

SUMMARY
Combinatorial interactions among transcription factors (TFs) play essential roles in generating gene expression specificity and diversity in metazoans. Using yeast 2-hybrid (Y2H) assays on nearly all sequence-specific Drosophila TFs, we identified 1,983 protein-protein interactions (PPIs), more than doubling the number of currently known PPIs among Drosophila TFs. For quality assessment, we validated a subset of our interactions using MITOMI and bimolecular fluorescence complementation assays. We combined our interactome with prior PPI data to generate an integrated Drosophila TF-TF binary interaction network. Our analysis of ChIP-seq data, integrating PPI and gene expression information, uncovered different modes by which interacting TFs are recruited to DNA. We further demonstrate the utility of our Drosophila interactome in shedding light on human TF-TF interactions. This study reveals how TFs interact to bind regulatory elements in vivo and serves as a resource of Drosophila TF-TF binary PPIs for understanding tissue-specific gene regulation.
Graphical Abstract
In Brief
Combinatorial regulation by transcription factors (TFs) is one mechanism for achieving condition and tissue-specific gene regulation. Shokri et al. mapped TF-TF interactions between most Drosophila TFs, reporting a comprehensive TF-TF network integrated with previously known interactions. They used this network to discern distinct TF-DNA binding modes.
INTRODUCTION
Transcription factors (TFs) are essential for the regulation of gene expression during development and in response to environmental perturbations. One mechanism for achieving gene expression specificity and diversity in metazoans is the binding of multiple TFs at cis-regulatory elements. Such combinatorial interactions provide a means to integrate information about cell identity, cell state, and extracellular signals into condition-specific transcriptional responses and are essential in specifying tissue-specific programs during development (Carroll et al., 2005; Levine and Tjian, 2003; Michelson and Bulyk, 2006; Spitz and Furlong, 2012). Mapping the network of binary protein-protein interactions (PPIs) among TFs is therefore critical for understanding the regulatory interactions through which the specificity of gene expression programs is determined.
Several genome-scale PPI studies have focused on TFs. For humans and mice, a mammalian 2-hybrid approach was used to screen approximately half of an established catalog of human and mouse TFs, identifying 762 and 877 high-stringency interactions, respectively (Ravasi et al., 2010). For Caenorhabditis elegans, a yeast 2-hybrid (Y2H) approach assayed PPIs among 834 (89%) TFs and detected 2,253 high-confidence interactions among 437 TFs (Reece-Hoyes et al., 2013). A study using affinity chromatography followed by mass spectrometry (AP-MS) assayed 647 Drosophila melanogaster proteins, 229 of which were sequence-specific TFs, in the S2R+ cell line; 624 high-confidence interactions were identified, with 9.5% of interactions constituting binary TF-TF interactions (Rhee et al., 2014). Other studies assayed only small subsets of TFs (Grigoryan et al., 2009; Grove et al., 2009), detected relatively few TF-TF interactions as part of a larger PPI screen (Formstecher et al., 2005; Giot et al., 2003; Stanyon et al., 2004), or inferred TF PPIs from co-expression data and require independent experimental support (Adryan and Teichmann, 2010). In total, prior studies have identified 1,161 interactions among 468 Drosophila TFs (Friedman et al., 2011) (Table 1). Considering that there are 755 predicted Drosophila TFs (Hens et al., 2011), a large fraction of the Drosophila binary TF interactome (“TF-TF interactome”) remains to be mapped.
Table 1.
Experimentally Identified Drosophila TF-TF PPIs
| Study | Method | Total no. PPIs Detected | No. of TF-TF Interactionsa |
|---|---|---|---|
| Giot et al., 2003 | Y2H | 20,405 | 341 |
| Stanyon et al., 2004 | Y2H | 1,814 | 19 |
| Formstecher et al., 2005 | Y2H | 2,338 | 53 |
| Guruharsha et al., 2011 | AP-MS | 209,912 | 221 |
| Lowe et al., 2014 | AP-MS | 14,932 | 21 |
| Rhee et al., 2014 | AP-MS | 174,561 | 23 |
| MasterNet (PPIs detected for Drosophila proteins) | compilation | – | 1,161 |
| MasterNet (including PPIs inferred from Interologs) | compilation | – | 4,606 |
| This study | Y2H | – | 1,983 |
Numbers retrieved from MasterNet.
D. melanogaster serves as a powerful metazoan model organism for studies of transcriptional regulation because of its complexity in spatiotemporal gene regulation and abundance of available genetic and genomic resources (Beckingham et al., 2005). Gene expression profiling has been performed across a wide range of developmental time points (Chintapalli et al., 2007; Graveley et al., 2011; Hammonds et al., 2013), and genomic occupancies of numerous TFs have been profiled by chromatin immunoprecipitation (ChIP) using DNA microarrays or sequencing (Iyer et al., 2001; Johnson et al., 2007; Lieb et al., 2001; Ren et al., 2000; Wei et al., 2006). The highly evolutionarily conserved nature of many Drosophila protein sequences and their regulatory roles (Shubin et al., 2009) makes Drosophila a compelling system in which a comprehensive TF-TF interactome would broadly facilitate studies of tissue-specific transcriptional regulation.
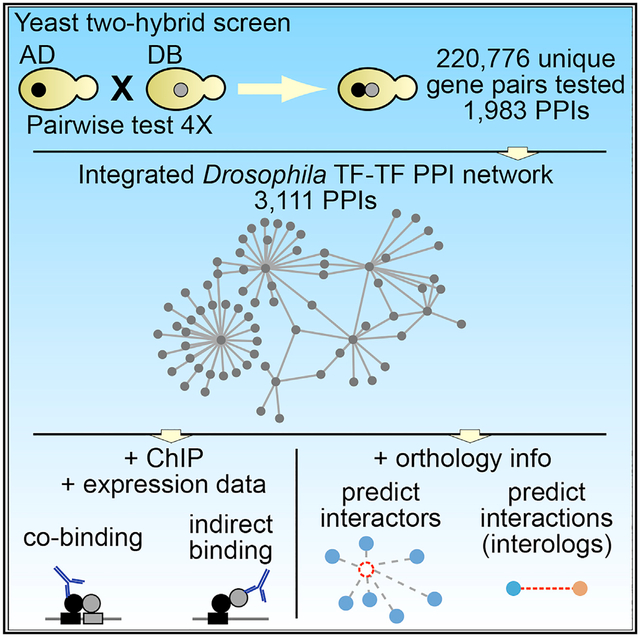
Here, we performed a high-throughput Y2H screen to map binary interactions between essentially all full-length Drosophila TFs. Y2H provides a more condition-independent survey of PPIs in a nuclear environment than does AP-MS and is not biased toward high-affinity, stable interactions. Furthermore, our approach individually tested binary TF pairs, in contrast to pooled approaches used by other Y2H screens, and offers the advantage of being able to distinguish between negative (i.e., no interaction) and untested interactions. Our Y2H screen resulted in 1,983 unique TF-TF interactions involving 584 TFs, representing a 168% increase in the number of known PPIs among Drosophila TFs. We found that the motifs of interacting TFs co-occur within ChIP followed by microarray hybridization and high-throughput sequencing (ChIP-chip/seq) peaks. We further integrated available ChIP-chip/seq and gene expression data to dissect the DNA recruitment mechanisms of interacting TF pairs and built a catalog of direct and indirect TF-DNA interactions in Drosophila development. Finally, we demonstrate how this Drosophila TF-TF interactome can be used to elucidate gene regulatory networks in humans.
RESULTS
A Binary Interaction Screen among Nearly All Drosophila TFs
We compiled a list of 755 predicted sequence-specific TFs in D. melanogaster using information from a prior cataloging of sequence-specific TFs for the FlyTF database (Adryan and Teichmann, 2006), as well as manual curation (Hens et al., 2011). For 720 (95%) of these TFs, Gateway-compatible Entry clones were available (Hens et al., 2011), and we generated Y2H-compatible AD (“prey”) and DB (“bait”) expression vectors, resulting in 695 and 575 successful clones, respectively (Table S1). We tested and analyzed a total of 385,431 AD-DB pairs in quadruplicate using our high-throughput Y2H assay (Walhout and Vidal, 2001) (see STAR Methods) (Figure 1A). These interactions corresponded to 220,776 unique TF pairs (Table S1). We detected 1,983 unique interactions between 584 TFs (Figure 1B; Table S2), including 26 putative homodimeric interactions. Of these interactions, 1,950 (98%) had not been previously identified experimentally.
Figure 1. Comprehensive Drosophila Y2H TF-TF Interactome.

(A) Overview of the Y2H screen.
(B) Network view of the Y2H TF-TF interactome.
(C) Degree distribution of the Y2H interactome. Median degree, 4; mean degree, 6.79. The red vertical line indicates the threshold for calling hub genes (degree, 50).
(D) Comparisons of identified interactions with those in MasterNet.
The frequency of interactions we detect here (1,983/220,776 =0.898%) is much higher than that obtained in other Y2H screens in flies (~0.004%, Giot et al., 2003), C. elegans (~0.002%, Simo-nis et al., 2009), or humans (~0.002%, Rual et al., 2005;~0.01%, Stelzl et al., 2005; or 0.15%, Rolland et al., 2014). Since those prior screens were performed on a wide range of genes not limited to TFs, our higher PPI rate may reflect a greater tendency of TFs to interact with one another, stemming from their often-combinatorial control of gene regulation. Among the 584 TFs represented in our interactome, the median number of interactions per TF was 4, and the network density was 0.011. These figures are comparable to those of TF-TF PPIs in other species (Ravasi et al., 2010; Reece-Hoyes et al., 2013). This network follows a scale-free degree distribution (power law fit with R2 = 0.81).
Based on the distribution of the numbers of interacting partners for each TF (Figure 1C), we chose 50 interactors as a threshold for identifying “hub” proteins that are particularly well connected in our Y2H network. Hub proteins are more likely to participate in a higher number of cellular processes and exhibit pleiotropy (Yu et al., 2008). Nine TFs met this criterion (Figure 1C): CG10654, Eip78C, Foxo, Drm, Irbp18, CG31955, Ets65A, Rel, and Hr78. The orthologs of these hub proteins are also highly enriched for being highly connected nodes in C. elegans and human TF-TF interaction networks (p < 2.2 × 10− 16, Wilcoxon rank sum test) (Figure S1) (Ravasi et al., 2010; Reece-Hoyes et al., 2013). This finding supports the conserved, regulatory importance of these TFs in TF-TF networks. For example, Hr78 is an essential nuclear receptor that is thought to function at the top of the ecdysteroid regulatory hierarchy, with null mutations leading to lethality during the third-instar larval stage (Fisk and Thummel, 1998). Irbp18 was recently identified as a critical gene for general DNA double-stranded break repair (Francis et al., 2016). The TFs interacting with Irbp18 in our Y2H network are enriched for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways relevant to DNA repair such as purine and pyrimidine metabolism and base excision repair (Fisher’s exact test, Benjamini-Hochberg adjusted p = 0.0122). We did not find enriched KEGG pathways or Gene Ontology (GO) annotation terms for TFs interacting with the other hub TFs, which may be due in part to the incomplete functional characterization of genes. Three of the hub proteins (Ets65A, CG10654, CG31955) are poorly characterized in Drosophila, but the key roles of their orthologs shed light on the possible importance of these genes; for example, the Ets65A ortholog Fli1 is one of the master regulators in blood and endothelial development in mouse and zebrafish (Kanki et al., 2017; Liu et al., 2008).
TF-TF Interactome Quality Assessment
As an initial quality assessment of our TF-TF interactome data, we mined the MasterNet database (Friedman et al., 2011), which is a compilation of experimentally determined Drosophila PPIs and predicted PPIs based on interactions detected in other species (interologs). We found support for 74 of our 1,983 Y2H PPIs (~4%): 33 had been previously determined experimentally (“MasterNet Fly PPIs”), and 51 were predicted interactions (“MasterNet Interologs”) (Figure 1D).
To assess the sensitivity and specificity of our Y2H assay, we assembled reference sets of positive and negative interaction pairs (see STAR Methods). To our knowledge, prior Drosophila PPI screens did not use such reference sets to evaluate the quality of their derived interaction data. Our positive reference set (PRS) represents a high-confidence set of 41 Drosophila TF-TF interactions supported by at least two pieces of evidence in the literature (Table S3). As it is not possible to obtain a high-confidence set of non-interacting pairs (i.e., true-negative interactions), we generated 1,000 random reference sets (RRSs) (Venkatesan et al., 2009) as proxies for non-interacting TFs. Each RRS comprised a set of 1,983 TF pairs that have not been described as interacting. Our Y2H data recovered 7 interactions in the PRS, yielding a sensitivity of 17% with a false-positive rate (here, the fraction of RRS pairs scoring positive) of1.2%. These rates are comparable to those of two prior human Y2H interactomes (Rual et al., 2005; Venkatesan et al., 2009), suggesting that our Drosophila TF–TF interactome map is of similar quality.
Next, we experimentally validated the quality of our Y2H data using two orthogonal binary interaction assays: an in vitro mechanically induced trapping of molecular interactions (MITOMI) assay adapted to identify PPIs (Gerber et al., 2009) and an in vivo bimolecular fluorescence complementation (BiFC) assay (Remy and Michnick, 2004) (Figure 2). Briefly, in MITOMI, TF pairs are expressed in vitro either as enhanced GFP (eGFP) or mCherry fusions (Isakova et al., 2016) and introduced into the MITOMI chip; TFs are defined as interacting if, after trapping in the MITOMI chip, the protein trapping area is positive for both eGFP and mCherry signal. In BiFC, two non-fluorescent fragments of yellow fluorescent protein (YFP) are fused to two proteins being tested for their ability to interact; if the two proteins interact, then YFP is reconstituted, and a detectable YFP fluorescence signal is emitted (Hu et al., 2002; Pusch et al., 2011).
Figure 2. Orthogonal Assays Validate Y2H Interactions.

Summary of Y2H interactions validated by MITOMI and BiFC (A). Representative examples of positive and negative interactions assayed by (B) MITOMI and (C) BiFC. The scale bars in (B) are 250 μm; the scale bars in (C) are 50 mm. The depicted BiFC interactions (C) were tested in Drosophila eye antennal discs.
We used MITOMI or BiFC to test PPIs among TF pairs that are (1) scored positive in our Y2H screen (“Y2H-positive pairs”) and not members of the PRS, (2) members of the PRS, or (3) scrambled pairings of TFs in the PRS (Figure 2; Table S4). Of 73 Y2H-positive pairs tested, 39 (53.4%) were positive by either MITOMI or BiFC (representative examples shown in Figures 2B and 2C; examples of different signal intensities shown in Figure S2). Of 12 tested TF pairs from the scrambled PRS pairings, none were positive by either MITOMI or BiFC, supporting the specificity of these assays. In comparison, 9 of 12 probed TF pairs in the PRS tested positive by either MITOMI or BiFC; notably, TF pairs in the PRS that were positive in our Y2H screen had a higher validation rate (6/7) than those that were negative in our Y2H screen (3/5). These validation rates, together with the sensitivity and specificity determined above, suggest that our Y2H PPI data are of high quality.
Assembly of an Integrated Drosophila TF-TF PPI Network
We integrated our Y2H results with data for 1,161 TF-TF interactions that we extracted from MasterNet (Friedman et al., 2011) to assemble a comprehensive network of known Drosophila TF-TF PPIs. While acknowledging that MasterNet-derived interactions may also include higher-order interactions, our integrated Drosophila TF-TF interactome (“integrated network”; Table S2) comprises 3,111 TF-TF PPIs, 1,950 of which were newly contributed by our Y2H data. This addition represents a 168% increase in the number of experimentally determined fly TF-TF PPIs.
Using this integrated network, we examined which TF families interact with one another (Table S2). Just over 3% (98/3,111) of interactions are homodimeric interactions. Many of these interactions are within basic leucine zipper (bZIP) TFs and helix-loop-helix (HLH) TFs, each of which are known to form homo- and het- erodimers. C2H2 zinc finger (ZF) TFs represent the largest group of homodimerizing TFs (20/98) and intrafamily heterodimerizing TFs (218/381) in the integrated network. We found that heterodimeric interactions occur primarily between different TF families (2,356/3,013); this figure is a conservative estimate due to the exclusion of 276 TF-TF interactions in which both TFs were classified as “other” for TF family membership. C2H2 ZF-homeodomain TFs are the most common type of interfamily het- erodimers (161/3,013), followed by C4 ZF-C2H2 ZF heterodimers (127/3,013). The large number of ZF TFs represented in these TF-TF interactions is consistent not only with the large number of ZFs represented in the integrated network but also with the known ability of ZFs to mediate PPIs (Brayer and Segal, 2008).
Utility of the TF-TF Interactome Network in the Analysis of cis-Regulatory Regions
To investigate whether Y2H-detected TF pairs may co-regulate genes in vivo, we investigated ChIP-chip/seq data on TF occupancies (Jakobsen et al., 2007; MacArthur et al., 2009; modENCODE Consortium et al., 2010). Interacting TFs can bind DNA through multiple different modes. In some cases, both partners bind to their cognate DNA binding site motif, and the extent of TF-TF cooperativity is influenced by the composition of the respective heterodimeric binding site (Isakova et al., 2016). In other cases, only one partner binds DNA directly and the other partner is recruited to DNA through PPIs. Such direct versus indirect DNA binding can occur in a cell-type-or condition-specific manner (Gordân et al., 2009, 2011; Mariani et al., 2017). To investigate whether the PPIs that we detected in vitro also exist in vivo, we inspected genomic regions occupied by a TF for the DNA binding site motif of itself and its partner TF in tissues in which the TFs are co-expressed.
Such an analysis of TF occupancy is dependent on the availability of high-quality TF DNA binding site motif data. To supplement publicly available TF binding motifs in FlyFactorSurvey (Zhu et al., 2011), we generated high-quality motifs for 44 TFs, including 9 TFs lacking prior motif data, by universal protein binding microarray (PBM) assays (Berger et al., 2006) (Table S5). In total, we compiled 307 motifs representing 304 TFs (Table S6) (see STAR Methods).
For the TF occupancy analysis, we filtered the integrated network to retain 333 TF pairs for which (1) the TF pair exhibits overlapping expression patterns, (2) DNA binding specificity data are available for both TFs, (3) ChIP-chip/seq data are available for at least one of the interacting TFs, (4) the expression patterns of the TFs overlapped with the time point(s) of the ChIP-chip/seq data, and (5) the DNA binding specificity motifs of the interacting TFs were dissimilar (using Pearson correlation coefficient <0.8 as a threshold, as in Kheradpour et al., 2007), to distinguish which TF motif was enriched. In all, we considered 57 ChIP-chip/seq datasets that represent these 333 TF pairs (Bradley et al., 2010; Harrison et al., 2011; Jakobsen et al., 2007; MacArthur et al., 2009; Meireles-Filho et al., 2014; modENCODE Consortium et al., 2010; Nevil et al., 2017; Paris et al., 2013; Ren et al., 2015; Shlyueva et al., 2014; Zinzen et al., 2009; Zolotarev et al., 2016). For each of these filtered TF pairs, we evaluated the enrichment of the binding motif of each TF within the ChIP “bound” regions as compared to background genomic regions (Tables 2 and S7) (Wilcoxon rank sum test, Benjamini-Hochberg false discovery rate q < 0.1) (Gordân et al., 2009; Mariani et al., 2017) (see STAR Methods).
Table 2.
Evidence of Interacting TFs Found in ChIP Data
| Binding Mode | PPI | TF1 (ChIP-Profiled TF) | Adjusted p | TF2 | Adjusted p | Overlapping Expression Patterns | ChIP Data Ref. | Time Point of ChIP Data |
|---|---|---|---|---|---|---|---|---|
| Co-binding | bcd Rel* | bcd | 1.01E-40 | Rel | 9.16E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, adult female | Paris et al., 2013 | E2–4 |
| Co-binding | bcd Rel* | bcd | 2.47E-7 | Rel | 4.72E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, adult female | MacArthur et al., 2009 | E2–3 |
| Co-binding | bcd ttk | bcd | 2.47E-7 | ttk | 4.29E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, adult female | MacArthur et al., 2009 | E2–3 |
| Co-binding | cyc foxo* | cyc | 1.74E-15 | foxo | 1.86E-4 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | Meireles-Filho et al., 2014 | adult female |
| Co-binding | cyc foxo* | cyc | 4.07E-12 | foxo | 3.44E-5 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | Meireles-Filho et al., 2014 | adult female |
| Co-binding | Dttk | D | 4.99E-5 | ttk | 6.78E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E1–12 |
| Co-binding | da caup* | da | 2.43E-2 | caup | 2.43E-2 | E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | MacArthur et al., 2009 | E2–3 |
| Co-binding | da foxo* | da | 2.43E-2 | foxo | 6.23E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | MacArthur et al., 2009 | E2–3 |
| Co-binding | da twi | da | 2.43E-2 | twi | 9.22E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | MacArthur et al., 2009 | E2–3 |
| Co-binding | hth exd* | hth | 1.81E-3 | exd | 1.86E-4 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E0–8 |
| Co-binding | hth Ubx | hth | 1.81E-3 | Ubx | 1.47E-4 | E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E0–8 |
| Co-binding | Kr Hr78* | Kr | 2.96E-45 | Hr78 | 8.30E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae | Paris et al., 2013 | E2–4 |
| Co-binding | Kr Hr78* | Kr | 4.16E-60 | Hr78 | 4.98E-4 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae | Paris et al., 2013 | E2–4 |
| Co-binding | Kr ovo | Kr | 4.16E-60 | ovo | 6.44E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae | Paris et al., 2013 | E2–4 |
| Co-binding | Kr Ptx1* | Kr | 4.16E-60 | Ptx1 | 6.08E-4 | E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae | Paris et al., 2013 | E2–4 |
| Co-binding | Trl foxo* | Trl | 3.93E-24 | foxo | 4.09E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E8–16 |
| Co-binding | Trl ken | Trl | 3.93E-24 | ken | 6.23E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E8–16 |
| Co-binding | zld Kah* | zld | 1.88E-118 | Kah | 4.79E-46 | E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | Harrison et al., 2011 | E2–2.5 |
| Co-binding | zld Kah* | zld | 9.75E-123 | Kah | 1.85E-41 | E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | Harrison et al., 2011 | E3–3.5 |
| Co-binding | zld mirr* | zld | 1.88E-118 | mirr | 6.23E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | Harrison et al., 2011 | E2–2.5 |
| Co-binding | zld mirr* | zld | 9.75E-123 | mirr | 4.09E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | Harrison et al., 2011 | E3–3.5 |
| Indirect | br Zif* | br | 1.00E+00 | Zif | 9.81E-2 | E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | WPP |
| Indirect | Dll CG3919* | Dll | 1.00E+00 | CG3919 | 3.63E-2 | E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female |
modENCODE Consortium et al., 2010 | WPP |
| Indirect | Dll mirr* | Dll | 1.00E+00 | mirr | 3.63E-2 | E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | WPP |
| Indirect | Dll rn* | Dll | 1.00E+00 | rn | 2.07E-7 | E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | WPP |
| Indirect | EcR Rel* | EcR | 8.34E-1 | Rel | 6.15E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | Shlyueva et al., 2014 | WPP |
| Indirect | gt lola | gt | 5.07E-1 | lola | 6.23E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | Paris et al., 2013 | E2–4 |
| Indirect | gt lola | gt | 7.22E-1 | lola | 8.89E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | Paris et al., 2013 | E2–4 |
| Indirect | Gtttk | gt | 5.07E-1 | ttk | 5.65E-3 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | Paris et al., 2013 | E2–4 |
| Indirect | Hr78 bowl* | Hr78 | 1.00E+00 | bowl | 6.81E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 disco* | Hr78 | 1.00E+00 | disco | 6.81E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 Doc2* | Hr78 | 1.00E+00 | Doc2 | 1.47E-4 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 E (spl)m3-HLH* | Hr78 | 1.00E+00 | E(spl) m3-HLH | 4.54E-2 | E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female |
modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 E (spl)mdelta-HLH* | Hr78 | 1.00E+00 | E(spl) mdelta-HLH | 9.14E-5 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E22–24, L3, WPP, pupae | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 ey* | Hr78 | 1.00E+00 | ey | 6.10E-5 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 Fer3* | Hr78 | 1.00E+00 | Fer3 | 3.25E-2 | E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, pupae, adult male | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 h* | Hr78 | 1.00E+00 | h | 1.31E-3 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 Max* | Hr78 | 1.00E+00 | Max | 4.72E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 Mondo | Hr78 | 1.00E+00 | Mondo | 9.16E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 sna | Hr78 | 1.00E+00 | sna | 1.31E-3 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 sv | Hr78 | 1.00E+00 | sv | 1.31E-3 | E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 Trl | Hr78 | 1.00E+00 | Trl | 7.55E-4 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | Hr78 zfh1 | Hr78 | 1.00E+00 | zfh1 | 3.25E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E8–16 |
| Indirect | kn Xbp1 | kn | 1.00E+00 | Xbp1 | 3.43E-2 | E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male | modENCODE Consortium et al., 2010 | E0–12 |
| Indirect | prd Hr78 | prd | 1.00E+00 | Hr78 | 6.23E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E22–24, L3, WPP, pupae, adult male | modENCODE Consortium et al., 2010 | E0–12 |
| Indirect | Su(H) da | Su(H) | 0.999999984 | da | 4.72E-2 | E0–2, E2–4, E4–6, E6–8, E8–10, E10–12, E12–14, E14–16, E16–18, E18–20, E20–22, E22–24, L1, L2, L3, WPP, pupae, adult male, adult female | modENCODE Consortium et al., 2010 | E0–8 |
Some interactions are listed twice because these pairs were found in multiple ChIP samples.
Interactions found in Y2H.
We found 16 unique TF co-binding pairs (10 from our Y2H interactions, 6 from previously known interactions), in which the motif of the ChIP-profiled TF was enriched along with the motif of at least one of its interacting partner TFs. Our analysis recovered well-known examples of interacting TFs. For example, in Hth (homothorax) ChIP-seq data obtained from stage E0–8 embryos (modENCODE Consortium et al., 2010), we observed enrichment of the Hth motif together with the Exd (extradenticle) and Ubx (ultrabithorax) motifs. Hox proteins such as Ubx are known to interact with Exd and Hth and bind DNA cooperatively in vivo as part of a trimeric complex (Ryoo et al., 1999). Interactions between Hth, Exd, and Ubx had been observed in a prior Hth ChIP-chip study of leg and haltere imaginal discs (Slattery et al., 2011).
In another co-binding interaction, we found the cycle (Cyc) motif to be co-enriched with the Foxo motif (Figures 3A and 3B). Cyc forms a heterodimer with clock (Clk) in circadian rhythm, and together activate key circadian rhythm genes (Al-lada et al., 2001). Foxo is the effector TF of the insulin signaling pathway (IIS), which is implicated in longevity and stress response (Tatar et al., 2003). While Cyc and Foxo were not known previously to physically interact, evidence for their over-lapping function exists in the literature. For example, under oxidative stress, Clk activity is repressed in cultured Drosophila cells, and foxo mutant flies become arrhythmic (Zheng et al., 2007). We observed Cyc and Foxo co-binding in the promoter region of vrille (Figure 3B), which has been shown to be a transcriptional target of Cyc-Clk in circadian rhythm (Cyran et al., 2003; Glossop et al., 2003) and of Foxo in the context of life-span extension (Bai et al., 2013). Thus, it is possible that Foxo and Cyc link IIS and circadian rhythm. Motif co-enrichment for physically interacting, co-binding TFs was also observed in human Encyclopedia of DNA Elements (ENCODE) ChIP-seq data using a similar analysis approach (Mariani et al., 2017).
Figure 3. Examples of TF-TF Co-binding and Indirect Binding in Genomic Regions.

(A) Schematic representation of Cyc-Foxo co-binding on DNA.
(B) Cyc-Foxo co-binding is observed in the promoter region of vrille. GOMER tracks indicate motif enrichment for each TF.
(C) Schematic representation of Hr78 indirect DNA binding through Hairy.
(D) Hr78 indirect binding through Hairy is observed in the promoter region of kayak. GOMER tracks indicate motif enrichment for each TF. “S14 Regions” track indicates open chromatin regions derived from DNase-seq experiments performed at 10–11 h post-hatch (Li et al., 2011) (retrieved from the University of California, Santa Cruz [UCSC] Genome Browser), corresponding to the co-expressed time points of the 2 TFs.
In 24 cases, the motif of the ChIP-profiled TF was not enriched, but instead the motif of at least one of its interacting TFs was enriched (21 from Y2H, 3 from previously known interactions). This finding suggests that, in those cellular conditions, the profiled factor associates with DNA indirectly through a recruiting TF. Such indirect binding was observed previously in analyses of Saccharomyces cerevisiae ChIP-chip (Gordân et al., 2009) and human ENCODE ChIP-seq datasets (Mariani et al., 2017; Wang et al., 2012). For most ChIP-profiled TFs in this category, we detected only 1–2 partner TF motifs. However, in the case of Hr78, 14 partner TF motifs were enriched, which is consistent with our finding that Hr78 is a hub protein in our Y2H network. Hr78 was shown to interact with >100 ecdysteroid-regulated chromosomal puff loci (Fisk and Thummel, 1998). One of the TFs that, based on our analyses, may recruit Hr78 is Hairy (Figure 3C), which is induced following steroid hormone 20-hy-droxyecdysone (20-HE) treatment of cultured larval organs (Beckstead et al., 2005) and plays a role in 20-HE-regulated cellular differentiation (Gauhar et al., 2009). While the other Hr78 interaction partners are not known to function in ecdysteroid signaling, our results suggest that partner TF-mediated recruitment of Hr78 to genomic loci could direct the transcriptional regulatory role of Hr78 in ecdysteroid signaling. For example, we observed the Hairy motif overlapping an Hr78 ChIP peak upstream of kayak, a putative transcriptional target of Hairy (Bianchi-Frias et al., 2004) (Figure 3D); this sequence falls within a chromosomal region previously identified as a 20-HE-regulated chromosomal puff (Fisk and Thummel, 1998).
Of the remaining TF-TF pairs, we observed the enrichment of only the ChIP-profiled TF for 113 pairs, and no enrichment of the ChIP-profiled or partner TF for 180 pairs. The lack of enrichment may be due to the profiled TF interacting with a factor that is absent from our TF interactome, incomplete motif models, or noise in the ChIP-chip/seq data. Alternatively, the profiled TF may bind DNA indirectly through interaction with a variety of different TFs, each of which recruits the profiled TF to only a small fraction of the regions occupied in vivo.
Analysis of Drosophila TF-TF Interactome Identifies Potential Human Disease TFs
Our integrated Drosophila TF PPI network can provide useful insights into human TF-TF interactions. We asked which new TFs and TF-TF interactions may be relevant for human congenital heart disease (CHD), as TFs involved in CHD have been studied extensively (McCulley and Black, 2012). There are 19 human TFs annotated in the Human Gene Mutation Database (HGMD) with the phenotype “congenital heart disease.” We identified fly orthologs of these TFs along with all of their immediately interacting TFs (“first neighbors”) in our fly network. We then restricted the selection to those TFs expressed in the Drosophila heart to select heart-relevant PPIs. The resulting network, composed of 28 nodes and 54 edges, is shown in Figure 4A. While we had started out with 12 CHD-associated TF fly orthologs, our approach identified 11 additional TFs that may play important roles in CHD based on interactions with known CHD-associated TFs (blue nodes in connected component). From a literature search, we found that human orthologs of 4 of these 11 fly TFs are important for normal development of the heart. We also found 4 fly TFs that are crucial for normal fly heart development, but their human orthologs are not well studied. The remaining three genes have not yet been implicated in CHD or heart development in any species. Thus, these TFs may be good candidates for future study into their potential roles in human heart development and disease.
Figure 4. Integrated Fly TF-TF Interactome Identifies Candidate Human TFs Involved in Congenital Heart Disease and Eye Development.

(A) The Drosophila TF-TF interactome can be used to identify candidate heart development and CHD genes. Nodes: Drosophila TFs; edges: PPIs. Nodes with human orthologs that are annotated as CHD associated in HGMD (pink nodes) and their first neighbors (blue nodes) that are expressed in Drosophila heart tissue are shown. Unconnected components arose due to this tissue expression requirement. Square: the Drosophila gene and its human ortholog(s) are involved in CHD and heart development based on a literature search; triangle: the Drosophila gene is involved in heart development; hexagon: the Drosophila gene has a human ortholog involved in heart development; circle: neither the Drosophila gene nor human ortholog(s) are known to be involved in heart development. Node sizes roughly reflect node degrees. Orange edges indicate newly discovered interactions in this study. Human ortholog gene names are given in parentheses below the Drosophila gene name.
(B) The Drosophila TF-TF interactome can be used to identify candidate eye development genes. Nodes: Drosophila TFs; edges: PPIs. Nodes associated with eye development in Drosophila (based on Kumar, 2009; purple nodes) and their first neighbors (green nodes) are shown. Triangle: the Drosophila gene has eye development-related GO terms; hexagon: human, rat, mouse, or zebrafish (henceforth, vertebrate) ortholog of the Drosophila gene shown has eye development-related GO terms; square: the Drosophila gene (shown) and its vertebrate ortholog(s) have eye development-relatedGO terms; circle: neither the Drosophilagene nor vertebrate ortholog(s) have GO terms pertaining to eye development. Orange edges indicate newly discovered interactions in this study. Node sizes roughly reflect node degrees.
As another example, we examined the Drosophila genes implicated in eye development. Although the eye architectures of Drosophila and vertebrates are very different, the set of genes that regulate eye development is well conserved (Kumar, 2009). We selected a set of 9 TFs that are key regulators in Drosophila eye development (Kumar, 2009) (henceforth, “eye regulators”) and included these genes’ first neighbors, resulting in a network comprising 115 nodes and 128 edges (Figure 4B). Most nodes were connected to only one of the eye regulators, but a handful of genes were connected to multiple eye regulators, potentially indicating their roles in this process. Of note, Hr78 is connected to four eye regulators (So, Eyg, Ey, and Optix). Although Hr78 was previously found to be dispensable for the progression of the morphogenetic furrow in the Drosophila eye (Brennan et al., 2001), it may have alternative functions in eye development. Notably, the Hr78 ortholog Nr2c1 has recently been found to regulate early retinal cell patterning in the mouse (Olivares et al., 2017). Exd interacts with Hth and two eye regulators, Tsh and Ey. Although Exd was not included in the original list of 9 eye regulators, it is known to be a negative regulator of eye development in the fly, functioning together with Hth (Pai et al., 1998). Exd orthologs Pbx2 and Pbx4 regulate eye-patterning genes in zebrafish (French et al., 2007). These findings suggest that other genes that interact with multiple eye regulators similarly may be good candidates for further study for their roles in eye development or function.
DISCUSSION
We report a Y2H-derived Drosophila TF-TF interactome representing 77% (584/755) of all Drosophila TFs. Our interactome adds 1,950 new interactions to the set of all of the known Drosophila TF-TF interactions (Friedman et al., 2011), increasing the total number by 168%. We combine our newly identified interactions with previously known interactions to present the integrated network, which serves as a comprehensive resource of all high-confidence Drosophila TF-TF interactions for studies of the TF co-regulation of gene expression (Table S2).
Our integrated Drosophila TF-TF network is likely incomplete. Prior studies have noted that there is only limited overlap between PPIs recovered by different methods, due at least in part to differences in the sensitivities of distinct assay methods that may be inherently better suited for detecting different kinds of biophysical interactions (Braun et al., 2009; Yu et al., 2008). For example, it has been suggested that AP-MS detects stable complexes, whereas Y2H detects more transient or condition-specific PPIs (Yu et al., 2008). Our BiFC experiments suggest that BiFC is a more sensitive assay compared to Y2H, as it is able to detect more PRS interactions.
Y2H sensitivity has been estimated to be ~20%, indicating that the same search space would need to be sampled multiple times to identify all of the biophysical interactions detectable by Y2H (Yu et al., 2008). While we conducted our screen in quadruplicate, additional replicates and the use of alternate Y2H vectors (Stellberger et al., 2010), yeast strains (Braun et al., 2009), or PPI screening technologies will likely improve the coverage of the Drosophila TF-TF interactome. Thus, it is important to keep this caveat in mind when interpreting negative Y2H interactions. Furthermore, a broader PPI screen including non-DNA-binding cofactors and chromatin-associated proteins may reveal additional co-regulatory interactions (Babb et al., 2001; Siggers et al., 2011) and higher-order TF complexes that may link transcriptional regulation to signaling pathways and networks.
A TF-TF interaction is not a prerequisite for cooperative DNA binding (Deplancke et al., 2016; Reiter et al., 2017). For example, THRA isoform α2 and RXRA TFs heterodimerize only upon binding to specific DNA sequences (Reginato et al., 1996). Furthermore, DNA-mediated TF-TF cooperativity between TFs that are not known to physically interact with one another may be common (Jolma et al., 2015). Such DNA-mediated or other ligand-dependent TF-TF interactions would not be detected in a conventional Y2H assay.
Interactions between TFs contribute to the spatiotemporal specificity of transcriptional regulation. Accurate models of cell-type- and condition-specific gene regulation will require an improved understanding of how TFs are recruited to DNA, such as via PPIs, and how such interactions affect the DNA binding specificities of the resulting TF complexes. Here, in Table 2, we provide a catalog of co-binding and indirect Drosophila TF-DNA interactions inferred from the analysis of ChIP-chip/seq, TF binding motif, gene expression, and PPI data.
Different DNA binding modes of TF partners (i.e., binding to DNA directly versus indirectly via a DNA-bound partner) may reflect condition-specific regulatory mechanisms; we found evidence for both modes of binding in our PPI network. Furthermore, we found that a TF can exhibit multiple DNA binding modes, depending on its interaction partner (Table 2). For example, Hr78 is recruited to DNA by multiple TFs in E8–16, but it recruits Prd to DNA in E0–12. Trl co-binds with Foxo and Ken in E8–16, but it recruits Hr78 at the same developmental stage. Different TF binding modes, including TF monomeric binding, can yield different DNA binding specificities, offering mechanisms by which a TF can regulate unique sets of target genes (Siggers and Gordân, 2014). Further studies are needed to determine whether different modes of TF dimer recruitment to DNA by TF-TF interactions found in our TF-TF interactome effect distinct transcriptional outputs or are used under different cellular conditions.
TF-TF interactions are important in effecting spatiotem-poral-specific transcription. Our systematic TF-TF interactome lays the groundwork for understanding the complexities of combinatorial transcriptional regulation in Drosophila. Future investigations are needed to elucidate how these physical interactions translate to dynamic transcriptional control.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Martha L. Bulyk. (mlbulyk@genetics.med.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila strains
For the BiFC analysis, select UAS-TF-3xHA fly strains generated earlier (Schertel et al., 2015) were subjected to C-terminal in vivo swapping to obtain the TF-VN and TF-VC strains. These strains contain either the N-terminal Venus YFP fragment VNm9 or the C-terminal Venus fragment VC155 at the C terminus of the TFs. After the swapping, strains were individually equipped with a GMR-GAL4 driver (second chromosome) for subsequent expression of the VN- or VC-tagged UAS-transgenes. TF combinations were generated by crossing pairs of GMR-GAL4;TF-VN and GMR-GAL4;TF-VC strains. All fly progeny from crosses were used without selecting a specific sex. The fly stocks were kept according to general fly husbandry conditions. As described in the “Bimolecular fluorescence complementation assays” section (see Method Details), BiFC analysis was performed in third instar eye-antennal discs.
Yeast strains
For the Y2H assay, AD (“prey”) and DB (“bait”) clones were transformed into the yeast strains MaV203 and MaV103 (Vidal et al., 1996a, 1996b), respectively. Growth conditions for Y2H assays are provided under the “Yeast two-hybrid screen” section (see Method Details).
METHOD DETAILS
Yeast two-hybrid screen
Preparation of TF clones and yeast strain transformation
A list of 755 predicted sequence-specific TFs in D. melanogaster was generated from a prior cataloguing of sequence-specific TFs for the FlyTF database (Adryan and Teichmann, 2006) and through manual curation (Hens et al., 2011). Our library consisted of 692 clones (of these 755 TFs) obtained from Berkeley Drosophila Genome Project (BDGP) cDNA clones (588 were full-length sequence-verified (Gold); 36 clones were end-sequence verified at both 5′ and 3′ ends (Silver); 68 were partially sequenced (Bronze)), plus five additional TFs that were cloned and full-length sequence-verified for this study. All TF names were updated to the October 2016 release of FlyBase (Gramates et al., 2017).
The Y2H assay utilizes the product of the yeast gene GAL4. The Gal4 TF has two separable domains: the DNA binding domain (DB) and the transcriptional activation domain (AD). Plasmids carrying sequences coding for DB and AD are fused in-frame to DNA sequences coding for proteins of interest and are introduced into yeast strains of opposite mating types. Pairwise combinations of TFs are generated by mating. A physical interaction between the tested proteins reconstitutes the Gal4 protein, resulting in transcription of reporter genes (Walhout et al., 2000; Walhout and Vidal, 2001).
We cloned our TF library into pAD-DEST and pDB-DEST vectors (Walhout and Vidal, 2001). We used low copy number yeast expression (ARS/CEN) vectors to control overexpression for low expression to minimize artifactual interactions. We transformed AD (“prey”) and DB (“bait”) clones into the yeast strains MaV203 and MaV103, respectively. A robotic platform automated the yeast transformation process in 384-well plate format (Hens et al., 2011). Briefly, yeast and TF ORF DNA were mixed using fixed pins and then incubated first at 30°C and then at 42°C. Transformed yeast cells were pelleted using an integrated centrifuge, resuspended, and then spotted on permissive medium. The resulting colonies were then robotically replica-stamped on selective medium (Sc-Trp for AD library, and Sc-Leu for DB library) to select for successful transformants. We successfully cloned and transformed 695 pAD clones and 575 pDB clones, corresponding to 689 and 569 unique TF genes, respectively (some TFs are represented by multiple sequences; see Table S1). The AD library was then replica-plated in 1536-well format and used for mating. DB-containing strains were individually spread out on plates, from which we robotically replicated 1536 colonies of a single DB strain on yeast extract-peptone-dextrose plates. Mating between the AD and DB strains was achieved by replica-plating the AD library onto the DB colonies on the YEPD plate and incubating overnight at 30°C. Colonies were selected for successful mating on Sc-Leu, -Trp plates. Empty AD vector served as a negative control.
Auto-activation detection
To detect PPIs, the colonies were replica plated on Sc-Leu, -Trp, -His plates containing two different concentrations of 3-aminotria-zole (3AT) (20 and 40 mM). 3AT is a competitive inhibitor of His3 and was used to titrate the minimum level of HIS3 expression required for growth on histidine-deficient medium. Thus, testing colony growth at various 3AT concentrations allowed us to make robust interaction calls using the automated detection protocol described below. Strong, uniform growth of all 1536 colonies on all 3-AT concentrations indicates auto-activation by the DB clone, which can be caused by the presence of an intrinsic transcriptional activation domain in some TFs. 37 pDB-DEST clones were flagged as auto-activating TFs (Table S1). 19 of these 37 flagged TFs could still be used for Y2H analysis by managing background levels and using our automated interaction detection software.
Omission of pseudogenes from analysis
Two of the 755 predicted TFs corresponded to pseudogenes (FBgn0053221 and FBgn0029920). Though these genes were included in the Y2H assay, they were omitted from further analysis.
Automated detection of interactions in Y2H screen
To process the yeast plate images and detect PPIs in an automated and objective manner, we adapted a MATLAB-based image analysis program, TIDY, which we previously used for the detection of yeast one-hybrid interactions (Hens et al., 2011). Briefly, the software semi-automatically calls interactions based on pattern detection of yeast colony images convoluted with a 2×2 spot pattern (corresponding to the spot pattern of the quadruplicate colonies), where the size of the spots matches the average size of the colonies. This approach is robust to differing background signals across the same plate. In addition, we computed a homogeneity score over the four replicate spots and penalized positions with 2 strong and 2 weak colonies. TIDY was improved from the original version (Hens et al., 2011) to provide a Z-score of a given interaction over one or more input images, for example, corresponding to plates screened at different 3AT concentrations (as described above and used here) or over multiple biological replicate experiments.
To calculate the Z-score, we first calculated the mean and standard deviation of intensities in each image after excluding outlier signals (corresponding to potential interactions) as defined by the Grubbs’ test; a normal distribution is expected for the background intensities (position with no interactions). We then normalized intensities across the entire image. As described previously, TIDY offers the option to “correct” for differences in colony growth which are sometimes observed between colonies at the interior versus the exterior regions of the plate. If this option is selected by the user, the normalization is performed independently for the interior and exterior colonies; we used this option as needed, based on visual inspection of the plate. The Z-score represents the average of the normalized intensities for a given interaction over multiple different analyzed images, excluding those images where the colonies are not homogeneous. All images are weighted equally, meaning that an interaction appearing in only one image could be averaged out and not recognized as a positive interaction, if the normalized intensity is not sufficiently strong in the other images.
Due to experimental dropout (e.g., systematic lack of colony growth), we tested and analyzed a total of 385,431 AD/DB pairs corresponding to 220,776 unique TF–TF interactions (Table S1), and detected 1,983 interactions in at least one direction, including 26 self-interactions (i.e., putative homodimers).
Experimental validation of Y2H interactions
MITOMI assays
A subset of our Y2H screen PPIs were tested by an independent experimental methodology using an adaptation of the MITOMI technology (Maerkl and Quake, 2007) to assay for PPIs (Gerber et al., 2009). Briefly, each TF was subcloned from the Entry vector into pF3A-eGFP and pF3A-mCherry vectors and arrayed in duplicate on a slide so that each eGFP-labeled TF was tested against all other TFs and itself as mCherry fusions. TFs were expressed by flowing an IVT reaction mixture over the flow cell. eGFP fusion proteins were trapped by an anti-GFP antibody coated on the slide. PPIs were determined by comparing the mCherry and eGFP signal intensities. We prioritized our Y2H PPIs for testing by MITOMI as follows: TFs resulting in many PPIs (‘sticky’ proteins), to investigate whether they were true hits or false positives; TF PPIs demonstrating an interaction in only one direction (AD/DB); TF PPIs spanning a wide range of protein domains; TF PPIs involving protein domains not previously known to interact.
Bimolecular fluorescence complementation assays
Briefly, two non-fluorescent fragments of YFP are fused to the ORFs of putatively interacting proteins. If the proteins interact, then YFP is reconstituted and the YFP fluorescent signal can be detected (Hu et al., 2002; Pusch et al., 2011). Selected UAS-TF-3xHA fly strains were subjected to C-terminal in vivo swapping to obtain the TF-VN and TF-VC strains, as described in the Drosophila strains section above. BiFC analysis of protein–protein interactions was performed in third instar eye-antennal discs. In brief, TF combinations were generated and tested by crossing pairs of GMR-Gal4;TF-VN and GMR-Gal4;TF-VC fly strains, leading to co-expression of the fusion proteins in the eye tissue. The crosses were kept at 21°C until dissection to saturate protein expression levels, which is a critical parameter for maximizing the specificity of BiFC. Eye-antennal discs were dissected from 5–6 third instar larvae and monitored for Venus YFP fluorescence signal with a Zeiss Lsm710 confocal microscope. (For further details, see (Bischof et al., 2013).) The BiFC signals were measured at identical microscope settings, and classified into signal intensity call categories based on visual assessment; for display, all images in Figure 2C and Figure S2 were identically brightness-adjusted (+150) in Adobe Photoshop. Examples of signal intensity calls are shown in Figure S2. “(+)” was not considered to be a positive interaction.
BiFC assays are expected to detect all PPIs in close proximity, including those that are mediated by a third protein. Thus, two TFs that are co-expressed in a given tissue might be more likely to be detected as a positive hit in a BiFC assay conducted in that tissue than two TFs that are not co-expressed. To explore this issue, we examined an RNA-seq dataset from third instar eye-antennal discs (Potier et al., 2014). This stage and tissue correspond to those used in our BiFC experiments. Of the 75 TF–TF pairs tested by BiFC, 48 pairs were co-expressed in the eye-antennal disc. The proportions of positive interactions in the co-expressed versus the non-co-expressed pairs were similar: 25/48 (52.1%) in co-expressed pairs, and 14/27 (51.9%) in the non-co-expressed pairs. Thus, we conclude that the likelihood of scoring a TF–TF positive interaction by BiFC is no higher among TFs that are normally co-expressed in third instar eye-antennal discs than TFs that are not co-expressed.
Computational validation of Y2H results
Comparison of Y2H results to MasterNet
We compared our Y2H results to published PPI data assembled in the MasterNet database (Friedman et al., 2011). Briefly, MasterNet is a compilation of databases, as follows: (i) fly binary PPI network constructed by integrating experimentally identified interactions from major PPI databases (e.g., BioGRID (Stark et al., 2011), IntAct (Aranda et al., 2010), MINT (Ceol et al., 2010), DIP (Salwinski et al., 2004), and DroID (Murali et al., 2011)), altogether comprising 65,754 interactions between 8,025 proteins. (ii) interolog binary PPI network predicted from experimentally identified binary PPIs for human, mouse, worm, and yeast; (iii) network of interolog protein complexes predicted from experimentally identified protein complexes for human, mouse, worm, and yeast compiled from the BioGrid, IntAct, MINT, DIP, and HPRD (Keshava Prasad et al., 2009) databases, with the caveat that not all proteins in a complex necessarily participate in binary interactions with each other; (iv) kinase-substrate network. There is evidence in MasterNet supporting 74 of our 1,983 Y2H PPIs.
Construction of Drosophila TF–TF PRS and RRSs
MasterNet (Freeze April 2015) was used as a reference for published PPIs. We constructed the PRS by selecting interactions published in at least two studies. In addition, we required that at least one of the methods used in these studies showed a binary interaction, in order to construct a PRS of direct PPIs. We individually examined and verified the evidence in the referenced literature supporting the interactions in the PRS. This yielded 41 high-confidence PPIs in the PRS. We constructed the RRSs by generating random sets of interactions from the same TF space, maintaining similar numbers of interactions and node degree distribution in each RRS as in our Y2H network, and removing all previously known interactions (i.e., those in MasterNet).
PBM assays
Protein expression
We generated N-terminal glutathione S-transferase (GST), HA, or FLAG fusion constructs of the DNA binding domain region or full-length TF clones by Gateway cloning into pDEST15, pT7CFE1-NHA (Thermo Fisher Scientific), or pT7CFE1-NFtag (Thermo Fisher Scientific) expression vectors; we Gateway-converted the latter two vectors. Sequences were full-length verified. In vitro transcription and translation (IVT) reactions were performed according to the manufacturer’s protocol (PURExpress IVT Kit (NEB, E6800) or 1-Step Coupled Human IVT Kit (Thermo Fisher Scientific, 88881)). Western blots were used to estimate molar concentrations of all in vitro translated proteins by utilizing a dilution series of recombinant GST (Sigma, G5663), FLAG (Sigma, P7582), or multi-tag protein including HA (Abcam, ab5395) as standard proteins. The following primary antibodies were used: rabbit anti-GST polyclonal primary antibody (Sigma, G7781; used at 20 ng/ml); mouse Alexa488-conjugated anti-HA monoclonal antibody (Thermo Fisher Scientific, A-21287; used at 1 μg/mL); mouse anti-FLAG M2-horse radish peroxidase (HRP)-conjugated antibody (Sigma, A8592; used at 1 μg/mL). The following secondary antibodies were used: goat HRP-conjugated anti-rabbit IgG (Pierce, 31460; added at 5 μg/ml); goat HRP-conjugated anti-mouse IgG (Thermo Fisher Scientific, 32430; used at 5 ng/mL). Glycerol was added to a final concentration of 30% to the completed IVT reactions samples prior to storage at 80°C.
PBM experiments using universal arrays
We employed an “all 10-mer” universal array design in 8 × 3 60K, GSE format (Agilent Technologies; AMADID #030236). Double-stranding of oligonucleotide arrays and PBM experiments were performed following previously described experimental protocols (Berger and Bulyk, 2006, 2009; Berger et al., 2006). Briefly, the TF of interest was expressed with an epitope tag (GST, HA, or FLAG), applied to the double-stranded DNA array, and detected with fluorescently labeled antibody specific for the tag. Experimental conditions used for all PBM experiments, including TF concentrations and buffers, are provided in Table S5. The following Alexa Fluor 488-conjugated antibodies were used: anti-GST (Thermo Fisher Scientific, A-11131), anti-HA (Thermo Fisher Scientific, A-21287), and anti-FLAG (Cell Signaling, 5407).
PBM data analysis
Microarray data quantification and normalization were performed as described before (Berger and Bulyk, 2006, 2009; Berger et al., 2006) using the Universal PBM Analysis Suite. TF DNA binding specificities were derived using the Seed-and-Wobble algorithm (Berger and Bulyk, 2009). Success in motif derivation from universal PBM data was assessed according to seed 8-mer enrichment score (E-score) and obtaining at least five 8-mers with E-score at least 0.45 matching the derived motif. The E-score is a modified form of the Wilcoxon Mann-Whitney rank-based statistic and is robust to technical variation across arrays. Larger E-score values reflect higher specificity of a TF for a particular 8-mer. Motif sequence logos representing the derived DNA binding specificity position weight matrices (PWMs) were generated using enoLOGOS (Workman et al., 2005).
Motif co-occurrence analysis
Motif compilation and data processing
We obtained 307 DNA binding site motifs (Table S6) as PWMs from FlyFactorSurvey (FFS) via the MEME motif database download (Zhu et al., 2011) or from our newly generated PBM data. For FFS-derived PWMs, we matched each PWM to a TF or to both TFs in a heterodimer pair and separated out those TFs relevant to our analysis (Tables S2) for further consideration. FFS curates PWMs generated from a variety of experimental methods and collected from multiple sources. If there were multiple PWMs associated with a TF, we preferentially chose a single PWM from PBMs performed in this study, then from FFS (Zhu et al., 2011) with the following evidence codes, in decreasing order of priority: SOLEXA, FlyReg, SANGER, Cell, NAR, and NBT. If there were multiple PWMs available from a chosen source, we manually chose a representative(s) PWM for each TF. Special cases included isoforms and dimers. For isoforms, we chose the PWM representing the assayed TF isoform, where available; for dimers, we chose PWMs representing homodimers, where available. We also added two motifs not found in FFS nor assayed by PBM in this study, but needed for the ChIP analysis (see below): Stat92E and gcm were taken from CIS-BP (Weirauch et al., 2014) with IDs M2327 and M2560, respectively. We trimmed both the 5′ and 3′ ends of all chosen PWMs until two consecutive positions with information content greater than 0.5 were obtained, as previously described (Barrera et al., 2016).
Assembling and processing ChIP-chip/seq data
ChIP-chip/seq datasets were gathered from modENCODE and a search of GEO DataSets for dm3 ChIP-seq datasets. Altogether, we assembled a total of 57 datasets applicable to our analysis (Table S7). Peaks were taken directly from the published analyses. For each ChIP dataset, we defined two sets of euchromatic regions: the foreground (i.e., ChIP ‘bound’ regions, as defined by the authors of each ChIP dataset) and the background (i.e., genomically matched regions not found in the foreground). We took the top 500 peaks of each set (as denoted by signal value) and trimmed them to 200 bp around the peak summit (i.e., peak summit ± 100 bp). If summit information was not available, then we trimmed to 200 bp around the center of the peak (i.e., peak center ± 100 bp). The liftOver tool (Hinrichs et al., 2006) was used to convert dm2 mappings to the dm3 genome, and any unmapped regions were discarded. To generate matched genomic background sequences, we used GENRE (Mariani et al., 2017) to match for promoter overlap, repeat overlap, and GC content specified for the dm3 genome and 200-bp regions.
Analysis of ChIP-chip/seq data
We analyzed publicly available in vivo TF genomic occupancy data to determine the extent of direct versus indirect TF–DNA interactions in vivo. Here, we filtered interactions in the Integrated Network (Table S2) for the following criteria: (a) the two interacting TFs exhibited overlapping expression patterns as defined by the modENCODE Temporal Expression Data (Graveley et al., 2011);(b) DNA binding specificity data were available for both interacting TFs (see above); (c) ChIP-chip/seq data were available for at least one of the interacting TFs (see above); (d) TF expression pattern overlapped with the time point of the ChIP-chip/seq data; and (e) the DNA binding specificity motifs of the interacting TFs were dissimilar (using Pearson correlation coefficient < 0.8 as a threshold as in (Kheradpour et al., 2007)) in order to distinguish which TF’s motif was enriched. For the 333 pairs that were left, we scored the foreground and background regions using a model similar to GOMER (generalizable occupancy model of expression regulation) (Granek and Clarke, 2005) to compute the probability that a TF binds a DNA sequence. We assessed the statistical significance of the area under the receiver operating characteristic curve (AUROC) value for a particular motif in a particular ChIP dataset by using a Wilcoxon rank sum test comparing the scores for foreground versus background sequences. We considered motif enrichment to be significant at Benjamini-Hochberg false discovery rate (FDR) q < 0.1.
Network analysis
Network visualization
We imported our Y2H network and the Integrated Network into CytoScape (version 3.4.0) (Shannon et al., 2003) for visualization.
Comparison with nematode and human TF–TF interactomes
We compared our Drosophila TF–TF interactome against similar interactomes in C. elegans and human (Ravasi et al., 2010; Reece-Hoyes et al., 2013). Interactions in the C. elegans network were restricted to those between TFs as it contained interactions with co-factors, based on a list published in the same study, using a custom Python script. The numbers of interacting genes and basic network statistics were retrieved using NetworkAnalyzer (Doncheva et al., 2012), as implemented in CytoScape.
To determine whether the hub TFs in our network were similarly highly connected TFs in the C. elegans and human networks, we compared the degree distribution of hub TF orthologs and that of the same number of randomly selected TFs in the respective species. We retrieved orthologs for our hub genes in the respective species through FlyBase. Because multiple orthologs exist for some Drosophila hub TFs, to avoid skewing the degree distribution, we randomly chose one ortholog to represent each hub TF in each of 10,000 samplings. The distributions of median node degree from each sampling were compared between the hub and random TFs using the Wilcoxon rank sum test; p < 0.001 was our threshold for significance.
Functional annotation retrieval
GO term annotation
GO Term annotations were retrieved from AmiGO 2 (http://amigo.geneontology.org/amigo/) (Balsa-Canto et al., 2016).
Ortholog annotation
We obtained human orthologs of Drosophila genes using the DRSC Integrative Ortholog Prediction Tool (DIOPT) (Hu et al., 2011). Because each Drosophila gene can have multiple human orthologs, we chose the top-scoring ortholog for the analysis shown in Figure 4, based on the DIOPT score, or based on what has been used as the ortholog in the literature. Additionally, human, mouse, rat, and zebrafish orthologs of fly genes were retrieved using Ensembl BioMart (Kinsella et al., 2011).
Tissue expression information
Drosophila tissue expression information was retrieved from the BDGP (Hammonds et al., 2013). The BDGP dataset includes tissue and developmental stage range information (henceforth, “expression term”). When one TF had a “ubiquitous” expression term, if the other TF was expressed in any tissue during the same developmental stage range as the ubiquitous term, these TFs were considered co-expressed and the expression term for the specific tissue only was taken as the co-expression term. If both TFs had ubiquitous terms at the same developmental stage range, the lesser ubiquitous term (e.g., “faint ubiquitous” as opposed to “strong ubiquitous”) was taken as the co-expression term.
Disease annotation
Human disease annotations for genes were retrieved from the Human Gene Mutation Database (Stenson et al., 2003).
TF family annotation
TF families were assigned based on domain annotations retrieved from (Hens et al., 2011). Sequence-specific DNA binding domains were manually curated based on annotations in Pfam (El-Gebali et al., 2019). If a TF did not have at least one curated DNA binding domain, it was classified into the “other” TF family.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of Y2H-based interactions
PPIs were called automatically by a modified version of the TIDY software which calculates a Z-score for a given interaction over multiple different analyzed images (see “Automated detection of interactions in Y2H screen” section under Method Details for detail).
Sensitivity and specificity of the Y2H assay
The sensitivity and specificity of our Y2H assay were calculated against the PRS and RRSs, respectively, as described in the Results section.
PBM data analysis
DNA binding specificities of PBM-assayed TFs are represented as E-scores for individual 8-mers. Details can be found in the “PBM data analysis” section under Method Details.
Statistical analysis for motif enrichment analysis
Details of this analysis can be found in the “Analysis of ChIP-chip/seq data” section under Method Details and are reported in Table 2.
DATA AND SOFTWARE AVAILABILITY
TF–TF PPI data resulting from our Y2H screen: Table S2.
The Integrated Drosophila TF–TF PPI Network: Table S2.
The catalog of TF pairs demonstrating direct and indirect DNA binding: Table 2.
The accession number for the universal PBM data reported in this paper is [UniPROBE (http://thebrain.bwh.harvard.edu/uniprobe/)]: [SHO18A].
Custom scripts and software were used for: automated Y2H PPI detection; processing Y2H data; extracting tissue expression information from BDGP; extracting TF family information; extracting MasterNet information; comparing the Drosophila interactome with C. elegans and human interactomes; hub protein analyses; and the construction of RRSs. All custom scripts and software used in this study are available upon request.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GST Tag Polyclonal Antibody, Alexa Fluor 488 | ThermoFisher SCIENTIFIC | A-11131; RRID: AB_2534137 |
| Anti-GST polyclonal primary antibody | SIGMA-ALDRICH | G7781; RRID: AB_259965 |
| Anti-HA antibody produced in rabbit | SIGMA-ALDRICH | H6908; RRID: AB_260070 |
| HA Tag Monoclonal Antibody (16B12), Alexa Fluor 488 | ThermoFisher SCIENTIFIC | A-21287; RRID: AB_2535829 |
| Monoclonal ANTI-FLAG® M2-Peroxidase (HRP) antibody produced in mouse | SIGMA-ALDRICH | A8592; RRID: AB_439702 |
| FLAG Tag Antibody, Alexa Fluor 488 | Cell Signaling TECHNOLOGY | 5407; RRID: AB_1950473 |
| Goat HRP-conjugated anti-rabbit IgG | ThermoFisher SCIENTIFIC | 31460; RRID: AB_228341 |
| Goat HRP-conjugated anti-mouse IgG | ThermoFisher SCIENTIFIC | 32430; RRID: AB_1185566 |
| Anti-GFP antibody (Biotin) | Abcam | ab6658; RRID: AB_305631 |
| Anti-RFP antibody (Biotin) | Abcam | ab34771; RRID: AB_777699 |
| Bacterial and Virus Strains | ||
| E. coli C41 DE3 cells | Lucigen | 60444 |
| Gateway Vector Conversion System with One Shot ccdB Survival Cells | ThermoFisher SCIENTIFIC | 11828029 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NeutrAvidin Protein | ThermoFisher SCIENTIFIC | 31000 |
| Glutathione S-Transferase from E. coli | SIGMA-ALDRICH | G5663 |
| Amino-terminal FLAG-BAP fusion protein | SIGMA-ALDRICH | P7582 |
| E. coli positive control whole cell lysate | Abcam | Ab5395 |
| Biotinylated BSA | SIGMA-ALDRICH | A8549 |
| Cy3-conjugated dUTP | GE Healthcare | PA53022 |
| Protease, from Streptomyces griseus | SIGMA-ALDRICH | P6911 |
| Thermo sequenase cycle sequencing kit | USB | 78500 |
| Gateway® Vector Conversion System with One Shot® ccdB Survival Cells | ThermoFisher SCIENTIFIC | 11828029 |
| Carboxy-terminal FLAG-BAP fusion protein | SIGMA-ALDRICH | p7457 |
| Critical Commercial Assays | ||
| PURExpress in vitro transcription and translation kit | NEB | E6800 |
| 1-Step Human Coupled IVT Kit - DNA | ThermoFisher SCIENTIFIC | 88881 |
| TnT® Coupled Wheat Germ Extract System | Promega | L3260 |
| Deposited Data | ||
| PBM data and derived DNA binding specificity motifs | This paper | http://the_brain.bwh.harvard.edu/uniprobe/ (Accession ID: SHO18A) |
| TF DNA binding specificity motifs | FlyFactorSurvey (Zhu et al., 2011); CIS-BP (Weirauch et al., 2014) |
http://mccb.umassmed.edu/ffs/; http://cisbp.ccbr.utoronto.ca/ |
| ChIP datasets | See Table S7 | N/A |
| Y2H TF-TF interactome data | This paper | Table S2 |
| Integrated TF-TF Interactome | This paper | Table S2 |
| MasterNet (Freeze April 2015) | Friedman et al., 2011 | N/A |
| C. elegans TF-TF interactome | Reece-Hoyes et al., 2013 | N/A |
| Human TF-TF interactome | Ravasi et al., 2010 | N/A |
| Human Gene Mutation Database (HGMD) | Stenson et al., 2003 | http://www.hgmd.cf.ac.uk/ |
| Berkeley Drosophila Genome Project (BDGP) | Hammonds et al., 2013 | http://www.fruitfly.org/ |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: UAS-TF-3xHA fly strains | Schertel et al., 2015 | https://flyorf.ch/ |
| S. cerevisiae: MaV203 and MaV103 strains | Vidal et al., 1996a, 1996b | N/A |
| Oligonucleotides | ||
| HPLC-purified primer (unmodified) for double-stranding of DNA oligonucleotide array 5’-CAGCACGGACAACGGAACACAGAC-3’ | Integrated DNA Technologies | http://www.idtdna.com/pages |
| Recombinant DNA | ||
| Plasmid: pTSVNm9.attB & pTSVC155.attB | Bischof et al., 2013 | https://flyorf.ch/ |
| Plasmid: pT7CFE1-NFtag Vector | ThermoFisher SCIENTIFIC | 88865 |
| Plasmid: pT7CFE1-NHA | ThermoFisher SCIENTIFIC | 88861 |
| Plasmid: pF3A-eGFP | Isakova et al., 2016 | N/A |
| Plasmid: pF3A-mCherry | Isakova et al., 2016 | N/A |
| Plasmid: pGW-HA.attB | Bischof et al., 2013 | https://flyorf.ch/ |
| Plasmid: pAD-DEST | Walhout and Vidal, 2001 | N/A |
| Plasmid: pDB-DEST | Walhout and Vidal, 2001 | N/A |
| Plasmids: Drosophila TF clones collection | Hens et al., 2011 | N/A |
| Software and Algorithms | ||
| Universal PBM Analysis Suite | Berger and Bulyk, 2009 | http://the_brain.bwh.harvard.edu/PBMAnalysisSuite/index.html |
| MATLAB | N/A | https://www.mathworks.com |
| TIDY | Hens et al., 2011 | Available by request |
| Python (2.7.12) | N/A | https://www.python.org/ |
| Perl | N/A | https://www.perl.org/ |
| AMIGO2 | Balsa-Canto et al., 2016 | http://amigo.geneontology.org/amigo/ |
| DRSC Integrative Ortholog Prediction Tool (DIOPT) | Hu et al. 2011 | https://www.fiyrnai.org/cgi-bin/DRSC_orthologs.pl |
| liftOver | Hinrichs et al., 2006 | https://genome.ucsc.edu/cgi-bin/hgLiftOver |
| GENRE | Mariani et al., 2017 | http://thebrain.bwh.harvard.edu/glossary-GENRE/download.html |
| GOMER | Granek and Clarke, 2005 | http://people.duke.edu/~josh/biophysicsweb/GOMER/index.html |
| arrayWorx | Applied Precision | (discontinued) |
| CytoScape (v 3.4.0) | Shannon et al., 2003 | https://cytoscape.org |
| NetworkAnalyzer | Doncheva et al., 2012 | Available in CytoScape |
| ImageJ | https://imagej.nih.gov/ij/ | |
| Other | ||
| Custom-designed “universal all 10-mer” oligonucleotide arrays | Agilent Technologies | AMADID #030236 |
Highlights.
Comprehensive yeast PPI screen among essentially all D. melanogaster TFs
A subset of Y2H results validated by in vitro and in vivo binary interaction assays
Compilation of an integrated fly TF-TF interactome
TF pairs can be recruited to DNA in different modes
ACKNOWLEDGMENTS
We thank R. Gordân, L. Barrera, B. Zhou, and H. Mulholland for technical assistance; A. Vinayagam for assistance with MasterNet; the Center for Cancer Systems Biology at Dana-Farber Cancer Institute for assistance with cloning for PBM experiments; M. Calderwood and D. Hill for helpful discussions; and M. Hafner for technical assistance with the image analysis software. This work was funded by NIH grant R01 GM104062 (to M.L.B.), American Heart Association Postdoctoral Fellowship 13POST17110016 (to L.S.), Boehringer Ingelheim Fonds PhD Fellowship (to A.H.), NIH Bioinformatics and Integrative Genomics training grant T32 HG002295 (to K.W.), SystemsX.ch (AgingX, to B.D.), the Swiss National Science Foundation (CRSI33_127485, to B.D.), and institutional support from the École Polytechnique Fédérale de Lausanne (EPFL) (to B.D.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.071.
DECLARATION OF INTERESTS
M.L.B. is a co-inventor on patents on PBM technology.
REFERENCES
- Adryan B, and Teichmann SA (2006). FlyTF: a systematic review of site-specific transcription factors in the fruit fly Drosophila melanogaster. Bioinformatics 22, 1532–1533. [DOI] [PubMed] [Google Scholar]
- Adryan B, and Teichmann SA (2010). The developmental expression dynamics of Drosophila melanogaster transcription factors. Genome Biol 11, R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allada R, Emery P, Takahashi JS, and Rosbash M (2001). Stopping time: the genetics of fly and mouse circadian clocks. Annu. Rev. Neurosci 24, 1091–1119. [DOI] [PubMed] [Google Scholar]
- Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, Derow C, Feuermann M, Ghanbarian AT, Kerrien S, Khadake J, et al. (2010). The IntAct molecular interaction database in 2010. Nucleic Acids Res 38, D525–D531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babb R, Huang CC, Aufiero DJ, and Herr W (2001). DNA recognition by the herpes simplex virus transactivator VP16: a novel DNA-binding structure. Mol. Cell. Biol 21, 4700–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H, Kang P, Hernandez AM, and Tatar M (2013). Activin signaling targeted by insulin/dFOXO regulates aging and muscle proteostasis in Drosophila. PLoS Genet 9, e1003941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsa-Canto E, Henriques D, Gábor A, and Banga JR (2016). AMIGO2, a toolbox for dynamic modeling, optimization and control in systems biology. Bioinformatics 32, 3357–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera LA, Vedenko A, Kurland JV, Rogers JM, Gisselbrecht SS, Rossin EJ, Woodard J, Mariani L, Kock KH, Inukai S, et al. (2016). Survey of variation in human transcription factors reveals prevalent DNA binding changes. Science 351, 1450–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckingham KM, Armstrong JD, Texada MJ, Munjaal R, and Baker DA (2005). Drosophila melanogaster–the model organism of choice for the complex biology of multi-cellular organisms. Gravit. Space Biol. Bull 18, 17–29. [PubMed] [Google Scholar]
- Beckstead RB, Lam G, and Thummel CS (2005). The genomic response to 20-hydroxyecdysone at the onset of Drosophila metamorphosis. Genome Biol 6, R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, and Bulyk ML (2006). Protein binding microarrays (PBMs) for rapid, high-throughput characterization of the sequence specificities of DNA binding proteins. Methods Mol. Biol 338, 245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, and Bulyk ML (2009). Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat. Protoc 4, 393–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW 3rd, and Bulyk ML (2006). Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol 24, 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi-Frias D, Orian A, Delrow JJ, Vazquez J, Rosales-Nieves AE, and Parkhurst SM (2004). Hairy transcriptional repression targets and cofactor recruitment in Drosophila. PLoS Biol 2, E178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Björklund M, Furger E, Schertel C, Taipale J, and Basler K (2013). A versatile platform for creating a comprehensive UAS-ORFeome library in Drosophila. Development 140, 2434–2442. [DOI] [PubMed] [Google Scholar]
- Bradley RK, Li XY, Trapnell C, Davidson S, Pachter L, Chu HC, Tonkin LA, Biggin MD, and Eisen MB (2010). Binding site turnover produces pervasive quantitative changes in transcription factor binding between closely related Drosophila species. PLoS Biol 8, e1000343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun P, Tasan M, Dreze M, Barrios-Rodiles M, Lemmens I, Yu H, Sahalie JM, Murray RR, Roncari L, de Smet AS, et al. (2009). An experimentally derived confidence score for binary protein-protein interactions. Nat. Methods 6, 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayer KJ, and Segal DJ (2008). Keep your fingers off my DNA: protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem. Biophys 50, 111–131. [DOI] [PubMed] [Google Scholar]
- Brennan CA, Li TR, Bender M, Hsiung F, and Moses K (2001). Broad-complex, but not ecdysone receptor, is required for progression of the morphogenetic furrow in the Drosophila eye. Development 128, 1–11. [DOI] [PubMed] [Google Scholar]
- Carroll SB, Grenier JK, and Weatherbee SD (2005). From DNA to Diversity: Molecular Genetics and the Evolution of Animal Design, Second Edition (Blackwell; ). [Google Scholar]
- Ceol A, Chatr Aryamontri A, Licata L, Peluso D, Briganti L, Perfetto L, Castagnoli L, and Cesareni G (2010). MINT, the molecular interaction database: 2009 update. Nucleic Acids Res 38, D532–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintapalli VR, Wang J, and Dow JA (2007). Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet 39, 715–720. [DOI] [PubMed] [Google Scholar]
- Cyran SA, Buchsbaum AM, Reddy KL, Lin MC, Glossop NR, Hardin PE, Young MW, Storti RV, and Blau J (2003). vrille, Pdp1, and dClock form a second feedback loop in the Drosophila circadian clock. Cell 112, 329–341. [DOI] [PubMed] [Google Scholar]
- Deplancke B, Alpern D, and Gardeux V (2016). The Genetics of Transcription Factor DNA Binding Variation. Cell 166, 538–554. [DOI] [PubMed] [Google Scholar]
- Doncheva NT, Assenov Y, Domingues FS, and Albrecht M (2012). Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc 7, 670–685. [DOI] [PubMed] [Google Scholar]
- El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res 47 (D1), D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk GJ, and Thummel CS (1998). The DHR78 nuclear receptor is required for ecdysteroid signaling during the onset of Drosophila metamorphosis. Cell 93, 543–555. [DOI] [PubMed] [Google Scholar]
- Formstecher E, Aresta S, Collura V, Hamburger A, Meil A, Trehin A, Reverdy C, Betin V, Maire S, Brun C, et al. (2005). Protein interaction mapping: a Drosophila case study. Genome Res 15, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis MJ, Roche S, Cho MJ, Beall E, Min B, Panganiban RP, and Rio DC (2016). Drosophila IRBP bZIP heterodimer binds P-element DNA and affects hybrid dysgenesis. Proc. Natl. Acad. Sci. USA 113, 13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CR, Erickson T, Callander D, Berry KM, Koss R, Hagey DW, Stout J, Wuennenberg-Stapleton K, Ngai J, Moens CB, and Waskiewicz AJ (2007). Pbx homeodomain proteins pattern both the zebrafish retina and tectum. BMC Dev. Biol 7, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AA, Tucker G, Singh R, Yan D, Vinayagam A, Hu Y, Binari R, Hong P, Sun X, Porto M, et al. (2011). Proteomic and functional genomic landscape of receptor tyrosine kinase and ras to extracellular signal-regulated kinase signaling. Sci. Signal 4, rs10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauhar Z, Sun LV, Hua S, Mason CE, Fuchs F, Li TR, Boutros M, and White KP (2009). Genomic mapping of binding regions for the Ecdysone receptor protein complex. Genome Res 19, 1006–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber D, Maerkl SJ, and Quake SR (2009). An in vitro microfluidic approach to generating protein-interaction networks. Nat. Methods 6, 71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, et al. (2003). A protein interaction map of Drosophila melanogaster. Science 302, 1727–1736. [DOI] [PubMed] [Google Scholar]
- Glossop NR, Houl JH, Zheng H, Ng FS, Dudek SM, and Hardin PE (2003). VRILLE feeds back to control circadian transcription of Clock in the Drosophila circadian oscillator. Neuron 37, 249–261. [DOI] [PubMed] [Google Scholar]
- Gordân R, Hartemink AJ, and Bulyk ML (2009). Distinguishing direct versus indirect transcription factor-DNA interactions. Genome Res 19, 2090–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordân R, Murphy KF, McCord RP, Zhu C, Vedenko A, and Bulyk ML (2011). Curated collection of yeast transcription factor DNA binding specificity data reveals novel structural and gene regulatory insights. Genome Biol 12, R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramates LS, Marygold SJ, Santos GD, Urbano JM, Antonazzo G, Matthews BB, Rey AJ, Tabone CJ, Crosby MA, Emmert DB, et al. ; the FlyBase Consortium (2017). FlyBase at 25: looking to the future. Nucleic Acids Res 45 (D1), D663–D671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granek JA, and Clarke ND (2005). Explicit equilibrium modeling of transcription-factor binding and gene regulation. Genome Biol 6, R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. (2011). The developmental transcriptome of Drosophila melanogaster. Nature 471, 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan G, Reinke AW, and Keating AE (2009). Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature 458, 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove CA, De Masi F, Barrasa MI, Newburger DE, Alkema MJ, Bulyk ML, and Walhout AJ (2009). A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell 138, 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruharsha KG, Rual JF, Zhai B, Mintseris J, Vaidya P, Vaidya N, Beekman C, Wong C, Rhee DY, Cenaj O, et al. (2011). A protein complex network of Drosophila melanogaster. Cell 147, 690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammonds AS, Bristow CA, Fisher WW, Weiszmann R, Wu S, Hartenstein V, Kellis M, Yu B, Frise E, and Celniker SE (2013). Spatial expression of transcription factors in Drosophila embryonic organ development. Genome Biol 14, R140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison MM, Li XY, Kaplan T, Botchan MR, and Eisen MB (2011). Zelda binding in the early Drosophila melanogaster embryo marks regions subsequently activated at the maternal-to-zygotic transition. PLoS Genet 7, e1002266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hens K, Feuz JD, Isakova A, Iagovitina A, Massouras A, Bryois J, Callaerts P, Celniker SE, and Deplancke B (2011). Automated protein-DNA interaction screening of Drosophila regulatory elements. Nat. Methods 8, 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, et al. (2006). The UCSC Genome Browser Database: update 2006. Nucleic Acids Res 34, D590–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CD, Chinenov Y, and Kerppola TK (2002). Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9, 789–798. [DOI] [PubMed] [Google Scholar]
- Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, and Mohr SE (2011). An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakova A, Berset Y, Hatzimanikatis V, and Deplancke B (2016). Quantification of Cooperativity in Heterodimer-DNA Binding Improves the Accuracy of Binding Specificity Models. J. Biol. Chem 291, 10293–10306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, and Brown PO (2001). Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature 409, 533–538. [DOI] [PubMed] [Google Scholar]
- Jakobsen JS, Braun M, Astorga J, Gustafson EH, Sandmann T, Karzynski M, Carlsson P, and Furlong EE (2007). Temporal ChIP-on-chip reveals Biniou as a universal regulator of the visceral muscle transcriptional network. Genes Dev 21, 2448–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, and Wold B (2007). Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502. [DOI] [PubMed] [Google Scholar]
- Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M, Enge M, Kivioja T, Morgunova E, and Taipale J (2015). DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527, 384–388. [DOI] [PubMed] [Google Scholar]
- Kanki Y, Nakaki R, Shimamura T, Matsunaga T, Yamamizu K, Katayama S, Suehiro JI, Osawa T, Aburatani H, Kodama T, et al. (2017). Dynamically and epigenetically coordinated GATA/ETS/SOX transcription factor expression is indispensable for endothelial cell differentiation. Nucleic Acids Res 45, 4344–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, et al. (2009). Human Protein Reference Database–2009 update. Nucleic Acids Res 37, D767–D772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradpour P, Stark A, Roy S, and Kellis M (2007). Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res 17, 1919–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella RJ, Kähäri A, Haider S, Zamora J, Proctor G, Spudich G, Almeida-King J, Staines D, Derwent P, Kerhornou A, et al. (2011). Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford) 2011, bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar JP (2009). The molecular circuitry governing retinal determination. Biochim. Biophys. Acta 1789, 306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, and Tjian R (2003). Transcription regulation and animal diversity. Nature 424, 147–151. [DOI] [PubMed] [Google Scholar]
- Li XY, Thomas S, Sabo PJ, Eisen MB, Stamatoyannopoulos JA, and Biggin MD (2011). The role of chromatin accessibility in directing the wide-spread, overlapping patterns of Drosophila transcription factor binding. Genome Biol 12, R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb JD, Liu X, Botstein D, and Brown PO (2001). Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat. Genet 28, 327–334. [DOI] [PubMed] [Google Scholar]
- Liu F, Walmsley M, Rodaway A, and Patient R (2008). Fli1 acts at the top of the transcriptional network driving blood and endothelial development. Curr. Biol 18, 1234–1240. [DOI] [PubMed] [Google Scholar]
- Lowe N, Rees JS, Roote J, Ryder E, Armean IM, Johnson G, Drummond E, Spriggs H, Drummond J, Magbanua JP, et al. (2014). Analysis of the expression patterns, subcellular localisations and interaction partners of Drosophila proteins using a pigP protein trap library. Development 141, 3994–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur S, Li XY, Li J, Brown JB, Chu HC, Zeng L, Grondona BP, Hechmer A, Simirenko L, Keränen SV, et al. (2009). Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol 10, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maerkl SJ, and Quake SR (2007). A systems approach to measuring the binding energy landscapes of transcription factors. Science 315, 233–237. [DOI] [PubMed] [Google Scholar]
- Mariani L, Weinand K, Vedenko A, Barrera LA, and Bulyk ML (2017). Identification of Human Lineage-Specific Transcriptional Coregulators Enabled by a Glossary of Binding Modules and Tunable Genomic Backgrounds. Cell Syst 5, 187–201.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulley DJ, and Black BL (2012). Transcription factor pathways and congenital heart disease. Curr. Top. Dev. Biol 100, 253–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meireles-Filho ACA, Bardet AF, Yáñez-Cuna JO, Stampfel G, and Stark A (2014). cis-regulatory requirements for tissue-specific programs of the circadian clock. Curr. Biol 24, 1–10. [DOI] [PubMed] [Google Scholar]
- Michelson AM, and Bulyk ML (2006). Biological code breaking in the 21st century. Mol. Syst. Biol 2, 2006.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- modENCODE Consortium; Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, et al. (2010). Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330, 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali T, Pacifico S, Yu J, Guest S, Roberts GG 3rd, and Finley RL Jr. (2011). DroID 2011: a comprehensive, integrated resource for protein, transcription factor, RNA and gene interactions for Drosophila. Nucleic Acids Res 39, D736–D743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevil M, Bondra ER, Schulz KN, Kaplan T, and Harrison MM (2017). Stable Binding of the Conserved Transcription Factor Grainy Head to its Target Genes Throughout Drosophila melanogaster Development. Genetics 205, 605–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares AM, Han Y, Soto D, Flattery K, Marini J, Mollema N, Haider A, Escher P, DeAngelis MM, and Haider NB (2017). The nuclear hormone receptor gene Nr2c1 (Tr2) is a critical regulator of early retina cell patterning. Dev. Biol 429, 343–355. [DOI] [PubMed] [Google Scholar]
- Pai CY, Kuo TS, Jaw TJ, Kurant E, Chen CT, Bessarab DA, Salz-berg A, and Sun YH (1998). The Homothorax homeoprotein activates the nuclear localization of another homeoprotein, extradenticle, and suppresses eye development in Drosophila. Genes Dev 12, 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris M, Kaplan T, Li XY, Villalta JE, Lott SE, and Eisen MB (2013). Extensive divergence of transcription factor binding in Drosophila embryos with highly conserved gene expression. PLoS Genet 9, e1003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potier D, Davie K, Hulselmans G, Naval Sanchez M, Haagen L, HuynhThu VA, Koldere D, Celik A, Geurts P, Christiaens V, and Aerts S (2014). Mapping gene regulatory networks in Drosophila eye development by large-scale transcriptome perturbations and motif inference. Cell Rep 9, 2290–2303. [DOI] [PubMed] [Google Scholar]
- Pusch S, Dissmeyer N, and Schnittger A (2011). Bimolecular-fluorescence complementation assay to monitor kinase-substrate interactions in vivo. Methods Mol. Biol 779, 245–257. [DOI] [PubMed] [Google Scholar]
- Ravasi T, Suzuki H, Cannistraci CV, Katayama S, Bajic VB, Tan K, Akalin A, Schmeier S, Kanamori-Katayama M, Bertin N, et al. (2010). An atlas of combinatorial transcriptional regulation in mouse and man. Cell 140, 744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece-Hoyes JS, Pons C, Diallo A, Mori A, Shrestha S, Kadreppa S, Nelson J, Diprima S, Dricot A, Lajoie BR, et al. (2013). Extensive rewiring and complex evolutionary dynamics in a C. elegans multiparameter transcription factor network. Mol. Cell 51, 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginato MJ, Zhang J, and Lazar MA (1996). DNA-independent and DNA-dependent mechanisms regulate the differential heterodimerization of the isoforms of the thyroid hormone receptor with retinoid X receptor. J. Biol. Chem 271, 28199–28205. [DOI] [PubMed] [Google Scholar]
- Reiter F, Wienerroither S, and Stark A (2017). Combinatorial function of transcription factors and cofactors. Curr. Opin. Genet. Dev 43, 73–81. [DOI] [PubMed] [Google Scholar]
- Remy I, and Michnick SW (2004). Mapping biochemical networks with protein-fragment complementation assays. Methods Mol. Biol 261, 411–426. [DOI] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeit-linger J, Schreiber J, Hannett N, Kanin E, et al. (2000). Genome-wide location and function of DNA binding proteins. Science 290, 2306–2309. [DOI] [PubMed] [Google Scholar]
- Ren W, Zhang Y, Li M, Wu L, Wang G, Baeg GH, You J, Li Z, and Lin X (2015). Windpipe controls Drosophila intestinal homeostasis by regulating JAK/STAT pathway via promoting receptor endocytosis and lysosomal degradation. PLoS Genet 11, e1005180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee DY, Cho DY, Zhai B, Slattery M, Ma L, Mintseris J, Wong CY, White KP, Celniker SE, Przytycka TM, et al. (2014). Transcription factor networks in Drosophila melanogaster. Cell Rep 8, 2031–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland T, Taşxan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, et al. (2014). A proteome-scale map of the human interactome network. Cell 159, 1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al. (2005). Towards a proteome-scale map of the human protein-protein interaction network. Nature 437, 1173–1178. [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Marty T, Casares F, Affolter M, and Mann RS (1999). Regulation of Hox target genes by a DNA bound Homothorax/Hox/Extradenticle complex. Development 126, 5137–5148. [DOI] [PubMed] [Google Scholar]
- Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, and Eisenberg D (2004). The Database of Interacting Proteins: 2004 update. Nucleic Acids Res 32, D449–D451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schertel C, Albarca M, Rockel-Bauer C, Kelley NW, Bischof J, Hens K, van Nimwegen E, Basler K, and Deplancke B (2015). A large-scale, in vivo transcription factor screen defines bivalent chromatin as a key property of regulatory factors mediating Drosophila wing development. Genome Res 25, 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D, Stelzer C, Gerlach D, Yáñez-Cuna JO, Rath M, Boryń LM, Arnold CD, and Stark A (2014). Hormone-responsive enhancer-activity maps reveal predictive motifs, indirect repression, and targeting of closed chromatin. Mol. Cell 54, 180–192. [DOI] [PubMed] [Google Scholar]
- Shubin N, Tabin C, and Carroll S (2009). Deep homology and the origins of evolutionary novelty. Nature 457, 818–823. [DOI] [PubMed] [Google Scholar]
- Siggers T, and Gordân R (2014). Protein-DNA binding: complexities and multi-protein codes. Nucleic Acids Res 42, 2099–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggers T, Duyzend MH, Reddy J, Khan S, and Bulyk ML (2011). Non-DNA-binding cofactors enhance DNA-binding specificity of a transcriptional regulatory complex. Mol. Syst. Biol 7, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonis N, Rual JF, Carvunis AR, Tasan M, Lemmens I, Hirozane-Kishikawa T, Hao T, Sahalie JM, Venkatesan K, Gebreab F, et al. (2009). Empirically controlled mapping of the Caenorhabditis elegans protein-protein interactome network. Nat. Methods 6, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery M, Ma L, Négre N, White KP, and Mann RS (2011). Genome-wide tissue-specific occupancy of the Hox protein Ultrabithorax and Hox cofactor Homothorax in Drosophila. PLoS One 6, e14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, and Furlong EE (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet 13, 613–626. [DOI] [PubMed] [Google Scholar]
- Stanyon CA, Liu G, Mangiola BA, Patel N, Giot L, Kuang B, Zhang H, Zhong J, and Finley RL Jr. (2004). A Drosophila protein-interaction map centered on cell-cycle regulators. Genome Biol 5, R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang X, Shi X, et al. (2011). The BioGRID Interaction Database: 2011 update. Nucleic Acids Res 39, D698–D704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellberger T, Häuser R, Baiker A, Pothineni VR, Haas J, and Uetz P (2010). Improving the yeast two-hybrid system with permutated fusions proteins: the Varicella Zoster Virus interactome. Proteome Sci 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, et al. (2005). A human protein-protein interaction network: a resource for annotating the proteome. Cell 122, 957–968. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, and Cooper DN (2003). Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat 21, 577–581. [DOI] [PubMed] [Google Scholar]
- Tatar M, Bartke A, and Antebi A (2003). The endocrine regulation of aging by insulin-like signals. Science 299, 1346–1351. [DOI] [PubMed] [Google Scholar]
- Venkatesan K, Rual JF, Vazquez A, Stelzl U, Lemmens I, Hirozane-Kishikawa T, Hao T, Zenkner M, Xin X, Goh KI, et al. (2009). An empirical framework for binary interactome mapping. Nat. Methods 6, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Brachmann RK, Fattaey A, Harlow E, and Boeke JD (1996a). Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc. Natl. Acad. Sci. USA 93, 10315–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Braun P, Chen E, Boeke JD, and Harlow E (1996b). Genetic characterization of a mammalian protein-protein interaction domain by using a yeast reverse two-hybrid system. Proc. Natl. Acad. Sci. USA 93, 10321–10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhout AJ, and Vidal M (2001). High-throughput yeast two-hybrid assays for large-scale protein interaction mapping. Methods 24, 297–306. [DOI] [PubMed] [Google Scholar]
- Walhout AJ, Boulton SJ, and Vidal M (2000). Yeast two-hybrid systems and protein interaction mapping projects for yeast and worm. Yeast 17, 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, et al. (2012). Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res 22, 1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. (2006). A global map of p53 transcription-factor binding sites in the human genome. Cell 124, 207–219. [DOI] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman CT, Yin Y, Corcoran DL, Ideker T, Stormo GD, and Benos PV (2005). enoLOGOS: a versatile web tool for energy normalized sequence logos. Nucleic Acids Res 33, W389–W392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, et al. (2008). High-quality binary protein interaction map of the yeast interactome network. Science 322, 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Yang Z, Yue Z, Alvarez JD, and Sehgal A (2007). FOXO and insulin signaling regulate sensitivity of the circadian clock to oxidative stress. Proc. Natl. Acad. Sci. USA 104, 15899–15904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Christensen RG, Kazemian M, Hull CJ, Enuameh MS, Basciotta MD, Brasefield JA, Zhu C, Asriyan Y, Lapointe DS, et al. (2011). FlyFactorSurvey: a database of Drosophila transcription factor binding specificities determined using the bacterial one-hybrid system. Nucleic Acids Res 39, D111–D117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzen RP, Girardot C, Gagneur J, Braun M, and Furlong EE (2009). Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature 462, 65–70. [DOI] [PubMed] [Google Scholar]
- Zolotarev N, Fedotova A, Kyrchanova O, Bonchuk A, Penin AA, Lando AS, Eliseeva IA, Kulakovskiy IV, Maksimenko O, and Georgiev P (2016). Architectural proteins Pita, Zw5, and ZIPIC contain homodimerization domain and support specific long-range interactions in Drosophila. Nucleic Acids Res 44, 7228–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
TF–TF PPI data resulting from our Y2H screen: Table S2.
The Integrated Drosophila TF–TF PPI Network: Table S2.
The catalog of TF pairs demonstrating direct and indirect DNA binding: Table 2.
The accession number for the universal PBM data reported in this paper is [UniPROBE (http://thebrain.bwh.harvard.edu/uniprobe/)]: [SHO18A].
Custom scripts and software were used for: automated Y2H PPI detection; processing Y2H data; extracting tissue expression information from BDGP; extracting TF family information; extracting MasterNet information; comparing the Drosophila interactome with C. elegans and human interactomes; hub protein analyses; and the construction of RRSs. All custom scripts and software used in this study are available upon request.