

Abstract
Chromosomal aberrations were comparatively assessed in nuclei extracted from synovial tissue, primary-culture (P-0) synovial cells, and early-passage synovial fibroblasts (SFB; 98% enrichment; P-1, P-4 [passage 1, passage 4]) from patients with rheumatoid arthritis (RA; n = 21), osteoarthritis (OA; n = 24), and other rheumatic diseases. Peripheral blood lymphocytes (PBL) and skin fibroblasts (FB) (P-1, P-4) from the same patients, as well as SFB from normal joints and patients with joint trauma (JT) (n = 4), were used as controls. Analyses proceeded by standard GTG-banding and interphase centromere fluorescence in situ hybridization. Structural chromosomal aberrations were observed in SFB (P-1 or P-4) from 4 of 21 RA patients (19%), with involvement of chromosome 1 [e.g. del(1)(q12)] in 3 of 4 cases. In 10 of the 21 RA cases (48%), polysomy 7 was observed in P-1 SFB. In addition, aneusomies of chromosomes 4, 6, 8, 9, 12, 18, and Y were present. The percentage of polysomies was increased in P-4. Similar chromosomal aberrations were detected in SFB of OA and spondylarthropathy patients. No aberrations were detected in i) PBL or skin FB from the same patients (except for one OA patient with a karyotype 45,X[10]/46,XX[17] in PBL and variable polysomies in long-term culture skin FB); or ii) synovial tissue and/or P-1 SFB of normal joints or of patients with joint trauma. In conclusion, qualitatively comparable chromosomal aberrations were observed in synovial tissue and early-passage SFB of patients with RA, OA, and other inflammatory joint diseases. Thus, although of possible functional relevance for the pathologic role of SFB in RA, these alterations probably reflect a common response to chronic inflammatory stress in rheumatic diseases.
Keywords: osteoarthritis, rheumatoid arthritis, spondylarthropathy, synovial fibroblasts, trisomy/polysomy 7
Human rheumatoid arthritis (RA) is characterized by chronic inflammation and destruction of multiple joints, perpetuated by an invasive pannus tissue. Activated synovial fibroblasts (SFB), whether reversibly stimulated by the inflammatory microenvironment [1,2,3,4,5,6,7,8] or irreversibly transformed [2,3,4,5,6,7,9,10,11], are a major component of the pannus and contribute to the joint destruction by secretion of proinflammatory cytokines and tissue-degrading enzymes [1].
While specific chromosomal aberrations (e.g. mosaic trisomy/polysomy 7; +7) are of particular interest with regard to malignant transformation and biological behavior of cells, (for example invasiveness, by analogy with observations in hematological malignancies and solid tumors [12]), recent evidence indicates that chromosomal abnormalities may also be of biological relevance in nontrans-formed cells, including synovial cells derived from patients with RA [13,14,15,16,17,18].
To date, analyses of +7 have been carried out either in nonseparated synovial cells (including contaminating inflammatory cells) or in passaged SFB [13,14,15,16,17,18,19]. Thus, the main goals of the present study were: i) to identify the full extent of structural and numerical chromosomal aberrations (see below) in primary-culture (P-0) synovial cells (collagenase digest), and early passages of isolated SFB (98% enrichment; P-1, P-4); ii) to compare these alterations in matched peripheral blood, synovial membrane, and skin of RA patients; iii) to analyze the specificity of the aberrations for RA by comparison with other rheumatic diseases (inflammatory/degenerative) and normal joints or joints that had undergone trauma; and iv) within synovial and skin fibroblasts (FB), to differentiate in vivo alteration from in vitro growth selection.
Materials and methods
Patients
Patients with RA (n = 21), OA (n = 24), or spondylarthropathies (n = 3; consisting of one case of ankylosing spondylitis and two of psoriatic arthritis), villonodular synovitis; systemic lupus erythematosus; juvenile rheumatoid arthritis; undifferentiated monoarthritis, and reactive arthritis (n = 1 each; Supplementary material) were classified according to criteria from the American College of Rheumatology/American Rheumatism Association or the European Spondylarthropathy Study Group [20,21,22,23,24]. Synovial tissue/cells from four patients with either no joint disease (postmortem samples) or recent joint trauma, and skin samples from four normal donors (derived from plastic surgery of the abdominal wall; mean sample size approximately 20 cm2), were used as controls (Supplementary material).
Inflamed synovial tissue, heparinized peripheral blood, and skin (from the edge of the surgical incision; approximately 0.3–0.6 cm2 in RA and OA) were obtained during open joint replacement surgery or arthroscopic synovectomy with the approval of the responsible ethics committees. Paired blood samples were immediately transferred to the Institutes of Human Genetics, Friedrich Schiller University Jena or University of Leipzig, for lymphocyte culture and karyotype/FISH analysis. Synovial tissue and skin were placed in cell culture medium at ambient temperature and subjected to tissue digestion within 2 h.
Tissue digestion, cell culture, and fibroblast isolation
Isolation/fluorocytometry of primary-culture SFB was performed as described elsewhere [25,26], resulting in enrichment of SFB (Thy-1+: RA 72.1%, n = 13; OA 71.5%, n = 15; and prolyl 4-hydroxylase+: RA 80.3%; n = 9; OA 93.1%, n = 9), with a contamination of <2% leukocytes or endothelial cells.
Primary-culture normal skin FB were prepared as published previously [25].
GTG-banding and fluorescence in situ hybridization
Peripheral blood lymphocytes (PBL) were analyzed using standard methods [19]. Synovial and skin FB were subjected to colcemid, hypotonic treatment, fixation with methanol/acetic acid, and air-drying. GTG-banding was performed according to standard protocols [27] on 10–50 metaphases/case. Karyotypes were described in accordance with the International System for Human Cytogenetic Nomenclature (ISCN) 1995 [28].
Nuclei were extracted from formalin-fixed/paraffin-embedded or cryofixed tissue by the method of Liehr et al [29,30]. Fluorescence in situ hybridization (FISH) with centromere probes was performed in interphase nuclei using standard protocols (VYSIS, Downers Grove, IL, USA). Four centromere probes were selected according to the results of GTG banding.
Data were analyzed and depicted either on the basis of the total polysomy of nuclei, i.e. focusing on the total gain of potential gene transcription units, or selectively on the basis of trisomic nuclei, focusing on mitotic nondisjunction as a possible underlying mechanism.
Statistical analysis
Data were analyzed using the multigroup Kruskal–Wallis test, the nonparametric Mann–Whitney U test, and the Spearman rank correlation test (SPSS 9.0™; Chicago, IL, USA; P ≤ 0.05).
Results
Structural chromosomal aberrations in RA
In 4 of 21 patients with RA (19%), structural chromosomal aberrations were observed in P-1 or P-4 SFB, with involvement of chromosome 1 in 3 of 4 cases (Supplementary material). Structural aberrations of the chromosomes 15 and X were also observed in one patient each. In P-1 skin FB, only a breakage of chromosome 5 was observed in one RA patient (Supplementary material). The enlarged secondary constriction observed on the long arm of chromosome 16 in all PBL and SFB metaphases of one RA patient is a known morphological variant without pathologic relevance (Supplementary material). In compliance with ISCN guidelines [28], only clonal structural aberrations occurring in more than one cell (i.e. the composite deletion in chromosome 15) were reported in the cytogenetic findings (Table 1).
Table 1.
Cytogenetic findings in various cells/nuclei from patients with rheumatoid arthritis
| Karyotype and FISH resultsa | |||||
| Peripheral blood lymphocytes | Nuclei extracted from synovial tissueb | Collagenase digest | Synovial fibroblasts | ||
| Patient | P-1 | P-4 | |||
| ES43 | 46,XX | n.a. | n.a. | 46,XX | n.a. |
| ES51 | 46,XX | n.a. | n.a. | 47,XX,+7[9]/46,XX [41] | n.a. |
| ES79 | 46,XY | n.a. | n.a. | 46,XY | n.a. |
| L07 | 46,XY | n.a. | n.a. | 46,XY | n.a. |
| L12 | 46,XY | n.a. | n.a. | 46,XY | n.a. |
| VS01 | 46,XY | n.a. | n.a. | 46,XY | n.a. |
| AA5 | n.a. | n.a. | n.a. | n.a. | n.a. |
| FISH:+6 [23%];+7 [19%];+8 [17%]; +9 [18%];incl. polyploid [11%] | |||||
| EB1 | 46,XY | n.a. | n.a. | 46,XY | n.a. |
| EB2 | 46,XX | n.a. | n.a. | 46,XX | n.a. |
| FISH:+6 [16%];+7 [31%];+8 [22%]; +9 [20%];incl. polyploid [14%] | |||||
| EB4 | 46,XX | n.a. | n.a. | n.a. | 46,XX |
| FISH:+7 [33%];+8 [12%]; -9 [15%];+18 [9%];-18 [11%]; incl. polyploid [4%] | |||||
| EB5 | 46,XY | n.a. | n.a. | 46,XY,del(15)(q22),-21,+mar [cp3]/46,XY[2] | 46,XY (Passage 6) |
| FISH:+6 [5%];+7 [11%];-8 [8%]; +9 [6%];incl. polyploid [2%] | FISH: +7 [6%];+8 [10%];+9 [6%]; incl. polyploid [4%] | ||||
| EB6 | 46,XX | n.a. | 47,XX,+76;46,XX[6] | n.a. | |
| FISH:+6 [7%];+7 [12%]; +8 [2%];-8 [0%];-18 [8%]; incl. polyploid [1%] | FISH:+6 [3%];+7 [40%];+8 [5%]; -8 [8%];-18 [9%] | FISH:+6 [13%];+7 [58%];+8 [8%]; -8 [4%];-18 [84%]; incl. polyploid [2%] | |||
| EB7 | n.a. | n.a. | 46,XY (1/12 metaphases 45,X,-Y | 46,XY (1/10 metaphases 45,X,-Y,+7?) | |
| FISH:+6 [6%];+7 [3%]; -18 [9%];+/-Y [n.a.]; incl. polyploid [1%] | FISH:+6 [n.a.];+7 [5%];-18 [8%]; -Y [5%];+Y [4%]; incl. polyploid [2%] | FISH: :+6 [n.a.];+7 [4%];-18 [n.a.]; -Y [30%];+Y [0%] | |||
| EB20 | 46,XY | 46,XY | 46,XY | n.a. | |
| FISH:normal | FISH:+7 [2%];+12 [5%] | FISH:+7 [4%]; +12 [5%] | FISH:+7 [9%];+12 [14%]; incl. polyploid [1%] | ||
| EB23 | 46,XX | n.a. | 46,XX | 46,XX | 46,XX |
| FISH:normal | FISH:+7 [4%]; +9 [3%];+18 [1%] | FISH:+7 [9%];+9 [5%];+18 [2%] | FISH:+7 [13%];+9 [11%];+18 [7%] | ||
| EB25 | 46,XX | n.a. | 46,XX | n.a. | |
| FISH:normal | FISH:+8 [5%] | FISH:normal | |||
| EB26 | 46,XY | 46,XY | 46,XY | 47,XY,+74/46,XY[4] | |
| FISH:normal | FISH:+6 [2%];+7 [8%]; +8 [n.a.];+9 [3%] | FISH:+6 [0%]; +7 [12%];+8 [3%]; +9 [0%] | FISH:+6 [3%];+7 [14%];+8 [5%]; +9 [2%];incl. polyploid [2%] | FISH:+6 [10%];+7 [9%];+8 [n.a.]; +9 [9%];incl. polyploid [1%] | |
| EB27 | 46,XX | n.a. | 47,XX,+7[2] | n.a. | |
| FISH:normal | FISH:+7 [0%]; +8 [8%];+18 [7%] | FISH:+7 [7%]; +8 [n.a.];+18 [n.a.] | FISH:+7 [13%];+8 [n.a.];+18 [n.a.] incl. polyploid [2%] | FISH:+7 [20%];+8 [n.a.];+18 [n.a.] incl. polyploid [4%] | |
| EB28 | 46,XY | n.a. | n.a. | n.a. | |
| FISH:normal | FISH:+4 [8%];+7 [12%] | ||||
| EB30 | 46,XX | n.a. | n.a. | n.a. | n.a. |
| FISH:normal | |||||
| EB31 | 46,XX,16qh+ | n.a. | 46,XX,16qh+ | 46,XX,16qh+ | n.a. |
| FISH:normal | FISH:+4 [1%]; +7 [4%] | FISH:+4 [5%];+7 [5%]; incl. polyploid [5%] | |||
aData sets are not complete for all patients due to limited sample availability and the paucity of mitoses in some cell cultures. bOnly FISH data are available. incl. = including; n.a. = not analyzed (Karyotype and/or FISH); P-1 = passage 1; P-4 = passage 4.
Numerical chromosomal aberrations in RA
Peripheral blood lymphocytes
Findings for PBL were normal in all the RA patients for whom karyotype analysis was performed (Table 1).
Synovial fibroblasts
First-passage SFB from 10 of the 18 RA patients for whom karyotype analysis was performed (56%) displayed numerical chromosomal aberrations involving chromosomes 4, 6, 7, 8, 9, 12, 18, and Y (Table 1). Until P-4, there was an increase of both the proportion of affected patients (8 of 8; 100%) and the percentage of SFB showing gains or losses of whole chromosomes (Table 1). Chromosome 7 was the chromosome predominantly affected (in 10 of 10 patients at P-1, and in 7 of 8 patients at P-4). (Note that data sets are not complete for all patients due to limited sample availability and the paucity of mitoses in some cell cultures.)
Quantitative comparison of the mean levels of polysomies or monosomies ex vivo and in vitro in RA
In synovial tissue of RA patients, the mean percentages for polysomies of chromosomes 4, 6, 8, 9, and 18 did not exceed threshold levels of 4% (Fig. 1) [31]. The mean percentages for monosomies of chromosomes 7 and 18 did not exceed threshold levels of 7% (Fig. 1) [31]. In P-1 SFB, threshold levels were exceeded for polysomies of chromosomes 6, 7, 8, and 9. In P-4 SFB, threshold levels were exceeded for monosomy 18 and for polysomies of chromosomes 6, 7, 8, 9, and 18 (Fig. 1). Isolated SFB strongly contributed to polysomy 7 in synovial tissue.
Figure 1.
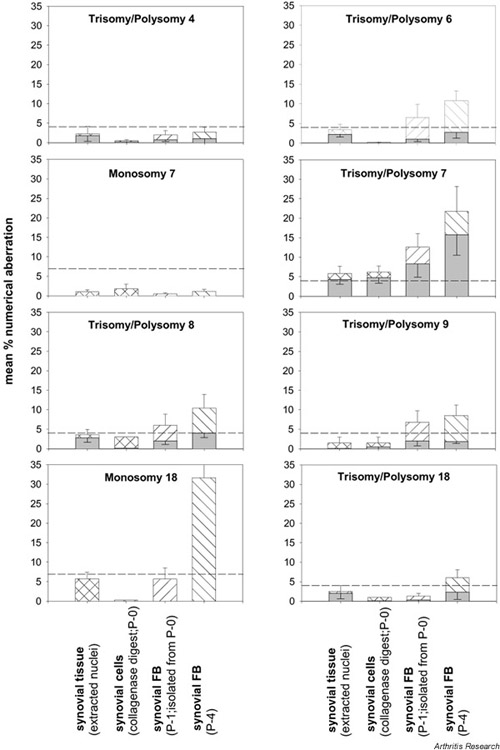
Numerical chromosomal aberrations in the RA synovial membrane. Polysomy (hatched bars) and trisomy (shaded bars) of the individual chromosomes are depicted as means ± SEM for n = 7–13 patients. Polysomies in synovial tissue of RA patients exceeded threshold levels (broken lines) only in the case of chromosome 7. In P-1 synovial fibroblasts (FB), threshold levels were exceeded for polysomies of chromosomes 6, 7, 8, and 9. In P-4 synovial FB, threshold levels were exceeded for polysomies of chromosomes 6, 7, 8, 9, and 18 and monosomy 18; there was a clear increase between P-1 and P-4. Considering only trisomic nuclei, only aberrations in the numbers of chromosome 7 exceeded threshold levels, even in P-4 synovial FB. P-0 = primary culture; P-1 = passage 1; P-4 = passage 4.
The percentage of +7 SFB increased between P-1 and P-4, indicating a selective growth advantage for 7-polysomic SFB in vitro (Fig. 1). The percentage of SFB monosomic and polysomic for chromosome 18 also increased until P-4 (5.0-fold and 4.6-fold, respectively). The percentage of monosomic SFB was always higher than that of polysomic SFB, indicating a growth advantage of 18-monosomic SFB in vivo and in vitro in RA (Fig. 1).
The percentage of cells with trisomic nuclei was generally lower than that with polysomic nuclei, indicating that both chromosomal nondisjunction and endoreduplication contribute to the ex vivo and in vitro aberrations (Fig. 1; shaded bars).
Structural chromosomal aberrations in OA
In 9 of 24 OA patients (38%), structural chromosomal aberrations were observed in P-0 synovial cells or P-1/P-4 SFB, with involvement of chromosome 1 in 4 of 9 cases (e.g. chrb(1)(q12)[2]; Supplementary material). Structural aberrations of chromosomes 8, 9, 11, and 12 were also observed in one patient each, and aberrations of chromosomes 5 and 7 were observed in two patients. In one OA patient, a deletion in the long arm of chromosome 1 was also detected in P-4 skin FB (Supplementary material). Two clonal aberrations (chromosomes 9 and 16) were observed in PBL of OA patients (Supplementary material). However, the clonal deletion in the long arm of chromosome 9 was not confirmed in a larger number of metaphases; also, the fragile site at q22.1 of chromosome 16 is a known morphological variant, without pathologic relevance.
Numerical chromosomal aberrations in OA
Peripheral blood lymphocytes
Except for one patient with karyotype 45,X[10]/46,XX[17], i.e. a mosaic form of the Turner syndrome, cytogenetic findings for PBL were normal in all OA patients (Table 2).
Table 2.
Cytogenetic findings in various cells/nuclei from patients with osteoarthritis
| Karyotype and FISH resultsa | |||||
| Peripheral | Nuclei | Synovial fibroblasts | |||
| blood | extracted from | Collagenase | |||
| Patient | lymphocytes | synovial tissueb | digest | P-1 | P-4 |
| ES37 | 46,XY | n.a. | n.a. | 47,XY,+7[1]/46,XY [49] | 47,XY,+7[2]/46, XY [48] |
| ES52 | 46,XX | n.a. | n.a. | 46,XX | n.a. |
| ES53 | 46,XY | n.a. | n.a. | 46,XY | n.a. |
| ES54 | 46,XX | n.a. | n.a. | 46,XX | n.a. |
| L09 | 46,XX | n.a. | n.a. | 47,XX,+7[12]/48,XX,+7,+8[5];46,XX[33] | n.a. |
| L11 | 46,XX | n.a. | n.a. | 46,XX | n.a. |
| EB8 | 46,XY | n.a. | 46,XY | n.a. | |
| FISH:normal | FISH:+7 [4%];+8 [4%]; +9 [3%];-18 [7%]; incl. polyploid [1%] | FISH:+7 [17%];+8 [7%];+9 [9%]; -18 [19%] | |||
| EB9 | 46,XX | n.a. | n.a. | 47,XX,+7[2] | n.a. |
| FISH:normal | FISH:+7 [52%]; -9 [4%] | FISH:+7 [n.a.]; -9 [25%] | |||
| EB10 | 46,XX | n.a. | n.a. | 46,XX | n.a. |
| FISH:normal | FISH:+6 [6%];+7 [20%];+8 [11%];-8 [2%];-18 [8%] | FISH:+7 [32%];-8 [10%];-18 [18%] | |||
| EB11 | 46,XX | n.a. | 47,XX,+7[3]/46,XX[7] | 47,XX,+7[3]/46,XX[6] | |
| FISH:normal | FISH:+4 [3%];+7 [8%];+18 [0%] | FISH:+4 [3%]; +7 [19%];+18 [2%] | FISH:+4 [7%];+7 [15%];+18 [8%]; incl. polyploid [7%] | ||
| EB12 | 45,X[10]/46,XX[17] | n.a. | 47,XX,+7,+4 or +5[3]/46,XX[4] | 46,XX | |
| FISH:-X [32%] | FISH:+4 [0%];+7 [12%];+8 [12%];+9 [4%] | FISH:+4 [4%];+7 [42%];+8 [0%];+9 [11%] | FISH:+4 [2%];+7 [36%];+8 [1%];+9 [6%] | ||
| EB13 | 46,XY | n.a. | n.a. | 46,XY | 46,XY |
| FISH:normal | FISH:+7 [15%];-9 [3%];+12 [0%] | FISH:+7 [37%];-9 [8%];+12 [11%] | FISH:+4 [8%];+7 [16%]; +9 [5%];-9 [25%];+12 [6%] | ||
| EB14 | 46,XX | n.a. | 47,XX,+7[3]/46,XX[7] | 46,XX,+7,+8,-15,-21[1]/48,XX,+7,+9[1]/47,XX,+7[1]/48,X,+7,+8,+9[1]/46,X,-X,+7,-17,+mar[1]/46,XX,t(5;11)(q23;q12)[1]/48,X,-X, +5,+7,+8[1]/46,XX[1] | |
| FISH:normal | FISH:+7 [14%];+8 [0%];+X [4%] | FISH:+7 [10%];+8 [0%];+X [6%] | FISH:+7 [36%];+8 [2%];+X [0%] | FISH:+7 [34%];+8 [8%];+X [6%] | |
| EB15 | 46,XX | n.a. | n.a. | 46,XX | n.a. |
| FISH:normal | FISH:+6 [7%];+7 [10%];+8 [7%];+9 [10%];incl. polyploid [7%] | FISH:+6 [12%];+7 [16%];+8 [7%];+9 [10%];incl. polyploid [4%] | |||
| EB16 | 46,XX | n.a. | n.a. | n.a. | n.a. |
| FISH:normal | FISH:+6 [5%]; +7 [18%];+8 [0%]; -X [18%] | FISH:+6 [26%];+7 [26%];+8 [35%];+X [17%];incl. polyploid [17%] | |||
| EB17 | 46,XX | n.a. | 46,XX,chrb(1)(q12)[2]/46,XX[4] | 46,XX | n.a. |
| FISH:normal | FISH:+4 [17%];+6 [3%];+12 [1%] | FISH:+4 [16%];+6 [6%];+12 [5%];incl. polyploid [1%] | |||
| EB18 | 46,XX | n.a. | 46,XX | 46,XX | n.a. |
| FISH:normal | FISH:+7 [1%];+8 [0%];+9 [2%] | FISH:+7 [5%];+8 [4%];+9 [1%] | FISH: +7 [18%];+8 [18%];+9 [18%];incl. polyploid [3%] | ||
| EB19 | 46,XX | 46,XX | 47,XX,+7[2]/46,XX[7] | 47,XX,+7[2]/46,XX[6] | |
| FISH:normal | FISH:+7 [18%];+8[2%];+9 [4%];+12 [4%];incl. polyploid [2%] | FISH:+7 [14%];+8 [0%];+9 [0%];+12 [0%] | FISH:+7 [10%];+8 [0%];+9 [4%];+12 [7%] | FISH:+7 [26%];+8 [18%];+9 [7%];+12 [8%];incl. polyploid [7%] | |
| EB21 | 46,XX | n.a. | 46,XX | 46,XX | n.a. |
| FISH:normal | FISH:+4 [17%];+6 [7%];+7 [11%];+9 [17%]; incl. polyploid [5%] | FISH:+4 [3%];+6 [3%];+7 [5%];+9 [3%]; incl. polyploid [1%] | FISH:+4 [3%];+6 [4%];+7 [11%];+9 [3%]; incl. polyploid [3%] | ||
| EB22 | 46,XY | n.a. | n.a. | 46,XY | n.a. |
| FISH:normal | FISH:+4 [4%]; +7 [7%];+9 [3%]; incl. polyploid [1%] | FISH:+4 [2%];+7 [6%];+9 [1%]; incl. polyploid [1%] | FISH:+4 [8%];+7 [8%]; +9 [7%] | ||
| EB24 | 46,XX | n.a. | 46,XX | n.a. | |
| FISH:normal | FISH:+4 [0%];+6 [3%];+7 [7%];+9 [3%] | FISH:+4 [8%];+6 [8%];+7 [25%];+9 [8%]; incl. polyploid [7%] | FISH:+4 [9%];+6 [2%];+7 [33%];+9 [3%]; incl. polyploid [2%] | FISH:+4 [2%];+6 [2%];+7 [24%];+9 [3%]; incl. polyploid [2%] | |
| EB29 | 46,XX | n.a. | n.a. | n.a. | n.a. |
| FISH:normal | |||||
| J4 | n.a. | n.a. | 46,XX | 46,XX | |
| FISH:+4 [5%];+7 [14%]; incl. polyploid [1%] | FISH:+4 [1%];7 [6%]; incl. polyploid [1%] | ||||
| J5 | n.a. | n.a. | n.a. | n.a. | |
| FISH:+4 [5%];+7 [13%] | |||||
aSee Table 1. b Only FISH data are available. incl. = including; n.a. = not analyzed (Karyotype and/or FISH); P-1 = passage 1; P-4 = passage 4.
Synovial fibroblasts
In 18 of 24 OA patients (75%), early-passage SFB showed numerical chromosomal aberrations involving chromosomes 4, 5, 6, 7, 8, 9, 12, 18, and X (Table 2). Until P-4, both the proportion of affected patients (12 of 13; 92%) and the percentage of synovial fibroblasts showing chromosome gains or losses increased (Table 2). As in RA, chromosome 7 was the one predominantly affected (in 17 of 18 patients in P-1, and 12 of 13 in P-4).
Quantitative comparison of the mean levels of polysomies or monosomies ex vivo and in vitro in OA
The mean percentages for polysomies of chromosomes 4, 6, 8, 9, and 18 and for monosomies of chromosomes 7 and 18 did not exceed (or only marginally exceeded) the threshold levels (Fig. 2). In P-1 SFB, threshold levels were exceeded for monosomy 18 and for polysomies of chromosomes 4 and 7; in P-4 SFB, threshold levels were exceeded for polysomies of chromosomes 4, 6, 7, 8, and 9 (Fig. 2). Isolated SFB (P-1 18.4%; n = 15) strongly contributed to polysomy 7 in synovial tissue.
Figure 2.
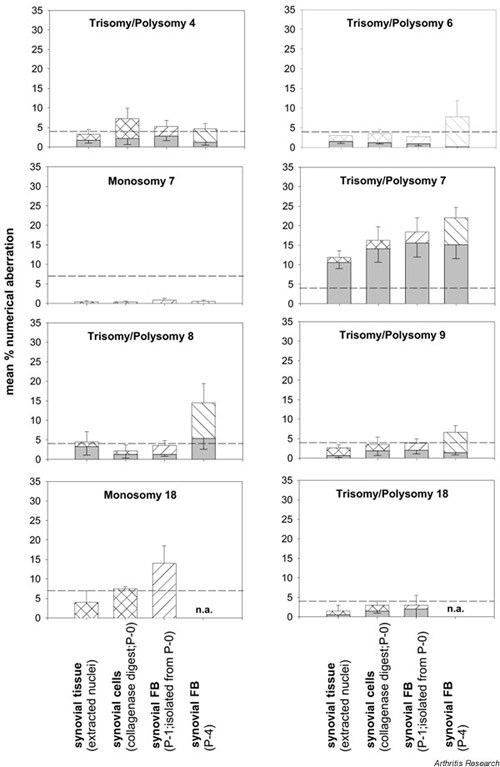
Numerical chromosomal aberrations in the OA synovial membrane. Bars as in Fig. 1 (means ± SEM for n = 10–16 patients). Polysomies (hatched bars) in OA synovial tissue only exceeded threshold levels (broken lines) in the case of chromosome 7, except for a very marginal increase for chromosome 8. In P-1 synovial FB, polysomies for chromosomes 4 and 7 and monosomy 18 were above threshold levels; in P-4 synovial FB, polysomies for chromosomes 4, 6, 7, 8, and 9 were above threshold levels. Considering only trisomic nuclei (shaded bars), numerical chromosomal aberrations were observed for chromosome 7 only, except for a limited elevation of trisomy 8 in P-4 synovial FB. P-0 = primary culture; P-1 = passage 1; P-4 = passage 4.
As in RA, the percentage of +7 SFB increased until P-4, and the percentage of SFB monosomic for chromosome 18 was always higher than the percentage of SFB polysomic for chromosome 18 (Fig. 2).
The percentage of cells with trisomic nuclei was generally lower than that with polysomic nuclei, indicating that both chromosomal nondisjunction and endoreduplication contribute to the ex vivo and in vitro aberrations (Fig. 2; shaded bars).
Comparison of ex vivo and in vitro findings for individual patients
Numerical chromosomal aberrations observed in RA and OA tissue as a whole (i.e. in nuclei extracted from the synovial membrane; Tables 1 and 2) showed good correspondence with the findings in the collagenase digest (i.e. in adherent synovial cells after in vitro culture for approximately 8–9 days), or in isolated, P-1 SFB (in vitro culture for approximately 14 days).
Numerical chromosomal aberrations in other joint diseases
Peripheral blood lymphocytes
PBL of patients with other rheumatic diseases showed no cytogenetic abnormalities (Supplementary material).
Synovial fibroblasts
Polysomy 7 was observed in P-1 (2%) and P-4 (4%) SFB of one patient with ankylosing spondylitis (see Supplementary material).
Limited degree of chromosomal aberrations in normal joints or patients with joint trauma
The cytogenetic findings for nuclei extracted from synovial tissue, mixed synovial cells (collagenase digest), or P-1 SFB of four control patients were normal. Upon extended in vitro culture, however, polysomies of several chromosomes exceeding the threshold levels appeared in SFB of two patients with joint trauma (JT) (in P-4 after 148 days of culture in one patient, and in P-1 after 21 days in the other). The mean percentage of trisomic nuclei/cells was well below the threshold levels for all samples (including P-4 SFB) and chromosomes (Supplementary material).
Numerical chromosomal aberrations in skin fibroblasts in RA and OA
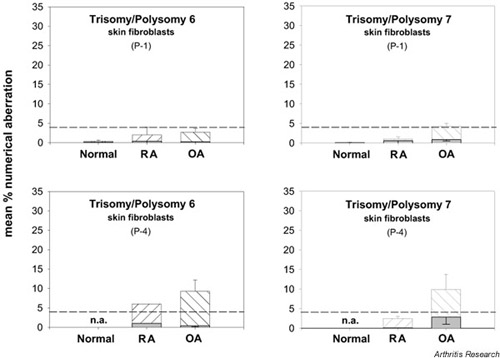
The mean percentages for polysomies of chromosomes 4, 6, 7, and 9 and for monosomy 7 in P-1 skin FB from normal subjects or RA and OA patients did not exceed threshold levels (Supplementary material). Until P-4, the mean percentage of polysomic skin FB increased (possibly reflecting culture artifacts), exceeding threshold levels for chromosome 6 in RA and for chromosomes 4, 6, 7, and 9 in OA. The mean percentages of nuclei trisomic for chromosomes 4, 6, 7, and 9 were well below threshold levels in P-1 or P-4 skin FB derived from normal subjects or RA and OA patients (Supplementary material).
Correlations among cytogenetic findings, and between cytogenetic findings and clinical parameters
A significant positive correlation between the percentages of polysomies for chromosome 7 in collagenase digest and P-1 SFB in both RA (n = 5; P = 0.028; ρ = 0.918) and OA (n = 11; P = 0.031; ρ = 0.648) underlined good correspondence of ex vivo and in vitro data (Tables 1 and 2).
Significant positive correlations between the disease duration in RA patients and the levels of polysomies in P-1 SFB for chromosomes 6 (n = 5; P = 0.014; ρ = 0.947) and 8 (n = 4; P = 0.050; ρ = 0.949) indicated a possible link between the chronicity of disease and the occurrence of numerical aberrations.
An influence of the inflammatory activity on the occurrence of polysomies was suggested by a significant positive correlation between levels of polysomy for chromosome 7 in RA collagenase digests and the serum concentrations of C-reactive protein (n = 5; P = 0.041; ρ = 0.894).
The lack of significant correlations between the levels of polysomy for any chromosome in RA SFB and concurrent treatment with methotrexate is in contrast to results in previous reports [32,33], thereby questioning a clear-cut influence of methotrexate treatment in this study.
None of the other clinical parameters, including the ages of patients with RA and OA, showed any correlation with the occurrence or the percentages of chromosomal aberrations (data not shown).
Discussion
Structural chromosomal aberrations
Structural aberrations of various chromosomes occur in isolated SFB from RA and OA patients, as reported in other chronic rheumatic disorders [34,35,36]. Although mostly nonclonal, a striking proportion of the structural chromosomal aberrations (del and chrb) in SFB of both RA (three of four) and OA (four of nine) patients was found on chromosome 1, affecting regions q12, q13, q21, and q31. Benign mesenchymal neoplasms do not show any unbalanced chromosomal abnormalities in these regions on chromosome 1 [34], whereas malignant mesenchymal neoplasms, e.g. chondrosarcoma, osteosarcoma, or pleomorphic sarcoma, carry such unbalanced chromosomal abnormalities (der or del), with a focus on q11 (26 cases), q12 (10 cases), and q21 (16 cases) [34]. Whether this is a pure coincidence and whether the location of genes (such as those encoding cathepsins S and K and the Fc receptor CD64) on 1q21 implies functional consequences for SFB remain matters for further investigation.
Specificity of chromosomal aberrations for RA
Numerical aberrations were observed in SFB of patients with RA, OA, or ankylosing spondylitis, with a similar spectrum of affected chromosomes. In RA and OA, however, chromosome 7 was the one predominantly affected. This observation expands and confirms similar findings in non-purified synovial cells in RA [12,13,14,15], villonodular synovitis [13], and OA [16,19]. As previously observed [19], the proportion of patients with +7 and the percentage of cells/nuclei with trisomy/polysomy 7 were significantly higher in OA than in RA, concerning both synovial tissue (11.9%; n = 7 versus 5.9%; n = 7; P ≤ 0.05) and collagenase digest (16.2%; n = 13 versus 6.2%; n = 5; P ≤ 0.05).
In addition, +7 (or general tetraploidy [37]) seems to be linked to a number of non-neoplastic diseases/tissues unrelated to joint inflammation, including chronic pyelonephritis and focal steatosis of the liver [12]. Thus, mosaic +7 likely does not bear any specificity for RA. On average, polysomy 7 was not observed in PBL or P-1 skin FB of the patients. This excludes that there was a generalized chromosomal mosaic in all somatic cells.
Functional aspects of trisomy 7
Chromosome 7 carries a number of potentially relevant genes (for example, cytokines/growth factors and their receptors, transcription factors, signal-transduction molecules, and molecules involved in matrix formation and cell-extracellular matrix [38]) implying functional consequences for +7 SFB. Indeed, both RA and OA SFB with +7 appeared to have a selective growth advantage (Tables 1 and 2; Figs 1 and 2), confirming previous findings [14,15]. Whether gains of chromosomes result in overexpression of target genes on these chromosomes apparently depends on individual cases ([39,40] and our own preliminary results on the expression of the proto-oncogene c-met [hepatocyte growth factor receptor encoded on 7q31]). The hypothesis that genes located on human chromosome 7 (or other chromosomes showing aberrations in the present study) may be linked to RA susceptibility is being addressed also by other groups, for example, by analyzing quantitative trait loci in the homologous chromosomal regions of intercrosses of inbred mouse or rat strains susceptible or resistant to the induction of experimental arthritis [41,42]. Also, potential susceptibility loci [43] or single nucleotide polymorphisms in potentially disease-relevant genes [44] are being assessed by genomewide screening in RA patients.
Parallel occurrence of different polysomies
This study also confirms the parallel presence of different trisomies, as has previously been described in villonodular synovitis (+5,+7) [13], RA (+7,+8) [14], OA (+5,+7) [19], and Dupuytren's contracture (+7,+8) [17]. While these findings may reflect a general chromosomal instability, the predominance of the effect in chromosome 7 argues for a biased instability.
Conclusion
The findings in RA patients were numerical chromosomal aberrations, i.e. trisomy or polysomy 7 and monosomy 18, and structural aberrations, particularly in chromosome 1. These alterations were limited to the synovial compartment: skin FB and blood lymphocytes were virtually normal. However, the alterations were not specific for RA; similar ones were seen in OA and spondylarthropathy. Thus, such alterations may not be functional to a particularly aggressive phenotype of RA SFB, but rather reflect a common response to inflammatory/microenvironmental stimuli in rheumatic diseases. Because JT/control samples were virtually normal, it appears that chromosomal alterations may be associated with chronic pathology of the joints.
Abbreviations
DMEM = Dulbecco's modified Eagle's medium; FB = fibroblasts; FCS = fetal calf serum; FISH = fluorescence in situ hybridization; HEPES = N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid; IL = interleukin; ISCN = International System for Human Cytogenetic Nomenclature; JT = joint trauma; OA = osteoarthritis; P-0 = primary culture; P-1 = passage 1; P-4 = passage 4; PBL = peripheral blood lymphocytes; PBS = phosphate-buffered saline; RA = rheumatoid arthritis; RPMI = Roswell Park Memorial Institute [medium]; SFB = synovial fibroblasts.
Supplementary materials and methods
Patients
Inflamed synovial tissue, heparinized peripheral blood, and skin samples from the edge of the surgical incision were obtained from RA patients (n = 21), OA patients (n = 24), and patients (n = 8) with various inflammatory/degenerative joint diseases (three spondylarthropathies, comprising one ankylosing spondylitis and two psoriatic arthritis; one villon-odular synovitis; one systemic lupus erythematosus; one juvenile rheumatoid arthritis; one undifferentiated monoarthritis; and one reactive arthritis) (Supplementary Table 1). The patients were classified according to the criteria of the American College of Rheumatology/American Rheumatism Association or the European Spondylarthropathy Study Group [20,21,22,23,24]. As controls, synovial tissue/cells from four patients with either no joint disease (postmortem samples) or recent joint trauma as well as skin samples (taken during plastic surgery) from four normal donors were studied (Supplementary Table 1). The groups were generally gender-matched, since no significant differences were observed among groups for gender distribution, except for a significant difference between the spondylarthropathy group (containing 3 male patients) and the OA group (containing 19 female and 5 male patients) (P ≤ 0.005). Because of limited availability of patient/donor material, groups were not strictly age-matched, with the result that the RA patients studied were significantly younger than OA patients, and that the patients in both of these groups were older (P ≤ 0.05) than patients with spondylarthropathy, JT/normals, and normal skin donors. Due to the lack of correlation between the age of the patients and the occurrence or the percentages of chromosomal aberrations, however, this was not considered to question the validity of the results. No significant differences were observed for the disease duration among RA, OA, and spondylarthropathy patients. As expected, serum concentrations of C-reactive protein were significantly higher in RA than in OA patients (Supplementary Table 1).
Supplementary Table 1.
Clinical characteristics of the subjects at the time of synovectomy/sampling
| Subjects (n) | Gender (M/F) | Age (years) | Disease duration (years) | RF (+/-) | ESR (mm/h) | CRPa (mg/l) | No. of ARA criteria (RA) | Concomitant medication (n) |
| Rheumatoid arthritis | ||||||||
| 21 | 10/11 | 63.1 ± 2.0 | 10.7 ± 2.1 (n.d. = 1) | 14/4 (n.d. = 3) | 36.6 ± 5.2 (n.d. = 3) | 41.4 ± 7.8 (n.d. = 1) | 5.1 ± 0.2 | Methotrexate (10), Prednisolone (16), Sulfasalazine (4), Gold salts (1), NSAIDs (14) |
| Osteoarthritis | ||||||||
| 24 | 5/19 | 69.5 ± 2.3 | 9.3 ± 3.0 (n.d. = 15) | 0/6 (n.d. = 18) | 18.4 ± 3.7 (n.d. = 2) | 6.4 ± 1.1 (n.d. = 1) | 0.3 ± 0.1 | Prednisolone(1), NSAIDs (11), none (13) |
| Spondylarthropathy | ||||||||
| 3 | 3/0 | 37.0 ± 12.5 | 3.8 ± 3.1 | 0/3 | 22.3 ± 12.8 | 24.4 ± 19.4 | 1.0 ± 1.0 | Sulfasalazine (1), NSAIDs (2), none (1) |
| Other arthritides | ||||||||
| L02 / VNS | F | 64 | 4 | - | 18 | <5 | 0 | Methotrexate, NSAIDs |
| L04 / SLE | F | 38 | 7 | - | 11 | 28.9 | 3 | Methotrexate, Prednisolone |
| ES36 / JRA | M | 18 | 14 | - | 4 | <5 | 2 | Chloroquine, NSAIDs |
| ES63 / UA | M | 18 | 1 | - | 2 | <5 | 0 | NSAIDs |
| EB32 / ReA | M | 43 | 8 | n.d. | 60 | 8.5 | 1 | none |
| Joint trauma/normals | ||||||||
| 4 | 2/2 | 49.3 ± 9.5 | 0.5 ± 0.5 | n.d. | n.d. | n.d. | 0.0 ± 0.0 | none (4) |
| Normal skin | ||||||||
| 4 | 1/3 | 20.3 ± 10.7 | 0.0 ± 0.0 | n.d. | n.d. | n.d. | 0.0 ± 0.0 | none (4) |
aNormal range, <5 mg/l. +/- = positive/negative; ARA = American Rheumatism Association (now American College of Rheumatology); AS = ankylosing spondylitis; CRP = C-reactive protein; ESR = erythrocyte sedimentation rate; JRA = juvenile rheumatoid arthritis; n.d. = not determined; NSAIDs = nonsteroidal anti-inflammatory drugs. RA = rheumatoid arthritis; ReA = reactive arthritis; RF = rheumatoid factor; SLE = systemic lupus erythematosus; UA = undifferentiated monoarthritis; VNS = villonodular synovitis. For the parameters age, disease duration, ESR, CRP, and number of ARA-Criteria (RA), values are means ± SEM; for the other parameters, values are numbers (n).
Synovial tissue, peripheral blood, and skin were obtained during open joint replacement/traumatology surgery or arthroscopic synovectomy at the Department of Orthopedics, University of Leipzig; the Clinic of Orthopedics, Bad Düben; the Clinic of Orthopedics, Friedrich Schiller University Jena (Eisenberg); the Department of Traumatology, Friedrich Schiller University Jena, Germany; and the Department of Orthopedic Surgery, University of Michigan, Ann Arbor, MI, USA. The study was approved by the ethics committees of the respective universities. Paired blood samples were immediately transferred to the Institutes of Human Genetics in Jena or Leipzig for lymphocyte culture and karyotype/FISH analysis. Synovial tissue and skin were placed in cell culture medium at ambient temperature and digested within 2 h.
Tissue digestion, cell culture, and fibroblast isolation
Synovectomy samples of synovial membranes were finely minced and digested for 30 min at 37°C in PBS containing 0.1% trypsin (Sigma, Deisenhofen, Germany), followed by digestion in 0.1% collagenase P (Boehringer Mannheim, Mannheim, Germany) in DMEM/10% FCS; (both Gibco BRL, Eggenstein, Germany) for 2 h at 37°C, in a 5% CO2 atmosphere. After two washes with serum- free DMEM, the cells were cultured in DMEM/10% FCS, 25 mM HEPES, penicillin (100 U/ml), streptomycin (100 μg/ml), amphotericin B (2.5 mg/ml), and gentamycin (0.1 mg/ml) (all Gibco BRL). The medium was changed after 1 day, and then every 2 or 3 days, to remove nonadherent cells from the culture. After 7 days of primary culture, the adherent cells were trypsinized (in 0.25% trypsin/0.2% EDTA; Gibco BRL), removed from the culture dish by mechanical dislocation, washed in PBS/2% FCS, and used for negative isolation. The samples were randomly tested to exclude Mycoplasma contamination using a commercially available ELISA kit (Boehringer Mannheim).
Negative isolation of FB was performed as described elsewhere [25]. Briefly, primary culture synovial cells were incubated with Dynabeads® M-450 CD14 (clone RMO52; Dynal, Hamburg, Germany) in PBS/2% FCS for 1 h at 4°C, under bidirectional rotation. Conjugated and unconjugated cells were separated using the magnetic particle concentrator Dynal® and, after two washes in PBS/2% FCS, either analyzed by fluorocytometry with the mono-clonal antibodies mentioned below or cultured to P-4 by 1:3 split at confluency.
Primary-culture skin FB from normal donors (kindly provided by Dr D Sauer, plastic surgeon, Leipzig, Germany) were prepared by first incubating skin samples with Dispase II (0.5 U/ml; Boehringer Mannheim) overnight at 4°C and, after removal of the epidermis, by digesting with 0.25% collagenase P (Boehringer Mannheim) in DMEM/1% FCS at 37°C and 5% CO2 for 4 h. The result-ing cell suspension was then cultured as above.
Fluorocytometry
FACS (fluorescence-activated cell sorting) analyses of isolated SFB were performed according to standard procedures as described elsewhere [26]. The specificity of staining was confirmed using isotype-matched control monoclonal antibodies at identical concentrations. Analyses were performed on a FACScan® (Becton Dickinson, San Jose, CA, USA). Gates were set to include all viable cells and to exclude 99% of the cells stained with control immunoglobulins. Unconjugated cells obtained from the primary culture of the synovial membrane by negative isolation with Dynabeads® M-450 CD14 showed an enrichment of SFB (Thy-1+: RA 72.1%, n = 13; OA 71.5%, n = 15; and prolyl 4-hydroxylase+: RA 80.3%, n = 9; OA 93.1%, n = 9), with a contamination of <2% macrophages (CD14+, CD68+, CD11b+) and <1% T cells, B cells, plasma cells, natural killer cells, dendritic cells, or polymorphonuclear neutrophil leucocytes [25].
GTG-banding and fluorescence in situ hybridization
Cytogenetic studies of PBL were performed according to standard methods (RPMI medium with 10% FCS and 1.2% phytohemagglutinin in a 72-h culture). For cytogenetic studies of primary-culture synovial cells (collagenase digest; i.e. adherent synovial cells), SFB (P-1; P-4) or skin FB (P-1 and P-4; for culture times, see Supplementary Table 2), cells were seeded at a density of 1 × 105 cells/glass slide in QuadriPERM wells (Heraeus, Hanau, Germany). At near confluency, cells were treated with colcemid (1 μg/ml) for 1.75 h, followed by hypotonic treatment with 0.2% MgCl2/0.4% sodium citrate solution for 10 min, both at 37°C. The cells were fixed with three changes of methanol/acetic acid (3:1; 10 min each; room temperature) and air-dried under laminar flow. GTG-banding was performed according to standard protocols [27] on 10–50 metaphases per case. Karyotypes were described in accordance with ISCN 1995 [28]. Structural aberrations occurring in one cell only, i.e. nonclonal events, are reported for the sake of general interest but are not included in the karyotype (Tables 1 and 2; Supplementary Tables 3 and 4).
Supplementary Table 2.
Culture time (days) of synovial cells in primary culture (collagenase digest), synovial fibroblasts, and skin fibroblasts from patients with rheumatoid arthritis, osteoarthritis, and joint trauma, or normal donors
| Synovial fibroblasts | Skin fibroblasts | ||||
| Synovial cells | |||||
| Source of cells | (collagenase digest) | Passage 1 | Passage 4 | Passage 1 | Passage 4 |
| Rheumatoid arthritis | 9.2 ± 3.3 (n = 5) | 13.8 ± 1.8 (n = 9) | 81.1 ± 9.9 (n = 7) | 46.2 ± 20.7 (n = 6) | 63.5 ± 19.5 (n = 2) |
| Osteoarthritis | 8.1 ± 1.2 (n = 14) | 13.9 ± 1.7 (n = 17) | 91.4 ± 9.0 (n = 11) | 18.0 ± 2.0 (n = 14) | 62.4 ± 5.8 (n = 13) |
| Joint trauma | n.a. | 28.0 ± 7.0 (n = 2) | n.a. | n.a. | n.a. |
| Normal subjects | n.a. | n.a. | n.a. | 20.0 ± 8.9 (n = 3) | n.a. |
Values are shown as means ± SEM. n.a. = not analyzed.
Supplementary Table 3.
Structural chromosomal aberrations in rheumatoid arthritis
| Karyotype | |||||
| Peripheral | Synovial fibroblasts | Skin fibroblasts | |||
| blood | |||||
| Chromosome | lymphocytes | Passage 1 | Passage 4 | Passage 1 | Passage 4 |
| 1 | 46,XY,del(1)(q31),t(5,8,6)(p11;p11.2;p11.1),del(11)(p14),-18,+20[1] 46,XY,chrb(1q?21)[1] | 46,XY,del(1)(q12 or q13),-3,-19,+mar1,+mar2[1] | |||
| 5 | 46,XY,chrb(5)(q10)[1] | ||||
| 15 | 46,XY,del(15)(q22),-21,+mar [cp3] | ||||
| 16 | 46,XX,16qh+ | 46,XX,16qh+ | |||
| X | 46,XX,chrb(X)(q11)[1] | ||||
Supplementary Table 4.
Structural chromosomal aberrations in osteoarthritis
| Karyotype | |||||
| Peripheral | Synovial fibroblasts | Skin fibroblasts | |||
| blood | |||||
| Chromosome | lymphocytes | Passage 1 | Passage 4 | Passage 1 | Passage 4 |
| 1 | 46,XX,del(1)(q23),+mar[1] | 46,XX,chrb(1)(q23)[1]t(3;7)(q23;q34),-21,(Collagenase digest) | 46,XX,del(1)(q11 or q12)[1] | ||
| 48,XX,+del(1),+7,+9[1] | |||||
| 46,XX,chrb(1)(q12)[2] (Collagenase digest) | |||||
| 46,X,-X,+7, chrb(1)(q12)[1] | |||||
| 2 | 46,XX,t(2;10)(q14;q22)[1] | ||||
| 5 | 46,XX,del(5)(p13)[1] | 46,XX,t(5;11)(q23;q12)[1] | |||
| 7 | 46,XX,t(7;12)(p14;q13)[1] | 46,XY,del(7)(q32)[1] | |||
| 47,XX,+del(7) (q11.1→ qter)[1] | 46,XX,-2,+4,-6,-8,+15,+add(7)(p22)[1] | ||||
| 8 | 46,XY,-4,+7,t(8;11) (p11.2;p11.2)[1] | ||||
| 9 | 46,XX,del(9)(q11)[2] | 46,XX,chrb(9)(q13)[1] | |||
| 46,XX,chrb(9)(q21)[1] | |||||
| 11 | 45,X-X,-10,inv(11) (p15.5;q23.3),+mar[1] | 46,XX,t(11;15)[1] (Collagenase-digest) | |||
| 12 | 46,XX,chrb(12q?13)[1] (Collagenase-digest) | ||||
| 16 | 46,XY,fra(16)(q22.1)[2] | ||||
Extraction of nuclei from formalin-fixed/paraffin-embedded or cryofixed tissue was performed in accordance with the methods of Liehr et al ([29] and [30], respectively). FISH for the analysis of the copy number of particular chromosomes in interphase nuclei was performed according to standard protocols (VYSIS, Downers Grove, IL, USA) by applying fluorescence-labeled, repetitive, α-satellite DNA probes (VYSIS) specific for the centromeric region of each chromosome. In most cases, centromere probes for four different chromosomes were used per case, selected on the basis of numerical aberrations detected by GTG banding. One hundred cells/sample were examined applying dual-color FISH. A cutoff gate of 4% for chromosome gains and of 7% for chromosome losses was set, i.e. higher than previously published for leukemia [31], in order to account for the unknown tissue investigated and to consider only those values abnormal, which exceeded variations in cells from normal tissue. In cases in which ex vivo data (i.e. nuclei extracted from synovial tissue) or data from mixed synovial cells (collagenase digest) could be compared with in vitro data of SFB (P-1 or P-4), paired values were included/reported even if they did not reach the cutoff level (Tables 1 and 2; Figs 1 and 2; Supplementary Figs 123).
Supplementary Figure 1.
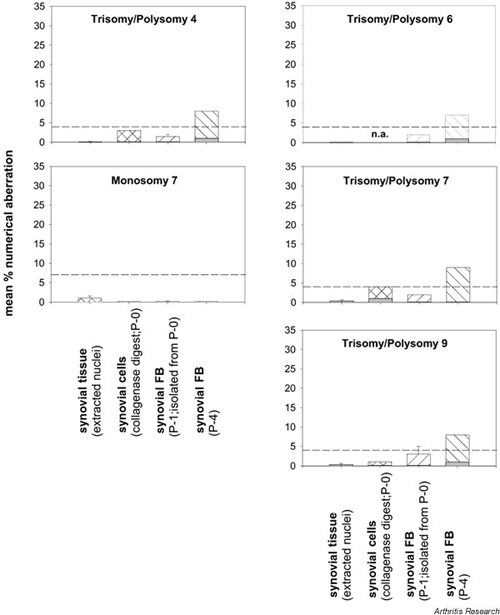
Numerical chromosomal aberrations in the normal/JT synovial membrane. Polysomy (hatched bars) and trisomy (shaded bars) of the individual chromosomes in extracted nuclei from synovial tissue, nonseparated synovial cells (collagenase digest), and isolated synovial fibroblasts (FB; P-1, P-4) are shown as means ± SEM of n = 1–3 patients. The mean percentages of polysomies in normal/JT synovial tissue, collagenase digest, and P-1 synovial FB did not exceed threshold levels (broken lines). In P-4, the mean percentages of synovial FB polysomic for chromosomes 4, 6, 7, and 9 were elevated to levels above threshold. If instead of all polysomic nuclei, only trisomic nuclei were considered, there was no elevation of chromosomal aberrations above threshold levels, not even in P-4 synovial FB. P-0 = primary culture; P-1 = passage 1; P-4 = passage 4.
Supplementary Figure 2.
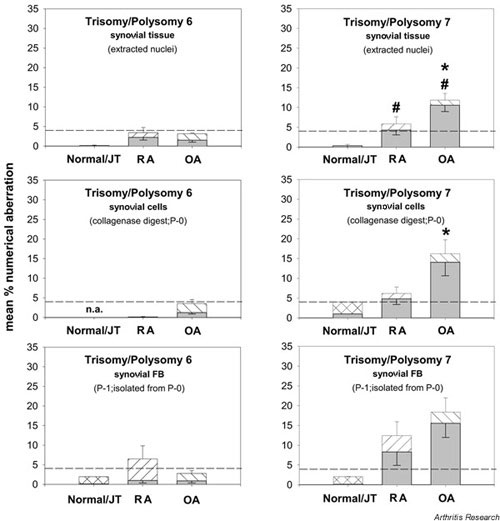
Comparison of numerical chromosomal aberrationsin the RA, OA, and normal/JT synovial membrane. Polysomy (hatched bars) and trisomy (shaded bars) of the individual chromosomes in extracted nuclei from synovial tissue, nonseparated synovial cells (collagenase digest), and isolated synovial fibroblasts (FB; P-1) are depicted as means ± SEM of n = 5–18 patients. #P ≤ 0.05 versus normal/JT for the comparison of polysomies; *P ≤ 0.05 versus RA for the comparison of polysomies. While chromosome 6 showed almost no numerical aberrations (with the exception of an increased level of polysomy 6 in P-1 RA synovial FB), both OA and RA synovial tissue showed a significant elevation of polysomy 7 in comparison with normal/JT tissue, with a significantly higher value in OA than in RA. A significant difference between RA and OA was also observed in nonseparated synovial cells (P-0).
Supplementary Figure 3.
Comparison of numerical chromosomal aberrations in skin fibroblasts of RA and OA patients and of normal donors. Polysomy (hatched bars) and trisomy (shaded bars) of the individual chromosomes in skin fibroblasts (P-1, P-4) are depicted as means ± SEM of n = 3–14 patients. On average, skin fibroblasts from RA and OA patients and from normal donors did not show any polysomies/trisomies above threshold levels, except for hyperdiploid nuclei upon extended culture (P-4), which may reflect culture artifacts.
Data were analyzed and depicted either on the basis of the total polysomy of nuclei, i.e. irrespective of the underlying mechanism, and pointed to the total gain of potential gene transcription units, or selectively, on the basis of trisomic nuclei, focusing on mitotic nondisjunction as a possible underlying mechanism.
Statistical analysis
Because of multiple comparisons, the data were first subjected to the multi-group Kruskal–Wallis test. The nonpara-metric Mann–Whitney U test was then applied to analyze differences between data of different disease groups. The Spearman rank correlation test was used to analyze correlations among parameters and between these parameters and the clinical status/treatment of individual patients. In all cases, differences were considered statistically significant for P ≤ 0.05. Analyses were performed using the SPSS 9.0™ program (SPSS Inc, Chicago, IL, USA).
Supplementary results
Numerical chromosomal aberrations in other joint diseases
Peripheral blood lymphocytes
PBL of patients with several other rheumatic diseases showed no cytogenetic abnormalities, as assessed by GTG-banding (Supplementary Table 5).
Supplementary Table 5.
Cytogenetic findings in various cells/nuclei from subjects with various joint disorders and normal joints
| Karyotype/FISH | ||||
| Peripheral | Nuclei | Synovial fibroblasts | ||
| Subject/ | blood | extracted from | ||
| diagnosis | lymphocytes | synovial tissue | Passage 1 | Passage 4 |
| ES45/AS | 46,XY | n.a. | 47,XY,+7[1]/46,XY [49] | 47,XY,+7[2]/46,XY [48] |
| L05/PsA | 46,XY | n.a. | 46,XY | n.a. |
| ES60/PsA | 46,XY | n.a. | 46,XY | n.a. |
| L02/VNS | 46,XX | n.a. | 46,XX | n.a. |
| L04/SLE | 46,XX | n.a. | 46,XX | n.a. |
| ES36/JRA | 46,XY | n.a. | 46,XY | 46,XY |
| ES63/UA | 46,XY | n.a. | 46,XY | n.a. |
| EB32/ReA | n.a. | n.a. | FISH:normal | n.a. |
| J1/JT (1 month culture) | n.a. | n.a. | 46,XX FISH:normal | 46,XX FISH:+4 [8%];+6 [7%];+7 [9%];+9 [8%];incl. polyploid [6%] |
| J2/JT (3 weeks culture) | n.a. | FISH:normal | 46,XX FISH:+9 [5%];+10 [5%];incl. polyploid [2%] | n.a. |
| FISH:normal (synovial cells in collagenase digest) | ||||
| SynS7/Nor. (paraffin sections) | n.a. | FISH:normal | n.a. | n.a. |
| B1/Nor. (cryostat sections) | n.a. | FISH:normal | n.a. | n.a. |
AS = ankylosing spondylitis; JRA = juvenile rheumatoid arthritis; JT = joint trauma; n.a. = not analyzed (Karyotype and/or FISH); Nor. = normal; PsA = psoriasis arthritis; ReA = reactive arthritis; SLE = systemic lupus erythematosus; UA = undifferentiated monoarthritis; VNS = villonodular synovitis.
Synovial fibroblasts
Numerical chromosomal aberrations were observed, i.e. polysomy 7 in P-1 (2%) and P-4 (4%) SFB from one patient with ankylosing spondylitis, indicating that the aberrations in SFB are not restricted to RA and OA but rather are a possible common feature of chronic rheumatic diseases (Supplementary Table 5).
Limited degree of chromosomal aberrations in normal joints or patients with joint trauma
Nuclei extracted from synovial tissue, collagenase digest, and synovial fibroblasts
In four control patients (two with JT and two with normal joints), the nuclei extracted from synovial tissue or P-1 SFB showed normal cytogenetic findings (Supplementary Table 5). Upon extended in vitro culture, however, polysomies of multiple chromosomes exceeding the threshold levels appeared in P-4 SFB from one JT patient (148 days culture) and in P-1 SFB from another JT patient (21 days culture; Supplementary Tables 2 and 5).
The mean percentages for polysomies of chromosomes 4, 6, 7, and 9 in normal/JT synovial tissue and collagenase digest did not exceed the threshold levels (4% for polysomies and 7% for monosomies). The same was true for monosomy of chromosome 7 (Supplementary Fig. 1).
The mean percentages of SFB polysomic for chromosomes 4, 6, 7, and 9 equally increased until P-4, leading to levels above threshold for these chromosomes in P-4 (Supplementary Fig. 1).
If instead of all polysomic nuclei only trisomic nuclei were considered, the mean percentage of nuclei/cells with aberrations was well below the threshold levels for all samples (including P-4 SFB) and chromosomes (Supplementary Fig. 1; shaded bars).
Direct comparison between numerical chromosomal aberrations in the synovial membrane of patients with RA, OA, and joint trauma or normal joints
While chromosome 6 showed almost no numerical aberrations (with the exception of an increased level of polysomy 6 in P-1 RA SFB), both OA and RA synovial tissue showed significant more polysomy 7 than did normal/JT tissue, with a significantly higher value in OA than in RA (11.9%; n = 7 versus 5.9%; n = 7; P ≤ 0.05; Supplementary Fig. 2). A significant difference between RA and OA was also observed in the collagenase digest (Supplementary Fig. 2).
Isolated SFB from RA patients (12.4%; n = 10) and OA patients (18.4%; n = 15) strongly contributed to the polysomy 7 observed in nuclei from synovial tissue and collagenase digest (containing a mixture of synovial cells). These findings were also confirmed if only trisomic nuclei were considered (Supplementary Fig. 2).
Numerical chromosomal aberrations in skin fibroblasts of patients with RA or OA and of normal controls
To answer the question of whether the cytogenetic aberrations observed in SFB were limited to the synovial compartment, paired skin FB from RA and OA patients and skin FB from normal donors were analyzed. In 2 of 5 RA patients analyzed and in 15 of 16 OA patients analyzed, polysomies above threshold were observed in P-1 or P-4 skin FB, with chromosomes 6, 9, and 12 being involved in RA, and chromosomes 4, 6, 7, 8, 9, 12, 18, and X in OA (GTG-banding/FISH; Supplementary Table 6). Until P-4, the percentage of skin FB showing chromosome gains or losses increased (Supplementary Table 6). Such numerical chromosomal aberrations were not observed in P-1 skin FB from normal donors.
Supplementary Table 6.
Cytogenetic findings in skin fibroblasts from patients with rheumatoid arthritis and osteoarthritis or unaffected normal controls
| Karyotype/FISH | ||
| Subject/diagnosis | Passage 1 | Passage 4 |
| EB20/RA | 46,XY; FISH:normal | 46,XY; FISH:+6 [6%];+12 [8%];incl. polyploid [2%] |
| EB25/RA | 46,XX; FISH:+6 [6%];+9 [6%];incl. polyploid [1%] | n.a. |
| EB26/RA | 46,XY; FISH:normal | n.a. |
| EB27/RA | 46,XX; FISH:normal | n.a.; FISH:normal |
| EB28/RA | 46,XY; FISH:normal | 46,XY |
| EB9/OA | 46,XX; FISH:+7 [6%];+8 [7%] | n.a. |
| EB10/OA | 46,XX: FISH:+7 [7%];+8 [7%] | 46,XX; FISH:+8 [9%];-18 [8%] |
| EB11/OA | 46,XX; FISH:+18 [8%] | 46,XX; FISH:normal |
| EB12/OA | 46,XX | 46,XX; FISH:+9 [6%]) |
| EB13/OA | 46,XY; FISH:normal | n.a.; FISH:+4 [18%];+7 [17%];+9 [9%];+12 [10%];incl. polyploid [8%] |
| EB14/OA | n.a. | 47,XX,+7[5]/46,XX[3]; FISH:+7 [5%];+8 [5%];+9 [6%];+X [8%];incl. polyploid [2%] |
| EB15/OA | 46,XX; FISH:+7 [6%];+9 [6%];incl. polyploid [4%] | 47,XX,+7[2]; FISH:+6 [16%];+7 [45%];+8 [15%];+9 [23%];incl. polyploid [15%] |
| EB16/OA | 46,XX; FISH:+6 [10%];+7 [10%];+8 [22%];+X [24%];incl. polyploid [8%] | n.a. |
| EB17/OA | 46,XX; FISH:normal | 46,XX; FISH:+4 [10%];+6 [10%];+8 [11%];+12 [11%];incl. polyploid [9%] |
| EB18/OA | 46,XX; FISH:+8 [5%] | 46,XX; FISH:normal |
| EB19/OA | n.a.; FISH:normal | 46,XX; FISH:+7 [7%];+8 [7%];+9 [6%];+12 [10%];incl. polyploid [3%] |
| EB21/OA | 46,XX; FISH:normal | 46,XX; FISH:+4 [5%];incl. polyploid [1%] |
| EB22/OA | 46,XY; FISH:normal | 46,XY; FISH:+4 [11%];+6 [13%];+7 [12%];+9 [11%];incl. polyploid [10%] |
| EB24/OA | n.a.; FISH:+4 [7%];+7 [7%];incl. polyploid [1%]. | 46,XX; FISH:+4 [23%];+6 [24%];+7 [22%];+9 [24%];incl. polyploid [22%] |
| EB29/OA | n.a.; FISH:normal | n.a. |
| J5/OA | n.a.; FISH:+4 [5%];incl. polyploid [3%] | n.a.; FISH:+4 [5%];+6 [7%];+7 [5%];+9 [7%];incl. polyploid [5%] |
| LZ1/Nor | n.a.; FISH:normal | n.a. |
| LZ2/Nor | 46,XX; FISH:normal | n.a. |
| LZ3/Nor | 46,XY; FISH:normal | n.a. |
| LZ5/Nor | 46,XY | n.a. |
Nor = normal; OA = osteoarthritis; RA = rheumatoid arthritis; n.a. = not analysed (Karyotype and/or FISH).
Although clearly above the threshold levels in individual patients (Supplementary Table 6), the mean percentages for polysomies of chromosomes 6 and 7 in P-1 skin FB from normal subjects or RA and OA patients did not exceed threshold levels (Supplementary Fig. 3). This was also the case for polysomy of chromosomes 4 and 9, as well as monosomy 7 (Supplementary Table 6).
Until P-4, the mean percentage of polysomic skin FB increased, exceeding threshold levels for chromosome 6 in RA and for chromosomes 4, 6, 7, and 9 in OA.
If, instead of all polysomic nuclei, only trisomic nuclei were considered, the mean percentages of numerical aberrations for chromosomes 6, 7 (Supplementary Fig. 3; shaded bars), 4, and 9 were well below threshold levels in P-1 or P-4 skin FB derived from RA patients, OA patients, or normal subjects.
On average, therefore, skin FB from RA and OA patients did not show any polysomies above threshold levels, except for hyperdiploid nuclei after extended culture (P-4), which probably reflects culture artifacts.
Supplementary discussion
Functional aspects of numerical chromosomal aberrations
Although not specific for RA, the occurrence of mosaic +7 in SFB may have functional consequences for rheumatic diseases, inasmuch as this chromosome carries a number of potentially relevant genes. These include cytokines/ growth factors and their receptors (e.g. platelet-derived growth factor-alpha chain, IL-6, epidermal growth factor, insulin-like growth factor, hepatocyte growth factor and its receptor); transcription factors (e.g. ETV1, SP4, cAMP-response element binding protein); signal-transduction molecules (e.g. the catalytic subunit of phospatidylinositol 3-kinase-gamma, protein-tyrosine phosphatase-zeta, human tyrosine kinase and its receptor); and molecules involved in matrix formation and cell-extracellular matrix contact (e.g. the α2-chain of collagen I and the beta-1 integrin subunit), among others [38]. Indeed, both RA and OA SFB with +7 appeared to have a selective growth advantage (as shown by the increase of the percentage of +7 SFB from P-1 to P-4; Tables 1 and 2; Figs 1 and 2). This finding is consistent with those in previous studies, in which up to 50% of the cells had +7 by P-10 to P-14 ([13,14]; in the present study, mean of 23% in P-10, n = 2 RA SFB).
Interestingly, a selective growth advantage seems to be conferred not only by +7, but also by several other numerical chromosomal aberrations (Tables 1 and 2). In RA, for example, an in vitro expansion of SFB with +6, +8, +9, +18, –18, and –Y was observed in individual cases (Table 1; Fig. 1). In OA, such expansion was noted in SFB with +4, +6, +8, +9, and –9 (Table 2; Fig. 2). Whether the presence/disruption of key regulatory genes of the cell cycle, such as oncogenes (homologues), transcription factors, and tumor-suppressor genes on these chromosomes [15,18,38] favors growth in vitro remains to be determined. Selective growth advantages of cells with chromosomal aberrations may therefore be relevant for the interpretation of future studies with SFB.
Whether gains of chromosomes result in overexpression of target genes on these chromosomes apparently depends on the individual case. For example, a coincidence of trisomy 7 and overexpression of epidermal growth factor has been reported in Barrett's esophagus [39]. On the other hand, lack of overexpression of the proto-oncogene ets-2, encoded on chromosome 21, in heart tissue of patients with Down syndrome provides evidence against a gene-dosage effect of polysomies [40]. Our own preliminary results from polymerase chain reactions show that the expression of the proto-oncogene c-met (hepatocyte growth factor receptor; encoded on 7q31) is considerably stronger in OA than in RA synovial tissue, providing further support for dissociation of polysomy and expression of a target gene (manuscript in preparation).
Extent of numerical chromosomal aberrations
Since it is technically impossible to perform GTG-banding on interphase nuclei extracted from synovial tissue and since normally centromere probes for only four different chromosomes were used per case (on the basis of numerical aberrations detected by GTG banding), the present study does not yet provide a complete assessment of chromosomal aberrations in synovial tissue and SFB from inflamed joints. Therefore, the extent of chromosomal aberrations may have been underestimated, leaving open the possibility of an even higher degree of aberrations in vivo.
Correlation of aberrations with clinical parameters and methotrexate treatment
Concurrent treatment may also play a role in the occurrence of chromosomal alterations. Indeed, repeated and/or high-dose application of methotrexate can induce hepatic fibrosis or cytogenetic damage (e.g. multinucle-ated binucleated cells and chromosomal aberrations [32,33]). In the present study, two of the four RA patients with structural chromosomal aberrations (irrespective of the affected chromosome), and two of the three RA patients with structural aberrations of chromosome 1, had been concurrently treated with methotrexate, raising the possibility that the treatment may have contributed to the occurrence of such alterations. On the other hand, there was no significant correlation between the levels of polysomy for any chromosome in RA SFB and concurrent treatment with methotrexate, raising the question of whether there was a clear-cut influence of the treatment.
Comparison of ex vivo and in vitro findings
The present study shows that limited cultivation of adherent cells for 7 days, subsequent isolation of SFB, and rapid analysis of chromosomal aberrations does not greatly alter the cytogenetic status of the cells. Therefore, this previously published technique of isolating SFB from primary culture [25], by analyzing cells with few replication cycles, proves helpful in limiting the influence of in vitro growth selection and/or exposure to culture media.
Acknowledgments
Acknowledgements
We are grateful to Prof Dr J Dippold and Prof Dr G von Salis-Soglio (Department of Orthopedics, University of Leipzig), Dr D Sauer (Leipzig), Prof Dr D Jungmichel (Clinic of Orthopedics, Bad Düben), and Dr J Liebau (Clinic of Orthopedics, Vogelsang, Germany), for providing patient material and to Bärbel Ukena, Babette Niescher, and Doris Claus for expert technical assistance. Dr Ernesta Palombo-Kinne is gratefully acknowledged for critical revision of the manuscript.
This work was supported by the Bundesministerium für Bildung und Forschung (BMBF; grants of the Interdisciplinary Center for Clinical Research (IZKF) at the University of Leipzig 01KS9504 to TZ, 01KS9504 project A1 to H-DS, and 01KS9504 project A11 to UGF and HH, as well as grants 01VM9311/3 and 01ZZ9602 to RWK, G Hein, and U Claussen).
References
- Kinne RW, Palombo-Kinne E, Emmrich F. Activation of synovial fibroblasts in rheumatoid arthritis. Ann Rheum Dis. 1995;54:501–504. doi: 10.1136/ard.54.6.501-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassbender HG. Histomorphological basis of articular cartilage destruction in rheumatoid arthritis. Coll Relat Res. 1983;3:141–155. doi: 10.1016/s0174-173x(83)80040-5. [DOI] [PubMed] [Google Scholar]
- Trabandt A, Aicher WK, Gay RE, Sukhatme VP, Nilson-Hamilton M, Hamilton RT, McGhee JR, Fassbender HG, Gay S. Expression of the collagenolytic and Ras-induced cysteine proteinase cathepsin L and proliferation-associated oncogenes in synovial cells of MRL/I mice and patients with rheumatoid arthritis. Matrix. 1990;10:349–361. doi: 10.1016/s0934-8832(11)80142-3. [DOI] [PubMed] [Google Scholar]
- Ritchlin CT, Winchester RJ. Potential mechanisms for coordinate gene activation in the rheumatoid synoviocyte: implications and hypotheses. Springer Semin Immunopathol. 1989;11:219–234. doi: 10.1007/BF00197304. [DOI] [PubMed] [Google Scholar]
- Case JP, Lafyatis R, Remmers EF, Kumkumian GK, Wilder RL. Transin/stromelysin expression in rheumatoid synovium. A transformation-associated metalloproteinase secreted by phenotypically invasive synoviocytes. Am J Pathol. 1989;135:1055–1064. [PMC free article] [PubMed] [Google Scholar]
- Trabandt A, Aicher WK, Gay RE, Sukhatme VP, Fassbender HG, Gay S. Spontaneous expression of immediately-early response genes c-fos and egr-1 in collagenase-producing rheumatoid synovial fibroblasts. Rheumatol Int. 1992;12:53–59. doi: 10.1007/BF00300977. [DOI] [PubMed] [Google Scholar]
- Aicher WK, Heer AH, Trabandt A, Bridges SLJ, Schroeder HWJ, Stransky G, Gay RE, Eibel H, Peter HH, Siebenlist U. Overex-pression of zinc-finger transcription factor Z-225/Egr-1 in synoviocytes from rheumatoid arthritis patients. J Immunol. 1994;152:5940–5948. [PubMed] [Google Scholar]
- Fox DA, Millard JA, Kan L, Zeldes WS, Davis W, Higgs J, Emmrich F, Kinne RW. Activation pathways of synovial T lymphocytes. Expression and function of the UM4D4/CDw60 antigen. J Clin Invest. 1990;86:1124–1136. doi: 10.1172/JCI114817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvaifler NJ, Firestein GS. Pannus and pannocytes. Alternative models of joint destruction in rheumatoid arthritis. Arthritis Rheum. 1994;37:783–789. doi: 10.1002/art.1780370601. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Hasunuma T, Kato T, Sumida T, Kobata T. Apoptosis in rheumatoid arthritis: a novel pathway in the regulation of synovial tissue. Arthritis Rheum. 1998;41:1–9. doi: 10.1002/1529-0131(199801)41:1<1::AID-ART1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Firestein GS. Novel therapeutic strategies involving animals, arthritis, and apoptosis. Curr Opin Rheumatol. 1998;10:236–241. doi: 10.1097/00002281-199805000-00012. [DOI] [PubMed] [Google Scholar]
- Johansson B, Heim S, Mandahl N, Mertens F, Mitelman F. Trisomy 7 in nonneoplastic cells. Genes Chromosomes Cancer. 1993;6:199–205. doi: 10.1002/gcc.2870060402. [DOI] [PubMed] [Google Scholar]
- Mertens F, Orndal C, Mandahl N, Heim S, Bauer HF, Rydholm A, Tufvesson A, Willen H, Mitelman F. Chromosome aberrations in tenosynovial giant cell tumors and nontumorous synovial tissue. Genes Chromosomes Cancer. 1993;6:212–217. doi: 10.1002/gcc.2870060404. [DOI] [PubMed] [Google Scholar]
- Ermis A, Hopf T, Hanselmann R, Remberger K, Welter C, Dooley S, Zang KD, Henn W. Clonal chromosome aberrations in cell cultures of synovial tissue from patients with rheumatoid arthritis. Genes Chromosomes Cancer. 1993;6:232–234. doi: 10.1002/gcc.2870060407. [DOI] [PubMed] [Google Scholar]
- Ermis A, Henn W, Remberger K, Hopf C, Hopf T, Zang KD. Proliferation enhancement by spontaneous multiplication of chromosome 7 in rheumatic synovial cells in vitro. Hum Genet. 1995;96:651–654. doi: 10.1007/BF00210293. [DOI] [PubMed] [Google Scholar]
- Weiss KR, Georgescu HI, Gollin SM, Kang R, Evans CH. Trisomy 7 in synovial fibroblasts obtained from arthritic joints. Inflamm Res. 1999;48 (suppl 2):S132–S133. doi: 10.1007/s000110050553. [DOI] [PubMed] [Google Scholar]
- Bonnici AV, Birjandi F, Spencer JD, Fox SP, Berry AC. Chromosomal abnormalities in Dupuytren's contracture and carpal tunnel syndrome. J Hand Surg [Br] 1992;17:349–355. doi: 10.1016/0266-7681(92)90128-o. [DOI] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Rock H, Gotz H, Siegel A, Krone W. Monosomy 6 in human cultured fibroblast-like cells permanently stimulated by fibroblast growth factor 1: evidence for selection. Cytogenet Cell Genet. 1999;86:28–33. doi: 10.1159/000015424. [DOI] [PubMed] [Google Scholar]
- Mertens F, Palsson E, Lindstrand A, Toksvig-Larsen S, Knuutila S, Larramendy ML, el-Rifai W, Limon J, Mitelman F, Mandahl N. Evidence of somatic mutations in osteoarthritis. Hum Genet. 1996;98:651–656. doi: 10.1007/s004390050278. [DOI] [PubMed] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- Altman R, Asch E, Bloch D, Bole G, Borenstein D, Brandt K, Christy W, Cooke TD, Greenwald R, Hochberg M. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 1986;29:1039–1049. doi: 10.1002/art.1780290816. [DOI] [PubMed] [Google Scholar]
- Dougados M, van der Linden S, Juhlin R, Huitfeldt B, Amor B, Calin A, Cats A, Dijkmans B, Olivieri I, Pasero G. The European Spondylarthropathy Study Group preliminary criteria for the classification of spondylarthropathy. Arthritis Rheum. 1991;34:1218–1227. doi: 10.1002/art.1780341003. [DOI] [PubMed] [Google Scholar]
- Cassidy JT, Levinson JE, Bass JC, Baum J, Brewer EJJ, Fink CW, Hanson V, Jacobs JC, Masi AT, Schaller JG. A study of classification criteria for a diagnosis of juvenile rheumatoid arthritis. Arthritis Rheum. 1986;29:274–281. doi: 10.1002/art.1780290216. [DOI] [PubMed] [Google Scholar]
- Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- Zimmermann T, Kunisch E, Pfeiffer R, Jüngel A, Stahl H-D, Sack U, Laube A, Liesaus , Roth A, Palombo-Kinne E, Emmrich F, Kinne RW. Isolation and characterization of rheumatoid arthritis synovial fibroblasts from primary culture - Primary-culture cells markedly differ from 4th passage cells. Arthritis Res. 2001;3:72–76. doi: 10.1186/ar142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrí C, Kühnlein P, Buchner E, Schmidt CB, Franch A, Castell M, Hünig T, Emmrich F, Kinne RW. Depletion of gamma/delta T cells does not prevent or ameliorate, but rather aggravates, rat adjuvant arthritis. Arthritis Rheum. 1996;39:204–215. doi: 10.1002/art.1780390206. [DOI] [PubMed] [Google Scholar]
- Seabright M. A rapid banding technique for human chromosomes. Lancet. 1971;2:971–972. doi: 10.1016/s0140-6736(71)90287-x. [DOI] [PubMed] [Google Scholar]
- Mitelmann F, (Ed) ISCN (1995): An International System for Human Cytogenetic Nomenclature Basel: S Karger, 1995.
- Liehr T, Grehl H, Rautenstrauss B. FISH analysis of interphase nuclei extracted from paraffin-embedded tissue. Trends Genetics. 1995;11:377–378. doi: 10.1016/s0168-9525(00)89113-1. [DOI] [PubMed] [Google Scholar]
- Liehr T, Grehl H, Rautenstrauss B. A rapid method for FISH analysis on interphase nuclei extracted from cryofixed tissue. Trends Genetics. 1996;12:505–506. doi: 10.1016/s0168-9525(96)90047-5. [DOI] [PubMed] [Google Scholar]
- Gebhart E, Trautmann U, Reichardt S, Liehr T. Chromosomal heterogeneity of aneuploid leukemic cell populations detected by conventional caryotyping and by fluorescence in situ hybridization (FISH). Anticancer Res. 1993;13:1857–1862. [PubMed] [Google Scholar]
- Hazleman BL. The comparative incidence of malignant disease in rheumatoid arthritics exposed to different treatment regimens. Ann Rheum Dis. 1982;41(suppl 1):12–17. doi: 10.1136/ard.41.suppl_1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshava C, Keshava N, Whong WZ, Nath J, Ong TM. Inhibition of methotrexate-induced chromosomal damage by vanillin and chlorophyllin in V79 cells. Teratog Carcinog Mutagen. 1997;17:313–326. [PubMed] [Google Scholar]
- Mitelman F, Mertens F, Johansson B. Breakpoint map of recurrent chromosome aberrations Bethesda, MD, USA: National Cancer Institute. http://www.ncbi.nlm.nih.gov/CCAP/mitelsum.cgi
- Broberg K, Hoglund M, Limon J, Lindstrand A, Toksvig-Larsen S, Mandahl N, Mertens F. Rearrangement of the neoplasia-associated gene HMGIC in synovia from patients with osteoarthritis. Genes Chromosomes Cancer. 1999;24:278–282. [PubMed] [Google Scholar]
- Emerit I. Chromosomal breakage in systemic sclerosis and related disorders. Dermatologica. 1976;153:145–156. doi: 10.1159/000251109. [DOI] [PubMed] [Google Scholar]
- Ermis A, Oberringer M, Wirbel R, Koschnick M, Mutschler W, Hanselmann RG. Tetraploidization is a physiological enhancer of wound healing. Eur Surg Res. 1998;30:385–392. doi: 10.1159/000008603. [DOI] [PubMed] [Google Scholar]
- Deloukas P, Schuler GD, Gyapay G, Beasley EM, Soderlund C, Rodriguez-Tome P, Hui L, Matise TC, McKusick KB, Beckmann JS, Bentolila S, Bihoreau M, Birren BB, Browne J, Butler A, Castle AB, Chiannilkulchai N, Clee C, Day PJR, Dehejia A, Dibling T, Drouot N, Duprat S, Fizames C, Fox S. A physical map of 30,000 human genes. Science. 1998;282:744–746. doi: 10.1126/science.282.5389.744. [DOI] [PubMed] [Google Scholar]
- Garewal H, Meltzer P, Trent J, Prabhala R, Sampliner R, Korc M. Epidermal growth factor overexpression and trisomy 7 in a case of Barrett's esophagus. Dig Dis Sci. 1990;35:1115–1120. doi: 10.1007/BF01537584. [DOI] [PubMed] [Google Scholar]
- Greber-Platzer S, Schatzmann-Turhani D, Wollenek G, Lubec G. Evidence against the current hypothesis of "gene dosage effects" of trisomy 21: ets-2, encoded on chromosome 21, is not overexpressed in the hearts of patients with Down Syndrome. Biochem Biophys Res Commun. 1999;254:395–399. doi: 10.1006/bbrc.1998.9743. [DOI] [PubMed] [Google Scholar]
- Otto JM, Chandrasekeran R, Vermes C, Mikecz K, Finnegan A, Rickert SE, Enders JT, Glant TT. A genome scan using a novel genetic cross identifies new susceptibility loci and traits in a mouse model of rheumatoid arthritis. J Immunol. 2000;165:5278–5286. doi: 10.4049/jimmunol.165.9.5278. [DOI] [PubMed] [Google Scholar]
- Furuya T, Salstrom JL, McCall-Vining S, Cannon GW, Joe B, Remmers EF, Griffiths MM, Wilder RL. Genetic dissection of a rat model for rheumatoid arthritis: Significant gender influences on autosomal modifier loci. Hum Mol Genet. 2000;9:2241–2250. doi: 10.1093/oxfordjournals.hmg.a018915. [DOI] [PubMed] [Google Scholar]
- Jawaheer D, Seldin MF, Amos CI, Chen WV, Shigeta R, Monteiro J, Kern M, Criswell LA, Albani S, Nelson JL, Clegg DO, Pope R, Schroeder HW, Jr, Bridges SL, Jr, Pisetsky DS, Ward R, Kastner DL, Wilder RL, Pincus T, Callahan LF, Flemming D, Wener MH, Gregersen PK. A genomewide screen in multiplex rheumatoid arthritis families suggests genetic overlap with other autoimmune diseases. Am J Hum Genet. 2001;68:927–936. doi: 10.1086/319518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada R, Tanaka T, Ohnishi Y, Suematsu K, Minami M, Seki T, Yukioka M, Maeda A, Murata N, Saiki O, Teshima R, Kudo O, Ishikawa K, Ueyosi A, Tateishi H, Inaba M, Goto H, Nishizawa Y, Tohma S, Ochi T, Yamamoto K, Nakamura Y. Identification of 142 single nucleotide polymorphisms in 41 candidate genes for rheumatoid arthritis in the Japanese population. Hum Genet. 2000;106:293–297. doi: 10.1007/s004390051040. [DOI] [PubMed] [Google Scholar]