

Abstract
We propose that the multiple enhancer elements associated with locus control regions and super-enhancers recruit RNA polymerase II and efficiently assemble elongation competent transcription complexes that are transferred to target genes by transcription termination and transient looping mechanisms. It is well established that transcription complexes are recruited not only to promoters but also to enhancers, where they generate enhancer RNAs. Recently, it was shown that transcription at enhancers is unstable and frequently aborted. Furthermore, the Integrator and WD-domain containing protein 82 were shown to mediate transcription termination at enhancers. Abortion and termination of transcription at the multiple enhancers of locus control regions and super-enhancers provides a large pool of elongation competent transcription complexes. These are efficiently captured by strong basal promoter elements at target genes during transient looping interactions.
Keywords: Mediator, phase separation, RNA polymerase II, super-enhancer, transcription regulation
Graphical Abstract
Super-enhancers and locus control regions recruit Mediator and RNA Polymerase II transcription complexes, which form phase separated domains. We propose that transcription abortion and termination at the super-enhancers provides a large pool of transcription competent Pol II that is efficiently captured by strong basal promoter elements at the target genes.
1. Introduction
RNA polymerase II (Pol II) is efficiently recruited to individual enhancer elements of locus control regions (LCRs) and super-enhancers (SEs), and initiates bi-directional transcription. The high concentration of transcription factors, Mediator, and other co-activators at LCRs and SEs establish an environment for the efficient assembly of transcription complexes. The essence of our hypothesis is that transcription termination and abortion releases elongation competent transcription complexes from LCRs and SEs, which are then transferred to high-affinity basal promoters at target genes (Figure 1). Important aspects of this hypothesis are introduced in the following sections, and we end this essay with a more detailed description of the Pol II transfer model using the human β-globin gene locus as an example.
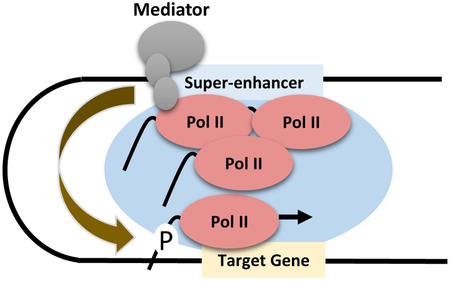
Figure 1: RNA polymerase II transfer model.

Depicted are steps involved in the recruitment of Pol II to SEs, assembly into elongation competent transcription complexes, transcription initiation and elongation, abortion and termination, and transfer to target genes. Transcription factors recruit Mediator and other co-regulators to SEs. Mediator recruits Pol II and assembles a fraction into elongation competent transcription complexes. Transcription is initiated by phosphorylation of the CTD. Early abortion and transcription termination conferred by Integrator releases Pol II, which is dephosphorylated and transferred to target gene promoters.
1.1. Enhancers and Promoters recruit transcription complexes and initiate transcription
Promoters are defined as DNA elements that recruit transcription complexes for the synthesis of coding and non-coding RNA [1,2]. Enhancers are defined as DNA elements that positively regulate transcription at promoters over long distances in a position- and orientation-independent manner [1,2]. However, high-throughput RNA- and ChIP-sequencing studies revealed that many enhancers recruit Pol II and initiate transcription of enhancer RNA (eRNA), thus blurring the functional distinction between enhancers and promoters [3–6]. Enhancers and promoters contain multiple binding sites for transcription factors that are either ubiquitously expressed or restricted in expression to specific cell types, developmental stages, or other stimuli [7]. The transcription factors bind to promoters and enhancers and establish regions of open chromatin, which are detected as heightened sensitivity to DNase I and referred to as hypersensitive sites (HSs) [8]. Enhancer and promoter associated HSs are nucleosome-free or are associated with unstable nucleosomes containing histone variants [1,2,7–9]. Nucleosomes flanking enhancers are characterized by the presence of specific histone marks, including mono-methylation at H3K4 (H3K4me) and acetylation at H3K27 (H3K27ac) [9]. The nucleosomes at active promoters contain elevated levels of tri-methylated H3K4 (H3K4me3), which are also found at highly transcribed enhancers [10].
Transcription at most, but not all, enhancers proceeds in both directions [3,11,12]. Bi-directional transcription also occurs at promoters but it is not as common as at enhancers [13]. Several new studies point to differences in promoter and enhancer associated transcription. Enhancer transcription produces relatively short noncoding RNA and is terminated by Integrator and/or the WD-domain containing protein 82 (WDR82), a component of the Set1A/Set1B histone H3K4 methyltransferase complex that is recruited to transcription start sites (TSSs) by phosphorylated Pol II [14,15]. Furthermore, transcription at enhancers is unstable and often leads to abortion of elongation [16,17]. In contrast, transcription initiation at most Pol II promoters is stable and produces long mRNAs. These are interesting observations indicating that the mechanisms conferring transcription initiation, elongation, and termination are different at enhancers and promoters.
Topological studies revealed that enhancers come in close proximity to target gene promoters during transcription activation [18–20]. In fact, forced looping between an enhancer and a promoter led to transcription activation [21]. According to current gene activation models, the Mediator complex, a transcriptional coactivator, forms a physical bridge between distant regulatory regions and promoters, thereby promoting looping [22]. Transcription of at least a subset of genes regulated by enhancers occurs in bursts indicating a discontinuous process of transcription complex recruitment, assembly, and/or conversion to elongation competent forms [23–25]. The bursting phenomenon suggests that enhancer/promoter contacts may be transient and infrequent. Indeed, recent high resolution imaging experiments provide evidence for transient encounters between enhancers and promoters [26]. The duration of these contacts may vary depending on the number of interacting molecules.
1.2. LCRs and SEs are centers of efficient Pol II recruitment and transcription complex assembly
LCRs are powerful, composite DNA-regulatory elements containing several HSs that function together to mediate high-level expression of target genes [27,28]. LCRs usually regulate multiple genes that are differentially expressed during development or cell-type specification. Transgenic studies have shown that LCRs activate transcription in a position-independent and copy-number dependent manner [27–29]. The LCRs associated with the human and murine β-globin gene loci have been intensively studied over the last 30 years. The β-globin LCR contains multiple HSs that confer extremely high expression levels to the linked globin genes during development [27,28,30,31]. Studies using transgenic mice demonstrated that deletion of individual HSs often resulted in a large but variable reduction in globin gene expression, suggesting that the HSs function synergistically [32–34]. This illustrates that a full complement of HSs is critical for globin activation at ectopic sites, which is significant with respect to using LCRs in gene therapy settings. In contrast, deletions of HS elements from the endogenous mouse globin gene locus revealed that they function additively [35,36]. The results from the transgenic studies suggested that the individual LCR HSs interact to form a large complex that contacts the globin gene promoters. Recent studies that examined the topology of the murine β-globin gene locus at single allele resolution also revealed interactions between the LCR HSs [37]. Thus, the β-globin LCR forms a large holocomplex, in which each HS element contributes more or less equally to LCR function in the native chromosomal environment [32,35–37].
Transcription originating from β-globin LCR HS2 was reported as early as 1992 by the Tuan laboratory [38]. Subsequent studies have shown that all β-globin LCR HSs recruit Pol II and exhibit promoter activity [39–41]. We previously proposed that the β-globin LCR serves as the primary site of transcription complex recruitment, and that transcription at the LCR HSs helps to maintain an accessible chromatin configuration during early stages of erythroid differentiation [42,43]. We further proposed that at later differentiation stages, transcription complexes are transferred from the LCR to the globin genes [42].
SEs are extended chromosomal regions, much longer than traditional enhancers, that are characterized by high accessibility (ATAC-seq or DNase I HSs) as well as by association with histone marks typical for enhancers, including H3K4me1 and H3K27ac [44–46]. It was shown that some SEs are organized like LCRs in that they contain several HSs that function together to mediate high level target gene activation [47,48]. It is not clear at this point whether there is a functional distinction between LCRs and SEs. As mentioned before, LCRs have been shown to confer position-independent expression [29], an activity that, as far as we know, has not been demonstrated for SEs. Like LCR associated HSs, SEs are bound by multiple transcription factors, recruit Pol II, and generate eRNAs. In fact, extremely high levels of Mediator, a large co-regulator complex, and Pol II, are found at SEs [45,46,49]. SEs are associated with highly expressed genes that play important roles in cell-identity during differentiation and development [44]. Recent evidence suggests that at least a subset of SEs is hierarchically organized with hub and non-hub enhancers [47]. Hub-enhancers associate with CTCF and cohesin and are involved in organizing other enhancer elements within SEs for optimal activity. This is true for the whey-acidic protein (WAP) SE in mammary glands [48], in which a distal enhancer is particularly critical for orchestrating SE activity, whereas the other HSs functionally contribute to the combined activating potential of the SE.
1.3. Mediator and the Pol II C-terminal domain (CTD) form phase separated condensates at SEs and LCRs
1.3.1. Mediator recruits the unphosphorylated form of Pol II to SEs and LCRs
Mediator is a large protein complex that functions as a co-regulator for Pol II [50]. It consists of tail, middle, head, and kinase domains that undergo conformational changes during transcriptional activation [51]. Mediator primarily associates with long-distance regulatory DNA elements, including enhancers and SEs [49,52]. Studies in yeast also demonstrated that Mediator primarily interacts with upstream activating sequences [53] and was only found at core promoters in cells in which Pol II activity was inhibited [53,54].
The function of Mediator is not completely understood, but it is involved in the recruitment and assembly of elongation competent Pol II transcription complexes [50]. Thus, while TFIID and other basal transcription factors recruit Pol II to promoters, which are characterized by the presence of consensus basal promoter elements (e.g. TATA, initiator, INR, and downstream promoter element, DPE), the Mediator may be the primary protein complex that governs recruitment and transcription initiation at enhancers. Mediator functionally links transcription factors with components of the basal Pol II transcription machinery [50]. Mediator interacts with the Pol II CTD as well as with basal transcription factors (e.g. TFIID, TFIIB, and TFIIH) and elongation factors (e.g. TFIIS) [50], and regulates enhancer/promoter contacts. Mediator regulated interactions between enhancers and promoters as well as the association of Mediator with components of the preinitiation complex (PIC) are transient [55]. Furthermore, for at least a subset of gene loci Mediator recruits a poised Pol II transcription complex that lacks Kin28, a subunit of TFIIH responsible for the phosphorylation of serine-5 of the CTD [56].
1.3.2. The Pol II CTD is phosphorylated during transcription and dephosphorylated after release from SEs and LCRs.
The CTD is a long, relatively unstructured scaffold of the largest Pol II subunit [57]. It consists of the heptad Tyr1Ser2Pro3Thr4Ser5Pro6Ser7, a motif repeated more than 50 times in humans. All the residues of the heptad except for the prolines are subject to phosphorylation. Ser2 and Ser5 phosphorylation has been extensively studied and shown to play important roles in the transcription cycle [57]. The unphosphorylated form of Pol II is recruited to promoters and interacts with Mediator and TFIID [50,58]. Transcription initiation is accompanied by Ser5 phosphorylation, which is carried out by the Kin28/CDK7 subunit of basal transcription factor TFIIH [59]. Ser5 phosphorylation contributes to the release of Pol II from the transcription start site (TSS) by disrupting interactions with the Mediator and provides a recognition motif for the recruitment of the negative transcription elongation factors DSIF (DRB sensitivity inhibitory factor) and NELF (negative elongation factor) [60]. These two proteins temporarily halt transcription to allow capping of the 5’end of the mRNA. Shortly thereafter, pTEFB (transcription elongation factor B) phosphorylates DSIF and Ser2 leading to the dissociation of NELF and the conversion of DSIF into a positive transcription elongation factor [60]. During transcription elongation there is increased phosphorylation of Ser2, which attracts mRNA processing activities, and decreased phosphorylation of Ser5 [57].
There are several phosphatases capable of removing phosphates from the CTD [61]. RNA Polymerase Associated Protein 2 (RPAP2), the human homologue of yeast Rtr1, is a Ser5 phosphatase that appears to play a role during the transition from transcription initiation to elongation [61]. There is an interesting connection between RPAP2 and SEs. A recent study found that depletion of the RNA Polymerase Associated Protein 1 (RPAP1) caused a reduction of total Pol II and Ser5 phosphorylation mainly at SE regulated genes [62]. RPAP1 is required for the interaction between Mediator and Pol II and for the recruitment of RPAP2. Because Mediator is found primarily at enhancers and prominently at SEs it is reasonable to speculate that RPAP1 and RPAP2 regulate the phosphorylation status of Pol II during early transcription initiation and perhaps after transcription abortion and termination at SEs and LCRs.
1.3.3. Phase separation at LCRs and SEs facilitates assembly of elongation competent transcription complexes.
Recent studies demonstrate that high concentrations of Pol II CTD and Mediator, through multivalent interactions, form condensates leading to the formation of liquid droplets, a process referred to as phase separation [49,63–66]. Formation of liquid droplets is mediated by intrinsically disordered low complexity sequence domains (LCDs) present in the Pol II CTD and in the Mediator subunit Brd4 [49,65]. Phase separation has been observed at SEs, a feature that is likely due to high levels of Pol II and Mediator recruited to these complex DNA-regulatory elements [49,64]. Cho et al. demonstrated that SE target genes associate with the condensates during activation of transcription [64]. Formation of Mediator and Pol II aggregates (condensates) is inhibited in cells exposed to 1,6-hexanediol or to an inhibitor of the Mediator subunit Brd4 [49,64]. Inhibition of phase separation/condensation reduced the levels of Pol II and Mediator at the SEs and diminished transcription of target genes. Interestingly, Boehning et al., [66] showed that phase separation occurred only with the unphosphorylated form of Pol II and that phosphorylation of the CTD releases Pol II and allows transcription elongation at target genes. This is consistent with previous studies demonstrating that unphosphorylated Pol II interacts with LCDs and that this interaction is disrupted by CTD phosphorylation [67]. These data support a previously published model suggesting that phase separation is an important part of SE mediated gene regulation [68].
2. Hypothesis
2.1. Elongation competent Pol II transcription complexes are assembled at LCRs and SEs and transferred to high affinity target promoters during transient looping interactions
We propose that the multiple HSs associated with LCRs and SEs efficiently recruit Pol II and other components of the transcription complex, which are then transferred to high affinity target promoters during transient looping interactions. Efficient recruitment of Pol II and assembly of transcription complexes is the result of two factors: high fractional accessibility at the HSs; and a large number of transcription factors and Mediator that bind to these sites and contact components of the Pol II transcription machinery. The high local concentration of transcription factors and co-regulators at LCRs and SEs provides an optimal environment for the assembly of elongation competent Pol II [49,68]. Transcription initiation within LCR or SE associated HSs occurs bi-directionally and due to instability leads to frequent abortion and release of elongation competent Pol II transcription complexes [11,16]. The initial transcribing complexes may not be phosphorylated at Ser5 because Mediator may recruit a Kin28 deficient TFIIH sub-complex [56]. However, Ser5 that is phosphorylated during initiation and early elongation steps would be dephosphorylated by RPAP2, which is recruited to SEs by Mediator and RPAP1 [62]. We propose that dephosphorylated but otherwise complete elongation competent transcription complexes are transferred to target promoters during transient looping interactions. Assembly and transfer of transcription complexes is facilitated by phase separation, which contributes to concentrating all activities at SEs and LCRs and may also facilitate the recruitment of target genes to the phase separated domains [66]. Because of the multiple HSs associated with SEs and LCRs, the transient interactions allow a large number of elongation competent transcription complexes to be transferred to the strong basal promoter elements (TATA, INR, and DPE) at the target genes, which is consistent with the bursting phenomenon [23–25]. It is notable that target genes of Mediator, and consequently of SEs, contain consensus TATA motifs [69,70]. It is interesting to note that the presence of strong basal promoter elements, especially the TATA box, increases the transcription burst size, while Mediator levels determine bursting frequencies [71–73].
An additional source of elongation competent Pol II may be provided by termination of eRNA transcription mediated by Integrator and other protein complexes [14,15]. The Integrator plays a role in 3’ end cleavage of eRNA transcripts thereby leading to termination of transcription and release of mature eRNA, Pol II, and Integrator complex [14]. The transcription complexes released by Integrator are characterized by the presence of phosphorylated Ser7 and Ser2, which have to be dephosphorylated before transfer to the genes [74]. It is possible that transcription termination of eRNAs by Integrator may prevent formation of long potentially harmful non-coding transcripts. Alternatively, the eRNAs that are released during transcription termination may play a role in the activation of the target genes. For example, the eRNAs could participate in phase separation proposed to be important for enhancer mediated transcription activation [68].
The prevailing model of LCR and SE function is that the distal regulatory elements recruit Mediator and other co-regulators, which either facilitate assembly of Pol II transcription complexes at target promoters and/or promote transcription elongation (“bridging or kissing” models) [64,75]. The recent data on phase separation appear to be consistent with this model. For example, Sabari et al. [49] found that inhibition of phase separation by 1,6-hexanediol led to a reduction of Pol II occupancy at the coding region of an SE target gene but not at the promoter. This is consistent with previous studies on the murine β-globin LCR showing that deletion of the LCR results in defects in transcription elongation [76]. However, we would argue that the data on Mediator and Pol II condensation are also consistent with a Pol II transfer model. In the absence of SE associated phase separation or after deletion of an LCR, the formation of elongation competent transcription complexes at the target promoters is likely very inefficient due to low concentration of the activities required for assembly. Slow assembly of elongation competent transcription complexes accumulates Pol II at the promoters. Elongation competent transcription complexes are efficiently assembled at LCRs and at SEs due to the high concentration of co-activators and Pol II. In contrast to the “bridging/kissing” model, the Pol II transfer model takes into account the fact that SEs and LCRs recruit high levels of Pol II. Especially the data from Boehning et al. suggest that SEs recruit and assemble unphosphorylated Pol II [66]. Transcription associated phosphorylation dissociates Pol II from the phase separated domain at SEs and allow recruitment to target gene promoters during transient looping interactions. This scenario is consistent with observations showing that Mediator recruits Kin28/CDK7 deficient TFIIH and unphosphorylated Pol II [56]. The published data suggest that there are three different forms of Pol II at SEs that are available for transfer to target gene promoters: unphosphorylated Pol II recruited by Mediator [66], Ser5 phosphorylated Pol II released during early abortion of transcription [16], and Ser2 phosphorylated Pol II released by Integrator [14]. As mentioned before, the phosphorylated forms will likely have to be dephosphorylated before they are recruited to target gene promoters.
2.1.1. The human β-globin LCR serves as a center for transcription complex assembly
The Pol II transfer model is summarized in Figure 2 using the human β-globin gene locus as an example. This complex gene locus consists of five genes that are expressed in a developmental stage specific manner [77]. The adult β-globin gene is expressed at extremely high levels in erythroid cells differentiating from stem cells in the bone marrow. The five genes are regulated by an LCR that harbors five HSs [30,31]. Pol II is recruited to all of the LCR HSs and initiates transcription of eRNA [38–41]. We propose that transcription complexes are efficiently recruited and assembled at the LCR HSs in the context of phase separated domains. Most of the recruited and assembled Pol II may be unphosphorylated and phosphorylation takes place after transfer to the globin gene promoters. Phosphorylated Pol II, especially Pol II released by integrator, will be de-phosphorylated before transfer to the globin gene promoter.
Figure 2: LCR mediated activation of β-globin gene transcription in erythroid cells.

The human β-globin gene locus contains five genes (ɛ, Gγ, Aγ, δ, and β) that are regulated by an LCR consisting of 5 HSs (numbered 1 to 5). In adult erythroid cells the LCR interacts with the β-globin gene to mediate high level transcription. We propose that Pol II is recruited to the LCR HSs by Mediator, which together with Pol II forms a phase separated domain optimal for transcription complex assembly. A fraction of the assembled transcription complexes initiate bidirectional transcription (small black arrows). Transcription abortion and Integrator mediated termination releases a large pool of elongation competent Pol II that is dephosphorylated by phosphatases and efficiently captured by basal transcription factors, including TFIID, TFIIB, and TFIIA, which are stably bound at the TATA box and Initiator (INR) of the adult β-globin gene promoter. TFIIH and pTEFB mediated phosphorylation of serine 5 and serine 2 residues of the CTD, respectively, converts Pol II into a high fidelity transcription elongation complex.
The human β-globin LCR has been extensively studied and the proposed model of LCR/SE function is consistent with many of the data accumulated over the last 30 years since its discovery. For example, studies in the endogenous murine β-globin gene locus demonstrated that the LCR HSs function additively and this is explained by each HS providing an equal number of transcription complexes that are transferred to the genes [35,36]. Moreover, inhibition of transcription elongation at the adult β-globin promoter led to increased Pol II levels at the LCR [78]. Finally, Pol II transfer to the β-globin promoter has been recapitulated in vitro using immobilized LCR constructs [79,80]. The Groudine laboratory demonstrated that there is a decrease in the recruitment of Pol II at the adult β-globin gene and an even more dramatic decrease in elongation activity of recruited transcription complexes in the absence of the murine β-globin LCR [76]. This is consistent with our proposed model and suggests that elongation competent Pol II transcription complexes are more efficiently assembled and primed at the LCR HSs, which may represent an erythroid-specific transcription factory [81,82]. Furthermore, the Grosveld laboratory demonstrated that transcription complexes associate with genes in the context of transcription factories and that transcription elongation occurs away from these factories [83]. This is consistent with transcription complex recruitment at SEs and LCRs, transient contact with the target genes, transfer of Pol II to the promoters, and transcription of the genes proceeding away from the phase separated domains [66,83,84]. Moreover, the observation that enhancer mediated target gene transcription occurs in bursts is consistent with transient enhancer/promoter interactions during which elongation competent Pol II is transferred to the genes [23,24].
3. Conclusion and Outlook
Accumulating evidence suggests that SEs and LCRs efficiently recruit and assemble elongation competent transcription complexes. We propose that abortion and termination of transcription at SEs/LCRs releases a large pool of transcription complexes that are efficiently captured by strong basal promoter elements at target gene promoters during transient interactions. The Pol II transfer model may also apply to single enhancers that are transcribed and activate transcription by transient looping interactions with target gene promoters. However, as mentioned previously, enhancers may activate transcription by different mechanisms. For example, enhancers located in close proximity to promoters are unlikely to function by the proposed Pol II transfer mechanism. In the future, it will be important to investigate in more detail the role of the Mediator and the Pol II CTD phosphorylation status at SEs and at promoters. Furthermore, it will be important to design additional experimental settings that will support or refute the Pol II transfer model. For example, inhibiting transcription termination at SEs and LCRs should reduce Pol II at target genes and accumulate it at the distal elements. Likewise, introducing mutations into basal promoter elements, particularly the TATA box, at target genes should prevent transfer to the genes and should increase the levels of Pol II at the SEs and LCRs. Another potential strategy to test the Pol II transfer model is to express a fluorescently labeled Pol II in an inducible manner. If combined with an inducible SE driven transcription system in which the SE and target gene are located far away from each other, Pol II could be tracked by combining fluorescent microscopy and super-resolution DNA-FISH during the activation process [85].
Acknowledgement
We thank our colleagues in the laboratory for stimulating discussions and Drs. Rolf Renne, Michael Kilberg, and Thomas Yang (all at UF) for helpful comments on the manuscript. This work was funded by the NIH (RO1DK52356 to J.B.).
Abbreviations:
- ATAC-seq
assay for transposase-accessible chromatin using sequencing
- ChIP
chromatin immunoprecipitation
- CTD
C-terminal domain
- DSIF
DRB-sensitive inhibitory factor
- eRNA
enhancer RNA
- HS
hypersensitive site
- INI
initiator
- LCD
low complexity sequence domain
- LCR
locus control region
- mRNA
messenger RNA
- NELF
negative elongation factor
- Pol II
RNA polymerase II
- pTEFB
transcription elongation factor B
- RPAP
RNA polymerase associated protein
- SE
super-enhancer
- TSS
transcription start site
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
4. References
- [1].Vernimmen D, Bickmore WA, Trends Genet 2015, 31, 696. [DOI] [PubMed] [Google Scholar]
- [2].Medina-Rivera A, Santiago-Algarra D, Puthier D, Spicuglia S, Trends Biochem Sci 2018, 43, 452. [DOI] [PubMed] [Google Scholar]
- [3].Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME, Nature 2010, 465, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, Natoli G, PLoS Biol 2010, 8, e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kim TK, Shiekhattar R, Cell 2015, 162, 948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Beagrie RA, Pombo A, Bioessays 2016, 38, 881. [DOI] [PubMed] [Google Scholar]
- [7].Bulger M, Groudine M, Cell 2011, 144, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Felsenfeld G, Groudine M, Nature 2003, 421, 448. [DOI] [PubMed] [Google Scholar]
- [9].Zentner GE, Scacheri PC, J Biol Chem 2012, 287, 30888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yang W, Ernst P, Curr Opin Hematol 2017, 24, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mikhaylichenko O, Bondarenko V, Harnett D, Schor IE, Males M, Viales RR, Furlong EEM, Genes Dev 2018, 32, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Andersson R, Refsing Andersen P, Valen E, Core LJ, Bornholdt J, Boyd M, Heick Jensen T, Sandelin A, Nat Commun 2014, 5, 5336. [DOI] [PubMed] [Google Scholar]
- [13].Grzechnik P, Tan-Wong SM, Proudfoot NJ, Trends Biochem Sci 2014, 39, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lai F, Gardini A, Zhang A, Shiekhattar R, Nature 2015, 525, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Austenaa LM, Barozzi I, Simonatto M, Masella S, Della Chiara G, Ghisletti S, Curina A, de Wit E, Bouwman BA, de Pretis S, Piccolo V, Termanini A, Prosperini E, Pelizzola M, de Laat W, Natoli G, Mol Cell 2015, 60, 460. [DOI] [PubMed] [Google Scholar]
- [16].Henriques T, Scruggs BS, Inouye MO, Muse GW, Williams LH, Burkholder AB, Lavender CA, Fargo DC, Adelman K, Genes Dev 2018, 32, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Price DH, Proc Natl Acad Sci U S A 2018, 115, 4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, de Laat W, Nat Genet 2003, 35, 190. [DOI] [PubMed] [Google Scholar]
- [19].Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P, Nat Genet 2002, 32, 623. [DOI] [PubMed] [Google Scholar]
- [20].Benabdallah NS, Bickmore WA, Cold Spring Harb Symp Quant Biol 2015, 80, 45. [DOI] [PubMed] [Google Scholar]
- [21].Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, Blobel GA, Cell 2012, 149, 1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, Taatjes DJ, Dekker J, Young RA, Nature 2010, 467, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fukaya T, Lim B, Levine M, Cell 2016, 166, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lim B, Curr Opin Biotechnol 2018, 52, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bartman CR, Hsu SC, Hsiung CC, Raj A, Blobel GA, Mol Cell 2016, 62, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gu B, Swigut T, Spencley A, Bauer MR, Chung M, Meyer T, Wysocka J, Science 2018, 359, 1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Engel JD, Tanimoto K, Cell 2000, 100, 499. [DOI] [PubMed] [Google Scholar]
- [28].Li Q, Peterson KR, Fang X, Stamatoyannopoulos G, Blood 2002, 100, 3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Grosveld F, van Assendelft GB, Greaves DR, Kollias G, Cell 1987, 51, 975. [DOI] [PubMed] [Google Scholar]
- [30].Tuan D, Solomon W, Li Q, London IM, Proc Natl Acad Sci U S A 1985, 82, 6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Forrester WC, Takegawa S, Papayannopoulou T, Stamatoyannopoulos G, Groudine M, Nucleic Acids Res 1987, 15, 10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bungert J, Davé U, Lim KC, Lieuw KH, Shavit JA, Liu Q, Engel JD, Genes Dev 1995, 9, 3083. [DOI] [PubMed] [Google Scholar]
- [33].Milot E, Strouboulis J, Trimborn T, Wijgerde M, de Boer E, Langeveld A, Tan-Un K, Vergeer W, Yannoutsos N, Grosveld F, Fraser P, Cell 1996, 87, 105. [DOI] [PubMed] [Google Scholar]
- [34].Navas PA, Peterson KR, Li Q, Skarpidi E, Rohde A, Shaw SE, Clegg CH, Asano H, Stamatoyannopoulos G, Mol Cell Biol 1998, 18, 4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bender MA, Ragoczy T, Lee J, Byron R, Telling A, Dean A, Groudine M, Blood 2012, 119, 3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hu X, Bulger M, Bender MA, Fields J, Groudine M, Fiering S, Blood 2006, 107, 821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Allahyar A, Vermeulen C, Bouwman BAM, Krijger PHL, Verstegen MJAM, Geeven G, van Kranenburg M, Pieterse M, Straver R, Haarhuis JHI, Jalink K, Teunissen H, Renkens IJ, Kloosterman WP, Rowland BD, de Wit E, de Ridder J, de Laat W, Nat Genet 2018, 50, 1151. [DOI] [PubMed] [Google Scholar]
- [38].Tuan D, Kong S, Hu K, Proc Natl Acad Sci U S A 1992, 89, 11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Leach KM, Nightingale K, Igarashi K, Levings PP, Engel JD, Becker PB, Bungert J, Mol Cell Biol 2001, 21, 2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Johnson KD, Christensen HM, Zhao B, Bresnick EH, Mol Cell 2001, 8, 465. [DOI] [PubMed] [Google Scholar]
- [41].Routledge SJ, Proudfoot NJ, J Mol Biol 2002, 323, 601. [DOI] [PubMed] [Google Scholar]
- [42].Levings PP, Bungert J, Eur J Biochem 2002, 269, 1589. [DOI] [PubMed] [Google Scholar]
- [43].Liang S, Moghimi B, Yang TP, Strouboulis J, Bungert J, J Cell Biochem 2008, 105, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pott S, Lieb JD, Nat Genet 2015, 47, 8. [DOI] [PubMed] [Google Scholar]
- [45].Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA, Cell 2013, 155, 934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA, Cell 2013, 153, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Huang J, Li K, Cai W, Liu X, Zhang Y, Orkin SH, Xu J, Yuan GC, Nat Commun 2018, 9, 943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shin HY, Willi M, HyunYoo K, Zeng X, Wang C, Metser G, Hennighausen L, Nat Genet 2016, 48, 904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, Li CH, Guo YE, Day DS, Schuijers J, Vasile E, Malik S, Hnisz D, Lee TI, Cisse II, Roeder RG, Sharp PA, Chakraborty AK, Young RA, Science 2018, 361, eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Soutourina J, Nat Rev Mol Cell Biol 2018, 19, 262. [DOI] [PubMed] [Google Scholar]
- [51].Nozawa K, Schneider TR, Cramer P, Nature 2017, 545, 248. [DOI] [PubMed] [Google Scholar]
- [52].Kuras L, Borggrefe T, Kornberg RD, Proc Natl Acad Sci U S A 2003, 100, 13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jeronimo C, Langelier MF, Bataille AR, Pascal JM, Pugh BF, Robert F, Mol Cell 2016, 64, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wong KH, Jin Y, Struhl K, Mol Cell 2014, 54, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jeronimo C, Robert F, Nat Struct Mol Biol 2014, 21, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].D’Urso A, Takahashi YH, Xiong B, Marone J, Coukos R, Randise-Hinchliff C, Wang JP, Shilatifard A, Brickner JH, Elife 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zaborowska J, Egloff S, Murphy S, Nat Struct Mol Biol 2016, 23, 771. [DOI] [PubMed] [Google Scholar]
- [58].Chesnut JD, Stephens JH, Dahmus ME, J Biol Chem 1992, 267, 10500. [PubMed] [Google Scholar]
- [59].Akoulitchev S, Mäkelä TP, Weinberg RA, Reinberg D, Nature 1995, 377, 557. [DOI] [PubMed] [Google Scholar]
- [60].Yamaguchi Y, Shibata H, Handa H, Biochim Biophys Acta 2013, 1829, 98. [DOI] [PubMed] [Google Scholar]
- [61].Mayfield JE, Burkholder NT, Zhang YJ, Biochim Biophys Acta 2016, 1864, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lynch CJ, Bernad R, Calvo I, Nóbrega-Pereira S, Ruiz S, Ibarz N, Martinez-Val A, Graña-Castro O, Gómez-López G, Andrés-León E, Espinosa Angarica V, Del Sol A, Ortega S, Fernandez-Capetillo O, Rojo E, Munoz J, Serrano M, Cell Rep 2018, 22, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chong S, Dugast-Darzacq C, Liu Z, Dong P, Dailey GM, Cattoglio C, Heckert A, Banala S, Lavis L, Darzacq X, Tjian R, Science 2018, 361, eaar2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cho WK, Spille JH, Hecht M, Lee C, Li C, Grube V, Cisse II, Science 2018, 361, 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lu H, Yu D, Hansen AS, Ganguly S, Liu R, Heckert A, Darzacq X, Zhou Q, Nature 2018, 558, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Boehning M, Dugast-Darzacq C, Rankovic M, Hansen AS, Yu T, Marie-Nelly H, McSwiggen DT, Kokic G, Dailey GM, Cramer P, Darzacq X, Zweckstetter M, Nat Struct Mol Biol 2018, 25, 833. [DOI] [PubMed] [Google Scholar]
- [67].Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, Han T, Xie S, Corden JL, McKnight SL, Cell 2013, 155, 1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Hnisz D, Shrinivas K, Young RA, Chakraborty AK, Sharp PA, Cell 2017, 169, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Eychenne T, Novikova E, Barrault MB, Alibert O, Boschiero C, Peixeiro N, Cornu D, Redeker V, Kuras L, Nicolas P, Werner M, Soutourina J, Genes Dev 2016, 30, 2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bruzzone MJ, Grünberg S, Kubik S, Zentner GE, Shore D, Genes Dev 2018, 32, 1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Catarino RR, Stark A, Genes Dev 2018, 32, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Carey LB, van Dijk D, Sloot PM, Kaandorp JA, Segal E, PLoS Biol 2013, 11, e1001528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Tantale K, Mueller F, Kozulic-Pirher A, Lesne A, Victor JM, Robert MC, Capozi S, Chouaib R, Bäcker V, Mateos-Langerak J, Darzacq X, Zimmer C, Basyuk E, Bertrand E, Nat Commun 2016, 7, 12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Rienzo M, Casamassimi A, Biochim Biophys Acta 2016, 1859, 1269. [DOI] [PubMed] [Google Scholar]
- [75].Carlsten JO, Zhu X, Gustafsson CM, Trends Biochem Sci 2013, 38, 531. [DOI] [PubMed] [Google Scholar]
- [76].Sawado T, Halow J, Bender MA, Groudine M, Genes Dev 2003, 17, 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Stamatoyannopoulos G, Exp Hematol 2005, 33, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Barrow JJ, Li Y, Hossain M, Huang S, Bungert J, Nucleic Acids Res 2014, 42, 4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Vieira KF, Levings PP, Hill MA, Crusselle VJ, Kang SH, Engel JD, Bungert J, J Biol Chem 2004, 279, 50350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhou Z, Li X, Deng C, Ney PA, Huang S, Bungert J, J Biol Chem 2010, 285, 15894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, Sim HS, Peh SQ, Mulawadi FH, Ong CT, Orlov YL, Hong S, Zhang Z, Landt S, Raha D, Euskirchen G, Wei CL, Ge W, Wang H, Davis C, Fisher-Aylor KI, Mortazavi A, Gerstein M, Gingeras T, Wold B, Sun Y, Fullwood MJ, Cheung E, Liu E, Sung WK, Snyder M, Ruan Y, Cell 2012, 148, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Cook PR, Science 1999, 284, 1790. [DOI] [PubMed] [Google Scholar]
- [83].Ghamari A, van de Corput MP, Thongjuea S, van Cappellen WA, van Ijcken W, van Haren J, Soler E, Eick D, Lenhard B, Grosveld FG, Genes Dev 2013, 27, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, Eskiw CH, Luo Y, Wei CL, Ruan Y, Bieker JJ, Fraser P, Nat Genet 2010, 42, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, Zhuang X, Science 2018, 362, eaau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]