

 Asiatic acid based 1,2,3-triazole derivatives as potential inhibitors of NF-κB with high binding affinity and apoptotic induction effects.
Asiatic acid based 1,2,3-triazole derivatives as potential inhibitors of NF-κB with high binding affinity and apoptotic induction effects.
Abstract
A series of asiatic acid (AA) based 1,2,3-triazole derivatives were designed, synthesized and subjected to a cell-based NF-κB inhibition screening assay. Among the tested compounds, compound 6k displayed impressive NF-κB inhibitory activity with an IC50 value in the low micromolar range. A molecular docking study was performed to reveal key interactions between 6k and NF-κB in which the 1,2,3-triazole moiety and the hydroxyl groups of the AA skeleton were important for improving the inhibitory activity. Subsequently, surface plasmon resonance analysis validated the high affinity between compound 6k and NF-κB protein with an equilibrium dissociation constant (KD) value of 0.36 μM. Further studies showed that compound 6k observably inhibited the NF-κB DNA binding, nuclear translocation and IκBα phosphorylation. Moreover, in vitro antitumor activity screening showed that compound 6k (IC50 = 2.67 ± 0.06 μM) exhibited the best anticancer activity against A549 cells, at least partly, by inhibition of the activity of NF-κB. Additionally, the treatment of A549 cells with compound 6k resulted in apoptosis induction potency and in vitro cell migration inhibition. Thus, we conclude that AA based 1,2,3-triazole derivatives may be potential NF-κB inhibitors with the ability to induce apoptosis and suppress cell migration.
Introduction
Nuclear factor kappa B (NF-κB) is a crucial transcription factor involved in regulating the transcription of multiple target genes included in tumor processes, innate and adaptive immune responses, inflammation, cellular growth and apoptosis.1–3 NF-κB is commonly over-expressed and constitutively activated in different types of cancers.4 It is well established that aberrant NF-κB activation has been associated with tumor cell proliferation, invasion, angiogenesis, and metastasis through its regulation of various gene products.5,6 Additionally, increasing resistance to chemotherapy is tightly linked to aberrant activation of NF-κB signaling.7–9 There are pieces of evidence which suggest that the inhibition of NF-κB activation can prevent tumor resistance to chemotherapeutic agents, shift the death–survival balance toward apoptosis, and improve the efficacy of current chemotherapeutic regimens.10,11 Because the activation of NF-κB is regarded as an essential feature of the survival of cancer cells during treatment, which contributes to cancer drug resistance, considerable efforts have been focused on targeting NF-κB for the development of therapeutic drugs for cancer therapy and overcoming drug resistance.
In previous work, we have reported that aniline-derived ursolic acid derivatives function as multidrug resistance reversers by blocking the NF-κB pathway.12,13 Triterpenoids are highly multifunctional agents owing to their ability to interact with multiple biological targets and have been reported to block NF-κB activation.14,15 Asiatic acid (AA), a pentacyclic triterpenoid isolated from the tropical medicinal plant, Centella asiatica (Apiaceae), has been found to possess numbers of pharmaceutical effects, including hepatoprotective,16 anti-inflammation,17 antidiabetic,18 and antitumor activities.19 In particular, AA exhibits potent anticancer efficacy in various types of cancer cells through its ability to inhibit NF-κB, modulate of the Bcl-2 family and activate p53.20,21 Furthermore, previous studies also reported that the modification of the C-28 position of AA has resulted in properties that increase the potency of anticancer drugs.22,23 Several groups were able to show that the introduction of a ketone group substituent on the C-11 position increased the biological activity of the compounds.22,24 Besides, it is generally acknowledged that 1,2,3-triazole derivatives showed remarkable binding affinity with a variety of enzymes and receptors by non-covalent interactions, hence resulting in versatile biological activities.25 In fact, many studies demonstrated that 1,2,3-triazole moieties were found to be highly useful as pharmacophores in medicinal chemistry and have great potential as therapeutic agents such as anticancer, anti-infective, antiviral, and antimicrobial agents.26–28 Due to their favourable therapeutic indexes, several 1,2,3-triazole containing drugs have been developed for application in clinical trials.29 Recently, several reports revealed that pentacyclic triterpene ursolic, betulinic and oleanolic acid based triazole derivatives exhibited cytotoxic activity and induced apoptosis towards a variety of cancer cell lines.30–32 However, the molecular target or the mechanism of action of these triazole derivative-mediated antitumor activities remains unclear and is yet to be elucidated.
In continuation of our studies aiming at the discovery of more potential compounds as NF-κB inhibitors, in the present study, we synthesized a series of 1,2,3-triazole derivatives based on the modification at the C-28 position of AA and assessed the potency of these compounds towards TNF-α-induced NF-κB activation and their anticancer activity against a number of cancer cell lines. The most active compound 6k was further docked with NF-κB protein to explore the possible binding interactions and its inhibitory potency for migration and drug resistance were investigated.
Results and discussion
Chemistry
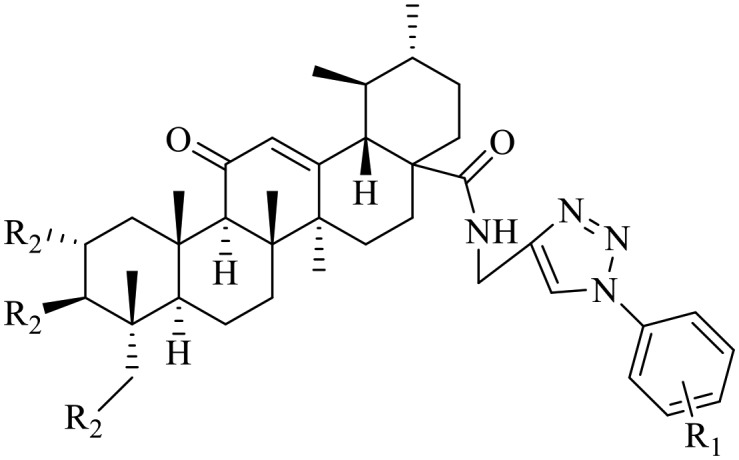
The general procedures for the synthesis of AA bearing 1,2,3-triazole derivatives are outlined in Scheme 1. Compound 2 was synthesized by the treatment of AA (1) with acetic anhydride in dry pyridine. Subsequently, compound 2 was treated with potassium dichromate in acetic acid to obtain compound 3, which was then reacted with propargyl amine and sodium hydride in the presence of THF at room temperature to afford compound 4. Compound 4 was reacted with different substituted aromatic azides (9), sodium ascorbate and CuSO4·5H2O in THF to yield compounds 5. The aromatic azides (9) were prepared by addition of substituted phenyl amine (7) in the presence of HCl with NaNO2; this intermediate was then reacted with sodium azide in DMF. The hydrolyzation reaction of compounds 5 with NaOH (aq) resulted in the formation of target compounds 6. All the target compounds (5 and 6) were confirmed by spectroscopic methods, including 1H NMR, 13C NMR and high resolution mass spectrometry (HR-MS).
Scheme 1. Synthetic pathway to target compounds 5 and 6. Reagents and conditions: (a) acetic anhydride, pyridine, 8 h, r.t, (b) acetic acid, K2CrO7, 70 °C, 5 h, r.t, (c) sodium hydride, THF, propargyl amine, Et3N, 8 h, r.t, (d) NaNO2, 10% HCl, 3 h, r.t, (e) sodium azide, DMF, r.t, (f) CuSO4, sodium-l-ascorbate, t-BuOH : H2O (1 : 1), 48 h, r.t, (g) NaOH, 2 h, r.t.

In vitro inhibition of TNF-α-induced NF-κB activation in the A549 lung cancer cell line
The inhibitory activity of all the synthesized compounds toward NF-κB was evaluated by a cell-based luciferase assay in A549 lung cancer cells, which specifically reported the ability of molecules to inhibit TNF-α-induced NF-κB transcriptional activity. The human lung cancer A549 cell line transiently co-transfected with NF-κB-luc was used to determine the effects of AA containing 1,2,3-triazole derivatives on TNF-α-induced NF-κB activation. IMD-0354, a well-characterized IKKβ inhibitor, was used as a positive control. The inhibitory activities of the tested compounds 5a–5h and 6a–6l from the TNF-α-induced NF-κB activation assay are presented in Table 1. As shown in Table 1, some of the synthesized AA containing 1,2,3-triazole derivatives display highly NF-κB inhibition potency, with IC50 values mostly in the micromolar range. In addition, the investigated compounds showed improved inhibition of TNF-α-induced NF-κB activation compared to the parent compound AA, indicating that the introduction the 1,2,3-triazole moieties resulted in an increase in the activity.
Table 1. In vitro inhibition of TNF-α-induced NF-κB activation in the A549 lung adenocarcinoma cell line.
| |||
| Compd. | R1 | R2 | IC50 a (μM) |
| 5a | 4-Cl | OAc | 10.61 ± 0.59 |
| 5b | 4-Br | OAc | 15.04 ± 1.21 |
| 5c | 4-F | OAc | 5.39 ± 0.71 |
| 5d | 3-F | OAc | 18.84 ± 1.13 |
| 5e | 3-Br | OAc | >20 |
| 5f | 2-Cl | OAc | 8.17 ± 0.68 |
| 5g | 2-F | OAc | 4.78 ± 0.32 |
| 5h | 2-Br | OAc | 12.15 ± 0.87 |
| 6a | 4-Br | OH | 7.86 ± 0.44 |
| 6b | 4-CH3 | OH | 13.28 ± 1.05 |
| 6c | 4-NO2 | OH | 0.92 ± 0.14 |
| 6d | 3-CH3 | OH | >20 |
| 6e | 3-Cl | OH | 17.26 ± 1.18 |
| 6f | 3-Br | OH | 19.79 ± 0.93 |
| 6g | 2-CH3 | OH | 3.32 ± 0.47 |
| 6h | 2-OCH3 | OH | >20 |
| 6i | 2-Cl | OH | 1.06 ± 0.26 |
| 6j | 2-Br | OH | 1.88 ± 0.39 |
| 6k | 2-F | OH | 0.14 ± 0.08 |
| 6l | 2-NO2 | OH | 0.23 ± 0.06 |
| AA | — | — | >20 |
| IMD-0354 | — | — | 1.27 ± 0.31 |
aIC50 values are presented as the mean ± SD (standard error of the mean) from three separate experiments.
At first, polar groups were investigated at R1 using halogen group substitution, of which the bromine group substitution (5b) yielded a moderate IC50 value of 15.04 μM. In contrast, in comparison to its analogue 5b, bearing the acetyl moiety at R2, compound 6a is about 2-fold more potent. Even higher activities were obtained by substitution of 4-fluorine (5c) and 2-fluorine (5g) at R1, with low IC50 values of 5.39 and 4.78 μM, respectively. The 2-fluorine derivative with hydroxyl moiety substitution at R2 (6k), which showed the highest activity among the investigated compounds, yielded a significantly lower IC50 of 0.14 μM. Similar activities resulted from a 2-bromine (6j, 1.88 μM) substituent. The resulting IC50 values of compounds 5a–5h, all containing an acetyl group at R2, were considerably higher than those of the corresponding analogues (6a–6l), lacking the acetyl function at R2, illustrating the beneficial impact of the hydroxyl group.22 Replacement of the bromine group at R1 of 6a with a 4-methyl group led to compound 6b, which resulted in decreased activity (IC50 = 13.28 μM), while an electron withdrawing functional group (–NO2) in 6c resulted in the recovery of activity (IC50 = 0.92 μM). Also, installation of a 2-chlorine group at R1 led to compound 6i (IC50 = 1.06 μM), which was 3 times more potent than its analogue 6g with a 2-methyl group (IC50 = 3.32 μM). In general, compounds containing electron withdrawing functional groups (–F, –Cl) displayed more potent inhibitory activity compared with compounds bearing electron donating functional groups (–CH3, –OCH3). A similar trend is observed for compound 6d, substituted in the meta-position with a methyl group, exhibiting weak potency. Additionally, by the comparison of IC50 values of para-, meta- and ortho-position substituents, it also suggested that an ortho-position substituent at R1 showed better inhibitory activity compared to a meta- or para-substituent. Extraordinarily high inhibitory activities resulted from substitutions at R1 with 2-nitro (IC50 = 0.23 μM), 2-fluorine (IC50 = 0.14 μM) and 2-chlorine (IC50 = 1.06 μM). Substitution with 2-bromine resulted in good activities compared with 3-bromine and 4-bromine substitution. Unfortunately, substitution with a bulky 2-methoxy group (6h) completely abolished the activity. In short, the investigation of AA containing 1,2,3-triazole derivatives implied that the introduction of 1,2,3-triazole moieties at the C-28 position and retaining the hydroxyl groups at the C-2, C-3, and C-23 positions in AA were beneficial for inhibition of NF-κB.
Molecular docking
As revealed in the SAR studies, 6k exhibited the most potent inhibitory activity towards TNF-α-induced NF-κB activation. To further elucidate the improved potency of 6k at the molecular level, molecular docking studies of 6k in the active site of NF-κB were performed using SYBYL-X 2.0 software. The docking score results are summarized in Table S1.† The best possible interacting mode of compound 6k (docking score 10.75) at the binding site of the NF-κB protein is shown in Fig. 1. As shown in Fig. 1, the docking model proposed that 6k is extended tightly into the cavity of the NF-κB active site, adopting an energetically stable conformation. The fluorine atom of the terminal aromatic group formed one hydrogen-bonding interaction with the DNA chain (DA6), while the benzene ring was appropriately positioned to form π–π interactions with PHE307. Remarkably, the 1,2,3-triazole moiety as an acceptor established four hydrogen bonds with the amino hydrogen of LYS272 and DA5, which confirmed that this moiety was also crucial for binding and contributed to the improvement of the potency of 6k. Moreover, the polar hydrogen of the C-2 and C-3 hydroxy moiety formed two hydrogen bonds with the DNA backbone P O of DG2. Also, the C-23 hydroxy of compound 6k formed two hydrogen bonds with the amine group of LYS249. Additionally, the pentacyclic ring skeleton of compound 6k was surrounded by LYS241, PRO243, SER246, ASN247, LYS249, ASP271 and LYS272 residues mainly through hydrophobic interactions. We also rechecked the docking study on the NF-κB structure without DNA fragments. As indicated in Fig. S1,† the C-2 and C-23 hydroxy moieties of 6k formed two hydrogen bonds with ASP291 and ARG295, respectively, and the terminal aromatic group extended into a hydrophobic groove.
Fig. 1. Binding modes of compound 6k in the active site of NF-κB (PDB: ; 1NFK). The ligands and the important residues for binding interactions are represented by stick and line models. The hydrogen bonds are shown as yellow dotted lines.

Surface plasmon resonance assay
The docking study revealed that compound 6k directly bonded to NF-κB. To further validate this possibility, a bio-specific interaction analysis was performed through a surface plasmon resonance (SPR) based binding assay using Biacore T200. This analytic technique allows the measurement of the kinetic and thermodynamic parameters of ligand–protein complex formation, and it is widely utilized to investigate several enzyme/inhibitor interactions.33 Association and dissociation measurements were taken and the binding affinity of compound 6k for NF-κB was determined using Biacore evaluation software. Results showed that compound 6k efficiently interacted with the immobilized NF-κB protein (Fig. 2), as demonstrated by the concentration-dependent responses for the association and dissociation, respectively, and by the clearly discernible exponential curves, indicative of the binding of compound 6k to and its dissociation from the NF-κB protein, with a binding affinity, equilibrium dissociation constant (KD) of 0.36 μM. These data provide the definitive evidence for the interaction of 6k with NF-κB protein.
Fig. 2. Biophysical binding data supporting the interaction of compound 6k with NF-κB protein. The dose response curve determined by SPR for the binding of NF-κB with 6k is shown. The measurement yielded an equilibrium dissociation constant (KD) of 0.36 μM.

Compound 6k inhibits NF-κB DNA binding
Since the docking experiments suggested that compound 6k bonded to NF-κB through alternating NF-κB/DNA interactions, we subsequently evaluated its effects on the NF-κB signaling pathway using an ELISA based DNA binding assay. As expected, we observed a significant inhibition of TNF-α-induced NF-κB activation in A549 cells treated with 6k (Fig. 3). Compound 6k reduced the TNF-α-induced NF-κB DNA binding activity by 18.2%, 34.1%, and 61.4% as compared with TNF-α at 5, 10, and 20 μM, respectively. These results indicated that 6k bonded to NF-κB and modulated the constitutive NF-κB activation by interfering with the NF-κB/DNA interaction.
Fig. 3. ELISA based DNA binding assay to evaluate the NF-κB DNA binding ability following compound 6k treatment. The data are expressed as mean ± SD, compared with the untreated control and TNF-α treated control (p < 0.05).

Compound 6k inhibits NF-κB translocation to the nucleus
Translocation to the nucleus is a symbol for TNF-α induced NF-κB activation. Recently, extensive research has reported that the anticancer activity of pentacyclic triterpenoids is associated with the suppression of NF-κB translocation to the nucleus.34 Mechanistically, NF-κB nuclear translocation was examined using immunofluorescence staining. A549 cells were treated for 24 h with 10 μM 6k, after which NF-κB translocation was stimulated by adding TNF-α for 30 minutes. As shown in Fig. 4, in A549 cells treated with TNF-α, an intense nuclear fluorescence was observed, showing the nuclear translocation of NF-κB. NF-κB nuclear translocation in the 6k-treated cells was inhibited in comparison with that in TNF-α-stimulated cells. Therefore, these results confirmed that 6k blocked the nuclear translocation, resulting in the inhibitory effect of 6k on NF-κB activation.
Fig. 4. The effects of compound 6k on the nuclear translocation of the NF-κB protein induced by cytokine TNF-α. After culturing overnight, A549 cells were pretreated with 6k (10 μM) for 24 hours, followed by TNF-α (10 ng mL–1) for 30 min, and then processed for NF-κB nuclear translocation detection by immunofluorescence staining.

Compound 6k inhibits the activity of IKKβ and IκBα phosphorylation
TNF-α triggers the nuclear translocation of NF-κB via the phosphorylation and activation of the IKK complex, which subsequently phosphorylates IκBα, resulting in the activation of NF-κB.35 To determine if the inhibition of TNF-α-induced NF-κB activation and nuclear translocation is caused by the inhibition of IκBα phosphorylation, the expression of phosphorylated IKKβ and IκBα were evaluated by a western blotting assay. As shown in Fig. 5, after treatment with TNF-α, the phosphorylated IκBα levels were markedly increased, while compound 6k significantly inhibited this phosphorylation in a concentration-dependent manner. Meanwhile, as illustrated in Fig. 5, a similar trend was also observed on p-IKKβ and NF-κB. The results revealed that compound 6k suppressed the IKKβ activation in a concentration-dependent manner stimulated by TNF-α. These data also demonstrated that compound 6k inhibits the TNF-α-induced NF-κB activity via impairment of IκBα phosphorylation mediated by the IKKβ phosphorylation.
Fig. 5. Suppression of the NF-κB signaling cascade by compound 6k on A549 cells. The effect of compound 6k on the expression of p-IκBα, p-IKKβ and NF-κB stimulated with TNF-α. β-Actin was used as a loading control. Means ± SD were from three separate experiments, P < 0.05.

In vitro cytotoxicity activity
NF-κB is a transcription factor involved in the control of various normal cellular and organism processes, such as immune and inflammatory responses, cellular proliferation and tumorigenesis. Due to the pivotal role of the transcription factor NF-κB in tumorigenesis and survival, we speculate whether the NF-κB inhibitory activity of AA containing 1,2,3-triazole derivatives is translated to anticancer activity. The in vitro cytotoxicity of the synthesized compounds was evaluated by the MTT assay against a panel of cancer cell lines including human lung cancer cells (NCI-H460), human liver cancer cells (HepG2), human non-small cell lung cancer cells (A549), human bladder cancer cells (T24), and human normal cells (HL-7702). AA and doxorubicin (DOX) were used as positive controls. As shown in Table 2, most of the newly synthesized AA derivatives exhibited a considerable growth inhibitory activity compared with AA against the tested cell lines, indicating that the introduction of 1,2,3-triazoles on AA could markedly increase the anticancer activity. Interestingly, compound 6k was the most potent compound in this series, with IC50 values of 5.24 μM, 4.46 μM, 2.67 μM and 3.96 μM against HepG2, NCI-H460, A549 and T24 cancer cell lines, respectively. Moreover, 6k showed lower toxicity toward to human normal live cells HL-7702, while it was weaker than DOX.
Table 2. Cytotoxicity of compounds 5a–5h and 6a–6l against different cancer cell lines.
| Compd. | IC50
a
(μM) |
RF c | |||||
| T24 | HepG2 | A549 | NCI-H460 | NCI-H460/DOX | HL-7702 | ||
| 5a | 27.39 ± 1.15 | 29.76 ± 0.57 | 26.71 ± 1.06 | 26.84 ± 0.36 | n.d. b | >50 | — |
| 5b | 33.82 ± 0.63 | 37.55 ± 0.88 | 33.47 ± 1.21 | 34.63 ± 0.99 | n.d. | >50 | — |
| 5c | 20.27 ± 0.54 | 25.12 ± 1.25 | 18.51 ± 1.34 | 20.78 ± 0.85 | >50 | >50 | — |
| 5d | 39.28 ± 1.26 | 43.75 ± 1.48 | 38.23 ± 1.26 | 40.91 ± 1.37 | n.d. | >50 | — |
| 5e | >50 | >50 | >50 | >50 | n.d. | >50 | — |
| 5f | 22.47 ± 1.23 | 26.93 ± 0.91 | 21.85 ± 0.68 | 23.50 ± 0.59 | n.d. | >50 | — |
| 5g | 17.33 ± 1.09 | 21.48 ± 0.78 | 16.74 ± 0.93 | 19.06 ± 0.76 | 27.11 ± 1.05 | >50 | 1.42 |
| 5h | 31.95 ± 0.43 | 36.84 ± 0.96 | 30.26 ± 0.87 | 34.49 ± 1.14 | n.d. | >50 | — |
| 6a | 23.62 ± 1.35 | 28.42 ± 1.03 | 20.36 ± 0.62 | 24.96 ± 1.46 | n.d. | >50 | — |
| 6b | 34.49 ± 1.12 | 37.29 ± 1.07 | 33.15 ± 0.98 | 34.88 ± 0.75 | n.d. | >50 | — |
| 6c | 9.10 ± 0.74 | 10.32 ± 0.45 | 8.98 ± 0.56 | 9.65 ± 0.29 | 14.43 ± 0.58 | >50 | 1.49 |
| 6d | >50 | >50 | >50 | >50 | n.d. | >50 | — |
| 6e | 37.69 ± 0.71 | 40.58 ± 0.87 | 35.68 ± 1.25 | 39.73 ± 0.79 | n.d. | >50 | — |
| 6f | 41.14 ± 2.58 | 45.80 ± 1.80 | 39.87 ± 1.55 | 43.57 ± 1.64 | n.d. | >50 | — |
| 6g | 14.95 ± 1.15 | 18.71 ± 0.90 | 14.78 ± 0.32 | 15.92 ± 0.57 | 17.41 ± 0.34 | >50 | 1.09 |
| 6h | >50 | >50 | >50 | >50 | n.d. | >50 | — |
| 6i | 9.61 ± 0.63 | 12.66 ± 0.86 | 9.24 ± 1.14 | 11.74 ± 0.37 | 13.99 ± 0.95 | >50 | 1.19 |
| 6j | 10.67 ± 0.72 | 14.87 ± 0.37 | 10.20 ± 0.49 | 12.05 ± 0.84 | 16.92 ± 1.01 | >50 | 1.40 |
| 6k | 3.96 ± 0.21 | 5.24 ± 1.34 | 2.67 ± 0.06 | 4.46 ± 0.15 | 4.84 ± 0.42 | >50 | 1.08 |
| 6l | 7.51 ± 0.54 | 8.36 ± 1.11 | 5.16 ± 0.51 | 7.85 ± 0.83 | 10.27 ± 0.35 | >50 | 1.31 |
| AA | 43.97 ± 1.36 | 35.37 ± 1.47 | 41.02 ± 0.78 | 44.82 ± 1.25 | n.d. | >50 | — |
| DOX | 0.96 ± 0.09 | 1.37 ± 0.24 | 1.07 ± 0.18 | 0.8 ± 0.06 | 30.99 ± 1.15 | 10.44 ± 0.5 | 38.74 |
aIC50 values are presented as the mean ± SD (standard error of the mean) from three separate experiments.
bn.d. = not detected.
cRF (resistant factor) is defined as IC50 in NCI-H460/DOX/IC50 in NCI-H460.
Drug resistance is a critical therapeutic problem that limits the efficacies of most anticancer drugs for a variety of human cancer cells. In terms of their strong inhibitory effects on the tested cancer cell lines, compounds 5c, 5g, 6c, 6g and 6i–6l were further evaluated against DOX-resistant NCI-H460 (NCI-H460/DOX). As shown in Table 2, DOX displayed low activity on NCI-H460/DOX resistant cell lines. It was significant to observe that compound 6k had a much lower resistance factor (1.08) for the resistant NCI-H460 cell line. Moreover, except for 5c, the other selected compounds showed potent cytotoxicity against the corresponding DOX resistant cells comparable to that of DOX. These results implied that these compounds might be useful in the treatment of drug refractory tumors resistant to DOX.
Compound 6k induces apoptosis in A549 cells
The ability of 6k to induce apoptosis was investigated by flow cytometry. A549 cells were allowed to grow in the presence or absence of 6k for 24 h, and co-stained with PI and Annexin-V/FITC. Four quadrant images, Q1, Q2, Q3, and Q4, represented four different cell states: necrotic cells, late apoptotic or necrotic cells, early apoptotic cells and living cells, respectively. In the cells treated with compound 6k, we observed a dose-dependent increase in the percentage of apoptotic cells, at concentrations of 5 μM, 10 μM and 20 μM for 24 h. As shown in Fig. 6, few (1.58%) apoptotic cells were present in the control panel; in contrast, the percentage rose to 11.80% at the concentration of 5 μM after treatment with 6k for 24 h. In the presence of 6k with concentrations of 10 μM and 20 μM, the percentage of apoptotic cells was further increased to 29.42% and 40.38%, respectively. These data suggested that compound 6k effectively induced apoptosis in A549 cells in a dose-dependent manner.
Fig. 6. Annexin V-FITC and PI staining to evaluate apoptosis in A549 cells following compound 6k treatment. A549 cells were treated with 6k (5, 10 and 20 μM, for 24 h), incubated with Annexin V-FITC and PI and analyzed using flow cytometry.

Apoptosis assay by acridine orange/ethidium bromide (AO/EB) staining
To further characterize the cell apoptosis induced by compound 6k, AO/EB staining was performed to evaluate the accompanying changes in morphology. A549 cells were exposed to compound 6k at concentrations of 5 μM, 10 μM and 20 μM for 24 h. The results (Fig. 7) showed that the morphology of A549 cells had changed significantly in the presence of 6k. The cell nuclei were stained yellow green or orange, and the morphology showed pycnosis, membrane blebbing and cell budding, characteristic of apoptosis. The nearly complete absence of red-stained cells showed that 6k treatment was associated with low toxicity. Hence, the observation results further demonstrated that compound 6k induced apoptosis in A549 cells.
Fig. 7. Compound 6k induced apoptosis in A549 cells was studied by AO/EB staining and photographed via fluorescence microscopy. A549 cells were treated with 6k (5, 10 and 20 μM, for 24 h), and incubated with AO/EB. Images were acquired using a Nikon Te2000 deconvolution microscope (magnification 200×).

Compound 6k inhibits the migration of A549 cells in vitro
NF-κB is constitutively activated in a number of cancer cells and promotes tumor proliferation, invasion, and metastasis.36 Increasing evidence has revealed that metastatic cancer cells exhibited great capability for migration and invasion.37 Migration plays a key role in the later period of cancer progression and is known to be related to tumor progression and metastatic cascade. Since cell migration is connected with the metastatic activity of cancer cells, transwell migration assays were carried out to investigate whether compound 6k could suppress the migration of A549 cells. The migration of A549 cells was recorded by microscopic observations at concentrations of 5 μM, 10 μM and 20 μM of compound 6k for 24 h. Results from Fig. 8 clearly showed that 6k strongly decrease the number of migrating cells in a dose-dependent manner after 24 h as compared with the control. This demonstrated that the NF-κB inhibitory functionality is critical for 6k to suppress migration.
Fig. 8. Transwell assay detected the migration inhibition of A549 cells after incubation with compound 6k at concentrations of 5, 10 and 20 μM for 24 h, respectively. The experiments were performed three times, and the results of the representative experiments are shown.

Conclusions
A series of AA containing 1,2,3-triazole derivatives were rationally designed and synthesized as anticancer agents which targeted NF-κB with good IC50 values in the low micromolar range. The cell-based luciferase assay led to the identification of compound 6k, possessing high affinity and potency, as a potent inhibitor of TNF-α-induced NF-κB activation. A docking study of the most active compound 6k revealed key interactions between 6k and NF-κB in which the 1,2,3-triazole moiety and the hydroxyl groups of the AA skeleton were important for improving the inhibitory activity. Subsequently, surface plasmon resonance analysis validated the interaction between compound 6k and NF-κB protein with an equilibrium dissociation constant (KD) value of 0.36 μM. Further studies revealed that compound 6k observably inhibited the NF-κB DNA binding, nuclear translocation and IκBα phosphorylation. Notably, further antitumor activity screening showed that compound 6k (IC50 = 2.67 ± 0.06 μM) exhibited the best anticancer activity against the A549 cell line, while displaying slightly weaker inhibitory activity than DOX. The exposure of compound 6k to A549 cells resulted in induction of apoptosis evidenced by flow cytometry and the AO/EB staining assay. In addition, transwell migration assays indicated that compound 6k inhibited in vitro cell migration by blockage of the NF-κB signaling pathway. Consequently, the rational design of AA containing 1,2,3-triazole derivatives offers significant potential for the discovery of a new class of NF-κB inhibitors with the ability to suppress cancer cell migration and induce apoptosis.
Experimental methods
General
Asiatic acid was purchased from Biological Technology of Wuhan, China. All the chemical reagents and solvents used were of analytical grade. Silica gel (200–300 mesh) used in column chromatography was provided by Tsingtao Marine Chemistry Co. Ltd. 1H NMR and 13C NMR spectra were recorded on a BRUKER AV-400 spectrometer with TMS as an internal standard in CDCl3. Mass spectra were determined on an FTMS ESI spectrometer.
Synthesis: general procedure for compounds 5a–5h
AA (1, 200 mg, 0.4 mmol) was added to pyridine (10 mL) followed by acetic anhydride (0.5 mL, 5.0 mmol) and the mixture was stirred at 20 °C for 8 h. After the reaction, the mixture was diluted with ethyl acetate (25 mL), washed with aqueous 1 M HCl (10 mL × 5), saturated CuSO4 (15 mL × 2) and saturated NaCl solution (20 mL). The organic phase was dried with anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel eluted with petroleum ether/ethyl acetate (V : V = 3 : 1) to give 2. Compound 2 (100 mg, 0.16 mmol) and K2Cr2O7·2H2O (150 g, 0.5 mmol) were added to acetic acid (20 mL) and refluxed for 5 h. The mixture was cooled to 20 °C and neutralized with 10% NaHCO3 solution to pH 7–8. Then, the mixture was diluted with ethyl acetate (20 mL) and washed with water (10 mL × 5). The organic phase was dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The crude product was purified by silica gel chromatography with a gradient elution of CH2Cl2/MeOH (V : V = 30 : 1) to yield compound 3. Compound 3 (125.6 mg, 0.2 mmol) was allowed to react with NaH (80% in oil, 0.019 g, 0.8 mmol) in dry THF (20 mL) under a nitrogen atmosphere for 8 h before a solution of propargyl amine (0.025 g; 0.45 mmol in 5 mL THF) was slowly added. After the reaction was completed, the reaction mixture was extracted with ethyl acetate. The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the crude product, which was purified by column chromatography using 5% EtOAc : hexane to give the propargylated product 4. The title compounds (5) were prepared by the reaction of substituted aryl azides (1.5 mmol), intermediate 4 (1.0 mmol), and CuSO4·5H2O (0.1 mmol) in a t-BuOH : H2O (1 : 1) mixture. Sodium ascorbate (0.1 mmol) was added to the reaction mixture and the contents were stirred vigorously at room temperature for 48 h till completion of the reaction (monitored by TLC analysis). The reaction was worked up by dilution of the contents with water and extraction with ethyl acetate (3 times). The combined ethyl acetate extract was washed with brine solution, dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude product was purified by chromatography on silica gel eluted with ethyl acetate/hexane (V : V = 3 : 1) to obtain compounds 5a–5k. The structures were confirmed by 1H NMR, 13C NMR and HR-MS (see the ESI†).
Compound 5a
Yield 88%, as a white solid. Mp: 164.7–165.8 °C. 1H NMR (400 MHz, CD3OD) δ 8.31 (s, 1H, H-33), 7.84–7.81 (m, 2H, Ar–H), 7.59–7.57 (m, 2H, Ar–H), 5.57 (s, 1H, H-12), 5.23–5.16 (m, 1H, H-2), 4.99 (d, J = 11.2 Hz, 1H, H-3), 4.49–4.37 (m, 2H, H-31), 3.79 (d, J = 9.6 Hz, 1H, H-23), 3.62 (d, J = 10.8 Hz, 1H, H-23), 3.07 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.46 (d, J = 10.8 Hz, 1H, H-16), 2.44 (s, 1H, H-9), 2.25–2.18 (m, 1H, H-20), 2.04 (s, 3H, CH3CO), 2.01 (s, 3H, CH3CO), 1.95 (s, 3H, CH3CO), 1.79–1.09 (m, 15H), 1.31 (s, 3H, CH3-27), 1.04 (s, 3H, CH3-24), 0.97 (d, J = 6.4 Hz, 3H, CH3-29), 0.87 (d, J = 6.4 Hz, 3H, CH3-30), 0.85 (s, 3H, CH3-25), 0.48 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 200.94, 178.95, 172.37, 172.19, 172.00, 166.45, 147.21, 136.96, 135.48, 131.17, 131.05, 123.04, 122.74, 76.11, 70.47, 66.17, 61.99, 54.04, 48.42, 48.31, 45.74, 45.04, 44.87, 43.12, 39.98, 39.86, 38.92, 38.01, 35.54, 33.39, 31.49, 29.18, 24.72, 21.34, 21.00, 20.76, 19.54, 18.24, 17.97, 17.64, 14.13. HR-MS (m/z) (ESI): calc for C45H59ClN4O8 [M + Na]+: 841.3914, found: 841.3893.
Compound 5b
Yield 85%, as a white solid. Mp: 177.1–179.0 °C. 1H NMR (400 MHz, CD3OD) δ 8.32 (s, 1H, H-33), 7.78–7.71 (m, 4H, Ar–H), 5.57 (s, 1H, H-12), 5.23–5.16 (m, 1H, H-2), 4.99 (d, J = 11.2 Hz, 1H, H-3), 4.49–4.37 (m, 2H, H-31), 3.79 (d, J = 9.6 Hz, 1H, H-23), 3.62 (d, J = 10.8 Hz, 1H, H-23), 3.07 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.45 (d, J = 10.8 Hz, 1H, H-16), 2.44 (s, 1H, H-9), 2.25–2.17 (m, 1H, H-20), 2.04 (s, 3H, CH3CO), 2.01 (s, 3H, CH3CO), 1.95 (s, 3H, CH3CO), 1.79–1.09 (m, 15H), 1.31 (s, 3H, CH3-27), 1.03 (s, 3H, CH3-24), 0.97 (d, J = 6.4 Hz, 3H, CH3-29), 0.87 (d, J = 6.4 Hz, 3H, CH3-30), 0.85 (s, 3H, CH3-25), 0.47 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 200.92, 178.93, 172.37, 172.19, 171.99, 166.45, 147.24, 137.40, 134.09, 131.16, 123.25, 123.00, 122.93, 76.11, 70.47, 66.17, 61.97, 54.03, 48.41, 48.30, 45.73, 45.03, 44.86, 43.12, 39.97, 39.84, 38.91, 38.01, 35.53, 33.38, 31.50, 29.17, 24.72, 21.34, 21.02, 20.80, 20.77, 19.54, 18.26, 17.97, 17.65, 14.17. HR-MS (m/z) (ESI): calc for C45H59BrN4O8 [M + Na]+: 885.3408, found: 885.3391.
Compound 5c
Yield 84%, as a white solid. Mp: 180.3–182.2 °C. 1H NMR (400 MHz, CD3OD) δ 8.27 (s, 1H, H-33), 7.85–7.82 (m, 2H, Ar–H), 7.35–7.31 (m, 2H, Ar–H), 5.58 (s, 1H, H-12), 5.23–5.17 (m, 1H, H-2), 5.00 (d, J = 11.2 Hz, 1H, H-3), 4.48–4.37 (m, 2H, H-31), 3.79 (d, J = 9.6 Hz, 1H, H-23), 3.62 (d, J = 10.8 Hz, 1H, H-23), 3.07 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.47 (d, J = 10.8 Hz, 1H, H-16), 2.46 (s, 1H, H-9), 2.26–2.19 (m, 1H, H-20), 2.05 (s, 3H, CH3CO), 2.01 (s, 3H, CH3CO), 1.96 (s, 3H, CH3CO), 1.81–1.12 (m, 15H), 1.33 (s, 3H, CH3-27), 1.07 (s, 3H, CH3-24), 0.98 (d, J = 6.4 Hz, 3H, CH3-29), 0.88 (d, J = 6.4 Hz, 3H, CH3-30), 0.85 (s, 3H, CH3-25), 0.52 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 200.99, 178.99, 172.40, 172.21, 172.03, 166.46, 163.87 (1J(C,F) = 246.0 Hz), 147.08, 134.73 (4J(C,F) = 4.0 Hz), 131.18, 123.59 (3J(C,F) = 9.0 Hz), 123.23, 117.72 (2J(C,F) = 23.0 Hz), 76.12, 70.49, 66.16, 62.03, 54.06, 48.44, 48.34, 45.78, 45.08, 44.88, 43.14, 40.00, 39.88, 38.94, 38.03, 35.55, 33.41, 31.50, 29.19, 24.71, 21.34, 21.31, 20.98, 20.75, 20.73, 19.52, 18.23, 17.99, 17.62, 14.10 (1J(C,F) = 249.3 Hz). HR-MS (m/z) (ESI): calc for C45H59FN4O8 [M + Na]+: 825.4209, found: 825.4191.
Compound 5d
Yield 86%, as a white solid. Mp: 182.4–183.1 °C. 1H NMR (400 MHz, CD3OD) δ 8.20 (s, 1H, H-33), 7.86–7.81 (m, 1H, Ar–H), 7.59–7.54 (m, 1H, Ar–H), 7.46–7.39 (m, 2H, Ar–H), 5.58 (s, 1H, H-12), 5.24–5.17 (m, 1H, H-2), 5.00 (d, J = 11.2 Hz, 1H, H-3), 4.52–4.38 (m, 2H, H-31), 3.80 (d, J = 9.6 Hz, 1H, H-23), 3.63 (d, J = 10.8 Hz, 1H, H-23), 3.08 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.47 (s, 1H, H-9), 2.46 (d, J = 10.8 Hz, 1H, H-16), 2.28–2.19 (m, 1H, H-20), 2.05 (s, 3H, CH3CO), 2.01 (s, 3H, CH3CO), 1.96 (s, 3H, CH3CO), 1.81–1.13 (m, 15H), 1.34 (s, 3H, CH3-27), 1.11 (s, 3H, CH3-24), 0.99 (d, J = 6.4 Hz, 3H, CH3-29), 0.89 (d, J = 6.4 Hz, 3H, CH3-30), 0.87 (s, 3H, CH3-25), 0.57 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.07, 179.09, 172.46, 172.27, 172.08, 166.51, 155.25 (1J(C,F) = 249.3 Hz), 146.75, 132.16 (1J(C,F) = 8.0 Hz), 131.20, 126.54 (1J(C,F) = 3.8 Hz), 126.35 (1J(C,F) = 9.0 Hz), 126.31, 126.05 (1J(C,F) = 6.4 Hz), 118.26 (1J(C,F) = 10.0 Hz), 76.15, 70.51, 66.17, 62.09, 54.03, 48.47, 48.38, 45.80, 45.11, 44.92, 43.16, 40.01, 39.90, 38.96, 38.07, 35.53, 33.44, 31.51, 29.21, 24.70, 21.33, 21.30, 20.96, 20.74, 20.72, 19.48, 18.23, 18.01, 17.60, 14.12. HR-MS (m/z) (ESI): calc for C45H59FN4O8 [M + Na]+: 825.4209, found: 825.4195.
Compound 5e
Yield 87%, as a white solid. Mp: 172.5–174.3 °C. 1H NMR (400 MHz, CDCl3) δ 7.97 (s, 1H, H-33), 7.91–7.90 (m, 1H, Ar–H), 7.62–7.60 (m, 1H, Ar–H), 7.56–7.54 (m, 1H, Ar–H), 7.39–7.35 (m, 1H, Ar–H), 5.63 (s, 1H, H-12), 5.27–5.20 (m, 1H, H-2), 5.01 (d, J = 11.2 Hz, 1H, H-3), 4.57–4.52 (m, 1H, H-31), 4.41–4.36 (m, 1H, H-31), 3.79 (d, J = 9.6 Hz, 1H, H-23), 3.55 (d, J = 10.8 Hz, 1H, H-23), 3.14 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.37 (d, J = 10.8 Hz, 1H, H-16), 2.32 (s, 1H, H-9), 2.19–2.10 (m, 1H, H-20), 2.05 (s, 3H, CH3CO), 1.99 (s, 3H, CH3CO), 1.93 (s, 3H, CH3CO), 1.75–1.10 (m, 15H), 1.25 (s, 3H, CH3-27), 1.14 (s, 3H, CH3-24), 0.94 (d, J = 6.4 Hz, 3H, CH3-29), 0.85 (s, 3H, CH3-25), 0.83 (d, J = 6.4 Hz, 3H, CH3-30), 0.62 (s, 3H, CH3-26). 13C NMR (100 MHz, CDCl3) δ 198.68, 176.78, 170.86, 170.53, 170.17, 163.40, 145.67, 137.88, 131.94, 131.22, 130.45, 123.67, 123.46, 121.32, 118.89, 74.92, 69.06, 65.30, 61.14, 52.99, 47.38, 47.22, 44.61, 44.20, 43.94, 41.96, 39.05, 38.68, 37.73, 37.09, 34.98, 32.52, 30.56, 28.14, 24.39, 21.09, 20.98, 20.95, 20.86, 18.73, 17.76, 17.18, 17.10, 13.95. HR-MS (m/z) (ESI): calc for C45H59BrN4O8 [M + Na]+: 885.3408, found: 885.3397.
Compound 5f
Yield 83%, as a white solid. Mp: 166.4–168.3 °C. 1H NMR (400 MHz, CD3OD) δ 8.14 (s, 1H, H-33), 7.70–7.67 (m, 2H, Ar–H), 7.60–7.52 (m, 2H, Ar–H), 5.59 (s, 1H, H-12), 5.26–5.19 (m, 1H, H-2), 5.02 (d, J = 11.2 Hz, 1H, H-3), 4.53–4.40 (m, 2H, H-31), 3.82 (d, J = 9.6 Hz, 1H, H-23), 3.63 (d, J = 10.8 Hz, 1H, H-23), 3.09 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.49 (s, 1H, H-9), 2.47 (d, J = 10.8 Hz, 1H, H-16), 2.27–2.19 (m, 1H, H-20), 2.05 (s, 3H, CH3CO), 2.01 (s, 3H, CH3CO), 1.96 (s, 3H, CH3CO), 1.83–1.12 (m, 15H), 1.35 (s, 3H, CH3-27), 1.19 (s, 3H, CH3-24), 0.98 (d, J = 6.4 Hz, 3H, CH3-29), 0.89 (s, 3H, CH3-25), 0.88 (d, J = 6.4 Hz, 3H, CH3-30), 0.70 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 200.98, 179.01, 172.39, 172.20, 172.01, 166.43, 146.27, 136.02, 132.52, 131.98, 131.18, 129.95, 129.34, 129.01, 126.67, 76.14, 70.47, 66.19, 62.09, 53.95, 48.45, 45.87, 45.12, 44.90, 43.15, 39.97, 39.86, 38.96, 38.02, 35.63, 33.47, 31.49, 29.23, 24.69, 21.39, 21.34, 21.01, 20.80, 20.77, 19.82, 18.36, 18.07, 17.66, 14.18. HR-MS (m/z) (ESI): calc for C45H59ClN4O8 [M + Na]+: 841.3914, found: 841.3895.
Compound 5g
Yield 86%, as a white solid. Mp: 187.8–189.6 °C. 1H NMR (400 MHz, CD3OD) δ 8.35 (s, 1H, H-33), 7.67–7.57 (m, 3H, Ar–H), 7.27–7.22 (m, 1H, Ar–H), 5.57 (s, 1H, H-12), 5.23–5.16 (m, 1H, H-2), 4.99 (d, J = 11.2 Hz, 1H, H-3), 4.49–4.37 (m, 2H, H-31), 3.78 (d, J = 9.6 Hz, 1H, H-23), 3.62 (d, J = 10.8 Hz, 1H, H-23), 3.07 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.46 (d, J = 10.8 Hz, 1H, H-16), 2.44 (s, 1H, H-9), 2.26–2.18 (m, 1H, H-20), 2.04 (s, 3H, CH3CO), 2.01 (s, 3H, CH3CO), 1.95 (s, 3H, CH3CO), 1.80–1.18 (m, 15H), 1.32 (s, 3H, CH3-27), 1.04 (s, 3H, CH3-24), 0.98 (d, J = 6.4 Hz, 3H, CH3-29), 0.88 (d, J = 6.4 Hz, 3H, CH3-30), 0.84 (s, 3H, CH3-25), 0.48 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 200.98, 179.00, 172.41, 172.22, 172.03, 166.49, 164.50 (1J(C,F) = 245.3 Hz), 147.20, 139.56 (3J(C,F) = 10.1 Hz), 132.81 (3J(C,F) = 9.0 Hz), 131.17, 123.21, 116.92 (4J(C,F) = 3.2 Hz), 116.58 (2J(C,F) = 21.3 Hz), 108.81 (2J(C,F) = 26.6 Hz), 76.12, 70.47, 66.17, 62.01, 54.05, 48.43, 48.33, 45.75, 45.06, 44.88, 43.13, 39.99, 39.87, 38.93, 38.04, 35.50, 33.40, 31.50, 29.18, 24.72, 21.33, 21.31, 20.98, 20.76, 20.74, 19.50, 18.21, 17.97, 17.62, 14.08. HR-MS (m/z) (ESI): calc for C45H59FN4O8 [M + Na]+: 825.4209, found: 825.4194.
Compound 5h
Yield 84%, as a white solid. Mp: 182.2–184.6 °C. 1H NMR (400 MHz, CD3OD) δ 8.10 (s, 1H, H-33), 7.87–7.84 (m, 1H, Ar–H), 7.61–7.56 (m, 1H, Ar–H), 7.54–7.49 (m, 2H, Ar–H), 5.60 (s, 1H, H-12), 5.26–5.20 (m, 1H, H-2), 5.02 (d, J = 11.2 Hz, 1H, H-3), 4.53–4.41 (m, 2H, H-31), 3.82 (d, J = 9.6 Hz, 1H, H-23), 3.64 (d, J = 10.8 Hz, 1H, H-23), 3.10 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.50 (s, 1H, H-9), 2.48 (d, J = 10.8 Hz, 1H, H-16), 2.28–2.20 (m, 1H, H-20), 2.06 (s, 3H, CH3CO), 2.01 (s, 3H, CH3CO), 1.96 (s, 3H, CH3CO), 1.83–1.12 (m, 15H), 1.36 (s, 3H, CH3-27), 1.21 (s, 3H, CH3-24), 0.99 (d, J = 6.4 Hz, 3H, CH3-29), 0.90 (s, 3H, CH3-25), 0.89 (d, J = 6.4 Hz, 3H, CH3-30), 0.74 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.08, 179.07, 172.44, 172.25, 172.06, 166.50, 146.21, 137.72, 135.17, 132.87, 131.19, 129.89, 129.38, 126.73, 119.74, 76.16, 70.51, 66.19, 62.13, 53.98, 48.48, 48.41, 45.92, 45.15, 44.92, 43.17, 39.99, 39.90, 38.99, 38.05, 35.65, 33.49, 31.49, 29.25, 24.69, 21.38, 21.32, 20.99, 20.78, 20.75, 19.92, 18.38, 18.09, 17.63, 14.18. HR-MS (m/z) (ESI): calc for C45H59BrN4O8 [M + Na]+: 885.3408, found: 885.3387.
Synthesis: general procedure for compounds 6a–6l
To a solution of 5 (0.12 mmol) in MeOH (4 mL) and THF (6 mL), 4 N NaOH (1.2 mL) was added dropwise, and this reaction mixture was stirred for 2 h at room temperature. The mixture was acidified with hydrochloric acid and extracted with ethyl acetate. The organic phase was washed with sodium bicarbonate solution and brine in sequence, and concentrated under reduced pressure. The crude product was purified column chromatography with a gradient elution of CH2Cl2/MeOH (V : V = 20 : 1) to give a white solid of 6a–6l. The structures were confirmed by 1H NMR, 13C NMR and HR-MS (see the ESI†).
Compound 6a
Yield 76%, as a white solid. Mp: 212.5–214.1 °C. 1H NMR (400 MHz, CD3OD) δ 8.31 (s, 1H, H-33), 7.78–7.72 (m, 4H, Ar–H), 5.56 (s, 1H, H-12), 4.48–4.36 (m, 2H, H-31), 3.73–3.66 (m, 1H, H-2), 3.49 (d, J = 11.2 Hz, 1H, H-3), 3.33 (d, J = 9.6 Hz, 1H, H-23), 3.22 (d, J = 10.8 Hz, 1H, H-23), 2.98 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.45 (d, J = 10.8 Hz, 1H, H-16), 2.37 (s, 1H, H-9), 2.26–2.18 (m, 1H, H-20), 1.79–1.14 (m, 15H), 1.31 (s, 3H, CH3-27), 0.99 (d, J = 6.4 Hz, 3H, CH3-29), 0.94 (s, 3H, CH3-24), 0.88 (d, J = 6.4 Hz, 3H, CH3-30), 0.63 (s, 3H, CH3-25), 0.43 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.85, 179.11, 166.47, 147.25, 137.45, 134.09, 131.27, 123.31, 123.11, 122.97, 77.59, 69.32, 65.76, 62.40, 54.09, 48.17, 47.66, 45.84, 45.15, 44.19, 40.03, 39.89, 38.96, 38.10, 35.50, 33.34, 31.52, 29.17, 24.75, 21.36, 21.29, 19.68, 18.21, 18.00, 17.60, 13.86. HR-MS (m/z) (ESI): calc for C39H53BrN4O5 [M + Na]+: 759.3092, found: 759.3082.
Compound 6b
Yield 72%, as a white solid. Mp: 224.8–226.4 °C. 1H NMR (400 MHz, CD3OD) δ 8.26 (s, 1H, H-33), 7.61–7.56 (m, 2H, Ar–H), 7.45–7.41 (m, 1H, Ar–H), 7.31–7.29 (m, 1H, Ar–H), 5.56 (s, 1H, H-12), 4.49–4.35 (m, 2H, H-31), 3.72–3.65 (m, 1H, H-2), 3.49 (d, J = 11.2 Hz, 1H, H-3), 3.33 (d, J = 9.6 Hz, 1H, H-23), 3.21 (d, J = 10.8 Hz, 1H, H-23), 2.99 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.46 (d, J = 10.8 Hz, 1H, H-16), 2.45 (s, 3H, Ar–CH3), 2.37 (s, 1H, H-9), 2.25–2.17 (m, 1H, H-20), 1.79–1.13 (m, 15H), 1.30 (s, 3H, CH3-27), 0.99 (d, J = 6.4 Hz, 3H, CH3-29), 0.95 (s, 3H, CH3-24), 0.88 (d, J = 6.4 Hz, 3H, CH3-30), 0.62 (s, 3H, CH3-25), 0.43 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.78, 179.07, 166.40, 146.88, 141.42, 138.27, 131.27, 130.76, 130.65, 123.26, 121.91, 118.52, 77.59, 69.30, 65.78, 62.40, 54.05, 48.16, 47.64, 45.81, 45.15, 44.17, 40.01, 39.87, 38.95, 38.12, 35.50, 33.35, 31.53, 29.15, 24.74, 21.45, 21.35, 21.30, 19.64, 18.24, 17.99, 17.60, 13.85. HR-MS (m/z) (ESI): calc for C40H56N4O5 [M + Na]+: 695.4143, found: 695.4133.
Compound 6c
Yield 74%, as a white solid. Mp: 228.3–230.2 °C. 1H NMR (400 MHz, CD3OD) δ 8.47 (s, 1H, H-33), 7.46–7.43 (m, 2H, Ar–H), 7.15–7.12 (m, 2H, Ar–H), 5.56 (s, 1H, H-12), 4.50–4.41 (m, 2H, H-31), 3.71–3.64 (m, 1H, H-2), 3.48 (d, J = 11.2 Hz, 1H, H-3), 3.31 (d, J = 9.6 Hz, 1H, H-23), 3.20 (d, J = 10.8 Hz, 1H, H-23), 2.97 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.45 (d, J = 10.8 Hz, 1H, H-16), 2.37 (s, 1H, H-9), 2.26–2.18 (m, 1H, H-20), 1.79–1.15 (m, 15H), 1.31 (s, 3H, CH3-27), 0.99 (d, J = 6.4 Hz, 3H, CH3-29), 0.92 (s, 3H, CH3-24), 0.89 (d, J = 6.4 Hz, 3H, CH3-30), 0.59 (s, 3H, CH3-25), 0.46 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.79, 179.12, 166.40, 148.68, 147.72, 142.54, 131.27, 126.53, 123.28, 121.72, 77.58, 69.28, 65.76, 62.38, 54.11, 48.39, 48.14, 47.63, 45.86, 45.16, 44.16, 40.03, 39.89, 38.94, 38.08, 35.51, 33.34, 31.51, 29.18, 24.74, 21.38, 21.29, 19.65, 18.19, 18.01, 17.60, 13.80. HR-MS (m/z) (ESI): calc for C39H53N5O7 [M + Na]+: 726.3837, found: 726.3826.
Compound 6d
Yield 78%, as a white solid. Mp: 224.8–226.5 °C. 1H NMR (400 MHz, CD3OD) δ 8.23 (s, 1H, H-33), 7.67–7.65 (m, 2H, Ar–H), 7.38–7.36 (m, 2H, Ar–H), 5.56 (s, 1H, H-12), 4.48–4.35 (m, 2H, H-31), 3.72–3.66 (m, 1H, H-2), 3.49 (d, J = 11.2 Hz, 1H, H-3), 3.33 (d, J = 9.6 Hz, 1H, H-23), 3.21 (d, J = 10.8 Hz, 1H, H-23), 2.99 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.45 (d, J = 10.8 Hz, 1H, H-16), 2.41 (s, 3H, Ar–CH3), 2.37 (s, 1H, H-9), 2.25–2.17 (m, 1H, H-20), 1.79–1.14 (m, 15H), 1.30 (s, 3H, CH3-27), 0.98 (d, J = 6.4 Hz, 3H, CH3-29), 0.95 (s, 3H, CH3-24), 0.88 (d, J = 6.4 Hz, 3H, CH3-30), 0.62 (s, 3H, CH3-25), 0.45 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.81, 179.07, 166.44, 146.87, 140.39, 136.05, 131.35, 131.27, 123.12, 121.30, 77.59, 69.31, 65.78, 62.40, 54.04, 48.17, 47.66, 45.82, 45.14, 44.17, 40.01, 39.87, 38.96, 38.09, 35.52, 33.34, 31.52, 29.16, 24.75, 21.37, 21.30, 21.06, 19.67, 18.22, 17.99, 17.60, 13.83. HR-MS (m/z) (ESI): calc for C40H56N4O5 [M + Na]+: 695.4143, found: 695.4136.
Compound 6e
Yield 80%, as a white solid. Mp: 219.7–220.9 °C. 1H NMR (400 MHz, CD3OD) δ 8.35 (s, 1H, H-33), 7.89–7.88 (m, 1H, Ar–H), 7.77–7.74 (m, 1H, Ar–H), 7.57–7.53 (m, 1H, Ar–H), 7.49–7.47 (m, 1H, Ar–H), 5.55 (s, 1H, H-12), 4.49–4.37 (m, 2H, H-31), 3.73–3.66 (m, 1H, H-2), 3.49 (d, J = 11.2 Hz, 1H, H-3), 3.33 (d, J = 9.6 Hz, 1H, H-23), 3.21 (d, J = 10.8 Hz, 1H, H-23), 2.99 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.44 (d, J = 10.8 Hz, 1H, H-16), 2.36 (s, 1H, H-9), 2.24–2.16 (m, 1H, H-20), 1.79–1.13 (m, 15H), 1.29 (s, 3H, CH3-27), 0.97 (d, J = 6.4 Hz, 3H, CH3-29), 0.96 (s, 3H, CH3-24), 0.87 (d, J = 6.4 Hz, 3H, CH3-30), 0.63 (s, 3H, CH3-25), 0.41 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.73, 179.03, 166.36, 147.22, 139.26, 136.53, 132.40, 131.23, 129.84, 123.30, 121.45, 119.55, 77.58, 69.28, 65.77, 62.35, 54.02, 48.32, 48.14, 47.63, 45.79, 45.11, 44.15, 39.98, 39.84, 38.93, 38.07, 35.46, 33.33, 31.52, 29.13, 24.75, 21.37, 21.33, 19.61, 18.29, 17.99, 17.62, 13.88. HR-MS (m/z) (ESI): calc for C39H53ClN4O5 [M + Na]+: 715.3597, found: 715.3586.
Compound 6f
Yield 78%, as a white solid. Mp: 223.8–225.3 °C. 1H NMR (400 MHz, CD3OD) δ 8.35 (s, 1H, H-33), 8.04–8.03 (m, 1H, Ar–H), 7.81–7.79 (m, 1H, Ar–H), 7.64–7.62 (m, 1H, Ar–H), 7.51–7.47 (m, 1H, Ar–H), 5.55 (s, 1H, H-12), 4.49–4.36 (m, 2H, H-31), 3.73–3.66 (m, 1H, H-2), 3.49 (d, J = 11.2 Hz, 1H, H-3), 3.33 (d, J = 9.6 Hz, 1H, H-23), 3.21 (d, J = 10.8 Hz, 1H, H-23), 2.99 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.44 (d, J = 10.8 Hz, 1H, H-16), 2.36 (s, 1H, H-9), 2.24–2.16 (m, 1H, H-20), 1.79–1.12 (m, 15H), 1.29 (s, 3H, CH3-27), 0.97 (d, J = 6.4 Hz, 3H, CH3-29), 0.96 (s, 3H, CH3-24), 0.87 (d, J = 6.4 Hz, 3H, CH3-30), 0.63 (s, 3H, CH3-25), 0.40 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.71, 179.02, 166.34, 147.21, 139.32, 132.85, 132.60, 131.23, 124.32, 124.18, 123.30, 120.00, 77.58, 69.28, 65.78, 62.35, 54.02, 48.32, 48.14, 47.63, 45.79, 45.11, 44.16, 39.98, 39.85, 38.93, 38.07, 35.46, 33.33, 31.53, 29.13, 24.77, 21.36, 21.33, 19.61, 18.34, 18.00, 17.63, 13.94. HR-MS (m/z) (ESI): calc for C39H53BrN4O5 [M + Na]+: 759.3092, found: 759.3080.
Compound 6g
Yield 77%, as a white solid. Mp: 213.5–215.5 °C. 1H NMR (400 MHz, CD3OD) δ 7.99 (s, 1H, H-33), 7.49–7.43 (m, 2H, Ar–H), 7.39–7.35 (m, 1H, Ar–H), 7.34–7.32 (m, 1H, Ar–H), 5.58 (s, 1H, H-12), 4.52–4.39 (m, 2H, H-31), 3.76–3.69 (m, 1H, H-2), 3.51 (d, J = 11.2 Hz, 1H, H-3), 3.35 (d, J = 9.6 Hz, 1H, H-23), 3.24 (d, J = 10.8 Hz, 1H, H-23), 3.02 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.47 (d, J = 10.8 Hz, 1H, H-16), 2.43 (s, 1H, H-9), 2.28–2.20 (m, 1H, H-20), 2.19 (s, 3H, Ar–CH3), 1.83–1.22 (m, 15H), 1.35 (s, 3H, CH3-27), 1.11 (s, 3H, CH3-24), 0.99 (d, J = 6.4 Hz, 3H, CH3-29), 0.89 (d, J = 6.4 Hz, 3H, CH3-30), 0.69 (s, 3H, CH3-25), 0.67 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.84, 179.17, 166.41, 146.27, 137.68, 134.75, 132.68, 131.30, 131.17, 128.03, 126.93, 126.29, 77.63, 69.32, 65.81, 62.51, 54.01, 48.45, 48.20, 47.70, 45.99, 45.23, 44.21, 40.02, 39.90, 39.01, 38.10, 35.67, 33.44, 31.52, 29.23, 24.73, 21.42, 21.31, 20.01, 18.33, 18.10, 17.99, 17.62, 13.89. HR-MS (m/z) (ESI): calc for C40H56N4O5 [M + Na]+: 695.4143, found: 695.4135.
Compound 6h
Yield 72%, as a white solid. Mp: 214.4–217.1 °C. 1H NMR (400 MHz, CD3OD) δ 8.19 (s, 1H, H-33), 7.67–7.64 (m, 1H, Ar–H), 7.51–7.47 (m, 1H, Ar–H), 7.28–7.25 (m, 1H, Ar–H), 7.13–7.09 (m, 1H, Ar–H), 5.57 (s, 1H, H-12), 4.51–4.35 (m, 2H, H-31), 3.91 (s, 3H, Ar–OCH3), 3.74–3.67 (m, 1H, H-2), 3.50 (d, J = 11.2 Hz, 1H, H-3), 3.33 (d, J = 9.6 Hz, 1H, H-23), 3.23 (d, J = 10.8 Hz, 1H, H-23), 3.00 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.46 (d, J = 10.8 Hz, 1H, H-16), 2.39 (s, 1H, H-9), 2.26–2.18 (m, 1H, H-20), 1.81–1.18 (m, 15H), 1.32 (s, 3H, CH3-27), 1.02 (s, 3H, CH3-24), 0.98 (d, J = 6.4 Hz, 3H, CH3-29), 0.88 (d, J = 6.4 Hz, 3H, CH3-30), 0.65 (s, 3H, CH3-25), 0.53 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.79, 179.09, 166.34, 152.78, 145.83, 131.72, 131.30, 127.23, 126.85, 126.21, 122.04, 113.87, 77.64, 69.31, 65.83, 62.45, 56.66, 53.99, 48.39, 48.18, 47.70, 45.85, 45.19, 44.18, 40.01, 39.87, 38.98, 38.12, 35.61, 33.39, 31.53, 29.18, 24.73, 21.38, 21.31, 19.70, 18.27, 18.06, 17.60, 13.86. HR-MS (m/z) (ESI): calc for C40H56ClN4O6 [M + Na]+: 711.4092, found: 711.4079.
Compound 6i
Yield 74%, as a white solid. Mp: 201.5–202.9 °C. 1H NMR (400 MHz, CD3OD) δ 8.31 (s, 1H, H-33), 7.84–7.81 (m, 2H, Ar–H), 7.60–7.56 (m, 2H, Ar–H), 5.56 (s, 1H, H-12), 4.48–4.37 (m, 2H, H-31), 3.73–3.67 (m, 1H, H-2), 3.49 (d, J = 11.2 Hz, 1H, H-3), 3.32 (d, J = 9.6 Hz, 1H, H-23), 3.26 (d, J = 10.8 Hz, 1H, H-23), 2.99 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.45 (d, J = 10.8 Hz, 1H, H-16), 2.37 (s, 1H, H-9), 2.25–2.17 (m, 1H, H-20), 1.79–1.14 (m, 15H), 1.31 (s, 3H, CH3-27), 0.98 (d, J = 6.4 Hz, 3H, CH3-29), 0.95 (s, 3H, CH3-24), 0.88 (d, J = 6.4 Hz, 3H, CH3-30), 0.63 (s, 3H, CH3-25), 0.45 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.80, 179.08, 166.42, 147.22, 136.98, 135.53, 131.27, 131.03, 123.12, 122.76, 77.58, 69.30, 65.77, 62.39, 54.07, 48.17, 47.65, 45.83, 45.14, 44.17, 40.02, 39.88, 38.95, 38.09, 35.51, 33.34, 31.52, 29.16, 24.74, 21.38, 21.30, 19.66, 18.21, 18.00, 17.61, 13.84. HR-MS (m/z) (ESI): calc for C39H53ClN4O5 [M + Na]+: 715.3597, found: 715.3602.
Compound 6j
Yield 79%, as a white solid. Mp: 203.7–205.6 °C. 1H NMR (400 MHz, CD3OD) δ 8.10 (s, 1H, H-33), 7.86–7.84 (m, 1H, Ar–H), 7.59–7.55 (m, 1H, Ar–H), 7.53–7.48 (m, 2H, Ar–H), 5.58 (s, 1H, H-12), 4.53–4.41 (m, 2H, H-31), 3.77–3.70 (m, 1H, H-2), 3.51 (d, J = 11.2 Hz, 1H, H-3), 3.45 (d, J = 9.6 Hz, 1H, H-23), 3.24 (d, J = 10.8 Hz, 1H, H-23), 3.02 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.46 (d, J = 10.8 Hz, 1H, H-16), 2.43 (s, 1H, H-9), 2.26–2.19 (m, 1H, H-20), 1.82–1.22 (m, 15H), 1.34 (s, 3H, CH3-27), 1.13 (s, 3H, CH3-24), 0.98 (d, J = 6.4 Hz, 3H, CH3-29), 0.88 (d, J = 6.4 Hz, 3H, CH3-30), 0.72 (s, 3H, CH3-25), 0.68 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.78, 179.09, 166.34, 146.22, 137.70, 135.16, 132.85, 131.26, 129.85, 129.36, 126.73, 119.74, 77.60, 69.28, 65.80, 62.47, 53.94, 48.40, 48.16, 47.67, 45.97, 45.20, 44.19, 39.98, 39.86, 38.98, 38.07, 35.64, 33.42, 31.50, 29.22, 24.70, 21.45, 21.34, 20.04, 18.40, 18.12, 17.64, 13.93. HR-MS (m/z) (ESI): calc for C39H53BrN4O5 [M + Na]+: 759.3092, found: 759.3081.
Compound 6k
Yield 73%, as a white solid. Mp: 212.4–214.6 °C. 1H NMR (400 MHz, CD3OD) δ 8.20 (s, 1H, H-33), 7.85–7.81 (m, 1H, Ar–H), 7.59–7.53 (m, 1H, Ar–H), 7.45–7.38 (m, 2H, Ar–H), 5.57 (s, 1H, H-12), 4.52–4.38 (m, 2H, H-31), 3.74–3.67 (m, 1H, H-2), 3.50 (d, J = 11.2 Hz, 1H, H-3), 3.33 (d, J = 9.6 Hz, 1H, H-23), 3.23 (d, J = 10.8 Hz, 1H, H-23), 3.00 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.46 (d, J = 10.8 Hz, 1H, H-16), 2.40 (s, 1H, H-9), 2.26–2.20 (m, 1H, H-20), 1.81–1.20 (m, 15H), 1.32 (s, 3H, CH3-27), 1.03 (s, 3H, CH3-24), 0.99 (d, J = 6.4 Hz, 3H, CH3-29), 0.89 (d, J = 6.4 Hz, 3H, CH3-30), 0.65 (s, 3H, CH3-25), 0.55 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.81, 179.14, 166.37, 155.25 (1J(C,F) = 249.2 Hz), 146.77, 132.13 (2J(C,F) = 7.9 Hz), 131.30, 126.50 (3J(C,F) = 3.8 Hz), 126.41, 126.30, 126.05 (3J(C,F) = 6.5 Hz), 118.25 (2J(C,F) = 19.9 Hz), 77.62, 69.31, 65.80, 62.46, 54.02, 48.40, 48.18, 47.68, 45.86, 45.19, 44.19, 40.02, 39.88, 38.97, 38.10, 35.53, 33.38, 31.52, 29.19, 24.73, 21.39, 21.31, 19.62, 18.23, 18.04, 17.61, 13.86. HR-MS (m/z) (ESI): calc for C39H53FN4O5 [M + H]+: 677.4073, found: 677.4078.
Compound 6l
Yield 76%, as a white solid. Mp: 218.3–219.1 °C. 1H NMR (400 MHz, CD3OD) δ 8.16 (s, 1H, H-33), 8.15–8.12 (m, 1H, Ar–H), 7.91–7.87 (m, 1H, Ar–H), 7.82–7.78 (m, 1H, Ar–H), 7.71–7.69 (m, 1H, Ar–H), 5.60 (s, 1H, H-12), 4.53–4.40 (m, 2H, H-31), 3.75–3.69 (m, 1H, H-2), 3.51 (d, J = 11.2 Hz, 1H, H-3), 3.35 (d, J = 9.6 Hz, 1H, H-23), 3.24 (d, J = 10.8 Hz, 1H, H-23), 3.02 (dd, J = 12.8, 4.4 Hz, 1H, H-19), 2.47 (d, J = 10.8 Hz, 1H, H-16), 2.44 (s, 1H, H-9), 2.28–2.20 (m, 1H, H-20), 1.82–1.21 (m, 15H), 1.35 (s, 3H, CH3-27), 1.11 (s, 3H, CH3-24), 0.99 (d, J = 6.4 Hz, 3H, CH3-29), 0.90 (d, J = 6.4 Hz, 3H, CH3-30), 0.71 (s, 3H, CH3-25), 0.66 (s, 3H, CH3-26). 13C NMR (100 MHz, CD3OD) δ 201.96, 179.20, 166.50, 146.99, 145.94, 135.11, 132.25, 131.30, 130.96, 128.53, 126.69, 125.85, 77.67, 69.33, 65.84, 62.54, 54.04, 48.45, 48.20, 47.73, 46.02, 45.25, 44.20, 40.03, 39.90, 39.01, 38.03, 35.55, 33.39, 31.52, 29.24, 24.73, 21.43, 21.31, 19.89, 18.32, 18.08, 17.62, 13.88. HR-MS (m/z) (ESI): calc for C39H53ClN5O7 [M + Na]+: 726.3837, found: 726.3826.
In vitro inhibition of TNF-a-induced NF-κB activation in A549 lung cells
The A549 cells were cultured in 12-well plates and transiently co-transfected with 0.2 mg of a pNF-κB-Luc vector (Stratagene, La Jolla, CA) and 0.2 mg of pSV-b-galactosidase dissolved in 3 ml lipofectamine or Lipofectamine 2000 (Invitrogen, Carlsbad, CA) as the internal control. The plasmids were transfected according to the manufacturer's instructions. After 6 h, the medium was changed and cells were cultured for 6 h. Cells were then treated with TNF-α (15 ng mL–1) and the test compounds simultaneously for 7 h. The A549 cells treated with TNF-α alone served as positive controls, while cells without TNF-α treatment served as negative controls. Luciferase activities from these cells were then measured by using the Bright-Glo Luciferase Assay kit from Promega (Madison, WI), following the manufacturer's protocol. The relative NF-κB activities of the cells treated with the test compounds were obtained as the ratio of their luciferase activity to that from the positive controls, both of which have been corrected with background (signals from negative controls) and cell viability. Under these experimental conditions, none of the test compounds induced significant toxicity to A549 cells (<5% reduction of cell viability). The IC50 of each fraction was determined by fitting the relative NF-κB activity to the drug concentration by using a sigmoidal dose response model of varied slope in GraphPad Prism 6. The IC50 reported herein is the average of at least three replicates.
SPR analysis
Surface plasmon resonance experiments were performed using a Biacore T200 instrument at 25 °C. Sensor chips, buffer stock solutions, and immobilization reagents were from GE Healthcare. Recombinant human NF-κB was Novoprotein. Other reagents were obtained from Sigma. Immobilization: PBS was used as the running buffer. The four flow cells were treated in the same way to optimize the throughput. In summary, using a CM5 chip, spots 1 and 2 were activated with the coupling reagents EDC and NHS for 10 min. NF-κB at a concentration of 20 μg mL–1 in 10 mM sodium acetate (pH 5) was injected onto the surface for 10 and 5 min in spots 1 and 2, respectively, to generate surfaces with high and low density. The unmodified spot 3 was used as a reference. Kinetics and affinity measurements: PBS (containing 0.05% DMSO) was used as the running buffer and sample dilution buffer. Dose–responses were obtained using a 2-fold sample dilution from 0 to 500 μM, using an injection time of 60 s. Data processing: binding curves were corrected for variations in DMSO concentration and normalized by molecular weight. KDs reported are derived from steady-state binding responses and therefore correspond to the equilibrium binding affinity of the compounds.
NF-κB DNA binding assay
To determine NF-κB activation, we performed a DNA binding assay using the TransAM NF-κB Kit according to the manufacturer's instructions and as previously described.38 Briefly, 20 μg of nuclear proteins was added into 96-well plates coated with an unlabeled oligonucleotide containing the consensus binding site for NF-κB (5′-GGGACTTTCC-3′) and incubated for 1 h. The wells were washed and incubated with antibodies against NF-κB. An HRP conjugated secondary antibody was then applied to detect the bound primary antibody and provided the basis for colorimetric quantification. The enzymatic product was measured at 450 nm using a microplate reader (TECAN Systems).
Molecular docking
To study the binding mode of the inhibitors in the active site of NF-κB protein, molecular docking was performed using the Surflex-Dock module in SYBYL-X 2.0. The crystal structure of the NF-κB complex was retrieved from the RCSB Protein Data Bank (PDB entry code: 1NFK, ; 1IKN).39,40 Ligands were docked in the corresponding protein's binding site by an empirical scoring function and a patented search engine in Surflex-Dock. Before the docking process, the natural ligand was extracted; the water molecules were removed from the crystal structure. Subsequently, the protein was prepared by using the Biopolymer module implemented in Sybyl. The polar hydrogen atoms were added. The automated docking manner was applied in the present work. Other parameters were established by default in the software. Surflex-Dock total scores, which were expressed in –log10(Kd) units to represent binding affinities, were applied to estimate the ligand–receptor interactions of newly designed molecules.
Immunofluorescence staining
Studies were performed as previously described.41 Briefly, A549 cells were cultured in six-well plates (2 × 105 cells per well) for 24 h and then pretreated with compound 6k at 10 μM for 24 h, followed by TNF-α for another 30 minutes. The cells were then washed and fixed with anhydrous methanol for 15 min at room temperature, permeabilized with 0.1% Triton X-100 for 15 min, and blocked with 1% BSA for 10 min at room temperature. Next, the cells were incubated with primary NF-κB antibody in PBS overnight at 4 °C, followed by FITC-conjugated secondary antibodies. After being washed with PBS three times, the cells were incubated in 10 μM DAPI for 15 min in the dark. The fluorescence of the NF-κB protein and nucleus was green and blue, respectively, and live cell imaging was simultaneously accomplished using a fluorescence microscope (Olympus BX-51, Tokyo, Japan).
Western blot assay
Total cell lysates from cultured A549 cells after compound 6k treatments as mentioned earlier were obtained by lysing the cells in ice-cold RIPA buffer with protease and phosphatase inhibitor and stored at –20 °C for future use. The protein concentrations were quantified by the Bradford method (BIO-RAD) using a multimode Varioskan instrument (Thermo Fisher Scientific). Equal amounts of protein per lane were applied in 12% SDS polyacrylamide gel for electrophoresis and transferred to the polyvinylidene difluoride (PVDF) membrane (Amersham Biosciences). After the membrane was blocked at room temperature for 2 h in blocking solution, the primary antibody was added and incubated at 4 °C overnight. IκBα, IKKβ and NF-κB antibodies were purchased from Imgenex, USA. After three TBST washes, the membrane was incubated with the corresponding horseradish peroxidase-labeled secondary antibody (1 : 2000) (Santa Cruz) at room temperature for 1 h. Membranes were washed with TBST three times for 15 min and the protein blots were detected with the chemiluminescence reagent (Thermo Fisher Scientific Ltd.). The X-ray films were developed with a developer and fixed with fixer solution.
Cytotoxicity assay
The human cell lines HepG2, NCI-H460, A549, T24, A549, A549/DOX and HL-7702 were obtained from the Shanghai Cell Bank in the Chinese Academy of Sciences. HepG2, NCI-H460, A549, T24, A549, A549/DOX and HL-7702 cell lines were grown on 96-well microtitre plates at a cell density of 10 × 105 cells per well in DMEM medium with 10% FBS. DMEM and FBS were obtained from Gibco-Thermo (BRL Co. Ltd., USA). The plates were incubated at 37 °C in a humidified atmosphere of 5% CO2/95% air for overnight. Therewith, the cells were exposed to different concentrations of the target compounds and DOX, and incubated for another 48 h. The cells were stained with 10 μl of MTT in an incubator for about 4 h. The medium was thrown away and replaced by 100 mL DMSO. The O.D. value was read at 570/630 nm using an enzyme labeling instrument.
Apoptosis analysis
A549 cells were seeded at a density of 2 × 106 cells per mL of the DMEM medium with 10% FBS on 6-well plates to a final volume of 2 mL. The plates were incubated overnight and then treated with different concentrations of compound 6k for 24 h. Briefly, after treatment with compound 6k for 24 h, cells were collected and washed with PBS twice, and then re-suspend cells in 1× binding buffer (0.1 M Hepes/NaOH (pH 7.4), 1.4 M NaCl, and 25 mM CaCl2) at a concentration of 1 × 106 cells per mL. The cells were subjected to 5 μL of FITC Annexin V and 5 μL propidium iodide (PI) staining using the Annexin-V FITC apoptosis kit (BD, Pharmingen) and then 100 μL of the solution was transferred to a 5 mL culture tube and incubated for 30 min at RT (25 °C) in the dark. The apoptosis ratio was quantified by system software (CellQuest; BD Biosciences).
AO/EB staining
The AO/EB molecular probes were also used to detect apoptotic cells. A549 cells (1 × 106 cells) were seeded in six-well tissue culture plates. Following incubation, the medium was removed and replaced with fresh medium plus 10% fetal bovine serum and treated with 5 μM, 10 μM and 20 μM compound 6k for 24 h. After the treatment period, briefly, the cells were harvested, suspended in PBS, and stained with 2 μL of AO/EB stain (100 mg mL–1) at room temperature for 20 minutes. Fluorescence was read on a Nikon ECLIPSETE2000-S fluorescence microscope (OLYMPUS Co., Japan).
Transwell migration assay
A transwell migration assay was performed to investigate the impact of compound 6k on cell migration by a modified Boyden's chamber method in a 24-well cell culture plate with an 8 mm pore. Chambers were washed three times with ice-PBS. Then the medium with the tested compound was placed in the lower chamber, and then the A549 cells were grown in the top chamber. Cells were treated with or without the tested compound 6k at the indicated concentrations at 37 °C in a humidified atmosphere of 5% CO2 for 24 h. After incubation for 24 h, the medium was removed and then the cells in the membrane were washed with ice-PBS three times. The migrated cells were fixed with 4% paraformaldehyde for 30 min and stained with 0.2% crystal violet. Images were observed by fluorescence microscopy.
Statistics
The data were processed by Student' s t-test with the significance level P ≤ 0.05 using SPSS.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Supplementary Material
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 21431001 and 81760626), the Qinghai key R & D and transformation project (Qinghai Science and Technology Department) (2017-NK-C25), the Project of State Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, Ministry of Science and Technology of China (No. CMEMR2016-B06), A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (No. 1107047002), The Open Project of Qinghai Key Laboratory of Qinghai-Tibet Plateau Biological Resources (2017-ZJ-Y10), the Ministry of Education Innovation Team Fund (IRT_16R15, 2016GXNSFGA380005), the Fundamental Research Funds for the Central Universities and the Innovation Program for Graduate Students in Jiangsu Province (KYCX17_0133), and the Scientific Research Foundation of Graduate School of Southeast University (YBJJ1786).
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00620b
References
- Xiao W. Cell. Mol. Immunol. 2004;1:425–435. [PubMed] [Google Scholar]
- Li Q., Verma I. M. Nat. Rev. Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Lenardo M. J., Baltimore D. Cell. 2017;168:37–57. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikarsky E., Porat R. M., Stein I., Abramovitch R., Amit S., Kasem S., Gutkovich-Pyest E., Urieli-Shoval S., Galun E., Ben-Neriah Y. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Karin M. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Liu B., Sun L., Liu Q., Gong C., Yao Y., Lv X., Lin L., Yao H., Su F., Li D., Zeng M., Song E. Cancer Cell. 2015;27:370–381. doi: 10.1016/j.ccell.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Shen H. M., Tergaonkar V. Apoptosis. 2009;14:348–363. doi: 10.1007/s10495-009-0315-0. [DOI] [PubMed] [Google Scholar]
- Li F., Sethi G. Biochim. Biophys. Acta. 2010;1805:167–180. doi: 10.1016/j.bbcan.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Wu D. W., Lee M. C., Hsu N. Y., Wu T. C., Wu J. Y., Wang Y. C., Cheng Y. W., Chen C. Y., Lee H. Oncogene. 2015;34:2505–2515. doi: 10.1038/onc.2014.184. [DOI] [PubMed] [Google Scholar]
- Baldwin S. J. Clin. Invest. 2001;107:241–246. doi: 10.1172/JCI11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Zhou Y., Li Y., Zhou J., Wu Y., Cui Y., Yang G., Hong Y. Cancer Lett. 2015;357:520–526. doi: 10.1016/j.canlet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Huang R. Z., Hua S. X., Liao Z. X., Huang X. C., Wang H. S. Med. Chem. Commun. 2017;8:1421–1434. doi: 10.1039/c7md00105c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W., Huang R. Z., Zhang J., Guo T., Zhang M. T., Huang X. C., Zhang B., Liao Z. X., Sun J., Wang H. S. Bioorg. Chem. 2018;79:265–276. doi: 10.1016/j.bioorg.2018.05.005. [DOI] [PubMed] [Google Scholar]
- Kong Y., Li F., Nian Y., Zhou Z., Yang R., Qiu M. H., Chen C. Theranostics. 2016;6:875–886. doi: 10.7150/thno.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthar S. K., Boon H. L., Sharma M. Eur. J. Med. Chem. 2014;74:135–144. doi: 10.1016/j.ejmech.2013.12.052. [DOI] [PubMed] [Google Scholar]
- Ma K., Zhang Y., Zhu D., Lou Y. Eur. J. Pharmacol. 2009;603:98–107. doi: 10.1016/j.ejphar.2008.11.054. [DOI] [PubMed] [Google Scholar]
- Hao C., Wu B., Hou Z., Xie Q., Liao T., Wang T., Ma D. Int. Immunopharmacol. 2017;50:313–318. doi: 10.1016/j.intimp.2017.07.005. [DOI] [PubMed] [Google Scholar]
- Ramachandran V., Saravanan R., Senthilraja P. Phytomedicine. 2014;21:225–232. doi: 10.1016/j.phymed.2013.08.027. [DOI] [PubMed] [Google Scholar]
- Gonçalves B. M. F., Salvador J. A. R., Marín S., Cascante M. Eur. J. Med. Chem. 2016;114:101–117. doi: 10.1016/j.ejmech.2016.02.057. [DOI] [PubMed] [Google Scholar]
- Qian Y., Xin Z., Lv Y., Wang Z., Zuo L., Huang X., Li Y., Xin H. B. Food Funct. 2018;9:1048–1057. doi: 10.1039/c7fo01442b. [DOI] [PubMed] [Google Scholar]
- Lee Y. S., Jin D. Q., Kwon E. J., Park S. H., Lee E. S., Jeong T. C., Nam D. H., Huh K., Kim J. A. Cancer Lett. 2002;186:83–91. doi: 10.1016/s0304-3835(02)00260-4. [DOI] [PubMed] [Google Scholar]
- Li J. F., Huang R. Z., Yao G. Y., Ye M. Y., Wang H. S., Pan Y. M., Xiao J. T. Eur. J. Med. Chem. 2014;86:175–188. doi: 10.1016/j.ejmech.2014.08.003. [DOI] [PubMed] [Google Scholar]
- Huang R. Z., Wang C. Y., Li J. F., Yao G. Y., Pan Y. M., Ye M. Y., Wang H. S., Zhang Y. RSC Adv. 2016;6:62890–62906. [Google Scholar]
- Siewert B., Wiemann J., Köwitsch A., Csuk R. Eur. J. Med. Chem. 2014;72:84–101. doi: 10.1016/j.ejmech.2013.11.025. [DOI] [PubMed] [Google Scholar]
- Mungalpara D., Stegmüller S., Kubik S. Chem. Commun. 2017;53:5095–5098. doi: 10.1039/c7cc02424j. [DOI] [PubMed] [Google Scholar]
- Chandrashekhar M., Nayak V. L., Ramakrishna S., Mallavadhani U. V. Eur. J. Med. Chem. 2016;114:293–307. doi: 10.1016/j.ejmech.2016.03.013. [DOI] [PubMed] [Google Scholar]
- Boechat N., Ferreira V. F., Ferreira S. B., Ferreira M. L. G., Silva F. C., Bastos M. M., Costa M. S., Lourenc-o M. C. S., Pinto A. C., Krettli A. U., Aguiar A. C., Teixeira B. M., Silva N. V., Martins P. R. C., Bezerra F. A. F. M., Camilo A. L. S., Silva G. P., Costa C. C. P. J. Med. Chem. 2011;54:5988–5999. doi: 10.1021/jm2003624. [DOI] [PubMed] [Google Scholar]
- Garudachari B., Isloor A. M., Satyanarayana M. N., Fun H. K., Hegde G. Eur. J. Med. Chem. 2014;74:324–332. doi: 10.1016/j.ejmech.2014.01.008. [DOI] [PubMed] [Google Scholar]
- Sheng C., Zhang W. Curr. Med. Chem. 2011;18:733–766. doi: 10.2174/092986711794480113. [DOI] [PubMed] [Google Scholar]
- Rashid S., Dar B. A., Majeed R., Hamid A., Bhat B. A. Eur. J. Med. Chem. 2013;66:238–245. doi: 10.1016/j.ejmech.2013.05.029. [DOI] [PubMed] [Google Scholar]
- Khan S. K., Guru S. K. Rath, Chinthakindi P. K., Singh B., Koul S., Bhushan S., Sangwan P. L. Eur. J. Med. Chem. 2016;108:104–116. doi: 10.1016/j.ejmech.2015.11.018. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Hernandez D., Demuner A. J., Barbosa L. C. A., Heller L., Csuk R. Eur. J. Med. Chem. 2016;115:257–267. doi: 10.1016/j.ejmech.2016.03.018. [DOI] [PubMed] [Google Scholar]
- Marzaro G., Guiotto A., Borgatti M., Finotti A., Gambari R., Breveglieri G., Chilin A. J. Med. Chem. 2013;56:1830–1842. doi: 10.1021/jm3009647. [DOI] [PubMed] [Google Scholar]
- Cheng Q. L., Li H. L., Huang Z. Q., Chen Y. J., Liu T. S. Chem.-Biol. Interact. 2015;240:1–11. doi: 10.1016/j.cbi.2015.06.038. [DOI] [PubMed] [Google Scholar]
- Rana S., Blowers E. C., Tebbe C., Contreras J. I., Radhakrishnan P., Kizhake S., Zhou T., Rajule R. N., Arnst J. L., Munkarah A. R., Rattan R., Natarajan A. J. Med. Chem. 2016;59:5121–5127. doi: 10.1021/acs.jmedchem.6b00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan A. E., Colleran A., O'Gorman A., O'Flynn L., Pindjacova J., Lohan P., O'Malley G., Nosov M., Mureau C., Egan L. J. Oncogene. 2015;34:1563–1574. doi: 10.1038/onc.2014.86. [DOI] [PubMed] [Google Scholar]
- Reddy T. S., Kulhari H., Reddy V. G., Bansal V., Kamal A., Shukla R. Eur. J. Med. Chem. 2015;101:790–805. doi: 10.1016/j.ejmech.2015.07.031. [DOI] [PubMed] [Google Scholar]
- Shanmugam M. K., Rajendran P., Li F., Nema T., Vali S., Abbasi T., Kapoor S., Sharma A., Kumar A. P., Ho P. C., Hui K. M., Sethi G. J. Mol. Med. 2011;89:713–727. doi: 10.1007/s00109-011-0746-2. [DOI] [PubMed] [Google Scholar]
- Ghosh G., Duyne G., Ghosh S., Sigler P. B. Nature. 1995;373:303–310. doi: 10.1038/373303a0. [DOI] [PubMed] [Google Scholar]
- Huxford T., Huang D. B., Malek S., Ghosh G. Cell. 1998;95:759–770. doi: 10.1016/s0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- Sankpal U. T., Nagaraju G. P., Gottipolu S. R., Hurtado M., Jordan C. G., Simecka J. W., Shoji M., El-Rayes B., Basha R. Oncotarget. 2015;7:3186–3200. doi: 10.18632/oncotarget.6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.