

Abstract
The enzyme-substrate complex is inherently transient, rendering its detection difficult. In our framework, IsoLAIT (Isotope-Labeled, Activity-based Identification and Tracking), designed for bisubstrate systems, the common substrate, such as S-adenosyl-L-methionine (AdoMet) for methyltransferases, is replaced by an analogue (e.g., S-adenosyl-L-vinthionine) that, as a probe, creates a tightly bound [enzyme•substrate-probe] complex upon catalysis by thiopurine-S-methyltransferase (TPMT, EC 2.1.1.67). Then, this persistent complex is identified by native mass spectrometry from the cellular milieu without separation. Furthermore, the probe’s isotope pattern flags even unknown substrates and enzymes. IsoLAIT can be broadly applicable for other enzyme systems, particularly those catalyzing group transfer and with multiple substrates, such as glycosyltransferases and kinases.
Keywords: Mass spectrometry, Bioorganic chemistry, Transferases, Enzymes, Substrate identification
Graphical Abstract
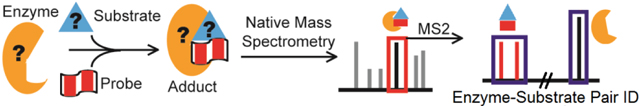
IsoLAIT (Isotope-Labeled, Activity-based Identification and Tracking) is a framework designed to identify enzyme-substrate pairs from complex matrices. By replacing a common enzyme substrate with an isotope-labeled, activity-based probe, detection of the [enzyme•substrate-probe] complex by native mass spectrometry is rendered facile, even if the components are of unknown chemical nature and mass.
In the complex cellular milieu, understanding which enzyme catalyzes which reaction is paramount for deciphering biology and disease. There are still many enzymes whose exact functions or substrates remain unknown. Conversely, we know the nature of some biotransformations, but not precisely the responsible enzymes or proteoforms[1] (specific genetic and splicing variations including any post-translational modifications). For example, more than 80 methyltransferases exist in humans; and while histone substrates are well characterized, non-histone substrates remain poorly understood.[2, 3]
Screening is a common approach for identifying enzyme-substrate pairs.[4–6] For enzyme families with common substrates, such as S-adenosyl-L-methionine (AdoMet or SAM) for methyltransferases or adenosine triphosphate (ATP) for kinases, radiolabeled or affinity-labeled substrates, such as biotin-labeled ATP, or bioorthogonal-tagged analogues, such as propargyl and ketone AdoMet analogues, have been used to screen for enzyme-substrate pairs.[7–14] However, this methodology fails to identify specific enzyme-substrate pairs, particularly from cellular contexts as multiple substrates can be tagged by multiple enzymes with no clear path for deconvolution.
Identifying enzyme-substrate pairs is hampered by the fact that their interactions are inherently transient. In a catalytic cycle (Figure 1A), the enzyme binds the substrates, converts to products, and releases them. With few exceptions,[15–18] ternary complexes are generally weakly bound, so cannot be directly observed by mass spectrometry (MS). However, non-catalytic complexes are observed by native MS. Enhancing the enzyme-substrate affinity is one key to successful detection.
Figure 1.

(A, inset) In the conventional catalytic cycle, an enzyme binds two substrates to create a ternary [enzyme•substrate•substrate] complex. After catalysis, the [enzyme•product•product] complex dissociates, products are released, and the apo-enzyme continues on to substrate binding. The transient enzyme-substrate interactions often do not have sufficient affinity to be analyzed by mass spectrometry. (B) Alternatively, the IsoLAIT platform uses a substrate analogue as a probe. An enzyme catalyzes bisubstrate adduct formation between the probe and the enzyme’s native substrate. The resulting tightly bound [enzyme•substrate-probe] complex can be analyzed by native mass spectrometry. Tandem mass spectrometry dissociates the enzyme from the adduct. The probe’s isotopic flag is easily identified, and serves as a reporter for such complexes; thereby, unknowns can be identified without a priori knowledge of the mass of the components involved. Further analysis can be used to elucidate the structures and sequences of the substrates and enzyme from the complex.
Towards this end, we envision a new enzyme-substrate pair detection platform, dubbed IsoLAIT (Isotope-Labeled, Activity-Based Identification and Tracking). IsoLAIT is a method that links solution-phase activity to gas-phase detection by using a probe to capture, and native MS to identify, enzyme-substrate pairs from a single sample run with minimal sample preparation (Figure 1B).
IsoLAIT has three keys: first, a probe that preserves the interaction between the enzyme and substrate for analysis; next, a detection method that retains enzyme-substrate interactions; and finally, a unique isotopic-flag that clearly identifies the complex, without a priori knowledge of its constituents.
First, the IsoLAIT probe forms an activity-based bisubstrate adduct (Scheme 1), which has sufficient affinity to the enzyme such that the [enzyme•substrate-probe] complex remains intact throughout subsequent analysis. Additionally, high affinity and low turnover enriches the [enzyme•substrate-probe] complex over the transient catalytic complex.
Scheme 1.

(A) S-adenosylmethionine (AdoMet or SAM), is used by thiopurine methyltransferase (TPMT) to methylate aromatic thiols and produces S-adenosylhomocysteine (AdoHcy or SAH). (B) S-Adenosylvinthionine (AdoVin), an AdoMet analogue and methyltransferase probe, forms a covalent bond with aromatic thiols under TPMT catalysis and results in a substrate-probe adduct that tightly binds to the enzyme.
Second, the complex is analyzed by native MS, which maintains protein complexation in the gas phase. Augmented with nano-electrospray ionization, native MS has emerged as a powerful way to characterize protein-ligand binding.[18–20] Upon dissociation, the enzyme and substrate-probe are separated and can be further interrogated, elucidating the identity and structure of each component. An added benefit, the complex is detected in multiple charge states (Figure 2), providing redundancy in complex systems where peaks overlap and helping to deduce the enzyme mass.
Figure 2.

(A) In vitro [TPMT•TNB-AdoVin] complex and apo-TPMT were detected in multiple charge states using native mass spectrometry. In this simple system, the bound and apo- forms of TPMT were easily distinguished (inset). (B) Collision induced dissociation (CID) at a collision energy of 500 eV of the 10+ charged precursor ion of the [TPMT•TNB-AdoVin] complex (*) in (A). This collision resulted in some apo-TPMT with the 10+ and 9+ charge states and the TNB-AdoVin adduct (highlighted and inset). Doublets observed in the TNB-AdoVin spectra are due to a mixture of natural and isotopically labeled AdoVin (+15 Da). (C) [TPMT•AMBA-AdoVin] complex was identified in multiple charge states from whole cells without purification. In this complex matrix, bound and apo- forms of TPMT were not resolved (inset). (D) CID at a collision energy of 500 eV of the 10+ charged precursor ion of the [TPMT•AMBA-AdoVin] complex (*) in (C). After collision, again, multiple charge states for the apo-enzyme were observed along with the AMBA-AdoVin adduct (highlighted and inset). In this case, isotopically labeled probe was not used, resulting in a single peak. Isotopically-labeled spectra were previously reported. [22]
Third, the unique isotopic-flag conferred by the probe is used as a telltale-sign for the [enzyme•substrate-probe] complex. Overcoming the limitation of typical workflows that require pre-defined masses, we are able to identify unknown substrates and enzyme proteoforms with unforeseen modifications.
Herein, the IsoLAIT framework is demonstrated with methyltransferases, a large family of enzymes with diverse substrates. In transmethylation, the enzyme transfers a methyl from common donor AdoMet to a nucleophilic substrate—such as thiopurine methyltransferase[21] (TPMT, EC 2.1.1.67) which methylates thiophenols (Scheme 1A). For this system the selected IsoLAIT probe was S-adenosyl-L-vinthionine (AdoVin).[22, 23]
We posited that TPMT would catalyze the formation of a substrate-probe adduct[7, 24–29] (Scheme 1B); indeed adduct formation between the substrate’s thiol and the vinyl sulfonium in AdoVin was confirmed.[22] However, it was not clear whether such an [enzyme•substrate-probe] complex would bind sufficiently to survive native MS. To investigate, we prepared both in vitro and ex vivo (i.e., E. coli cell lysate) samples (Figure S1). AdoVin was prepared enzymatically[22] with S-adenosyl-methionine synthetase (MAT, EC 2.5.1.6) using both labeled (+15 Da) and unlabeled ATP along with vinthionine (Figure S1).
For the IsoLAIT platform, minimal sample preparation is required. The reaction mixtures were first exchanged into a volatile buffer near physiological pH (e.g., ammonium acetate adjusted to pH 8.0 with ammonium hydroxide) to maintain native conformations and enhance ionization, and then directly infused into the mass spectrometer.
From the in vitro samples, the [enzyme•substrate-probe] complex and apo-enzyme were readily detected and also resolved from each other (Figure 2A, inset). Tandem MS (e.g., collision induced dissociation (CID) at a collision energy of 500 eV) resulted in the apo-enzyme and the substrate-probe adduct (e.g., TNB-AdoVin) (Figure 2B). The latter was easily spotted from its signature 15 Da doublet imparted by the isotopically labeled AdoVin probe (Figure 2B, inset).
Further fragmentation of the TNB-AdoVin adduct was achieved by using higher collision energies for CID (1200 eV) (Figure S2) or quasi-MS3 analysis (Figure S3). The resulting fragments confirmed the identity of the TNB substrate.
As compared to in vitro results, it is not clear whether both adduct-bound and apo-forms of the enzyme from the ex vivo sample were present (Figure 2C, inset). This could be due to peak overlap, a common issue for the analysis of complex biological samples, as well as overall signal strength.
It is worth noting, the mass charge ratio for the ex vivo sample differs from that of the in vitro sample. Tandem MS of the ex vivo samples revealed the substrate-probe adduct to be unexpectedly modified. The nitro group of the TNB substrate was reduced to an amine (2-amino-5-mercaptobenzoic acid or AMBA) in the cell lysate, likely due to endogenous nitroreductases,[22, 30] and the corresponding amine-containing substrate-probe adduct (i.e., AMBA-AdoVin) was formed (Figure 2D). Conventional workflows would likely miss this species as the mass changes were unexpected; however, IsoLAIT is not bound by predefined masses, but screens for mass patterns imparted by the probe and thus will identify substrates no matter their modifications.
Equally interesting is the ability to identify the functional enzyme proteoform. For tandem mass spectra, identifying the apo- and adduct-bound-enzyme peaks was straightforward. Upon closer analysis of apo-TPMT, the observed and theoretical masses differed—the enzyme underwent methionine cleavage when expressed in E. coli (Figure S4). These results highlight IsoLAIT’s utility in identifying an unknown, biologically-relevant, active proteoform.
Complex dissociation using surface induced dissociation (SID) instead of CID yielded similar results (Figure S5). Though redundant in the case of the monomeric TPMT enzyme, as interest in characterizing multimers or protein complexes increases,[20] SID’s utility is significant.[31, 32]
Throughout screening, we attempted to identify the enzyme-substrate complex without the use of a probe. Though TPMT was observed in complex with the common substrate (AdoMet) and byproduct S-adenosylhomocysteine (AdoHcy or SAH) (Figures S6, S7), we did not observe the TPMT enzyme bound to either TNB or AMBA substrates without the use of the AdoVin probe, again highlighting the transient nature of substrate-enzyme interactions.
Through the given examples, IsoLAIT demonstrates its robustness as a general platform, succeeding in contexts where conventional workflows may fail. Peak overlap in complex matrices is overcome through using the probe as a flag in the tandem mass spectra. Importantly, this method requires no prior knowledge of the specific enzyme or substrate for identification. The unexpected alteration of substrate observed in the ex vivo samples perfectly illustrate both the challenges associated with biological systems and the utility of our methods. Had the ex vivo screening been based on the in vitro results, we would have had completely missed the [enzyme•substrate-probe] complex from ex vivo samples due to modifications to both the enzyme and substrate.
What we have demonstrated is a streamlined version of IsoLAIT. The framework could be augmented by including other compatible separation techniques providing even greater detail. Upstream sample purification, inline liquid chromatography methods, and downstream separation, such as ion mobility, which we have successfully used, could further enhance the method.
The IsoLAIT framework solves two key challenges in detecting enzyme-substrate pairs: first, how to overcome the transient nature of enzyme-substrate interactions, and second, how to identify components from a complex mixture with unexpected modifications. The former—transient interaction—is overcome by the combination of a probe that perpetuates enzyme-substrate interactions and native MS that retains binding in the gas phase. The latter—identification—is accomplished by using an activity-based probe and monitoring the telltale isotopic pattern.
This appears to be the first reported example of mass spectrometric identification of enzyme-substrate complexes from the cellular milieu, but it need not be the last. In this case, we used a TPMT specific probe (i.e., AdoVin), but IsoLAIT probes can be tailored for other systems. More promiscuous probes could be used to screen for enzyme-substrate pairs more broadly. For example, aziridinoadenosines have been shown to work as activity-based probes for a range of methyltransferases and form stable, tight-binding bisubstrate adducts.[26, 33–35] Previous studies using chemical tagging and shotgun proteomics may be amenable for IsoLAIT adaptation.[36, 37] IsoLAIT can be broadly applicable and similarly successful for other enzyme systems, particularly those catalyzing group transfer and with multiple substrates, such as glycosyltransferases and kinases.
Experimental Section
General Procedures
All chemicals with reagent purity or above were purchased from Sigma (St. Louis, MO) and Fisher (Pittsburgh, PA) unless otherwise noted. Immobilized metal ion affinity chromatography (IMAC) was performed on HisTrap HP columns. Ultrafiltration was carried out using filters with 10,000 molecular weight cut off (MWCO).
Preparation of S-adenosylvinthionine (AdoVin)
S-Adenosyl-L-vinthionine [CAS 83768-89-2] was prepared enzymatically[22] using S-adenosyl-methionine synthetase (MAT, EC 2.5.1.6) using both labeled (+15 Da) and unlabeled ATP (10 mM) along with vinthionine (1 mM). The reaction proceeded in a 50 mM potassium phosphate buffer (pH 8.0), 5 mM KCl, 2.5 mM MgCl2, and was initiated with 50 μM MAT and incubated at 37 °C. After 2–4 hours incubation, this mixture was used as the in situ probe. AdoVin has similar stability to AdoMet, thus the samples were used immediately and without freezing.
In Vitro Reactions
For the in vitro samples, His-tagged TPMT was grown in transformed E. coli and purified using an IMAC column.[38] Adduct formation was performed in 50 mM potassium phosphate (pH 8.0). The reaction solution contained the in situ AdoVin probe at 1 mM, 500 μM 2-nitro-5-thiobenzoic acid (TNB), 2 mM tris(2-carboxyethyl)phosphine (TCEP), 3.5 μM MTAN and 100 μM TPMT. The reaction was incubated at 37 °C for 3 hours.
Ex Vivo Reactions
For ex vivo reactions, TPMT-transformed E. coli were grown at 37 °C in LB broth, stimulated with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 12 hours, then washed three times with lysis buffer (50 mM potassium phosphate, 0.5 M NaCl, pH 8.0). Cells were lysed via sonication on ice, and the cell debris and unbroken cells were removed by centrifugation (8000 xg, 4 °C, and 60 minutes). To the supernatant, exogenous reagents were added: in situ AdoVin probe at 1 mM, 500 μM TNB, 2 mM TCEP, 3.5 μM MTAN. The reaction was allowed to react at 37 °C for 3 hours.
Sample Preparation
Prior to analysis by MS, all samples were buffer exchanged into 20 mM ammonium acetate (adjusted to pH 8.0 with ammonium hydroxide) using at least ten cycles of concentration and dilution in a 10 kDa MWCO centrifuge, ultrafiltration concentrator. Samples were then frozen at −80 °C and thawed immediately prior to analysis. Note: in the case where freezing denatures or unfolds the proteins, samples should be stored at 4 °C or analyzed immediately. Further sample dilution was done in 20 mM ammonium acetate (pH 8). The concentration of ex vivo [TPMT•AMBA-AdoVin] was estimated at 2 μM by SDS-PAGE. The concentration of the in vitro [TPMT•TNB-AdoVin] was 10 μM as determined by A280nm.
Native Mass Spectrometry
The nanoelectrospray experiments were performed on a Synapt G2S HDMS (Waters Corp., Wilmslow, UK) with a customized surface-induced dissociation (SID) device installed before the ion mobility cell as previously described.[32] Each sample was filled into a glass capillary pulled using a Sutter Instruments P-97 micropipette puller (Novato, CA) and electrically connected to high voltage with a platinum wire. The nanoelectrospray source was at a voltage of 1.2–1.5 kV. The sampling cone voltage was set to 20 V and the source offset voltage was set to 20 V to avoid source activation of the complex. Other instrument conditions were 5 × 10−3 mbar for the source pressure, 2.0 mL/min gas flow rate to the trap cell, 120 mL/min gas flow to the helium cell and 60 mL/min gas flow to the ion mobility cell. The ion mobility wave velocity was 200 m/s and the wave height was 16 V. The TOF analyzer pressure was 1.2 × 10−6 mbar.
Tandem Mass Spectrometry
Tandem mass spectrometry experiments were performed via dissociation of the selected ions with collision induced dissociation (CID) and SID. CID experiments were conducted with a trap gas flow rate of 4.0 mL/min and SID was conducted with a trap gas flow rate of 2.0 mL/min. The CID and SID (MS2) experiments were conducted in the trap travelling wave ion guide region before the ion mobility cell. The acceleration voltage in CID and SID was obtained as described previously.[31, 32] The collision energy in eV was calculated via multiplying the acceleration voltage by the charge state of the precursor ion. The fragment ions generated from MS2 experiment were separated in the ion mobility cell. The quasi MS3 experiment was conducted by selection in the quadrupole, activation in the trap CID cell and separation in the ion mobility cell and activation in the transfer travelling wave ion guide.
Supplementary Material
Acknowledgements
We are grateful for the financial support from NSF DBI-0923551 (to V.H.W.) and NSF DBI-1455654 (to V.H.W.) and NIH NIGMS 1R01GM101396 (to Z.S.Z.). Additionally, K.C.C. would like to thank the ACS WCC for a Fall 2015 WCC / Lilly Travel Grant.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Smith LM, Kelleher NL and P. Consortium for Top Down, Nat Methods 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schubert HL, Blumenthal RM and Cheng in X 1 Protein Methyltransferases: Their Distribution Among the Five Structural Classes of AdoMet-Dependent Methyltransferases, Volume 24 (Eds.: Academic Press, 2006, pp. [DOI] [PubMed] [Google Scholar]
- [3].Tamanoi F and Clarke SG in The Enzymes: Protein Methyltransferases, (Eds.: Elsevier Science, 2011, pp. [Google Scholar]
- [4].Kuhn ML, Majorek KA, Minor W and Anderson WF, Protein Sci 2013, 22, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Levy D, Liu CL, Yang Z, Newman AM, Alizadeh AA, Utz PJ and Gozani O, Epigenetics Chromatin 2011, 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Xu T, Zhang L, Wang X, Wei D and Li T, BMC Bioinformatics 2009, 10, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Peters W, Willnow S, Duisken M, Kleine H, Macherey T, Duncan KE, Litchfield DW, Luscher B and Weinhold E, Angew Chem Int Ed Engl 2010, 49, 5170–5173. [DOI] [PubMed] [Google Scholar]
- [8].Willnow S, Martin M, Luscher B and Weinhold E, Chembiochem 2012, 13, 1167–1173. [DOI] [PubMed] [Google Scholar]
- [9].Lee BW, Sun HG, Zang T, Kim BJ, Alfaro JF and Zhou ZS, J Am Chem Soc 2010, 132, 3642–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jiang H, Kim JH, Frizzell KM, Kraus WL and Lin H, J Am Chem Soc 2010, 132, 9363–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Islam K, Bothwell I, Chen Y, Sengelaub C, Wang R, Deng H and Luo M, J Am Chem Soc 2012, 134, 5909–5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shah K, Liu Y, Deirmengian C and Shokat KM, Proc Natl Acad Sci U S A 1997, 94, 3565–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Senevirathne C, Embogama DM, Anthony TA, Fouda AE and Pflum MK, Bioorg Med Chem 2016, 24, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Green KD and Pflum MK, J Am Chem Soc 2007, 129, 10–11. [DOI] [PubMed] [Google Scholar]
- [15].Yu Y, Kirkup CE, Pi N and Leary JA, J Am Soc Mass Spectrom 2004, 15, 1400–1407. [DOI] [PubMed] [Google Scholar]
- [16].Yin S, Xie Y and Loo JA, J Am Soc Mass Spectrom 2008, 19, 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ganem B, Li YT and Henion JD, Journal of the American Chemical Society 1991, 113, 7818–7819. [Google Scholar]
- [18].Wiseman JM, Takats Z, Gologan B, Davisson VJ and Cooks RG, Angew Chem Int Ed Engl 2005, 44, 913–916. [DOI] [PubMed] [Google Scholar]
- [19].Sun J, Kitova EN, Wang W and Klassen JS, Anal Chem 2006, 78, 3010–3018. [DOI] [PubMed] [Google Scholar]
- [20].Catherman AD, Skinner OS and Kelleher NL, Biochem Biophys Res Commun 2014, 445, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Woodson LC and Weinshilboum RM, Biochem Pharmacol 1983, 32, 819–826. [DOI] [PubMed] [Google Scholar]
- [22].Qu W, Catcott KC, Zhang K, Liu S, Guo JJ, Ma J, Pablo M, Glick J, Xiu Y, Kenton N, Ma X, Duclos RI Jr. and Zhou ZS, J Am Chem Soc 2016, 138, 2877–2880. [DOI] [PubMed] [Google Scholar]
- [23].Zhao G and Zhou ZS, Bioorg Med Chem Lett 2001, 11, 2331–2335. [DOI] [PubMed] [Google Scholar]
- [24].Anderson GL, Bussolotti DL and Coward JK, J Med Chem 1981, 24, 1271–1277. [DOI] [PubMed] [Google Scholar]
- [25].Osborne T, Roska RL, Rajski SR and Thompson PR, J Am Chem Soc 2008, 130, 4574–4575. [DOI] [PubMed] [Google Scholar]
- [26].Comstock LR and Rajski SR, Nucleic Acids Res 2005, 33, 1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang C, Weller RL, Thorson JS and Rajski SR, J Am Chem Soc 2006, 128, 2760–2761. [DOI] [PubMed] [Google Scholar]
- [28].Pljevaljcic G, Pignot M and Weinhold E, J Am Chem Soc 2003, 125, 3486–3492. [DOI] [PubMed] [Google Scholar]
- [29].Gottfried A and Weinhold E, Biochemical Society Transactions 2011, 39, 623–628. [DOI] [PubMed] [Google Scholar]
- [30].Spain JC, Annu Rev Microbiol 1995, 49, 523–555. [DOI] [PubMed] [Google Scholar]
- [31].Zhou M, Jones CM and Wysocki VH, Anal Chem 2013, 85, 8262–8267. [DOI] [PubMed] [Google Scholar]
- [32].Zhou M, Dagan S and Wysocki VH, Angew Chem Int Ed Engl 2012, 51, 4336–4339. [DOI] [PubMed] [Google Scholar]
- [33].Hymbaugh Bergman SJ and Comstock LR, Bioorg Med Chem 2015, 23, 5050–5055. [DOI] [PubMed] [Google Scholar]
- [34].Weller RL and Rajski SR, Chembiochem 2006, 7, 243–245. [DOI] [PubMed] [Google Scholar]
- [35].Weller RL and Rajski SR, Org Lett 2005, 7, 2141–2144. [DOI] [PubMed] [Google Scholar]
- [36].Dugan A, Majmudar CY, Pricer R, Niessen S, Lancia JK, Fung HY, Cravatt BF and Mapp AK, J Am Chem Soc 2016, 138, 12629–12635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jessani N and Cravatt BF, Curr Opin Chem Biol 2004, 8, 54–59. [DOI] [PubMed] [Google Scholar]
- [38].Cannon LM, Butler FN, Wan W and Zhou ZS, Anal Biochem 2002, 308, 358–363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.