x 2 daily, n = 58 Concomitant medication, NSAIDs, paracetamol continued. Prophylactic laxatives, antiemetics as required |
|
| Outcomes | PI: 100 mm VAS Sleep: 4‐point VRS Adverse events: alternate day telephone interviews Global assessment: 4‐point categorical scale Treatment preference |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "patients were randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not reported as blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not reported as blinded |
| Incomplete adverse event outcome data‐ patient level | Low risk | <90% participants included |
| Selective reporting bias for adverse events | High risk | Selective reporting of events |
| Size | Unclear risk | 50 ‐ 200 participants per treatment arm |
Walsh 1985.
| Methods | Design: randomised, double blind (double dummy), cross‐over study comparing different formulations of morphine. Participants stabilised on MIR pre study Duration: 10 days, with cross‐overs at 3, 5, and 8 days Setting: hospice inpatients |
|
| Participants | Cancer pain, adequately controlled with stable doses of oral morphine N = 36 (30 completed) Mean age 67 ± 8 years |
|
| Interventions |
The same total daily dose was used for both formulations Other medication at discretion of physician, including sedatives Rescue medication: "agreed scheme" |
|
| Outcomes | PI: 100 mm VAS Mood Nurse‐reported: pain, sedation; nausea and vomiting; constipation; orientated; pain breakthrough |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 0. Total = 3/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "identical SRM placebo/pills were used" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "identical SRM placebo/pills were used" |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | Data not reported |
| Selective reporting bias for adverse events | High risk | Selective reporting of AEs |
| Size | High risk | < 50 participants per treatment arm |
Walsh 1992.
| Methods | Design: randomised, double blind (double dummy), two phase cross‐over study. Stable dose of MIR with < 2 doses of rescue medication for ≥ 24 hours before randomisation Duration: 5 days, with cross‐over at 3 days Setting: inpatient |
|
| Participants | Advanced cancer, requiring > 60 mg IR morphine/day to treat pain N = 33 (27 competed) M 12, F 15 Mean age 61 years (SD 2) |
|
| Interventions |
The same total daily dose was used for both formulations Pre‐study non‐opioids allowed Rescue medication: morphine (IR), paracetamol, IM/SC morphine |
|
| Outcomes | PI: VAS Anxiety, depression, sedation, nausea, constipation and confusion: VAS Participant preference Breakthrough pain Adverse events |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1. Total = 5/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomisation was performed by pharmacist using a random number table" |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double dummy ....with identical placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "double dummy ....with identical placebo" |
| Incomplete adverse event outcome data‐ patient level | Low risk | <90% participants included |
| Selective reporting bias for adverse events | High risk | Mean VAS scores for selected AEs |
| Size | High risk | < 50 participants per treatment arm |
Wilder‐Smith1994.
| Methods | Design: randomised, double blind, two‐phase cross‐over study. Dose titrated daily, according to need for rescue medication Duration: 2 x 4 days, no washout Setting: hospital, two centres |
|
| Participants | Cancer pain, severe and not responsive to previous treatment N = 20 M 11, F 9 Mean age 55 years (range 26 ‐ 75) |
|
| Interventions |
Non‐opioids stopped where possible; prophylactic laxatives and antiemetics given Rescue medication: additional dose of same size for breakthrough pain |
|
| Outcomes | PI: 5‐point VRS, daily Adverse events: 5‐point VRS Participant preference |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1. Total = 4/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double blind fashion. . . . .to taste, smell and look identical" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "double blind fashion. . . . .to taste, smell and look identical" |
| Incomplete adverse event outcome data‐ patient level | Low risk | <90% participants included |
| Selective reporting bias for adverse events | High risk | Mean VAS scores for selected AEs, miscellaneous AEs as events |
| Size | High risk | < 50 participants per treatment arm |
Wilkinson 1992.
| Methods | Design: randomised, open label, 4‐phase cross‐over comparing oral and rectal routes of administration for Mm/r. Duration: 4 days Setting: hospital inpatients |
|
| Participants | Cancer inpatients on stable doses of morphine to control pain N = 11 (10 completed) M 6, F 4 Age 70 years (range 40 ‐ 83) |
|
| Interventions |
Rescue medication: paracetamol or pethidine (oral) |
|
| Outcomes | PI: VAS every 12 h Adverse events: VAS Pharmacokinetic measurement after 4th dose each treatment Participant preference |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 0. Total = 1/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised". Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | Unclear risk | <90% participants included |
| Selective reporting bias for adverse events | High risk | 'No difference between groups' |
| Size | High risk | < 50 participants per treatment group |
Wong 1997.
| Methods | Design: randomised, open label, parallel group study. Prestudy stabilisation phase with MIR Assessment during stabilisation, at start of treatment phase, and in immediate and final phases of treatment Duration: 14 days + 7 day stabilisation phase (if necessary) Setting: not stated |
|
| Participants | Adult terminal cancer patients with estimated survival time ≥ 2 months, and pain requiring oral morphine or equivalent ≤ 404 mg per day N = 47 (40 completed) M 29, F 11 (completers) Mean age 59 years (range 30 to 79) |
|
| Interventions |
Previous opioid treatment converted to MIR during stabilisation phase Rescue medication: MIR |
|
| Outcomes | PI: 5‐point VRS (no pain, mild, moderate, severe, excruciating) Frequency of pain: 4‐point VRS (no pain, occasional, always, persistent) Degree of pain improvement: 5‐point VRS (no pain, obvious, moderate, little, no improvement) Profile of mood state as effected by the pain: 4‐point VRS (no, mild, moderate, severe interference) Quality of sleep: 4‐point VRS (normal, occasionally awakened by pain, always awakened by pain, insomnia) Activity status: Eastern Cooperative oncology group (ECOG) 5‐point VRS (0 = fully active, 4 = completely disabled) Use of rescue medication Patient satisfaction Treatment preference Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 0, W = 1. Total = 2/5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method used to generate sequence not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete adverse event outcome data‐ patient level | Low risk | <90% participants included |
| Selective reporting bias for adverse events | Low risk | All AEs reported |
| Size | High risk | < 50 participants per treatment arm |
AE: adverse event; BPI: Brief Pain Inventory; CR: controlled release; DB: double blind; F: female; h:hour; M: male; min: minute; MIR: immediate release morphine; m/r: modified release; Mm/r: modified release morphine; N: number of participants in study; n: number of participants in treatment arm; NRS: numerical rating scale; NSAID: nonsteroidal anti‐inflammatory drug; PGIC: Patient Global Imporession of Change; PI: pain intensity; PPI: present pain intensity; R: randomised; QoL: quality of life; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant, VAS: visual analogue scale; VRS: verbal rating scale; W: withdrawals; WHO: World Health Organisation
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Beaver 1978 I | Included in 'Codeine, alone and with paracetamol (acetaminophen), for cancer pain' but excluded here as it is a single dose study |
| Beaver 1978 II | Included in 'Codeine, alone and with paracetamol (acetaminophen), for cancer pain' but excluded here as it is a single dose study |
| Capretti 1970 | Included in 'Codeine, alone and with paracetamol (acetaminophen), for cancer pain' but excluded here as it is a single dose study |
| Chen 2003 | Included in 'Codeine, alone and with paracetamol (acetaminophen), for cancer pain' but excluded here as it is a single dose study |
| Coluzzi 2001 | Study of breakthrough pain |
| Jochimsen 1978 I | Included in 'Codeine, alone and with paracetamol (acetaminophen), for cancer pain' but excluded here as it is a single dose study |
| Leow 1995 | Single dose fentanyl |
| Moertel 1971 | Included in 'Codeine, alone and with paracetamol (acetaminophen), for cancer pain' but excluded here as it is a single dose study |
| Noyes 1975 | Included in 'Codeine, alone and with paracetamol (acetaminophen), for cancer pain' but excluded here as it is a single dose study |
| Stambaugh 1987 | Included in 'Codeine, alone and with paracetamol (acetaminophen), for cancer pain' but excluded here as it is a single dose study |
| Staquet 1971 | Not all patients had cancer pain |
| Staquet 1978 | Included in 'Codeine, alone and with paracetamol (acetaminophen), for cancer pain' but excluded here as it is a single dose study |
| Staquet 1993 | Included in 'Codeine, alone and with paracetamol (acetaminophen), for cancer pain' but excluded here as it is a single dose study |
| Twycross 1977 | Morphine and cocaine mixture |
Differences between protocol and review
The protocol had double‐blinding as an inclusion criterion. This was removed at the review stage, because otherwise there would probably have been too few studies. Blinding is known to affect efficacy estimates, but effect on adverse event reporting is not clear. A simple PubMed search was added as the Oxycodone review was not yet completed.
Contributions of authors
PW wrote the first draft of the protocol. All authors contributed to the concept, amended subsequent versions and approved the final version for publication.
PW and SD carried out data extraction for the full review and RAM carried out analyses. All authors were involved in writing the full review.
Sources of support
Internal sources
-
Oxford Pain Relief Trust, UK.
General institutional support
External sources
NIHR Incentive Award (Reference: 13/180/08), UK.
Declarations of interest
SD, RAM, and PW have received research support from charities, government, and industry sources at various times. RAM, and PW have consulted for various pharmaceutical companies. RAM has received lecture fees from pharmaceutical companies related to analgesics and other healthcare interventions.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Ahmedzai 1997 {published data only}
- Ahmedzai S, Brooks D. Transdermal fentanyl versus sustained‐release oral morphine in cancer pain: preference, efficacy, and quality of life. The TTS‐Fentanyl Comparative Trial Group. Journal of Pain and Symptom Management 1997;13(5):254‐61. [DOI: 10.1016/S0885-3924(97)00082-1] [DOI] [PubMed] [Google Scholar]
Ahmedzai 2012 {published data only}
- Ahmedzai SH, Nauck F, Bar‐Sela G, Bosse B, Leyendecker P, Hopp M. A randomized, double‐blind, active‐controlled, double‐dummy, parallel‐group study to determine the safety and efficacy of oxycodone/naloxone prolonged‐release tablets in patients with moderate/severe, chronic cancer pain. Palliative Medicine 2012;26:50‐60. [DOI: 10.1177/0269216311418869] [DOI] [PMC free article] [PubMed] [Google Scholar]
Arkinstall 1989 {published data only}
- Arkinstall WW, Goughnour BR, White JA, Stewart JH. Control of severe pain with sustained‐release morphine tablets versus oral morphine solution. Canadian Medical Association Journal 1989;140(6):653‐7. [PMC free article] [PubMed] [Google Scholar]
- Goughnour BR, Arkinstall WW, Stewart JH. Analgesic response to single and multiple doses of controlled release morphine tablets and morphine oral solution in cancer patients. Cancer 1989;63(Suppl 11):2294‐7. [DOI] [PubMed] [Google Scholar]
Babul 1998 {published data only}
- Babul N, Provencher L, Laberge F, Harsanyi Z, Moulin D. Comparative efficacy and safety of controlled‐release morphine suppositories and tablets in cancer pain. Journal of Clinical Pharmacology 1998;38(1):74‐81. [DOI] [PubMed] [Google Scholar]
Boureau 1992 {published data only}
- Boureau F, Saudubray F, D'Arnoux C, Vedrenne J, Estève M, Roquefeuil B, et al. A comparative study of controlled‐release morphine (CRM) suspension and CRM tablets in chronic cancer pain. Journal of Pain and Symptom Management 1992;7(7):393‐9. [DOI] [PubMed] [Google Scholar]
Broomhead 1997 {published data only}
- Broomhead A, Kerr R, Tester W, O'Meara P, Maccarrone C, Bowles R, et al. Comparison of a once‐a‐day sustained‐release morphine formulation with standard oral morphine treatment for cancer pain. Journal of Pain and Symptom Management 1997;14(2):63‐73. [DOI] [PubMed] [Google Scholar]
Bruera 1998 {published data only}
- Bruera E, Belzile M, Pituskin E, Fainsinger R, Darke A, Harsanyi Z, et al. Randomized, double‐blind, cross‐over trial comparing safety and efficacy of oral controlled‐release oxycodone with controlled‐release morphine in patients with cancer pain. Journal of Clinical Oncology 1998;16(10):3222‐9. [DOI] [PubMed] [Google Scholar]
Bruera 2004 {published data only}
- Bruera E, Palmer JL, Bosnjak S, Rico MA, Moyano J, Sweeney C, et al. Methadone versus morphine as a first‐line strong opioid for cancer pain: a randomized, double‐blind study. Journal of Clinical Oncology 2004;22(1):185‐92. [DOI] [PubMed] [Google Scholar]
Carlson 1990 {published data only}
- Carlson RW, Borrison RA, Sher HB, Eisenberg PD, Mowry PA, Wolin EM. A multiinstitutional evaluation of the analgesic efficacy and safety of ketorolac tromethamine, acetaminophen plus codeine, and placebo in cancer pain. Pharmacotherapy 1990;10(3):211‐6. [PubMed] [Google Scholar]
Cundiff 1989 {published data only}
- Cundiff D, McCarthy K, Savarese JJ, Kaiko R, Thomas G, Grandy R, et al. Evaluation of a cancer pain model for the testing of long‐acting analgesics. The effect of MS Contin in a double‐blind, randomized crossover design. Cancer 1989;63(11 Suppl):2355‐9. [DOI] [PubMed] [Google Scholar]
- Cundiff D, Savarese J, Grandy R, MacCarthy K, R. Kaiko R, G. Thomas G, et al. Evaluation of a controlled‐ and immediate release morphine and a unique well‐controlled study design in cancer pain. Journal of Pain and Symptom Management 1988;3((Suppl)):18. [Google Scholar]
Currow 2007 {published data only}
- Currow DC, Plummer JL, Cooney NJ, Gorman D, Glare PA. A randomized, double‐blind, multi‐site, crossover, placebo‐controlled equivalence study of morning versus evening once‐daily sustained‐release morphine sulfate in people with pain from advanced cancer. Journal of Pain and Symptom Management 2007;34:17‐23. [DOI] [PubMed] [Google Scholar]
Dale 2009 {published data only}
- Dale O, Piribauer M, Kaasa S, Moksnes K, Knobel H, Klepstad P. A double‐blind, randomized, crossover comparison between single‐dose and double‐dose immediate‐release oral morphine at bedtime in cancer patients. Journal of Pain and Symptom Management 2009;37:68‐76. [DOI] [PubMed] [Google Scholar]
De Conno 1995 {published data only}
- Conno F, Ripamonti C, Saita L, MacEachern T, Hanson J, Bruera E. Role of rectal route in treating cancer pain: a randomized crossover clinical trial of oral versus rectal morphine administration in opioid‐naive cancer patients with pain. Journal of Clinical Oncology 1995;13(4):1004‐8. [DOI] [PubMed] [Google Scholar]
Dellemijn 1994 {published data only}
- Dellemijn PL, Verbiest HB, Vliet JJ, Roos PJ, Vecht CJ. Medical therapy of malignant nerve pain. A randomised double‐blind explanatory trial with naproxen versus slow‐release morphine. European Journal of Cancer 1994;30A(9):1244‐50. [DOI] [PubMed] [Google Scholar]
Deschamps 1992 {published data only}
- Deschamps M, Band PR, Hislop TG, Rusthoven J, Iscoe N, Warr D. The evaluation of analgesic effects in cancer patients as exemplified by a double‐blind, crossover study of immediate‐release versus controlled‐release morphine. Journal of Pain and Symptom Management 1992;7(7):384‐92. [DOI] [PubMed] [Google Scholar]
Dhaliwal 1995 {published data only}
- Dhaliwal HS, Sloan P, Arkinstall WW, Thirlwell MP, Babul N, Harsanyi Z, et al. Randomized evaluation of controlled‐release codeine and placebo in chronic cancer pain. Journal of Pain and Symptom Management 1995;10(8):612‐23. [DOI] [PubMed] [Google Scholar]
Ferrell 1989 {published data only}
- Ferrell B, Wisdom C, Wenzl C, Brown J. Effects of controlled released morphine on quality of life for cancer pain. Oncology Nursing Forum 1989;16(4):521‐6. [PubMed] [Google Scholar]
Finn 1993 {published data only}
- Finn JW, Walsh TD, MacDonald N, Bruera E, Krebs LU, Shepard KV. Placebo‐blinded study of morphine sulfate sustained‐release tablets and immediate‐release morphine sulfate solution in outpatients with chronic pain due to advanced cancer. Journal of Clinical Oncology 1993;11(5):967‐72. [DOI] [PubMed] [Google Scholar]
Flöter 1997 {published data only}
- Flöter T, Koch EMW, and the Kap‐Cas study group. Comparison of two oral morphine formulations for chronic severe pain of malignant and non malignant origin Kapanol (R) vs MST (R). Clinical Drug Investigation 1997;14(3):183‐91. [Google Scholar]
Gabrail 2004 {published data only}
- Gabrail NY, Dvergsten C, Ma T, Frailey A, Ahdieh H. Oxymorphone extended‐release (ER) provides safe, effective, and rapid analgesia during opioid radiation: results of a randomized, double‐blind, crossover, comparative study with oxycodone controlled‐release (CR) [abstract]. 2003; Vol. 22:737.
- Gabrail Nashat Y, Dvergsten Chris, Ahdieh Harry. Establishing the dosage equivalency of oxymorphone extended release and oxycodone controlled release in patients with cancer pain: a randomized controlled study. Current Medical Research & Opinion 2004;20(6):911‐8. [DOI] [PubMed] [Google Scholar]
Gillette 1997 {published data only}
- Gillette JF, Ferme C, Moisy N, Mignot L, Schach R, Vignaux J, et al. Double‐blind crossover clinical and pharmacokinetic comparison of oral morphine syrup and sustained release morphine sulfate capsules in patients with cancer‐related pain. Clinical Drug Investigation 1997;14(Suppl 1):22‐7. [Google Scholar]
Gourlay 1997 {published data only}
- Gourlay GK, Cherry DA, Onley MM, Tordoff SG, Conn DA, Hood GM, et al. Pharmacokinetics and pharmacodynamics of twenty‐four‐hourly Kapanol compared to twelve‐hourly MS Contin in the treatment of severe cancer pain. Pain 1997;69(3):295‐302. [DOI] [PubMed] [Google Scholar]
Guo‐Zhu 1997 {published data only}
- Guo‐Zhu Xu, Zhi‐Ji Cai, Yan‐Ping Deng, Jun Hou, Guang‐Ru Xie, Shu‐Jun Liu. Clinical Evaluation of the analgesic effect of sustained release morphine sulfate microgranules in patients with terminal cancer. Clinical Drug Investigation 1997;Suppl: 14(1):34‐42. [Google Scholar]
Hagen 1997 {published data only}
- Hagen NA, Babul N. Comparative clinical efficacy and safety of a novel controlled‐release oxycodone formulation and controlled‐release hydromorphone in the treatment of cancer pain. Cancer 1997;79(7):1428‐37. [PubMed] [Google Scholar]
Hagen 2005 {published data only}
- Hagen NA, Thirlwell M, Eisenhoffer J, Quiqley P, Harsanyi Z, Darke A. Efficacy, safety, and steady state pharmacokinetics of once‐a ‐day controlled release morphine (MS Contin XL) in cancer pain. Journal of Pain and Symptom Management 2005;29(1):80‐90. [DOI] [PubMed] [Google Scholar]
Hanks 1987 {published data only}
- Hanks GW, Twycross RG, Bliss JM. Controlled release morphine tablets: a double blind trial in patients with advanced cancer. Anaesthesia 1987;42(8):840‐4. [DOI] [PubMed] [Google Scholar]
Hanks 1995 {published data only}
- Hanks GW, Hanna M, Finlay I, Radstone DJ, Keeble T. Efficacy and pharmacokinetics of a new controlled‐release morphine sulfate 200‐mg tablet. Journal of Pain and Symptom Management 1995;10(1):6‐12. [DOI] [PubMed] [Google Scholar]
Hanna 2008 {published data only}
- Hanna M, Thipphawong J. A randomized, double‐blind comparison of OROS hydromorphone and controlled‐release morphine for the control of chronic cancer pain. BMC Palliative Care 2008;7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Harris 2003 {published data only}
- Harris JT, Suresh Kumar K, Rajagopal MR. Intravenous morphine for rapid control of severe cancer pain. Palliative Medicine 2003;17:248‐56. [DOI] [PubMed] [Google Scholar]
Heiskanen 1997 {published data only}
- Heiskanen T, Kalso E. Controlled‐release oxycodone and morphine in cancer related pain. Pain 1997;73(1):37‐45. [DOI] [PubMed] [Google Scholar]
- Heiskanen TE, Ruismaki PM, Seppala TA, Kalso EA. Morphine or oxycodone in cancer pain?. Acta Oncologica 2000;39(8):941‐7. [DOI] [PubMed] [Google Scholar]
Homsi 2010 {published data only}
- Homsi J, Walsh D, Lasheen W, Nelson K A, Rybicki L A, Bast J, et al. A comparative study of 2 sustained‐release morphine preparations for pain in advanced cancer. American Journal of Hospice and Palliative Care 2010;27:99‐105. [DOI] [PubMed] [Google Scholar]
Hoskin 1989 {published data only}
- Hoskin PJ, Poulain P, Hanks GW. Controlled release morphine in cancer pain. Is a loading dose required when the formulation is changed?. Anaesthesia 1989;44(11):897‐901. [DOI] [PubMed] [Google Scholar]
Imanaka 2013 {published data only}
- Imanaka K, Tominaga Y, Etropolski M, Hove I, Ohsaka M, Wanibe M. Efficacy and safety of oral tapentadol extended release in Japanese and Korean patients with moderate to severe, chronic malignant tumor‐related pain. Current Medical Research and Opinion 2013;29(10):1399‐409. [DOI: 10.1185/03007995.2013.831816] [DOI] [PubMed] [Google Scholar]
Kalso 1990 {published data only}
- Kalso E, Vainio A. Morphine and oxycodone hydrochloride in the management of cancer pain. Clinical Pharmacology and Therapeutics 1990;47(5):639‐46. [DOI] [PubMed] [Google Scholar]
Kaplan 1998 {published data only}
- Kaplan R, Parris WC, Citron ML, Zhukovsky D, Reder RF, Buckley BJ, et al. Comparison of controlled‐release and immediate‐release oxycodone tablets in patients with cancer pain. Journal of Clinical Oncology 1998;16(10):3230‐7. [DOI] [PubMed] [Google Scholar]
Kerr 2000 {published data only}
- Kerr RO, Tester WJ. A patient preference study comparing two extended‐release morphine sulfate formulations (once‐daily Kadian™ versus twice‐daily MS Contin™ for cancer pain. Clinical Drug Investigation 2000;19(1):25‐32. [Google Scholar]
Klepstad 2003 {published data only}
- Klepstad P, Kaasa S, Jystad A, Hval B, Borchgrevink PC. Immediate‐ or sustained‐release morphine for dose finding during start of morphine to cancer patients: a randomized, double‐blind trial. Pain 2003;101(1‐2):193‐8. [DOI] [PubMed] [Google Scholar]
Knudsen 1985 {published data only}
- Henriksen H, Knudsen J. Controlled evaluation and ongoing experience with sustained release morphine in patients with advanced cancer pain. Advances in Cancer Pain Management. The International Symposium on Pain Control. Toronto: Purdue Frederick Inc, 1986.
- Knudsen J, Mortensen SM, Eikard B, Henriksen H. Slow‐release morphine tablets compared with conventional morphine tablets in the treatment of cancer pain [Morfin‐depottabletter og konventionelle morfintabletter ved cancersmerter]. Ugeskr Læger 1985;147(9):780‐4. [PubMed] [Google Scholar]
Kongsgaard 1998 {published data only}
- Kongsgaard UE, Poulain P. Transdermal fentanyl for pain control in adults with chronic cancer pain. European Journal of Pain 1998;2(1):53‐62. [DOI] [PubMed] [Google Scholar]
Kossman 1983 {published data only}
- Kossman B, Dick W, Bowdler I, Kilian J, Hecht M. Modern aspects of morphine therapy. Advances in Morphine Therapy. The 1983 International Symposium on Pain Control. Royal Society of Medicine International Congress and Symposium Series no 64. London: Royal Society of Medicine, 1983:73‐81. [Google Scholar]
Kress 2008 {published data only}
- Kress HG, Laage D, Hoerauf KH, Nolte T, Heiskanen T, Petersen R, et al. A randomized, open, parallel group, multicenter trial to investigate analgesic efficacy and safety of a new transdermal fentanyl patch compared to standard opioid treatment in cancer pain. Journal of Pain and Symptom Management 2008;36(3):268‐79. [DOI: 10.1016/j.jpainsymman.2007.10.023] [DOI] [PubMed] [Google Scholar]
Lauretti 2003 {published data only}
- Lauretti GR, Oliveira GM, Pereira NL. Comparison of sustained‐release morphine with sustained‐release oxycodone in advanced cancer patients. British Journal of Cancer 2003;89(11):2027‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Leppart 2001 {published data only}
- Leppart W. Analgesic efficacy and side effects of oral tramadol and morphine administered orally in the treatment of cancer pain. Nowotwory 2001;51(3):257‐66. [Google Scholar]
Melzack 1979 {published data only}
- Melzack R, Mount BM, Gordon JM. The Brompton mixture versus morphine solution given orally: effects on pain. Canadian Medical Association Journal 1979;120(4):435‐8. [PMC free article] [PubMed] [Google Scholar]
Mercadante 1998 {published data only}
- Mercadante S, Salvaggio L, Dardanoni G, Agnello A, Garofalo S. Dextropropoxyphene versus morphine in opioid‐naive cancer patients with pain. Journal of Pain and Symptom Management 1998;15(2):76‐81. [PubMed] [Google Scholar]
Mercadante 2008 {published data only}
- Mercadante S, Porzio G, Ferrera P, Fulfaro F, Aielli F, Verna L, et al. Sustained‐release oral morphine versus transdermal fentanyl and oral methadone in cancer pain management. European Journal of Pain 2008;12(8):1040‐6. [DOI: 10.1016/j.ejpain.2008.01.013] [DOI] [PubMed] [Google Scholar]
Mercadante 2010 {published data only}
- Mercadante S, Tirelli W, David F, Arcara C, Fulfaro F, Casuccio A, et al. Morphine versus oxycodone in pancreatic cancer pain: a randomized controlled study. Clinical Journal of Pain 2010;26(9):294‐7. [DOI] [PubMed] [Google Scholar]
Mignault 1995 {published data only}
- Mignault GG, Latreille J, Viguie F, Richer P, Lemire F, Harsanyi Z, et al. Control of cancer‐related pain with MS Contin: a comparison between 12‐hourly and 8‐hourly administration. Journal of Pain and Symptom Management 1995;10(6):416‐22. [DOI] [PubMed] [Google Scholar]
Mizuguchi 1990 {published data only}
- Mizuguchi K, Takeda F, Hiraga K, Nakashima M. Utility evaluation of morphine hydrochloride suppository, AN‐631, in the treatment of cancer pain. Rinshou Igaku 1990;6(11):2357‐76. [Google Scholar]
Moriarty 1999 {published data only}
- Moriarty M, McDonald CJ, Miller AJ. A randomised crossover comparison of controlled release hydromorphone tablets with controlled release morphine tablets in patients with cancer pain. Journal of Clinical Research 1999;2:1‐8. [Google Scholar]
Mucci LoRusso 1998 {published data only}
- Mucci LoRusso P, Berman BS, Silberstein PT, Citron ML, Bressler L, Weinstein SM, et al. Controlled‐release oxycodone compared with controlled‐release morphine in the treatment of cancer pain: A randomized, double‐blind, parallel‐group study. European Journal of Pain 1998;2:239‐49. [DOI] [PubMed] [Google Scholar]
Mystakidou 2005 {published data only}
- Mystakidou K, Katsouda E, Kouloulias V, Kouvaris J, Tsiatas M, Vlahos L. Comparison of transdermal fentanyl with codeine/paracetamol, in combination with radiotherapy, for the management of metastatic bone pain. Journal of Opioid Management 2005;1(4):204‐10. [DOI] [PubMed] [Google Scholar]
O'Brien 1997 {published data only}
- O'Brien T, Mortimer PG, McDonald CJ, Miller AJ. A randomized crossover study comparing the efficacy and tolerability of a novel once‐daily morphine preparation (MXL capsules) with MST Continus tablets in cancer patients with severe pain. Palliative Medicine 1997;11(6):475‐82. [DOI] [PubMed] [Google Scholar]
Oztürk 2008 {published data only}
- Oztürk T, Karadibak K, Catal D, Cakan A, Tugsavul F, Cirak K. Comparison of TD‐fentanyl with sustained‐release morphine in the pain treatment of patients with lung cancer. Agri 2008;20(3):20‐5. [PubMed] [Google Scholar]
Panich 1993 {published data only}
- Panich A, Charnvej L. Comparison of morphine slow release tablet (MST) and morphine sulphate solution (MSS) in the treatment of cancer pain. Journal of Medical Association of Thailand 1993;76(12):672‐6. [PubMed] [Google Scholar]
Parris 1998 {published data only}
- Parris WC, Johnson BW Jr, Croghan MK, Moore MR, Khojasteh A, Reder RF, et al. The use of controlled‐release oxycodone for the treatment of chronic cancer pain: a randomized, double‐blind study. Journal of Pain & Symptom Management 1998;16(4):205‐11. [DOI] [PubMed] [Google Scholar]
Pistevou‐Gompaki 2004 {published data only}
- Pistevou‐Gompaki K, Kouloulias VE, Varveris C, Mystakidou K, Georgakopoulos G, Eleftheriadis N, et al. Radiotherapy plus either transdermal fentanyl or paracetamol and codeine for painful bone metastases: a randomised study of pain relief and quality of life. Current Medical Research and Opinion 2004;20(2):159‐63. [DOI: 10.1185/030079903125002829] [DOI] [PubMed] [Google Scholar]
Portenoy 1989 {published data only}
- Portenoy R, Maldonado M, Fitzmartin R, Kaiko R, Kanner R. Oral controlled release morphine sulfate. Analgesic efficacy and side effects of a 100 mg tablet in cancer pain patients. Cancer 1989;63(Suppl: 11):2284‐8. [DOI] [PubMed] [Google Scholar]
- Portenoy RK, Maldonado M, Fitzmartin R, Kaiko R, Kanner R. Controlled‐release morphine sulfate: analgesic efficacy and side effects of a 100 mg tablet. Journal of Pain and Symptom Management 1988;3(3):16 (supplement). [DOI] [PubMed] [Google Scholar]
Rico 2000 {published data only}
- Rico MA, Cura MA, Harbst H, Palominos A, Figueroa M, Kramer V. Assessment of tramadol as an alternative opioid instead of codeine at the second step of the WHO analgesic scale [Evaluación de tramadol como un opioide alternativoa la codeína en el segundo peldaño de la escalera analgésica de la OMS]. Revista de la Sociedad Espanola del Dolor 2000;7:345‐53. [Google Scholar]
Ridgway 2010 {published data only}
- Ridgway D, Sopata M, Burneckis A, Jespersen L, Andersen C. Clinical efficacy and safety of once‐daily dosing of a novel, prolonged‐release oral morphine tablet compared with twice‐daily dosing of a standard controlled‐release morphine tablet in patients with cancer pain: a randomized, double‐blind, exploratory crossover study. Journal of Pain and Symptom Management 2010;39:712‐20. [DOI] [PubMed] [Google Scholar]
Rodriguez 1994 {published data only}
- Rodriguez M, Barutell C, Rull M, Gálvez R, Pállares J, Vidal F, et al. Efficacy and tolerance of oral dipyrone versus oral morphine for cancer pain. European Journal of Cancer 1994;30A(5):584‐7. [DOI] [PubMed] [Google Scholar]
Rodriguez 2007 {published data only}
- Rodriguez RF, Bravo LE, Castro F, Montoya O, Castillo JM, Castillo MP, Daza P, Restrepo JM, Rodriguez MF. Incidence of weak opioids adverse events in the management of cancer pain: a double‐blind comparative trial. Journal of Palliative Medicine 2007;10(1):56‐60. [DOI] [PubMed] [Google Scholar]
- Rodriguez RF, Castillo JM, Castillo MP, Montoya O, Daza P, Rodríguez MF, et al. Hydrocodone/acetaminophen and tramadol chlorhydrate combination tablets for the management of chronic cancer pain: a double‐blind comparative trial. Clinical Journal of Pain 2008;24(1):1‐4. [DOI: 10.1097/AJP.0b013e318156ca4d] [DOI] [PubMed] [Google Scholar]
- Rodriguez RF, Castillo JM, Pilar Castillo M, Nuñez PD, Rodriguez MF, Restrepo JM, et al. Codeine/acetaminophen and hydrocodone/acetaminophen combination tablets for the management of chronic cancer pain in adults: a 23‐day, prospective, double‐blind, randomized, parallel‐group study. Clinical Therapeutics 2007;29(4):581‐7. [DOI] [PubMed] [Google Scholar]
Salzman 1999 {published data only}
- Salzman RT, Roberts MS, Wild J, Fabian C, Reder RF, Goldenheim PD. Can a controlled‐release oral dose form of oxycodone be used as readily as an immediate‐release form for the purpose of titrating to stable pain control?. Journal of Pain & Symptom Management 1999;18(4):271‐9. [DOI] [PubMed] [Google Scholar]
Smith 1991 {published data only}
- Smith KJ, Miller AJ, McKellar J, Court M. Morphine at gramme doses: kinetics, dynamics and clinical need. Postgraduate Medical Journal 1991;67(Suppl 2):S55‐9. [PubMed] [Google Scholar]
Stambaugh 2001 {published data only}
- Stambaugh JE, Reder RF, Stambaugh M. Double‐blind, randomized, two‐period crossover efficacy and pharmacokinetic comparison of immediate‐release oxycodone (IR) and controlled‐release oxycodone (CR) in cancer patients with pain. Clinical Pharmacology & Therapeutics 1997;61(2):197. [Google Scholar]
- Stambaugh JE, Reder RF, Stambaugh MD, Stambaugh H, Davis M. Double‐blind, randomized comparison of the analgesic and pharmacokinetic profiles of controlled‐ and immediate‐release oral oxycodone in cancer pain patients. Journal of Clinical Pharmacology 2001;41(5):500‐6. [DOI] [PubMed] [Google Scholar]
Thirlwell 1989 {published data only}
- Thirlwell MP, Sloan PA, Maroun JA, Boos GJ, Besner JG, Stewart JH, et al. Pharmacokinetics and clinical efficacy of oral morphine solution and controlled‐release morphine tablets in cancer patients. Cancer 1989;63(Suppl 11):2275‐83. [DOI] [PubMed] [Google Scholar]
Todd 2002 {published data only}
- Todd J, Rees E, Gwilliam B, Davies A. An assessment of the efficacy and tolerability of a 'double dose' of normal‐release morphine sulphate at bedtime.. Palliative Medicine 2002;16:507‐12. [DOI] [PubMed] [Google Scholar]
Vainio 1988 {published data only}
- Vainio A, Tigerstedt I. Opioid treatment for radiating cancer pain: oral administration vs. epidural techniques. Acta Anaesthesiologica Scandinavica 1988;32(3):179‐85. [DOI] [PubMed] [Google Scholar]
van Seventer 2003 {published data only}
- Seventer R, Smit JM, Schipper RM, Wicks MA, Zuurmond WW. Comparison of TTS‐fentanyl with sustained‐release oral morphine in the treatment of patients not using opioids for mild‐to‐moderate pain. Current Medical Research and Opinion 2003;19(6):457‐69. [DOI: 10.1185/030079903125002045] [DOI] [PubMed] [Google Scholar]
Ventafridda 1986 {published data only}
- Ventafridda V, Ripamonti C, Bianchi M, Sbanotto A, Conno F. A randomized study on oral administration of morphine and methadone in the treatment of cancer pain. Journal of Pain and Symptom Management 1986;1(4):203‐7. [DOI] [PubMed] [Google Scholar]
Ventafridda 1989 {published data only}
- Ventafridda V, Saita L, Barletta L, Sbanotto A, Conno F. Clinical observations on controlled release morphine in cancer pain. Journal of Pain and Symptom Management 1989;4(3):124‐9. [DOI] [PubMed] [Google Scholar]
Vielvoye‐Kerkmeer 2002 {published data only}
- Vielvoye‐Kerkmeer A, Tinteren H, Mattern C, Schüller J, Farnell A. Sustained release morphine in cancer pain. European Journal of Palliative Care 2002;9(4):137‐40. [Google Scholar]
Walsh 1985 {published data only}
- Walsh TD. A controlled study of MST Continus tablets for chronic pain in advanced cancer. In: Wilkes E editor(s). Advances in Morphine Therapy: International Symposium on Pain Control. London: Royal Society of Medicine, 1984:99‐102. [Google Scholar]
- Walsh TD. Clinical evaluation of slow release morphine tablets. Advances in Pain Research and Therapy 1985;9:727‐31. [Google Scholar]
Walsh 1992 {published data only}
- Walsh TD, MacDonald N, Bruera E, Shepard KV, Michaud M, Zanes R. A controlled study of sustained‐release morphine sulfate tablets in chronic pain from advanced cancer. American Journal of Clinical Oncology 1992;15(3):268‐72. [DOI] [PubMed] [Google Scholar]
Wilder‐Smith1994 {published data only}
- Wilder‐Smith CH, Schimke J, Osterwalder B, Senn HJ. Oral tramadol, a mu‐opioid agonist and monoamine reuptake‐blocker, and morphine for strong cancer‐related pain. Annals of Oncology 1994;5(2):141‐6. [DOI] [PubMed] [Google Scholar]
Wilkinson 1992 {published data only}
- Wilkinson TJ, Robinson BA, Begg EJ, Duffull SB, Ravenscroft PJ, Schneider JJ. Pharmacokinetics and efficacy of rectal versus oral sustained‐release morphine in cancer patients. Cancer Chemotherapy and Pharmacology 1992;31(3):251‐4. [DOI] [PubMed] [Google Scholar]
Wong 1997 {published data only}
- Wong JO, Chiu GL, Tsao CJ, Chang CL. Comparison of oral controlled‐release morphine with transdermal fentanyl in terminal cancer pain. Acta Anaesthesiologica Sinica 1997;35(3):25‐32. [PubMed] [Google Scholar]
References to studies excluded from this review
Beaver 1978 I {published data only}
- Beaver WT, Wallenstein SL, Rogers A, Houde RW. Analgesic studies of codeine and oxycodone in patients with cancer. I. Comparisons of oral with intramuscular codeine and of oral with intramuscular oxycodone. Journal of Pharmacology and Experimental Therapeutics 1978;207(1):92‐100. [PubMed] [Google Scholar]
Beaver 1978 II {published data only}
- Beaver WT, Wallenstein SL, Rogers A, Houde RW. Analgesic studies of codeine and oxycodone in patients with cancer. II. Comparisons of intramuscular oxycodone with intramuscular morphine and codeine. Journal of Pharmacology and Experimental Therapeutics 1978;207(1):101‐8. [PubMed] [Google Scholar]
Capretti 1970 {published data only}
- Capretti G, Frigerio G. Controlled clinical study of the analgesic activity of a new drug (Z. 424) in pain caused by neoplasms [Studio clinico controllato dell'attivita analgesica di un nuovo farmaco (Z. 424) nel dolore da neoplasie]. La Clinica Terapeutica 1970;52(4):361‐9. [PubMed] [Google Scholar]
Chen 2003 {published data only}
- Chen Y, Zhu W, Liang H, Wu G. The analgesic effect of ibuprofen‐codeine sustained release tablets on postoperative and cancer pain. Chinese Journal of Clinical Rehabilitation 2003;7(8):1290‐1. [Google Scholar]
Coluzzi 2001 {published data only}
- Coluzzi PH, Schwartzberg L, Conroy JD, Charapata S, Gay M, Busch MA, et al. Breakthrough cancer pain: a randomized trial comparing oral transmucosal fentanyl citrate (OTFC) and morphine sulfate immediate release (MSIR). Pain 2001;91(1‐2):123‐30. [DOI: 10.1016/S0304-3959(00)00427-9] [DOI] [PubMed] [Google Scholar]
Jochimsen 1978 I {published data only}
- Jochimsen PR, Lawton RL, VerSteeg K, Noyes R Jr. Effect of benzopyranoperidine, a delta‐9‐THC congener, on pain. Clinical Pharmacology and Therapeutics 1978;24(2):223‐7. [DOI] [PubMed] [Google Scholar]
Leow 1995 {published data only}
- Leow KP, Cramond T, Smith MT. Pharmacokinetics and pharmacodynamics of oxycodone when given intravenously and rectally to adult patients with cancer pain. Anesthesia & Analgesia 1995;80(2):296‐302. [DOI] [PubMed] [Google Scholar]
Moertel 1971 {published data only}
- Moertel CG, Ahmann DL, Taylor WF, Schwartau N. Aspirin and pancreatic cancer pain. Gastroenterology 1971;60(4):552‐3. [PubMed] [Google Scholar]
Noyes 1975 {published data only}
- Noyes R Jr, Brunk SF, Avery DA, Canter AC. The analgesic properties of delta‐9‐tetrahydrocannabinol and codeine. Clinical Pharmacology and Therapeutics 1975;18(1):84‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stambaugh 1987 {published data only}
- Stambaugh JE Jr, McAdams J. Comparison of the analgesic efficacy and safety oral ciramadol, codeine, and placebo in patients with chronic cancer pain. Journal of Clinical Pharmacology 1987;27(2):162‐6. [DOI] [PubMed] [Google Scholar]
Staquet 1971 {published data only}
- Staquet M, Luyckx A, Cauwenberge H. A double‐blind comparison of alclofenac, pentazocine, and codeine with placebo control in pathologic pain. Journal of clinical pharmacology and new drugs 1971;11(6):450‐5. [PubMed] [Google Scholar]
Staquet 1978 {published data only}
- Staquet M, Gantt C, Machin D. Effect of a nitrogen analog of tetrahydrocannabinol on cancer pain. Clinical Pharmacology and Therapeutics 1978;23(4):397‐401. [DOI] [PubMed] [Google Scholar]
Staquet 1993 {published data only}
- Staquet M, Renaud A. Double‐blind randomized trial of piroxicam and codeine in cancer pain. Current Theraputics and Research 1993;53(4):435‐40. [Google Scholar]
Twycross 1977 {published data only}
- Twycross R. The measurement of pain in terminal carcinoma. Journal of International Medical Research 1976;4(Suppl 2):58‐67. [PubMed] [Google Scholar]
- Twycross RG. Choice of strong analgesic in terminal cancer: diamorphine or morphine?. Pain 1977;3:93‐104. [DOI] [PubMed] [Google Scholar]
Additional references
AUREF 2012
- PaPaS author and referee guidance. http://papas.cochrane.org/papas‐documents (accessed 14 March 2014).
Avorn 2008
- Avorn J, Shrank WH. Adverse Drug Reactions in Elderly People: A substantial cause of preventable illness. BMJ 2008;336(7650):956‐7. [DOI: 10.1136/bmj.39520.671053.94] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bailey 2005
- Bailey FA, Burgio KL, Woodby LL, Williams BR, Redden DT, Kovac SH, et al. Improving processes of hospital care during the last hours of life. Arch Intern Med 2005;165:1722‐27. [DOI] [PubMed] [Google Scholar]
Bookbinder 2005
- Bookbinder M, Blank A, Arney E, Wollner D, Lesage P, McHugh M, et al. Improving end of life care: development and pilot test of a clinical pathway. J Pain Symptom Manage 2005;29:529‐43. [DOI] [PubMed] [Google Scholar]
Chan 2013
- Chan RJ, Webster J. End‐of‐life care pathways for improving outcomes in caring for the dying. Cochrane Database of Systematic Reviews 2013, Issue 11. [DOI: 10.1002/14651858.CD008006.pub3] [DOI] [PubMed] [Google Scholar]
Cook 1995
- Cook RJ, Sackett DL. The number needed to treat: a clinically useful measure of treatment effect. BMJ 1995;310(6977):452‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Costantini 2014
- Costantini M, Romoli V, Leo SD, Beccaro M, Bono L, Pilastri P, et al. Liverpool Care Pathway for patients with cancer in hospital: a cluster randomised trial. Lancet 2014;383(9913):226‐37. [DOI: 10.1016/S0140-6736(13)61725-0] [DOI] [PubMed] [Google Scholar]
Derry 2008
- Derry S. Adverse events. In: McQuay HJ, Kalso E, Moore RA editor(s). Systematic Reviews in Pain Research: Methodology Refined. Seattle: IASP Press, 2008:67‐84. [ISBN: 978‐0‐931092‐69‐5] [Google Scholar]
DH 2013
- Anon. More care, less pathway. A review of the Liverpool Care Pathway. Report by Baroness Julia Neuberger (Chair) 2013.
Edwards 1999
- Edwards JE, McQuay HJ, Moore RA, Collins SL. Reporting of adverse effects in clinical trials should be improved: lessons from acute postoperative pain. Journal of Pain and Symptom Management 1999;18(6):427‐37. [DOI: 10.1016/S0885-3924(99)00093-7] [DOI] [PubMed] [Google Scholar]
Ellershaw 2003
- Ellershaw J, Ward C. Care of the dying: the last hours or days of life. BMJ 2003;326:30‐34. [PMC free article] [PubMed] [Google Scholar]
George 2014
- George R, Martin J, Robinson V. The Liverpool care pathway for the dying (LCP): lost in translation and a tale of elephants, men, myopia ‐ and a horse. Palliative Medicine 2014;28(1):3‐7. [DOI: 10.1177/0269216313514706] [DOI] [PubMed] [Google Scholar]
Hadley 2012
- Hadley G, Derry S, Moore RA, Wiffen PJ. Transdermal fentanyl for cancer pain. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.CD010270] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Altman DG, Sterne JAC editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. [Google Scholar]
Ioannidis 2001
- Ioannidis JP, Lau J. Completeness of safety reporting in randomized trials: an evaluation of 7 medical areas. JAMA 2001;285(4):437‐43. [DOI: 10.1001/jama.285.4.437] [DOI] [PubMed] [Google Scholar]
Ioannidis 2004
- Ioannidis JP, Evans SJ, Gotzsche PC, O'Neill RT, Altman DG, Schulz K, et al. Better reporting of harms in randomized trials: an extension of the CONSORT statement. Annals of Internal Medicine 2004;141(10):781‐8. [DOI] [PubMed] [Google Scholar]
Jadad 1996
- Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJM, Gavaghan DJ, McQuay HJ. Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Controlled Clinical Trials 1996;17:1‐12. [DOI] [PubMed] [Google Scholar]
Knights 2013
- Knights D, Wood D, Barclay S. The Liverpool Care Pathway for the dying: what went wrong?. British Journal of General Practice 2013;63(615):509‐10. [DOI: 10.3399/bjgp13X673559] [DOI] [PMC free article] [PubMed] [Google Scholar]
Loke 2001
- Loke YK, Derry S. Reporting of adverse drug reactions in randomised controlled trials ‐ a systematic survey. BMC Clinical Pharmacology 2001;1:13. [DOI: 10.1186/1472-6904-1-3] [DOI] [PMC free article] [PubMed] [Google Scholar]
McQuay 1997
- McQuay HJ, Moore RA. Using numerical results from systematic reviews in clinical practice. Annals of Internal Medicine 1997;126(9):712‐20. [DOI: 10.7326/0003-4819-126-9-199705010-00007] [DOI] [PubMed] [Google Scholar]
Meyer 1996
- Meyer FP, Tröger U, Röhl FW. Adverse nondrug reactions: an update. Clinical Pharmacology and Therapeutics 1996;60(3):347‐52. [DOI] [PubMed] [Google Scholar]
Moore 1998
- Moore RA, Gavaghan D, Tramer MR, Collins SL, McQuay HJ. Size is everything‐‐large amounts of information are needed to overcome random effects in estimating direction and magnitude of treatment effects. Pain 1998;78(3):209‐16. [DOI] [PubMed] [Google Scholar]
Moore 2005
- Moore RA, McQuay HJ. Prevalence of opioid adverse events in chronic non‐malignant pain: systematic review of randomised trials of oral opioids. Arthritis Research and Therapy 2005;7(5):R1046‐51. [DOI: 10.1186/ar1782] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2009
- Moore A, Bjarnason I, Cryer B, Garcia‐Rodriguez L, Goldkind L, Lanas A, et al. Evidence for endoscopic ulcers as meaningful surrogate endpoint for clinically significant upper gastrointestinal harm. Clinical Gastroenterology and Hepatology 2009;7(11):1156‐63. [DOI: 10.1016/j.cgh.2009.03.032.] [DOI] [PubMed] [Google Scholar]
Moore 2010
- Andrew Moore R, Eccleston C, Derry S, Wiffen P, Bell RF, Straube S, McQuay H, ACTINPAIN Writing Group of the IASP Special Interest Group on Systematic Reviews in Pain Relief, Cochrane Pain, Palliative and Supportive Care Systematic Review Group Editors. "Evidence" in chronic pain ‐ establishing best practice in the reporting of systematic reviews. Pain 2010;150(3):386‐9. [DOI: 10.1016/j.pain.2010.05.011] [DOI] [PubMed] [Google Scholar]
Moore 2013
- Moore RA. Endoscopic ulcers as a surrogate marker of NSAID‐induced mucosal damage. Arthritis Research & Therapy 2013;15(Suppl 3):S4. [DOI: 10.1186/ar4176] [DOI] [PMC free article] [PubMed] [Google Scholar]
Murray 2008
- Murray SA, Sheikh A. Care for all at the end of life. BMJ 2008;336(7650):958‐9. [DOI: 10.1136/bmj.39535.491238.94] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nuovo 2007
- Nuovo J, Sather C. Reporting adverse events in randomized controlled trials. Pharmacoepidemiology and Drug Safety 2007;16(3):349‐51. [DOI: 10.1002/pds.1310] [DOI] [PubMed] [Google Scholar]
Nüesch 2010
- Nüesch E, Trelle S, Reichenbach S, Rutjes AW, Tschannen B, Altman DG, et al. Small study effects in meta‐analyses of osteoarthritis trials: meta‐epidemiological study. BMJ 2010;341:c3515. [DOI: 10.1136/bmj.c3515] [DOI] [PMC free article] [PubMed] [Google Scholar]
Olsen 1999
- Olsen H, Klemetsrud T, Stokke HP, Tretli S, Westheim A. Adverse drug reactions in current antihypertensive therapy: a general practice survey of 2586 patients in Norway. Blood Pressure 1999;8(2):94‐101. [DOI] [PubMed] [Google Scholar]
Portenoy 1999
- Portenoy RK, Lesage P. Management of cancer pain. Lancet 1999;353:1695‐1700. [DOI] [PubMed] [Google Scholar]
Regnard 2014
- Regnard C. The demise of the Liverpool Care Pathway: should we ban the highway code because of bad drivers?. Age Ageing 2014;43(2):171‐3. [DOI: 10.1093/ageing/aft195] [DOI] [PubMed] [Google Scholar]
Reidenberg 1968
- Reidenberg MM, Lowenthal DT. Adverse nondrug reactions. New England Journal of Medicine 1968;279(13):678‐9. [DOI] [PubMed] [Google Scholar]
RevMan 2012 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2012.
Rief 2006
- Rief W, Avorn J, Barsky AJ. Medication‐attributed adverse effects in placebo groups: implications for assessment of adverse effects. Archives of Internal Medicine 2006;166(2):155‐60. [DOI: 10.1001/archinte.166.2.155] [DOI] [PubMed] [Google Scholar]
Rief 2009
- Rief W, Nestoriuc Y, Lilienfeld‐Toal A, Dogan I, Schreiber F, Hofmann SG. Differences in adverse effect reporting in placebo groups in SSRI and tricyclic antidepressant trials: a systematic review and meta‐analysis. Drug Safety 2009;32(11):1041‐56. [DOI: 10.2165/11316580-000000000-00000] [DOI] [PubMed] [Google Scholar]
Schmidt‐Hansen 2010
- Schmidt‐Hansen M, Bennett MI, Arnold S. Oxycodone for cancer‐related pain. Cochrane Database of Systematic Reviews 2010, Issue 3. [DOI: 10.1002/14651858.CD003870.pub3] [DOI] [Google Scholar]
Schremmer 2013
- Schremmer C, Derry S, Jackson KC, Wiffen PJ, Bell RF, Strassels S, Straube S. Codeine, alone and with paracetamol (acetaminophen), for cancer pain. Cochrane Database of Systematic Reviews 2013, Issue 5. [DOI: 10.1002/14651858.CD006601.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tassi 2001
- Tassi P, Muzet A. Defining the states of consciousness. Neuroscience and Biobehavioral Reviews 2001;25(2):175‐91. [DOI] [PubMed] [Google Scholar]
Tramèr 1997
- Tramèr MR, Moore RA, McQuay HJ. Propofol and bradycardia: causation, frequency and severity. British Journal of Anaesthesia 1997;76(6):642‐51. [DOI: 10.1093/bja/78.6.642] [DOI] [PubMed] [Google Scholar]
Tramèr 2000
- Tramèr MR, Moore RA, Reynolds DJ, McQuay HJ. 13.Quantitative estimation of rare adverse events which follow a biological progression: a new model applied to chronic NSAID use. Pain 2000;85(1‐2):169‐82. [DOI: 10.1016/S0304-3959(99)00267-5] [DOI] [PubMed] [Google Scholar]
Wiffen 2013
- Wiffen PJ, Wee B, Moore RA. Oral morphine for cancer pain. Cochrane Database of Systematic Reviews 2013, Issue 7. [DOI: 10.1002/14651858.CD003868.pub3] [DOI] [PubMed] [Google Scholar]