

Abstract
Background
Detection of changes in a protein's evolutionary rate may reveal cases of change in that protein's function. We developed and implemented a simple relative rates test in an attempt to assess the rate constancy of protein evolution and to detect cases of functional diversification between orthologous proteins. The test was performed on clusters of orthologous protein sequences from complete bacterial genomes (Chlamydia trachomatis, C. muridarum and Chlamydophila pneumoniae), complete archaeal genomes (Pyrococcus horikoshii, P. abyssi and P. furiosus) and partially sequenced mammalian genomes (human, mouse and rat).
Results
Amino-acid sequence evolution rates are significantly correlated on different branches of phylogenetic trees representing the great majority of analyzed orthologous protein sets from all three domains of life. However, approximately 1% of the proteins from each group of species deviates from this pattern and instead shows variation that is consistent with an acceleration of the rate of amino-acid substitution, which may be due to functional diversification. Most of the putative functionally diversified proteins from all three species groups are predicted to function at the periphery of the cells and mediate their interaction with the environment.
Conclusions
Relative rates of protein evolution are remarkably constant for the three species groups analyzed here. Deviations from this rate constancy are probably due to changes in selective constraints associated with diversification between orthologs. Functional diversification between orthologs is thought to be a relatively rare event. However, the resolution afforded by the test designed specifically for genomic-scale datasets allowed us to identify numerous cases of possible functional diversification between orthologous proteins.
Background
In an early attempt to relate theoretical concepts and empirical results bearing on the molecular basis of evolution, Kimura and Ohta listed five principles of molecular evolution [1,2]. The first of these deals with the concept of the molecular clock and predicts that, as long as a protein's function remains unaltered, the protein's rate of evolution will be approximately constant within different phylogenetic lineages. From this principle, it follows that detection of changes in a protein's rate of evolution may reveal functional changes [3]. This rate-constancy prediction is tested here by studying the relative rates of protein evolution in a simple phylogenetic context. The test relies on the simultaneous analysis of numerous protein sequences that represent complete protein sets from sequenced genomes or partial protein sets from genomes of which a significant fraction has been sequenced. Consistent with the rate-constancy prediction, given a phylogenetic tree topology, the ratio of evolutionary distances along different branches of the tree should be approximately constant for all proteins from an analyzed set of species.
Clusters of orthologous proteins from bacterial, archaeal and eukaryotic species sets of size three were analyzed with respect to this prediction. Specifically, for each protein phylogeny, the length of the longest branch was compared to the sum of the two shorter branches (Figure 1). Such a genome-scale test allowed for a robust assessment of the rate constancy of protein evolution, as well as the detection of multiple cases of accelerated evolutionary change and a quantitative assessment of the fraction of genes that undergo such acceleration.
Figure 1.
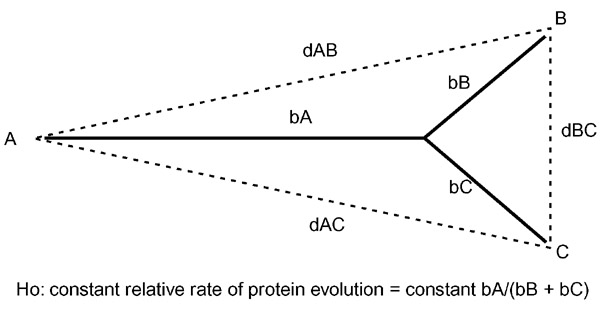
Schematic of an orthologous protein phylogeny and the null hypothesis (Ho) of a constant relative rate of protein evolution. Comparison of multiple orthologous proteins is predicted to reveal a constant relative rate of evolution. This should be manifest as an approximately constant ratio of phylogenetic branch lengths (bold lines) bA/(bB + bC). The branch lengths were calculated using the evolutionary distances (dashed lines) between the proteins from three species (dAB, dAC, dBC) as described in Materials and methods.
Results and discussion
Genomic-scale relative rates test
Three genomes of intracellular pathogenic bacteria of the family Chlamydiaceae (Chlamydophila pneumoniae, Chlamydia trachomatis and Chlamydia muridarum) and three genomes of archaeal hyperthermophiles of the genus Pyrococcus (Pyrococcus abyssi, P. horikoshii and P. furiosus) were chosen for analysis. Orthologs were identified using the rationale and procedure that underlie the construction of clusters of orthologous groups of proteins (COGs) [4,5]. This approach has been designed to maximize the likelihood of identification of orthologs when complete genomes are compared. A modified approach involving two additional steps was used to identify probable orthologs among the encoded proteins from three partially completed mammalian genomes (Homo sapiens (human), Mus musculus (mouse) and Rattus norvegicus (rat).
These procedures resulted in a total of 703 Chlamydiaceae, 1,305 Pyrococcus and 2,112 mammalian orthologous protein sets. Each orthologous protein-sequence set was aligned and the evolutionary distances between sequences within the sets were computed. These distances were used in phylogenetic reconstructions for all orthologous protein sets. For each species group, branch lengths were averaged to produce the trees shown (Figure 2). Each of the trees contains two relatively short branches of virtually identical length and one long branch, forming two main phylogenetic partitions. The rate-constancy prediction holds that the evolutionary rates along these phylogenetic branches should be correlated. In other words, the length of branch A (bA) should be correlated with the length of branches B and C (bB + bC) (Figures 1,2) resulting, in an ideal case, in a constant bA/(bB+bC) ratio.
Figure 2.
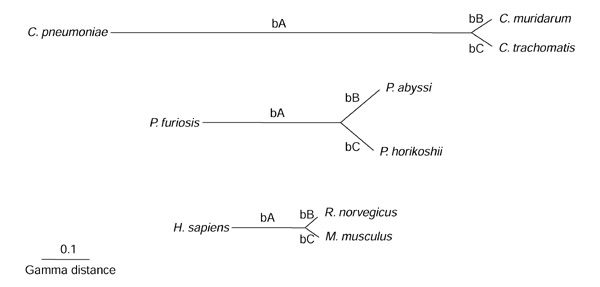
Phylogenies of the three analyzed species groups. Branch lengths are the average of all branch lengths for a given species group. The different branches bA, bB and bC are indicated.
This prediction was tested by evaluating the branch lengths estimated from alignments of orthologous protein sets of the three species groups. Consistent with the prediction, there is a highly significant positive correlation between bA and bB + bC for all three species groups (Figure 3). Thus, the vast majority of proteins in each analyzed group of species seem to evolve in accord with the rate-constancy prediction, suggesting that the functional constraints among orthologous proteins remain largely unchanged. However, in each group of species there are outliers (Figure 3) that do not conform to the expected pattern of variation. For each of the three groups of orthologous protein sets, upper and lower limits of the distance from the linear trend line that correspond to an expectation value of 0.05 were determined. All points above and below these lines (Figure 3) represent statistically significant deviants from the linear trend, given the standard error and the number of orthologous protein sets analyzed for each group of species. Points above the upper limit line show more variation between the phylogenetic partitions (that is, longer bA) than expected by chance, and points below the lower limit line show more variation within one partition (that is, longer bB and/or bC) than expected. The majority of the deviants fall above the upper limit. These points represent proteins with an accelerated rate of amino-acid substitution between phylogenetic partitions. The less frequent outlier points below the lower line represent proteins that have undergone accelerated evolution in one or both of the two more closely related species in the corresponding group. All of the orthologous protein sets that showed significant deviations from the linear trend were re-analyzed using BLAST similarity searches followed by phylogenetic analysis. This was done to verify that these sets contain orthologous proteins. For orthologous proteins, it is expected that they will form monophyletic clades without any intervening paralogs or proteins from distantly related species. For all Chlamydiaceae and mammalian data, the phylogenetic analysis revealed that these sets do in fact consist of orthologous proteins. However, the Pyrococcus analysis yielded five cases where orthology could not be ascertained. These ambiguous Pyrococcus sets were removed and the linear relationship between bA and bB +bC was re-assessed. The total number of orthologous protein sets that show evidence of accelerated evolution suggestive of functional diversification is between 0.76% and 1% of the total number of such sets analyzed for each species (Chlamydiaceae = 7/703, Pyrococcus = 11/1305, human-mouse-rat= 16/2112).
Figure 3.
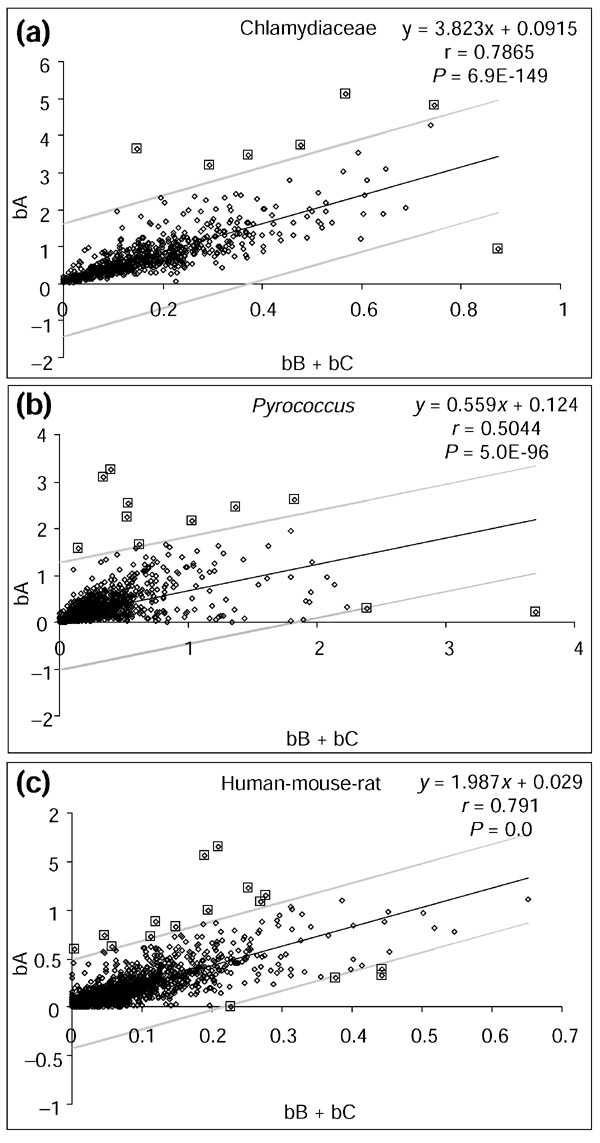
Correlation between the branch lengths in different phylogenetic partitions. Linear regression where the bA branch length (y-axis) is plotted against the sum of bB + bC branch lengths (x-axis) for all orthologous protein sets of each species group. (a) Chlamydiaceae species group including C. pneumoniae, C. muridarum and C. trachomatis. (b)Pyrococcus species group including P. furiosus, P. abyssi and P. horikoshii. (c) Human-mouse-rat species group including H. sapiens, M. musculus and R. norvegicus. The equation for the linear regression trend line (y = mx + b), the correlation coefficient (r) and the level of significance for the correlation (P) are shown on each plot. The linear regression trend line is shown in bold black and the upper and lower limits, corresponding to an expectation value of 0.05, are shown in light gray. For each plot, only 0.05 points are expected to fall outside of these limits by chance. All values are shown as diamonds and the values outside the upper and lower limits that represent functionally diversified orthologous protein sets are indicated by larger squares.
Nucleotide-substitution rates
A number of cases of positive selection at the molecular level have been supported by comparison of the rates of synonymous (Ks) and non-synonymous (Ka) substitution [3]. Ka > Ks is considered unequivocal evidence of positive selection [2]. With this in mind, comparisons of Ks and Ka were carried out for the orthologous protein sets that showed a pattern of variation consistent with functional diversification in the whole-genome test (Figure 3). For the bacterial and archaeal species groups, the vast majority of these comparisons were inconclusive because the evolutionary distance between phylogenetic partitions resulted in a saturation of Ks. The more closely related human-mouse-rat group did not show saturation of Ks. Comparisons of Ks and Ka for functionally diversified mammalian protein sets did not reveal any cases with Ka > Ks. This is probably due to the fact that, if directional selection led to the acceleration of the amino-acid substitution rate in these proteins, it acted in an episodic manner [6,7] and on a minority of residues against a constant background of purifying selection [8,9]. This is consistent with previous work suggesting that adaptive changes are difficult to find using the strict Ka > Kscriterion and that even documented cases of positive selection can be missed using this technique alone [6,10,11,12,13]. The average Ka is, however, significantly higher for the putative functionally diversified proteins than for the total dataset (Table 1, Figure 4). The mammalian proteins with an increased rate of amino-acid substitution between the human and rodent lineages also show a level of Ks significantly higher than that of the total dataset (Table 1). This can be explained by the fact that, consistent with previous reports [10,14,15,16,17], we observe a positive correlation between Ks and Ka (r = 0.556, P < 10-10). This correlation could reflect a mechanistic bias in mutation [10,18,19] or synonymous sites may be subject to some degree of selection [20,21,22,23] (or both). However, the Ka/Ks ratio is also significantly higher for the sets of putative functionally diversified proteins (Table 1), indicating a relative acceleration of amino-acid substitution, which could be due to a relaxation of functional constraints and/or directional selection.
Table 1.
Synonymous (Ks) and non-synonymous (Ka) substitution rates for the human-mouse-rat orthologous protein sets
| All† | Selected‡ | P§ | |
| Human versus rodent* | |||
| Ks | 0.496 ± 0.14 | 0.692 ± 0.19 | 4.5 × 10-3 |
| Ka | 0.079 ± 0.08 | 0.349 ± 0.08 | 2.4 × 10-7 |
| Ka/Ks | 0.147 ± 0.13 | 0.537 ± 0.18 | 1.3 × 10-5 |
| Mouse versus rat¶ | |||
| Ks | 0.172 ± 0.06 | 0.193 ± 0.07 | 0.57 |
| Ka | 0.029 ± 0.03 | 0.148 ± 0.04 | 5.7 × 10-3 |
| Ka/Ks | 0.161 ± 0.16 | 0.787 ± 0.12 | 2.0 × 10-3 |
*Average Ks and Ka for the human-mouse and human-rat pairwise comparisons. †Average and standard deviation values for all protein sets. ‡Average and standard deviation values for the protein sets identified as having accelerated rates of amino-acid substitution. §P value associated with a t-test comparing the means of the all versus selected protein sets. ¶ Average Ks and Ka for the mouse-rat pairwise comparisons.
Figure 4.
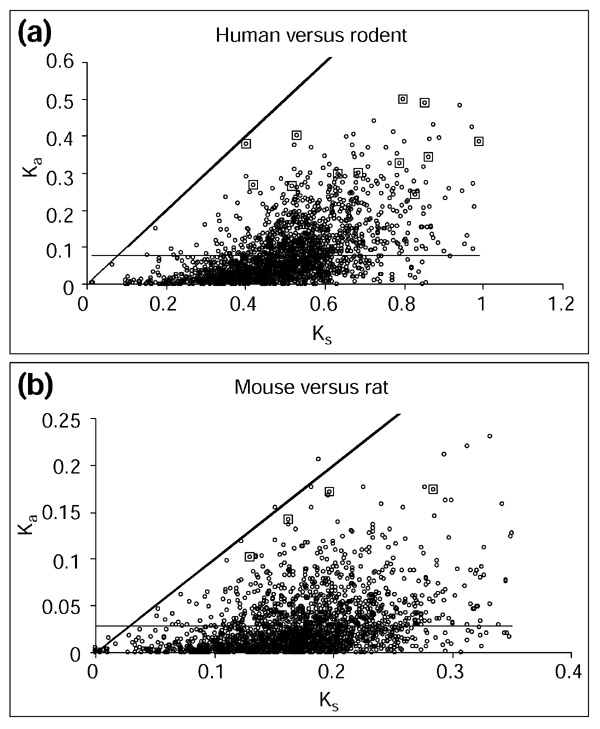
Non-synonymous (Ka) versus synonymous (Ks) substitution rates for the human-mouse-rat orthologous protein sets. (a) Average Ks and Ka for the human-mouse and human-rat pairwise comparisons. (b) Ks and Ka for the mouse-rat pairwise comparisons. Thick diagonal line, Ks = Ka; thin horizontal line, average Ka. All values are shown with circles and the values corresponding to the functionally diversified proteins are indicated by larger squares.
Functional prediction and phyletic distribution
There are some notable similarities in the (predicted) functional identities and the phyletic distributions of the putative functionally diversified proteins (Table 2). The sets from all three species groups are enriched for proteins that appear to function at the periphery of the cells and mediate their interaction with the environment. Specifically, 5 of the 7 cases in Chlamydiaceae, 7 of the 11 Pyrococcus cases and 13 of the 16 mammalian cases are (predicted) membrane or secreted proteins (Table 2). Some of the remaining proteins are completely uncharacterized and could associate with membranes by other mechanisms that do not involve recognizable structural features such as signal peptides or transmembrane segments. The mammalian set of functionally diversified proteins, for which much more experimental information is available than for the bacterial and archaeal sets, is particularly enriched for secreted proteins, including several proteins involved in immune surveillance and defense (Table 2). An example of a protein that is implicated in a different class of functions, but is still related to defense mechanisms is the predicted restriction endonuclease from Pyrococcus. Generally, the functions of proteins for which accelerated evolution was detected and functional diversification inferred are compatible with the previously reported cases of accelerated evolution which is particularly prominent, for example, in the immune system [3].
Table 2.
Domain architecture and functional predictions for functionally diversified proteins
| Gene name (GI numbers)* | Domain organization† | Phyletic distribution‡ | Predicted function§ |
| Chlamydia trachomatis-Chlamydia muridarum-Chlamydophila pneumoniae | |||
| CT079/CP0424/TC0351 (4376613, 3328474, 7190393) | Signal peptide, four transmembrane regions (TMs) | Aquifex aeolicus | Membrane protein, potential receptor |
| CT288/CP0709/TC0561 (4376321, 3328702, 7190602) | Four TMs, coiled coil | Chlamydia-specific, distant additional paralogs | Membrane protein, potential receptor |
| CT656/CP0029/TC0027 (4377021, 3329106, 7190067) | NA | Chlamydia-specific | Unknown |
| CT006/CP0311/TC0274 (4376725, 3328394, 7190315) | Three TMs | Chlamydia-specific | Membrane protein, potential receptor |
| CT036/CP0642/TC0306 (4376393, 3328427, 7190347) | Two TMs | Chlamydia-specific | Membrane protein, potential receptor |
| CT147/CP0623/TC0424 (4376417, 3328548, 7190467) | Three-four TMs, coiled coil | Chlamydia-specific | Membrane protein, potential receptor |
| CT695/CP0071/TC0067 (4376977, 3329149, 7190102) | Low sequence complexity | Chlamydia-specific | Non-globular protein of unknown function |
| Pyrococcus horikoshii-abyssi-furiosus | |||
| PH0310 (301754, 3256700, 5459075) | Cathepsin-like cysteine protease, signal peptide, three to seven TMs | Orthologs only in pyrococci; distantly related cathepsins in animals | Pyrolysin - hyperthermostable membrane protease |
| PH1993, PAB1163 (1849588, 3258437, 5459195) | Signal peptide, deacetylase superfamily hydrolase domain | Methanococcus jannaschii, Pseudomonas aeruginosa, Mycobacterium tuberculosis (COG3233) | Predicted secreted deacetylase |
| PH1708, PAB2041 (76490, 3258139, 5457898) | Signal peptide, low-sequence-complexity regions | Pyrococcus-specific | Secreted, non-globular protein |
| PH0103, PAB0064 (693117, 3256489, 5457538) | Type IV restriction endonuclease, Eco57I homolog | Sporadic distribution in archaea and bacteria (COG2810) | Predicted restriction endonuclease |
| PH1340, PAB1824 (1411868, 3257763, 5458235) | NA | Pyrococcus-specific | Unknown |
| PH0996, PAB0660 (984613, 3257410, 5458405) | Signal peptide, coiled coil, low complexity | Pyrococcus-specific | Secreted, non-globular protein |
| PH0617, PAB1428 (506618, 3257023, 5458851) | Signal peptide, seven TMs | Pyrococcus-specific | Integral membrane protein |
| PH0692, PAB0621 (917540, 3257100, 5458351) | Signal peptide, low sequence complexity regions | Pyrococcus-specific | Secreted, non-globular protein |
| PH0228, PAB0142 (1132938, 3256617, 5457641) | Acetate/butyrate kinase domain | Conserved orthologs in some archaea (Archaeoglobus, Aeropyrum); poorly conserved orthologs in most bacteria (COG2441) | Predicted butyrate kinase |
| PH1703, PAB0312(1599997, 3258134, 5457902) | Signal peptide, low sequence complexity regions | Pyrococcus-specific | Secreted small protein |
| PH0538, PAB0257 (396645, 3256944, 5457867) | Signal peptide, four TMs | Pyrococcus-specific (distant homologs in some archaea and bacteria) | Integral membrane protein |
| Homo sapiens-Mus musculus-Rattus norvegicus | |||
| Interferon precursor (32680, 309328, 2317784) | Signal peptide, interferon gamma domain | Vertebrate-specific | Interferon gamma |
| Interferon-beta-2 (32674, 52702, 204926) | Signal peptide, interleukin 6 domain | Vertebrate-specific | Interleukin 6 (interferon-beta-2) |
| Glycoprotein 34 (219666, 551081, 3779224) | Tumor necrosis factor domain, one TM | Mammal-specific | OX40 ligand (membrane-associated cytokine) |
| Uteroglobin (23132, 49691, 206040) | Signal peptide, uteroglobin domain | Mammal-specific | Secreted phospholipid-binding protein |
| Eotaxin (1280141, 995911, 1707665) | Signal peptide, interleukin 8 domain | Vertebrate-specific | Eotaxin (small inducible CxC cytokine) |
| Taste receptor T2R1 (9625043, 10048430, 7262627) | Signal peptide, seven TMs | Mammal-specific | Taste receptor |
| Relaxin, H2 (35927, 414781, 57044) | Signal peptide, insulin-like growth factor/relaxin family domain | Mammal-specific | Relaxin |
| IgE receptor (34003, 193246, 313673) | Lectin C-type domain (CTL), one transmembrane segment, coiled coil domain | Orthologs only in mammals, lectin domain in all animals | Immunoglobulin E receptor |
| Lactadherin (1381162, 4586464, 1620007) | Signal peptide, epidermal growth factor-like domains, coagulation factor 5/8 C-terminal (discoidin) domain (2) | Orthologs in mammals only, discoidin domain animal-specific | Lactadherin (secreted integrin- and phospholipid-binding protein, involved in antimicrobial defense) |
| Secretin preproprotein (11345450, 313711, 206888) | Glucagon-like hormone domain | Mammal-specific | Secretin |
| C-reactive protein (30213, 50564, 203592) | Signal peptide, pentraxin/C-reactive protein family domain | Vertebrate-specific | C-reactive protein, phosphorylcholine-binding, involved in host defense against bacterial infection |
| Preproapolipoprotein AI (28772, 50015, 202945) | Signal peptide, apolipoprotein A1/A4/E family domain | Vertebrate-specific | Preproapolipoprotein AI |
| EDAG-1 (7677357, 11244774, 11140172) | Regions of low sequence complexity | Mammal-specific | Unknown |
| Deoxyribonuclease II beta (11427442, 6175550, 6470131) | Signal peptide, deoxyribonuclease | Mammals, nematodes, insects (animal-specific) | Deoxyribonuclease (lysosomal enzyme, implicated in apoptosis) |
| Hydroxysteroid sulfotransferase (306702, 496152, 2104492) | Sulfotransferase domain | Animals, plants, mycobacteria | Hydroxysteroid sulfotransferase |
| Hydroxy acid oxidase 3 (7208440, 8926328, 311833) | FMN-dependent oxidoreductase | All eukaryotes, many bacteria | Hydroxy acid oxidase (peroxisomal enzyme) |
*Global identifiers (Gis) for the selected orthologous protein sets. The first GI of each Pyrococcus orthologous set corresponds to the P. furiosus identifiers used at the sequencers' site. †Domain organization was assessed using the SMART and CD-Search servers, and PSI-BLAST search results. TM, predicted transmembrane α-helix; NA (not applicable) indicates that no distinct domains could be identified. ‡ Species or taxa in which homologs were detected in BLAST or PSI-BLAST searches. §Functional prediction was based on the domain architecture and comparison of the results of BLAST searches with the protein annotation in Entrez. The COGnitor server was used to query the COG database for Chlamydiaceae and Pyrococcus proteins. Only Pyrococcus proteins grouped into any existing COGs.
The phyletic distribution of the functionally diversified proteins is typically narrow, particularly in Chlamydiaceae (six of the seven proteins have no homologs outside this bacterial family). Although the selected Pyrococcus proteins showed more evolutionary conservation, the majority had orthologs only in other archaeal genomes, if not only in Pyrococcus. Similarly, 13 of the 16 selected mammalian proteins were either mammal-specific or vertebrate-specific (Table 2). The only two clear exceptions to this pattern of poorly conserved proteins primarily involved in defense and extracellular signaling were the mammalian enzymes hydroxysteroid sulfotransferase and hydroxy acid oxidase. Accelerated evolution in proteins with a narrow phyletic distribution may represent species-specific adaptations in how each organism responds to and processes unique environmental cues.
Conclusions
The present results show that the mode of evolution of the vast majority of the analyzed proteins from all three domains of life - bacteria, archaea and eukaryotes is compatible with the rate-constancy prediction of molecular evolution. A recent analysis of the genome-wide distribution of the evolutionary rates of prokaryotic genes suggested that the shape of this distribution does not significantly change during evolution, although the rates themselves may change [24]. This is consistent with the present finding of a strong correlation between evolutionary rates in different phylogenetic partitions. However, with remarkable consistency, for all three species groups analyzed here, approximately 1% of orthologous genes seem to have experienced accelerated evolution, suggesting functional and potentially adaptive diversification that is conspicuous enough to be detected at a statistically significant level.
It is also a formal possibility that some of the genes identified as functionally diversified may be losing their function. As this would entail a change in function, it can be considered as a special type of functional diversification. However, two points argue against this possibility. First, the Ks/Ka values for mammalian genes are all < 1 (including the putative diversified genes) and thus do not show evidence of loss of function or of pseudogenes. Second, amino-acid sequence alignments of the diversified proteins show that these proteins still retain the identity of critical domains, such as signal peptides and transmembrane domains, as well as critical residues in the active sites of enzymes (Table 2 and data not shown).
Accelerated evolution and functional diversification between paralogs is thought to be common in evolution and is posited as a major mechanism for the generation of new genes and new biochemical functions [25]. Functional diversification between orthologs is considered to be much less common, however. In fact, directional selection leading to functional diversification between orthologs is the least common form of natural selection observed at the molecular level [3]. In the present study, the implementation of a simple test designed specifically to exploit the availability of numerous complete and partially complete genome sequences facilitated the detection of multiple cases of probable functional diversification between orthologs. Application of this method to the growing set of complete genomes is expected to yield similar results and may reveal numerous additional cases of functionally diversified orthologs.
Materials and methods
Sequence analysis
Three closely related Chlamydiaceae genomes and three closely related Pyrococcus genomes were chosen for analysis. These genomes were chosen on the basis of three criteria. First, there were three or more closely related complete genome sequences available; second, the genomes were closely enough related to avoid saturation of amino-acid changes and to ensure unambiguous identification of orthologs; and third, the genomes were distantly enough related to provide meaningful information on amino-acid variation. Predicted proteomes for three Chlamydiaceae genomes (Chlamydophila pneumoniae - CWL029, Chlamydia muridarum and Chlamydia trachomatis) and two Pyrococcus genomes (P. abyssi and P. horikoshii) were obtained from the National Center for Biotechnology Information (NCBI) public server [26]. The predicted proteome for the P. furiosus genome was obtained from the sequencer's site [27]. The method for identifying probable orthologs is based on the genome-specific best hits (BeTs) rationale that underlies the construction of the COGs database [4,5]. For each group of genomes, all-against-all BLAST [28] comparisons were done using the predicted protein sequences. For each query sequence, the BeT (highest score or lowest expectation value) to each of the other proteomes was recorded. Triangles of mutually consistent symmetrical BeTs were detected to form COGs. Only COGs with one representative (ortholog) from each genome (COGs of size 3) were considered for further analysis. This sequence-similarity-based approach is generally not dependent on constant rates of evolution and thus is not misled by rate variation. However, it is a formal possibility that misidentification of orthologs could result from cases of extreme rate variations that would make the orthologs less similar to each other than to some paralogs. This possibility was examined using phylogenetic analysis, as described below.
Messenger RNA sequences from R. norvegicus, Mus musculus and Homo sapiens were obtained using NCBI's batch Entrez server [29]. The encoded amino-acid sequences were clustered at 99% similarity threshold using BLASTCLUST [30] to remove redundant sequences. The BeT procedure was used to identify perfectly symmetrical COGs of size 3 for this species group. Because these mammalian genomes are not complete, a formal possibility exists that the COGs procedure will isolate non-orthologous protein sets. For this reason, two additional steps were added to the ortholog identification procedure. Subsequent to the construction of human-mouse-rat COGs and phylogenetic trees, the 'triplet test' [17] was used to further ensure that the COGs of size 3 contained only orthologs. The triplet test verifies that the mouse and rat members of the COGs are approximately equidistant from the human member. In addition to the triplet test, the distributions of Ks between human and rodents and Ks between mouse and rat was used to eliminate potentially non-orthologous sequence sets. In both cases, the distribution of Ks was approximately normal, with the exception of long and sparsely populated tails. Sequence sets that fell in the tails of the Ks distribution (Ks > 0.35 for mouse-rat and Ks > 1.0 for human-rodent) were removed before analysis to eliminate potentially non-orthologous protein sets. This manual selection process does not appear to bias the results in any way (for example, by eliminating a certain functional class of genes). Omitting this step resulted in the inclusion of a number of distantly related and poorly aligned sequence sets that may not actually include orthologs. Thus, the relative rates test as implemented for the mammalian dataset is a conservative one. Furthermore, the most rapidly evolving genes will not be included in the analysis as many of them will not have detectable orthologs even in closely related genomes.
Orthologous protein sequence sets were aligned using CLUSTALW [31] with default options. Alignments that resulted in any branch lengths (based on the corrected gamma distance, see below) longer than 2 in the case of the Chlamydiaceae and Pyrococcus species groups or longer than 1 in the case of the human-mouse-rat species group were visually inspected and adjusted manually. Manual adjustment included removal of poorly aligning and/or low-sequence-complexity regions identified by BLAST. These adjustments did not result in any qualitative differences in the results of subsequent analyses.
For each orthologous protein set of size 3, evolutionary distances between the sequences were calculated using 3BRANCH program (Y.I.W., unpublished data, available from the author on request) that uses the distance correction based on the gamma distribution of site rate variation [32,33] with an α parameter of 1.0. Phylogenetic branch lengths were calculated from distances by solving the following set of equations (Figure 1):
distance from species A to species B (dAB) = branch length A (bA) + bB,
dAC = bA + bC,
dBC = bB + bC.
For example, solving for bA yields bA = (dAB + dAC - dBC)/2.
For each orthologous gene set, pairwise estimates of the synonymous (Ks) and non-synonymous (Ka) substitution rates were obtained from nucleotide-coding sequence alignments using the Pamilo-Bianchi-Li method [34,35] implemented in the PBLtest program (I.B.R., unpublished data, available from the author on request). Pairwise estimates were averaged to obtain the data presented in Table 1.
Domain architectures of the functionally diversified proteins were assessed by using the SMART server [36,37] and the NCBI's CD-Search server [28,38]. Domain architectures together with the results of PSI-BLAST [28] searches that were run for three iterations, Entrez protein annotation [39] and protein annotations in the COG database [5,40], were used to assign (probable) functions to the selected proteins. The phyletic distribution of these proteins was determined from the PSI-BLAST search results using the TAX_COLLECTOR program of the SEALS package [41]. Additional case-by-case inspection of these results was conducted and neighbor-joining phylogenetic trees [42,43] were constructed to verify the orthologous relationships between the selected proteins. This was done by ensuring that putative orthologous proteins did not show a greater similarity to any paralogs or proteins from more distant species than they did to the proteins included in the analyzed COGs. As a result of this inspection, five Pyrococcus COGs were removed from the original set of selected proteins.
Statistical analysis
For each species set, the standard error of the predicted y-value for each x-value in the regression (Figure 3) was determined. The number of standard errors away from the linear trend line that corresponds to an expectation value of 0.05 was calculated using the inverse of the standard normal cumulative distribution for a probability equal to 1 - (0.05/number of values in plot/2).
Acknowledgments
Acknowledgements
We thank Alexey Kondrashov for many helpful discussions and constructive criticisms, Charles DeLisi, David Landsman, Detlef Leipe, Wojciech Makalowski and Itai Yanai for critical reading of the manuscript and constructive comments and L. Aravind for advice on protein function prediction. The release of the unpublished P. furiosus genome sequence by the Utah Genome Center at the University of Utah is acknowledged and appreciated.
References
- Kimura M, Ohta T. On some principles governing molecular evolution. Proc Natl Acad Sci USA. 1974;71:2848–2852. doi: 10.1073/pnas.71.7.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The Neutral Theory of Molecular Evolution, New York: Cambridge University Press; 1983.
- Hughes AL. Adaptive Evolution of Genes and Genomes, Oxford: Oxford University Press; 1999.
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messier W, Stewart CB. Episodic adaptive evolution of primate lysozymes. Nature. 1997;385:151–154. doi: 10.1038/385151a0. [DOI] [PubMed] [Google Scholar]
- Zhang J, Rosenberg HF, Nei M. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc Natl Acad Sci USA. 1998;95:3708–3713. doi: 10.1073/pnas.95.7.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M, Moore GW, Matsuda G. Darwinian evolution in the genealogy of haemoglobin. Nature. 1975;253:603–608. doi: 10.1038/253603a0. [DOI] [PubMed] [Google Scholar]
- Gonzalez DS, Jordan IK. The alpha-mannosidases: phylogeny and adaptive diversification. Mol Biol Evol. 2000;17:292–300. doi: 10.1093/oxfordjournals.molbev.a026309. [DOI] [PubMed] [Google Scholar]
- Wolfe KH, Sharp PM. Mammalian gene evolution: nucleotide sequence divergence between mouse and rat. J Mol Evol. 1993;37:441–456. doi: 10.1007/BF00178874. [DOI] [PubMed] [Google Scholar]
- Endo T, Ikeo K, Gojobori T. Large-scale search for genes on which positive selection may operate. Mol Biol Evol. 1996;13:685–690. doi: 10.1093/oxfordjournals.molbev.a025629. [DOI] [PubMed] [Google Scholar]
- Sharp PM. In search of molecular Darwinism. Nature. 1997;385:111–112. doi: 10.1038/385111a0. [DOI] [PubMed] [Google Scholar]
- Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graur D. Amino acid composition and the evolutionary rates of protein-coding genes. J Mol Evol. 1985;22:53–62. doi: 10.1007/BF02105805. [DOI] [PubMed] [Google Scholar]
- Mouchiroud D, Gautier C, Bernardi G. Frequencies of synonymous substitutions in mammals are gene-specific and correlated with frequencies of nonsynonymous substitutions. J Mol Evol. 1995;40:107–113. doi: 10.1007/BF00166602. [DOI] [PubMed] [Google Scholar]
- Ohta T, Ina Y. Variation in synonymous substitution rates among mammalian genes and the correlation between synonymous and nonsynonymous divergences. J Mol Evol. 1995;41:717–720. doi: 10.1007/BF00173150. [DOI] [PubMed] [Google Scholar]
- Makalowski W, Boguski MS. Evolutionary parameters of the transcribed mammalian genome: an analysis of 2,820 orthologous rodent and human sequences. Proc Natl Acad Sci USA. 1998;95:9407–9412. doi: 10.1073/pnas.95.16.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averof M, Rokas A, Wolfe KH, Sharp PM. Evidence for a high frequency of simultaneous double-nucleotide substitutions. Science. 2000;287:1283–1286. doi: 10.1126/science.287.5456.1283. [DOI] [PubMed] [Google Scholar]
- Williams EJ, Hurst LD. The proteins of linked genes evolve at similar rates. Nature. 2000;407:900–903. doi: 10.1038/35038066. [DOI] [PubMed] [Google Scholar]
- Miyata T, Hayashida H. Extraordinarily high evolutionary rate of pseudogenes: evidence for the presence of selective pressure against changes between synonymous codons. Proc Natl Acad Sci USA. 1981;78:5739–5743. doi: 10.1073/pnas.78.9.5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipman DJ, Wilbur WJ. Interaction of silent and replacement changes in eukaryotic coding sequences. J Mol Evol. 1984;21:161–167. doi: 10.1007/BF02100090. [DOI] [PubMed] [Google Scholar]
- Ticher A, Graur D. Nucleic acid composition, codon usage, and the rate of synonymous substitution in protein-coding genes. J Mol Evol. 1989;28:286–298. doi: 10.1007/BF02103424. [DOI] [PubMed] [Google Scholar]
- Akashi H. Synonymous codon usage in Drosophila melanogaster : natural selection and translational accuracy. Genetics. 1994;136:927–935. doi: 10.1093/genetics/136.3.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin NV, Wolf YI, Koonin EV. From complete genomes to measures of substitution rate variability within and between proteins. Genome Res. 2000;10:991–1000. doi: 10.1101/gr.10.7.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WH. Molecular Evolution, Sunderland, MA: Sinauer Associates; 1997.
- National Center for Biotechnology Information Bacterial Genomes ftp://ncbi.nlm.nih.gov/genomes/Bacteria/
- Utah Genome Center: P. furiosus ORF sequences http://www.genome.utah.edu/pfu599.pep
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batch Entrez http://www.ncbi.nlm.nih.gov:80/Entrez/batch.html
- NCBI BLAST download ftp://ncbi.nlm.nih.gov/blast/executables/
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota T, Nei M. Estimation of the number of amino acid substitutions per site when the substitution rate varies among sites. J Mol Evol. 1994;38:469–482. doi: 10.1007/BF00175826. [DOI] [PubMed] [Google Scholar]
- Grishin NV. Estimation of the number of amino-acid substitutions per site when the substitution rate varies among sites. J Mol Evol. 1995;41:675–679. doi: 10.1007/BF00175826. [DOI] [PubMed] [Google Scholar]
- Li WH. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol. 1993;36:96–99. doi: 10.1007/BF02407308. [DOI] [PubMed] [Google Scholar]
- Pamilo P, Bianchi NO. Evolution of the Zfx and Zfy genes: rates and interdependence between the genes. Mol Biol Evol. 1993;10:271–281. doi: 10.1093/oxfordjournals.molbev.a040003. [DOI] [PubMed] [Google Scholar]
- Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28:231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMART http://smart.embl-heidelberg.de/
- CD-search http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
- Entrez protein http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Protein
- COGs: Phylogenetic classification of proteins encoded in complete genomes database http://www.ncbi.nlm.nih.gov/COG/
- Walker DR, Koonin EV. SEALS: a system for easy analysis of lots of sequences. Proc Int Conf Intell Syst Mol Biol. 1997;5:333–339. [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]