

Light field microscopy through bare optical fiber bundles paves the way for depth-resolved fluorescence microendoscopy.
Abstract
Optical fiber bundle microendoscopes are widely used for visualizing hard-to-reach areas of the human body. These ultrathin devices often forgo tunable focusing optics because of size constraints and are therefore limited to two-dimensional (2D) imaging modalities. Ideally, microendoscopes would record 3D information for accurate clinical and biological interpretation, without bulky optomechanical parts. Here, we demonstrate that the optical fiber bundles commonly used in microendoscopy are inherently sensitive to depth information. We use the mode structure within fiber bundle cores to extract the spatio-angular description of captured light rays—the light field—enabling digital refocusing, stereo visualization, and surface and depth mapping of microscopic scenes at the distal fiber tip. Our work opens a route for minimally invasive clinical microendoscopy using standard bare fiber bundle probes. Unlike coherent 3D multimode fiber imaging techniques, our incoherent approach is single shot and resilient to fiber bending, making it attractive for clinical adoption.
INTRODUCTION
In ray optics, the light field is a spatio-angular description of light rays emanating from a scene. Light field imaging systems give the user the ability to computationally refocus, change viewpoint, and quantify scene depth, all from a single exposure (1–5). These systems are particularly advantageous when the ability to capture three-dimensional (3D) image data are compromised because of experimental constraints. For example, it is challenging to record 3D volumes at high speed because of the inertia involved with mechanical focusing. In this case, light field imaging provides an elegant inertia-free, single-shot alternative for 3D fluorescence microscopy (3). In principle, light field architectures can also be compact, since focusing is achieved computationally instead of mechanically. As a result, electromechanical objective lens systems can be simplified or eliminated—a fixed lens can provide ample functionality in a light field system. This is particularly attractive for fluorescence microendoscopy, where millimeter-scale diameter fiber optic imaging probes (6–8) are currently used to reach convoluted regions of the body (e.g., the distal lung) (9, 10). Engineering and powering remote microelectromechanical optical systems remain elusive at this length scale, and as such, clinical fluorescence microendoscopes concede focusing and depth-resolved imaging capabilities in favor of a slim cross section. Consequently, acquiring consistently in-focus images of microscopic structures is challenging, and crucial depth information is lost. A slim light field microendoscope would enable clinicians to capture crucial depth-resolved tissue structure, yielding more informative optical biopsies with improved ease of operation.
Despite the potential utility of a fiber optic light field microendoscope, one has yet to be realized because the light field is scrambled upon optical fiber propagation. In principle, 3D image information can be unscrambled for coherent light in single-core multimode fibers via transmission matrix or wavefront shaping techniques (11–14). In practice, these approaches are incredibly sensitive to dynamic fiber bending, and the required beam scanning can be slow, making it inappropriate for clinical use in its current form. Moreover, these techniques are ineffective for incoherent light such as fluorescence. Optical fiber bundles solve the problem of incoherent image transmission by subdividing the image-relaying task to thousands of small cores with relatively little cross-talk (6, 15, 16). The convenience of the fiber bundle approach has made it the dominant microendoscopic solution, although it still lacks depth-resolved imaging capability.
In this work, we show that fiber bundles do, in fact, transmit depth information in the form of a light field. Our key observation is that the light field’s angular dimension is contained within the fiber bundle’s intracore intensity patterns, which have traditionally been ignored. We quantitatively relate these intensity patterns, which arise because of angle-dependent modal coupling, to the angular structure of the light field. Our work establishes optical fiber bundles as a new class of light field sensor, alongside microlens arrays (1–4), aperture masks (17), angle-sensitive pixels (18), and camera arrays (19). We demonstrate that optical fiber bundles can perform single-shot surface and depth mapping with accuracy better than 10 μm at up to ~80 μm from the fiber bundle facet. For context, we note that this depth ranging resolution is better than the confocal slice thickness of commercial bare fiber, fixed-focus microendoscopes (7), which are not capable of resolving features in depth.
RESULTS
Principle of operation
Consider a fluorescent point source (a fluorescent bead) imaged through an optical fiber bundle, as shown schematically in Fig. 1 (A and B) (full optical setup shown in fig. S1). The raw output image of the bead at an axial distance of z = 26 μm as seen through the fiber bundle is shown in Fig. 1C. A radially symmetric pattern of fiber modes is easily visible because of the relationship between modal coupling efficiency and input ray angle (15, 20). The fiber bundle used in this work (Fig. 1D) has an outer diameter of 750 μm and contains ~30,000 roughly circular cores with 3.2-μm average center-to-center spacing, average core radius a = 1 μm, and a numerical aperture (NA) of 0.39 (16). On average, each core in this fiber bundle will support approximately modes at λ = 550 nm (21).
Fig. 1. Imaging a point source with an optical fiber bundle.
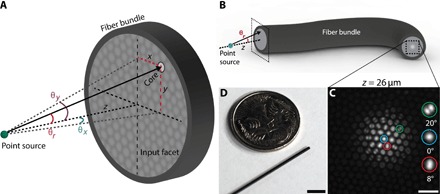
(A) Schematic of point source placed a distance z from the input (distal) facet of a bare optical fiber bundle. A ray traveling from the point emitter to an arbitrary core on a fiber facet with an angle of incidence of θr is shown. Angles θx and θy are also defined here. (B) A schematic of a fiber bundle showing its input (left side) and output facets (right side). The image of a point source is transmitted through the bundle cores to the output facet. A zoomed-in view of the input facet geometry is shown in (A). (C) Example raw image of the distal facet when observing a fluorescent bead at a depth of z = 26 μm from the input facet. The bead diameter is smaller than the core diameter, and therefore, the bead approximates a fluorescent point source. Three example cores are expanded in the inset and displayed along with the angle of incidence of input light, calculated from the known bead depth (see “Beads” section) and location of the core on the facet. Scale bar, 10 μm. (D) Photograph of the optical fiber bundle used in this work (diameter, 750 μm; 30,000 cores) next to an Australian five-cent coin. Scale bar, approximately 5 mm.
Under typical operation, the modal information in Fig. 1C is lost because the raw image from fiber bundle output is downsampled to remove pixelation from the fiber cores. Recently, however, we showed that the intracore intensity structure of this output light carries information about the angular distribution of the input light, enabling digital manipulation of the fiber’s NA in postprocessing (15). This is possible because the higher-order modes, which are preferentially excited at larger angles of incidence (20, 22), carry more energy near the core-cladding interface than the lower-order modes. Light is effectively pushed toward the edge of each core with increasing ray angle (Fig. 1C and fig. S2). When using incoherent light (i.e., fluorescence), this modal information is conveniently robust against fiber bending (figs. S3 and S4). By selectively removing light near the periphery of each core (Fig. 2, A and B), a process we call digital aperture filtering, one can achieve a synthetically constricted NA (see Fig. 2, C and D, and “Digital aperture filtering” section).
Fig. 2. Digital aperture filtering.
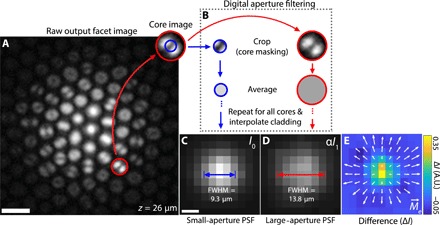
(A) Example raw output facet image from an optical fiber bundle for a fluorescent bead at a distance of z = 26 μm from the input facet. Scale bar, 5 μm. (B) Image of the mode pattern within a single core. The red and blue circles indicate example averaging regions for large- (full) and small-aperture images, respectively. Traditional large-aperture images are created by averaging the entire region circled in red for each core and then filling in the missing region between cores [dark cladding in (A)] via linear interpolation. A large-aperture image is what is traditionally obtained using fiber bundle depixelation techniques. Small-aperture images are created in the same way, with an average taken over the smaller blue circle. (C) Small-aperture image (I0) of a fluorescent bead at z = 26 μm after cladding interpolation. Scale bar, 5 μm. (D) Intensity-scaled large-aperture image (αI1) of a fluorescent bead at z = 26 μm after cladding interpolation. The scaling constant α is chosen such that the total intensity of (C) and (D) are equal. See section S1 for details. PSF full width at half maximum (FWHM) for small and large apertures are indicated in (C) and (D), respectively. (E) Difference between small-aperture and (intensity-scaled) large-aperture images. Arrows show the effective light field moment vector field . A.U., arbitrary units.
The full orientation of input light cannot be ascertained from this observation alone because of azimuthal degeneracies of the core’s modes (Fig. 2A). To circumvent this issue, we use “light field moment imaging” (LMI) (23) to extract the missing azimuthal information. In LMI, a continuity equation describing conservation of energy between two image planes is used to calculate the average ray direction (represented by the light field moment vector ) at a given point in the image I (23)
| (1) |
where . From this information, a light field L(x, y, u, v) can be constructed assuming a Gaussian distribution in (angular) uv space around this average ray angle
| (2) |
Here, angular ray space is parametrized by u = tan θx and v = tan θy, where tan θx,y are the angles of inclination of rays from the yz and xz planes, respectively (Fig. 1A). In this notation, , and σ is an adjustable parameter that we will soon address. This Gaussian assumption is based on the fact that a finely spatially sampled light field loses all structure in the angular domain (2, 5), similar to a Shack-Hartmann wavefront sensor (24). The resulting light field reveals depth information via lateral motion of objects when changing viewpoint and can be processed into stereographs, full-parallax animations, refocused images, and depth maps (1, 2, 23, 25).
Conventional LMI (Eq. 1) requires a pair of input images at different focus positions. However, fine focus control is not available on most microendoscopes, and even if it were, traditional LMI is not single shot. Instead, we modify Eq. 1 so that it can be used with pairs of images at the same focus position but with different collection apertures.
Imaging model
Consider a point source a distance z from the bare fiber facet. This source is out of focus since there is no imaging lens on the fiber facet. Thus, the apparent size of the point source as viewed from the output facet will grow with increasing acceptance angle (i.e., NA) of the fiber. When the fiber NA is computationally reduced from a large (full) aperture (Fig. 2B, red) to a smaller aperture (Fig. 2B, blue) by core masking, the width of the point-spread function (PSF) also decreases (Fig. 2, C and D) because of the increased depth of field (15). This behavior is opposite to diffraction-limited systems where the PSF shrinks with increasing NA. This is because the resolution in our case is limited by defocus, not by diffraction.
We model the PSF of the system as a 2D Gaussian with width proportional to tan θ (26), where θ is the maximum ray angle collected by the fiber (to be computationally adjusted after capture)
| (3) |
By considering a collection of j point sources, we arrive at the following modified LMI equation that depends on two images, I0 and I1, with maximum collection angles (apertures) θ0 and θ1 (see section S1)
| (4) |
where , is the effective light field moment vector, zj is the depth of point source j, BjPSFj is the intensity at position (x, y) due to point source j, and . Equation 4 is convenient since we can obtain both I0 and I1 in a single shot via digital aperture filtering. We subsequently solve for in the Fourier domain; the resulting for a fluorescent bead at z = 26 μm is superimposed over the image ΔI = I0 − αI1 in Fig. 2E. Last, we construct a light field as in Eq. 2, with . This substitution alters the parallax behavior of the light field such that the centroid shift of point source is not linear in z, as would be the case with a standard light field (see section S2) (25)
| (5) |
where h ≝ σ/tan θ0 is an adjustable reconstruction parameter and σ0 is the full width at half maximum (FWHM) of the PSF at z = 0. We obtain tan θ0, tan θ1, and σ0 experimentally by fitting a 2D Gaussian to images of isolated beads at a series of depths for large and small apertures (Fig. 3A). Figure 3B shows experimentally measured values for fluorescent beads at 1 to 101 μm from the fiber facet, along with simulated and theoretical results. Both simulation and theory show very good agreement with experimental data for a range of h values. The theoretical curves use known physical (z, tan θ0) and reconstruction quantities (u, v, h); no fitting parameters are used. The lateral shift of a fluorescent bead as a function of bead depth is shown in Fig. 3B, where left and right viewpoint images are shown in cyan and red, respectively (viewable with red-cyan stereo glasses). The characteristic slanted lines of point sources at different depths in the epipolar plane (27) (here, a yv slice of the light field) can be observed in Fig. 3C. The linearity of with [u, v] is verified in fig. S5.
Fig. 3. Imaging model quantification.
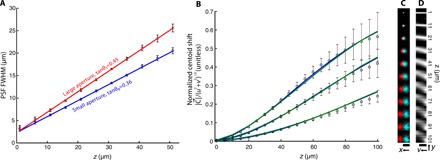
(A) PSF full width at half maximum (FWHM) for fluorescent beads located 1 to 51 μm from the fiber facet. Square (red) and circle (blue) datapoints are obtained via 2D Gaussian fits to the full aperture of the fiber bundle tanθ1 and the synthetically reduced fiber bundle collection aperture tanθ0, respectively. Error bars correspond to the FWHM SD over five beads. Red and blue lines are linear fits to the large- and small-aperture datasets, respectively. The slope of the fits represents the effective aperture of the fiber bundle: tanθ1 = 0.4534 (95% confidence bounds, 0.4436 to 0.4632) and tanθ0 = 0.3574 (95% confidence bounds, 0.3496 to 0.3652). The y intercept (denoted by σ0) is the FWHM of a bead located on the fiber facet due to finite sampling density of the fiber cores: σ0 = 2.322 μm (95% confidence bounds, 2.095 to 2.517 μm) and σ0 = 2.281 μm (95% confidence bounds, 2.027 to 2.534 μm) for large and small apertures, respectively. (B) Fluorescent bead centroid shift (disparity) as a function of bead depth for h = 75, 100, and 150 μm. Centroid shift is reported as the magnitude of the centroid shift in xy-space per unit displacement in uv-space (see Eq. 5). Datapoints are experimentally measured values, and error bars represent the SD over five beads. Blue and green curves are theoretical (following Eq. 5) and simulated centroid shifts, respectively. The SE in depth as a function of true depth is shown in fig. S6. (C) Extreme left- and right-viewpoint images of fluorescent beads at increasing depths. Image is viewable with red-cyan 3D glasses. Scale bar, 25 μm. (D) Central yv epipolar slice (x = 0, u = 0) of the light field for each bead depth. Scale bar, 25 μm.
Refocusing
The parallax contained in light fields can be used to refocus images of the scene. We demonstrate this capability by imaging a collection of fluorescent beads randomly distributed on a glass slide (Fig. 4). The resolution of the traditional large-aperture 2D fiber image (Fig. 4, A to C) is enhanced using the standard shift-and-add light field refocusing technique (Fig. 4, D to I) (1) and further refined using a 3D deconvolution technique similar to Broxton et al. (28) (Fig. 4, J to O). Focal stack deconvolution also notably increases axial localization as shown by the xz plane maximum intensity projections (MIPs) in Fig. 4 (E, G, and I) (refocused light field focal stack), Fig. 4 (K, M, and O) (deconvolved light field focal stack), and the axial profiles in fig. S7.
Fig. 4. Light field refocusing and deconvolution.

(A to C) Traditional large-aperture 2D images for a planar fluorescent sample consisting of fluorescent beads. (D to I) Refocused light field images of the same sample. (J to O) Maximum intensity projections (MIPs) of deconvolved light field focal stack, for the same sample. The sample is placed at a distance of z = 11, 31, and 51 μm from the fiber facet in the first, second, and third rows, respectively. xz MIPs for the light field and deconvolved light field are shown for each object depth in (E), (G), (I), (K), (M), and (O), respectively. Red arrows indicate the ground truth z-position of the bead sample. See fig. S7 for horizontal projections of (E), (G), (I), (K), (M), and (O). Scale bars, 50 μm (J and K). Insets I to IV for (J), (L), and (N) show zoomed-in images of the boxed regions of (J), (L), and (N) for the deconvolved light field focal stack near the ground truth object plane. Some features that are blended together in the MIP image are resolvable in deconvolved refocused planes near the true object plane. All images are intensity-normalized for visibility. Inset scale bar, 25 μm (J, inset IV).
3D sample visualization
A scene’s 3D structure can be directly observed by stereo images and perspective shifting animations. To demonstrate this effect, we image two 3D samples, fluorescent beads within polydimethylsiloxane (PDMS) (Fig. 5, A and B) and lens tissue with a fluorescent highlighter (Fig. 5, C and D). Figure 5 (A and C) shows red-cyan stereo images composed of diametrically opposed horizontal viewpoints. As in Fig. 3C, parallax is manifested as a lateral displacement between structures in the red and cyan images, resulting in 3D images when wearing red-cyan glasses. Although these stereographs use only two viewpoints, the light field contains images of the scene with disparity along any arbitrary direction (movies S1 to S6). We use this viewpoint information along with a least-squares depth mapping method (25) to create the depth map shown Fig. 5B, where depth is indicated by hue. Alternatively, we construct a depth map in Fig. 5D by an MIP of the deconvolved light field focal stack. Here, the hue corresponds to the depth of the maximum intensity. The depth structure in Fig. 5 (C and D) is alternatively visualized by selected slices of the deconvolved light field focal stack shown in Fig. 5 (E to H).
Fig. 5. Visualizing depth in 3D samples.
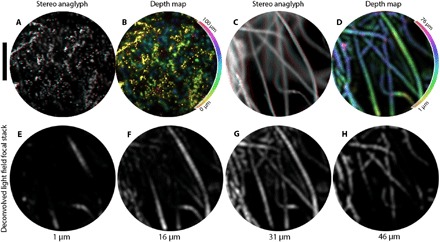
(A) Red-cyan stereo anaglyph of a 3D sample of fluorescent beads, viewable with red-cyan stereo glasses. The sample has a thickness of ~55 μm as verified by confocal microscopy and is placed 5 μm from the fiber facet. (B) Calculated depth map for the sample in (A), with depth color-coded by hue (see colorbar); pixel brightness is set to the pixel brightness in the [u, v] = [0, 0] viewpoint image. For a comparison between the ground truth confocal image and this depth map, see fig. S8. (C) Red-cyan stereo anaglyph of lens paper tissue with highlighter ink. (D) MIP depth map of the sample in (C). The depth of maximum intensity is color-coded by hue. Virtual reality goggle compatible stereo-pairs of (A) and (C) are available in fig. S9. (E to H) Slices of the deconvolved light field focal stack for fluorescent lens paper at depths of 1, 16, 31, and 46 μm, respectively. Scale bar, 200 μm.
We applied our fiber bundle light field technique to tissue imaging by staining a thick (~5 mm) slice of mouse brain with proflavine. Proflavine and acriflavine are commonly used fluorescent markers, which specifically target nuclear structures in vivo (29, 30). Cell nuclei are visible in the stereo image (Fig. 6A) obtained by imaging the proflavine-stained brain sample through the fiber bundle. A viewpoint shifting animation is shown in fig. S7. The corresponding depth map is shown in Fig. 6B, indicating cells located at a variety of depths up to ~50 μm from the fiber surface. This is consistent with the known finite penetration depth (~50 μm) of proflavine (30). To verify that our light field technique reports the correct depth distribution of cells in tissue, we record a 3D confocal stack of the top 75 μm of the brain slice for comparison to fiber bundle light field data. Because of the experimental difficulty in capturing matching fields of view with both confocal and fiber bundle, we instead compare the proflavine depth distribution, which is a result of the finite penetration depth and the physical structure of the tissue. We find excellent quantitative agreement between the proflavine depth distributions as measured by our light field approach (Fig. 6C, solid black curve) compared with a benchtop confocal microscope (Fig. 6C, dashed blue curve; see fig. S10 for confocal images). This confirms that our fluorescence light field imaging technique returns quantitative data in scattering tissue.
Fig. 6. Depth mapping of biological tissue.
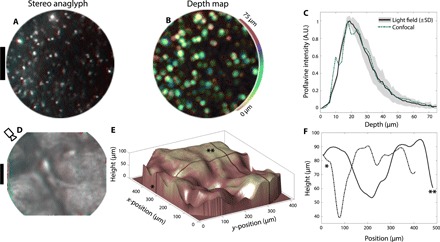
(A) Red-cyan stereo anaglyph of a proflavine-stained brain slice with a thickness of ~5 mm. Topically applied proflavine highlights cell nuclei. A viewpoint shifting animation along the x axis is shown in movie S7. Scale bar, 100 μm. (B) Light field depth map for the field of view in (A), with depth color-coded as hue (see colorbar). Pixel brightness is set to the depth confidence at each pixel. (C) Proflavine intensity distribution as a function of depth, measured from light field data (solid black curve), and from confocal data (blue dashed curve). Light field data are averaged over 25 separate fiber bundle fields of view. The gray area above and below the black curve indicates 1 SD above and below the mean proflavine intensity depth distribution, taken over 25 fiber bundle fields of view. The finite penetration depth of topically applied proflavine is apparent in both light field and confocal datasets, with good agreement in the distribution shape between the two imaging approaches. (D) Red-cyan stereo anaglyph of skin surface autofluorescence (thumb, near the knuckle). The camera icon indicates viewing orientation in (E). A viewpoint shifting animation along the x axis is shown in movie S8. Scale bar, 100 μm. (E) 3D surface plot of data from (D), using shape from focus (31). Dark lines with “*” and “**” symbols indicate positions for height plots in (F). (F) Height along lines designated by (*, dashed) and (**, solid) in (E).
Surface topography is also accessible with our light field imaging approach. To demonstrate this capability, we image skin autofluorescence of the backside of the thumb near the knuckle. 3D skin fold microstructure is readily visible in the red-cyan anaglyph of Fig. 6D and the corresponding perspective shifting animation (movie S8). The surface topography in Fig. 6 (E and F) is calculated via a simple shape from focus algorithm where a focus metric (modified Laplacian) is calculated at each pixel of each slice of the light field focal stack (31). The focus position containing the largest focus metric value yields the surface height at a given position in the image.
DISCUSSION
The ultimate form factor limit of our approach is reached when cores are shrunk until they are single mode, at which point no angular information can be obtained. For these step-index, 0.39-NA fiber bundles operating at 550 nm, this is achieved when the core radius is , only a factor of ~1.9 smaller than the cores in our fiber bundles (a ≈ 1 μm) (16). As previously pointed out (2), a similar limit applies regardless of the manner in which the light field is measured [e.g., lenslet array (2, 4), aperture mask (17), and angle-sensitive pixel (18)] because photon direction and position cannot be sampled simultaneously to arbitrary precision. Thus, these fiber bundles approach the fundamental limit for the physical size of a light field/stereo imaging device. Related considerations apply in the context of speckle correlation 2D imaging through fiber bundles (32) and wavefront shaping approaches (11), where the information throughput ultimately depends on the number of available modes. The angular information carried by these modes in our system requires finer spatial sampling (~2× per linear dimension) at the image sensor compared to standard systems (8). Consequently, roughly 4× more photons are needed to maintain the same signal-to-noise ratio because of the increased read noise. This is the natural trade-off for the angle- and depth-resolved information that we obtain via our light field approach. The Fourier filter approach that we use to obtain the light field moments can also introduce low–spatial frequency noise into the solution. Here, our work parallels a technique called transport of intensity equation-phase imaging, where this unusual noise behavior has been addressed in detail (33). In our work, we use a regularization parameter to mitigate this low-frequency noise (see “Effective light field moment calculation” section).
The Gaussian light field assumption in Eq. 2 prescribes a recovered light field that is determined by a single cone of rays arriving at each pixel (in this case, a fiber bundle core). If this cone of rays happens to be created by multiple sources at different depths, then the LIM depth map will contain a virtual object at the intensity-weighted depth of the sources visible at that pixel. Here, light field focal stack deconvolution can be used to separate the two objects in the axial direction (fig. S8C), at the expense of computational overhead on the order of seconds to minutes. In contrast, light field moment calculation, depth mapping, and stereo image construction can be achieved in real time (~10 frames/s) on a standard desktop computer (3.6-GHz Intel Core i7, 16-GB RAM). These simpler calculations apply to imaging of tissue surfaces or tissues containing mostly nonoverlapping fluorescent objects. These situations are often applicable to fluorescence microendoscopy, where fluorescent contrast agents (34–36) are applied topically to highlight surface features. In this context, we have demonstrated surface topography measurement of skin via autofluorescence; the same approach should find application for surface topography measurement in the gastrointestinal (34) and urinary tracts (35) stained with fluorescein, for example. We have also shown that the depth of cell nuclei stained with proflavine—a collection of sparse, mostly nonoverlapping objects near the tissue surface—can be reliably extracted using our light field approach. This could potentially aid in grading and classifying dysplasias where progression is related to depth-dependent changes in epithelial tissues (37).
Light field imaging improves image sharpness for out-of-focus objects, but it is not capable of reversing image degradation due to scattering. While our approach enables imaging of recorded objects at an extended depth into the sample, it cannot unveil objects that are fundamentally unobservable or scrambled because of light scattering and attenuation in the sample. More complex techniques such as multiphoton imaging or optical coherence tomography are required to achieve this goal, although working at longer wavelengths should improve light field imaging depth into scattering samples. A practical limitation to the axial dynamic range of our approach is dictated by the dynamic range of the camera. The total excitation power experienced by a fluorophore and the emission intensity collected by a fiber core from the said fluorophore combine for a steeper than 1/z2 drop-off in apparent object intensity. At some distance, fluorescing objects disappear below the noise floor, even in a nonscattering sample. While integration time and/or excitation intensity can be increased, this would cause objects close to the fiber facet to saturate the camera, rendering Eq. 4 invalid. In practice, we found that this consideration limits imaging depth to the first ~80 μm. This effect could be mitigated by using a larger dynamic range camera or by combining multiple images with different integration times for a high–dynamic range image.
We have shown that the fiber bundles widely used for microendoscopy are capable of recording depth information in a single shot. To our knowledge, this is the thinnest light field imaging device reported to date. Our technique is camera frame rate limited, does not require calibration, and is not perturbed by moderate fiber bending, making it robust for potential clinical applications. Other incoherent imaging modalities such as brightfield are also amenable to our approach, as are fiber bundles using distal lenses.
MATERIALS AND METHODS
Fiber
For all datasets expect Fig. 6 and figs. S3 and S4, we used a 30-cm-long Fujikura FIGH-30-650S fiber bundle with the following nominal manufacturer specs: 30,000 cores; average pixel-to-pixel spacing, 3.2 μm; image circle diameter, 600 μm; fiber diameter, 650 μm; coating diameter, 750 μm. For Fig. 6 (D to F), we used a 150-cm-long FIGH-30-650S fiber bundle. For Fig. 6A and figs. S3 and S4, we used a 1-m-long Fujikura FIGH-10-350S with the following nominal manufacturer specs: 10,000 cores; average pixel-to-pixel spacing, 3.2 μm; image circle diameter, 325 μm; fiber diameter, 350 μm; coating diameter, 450 μm.
Beads
Single-layer sample
For Figs. 1 to 5 (A and B), we used “Yellow” fluorescent beads from Spherotech with nominal diameters ranging from 1.7 to 2.2 μm. For Figs. 1 to 4, beads were first dried onto a 1-cm-thick piece of cured PDMS (later referred to as the microbead stamp). This piece of PDMS was then pressed firmly onto a clean microscope slide for 1 to 3 s to transfer a sparse collection of beads to the glass slide. The sample was placed on the sample stage and moved to within a known distance from the distal fiber facet using a linear manual micrometer stage (see “Optical setup” section).
Multilayer sample
The 3D sample in Fig. 5 (A and B) consists of six layers, the first layer of which was prepared in the same way as the sample in Figs. 1 to 4. After transferring beads to the glass slide, a layer of PDMS (10:1 precursor to curing agent ratio) was spin-coated at 5000 rpm for 60 s to reach an approximate thickness of 11 μm (verified by confocal microscopy). The PDMS was then cured by placing the glass slide on a hot plate at 75°C for 10 min. After curing, the microbead stamp was firmly pressed onto this thin layer of PDMS to transfer the next layer of microbeads, followed by spin coating of PDMS and subsequent hot plate baking. This procedure was repeated until the six layers of fluorescent microbeads were deposited onto the microscope slide. Each layer was embedded in PDMS, except the final layer, which was deposited onto the last PDMS surface without subsequent PDMS spin coating. See fig. S6D for the schematic.
Highlighter paper
For Fig. 5 (C to H), we used a piece of Thorlabs lens cleaning paper. The paper was made fluorescent by drawing on it with a yellow office highlighter (Staples Hype!).
Mouse brain tissue preparation and storage
The mouse brain sample (Fig. 6, A to C) was gifted from the Royal Melbourne Institute of Technology (RMIT) University animal facility as scavenged tissues. The tissues were placed in a cryoprotective solution containing 30% sucrose (Sigma-Aldrich, USA) in 0.01 M phosphate-buffered saline (1× PBS; Gibco, USA) and kept at 4°C for 3 days with gentle rotation (200 rpm) to allow the solution to suffuse through the whole brain. The brains were then snap-frozen, then placed in 2-ml cryotubes, and stored in liquid nitrogen storage until needed.
Proflavine staining
The brains were removed from frozen storage and sliced into ~1-mm sections, and the sections were placed on glass slides for staining and allowed to thaw at 4°C. A 0.01% (w/v) solution of proflavine (proflavine hemisulfate salt hydrate, Sigma-Aldrich) was prepared in sterile 1× PBS and applied topically to the brain slice before imaging. Fiber bundle imaging was performed within 15 min of staining. The sample was restained for confocal imaging.
Confocal imaging
Confocal imaging was used to provide ground truth quantification of samples in Figs. 5 (A and B) and 6 (A to C). In both cases, we used an Olympus FV1200 confocal microscope, with either a 0.4 NA, 10× objective (Fig. 5, A and B) or a 0.75 NA, 20× objective (Fig. 6, A to C). In both cases, the 473-nm laser was used to excite the sample, pixel size was set to 0.621 μm, dwell time was 2 μs per pixel, and z-slices were recorded each as 1.98 μm.
Optical setup
Excitation of fluorescent samples was achieved by focusing light from a blue light-emitting diode (Thorlabs M455L3; center wavelength, 455 nm) on the proximal facet of an optical fiber bundle (Fujikura FIGH 30-650S). Before reaching the proximal facet, the excitation light was filtered with a band-pass filter with a center wavelength of 465 nm and an FWHM of 40 nm (Semrock). Filtered excitation light was then relayed by a pair of singlet condenser lenses (f = 30 and 100 mm, Thorlabs AC254-30-A and AC254-100-A, respectively) and reflected into the optical path by a long-pass dichroic filter (Semrock FF509-Di01-25x36) with a cut-on wavelength at 509 nm. Focusing was achieved by an infinite conjugate 10×, 0.4 NA, objective lens (Olympus UPLSAPO10X), which also serves to image the fluorescence output at the proximal facet onto a monochrome camera [Point Grey Grasshopper 3 GS-U3-41S4M-C or Point Grey Grasshopper 3 GS-U3-51S5M-C for Fig. 6 (D to F)], via a tube lens (Thorlabs TTL200). Fluorescence emission was filtered immediately before the tube lens with a 600-nm short-pass filter (Edmund Optics #84-710) and a 532-nm long-pass filter (Semrock BLP01-532R-25). The 600-nm short-pass filter was used to eliminate fiber autofluorescence (38). Both ends of the fiber were fixed onto three-axis manual micrometer stages for fine adjustment (Thorlabs MBT616). The objective was mounted on a separate one-axis linear stage (Thorlabs PT1) for focusing at the proximal facet. The sample was mounted on a three-axis stage (Thorlabs NanoMax 300) for focusing at the distal facet. See fig. S1 for an optical setup diagram.
Digital aperture filtering
First, a reference image of the cores was acquired with a uniform fluorescent background, provided by ink on a standard business card placed ~0.5 mm from the fiber facet. This image was then thresholded using adaptive thresholding with a window size of 7 × 7 pixels. Pixels with a value greater than the mean pixel value within the averaging window were identified and set to 1. The remaining pixels were set to 0. From this binary image, a region corresponding to the area of each core was identified (those regions where the binary image = 1). The effective radius was then calculated for each core, where A is the core area as measured in the binary thresholded image. A new blank image with 8× fewer pixels than the raw image along each dimension was initialized. In this new blank image, the pixel corresponding to the center of each core was then set to the mean of all the pixels within R/3 pixels from the core center (blue circle in Fig. 2A) in the raw image of the fiber facet (i.e., raw image of the fiber facet when the sample is in view). The remaining blank pixels were then filled in via linear interpolation (MATLAB scatteredInterpolant function). Large-aperture images are constructed in the same manner, except that all pixels within R pixels of the core centers (red circles in Fig. 2B) are averaged (instead of R/3). This is akin to the usual method of fiber bundle pixelation removal.
Background subtraction
After aperture filtering, background subtraction was achieved by manually identifying an empty 5 × 5 pixel region of the image and subtracting the mean value within this region from the image.
Flat-field correction
Before each imaging experiment, a flat-field image was acquired by imaging a piece of paper at a distance of ~2 mm from the fiber endface. The resulting image was used as a flat field reference—subsequent images were divided by this flat field reference to eliminate inhomogeneities in core throughput.
Effective aperture measurement
To measure the effective collection aperture angle of full and small aperture, we fit a 2D Gaussian PSF to images of five isolated fluorescent beads at distances from 1 to 51 μm from the fiber facet. The FWHM of the bead images at each depth was extracted from the fit and averaged. We subsequently applied a linear fit to this FWHM as a function of depth (see Fig. 3A). The slope of this line is the effective aperture (tan θ0 and tan θ1 for small and large apertures, respectively). The y intercept is the FWHM at zero depth, which we denote by σ0. Both of these parameters are used to verify the model of Eq. 5 in Fig. 3B.
Effective light field moment calculation
Effective light field moment calculation is done in the same manner to that in LMI (23). Equation 4 is solved in Fourier space using the difference image ΔI = I0 − αI1 as input. Here, I0 is the small-aperture image, I1 is the large-aperture image, and α is the ratio of the total intensity of the two images: α = ∑I0/∑I1. To solve Eq. 4, we first recast the equation in terms of a scalar potential U, defined by , with I = (I0 + αI1)/2 (see section S1 for details). Next, we take the Fourier transform of the resulting equation, resulting in an equation for the Fourier transform of the scalar potential F(U): , where denotes the magnitude of the spatial frequency vector. In practice for 3D samples, we regularize the calculation of F(U) with a small term ϵ to limit noise amplification at low spatial frequencies: . For both datasets in Fig. 5, . The real-space scalar potential U is then obtained via inverse Fourier transform. Last, the effective light field moment vector field is calculated as . Note that solving for via a scalar potential assumes that . From the definition of the effective light field moment vector, we have for an isolated point source located at the center of the xy plane, regardless of the depth of the point source (see section S2). Therefore, is satisfied for any point source and moreover for any linear combination of point sources. Thus, the use of a scalar potential method to calculate is physically justified.
3D stereographs
The stereographs in Figs. 3C, 5 (A and C), and 6 (A and D) were constructed using the extreme horizontal viewpoints of the light field as inputs to the MATLAB function stereoAnaglyph. For Figs. 5 (A and C) and 6 (A and D), the left viewpoint was shifted by two pixels horizontally to move the point of fixation to the approximate middle depth of the sample.
Depth and surface topography map calculation
The plenoptic depth mapping procedure outlined by Adelson and Wang (25) is used to create the depth maps in Figs. 5B and 6B. This approach estimates the disparity at each point in the image as , where Lk is the partial derivative of the light field in the k direction. The summation is performed within a sliding patch p centered at each spatial pixel in the light field. The patch p covers 5× 5 pixels in the spatial (xy) dimensions and the entirety of the angular dimensions. The disparity can then be converted to depth using the relationship derived in Eq. 5 (Fig. 3B). For this specific dataset, we used the regularization parameter ϵ in the calculation of the effective light field moments. The use of ϵ modifies the analytic relationship in Eq. 5. To correct for this effect, we performed the effective light field moment calculation on simulated point sources at distances of 1 to 101 μm from the fiber facet, using for regularization (see “Effective light field moment calculation” section). We relate the resulting simulated PSF centroid shift to the true simulated depth via a calibration curve. The calibration curve is subsequently used to quantify depth from the disparity calculated via the Adelson method. This depth is then assigned to the hue dimension (H) of a hue-saturation-value (HSV) image. The saturation dimension (S) is set to a constant value of 0.65 across the entire image, and the value dimension (V) is set to the intensity image at the center viewpoint (Fig. 5B) or the confidence value at each point [see Fig. 6B and (25)].
The depth map in Fig. 5D is created by color-coding the depth of maximum intensity in the MIP of the deconvolved light field focal stack. See below for light field focal stack deconvolution information.
The surface topography map in Fig. 6E is calculated via shape from focus. Here, the “sum-modified Laplacian” is calculated for each pixel in each slice of the light field focal stack (31) (created using the standard shift-and-add technique without deconvolution; see “Light field focal stack deconvolution” section). We define the sum-modified Laplacian as . The surface height value is then extracted by finding the maximum of ML(x, y, z) over all z values at each (x, y) position. This surface height map is then convolved with a 2D Gaussian (SD, 15 μm) to obtain a smoothed height estimate (Fig. 6E).
Light field focal stack deconvolution
First, focal stacks are created directly from light fields using the standard shift-and-add technique. This results in a focal stack with poor axial localization, as can be seen in the xz-plane MIPs in Fig. 4. Because the resulting focal stack has a PSF that varies with depth, it cannot be treated with standard spatially invariant deconvolution techniques. The matrix equation governing focal stack image formation can be written as , where is the shift-and-add focal stack, is the true 3D distribution of fluorophores in the scene (i.e., the “deconvolved” focal stack), and W is a matrix consisting of PSFs placed at every voxel in the scene volume (i.e., each column of W represents the local PSF at a given voxel in the shift-and-add light field focal stack) but sampled only at each voxel in the target deconvolved focal stack volume. We purposefully chose the target volume to have fewer samples, making the matrix equation overdetermined. For , we used a refocused light field stack with refocus planes at every 7.3 μm from 0 to 226.3 μm (only the first 95 μm shown in Fig. 4). The target volume for has refocus planes at every 5 μm from 0 to 95 μm. We used simulated Gaussian PSFs (with the experimentally measured parameters) to populate the W matrix. This matrix equation has an extremely large number of elements (> 1012) when considering the entire volume. To make this problem tractable, we solved this equation locally in blocks of 17 × 17 pixels in xy (and the full depth in z). To further restrict the solution, we used a non-negative least-squares algorithm (MATLAB lsqnonneg function) (39) to solve for . To avoid edge effects, we discard the solution results for all but the central 3 × 3 sub-block. Neighboring blocks are shifted by three pixels with respect to each other to fill the entire image space. The resulting focal stacks are convolved with a 3D Gaussian to smooth out noise. The 3D Gaussian used for smoothing has an SD of 10.6 μm in the x- and y directions and 10 μm in the z direction. Deconvolution was performed in the same manner for Figs. 4 and 5 (D to H).
Centroid shift simulation
The green curves in Fig. 3B were obtained computationally by simulating I0 and I1 for isolated point emitters in MATLAB. First, the intensity distribution of a point source imaged with the fiber bundle using the large aperture setting (tan θ1 = 0.4534, σ0 = 2.30 μm) was calculated using the assumed PSF in Eq. 3. This calculation was performed for point emitters at depths z = 1, 11, …, 101 μm and then repeated for a small-aperture setting (tan θ0 = 0.3574, σ0 = 2.30 μm). This yielded simulated images I1 and I0 for each bead depth. Light field calculation was then performed exactly as it is in the case with experimental data (see “Effective light field moment calculation” section).
Centroid shift calculation
After calculation of the estimated light field, we obtained images of point emitters over a 7 × 7 grid in uv space for both experimental and simulated datasets. Both datasets are subsequently treated in the exact same way to extract centroid shifts. The centroid of a given point emitter at a given viewpoint along the x and y axes is obtained by fitting a 2D Gaussian to the image. The distance between the emitter’s centroid in the [u, v] = [u1, v1] and [u, v] = [0, 0] images is calculated by taking the magnitude of the vector difference of the two centroids. Because of the linear dependence between centroid shift and the [u, v] vector (Eq. 5), we then normalized the centroid shift by the magnitude of the [u, v] vector. For each emitter at a given depth, repeating this process over the x and y axes yielded 7 × 2 = 14 measurements of the centroid shift. For experimental data, we averaged centroid shift measurements over five isolated beads in the same sample. The error bars in Fig. 3B represent the SD of all these measurements.
Supplementary Material
Acknowledgments
We wish to thank G. Wetzstein, K. Dholakia, B. Wilson, and A. Greentree for stimulating discussions regarding this work. We also wish to thank E. Yang for advice on proflavine staining. Funding: This work was funded by the Australian Research Council (ARC) Centre of Excellence for Nanoscale BioPhotonics (CE140100003). M.P. was supported by an ARC DECRA Fellowship (DE170100241). I.S.M. was supported by an ARC Future Fellowship (FT108100343). Author contributions: A.O. conceived, designed, and performed the experiments. A.O., M.P., and I.S.M. developed the theoretical imaging model. E.R.W. performed sample preparation and aided in brain tissue experiments. B.C.G. supervised the research. A.O. wrote the manuscript, and all authors contributed to editing the manuscript. Competing interests: A.O. is an inventor on an Australian patent application related to this work filed by RMIT University (no. 2018900267, filed 29 January 2018). A.O. is also an inventor on an international patent application related to this work filed by RMIT University (no. PCT/AU2019/050055, filed 25 January 2019). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaav1555/DC1
Section S1. Derivation of Eq. 4
Section S2. Derivation of Eq. 5
Section S3. Resilience to bending
Section S4. Sampling considerations
Section S5. Light field imaging considerations: Axial direction
Fig. S1. Optical setup for fluorescence imaging through optical fiber bundles.
Fig. S2. Correlation between ray angle and intracore light distribution.
Fig. S3. Depth mapping is robust to dynamic fiber bending.
Fig. S4. Effect of dynamic core-to-core coupling during bending.
Fig. S5. Centroid shift of a fluorescent bead viewed from different u positions (viewpoints).
Fig. S6. SE in depth measurements from Fig. 3B.
Fig. S7. Effect of light field focal stack deconvolution on axial localization.
Fig. S8. Confocal versus light field depth maps.
Fig. S9. Stereo pair for the 3D samples from Fig. 5.
Fig. S10. Confocal stack of proflavine-stained mouse brain.
Movie S1. Viewpoint shifting animation of multilayered bead sample (Fig. 5, A and B) along the x axis.
Movie S2. Viewpoint shifting animation of multilayered bead sample (Fig. 5, A and B) along the y axis.
Movie S3. Viewpoint shifting animation of multilayered bead sample (Fig. 5, A and B) along a circular trajectory.
Movie S4. Viewpoint shifting animation of fluorescent paper sample (Fig. 5, C and D) along the x axis.
Movie S5. Viewpoint shifting animation of fluorescent paper sample (Fig. 5, C and D) along the y axis.
Movie S6. Viewpoint shifting animation of fluorescent paper sample (Fig. 5, C and D) along a circular trajectory.
Movie S7. Viewpoint shifting animation of proflavine-stained mouse brain sample (Fig. 6, A to C) along the x axis.
Movie S8. Viewpoint shifting animation of skin autofluorescence (Fig. 6, D to F) along the x axis.
REFERENCES AND NOTES
- 1.R. Ng, M. Levoy, M. Brédif, G. Duval, M. Horowitz, P. Hanrahan, Light field photography with a hand-held plenoptic camera. Computer Science Technical Report 2005-02 (2005).
- 2.Levoy M., Ng R., Adams A., Footer M., Horowitz M., Light field microscopy. ACM Transact. Graph. 25, 924–934 (2006). [Google Scholar]
- 3.Prevedel R., Yoon Y.-G., Hoffmann M., Pak N., Wetzstein G., Kato S., Schrödel T., Raskar R., Zimmer M., Boyden E. S., Vaziri A., Simultaneous whole-animal 3D imaging of neuronal activity using light-field microscopy. Nat. Methods 11, 727–730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orth A., Crozier K., Microscopy with microlens arrays: High throughput, high resolution and light-field imaging. Opt. Express 20, 13522–13531 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Levoy M., Zhang Z., McDowall I., Recording and controlling the 4D light field in a microscope using microlens arrays. J. Microsc. 235, 144–162 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Hughes M., Chang T. P., Yang G.-Z., Fiber bundle endocytoscopy. Biomed. Opt. Express 4, 2781–2794 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.A. Osdoit, F. Lacombe, C. Cavé, S. Loiseau, E. Peltier, in Endoscopic Microscopy II (International Society for Optics and Photonics, 2007), vol. 6432, pp. 64320F.
- 8.Muldoon T. J., Pierce M. C., Nida D. L., Williams M. D., Gillenwater A., Richards-Kortum R., Subcellular-resolution molecular imaging within living tissue by fiber microendoscopy. Opt. Express 15, 16413–16423 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krstajić N., Akram A. R., Choudhary T. R., McDonald N., Tanner M. G., Pedretti E., Dalgarno P. A., Scholefield E., Girkin J. M., Moore A., Bradley M., Dhaliwal K., Two-color widefield fluorescence microendoscopy enables multiplexed molecular imaging in the alveolar space of human lung tissue. J. Biomed. Opt. 21, 046009 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Thiberville L., Salaün M., Lachkar S., Dominique S., Moreno-Swirc S., Vever-Bizet C., Bourg-Heckly G., Human in-vivo fluorescence microimaging of the alveolar ducts and sacs during bronchoscopy. Eur. Respir. J. 33, 974–985 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Plöschner M., Tyc T., Čižmár T., Seeing through chaos in multimode fibres. Nat. Photonics 9, 529–535 (2015). [Google Scholar]
- 12.Choi Y., Yoon C., Kim M., Yang T. D., Fang-Yen C., Dasari R. R., Lee K. J., Choi W., Scanner-free and wide-field endoscopic imaging by using a single multimode optical fiber. Phys. Rev. Lett. 109, 203901 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papadopoulos I. N., Farahi S., Moser C., Psaltis D., High-resolution, lensless endoscope based on digital scanning through a multimode optical fiber. Biomed. Opt. Express 4, 260–270 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohayon S., Caravaca-Aguirre A., Piestun R., DiCarlo J. J., Minimally invasive multimode optical fiber microendoscope for deep brain fluorescence imaging. Biomed. Opt. Express 9, 1492–1509 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orth A., Ploschner M., Maksymov I. S., Gibson B. C., Extended depth of field imaging through multicore optical fibers. Opt. Express 26, 6407–6419 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Chen X., Reichenbach K. L., Xu C., Experimental and theoretical analysis of core-to-core coupling on fiber bundle imaging. Opt. Express 16, 21598–21607 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Veeraraghavan A., Raskar R., Agrawal A., Mohan A., Tumblin J., Dappled photography: Mask enhanced cameras for heterodyned light fields and coded aperture refocusing. ACM Trans. Graph. 26, 69 (2007). [Google Scholar]
- 18.Wang A., Gill P., Molnar A., Light field image sensors based on the Talbot effect. Appl. Optics 48, 5897–5905 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilburn B., Joshi N., Vaish V., Talvala E.-V., Antunez E., Barth A., Adams A., Horowitz M., Levoy M., High performance imaging using large camera arrays. ACM Transact. Graph. 24, 765–776 (2005). [Google Scholar]
- 20.A. W. Snyder, J. Love, Optical Waveguide Theory (Springer Science & Business Media, 2012). [Google Scholar]
- 21.A. Yariv, P. Yeh, Photonics (Oxford Univ. Press, 2007). [Google Scholar]
- 22.Eugui P., Lichtenegger A., Augustin M., Harper D. J., Muck M., Roetzer T., Wartak A., Konegger T., Widhalm G., Hitzenberger C. K., Woehrer A., Baumann B., Beyond backscattering: Optical neuroimaging by BRAD. Biomed. Opt. Express 9, 2476–2494 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orth A., Crozier K. B., Light field moment imaging. Opt. Lett. 38, 2666–2668 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Primot J., Theoretical description of Shack–Hartmann wave-front sensor. Opt. Commun. 222, 81–92 (2003). [Google Scholar]
- 25.Adelson E. H., Wang J. Y. A., Single lens stereo with a plenoptic camera. IEEE Trans. Pattern Anal. Mach. Intell. 14, 99–106 (1992). [Google Scholar]
- 26.G. Surya, M. Subbarao, in Proceedings of the 1993 IEEE Computer Society Conference on Computer Vision and Pattern Recognition (CVPR’93) (IEEE, 1993), pp. 61–67. [Google Scholar]
- 27.Bolles R. C., Baker H. H., Marimont D. H., Epipolar-plane image analysis: An approach to determining structure from motion. Int. J. Comput. Vis. 1, 7–55 (1987). [Google Scholar]
- 28.Broxton M., Grosenick L., Yang S., Cohen N., Andalman A., Deisseroth K., Levoy M., Wave optics theory and 3-D deconvolution for the light field microscope. Opt. Express 21, 25418–25439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bodenschatz N., Poh C. F., Lam S., Lane P. M., Guillaud M., MacAulay C. E., Dual-mode endomicroscopy for detection of epithelial dysplasia in the mouth: A descriptive pilot study. J. Biomed. Optics 22, 086005 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Schlosser C. L., Bodenschatz N., Lam S. F., Lee M., McAlpine J. N., Miller D. M., Van Niekerk D. J. T., Follen M., Guillaud M., MacAulay C. E., Lane P. M., Fluorescence confocal endomicroscopy of the cervix: Pilot study on the potential and limitations for clinical implementation. J. Biomed. Optics 21, 126011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nayar S. K., Nakagawa Y., Shape from focus. IEEE Trans. Pattern Anal. Mach. Intell. 16, 824–831 (1994). [Google Scholar]
- 32.Porat A., Andresen E. R., Rigneault H., Oron D., Gigan S., Katz O., Widefield lensless imaging through a fiber bundle via speckle correlations. Opt. Express 24, 16835–16855 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Paganin D., Barty A., McMahon P. J., Nugent K. A., Quantitative phase-amplitude microscopy. III. The effects of noise. J. Microsc. 214, 51–61 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Neumann H., Kiesslich R., Wallace M. B., Neurath M. F., Confocal laser endomicroscopy: Technical advances and clinical applications. Gastroenterology 139, 388–392.e2 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Chen S. P., Liao J. C., Confocal laser endomicroscopy of bladder and upper tract urothelial carcinoma: A new era of optical diagnosis? Curr. Urol. Rep. 15, 437 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su L.-M., Kuo J., Allan R. W., Liao J. C., Ritari K. L., Tomeny P. E., Carter C. M., Fiber-optic confocal laser endomicroscopy of small renal masses: Toward real-time optical diagnostic biopsy. J. Urol. 195, 486–492 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Pavlova I., Weber C. R., Schwarz R. A., Williams M., El-Naggar A., Gillenwater A., Richards-Kortum R., Monte Carlo model to describe depth selective fluorescence spectra of epithelial tissue: Applications for diagnosis of oral precancer. J. Biomed. Opt. 13, 064012 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Udovich J. A., Kirkpatrick N. D., Kano A., Tanbakuchi A., Utzinger U., Gmitro A. F., Spectral background and transmission characteristics of fiber optic imaging bundles. Appl. Optics 47, 4560–4568 (2008). [DOI] [PubMed] [Google Scholar]
- 39.C. L. Lawson, R. J. Hanson, Solving Least Squares Problems (Society for Industrial and Applied Mathematics, 1995). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaav1555/DC1
Section S1. Derivation of Eq. 4
Section S2. Derivation of Eq. 5
Section S3. Resilience to bending
Section S4. Sampling considerations
Section S5. Light field imaging considerations: Axial direction
Fig. S1. Optical setup for fluorescence imaging through optical fiber bundles.
Fig. S2. Correlation between ray angle and intracore light distribution.
Fig. S3. Depth mapping is robust to dynamic fiber bending.
Fig. S4. Effect of dynamic core-to-core coupling during bending.
Fig. S5. Centroid shift of a fluorescent bead viewed from different u positions (viewpoints).
Fig. S6. SE in depth measurements from Fig. 3B.
Fig. S7. Effect of light field focal stack deconvolution on axial localization.
Fig. S8. Confocal versus light field depth maps.
Fig. S9. Stereo pair for the 3D samples from Fig. 5.
Fig. S10. Confocal stack of proflavine-stained mouse brain.
Movie S1. Viewpoint shifting animation of multilayered bead sample (Fig. 5, A and B) along the x axis.
Movie S2. Viewpoint shifting animation of multilayered bead sample (Fig. 5, A and B) along the y axis.
Movie S3. Viewpoint shifting animation of multilayered bead sample (Fig. 5, A and B) along a circular trajectory.
Movie S4. Viewpoint shifting animation of fluorescent paper sample (Fig. 5, C and D) along the x axis.
Movie S5. Viewpoint shifting animation of fluorescent paper sample (Fig. 5, C and D) along the y axis.
Movie S6. Viewpoint shifting animation of fluorescent paper sample (Fig. 5, C and D) along a circular trajectory.
Movie S7. Viewpoint shifting animation of proflavine-stained mouse brain sample (Fig. 6, A to C) along the x axis.
Movie S8. Viewpoint shifting animation of skin autofluorescence (Fig. 6, D to F) along the x axis.