

Abstract
High-risk human papillomaviruses (HPVs) constitutively activate the ataxia telangiectasia mutated (ATM) and the ataxia telangiectasia and Rad3-related (ATR) DNA damage repair pathways for viral genome amplification. HPVs activate these pathways through the immune regulator STAT-5. For the ATR pathway, STAT-5 increases expression of the topoisomerase IIβ-binding protein 1 (TopBP1), a scaffold protein that binds ATR and recruits it to sites of DNA damage. TopBP1 also acts as a transcriptional regulator and we investigated how this activity influenced the HPV life cycle. We determined that TopBP1 levels are increased in cervical intraepithelial neoplasias as well as cervical carcinomas, consistent with studies in HPV-positive cell lines. Suppression of TopBP1 by shRNAs impairs HPV genome amplification and activation of the ATR pathway but does not affect the total levels of ATR and CHK1. In contrast, knockdown reduces the expression of other DNA damage factors such as RAD51 and Mre11 but not BRCA2 or NBS1. Interestingly, TopBP1 positively regulates the expression of E2F1, a TopBP1 binding partner, and p73, in HPV positive cells in contrast to effects in other cell types. TopBP1 transcriptional activity is regulated by AKT and treatment with AKT inhibitors suppresses expression of E2F1 and p73 without interfering with ATR signaling. Importantly, the levels of p73 are elevated in HPV-positive cells and knockdown impairs HPV genome amplification. This demonstrates that p73, like p63 and p53, is an important regulator of the HPV life cycle that is controlled by the transcriptional activating properties of the multifunctional TopBP1 protein.
Introduction
Human papillomaviruses (HPVs) are small DNA viruses that are the causative agents of cervical and many oropharyngeal cancers1–3. The life cycle of HPV is dependent on the differentiation of the infected epithelial host cells with productive replication restricted to highly differentiated suprabasal cells4. HPVs infect cells in the basal layer of stratified epithelia and establish persistent infections that maintain low copies of HPV episomes in basal cells. The activation of late gene expression and induction of productive replication of viral episomes, which is referred to as amplification, is restricted to differentiated suprabasal cells5. The HPV early proteins include E1 and E2 which are responsible for initiation of HPV replication 6,7 while E4 and E5 are involved in regulation of late gene expression 8,9. The E6 and E7 proteins act to immortalize infected cells and are necessary for HPV genome maintenance in undifferentiated cells as well as amplification upon differentiation 10–13. The L1 and L2 late proteins encode the viral capsid proteins 2,14,15.
The differentiation-dependent amplification of viral genomes is regulated by cellular transcription factors that bind to the early promoter in the upstream regulatory region (URR) as well as the late promoter located in the middle of the E7 ORF16. YY-1, TBP, AP-1, Oct-1 and Sp1 have been shown to regulate HPV early promoters 17–21whereas C/EBPβ, LIP, and LAP act on HPV late promoters 22. Recent studies have shown that both the Kruppel-like factor 4 (KLF4) and KLF13 can regulate HPV amplification but by different mechanisms23,24. KLF4 forms complexes with Blimp1 to activate HPV late transcription while KLF13 regulates STAT-5 expression. STAT-5 promotes HPV genome amplification by activating the ataxia telangiectasia mutated (ATM) as well as the ataxia telangiectasia and Rad3-related (ATR) DNA damage repair pathways25,26. Inhibition of either ATM or ATR signaling by pharmacological inhibitors or knockdown with shRNAs suppresses HPV genome maintenance 27or amplification26,28. The ATR pathway is important for homologous recombination repair in response to single strand DNA breaks while ATM acts to repair double strand breaks. ATR is recruited to sites of single strand breaks in complexes with the ATR-interacting protein (ATRIP), claspin and the topoisomerase IIβ-binding protein 1 (TopBP1) which results in its phosphorylation29. TopBP1 facilitates activation of ATR through direct binding, however, it can also associate with several transcription factors such as E2F1 to regulate gene expression30–33. TopBP1’s role in transcriptional activation is regulated by phosphorylation and this distinguishes its transcriptional activities from effects on ATR activation34. It is, however, unclear what role TopBP1’s transcriptional effects have on the HPV life cycle and what are the critical targets.
Our studies show that TopBP1 expression is increased in cervical intraepithelial neoplasia and cervical squamous cell carcinoma as well as in HPV-positive cell lines in culture. Knockdown of TopBP1 with shRNAs blocks ATR and CHK1 activation but has no effect on total levels of these two factors. In contrast, TopBP1 knockdown greatly reduces the levels of other DNA damage factors such as RAD51 and Mre11 with minimal effects on other factors such as BRCA2 and NBS1. Interestingly transcriptional analysis of TopBP1 regulated genes in HPV-positive cells identifies both p73 and E2F1 as positive targets. This contrasts with studies using other cell types that indicate TopBP1 is a repressor of E2F1 expression 30,31. Importantly TopBP1 mediated increases in the levels of p73 are shown to be important for genome amplification. This analysis demonstrates that TopBP1’s ability to regulate transcription of cellular genes plays a critical role in the HPV life cycle.
Results
TopBP1 levels are increased in HPV-positive cervical cancers
To investigate which activities of TopBP1 are critical for the HPV life cycle it was first important to demonstrate that the increased levels of TopBP1 seen in HPV-positive cell lines in tissue culture are also observed in clinical samples of varying degrees of cervical diseases. For this analysis RT-PCR analysis of TopBP1 mRNA levels was performed using 90 human cervical tissue samples, including 29 normal cervical tissues, 31 HPV16-positive cervical intraepithelial neoplasias (CIN 2–3), and 30 HPV16-positive cervical cancers. As shown in Figure 1A there is an increase in the expression of TopBP1 in CIN 2–3 lesions as compared to normal tissues that is further enhanced upon progression to cervical cancer. Similar increases in TopBP1 levels are seen in cell lines that contain HPV16, HPV18, or HPV31 episomes (HFK-16, HFK-18, HFK-31) that were generated by transfection of normal keratinocytes. We next screened for the distribution of TopBP1 protein by immunohistochemistry in an additional panel of 30 human cervical tissue samples, including 10 normal, 10 HPV16-positive CIN 2–3, and 10 HPV16-positive cervical cancer tissues (Figure 1B). In normal tissues, TopBP1 is mainly detected at low levels in the nuclei of basal layer cells with a further decrease in differentiated spinous layer cells and granular layer cells. In high-grade squamous intraepithelial lesions, the level of TopBP1 proteins is increased over that seen in normal tissues and is detected in all layers. Upon calcium-induced differentiation of both HFK-31 cells and CIN612 cells, increased levels of TopBP1 are observed in comparison to HFKs (Figure 2A). These studies demonstrate that the levels of TopBP1 are increased in both low-grade HPV cervical lesions and cancers, as well as in tissue culture cell lines (Figure 2B). We also generated keratinocyte cell lines following infection with retroviruses expressing either E6 or E7 and confirmed that the E7 protein but not E6 is responsible for the increases in TopBP126 (Figure 2C).
Figure 1.
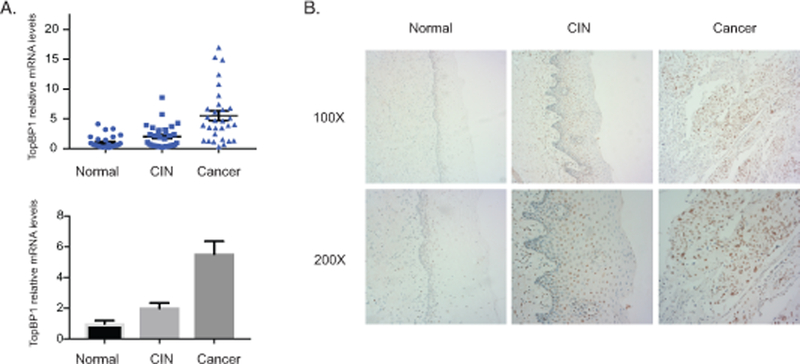
Increased levels of TopBP1 in HPV-positive cervical cancer tissues. A). RT-PCR analysis of TopBP1 mRNA levels in 29 normal tissues (Normal), 31 cervical intraepithelial neoplasias (CIN), and 30 cervical cancer (CA). The TopBP1 level was shown as as 2-ΔΔCT in biopsies of cancer tissues versus normal tissues. GAPDH was used as internal control and for normalization of the data. The results were shown in both scatter plot figure format and bar figure format. B). Representative immunohistochemical staining of TopBP1 in 10 normal tissues, 10 cervical intraepithelial neoplasias, and 10 cervical cancers.
Figure 2.
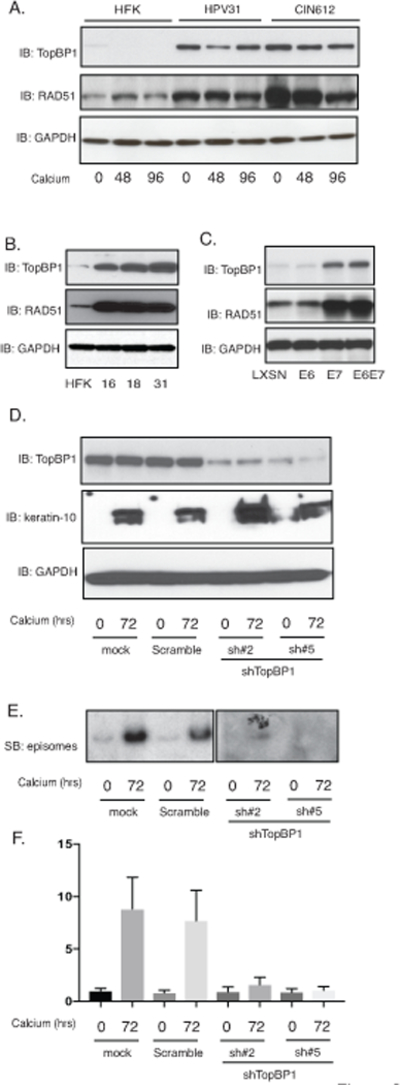
Expression of TopBP1 is necessary for HPV genome amplification upon keratinocyte differentiation. A). Western blot analysis of TopBP1, RAD51 and GAPDH levels in HFK, HFK-31 and HPV31 positive CIN612 cells differentiated in high calcium media for indicated times (hrs). B). Western blot analysis of TopBP1, RAD51 and GAPDH levels in HFK, HFK-16, HFK-18, and HFK-31 cells grown in monolayer cultures. C). Western blot analysis of TopBP1, RAD51 and GAPDH levels in HFK cells expressing LXSN vector, HPV31E6, HPV31E7, and HPV31E6E7. D). Western blot analysis of TopBP1, keratin-10 and GAPDH protein levels in CIN612 cells infected with lentiviruses expressing shRNA scramble control and two different sets of lentiviruses expressing shRNAs against TopBP1 upon differentiation in high-calcium media for 72 hours. E). Southern blot analysis for HPV31 episomes in CIN612 cells with TopBP1 knockdown following differentiation in high calcium media for indicated times (hrs). F) Quantification of band intensity of the Southern blot from (E) shown in bar figure format. All results are representative of observations from 2–3 independent experiments.
TopBP1 regulates expression of DNA damage factors and cell cycle factors
TopBP1 is a scaffold protein that forms complexes with ATR and ATRIP which are recruited to sites of DNA breaks resulting in phosphorylation of ATR35. Knockdown of TopBP1 in cells that stably maintain HPV31 episomes using two different sets of shRNAs blocks ATR signaling and HPV genome replication (Figure 2D–2F), which is consistent with previous observations using transient assays26. TopBP1 knockdown does not affect the differentiation process as the expression of keratin 10, a marker of differentiation, is unchanged. We previously demonstrated that viral proteins induce breaks into both cellular and viral DNAs 36. Consistent with TopBP1’s recruitment to sites of DNA breaks we observed using chromatin immunoprecipitation assays that it binds to multiple regions of the HPV genomes in both undifferentiated and differentiated states (Supplemental Figure 1).
TopBP1 also has transcriptional regulatory functions that require complex formation with factors such as E2F1, p53 or Miz130,33,37 and we investigated how TopBP1 transcriptional activities affected the HPV life cycle. For this analysis, keratinocytes that stably maintain HPV31 episomes (HFK-31 and CIN612) were infected with lentiviruses expressing shRNAs against TopBP1 and stable cell lines were selected by resistance to a co-expressed puromycin gene. Stable knockdown of TopBP1 was found to block ATR activation by phosphorylation but did not alter the total levels of ATR as determined by western analysis (Figure 3A). This suggests that the increased ATR levels in HPV-positive cells are not due to TopBP1 but to other factors. Similarly, knockdown of TopBP1 had no effect on the levels of the downstream effector kinase, CHK1 but did prevent its activation by phosphorylation. We next investigated if TopBP1 knockdown affected the levels of other members of the DNA damage repair pathways. Interestingly, our studies show that TopBP1 knockdown substantially reduced the levels of RAD51, Mre11 and to a lesser extent BRCA1 with minimal effect on the levels of BRCA2 or NBS1 (Figure 3B). This indicates that TopBP1 can control expression of a number of DNA damage factors that are critical for HPV genome amplification. We next examined if the mRNA levels of RAD51, one of the DNA damage factors we identified as regulated by TopBP1, exhibited similar correlations with TopBP1 levels in CIN and cervical cancer biopsy specimens. Thirty biopsy samples of varying degrees of cervical disease were examined by RT-PCR and RAD51 expression was found to exhibit similar increases as seen with TopBP1 (Figure 3C). Additionally, we screened the expression of RAD51 in HPV-positive cells and found the levels of RAD51 are greatly increased in HFKs that stably maintain HPV genomes (Figure 2A). The E7 protein is responsible for the increase of RAD51 levels (Figure 2B). This indicates that in addition to regulating ATR activation TopBP1 also contributes to regulating expression of a subset of DNA damage repair factors.
Figure 3.
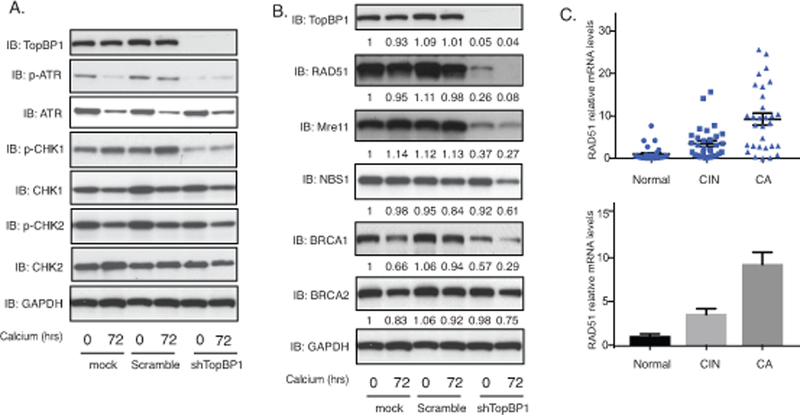
TopBP1 is necessary for ATR activation and increased expression of DNA damage factors upon keratinocyte differentiation. A). Western blot analysis of p-ATR, ATR, p-CHK1, CHK1, p-CHK2, CHK2 and GAPDH levels in CIN612 cells stably infected with lentiviruses expressing shRNAs against TopBP1 differentiated in high calcium media for indicated times. The experiments were repeated using two sets of TopBP1 shRNAs and only one was shown as a representative. All results are representative of observations from 3 independent experiments. B). Western blot analysis of TopBP1, RAD51, Mre11, NBS1, BRCA1, BRCA2 and GAPDH levels in CIN612 cells with TopBP1 knockdown in high calcium media for indicated times. The band intensities were quantified using ImageJ software and labelled accordingly under each panel. C). RT-PCR analysis of RAD51 mRNA levels in 29 normal tissues (Normal), 31 cervical intraepithelial neoplasias (CIN), and 30 cervical cancer (CA). The RAD51 levels are shown as as 2-ΔΔCT of cancer versus normal tissues. GAPDH was used as internal control and for normalization of the data. The results are shown in both spot figure format and bar figure format.
TopBP1 has been reported to bind to the E2F1 transcription factor and repress its ability to activate downstream genes30. We therefore investigated how TopBP1 affected E2F1 activities in HPV-positive cells. Our data show that the levels of E2F1 are increased in HPV-positive cells of three different high-risk types (HPV 16, 18 and 31) as compared to normal keratinocytes (Figure 4A). Similarly, we observe a substantial increase in E2F1 levels in HFKs that express HPV E7 (Figure 4B). E6 was also found to modestly increase levels of E2F1 and this is due to TopBP1 independent effects. Upon epithelial differentiation, the levels of E2F1 expression are higher in HPV-positive cells than HFKs (Figure 4C). Importantly TopBP1 knockdown with shRNAs significantly reduces E2F1 levels (Figure 4D) but does not completely abrogate these levels. This residual level is likely due to E6’s ability to activate E2F1 independent of TopBP1. RT-PCR analysis shows that the effect of TopBP1 on E2F1 is mediated at the level of transcription as mRNA levels are reduced upon shRNA knockdown and mimics effects seen at the protein level (Figure 4E). This indicates that increased expression of E2F1 in HPV-positive cells is largely dependent upon enhanced levels of TopBP1 though other mechanisms may also contribute. One downstream transcriptional target of E2F1 is p73 and we investigated if its levels were altered due to TopBP1 knockdown in HPV-positive cells. Our studies show the levels of p73 are increased in HPV-positive cells in comparison to normal keratinocytes (Figure 4A) and this is abrogated upon TopBP1 knockdown with shRNAs (Figure 4D&4E). RAD51 has also been suggested to be a downstream target of E2F1 38,39 and the reductions we observe in TopBP1 knockdowns are likely the result of decreased E2F1 levels. Rad51 has been shown to be recruited to viral genomes and is a critical regulator of viral amplification 36,40. These studies indicate that in HPV-positive cells TopBP1 positively increases E2F1 levels, which is in contrast to its effects reported in other cell types. Furthermore, TopBP1 does not act a repressor of E2F1 action on downstream genes in HPV-positive cells but rather contributes to their activation.
Figure 4.
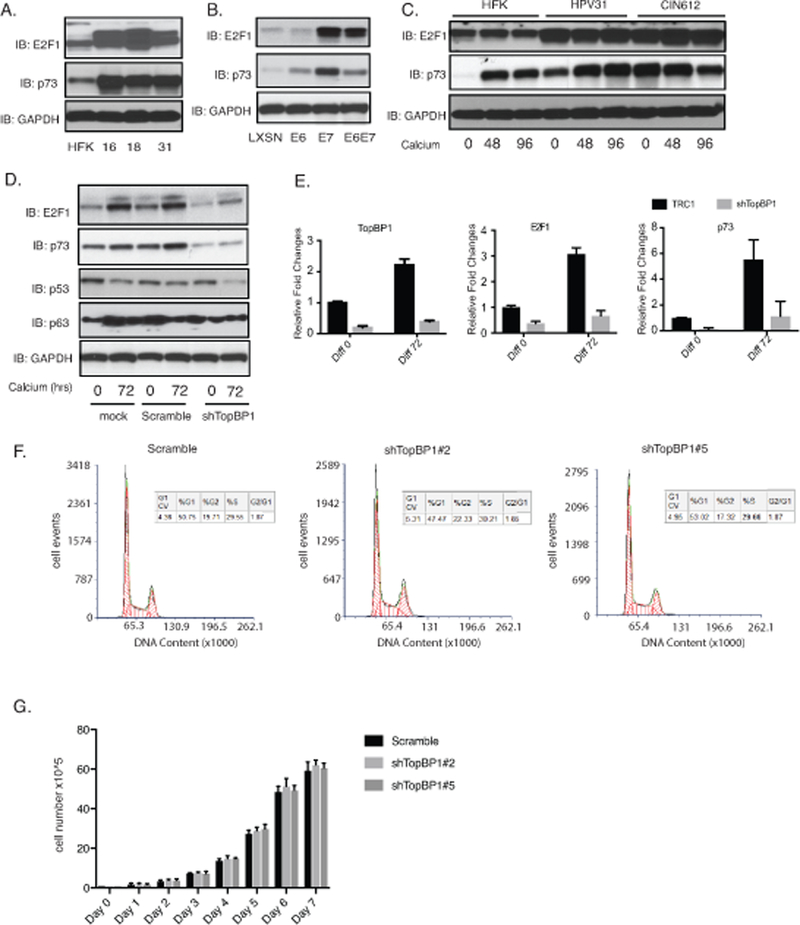
TopBP1 activates expression of E2F1 and p73 in HPV positive cells. A). Western blot analysis of E2F1, p73 and GAPDH levels in HFK, HFK-16, HFK-18, and HFK-31 cells grown in monolayer cultures. B). Western blot analysis of E2F1, p73 and GAPDH levels in HFK cells stably infected with LXSN vector control or retroviruses expressing HPV31E6, HPV31E7, and HPV31E6E7. C) Western blot analysis of E2F1, p73 and GAPDH levels in in HFK, HFK-31 and HPV31-positive CIN612 cells differentiated in high calcium media for indicated times (hrs). D) Western blot analysis of E2F1, p73, p53, p63 and GAPDH levels in CIN612 cells stably infected with lentiviruses expressing shRNAs against TopBP1 following differentiation in high calcium media for indicated times (hrs). E) RT-PCR analysis of TopBP1, E2F1, and p73 levels in CIN612 cells with TopBP1 knockdown in high calcium media for indicated times (hrs). GAPDH was used as internal control and for normalization of the data. Data=mean +/− standard error. p-value <0.05. The experiments were repeated using two sets of TopBP1 shRNAs and only one was shown as a representative. F) The cell cycle distribution of the scramble CIN612 cells and the CIN612 cells with two different TopBP1 knockdown was measured by flow cytometry following propidium iodide staining. G) Comparison of the cell growth rates of CIN612 cells expressing scramble control or two different TopBP1 knockdown. All results are representative of observations from 2–3 independent experiments.
To test whether inhibition of HPV genome amplification upon TopBP1 knockdown is due to cell cycle arrest, we examined the effect of TopBP1 knockdown on cell cycle distribution of control and knockdown cells by flow cytometry analysis after propidium iodide (PI) staining. The scramble and TopBP1 knockdown CIN 612 cells were fixed and permeabilized by 70% ethanol, stained with PI and analyzed by flow cytometry. Both knockdown and control cell populations exhibit similar cell cycle distributions with about 50% of cell in G1, 20% in G2, and 30% in S phase (Figure 4F). In addition, we examined the growth rates of the stable scramble and TopBP1 knockdown cells and found them to be comparable (Figure 4G). We conclude that the inhibition of amplification by TopBP1 following knockdown is not due to cell cycle arrest in G1.
AKT regulates for the transcription functions of TopBP1
The ability of TopBP1 to regulate transcription is dependent upon its phosphorylation, which is controlled, in large part by AKT kinase34. Phosphorylation of TopBP1 by AKT is necessary for TopBP1 repression of E2F1 activation but does not inhibit its binding to ATR. We first investigated whether AKT was activated in HPV-positive cells by western analysis using three HPV-positive cell lines that stably maintain viral episomes (HFK-16, −18, −31) together with HFKs. All three HPV-positive lines exhibit high levels activation of AKT in comparison to normal keratinocytes while the total levels of AKT are similar (Figure 5A). To determine which viral proteins were responsible for increased levels of p-AKT, we infected HFKs with retroviruses expressing either E6 or E7 alone as well as the combination of both genes. In HFKs that express either E6 or E7 from retroviral vectors, the levels of phosphorylated AKT are increased (Figure 5B). To investigate how AKT influenced TopBP1 phosphorylation, we first examined E7 alone expressing cells as it is the primary factor responsible for increased levels of TopBP1. Upon differentiation, E7-expressing cells maintain high levels of phosphorylated AKT, phosphorylated TopBP1, p73 and E2F1. To test whether AKT activation affects TopBP1-dependent E2F1 and p73 expression, E7-expressing cells were treated with MK2206 (Figure 5C) that blocks AKT phosphorylation but does not alter the total levels of AKT proteins. Our studies show that MK2206 treatment severely blocks phosphorylation of TopBP1 in E7-expressing cells and substantially reduces the levels of p73 and E2F1 but not activation of CHK1 phosphorylation. We next treated cells that stably maintain HPV episomes with MK2206 and observed decreases in the levels of p-AKT, p-TopBP1, p73 and E2F1, with no change in p-CHK1 activation (Figure 5D). To test whether AKT activation is required for HPV genome amplification, CIN612 cells were treated with two different AKT inhibitors MK2206 and LY294002. The DNA from these cells was examined by Southern blot analysis following calcium induced differentiation and results indicate that both inhibitors suppress HPV genome amplification (Figure 5E&5F). We conclude that inhibiting TopBP1 phosphorylation by AKT reduces expression of E2F1 and p73 levels, which is consistent with our knockdown studies. This also demonstrates novel effects of TopBP1 on E2F1 activities are specific to HPV-positive cells. Interestingly, we also observed a reduction in TopBP1 levels in p73 knockdown cells indicating a feedback loop may exist between the two factors (Supplemental Figure 2).
Figure 5.
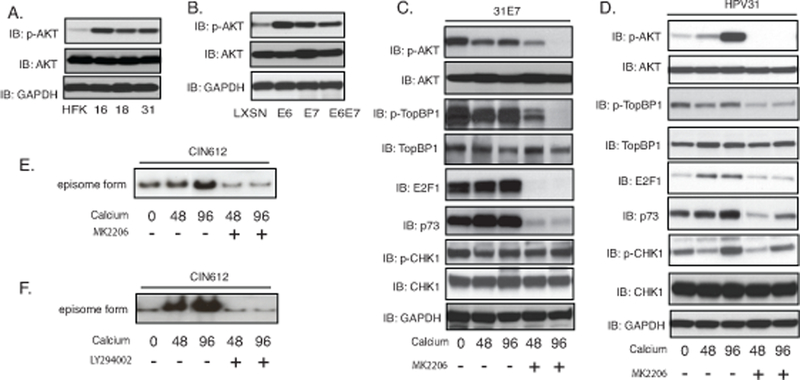
TopBP1 activation of E2F1 and p73 expression is dependent upon AKT. A). Western blot analysis of p-AKT, AKT and GAPDH levels in HFK, HFK-16, HFK-18, and HFK-31 grown in monolayer cultures. B). Western blot analysis for p-AKT, AKT and GAPDH levels in HFK cells stably infected with LXSN vector control, HPV31E6, HPV31E7, and HPV31E6E7. C). Western blot analysis of p-AKT, AKT, p-TopBP1, TopBP1, E2F1, p73, p-CHK1, CHK1 and GAPDH protein levels in 31E7-expressing cells upon differentiation in high-calcium media for 72 hours in the presence of the AKT inhibitor MK2206. D). Western blot analysis of p-AKT, AKT, p-TopBP1, TopBP1, E2F1, p73, p-CHK1, CHK1 and GAPDH protein levels in HPV31 cells upon differentiation in high-calcium media for 72 hours in the presence of the AKT inhibitor MK2206. E) Southern blot analysis for HPV31 episomes in CIN612 cells with MK2206 treatment following differentiation in high calcium media for indicated times (hrs). F) Southern blot analysis for HPV31 episomes in CIN612 cells treated with LY294002 following differentiation in high calcium media for indicated times (hrs). All results are representative of observations from 3 independent experiments.
E2F1 and p73 are downstream targets of TopBP1 transcriptional activities
The increased levels of p73 detected in HPV-positive is interesting since two other members of this family p63 41 and p53 42–44 have been shown to be critical for regulating the life cycle of high-risk HPVs. We next investigated whether p73 also provided an important function in the viral life cycle. First the levels of p73 were examined in two additional cell lines that are positive for HPV16 or 18 (HFK-16, −18 cells) to ensure increases are seen with other high-risk HPV types. We found all three high-risk types of HPV cells express high levels of p73 (Figure 4A). Figure 4B shows that E7 substantially increases the levels of E2F1 and p73 while E6 also can activate expression but to a lesser degree than E7 does. Furthermore, following calcium induced differentiation, the levels of p73 were also maintained at higher levels in HPV-positive cells as compared to HFKs (Figure 4C). To investigate if p73 has any effect on the HPV life cycle we screened for effects on viral replication. CIN612 cells were stably transduced with p73-specific shRNA lentiviruses, expanded and examined for amplification following differentiation in high calcium media. Figure 6A shows that 2 out of 3 p73 shRNAs examined are able to suppress p73 levels in comparison to cells transduced with lentiviruses expressing scrambled control shRNAs. We next examined the effect of p73 knockdown on amplification and found that this resulted in impaired genome amplification upon differentiation (Figure 6B). We conclude that p73 like p63, has critical activities for regulating viral amplification in differentiating cells.6
Figure 6.
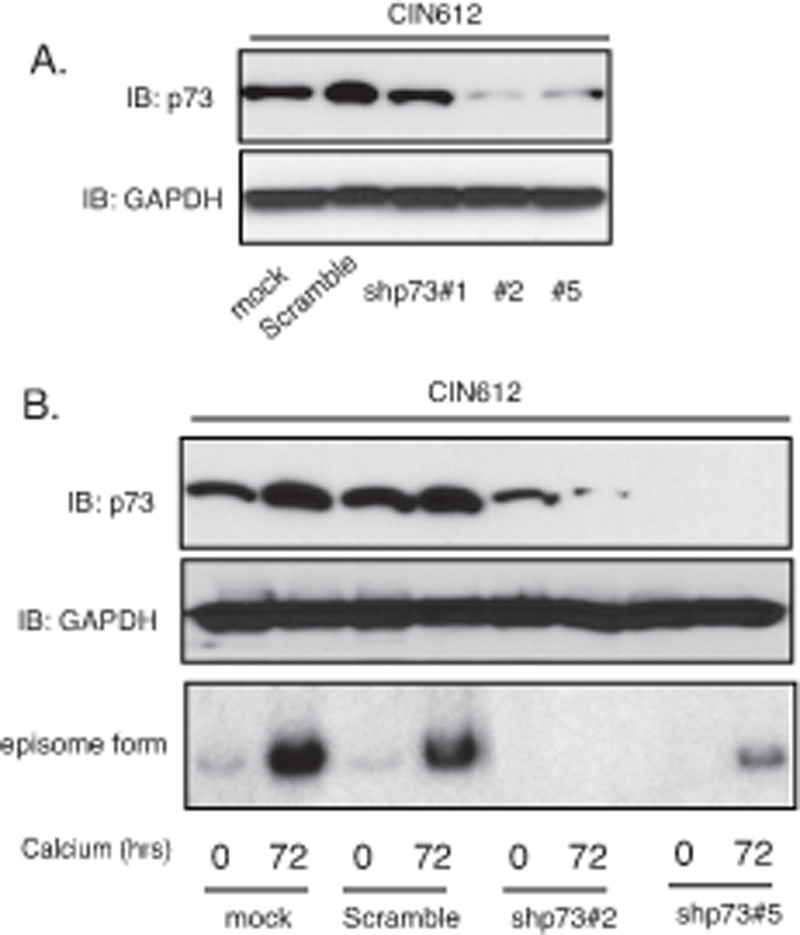
p73 is necessary for differentiation-dependent HPV genome amplification. A). Western blot analysis of p73 and GAPDH proteins in monolayer CIN612 cells infected with multiple shRNA lentiviruses targeting p73. B). Western blot analysis of p73 and GAPDH proteins in CIN612 cells with TopBP1 knockdown upon differentiation in high-calcium media for 72 hours. Southern blot analysis of HPV31 episomes in CIN612 cells with TopBP1 knockdown following differentiation in high calcium media for 72 hrs. All results are representative of observations from 2 or more independent experiments.
Additional downstream targets of TopBP1 transcriptional regulation
Our studies indicate that the transcriptional regulatory effects of TopBP1 are important for the HPV life cycle. We next sought to identify additional downstream targets of TopBP1 in HPV-positive cells by performing an unbiased global RNA-seq analysis with CIN612 cells in which TopBP1 was stably knocked down with shRNAs and compared effects to cells treated with a scramble shRNA control. Both TopBP1 knockdown and scramble control cells were differentiated in high calcium media for 72 hours and RNAs isolated for RNA-seq analysis. To identify genes regulated by TopBP1, we set a threshold criteria of at least a 1.5 fold change and a minimum of 40–45 reads to eliminate genes that are expressed at low levels. The genes that were found to be regulated by TopBP1 were categorized based on their functions and grouped using Metascape online tools. The top 20 pathways regulated by TopBP1 are shown in the figure 7A and the complete list of genes is included in supplemental table 1. The largest group of genes include those regulating interferon signaling and the innate immune defense response. This indicates that TopBP1 may play a significant role in regulating the innate immune response (Figure 7B). We next sought to confirm the effect of TopBP1 on a small number of specific interferon and innate immune regulatory genes by RT-PCR in TopBP1 knockdown cells. Previous studies have shown that interferon and interferon stimulated gene expression is suppressed in HPV positive cells 45 and our studies indicate TopBP1 contributes to this regulation. Following TopBP1 knockdown IFN-β, IFN-κ, CXCL2, CXCL10, IL-6, IL-8, and IL-18 are increased as determined by RT-PCR (Figure 7C). Similar effects of TopBP1 knockdown on these factors was observed following differentiation following suspension in methylcellulose (Supplemental Figure 3). We conclude that in addition to its positive effects on the activation of E2F1 and p73, TopBP1 also acts as a negative regulator of interferons, chemokines and some interleukins in HPV positive cells.
Figure 7.
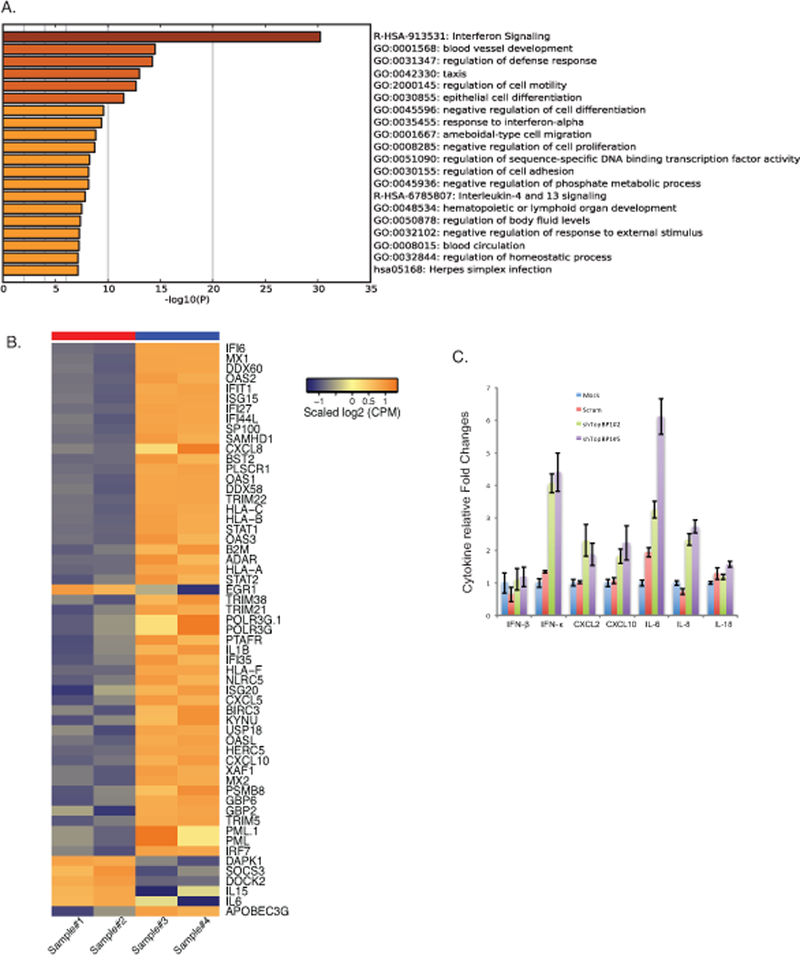
Downstream transcriptional targets of TopBP1 in HPV positive cells. A). RNA seq analysis was performed on CIN612 cells infected with scrambled shRNA control or shRNAs against TopBP1 following differentiation. Top 20 pathways regulated by TopBP1 was listed with Metascape online analysis tools. B). Heat maps of immune regulatory genes modulated by TopBP1 knockdown upon differentiation. Sample#1 and #2 are replicates of scramble CIN612 cells, while Sample #3 and #4 are replicates of CIN612 cells with TopBP1 knockdown. C) RT-PCR analysis of IFN-β, IFN-κ, CXCL2, CXCL10, IL-6, IL-8, and IL-18 in undifferentiated CIN612 cells with TopBP1 knockdown. GAPDH was used as internal control and for normalization of the data. Data=mean +/− standard error. p-value <0.05. All results are representative of observations from 2 or more independent experiments.
Discussion
TopBP1 is a multifunctional protein that is critical for activation of the ATR DNA damage repair pathway as well as for the regulation of transcription. Our studies show that TopBP1 levels are increased in low grade HPV16-positive lesions and this is further enhanced upon progression to cancer. These increases are similar to those seen in our tissue culture models of precancerous lesions that are able to produce viruses upon differentiation. The increased levels of TopBP1 in HPV-positive cells are important for both the increased expression of a novel set of genes which are critical for the viral life cycle and activation of the ATR kinase. The ability to bind and activate ATR is separable from TopBP1’s role in regulating transcription and our studies show that each provides distinct critical functions for HPV pathogenesis. The levels of TopBP1 are similar among HFK-16 cells, HFK-31 cells, and CIN612 cells, which suggests that this increase do not depend on HPV type. Interestingly the levels of TopBP1 were found to increase with progression from CIN to cancer. Many cancers contain integrated copies of HPV genomes which often results in increased levels of E7 expression 46 and this increase likely contributes to the higher levels of TopBP1. What role, if any, increased levels of TopBP1 might play in progression is still unclear.
The increased levels of TopBP1 in HPV-positive cells has no effect on the levels of ATR, but is necessary for enhanced expression of other members of the DNA damage repair pathway including RAD51 and Mre11 with little to no effects on NBS1 or BRCA2. Interestingly RAD51 and Mre11 are primarily associated with the double strand DNA break repair ATM pathway and less so for the ATR pathway 47,48, which TopBP1 activates through direct binding to ATR 35. TopBP1 acts to regulate gene expression by associating and modifying the activities of a series of transcription factors including E2F1, p53 and Miz1 30,33,37. Binding of TopBP1 to E2F1 normally represses its transcriptional activating ability 30 but in HPV-positive cells we see the opposite effect. Interestingly our studies indicate that TopBP1 also regulates expression of E2F1 itself as knockdown inhibits its transcription. While stable knockdowns of TopBP1 in many cell types have been difficult to generate as most undergo apoptosis, the isolation of stable HPV-positive cells infected with lentiviruses expressing shRNAs against TopBP1 is readily achievable. In our TopBP1 knockdown cells E2F1 expression is reduced in a process controlled by E7. This indicates that changes in the cellular environment induced by E7 action alter the requirements for TopBP1 functions so that cells no longer undergo cell death through apoptosis. Importantly, our studies show that in HPV-positive cells TopBP1 is a positive regulator of E2F1 expression.
In addition to activating the expression of E2F1 itself, high levels of TopBP1 also activate expression of downstream targets such as p73 and RAD51 in HPV-positive cells. TopBP1 has also been shown to bind to the HPV E2 protein which can modulate the levels of viral replication 49, however, the novel properties of TopBP1 observed in our studies can not be explained by its binding to E2 as we observed similar effects on E2F1 and p73 in cells expressing E7 alone. This indicates that factors other than E2 are responsible for the positive effects of TopBP1 on transcription but whether this is due to a novel binding partner or an upstream regulatory factor is unclear. While expression of p73 and RAD51 are regulated by E2F1, it is not clear whether Mre11 is similarly controlled but possible. High level expression of E2F1 has been shown to be sufficient to activate DNA damage repair pathways and this may in part be mediated by regulating the transcription of these factors. The question arises as to how TopBP1 can act as a repressor of E2F1 functions in HEK 293 and human fibroblasts but act as an activator in HPV-positive cells. TopBP1 associates with E2F1 in large complexes together with other factors and we believe additional regulators may be recruited to this complex in HPV-positive cells or alternatively the repressive complexes are prevented from forming.
The ability of TopBP1 to activate ATR can be distinguished from its role in regulating gene expression through phosphorylation by the AKT kinase 34. Phosphorylation of TopBP1 by AKT induces its oligomerization resulting in the ability to associate with a series of transcription factors while blocking binding to ATR 31,34. Since both activation of ATR and increased expression of TopBP1 regulated genes are observed in our studies, a mixed population of phosphorylated and unphosphorylated forms must exist in HPV positive cells. AKT is responsible for TopBP1 phosphorylation and our data show that both E6 and E7 can increase p-AKT levels in stable cells lines. A previous study from the Vande Pol group showed that upon serum starvation followed by stimulation a transient decrease in p-AKT levels is observed in cells expressed HPV 16E7 as compared to normal immortal keratinocytes (NIKs), This transient decrease is in contrast to our findings using steady-state analysis. In addition while both E6 and E7 are able to activate the AKT pathway, our studies indicate that only E7 activates TopBP1. This could be due to our previous observation that E7 but not E6 acts to activate the innate immune regulator STAT-5 which in turn directly regulates high-levels of transcription of TopBP1 26. It is also possible that additional regulatory factors upstream of STAT-5 contribute to the increased levels of TopBP1 in E7 cells. In cells that express only E7, inhibition of TopBP1 phosphorylation decreases E2F1 and p73 expression. In contrast in cells that express both E6 and E7 including those that harbor complete viral genomes, inhibition of TopBP1 phosphorylation substantially reduces but does not completely abrogate E2F1 and p73 transcription. We believe this indicates that while TopBP1 is a major positive regulator of E2F1 in HPV-positive cells, additional TopBP1 independent mechanisms exist that are likely regulated by E6.
One target of E2F1 that is increased in HPV-positive cells is p73 and by knockdown we show that it is critical for the HPV life cycle. The other members of this family, p53 42 and p63 41, also play critical roles in the life cycle. p53 levels are decreased in HPV positive cells through the action of E6/E6AP while p63 levels are increased through the suppression of miR-203 50 that normally inhibits p63 translation. Furthermore, p63 functions by activating expression of KLF-4 23 in suprabasal layers to control expression of genes regulating the cell cycle and differentiation. Which specific downstream targets of p73 are important for regulating the HPV life cycle are still unclear and subject to future analysis. Multiple isoforms of p73 exist in cells including full-length activator forms and N-terminal truncated forms ΔNp73 that is generated by alternative promoter usage. The activator TAp73 has been reported to increase expression of p53 responsive genes while ΔNp73 acts as a repressor and both are likely important for HPV pathogenesis 51–53. Interestingly p73 proteins have been described as primarily expressed in brain and play roles in neuronal development as well as differentiation 54. Furthermore, high levels of p73 proteins have been reported in HPV-positive oropharyngeal cancers 55 as well as in cells that contain the cutaneous HPV 38 56. The HPV 38 E6 and E7 proteins act to increase levels of p73 and this is important for repression of Toll like receptor 9 that functions to restore UV-activated cell cycle checkpoint arrest 57.
Our RNA –seq analysis identified a broad spectrum of genes whose expression is altered by TopBP1 in HPV-positive cells. In addition to activating expression of E2F1, p73, RAD51 and Mre11, our studies show that TopBP1 can also act as a repressor of one type of interferon specific to keratinocytes, IFN-κ as well as cytokines IL-6 and IL-8. These genes have not been characterized as E2F1 regulated genes and the mechanism by which TopBP1 acts to repress expression remains unclear but IFN-κ suppression has been shown to be important for HPV replication 58. Overall, our studies show that TopBP1’s transcriptional regulatory activities are critical for HPV life cycle.
Materials and Methods
Cell culture:
Human foreskin keratinocytes (HFKs) were isolated from neonatal foreskins from 3 different individuals as previously described 59. HFK-16, 18, and 31 cell lines that maintain viral episomes, as well as E6- or E7-expressing cells, were generated by transfection of HFKs with HPV genomes, or retroviruses expressing E6 or E7 as previously described 60. CIN612 cells stably maintains HPV31 episomes and are derived from a cervical intraepithelial neoplasia (CIN) II biopsy. All HPV-positive cell culture requires a coculture with mitomycin-treated NIH 3T3 J2 fibroblasts in E media. To induce differentiation, HPV-positive cells were cultured in keratinocyte basal medium without supplements and containing 1.5 mM CaCl2 for up to 96 hours as described 60.
Clinical samples:
The human cervical biopsy tissue samples assayed in this study were collected from Women’s Hospital, Zhejiang University School of Medicine, China. The samples were obtained with written informed consent. All samples were immediately snap-frozen in liquid nitrogen and stored at −80°C until use. The sample usage was approved by the Women’s Hospital Research Ethical Committee of Zhejiang University.
Immunohistochemistry:
The expression of TopBP1 protein was detected in 90 human cervical tissue samples using anti-TopBP1 antibody (Abcam). Tissue sections (4 μm) were obtained from Formalin Fixed Paraffin Embedded blocks and IHC staining was performed using the Envision method 61. In brief, Nuclear staining within the squamous epithelium was assessed. IHC scoring analyses of TopBP1 expression were evaluated by Image pro-plus 6.0 (Media Cybernetics, Bethesda, MD, USA).
Antibodies and western blot analysis:
The antibodies from this study were as follows: anti-keratin-10, anti-loricrin, anti-TopBP1, and anti-GAPDH (Santa Cruz, Santa Cruz, CA); anti-E2F1, anti-p63, anti-ATR, anti-p-ATR, anti-CHK1, anti-p-CHK1, anti-CHK2, anti-p-CHK2, anti-RAD51, anti-Mre11, anti-NBS1, anti-BRCA1, and anti-BRCA2 (Cell Signaling, Danvers, MA); anti-p73 (Abcam, Cambridge, MA); anti-p53 (Millipore, Burlington, MA); anti-p-TopBP1 (Abgent, San Diego, CA). AKT inhibitors MK2206 (2μM) and LY294002 (25μM) were purchased from SelleckChem (Houston, TX). Cell lysates were processed and assayed by western blot analysis as previously described 60. Briefly, J2 feeders were firstly removed by Versene (PBS containing 0.5 mM EDTA) treatment and keratinocytes were collected and lysed in RIPA lysis buffer on ice for 30 minutes. The samples were then electrophoresed on SDS-page gels and transferred to PVDF membranes. At last the membranes were developed using ECL prime or ECL reagents (Amersham, Pittsburgh, PA) and chemiluminescence signals were detected using Eastman Kodak x-ray films.
Total RNA isolation and quantitative real-time PCR:
Total RNA was extracted using Trizol reagent (Invitrogen) or RNeasy mini kit (Qiagen) according to the manufacturer’s instructions. Tissue cDNA was generated using the PrimeScript RT reagent Kit (TaKaRa) and quantitative real-time PCR (qPCR) was performed using SYBR Premix Ex Taq (TaKaRa). The cDNA products from cell culture were transcribed using an iScript cDNA synthesis kit (Bio-Rad). The real-time PCR was performed using a LightCycler 480 with LightCycler 480 SYBR green I master mix (Roche, Indianapolis, IN) The specific primers were designed as follows: TopBP1 (5’-ATTACTCCAGGCCAAAGGAAGCAC-3’ (forward); 5’-AGCAAGGCTGGCTTGAGGTTTG-3’ (reverse)); RAD51(5’-GATGGAGCAGCGATGTTT-3’ (forward); 5’-GGTTTCCCCTCTTCCTTTC-3’ (reverse)); E2F1(5’-ACGTGACGTGTCAGGACCT-3’ (forward); 5’-GATCGGGCCTTGTTTGCTCTT-3’ (reverse)); p73(5’-ACGCAGCGAAACCGGGGCCCG-3’ (forward); 5’-GCCGCGCGGCTGCTCATCTGG-3’ (reverse)); GAPDH (5’-GAGGACAGAGACCCAGCTGCC-3’ (forward); 5’-TGGAATTTGCCATGGGTG-3’ (reverse)). The results are normalized to those for GAPDH, and the data shown is representative of observations from 3 independent experiments. Significance was determined using Student’s t test, and a P value of <0.05 was considered significant.
Southern blot analysis:
Total DNA was exacted according to the standard protocol and the DNA samples were then assayed by Southern blot analysis as previously described 26. Multiple nanodrop measurements were used to determine total DNA levels. Equal loading of gels was confirmed following electrophoresis by ethidium bromide staining.
Lentiviral virion production and transduction:
MISSION short hairpin RNA (shRNA) lentiviral vectors (Sigma-Aldrich, St. Louis, MO) expressing five TopBP1-specific shRNAs (constructs 1–5) were transfected into 293T cells and lentiviral particles were prepared as previously described 25. CIN612 cells were then incubated with concentrated TopBP1 shRNA or scramble shRNA lentiviral soup with 4 μg/ml hexadimethrine bromide (Polybrene; Sigma-Aldrich, St. Louis, MO) in 5ml total volume E media overnight at 37°C. The cells were washed the next day, and maintained in fresh E media for an additional 48 hours before further processing or analysis. Similar processes applied to the preparation of p73 knockdown cells.
RNA-seq analysis:
RNA-sequencing was performed at University of Chicago using Illumina HiSeq 2500 NGS platform. Sequencing data were used as input to CRI Illumina RNA-seq pipeline for reads mapping, post-alignment QC, and expression quantification, as well as DEGs identification. The quality of raw sequencing reads was assessed using FastQC v0.11.5 62. And the post-alignment QC was evaluated with RSeQC 63 and Picard tools v2.9.0 (http://broadinstitute.github.io/picard/). Reads were mapped to GENCODE human genome model (GRCh38) using STAR v2.5.3a 64. Gene transcripts were assembled and quantified on their corresponding human genome using count-based method featureCounts 65. Differentially expressed genes were identified with edgeR 66,67 by comparing the duplicate samples in the treatment group, versus the duplicate scramble control samples. Finally, functional enrichment analysis was performed with Bioconductor package clusterProfiler 68 in R (R Core Team, 2014). The raw sequencing data will be uploaded to NIH GEO database and will be accessible to the public once the manuscript is accepted.
Supplementary Material
Acknowledgement
This work is supported by grants from the NCI (CA059655 and CA142861) to L.A.L, ACS grant IRG-15–173-21 to S.H., and National Natural Scientific Foundation of China (No. 81702552) to J.X.
References
- 1.Hausen zur H Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2002; 2: 342–350. [DOI] [PubMed] [Google Scholar]
- 2.Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol 2006; 16: 83–97. [DOI] [PubMed] [Google Scholar]
- 3.Spurgeon ME, Lambert PF. Human Papillomavirus and the Stroma: Bidirectional Crosstalk during the Virus Life Cycle and Carcinogenesis. Viruses 2017; 9: 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR et al. The biology and life-cycle of human papillomaviruses. Vaccine 2012; 30 Suppl 5: F55–70. [DOI] [PubMed] [Google Scholar]
- 5.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer 2010; 10: 550–560. [DOI] [PubMed] [Google Scholar]
- 6.Sedman J, Stenlund A. Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J 1995; 14: 6218–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McBride AA. The papillomavirus E2 proteins. Virology 2013; 445: 57–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiMaio D, Petti LM. The E5 proteins. Virology 2013; 445: 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doorbar J The E4 protein; structure, function and patterns of expression. Virology 2013; 445: 80–98. [DOI] [PubMed] [Google Scholar]
- 10.Munger K, Howley PM. Human papillomavirus immortalization and transformation functions. Virus Res 2002; 89: 213–228. [DOI] [PubMed] [Google Scholar]
- 11.Wise-Draper TM, Wells SI. Papillomavirus E6 and E7 proteins and their cellular targets. Frontiers in bioscience : a journal and virtual library 2008; 13: 1003–1017. [DOI] [PubMed] [Google Scholar]
- 12.Wallace NA, Galloway DA. Novel Functions of the Human Papillomavirus E6 Oncoproteins. Annu Rev Virol 2015; 2: 403–423. [DOI] [PubMed] [Google Scholar]
- 13.Thomas JT, Hubert WG, Ruesch MN, Laimins LA. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proceedings of the National Academy of Sciences of the United States of America 1999; 96: 8449–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas M, Narayan N, Pim D, Tomaic V, Massimi P, Nagasaka K et al. Human papillomaviruses, cervical cancer and cell polarity. Oncogene 2008; 27: 7018–7030. [DOI] [PubMed] [Google Scholar]
- 15.Conway MJ, Meyers C. Replication and assembly of human papillomaviruses. Journal of dental research 2009; 88: 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong S-Y. DNA damage response is hijacked by human papillomaviruses to complete their life cycle. J Zhejiang Univ Sci B 2017; 18: 215–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ai W, Narahari J, Roman A. Yin yang 1 negatively regulates the differentiation-specific E1 promoter of human papillomavirus type 6. J Virol 2000; 74: 5198–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartley KA, Alexander KA. Human TATA binding protein inhibits human papillomavirus type 11 DNA replication by antagonizing E1-E2 protein complex formation on the viral origin of replication. J Virol 2002; 76: 5014–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Offord EA, Beard P. A member of the activator protein 1 family found in keratinocytes but not in fibroblasts required for transcription from a human papillomavirus type 18 promoter. J Virol 1990; 64: 4792–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Connor M, Bernard HU. Oct-1 activates the epithelial-specific enhancer of human papillomavirus type 16 via a synergistic interaction with NFI at a conserved composite regulatory element. Virology 1995; 207: 77–88. [DOI] [PubMed] [Google Scholar]
- 21.Stunkel W, Bernard HU. The chromatin structure of the long control region of human papillomavirus type 16 represses viral oncoprotein expression. J Virol 1999; 73: 1918–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gunasekharan V, Hache G, Laimins L. Differentiation-dependent changes in levels of C/EBPbeta repressors and activators regulate human papillomavirus type 31 late gene expression. J Virol 2012; 86: 5393–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gunasekharan VK, Li Y, Andrade J, Laimins LA. Post-Transcriptional Regulation of KLF4 by High-Risk Human Papillomaviruses Is Necessary for the Differentiation-Dependent Viral Life Cycle. PLoS Pathog 2016; 12: e1005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang W, Hong S, Maniar KP, Cheng S, Jie C, Rademaker AW et al. KLF13 regulates the differentiation-dependent human papillomavirus life cycle in keratinocytes through STAT5 and IL-8. Oncogene 2016. 10.1038/onc.2016.97. [DOI] [PubMed]
- 25.Hong S, Laimins LA. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog 2013; 9: e1003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong S, Cheng S, Iovane A, Laimins LA. STAT-5 Regulates Transcription of the Topoisomerase IIβ-Binding Protein 1 (TopBP1) Gene To Activate the ATR Pathway and Promote Human Papillomavirus Replication. MBio 2015; 6: e02006–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edwards TG, Helmus MJ, Koeller K, Bashkin JK, Fisher C. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J Virol 2013; 87: 3979–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moody CA, Laimins LA. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 2009; 5: e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kidiyoor GR, Kumar A, Foiani M. ATR-mediated regulation of nuclear and cellular plasticity. DNA Repair (Amst) 2016. 10.1016/j.dnarep.2016.05.020. [DOI] [PMC free article] [PubMed]
- 30.Liu K, Lin FT, Ruppert JM, Lin WC. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol Cell Biol 2003; 23: 3287–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu K, Paik JC, Wang B, Lin F-T, Lin W-C. Regulation of TopBP1 oligomerization by Akt/PKB for cell survival. EMBO J 2006; 25: 4795–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu K, Ling S, Lin W-C. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol Cell Biol 2011; 31: 4464–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herold S, Hock A, Herkert B, Berns K, Mullenders J, Beijersbergen R et al. Miz1 and HectH9 regulate the stability of the checkpoint protein, TopBP1. EMBO J 2008; 27: 2851–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu K, Graves JD, Scott JD, Li R, Lin W-C. Akt switches TopBP1 function from checkpoint activation to transcriptional regulation through phosphoserine binding-mediated oligomerization. Mol Cell Biol 2013; 33: 4685–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell 2006; 124: 943–955. [DOI] [PubMed] [Google Scholar]
- 36.Mehta K, Laimins L. Human Papillomaviruses Preferentially Recruit DNA Repair Factors to Viral Genomes for Rapid Repair and Amplification. MBio 2018; 9: e00064–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu K, Bellam N, Lin H-Y, Wang B, Stockard CR, Grizzle WE et al. Regulation of p53 by TopBP1: a potential mechanism for p53 inactivation in cancer. Mol Cell Biol 2009; 29: 2673–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA et al. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes Dev 2002; 16: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M et al. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol 2001; 21: 4684–4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chappell WH, Gautam D, Ok ST, Johnson BA, Anacker DC, Moody CA. Homologous Recombination Repair Factors Rad51 and BRCA1 Are Necessary for Productive Replication of Human Papillomavirus 31. J Virol 2015; 90: 2639–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mighty KK, Laimins LA. p63 is necessary for the activation of human papillomavirus late viral functions upon epithelial differentiation. J Virol 2011; 85: 8863–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993; 75: 495–505. [DOI] [PubMed] [Google Scholar]
- 43.Lechner MS, Mack DH, Finicle AB, Crook T, Vousden KH, Laimins LA. Human papillomavirus E6 proteins bind p53 in vivo and abrogate p53-mediated repression of transcription. EMBO J 1992; 11: 3045–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lorenz LD, Rivera Cardona J, Lambert PF. Inactivation of p53 rescues the maintenance of high risk HPV DNA genomes deficient in expression of E6. PLoS Pathog 2013; 9: e1003717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reiser J, Hurst J, Voges M, Krauss P, Münch P, Iftner T et al. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J Virol 2011; 85: 11372–11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeon S, Lambert PF. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America 1995; 92: 1654–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morrison C, Sonoda E, Takao N, Shinohara A, Yamamoto K, Takeda S. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J 2000; 19: 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005; 308: 551–554. [DOI] [PubMed] [Google Scholar]
- 49.Donaldson MM, Mackintosh LJ, Bodily JM, Dornan ES, Laimins LA, Morgan IM. An interaction between human papillomavirus 16 E2 and TopBP1is required for optimum viral DNA replication and episomal genome establishment. J Virol 2012. 10.1128/JVI.01002-12. [DOI] [PMC free article] [PubMed]
- 50.Melar-New M, Laimins LA. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J Virol 2010; 84: 5212–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stiewe T, Pützer BM. Role of p73 in malignancy: tumor suppressor or oncogene? Cell Death Differ 2002; 9: 237–245. [DOI] [PubMed] [Google Scholar]
- 52.Fontemaggi G, Kela I, Amariglio N, Rechavi G, Krishnamurthy J, Strano S et al. Identification of direct p73 target genes combining DNA microarray and chromatin immunoprecipitation analyses. J Biol Chem 2002; 277: 43359–43368. [DOI] [PubMed] [Google Scholar]
- 53.Concin N, Becker K, Slade N, Erster S, Mueller-Holzner E, Ulmer H et al. Transdominant DeltaTAp73 isoforms are frequently up-regulated in ovarian cancer. Evidence for their role as epigenetic p53 inhibitors in vivo. Cancer Res 2004; 64: 2449–2460. [DOI] [PubMed] [Google Scholar]
- 54.Niklison-Chirou MV, Killick R, Knight RA, Nicotera P, Melino G, Agostini M. How Does p73 Cause Neuronal Defects? Mol Neurobiol 2016; 53: 4509–4520. [DOI] [PubMed] [Google Scholar]
- 55.Wang Z, Sturgis EM, Guo W, Song X, Zhang F, Xu L et al. Association of combined p73 and p53 genetic variants with tumor HPV16-positive oropharyngeal cancer. PLoS ONE 2012; 7: e35522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Accardi R, Dong W, Smet A, Cui R, Hautefeuille A, Gabet A-S et al. Skin human papillomavirus type 38 alters p53 functions by accumulation of deltaNp73. EMBO Rep 2006; 7: 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pacini L, Savini C, Ghittoni R, Saidj D, Lamartine J, Hasan UA et al. Downregulation of Toll-Like Receptor 9 Expression by Beta Human Papillomavirus 38 and Implications for Cell Cycle Control. J Virol 2015; 89: 11396–11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Habiger C, Jäger G, Walter M, Iftner T, Stubenrauch F. Interferon Kappa Inhibits Human Papillomavirus 31 Transcription by Inducing Sp100 Proteins. J Virol 2016; 90: 694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fehrmann F, Laimins LA. Human papillomavirus type 31 life cycle: methods for study using tissue culture models. Methods Mol Biol 2005; 292: 317–330. [DOI] [PubMed] [Google Scholar]
- 60.Hong S, Mehta KP, Laimins LA. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J Virol 2011; 85: 9486–9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fang Y, Yu H, Liang X, Xu J, Cai X. Chk1-induced CCNB1 overexpression promotes cell proliferation and tumor growth in human colorectal cancer. Cancer Biol Ther 2014; 15: 1268–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andrews S FastQC: a quality control tool for high throughput sequence data 2010.
- 63.Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 2012; 28: 2184–2185. [DOI] [PubMed] [Google Scholar]
- 64.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014; 30: 923–930. [DOI] [PubMed] [Google Scholar]
- 66.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 2012; 40: 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters https://homeliebertpubcom/omi 2012; 16: 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.