

Abstract
Natural extracts are complex mixtures that may be rich in useful bioactive compounds and therefore are attractive sources for new leads in drug discovery. This review describes drug discovery from natural products and in explaining this process puts the focus on ion-channel drug discovery. In particular, the identification of bioactives from natural products targeting nicotinic acetylcholine receptors (nAChRs) and serotonin type 3 receptors (5-HT3Rs) is discussed. The review is divided into three parts: “Targets,” “Sources,” and “Approaches.” The “Targets” part will discuss the importance of ion-channel drug targets in general, and the α7-nAChR and 5-HT3Rs in particular. The “Sources” part will discuss the relevance for drug discovery of finding bioactive compounds from various natural sources such as venoms and plant extracts. The “Approaches” part will give an overview of classical and new analytical approaches that are used for the identification of new bioactive compounds with the focus on targeting ion channels. In addition, a selected overview is given of traditional venom-based drug discovery approaches and of diverse hyphenated analytical systems used for screening complex bioactive mixtures including venoms.
Keywords: nicotinic acetylcholine receptors (nAChRs), 5-hydroxytryptamine receptors (5-HT3Rs), bioactive mixture profiling, venoms and natural extracts drug discovery
Introduction
Natural extracts are complex mixtures that may be rich in useful bioactive compounds with medicinal use. Natural extracts are therefore attractive sources for new leads in drug discovery. The work presented here mainly aims to review recent (technological) advances in the screening and identification of novel bioactive compounds targeting brain receptors. The focus is on the alpha-7 nicotinic acetylcholine receptor (α7-naChR) and the serotonin type 3 receptor (5-HT3R). In recent years many advances have been made in the development of analytical techniques facilitating improved identification of bioactive compounds from these natural extracts. This work is divided into three main parts: “Targets,” “Sources,” and “Approaches.” The “Targets” part discusses neurotransmitters, neurotransmitter receptors, and their function. Attention is given to the α7-naChR and 5-HT3R, their general relevance, and their relevance as a target in central nervous system (CNS) diseases specifically. In “Sources,” the importance of the environment in finding new drug leads is discussed. The use of various natural extracts, through history and their ongoing importance for current and future drug discovery purposes, is reviewed. A division is made between plant and animal extracts. The “Approaches” part gives an overview of the invaluable set of classical and novel techniques for the screening, detection, identification, and characterization of drug leads from natural extracts targeting ion channels. Included here is an overview of the venom-based drug discovery approach and the diverse hyphenated analytical systems used for complex-mixture screening.
Targets
Ligand-Gated Ion Channels in the Nervous System
The brain consists of hundreds of billions of neurons (nerve cells), which communicate with each other predominantly via chemical signaling in specialized parts of the neurons, so-called synapses. In synapses the presynaptic neurons can release endogenous compounds, neurotransmitters, which can bind to specific neurotransmitter receptors that are present on the postsynaptic neurons, or alternatively presynaptic terminals, resulting in modulation of synaptic activity.1 The neurotransmitters can be categorized in small-molecule-type transmitters that are enzymatically synthesized (e.g., acetylcholine, GABA, or serotonin) and peptide-type neurotransmitters encoded by the genome (e.g., the opioid peptides).2 The neurotransmitter receptors are categorized in two major groups: G-protein-coupled receptors (GPCRs) and ligand-gated ion channels (LGICs). Ion channels can be gated (i.e., opened or closed) by voltage (voltage-gated ion channels) or by a specific ligand binding to the channel (LGICs). The main role of ion channels is to generate ion concentration gradients that change the membrane potential, which may result in the propagation of electrical signals in the neurons.3 The LGICs can be divided in subcategories based on their structure: the Cys-loop-type LGICs consist of five subunits, the ionotropic glutamate receptors (iGluRs) are formed from four subunits, and the ionotopic ATP receptors (P2X-Rs), which consist of three subunits. The categorization of LGICs is shown in Table 1 .
Table 1.
Classification of the LGIC Superfamily.
| Type | Subtypes/Subunits | Main Functions in the CNS | Involvement in CNS Diseases |
|---|---|---|---|
| Anionic Cys-loop receptors | |||
| GABAA | α1–6, β1–3, γ1–3, δ ε π θ | Neuronal hyperpolarization, resulting in inhibitory effect on neuronal activity | Anxiety, insomnia, agitation219 |
| Glycine | α1–4, β | Inhibitory neurotransmission | Hyperekplexia220 |
| Cationic Cys-loop receptors | |||
| Serotonin (5-HT3) | 5-HT3A–E | Modulation of neurotransmitter release in interneurons, regulating the nausea-vomiting system in the CNS | Schizophrenia, addiction, anxiety and cognitive dysfunctions, emesis6,62 |
| Nicotinic acetylcholine receptors (nAChRs) | Muscle type: α1, β1, γ, δ, ε Neuronal type: α2–10, β2–4 |
Large diversity in roles, depending on subtype Mainly regulate presynaptic neurotransmitter release Excitatory postsynaptic potential (muscle type and ganglional), post- and presynaptic excitation Involved in memory and cognitive functions |
Alzheimer’s disease, Parkinson’s disease, epilepsy, schizophrenia, dementia, attention deficit, pain, depression, anxiety, and depression31,59 |
| Zinc-activated ion channel (ZAC) | Function not yet elucidated221 | ||
| Ionotropic glutamate receptors (iGluRs) | |||
| AMPA | GluA1–4 | Fast synaptic transmission, synaptic plasticity | Epilepsy222 |
| Kainate | GluK1–5 | Postsynaptic kainate receptors: excitatory neurotransmission Presynaptic kainate receptors: modulating GABA release |
Epilepsy223 |
| NMDA | GluN1, GluN2A–D, GluN3A–B |
Synaptic plasticity; learning and memory | Alzheimer’s disease, Parkinson’s disease, depression, and schizophrenia224 |
| Orphan | GluD1–2 | Synaptogenesis, synaptic plasticity and motor coordination in cerebellum | Ataxia, dementia, and schizophrenia225 |
| ATP-gated channels | |||
| P2X purinoreceptor | P2X1–7 | Nociception and modulation of synaptic transmission | Chronic pain226 |
Many CNS-related diseases are caused by the altered regulation, function, or expression of neurotransmitter receptors. This review focuses on two of these receptors that are involved in several CNS diseases and therefore comprise important therapeutic targets: the α7-nAChR and the 5-HT3R (Fig. 1A,B). However, it has to be noted that when reviewing signal transduction related to the CNS, gut-brain axis signaling cannot be ignored.4,5 Receptors, including 5-HT3Rs, are in some cases expressed in the gut and can play a role in signaling.6 As such, the microbiome also plays a role in brain and mental health and is implicated in CNS diseases.7,8 The microbiome, in this regard, would be interesting to include further. However, it is considered out of the scope of this review.
Figure 1.

(A) Structure of the α7-nAChR. A comparative model based on the homologue protein from Erwinia chrysanthemi (Protein Data Bank code 2VL0), side view. Figure adapted from Taly et al.24 (B) X-ray structure of mouse 5-HT3R in complex with the VHH15 stabilizing nanobody (Protein Data Bank code 4PIR, 3.50 Å resolution). Side view picture is shown. Figure adapted from Hassaine et al.214 (C,D) From x-ray structure of Lymnea stagnalis AChBP (Protein Data Bank code 1I9B, 2.7 Å resolution). (C) Top view, five subunits displayed. (D) Side view, displaying the ligand binding site between two subunits. Figures adapted from Brejc et al.215 License agreements for using these figures (A–D) were provided by the Copyright Clearance Center (CCC).
nAChRs and the α7-nAChR
The nAChRs belong to the Cys-loop receptor superfamily of the LGICs. The Cys-loop receptor family is named after a 13-amino-acid loop present in these receptors formed by a disulfide bridge. The members of this receptor family are the nAChRs, the GABAA receptors, the 5-HT3Rs, and the glycine receptors (GlyRs).9–12 The nAChRs can be divided into two groups: the muscle-type nAChRs and the neuronal-type nAChRs.13,14 The muscle-type nAChRs are found in neuromuscular junctions of the peripheral nervous system (PNS), whereas the neuronal types are found in the CNS, but are also expressed in non-neuronal tissues and organs, for example, in macrophages, lung, or skin. The nAChR subunits are classified as α subunits when the C loop of the receptor contains two adjacent cysteine residues, whereas in the β subunits these cysteine residues are absent. Up to now there are nine neuronal α subunits (α2–10) and three β subunits (β2–4) identified.15 Whereas some of the α subunits can form so-called homomeric receptors consisting of five homologous α subunits (the α7- and the α9-nAChR), the other neuronal α subunits form heteromers consisting of a combination of α and β subunits (e.g., α4β2 and α3β4). Crystal structure studies initially using the acetylcholine binding protein (AChBP) provided detailed information regarding the structure of nAChRs specifically and LGICs in general16 (Fig. 1C,D). AChBPs are soluble proteins expressed in glia cells of molluscan species, and they are homologous to the extracellular ligand recognition domain of nAChRs.16 These studies led to breakthrough discoveries in the understanding of the functioning and ligand recognition properties of the nAChRs.17–19
This review focuses on the homopentameric α7-nAChR, which has been implicated in CNS diseases. However, other subtypes of nAChR also have high clinical relevance. For example, the α4β2-nAChR is the predominant nAChR subtype in the brain and it is known to be involved in addiction to tobacco/smoking. For treatment of tobacco addiction, varenicline (Champix) is an approved drug targeting the α4β2-nAChR. Besides tobacco addiction, α4β2-nAChRs are also involved in cognitive disorders and in pain, and there are several compounds targeting α4β2-nAChR in clinical trials for the treatment of these.
In the brain the α7-nAChR is localized mainly in various brain regions involved in cognitive function, learning, and memory. α7-nAChRs were found in the cerebral cortex, hypothalamus, ventral tegmental area, substantia nigra, hippocampus, pineal gland, amygdala, medial habenula, olfactory bulb, and cerebellum.20–24 The α7-nAChR is also expressed in nonneuronal tissues, such as in macrophages, lymphocytes, skin, and kidney.25–30
Typical characteristics of the α7-nAChR are its high desensitization rate, calcium permeability, and the relatively low affinity of acetylcholine and nicotine toward the receptor.31,32 The most common functions awarded to the α7-nAChR are modulation of the other neurotransmitter systems, for example, modulation of synaptic plasticity in the brain (glutamate, dopamine, serotonin, GABA, and norephineprine), and the activation of messenger pathways (e.g., gene expression or neuronal survival) on postsynaptic neurons by changes in the intracellular Ca2+ concentration.33–35 The abnormal functioning or loss of nAChRs has been associated with many CNS diseases, such as to Alzheimer’s disease,36 Parkinson’s disease,37 epilepsy,38 schizophrenia,39 attention deficit hyperactivity disorder (ADHD),40 pain,41 anxiety,42 and depression.42
In schizophrenia patients, the expression level of the α7-nAChR is reduced in many brain regions compared with healthy subjects.43,44 There have been several efforts for developing drugs targeting the α7-nAChR. The partial α7-nAChR agonist Encenicline (EVP-6124), which showed promise in both Alzheimer’s disease and schizophrenia, successfully came through phase II clinical studies;45,46 however, due to severe adverse gastrointestinal effects in some patients during phase III trials, this compound has been put on hold for further studies.47,48 ATA-101, formally known as TC-5619, is a full α7-nAChR agonist that showed promise as a drug candidate in clinical phase I trials for improving the cognitive functions in schizophrenic patients,49 and also showed promise in the treatment of ADHD and potentially Alzheimer’s disease. However, in the phase II trial for schizophrenia50 the drug showed lack of efficacy and further development was halted for treatment of schizophrenia. Since then the manufacturer has also stated the drug candidate to be unsuccessful for treatment of ADHD; however, the compound was recently proposed as a drug candidate for treatment of acute and chronic cough.51
The role of the α7-nAChR in Alzheimer’s disease and in its treatment has been extensively studied, as discussed in recent reviews in the literature.36,52 In Alzheimer’s disease, the loss of α7-nAChRs in the hippocampus correlates with decreased cognitive functioning.53 Also, recent studies show that amyloid beta (Aβ) binds with high affinity to the α7-nAChR, and by this disrupts its normal functioning.54,55 However, there are still different opinions in the literature regarding the type of interaction and whether Aβ acts as an agonist or an antagonist on the α7-nAChR, and some publications state that there is no interaction for Aβ and the α7-nAChR. Nevertheless, there are α7-nAChR agonists, which are in phase I and phase II studies, that are potential new drug candidates for the treatment of the cognitive symptoms of Alzheimer’s disease.
Next to the possible involvement in CNS-related diseases, the α7-nAChR might be involved in non-CNS-related processes. Recently, the immunomodulatory and anti-inflammatory effects of the α7-nAChR were under investigation.25,56 Another recently discovered potential of non-CNS pharmaceutical targeting of the α7-nAChR is in cancer treatment. The α7-nAChR was found to modulate the chemosensitivity of gastric cancer cells, and recent studies showed that silencing α7-nAChR levels increased the sensitivity of gastric cancer cells to chemotherapy drugs.57,58 In a recent review, the role of nAChRs in disease is discussed in more detail, and an overview of the different nAChR-targeting drugs that are in clinical studies is provided.59 As a conclusion, the α7-nAChR is potentially an important therapeutic target against several medical conditions. Studies toward further understanding the biological functioning of this receptor are ongoing;60 however, there are still many remaining questions that need to be answered regarding the precise functions and roles of this receptor.
5-HT3R
The 5-HT3R ( Fig. 1B ) is the only serotonin receptor that belongs to the LGIC family, whereas all others are GPCRs.61 The 5-HT3R, like the α7-nAChR, is assembled from five subunits: either a homomeric receptor composed of five 5-HT3A subunits or a heteromeric receptor composed of 5-HT3A and 5-HT3B subunits.62–64 The expression of the subunits in the CNS or PNS has not yet been completely elucidated. Also, the genes of the 5-HT3C, 5-HT3D, and 5-HT3E subunits have been described, but their function and localization has not been characterized yet.65 The 5-HT3R has high similarity to the α7-nAChR. The two receptors share high sequence (30%) and structural homology, and therefore many ligands binding to the α7-nAChR are also interacting with the 5-HT3R.66 A few examples of ligands that show orthosteric binding to both the 5-HT3R and α7-nAChR are varenicline, epibatidine, and tropisetron.67–69 5-HT3Rs are expressed in both the CNS and PNS, and they are expressed in different brain locations, like in the cortex, amygdala, sustantia nigra, and brain areas involved in the vomiting reflex, such as the nucleus tractus solitarius and the area postrema.
The 5-HT3R has been associated with CNS diseases, such as schizophrenia, addiction, anxiety, and cognitive dysfunctions; however, there are currently no 5-HT3R-targeting drugs used clinically for the treatment of these diseases. In the clinic, the two main applications for 5-HT3R antagonists are the prevention of chemotherapy- and radiotherapy-induced nausea and vomiting, and the treatment of irritable bowel syndrome (IBS). 5-HT3Rs are expressed in both the PNS and CNS at the chemoreceptor trigger zone of the area postrema. 5-HT3R antagonists, such as ondansetron and granisetron, are used clinically to block these receptors; however, it is not certain yet if their action is mediated peripherally, centrally, or both. The 5-HT3R antagonist alosetron is widely used in the clinic for the treatment of diarrhea-predominant IBS, targeting the 5-HT3Rs of the enteric nervous system in the gastrointestinal track.
Since the α7-nAChR and the 5-HT3R are relevant drug targets, there is a high need for identifying new ligands that modulate these receptors, which can be developed further as new drug leads.
Sources
Natural Extracts as Sources of New Drug Leads
Nature has provided a large number of medicinal products for treating diseases over the course of history. The first medications known in many cultures and civilizations were made from plant extracts. The first examples of the use of natural extracts for medication are the Ebers Papyrus (Egyptian civilization, BCE 1500, a record of more than 700 natural extracts with medicinal properties), the Chinese Materia Medica (China, BCE 1100, 52 prescriptions),70 and the Ayurvedic (India, BCE 1000, more than 300 medicinal natural extracts described) systems.71 Besides the Middle and Far Eastern cultures, the ancient Greeks and Romans also used natural products as medicines. Hippocrates, who is considered the father of modern medicine, built the foundation of the Western medication system and applied phytotherapy, or healing with herbs, in his treatments. In the last century, many important medicines have been derived from natural sources.72 Well-known examples of medicines discovered from natural sources are aspirin from the Salix alba tree, digoxin from Digitalis purpurea, and penicillin from Penicillium notatum. Evidently, nature is a very important source for finding new drugs and drug leads for the treatment of disease.72–75 In the following paragraphs, examples of bioactive compounds derived from natural extracts targeting ion channels, with a focus on the α7-nAChR and the 5-HT3R, are overviewed.
Active Compounds Identified from Microbes, Plants, and Mushrooms
Today’s pharmacology would not be the same without the discovery of nicotine from the Nicotiana tabacum plant ( Fig. 2A ) and muscarine from the Amanita muscaria mushroom. These two specific agonists allowed researchers to discover and investigate the main two subclasses of AChRs (nicotinic and muscarinic AChRs). In nature, many examples can be found of plants that produce small molecular compounds that target nAChRs.76 Known examples of active compounds acting on nAChRs isolated from plants are cytisine from the golden chain tree (Laburnum anagyroides), the potent and selective α7-nAChR antagonist methyllylcaconitine (MLA) isolated from Delphinium brownii plant seeds ( Fig. 2B ), and the positive allosteric modulator galanthamine isolated from Galanthus woronowii (green snowdrop). Thus, natural sources are of great value for the discovery of bioactive compounds. These bioactives are often used as initial templates for structural drug lead optimization in which the original molecules are chemically modified by medicinal/synthetic chemical approaches to become final drug leads. Many new nAChR-targeting drugs, now being tested in preclinical and clinical phases for the treatment of Alzheimer’s disease, were initially derived from compounds of natural extracts. Hallucinogenic mushrooms, such as the Psilocybe mushrooms, contain many tryptamine-like small molecules that are agonists of the serotonin receptors. Next to plants and mushrooms, microbes and bacteria are also rich sources for discovering new bioactive compounds. Over the course of history, many life-saving medicines have been derived from microbial sources, of which penicillin and other antibacterial medicines are examples. Examples of active compounds from bacteria are anatoxin-a, which is a potent α7-nAChR agonist derived from a cyanobacterium,77 and ivermectin, an α7-nAChR-positive allosteric modulator produced by Streptomyces bacteria. A comprehensive overview of nAChR ligands discovered from natural sources can be found in Daly.76
Figure 2.

Examples of compounds targeting the α7-nAChR from natural sources. (A) Nicotine from the Nicotiana tabacum plant.76 (B) MLA from the Delphinium brownie plant.76 (C) Epibatidine from the Epipedobates tricolor frog.76 (D) α-Conotoxin ImI216 from the Conus imperalis cone snail venom. (E) α-Bungarotoxin217 from the Bungarus multicintus snake venom. (F) From the estimated 2 million species of the biodiversity (including plants, animals, fungi, and microorganisms, with still continuing discovery of new species), more than 95% have not been evaluated before for biological activity.218 License agreements for using these figures (A–F) were provided by the Copyright Clearance Center (CCC).
Active Compounds Discovered from Animal Venoms
Animal venoms are complex mixtures of biologically active compounds, which can cause various physiological effects, such as neurotoxicity and effects on the blood coagulation system, and are tissue necrotizing, when injected into a prey during the process of envenomation. The composition of venoms can range from small organic molecules to peptides and proteins, such as enzymes. Venoms can also contain metal ions, lipids, carbohydrates, and nucleosides. Diverse animal species such as the invertebrates (e.g., sea anemones, corals, mollusks, and arthropods) and vertebrates (e.g., reptiles, fish, and even mammals) produce venoms. These venoms contain a large variety of bioactive compounds, of which some are of interest as pharmacological tools and/or as new lead molecules in drug discovery.78–81
The diversity of toxins in venoms often targets a large variety of different types of ion channels, among which are the nAChRs. Venoms from marine animals in this regard are a rich source for discovering new bioactive compounds. For example, the cone snails, which are predatory sea snails, produce very potent neurotoxic venoms, each of which is a cocktail of tenths of different bioactive compounds.82–84 For instance, the α-conotoxin IMI ( Fig. 2D ) isolated from Conus imperalis cone snail venom is a potent and highly selective antagonist of the α7-nAChR. ω-Conotoxin isolated from the venom of Conus magus85,86 is a novel painkiller used in the clinic under the name Prialt (ziconotide). Neuroactive compounds isolated from different marine species besides cone snails are overviewed in a recent review from Sakai and Swanson (2014).87
The venoms of various types of arthropods, such as centipedes, spiders, and scorpions, contain a large variety of toxins acting mainly on different voltage-gated ion channels, but also on LGICs. Argiotoxin is an AMPA receptor blocker from the venom of the Argiope lobata. An example of an α7-nAChR ligand from arthropods is the alkaloid anabaseine, which is a potent agonist isolated from the venom of Aphaenogaster ants. Also, robber flies have been found to contain neurotoxic compounds in their venom. Drukewitz et al. (2018) characterized these through comparative transcriptomics, proteomics, and functional morphology.88
Frogs are not typically known for injecting venoms into their prey, and therefore they are considered poisonous (the toxin acts when it is eaten, inhaled, or digested). The high-affinity nAChR agonist epibatidine was discovered from the poisonous dendrobatid frog Epipedobates tricolor ( Fig. 2C ). Examples of noncompetitive nAChR antagonists (targeting mainly the α3β4-nAChR) are isodihydrohistrionicotoxin, isolated from the skin extract of Dendobatid frogs,89,90 and pseudophrynaminol, discovered from Australian Pseudophryne frogs.91
Snake venoms are one of the richest sources of toxins targeting the α7-nAChR. Based on their structure and function, the snake venom proteins can be divided into the following protein families: three-finger toxins, phospholipase A2s, proteinase inhibitors, serine proteinases, lectines, and metalloproteinases.92 Most of the snake venom toxins targeting the α7-nAChR belong to the family of the three-finger toxins. α-Bungarotoxin was isolated from Bungarus multicinctus (Taiwanese banded krait) venom ( Fig. 2E ) and is an irreversible antagonist of the α-nAChR. Used as a pharmacological tool, this toxin allowed identification of the α7-nAChR. Successful examples can be mentioned for the therapeutic use of snake venom toxins. One of the best-known examples of a successful drug on the market is the antihypertensive drug captopril, which is an angiotensin-converting enzyme (ACE) inhibitor derived from a peptide discovered in the venom of the lancehead viper (Bothrops jararaca).93 Another example of an approved medicine from snake venom is the antiplatelet drug epifibatide (Integrillin), which was derived from a peptide toxin found in the southeastern pygmy rattlesnake (Sistrurus miliarius barbouri).94 Next to compounds from snake venoms targeting the cardiovascular and neuromuscular system, compounds with antibacterial, antitumor, immunomodulator, and analgesic effects have also been discovered and are being extensively studied as drug leads.95
Many novel bioactive compounds discovered in venoms do not pass clinical trials and never make it to a new medicine.96 This may be due to the fact that compounds identified from natural sources often have high affinity toward target receptors and often are antagonists/inhibitors, a combination that is in most cases not an advantage. Moreover, venom-based peptides and proteins often cannot pass the blood–brain barrier (BBB) of mammals, so that they cannot reach their target in the CNS.79 In addition, peptide and protein drugs commonly have to be administered parenterally, encompassing an increased risk on adverse immunogenic responses. A recent review on peptide therapeutics from venoms states that successfulness looks more optimistic as structure elucidation’s successes increase due to advances in proteomic and transcriptomic approaches.96 Among others, these advances increase the potential of finding new bioactive peptides from venoms as potential therapeutics. Even if newly discovered bioactive peptides and proteins from venoms may not reach the market as medicines, they still can provide new relevant information for a better understanding of receptor expression and binding site information, and of receptor functions (physiology and structure). In addition, this information can find its way into diagnostics97,98 and allow for more efficient ways of the recombinant expression of snake venom toxins, such as recombinant viper three-finger toxins studied as potent inhibitors of nAChRs.99 Furthermore, other applications for these compounds are found, such as in cosmetics100,101 and pesticides.102–104
Approaches
Approaches to Find New Compounds Targeting Ion Channels
Ion channels are important targets for the development of new drug lead molecules. Several “classical” approaches are used in the drug discovery process for ion-channel targets. More advanced and faster screening techniques have emerged over the last years.105–109 Classical approaches in the initial screening process and lead optimization phase include binding assays and assays measuring ion fluxes across the cell membrane (e.g., monitoring the membrane potential). More recently developed screening technologies involve automation and increase in the speed of screening. The newest assay formats can be considered high throughput, such as the fluorescence imaging plate reader (FLIPR) and calcium-flux assay.110 Besides these high-throughput screening (HTS) assay formats, nowadays medium/high throughput can be achieved with automated patch-clamp technologies and with the newest HTS instrumentation for automated electrophysiology ion-flux assays.111–113 The following section discusses in detail the different assay formats and instrumental setups used in venom and ion-channel drug discovery, with a focus on the α7-nAChR and the 5-HT3R.
Binding Assays
Binding assays are used to detect binding of a ligand to a receptor, enzyme, macromolecule, or antibody. In order to measure the interaction between a ligand and a receptor, in most of the assay formats labeling of the ligand or the receptor is required. Depending on the type of labeling, the receptor–ligand binding assays can be classified into radioactive and nonradioactive labeling formats.114 Assays measuring the displacement of a radioactively labeled ligand are widely used screening techniques due to their sensitivity, general use, straightforward synthesis of radioactive ligands, and easy automation in HTS. The radioactively labeled ligand assays can be performed in heterogeneous and homogeneous assay formats. In heterogeneous assays, the bound ligand needs to be separated from the nonbound ligand by filtration, dialysis, or centrifugation. Due to the labor-intensiveness of this separation step, homogeneous scintillation proximity assays (SPAs) have also been developed. In SPAs, the receptor is immobilized on a scintillation bead or scintillation well, which emits light via energy transfer when a beta-radiating radioactive ligand molecule is bound to the immobilized receptor.115 This mechanism is based on close proximity. The beta radiation travels a short distance in water and decay is thus fast. Close to the scintillation material on a bead or coated well, the radiation is transferred to the scintillation material, which converts it into light. More recently, a study by Wittmann and Strasser (2017) investigated kinetic radioligand competition binding assays as a technique.116 One-state models, to date, are not applicable when radioligands display biphasic binding kinetics to a receptor. In his study, Wittmann and Strasser (2017) developed a molecular model for kinetic competitive radioligand binding assays to detect two different binding orientations. In this study they targeted the GPCRs. Possible limitations of this model were studied by comparison with different traditional models targeting only one binding orientation. However, the developed model, successful as an alternative experimental method for the detection of two different binding orientations of a ligand, had some limitations as the detection of the different binding orientations was only achieved under distinct conditions.116 Another study, by Guo et al. (2018), developed a mathematical model for determination of the binding kinetics of unlabeled ligands. A so-called two-state model for the binding kinetics determination was developed for the assessment of competitive radioligands showing biphasic binding characteristics to a receptor.117 Radioligand binding assays for screening ligands for binding to the α7-nAChR and the 5-HT3R were extensively used in academia and industry and allowed the identification of many nAChR ligands.118–122 The disadvantages of radioligand binding assays are the high costs (especially the SPAs) and aspects related to safety, health, and disposal of radioactive waste. The filtration assays are also lower in throughput and more labor-intensive. Therefore, many efforts have been made to develop nonradioactive methods.
Nonradioactive binding assays use various types of assay readout strategies, in most cases based on fluorescence, chemo-/bioluminescence, or colorimetric detection. More specifically, fluorescence/bioluminescence resonance energy transfer (FRET/BRET),123–125 fluorescence polarization (FP),126,127 total internal reflection fluorescence (TIRF),128,129 and fluorescence enhancement assay principles can be named.130 Fluorescence detection is often nondestructive and, due to its noninvasive character, enables live cell studies as shown by Hovius (2013). In this study, the compound GR-186741X was evaluated as a high-affinity fluorescent agonist for the 5-HT3R.131
Nonradioactive binding assays can be performed in both homogeneous and heterogeneous assay formats, based on the different types of assay component interactions. There are various types of instrumentation for measuring binding assays, such as plate reader formats, flow cytometry,132,133 chromatographic separation formats (frontal chromatography, quantitative affinity chromatography),134–136 and surface plasmon resonance (SPR) analysis.137,138 Nonradioactive binding assays developed for screening α7-nAChR and 5-HT3R ligands include SPR-based AChBP ligand binding assays,138 TIRF assays for studying the 5-HT3R,128,129 and fluorescence enhancement assays using AChBP as a soluble binding protein that mimics the extracellular domain of the α7-nAChR.130,139 Fluorescent ligands need sufficient affinity toward the protein target and need to give significantly high fluorescent signals for optimal in vitro (and possibly in vivo) pharmacology assessment. Often, high-affinity ligands are synthesized or modified with fluorophores to achieve this goal, as is done for 5-HT3 ligands, as discussed by the studies of Lochner et al. (2015)140 and Jack et al. (2015).141
Ligand binding assays in general have the limitation that only information on the binding affinity toward a receptor or ion channel is measured, and the mechanism of action (e.g., if a ligand is an agonist, antagonist, or a modulator) is not known. Also, ligand binding assays measure the interaction with a specific binding site of the receptor, and therefore ligands interacting with other sites of the receptor (allosteric modulators) are not detected. Therefore, functional cell-based assays, such as ion-flux assays and electrophysiology, are widely used for screening purposes.107,142
Ion-Flux and Membrane Potential Assays
Cell-based ion-flux assays measure the functioning, activation, and blocking of ion channels by monitoring the flux of ions passing through the channel. There are several approaches described in the literature for HTS ion-flux assays, such as flux assays based on radioactive isotopes using, for example, 86Rb+ for studying potassium channels and nonselective cation channels,143,144 and nonradioactive Rb+-flux assays using atomic absorption spectroscopy.145 Since only an endpoint can be measured, the kinetics of the ion-channel activation/inactivation cannot be assessed. More extensively used ion-flux assays are based on using voltage-sensitive FRET dyes or ion-selective fluorescence dyes.146 From the ion-selective fluorescence dyes, the calcium-sensitive fluorescence dyes, such as the fluo-4 and FLIPR calcium dyes, are most widely used.147,148 These dyes, which can monitor the changes in intracellular Ca2+ ion concentrations caused by ion influxes, are highly sensitive and provide a high dynamic range. Furthermore, they provide a real-time readout (i.e., they measure calcium fluxes in time upon ligand stimulation). The disadvantage of these assays is that they can only be used for measuring calcium fluxes. After the introduction of these calcium-flux assays in high-throughput plate readers, such as the FLIPR,110 calcium-flux assays became one of the most widely used screening techniques in the pharmaceutical industry and academic research for this type of assaying, as well as for the initial identification of new α7-nAChR ligands.149–153
A more traditional, but high-content information-providing technique to measure ion fluxes (ion currents) is electrophysiology. The most commonly used approaches for this type of measurement are the two-electrode voltage clamp (TEVC)154 in Xenopus oocytes and the patch-clamp techniques155 using mammalian cellular systems with the ion channel of interest overexpressed. Using electrophysiology in ion-channel drug discovery, we can fully characterize the mechanism of action of a ligand, including the amplitude of a current or voltage change evoked by the ligand on the ion channel and the duration of the signal. Using electrophysiology techniques, we can record millisecond-range signals, which is advantageous when an ion channel is desensitized in milliseconds, as is the case for the α7-nAChR. In contrast, techniques such as FLIPR and other ion-flux assays cannot measure these fast current changes. Moreover, electrophysiology does not depend on an ion-selective fluorescence dye, and therefore each type of ion channel can be measured by electrophysiology. A general disadvantage of electrophysiology techniques is the low throughput, labor-intensiveness, and requirement of highly skilled staff. In recent years, however, several efforts have been made toward automation to improve the throughput and complexity of these electrophysiology technologies.107,111,112
The approaches discussed in “Binding Assays” and “Ion-Flux and Membrane Potential Assays” are summarized and compared in Table 2 .
Table 2.
Drug Discovery Approaches Used for Ion-Channel Targets.
| Approach | Advantages | Disadvantages |
|---|---|---|
| Radioligand binding assays (homogenous [e.g., SPA] or heterogeneous [filtration, dialysis, centrifugation]) | - Sensitivity - General use - Straightforward synthesis of radioactive ligands - Easy automation in HTS |
- High costs - Aspects related to safety, health, and disposal of radioactive waste - Low-throughput and labor-intensive (filtration assays) - Only binding affinity information obtained |
| Nonradioactive binding assays (homogenous and heterogenous) Different mechanisms (e.g., FRET, BRET, FP, TIRF) Different instrumentation (e.g., plate reader, flow cytometry, chromatographic separation formats) |
- Fluorescent/bioluminescent: health, safety, and waste aspects - Easy automation in HTS - Possible multiplexing (e.g., with FC) |
- Information only on binding affinity |
| Nonradioactive binding assays using SPR | - No labeling is required, no interference with light scattering - Often binding kinetics can be measured |
- Receptor or ligand immobilization is required |
| Radioactive ion-flux assays | - Functional cell-based assay, information on activation/inactivation of receptor | - Endpoint can be measured (kinetics of the ion-channel activation/inactivation cannot be assessed) - Aspects related to safety, health, and disposal of radioactive waste |
| Nonradioactive ion-flux assays (voltage-sensitive FRET dyes, ion-selective fluorescence dyes) | - Functional cell-based assay, information on the function of the ligand (agonist, antagonist, or modulator) - Sensitivity, high dynamic range - Kinetic measurement possible (e.g., using FLIPR instrument) - Fluorescent: health, safety benefits compared with radioactive ion-flux assays - Easy automation in HTS |
- Ion-selective dyes (e.g., using calcium-dependent dyes only, calcium-dependent ion-channel function can be assessed) |
| Electrophysiology (e.g., patch-clamp, TEVC) | - Full functional characterization is possible (amplitude of a current or voltage change evoked by the ligand on the ion channel, duration of the signal) - Millisecond-range signals can also be recorded - Does not depend on an ion-selective fluorescence dye and every type of ion channel can be measured by electrophysiology |
- Low-throughput (however, in recent years several efforts were made for increasing the throughput and automation of electrophysiology technologies) - Labor-intensive - Requirement for highly skilled staff |
Classical Approaches for Screening of Mixtures: Bioassay-Guided Fractionation
A limitation of standard HTS approaches, which were discussed in “Binding Assays” and “Ion-Flux and Membrane Potential Assays,” is that they can be used only for the screening of libraries of pure compounds. When complex mixtures, such as natural extracts from plants or animal venoms, have to be screened for bioactive compounds, fractionation of the sample needs to be performed in order to allow for proper screening, detection, and identification of the bioactive compounds. This is important for the isolation of bioactive compounds in general; however, it is crucial when bioactive compounds relevant for signal transduction have to be distinguished from other compounds with other types of bioactivity. Commonly, multiple orthogonal separation steps are required in order to obtain sufficiently pure fractions to unambiguously assign the activity monitored by the applied screening assay to an individual bioactive. Also, the choice of bioassay is crucial for singling out bioactive compounds with desired activity. This traditional approach of natural extract screening is called bioassay-guided fractionation (BGF)156 and might also be used in environmental and food analysis, where it is called effect-directed analysis (EDA).157,158 BGF screens for medicinally relevant bioactives, whereas screening for toxicants (such as endocrine disruptors) is the focus of EDA. Prior to BGF, for many sample types, such as plants and mushrooms, a pretreatment is needed in order to remove interfering matrix constituents and transfer the bioactive compounds to a solution that can be analyzed. This sample pretreatment may involve steps of prewashing, drying, or freeze-drying of the material, grinding the material to obtain a homogeneous sample, and extracting the active compounds with an adequate extraction technique (e.g., pressurized-liquid extraction, solid-phase extraction, solid-phase microextraction, supercritical fluid extraction, or a surfactant-mediated technique).159
The isolation of bioactive compounds during the BGF process is usually performed using different separation techniques, including liquid chromatography (LC) on columns with different stationary phases, and by using thin-layer chromatography (TLC). With TLC, small quantities of complex mixtures can be separated while at the same time active compounds can be localized on the plate.158,160,161 Moreover, BGF approaches traditionally use low-resolution fractionation (fraction sizes of minutes or milliliters) after LC separation of a mixture (snake venom, plant extract, etc.), which showed bioactivity in an initial crude mixture screening. More specifically, the approach usually starts with a microfractionation step, commonly using ion-exchange chromatography (IEC) or size-exclusion chromatography (SEC) for the first separation round, after which each fraction is subjected to a bioassay of choice in order to select the bioactive fractions of interest.162,163 An orthogonal LC (often reverse-phase [RP]) analysis is then performed, including further microfractionation and bioassaying. When a bioactive compound is successfully isolated after repeated separation and bioassay steps, identification or confirmation of its chemical structure is attempted by mass spectrometry (MS), MS/MS, and/or nuclear magnetic resonance (NMR) analysis. However, even when using 2D LC, due to the low-resolution nature of the microfractionation approach, fractions collected remain complex mixtures. Consequently, this process has to be continued until eventually pure bioactive fractions (i.e., containing only one compound) are obtained for compound identification.164 However, during these procedures bioactive compounds may get lost in the process for various reasons, such as adsorption to LC tubing or to the walls of the collection vials, precipitation, degradation/oxidation, and/or denaturation in the case of peptides/proteins. Moreover, BGF approaches are very time-consuming and labor-intensive. In addition, BGF approaches often require large quantities of initial sample. As an exception, animal venoms typically require much less sample as venom peptides and proteins often can be fully identified using LC-MS only (i.e., proteomics approaches), while for small molecular bioactives, commonly NMR is required for full structural identification. Despite these drawbacks, there are still many successful examples to be named in which bioactive compounds were discovered using BGF, and development in BGF research is ongoing.
Recently, Nothias et al. (2018) developed a workflow procedure called “bioactive molecular networking” to help in cases where BGF is not able to isolate the active compound accordingly.165 In short, the workflow consists of the integration of MS/MS molecular networking and bioactivity scoring to assist in BGF. This would enable active compound detection directly from fractionated bioactive extracts and would make it possible to accelerate the dereplication of molecules using molecular networking prior to subsequent isolation of the compounds. Further development entails using bioactivity score prediction to help expose additional potentially bioactive molecules.
In a recent study by Lee et al. (2017), BGF was applied for the isolation of compounds from Morinda officinalis for their application as acetylcholinesterase (AChE), butyrylcholinesterase (BChE), and β-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitors as leads for Alzheimer’s treatment.166 Using LC in combination with ESI-MS/MS and NMR, the approach led to the isolation of 10 bioactive compounds, of which 5 were strong inhibitors of AChE.
Plant-derived bioactives, which mostly are low-molecular-weight compounds, may be a starting point for medicinal chemistry approaches to yield optimized leads with desired pharmacodynamic and pharmacokinetic profiles. Bioactives from venoms often are peptides or (small) proteins, and usually proteomics and biochemical approaches are needed for their structural and biological characterization. Structure optimization steps might include peptide synthesis of derivatives in the case of bioactive peptides and overexpression in suitable expression systems in the case of protein bioactives.
Hyphenated Approaches for Complex-Mixture Screening
Recently, BGF protocols have been redesigned in order to deal with some of the bottlenecks during the traditional BGF approach. As an example, the so-called at-line nanofractionation approaches can be mentioned. This workflow can be used for the rapid screening of medicinally relevant compounds in complex matrices, such as snake venom, and other complex mixtures. It can be applied using many different types of off-line assays, such as enzymatic and/or cell-based assays, for bioactivity assessment.167–170 In this setup, the separation column effluent is split to provide parallel MS detection and high-resolution (second-range) fraction collection. Recently, a fraction collection machine (FractioMate) was developed by the Vrije Universiteit and Spark Holland and is now commercially available.171 A typical at-line nanofractionation setup is shown in Figure 3 . The small second-range fractions are continuously collected onto 96-, 384-, or 1536-well plates. Usually, the plates are then vacuum-centrifuged to dryness, followed by direct on-plate pipetting of the bioassay reagents using pipetting robotics, followed by incubation and plate reader readout. In this setup, assays are developed post-LC separation toward a bioactivity of choice with the possibility of additionally adding test compounds postfractionation in order to induce targeted inhibition, activation, and/or quenching of the biochemical reaction toward obtaining more high-content information on bioactivities observed. Furthermore, by applying the high-resolution fraction collection, cleaner samples can be obtained and the parallel MS data gives the advantage of directly assigning an accurate mass to a bioactive compound. In the case of profiling for bioactive peptides and proteins from mixtures (e.g., venoms), the method also allows straightforward subsequent on-plate proteomics approaches (i.e., using the fractionated well plate) toward structure elucidation. This redesigned BGF approach further increases the possibility of successfully isolating bioactive compounds relevant in signal transduction. An example of this approach includes the development of an at-line cell-based screening methodology that combines LC followed by at-line nanofractionation and parallel MS with a functional fluorescence-based calcium-flux assay.172 This calcium-flux assay was performed with mammalian cells stably overexpressing the α7-nAChR. This nanofractionation system was developed and optimized in assay modes for screening agonists and for allosteric modulators of the α7-nAChR. The application of this methodology was demonstrated by the screening of a hallucinogenic mushroom extract (from Psilocybe mckennaii). In this study, the new approach of two orthogonal separations of the crude extract performed after each other using the same crude extract for each separation was used for precise bioactive compound identification from multiple co-eluting nonbioactive compounds. A potential drawback of nanofractionation is that it usually requires a vacuum-centrifugation step in which bioactive compounds can degrade or evaporate.
Figure 3.

Principle of the at-line nanofractionation approach. After LC separation (1) of a complex mixture, the flow is split in two. One part of the flow is directed to a UV detector followed by MS (2), while the other part (usually the larger part, like 90%) of the flow is collected as nanofractions into well plates by a nanofraction collector (3). The well plates are usually vacuum-centrifuged after nanofractionation and followed by a bioassay that is performed off-line on the nanofractionated well plates.
Other more automated analytical techniques have been developed for the screening and identification of bioactive compounds in complex mixtures. High-resolution screening (HRS) is based on the coupling of an LC separation with a continuous-flow bioassay for direct assessment of the biological activity of eluting compounds.173,174 The assay reagent mixture is continuously mixed with the LC effluent and directed to a coil providing incubation and reaction time. HRS also commonly encompasses parallel MS detection using a postcolumn split to yield accurate mass information on eluting compounds, including the bioactive ones. A typical HRS system is depicted in Figure 4 . Compared with the nanofractionation approach, HRS is far less time-consuming and the obtained bioassay readout has a higher time resolution. HRS is fast and allows the screening of many samples per day. Various types of biochemical assays have been successfully applied in HRS systems, although mostly enzymatic and binding assays.130,175,176 Normal bore chromatography HRS approaches require relatively large quantities of sample and biological reagents. This can be circumvented by employing miniaturized HRS systems, which consume much lower (bio)reagent amounts, and analyzing very low sample volumes. These so-called microfluidic HRS systems have been developed for profiling biologically active mixtures toward different targets including the AChBP. Using a postcolumn split, nano-LC is coupled in parallel to MS, via nano-electrospray ionization (nano-ESI), and to a microchip bioassay that utilizes a capillary bubble-cell LED-induced fluorescence detector for readout. The platform is operated in the range of nanoliter-per-minute flow rates and with nanoliter-range sample injection. The homogeneous bioassay is based on fluorescence enhancement caused by a tracer molecule bound to the AChBP. A decrease in fluorescence when the tracer is displaced by an eluting ligand binding to AChBP is used as bioassay readout. In a typical example, microfluidic HRS was developed and applied to the identification of AChBP bioactives in venoms.177 Subsequent work demonstrated both microfluidic HRS identification and follow-up purification of the bioactive compounds identified using straightforward MS-guided isolation.178 Subsequent bottom-up proteomics approaches were then used for chemical characterization of the bioactives. Next to snake venom profiling, other venoms and toxins were also screened using this technique.167 In the case of screening Conus textile cone snail venoms, a bioactive peptide was pinpointed among >1000 other peptides. The analytical workflow was also demonstrated to be able to screen and rapidly purify small-molecule bioactives from Bufo alvarius and Bufo marinus toad skin extracts. Tryptamine-like and steroidal-like binders of the AChBP were found in the crude skin extracts and subsequently purified using MS-guided isolation. Full structural identity of the compounds was eventually assessed by NMR and MS/MS, and their biological activity was tested and confirmed using conventional radioligand binding assays.
Figure 4.

Principle of an on-line postcolumn HRS system. After LC (or nano-LC) separation (1), the eluent flow is split into two parts. One part of the flow is directed to MS (2) for the identification of the eluting compounds, and the rest of the flow is directed to a continuous-flow incubation coil (3), in which eluting compounds are incubated with bioassay reagents, followed by a fluorescence detector for bioassay readout (4) measuring the biological activity (in the case of fluorescence assays).
Next to the fluorescence enhancement assay for the AChBP, a fluorescence enhancement assay has also been developed for the serotonin binding protein (5HTBP). This was first achieved in plate reader format and then implemented in the microfluidic HRS platform.167 The 5HTBP is an engineered binding protein that has the protein scaffold of the AChBP with the ligand recognition properties of the 5-HT3R. This fluorescence enhancement assay is a good initial screening technique for finding novel bioactives targeting the 5-HT3R. The applicability of the new technique was demonstrated by the screening of different snake venoms for compounds binding to 5HTBP.
Cell-based assays unfortunately in most cases cannot be used in HRS systems, primarily due to the technical difficulty of combining living cells with chromatographic eluents. However, for some cell lines overexpressing ion channels, this approach is feasible although technically and biologically extremely challenging. Heus et al. (2014) describe the analytical advancement to such a technique in which a Ca2+-flux assay is performed in postcolumn continuous-flow format using a flow cytometer (FC) as the readout device.167 The screening system consisted of an LC system coupled on-line to FC and MS using PEEK tubing and so-called superloops for the infusion and mixing of the bioassay components. The bioassay applied in this system was based on a calcium-flux assay using human α7-nAChR expressing SH-SY5Y neuroblastoma cells. This LC-FC-MS screening system was developed in agonist and in mixed antagonist–agonist assay modes. The latter assay mode allows the simultaneous detection of agonists and antagonists. In proof-of-principle experiments, the application of the screening system was demonstrated by the screening of tobacco plant leaf extract in agonist mode, and snake venoms in mixed antagonist–agonist assay mode.
A disadvantage of the on-line HRS approaches is that assays have to be relatively fast (incubation times of seconds to a maximum of a few minutes) in order to obtain a measurable response. Assays with long incubation and/or assay preparation times, such as radioligand binding assays, are out of scope and need to be addressed by the more traditional BGF or recently developed nanofractionation approach.
Next to the postcolumn HRS techniques and the nanofractionation approach, which both apply bioassays after chromatographic separation, other bioactivity screening approaches of mixtures are available. These can be divided in precolumn and on-column bioactivity profiling methods. These methods in general perform either an affinity extraction of ligands from a complex mixture using a selective precolumn, or a chromatographic separation using an affinity column.179 Using these precolumn and on-column approaches, bioactives bound to the affinity stationary phases can be separated from nonbinders before detection. Alternatively, the affinity proteins with ligands bound are separated from nonbinders predetection using ultrafiltration or size-exclusion separations. Techniques measuring protein–protein affinity and immobilized ligand–protein affinity interactions,180 MS binding assay approaches,181 and coupling ultrafiltration182 or size-exclusion separations183 to LC-MS can be named in this regard. Recently, Sichler et al. (2018) developed an MS-based binding assay for nAChRs using the ligand MB327, where a deuterated MB327 analogue was used as reporter ligand.184 The principle of MS binding assays is similar to radio ligand binding assays; however, the readout is based on mass spectrometric detection, instead of a radiolabeled (or fluorescent) reporter ligand.
Precolumn and on-column bioactivity profiling methods are comprehensively described in reviews.173,180,185 An example of yet another precolumn bioactivity profiling methodology used in nAChR drug discovery is a magnetic bead-based affinity-selection methodology for the identification of AChBP ligands in mixtures.135 A summary and comparison of complex-mixture screening techniques used in drug discovery approaches is given in Table 3 .
Table 3.
Summary and Comparison of Complex-Mixture Screening Techniques Used in Drug Discovery Approaches Discussed in This Review.
| Name of Approach | Advantages | Disadvantages |
|---|---|---|
| Bioassay-guided fractionation (BGF) | - Generically applicable approach for the identification of bioactives from complex mixtures | - Bioactive compounds often get lost in the process - Extremely time-consuming and labor-intensive - Requires large quantities of initial sample |
| At-line nanofractionation approach | - Most types of bioassays can be applied (also with longer incubation and preparation time) - Direct applicability on existing plate reader assays - Higher-resolution fractions are collected than in BGF - Direct correlation with MS |
- Time-consuming, lower resolution compared with on-line HRS - Commonly requires a freeze-drying or vacuum-centrifuge step of fractionated well plates prior to assaying (in which active compounds can get lost) |
| On-line high-resolution screening (HRS) and microfluidic HRS |
- Biological activity assessment is directly integrated after chromatographic separation - Rapidly obtained high-resolution chromatographic, mass spectrometric, and bioassay data |
- Only assays with short incubation times (i.e., seconds to few minutes range) are applicable - Only efficiently applicable to homogeneous add-only assays |
| Precolumn and on-column hyphenated screening approaches | - Rapid screening techniques - Wide applicability |
- Extended controls must be performed to validate the functionality of immobilized targets and avoid nonspecific binding - Only suitable for binding assays - Column preparation requires high expertise - Limited stability and memory effects from high-affinity ligands |
Drug Discovery Workflows toward Identification and Characterization of Venom Peptide-Derived Leads
In the previous part of this study, strategies for the screening and detection of components in natural extracts with relevant bioactivity were discussed. The focus in the following part of this review is the characterization of possible drug leads from natural extracts. The main focus is on snake venom as a matrix for novel drug leads using MS. In the previous part, MS was often mentioned as a popular, fast-growing technique that is gaining in importance in this field. Classical as well as novel approaches are also briefly discussed. Identifying and studying complete venom proteomes (i.e., all proteins and peptides in a venom and venom glands) comprehensively, by proteomics, transcriptomics, and genomics, is called venomics, an approach recently reviewed by Oldrati et al. (2016).186 MS is a very important technique in this field.187,188 Venomics can be used to better understand venom biodiversity and therewith functioning and evolution, and also for the identification of new lead compounds in drug discovery.80,189 When applying venomics for drug discovery, as described in “Classical Approaches for Screening of Mixtures: Bioassay-Guided Fractionation,” in most cases subsequent BGF purification and characterization of toxins is of interest and often performed by using a two-step purification involving successive SEC and reverse-phase liquid chromatography (RPLC).190 These are crucial steps for decreasing sample complexity toward successful MS measurements, by drastically decreasing ion suppression and improving the detection of low-abundance peptides and proteins. The mentioned nanofractionation with subsequent MS analysis is also a relevant technique in this field by the efficient combination of bioassaying and MS, directly after chromatographic separation. The introduction of nano-LC into the venomics workflow nowadays allows for highly reduced injection volumes, enabling the study of venoms available only in low amounts.
The most popular MS ionization approaches for life sciences research are ESI, easily combined with standard on-line high-performance liquid chromatography (HPLC) methodologies, and matrix-assisted laser desorption ionization (MALDI), where fractions are collected for subsequent off-line MALDI-MS/MS measurement. Another use here is in situ MALDI imaging-MS, as by Brunetti et al. (2018), for example, where peptides of skin tissue of a hylid tree frog were characterized, studying the difference in postsecretory peptide cleavage. This information can be valuable toward unraveling information on peptide function and mechanism of action, as some of these peptides show neuroactive and antimicrobial activities.191 MALDI imaging-MS can also be used for neurotransmitter distribution visualization/mapping of brain tissue, specifically focused on ACh192 and reviewed by Romero-Perez et al. (2015).193 Several studies have suggested that using MALDI-MS/MS and ESI-MS/MS in parallel for the study of venomics gives rise to complementary mass spectral data of venom compounds and therewith a more complete view of the venom profile.186,194,195 The most widely used approach for peptide and protein identification, into which many of the relevant drug leads fall, is the bottom-up approach, using amino acid sequence determination of the peptide/protein toxins after enzymatic digestion, followed by LC-MS and MS/MS (using either crude venom samples or prefractionated venom samples) or 2D sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).196,197 SDS-PAGE comprises separation of the venom proteins by one gel electrophoresis and subsequent staining for visualization. Alternatively, 2D gel electrophoresis of complex venom samples can be achieved in which separation occurs by isoelectric focusing (IEF) for isoelectric point separation (first dimension) and by SDS-PAGE for molecular weight separation (second dimension). After separation, individual spots can be excised and in-gel digestion can be performed, followed by LC-MS/MS analysis. Unfortunately, SDS-PAGE does not work well for low-molecular-weight peptides.187,196 After digestion of the proteins/peptides, the exact mass and fragmentation patterns determined are used for the identification of the peptides/proteins present in a sample using databases. Alternatively, N-terminal Edman sequencing can be used, but the peptides have to be analytically pure for this approach.
Identification resulting from the bottom-up approaches is nowadays reasonably automated; however, due to the absence of specific species information (e.g., genomes) in the available databases, in some cases the only possibility for identification is to assign found peptides to known protein families on the basis of similarity with existing sequence entries of other similar species for which information is available in a database. This results in incomplete sequence coverages, and generally it is difficult to differentiate between proteoforms.198 Recently, advances were made toward top-down proteomics (TDP) approaches, so-called top-down venomics. In these approaches, intact peptides and proteins are measured by MS. When leaving the molecules intact, ideally, complete protein masses and their fragment ions can be determined. More complete information on the identification and quantification of the toxin analyzed can be achieved compared with bottom-up approaches.198–200 A successful TDP approach, in this field, includes a denaturation step and is therefore called denaturing top-down proteomics (dTDP). In this approach, denaturation is performed by either a denaturing substance (i.e., reducing agents optionally assisted by detergents and/or organic modifiers, etc.) or sometimes physical methods, toward disrupting protein interactions and quaternary conformations (e.g., by heat, pressure, etc.).199 The dTDP approach usually consists of a prefractionation step such as IEF to reduce the complexity of the sample. The fractions are then measured with RPLC-MS/MS at low pH. To date, qualitative and quantitative analysis of intact peptides and proteins up to ~30 kDa can be achieved using this methodology.199 Native TDP is a similar approach and is used, as by Pla et al. (2018), after reduction of the disulfide bonds.200 However, it is not yet widely used and advances have to be made in both MS instrumentation, in order to retain potential biologically relevant noncovalent protein–protein and protein–ligand interactions, and database quality.201
One of the challenges in these approaches is considering posttranslational modifications (PTMs), such as amidations (e.g., for scorpion and spider peptide toxins) and glycosylations (mainly venom enzymes) on the toxins present in venoms. Some toxin classes may carry PTMs that can have a great influence on their biological and pharmacological functioning, as well as on their proteoform diversity. MS analysis approaches in this regard are valuable for unraveling these PTMs toward complete characterization.186
A relatively new technique for the analysis of binding interactions is ion mobility–mass spectrometry (IM-MS), by using ESI-IM-MS.202 Young et al. (2015) developed an HTS approach for the screening and identification of amyloid assembly inhibitors.202 Enabling the identification of protein–ligand interactions and providing information on the binding nature of the interacting species, this technique can be used as a tool for the identification of ligands in the drug discovery pipeline.
One of the growing research areas aiding in improved protein identification and automated MS identification is the development and advancement of comprehensive reference databases and bioinformatics algorithms/processing software. Databases are available for both bottom-up and TDP proteomics approaches. Databases exist containing, for example, genomic and transcriptomic data for the bottom-up approaches (e.g., Uniprot, National Center for Biotechnology Information [NCBI], and Indigenous Snake Species of Bangladesh [ISOB] [snake venom]). Regular updating of, for example, Arachnoserver (spider venoms) and Conoserver (cone snail venoms) will improve protein identification further.186,196
After bioactive peptide or protein screening, detection, and identification, subsequent large-quantity production can be performed. This is often done using solid-phase peptide synthesis or recombinant expression with a suitable expression system, often Escherichia coli. E. coli expression systems are a robust and cheap biological means for producing high yields of the protein of interest with the help of a genetically modified organism. In general, peptides larger than 60-amino-acid residues cannot be synthesized by solid-phase peptide synthesis, making overexpression in these cases necessary. The synthetic toxin expressed and purified is then analyzed by crystallography and/or NMR studies for determining its 3D structure, followed by docking studies and structure–activity relation (SAR) studies to determine the amino acids involved in its interaction with the drug target. The biological activity of the synthetic peptide or protein is usually at a later stage also characterized for binding interaction with different receptors to assess selectivity, and characterized with other off-target bioassays to assess toxicity. The biological activity of the peptide or protein is often improved by site-specific mutagenesis or by synthesis of different peptide derivates (i.e., rational changes or deletions of amino acids) compared with the original toxin. Often, this results in smaller derivates of the original toxin with enhanced pharmacodynamic and pharmacokinetic properties. A successful example in this regard is the peptide prohanin, which was derived from an analgesic, 72-amino-acid alpha-neurotoxin type protein, hannalgesin, from Ophiophagus hannah (king cobra) venom.203,204 Kini (2002) determined that the amino acids on the carboxy terminal end of hannalgesin are involved in the analgesic effect of the toxin.203 For this they used the proline bracket method, which is a prediction model for the active sites based on the presence of proline residues.205 Based on the information on the active site, they eventually synthesized the short, water-soluble, 11-amino-acid peptide prohanin, which showed a selective analgesic effect in vivo, and which did not cause neurotoxicity.203,206
The above-mentioned approach was used for peptide and protein bioactives from animal venoms. However, examples of extracting small molecular bioactive compounds can also be found from other animal sources, for example, epibatidine from the Epipedobates tricolor frog skin extract or tryptamine-like compounds from the skin extract of Bufo toads.207,208
A few case studies of the traditional approach of isolation and characterization of toxins from animal venom sources are given next. The first example deals with the isolation and characterization of an anticoagulant protein, fasxiator, from Bungarus fasciatus snake venom.163 In this endeavor, bioactive proteins were isolated from the crude venom (100 mg) using a two-step fractionation. First, SEC was used, which was followed by RPLC of the fractions that were tested to be bioactive. One-milliliter fractions were collected and pooled for bioactivity testing (effect on prothrombin time and activated partial thromboplastin time). The amino acid sequences of the purified bioactives were determined by MS/MS after pyridylethylation and Lys-C/Arg-C digestion, and by Edman degradation. Next, the protein identified as the best candidate was overexpressed in an E. coli system. The obtained recombinant protein showed good anticoagulant activity and protease specificity toward factor XIa (FXIa). Finally, after elucidating the structure–function relationship and the amino acid residues involved in the interaction with the target FXIa, the potency of the protein was improved by site-directed point mutagenesis.
The isolation and pharmacological characterization of a neurotoxic three-finger toxin targeting the α7-nAChR from black mamba (Dendroapsis polylepis polylepis) snake venom, is described by Wang et al. (2014).209 The aim of this work was to isolate and characterize three-finger toxins with unusual PTMs. The isolated toxin α-elapitoxin-Dpp2d had an amidated C-terminal arginine. The α-elapitoxin-Dpp2d was first purified from crude venom (30 mg) by IEC followed by RPLC of the fraction containing three-finger toxins including elapitoxin-Dpp2d. The amino acid sequence of this toxin was then determined by de novo peptide sequencing using reduction/alkylation procedures, parallel enzymatic digestion by trypsin and by Glu-C, and MS/MS analysis. The crystal structure of the toxin was determined using the hanging drop vapor diffusion crystallization technique. See Figure 5 for the chemical structure determined by Wang et al.209 Using docking studies, we modeled the interaction of the toxin and the pharmacophore regions with Lymnea stagnalis AChBP. After expressing the toxin in a larger quantity using the E. coli heterologous expression system and subsequent purification, the pharmacological activity of the toxin was characterized using radioligand binding assays and functional calcium-flux assays on the human α7-, α3β2-, α3β4-, and α1β1γδ-nAChRs. From the functional cell-based assays α-EPTX-Dpp2d was found to selectively inhibit α7-nAChR, indicating its potential as a pharmacological tool for studying the α7-nAChR.
Figure 5.
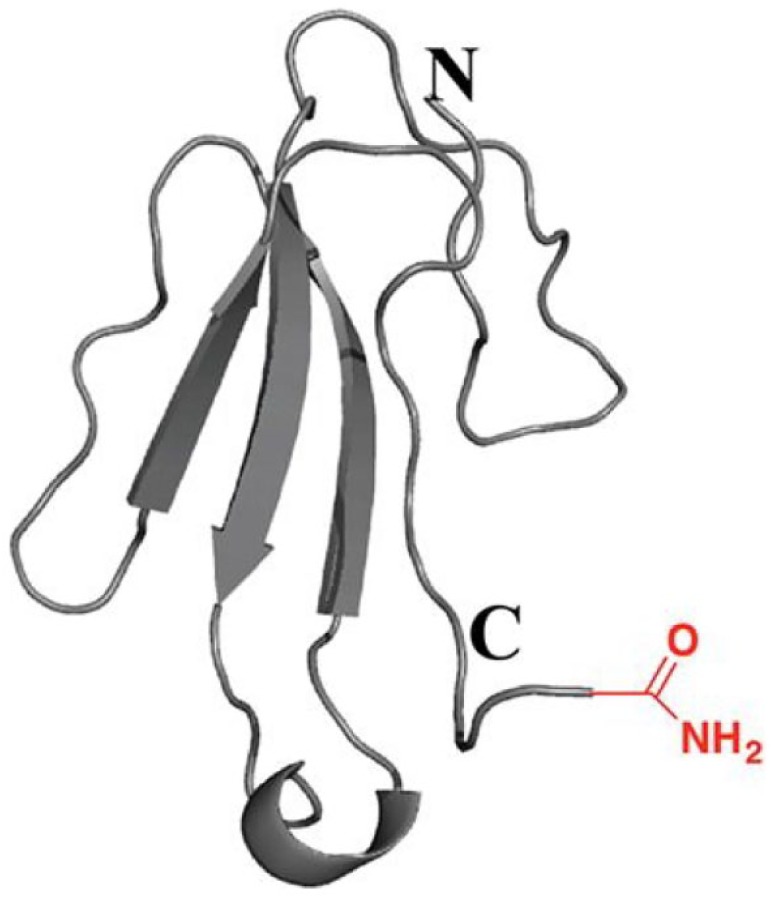
An example of a chemical structure that was isolated from natural extracts, targeting the α7-nAChR, presented in this paper as a case study example. The peptides were produced using an E. coli expression system. This figure shows the isolated and pharmacologically characterized neurotoxic three-finger toxin targeting the α7-nAChR from black mamba (Dendroapsis polylepis polylepis) snake venom. Wang et al. (2014)209 determined the isolated toxin to be an α-elapitoxin-Dpp2d with an amidated C-terminal arginine. The given figure represents a ribbon representation of the structure of the dimeric α-EPTX-Dpp2d isolated compound. Roman and Arabic numerals label the fingers and β-strands, respectively.
Cardoso et al. isolated an inhibiting peptide of the voltage-gated sodium channel hNav1.7, which is a potential drug target for the treatment of pain disorders, from a spider venom.210 Crude venom (1 mg) of Tarantula pruriens was centrifuged and fractionated by RPLC. Obtained fractions were screened for hNav1.7 inhibition. The masses of the bioactive peptides were then determined using matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) MS, and the amino acid sequence was determined by Edman N-terminal sequencing. The peptide was produced using an E. coli expression system. Next to recombinant expression, the toxin was also synthetized using Fmoc solid-phase peptide synthesis. Cardoso et al. provide the chemical structure, which can be found in their manuscript.210 The biological activity of the toxin was tested using membrane potential assays and patch-clamp electrophysiology. The biological activity of the toxin was subsequently characterized on different receptors, such as voltage-gated calcium channels and nAChRs, in order to assess selectivity. This was followed by in vivo pain behavior assessment using a mouse model. Finally, the 3D structure of the toxin was determined by 2D-NMR studies.
The ω-conotoxin MVIIA, which was the template of the approved painkiller drug ziconotide (Prialt), was discovered from the venom of the Conus magus cone snail.211,212 The purification of ω-conotoxin MVIIA was performed by three chromatographic steps: First, the crude venom (87 mg) was fractionated on a Sephadex gel filtration column. The activity of the fractions was tested using an intracerebral bioassay in mice. In this assay positive fractions elicited a characteristic “shaker” activity in mice. One active fraction was further fractionated on a semipreparative C18 column, followed by an analytical C18 separation in order to obtain the pure peptide. In between each fractionation step the activity was monitored with the mouse intracerebral bioassay. Amino acid composition analysis was performed after hydrolysis with an automated analyzer. The amino acid sequence was determined by Edman sequencing. The peptide was synthetized using solid-phase peptide synthesis. Identification and biological characterization of this ω-conotoxin led to the discrimination between calcium channel subtypes and to ziconotide, the synthetic version of ω-conotoxin. Ziconotide eventually became the painkiller drug Prialt used in the clinic. To summarize, venom-based drug discovery can be a successful endeavor to find new bioactive peptide and protein-based drug leads.
A recent approach in the venom-based drug discovery field is the integration of traditional BGF with proteomics, transcriptomics (all mRNA present in the venom gland), and bioinformatics approaches.213 For this approach, evidently proteome and transcriptome databases need to be available. This approach can provide efficient assignment of masses of bioactives and comprehensive elucidation of sequences of bioactive peptides and proteins.
Summary
As discussed in the “Targets” part of this review, there is a need for new compounds targeting ion channels. Besides being of interest as new drug lead molecules for CNS diseases, these compounds are also of interest as potential pharmacological and diagnostic tools. Nature is a rich source of compounds targeting ion channels such as the α7-nAChR and the 5-HT3R. There are multiple ligands of these receptors that have been identified from natural sources, as overviewed in the “Sources” part of this review. As natural extracts are complex mixtures, this makes the finding and identification of unknown bioactive components very challenging. There is thus an essential need for the development of new analytical approaches that allow for faster and more efficient screening of natural extracts for bioactives targeting ion channels. The “Approaches” part of this review aimed to discuss and review analytical techniques for the identification of bioactives from complex mixtures (such as natural extracts) acting on ion channels, with a focus on the α7-nAChR and the 5-HT3R. In this discussion, traditional and new analytical techniques were compared and evaluated for their potential and value in drug discovery from natural products in general, and for ion-channel drug discovery specifically.
Acknowledgments
The authors would like to thank Alexander Medvedev and Thomas Brown for providing us with Figure 2D,E, respectively.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work of Reka A. Otvos was supported by the AIMMS Bridging PhD project “Identification of Novel Bioactive Substances on Brain Receptors” (project no. 10-001-203).
References
- 1. Lovinger D. M. Communication Networks in the Brain: Neurons, Receptors, Neurotransmitters, and Alcohol. Alcohol Res. Health 2008, 31, 196–214. [PMC free article] [PubMed] [Google Scholar]
- 2. Lodish H., Berk A., Zipursky S. L. Neurotransmitters, Synapses, and Impulse Transmission—Molecular Cell Biology—NCBI Bookshelf. In Molecular Cell Biology; W. H. Freeman: New York; Section; 21.4. [Google Scholar]
- 3. Hille B. Ion Channels of Excitable Membranes Sinauer Associates: Sunderland, MA, 2001; Vol. 507. [Google Scholar]
- 4. Carabotti M., Scirocco A., Antonietta M., et al. The Gut-Brain Axis: Interactions between Enteric Microbiota, Central and Enteric Nervous Systems. Ann. Gastroenterol. 2015, 28, 203–209. [PMC free article] [PubMed] [Google Scholar]
- 5. Martin C. R., Osadchiy V., Kalani A., et al. The Brain-Gut-Microbiome Axis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thompson A. J., Lummis S. C. R. The 5-HT3 Receptor as a Therapeutic Target. Expert Opin. Ther. Targets 2007, 11, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Westfall S., Lomis N., Kahouli I., et al. Microbiome, Probiotics and Neurodegenerative Diseases: Deciphering the Gut Brain Axis. Cell. Mol. Life Sci. 2017, 74, 3769–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim D. S., Choi H.-I., Wang Y., et al. A New Treatment Strategy for Parkinson’s Disease through the Gut–Brain Axis: The Glucagon-Like Peptide-1 Receptor Pathway. Cell Transplant. 2017, 26, 1560–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Albuquerque E., Alkondon M., Pereira E., et al. Properties of Neuronal Nicotinic Acetylcholine Receptors: Pharmacological Characterization and Modulation of Synaptic Function. J. Pharmacol. Exp. Ther. 1997, 280, 1117–1136. [PubMed] [Google Scholar]
- 10. Jones S., Sudweeks S., Yakel J. L. Nicotinic Receptors in the Brain: Correlating Physiology with Function. Trends Neurosci. 1999, 22, 555–561. [DOI] [PubMed] [Google Scholar]
- 11. Dani J. A. Overview of Nicotinic Receptors and Their Roles in the Central Nervous System. Biol. Psychiatry 2001, 49, 166–174. [DOI] [PubMed] [Google Scholar]
- 12. Hogg R. C., Raggenbass M., Bertrand D. Nicotinic Acetylcholine Receptors: From Structure to Brain Function. Rev. Physiol. Biochem. Pharmacol. 2003, 147, 1–46. [DOI] [PubMed] [Google Scholar]
- 13. McGehee D. S., Role L. W. Physiological Diversity of Nicotinic Acetylcholine Receptors Expressed by Vertebrate Neurons. Annu. Rev. Physiol. 1995, 57, 521–546. [DOI] [PubMed] [Google Scholar]
- 14. Karlin A. Emerging Structure of the Nicotinic Acetylcholine Receptors. Nat. Rev. Neurosci. 2002, 3, 102–114. [DOI] [PubMed] [Google Scholar]
- 15. Cooper E., Couturier S., Ballivet M. Pentameric Structure and Subunit Stoichiometry of a Neuronal Nicotinic Acetylcholine Receptor. Nature 1991, 350, 235–238. [DOI] [PubMed] [Google Scholar]
- 16. Smit A. B., Syed N. I., Schaap D., et al. A Glia-Derived Acetylcholine-Binding Protein That Modulates Synaptic Transmission. Nature 2001, 411, 261–268. [DOI] [PubMed] [Google Scholar]
- 17. Brejc K., van Dijk W., Smit A., et al. The 2.7 A Structure of AChBP, Homologue of the Ligand-Binding Domain of the Nicotinic Acetylcholine Receptor. Novartis Found. Symp. 2002, 245, 22–32. [PubMed] [Google Scholar]
- 18. Celie P. H. N., Kasheverov I. E., Mordvintsev D. Y., et al. Crystal Structure of Nicotinic Acetylcholine Receptor Homolog AChBP in Complex with an α-Conotoxin PnIA Variant. Nat. Struct. Mol. Biol. 2005, 12, 582–588. [DOI] [PubMed] [Google Scholar]
- 19. Rucktooa P., Smit A. B., Sixma T. K. Insight in NAChR Subtype Selectivity from AChBP Crystal Structures. Biochem. Pharmacol. 2009, 78, 777–787. [DOI] [PubMed] [Google Scholar]
- 20. Clarke P. B., Schwartz R. D., Paul S. M., et al. Nicotinic Binding in Rat Brain: Autoradiographic Comparison of [3H]Acetylcholine, [3H]Nicotine, and [125I]-Alpha-Bungarotoxin. J. Neurosci. 1985, 5, 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Breese C. R., Adams C., Logel J., et al. Comparison of the Regional Expression of Nicotinic Acetylcholine Receptor Alpha7 MRNA and [125I]-Alpha-Bungarotoxin Binding in Human Postmortem Brain. J. Comp. Neurol. 1997, 387, 385–398. [DOI] [PubMed] [Google Scholar]
- 22. Tribollet E., Bertrand D., Marguerat A., et al. Comparative Distribution of Nicotinic Receptor Subtypes during Development, Adulthood and Aging: An Autoradiographic Study in the Rat Brain. Neuroscience 2004, 124, 405–420. [DOI] [PubMed] [Google Scholar]
- 23. Gotti C., Zoli M., Clementi F. Brain Nicotinic Acetylcholine Receptors: Native Subtypes and Their Relevance. Trends Pharmacol. Sci. 2006, 27, 482–491. [DOI] [PubMed] [Google Scholar]
- 24. Taly A., Corringer P. J., Guedin D., et al. Nicotinic Receptors: Allosteric Transitions and Therapeutic Targets in the Nervous System. Nat. Rev. Drug Discov. 2009, 8, 733–750. [DOI] [PubMed] [Google Scholar]
- 25. Borovikova L. V, Ivanova S., Zhang M., et al. Vagus Nerve Stimulation Attenuates the Systemic Inflammatory Response to Endotoxin. Nature 2000, 405, 458–462. [DOI] [PubMed] [Google Scholar]
- 26. Kurzen H., Berger H., Jäger C., et al. Phenotypical and Molecular Profiling of the Extraneuronal Cholinergic System of the Skin. J. Invest. Dermatol. 2004, 123, 937–949. [DOI] [PubMed] [Google Scholar]
- 27. Beckel J. M., Kanai A., Lee S., et al. Expression of Functional Nicotinic Acetylcholine Receptors in Rat Urinary Bladder Epithelial Cells. Am. J. Physiol. Physiol. 2006, 290, F103–F110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gwilt C. R., Donnelly L. E., Rogers D. F. The Non-Neuronal Cholinergic System in the Airways: An Unappreciated Regulatory Role in Pulmonary Inflammation? Pharmacol. Ther. 2007, 115, 208–222. [DOI] [PubMed] [Google Scholar]
- 29. Arredondo J., Chernyavsky A. I., Jolkovsky D. L., et al. Receptor-Mediated Tobacco Toxicity: Acceleration of Sequential Expression of 5 and 7 Nicotinic Receptor Subunits in Oral Keratinocytes Exposed to Cigarette Smoke. FASEB J. 2007, 22, 1356–1368. [DOI] [PubMed] [Google Scholar]
- 30. Wongsriraksa A., Parsons M. E., Whelan C. J. Characterisation of Nicotine Receptors on Human Peripheral Blood Mononuclear Cells (PBMC). Inflamm. Res. 2009, 58, 38–44. [DOI] [PubMed] [Google Scholar]
- 31. Dani J. A., Bertrand D. Nicotinic Acetylcholine Receptors and Nicotinic Cholinergic Mechanisms of the Central Nervous System. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 699–729. [DOI] [PubMed] [Google Scholar]
- 32. Quick M. W., Lester R. A. J. Desensitization of Neuronal Nicotinic Receptors. J. Neurobiol. 2002, 53, 457–478. [DOI] [PubMed] [Google Scholar]
- 33. Messi M., Renganathan M., Grigorenko E., et al. Activation of α 7 Nicotinic Acetylcholine Receptor Promotes Survival of Spinal Cord Motoneurons. FEBS Lett. 1997, 411, 32–38. [DOI] [PubMed] [Google Scholar]
- 34. Berg D. K., Conroy W. G. Nicotinic Α7 Receptors: Synaptic Options and Downstream Signaling in Neurons. J. Neurobiol. 2002, 53, 512–523. [DOI] [PubMed] [Google Scholar]
- 35. Wonnacott S., Barik J., Dickinson J., et al. Nicotinic Receptors Modulate Transmitter Cross Talk in the CNS. J. Mol. Neurosci. 2006, 30, 137–140. [DOI] [PubMed] [Google Scholar]
- 36. Parri H. R., Hernandez C. M., Dineley K. T. Research Update: Alpha7 Nicotinic Acetylcholine Receptor Mechanisms in Alzheimer’s Disease. Biochem. Pharmacol. 2011, 82, 931–942. [DOI] [PubMed] [Google Scholar]
- 37. Quik M., Kulak J. M. Nicotine and Nicotinic Receptors; Relevance to Parkinson’s Disease. Neurotoxicology 2002, 23, 581–594. [DOI] [PubMed] [Google Scholar]
- 38. Raggenbass M., Bertrand D. Nicotinic Receptors in Circuit Excitability and Epilepsy. J. Neurobiol. 2002, 53, 580–589. [DOI] [PubMed] [Google Scholar]
- 39. Young J. W., Geyer M. A. Evaluating the Role of the Alpha-7 Nicotinic Acetylcholine Receptor in the Pathophysiology and Treatment of Schizophrenia. Biochem. Pharmacol. 2013, 86, 1122–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilens T. E., Decker M. W. Neuronal Nicotinic Receptor Agonists for the Treatment of Attention-Deficit/Hyperactivity Disorder: Focus on Cognition. Biochem. Pharmacol. 2007, 74, 1212–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Decker M. W., Meyer M. D., Sullivan J. P. The Therapeutic Potential of Nicotinic Acetylcholine Receptor Agonists for Pain Control. Expert Opin. Investig. Drugs 2001, 10, 1819–1830. [DOI] [PubMed] [Google Scholar]
- 42. Picciotto M. R., Brunzell D. H., Caldarone B. J. Effect of Nicotine and Nicotinic Receptors on Anxiety and Depression. Neuroreport Rapid Commun. Neurosci. Res. 2002, 13, 1097–1106. [DOI] [PubMed] [Google Scholar]
- 43. Freedman R., Hall M., Adler L. E., et al. Evidence in Postmortem Brain Tissue for Decreased Numbers of Hippocampal Nicotinic Receptors in Schizophrenia. Biol. Psychiatry 1995, 38, 22–33. [DOI] [PubMed] [Google Scholar]
- 44. Court J., Spurden D., Lloyd S., et al. Neuronal Nicotinic Receptors in Dementia with Lewy Bodies and Schizophrenia: Alpha-Bungarotoxin and Nicotine Binding in the Thalamus. J. Neurochem. 1999, 73, 1590–1597. [DOI] [PubMed] [Google Scholar]
- 45. Preskorn S. H., Gawryl M., Dgetluck N., et al. Normalizing Effects of EVP-6124, an Alpha-7 Nicotinic Partial Agonist, on Event-Related Potentials and Cognition: A Proof of Concept, Randomized Trial in Patients with Schizophrenia. J. Psychiatr. Pract. 2018, 20, 12–24. [DOI] [PubMed] [Google Scholar]
- 46. Keefe R. S. E., Meltzer H. A., Dgetluck N., et al. Randomized, Double-Blind, Placebo-Controlled Study of Encenicline, an Α7 Nicotinic Acetylcholine Receptor Agonist, as a Treatment for Cognitive Impairment in Schizophrenia. Neuropsychopharmacology 2015, 40, 3053–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Garay R. P., Citrome L., Samalin L., et al. Therapeutic Improvements Expected in the Near Future for Schizophrenia and Schizoaffective Disorder: An Appraisal of Phase III Clinical Trials of Schizophrenia-Targeted Therapies as Found in US and EU Clinical Trial Registries. Expert Opin. Pharmacother. 2016, 17, 921–936. [DOI] [PubMed] [Google Scholar]
- 48. ALZFORUM. Rare but Severe Side Effects Sideline Some Phase 3 Encenicline Trials https://www.alzforum.org/news/research-news/rare-severe-side-effects-sideline-some-phase-3-encenicline-trials (accessed Dec 20, 2018).
- 49. Lieberman J. A., Dunbar G., Segreti A. C., et al. A Randomized Exploratory Trial of an Alpha-7 Nicotinic Receptor Agonist (TC-5619) for Cognitive Enhancement in Schizophrenia. Neuropsychopharmacology 2013, 38, 968–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Walling D., Marder S. R., Kane J., et al. Phase 2 Trial of an Alpha-7 Nicotinic Receptor Agonist (TC-5619) in Negative and Cognitive Symptoms of Schizophrenia. Schizophr. Bull. 2016, 42, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dicpinigaitis P. V, Canning B. J., DeVita R., et al. The Antitussive Effects of Alpha7 (Α7) Nicotinic Receptor Agonists. Eur. Respir. J. 2017, 50. [Google Scholar]
- 52. Lombardo S., Maskos U. Role of the Nicotinic Acetylcholine Receptor in Alzheimer’s Disease Pathology and Treatment. Neuropharmacology 2015, 96, 255–262. [DOI] [PubMed] [Google Scholar]
- 53. Perry E. K., Perry R. H., Smith C. J., et al. Nicotinic Receptor Abnormalities in Alzheimer’s and Parkinson’s Diseases. J. Neurol. Neurosurg. Psychiatry 1987, 50, 806–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Parri H., Dineley K. Nicotinic Acetylcholine Receptor Interaction with β-Amyloid: Molecular, Cellular, and Physiological Consequences. Curr. Alzheimer Res. 2010, 7, 27–39. [DOI] [PubMed] [Google Scholar]
- 55. Wang H. Y., Lee D. H., Davis C. B., et al. Amyloid Peptide Aβ1-42 Binds Selectively and with Picomolar Affinity to Α7 Nicotinic Acetylcholine Receptors. J. Neurochem. 2000, 75, 1155–1161. [DOI] [PubMed] [Google Scholar]
- 56. Bencherif M. Neuronal Nicotinic Receptors as Novel Targets for Inflammation and Neuroprotection: Mechanistic Considerations and Clinical Relevance. Acta Pharmacol. Sin. 2009, 30, 702–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang C.-Y., Chang Y.-Ji. α7 Nicotinic Acetylcholine Receptor Mediated the Chemosensitivity of Gastric Cancer Cells. Cancer Res. 2015, 75, 4988–4988. [Google Scholar]
- 58. Tu C. C., Huang C. Y., Cheng W. L., et al. The Α7-Nicotinic Acetylcholine Receptor Mediates the Sensitivity of Gastric Cancer Cells to Taxanes. Tumor Biol. 2016, 37, 4421–4428. [DOI] [PubMed] [Google Scholar]
- 59. Hurst R., Rollema H., Bertrand D. Nicotinic Acetylcholine Receptors: From Basic Science to Therapeutics. Pharmacol. Ther. 2013, 137, 22–54. [DOI] [PubMed] [Google Scholar]
- 60. Corradi J., Bouzat C. Understanding the Bases of Function and Modulation of α7 Nicotinic Receptors: Implications for Drug Discovery. Mol. Pharmacol. 2016, 90, 288–299. [DOI] [PubMed] [Google Scholar]
- 61. Maricq A. V., Peterson A. S., Brake A. J., et al. Primary Structure and Functional Expression of the 5HT3 Receptor, a Serotonin-Gated Ion Channel. Science 1991, 254, 432–437. [DOI] [PubMed] [Google Scholar]
- 62. Thompson A. J., Lummis S. C. 5-HT3 Receptors. Curr. Pharm. Des. 2006, 12, 3615–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dubin A. E., Huvar R., Andrea M. R. D., et al. The Pharmacological and Functional Characteristics of the Serotonin 5-HT 3A Receptor Are Specifically Modified by a 5-HT 3B Receptor Subunit. J. Biol. Chem. 1999, 274, 30799–30810. [DOI] [PubMed] [Google Scholar]
- 64. Davies P. A., Pistis M., Hanna M. C., et al. The S-HT3B Subunit Is a Major Determinant of Serotonin-Receptor Function. Nature 1999, 397, 359–363. [DOI] [PubMed] [Google Scholar]
- 65. Niesler B., Frank B., Kapeller J., et al. Cloning, Physical Mapping and Expression Analysis of the Human 5-HT3 Serotonin Receptor-Like Genes HTR3C, HTR3D and HTR3E. Gene 2003, 310, 101–111. [DOI] [PubMed] [Google Scholar]
- 66. Werner P., Kawashima E., Reid J., et al. Organization of the Mouse 5-HT3 Receptor Gene and Functional Expression of Two Splice Variants. Mol. Brain Res. 1994, 26, 233–241. [DOI] [PubMed] [Google Scholar]
- 67. Macor J. E., Gurley D., Lanthorn T., et al. The 5-HT 3 Antagonist Tropisetron (ICS 205-930) Is a Potent and Selective 7 Nicotinic Receptor Partial Agonist. Bioorg. Med. Chem. Lett. 2001, 11, 319–321. [DOI] [PubMed] [Google Scholar]
- 68. Drisdel R. C., Sharp D., Henderson T., et al. High Affinity Binding of Epibatidine to Serotonin Type 3 Receptors. J. Biol. Chem. 2008, 283, 9659–9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lummis S. C. R., Thompson A. J., Bencherif M., et al. Varenicline Is a Potent Agonist of the Human 5-Hydroxytryptamine 3 Receptor. J. Pharmacol. Exp. Ther. 2011, 339, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Huang K. C. The Pharmacology of Chinese Herbs; CRC Press: New York, 1998. [Google Scholar]
- 71. Dev S. Ancient-Modern Concordance in Ayurvedic Plants: Some Examples. In Development of Plant-Based Medicines: Conservation, Efficacy and Safety; Saxena P. K. Ed.; Kluwer Academic Publishers, Amsterdam, 2001; pp 47–67. [Google Scholar]
- 72. Cragg G. M., Newman D. J. Natural Products: A Continuing Source of Novel Drug Leads. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3670–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Harvey A. Strategies for Discovering Drugs from Previously Unexplored Natural Products. Drug Discov. Today 2000, 5, 294–300. [DOI] [PubMed] [Google Scholar]
- 74. Lam K. S. New Aspects of Natural Products in Drug Discovery. Trends Microbiol. 2007, 15, 279–289. [DOI] [PubMed] [Google Scholar]
- 75. Camp D., Davis R. A., Evans-Illidge E. A., et al. Guiding Principles for Natural Product Drug Discovery. Future Med. Chem. 2012, 4, 1067–1084. [DOI] [PubMed] [Google Scholar]
- 76. Daly J. W. Nicotinic Agonists, Antagonists, and Modulators from Natural Sources. Cell. Mol. Neurobiol. 2005, 25, 513–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Aráoz R., Molgó J., Tandeau de, Marsac N. Neurotoxic Cyanobacterial Toxins. Toxicon 2010, 56, 813–828. [DOI] [PubMed] [Google Scholar]
- 78. Calvete J. J. Snake Venomics: From the Inventory of Toxins to Biology. Toxicon 2013, 75, 44–62. [DOI] [PubMed] [Google Scholar]
- 79. King G. F. Venoms as a Platform for Human Drugs: Translating Toxins into Therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [DOI] [PubMed] [Google Scholar]
- 80. Escoubas P., King G. F. Venomics as a Drug Discovery Platform. Expert Rev. Proteomics 2009, 6, 221–224. [DOI] [PubMed] [Google Scholar]
- 81. Fox J. W., Serrano S. M. T. Exploring Snake Venom Proteomes: Multifaceted Analyses for Complex Toxin Mixtures. Proteomics 2008, 8, 909–920. [DOI] [PubMed] [Google Scholar]
- 82. Lewis R. J., Dutertre S., Vetter I., et al. Conus Venom Peptide Pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [DOI] [PubMed] [Google Scholar]
- 83. Vetter I.; J., Lewis R. Therapeutic Potential of Cone Snail Venom Peptides (Conopeptides). Curr. Top. Med. Chem. 2012, 12, 1546–1552. [DOI] [PubMed] [Google Scholar]
- 84. Gao B., Peng C., Yang J., et al. Cone Snails: A Big Store of Conotoxins for Novel Drug Discovery. Toxins (Basel) 2017, 9, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Miljanich G. Ziconotide: Neuronal Calcium Channel Blocker for Treating Severe Chronic Pain. Curr. Med. Chem. 2004, 11, 3029–3040. [DOI] [PubMed] [Google Scholar]
- 86. Pope J. E., Deer T. R. Ziconotide: A Clinical Update and Pharmacologic Review. Expert Opin. Pharmacother. 2013, 14, 957–966. [DOI] [PubMed] [Google Scholar]
- 87. Sakai R., Swanson G. T. Recent Progress in Neuroactive Marine Natural Products. Nat. Prod. Rep. 2014, 31, 273–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Drukewitz S. H., Fuhrmann N., Undheim E. A. B., et al. A Dipteran’s Novel Sucker Punch: Evolution of Arthropod Atypical Venom with a Neurotoxic Component in Robber Flies (Asilidae, Diptera). Toxins (Basel) 2018, 10, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Daly J. W., Brown G. B., Mensah-Dwumah M., et al. Classification of Skin Alkaloids from Neotropical Poison-Dart Frogs (Dendrobatidae). Toxicon 1978, 16, 163–188. [DOI] [PubMed] [Google Scholar]
- 90. Daly J. W., Myers C. W., Whittaker N. Further Classification of Skin Alkaloids from Neotropical Poison Frogs (Dendrobatidae), with a General Survey of Toxic/Noxious Substances in the Amphibia. Toxicon 1987, 25, 1023–1095. [DOI] [PubMed] [Google Scholar]
- 91. Badio B., Garraffo H. M., Padgett W. L., et al. Pseudophrynaminol: A Potent Noncompetitive Blocker of Nicotinic Receptor-Channels. Biochem. Pharmacol. 1997, 53, 671–676. [DOI] [PubMed] [Google Scholar]
- 92. Kini R. M., Doley R. Structure, Function and Evolution of Three-Finger Toxins: Mini Proteins with Multiple Targets. Toxicon 2010, 56, 855–867. [DOI] [PubMed] [Google Scholar]
- 93. Cushman D. W., Ondetti M. A. History of the Design of Captopril and Related Inhibitors of Angiotensin Converting Enzyme. Hypertension 1991, 17, 589–592. [DOI] [PubMed] [Google Scholar]
- 94. Tcheng J. E., O’Shea J. C. Eptifibatide: A Potent Inhibitor of the Platelet Receptor Integrin Glycoprotein IIb/IIIa. Expert Opin. Pharmacother. 2002, 3, 1199–1210. [DOI] [PubMed] [Google Scholar]
- 95. Vonk F. J., Jackson K., Doley R., et al. Snake Venom: From Fieldwork to the Clinic: Recent Insights into Snake Biology, Together with New Technology Allowing High-Throughput Screening of Venom, Bring New Hope for Drug Discovery. Bioessays 2011, 33, 269–279. [DOI] [PubMed] [Google Scholar]
- 96. Pennington M. W., Czerwinski A., Norton R. S. Peptide Therapeutics from Venom: Current Status and Potential. Bioorg. Med. Chem. 2018, 26, 2738–2758. [DOI] [PubMed] [Google Scholar]
- 97. Dutertre S., Nicke A., Tsetlin V. I. Nicotinic Acetylcholine Receptor Inhibitors Derived from Snake and Snail Venoms. Neuropharmacology 2017, 127, 196–223. [DOI] [PubMed] [Google Scholar]
- 98. Cremonez C. M., Maiti M., Peigneur S., et al. Structural and Functional Elucidation of Peptide Ts11 Shows Evidence of a Novel Subfamily of Scorpion Venom Toxins. Toxins (Basel) 2016, 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Makarova Y. V, Kryukova E. V, Shelukhina I. V. The First Recombinant Viper Three-Finger Toxins: Inhibition of Muscle and Neuronal Nicotinic Acetylcholine Receptors. Dokl. Biochem. Biophys. 2018, 479, 127–130. [DOI] [PubMed] [Google Scholar]
- 100. Prokopowicz M., Różycki K. M. Innovation in Cosmetics. World Sci. News 2017, 72, 448–456. [Google Scholar]
- 101. Debono J., Xie B., Violette A., et al. Viper Venom Botox: The Molecular Origin and Evolution of the Waglerin Peptides Used in Anti-Wrinkle Skin Cream. J. Mol. Evol. 2017, 84, 8–11. [DOI] [PubMed] [Google Scholar]
- 102. Jin L., Fang M., Chen M., et al. An Insecticidal Toxin from Nephila clavata Spider Venom. Amino Acids 2017, 49, 1237–1245. [DOI] [PubMed] [Google Scholar]
- 103. Ikonomopoulou M. P., Smith J. J., Herzig V., et al. Toxicon Isolation of Two Insecticidal Toxins from Venom of the Australian Theraphosid Spider Coremiocnemis tropix. Toxicon 2016, 123, 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Matsubara F. H., Meissner G. O., Herzig V. Insecticidal Activity of a Recombinant Knottin Peptide from Loxosceles intermedia Venom and Recognition of These Peptides as a Conserved Family in the Genus. Insect Mol. Biol. 2017, 26, 25–34. [DOI] [PubMed] [Google Scholar]
- 105. Worley J. F., Main M. J. An Industrial Perspective on Utilizing Functional Ion Channel Assays for High Throughput Screening. Receptors and Channels. Recept. Channels 2002, 8, 269–282. [PubMed] [Google Scholar]
- 106. Netzer R., Bischoff U., Ebneth A. HTS Techniques to Investigate the Potential Effects of Compounds on Cardiac Ion Channels at Early-Stages of Drug Discovery. Curr. Opin. Drug Discov. Devel. 2003, 6, 462–469. [PubMed] [Google Scholar]
- 107. Bennett P. B., Guthrie H. R. E. Trends in Ion Channel Drug Discovery: Advances in Screening Technologies. Trends Biotechnol. 2003, 21, 563–569. [DOI] [PubMed] [Google Scholar]
- 108. Zheng W., Spencer R. H., Kiss L. High Throughput Assay Technologies for Ion Channel Drug Discovery. Assay Drug Dev. Technol. 2004, 2, 543–552. [DOI] [PubMed] [Google Scholar]
- 109. Dunlop J., Bowlby M., Peri R., et al. High-Throughput Electrophysiology: An Emerging Paradigm for Ion-Channel Screening and Physiology. Nat. Rev. Drug Discov. 2008, 7, 358–368. [DOI] [PubMed] [Google Scholar]
- 110. Schroeder K. S., Neagle B. D. FLIPR: A New Instrument for Accurate, High Throughput Optical Screening. J. Biomol. Screen. 1996, 1, 75–80. [Google Scholar]
- 111. Mathes C., Friis S., Finley M., et al. QPatch: The Missing Link between HTS and Ion Channel Drug Discovery. Comb. Chem. High Throughput Screen. 2009, 12, 78–95. [DOI] [PubMed] [Google Scholar]
- 112. Mathes C. QPatch: The Past, Present and Future of Automated Patch Clamp. Expert Opin. Ther. Targets 2006, 10, 319–327. [DOI] [PubMed] [Google Scholar]
- 113. Willumsen N. J., Bech M., Olesen S. P., et al. High Throughput Electrophysiology: New Perspectives for Ion Channel Drug Discovery. Recept. Channels 2003, 9, 3–12. [PubMed] [Google Scholar]
- 114. De Jong L. A. A., Uges D. R. A., Franke J. P., et al. Receptor-Ligand Binding Assays: Technologies and Applications. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005, 829, 1–25. [DOI] [PubMed] [Google Scholar]
- 115. Glickman J. F., Schmid A., Ferrand S. Scintillation Proximity Assays in High-Throughput Screening. Assay Drug Dev. Technol. 2008, 6, 433–455. [DOI] [PubMed] [Google Scholar]
- 116. Wittmann H. J., Strasser A. Competitive Association Binding Kinetic Assays: A New Tool to Detect Two Different Binding Orientations of a Ligand to Its Target Protein under Distinct Conditions? Naunyn. Schmiedebergs. Arch. Pharmacol. 2017, 390, 595–612. [DOI] [PubMed] [Google Scholar]
- 117. Guo D., Peletier L. A., Bridge L., et al. A Two-State Model for the Kinetics of Competitive Radioligand Binding. Br. J. Pharmacol. 2018, 175, 1719–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bencherif M., Bane A. J., Miller C. H., et al. TC-2559: A Novel Orally Active Ligand Selective at Neuronal Acetylcholine Receptors. Eur. J. Pharmacol. 2000, 409, 45–55. [DOI] [PubMed] [Google Scholar]
- 119. Jensen A. A., Frølund B., Liljefors T., et al. Neuronal Nicotinic Acetylcholine Receptors: Structural Revelations, Target Identifications, and Therapeutic Inspirations. J. Med. Chem. 2005, 48, 4705–4745. [DOI] [PubMed] [Google Scholar]
- 120. Terstappen G. C. Ion Flux and Ligand Binding Assays for Analysis of Ion Channels. In Expression and Analysis of Recombinant Ion Channels: From Structural Studies to Pharmacological Screening; Clare J. J., Treise D. J. Eds.; Wiley-VCH: Weinheim, 2006; pp 165–186. [Google Scholar]
- 121. Akdemir A., Rucktooa P., Jongejan A., et al. Acetylcholine Binding Protein (AChBP) as Template for Hierarchical In Silico Screening Procedures to Identify Structurally Novel Ligands for the Nicotinic Receptors. Bioorg. Med. Chem. 2011, 19, 6107–6119. [DOI] [PubMed] [Google Scholar]
- 122. Rucktooa P., Haseler C. A., Van Elk R., et al. Structural Characterization of Binding Mode of Smoking Cessation Drugs to Nicotinic Acetylcholine Receptors through Study of Ligand Complexes with Acetylcholine-Binding Protein. J. Biol. Chem. 2012, 287, 23283–23293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Milligan G. Applications of Bioluminescence- and Fluorescence Resonance Energy Transfer to Drug Discovery at G Protein-Coupled Receptors. Eur. J. Pharm. Sci. 2004, 21, 397–405. [DOI] [PubMed] [Google Scholar]
- 124. Pfleger K. D. G., Eidne K. A. Illuminating Insights into Protein-Protein Interactions Using Bioluminescence Resonance Energy Transfer (BRET). Nat. Methods 2006, 3, 165–174. [DOI] [PubMed] [Google Scholar]
- 125. Srinivasan R., Richards C. I., Dilworth C., et al. Förster Resonance Energy Transfer (FRET) Correlates of Altered Subunit Stoichiometry in Cys-Loop Receptors, Exemplified by Nicotinic Α4β2. Int. J. Mol. Sci. 2012, 13, 10022–10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Burke T., Loniello K., Beebe J., et al. Development and Application of Fluorescence Polarization Assays in Drug Discovery. Comb. Chem. High Throughput Screen. 2003, 6, 183–194. [DOI] [PubMed] [Google Scholar]
- 127. Jameson D. M., Croney J. C. Fluorescence Polarization: Past, Present and Future. Comb. Chem. High Throughput Screen. 2003, 6, 167–176. [DOI] [PubMed] [Google Scholar]
- 128. Schmid E. L., Tairi A. P., Hovius R., et al. Screening Ligands for Membrane Protein Receptors by Total Internal Reflection Fluorescence: The 5-HT Serotonin Receptor. Anal. Chem. 1998, 70, 1331–1338. [DOI] [PubMed] [Google Scholar]
- 129. Hovius R., Schmid E. L., Tairi A.-P., et al. Fluorescence Techniques for Fundamental and Applied Studies of Membrane Protein Receptors: The 5-HT, Serotonin Receptor. J. Recept. Signal Transduct. 1999, 19, 533–545. [DOI] [PubMed] [Google Scholar]
- 130. Kool J., De Kloe G. E., Bruyneel B., et al. Online Fluorescence Enhancement Assay for the Acetylcholine Binding Protein with Parallel Mass Spectrometric Identification. J. Med. Chem. 2010, 53, 4720–4730. [DOI] [PubMed] [Google Scholar]
- 131. Hovius R. Characterization and Validation of Fluorescent Receptor Ligands: A Case Study of the Ionotropic Serotonin Receptor. Methods Mol. Biol. 2013, 995, 161–178. [DOI] [PubMed] [Google Scholar]
- 132. Vignali D. A. A. Multiplexed Particle-Based Flow Cytometric Assays. J. Immunol. Methods 2000, 243, 243–255. [DOI] [PubMed] [Google Scholar]
- 133. Edwards B. S., Oprea T., Prossnitz E. R., et al. Flow Cytometry for High-Throughput, High-Content Screening. Curr. Opin. Chem. Biol. 2004, 8, 392–398. [DOI] [PubMed] [Google Scholar]
- 134. Jozwiak K., Ravichandran S., Collins J. R., et al. Interaction of Noncompetitive Inhibitors with the Α3β2 Nicotinic Acetylcholine Receptor Investigated by Affinity Chromatography and Molecular Docking. J. Med. Chem. 2007, 50, 6279–6283. [DOI] [PubMed] [Google Scholar]
- 135. Pochet L., Heus F., Jonker N., et al. Online Magnetic Bead Based Dynamic Protein Affinity Selection Coupled to LC-MS for the Screening of Acetylcholine Binding Protein Ligands. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011, 879, 1781–1788. [DOI] [PubMed] [Google Scholar]
- 136. de Moraes M. C., Vanzolini K. L., Cardoso C. L., et al. New Trends in LC Protein Ligand Screening. J. Pharm. Biomed. Anal. 2014, 87, 155–166. [DOI] [PubMed] [Google Scholar]
- 137. Zimmerer C., Braun H.-G., Kitsche M., et al. Optical Biosensor Array Based on Natural Ion Channels. Proc. SPIE Int. Soc. Opt. Eng. 2003, 5047, 403–409. [Google Scholar]
- 138. Retra K., Geitmann M., Kool J., et al. Development of Surface Plasmon Resonance Biosensor Assays for Primary and Secondary Screening of Acetylcholine Binding Protein Ligands. Anal. Biochem. 2010, 407, 58–64. [DOI] [PubMed] [Google Scholar]
- 139. Kool J., Heus F., De Kloe G., et al. High-Resolution Bioactivity Profiling of Mixtures toward the Acetylcholine Binding Protein Using a Nanofractionation Spotter Technology. J. Biomol. Screen. 2011, 16, 917–924. [DOI] [PubMed] [Google Scholar]
- 140. Lochner M., Thompson A. J. A Review of Fluorescent Ligands for Studying 5-HT3 Receptors. Neuropharmacology 2015, 98, 31–40. [DOI] [PubMed] [Google Scholar]
- 141. Jack T., Simonin J., Ruepp M. D., et al. Characterizing New Fluorescent Tools for Studying 5-HT3 Receptor Pharmacology. Neuropharmacology 2015, 90, 63–73. [DOI] [PubMed] [Google Scholar]
- 142. Terstappen G. C. Ion Channel Screening Technologies Today. Drug Discov. Today Technol. 2005, 2, 133–140. [DOI] [PubMed] [Google Scholar]
- 143. Cheng C. S., Alderman D., Kwash J., et al. A High-Throughput HERG Potassium Channel Function Assay: An Old Assay with a New Look. Drug Dev. Ind. Pharm. 2002, 28, 177–191. [DOI] [PubMed] [Google Scholar]
- 144. Lukas R. J., Cullen M. J. An Isotopic Rubidium Ion Efflux Assay for the Functional Characterization of Nicotinic Acetylcholine Receptors on Clonal Cell Lines. Anal. Biochem. 1988, 175, 212–218. [DOI] [PubMed] [Google Scholar]
- 145. Terstappen G. C. Functional Analysis of Native and Recombinant Ion Channels Using a High-Capacity Nonradioactive Rubidium Efflux Assay. Anal. Biochem. 1999, 272, 149–155. [DOI] [PubMed] [Google Scholar]
- 146. González J. E., Tsien R. Y. Voltage Sensing by Fluorescence Resonance Energy Transfer in Single Cells. Biophys. J. 1995, 69, 1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Zhang Y., Kowal D., Kramer A., et al. Evaluation of FLIPR Calcium 3 Assay Kit—A New No-Wash Fluorescence Calcium Indicator Reagent. J. Biomol. Screen. 2003, 8, 571–577. [DOI] [PubMed] [Google Scholar]
- 148. Gee K. R., Brown K. A., Chen W. N. U., et al. Chemical and Physiological Characterization of Fluo-4 Ca2+-Indicator Dyes. Cell Calcium 2000, 27, 97–106. [DOI] [PubMed] [Google Scholar]
- 149. Hurst R. S., Hajós M., Raggenbass M., et al. A Novel Positive Allosteric Modulator of the 7 Neuronal Nicotinic Acetylcholine Receptor: In Vitro and In Vivo Characterization. J. Neurosci. 2005, 25, 4396–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Mazurov A., Hauser T., Miller C. H. Selective Α7 Nicotinic Acetylcholine Receptor Ligands. Curr. Med. Chem. 2006, 13, 1567–1584. [DOI] [PubMed] [Google Scholar]
- 151. Wishka D. G., Walker D. P., Yates K. M., et al. Discovery of N-[(3R)-1-Azabicyclo[2.2.2]oct-3-Yl]furo[2,3-c]pyridine-5-carboxamide, an Agonist of the Α7 Nicotinic Acetylcholine Receptor, for the Potential Treatment of Cognitive Deficits in Schizophrenia: Synthesis and Structure-Activity Relation. J. Med. Chem. 2006, 49, 4425–4436. [DOI] [PubMed] [Google Scholar]
- 152. Vetter I., Lewis R. J. Characterization of Endogenous Calcium Responses in Neuronal Cell Lines. Biochem. Pharmacol. 2010, 79, 908–920. [DOI] [PubMed] [Google Scholar]
- 153. Vetter I. Development and Optimization of FLIPR High Throughput Calcium Assays for Ion Channels and GPCRs. Adv. Exp. Med. Biol. 2012, 740, 45–82. [DOI] [PubMed] [Google Scholar]
- 154. Guan B., Chen X., Zhang H. Two-Electrode Voltage Clamp. Ion Channels 2013, 998, 4–8. [DOI] [PubMed] [Google Scholar]
- 155. Sakmann B., Neher E. Patch Clamp Techniques for Studying Ionic Channels in Excitable Membranes. Annu. Rev. Physiol. 1984, 46, 455–472. [DOI] [PubMed] [Google Scholar]
- 156. Weller M. G. A Unifying Review of Bioassay-Guided Fractionation, Effect-Directed Analysis and Related Techniques. Sensors (Switzerland) 2012, 12, 9181–9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Jonker W., Lamoree M. H., Houtman C. J., et al. Methodologies for Effect-Directed Analysis: Environmental Applications, Food Analysis, and Drug Discovery. In Analyzing Biomolecular Interactions by Mass Spectrometry; Niessen W. M. A., Falck D. Eds.; Wiley-VCH: Weinheim, 2015; pp 111–164. [Google Scholar]
- 158. Brack W., Ait-Aissa S., Burgess R. M., et al. Effect-Directed Analysis Supporting Monitoring of Aquatic Environments—An In-Depth Overview. Sci. Total Environ. 2016, 544, 1073–1118. [DOI] [PubMed] [Google Scholar]
- 159. Sasidharan S., Chen Y., Saravanan D., et al. Extraction, Isolation and Characterization of Bioactive Compounds from Plants’ Extracts. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 1–10. [PMC free article] [PubMed] [Google Scholar]
- 160. Balouiri M., Sadiki M., Ibnsouda S. K. Methods for In Vitro Evaluating Antimicrobial Activity: A Review. J. Pharm. Anal. 2016, 6, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Rakshith D., Santosh P., Pradeep T. P., et al. Application of Bioassay-Guided Fractionation Coupled with a Molecular Approach for the Dereplication of Antimicrobial Metabolites. Chromatographia 2016, 79, 1625–1642. [Google Scholar]
- 162. Graudins A., Little M. J., Pineda S. S., et al. Cloning and Activity of a Novel α-Latrotoxin from Red-Back Spider Venom. Biochem. Pharmacol. 2012, 83, 170–183. [DOI] [PubMed] [Google Scholar]
- 163. Chen W., Carvalho L. P. D., Chan M. Y., et al. Fasxiator, a Novel Factor XIa Inhibitor from Snake Venom, and Its Site-Specific Mutagenesis to Improve Potency and Selectivity. J. Thromb. Haemost. 2015, 13, 248–261. [DOI] [PubMed] [Google Scholar]
- 164. Mladic M., Zietek B. M., Iyer J. K., et al. At-Line Nanofractionation with Parallel Mass Spectrometry and Bioactivity Assessment for the Rapid Screening of Thrombin and Factor Xa Inhibitors in Snake Venoms. Toxicon 2016, 110, 79–89. [DOI] [PubMed] [Google Scholar]
- 165. Nothias L. F., Nothias-Esposito M., Da Silva R., et al. Bioactivity-Based Molecular Networking for the Discovery of Drug Leads in Natural Product Bioassay-Guided Fractionation. J. Nat. Prod. 2018, 81, 758–767. [DOI] [PubMed] [Google Scholar]
- 166. Lee Y. K., Bang H. J., Oh J. Bin, et al. Bioassay-Guided Isolated Compounds from Morinda officinalis Inhibit Alzheimer’s Disease Pathologies. Molecules 2017, 22, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Heus F., Otvos R., Aspers R., et al. Miniaturized Bioaffinity Assessment Coupled to Mass Spectrometry for Guided Purification of Bioactives from Toad and Cone Snail. Biology (Basel) 2014, 3, 139–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Mladic M., de Waal T., Burggraaff L., et al. Rapid Screening and Identification of ACE Inhibitors in Snake Venoms Using At-Line Nanofractionation LC-MS. Anal. Bioanal. Chem. 2017, 409, 5987–5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Mladic M., Scholten D. J., Niessen W. M. A., et al. At-Line Coupling of LC-MS to Bioaffinity and Selectivity Assessment for Metabolic Profiling of Ligands towards Chemokine Receptors CXCR1 and CXCR2. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015, 1002, 42–53. [DOI] [PubMed] [Google Scholar]
- 170. Still K. B. M., Nandlal R. S. S., Slagboom J., et al. Multipurpose HTS Coagulation Analysis: Assay Development and Assessment of Coagulopathic Snake Venoms. Toxins (Basel) 2017, 9, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Vrije Universiteit Amsterdam; Spark Holland. FractioMate™—Fastest Identification of Water Hazards. https://science.vu.nl/en/Images/FractioMateFlyer_tcm296-884745.pdf (accessed Dec 20, 2017).
- 172. Otvos R. A., Mladic M., Arias-Alpizar G., et al. At-Line Cellular Screening Methodology for Bioactives in Mixtures Targeting the Α7-Nicotinic Acetylcholine Receptor. J. Biomol. Screen. 2015, 21, 459–467. [DOI] [PubMed] [Google Scholar]
- 173. Kool J., Giera M., Irth H., et al. Advances in Mass Spectrometry-Based Post-Column Bioaffinity Profiling of Mixtures. Anal. Bioanal. Chem. 2011, 399, 2655–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Shi S. Y., Zhang Y. P., Jiang X. Y., et al. Coupling HPLC to On-Line, Post-Column (Bio)Chemical Assays for High-Resolution Screening of Bioactive Compounds from Complex Mixtures. Trends Analyt. Chem. 2009, 28, 865–877. [Google Scholar]
- 175. Falck D., de Vlieger J. S. B., Niessen W. M. A., et al. Development of an Online P38α Mitogen-Activated Protein Kinase Binding Assay and Integration of LC–HR-MS. Anal. Bioanal. Chem. 2010, 398, 1771–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Van Liempd S. M., Kool J., Niessen W. M. A., et al. On-Line Formation, Separation, and Estrogen Receptor Affinity Screening of Cytochrome P450-Derived Metabolites of Selective Estrogen Receptor Modulators. Drug Metab. Dispos. 2006, 34, 1640–1649. [DOI] [PubMed] [Google Scholar]
- 177. Heus F., Vonk F., Otvos R. A., et al. An Efficient Analytical Platform for On-Line Microfluidic Profiling of Neuroactive Snake Venoms towards Nicotinic Receptor Affinity. Toxicon 2013, 61, 112–124. [DOI] [PubMed] [Google Scholar]
- 178. Otvos R. A., Heus F., Vonk F. J., et al. Analytical Workflow for Rapid Screening and Purification of Bioactives from Venom Proteomes. Toxicon 2013, 76, 270–281. [DOI] [PubMed] [Google Scholar]
- 179. Hage D. S., Cazes J., Eds. Handbook of Affinity Chromatography; Taylor & Francis: Boca Raton, FL, 2005; 2nd ed. [Google Scholar]
- 180. Kool J., Jonker N., Irth H., et al. Studying Protein-Protein Affinity and Immobilized Ligand-Protein Affinity Interactions Using MS-Based Methods. Anal. Bioanal. Chem. 2011, 401, 1109–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Höfner G., Wanner K. T. Using Short Columns to Speed Up LC-MS Quantification in MS Binding Assays. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2010, 878, 1356–1364. [DOI] [PubMed] [Google Scholar]
- 182. Sun Y., Gu C., Liu X., et al. Ultrafiltration Tandem Mass Spectrometry of Estrogens for Characterization of Structure and Affinity for Human Estrogen Receptors. J. Am. Soc. Mass Spectrom. 2005, 16, 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Muckenschnabel I., Falchetto R., Mayr L. M., et al. SpeedScreen: Label-Free Liquid Chromatography-Mass Spectrometry-Based High-Throughput Screening for the Discovery of Orphan Protein Ligands. Anal. Biochem. 2004, 324, 241–249. [DOI] [PubMed] [Google Scholar]
- 184. Sichler S., Höfner G., Rappenglück S., et al. Development of MS Binding Assays Targeting the Binding Site of MB327 at the Nicotinic Acetylcholine Receptor. Toxicol. Lett. 2018, 293, 172–183. [DOI] [PubMed] [Google Scholar]
- 185. Jonker N., Kool J., Irth H., et al. Recent Developments in Protein-Ligand Affinity Mass Spectrometry. Anal. Bioanal. Chem. 2011, 399, 2669–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Oldrati V., Arrell M., Violette A., et al. Advances in Venomics. Mol. Biosyst. 2016, 12, 3530–3543. [DOI] [PubMed] [Google Scholar]
- 187. Calvete J. J., Sanz L., Angulo Y., et al. Venoms, Venomics, Antivenomics. FEBS Lett. 2009, 583, 1736–1743. [DOI] [PubMed] [Google Scholar]
- 188. Calvete J. J., Juarez P., Sanz L. Snake Venomics. Strategy and Applications. J. Mass Spectrom. 2007, 42, 1405–1414. [DOI] [PubMed] [Google Scholar]
- 189. Vetter I., Davis J. L., Rash L. D., et al. Venomics: A New Paradigm for Natural Products-Based Drug Discovery. Amino Acids 2011, 40, 15–28. [DOI] [PubMed] [Google Scholar]
- 190. Harrison J. A., Aquilina J. A. Mass Spectrometry Data and Size Exclusion Chromatography Profiles of Australian Taipan Venom Toxins. Data Brief 2016, 9, 501–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Brunetti A. E., Marani M. M., Soldi R. A., et al. Cleavage of Peptides from Amphibian Skin Revealed by Combining Analysis of Gland Secretion and In Situ MALDI Imaging Mass Spectrometry. ACS Omega 2018, 3, 5426–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Sugiura Y., Zaima N., Setou M., et al. Visualization of Acetylcholine Distribution in Central Nervous System Tissue Sections by Tandem Imaging Mass Spectrometry. Anal. Bioanal. Chem. 2012, 403, 1851–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Romero-Perez G. A., Takei S., Yao I. Imaging Mass Spectrometric Analysis of Neurotransmitters. Mass Spectrom. 2015, S0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Rodriguez A. M., Dutertre S., Lewis R. J., et al. Intraspecific Variations in Conus purpurascens Injected Venom Using LC/MALDI-TOF-MS and LC-ESI-TripleTOF-MS. Anal. Bioanal. Chem. 2015, 407, 6105–6116. [DOI] [PubMed] [Google Scholar]
- 195. Chapeaurouge A., Reza A., Mackessy S. P., et al. Interrogating the Venom of the Viperid Snake Sistrurus catenatus edwardsii by a Combined Approach of Electrospray and MALDI Mass Spectrometry. PLoS One 2015, 10, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Lomonte B., Calvete J. J. Strategies in “Snake Venomics” Aiming at an Integrative View of Compositional, Functional, and Immunological Characteristics of Venoms. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Juárez P., Sanz L., Calvete J. J. Snake Venomics: Characterization of Protein Families in Sistrurus Barbouri Venom by Cysteine Mapping, N-Terminal Sequencing, and Tandem Mass Spectrometry Analysis. Proteomics 2004, 4, 327–338. [DOI] [PubMed] [Google Scholar]
- 198. Petras D., Heiss P., Harrison R. A., et al. Top-Down Venomics of the East African Green Mamba, Dendroaspis angusticeps, and the Black Mamba, Dendroaspis polylepis, Highlight the Complexity of Their Toxin Arsenals. J. Proteomics 2016, 146, 148–164. [DOI] [PubMed] [Google Scholar]
- 199. Melani R. D., Nogueira F. C. S., Domont G. B. It Is Time for Top-Down Venomics. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Pla D., Petras D., Saviola A. J., et al. Transcriptomics-Guided Bottom-Up and Top-Down Venomics of Neonate and Adult Specimens of the Arboreal Rear-Fanged Brown Treesnake, Boiga irregularis, from Guam. J. Proteomics 2018, 174, 71–84. [DOI] [PubMed] [Google Scholar]
- 201. Melani R. D., Skinner O. S., Fornelli L., et al. Mapping Proteoforms and Protein Complexes from King Cobra Venom Using Both Denaturing and Native Top-Down Proteomics. Mol. Cell. Proteomics 2016, 15, 2423–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Young L. M., Saunders J. C., Mahood R. A., et al. Screening and Classifying Small-Molecule Inhibitors of Amyloid Formation Using Ion Mobility Spectrometry-Mass Spectrometry. Nat. Chem. 2015, 7, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Kini R. M. Molecular Moulds with Multiple Missions: Functional Sites in Three-Finger Toxins. Clin. Exp. Pharmacol. Physiol. 2002, 29, 815–822. [DOI] [PubMed] [Google Scholar]
- 204. Pu X. C., Wong P. T. H., Gopalakrishnakone P. A Novel Analgesic Toxin (Hannalgesin) from the Venom of King Cobra (Ophiophagus hannah). Toxicon 1995, 33, 1425–1431. [DOI] [PubMed] [Google Scholar]
- 205. Kini R. M. Proline Brackets and Identification of Potential Functional Sites in Proteins: Toxins to Therapeutics. Toxicon 1998, 36, 1659–1670. [DOI] [PubMed] [Google Scholar]
- 206. Lavergne V., King G. F., Lewis R. J., et al. Peptide Therapeutics from Venomous Creatures. In Pain Therapeutics: Current and Future Treatment Paradigms; Allerton, C., Ed.; Royal Society of Chemistry: London, 2013; pp 217–246. [Google Scholar]
- 207. Erspamer V., Vitali M., Roseghini M., et al. 5-Methoxy- and 5-Hydrixyindoles in the Skin of Bufo alvarius*. Biochem. Pharmacol. 1967, 16, 1149–1164. [DOI] [PubMed] [Google Scholar]
- 208. Daly J. W., Garraffo H. M., Spande T. F., et al. Alkaloids from Frog Skin: The Discovery of Epibatidine and the Potential for Developing Novel Non-Opioid Analgesics. Nat. Prod. Rep. 2000, 17, 131–135. [DOI] [PubMed] [Google Scholar]
- 209. Wang C. I. A., Reeks T., Vetter I., et al. Isolation and Structural and Pharmacological Characterization of α-Elapitoxin-Dpp2d, an Amidated Three Finger Toxin from Black Mamba Venom. Biochemistry 2014, 53, 3758–3766. [DOI] [PubMed] [Google Scholar]
- 210. Cardoso F. C., Dekan Z., Rosengren K. J., et al. Identification and Characterization of ProTx-III [µ-TRTX-Tp1a], a New Voltage-Gated Sodium Channel Inhibitor from Venom of the Tarantula Thrixopelma Pruriens. Mol. Pharmacol. 2015, 88, 291–303. [DOI] [PubMed] [Google Scholar]
- 211. Olivera B. M., Cruz L. J., Santos V., et al. Neuronal Calcium Channel Antagonists. Discrimination between Calcium Channel Subtypes Using Co-Conotoxin from Conus Magus Venom. Biochemistry 1987, 26, 2086–2090. [DOI] [PubMed] [Google Scholar]
- 212. Olivera B. M., McIntosh J. M., Cruz L. J., et al. Purification and Sequence of a Presynaptic Peptide Toxin from Conus Geographus Venom. Biochemistry 1984, 23, 5087–5090. [DOI] [PubMed] [Google Scholar]
- 213. Prashanth J. R., Lewis R. J., Dutertre S. Towards an Integrated Venomics Approach for Accelerated Conopeptide Discovery. Toxicon 2012, 60, 470–477. [DOI] [PubMed] [Google Scholar]
- 214. Hassaine G., Deluz C., Grasso L., et al. X-Ray Structure of the Mouse Serotonin 5-HT3 Receptor. Nature 2014, 512, 276–281. [DOI] [PubMed] [Google Scholar]
- 215. Brejc K., Van Dijk W. J., Klaassen R. V., et al. Crystal Structure of an ACh-Binding Protein Reveals the Ligand-Binding Domain of Nicotinic Receptors. Nature 2001, 411, 269–276. [DOI] [PubMed] [Google Scholar]
- 216. Rilei Y., Craik D. J., Kaas Q. Blockade of Neuronal α7-NAChR by α-Conotoxin ImI Explained by Computational Scanning and Energy Calculations. PLoS Comput. Biol. 2012, 7, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Tsetlin V. I. Three-Finger Snake Neurotoxins and Ly6 Proteins Targeting Nicotinic Acetylcholine Receptors: Pharmacological Tools and Endogenous Modulators. Trends Pharmacol. Sci. 2015, 36, 109–123. [DOI] [PubMed] [Google Scholar]
- 218. David B., Wolfender J. L., Dias D. A. The Pharmaceutical Industry and Natural Products: Historical Status and New Trends. Phytochem. Rev. 2015, 14, 299–315. [Google Scholar]
- 219. Rudolph U., Crestani F., Möhler H. GABAA Receptor Subtypes: Dissecting Their Pharmacological Functions. Trends Pharmacol. Sci. 2001, 22, 188–194. [DOI] [PubMed] [Google Scholar]
- 220. Lynch J. W. Molecular Structure and Function of the Glycine Receptor Chloride Channel. Physiol. Rev. 2004, 84, 1051–1095. [DOI] [PubMed] [Google Scholar]
- 221. Davies P. A., Wang W., Hales T. G., et al. A Novel Class of Ligand-Gated Ion Channel Is Activated by Zn2+. J. Biol. Chem. 2003, 278, 712–717. [DOI] [PubMed] [Google Scholar]
- 222. Malinow R., Malenka R. C. AMPA Receptor Trafficking and Synaptic Plasticity. Annu. Rev. Neurosci. 2002, 25, 103–126. [DOI] [PubMed] [Google Scholar]
- 223. Contractor A., Mulle C., Swanson G. T. Kainate Receptors Coming of Age: Milestones of Two Decades of Research. Trends Neurosci. 2011, 34, 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Paoletti P., Bellone C., Zhou Q. NMDA Receptor Subunit Diversity: Impact on Receptor Properties, Synaptic Plasticity and Disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [DOI] [PubMed] [Google Scholar]
- 225. Treutlein J., Mühleisen T. W., Frank J., et al. Dissection of Phenotype Reveals Possible Association between Schizophrenia and Glutamate Receptor Delta 1 (GRID1) Gene Promoter. Schizophr. Res. 2009, 111, 123–130. [DOI] [PubMed] [Google Scholar]
- 226. Abbracchio M. P., Burnstock G., Verkhratsky A., et al. Purinergic Signalling in the Nervous System: An Overview. Trends Neurosci. 2009, 32, 19–29. [DOI] [PubMed] [Google Scholar]