

Abstract
Background
Bisphosphonates are considered to be the treatment of choice for people with Paget's disease of bone. However, the effects of bisphosphonates on patient‐centred outcomes have not been extensively studied. There are insufficient data to determine whether reducing and maintaining biochemical markers of bone turnover to within the normal range improves quality of life and reduces the risk of complications.
Objectives
To assess the benefits and harms of bisphosphonates for adult patients with Paget's disease of bone.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, Embase, ISI Web of Knowledge and trials registers up to March 2017. We searched regulatory agency published information for rare adverse events.
Selection criteria
Randomised controlled trials (RCTs) of bisphosphonates as treatment for Paget's disease in adults.
Data collection and analysis
Two review authors independently screened search results, extracted data and assessed studies for risk of bias. We used standard methodological procedures expected by The Cochrane Collaboration.
Main results
We included 20 trials (25 reports, 3168 participants). Of these, 10 trials (801 participants) compared bisphosphonates (etidronate, tiludronate, ibandronate, pamidronate, olpadronate, alendronate, risedronate, zoledronate) versus placebo, seven compared two bisphosphonates (992 participants), one trial compared a bisphosphonates with a bisphosphonate plus calcitonin (44 participants), and two studies, the largest trial (1331 participants) and its interventional extension study (502 participants), compared symptomatic treatment and intensive treatment where the goal was to normalise alkaline phosphatase.
Most studies were assessed at low or unclear risk of bias. Six of 10 studies comparing bisphosphonates versus placebo were assessed at high risk of bias, mainly around incomplete outcome data and selective outcome reporting.
Participant populations were reasonably homogeneous in terms of age (mean age 66 to 74 years) and sex (51% to 74% male). Most studies included participants who had elevated alkaline phosphatase levels whether or not bone pain was present. Mean follow‐up was six months.
Bisphosphonates versus placebo
Bisphosphonates tripled the proportion (31% versus 9%) of participants whose bone pain disappeared (RR 3.42, 95% confidence interval (CI) 1.31 to 8.90; 2 studies, 205 participants; NNT 5, 95% CI 1 to 31; moderate‐quality evidence). This result is clinically important. Data were consistent when pain change was measured as any reduction (RR 1.97, 95% CI 1.29 to 3.01; 7 studies, 481 participants).
There was uncertainty about differences in incident fractures: 1.4% fractures occurred in the bisphosphonates group and none in the placebo group (RR 0.89, 95% CI 0.18 to 4.31; 4 studies, 356 participants; very low‐quality evidence).
None of the studies reported data on orthopaedic surgery, quality of life or hearing thresholds.
Results regarding adverse effects and treatment discontinuation were uncertain. There was a 64% risk of mild gastrointestinal adverse events in intervention group participants and 48% in the control group (RR 1.32, 95% CI 0.91 to 1.92; 6 studies, 376 participants; low‐quality evidence). The likelihood of study participants discontinuing due to adverse effects was slightly higher in intervention group participants (4.4%) than the control group (4.1%) (RR 1.01, 95% CI 0.41 to 2.52; 6 studies, 517 participants; low‐quality evidence). Zoledronate was associated with an increased risk of transient fever or fatigue (RR 2.57, 95% CI 1.21 to 5.44; 1 study, 176 participants; moderate‐quality evidence).
Bisphosphonates versus active comparator
More participants reported pain relief with zoledronate than pamidronate (RR 1.30, 95% CI 1.10 to 1.53; 1 study, 89 participants; NNT 5, 95% CI 3 to 11) or risedronate (RR 1.36, 95% CI 1.06 to 1.74; 1 study, 347 participants; NNT 7, 95% CI 4 to 24; very low quality evidence). This result is clinically important.
There was insufficient evidence to confirm or exclude differences in adverse effects of bisphosphonates (RR 1.05, 95% CI 0.95 to 1.76; 2 studies, 437 participants; low‐quality evidence) and treatment discontinuation (2 studies, 437 participants) (RR 2.04, 95% CI 0.43 to 9.59; 2 studies, 437 participants; very low‐quality evidence).
Intensive versus symptomatic treatment
There was no consistent evidence of difference to response in bone pain, bodily pain or quality of life in participants who received intensive versus symptomatic treatment.
Inconclusive results were observed regarding fractures and orthopaedic procedures for intensive versus symptomatic treatment (intensive treatment for fracture: RR 1.84, 95% CI 0.76 to 4.44; absolute risk 8.1% versus 5.2%; orthopaedic procedures: RR 1.58, 95% CI 0.80 to 3.11; absolute risk 5.6% versus 3.0%; 1 study, 502 participants; low‐quality evidence).
There was insufficient evidence to confirm or exclude an important difference in adverse effects between intensive and symptomatic treatment (RR 1.05, 95% CI 0.79 to 1.41; low‐quality evidence).
There was insufficient evidence to confirm or exclude an important difference of risk of rare adverse events (including osteonecrosis of the jaw) from the regulatory agencies databases.
Authors' conclusions
We found moderate‐quality evidence that bisphosphonates improved pain in people with Paget's disease of bone when compared with placebo. We are uncertain about the results of head‐to‐head studies investigating bisphosphonates. We found insufficient evidence of benefit in terms of pain or quality of life from intensive treatment. Information about adverse effects was limited, but serious side effects were rare, and rate of withdrawals due to side effects was low.
Plain language summary
Bisphosphonates for Paget's disease of bone in adults
Review question
We wanted to find out if using bisphosphonate treatment was better or worse than dummy treatment (placebo) to relieve bone pain in people with Paget's disease of bone and determine if treatment could prevent complications. We also wanted to discover which bisphosphonates were better.
What is Paget's disease of bone and what are bisphosphonates?
Paget's disease of bone is a chronic problem which usually affects one or a few bones. Paget's disease causes bone renewal and repair to become abnormal; bones become weak enlarged and misshapen, leading to pain, fractures and arthritis in joints close to affected bones.
Bisphosphonates are medications that slow down the bone remodelling process.
Search date
March 2017.
Study characteristics
We included 20 studies that involved 3168 people. Of these, 10 studies (801 people) compared bisphosphonates with placebo. Studies included elderly people; slightly more participants were men; and nearly all had raised blood serum markers of bone turnover. Fourteen studies recruited participants from hospitals, outpatient and general practitioner clinics. Studies were performed in USA, Canada, UK, Europe; Australia, New Zealand and Argentina.
What are the effects of bisphosphonates in people with Paget's disease of bone?
Bisphosphonates probably help to relieve bone pain. We are uncertain which bisphosphonate is better. Results were similar across studies.
We are uncertain if bisphosphonates can prevent bone fractures.
Effects on quality of life, need for orthopaedic surgery and hearing loss prevention were not reported in studies that compared bisphosphonates with placebo.
What are the side effects of bisphosphonates in people with Paget's disease of bone?
Most studies did not report details about drug‐related side effects and complications. Bisphosphonates may make little or no difference in side effects except for temporary fever or tiredness with intravenous treatments and mild gastrointestinal side effects with oral medications. Severe side effects causing treatment discontinuation were rare.
What happens to people with Paget's disease of bone treated with bisphosphonates?
Pain
We found that of 100 adults with Paget's disease of bone, 31 would experience complete pain relief if they took bisphosphonates for six months compared with 9 people not taking bisphosphonates.
Side effects
We found that of 100 adults with Paget's disease of bone, 64 would experience side effects if they took bisphosphonates for six months compared with 48 not taking bisphosphonates.
The number of people who stopped treatment due to side effects was the same for the bisphosphonate and placebo groups (4 out of 100).
Quality of evidence
Pain relief data provided moderate‐quality evidence, but data on fractures was assessed as providing very low‐quality evidence. Data on side effects provided low‐quality evidence; treatment discontinuation data provided moderate‐quality evidence. Evidence was downgraded mainly due to limited data and concerns about study design.
Study funding sources
Eleven studies were funded by drug manufacturers. Four studies were funded by government agencies or charities. Data on funding sources were not provided or unclear in five studies.
Summary of findings
Summary of findings for the main comparison. Bisphosphonates versus placebo for Paget's disease of bone.
| Bisphosphonates versus placebo for Paget's disease of bone | ||||||
|
Patient or population: Paget's disease of bone
Settings: Inpatients and outpatients in America, Europe, Australasia and Africa
Intervention: Bisphosphonates Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with bisphosphonates | |||||
| Number of participants with change in bone pain (disappearance of pain)¹ assessed on VAS Follow up: mean 6 months | Study population | RR 3.42 (1.31 to 8.90) | 205 (2 RCTs) | ⊕⊕⊕⊝ MODERATE² | NNTB 5 (1 to 35) Absolute risk difference: 23% more (12% to 34%) Relative percent change: 242 % (31% to 790%) (Improvement) |
|
| 91 per 1000 | 311 per 1000 (119 to 809) | |||||
| Number of participants who experienced radiologically‐confirmed fractures Follow up: mean 6 months | Low (study population)³ | RR 0.89 (0.18 to 4.31) | 356 (4 RCTs) | ⊕⊝⊝⊝ VERY LOW² ⁴ ⁵ | Absolute risk difference: 1% more (‐2% to 5%) Relative percentage change: 11% (‐82% to 331%) (improvement) Effect is uncertain due to very low quality evidence |
|
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate³ | ||||||
| 9 per 1000 | 8 per 1000 (2 to 39) | |||||
| High³ | ||||||
| 52 per 1000 | 46 per 1000 (9 to 224) | |||||
| Number of participants who needed orthopaedic surgery (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) |
See comments | Outcome not reported in the included studies |
| Number of participants with change in quality of life measures (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) |
See comments | Outcome not reported in the included studies |
| Number of participants with change in hearing thresholds (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) |
See comments | Outcome not reported in the included studies |
| Number of participants who experienced side effects related to use of bisphosphonates Follow up: mean 6 months | Study population | RR 1.32 (0.91 to 1.92) | 678 (6 RCTs) | ⊕⊕⊝⊝ LOW⁶ ⁷ | Absolute risk difference: 11% more (0% to 22%) Relative percent change: 32 % (‐10% to 92%) (worsening) Gastrointestinal side effects (diarrhoea, dyspepsia, vomiting, nausea, oesophagitis or gastritis) were the most common |
|
| 483 per 1000 | 638 per 1000 (440 to 928) | |||||
| Number of participants who withdrew due to adverse events Follow up: mean 6 months | Study population | RR 1.01 (0.41 to 2.52) | 517 (6 RCTs) | ⊕⊕⊝⊝ LOW⁸ ⁹ | Absolute risk difference: 0% (‐4% to 3%) Relative percent change: 1% (‐59% to 152%) (worsening) |
|
| 41 per 1000 | 41 per 1000 (17 to 102) | |||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; NNTB: Number needed to treat for an additional beneficial outcome; OR: odds ratio; RR: risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
¹ When pain was assessed as any pain reduction instead of disappearance of pain, the outcomes were consistent: 227 per 1000 in placebo vs. 446 per 1000 (292 to 682) in bisphosphonates group (RR 1.97, 95% CI 1.29 to 3.01), NNTD 4 (2 to 13), absolute risk difference 33% more (18% to 49%), relative percentage change 97% (29% to 201% improvement), based on results from seven RCTs (481 participants). Visual analogue scale ranging from 0 to 10 was used in four of the seven studies. One study classified pain in three groups; the tool used for pain assessment was not detailed in the other two studies. Moderate quality evidence: downgraded by one level; there was high risk for attrition bias in three studies and high risk for reporting bias in three studies. The outcome did not change in a sensitive analysis excluding high risk of bias studies.
² Downgraded by one level (imprecision). Few events, resulting in wide CI.
³ The 0% calculated assumed risk in the control group (no fractures in placebo group) is misleading. This outcome is likely due to the short follow‐up period of the studies. To give a more accurate data we have added two scenarios of moderate and high prevalence using data from a study with a longest follow‐up period, the PRISM‐EZ trial (Tan 2017). In summary there are three scenarios to calculate the assumed risk in the control group: 1) To calculate low prevalence we used data from the included studies (placebo groups). 2) To calculate moderate prevalence we used the percentage of fractures in bones affected by Paget's disease of bone in the symptomatic treatment arm of the PRISM‐EZ trial. 3) To calculate high prevalence we used the percentage of fractures in both affected and unaffected bones in the symptomatic treatment arm of the PRISM‐EZ trial.
⁴ Downgraded by two levels (limitation of studies). Most data were from studies assessed at high risk of bias; there was high risk for attrition bias in two studies and high risk for reporting bias in one study.
⁵ Downgraded by one level (indirectness). Long‐term impact on fractures was not assessed.
⁶ Downgraded by one level (limitation of studies). High risk for attrition bias in two studies.
⁷ Downgraded by one level (inconsistency). The side effects considered in the studies were heterogeneous. In addition, considerable heterogeneity was found when the six studies were meta‐analysed (I² = 75%, P = 0.001). However, only one study (McClung 1995) showed more adverse events in the placebo group than the bisphosphonates group. The heterogeneity could not be explained by differences in design of this study since it was similar to other studies. A sensitivity analysis excluding this study found low heterogeneity I² = 6% (RR 1.38, 95% CI 1.08 to 1.78).
⁸ Downgraded by two levels. Half of the included studies were assessed at high risk of bias; there was high risk for attrition bias in three studies and high risk for reporting bias in one study.
⁹ Rated down for imprecision. Optimal information size criterion was not met. The 95% CI is too wide.
Summary of findings 2. Zoledronate versus pamidronate or risedronate for Paget's disease of bone.
| Zoledronate versus pamidronate or risedronate for Paget's disease of bone | ||||||
| Patient or population: Paget's disease of bone Settings: Inpatients and outpatients in America, Europe, Australasia and Africa Intervention: Zoledronate Comparison: Pamidronate or risedronate | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with pamidronate or risedronate | Risk with zoledronate | |||||
| Number of participants with change in bone pain¹ assessed on VAS. Follow up: mean 6 months | Study population | RR 1.31 (1.15 to 1.51) | 436 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW² ³ | NNTB: 7 (4 to 14) Absolute risk difference: 17% (8% to 26%) Relative percent change: 31% (15% to 51%) (improvement) Not pooled effects: Zoledronate vs. pamidronate (89 participants); RR 1.30 (1.10 to 1.53), NNTB 4 (3 to 13). Zoledronate vs. risedronate (347 participants); RR 1.36 (1.06 to 1.74), NNTB 8 (4 to 45). |
|
| 465 per 1000 | 609 per 1000 (535 to 702) | |||||
| Number of participants who experienced fractures (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) |
See comments | Outcome not reported in the included studies |
| Number of participants who needed orthopaedic surgery (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) |
See comments | Outcome not reported in the included studies |
| Number of participants with change in quality of life measures (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) |
See comments | Effect is uncertain. Zoledronate showed a marginal improvement at 6 months in QoL when compared with risedronate. The physical component summary score of SF‐36 improved with zoledronate compared to risedronate (1.6 vs. 0.3 change from baseline score, on a 0 to 100 scale). This result is unlikely to be of clinical importance |
| Number of participants with change in hearing thresholds (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) |
See comments | Outcome not reported in the included studies |
| Number of participants who experienced side effects related to use of bisphosphonates. Follow up: mean 6 months | Study population | RR 1.05 (0.95 to 1.16) | 437 (2 RCTs) | ⊕⊕⊝⊝ LOW² | Absolute risk difference: 4% (‐4% to 12%) Relative percent change: 5% (‐5% to 16%) (worsening) |
|
| 745 per 1000 | 782 per 1000 (707 to 864) | |||||
| Number of participants who withdrew due to adverse events (withdrawals) follow up: mean 6 months | Study population | RR 2.04 (0.43 to 9.59) | 437 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW² ⁴ | Absolute risk difference: 1% (‐2% to 3%) Relative percent change: 104% (‐57% to 859%) (worsening) |
|
| 9 per 1000 | 18 per 1000 (4 to 83) | |||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; NNTB: Number needed to treat for an additional beneficial outcome; OR: odds ratio; QoL: quality of life; RCT: randomised controlled trial; RR: risk ratio; VAS: visual analogue scale | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
¹Unlike Table 1 (comparing bisphosphonates vs. placebo) when compared zoledronate vs pamidronate or risedronate we used any change of pain instead of disappearance of pain because data were not available. However, readers could find data on disappearance of pain in the original zoledronate vs. pamidronate manuscript (Merlotti 2007) [10/47 vs. 6/60, RR 2.12 95% IC 0.83‐5.43]. We did not include these data because we think they are misleading. They come from accumulate zoledronate effects in two different study phases (30 zoledronate patients from phase 1 + 17 patients no responders to pamidronate in phase 1 treated with zoledronate in phase 2) vs. pamidronate in only one study phase (60 patients).
² Downgraded by two levels (limitation of studies). Information is from studies at high risk of bias. High risk for performance bias in 1 study and high risk for reporting bias in 1 study.
³ Downgraded by one level (indirectness). In the risedronate study, the author assessed bodily pain but not bone pain associated directly with Paget's bone lesions (Change in bone pain was defined as"5‐point improvement from baseline" in SF‐36 bodily pain item). In the pamidronate study change in bone pain was defined as "subjects reported disappearance or decrease in pain".
⁴Downgraded by one level (imprecision). There were few events, resulting in wide CI,
Background
Description of the condition
Paget's disease of bone is a common disorder characterised by focal areas of increased and disorganised bone remodeling affecting one or more bones throughout the skeleton. Paget's disease preferentially targets the axial skeleton, most frequently affecting the pelvis (70% of cases), femur (55%), lumbar spine (53%), skull (42%) and tibia (32%) (Ralston 2013). Paget's disease of bone is rare before the age of 55 years, but increases in prevalence thereafter, affecting about 5% of women and 8% of men by the eighth decade of life in the United Kingdom (Van Staa 2002). The incidence and prevalence rates of Paget's disease of bone vary widely, but both have decreased in most regions over recent years. The changes are heterogeneous among and within countries; the largest changes have occurred in areas that previously had high prevalence (Corral‐Gudino 2013).
Paget's disease of bone is a cause of substantial morbidity in people who come to medical attention. Up to 50% of these people experience complications such as bone pain, bone deformity, pathologic fracture, deafness and secondary osteoarthritis that adversely affect quality of life (Tan 2014; Van Staa 2002). People with Paget's disease of bone run a significantly increased risk of developing osteoarthritis (Van Staa 2002) and their need for hip replacement is substantially higher than among age‐matched controls (Van Staa 2002). Osteosarcoma (malignant bone tumour) is a rare complication; however, virtually all osteosarcomas found in adults aged over 60 years occur in people with Paget's disease of bone (Sandberg 2003). Other uncommon complications of Paget's disease of bone include spinal stenosis (narrowing of the spinal canal), internal hydrocephalus (raised pressure within the brain), basilar impression (compression at the base of the skull affecting the spinal cord and blood flow to the brain) or cranial nerve deficits and other nerve compression syndromes (Selby 2002).
Description of the intervention
Medical therapy is based on drugs that inhibit the increased osteoclastic bone resorption that characterises Paget's disease of bone. Bisphosphonates are considered to be the treatment of choice for people with Paget's disease of bone because they are highly effective in suppressing elevated bone turnover. The principal indication for anti‐resorptive therapy is localised bone pain thought to be caused by increased metabolic activity (Selby 2002).
How the intervention might work
Bisphosphonates share a common phosphorous‐carbon‐phosphorous core to which various chemical side chains are attached. These side chains have a profound effect on the anti‐resorptive potency of bisphosphonates and on the mechanism by which osteoclast inhibition occurs. Non‐aminobisphosphonates, such as etidronate, clodronate or tiludronate, are relatively weak anti‐resorptive agents; nitrogen‐containing bisphosphonates (aminobisphosphonates) are much more powerful.
Bisphosphonates selectively bind to mineral surfaces in bone. During the process of bone resorption, bisphosphonates are internalised by osteoclasts and interfere with osteoclast survival and bone resorptive function. Non‐aminobisphosphonates are metabolically incorporated into non‐hydrolysable analogues of adenosine triphosphate, which interfere with adenosine triphosphate‐dependent intracellular pathways. These compounds inhibit formation of adenosine triphosphate in the osteoclast, thereby depleting intracellular energy stores and promoting apoptosis (programmed cell death) (Frith 2001). Aminobisphosphonates such as alendronate, neridronate, pamidronate, olpadronate, ibandronate, risedronate, or zoledronate, are much more potent than non‐aminobisphosphonates and act by inhibiting the enzyme farnesyl pyrophosphate synthase. Aminobisphosphonate‐mediated inhibition of this enzyme disrupts signalling pathways in osteoclasts, leading to failure of their resorptive function and cell death.
Aminobisphosphonates have greater inhibitory effects on bone turnover in Paget's disease of bone than non‐aminobisphosphonates and offer the prospect of reducing bone turnover to normal levels in a greater proportion of people than is possible with non‐aminobisphosphonates (Miller 1999; Siris 1996). Bone biopsy studies have shown that aminobisphosphonates therapy can restore the architecture of newly formed bone to normal (Siris 1996b). This raises the possibility that the more potent bisphosphonates, by giving greater inhibitor of bone turnover, may be able to prevent long‐term complications of the disease such as progression of deafness, bone deformity, fractures and progression of osteoarthritis.
Why it is important to do this review
Many experts believe that bisphosphonate therapy should be administered with the aim of normalising bone turnover in the hope this will arrest disease progression and prevent the development of some complications such as facial deformity, deafness when the skull base is affected, and spinal cord dysfunction in people with spinal Paget's disease of bone (Selby 2002). However, there are insufficient data to determine whether maintaining alkaline phosphatase levels within the normal range reduces the risk of complications. There is no evidence that people who are asymptomatic benefit from anti‐resorptive therapy (Langston 2010).
This review aimed to gather published evidence to inform us if bisphosphonate therapy improves clinical outcomes or prevents complications, apart from the known effect, over the biochemical markers of bone turnover.
Objectives
To assess the benefits and harms of bisphosphonates for adult patients with Paget's disease of bone.
Methods
Criteria for considering studies for this review
Types of studies
We included all randomised controlled trials (RCTs) or controlled clinical trials (CCTs) comparing bisphosphonate versus placebo or other active treatment for Paget's disease of bone in adults, including those that compared regimens of bisphosphonates aimed to normalise biochemical markers of bone turnover with those that did not.
Types of participants
Participants aged 18 years and over with Paget's disease of bone confirmed by plain radiographs or isotope bone scintigraphy (Selby 2002).
Types of interventions
The experimental intervention was defined as the use of bisphosphonates including non‐aminobisphosphonates (etidronate, clodronate or tiludronate) or aminobisphosphonates (alendronate, neridronate, pamidronate, olpadronate, ibandronate, risedronate or zoledronate).
The comparators were defined as placebo or other interventions including calcitonin, comparisons between non‐aminobisphosphonates and aminobisphosphonates; comparisons between different aminobisphosphonates, or comparisons among different treatment strategies using bisphosphonates.
We compared:
bisphosphonates versus placebo;
bisphosphonates versus calcitonin;
-
bisphosphonates versus bisphosphonates:
non‐aminobisphosphonates;
aminobisphosphonates and non‐aminobisphosphonates;
different aminobisphosphonates;
bisphosphonates regimens that aimed to normalise elevated bone turnover with those that did not (intensive versus non‐intensive treatment); and
bisphosphonates versus bisphosphonates plus calcitonin.
We considered calcium, vitamin D supplementation and analgesics as co‐interventions if use was equally available to all treatment groups.
The planned analysis according time points (3 or 6 months from the start of treatment; 1, 2, 3, 4 or more years from the start of treatment; and at the end of the trial) were not performed because nearly all included trials had only six months follow‐up.
Types of outcome measures
Major outcomes
We included assessment of the following patient‐oriented evidence.
-
Change in pain:
we included studies reporting any of the following tools for measuring pain: visual analogue scales (VAS), nominal scales or pain domains in generic quality of life measures (e.g. 'bodily pain' in the Short‐Form Health Survey (SF‐36));
we included only evaluations of pain rated by the participant and excluded physician‐rated evaluations;
-
we measured change in pain as a continuous variable whenever possible. Whenever pain was assessed as the number of participants with improved bone pain from baseline, we considered the following categories:
complete reduction of pain (reduction from baseline is 100%);
complete/partial reduction of pain (reduction from baseline ≥ 50%, up to and including complete reduction);
any reduction in pain.
Number of participants experiencing radiologically‐confirmed clinical fractures. Pathological fractures are uncommon (5.7%) and need long follow‐up to be assessed adequately (Tan 2014). We considered follow‐up periods longer than one year to properly assess this outcome.
Number of participants who underwent orthopaedic surgery.
-
Change in quality of life:
we included studies reporting use of tools for measuring generic quality of life including the Medical Outcomes Study (MOS), SF‐36, EuroQoL five dimensions questionnaire (EQ‐5D) and tools for measuring arthritis‐specific quality of life including the arthritis‐specific version of the SF‐36, and Stanford Health Assessment Questionnaire (HAQ) disability Index.
we measured change in quality of life as a continuous variable whenever possible. Whenever quality of life was assessed as the number of participants with improved quality of life from baseline, we considered any improvement in quality of life.
-
Change in hearing thresholds or degree of deafness:
we included studies reporting audiometric assessment and hearing threshold examinations;
we measured change in hearing thresholds or degree of deafness measured as a continuous variable whenever possible;
we also considered the number of participants who received hearing aids as an assessment of degree of deafness.
Number of participants experiencing adverse events related to use of bisphosphonates. We considered the following as serious adverse events: symptomatic hypocalcaemia, oesophagitis, oesophageal cancer, osteonecrosis of the jaw, osteonecrosis of the external auditory canal, uveitis, arrhythmias, atypical fracture and renal failure. We considered influenza‐like symptoms as a mild adverse events. Influenza‐like symptoms include myalgia, pyrexia, nausea, diarrhoea, dyspepsia, abdominal pain, headache, bone pain and fatigue.
Number of participants who withdrew due to adverse events.
Minor outcomes
For patient‐oriented evidence, we considered numbers of participants who relapsed due to recurrence of bone pain. For disease‐oriented evidence, we included:
mean percentage change from baseline in serum total alkaline phosphatase activity. We also recorded the number of participants who achieved normalised alkaline phosphatase levels; and
number of participants who relapsed due to recurrence of increased alkaline phosphatase level.
We did not consider data on radiographic changes or microscopic structure changes as outcomes following bisphosphonate therapy.
Search methods for identification of studies
Electronic searches
We searched:
MEDLINE (1946 to 3 March 2017) (Appendix 1);
Embase (1980 to 3 March 2017) (Appendix 2);
Cochrane Central Register of Controlled Trials (CENTRAL) (all issues to 3 March 2017) (Appendix 3);
ISI Web of Knowledge (all years to 8 March 2017) (Appendix 4).
We used the 'Cochrane Highly Sensitive Search Strategy for identifying randomised trials in MEDLINE: sensitivity‐maximising version (2008 revision); Ovid format' for identifying randomised trials as proposed in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). For Embase, we used a combination of the search filters for identifying randomised trials listed in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We set no year, language or country of publication restrictions.
Searching other resources
We screened the reference lists of all included articles to identify additional eligible studies. We focused on systematic reviews and meta‐analyses and reference lists from identified RCTs to identify further relevant studies.
We searched the ISRCTN registry on metaRegister of Controlled Trials (mRCT) and ClinicalTrials.gov to identify ongoing trials (Date of last search: 20 February 2017). The mRCT includes the International Clinical Trials Registry Platform (ICTRP) of the World Health Organization (WHO) and the United Kingdom Clinical Trials Gateway (UKCTG).
We also handsearched for abstracts presented from 2010 to 2016 at scientific meetings from the following societies and included those with sufficient information in the body of the abstract:
American Society for Bone and Mineral Research;
International Bone and Mineral Society;
International Osteoporosis Foundation/European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis; and
European Calcified Tissue Society.
In accordance with Cochrane Musculoskeletal Group recommendations we searched the websites of the regulatory agencies to broaden our search for specific and rare adverse events: USA Food and Drug Administration MedWatch (FDA), European Medicines Agency (EMA), and the UK Medicines and Healthcare products Regulatory Agency pharmacovigilance and drug safety updates (MHRA) using the terms 'Paget's' and 'bisphosphonate' (Date of last search for the regulatory agencies websites: 22 February 2017). We also searched in the Australian Adverse Drug Reactions Bulletin (AADRB) [published from 1995 to 2009 and available at https://www.tga.gov.au/publication/australian‐adverse‐drug‐reactions‐bulletin] and in the Australian Therapeutic Goods Administration website (Date of last search: 22 February 2017).
Data collection and analysis
Selection of studies
Three review authors (AT, LCG, JdPM) independently assessed results from searches of electronic databases to identify potentially‐relevant articles based on title or title and abstract. We retrieved the full manuscripts of potentially‐relevant articles for further assessment. Review authors (AT and LCG; JdPM and LCG) screened the selected articles independently against the inclusion criteria. We resolved any differences of opinion during the selection process by discussion and consensus, or by consulting a third review author (SHR) if needed.
Data extraction and management
Two review authors (AT, LCG) extracted relevant data using a pre‐defined data collection form designed according to guidelines from the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). One review author (AT) who was involved in data extraction was also an author of a primary study (PRISM‐EZ trial Tan 2017). Because authors of primary studies should not extract data from their own studies, we recruited an additional reviewer (JdPM) who extracted trial data from Tan 2017.
We extracted the following data:
Study identification: author, year of publication, journal.
Characteristics of the trial: study design, calculation of sample size before the study, use of intention‐to‐treat analysis, setting or location, number of centres, country, period of study, follow‐up period, outcomes, methods of randomisation, allocation concealment and blinding.
Study inclusion and exclusion criteria.
Participants' characteristics: age, gender, presence of monostotic disease, whether treated previously for Paget's disease of bone, symptomatic participants, numbers who were randomised and excluded (post‐randomisation), reasons for exclusion, participants assessed, withdrawals and reasons for withdrawals.
Characteristics of intervention: drug analysed, comparator, dosage, duration of treatment, co‐interventions.
Risk of bias assessment.
Outcome data (Types of outcome measures).
Source of funding.
Conflicts of interest.
We created a specific database to carry out the data extraction process. We resolved any differences of opinion during the data extraction process by discussion and consensus or discussion with a third author (SHR).
When data were neither available from the original manuscript, nor following requests to authors, data were directly extracted from figures in the manuscript using a vector graphics editor.
Assessment of risk of bias in included studies
Two authors (AT, LCG) assessed the risk of bias in included studies independently as recommended by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
We assessed the following methodological domains.
Adequate sequence generation (checking for possible selection bias): were methods used to generate the allocation sequence for each included study reported in sufficient detail to enable assessment of whether groups were comparable.
Adequate measures to conceal allocation (checking for possible selection bias): were methods used to conceal the allocation sequence for each included study reported in sufficient detail and we could determine whether intervention allocation could have been foreseen in advance or during recruitment, or changed after assignment.
Blinding of study participants and study researchers (checking for possible performance bias): were methods used to blind participants and study researchers from knowledge of which intervention a participant received for each included study reported.
Blinding of outcome assessors (checking for possible detection bias): were methods used to blind outcome assessors from knowledge of which intervention a participant received for each included study reported.
Incomplete outcome data (checking for possible attrition bias through withdrawals, dropouts, protocol deviations): the completeness of data including attrition and exclusions from the analysis was assessed. We stated whether attrition and exclusions were reported; the numbers included in the analysis at each stage (compared with the total randomised participants); reasons for attrition or exclusion where reported; and whether missing data were balanced across groups or were related to outcomes. We did not consider that a high attrition rate was a source of bias if an intention‐to‐treat (ITT) analysis was performed. Where sufficient information was reported or could be supplied by the study authors, we re‐included missing data in the analyses which we undertook.
Selective outcome reporting (checking for possible reporting bias): differences, if any, between the planned protocol analysis data and the reported data for each included study, to look for unreported findings.
Other potential threats to validity (considering external validity, e.g. relevant use of co‐intervention).
Each of these criteria were explicitly judged as 'low risk, 'high risk or 'unclear risk' of bias.
We assessed the likely overall magnitude of the bias for each included study and whether it was likely to impact on the findings. We elaborated a summary assessment of the risk of bias for each outcome. In order to assess the risk of bias within a study, we evaluated adequate sequence generation, allocation sequence concealment and incomplete outcome data as key domains for all the outcomes. We evaluated blinding of study participants and study researchers as key domains for change in pain, reporting of adverse events and change in quality of life. We did not consider blinding as a key domain for assessing the risk of bias for fractures, receiving orthopaedic surgery, change in hearing thresholds and total alkaline phosphate activity. Other potential threats to validity such as the use of analgesics as co‐interventions were considered a key domain for changes in pain and quality of life.
For each outcome, we considered low risk of bias to be present across studies when most information was from studies assessed at low risk. We considered high risk of bias across studies when the proportion of information from studies at high risk of bias was sufficient to affect the interpretation of results. Lastly, we considered unclear risk of bias when most information was from studies at low or unclear risk of bias.
We resolved any difference of opinion during the assessment of risk of bias process by discussion and achieving consensus. For disagreements not resolved by consensus, we consulted a third review author (SHR).
Measures of treatment effect
For dichotomous outcomes, we estimated the effect of treatment across trials using the risk ratios (RRs) with the corresponding 95% confidence intervals (CIs). For significant outcomes, we computed the number needed to treat to benefit (NNTB) one participant or the number needed to treat to harm (NNTH) one participant as the inverse of the pooled risk differences (RDs).
Our dichotomous outcomes were numbers of participants who:
experienced adverse events;
experienced radiologically‐confirmed clinical fractures;
received orthopaedic surgery;
withdrew due to adverse events;
relapsed due to recurrence of bone pain; and
relapsed due to recurrence of increased serum alkaline phosphatase level.
Where continuous scales of measurement were used to assess the effects of treatment, we used the mean difference (MD) with the corresponding 95% CI. If different scales were used to measure an outcome, we calculate the standardised mean difference (SMD).
Our continuous outcomes were:
change in pain;
change in quality of life;
change in hearing thresholds; and
mean percentage change from baseline in serum total alkaline phosphatase activity.
Unit of analysis issues
The unit of analysis was individual people undergoing treatment for Paget's disease of bone. We included only RCTs and CCTs, and we were not expecting studies with non‐standard designs such as cross‐over trials.
For studies with more than one intervention group, such as different doses of the same bisphosphonate, we combined data from the experimental intervention groups to create a single pair‐wise comparison versus the control group.
Dealing with missing data
When outcome data were not available from trials, we contacted the primary investigators of the eligible trials. We contacted study authors using email addresses published in the studies. When there was no response from the study author, we analysed the available data only, ignoring missing data, because we assumed that data were missing at random.
We performed a sensitivity analysis according to the missing data to assess the robustness of our assumption that data were missing data at random (Sensitivity analysis). We addressed the potential impact of missing data on findings in the Discussion.
Assessment of heterogeneity
We computed global estimates for each variable effect by meta‐analysing study single effect measures (RR for dichotomous variables and MD or SMD for continuous variables) using Review Manager 2014. Before calculating estimates of effect, we assessed the presence and degree of heterogeneity using the I² statistic to describe the percentage of variability in effect estimates due to heterogeneity rather than chance. We categorised I² values greater than 75% as considerable heterogeneity (Higgins 2011).
We undertook a narrative review of potential heterogeneity according to variability in populations, interventions, outcomes and settings.
Assessment of reporting biases
Because we anticipated inclusion of several studies with small sample sizes, we assessed the likely impact on our findings of small‐study effects (tendency for estimates of the intervention effect to be more beneficial in smaller studies) by using inverted funnel plot techniques. We planned to construct funnel plots when there were at least 10 included studies in comparisons. However, none of the comparisons included 10 studies, so funnel plots could not be constructed. We provided a narrative summary of potential small‐study effect in the Discussion.
Data synthesis
We used Review Manager 2014 for data synthesis. Quantitative synthesis was planned if more than one eligible study was identified. Where appropriate, we calculated a pooled estimate of treatment effect across similar studies for each pre‐specified outcome. We estimated overall effect by meta‐analysis using a random‐effects model. We did not conduct meta‐analysis if there was considerable heterogeneity (I² > 75%) (Higgins 2011). We calculated RRs and 95% CIs for dichotomous data. We calculated the number needed to treat to provide an indication for each dichotomous outcome, reflecting the number of participants required to obtain a beneficial outcome with the intervention. For continuous data measured on the same scale, we calculated MDs. When different scales were used, we calculated SMDs. When possible, we analysed data using an intention‐to‐treat (ITT) model.
We used the mean and standard deviation when available. If only median and interquartile ranges were reported, we followed guidance from the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), where the median was used as the mean and the standard deviation was set as 1.35. If no standard deviation was given at the end of the study, we used the baseline standard deviation at the end as well.
We undertook a narrative review of eligible studies where statistical synthesis of data from more than one study was not possible or appropriate.
Although we included different bisphosphonates whose comparisons against placebo which together create a network (Figure 1), we did not perform a network meta‐analysis. The reason for this was that tor the major outcome of change in pain, heterogeneity within studies in the definition of 'change in pain' did not allow us to conduct a meaningful network meta‐analysis. For the other major outcomes, such as fractures or orthopaedic surgery, there were few comparisons between bisphosphonates.
1.
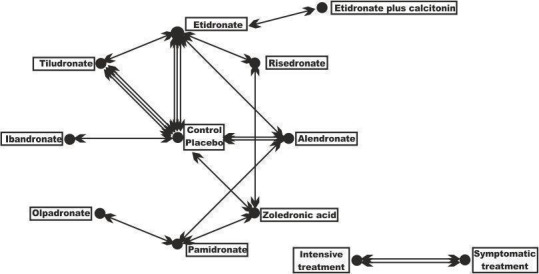
Geometry of the network of randomised trials of bisphosphonates for Paget's disease of bone. The nodes of the network represent the treatments compared. The links reflect comparisons and the number of links is proportional to the number of comparisons
Subgroup analysis and investigation of heterogeneity
Neither of the planned subgroup analyses: symptomatic bone pain versus those who were asymptomatic; and biochemically active (raised alkaline phosphatase) Paget's disease of bone versus non‐biochemically active Paget's disease of bone (normal alkaline phosphatase) were possible.
Sensitivity analysis
We planned to perform the following sensitivity analyses:
To determine the robustness of pooled effect estimate in terms of risk of bias by including or excluding studies with high or unclear risk of bias from the comparative analysis.
To determine the robustness of the pooled effect estimate in terms of missing data by including or excluding studies with high levels of missing data, more than 20% of missing data for the overall trial population, or for any of the trial arms from the comparative analysis.
To determine the robustness of the pooled effect estimate in terms of withdrawals by performing either worst‐case scenario sensitivity analysis (all withdrawal data treated as negative events) or the same‐as‐control scenario sensitivity analysis, where the rate of negative events in the withdrawal data were the same as in the control group.
'Summary of findings' table
We evaluated the quality of the evidence using the GRADE approach and developed 'Summary of findings' tables (Guyatt 2008). These tables have the following three elements:
the outcomes most relevant to people (critical and important outcomes according to GRADE);
a summary measure for the quality of the evidence (confidence in estimate of a treatment effect); and
a summary estimate for the RR and absolute effect for the interventions of interest.
The seven important outcomes that we considered for the 'Summary of findings' tables were numbers of participants:
with change in bone pain;
who experienced severe side effects related to use of aminobisphosphonates;
who experienced fractures;
who needed orthopaedic surgery;
with change in quality of life measures including bodily pain;
with change in hearing thresholds; and
who withdrew due to adverse events.
We used GRADEpro GDT 2015 to create 'Summary of findings' tables.
Results
Description of studies
See: Characteristics of included studies; Characteristics of excluded studies; Characteristics of ongoing studies.
Results of the search
Quantity of research available
The literature search identified 2781 potentially‐relevant records (Figure 2). Of these, 63 full text articles were retrieved for further assessment for inclusion.
2.

Study flow diagram
Inconsistent or missing information
We attempted to contact most study authors to request additional information or to clarify inconsistencies. Appendix 5 presents the status of requests to study authors for further information or clarification.
Included studies
We included 20 trials (25 references) that met our selection criteria. Sixteen individual studies with a single reference, (Altman 1973; Canfield 1977; Fraser 1997; Langston 2010; McClung 1995; Merlotti 2007; Miller 1999; O'Doherty 1992; O'Donoghue 1987; Ralston 1987; Reginster 1992; Reid 1996; Roux 1995; Siris 1996; Tan 2017Walsh 2004) 3 studies with two references (Barreira 2009; Buckler 1999; Reid 2004) and 1 study with 3 references (Reid 2005).
The trials involved a total of 3168 participants. All included studies were published in English language. One trial was published only as an abstract (Barreira 2009).
Characteristics of included studies
See Characteristics of included studies.
We included 10 studies (801 participants) that compared bisphosphonates and placebo (Altman 1973; Buckler 1999; Canfield 1977; Fraser 1997; McClung 1995; O'Doherty 1992; Ralston 1987; Reginster 1992; Reid 1996; Reid 2004). One study (234 participants) compared two non‐aminobisphosphonates (Roux 1995). Two studies (212 participants) compared non‐aminobisphosphonates and aminobisphosphonates (Miller 1999; Siris 1996). Four studies (546 participants) compared two aminobisphosphonates (Barreira 2009; Merlotti 2007; Reid 2005; Walsh 2004). One study (44 participants) compared etidronate and etidronate plus calcitonin (O'Donoghue 1987). One study compared intensive treatment (aimed at normalising alkaline phosphatase) and symptomatic treatment (aimed at treating bone pain) (PRISM trial Langston 2010; 1331 participants). An extension of Langston 2010 (PRISM‐EZ trial Tan 2017; 502 participants) investigated the effects of these treatment strategies for up to 7.3 years. PRISM‐EZ is described separately in Characteristics of included studies because although the study was defined by the authors as an extension study, it was an interventional extension study (rather than an observational extension study) where the researchers continued intensive or symptomatic treatment as in the core study. In this case, zoledronate was used as the treatment of first choice in the extension as compared with risedronate in the PRISM study.
None of the included studies used a run in period.
The different comparisons investigated by the studies are represented in Figure 1. The sample sizes ranged from 15 to 1331 participants; 11 studies included fewer than 100 participants. Only Langston 2010 included more than 1000 participants. Sample size calculations were performed in advance of the study for only five trials (Fraser 1997; Langston 2010; Reid 2005; Roux 1995; Walsh 2004).
The study populations were reasonably homogeneous in terms of participants' age and gender. A summary of the principal characteristics of trial samples is provided in Table 3. Participants' mean age ranged from 66 years to 74 years, the percentage of males ranged from 51% to 74%, and the percentage of symptomatic participants ranged from 63% to 100%. Nearly all participants had raised serum total alkaline phosphatase, but one study (Langston 2010) included participants with normal alkaline phosphatase at baseline (707/1324; 53%). Alkaline phosphatase was normal at baseline in participants who were enrolled into an extension of the Langston 2010 study (Tan 2017) (355/502; 71%). The percentage of symptomatic participants (defined as those with pain due to Pagetic bone lesions) ranged from 66% to 100%; this information was not recorded for about half the included studies.
1. Principal study characteristics.
| Study ID | Intervention | Comparator | Alkaline phosphatase | Follow‐up | N | Age | Male | Symptomatic | Previously treated |
| Altman 1973 | Etidronate | Placebo | Yes | 6 m | 50 | 67 y | 60% | NA | NA |
| Canfield 1977 | Etidronate | Placebo | Yes | 6 m | 48 | NA | 58% | NA | NA |
| Ralston 1987 | Etidronate | Placebo | No | 3 m | 32 | NA | NA | 100% | 38% |
| Fraser 1997 | Tiludronate | Placebo | Yes | 6 m (18 m) | 112 | 70 y | 54% | 63% | NA |
| McClung 1995 | Tiludronate | Placebo | Yes | 6 m | 139 | 70 y | 54% | NA | NA |
| Reginster 1992 | Tiludronate | Placebo | Yes | 6 m | 149 | 69 y | 54% | NA | 82% |
| O'Doherty 1992 | Alendronate | Placebo | Yes | 6 m | 15 | 67 y | 60% | 87% | 66% |
| Reid 1996 | Alendronate | Placebo | Yes | 6 m | 55 | 70 y | 56% | NA | 35% |
| Buckler 1999 | Zoledronate | Placebo | Yes | 3 m | 176 | 71 y | 61% | NA | NA |
| Reid 2004 | Ibandronate | Placebo | Yes | 6 m (12 m) | 25 | 73 y | 74% | NA | 64% |
| Roux 1995 | Tiludronate | Etidronate | Yes | 6m | 234 | 69y | 59% | 74% | 71% |
| Siris 1996 | Alendronate | Etidronate | Yes | 6m | 89 | 69y | 67% | NA | 25% |
| Miller 1999 | Risedronate | Etidronate | Yes | 12 m (18 m) | 123 | 66 y | 69% | 91% | 72% |
| Walsh 2004 | Alendronate | Pamidronate | Yes | 12 m (24 m) | 72 | 70 y | 58% | 94% | 39% |
| Barreira 2009 | Olpadronate | Pamidronate | Yes | 6 m | 27 | NA | NA | NA | NA |
| Merlotti 2007 | Zoledronate | Pamidronate | Yes | 6 m | 90 | 70 y | 69% | 99% | 67% |
| Reid 2005 | Zoledronate | Risedronate | Yes | 6 m (6.5 y) | 357 | 70 y | 68% | NA | 54% |
| O'Donoghue 1987 | Etidronate + calcitonin |
Etidronate | Yes | 12 m | 44 | NA | NA | 100% | 10% |
| Langston 2010 | Intensive | Symptomatic | No | 3 y | 1331 | 74 y | 51% | 69% | NA |
| Tan 2017 | Intensive | Symptomatic | No | 3 y | 502 | 76 y | 55% | 63% | 70% |
Alkaline phosphatase: Serum total alkaline phosphatase above the upper limit of normal as an inclusion criterion. Follow‐up: Extended follow‐up periods are shown in parentheses. N: Number of randomised participants. NA: Not available. SC: Sample size calculated before study.
For all but one study (Langston 2010) and its extension (Tan 2017) the primary outcome was change in serum total alkaline phosphatase activity. Most studies did not include the major outcomes (bone fracture, need for orthopaedic surgery, change in quality of life or hearing thresholds) of primary interest in this review.
Thirteen studies had six months of follow‐up; two studies (Buckler 1999; Ralston 1987) had only three months follow‐up. Another three studies (Miller 1999; O'Donoghue 1987; Walsh 2004) had 12 months of follow up. The authors of the Reid 2004 study published long‐term follow‐up data (nearly 10 years), but analysis of these data was limited due to change in the initial randomisation allocation (see Characteristics of included studies). The PRISM study (intensive versus symptomatic treatment, Langston 2010) was an event‐driven trial with an average of three years follow‐up (range 2 years to 5 years). In PRISM‐EZ, Tan 2017 followed up participants for an additional three years (total of 7.3 years follow‐up). An extension to the comparison of zoledronate and risedronate followed up participants for up to 6.5 years but these were a select group who had normalised alkaline phosphatase during the core study (Reid 2005).
Study funding sources
Eleven included studies were funded by drug manufacturers. Of these, four were co‐funded by government agencies or charities. Although a further three studies reported funding by government agencies or charities, the study drugs were supplied by drug manufacturers. One article was funded only by government agencies. Funding sources were not mentioned or unclear in five studies.
Excluded studies
We excluded 35 studies: 17 were non‐randomised clinical trials (Adami 1994; Altman 1985; Arlot 1981; Atkins 1987; Cundy 2016; Dewis 1985; Filipponi 1994; Gallacher 1991; Grauer 1999; Gutteridge 1996; Hosking 1976; Khairi 1977; Mazeries 1996; O'Doherty 1995; Pepersack 1994; Russell 1974; Stone 1990); nine were RCTs that lacked comparisons of interest for this review (Adami 2002; Buckler 1998; Delmas 1982; Hooper 2009; Khan 1997; Merlotti 2011; Reginster 1988; Reginster 1993; Vega 1994); two were non‐randomised extension studies (Khairi 1974; Siris 1980); four were non‐randomised samples of participants from clinical trials (Devogelaer 1997; Garnero 1998; Garnero 2001; Goldman 1975); one was a case control study (Donáth 2004); one provided data for a patient registry (Devogelaer 2014); and one was a review (Lombardi 1999).
Ongoing studies
We identified two ongoing studies (NCT02106455; ISRCTN11616770). These studies will be assessed for inclusion in a future update of this review.
Risk of bias in included studies
For details on the risk of bias of included studies see Characteristics of included studies. For an overview of review authors' judgements about each risk of bias item for individual and across all studies see Figure 3 and Figure 4.
3.
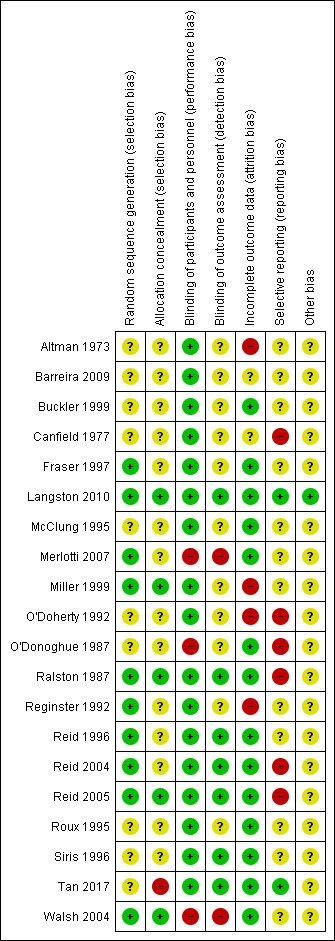
Risk of bias summary: review authors' judgements about each risk of bias item for each included study
4.
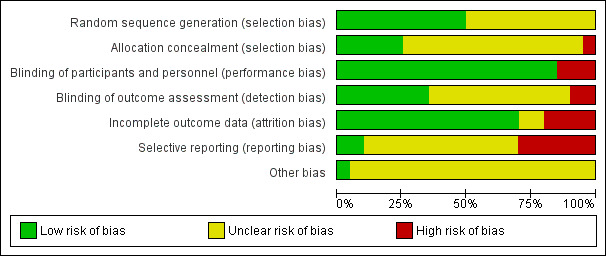
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies
Allocation
The generation of the random sequence for allocation was considered to be adequate in 10 studies (low risk of bias; Fraser 1997; Langston 2010; Merlotti 2007; Miller 1999; Ralston 1987; Reginster 1992; Reid 1996; Reid 2004; Reid 2005; Walsh 2004). Risk of bias was judged as unclear in the remainder (Altman 1973; Barreira 2009; Buckler 1999; Canfield 1977; McClung 1995; O'Doherty 1992; O'Donoghue 1987; Roux 1995; Siris 1996; Tan 2017). Allocation concealment was considered adequate in only six studies (low risk of bias; Langston 2010; Miller 1999; Ralston 1987; Reid 2005; Tan 2017; Walsh 2004) and judged as unclear in the remainder (Altman 1973; Barreira 2009; Buckler 1999; Canfield 1977; Fraser 1997; McClung 1995; Merlotti 2007; O'Doherty 1992; O'Donoghue 1987; Reginster 1992; Reid 1996; Reid 2004; Roux 1995; Siris 1996). No included studies were considered to be at high risk of bias for this domain.
Blinding
Fifteen studies were double‐blinded (Altman 1973; Barreira 2009; Buckler 1999; Canfield 1977; Fraser 1997; McClung 1995; Miller 1999; O'Doherty 1992; Ralston 1987; Reginster 1992; Reid 1996; Reid 2004; Reid 2005; Roux 1995; Siris 1996) and their risk of bias was judged as low. Five studies were open‐label (Langston 2010; Merlotti 2007; O'Donoghue 1987; Walsh 2004; Tan 2017). The performance risk of bias was considered as high for subjective outcomes as pain or quality of life for all of them. The performance risk of bias was considered low for two of the open‐label studies (Langston 2010; Tan 2017) because the main outcome (radiologically‐confirmed clinical fracture) was not likely to be influenced by lack of blinding. The outcome assessment was not blinded in another study (Miller 1999).
Incomplete outcome data
The risk of bias due to incomplete outcome data was considered to be low in fourteen studies (Buckler 1999; Fraser 1997; Langston 2010; McClung 1995; Merlotti 2007; O'Donoghue 1987; Ralston 1987; Reid 1996; Reid 2004; Reid 2005; Roux 1995; Siris 1996; Tan 2017; Walsh 2004). Four studies were judged at high risk of bias because of significant missing data (Reginster 1992), differences between the follow‐up of the different control and experimental group (O'Doherty 1992) or data inconsistencies in the manuscript (Altman 1973; Miller 1999). Risk of bias was unclear in two studies (Barreira 2009; Canfield 1977).
Selective reporting
Selective reporting bias was difficult to evaluate because study protocols were available for only four studies (Langston 2010; Reid 2004; Reid 2005; Tan 2017). Five studies were judged at high risk of bias due to selective reporting because data on adverse events were not clearly detailed (Canfield 1977); there were no data on adverse events (O'Doherty 1992; O'Donoghue 1987); or because adverse events were not systematically recorded in the trials (Ralston 1987; Reid 2004). Two studies were judged at high risk of bias due to selective reporting because details of assessment for some outcomes included in the results sections (pain, fractures) were not detailed in methods sections (O'Doherty 1992; O'Donoghue 1987). Conversely in another study, some outcomes mentioned in the methods section (pain, fractures) were not provided in the results section (high risk of bias) (Reid 2005). Risk of bias was judged as unclear for the remainder of the included studies. No studies were assessed as at low risk of bias.
Other potential sources of bias
Langston 2010 and its extension study (Tan 2017) were assessed with potential risk of bias for subjective outcomes due to their open‐label designs. In these studies, the attending clinician was able to choose which bisphosphonate should be prescribed, resulting in heterogeneity between groups in numbers and types of bisphosphonate used. However, adherence to the randomised treatment strategy was confirmed by the fact that alkaline phosphatase values were significantly lower in the intensive group as compared with the symptomatic group for both trials.
Effects of interventions
Table 1 presents findings for bisphosphonates versus placebo; Table 2 presents findings for bisphosphonates versus bisphosphonates.
Major outcomes
Effect on bone pain
Bone pain was not considered as a primary outcome by any of the included studies, although 15 reported change in bone pain as secondary outcomes. Eight studies assessed pain on visual analogue scales (VAS; range 0 to 10) (Fraser 1997; McClung 1995; Ralston 1987; Reid 2005; Reginster 1992; Roux 1995; Siris 1996; Walsh 2004).
Studies classified pain using different categories. Merlotti 2007 classified pain as: never in pain, pain disappeared, pain decreased or no change in pain. Altman 1973 applied three categories: mild pain, moderate pain or severe pain. Langston 2010 and Tan 2017 categorised pain according to whether or not participants had bone pain. Three studies measured bone pain using the SF‐36 bodily pain domain (Langston 2010; Miller 1999; Tan 2017). Three studies did not report the tool used for pain assessment (Canfield 1977; O'Doherty 1992; O'Donoghue 1987). Analyses were based on any bone pain reduction and disappearance of bone pain to assess changes in bone pain.
Bisphosphonates versus placebo
Two studies (Fraser 1997; Reginster 1992) compared tiludronate versus placebo. The overall effect on disappearance of pain (defined as complete reduction of pain) favoured bisphosphonates (RR 3.42, 95% CI 1.31 to 8.90; NNTB 5, 95% CI 1 to 35; absolute risk of event 31% versus 9%; Analysis 1.1).
1.1. Analysis.

Comparison 1 Bisphosphonates versus placebo, Outcome 1 Number of participants whose bone pain disappeared completely.
When effect on bone pain was defined as any reduction in pain, the overall effect favoured bisphosphonates (RR 1.97, 95% CI 1.29 to 3.01; RD 0.33, 95% CI 0.18 to 0.49; NNTB 4, 95% CI 2 to 13; absolute risk of event 45% versus 23%). Results were consistent across the trials; all studies included in the meta‐analysis showed a favourable effect (RR range from 1.20 to 10.00; Analysis 1.2; Figure 5).
1.2. Analysis.
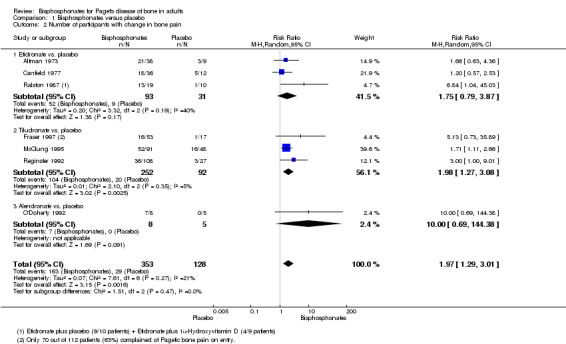
Comparison 1 Bisphosphonates versus placebo, Outcome 2 Number of participants with change in bone pain.
5.
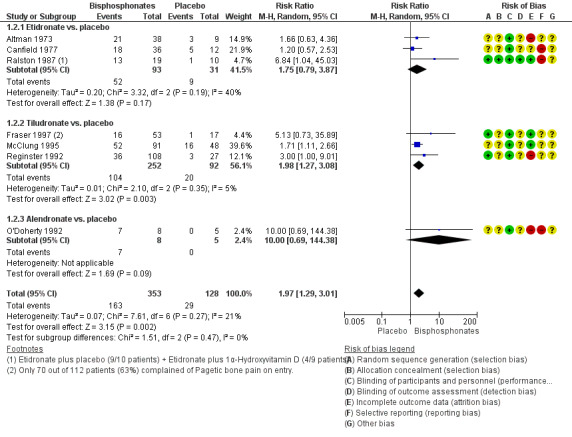
Forest plot of comparison: 1 Bisphosphonates versus placebo, outcome: 1.2 Number of participants with change in bone pain
Results for effect on bone pain (defined as pain disappearance or any reduction in pain) were consistent.
When change in bone pain was assessed as a continuous variable, data were heterogeneous. Ralston 1987 used a linear analogue scale and reported that etidronate was better than placebo. McClung 1995 measured bone pain using the Huskisson pain severity score instrument and reported no detectable difference between bisphosphonates and placebo in average pain scores. Reid 1996 used the Brief Pain Inventory and reported detectable differences favouring placebo when compared with alendronate (placebo ‐1.4 ± 0.3 versus alendronate ‐0.7 ± 0.5, difference favoured placebo ‐0.7, 95% CI 0.41 to 0.99).
Bisphosphonates versus active comparator
There was insufficient evidence to confirm or exclude an important difference (MD ‐2.60, 95% CI ‐5.03 to ‐0.17) in pain scores (0 to 100) when etidronate was compared with risedronate (measured on SF‐36) (Miller 1999) or alendronate (measured on the Brief Pain Inventory slightly modified for use in Paget's disease of bone) (Siris 1996) (Analysis 2.1). Pain assessment as a dichotomous variable was not made for this comparison.
2.1. Analysis.

Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 1 Mean change from baseline in pain.
Zoledronate showed a statistically‐ and clinically‐significantly greater effect on partial or total pain relief (binary outcome) when compared with the aminobisphosphonates pamidronate (RR 1.30, 95% CI 1.10 to 1.53; Merlotti 2007; 89 participants; NNTB 4, 95% CI 3 to 13; absolute risk of event 97% versus 75%) and risedronate (RR 1.36 95% CI 1.06 to 1.74; Reid 2005; 347 participants; NNTB 8, 95% CI 4 to 45, absolute risk of event 50% versus 37%; Analysis 3.1). However, there was no evidence of an effect when bone pain was assessed as a continuous outcome (scores) between zoledronate and risedronate (Reid 2005) (Table 4). Results were inconclusive between treatments for Pagetic bone pain, Pagetic joint pain, or non‐Pagetic pain when alendronate was compared with pamidronate (Walsh 2004). Scores for bone pain, joint pain and non‐Pagetic pain all fell significantly (P < 0.05) from baseline during the first 12 months of the study in both treatment groups (Walsh 2004). Between‐group numerical results were not provided in Walsh 2004.
3.1. Analysis.
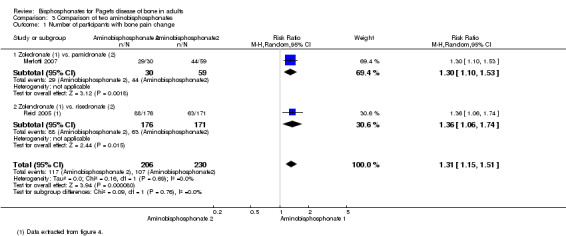
Comparison 3 Comparison of two aminobisphosphonates, Outcome 1 Number of participants with bone pain change.
2. Comparison of two aminobisphosphonates (Reid 2005).
| Study ID | Outcome | Zoledronate | Risedronate | RR (95% IC) | ||
| Events | N | Events | N | |||
| Reid 2005 | Radiologically‐confirmed clinical fracture | 2 | 177 | 2 | 172 | 0.97 (0.14 to 6.82) |
| Reid 2011 (extension) | Radiologically‐confirmed clinical fracture | 3 | 152 | 1 | 115 | 2.30 (0.24 to 22.36) |
| Reid 2005 | Quality of life change from baseline | 48 | 176 | 36 | 171 | 1.30 (0.89 to 1.89) |
| Reid 2011 (extension) | Clinical relapse | 14 | 152 | 29 | 115 | 0.30 (0.15 to 0.60) |
| Study ID | Outcome | Mean (SD) | N | Mean (SD) | N | Mean difference |
| Reid 2005 | Mean change from baseline in pain | ‐0.5 (1.75) | 101 | ‐0.4 (2.13) | 92 | ‐0.10 (‐0.65 to 0.45) |
| Reid 2005 | Mean change from baseline in quality of life¹ | 1.5 (0.5) | 176 | 0.2 (0.6) | 171 | 1.30 (1.18 to 1.42) |
| Reid 2011 (extension) | Mean change from baseline in total SF‐36 score² | 1.3 (3.1) | 152 | ‐2.5 (2.6) | 115 | 3.8 (3.12 to 4.49) |
¹Physical‐component summary (data extracted from Figure 4 in Reid 2005).
²Total SF‐36 scores to 54 months (data extracted from Figure 6 in Reid 2011 extension) (+1.3 ± 3.1 versus ‐2.5 ± 2.6) [D]
Adding calcitonin to etidronate (O'Donoghue 1987) reduced the proportion of participants experiencing partial or total pain relief as compared with etidronate alone (RR 0.73, 95% CI 0.43 to 1.25; Table 5).
3. Bisphosphonates vs. bisphosphonates plus calcitonin (O'Donoghue 1987).
| Ouctome | Etidronate plus calcitonin | Etidronate | RR (95% IC) | ||
| Events | N | Events | N | ||
| Change in bone pain | 10 | 21 | 15 | 23 | 0.73 (0.43 to 1.25) |
Intensive versus symptomatic treatment
In Langston 2010 and the extension study Tan 2017, there was insufficient evidence to confirm or exclude a important difference for partial or total pain relief or change in bodily pain measured using the SF‐36 between symptomatic and intensive treatment groups (improvement in bone pain RR 0.86, 95% CI 0.67 to 1.10) (Table 6).
4. Intensive versus symptomatic treatment.
| Study ID | Outcome | Intensive | Symptomatic | RR (95% IC) | ||
| Events | N | Events | N | |||
| Langston 2010 | Improvement in bone pain | 78 | 295 | 96 | 311 | 0.86 (0.67 to 1.10) |
| Langston 2010 | Radiologically‐confirmed fractures | 46 | 661 | 49 | 663 | 0.94 (0.64 to 1.39) |
| Tan 2017 | Radiologically‐confirmed fractures*¹ | 22 | 270 | 12 | 232 | 1.58 (0.80 to 3.11) |
| Langston 2010 | Radiologically‐confirmed fractures (pagetic bone) | 8 | 661 | 13 | 663 | 0.62 (0.25 to 1.49) |
| Tan 2017 | Radiologically‐confirmed fractures (pagetic bone)¹ | 5 | 270 | 2 | 232 | 2.15 (0.42 to 10.96) |
| Langston 2010 | Number of orthopaedic surgeries | 48 | 661 | 55 | 663 | 0.88 (0.60 to 1.27) |
| Tan 2017 | Number of orthopaedic surgeries¹ | 15 | 270 | 7 | 232 | 1.84 (0.76 to 4.44) |
| Langston 2010 | Number of orthopaedic procedures | 50 | 661 | 63 | 663 | 0.78 (0.53 to 1.15) |
| Tan 2017 | Number of orthopaedic procedures | 16 | 270 | 9 | 232 | 1.52 (0.69 to 3.39) |
| Langston 2010 | Change in hearing thresholds² | 134 | 505 | 133 | 486 | 0.97 (0.79 to 1.19) |
| Langston 2010 | Hearing classification worse at study end (left ear)³ | 6 | 50 | 8 | 63 | 0.95 (0.35 to 2.55) |
| Langston 2010 | Hearing classification worse at study end (right ear)³ | 4 | 51 | 8 | 60 | 0.58 (0.19 to 1.84) |
| Langston 2010 | Serious adverse events | 345 | 661 | 359 | 663 | 0.96 (0.87 to 1.07) |
| Tan 2017 | Serious adverse events | 87 | 270 | 66 | 232 | 1.13 (0.87 to 1.48) |
| Langston 2010 | Withdrawal due to adverse events⁴ | 83 | 661 | 79 | 663 | 1.05 (0.79 to 1.41) |
| Langston 2010 | Normalised alkaline phosphatase levels | 512 | 661 | 406 | 663 | 1.26 (1.18 to 1.36) |
| Study ID | Outcome | Mean (SD) | N | Mean (SD) | N | Mean difference |
| Langston 2010 | Mean change from baseline in quality of life (bodily pain SF‐36)⁵ | ‐0.4 (8.9) | 479 | 0.3 (9.4) | 477 | ‐0.7 (‐1.8 to 0.5) |
| Tan 2017 | Mean change from baseline in quality of life (bodily pain SF‐36)⁶ | 0.1 (9.3) | 149 | ‐1.0 (9.1) | 138 | ‐1.0 (‐3.0 to 1.1) |
| Langston 2010 | Mean change from baseline in quality of life (physical summary SF‐36)*⁵ | ‐1.2 (8.1) | 408 | ‐1.1 (8.2) | 396 | ‐0.1 (‐1.3 to 1.0) |
| Tan 2017 | Mean change from baseline in quality of life (physical summary SF‐36)⁶ | ‐1.0 (7.7) | 144 | ‐2.7 (7.7) | 126 | ‐1.6 (‐3.4 to 0.3) |
| Langston 2010 | Mean change from baseline in quality of life (mental summary SF‐36)*⁵ | ‐1.7 (10.2) | 408 | ‐2.6 (10.9) | 396 | 0.9 (‐0.6 to 2.3) |
| Tan 2017 | Mean change from baseline in quality of life (mental summary SF‐36)⁶ | ‐1.0 (10.0) | 144 | ‐0.4 (9.9) | 126 | ‐0.6 (‐1.7 to 3.1) |
| Langston 2010 | Mean hearing loss (left ear)³ | 1.8 (14.6) | 50 | 0.0 (12.6) | 63 | 1.8 (‐3.4 to 7.0) |
| Langston 2010 | Mean hearing loss (right ear)³ | 2.5 (5.7) | 51 | 2.1 (9.4) | 60 | 0.5 (‐2.4 to 3.3) |
| Langston 2010 | Mean percentage change from baseline in serum total alkaline phosphatase activity | ‐40.5 (23.7) | 430 | ‐18 (71.2) | 424 | ‐22.5 (‐29.6 to ‐15.4) |
| Tan 2017 | Mean percentage change from baseline in adjusted serum total alkaline phosphatase activity | ‐0.15 (0.72) | 203 | ‐0.05 (0.75) | 181 | ‐0.11 (‐0.03 to 0.25) |
Data at 24 months for Langston 2010.
¹Data shown for these outcomes are number of events, patient years of follow up, rate ratios and 95% CI calculated using the method described by Cohen 2011.
²Number of participants using hearing aids at the end of the study.
³Patients with baseline and end of the trial measurements.
⁴Serious adverse event: any untoward medical occurrence that: 1) results in death, 2) is life‐threatening, 3) requires inpatient hospitalisation or prolongation of existing hospitalisation, 4) results in persistent or significant disability/incapacity, or 5) is a congenital anomaly/birth defect.
⁵Data at 24 months.
⁶Difference between baseline and 36 months.
Effect on fractures
Radiologically‐confirmed clinical fracture was the primary outcome in Langston 2010 and its extension study Tan 2017, and a secondary outcome in six studies (Altman 1973; Canfield 1977; Fraser 1997; Reid 2005; Reginster 1992; Roux 1995). The number of new fractures were extremely low in all studies except Langston 2010 and its extension Tan 2017.
Bisphosphonates versus placebo
We could not determine whether the intervention had an important effect on fractures because the sample size was small and the long‐term impact on fractures was not assessed. Etidronate (Altman 1973; Canfield 1977) and tiludronate (Fraser 1997; Reginster 1992) were compared to placebo (pooled RR, 0.89 95% CI 0.18 to 4.31; 0 fractures in the placebo group; Analysis 1.3). The mean follow‐up period was six months (Altman 1973; Canfield 1977; Fraser 1997; Reginster 1992).
1.3. Analysis.
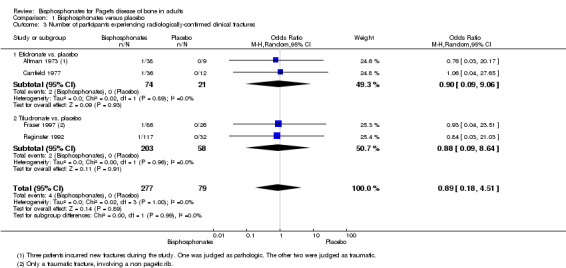
Comparison 1 Bisphosphonates versus placebo, Outcome 3 Number of participants experiencing radiologically‐confirmed clinical fractures.
Bisphosphonates versus active comparator
Tiludronate was compared to etidronate (Roux 1995) and zoledronate to risedronate (Reid 2005) with no evidence of a difference between these bisphosphonates (Table 7; Table 4). The results were inconclusive because long‐term impact on fractures was not assessed and samples sizes were small.
5. Comparison of two non‐aminobisphosphonates: Roux 1995.
| Outcome | Tiludronate | Etidronate | RR (95% IC) | ||
| Events | N | Events | N | ||
| Number of participants with change in bone pain | 32 | 120 | 10 | 52 | 1.39 (0.74 to 2.61) |
| Number of participants with radiologically‐confirmed fractures | 1 | 155 | 2 | 79 | 0.25 (0.02 to 2.77) |
| Number of participants with severe side effects | 75 | 155 | 27 | 79 | 1.42 (1.00 to 2.00) |
| Number of participants who withdrew due to adverse events | 10 | 155 | 2 | 79 | 2.55 (0.57 to 11.35) |
| Number of participants who normalised alkaline phosphatase levels | 40 | 155 | 9 | 79 | 2.27 (1.16 to 4.43) |
Intensive versus symptomatic treatment
The Langston 2010 study of intensive bisphosphonate treatment versus symptomatic treatment showed no evidence of an effect in the number of new fractures between treatment approaches (6.7% versus 7.4%, RR 0.94, 95% CI 0.64 to 1.39; Table 6). Follow‐up was long enough duration to assess long‐term impact on fractures (3 years mean follow‐up). In the extension study by Tan 2017, all fractures and fractures in Pagetic bone were more common in the intensive treatment group (8.1% versus 5.2%, RR 1.58, 95% CI 0.80 to 3.11; 1.9% versus 0.9%, RR 2.15, 95% CI 0.42 to 10.96 respectively). However, the confidence intervals were wide. It should be noted that Tan 2017 reported hazard ratio for fractures in the intensive versus symptomatic group when corrected for baseline differences between groups using propensity scoring. This showed a hazard ratio for fracture in the intensive versus symptomatic group of 1.90 (95% CI 0.91 to 3.98).
Effect on need for orthopaedic surgery
The need for orthopaedic surgery was not considered as a primary outcome by any of the included studies; only Langston 2010 and Tan 2017 included this outcome.
Bisphosphonates versus placebo
No included studies addressed need for orthopaedic surgery for bisphosphonates versus placebo.
Bisphosphonates versus active comparator
No included studies addressed need for orthopaedic surgery for comparison between bisphosphonates.
Intensive versus symptomatic treatment
There was no evidence of a difference between intensive and symptomatic treatment; 7.3% of participants in the intensive treatment group and 8.3% participants in the symptomatic treatment group needed orthopaedic surgery (RR 0.88, 95% CI 0.60 to 1.27; Table 6). In Tan 2017, all orthopaedic procedures were more common in intensive group participants (5.6% versus 3.0%, RR 1.84, 95% CI 0.76 to 4.44) as were procedures in Pagetic bone; 2.6% versus 1.7% (RR 1.50, 95% CI 0.45 to 5.07). However, the confidence intervals were wide. It should be noted that Tan 2017 reported hazard ratio for orthopaedic procedures in the intensive versus symptomatic group when corrected for baseline differences between groups using propensity scoring. This showed a hazard ratio for orthopaedic procedures in the intensive versus symptomatic group of 1.81 (95% CI 0.71 to 4.61).
Effect on quality of life
The impact of bisphosphonates on quality of life was included as a secondary outcome by six studies (Langston 2010; Merlotti 2007; Miller 1999; Reid 2005; Tan 2017; Walsh 2004).
Bisphosphonates versus placebo
No studies addressed effects on quality of life for the comparison of bisphosphonates versus placebo.
Bisphosphonates versus active comparator
There was no evidence of an effect on quality of life when zoledronate was compared with risedronate (Reid 2005), although zoledronate showed a marginal improvement at six months, with a mean difference in change on the physical component summary score of 1.30 (95% CI 1.18 to 1.42: Table 4). Furthermore, in the extension study (Reid 2011), participants who attained normal alkaline phosphatase levels following zoledronate maintained lower levels of total SF‐36 score when compared with risedronate for a period of up to 54 months (Table 4). For the pain domain of the SF‐36, changes from baseline were significant in the zoledronate group at 24 months (7.5 ± 2.6) and 36 months (5.6 ± 2.4); whereas no significant changes were observed for the risedronate group at any time point.
General health score on the SF‐36 was higher when alendronate was compared to pamidronate according to Walsh 2004. There was no evidence of a difference between treatments in the other seven domains of the SF‐36 or in the mental and physical component summary scores. Numeric data were not provided in the study report (Walsh 2004). Merlotti 2007 did not detect a difference when zoledronate was compared to pamidronate. The authors of both Walsh 2004 and Merlotti 2007 reported there were no differences, but numeric data were not provided.
Intensive versus symptomatic treatment
A comparison of intensive and symptomatic treatment did not detect a difference in quality of life between treatment groups (Langston 2010). Mean changes in bodily pain, physical and mental summaries of SF‐36 are shown in Table 6. In the extension study to Langston 2010, Tan 2017 reported small differences between groups at Year 1 (physical component summary scores and arthritis‐specific health index) which favoured the intensive treatment group. However, there was no sustained difference in quality of life at Year 2 or Year 3.
Effect on deafness and hearing thresholds
Deafness and hearing thresholds were not considered as a primary outcome by any of the included studies; only Langston 2010 included data on these outcomes.
Bisphosphonates versus placebo
No included studies addressed effects on deafness and hearing thresholds for the comparison of bisphosphonates versus placebo.
Bisphosphonates versus active comparator
No included studies addressed effects on deafness and hearing thresholds for the comparison of bisphosphonates with an active comparator.
Intensive versus symptomatic treatment
A comparison of intensive and symptomatic treatment did not detect a difference between groups in deafness or hearing thresholds. Changes in hearing thresholds were recorded in 26.7% of participants in the intensive treatment group versus 27.4% of participants in the symptomatic group (RR 0.97, 95% CI 0.79 to 1.19). Changes in hearing loss and hearing classification data are provided in Table 6.
Adverse events
Four trials (Canfield 1977; O'Doherty 1992; O'Donoghue 1987; Ralston 1987) did not report on adverse events related to bisphosphonates. Data from other trials on adverse events were heterogeneous; adverse events reporting was not comprehensive. Furthermore, assessment of adverse events severity varied among studies. Adverse events reported in trials comparing bisphosphonates versus placebo (Altman 1973; Buckler 1999; Fraser 1997; McClung 1995; Reginster 1992; Reid 1996), bisphosphonates versus bisphosphonates (Barreira 2009; Merlotti 2007; Miller 1999; Reid 2005; Roux 1995; Siris 1996; Walsh 2004) or intensive versus symptomatic treatment (Langston 2010) are provided in Table 8, Table 9 and Table 10 respectively.
6. Drug‐related adverse events reported in randomised placebo‐controlled trials.
| Study ID | Comparison | Side effect | Bisphosphonate | Placebo | RR (95% CI) |
| Altman 1973 | Etidronate (38) vs. placebo (9) | Diarrhoea | 5 (13%) | 1 (11%) | 1.18 (0.16 to 8.93) |
| Buckler 1999 | Zoledronate (141) vs. placebo (35) | Fatigue Fever Arthralgia Pain, back Pain, skeletal Hypocalcaemia |
12 (9%) 7 (5%) 15 (11%) 14 (10%) 11 (8%) 3 (2%) |
0 (0%) 0 (0%) 3 (9%) 1 (3%) 2 (6%) 0 (0%) |
6.34 (0.38 to 104.5) 3.80 (0.50 to 28.88) 1.24 (0.89 to 1.77) 3.47 (0.53 to 23.02) 1.37 (0.85 to 2.19) 1.78 (0.74 to 4.24) |
| Fraser 1997 | Tiludronate (86) vs. placebo (26) | Nausea Vomiting Dyspepsia Diarrhoea Arthralgia Skeletal pain Raised liver enzymes Eosinophilia |
15 (17%) 7 (7%) 9 (10%) 14 (16%) 8 (9%) 5 (6%) 1 (1%) 0 (0%) |
2 (8%) 0 (0%) 0 (0%) 0 (0%) 2 (8%) 3 (12%) 0 (0%) 1 (4%) |
2.27 (0.65 to 7.86) 4.66 (0.45 to 48.06) 5.90 (0.40 to 87.16) 9.00 (0.32 to 252.8) 1.21 (0.91 to 1.61) 0.50 (0.13 to 1.97) 0.93 (0.04 to 22.20) 0.10 (0.01 to 2.47) |
| McClung 1995 | Tiludronate (91) vs. placebo (48) | Gastrointestinal | 31 (34%) | 15 (31%) | 1.09 (0.66 to 1.81) |
| Reginster 1992 | Tiludronate (117) vs. placebo (32) | Gastralgia Nausea |
20 (17.1%) 11 (9.4%) |
5 (16.1%) 3 (9.6%) |
1.09 (0.45 to 2.69) 1.00 (0.30 to 3.38) |
| Reid 1996 | Alendronate (27) vs. placebo (28) | Gastrointestinal Gastritis Duodenal ulcer Oesophagitis |
2 (7%) 0 (0%) 0 (0%) 1 (4%) |
5 (18%) 1 (4%) 1 (4%) 0 (0%) |
0.42 (0.09 to 2.00) 0.35 (0.02 to 8.13) 0.35 (0.02 to 8.13) 3.10 (0.13 to 73.10) |
7. Drug‐related adverse effects reported in randomised versus non‐randomised bisphosphonates trials.
| Study ID | Comparison | Adverse effect | Bisphosphonate 1 | Bisphosphonate 2 | RR (95% CI) |
| Roux 1995 | Tiludronate (155) vs. etidronate (79) | Gastrointestinal | 32 (20.8%) | 10 (12.7%) | 1.63 (0.85 to 3.14) |
| Abdominal pain | 10 (6.5%) | 2 (2.5%) | 2.55 (0.57 to 11.35) | ||
| Nausea, vomiting | 8 (5.2%) | 2 (2.5%) | 2.04 (0.44 to 9.37) | ||
| Fracture | 1 (1%) | 2 (3%) | 0.25 (0.02 to 2.77) | ||
| Siris 1996 | Alendronate (42) vs. etidronate (47) |
Gastrointestinal | 11 (26%) | 10 (21%) | 1.23 (0.58 to 2.60) |
| Abdominal distention | 0 (%) | 1 (2%) | 0.37 (0.02 to 8.90) | ||
| Abdominal pain | 3 (7%) | 4 (9%) | 0.84 (0.2 to 3.54) | ||
| Acid regurgitation | 1 (2%) | 1 (2%) | 1.12 (0.07 to 17.34) | ||
| Dyspepsia | 0 (0%) | 1 (2%) | 0.37 (0.02 to 8.90) | ||
| Melena | 1 (2%) | 0 (0%) | 3.35 (0.14 to 80.05) | ||
| Nausea | 2 (5%) | 3 (6%) | 0.75 (0.13 to 4.25) | ||
| Leg pain | 1 (2%) | 9 (19%) | 0.12 (0.02 to 0.94) | ||
| Laboratory adverse experiences | 9 (21%) | 6 (13%) | 1.68 (0.65 to 4.32) | ||
| Miller 1999 | Risedronate (62) vs. etidronate (61) |
Upper gastrointestinal | 12 (19%) | 12 (20%) | 0.98 (0.48 to 2.02) |
| Barreira 2009 | Olpadronate (14) vs. pamidronate (7) |
Digestive | 9 (64%) | 7 (100%) | 0.68 (0.44 to 1.03) |
| Merlotti 2007* | Zoledronate (47)* vs. pamidronate (60) |
Influenza‐like illness | 4 (9%) | 5 (8%) | 1.02 (0.29 to 3.59) |
| Myalgia | 3 (6%) | 4 (7%) | 0.96 (0.23 to 4.07) | ||
| Pyrexia | 3 (6%) | 4 (7%) | 0.96 (0.23 to 4.07) | ||
| Fatigue | 3 (6%) | 8 (13%) | 0.48 (0.13 to 1.71) | ||
| Headache | 4 (9%) | 5 (8%) | 1.02 (0.29 to 3.59) | ||
| Diarrhoea | 1 (2%) | 2 (3%) | 0.64 (0.06 to 6.83) | ||
| Bone pain | 3 (6%) | 6 (10%) | 0.64 (0.17 to 2.42) | ||
| Pain in arm or leg | 3 (6%) | 4 (7%) | 0.96 (0.23 to 4.07) | ||
| Hypocalcaemia | 3 (6%) | 1 (2%) | 3.83 (0.41 to 35.64) | ||
| Dermatitis | 1 (2%) | 0 (0%) | 0.42 (0.02 to 10.17) | ||
| Reid 2005 | Zoledronate (177) vs. risedronate (172) |
Study days 1 to 3 | |||
| Influenza‐like illness | 17 (9.6%) | 7 (4.1%) | 2.36 (1 to 5.55) | ||
| Myalgia | 13 (7.3%) | 6 (3.5%) | 2.11 (0.82 to 5.41) | ||
| Pyrexia | 13 (7.3%) | 1 (0.6%) | 12.63 (1.67 to 95.53) | ||
| Fatigue | 12 (6.8%) | 4 (2.3%) | 2.92 (0.96 to 8.86) | ||
| Headache | 12 (6.8%) | 7 (4.1%) | 1.67 (0.67 to 4.13) | ||
| Rigor | 12 (6.8%) | 1 (0.6%) | 11.66 (1.53 to 88.72) | ||
| Nausea | 11 (6.2%) | 3 (1.7%) | 3.56 (1.01 to 12.55) | ||
| Bone pain | 9 (5.1%) | 2 (1.2%) | 4.37 (0.96 to 19.95) | ||
| After study day 3 | |||||
| Pain in an arm or leg | 13 (7.3%) | 12 (7%) | 1.05 (0.49 to 2.24) | ||
| Arthralgia | 9 (5.1%) | 19 (11%) | 0.46 (0.21 to 0.99) | ||
| Dizziness | 9 (5.1%) | 5 (2.9%) | 1.75 (0.6 to 5.11) | ||
| Nasopharyngitis | 9 (5.1%) | 14 (8.1%) | 0.62 (0.28 to 1.41) | ||
| Diarrhoea | 8 (4.5%) | 9 (5.2%) | 0.86 (0.34 to 2.19) | ||
| Headache | 7 (4%) | 10 (5.8%) | 0.68 (0.26 to 1.75) | ||
| Back pain | 4 (2.3%) | 12 (7.0%) | 0.32 (0.11 to 0.98) | ||
| Symptomatic hypocalcaemia | 2 (1.1%) | 1 (0.6%) | 1.94 (0.18 to 21.24) | ||
| Reid 2011 (extension) | Zoledronate (152) vs. risedronate (115) |
Atrial fibrillation | 1 (0.7%) | 1 (0.9%) | 0.76 (0.05 to 12.20) |
| Atrial flutter | 0 (0%) | 2 (1.7%) | 0.15 (0.01 to 3.13) | ||
| Osteonecrosis jaw | 0 (0%) | 0 (0%) | ‐ | ||
| Walsh 2004 | Alendronate (36) vs. pamidronate (36) |
Gastrointestinal | 16 (44%) | 4 (11%) | 4 (1.48 to 10.80) |
| Fatigue | 0 (0%) | 23 (64%) | 0.02 (0.00 to 0.34) | ||
| General aches/pain | 4 (11%) | 16 (44%) | 0.25 (0.09 to 0.68) | ||
| Deteriorating kidney failure | 1 (3%) | 0 (0%) | 3.00 (0.12 to 71.28) | ||
*Zoledronate group data were extracted from Table 3 in Merlotti 2007. In this table, the authors presented data from 30 participants who took part in the first part of the study (which was included in our systematic review) plus 17 participants from the second part of the study (which was not included in our systematic review).
8. Drug‐related adverse events reported in randomised trials comparing regimens aimed to normalise elevated bone turnover (intensive) versus regimens aimed to control bone pain referable to Paget's disease of bone (symptomatic).
| Study ID | Comparison | Side effect | Intensive | Symptomatic | RR (95% CI) |
| Langston 2010* | Intensive (661) vs. symptomatic (663) |
All adverse events Serious adverse events Musculoskeletal Sensory Gastrointestinal Cardiovascular Arrythmia Cancer Renal Other |
3429 345 691 203 172 360 13 (1.9%) 55 98 1850 |
3471 359 734 196 157 327 7 (1%) 47 78 1932 |
‐ ‐ ‐ ‐ ‐ ‐ 1.86 (0.75 to 4.64) ‐ ‐ ‐ |
| Tan 2017 | Intensive (270) vs. symptomatic (232) |
All adverse events Serious adverse events Musculoskeletal Osteonecrosis of the jaw Delayed union of fracture Ophthalmic Uveitis Gastrointestinal Cardiovascular Arrythmia Cerebrovascular Central nervous system Endocrine Ear, nose or throat Genitourinary Haematological Respiratory Skin Miscellaneous |
226 87 123 1 2 34 1 54 67 14 4 28 28 28 41 10 48 41 33 |
196 66 104 0 1 41 0 46 49 8 3 28 21 26 39 9 43 33 32 |
0.99 (0.91 to 1.07) 1.13 (0.87 to 1.48) 1.02 (0.84 to 1.23) 2.58 (0.11 to 63.01) 1.72 (0.16 to 18.83) 0.71 (0.47 to 1.08) 2.58 (0.11 to 63.01) 1.01 (0.71 to 1.43) 1.18 (0.85 to 1.62) 1.50 (0.64 to 3.52) 1.14 (0.26 to 5.07) 0.86 (0.52 to 1.41) 1.15 (0.68 to 1.96) 0.93 (0.56 to 1.53) 0.90 (0.61 to 1.35) 0.96 (0.40 to 2.31) 0.95 (0.66 to 1.39) 1.07 (0.70 to 1.63) 0.89 (0.56 to 1.40) |
* Data represent total numbers of reported side effects regardless of numbers of participants who experienced them. The authors reported numbers of participants only for arrhythmia.
Bisphosphonates versus placebo
When we considered studies of all bisphosphates versus placebo we did not detect a difference in adverse events although the RR was not equal to one (RR 1.32, 95% CI 0.91 to 1.92; Analysis 1.4; Figure 6). When we considered studies of zoledronate we found that this bisphosphonate had a significantly increased risk of adverse events when compared with placebo (RR 2.57, 95% CI 1.21 to 5.44). Gastrointestinal side effects were common for oral bisphosphonates and fatigue or fever for intravenous bisphosphonates (Table 8).
1.4. Analysis.
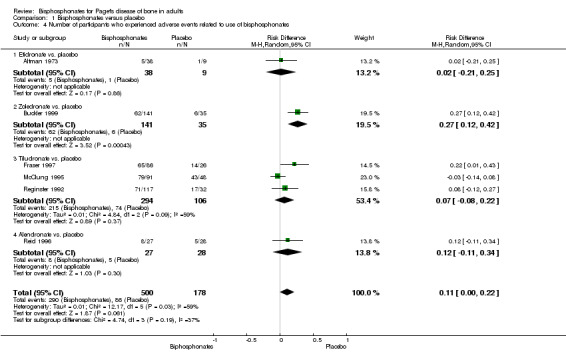
Comparison 1 Bisphosphonates versus placebo, Outcome 4 Number of participants who experienced adverse events related to use of bisphosphonates.
6.

Forest plot of comparison: 1 Bisphosphonates versus placebo, outcome: 1.4 Number of participants who experienced adverse events related to use of bisphosphonates
Bisphosphonates versus active comparator
We did not detect a difference in occurrence of adverse events among bisphosphonates, nor when compared risedronate or alendronate was compared to etidronate (RR 0.98, 95% CI 0.72 to 1.35; Analysis 2.2). There was no evidence of a difference when newer aminobisphosphonates (zoledronate, olpadronate) were compared to older aminobisphosphonates (pamidronate, risedronate) (RR 0.93, 95% CI 0.73 to 1.19; Analysis 3.2). Influenza‐like illnesses were common among participants who received intravenous bisphosphonates. Symptomatic hypocalcaemia was uncommon (< 1%).
2.2. Analysis.

Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 2 Number of participants who experienced adverse events related to use of bisphosphonates.
3.2. Analysis.
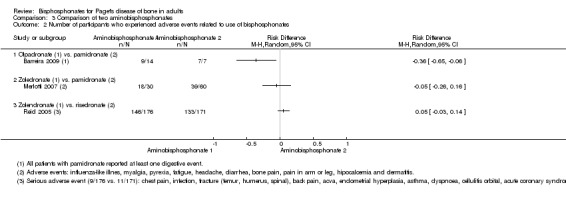
Comparison 3 Comparison of two aminobisphosphonates, Outcome 2 Number of participants who experienced adverse events related to use of bisphosphonates.
Intensive versus symptomatic treatment
No difference was detected for total number of adverse effects when intensive treatment was compared with symptomatic treatment (RR 1.05, 95% CI 0.79 to 1.41; Table 6). Serious adverse effects were more frequent with intensive treatment; 32.2% versus 28.4%, (RR 1.28, 95% CI 0.96 to 1.72). One case each of osteonecrosis of the jaw and uveitis (both in intensive group participants) and three delayed unions of fracture (2 in the intensive group, 1 in the symptomatic group) were recorded in the PRISM‐EZ trial (Tan 2017).
Rare adverse events
In addition to data from the included trials, we reviewed data on six specific rare events obtained from the websites of three regulatory agencies (FDA, EMA, and MHRA) and the AADRB / Australian TGA.
Osteonecrosis of the jaw
The risk of developing osteonecrosis of the jaw is substantially greater for people receiving intravenous bisphosphonates for cancer indications (metastatic cancer of bone, tumour‐associated hypercalcaemia and multiple myeloma) than for those with Paget’s disease of bone or osteoporosis. According to the EMA 2009 Committee for Medicinal Products for Human Use (CHMP) assessment report on bisphosphonates and osteonecrosis of the jaw, people receiving oral bisphosphonates for Paget’s disease of bone are considered to be at much lower risk for developing osteonecrosis of the jaw. Reported incidence ranged from 0.0004% to 0.06%. There is no clear evidence regarding the risk of osteonecrosis of the jaw following use of intravenous bisphosphonates for Paget's disease of bone. The evidence further suggests that the cumulative dose is a risk factor for osteonecrosis of the jaw, so the very low number of reports of osteonecrosis of the jaw in people with Paget’s disease could be explained by the short courses of bisphosphonates used (EMA, FDA, MHRA, TGA).
Osteonecrosis of the external auditory canal
Oseonecrosis of the external auditory canal has been reported with use of bisphosphonates both for cancer‐related bone disease and osteoporosis, but not for Paget’s disease of bone (MHRA).
Ocular inflammation
A small number of reports describe inflammatory eye disorders such as uveitis, iritis, scleritis/episcleritis, haemorrhage and optic neuritis. Inflammatory ocular disorders appear to be a rare effect of all bisphosphonates. There is no specific evidence about the risk of ocular inflammation in people with Paget’s disease of bone after bisphosphonate use (AADRB).
Atrial fibrillation
Evidence regarding atrial fibrillation suggests an increased risk of atrial fibrillation for zoledronate, pamidronate and possibly for alendronate in people treated for osteoporosis and cancer‐related bone disease. There is no specific evidence about the risk of atrial fibrillation in Paget’s disease of bone following bisphosphonate treatment (EMA, MHRA).
Atypical femoral fracture
Atypical femoral fracture is considered to be a class effect of bisphosphonates. These fractures have been seldom reported and mainly among those receiving long‐term treatment for osteoporosis. Only one post‐marketing report of possible atypical femoral fracture with zoledronate in a patient with Paget’s disease has been received according EMA (Assessment report for bisphosphonates containing medicinal products 2011). The very low number of reports of atypical fractures in Paget’s disease of bone patients could be explained by the short bisphosphonates regimens used for this disease compared osteoporosis (EMA, MHRA).
Oesophageal cancer risk
There was insufficient evidence of a link between the risk of oesophageal cancer and the use of oral bisphosphonates. This suggested association has been proposed for people taking oral bisphosphonate for more than five years, a therapeutic schedule that is not used for people with Paget’s disease of bone (EMA, MHRA).
Withdrawals due to adverse events
Discontinuations due to adverse events were reported and analysed for 13 trials.
Bisphosphonates versus placebo
The pooled estimate for the comparison between bisphosphonates and placebo demonstrated no evidence of effect in the risk of discontinuing medication due to adverse events (RR 1.01, 95% CI 0.41 to 2.52; 6 studies, 517 participants; Analysis 1.5). The results were consistent among trials, with low rates of withdrawals in both treatment groups.
1.5. Analysis.
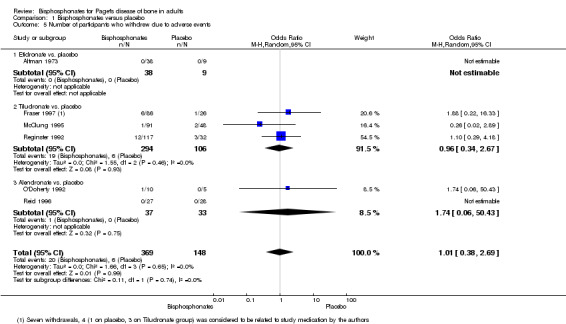
Comparison 1 Bisphosphonates versus placebo, Outcome 5 Number of participants who withdrew due to adverse events.
Bisphosphonates versus active comparator
Results were inconclusive for the comparison between two non‐aminobisphosphonates (Roux 1995) (RR 2.55, 95% CI 0.57 to 11.35; Table 7). There was no evidence of a difference between aminobisphosphonates and non‐aminobisphosphonates (RR 0.69, 95% CI 0.25 to 1.89; 2 studies, 212 participants; Analysis 2.3) or between aminobisphosphonates (RR 0.93, 95% CI 0.37 to 2.35; 3 studies; Analysis 3.3).
2.3. Analysis.
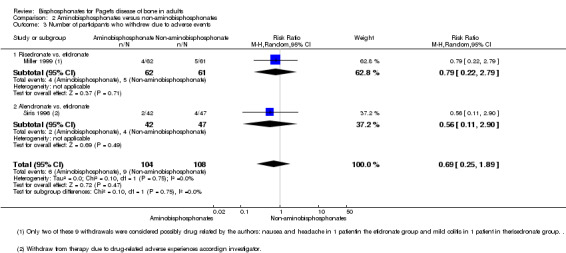
Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 3 Number of participants who withdrew due to adverse events.
3.3. Analysis.
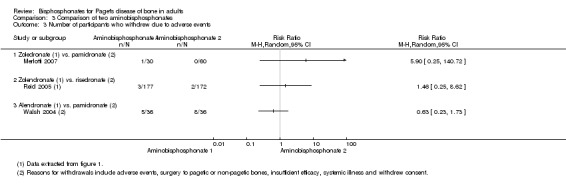
Comparison 3 Comparison of two aminobisphosphonates, Outcome 3 Number of participants who withdrew due to adverse events.
Intensive versus symptomatic treatment
No difference was detected in the risk of discontinuing medication due to adverse events between intensive and symptomatic treatment (Langston 2010) (RR 1.05, 95% CI 0.79 to 1.41; Table 6).
Minor outcomes
Relapses due to recurrence of bone pain
Relapses due to recurrence of bone pain were analysed only by Fraser 1997. When compared to placebo, tiludronate achieved a significant reduction in the number of participants who relapsed due to bone pain (RR 0.51, 95% CI 0.32 to 0.80; Table 11).
9. Bisphosphonates versus placebo (Fraser 1997).
| Study ID | Outcome | Tiludronate | Placebo | RR (95% IC) | ||
| Events | N | Events | N | |||
| Fraser 1997 | Number of participants who relapsed due to bone pain recurrence | 23 | 66 | 13 | 19 | 0.51 (0.32 to 0.80) |
Effect on serum total alkaline phosphatase activity
Changes in serum bone markers, especially changes in serum total alkaline phosphatase activity, was considered as the primary outcome in all but two included trials (Langston 2010; Tan 2017).
Bisphosphonates versus placebo
Bisphosphonates achieved a significantly greater reduction in serum total alkaline phosphatase activity when compared to placebo (MD 50.1%, 95% CI 32.5 to 67.7; Analysis 1.6). In line with the greater reduction in serum alkaline phosphatase activity, a high proportion of participants normalised their alkaline phosphatase levels (RR 9.96, 95% CI 3.74 to 26.58; Analysis 1.7).
1.6. Analysis.
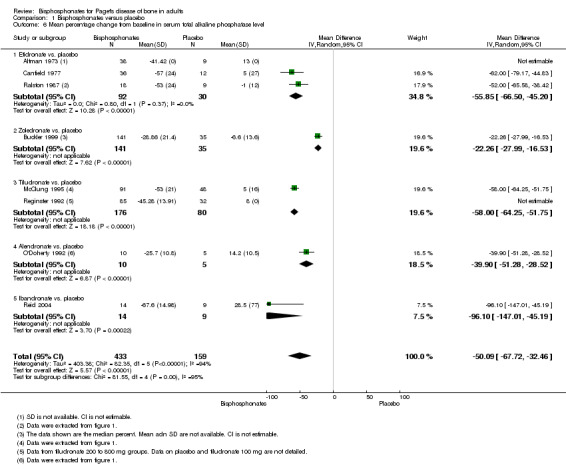
Comparison 1 Bisphosphonates versus placebo, Outcome 6 Mean percentage change from baseline in serum total alkaline phosphatase level.
1.7. Analysis.
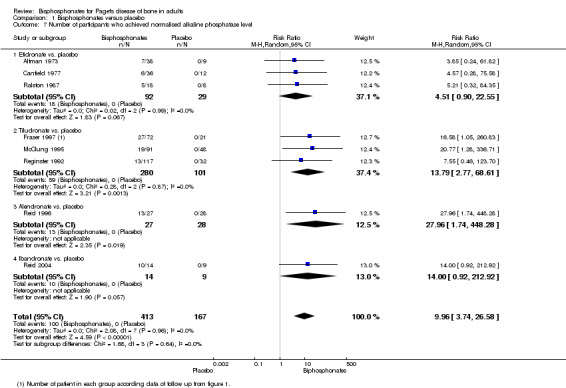
Comparison 1 Bisphosphonates versus placebo, Outcome 7 Number of participants who achieved normalised alkaline phosphatase level.
Bisphosphonates versus active comparator
Among non‐aminobisphosphonates, tiludronate was more effective than etidronate (Roux 1995) (RR 2.27, 95% CI 1.16 to 4.43; Table 7) for normalisation of alkaline phosphatase levels.
Aminobisphosphonates had greater effect on serum biochemical markers of bone turnover than non‐aminobisphosphonates. This was shown in the comparisons between risedronate and etidronate (Miller 1999) or alendronate and etidronate (Siris 1996). The pooled mean difference in the reduction in alkaline phosphatase activity was 41.0% (95% CI 32.8 to 49.1; Analysis 2.4). The pooled RR for normalisation of alkaline phosphatase levels was 4.30 (95% CI 2.72 to 6.79; NNTB 2, 1 to 4; Analysis 2.5).
2.4. Analysis.
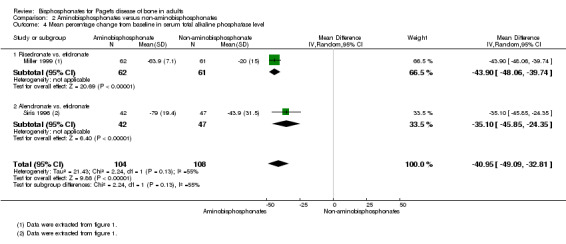
Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 4 Mean percentage change from baseline in serum total alkaline phosphatase level.
2.5. Analysis.
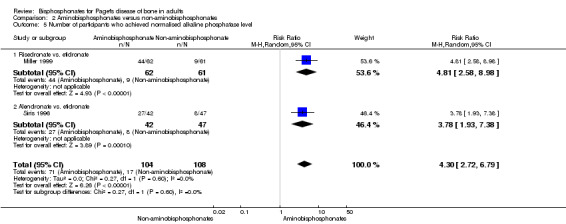
Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 5 Number of participants who achieved normalised alkaline phosphatase level.
Zoledronate was the most effective of the aminobisphosphonates. The mean difference in reduction of alkaline phosphatase activity for zoledronate compared to risedronate was 22.7% (95% CI 19.3 to 26.1; Analysis 3.4); RR for normalisation of alkaline phosphatase levels was 1.53 (95% CI 1.33 to 1.76; NNTB 3, 3 to 5). Alendronate was more effective than pamidronate (Walsh 2004) in normalising alkaline phosphatase levels (RR 1.48, 95% CI 1.09 to 2.00; Analysis 3.5).
3.4. Analysis.
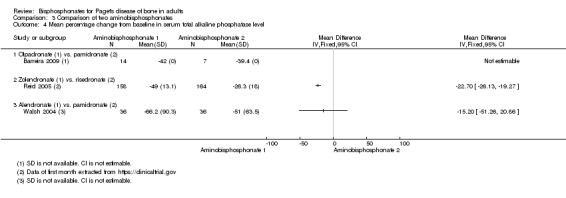
Comparison 3 Comparison of two aminobisphosphonates, Outcome 4 Mean percentage change from baseline in serum total alkaline phosphatase level.
3.5. Analysis.
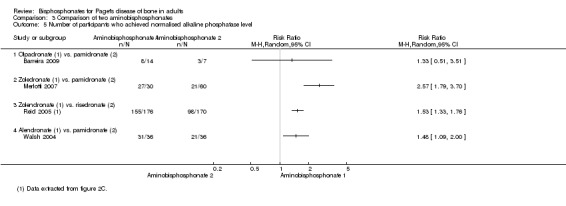
Comparison 3 Comparison of two aminobisphosphonates, Outcome 5 Number of participants who achieved normalised alkaline phosphatase level.
When etidronate was compared with etidronate plus calcitonin, the combination showed a greater reduction in alkaline phosphatase than etidronate alone: 71% at 6 months versus 56% (CI not reported) (O'Donoghue 1987).
Intensive versus symptomatic treatment
Intensive treatment had a greater effect than symptomatic treatment (Langston 2010) in reducing mean alkaline phosphatase level (22.5%, 95% CI 15.4 to 29.6) and in the percentage of participants whose alkaline phosphatase level was normalised (RR 1.26, 95% CI 1.18 to 1.36; Table 6).
Relapse due to recurrence of increased serum alkaline phosphatase level
Bisphosphonates versus placebo
No included studies evaluated the recurrence of increased serum alkaline phosphatase level following treatment for bisphosphonates versus placebo.
Bisphosphonates versus active comparator
Three trials evaluated relapse due to recurrence of increased serum alkaline phosphatase level following treatment. Miller 1999 compared aminobisphosphonates and non‐aminobisphosphonates and reported that risedronate was more effective than etidronate (RR 0.25, 95% CI 0.05 to 1.11; Table 12). Among aminobisphosphonates, zoledronate was more effective than pamidronate (Merlotti 2007) or risedronate (Reid 2005) (RR 0.06, 95% CI 0.01 to 0.42; NNTB 3, 95% CI 3 to 5; Analysis 3.6).
10. Aminobisphosphonates versus non‐aminobisphosphonates (Miller 1999).
| Study ID | Outcome | Risedronate | Tiludronate | RR (95% IC) | ||
| Events | N | Events | N | |||
| Miller 1999 | Number of participants who relapsed due to recurrence of increased serum alkaline phosphatase level |
2 | 62 | 8 | 61 | 0.25 (0.05 to 1.11) |
3.6. Analysis.

Comparison 3 Comparison of two aminobisphosphonates, Outcome 6 Number of participants who experienced biochemical relapse with increased alkaline phosphatase level.
Discussion
Summary of main results
Evidence from two studies (205 participants) indicated that compared with placebo bisphosphonates helped to relieve bone pain (disappearance of pain) (RR 3.42 95% CI 1.31 to 8.90, NNTB 5 95% CI 1 to 35%; moderate‐quality evidence). Data on any pain reduction were in accordance with complete disappearance of pain (RR 1.97, 95% CI 1.29 to 3.01; 7 studies, 481 participants; NNTB 5 95% CI 2 to 15; moderate‐quality evidence). Zoledronate provided better pain relief than pamidronate (RR 1.30, 95% CI 1.10 to 1.53) or risedronate (RR 1.36, 95% CI 1.06 to 1.74; very low‐quality evidence).
There was insufficient evidence to evaluate whether bisphosphonate therapy prevented fractures compared with placebo; few studies were designed to study the effects of treatment on the occurrence of fractures (bisphosphonates versus placebo: RR 0.89, 95% CI 0.18 to 4.3; very low‐quality evidence).
There was limited evidence to make firm conclusions about the impact of bisphosphonate on hearing thresholds, deafness, bone deformity, fractures or need for orthopaedic surgery. When zoledronate was compared with risedronate, a marginal improvement in some aspects of quality of life were reported. Differences were also observed in quality of life with long‐term follow‐up but this was restricted to the subgroup of participants who had normal levels of alkaline phosphatase after the original study comparing zoledronate with risedronate.
Bisphosphonates were highly effective in reducing biochemical markers of bone turnover. Bisphosphonates achieved a greater reduction in alkaline phosphatase values compared with placebo (50.1%, 95% CI 32.5 to 67.7; moderate‐quality evidence). Aminobisphosphonates were more effective than non‐aminobisphosphonates in reducing alkaline phosphatase (RR 4.3, 95% CI 2.72 to 6.79, moderate‐quality evidence). Among aminobisphosphonates, zoledronate was more effective than pamidronate (RR 2.57, 95% CI 1.79 to 3.70; moderate‐quality evidence) and risedronate (RR 1.53, 95% CI 1.33 to 1.76; moderate‐quality evidence) in normalising alkaline phosphatase levels.
Adverse events were numerically more common with bisphosphonate treatment than placebo, but differences were not significant, yielding results that were inconclusive (RR 1.32, 95% CI 0.91 to 1.92; risk of event 64% versus 48%; low‐quality evidence). Zoledronate had a significantly increased risk of adverse events than placebo (RR 2.57, 95% CI 1.21 to 5.44). Results were inconclusive for other bisphosphonates. Gastrointestinal side effects were common among participants in oral bisphosphonates trials, whereas transitory influenza‐like illness or pyrexia (3 days or fewer) were common with intravenous bisphosphonates. Serious adverse events attributable to bisphosphonates were rare. The rate of withdrawals due to serious adverse events was low. There were no clinically significant differences in adverse events when different bisphosphonates were compared.
The PRISM trial, which involved 1331 participants, addressed the issue of a 'treat to target' design using bisphosphonates in participants with Paget's disease of bone with fractures as the primary endpoint (Langston 2010). Langston 2010 compared two strategies; in the symptomatic group, bisphosphonates and other treatments were administered with the aim of treating bone pain. In the intensive group, bisphosphonates were administered with the aim of normalising alkaline phosphatase levels. Langston 2010 reported no differences in fractures between treatment groups, or for secondary outcomes, including pain relief, hearing thresholds, need for orthopaedic surgery or quality of life. An extension study by Tan 2017, which involved 502 participants, reported no benefits from intensive treatment on quality of life. There was a non‐significant trend for increased risk of fractures and need for orthopaedic procedures in the intensive treatment group (low‐quality evidence).
Overall completeness and applicability of evidence
The review process included an extensive and systematic literature search that included articles published in all languages. We also searched trials registers to identify potentially relevant, but not yet published, studies.
The included trials covered the usual spectrum of older people diagnosed with Paget's disease of bone. Many studies focused on participants with raised serum biochemical markers of bone turnover, but the PRISM (Langston 2010) and PRISM‐EZ (Tan 2017) studies also included participants with normal serum biomarkers, some with no bone pain. Therefore, the included trials included covered a wide range of participants with differing degrees of metabolic activity and symptoms.
We found no evidence that bisphosphonates could prevent bone deformity or long‐term complications such as bone fractures, need for orthopaedic surgery or progression of deafness in participants with skull involvement. However, few studies were conducted that evaluated effects on these outcomes.
Most studies planned to analyse change in serum bone turnover markers as the primary outcome. While the treatment group always achieved a reduction on serum bone turnover markers compared to control groups, it is noteworthy that the biochemical improvements were not necessarily associated with similar improvements in patient‐oriented outcomes. Only pain relief, as a short‐term patient‐oriented outcome, was addressed in most trials.
Based on available data, there was no clinical benefit from adopting a strategy of administering bisphosphonates with the aim of normalising alkaline phosphatase in people with Paget's disease of bone. A long‐term study of intensive bisphosphonate treatment reported no benefit in quality of life compared with symptomatic treatment and a trend toward increased risk of fractures with intensive treatment, although the difference between groups was not significant.
Quality of the evidence
The review includes 20 trials (3168 participants). Of these, ten trials (801 participants) compared bisphosphonates versus placebo. Seven trials compared two bisphosphonates (992 participants). One trial compared a bisphosphonates with a bisphosphonate plus calcitonin (44 participants). One trial (1331 participants) and its interventional extension study (502 participants) compared symptomatic treatment and intensive treatment where the goal was to normalise alkaline phosphatase.
'Summary of findings' tables are presented that compare bisphosphonates and placebo (Table 1) and zoledronate and risedronate or pamidronate (Table 2). Evidence quality for effects on seven patient‐relevant outcomes ranged from very low to moderate (4 outcomes); several outcomes could not be assessed because of lack of reporting in the included studies.
Evidence quality was assessed as moderate for bone pain relief (7 studies, 481 participants). Evidence was downgraded by one level due to high risk for attrition bias in three studies and a high risk for reporting bias in another three studies. None of the included studies were assessed at high risk for performance bias for bisphosphonates compared with placebo. The heterogeneity of methods for assessing and defining Pagetic bone pain and lack of a standard way to assess changes in bone pain made comparisons between studies difficult. Differentiation of the cause of pain was also difficult because pain could be related to increased metabolic activity in Pagetic bone, co‐existing arthritis, nerve compression syndromes from misshapen bones or other causes.
Despite the heterogeneity of definitions, the conclusion regarding the role of bisphosphonates to improve pain in people with Paget's disease of bone is robust. Results were consistent among trials, sensitivity analyses excluding studies at high risk of bias or including only studies that used total disappearance of pain as response definition did not alter findings.
We found low‐quality evidence for effects of bisphosphonates on fractures. Evidence was downgraded by three levels because most data were from studies at risk of bias (2 studies with high risk for attrition bias and 1 study with high risk for reporting bias). The design of most studies was not suitable to study the long‐term impact on fractures because the mean follow‐up period was six months, and there were few events, resulting in wide confidence intervals.
We found low‐quality evidence for adverse events. Evidence was downgraded by one level because two studies were assessed at high risk of attrition bias and downgraded another level because of inconsistency. A potential source of heterogeneity was lack of uniform definitions of adverse events reported in included studies. Adverse events were not assessed as a primary outcome in any included study, and in many studies, was not assessed at all. Furthermore, reporting of adverse events was frequently incomplete and studies were not sufficiently powered to address rare adverse effects. We tried to avoid outcome reporting bias by contacting study authors for additional data on adverse events. However, since the included studies were carried out many years ago, requests for further information frequently remained unanswered, or study authors confirmed that original study data were no longer available. Another limitation of evaluating data on adverse events from summary meta‐analyses is that study participants tend to be healthier, with less comorbid disease. Therefore, the results may not be generalisable to routine clinical practice. Furthermore, five included studies (Fraser 1997; Reid 1996; Reid 2005; Siris 1996; Walsh 2004) excluded participants with pre‐existing gastrointestinal disease. Considerable heterogeneity was found in the meta‐analysis. However, only one study reported more adverse events in placebo than bisphosphonates group participants. A sensitivity analysis excluding this study had no heterogeneity.
The quality of the evidence for withdrawals due to adverse events was assessed as moderate. Evidence was downgraded by one level because four of the studies were assessed at high risk of bias: three for attrition bias (Altman 1973; O'Doherty 1992; Reginster 1992); and one for reporting bias in (O'Doherty 1992). Although there was a wide confidence interval due to few events, this outcome was not downgraded because the relative risk was close to one.
Another methodological limitation concerned concealment of treatment allocation methods which were unclear in 15 of 20 included studies.
Another limitation was the length of follow‐up. Most studies were short‐term with duration of six months or fewer. It was difficult to extrapolate beyond the duration of follow‐up studies in this review with respect to long‐term impact on fractures and other complications of Paget's disease of bone.
Only a few included studies provided data for comparisons between bisphosphonates. The quality of evidence for comparisons between two non‐aminobisphosphonates was low; only one trial compared tiludronate versus etidronate. The quality of evidence for aminobisphosphonates versus non‐aminobisphosphonates was low; only two small trials provided data. Evidence for comparison among aminobisphosphonates was also low because only four studies provided data; of these two were open label, and one was published only in abstract form. The quality of evidence for the comparison between zoledronate and risedronate was drawn from a trial that compared these bisphosphonates (357 participants), with a low risk of bias for most of the endpoints assessed.
Evidence from PRISM trial (Langston 2010), and its extension study which compared two treatment strategies (Tan 2017), was limited by assessment of high risk of bias for patient‐reported outcomes due to its open‐label design. However the PRISM and PRISM‐EZ studies were the only trials that collected data on fractures and need for orthopaedic procedures.
Potential biases in the review process
The principal limitation of this review was the limited evidence provided by the included studies and low‐quality evidence for patient‐related outcomes that were the focus of the review, such as fractures, need for orthopaedic procedures, bone deformity and progression of deafness. Many included studies were assessed at high risk of bias or were relatively small and underpowered. Reporting of study design and methods was generally poor, especially among the older trials. We found protocols or trial registration records for four included studies (Langston 2010; Reid 2004; Reid 2005; Tan 2017).
Reporting bias was difficult to evaluate since the planned funnel plots for all analysed outcomes could not be constructed due to there being few included studies for each outcome. We made attempts to reduce reporting bias and to find unpublished studies by gathering information in clinical trial registers, contacting experts in the field and checking for abstracts presented at scientific meetings. It is therefore unlikely that reporting was high for aminobisphosphonates since these drugs' trials were performed from the 1990s to the first decade of the 21st century when nearly all studies should be registered. Nonetheless, it is possible that we have missed some old studies from the 1970s to the 1990s when there were no systematic registers of clinical trials.
We conducted an extensive search of the literature in all relevant databases and identified two ongoing trials. For rare adverse events we searched in the websites of four regulatory agencies (FDA, EMA, AADRB and MHRA). We attempted to obtain missing data by contacting study authors or to extract data from figures in reports. However, few data were obtained from attempts to contact study authors.
Agreements and disagreements with other studies or reviews
We summarised the evidence from randomised controlled trials designed to test bisphosphonates as a treatment for people with Paget's disease of bone. We did not find any other systematic reviews that analysed bisphosphonates in Paget's disease of bone. We found four relevant clinical guidelines: the Bone and Tooth Society of Great Britain and the National Association for the Relief of Paget’s Disease guideline (Selby 2002), the Belgian guideline (Devogelaer 2008); the Japanese guideline (Takata 2006) and the Endocrine Society Clinical Practice Guideline (Singer 2015).
Findings from our review agree with the British guideline (Selby 2002) which stated that "...bone pain is the only complication of Paget's disease of bone for which there is firm evidence that specific anti pagetic therapy is associated with clinical benefit". Four of the five studies cited in support of this statement were included in our review (Altman 1973; Ralston 1987; Reginster 1992; Reid 1996); one was excluded (Khairi 1974; Characteristics of excluded studies). Selby 2002 also stated there was no evidence to support the use of specific treatment to prevent deafness, fractures, osteoarthritis, progression of bone deformity or spinal cord compression.
Other guidelines support pain as the only evidence‐based indication for the treatment of Paget's disease of bone.
Ralston 2013 highlighted inclusion of zoledronate versus risedronate trials (Reid 2005) which reported a better profile for zoledronate for normalising alkaline phosphatase and evidence for benefit in some aspects of quality of life.
Our review findings disagreed with certain aspects of the Endocrine Society guideline which suggested that bisphosphonate therapy should be given to normalise biochemical markers of bone turnover with the aim of preventing complications (Singer 2015). We found no evidence of benefit for this strategy in participants with established Paget's disease of bone and there was some evidence that it may even be harmful (Tan 2017).
Authors' conclusions
Implications for practice.
Based on the included studies, there is moderate‐quality evidence that bisphosphonates improve pain in people with Paget's disease of bone. There was insufficient evidence to determine the effects of bisphosphonates for other complications or impact on quality of life. There was limited evidence on the effects of long‐term treatment on complications such fractures, deformity, progression of deafness or need for orthopaedic surgery. Among the bisphosphonates, there was low‐quality evidence that zoledronate may have a better balance between benefit and harm for the treatment of Paget's disease of bone compared with risedronate or pamidronate. However, there was evidence of increased risk of adverse events with zoledronate.
The body of evidence did not enable conclusions to be drawn about the risk of rare adverse events with bisphosphonates treatment. Lastly, based on our review which included one study of intensive versus symptomatic treatment, we found insufficient evidence to establish the benefits and harms of intensive versus symptomatic treatment strategies.
Implications for research.
The currently available data did not resolve the question of whether bisphosphonates could prevent long‐term outcomes such as fractures, deafness progression or need for orthopaedic surgery. Appropriately designed clinical trials including long‐term evaluations and larger sample sizes are needed. Additional research is needed to clarify the impact of bisphosphonates in quality of life of people with Paget's disease of bone; the current evidence indicates these drugs have limited impact on quality of life (evidence from one study comparing zoledronate versus risedronate). The role of bisphosphonates for people who are asymptomatic with biochemical markers of bone turnover within normal range also merits further investigation.
What's new
| Date | Event | Description |
|---|---|---|
| 11 December 2017 | Amended | The results for the side effects and pain have been edited to remove the absolute risk difference to avoid misinterpretation. |
History
Protocol first published: Issue 4, 2004 Review first published: Issue 11, 2017
| Date | Event | Description |
|---|---|---|
| 5 December 2017 | Amended | The plain language summary included a mistake in the number of people experiencing pain relief and the number of people experiencing side effects: the results for the placebo and treatment groups were reversed. The labels on Analysis 1.1 were also inverted. These errors have been corrected in this amendment. |
| 4 September 2008 | Amended | converted to new review format. CMSG ID: C106‐P |
Acknowledgements
We wish to acknowledge the hard work that went into the protocol by AL Langston, MK Campbell and C Roberston (Langston 2004).
Appendices
Appendix 1. MEDLINE search strategy
1. exp bone diseases/
2. 1 and paget$.mp.
3. (paget$ adj10 bone$).mp.
4. exp Osteitis Deformans/
5. osteitis deformans.mp.
6. ostitis deformans.mp.
7. or/2‐6
8. randomised controlled trial.pt.
9. controlled clinical trial.pt.
10. randomized.ab.
11. placebo.ab.
12. drug therapy.fs.
13. randomly.ab.
14. trial.ab.
15. groups.ab.
16. 8 or 9 or 10 or 11 or 12 or 13 or 14 or 15
17. exp animals/ not humans.sh.
18. 16 not 17
19. 7 and 18
Appendix 2. Embase search strategy
1. exp bone diseases/
2. 1 and paget$.mp.
3. (paget$ adj10 bone$).mp.
4. exp Paget Bone Disease/
5. osteitis deformans.mp.
6. ostitis deformans.mp.
7. or/2‐6
8. random$.ti,ab.
9. factorial$.ti,ab.
10. (crossover$ or cross over$ or cross‐over$).ti,ab.
11. placebo$.ti,ab.
12. (doubl$ adj blind$).ti,ab.
13. (singl$ adj blind$).ti,ab.
14. assign$.ti,ab.
15. allocat$.ti,ab.
16. volunteer$.ti,ab.
17. crossover procedure.sh.
18. double blind procedure.sh.
19. randomised controlled trial.sh.
20. single blind procedure.sh.
21. or/8‐20
22. exp animal/ or nonhuman/ or exp animal experiment/
23. exp human/
24. 22 and 23
25. 22 not 24
26. 21 not 25
27. 7 and 26
Appendix 3. CENTRAL search strategy
#1MeSH descriptor Osteitis Deformans explode all trees in MeSH products
#2 paget near/10 bone in All Fields in all products
#3 (#1 OR #2)
Appendix 4. Web of Knowlegde/Web of Science search strategy
#1 osteitis deformans
#2 paget bone
#3 #1 OR #2
#4 trial* or random* or placebo* or control* or double or treble or triple or blind* or mask* or allocat* or prospective* or volunteer*or comparative or evaluation or follow‐up or followup
#5 #3 AND #4
Appendix 5. Summary of approaches to study authors for further information on trials
| Study ID | Study author contacted | Study author replied | Additional data provided |
| Altman 1973 | No | ‐ | Not applicable |
| Barreira 2009 | 01/12/2014 | No | Not applicable |
| Buckler 1999 (+ Schaffer 1996) | 30/06/2016 | No | Not applicable |
| Canfield 1977 | No | ‐ | Not applicable |
| Fraser 1997 | 30/6/2014 | No | Not applicable |
| Langston 2010 (+ Ralston 1987) | 30/6/2014 | 29/12/2014 | Provided more information on co‐interventions and blinding (Langston 2010; Ralston 1987), data on pain, QoL, hearing and mean alkaline phosphatase and SD change (Langston 2010) |
| McClung 1995 | 30/6/2014 | 02/07/2014 | Original study data no longer accessible |
| Merlotti 2007 | 30/6/2014 | No | Not applicable |
| Miller 1999 | 30/6/2014 | No | Not applicable |
| O'Doherty 1992 | 30/6/2014 | No | Not applicable |
| O'Donoghue 1987 | No | ‐ | Not applicable |
| Reginster 1992 | 30/06/2014 | 30/06/2014 | Original study data no longer accessible |
| Reid 1996; Reid 2004; Reid 2005 | 30/6/2014 | 02/09/2014 | Provided more information on concealment (Reid 1996; Reid 2004), mean alkaline phosphatase and SD change (Reid 2004), blinding (Reid 2005) and data on adverse events (Reid 2004) |
| Roux 1995 | 30/6/2014 | 08/07/2014 | Original study data no longer accessible |
| Siris 1996 | 30/06/2014 | 01/07/2014 | Original study data no longer accessible |
| Tan 2017 | 28/02/2015 | 22/06/2015; 10/12/2015 | Unpublished data provided. Provided more information on QoL and adverse events |
| Walsh 2004 | 30/6/2014 | 19/08/2014 | Provided more information on concealment, mean alkaline phosphatase and SD change, data on pain scores, QoL and adverse events |
Abbreviations: QoL ‐ quality of life; SD ‐ standard deviation
Data and analyses
Comparison 1. Bisphosphonates versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number of participants whose bone pain disappeared completely | 2 | 205 | Risk Ratio (M‐H, Random, 95% CI) | 3.42 [1.31, 8.90] |
| 2 Number of participants with change in bone pain | 7 | 481 | Risk Ratio (M‐H, Random, 95% CI) | 1.97 [1.29, 3.01] |
| 2.1 Etidronate vs. placebo | 3 | 124 | Risk Ratio (M‐H, Random, 95% CI) | 1.75 [0.79, 3.87] |
| 2.2 Tiludronate vs. placebo | 3 | 344 | Risk Ratio (M‐H, Random, 95% CI) | 1.98 [1.27, 3.08] |
| 2.3 Alendronate vs. placebo | 1 | 13 | Risk Ratio (M‐H, Random, 95% CI) | 10.00 [0.69, 144.38] |
| 3 Number of participants experiencing radiologically‐confirmed clinical fractures | 4 | 356 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.18, 4.51] |
| 3.1 Etidronate vs. placebo | 2 | 95 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.09, 9.06] |
| 3.2 Tiludronate vs. placebo | 2 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 0.88 [0.09, 8.64] |
| 4 Number of participants who experienced adverse events related to use of bisphosphonates | 6 | 678 | Risk Difference (M‐H, Random, 95% CI) | 0.11 [‐0.00, 0.22] |
| 4.1 Etidronate vs. placebo | 1 | 47 | Risk Difference (M‐H, Random, 95% CI) | 0.02 [‐0.21, 0.25] |
| 4.2 Zoledronate vs. placebo | 1 | 176 | Risk Difference (M‐H, Random, 95% CI) | 0.27 [0.12, 0.42] |
| 4.3 Tiludronate vs. placebo | 3 | 400 | Risk Difference (M‐H, Random, 95% CI) | 0.07 [‐0.08, 0.22] |
| 4.4 Alendronate vs. placebo | 1 | 55 | Risk Difference (M‐H, Random, 95% CI) | 0.12 [‐0.11, 0.34] |
| 5 Number of participants who withdrew due to adverse events | 6 | 517 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.38, 2.69] |
| 5.1 Etidronate vs. placebo | 1 | 47 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 Tiludronate vs. placebo | 3 | 400 | Odds Ratio (M‐H, Random, 95% CI) | 0.96 [0.34, 2.67] |
| 5.3 Alendronate vs. placebo | 2 | 70 | Odds Ratio (M‐H, Random, 95% CI) | 1.74 [0.06, 50.43] |
| 6 Mean percentage change from baseline in serum total alkaline phosphatase level | 8 | 592 | Mean Difference (IV, Random, 95% CI) | ‐50.09 [‐67.72, ‐32.46] |
| 6.1 Etidronate vs. placebo | 3 | 122 | Mean Difference (IV, Random, 95% CI) | ‐55.85 [‐66.50, ‐45.20] |
| 6.2 Zoledronate vs. placebo | 1 | 176 | Mean Difference (IV, Random, 95% CI) | ‐22.26 [‐27.99, ‐16.53] |
| 6.3 Tiludronate vs. placebo | 2 | 256 | Mean Difference (IV, Random, 95% CI) | ‐58.0 [‐64.25, ‐51.75] |
| 6.4 Alendronate vs. placebo | 1 | 15 | Mean Difference (IV, Random, 95% CI) | ‐39.9 [‐51.28, ‐28.52] |
| 6.5 Ibandronate vs. placebo | 1 | 23 | Mean Difference (IV, Random, 95% CI) | ‐96.1 [‐147.01, ‐45.19] |
| 7 Number of participants who achieved normalised alkaline phosphatase level | 8 | 580 | Risk Ratio (M‐H, Random, 95% CI) | 9.96 [3.74, 26.58] |
| 7.1 Etidronate vs. placebo | 3 | 121 | Risk Ratio (M‐H, Random, 95% CI) | 4.51 [0.90, 22.55] |
| 7.2 Tiludronate vs. placebo | 3 | 381 | Risk Ratio (M‐H, Random, 95% CI) | 13.79 [2.77, 68.61] |
| 7.3 Alendronate vs. placebo | 1 | 55 | Risk Ratio (M‐H, Random, 95% CI) | 27.96 [1.74, 448.28] |
| 7.4 Ibandronate vs. placebo | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 14.00 [0.92, 212.92] |
Comparison 2. Aminobisphosphonates versus non‐aminobisphosphonates.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean change from baseline in pain | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 1.1 Risedronate vs. etidronate | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Number of participants who experienced adverse events related to use of bisphosphonates | 2 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.72, 1.35] |
| 2.1 Risedronate vs. etidronate | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.68, 1.43] |
| 2.2 Alendronate vs. etidronate | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.55, 1.76] |
| 3 Number of participants who withdrew due to adverse events | 2 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.25, 1.89] |
| 3.1 Risedronate vs. etidronate | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.22, 2.79] |
| 3.2 Alendronate vs. etidronate | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.11, 2.90] |
| 4 Mean percentage change from baseline in serum total alkaline phosphatase level | 2 | 212 | Mean Difference (IV, Random, 95% CI) | ‐40.95 [‐49.09, ‐32.81] |
| 4.1 Risedronate vs. etidronate | 1 | 123 | Mean Difference (IV, Random, 95% CI) | ‐43.9 [‐48.06, ‐39.74] |
| 4.2 Alendronate vs. etidronate | 1 | 89 | Mean Difference (IV, Random, 95% CI) | ‐35.1 [‐45.85, ‐24.35] |
| 5 Number of participants who achieved normalised alkaline phosphatase level | 2 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 4.30 [2.72, 6.79] |
| 5.1 Risedronate vs. etidronate | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 4.81 [2.58, 8.98] |
| 5.2 Alendronate vs. etidronate | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 3.78 [1.93, 7.38] |
Comparison 3. Comparison of two aminobisphosphonates.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number of participants with bone pain change | 2 | 436 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [1.15, 1.51] |
| 1.1 Zoledronate (1) vs. pamidronate (2) | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [1.10, 1.53] |
| 1.2 Zolendronate (1) vs. risedronate (2) | 1 | 347 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.06, 1.74] |
| 2 Number of participants who experienced adverse events related to use of bisphosphonates | 3 | Risk Difference (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Olpadronate (1) vs. pamidronate (2) | 1 | Risk Difference (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Zoledronate (1) vs. pamidronate (2) | 1 | Risk Difference (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Zolendronate (1) vs. risedronate (2) | 1 | Risk Difference (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Number of participants who withdrew due to adverse events | 3 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Zoledronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Zolendronate (1) vs. risedronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Alendronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Mean percentage change from baseline in serum total alkaline phosphatase level | 3 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Olpadronate (1) vs. pamidronate (2) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Zolendronate (1) vs. risedronate (2) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Alendronate (1) vs. pamidronate (2) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Number of participants who achieved normalised alkaline phosphatase level | 4 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Olpadronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Zoledronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Zolendronate (1) vs. risedronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 Alendronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Number of participants who experienced biochemical relapse with increased alkaline phosphatase level | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 Zoledronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Zoledronate (1) vs. risedronate (2) | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Alendronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Altman 1973.
| Methods | RCT. Randomisation ratio: 9 (placebo): 7 (etidronate 1 mg/kg/day): 7 (etidronate 2.5 mg/kg/day): 9 (etidronate 5 mg/kg/day): 8 (etidronate 10 mg/kg/day): 7 (etidronate 20 mg/kg/day). Superiority design |
|
| Participants |
Diagnostic criteria: x‐ray evidence of Paget's disease of bone. Inclusion criteria: Pain at one or more sites of Paget's bone involvement and elevated serum alkaline phosphatase or urinary hydroxyproline at least twice the upper limit of normal (ULN) range. Exclusion criteria: not stated. Number screened: not stated. Number randomised: 50. Number analysed: 47 (65 years, 60% male; percentages monostotic, symptomatic and previously treated for Paget's disease of bone not stated) |
|
| Interventions | 6 parallel treatment groups; placebo and etidronate in 5 different doses (1 mg, 2.5 mg, 5 mg, 10 mg and 20 mg/kg/day). Co‐interventions: not stated |
|
| Outcomes |
Outcomes reported in abstract:
Time points for measurement: 6 months. How were the outcomes measured: prospectively |
|
| Setting and date | One hospital bone unit (Miami, FL, USA). Period when the study was conducted: Not stated |
|
| Follow up period | 6 months | |
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article Funding source: Supported in part by the General Clinical Research Center and a grant from the National Institute of Health (5MO1‐RR‐261‐08). Etidronate was supplied by the Procter and Gamble Company, Cincinnati, OH, USA Declarations of interest among primary researchers: Not stated |
|
| Notes | Etidronate groups data were pooled for meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | There were no data about how participants were selected. The authors did not use the term 'randomization', but the study was double‐blinded, so the study design should have had some way of randomising participants to administer treatment. However, the risk of bias should be likely high because no information was provided for the clinical characteristics of groups before starting the study (gender, age…) and the data on mean alkaline phosphatase or urinary hydroxyproline are clearly different at the start of the study for all groups |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind study". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Scans were coded and read blind for qualitative change by at least two observers". Comment: Outcome assessment was blinded for scans, but there was insufficient information to permit judgement about other outcomes |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Data from the original manuscript (Altman 1973) did not agree with data in the extension study (Khairi 1974). Altman 1973: "Fifty patients entered the study". Khairi 1974: "Fifty‐four patients started in this study". Numbers of participants included in the treatment groups differed for Altman 1973 and Khairi 1974. In Altman 1973 23 participants were randomised to placebo plus 1 mg/kg and 2.5 mg/kg dose groups; this number was 24 in Khairi 1974. Altman 1973 reported randomising 9, 8 and 7 participants respectively to 5 mg/kg, 10 mg/kg and 10 mg/kg groups; in Khairi 1974 8, 10 and 8 participants were reported to have been randomised to these respective treatment arms |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement. Methods and results sections were consistent. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting was insufficient to permit judgement. |
Barreira 2009.
| Methods | RCT. Randomisation ratio: 1:2 (pamidronate: olpadronate). Superiority design |
|
| Participants |
Diagnostic criteria: Not stated. Inclusion criteria: active Paget's disease of bone despite previous therapies. Exclusion criteria: not stated. Number screened: not stated. Number randomised: 27. Number analysed: 21 (age and sex not stated; percentages monostotic, symptomatic and previously treated for Paget's disease of bone were not stated) |
|
| Interventions | Two parallel treatment groups; pamidronate 400 mg/day for 4 months vs. olpadronate 200 mg/day for 12 days. Co‐interventions: Not reported |
|
| Outcomes |
Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints: Number of participants who experienced adverse events related to use of bisphosphonates. Time points for measurement: 6 months. How were the outcomes were measured: Insufficient information. Likely prospectively |
|
| Setting and date | Multicentre; Argentina. Period when the study was conducted: Not stated |
|
| Follow up period | 6 months | |
| Publication details and funding source |
Language of publication: English.
Publication status: conference abstracts only. Funding source: Not stated. Declarations of interest among primary researchers: Not stated |
|
| Notes | Data only from abstracts | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized in a 1:2 schedule." Randomisation method not reported |
| Allocation concealment (selection bias) | Unclear risk | Randomisation method not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Both formulations were double dummy with placebos" Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement. Only an abstract of the trials was published. Many data were lacking; there were no data on attrition, exclusions or adverse events |
| Selective reporting (reporting bias) | Unclear risk | Insufficient reporting to permit judgement. Methods and results sections were poorly described. It was not possible to assess if they were consistent. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting was insufficient to permit judgement. |
Buckler 1999.
| Methods | RCT. Randomisation ratio: 1:1:1:1:1 (placebo: zoledronate 50 µg: 100 µg; 200 µg; 400 µg) Superiority design |
|
| Participants |
Diagnostic criteria: x‐ray evidence of Paget's disease of bone Inclusion criteria: Paget's disease of bone and serum alkaline phosphatase concentrations at least twice the ULN Exclusion criteria: Treatment with bisphosphonates in the previous 6 months or calcitonin or plicamycin in the previous 3 months. Creatinine clearance < 60 mL/min. Abnormal liver function Number screened: not stated Number randomised: 176 Number analysed: 172 (71 years, 61% male; percentages monostotic, symptomatic or previously treated for Paget's disease of bone were not stated) |
|
| Interventions | 5 parallel treatment groups; placebo and zoledronate in 4 doses (50 µg, 100 µg, 200 µg and 400 µg in a single intravenous infusion) Co‐interventions: Not reported |
|
| Outcomes |
Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity Secondary endpoints: Numbers of participants who experienced adverse events related to use of bisphosphonates Time points for measurement: Baseline, 5 days, 10 days, 30 days, 45 days, 60 days and 90 days. How were the outcomes measured: prospectively |
|
| Setting and date | 20 sites in USA and UK. Period when the study was conducted: Not stated |
|
| Follow up period | 3 months | |
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article. Funding source: Funding source not reported in the manuscript, but one author, P Richardson, indicated "Ciba Pharmaceuticals" (abstract) and "Clinical Research, Novartis Pharmaceuticals, East Hanover, NJ, USA" (primary reference) affiliations Declarations of interest among primary researchers: Not stated |
|
| Notes | Zoledronate groups data were pooled for meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were randomized in a double‐blind manner". The allocation method was not described |
| Allocation concealment (selection bias) | Unclear risk | The allocation method was not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Randomized in a double blind manner". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement. The risk of bias was likely to be low for serum data because assays for bone‐specific alkaline phosphatase, urinary calcium, creatinine, hydroxyproline, pyridinoline and deoxypiridinoline were conducted in a central laboratory (Nichols Institute, San Juan Capistrano, CA, USA) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "All efficacy analyses were based on all randomised patients who had at least one post baseline measurement (intention‐to‐treat analysis). Four of the 176 patients did not complete all post baseline evaluations". Comment: Attrition data were provided |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement. Methods and results sections were consistent. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting was insufficient to permit judgement |
Canfield 1977.
| Methods | RCT. Randomisation ratio: 12 (placebo): 7 (etidronate 2.5 mg/kg/day): 8 (etidronate 5 mg/kg/day): 11 (etidronate 10 mg/kg/day): 10 (etidronate 20 mg/kg/day). Superiority design |
|
| Participants |
Diagnostic criteria: x‐ray evidence of Paget's disease of bone. Inclusion criteria: symptoms referable to the disorder, elevation of serum alkaline phosphatase and urinary hydroxyproline (with at least twice the normal value of one these indices). Exclusion criteria: No co‐existing condition that would complicate the interpretation of therapeutic results. No other specific therapy for Paget's disease for at least 6 months before entry into the study. Number screened: not stated. Number randomised: 48. Number analysed: 48 (age not stated, 58% male; percentages monostotic, symptomatic or previously treated for Paget's disease of bone were not stated) |
|
| Interventions | 5 parallel treatment groups; placebo, and etidronate in 4 doses (2.5 mg, 5 mg, 10 mg and 20 mg/kg/day). Co‐interventions: Not reported |
|
| Outcomes |
Outcomes reported in abstract Primary endpoint: mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints: change in bone pain (assessment tool used not reported); radiologically‐confirmed clinical fracture. Time points for measurement: Baseline, 1 month, 2 months, 3 months, 4.5 months, 6 months. How were the outcomes measured: prospectively |
|
| Setting and date | Participants admitted to 4 New York City Hospitals. Period when the study was conducted: Not stated |
|
| Follow up period | 6 months | |
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article. Funding source: Supported by National Institute of Health grants (MO1.RR00645, 5.MO1‐RR‐96, AM‐09579, TIAM‐05397 and TIAM‐05531). Etidronate was supplied by the Procter and Gamble Company, Cincinnati, OH, USA. Declarations of interest among primary researchers: Not stated |
|
| Notes | Etidronate groups data were pooled for meta‐analysis. Data from 27 additional participants treated with etidronate in a non‐blinded, non‐randomised basis were included in the manuscript |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "each patient… was assigned randomly" Comment: Randomisation method was not reported |
| Allocation concealment (selection bias) | Unclear risk | Randomisation method was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "for all the patients in treatment group A neither the investigator nor the patient knew the identity of the treatment". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided to permit judgement |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No losses to follow‐up reported. However, it was not clear how many participants were included |
| Selective reporting (reporting bias) | High risk | We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. Likely selective reporting risk is high due to vague data on adverse events. There are no specific number of patients but only sentences as "some patients… showed a rise in serum phosphate" |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting was insufficient to permit judgement |
Fraser 1997.
| Methods | RCT. Randomisation ratio: 1:1:1:1 (placebo: tiludronate 200 mg, 400 mg, 600 mg). Superiority design |
|
| Participants |
Diagnostic criteria: Paget's disease confirmed by scintigraphy or radiography or both Inclusion criteria: Age ≥ 18 years; serum alkaline phosphate concentration at least twice the ULN Exclusion criteria:
Number screened: not stated. Number randomised: 112. Number analysed: 112 (70 years, 54% male, 30% monostotic, 63% symptomatic, percentage previously treated for Paget's disease of bone not stated) |
|
| Interventions | 4 parallel treatment groups; placebo and tiludronate in 3 doses (200 mg, 400 mg, 600 mg/day) for 12 weeks. Co‐interventions: Not reported |
|
| Outcomes |
Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Treatment success was defined as 50% reduction in serum alkaline phosphatase concentration compared with baseline after 12 weeks' treatment. (Concentration decreased by 25% or less compared with week 0 reading was defined as resistant). Secondary endpoints
Time points for measurement: Baseline, 2 weeks, 8 weeks, 12 weeks, 24 weeks. How were the outcomes measured: prospectively |
|
| Setting and date | 16 hospitals in the UK. Period when the study was conducted: Not stated |
|
| Follow up period | 6 months. An additional follow‐up to assess the need for re‐treatment was carried out 18 months after the last participant had completed the 6 month trial. Follow‐up was via a postal questionnaire with participants who had completed at least 11 weeks of treatment | |
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article. Funding source: Research supported by Sanofi Winthrop Ltd. Declarations of interest among primary researchers: Not stated |
|
| Notes | Tiludronate groups data were pooled for meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were allocated sequential study numbers and randomly assigned to one of the four treatment groups". Comment: Likely there was a central allocation because 16 hospitals recruited participants in the UK |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind randomised study". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided to permit judgement |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Data were analysed on an 'intention to treat' basis". Comment: Reasons for missing outcome data (lost to follow‐up and withdrawals) are reported |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. Quote: "A visual analogue scale was used for patients to assess their Pagetic pain". Comment: Although stated that "there were no significant differences between any of the treatment groups" data for pain assessment were only provided for placebo and tiludronate higher dose groups |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting was insufficient to permit judgement |
Langston 2010.
| Methods | RCT. Randomisation ratio: 1:1 (symptomatic treatment: intensive treatment). Superiority design |
|
| Participants |
Diagnostic criteria: diagnosis confirmed on plain radiograph of at least one skeletal site according standard criteria from UK guidelines (Selby 2002). Inclusion criteria: Paget's disease of bone and life expectancy > 1 year. Exclusion criteria: No specific exclusion criteria were applied on the basis of treatment history, baseline alkaline phosphatase or co‐existing diseases. Number assessed: 2110. Number randomised: 1331. Number analysed: 1159 (74 years, 51% male, 35% monostotic, 69% symptomatic, 90% previously treated with bisphosphonates for Paget's disease, 52% had normal alkaline phosphatase at baseline) |
|
| Interventions | Two parallel treatment groups; symptomatic vs. intensive treatment. Symptomatic treatment: Philosophy; treat bone pain, not alkaline phosphatase.
Intensive treatment: Philosophy; maintain normal alkaline phosphatase.
Co‐interventions: Analgesics and nonsteroidal anti‐inflammatory drugs for symptomatic and intensive treatment group |
|
| Outcomes |
Outcomes reported in abstract Primary endpoint: Radiologically‐confirmed clinical fracture. Secondary endpoints:
Time points for measurement: Visits scheduled at 4 monthly intervals. How were the outcomes measured: prospectively |
|
| Setting and date | 39 secondary referral centres in the UK. Period when the study was conducted: December 2001 to June 2004 |
|
| Follow up period | Median 3 years (range 2 to 5 years) | |
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article. Funding source: Supported by grants from the Arthritis Research Campaign UK (Ref. 13627), National Association for Relief of Paget’s Disease, Procter & Gamble Pharmaceuticals and Sanofi‐Aventis. Declarations of interest among primary researchers:
|
|
| Notes | The study was described by the authors as a "pragmatic randomised controlled trial designed to compare the effects of two management strategies". The study was not blinded | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were assigned to the treatment groups by an integrated telephone and web‐based randomization system" |
| Allocation concealment (selection bias) | Low risk | Quote: "Treatment allocation employed minimization to ensure that the treatment group were balanced with respect to key prognostic variables" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study was not blinded". Comment: |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Information provided by authors: The assessments of events were performed by individuals who were unaware of the treatment allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Information provided by authors: The study was an event‐driven trial, with fracture as the primary endpoint. Data on all fracture events were available even in subjects who had withdrawn from the study. Comment: The risk of attrition bias was low for data at 2 years (minimum duration of follow‐up). The rate of losses to follow‐up at this point was 12% and 13% respectively. The risk of attrition bias is high for 36 or 48 months |
| Selective reporting (reporting bias) | Low risk | Study protocol available. All proposed outcomes were reported |
| Other bias | Low risk | Comment: The study design allowed the attending clinicians to choose different drugs within a specified treatment strategy. Evidence that the attending clinicians adhered to the allocated strategy came from the observation that alkaline phosphatase levels differed significantly between groups |
McClung 1995.
| Methods | RCT. Randomisation ratio: 1:1:1 (placebo: tiludronate 200 mg: tiludronate 400 mg). Superiority design |
|
| Participants |
Diagnostic criteria: Diagnosis confirmed by radiographic and clinical criteria. Inclusion criteria: participants aged > 32 years and with serum alkaline phosphatase values at least twice ULN. Exclusion criteria:
Number screened: not stated. Number randomised: 139. Number analysed: 134 (70 years, 54% male; percentages monostotic, symptomatic or previously treated for Paget's disease of bone not stated) |
|
| Interventions | Three parallel treatment groups; placebo and tiludronate in two different doses (200 mg, 400 mg) nightly for 12 weeks. Co‐interventions: Calcium‐containing food and supplements were avoided around the time the study medication was taken |
|
| Outcomes |
Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: Baseline, 2 weeks, 4 weeks, 8 weeks, 12 weeks, 16 weeks, 20 weeks, 24 weeks. How were the outcomes measured: prospectively |
|
| Setting and date | 20 centres in the USA and Canada. Period when the study was conducted: Not stated |
|
| Follow up period | 6 months | |
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article. Funding source: Research supported by Sanofi Winthrop Ltd. Declarations of interest among primary researchers: Not stated |
|
| Notes | Data on the tiludronate groups were pooled for meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "In a randomized, double‐blind, placebo‐controlled study... were enrolled at 20 centers throughout the USA and Canada" Comment: Likely that the risk of bias was low because for a multicentre design, the allocation process should be centralized |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Double‐blind" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement. The risk is probably low for laboratory data because two laboratories were involved:
|
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reasons for attrition and exclusion were reported. 2/91 (2%) participants were lost from the interventions group and 3/48 (65) participants from the placebo group |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. Comment: Data for pain assessment were poorly reported |
| Other bias | Unclear risk | No other risks of bias found, but reporting was insufficient to permit judgement. |
Merlotti 2007.
| Methods | RCT. Randomisation ratio: 2:1 (pamidronate: zoledronate). Superiority design |
|
| Participants |
Diagnostic criteria: confirmed by bone scintigraphy and x‐ray of areas of increased isotope uptake. Inclusion criteria: Presence of serum total alkaline phosphatase above ULN (298 IU/L) on 2 consecutive measurements and no treatment with bisphosphonates or other drugs affecting bone metabolism for at least 6 months before the study. Exclusion criteria:
Number screened: not stated. Number randomised: 90. Number analysed: 89 (70 years, 64% male; percentage monostotic not stated, 99% symptomatic, 66.7% previously treated for Paget's disease of bone) |
|
| Interventions | 2 parallel treatment groups; 4 mg single infusion of zoledronate or 30 mg infusion of pamidronate for 2 consecutive days every 3 months. Co‐interventions: 1 g calcium and 800 IU cholecalciferol per day |
|
| Outcomes |
Outcomes reported in abstract Primary endpoint: Rate of therapeutic response (defined as normalisation of alkaline phosphatase levels or a reduction of at least 75% in total alkaline phosphatase excess). Secondary endpoints:
Time points for measurement: Baseline and 6 months. How were the outcomes measured: prospectively |
|
| Setting and date | One centre in Siena, Italy. Period when the study was conducted: Not stated |
|
| Follow up period | 6 months | |
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article. Funding source: Not stated. Declarations of interest among primary researchers: No authors had conflicts of interest |
|
| Notes | The study was subdivided:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomized study... randomization was stratified according to baseline alkaline phosphatase levels and previous bisphosphonate treatment (as a binary variable: yes or no)". Comment: Random sequencing was probably computer‐generated; the randomisation was stratified |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "29 of 30 patients receiving zoledronate and 60 of 60 patients receiving pamidronate completed the follow‐up at 6 mo". Comment: Only one participant was lost to follow‐up at 6 months |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | No other risks of bias found, but reporting insufficient to permit judgement |
Miller 1999.
| Methods | RCT. Randomisation ratio: 1:1 (risedronate: etidronate). Superiority design |
|
| Participants |
Diagnostic criteria: confirmed by bone scintigraphy or x‐ray. Inclusion criteria: participants aged 18 to 85 years, with serum alkaline phosphatase concentration of at least twice the ULN. Women had to be at least 1 year postmenopause or using contraception. Exclusion criteria:
Number screened: 179. Number randomised: 123. Number analysed: 103 (66 years, 69% males, 24% monostotic, 91% symptomatic, 72% previously treated for Paget's disease of bone) |
|
| Interventions | Two parallel treatment groups; oral risedronate 30 mg daily for 2 months and oral etidronate 400 mg for 6 months. Co‐interventions: Not reported |
|
| Outcomes |
Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: Baseline and 1 through 6, 8, 10, 12, and 18 months for laboratory measures. Baseline, 2, 6 and 12 months for quality of life (including pain). How were the outcomes measured: prospectively |
|
| Setting and date | 12 study centres in the USA and Canada. Period when the study was conducted: Not stated |
|
| Follow up period | 12 months plus an optional 6 month extended follow‐up | |
| Publication details and funding source |
Language of publication: English
Publication status: peer‐reviewed journal; full article. Funding source: Supported by Procter & Gamble Pharmaceuticals, Cincinnati, OH, USA. Declarations of interest among primary researchers: Not stated |
|
| Notes | None | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were assigned unique, sequential identification numbers according to the chronological order of entry at each of the 12 centers and were stratified into those who had, and those who had not, received previous etidronate treatment. Within each center, and within each stratum of previous etidronate use, patients were assigned to one of the two treatment groups according to a randomisation schedule generated using SAS Version 6.07 PLAN procedure (SAS Institute, Cary, NC)". |
| Allocation concealment (selection bias) | Low risk | Computerised random number generator was used |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blind design". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided to permit judgement |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Although the authors stated that the study included an intention‐to‐treat analysis:
|
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting insufficient to make judgement |
O'Doherty 1992.
| Methods | RCT. Randomisation ratio: 2:1 (alendronate: placebo). Superiority design |
|
| Participants |
Diagnostic criteria: diagnosis established by finding hyperphosphatasia and characteristic radiographic and scintigraphic features. Inclusion criteria: participants diagnosed as active Paget's disease of bone. Exclusion criteria:
Number screened: not stated. Number randomised: 15. Number analysed: 15 (67 years, 60% male, % monostotic not stated, 87% symptomatic, 66% previously treated for Paget's disease of bone) |
|
| Interventions | Two parallel treatment groups: intravenous alendronate 10 mg/day for 5 days and placebo. Co‐interventions: Not reported |
|
| Outcomes |
Outcomes reported in abstract: Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: Baseline and 2, 3 and 4 weeks from the start of treatment and monthly thereafter for 6 months. How were the outcomes measured: prospectively |
|
| Setting and date | One centre in Sheffield, UK. Period when the study was conducted: Not stated |
|
| Follow up period | 6 months. Comment: The blind was broken 4 weeks after the start of treatment. The 10 participants who received treatment with alendronate were additionally followed for 6 months. The 5 participants who received placebo were allowed to withdraw from the study after the blind was broken |
|
| Publication details and funding source |
Language of publication: English
Publication status: peer‐reviewed journal; full article. Funding source: Supported by a Programme Grant from the Medical Research Council and by Merck, Sharp and Dohme Research Laboratories, Woodbridge, NJ, USA and Harlow, UK. Declarations of interest among primary researchers: Not stated |
|
| Notes | None | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "A group of 15 patients with active Paget’s disease of bone were randomised prospectively". Comment: Insufficient information provided to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "receive 5 days of treatment with either alendronate (10 mg/day) or placebo under double‐blind conditions". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided to permit judgement |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "The blind was broken 4 weeks after the start of treatment. The 10 participants who received treatment with alendronate were additionally followed for 6 months. The 5 participants who received placebo were allowed to withdraw from the study after the blind was broken." Comment: The follow‐up was different for participants in the placebo and experimental groups (e.g. data on urinary hydroxyproline and serum alkaline phosphatase (Figure 1) were registered to different weeks from the start of treatment for alendronate or placebo) |
| Selective reporting (reporting bias) | High risk | We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. Comment: Pain data were reported, but pain was not defined as an outcome. There was no reporting of how pain was assessed. There were no data reported on adverse effects. |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting was insufficient to permit judgement |
O'Donoghue 1987.
| Methods | RCT. Randomisation ratio: 1:1 (etidronate: etidronate plus calcitonin). Superiority design |
|
| Participants |
Diagnostic criteria: Iliac crest bony biopsy, skeletal radiography and scintigraphy were performed to confirm the diagnosis of Paget’s disease. Inclusion criteria: participants with biochemically active Paget’s disease whose bone pain was unresponsive to simple analgesia or nonsteroidal anti‐inflammatory agents. Exclusion criteria: Not stated. Number screened: not stated. Number randomised: 44. Number analysed: 44 (age and sex not stated; percentage monostotic not stated, 100% symptomatic, 10% previously treated for Paget's disease of bone) |
|
| Interventions | Two parallel treatment groups: oral etidronate 400 mg and oral etidronate 400 mg in combination with subcutaneous calcitonin 100 MRC units thrice weekly for six months. Co‐interventions: Participants were on a constant calcium low gelatin diet. |
|
| Outcomes |
Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: Baseline, monthly during treatment, and every 2 to 3 months for an additional 6 months period. How were the outcomes measured: prospectively |
|
| Setting and date | One centre in Nottingham, UK. Period when the study was conducted: Not stated |
|
| Follow up period | 12 months | |
| Publication details and funding source |
Language of publication: English
Publication status: peer‐reviewed journal; full article. Funding source: Not stated. Declarations of interest among primary researchers: Not stated |
|
| Notes | The study randomised a group of participants to be treated with etidronate or etidronate plus calcitonin. The authors also compared these two groups with data accumulated from participants who received calcitonin alone prior to the introduction of etidronate in the metabolic unit (28 participants); this group was excluded because participants were not randomised | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Forty‐four sequential patients were randomised to treatment". Comment: Insufficient information provided to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "patients were randomised to treatment with either EHDP 400 mg daily alone (21 patients) or in combination with SCT 100 MRC units thrice weekly (23 patients)". Comment: Because there was no mention of placebo, it is likely that one group was treated with oral drugs alone and the other with a combination of oral and subcutaneous drugs; hence, blinding was not possible |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided to permit judgement |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Two patients treated with EHDP defaulted from follow‐up but the other 70 were reevaluated in the metabolic unit after 6 months". Comment: Attrition rate was low; only two participants were lost to follow‐up. The proportion of missing outcomes compared with observed event risk was insufficient to have a clinically‐relevant impact on the intervention effect estimate |
| Selective reporting (reporting bias) | High risk | We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. Comment: There are some contradictions between methods and results sections.
|
| Other bias | Unclear risk | No other risks of bias found, but reporting insufficient to make judgement |
Ralston 1987.
| Methods | RCT. Randomisation ratio: 1:1:1 (etidronate plus 1α‐hydroxyvitamin D: etidronate plus placebo: placebo plus placebo). Superiority design |
|
| Participants |
Diagnostic criteria: Radiological and bone scan evidence of Paget's disease of bone. Inclusion criteria: Symptomatic participants. Exclusion criteria: Treatment for Paget's disease of bone in the 12 months before study commencement. Number screened: not stated. Number randomised: 32. Number analysed: 29 (age and sex not reported; 31% monostotic, 100% symptomatic, 37.5% previously treated for Paget's disease of bone) |
|
| Interventions | Three parallel treatment groups: oral etidronate 400 mg plus 1α‐hydroxyvitamin D 0.5 μg, oral etidronate 400 mg plus placebo and placebo plus placebo for three months. Co‐interventions: participants received other medications according to routine practice (e.g. analgesics, nonsteroidal anti‐inflammatory drugs) (information provided by authors). |
|
| Outcomes |
Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity Secondary endpoints: change in bone pain (linear analogue technique with a scale ranging from 0 (no pain) to 30 (very severe pain)). Time points for measurement: Baseline, and after 2, 8 and 12 weeks on drug therapy. How were the outcomes measured: prospectively |
|
| Setting and date | One centre in Glasgow, UK. Period when the study was conducted: not stated |
|
| Follow up period | 3 months | |
| Publication details and funding source |
Language of publication: English
Publication status: peer‐reviewed journal; full article. Funding source: Partially supported by a grant to BFB from the Scottish Hospital Endowments Research Trust and a grant to ITB from Brocades (UK) Ltd. The 1α‐hydroxyvitamin D and placebo tablets were supplied by Leo Laboratories Ltd. Declarations of interest among primary researchers: not stated |
|
| Notes | Data on etidronate plus 1α‐hydroxyvitamin D and etidronate plus placebo groups were pooled for meta‐analysis. Transiliac bone biopsies were obtained before therapy in all cases and after therapy in 28 participants. Bone histomorphometry data were reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "On entry to the trial patients were randomised". Information provided by authors: Allocation by referring to random numbers |
| Allocation concealment (selection bias) | Low risk | Information provided by authors: Investigators were blinded to treatment allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double blind" Information provided by authors: A double dummy was used with placebo/placebo, etidronate/placebo and etidronate/1α‐hydroxyvitamin D |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Information provided by authors: Investigators were blinded to treatment when assessing outcome |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "There were three withdrawals from the trial after randomisation". Comment: The rate of withdrawals (and reasons) were reported |
| Selective reporting (reporting bias) | High risk | We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. The authors reported symptoms and side effects and described only one adverse event ("one patient who developed increasing pain and radiological deterioration at the site of an asymptomatic pseudofracture of the tibia"). However, at the start of the results section the authors described another adverse event which was not included in the "symptoms and side effects" paragraph ("1 patient in Group A stopped medication because of dyspepsia"). Data on hypercalcaemia are on biochemistry ("One patient... developed mild hypercalcaemia"). There were no other data on adverse events. Information provided by authors: Adverse events were not systematically recorded |
| Other bias | Unclear risk | No other risks of bias found, but reporting insufficient to make judgement |
Reginster 1992.
| Methods | RCT. Randomisation ratio: 1:1:1:1:1 (placebo: tiludronate 100 mg: tiludronate 200 mg: tiludronate 400 mg: tiludronate 800 mg). Superiority design |
|
| Participants |
Diagnostic criteria: Paget's disease of bone with characteristic radiologic lesions and/or with 99mTc bisphosphonates scintigraphic evidence of local increase in bone turnover. Inclusion criteria: Serum alkaline phosphatase levels were at least twice the ULN. Exclusion criteria:
Number screened: not stated. Number randomised: 149. Number analysed: 149 (69 years, 54 % male, % monostotic not stated, % symptomatic participants not stated, 82% previously treated for Paget's disease of bone) |
|
| Interventions | The study was divided into 2 periods. In the first period there were five parallel treatment groups: placebo (4 x placebo capsules), tiludronate 100 mg (2 x placebo and 2 x 50 mg tiludronate capsules), tiludronate 200 mg (4 x capsules 50 mg tiludronate), tiludronate 400 mg (4 x capsules 100 mg), tiludronate 800 mg (4 x capsules 200 mg tiludronate). All participants took 2 capsules at 10.00 am and 2 capsules at 4.00 pm daily for 3 months. In the second period all participants received 4 placebo capsules. The investigators initiated tiludronate therapy during the second period for participants experiencing a lack of efficacy (participants were considered discontinued from the study). Co‐interventions: Not reported |
|
| Outcomes |
Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: Baseline and monthly intervals. How were the outcomes measured: prospectively |
|
| Setting and date | 29 centres in Belgium, France and USA. Period when the study was conducted: Not stated |
|
| Follow up period | 6 months | |
| Publication details and funding source |
Language of publication: English
Publication status: peer‐reviewed journal; full article. Funding source: Supported by Sanofi Research, Montpellier, France. Declarations of interest among primary researchers: Not stated |
|
| Notes | Data on tiludronate groups were pooled for meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "double‐blind, randomised, placebo‐controlled study". Comment: Insufficient information provided to permit judgement, but probably low risk because the allocation process should have been centralized because the study involved 29 centres in different countries |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind, randomised, placebo‐controlled study". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "All biochemical determinations were performed at
a central location throughout the period of study." Comment: Insufficient information provided, but likely low risk because it is unlikely that the blinding was broken in a central location |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "Follow up assessments were available for all 149 patients." Quote: "Thirty‐five patients withdrew prematurely from the study. The reasons were lack of efficacy in 9, adverse events in 15, at the patient’s request in 4, lost to follow‐up in 4, and miscellaneous reasons unrelated to treatment in 3. There was no relationship noted between the dosage of tiludronate and the number of study withdrawals." Comment: Although the reasons for missing outcome data were explained and withdrawals balanced between groups, the proportion of missing outcomes (25%) compared with observed event risk is enough to induce clinically‐relevant bias in intervention effect estimate |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting insufficient to permit judgement |
Reid 1996.
| Methods | RCT. Randomisation ratio: 1:1 (placebo: alendronate 40 mg). Superiority design |
|
| Participants |
Diagnostic criteria: Paget’s disease of bone documented by standard clinical, radiological or scintigraphic methods or both. Inclusion criteria:
Exclusion criteria:
Number screened: not stated. Number randomised: 55. Number analysed: 53 (70 years, 56 % male, percentage monostotic or symptomatic not stated, 35% previously treated for Paget's disease of bone) |
|
| Interventions | Two parallel treatment groups: placebo and oral alendronate 40 mg daily for 6 months. Co‐interventions: Calcium (450 mg to 500 mg) and vitamin D (400 IU to 600 IU) |
|
| Outcomes |
Outcomes reported in abstract: Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: baseline, 3 and 6 months. How were the outcomes measured: prospectively |
|
| Setting and date | Three sites in Australia, New Zealand, and UK. Period when the study was conducted: Not stated |
|
| Follow up period | 6 months | |
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article. Funding source: Supported by Merck Research Laboratories, Rahway, NJ, USA. Declarations of interest among primary researchers: Not stated |
|
| Notes | Bone biopsy conducted for 28 participants. Bone histomorphometry data reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Probably done because the study was multicentre, involving 3 countries |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study was double‐blinded". Comment: Insufficient information provided to permit judgement, but probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Radiographs were all evaluated by one skeletal radiologist, who was blinded with respect to film sequence and treatment allocation". "Bone biopsy: Each specimen was coded, so that the reader was unaware of the subjects' drug therapy..." Adverse events that were considered by the blinded investigator to be possibly treatment related occurred in..." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "No subjects withdrew from the study because of drug side effects. Comment: Attrition rate was low because only two participants did not complete the study in the placebo group but none in the alendronate group. The authors did not explain the reasons for not completing the trial for these two participants. However, both participants were from the placebo group and the proportion of missing outcomes compared with observed event risk were insufficient to have a clinically relevant impact on the intervention effect estimate |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. |
| Other bias | Unclear risk | No other risks of bias found, but reporting was insufficient to permit judgement |
Reid 2004.
| Methods | RCT: Randomisation ratio: 1:1:1 (placebo: ibandronate 6 mg: ibandronate 12 mg). Superiority design |
|
| Participants |
Diagnostic criteria: Paget’s disease of bone confirmed radiologically. Inclusion criteria: Serum alkaline phosphatase activities at baseline at least twice the upper limit of the reference range. Exclusion criteria: Other medical conditions, or on medications that affect bone or calcium metabolism. Number screened: not stated. Number randomised: 25. Number analysed: 23 (73 years, 74 % male, percentages monostotic and symptomatic not stated, 64% previously treated for Paget's disease of bone) |
|
| Interventions | Three parallel treatment groups: placebo (infusions of normal saline at baseline and at 1 month), ibandronate 6 mg (infusions of 6 mg of ibandronate at baseline and normal saline at 1 month), ibandronate 12 mg (infusions of 6 mg of ibandronate at both baseline and 1 month). Co‐interventions: None (information provided by authors) |
|
| Outcomes |
Outcomes reported in abstract: Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints: Number who achieved normalised alkaline phosphatase level. Time points for measurement: Not reported. At least at baseline, 3, 6 and 12 months. How were the outcomes measured: prospectively |
|
| Setting and date | One centre in Auckland, New Zealand. Period when the study was conducted: Not stated |
|
| Follow up period | 6 months (12 months, see notes) | |
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article. Funding source: Supported by the Health Research Council of New Zealand and Roche Products (NZ) Ltd. Declarations of interest among primary researchers: not stated |
|
| Notes | Ibandronate group data were pooled for meta‐analysis. Participants were followed for an additional 6 month period after finishing the first 6 months of the study. In this second part of the study, 6 placebo group participants (67%) were given ibandronate in a non‐randomised way. Data from this second part of the study were not included in this review. In 2017 the authors published a manuscript with data from original participants after 10 years follow up |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly allocated to one of three groups using a minimization algorithm to ensure balance between groups for alkaline phosphatase level". Quote: "Six subjects who had placebo at both of these time points subsequently were given ibandronate 6 mg at 6 months, so posttreatment data are shown for 20 subjects" Quote: "Because the focus of this study was on relative response of markers to ibandronate, the pre‐ and posttreatment data in the two ibandronate groups and in those patients from the ‘placebo’ group who went on to have ibandronate have been pooled, so that every subject in the study provides a pre‐ and 6 months post‐ibandronate value". Comment: Although initially participants were randomised, the authors pooled data from participants who were initially randomised to ibandronate with data from placebo participants who received ibandronate in a 6 month study extension. Note: Although the study general assessment of risk of bias for random sequence generation is high, we assessed risk as unclear because on we included data from the first part of the study only in the meta‐analysis |
| Allocation concealment (selection bias) | Unclear risk | Comment: As for assessment of random sequence generation, although for the first 6 months the authors probably concealed allocation, when finishing the first part of the study, they treated two thirds of placebo participants with ibandronate so they knew participant allocation at that time. Note: Although the study general assessment of risk of bias for allocation concealment is high, we assess the risk as unclear because, on meta‐analysis, we introduce only data from the first part of the study. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Information provided by authors: Infusions were prepared by staff members who had no contact with the participants, so that study staff and participants were blinded to treatment received |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Information provided by authors: Outcome assessment was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "'Placebo' group (nine patients), received infusions of normal saline at baseline and at 1 month, the ‘6 mg’ group (nine patients, eight with follow‐up data) received ibandronate 6 mg at baseline and placebo at 1 month, and the ‘12 mg’ group (seven patients, six with follow‐up data) received ibandronate 6 mg at both baseline and 1 month". Comment: Follow‐up data were provided and missing outcome data are likely balanced; however, reasons for missing outcomes were not explained. The proportion of missing outcomes compared with observed event risk is not enough to have a clinically‐relevant impact on the intervention effect estimate |
| Selective reporting (reporting bias) | High risk | Information provided by authors: Adverse events were not systematically recorded |
| Other bias | Unclear risk | No other risks of bias found, but reporting was insufficient to permit judgement |
Reid 2005.
| Methods | RCT. Randomisation ratio: 1:1 (zoledronate acid: risedronate). Non inferiority design |
|
| Participants |
Diagnostic criteria: Paget’s disease of bone was confirmed by x‐ray, magnetic resonance imaging, computerized tomography, radio‐isotope imaging, etc. Inclusion criteria:
Exclusion criteria:
Number screened: 688 (371 + 317). Number randomised: 357 (185 + 172). Number analysed: 349 (70 years, 67.7% male; percentages monostotic or symptomatic not stated; 54% previously treated for Paget's disease of bone). An extension study of the core trial was published including participants who had therapeutic response defined as normalisation of the alkaline phosphatase level or a reduction of at least 75% in alkaline phosphatase excess (the difference from the midpoint of the reference range) at 6 months of treatment. Number screened for the extension study: 296 (169 responders from 182 participants from the zoledronate group + 127 responders from 175 participants from the risedronate group). Number analysed for the extension study: 267 (152 responders from the zoledronate group + 115 responders from the risedronate group) (70 years; sex, monostotic status and symptomatic participants not stated; 100% previously treated for Paget's disease of bone) |
|
| Interventions | Two parallel treatment groups: zoledronate (single 5 mg infusion zoledronate at baseline followed by placebo tablets daily for 60 days), risedronate (saline infusion followed by 30 mg tables of risedronate daily for 60 days). Co‐interventions: All participants received 1 g calcium per day and 400 IU to 1000 IU calciferol per day. No further interventions were added after treatment in the core trial for the extension study |
|
| Outcomes |
Outcomes reported in abstract: Primary endpoint: Proportion of participants who had therapeutic response. Therapeutic response was defined as normalisation of the alkaline phosphatase level or a reduction of at least 75% in the alkaline phosphatase excess (the difference from the midpoint of the reference range) at 6 months. Secondary endpoints:
Time points for measurement: Not reported. Probably at baseline, 10 days, 1, 2, 3 and 6 months according to x‐axis graph data. Quote: "Physical examinations, hematologic tests, and serum chemical tests were performed regularly throughout the six‐month study. Serum creatinine and urinary protein were measured 9 to 11 days after intravenous dosing". How were the outcomes measured: prospectively. Relapse rate was the primary endpoint for the extension study. Relapse was defined as a return of alkaline phosphatase to within 20% of the pre‐treatment baseline value. Partial relapse was defined as an alkaline phosphatase level that was both 50% above its 6 month value and 1.25 times the ULN range. Outcomes were monitored at 6 monthly visits |
|
| Setting and date | 76 centres in 10 countries (Australia, Belgium, Canada, France, Germany, New Zealand, South Africa, Spain, UK, USA). Period when the study was conducted: January 2002 to March 2004. Period when the extension study was conducted: Follow up covers the period from the end of the core trials to 29 January 2009 |
|
| Follow up period | 6 months. 6.5 years observation period in the extension study: median follow‐up time from the beginning of the core study was 5.0 years for zoledronate and 3.3 years for risedronate (patient‐years of follow‐up were 574 years and 340 years respectively) |
|
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article. Funding source: Supported by a research grant from Novartis Pharma AG, Basel, Switzerland. Declarations of interest among primary researchers:
|
|
| Notes | The authors reported pooled data from two identical RCTs. Participants identified as treatment responders were entered to an extended observation period including only those whose alkaline phosphatase was normal at completion of the core study. Bone biopsies were performed for 22 participants (12 zoledronate acid and 10 risedronate). Bone histomorphometry data were reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned in a double‐blind fashion through an interactive voice‐response system". Comment: Probably done; authors describe a telephone system for sequence generation |
| Allocation concealment (selection bias) | Low risk | Study was conducted using a central allocation system (telephone) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients were randomly assigned in a double‐blind fashion". Comment: Probably done; placebo treatment is reported. The risk of bias was judged as high risk for the extension studies because much of the extended follow‐up was conducted with participants and doctors knowing what treatment they received in the core trial (information provided by authors). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Measurements were made by Covance Central Laboratory Services" Information provided by authors: Outcome assessment was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "All efficacy variables, except the time to a therapeutic response, were analysed according to the modified intention‐to‐treat principle, which required patients to have a baseline and at least one post‐baseline measurement of alkaline phosphatase." Comment: Participant flow diagram including reasons for losses to follow‐up was included. MIssing outcome data were balanced between interventions and the proportion of missing outcomes compared with observed event risk was insufficient to have a clinically relevant impact on the intervention effect estimate. The risk of bias was judged as high risk for the extension studies because only participants who responded to treatment were included in the follow‐up. Participants who relapsed or whose physicians provided further treatment for Paget's disease were discontinued from the study during the follow‐up. |
| Selective reporting (reporting bias) | High risk | Comment: Brief Pain Inventory‐Short Form (BPI‐SF) was stated as a secondary outcome measure in the study characteristics in the trial registry record, but was not reported in the study publication. Data on fractures were not reported in the study publication but were proposed in the trial registry record |
| Other bias | Unclear risk | No other risks of bias found, but reporting was insufficient to permit judgement |
Roux 1995.
| Methods | RCT. Randomisation ratio: 1:1:1 (tiludronate 400 mg 3 months, tiludronate 400 mg 6 months; etidronate 400 mg 6 months). Superiority design |
|
| Participants |
Diagnostic criteria: Paget's disease of bone was confirmed by the presence of radiologically evident lesions characteristic of the disease. Inclusion criteria: Serum alkaline phosphatase concentration at least twice the ULN range. Exclusion criteria:
Number screened: Not stated. Number randomised: 234. Number analysed: 234 (69 years, 59% males, % monostotic not stated, 74% symptomatic participants, 71% previously treated for Paget's disease of bone) |
|
| Interventions | Three parallel treatment groups: tiludronate 400 mg 3 months (2 x 200 mg tiludronate tablets and 2 x etidronate placebo capsules during the first 3 months followed by 2 x tiludronate placebo tablets and 2 x etidronate placebo capsules during the second 3 months), tiludronate 400 mg 6 months (2 x 200 mg tiludronate tablets and 2 x etidronate placebo capsules for 6 months) and etidronate 400 mg 6 months (2 x 200 mg etidronate capsules and 2 tiludronate placebo tablets for 6 months). Co‐interventions: Not reported |
|
| Outcomes |
Outcomes reported in abstract: Primary endpoint: Proportion of participants who had a response to treatment. Response to treatment was defined as a reduction of at least 50% in serum alkaline phosphatase concentration after a 3 month treatment period. Secondary endpoints:
Time points for measurement: Baseline, 3 months, 6 months. How were the outcomes measured: prospectively |
|
| Setting and date | 85 centres in 6 countries (Belgium, France, Germany, Italy, Netherlands, Spain). Period when the study was conducted: February 1991 to September 1992 |
|
| Follow up period | 6 months | |
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article. Funding source: Supported by Sanofi Recherche, Montpellier, France. Declarations of interest among primary researchers: Not stated |
|
| Notes | Data on tiludronate groups were pooled for meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomly allocated into 1 of 3 treatment groups." Comment: Insufficient information provided. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study was a double‐blind, randomized, multicenter, parallel‐group comparison." "According to the randomisation, patients received 4 doses daily: 2 200 mg tiludronate tablets and 2 etidronate placebo capsules during the first 3 months followed by 2 tiludronate placebo tablets and 2 etidronate placebo capsules during the second 3 months, 2 200 mg tiludronate tablets and 2 etidronate placebo capsules for 6 months, or 2 200‐mg etidronate capsules and 2 tiludronate placebo tablets for 6 months." Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "All samples were analysed at a central laboratory (Cerba Laboratories, Cergy‐Pontoise, France)." Comment: Probably done for laboratory assessment. The information provided was insufficient to judge the blinding of outcome for clinical assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of the 234 patients enrolled, 30 (12.8%) discontinued treatment before completing the 6 months of the study: 14 in the tiludronate 3‐month group, 11 in the tiludronate 6‐month group, and 5 in the etidronate group." "Intent‐to‐treat analyses were conducted." "For dichotomous variables, cases with missing data were considered as treatment failures." Comment: Proportion of missing data were reported. Although the number of participants who discontinued treatment from both tiludronate groups was twice the number in the etidronate group, the proportion of missing outcomes compared with observed event risk was insufficient to have a relevant impact of the effect estimate |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | No other risks of bias found, but reporting was insufficient to permit judgement |
Siris 1996.
| Methods | RCT: Randomisation ratio: 1:1 (alendronate 40 mg: etidronate 400 mg). Superiority design |
|
| Participants |
Diagnostic criteria: Paget's disease of bone was confirmed by standard clinical, radiological or scintigraphic imaging or both methods Inclusion criteria: Serum alkaline phosphatase at least twice the ULN if the participant had never been treated with bisphosphonates or plicamycin, or at least 4 times the ULN if the participant had received such therapy at any time in the past. Exclusion criteria:
Number screened: Not stated. Number randomised: 89. Number analysed: 88 (69 years, 67% male, percentages of monostotic and symptomatic participants not stated, 25% previously treated for Paget's disease of bone) |
|
| Interventions | Two parallel treatment groups: alendronate 40 mg (1 x etidronate placebo tablet and 1 x 40 mg alendronate tablet for 6 months) and etidronate 400 mg (1 x 400 mg etidronate tablet and 1 x alendronate placebo tablet for 6 months). Co‐interventions: All participants received daily vitamin supplements containing 450 mg calcium carbonate and 400 IU vitamin D. Analgesics were available on demand and use was not balanced ("the regression analysis with adjustment for analgesics use at month 6 between the two treatment groups approached significance") |
|
| Outcomes |
Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: Baseline, 1 month, 3 months, 6 months. How were the outcomes measured: prospectively |
|
| Setting and date | 11 centres in USA. Period when the study was conducted: Not reported |
|
| Follow up period | 6 months | |
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article. Funding source: Not reported. Declarations of interest among primary researchers: Not reported |
|
| Notes | Transiliac bone biopsy was conducted for 43 participants at month 6. Bone biopsies were obtained from 25 healthy volunteers as control | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "the study was randomised". Comment: insufficient information provided to permit judgement. Allocation was probably centralized because this was a multicentre study |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study was randomised and double blind", "The etidronate tablets were purchased as Didronel, crushed, and recompressed into tablets identical to placebo for etidronate". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Baseline and 6 months radiographs of one or more sites were read by one radiologist, who remained blind with respect to both treatment allocation and sequence of films". "Each specimen (bone biopsy) was blinded, so that the reader was unaware of the subjects' drug therapy." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Only one participant was excluded from the analysis (in the alendronate group). Reason for exclusion was reported. The proportion of missing outcomes compared with observed event risk was insufficient to have a relevant impact of the effect estimate |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | There were differences between groups in co‐interventions because analgesics were taken on demand and use was not balanced. However this was thought unlikely to have resulted in bias. No other risks of bias found, but reporting was insufficient to permit judgement |
Tan 2017.
| Methods | Interventional extension study including participants who complete a previous RCT (Langston 2010). The bisphosphonate of first choice in the intensive treatment arm was different from the original trial. Randomisation ratio: 1:1 (symptomatic treatment: intensive treatment). Superiority design |
|
| Participants |
Diagnostic criteria: participants with Paget's disease of bone confirmed by plain radiology of at least one skeletal site according standard criteria from UK guidelines (Selby 2002) Inclusion criteria: Participants who completed the PRISM trial (Langston 2010). Exclusion criteria: No specific exclusion criteria were applied on the basis of treatment history, baseline alkaline phosphatase or co‐existing diseases. Number invited to participate: 2110 Number enrolled: 502 Number analysed: 404 (76 years, 54% male, 62% symptomatic, 91% previously treated with bisphosphonates for Paget's disease, 70% had normal alkaline phosphatase at baseline) |
|
| Interventions | Two parallel treatment groups; symptomatic vs. intensive treatment. Symptomatic treatment: Philosophy; treat bone pain, not alkaline phosphatase.
Intensive treatment: Philosophy; maintain normal alkaline phosphatase.
Co‐interventions: Analgesics and nonsteroidal anti‐inflammatory drugs |
|
| Outcomes |
Primary outcome: radiologically‐confirmed clinical fracture. Secondary outcomes:
Time points for measurement: Data on fractures, orthopaedic procedures and serious adverse events were collected on a continuous basis. Laboratory data, quality of life, bone pain and adverse events (based on participant diaries) data were measured annually. How were the outcomes measured: prospectively |
|
| Setting and date | 30 secondary referral centres in the UK. Period the study was conducted: January 2007 to January 2012 |
|
| Follow up period | 3 years | |
| Publication details and funding source |
Language of publication: English.
Publication status: Abstract. Funding source: The study was supported by grants from the Arthritis Research Campaign UK (Ref. 13627) and the National Association for Relief of Paget’s Disease. Declarations of interest among primary researchers:
|
|
| Notes | The study was described by the authors as a "pragmatic randomised controlled trial designed to compare the effects of two management strategies". The study was not blinded | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "At the end of PRISM, patients were asked if they wanted to continue in the study for a further three years". Comment: The participants included in this trial had participated in a previous trial (Langston 2010). The risk of bias assessment should be the same as for the previous trial. However, as only participants who voluntarily agreed to continue in the study were included, it is difficult to verify if the balance among the trial groups created in the original trial by randomisation was kept in this extension study. At baseline serum alkaline phosphatase levels were lower in the intensive versus symptomatic group reflecting the fact that these participants already had been subjected to intensive treatment |
| Allocation concealment (selection bias) | High risk | Comment: The participants included in this trial had participated in a previous trial (Langston 2010). The risk of bias assessment is the same as for the previous trial |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: The study was not blinded. The main outcome (fractures) was unlikely to be influenced by lack of blinding. The risk of bias was high for quality of life, adverse events or bone pain, but low for other secondary endpoints as orthopaedic procedures or alkaline phosphatase concentrations |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Patient‐reported fractures and orthopaedic procedures were validated against medical records and x‐ray reports at participating centres by assessors blinded to treatment allocation" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Although a similar proportion of participants were deceased, withdrew from the study, declined to participate or were lost to follow up in each group (41/232 (18%) in the symptomatic versus 57/270 (21%) in the intensive group) at 3 years time, the proportion of missing outcomes compared with observed event risk is enough to induce clinically relevant bias in intervention effect estimate (fracture). However, the assessment of attrition bias is not assessed as high but low as, according to authors, the study was an event driven trial, with fracture as the primary endpoint. Data on all fracture events were available even in subjects who had withdrawn from the study |
| Selective reporting (reporting bias) | Low risk | Study protocol available. There was a prespecified record of the studies outcomes and all of them were included in published manuscript |
| Other bias | Unclear risk | The study design permitted attending clinicians to choose between different drugs within a specified treatment strategy. Evidence that the attending clinicians adhered to this strategy was confirmed by the observed difference in alkaline phosphatase levels between groups |
Walsh 2004.
| Methods | RCT. Randomisation ratio: 1:1 (alendronate 40 mg: pamidronate 60 mg). Superiority design |
|
| Participants |
Diagnostic criteria: Paget's disease of bone was confirmed by the presence of typical lesions of Paget’s disease on isotope bone scanning and radiographs. Inclusion criteria: Serum alkaline phosphatase above the upper limit of laboratory reference range. Exclusion criteria:
Number screened: 139. Number randomised: 72. Number analysed: 72 (70 years, 58% male, % monostotic not stated, 94% symptomatic participants, 39% participants previously treated for Paget's disease of bone) |
|
| Interventions | Two parallel treatment groups: alendronate 40 mg daily in 3 months blocks and pamidronate four x 60 mg IV infusions a year (once every 3 months). Treatment was continued until biochemical remission was achieved or there were a no significant reduction on two consecutives measurements. Biochemical remission was defined as both, serum total alkaline phosphatase activity and urine deoxypyridinoline/creatinine ratio within the reference range. Co‐interventions: All participants with plasma baseline total alkaline phosphatase > 675 U/L were prescribed ergocalciferol 30,000 U weekly for 3 months and calcium carbonate 600 mg daily for 8 months to minimize bisphosphonate‐induced secondary hyperparathyroidism. Other participants were treated with calcium and vitamin D supplements at the treating clinician’s discretion |
|
| Outcomes |
Outcomes reported in abstract: Primary endpoint: Proportion of participants achieving biochemical remission (see above). Secondary endpoints: Numbers of participants; with change in bone pain (measured on VAS); with change on quality of life from baseline (assessed using the SF‐36 Australian version); who experienced severe side effects related to use of bisphosphonates; who withdrew due to adverse events, mean percentage change from baseline in serum total alkaline phosphatase activity; who normalised alkaline phosphatase level; and who relapsed due to recurrence of increased serum alkaline phosphatase level. Time points for measurement: Baseline, 3, 6, 9, 12, 18 and 24 months. How were the outcome measured: Prospectively |
|
| Setting and date | Three centres in Western Australia. Period when the study was conducted: From May 1997 to October 2001 |
|
| Follow up period | 1 year without protocol amendment (2 year with protocol amendment) | |
| Publication details and funding source |
Language of publication: English.
Publication status: peer‐reviewed journal; full article. Funding source: Although the authors described the trial as an "investigator‐initiated study", the study was supported by research grants from Merck, Sharp & Dohme (Australia), Novartis Pharmaceuticals Australia and the Arthritis Foundation of Western Australia. Declarations of interest among primary researchers: Not stated |
|
| Notes | The initial protocol was amended to cross participants over from pamidronate to alendronate treatment at 12 months. According to the author, it was apparent that some participants randomised to pamidronate were showing little or no biochemical response to treatment and for "ethical" reasons, they amended the protocol so that participants randomised to pamidronate who did not achieve biochemical remission at 12 months were crossed over to alendronate treatment for the second year of the study. We included data from the first year only in the meta‐analysis. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "To ensure that the two treatment groups were well matched, randomisation was stratified (using an in‐house computer program) by two variables: baseline plasma alkaline phosphatase (in three strata: 136–270, 271–675, > 675 U/L) and previous bisphosphonate treatment (as a binary variable: yes or no)." "In the course of the study, it was apparent that some patients randomised to pamidronate (predominantly in the previously treated subgroup) were showing little or no biochemical response to treatment. For ethical reasons, the protocol was amended so that patients randomised to pamidronate who did not achieve biochemical remission at 12 months were crossed over to alendronate treatment for the second year of the study". Comment: The risk assessment was judged as low risk for first year data. For the second year, allocation was broken, and the risk of bias was assessed as high |
| Allocation concealment (selection bias) | Low risk | Authors describe using a computer program to generate the allocation |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Proportions of patients achieving remission for each treatment (on an intention to treat basis) were compared by Fisher’s exact test". Comment: Data on screened participants, randomised participants and withdrawals (with reasons) are reported in a flow chart (Figure 1) |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | Likely high at 12 months due to amendment of the initial protocol. No other risks of bias found, but reporting was insufficient to permit judgement |
Abbreviations: IV ‐ intravenous; RCT ‐ randomised controlled trial; ULN ‐ upper limit of normal
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adami 1994 | Not a randomised clinical study. Study without a comparison of interest. The study compared alendronate (20 mg or 40 mg daily) for 3 to 6 months |
| Adami 2002 | Randomised clinical study. Study without a comparison of interest. The study compared three doses of neridronate (12.5 mg, 25 mg, 50 mg or 100 mg daily for two consecutive days) |
| Altman 1985 | Not a randomised clinical study. Study without a comparison of interest. The study describes the outcomes in a series of participants treated with different regimes of oral Etidronate (5 mg/kg daily for 6 months, 10 mg/kg daily for 3 months, or 20 mg/kg daily for 3 moths). |
| Arlot 1981 | Not a randomised clinical study. Study without an outcome of interest. The study was designed to analyse the effect of different doses of Etidronate (5 mg/kg or 20 mg/kg for 6 months) or clodronate (400 mg/day or 1600 mg/day for 6 months) over the serum acid level. |
| Atkins 1987 | Not a randomised clinical study. Study without a comparison of interest. The study compares three doses of the Neridronate (400 mg oral for one month, 25 mg daily infusion for 5 days and 50 mg daily infusion for 5 days). |
| Buckler 1998 | Randomised clinical study. Study without a comparison of interest. The study compared two regimes of administration of pamidronate (initial 30 mg infusion followed by three infusion of 60 mg and a final placebo infusion at fortnightly intervals or initial placebo infusion followed by three infusion of 60 mg and a final 30 mg infusion at fortnightly intervals), with the same total dose (210 mg). |
| Cundy 2016 | Not a randomised clinical study. Study without a comparison of interest. The study shows data from a cohort of 107 elderly participants (mean age 76 years) treated with intravenous zoledronate followed‐up for 10 years. |
| Delmas 1982 | Randomised clinical study. Study without a comparison of interest. The study compared four doses of clodronate (400, 800, 1600 or 2400 mg daily), with (the four doses) or without (only 400 and 1600 mg doses) vitamin D and calcium supplementation (elemental calcium 1g/day and vitamin D2 8000 IU/day). |
| Devogelaer 1997 | Data on a sample of participants diagnosed with Paget's disease of bone from a randomised clinical trial (Roux 1995). The study was designed to analyse radiological changes during treatment with bisphosphonates. |
| Devogelaer 2014 | Not a randomised clinical study. Study without a comparison of interest. Data from the Belgian Paget's Disease Registry of patients diagnosed as Paget's disease of bone and treated with a 5 mg intravenous infusion of zoledronate. |
| Dewis 1985 | Not a randomised clinical study. Study with a comparison of interest. "Open trial" (9 participants were randomised and 8 participants were put directly into one of the two treatment groups) comparing the effectiveness of two bisphosphonates (etidronate 20 mg/kg once daily for 3 months vs. pamidronate 4 to 5 mg/kg twice daily for 3 months). |
| Donáth 2004 | Case‐control study. Study without a comparison of interest. Cases were patients diagnosed as Paget's disease of bone with temporal bone involvement. Control were healthy individuals matched for age and sex. The study was designed to analyse the effectiveness of two bisphosphonates (pamidronate 30 mg daily infusion for 6 days or oral tiludronate 400 mg daily for 3 months). The study included audiometric assessment and hearing threshold examination. |
| Filipponi 1994 | Not a randomised clinical study. Study with a comparison of interest. The study compares the effectiveness of two bisphosphonates (Clodronate 300 mg/daily intravenous infusion for 5 consecutive days vs. Alendronate 5 mg/ daily intravenous infusion for 5 consecutive days). |
| Gallacher 1991 | Not a randomised clinical study. Study without a comparison of interest. The study compares three regimes of administration of pamidronate, two with a total dose of 180 mg (30 mg weekly infusions for 6 weeks or 45 mg infusions every 3 months for one year) and one with a total dose of 360 mg (30 mg weekly infusions for 6 weeks and 60 mg weekly infusions for three additional weeks). A random subgroup of 6 participants were given placebo infusions of 0.9% saline weekly for three weeks at the start of treatment in a single‐blind study in the group of 30 mg weekly doses for 6 weeks (data are not shown on manuscript). |
| Garnero 1998 | Data on a sample of participants diagnosed as Paget's disease of bone from a randomised clinical trial, and a case‐control study. For the case‐control study, cases were selected from three of five groups of a clinical trial on Paget's disease of bone (Schaffer 1996). Controls were healthy individuals matched for age and sex. The clinical trial included the cases and was designed to analyse the effect of zoledronate on bone turnover markers. |
| Garnero 2001 | Data on a sample of participants diagnosed with Paget's disease of bone from a randomised clinical trial and a case‐control study (selected from a clinical trial on Paget's disease of bone, Schaffer 1996). Controls were healthy age‐matched individuals. The clinical trial included the cases and was designed to analyse the effect of zoledronate on bone turnover markers |
| Goldman 1975 | Data from a sample of participants diagnosed with Paget's disease of bone from a randomised clinical trial (Altman 1973). The study was designed to assess the changes in radionuclide uptake after bisphosphonate treatment |
| Grauer 1999 | Not a randomised clinical study. Study without a comparison of interest. The study compared three different doses of ibandronate (2 mg, 4 mg or 6 mg intravenous infusions) for re‐treatment of participants previously treated with ibandronate after biochemical relapse (increase of alkaline phosphatase). |
| Gutteridge 1996 | Not a randomised clinical study. Study without a comparison of interest. The study compared three different doses of pamidronate. Participation allocation had two steps: three groups according to index of bone resorption (urinary hydroxyproline x plasma creatinine/urinary creatinine). The higher the index (< 5, 5 to 9.99, ≥ 10), the higher the pamidronate dose (120 mg, 180 mg, 240 mg). The three groups were then randomised to two subgroups; total dose in 30 mg infusions or total dose in 60 mg infusions |
| Hooper 2009 | Randomised clinical study. Study without a comparison of interest. The study compared administration of alendronate 280 mg weekly vs. 40 mg daily for 6 months |
| Hosking 1976 | Not a randomised clinical study. Study without a comparison of interest. The study compared administration of etidronate (7.5 mg/kg or 15 mg/kg daily and etidronate plus calcitonin simultaneously (etidronate 7.5 mg/kg daily together with calcitonin 0.5 mg once daily subcutaneous for 6 months) or sequentially (etidronate 15 mg/kg daily for 6 months followed by calcitonin 0.5 mg twice daily subcutaneous for 6 months) |
| Khairi 1974 | Extension study (Altman 1973) with a non‐randomised crossover design. The groups from the original study (placebo and etidronate doses of 1 mg/kg, 2.5 mg/kg, 5 mg/kg, 10 mg/kg, 20 mg/kg) were reassigned to different doses of etidronate for six additional months. Participants from placebo and 1 mg/kg and 2.5 mg/kg groups were reassigned to etidronate on doses of 5 mg/kg, 10 mg/kg or 20 mg/kg. Participants from the 5 mg/kg group were kept on the same dose. Participants from the 10 mg/kg or 20 mg/kg groups were reassigned to placebo or to remain on the same etidronate dose |
| Khairi 1977 | Not a randomised clinical study. Study without a comparison of interest. Data from a cohort of participants diagnosed with Paget's disease of bone who were treated with etidronate; 60/116 participants took part in a previous study (Khairi 1974) |
| Khan 1997 | Randomised clinical study. Study without a comparison of interest. The study compared different doses (40 or 80 mg/daily) and regimens (3 or 6 months) of oral alendronate |
| Lombardi 1999 | Review. Compilation of the results of two RCTs on alendronate for Paget's disease of bone (Reid 1996 and Siris 1996). |
| Mazeries 1996 | Not a randomised clinical study. Study without a comparison of interest. The study compared two regimens of pamidronate infusion (1 infusion of 60 mg vs. 2 infusions of 60 mg over 24 hours) |
| Merlotti 2011 | Randomised clinical study. Study without a comparison of interest. The study compared two regimens of neridronate (100 mg intravenous infusion for 2 consecutive days vs. 25 mg intramuscular infusion once a week for 2 months) |
| O'Doherty 1995 | Not a randomised clinical study. Study without a comparison of interest. The study compared three doses of intravenous alendronate (2.5 mg, 5 mg and 10 mg daily) for 5 consecutive days |
| Pepersack 1994 | Not a randomised clinical study. Study without a comparison of interest. The study compared one oral and four intravenous regimens of pamidronate (600 mg daily for 6 months, 40 mg daily for five days, 20 mg daily for 10 days, 10 mg daily for 4 days and a single dose of 10 mg) |
| Reginster 1988 | Randomised clinical study. Study without a comparison of interest. The study compared three regimens of tiludronate (200 mg daily for 6 months, 400 mg daily for 6 months, 200 mg daily for 3 months and 400 mg daily for 3 additional months) |
| Reginster 1993 | Randomised clinical study. Study without a comparison of interest. The study compared three regimens of tiludronate (600 mg, 800 mg and 1200 mg daily) for five days |
| Russell 1974 | Not a randomised clinical study. Study without a comparison of interest. The study compared four doses of etidronate (1 mg, 5 mg, 10 mg and 20 mg) for 6 months and placebo |
| Siris 1980 | Extension study (of Canfield 1977). Only participants from the Columbia Presbyterian Medical Center (1 of the 4 hospitals in the original trial) were included in the study. All participants were treated with etidronate |
| Stone 1990 | Not a randomised clinical study. Study without a comparison of interest. The study compared three regimens of pamidronate (15 mg, 30 mg, or 45 mg infusions) at 6 week intervals |
| Vega 1994 | Randomised clinical study. Study without a comparison of interest. The study compared seven regimens of pamidronate in two formulations (3 dose levels of oral capsules of pamidronate; 300 mg, 600 mg and 900 mg and four dose levels of oral tables of dimethyl pamidronate; 50 mg, 100 mg, 200 mg and 400 mg). Each dose was administered for 15 days |
Characteristics of ongoing studies [ordered by study ID]
ISRCTN11616770.
| Trial name or title | Zoledronate in the Prevention of Paget's: the ZiPP study |
| Methods | Multisite double blind placebo controlled randomised trial. Two sub‐studies:
Follow up: 5 years. |
| Participants |
Participant inclusion criteria Interventional study: Relatives of patients with SQSTM1 mutations, aged 30 years old or greater, who carry SQSTM1 mutations and who have not already diagnosed with PDB at study entry Observational study: Relatives of patients with SQSTM1 mutations, aged between 30 years old or greater who on screening are found NOT to have SQSTM1 mutations Countries of recruitment: Australia, Belgium, Ireland, Italy, Spain, United Kingdom. |
| Interventions | Interventional study: Participants will be randomised to either infusions of zoledronate (Aclasta®) 5 mg by intravenous infusion over 15 minutes or placebo (0.9% saline) at baseline. |
| Outcomes |
Interventional study: Primary outcome: Total number of subjects who develop new bone lesions between the baseline visit and the final follow up visit. Observational study: Primary outcome: Anxiety/depression, measured using the HADS scale. Both studies: Secondary outcomes: 1. Development of elevated bone turnover, as measured by alkaline phosphatase and other biochemical markers of bone turnover. 2. Quality of life, and anxiety and depression assessed by the SF‐36, BPI and HADS questionnaires. |
| Starting date | 12/01/2009 |
| Contact information | Mr Adam Wilson Clinical Trials Manager e‐mail: zipptri1@exseed.ed.ac.uk |
| Ending date | 31/01/2020 |
| Target number of participants |
Intervention study: 188 participants Observational study: 125 participants |
| Identifier | ISRCTN11616770 DOI 10.1186/ISRCTN11616770 |
| Notes | Sponsor information: University of Edinburgh (UK) Funder name: Medical Research Council (MRC) (UK) (ref: G0701625; 85281) |
NCT02106455.
| Trial name or title | Takeda Study Registration Call Center, post marketing Group Manager |
| Methods | Prospective cohort |
| Participants |
Inclusion criteria: participants diagnosed with Paget's disease of bone treated with sodium risedronate tablets Exclusion criteria: No exclusions by age or gender Countries of recruitment: Japan |
| Interventions | Sodium risedronate tablets (Benet 17.5 mg) administered orally with a sufficient volume (approximately 180 mL) of water once daily after waking for 8 consecutive weeks. |
| Outcomes | Primary outcome:
Secondary outcomes:
|
| Starting date | September 2008 |
| Contact information | Telephone: 1‐800‐778‐2860 (USA & EU), email: medicalinformation@tpna.com |
| Ending date | Estimated study completion date: July 2017. |
| Target number of participants | 2500 |
| Identifier | ClinicalTrials.gov Identifier: NCT02106455 |
| Notes | Sponsor information: Takeda |
Differences between protocol and review
There are nine differences between protocol and review:
The objective in the original protocol: "To assess the benefits to improve clinical outcomes or prevent complications and the harms of bisphosphonate therapy on patients with Paget's disease of bone in adults", was rewritten as "To assess the benefits and harms of bisphosphonates for adult patients with Paget's disease of bone" according to Cochrane Musculoskeletal Group recommendation.
Comparison between bisphosphonates was subdivided. In addition, two comparisons were added: comparison of two non‐aminobisphosphonates and bisphosphonates versus bisphosphonates plus calcitonin.
A minor outcome (mean reduction in serum total alkaline phosphatase activity) was renamed: mean percentage change from baseline in serum total alkaline phosphatase activity. A sub outcome: number of participants who achieved normalised alkaline phosphatase level, was added.
For trials where there were multiple arms with several doses of the same bisphosphonates, we combined the experimental intervention groups to create a single pair‐wise comparison versus the control group. Experimental group data were combined as a single group, instead of the planned high dose and low dose groups.
We estimated overall effect by performing meta‐analyses using a random‐effect model in all cases. The fixed‐effect model was not performed when I² < 40% as planned.
Some data were extracted directly from figures. (See last paragraph in Data extraction and management).
We added a specific search for specific rare events found from searches of websites of four regulatory agencies.
We added a new co‐author in March 2017. JdPM was recruited to assess results from the most recent search and analyse the PRISM‐EZ trial (Tan 2017).
We considered one year of follow‐up to properly assess the outcome "Number of participants experiencing radiologically‐confirmed clinical fractures" that does not have a predefined follow‐up period in the protocol.
Contributions of authors
Draft the protocol: LCG, SHR Study selection: LCG, AT, JdPM Extract data from studies: LCG, AT, JdPM Enter data into Review Manager 2014: LCG Carry out the analysis: LCG Interpret the analysis: LCG, AT, SHR, JdPM Draft the final review: LCG, AT, SHR, JdPM Disagreement resolution: SHR Update the review: LCG, AT, SHR, JdPM
Sources of support
Internal sources
No sources of support supplied
External sources
-
Arthritis Research, UK.
The review was supported in part by a grant from Arthritis Research UK to SHR (18304)
-
Instituto de Salud Carlos III, Spain.
The review was supported in part by a grant from Carlos III
Declarations of interest
Luis Corral‐Gudino: none known.
Adrian JH Tan: was co‐investigator on the PRISM‐EZ study (Tan 2017).
Javier del Pino‐Montes: was co‐investigator on zoledronate versus risedronate trial (Reid 2005). He has received grants from the research Spanish agencies SACYL and Institute of Health Carlos III and payment for lectures and meeting expenses from Merck Sharp Dohme.
Stuart H Ralston: previously acted as a consultant for Novartis and Merck on behalf of his institution (the University of Edinburgh) and was the principal investigator on three included primary studies (Langston 2010; Ralston 1987; Tan 2017).
Edited (no change to conclusions)
References
References to studies included in this review
Altman 1973 {published data only}
- Altman RD, Johnston CC, Khairi MR, Wellman H, Serafini AN, Sankey RR. Influence of disodium etidronate on clinical and laboratory manifestations of Paget's disease of bone (osteitis deformans). New England Journal of Medicine 1973;289(26):1379‐84. [DOI] [PubMed] [Google Scholar]
Barreira 2009 {published data only}
- Barreira JC, Man Z, Acotto CG, Roldan E. Double blind, double dummy, randomized controlled trial of oral bisphosphonates (OB) in the treatment of active paget's bone disease (PBD). Journal of Bone and Mineral Research 2011;26(Suppl 1):SU0164. [Google Scholar]
- Barreira JC, Man Z, Plantalech L, Gomez Acotto C, Roldán EJA. Double blind, double dummy, randomized controlled trial of oral bisphosphonates (OB) in the treatment of active Paget's bone disease (PBD). Osteoporosis International 2009;20(1):179‐80. [Google Scholar]
Buckler 1999 {published data only (unpublished sought but not used)}
- Buckler H, Fraser W, Hosking D, Ryan W, Maricic MJ, Singer F, et al. Single infusion of zoledronate in Paget's disease of bone: a placebo‐controlled, dose‐ranging study. Bone 1999;24(Suppl 5):81‐5S. [DOI] [PubMed] [Google Scholar]
- Schaffer AV, Buckler H, Ryan W, Richardson P. A double blind placebo‐controlled trial using intravenous zoledronate (CGP 42446) in patients with Paget's disease of bone. Journal of Bone and Mineral Research 1996;11(Suppl 1):S373. [Google Scholar]
Canfield 1977 {published data only}
- Canfield R, Rosner W, Skinner J, McWhorter J, Resnick L, Feldman F, et al. Diphosphonate therapy of Paget's disease of bone. Journal of Clinical Endocrinology and Metabolism 1977;44(1):96‐106. [DOI] [PubMed] [Google Scholar]
Fraser 1997 {published data only (unpublished sought but not used)}
- Fraser WD, Stamp TC, Creek RA, Sawyer JP, Picot C. A double‐blind, multicentre, placebo‐controlled study of tiludronate in Paget's disease of bone. Postgraduate Medical Journal 1997;73(862):496‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Langston 2010 {published data only}
- Langston AL, Campbell MK, Fraser WD, MacLennan GS, Selby PL, Ralston SH. PRISM Trial Group. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget's disease of bone. Journal of Bone and Mineral Research 2010;25(1):20‐31. [DOI] [PubMed] [Google Scholar]
McClung 1995 {published data only}
- McClung MR, Tou CKP, Goldstein NH, Picot C. Tiludronate therapy for Paget's disease of bone. Bone 1995;17(5 Suppl):493‐6S. [DOI] [PubMed] [Google Scholar]
Merlotti 2007 {published data only (unpublished sought but not used)}
- Merlotti D, Gennari L, Martini G, Valleggi F, Paola V, Avanzati A, et al. Comparison of different intravenous bisphosphonate regimens for Paget's disease of bone. Journal of Bone and Mineral Research 2007;22(10):1510‐7. [DOI] [PubMed] [Google Scholar]
Miller 1999 {published data only (unpublished sought but not used)}
- Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ. A randomized, double‐blind comparison of risedronate and etidronate in the treatment of Paget's disease of bone. Paget's Risedronate/Etidronate Study Group. American Journal of Medicine 1999;106(5):513‐20. [DOI] [PubMed] [Google Scholar]
O'Doherty 1992 {published data only}
- O'Doherty DP, Gertz BJ, Tindale W, Sciberras DG, Survill TT, Kanis JA. Effects of five daily 1 h infusions of alendronate in Paget's disease of bone. Journal of Bone and Mineral Research 1992;7(1):81‐7. [DOI] [PubMed] [Google Scholar]
O'Donoghue 1987 {published data only}
- O'Donoghue DJ, Hosking DJ. Biochemical response to combination of disodium etidronate with calcitonin in Paget’s disease. Bone 1987;8(4):219‐25. [DOI] [PubMed] [Google Scholar]
Ralston 1987 {published and unpublished data}
- Ralston SH, Boyce BF, Cowan RA, Fogelman I, Smith ML, Jenkins A, et al. The effect of 1 alpha‐hydroxyvitamin D3 on the mineralization defect in disodium etidronate‐treated Paget's disease ‐ a double‐blind randomized clinical study. Journal of Bone and Mineral Research 1987;2(1):5‐12. [DOI] [PubMed] [Google Scholar]
Reginster 1992 {published data only (unpublished sought but not used)}
- Reginster JY, Colson F, Morlock G, Combe B, Ethgen D, Geusens P. Evaluation of the efficacy and safety of oral tiludronate in Paget's disease of bone. A double‐blind, multiple‐dosage, placebo‐controlled study. Arthritis and Rheumatism 1992;35(8):967‐74. [DOI] [PubMed] [Google Scholar]
Reid 1996 {published data only (unpublished sought but not used)}
- Reid IR, Nicholson GC, Weinstein RS, Hosking DJ, Cundy T, Kotowicz MA, et al. Biochemical and radiologic improvement in Paget's disease of bone treated with alendronate: a randomized, placebo‐controlled trial. American Journal of Medicine 1996;101(4):341‐8. [DOI] [PubMed] [Google Scholar]
Reid 2004 {published and unpublished data}
- Reid IR, Davidson JS, Wattie D, Wu F, Lucas J, Gamble GD, et al. Comparative responses of bone turnover markers to bisphosphonate therapy in Paget's disease of bone. Bone 2004;35(1):224‐30. [DOI] [PubMed] [Google Scholar]
- Reid IR, Wattie D, Gamble GD, Kalluru R, Cundy T. Long‐term effects of intravenous ibandronate in Paget’s disease of bone. Calcified Tissue International 2017;100(3):250‐4. [DOI: 10.1007/s00223-016-0214-7] [DOI] [PubMed] [Google Scholar]
Reid 2005 {published and unpublished data}
- Hosking D, Lyles K, Brown JP, Fraser WD, Miller P, Curiel MD, et al. Long‐term control of bone turnover in Paget's disease with zoledronic acid and risedronate. Journal of Bone and Mineral Research 2007;22(1):142‐8. [DOI] [PubMed] [Google Scholar]
- Reid IR, Lyles K, Su G, Brown JP, Walsh JP, Pino‐Montes J, et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. Journal of Bone and Mineral Research 2011;26(9):2261‐70. [DOI: 10.1002/jbmr.438] [DOI] [PubMed] [Google Scholar]
- Reid IR, Miller P, Lyles K, Fraser W, Brown JP, Saidi Y, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget's disease. New England Journal of Medicine 2005;353(9):898‐908. [DOI] [PubMed] [Google Scholar]
Roux 1995 {published data only (unpublished sought but not used)}
- Roux C, Gennari C, Farrerons J, Devogelaer JP, Mulder H, Kruse HP, et al. Comparative prospective, double‐blind, multicenter study of the efficacy of tiludronate and etidronate in the treatment of Paget's disease of bone. Arthritis and Rheumatism 1995;38(6):851‐8. [DOI] [PubMed] [Google Scholar]
Siris 1996 {published data only (unpublished sought but not used)}
- Siris E, Weinstein RS, Altman R, Conte JM, Favus M, Lombardi A, et al. Comparative study of alendronate versus etidronate for the treatment of Paget's disease of bone. Journal of Clinical Endocrinology and Metabolism 1996;81(3):961‐7. [DOI] [PubMed] [Google Scholar]
Tan 2017 {published and unpublished data}
- Tan A, Goodman K, Walker A, Hudson J, MacLennan GS, Selby PL, et al. PRISM‐EZ Trial Group. Long‐term randomized trial of intensive versus symptomatic management in Paget's disease of bone: The PRISM‐EZ Study. Journal of Bone and Mineral Research 2017;32(6):1165‐73. [DOI: 10.1002/jbmr.3066] [DOI] [PubMed] [Google Scholar]
Walsh 2004 {published data only (unpublished sought but not used)}
- Walsh JP, Ward LC, Stewart GO, Will RK, Criddle RA, Prince RL, et al. A randomized clinical trial comparing oral alendronate and intravenous pamidronate for the treatment of Paget's disease of bone. Bone 2004;34(4):747‐54. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Adami 1994 {published data only}
- Adami S, Mian M, Gatti P, Rossini M, Zamberlan N, Bertoldo F, et al. Effects of two oral doses of alendronate in the treatment of Paget's disease of bone. Bone 1994;15(4):415‐7. [DOI] [PubMed] [Google Scholar]
Adami 2002 {published data only}
- Adami S, Bevilacqua M, Broggini M, Filipponi P, Ortolani S, Palummeri E, et al. Short‐term intravenous therapy with neridronate in Paget's disease. Clinical and Experimental Rheumatology 2002;20(1):55‐8. [PubMed] [Google Scholar]
Altman 1985 {published data only}
- Altman RD. Long‐term follow‐up of therapy with intermittent etidronate disodium in Paget's disease of bone. American Journal of Medicine 1985;79(5):583‐90. [DOI] [PubMed] [Google Scholar]
Arlot 1981 {published data only}
- Arlot ME, Meunier PJ. Effects of two diphosphonates (EHDP and Cl2MDP) on serum uric acid in pagetic patients. Calcified Tissue International 1981;33(3):195‐8. [DOI] [PubMed] [Google Scholar]
Atkins 1987 {published data only}
- Atkins RM, Yates AJ, Gray RE, Urwin GH, Hamdy NA, Beneton MN, et al. Aminohexane diphosphonate in the treatment of Paget's disease of bone. Journal of Bone and Mineral Research 1987;2(4):273‐9. [DOI] [PubMed] [Google Scholar]
Buckler 1998 {published data only}
- Buckler HM, Mercer SJ, Davison CE, Hollis S, Richardson PC, Anderson DG. Evaluation of adverse experiences related to pamidronate infusion in Paget's disease of bone. Annals of the Rheumatic Diseases 1998;57(9):572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cundy 2016 {published data only}
- Cundy T, Maslowski K, Grey A, Reid IR. Durability of response to zoledronate treatment and competing mortality in Paget’s disease of bone. Journal of Bone and Mineral Research 2016;32(4):753‐6. [DOI] [PubMed] [Google Scholar]
Delmas 1982 {published data only}
- Delmas PD, Chapuy MC, Vignon E, Charhon S, Briancon D, Alexandre C, et al. Long term effects of dichloromethylene diphosphonate in Paget's disease of bone. Journal of Clinical Endocrinology and Metabolism 1982;54(4):837‐44. [DOI] [PubMed] [Google Scholar]
Devogelaer 1997 {published data only}
- Devogelaer JP, Malghem J, Stasse P, Nagant de Deuxchaisnes C. Biological and radiological responses to oral etidronate and tiludronate in Paget's disease of bone. Bone 1997;20(3):259‐61. [DOI] [PubMed] [Google Scholar]
Devogelaer 2014 {published data only}
- Devogelaer JP, Geusens P, Daci E, Gielen E, Denhaerynck K, Macdonald K, et al. Remission over 3 years in patients with Paget disease of bone treated with a single intravenous infusion of 5 mg zoledronic acid. Calcified Tissue International 2014;94(3):311‐8. [DOI] [PubMed] [Google Scholar]
Dewis 1985 {published data only}
- Dewis P, Prasad BK, Anderson DC, Willets S. Clinical experience with the use of two diphosphonates in the treatment of Paget's disease. Annals of the Rheumatic Diseases 1985;44(1):34‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Donáth 2004 {published data only}
- Donáth J, Krasznai M, Fornet B, Gergely P Jr, Poór G. Effect of bisphosphonate treatment in patients with Paget's disease of the skull. Rheumatology (Oxford, England) 2004;43(1):89‐94. [DOI] [PubMed] [Google Scholar]
Filipponi 1994 {published data only}
- Filipponi P, Pedetti M, Beghe F, Giovagnini B, Miam M, Cristallini S. Effects of two different bisphosphonates on Paget's disease of bone: ICTP assessed. Bone 1994;15(3):261‐7. [DOI] [PubMed] [Google Scholar]
Gallacher 1991 {published data only}
- Gallacher SJ, Boyce BF, Patel U, Jenkins A, Ralston SH, Boyle IT. Clinical experience with pamidronate in the treatment of Paget's disease of bone. Annals of the Rheumatic Diseases 1991;50(12):930‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Garnero 1998 {published data only}
- Garnero P, Gineyts E, Schaffer AV, Seaman J, Delmas PD. Measurement of urinary excretion of nonisomerized and beta‐isomerized forms of type I collagen breakdown products to monitor the effects of the bisphosphonate zoledronate in Paget's disease. Arthritis and Rheumatism 1998;41(2):354‐60. [DOI] [PubMed] [Google Scholar]
Garnero 2001 {published data only}
- Garnero P, Christgau S, Delmas PD. The bisphosphonate zoledronate decreases type II collagen breakdown in patients with Paget's disease of bone. Bone 2001;28(5):461‐4. [DOI] [PubMed] [Google Scholar]
Goldman 1975 {published data only}
- Goldman AB, Braunstein P, Wilkinson D, Kammerman S. Radionuclide uptake studies of bone: a quantitative method of evaluating the response of patients with Paget's disease to diphosphonate therapy. Radiology 1975;117(2):365‐9. [DOI] [PubMed] [Google Scholar]
Grauer 1999 {published data only}
- Grauer A, Heichel S, Knaus J, Dosch E, Ziegler R. Ibandronate treatment in Paget's disease of bone. Bone 1999;24(5 Suppl):87‐9S. [DOI] [PubMed] [Google Scholar]
Gutteridge 1996 {published data only}
- Gutteridge DH, Retallack RW, Ward LC, Stuckey BGA, Stewart GO, Prince RL, et al. Clinical, biochemical, hematologic, and radiographic responses in Paget's disease following intravenous pamidronate disodium: a 2‐year study. Bone 1996;19(4):387‐94. [DOI] [PubMed] [Google Scholar]
Hooper 2009 {published data only}
- Hooper M, Faustino A, Reid IR, Hosking D, Gilchrist NL, Selby P, et al. Randomized, active‐controlled study of once‐weekly alendronate 280 mg high dose oral buffered solution for treatment of Paget's disease. Osteoporosis International 2009;20(1):141‐50. [DOI] [PubMed] [Google Scholar]
Hosking 1976 {published data only}
- Hosking DJ, Bijvoet OL. Combined diphosphonate and calcitonin therapy for Paget's disease of bone. Calcified Tissue Research 1976;21(Suppl):321‐6. [PubMed] [Google Scholar]
Khairi 1974 {published data only}
- Khairi MR, Johnston CC Jr, Altman RD, Wellman HN, Serafini AN, Sankey RR. Treatment of Paget disease of bone (osteitis deformans). Results of a one‐year study with sodium etidronate. JAMA 1974;230(4):562‐7. [PubMed] [Google Scholar]
Khairi 1977 {published data only}
- Khairi MR, Altman RD, DeRosa GP, Zimmermann J, Schenk RK, Johnston CC. Sodium etidronate in the treatment of Paget's disease of bone. A study of long‐term results. Annals of Internal Medicine 1997;87(6):656‐63. [DOI] [PubMed] [Google Scholar]
Khan 1997 {published data only}
- Khan SA, Vasikaran S, McCloskey EV, Benéton MN, Rogers S, Coulton L, et al. Alendronate in the treatment of Paget's disease of bone. Bone 1997;20(3):263‐71. [DOI] [PubMed] [Google Scholar]
Lombardi 1999 {published data only}
- Lombardi A. Treatment of Paget's disease of bone with alendronate. Bone 1999;24(5 Suppl):59‐61S. [DOI] [PubMed] [Google Scholar]
Mazeries 1996 {published data only}
- Mazieres B, Ahmed I, Moulinier L, Bon E, Cantagrel A, Laroche M. Pamidronate infusions for the treatment of Paget's disease of bone. Review du Rhumatisme (English Edition) 1996;63(1):36‐43. [PubMed] [Google Scholar]
Merlotti 2011 {published data only}
- Merlotti D, Rendina D, Gennari L, Mossetti G, Gianfrancesco F, Martini G, et al. Comparison of intravenous and intramuscular neridronate regimens for the treatment of Paget disease of bone. Journal of Bone and Mineral Research 2011;26(3):512‐8. [DOI: 10.1002/jbmr.237] [DOI] [PubMed] [Google Scholar]
O'Doherty 1995 {published data only}
- O'Doherty DP, McCloskey EV, Vasikaran S, Khan S, Kanis JA. The effects of intravenous alendronate in Paget's disease of bone. Journal of Bone and Mineral Research 1995;10(7):1094‐100. [DOI] [PubMed] [Google Scholar]
Pepersack 1994 {published data only}
- Pepersack T, Karmali R, Gillet C, François D, Fuss M. Paget's disease of bone: five regimens of pamidronate treatment. Clinical Rheumatology 1994;13(1):39‐44. [DOI] [PubMed] [Google Scholar]
Reginster 1988 {published data only}
- Reginster JY, Jeugmans‐Huynen AM, Albert A, Denis D, Deroisy R, Lecart MP, et al. Biological and clinical assessment of a new bisphosphonate, (chloro‐4 phenyl) thiomethylene bisphosphonate, in the treatment of Paget's disease of bone. Bone 1988;9(6):349‐54. [DOI] [PubMed] [Google Scholar]
Reginster 1993 {published data only}
- Reginster JY, Lecart MP, Deroisy R, Ethgen D, Zegels B, Franchimont P. Paget's disease of bone treated with a five day course of oral tiludronate. Annals of the Rheumatic Diseases 1993;52(1):54‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Russell 1974 {published data only}
- Russell RG, Smith R, Preston C, Walton RJ, Woods CG. Diphosphonates in Paget's disease. Lancet 1974;1(7863):894‐8. [DOI] [PubMed] [Google Scholar]
Siris 1980 {published data only}
- Siris ES, Canfield RE, Jacobs TP, Baquiran DC. Long‐term therapy of Paget's disease of bone with EHDP. Arthritis and Rheumatism 1980;23(10):1177‐84. [DOI] [PubMed] [Google Scholar]
Stone 1990 {published data only}
- Stone MD, Hawthorne AB, Kerr D, Webster G, Hosking DJ. Treatment of Paget's disease with intermittent low‐dose infusions of disodium pamidronate (APD). Journal of Bone and Mineral Research 1990;5(12):1231‐5. [DOI] [PubMed] [Google Scholar]
Vega 1994 {published data only}
- Vega E, Mautalen C, Roldán EJ, Pérez Lloret A. Preliminary study of multiple increasing oral doses of dimethyl‐APD on bone metabolism dynamics and safety profile. Drugs under Experimental and Clinical Research 1994;20(3):103‐8. [PubMed] [Google Scholar]
References to ongoing studies
ISRCTN11616770 {published data only}
- ISRCTN11616770. Zoledronate in the Prevention of Paget's: the ZiPP study. isrctn.com/show/ISRCTN11616770 (first received 16 September 2008). [DOI 10.1186/ISRCTN11616770]
NCT02106455 {published data only}
- NCT02106455. Sodium Risedronate Tablets ‐ Special Drug Use Surveillance in Patients With Osseous Paget's Disease (All‐case Surveillance) ‐48‐week Surveillance −. clinicaltrials.gov/show/NCT02106455 (first received 3 April 2014).
Additional references
Cohen 2011
- Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, et al. Prevention of HIV‐1 infection with early retroviral therapy. New England Journal of Medicine 2011;365(6):493‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Corral‐Gudino 2013
- Corral‐Gudino L, Borao‐Cengotita‐Bengoa M, Pino‐Montes J, Ralston S. Epidemiology of Paget's disease of bone: a systematic review and meta‐analysis of secular changes. Bone 2013;55(2):347‐52. [DOI] [PubMed] [Google Scholar]
Devogelaer 2008
- Devogelaer JP, Bergmann P, Body JJ, Boutsen Y, Goemaere S, Kaufman JM, et al. Belgian Bone Club. Management of patients with Paget's disease: a consensus document of the Belgian Bone Club. Osteoporosis International 2008;19(8):1109‐17. [DOI: 10.1007/s00198-008-0629-8] [DOI] [PubMed] [Google Scholar]
Frith 2001
- Frith JC, Mönkkönen J, Auriola S, Mönkkönen H, Rogers MJ. The molecular mechanism of action of the antiresorptive and anti‐inflammatory drug clodronate: evidence for the formation in vivo of a metabolite that inhibits bone resorption and causes osteoclast and macrophage apoptosis. Arthritis and Rheumatism 2001;44(9):2201‐10. [DOI] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro GDT. Version accessed prior to 7 November 2017. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Ralston 2013
- Ralston SH. Paget's disease of bone. New England Journal of Medicine 2013;368(7):644‐50. [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Sandberg 2003
- Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: osteosarcoma and related tumors. Cancer Genetics and Cytogenetics 2003;145(1):1‐30. [PubMed] [Google Scholar]
Schaffer 1996
- Schaffer, V, Buckler, H, Ryaa, W, Richardson, P. Double blind placebo‐controlled trial using intraveonous zoledronate (CGP.42446) in patients with Paget´s diasese of bone. [M766]. ASBMR 18th Annual Meeting book of abstract August 1996:S373.
Selby 2002
- Selby PL, Davie MWJ, Ralston SH, Stone MD. Bone and Tooth Society of Great Britain, National Association for the Relief of Paget's Disease. Guidelines on the management of Paget's disease of bone. Bone 2002;31(3):366‐73. [DOI] [PubMed] [Google Scholar]
Singer 2015
- Singer FR, Bone HG III, Hosking DH, Lyles KW, Murad MH, Reid IR, et al. Endocrine Society. Paget's disease of bone: an Endocrine Society Clinical Practice Guideline. Journal of Clinical Endocrinology and Metabolism 2015;99(12):4408‐22. [DOI] [PubMed] [Google Scholar]
Siris 1996b
- Siris ES. A potent new bisphosphonate for Paget's disease of bone. American Journal of Medicine 1996;101(4):339‐40. [DOI] [PubMed] [Google Scholar]
Takata 2006
- Takata S, Hashimoto J, Nakatsuka K, Yoshimura N, Yoh K, Ohno I, et al. Guidelines for diagnosis and management of Paget's disease of bone in Japan. Journal of Bone and Mineral Metabolism 2006;24(5):359‐67. [DOI] [PubMed] [Google Scholar]
Tan 2014
- Tan A, Ralston SH. Clinical presentation of Paget’s disease: evaluation of a contemporary cohort and systematic review. Calcified Tissue International 2014;95(5):385‐92. [DOI: 10.1007/s00223-014-9904-1] [DOI] [PubMed] [Google Scholar]
Van Staa 2002
- Staa TP, Selby P, Leufkans HG, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget's disease of bone in England and Wales. Journal of Bone and Mineral Research 2002;17(3):465‐71. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Langston 2004
- Langston AL, Campbell MK, Ralston S, Robertson C. Aminobisphosphonates versus other active treatment for Paget's disease of the bone in adults. Cochrane Database of Systematic Reviews 2004, Issue 4. [DOI: 10.1002/14651858.CD004956] [DOI] [Google Scholar]