

Abstract
The HPV life cycle is differentiation-dependent, with cellular differentiation driving initiation of the late, productive stage of the viral life cycle. Here, we identify a role for the protein NFX1-123 in regulating keratinocyte differentiation and events of the late HPV life cycle. NFX1-123 itself increased with differentiation of epithelial cells. Greater NFX1-123 augmented differentiation marker expression and JNK phosphorylation in differentiating 16E6-expressing human foreskin keratinocytes (16E6 HFKs). This was associated with altered expression of MKK4 and MKK7, upstream kinase regulators of JNK phosphorylation. Modulating levels of NFX1-123 in HPV16-positive W12E cells recapitulated the effects on differentiation markers, JNK phosphorylation, and MKK4/7 seen in 16E6 HFKs. Crucially, levels of NFX1-123 also correlated with expression of L1, the capsid protein of HPV. Altogether, these studies define a role for NFX1-123 in mediating epithelial differentiation through the JNK signaling pathway, potentially linking expression of cellular genes and HPV genes during differentiation.
Keywords: NFX1-123, HR HPV, HR E6, Differentiation, Keratinocyte
INTRODUCTION
The human papillomaviruses (HPVs) are small, non-enveloped, double-stranded DNA viruses that infect keratinocytes of cutaneous and mucosal stratified squamous epithelium (1, 2). There are over 200 types of HPV, and those that specifically target mucosal epithelium are further categorized as low-risk HPV (LR HPV) or high-risk HPV (HR HPV) based on their epidemiologic association with cancer (3–5). HR HPV cause nearly all cervical cancer, the fourth most common cancer in women, in addition to other anogenital and oropharyngeal cancers (3, 6–9). Altogether, cancers caused by HR HPV account for approximately 5% of the global burden of cancer (10).
HPV has a unique life cycle that is tied to the differentiation programming of its target cell, the keratinocyte (11, 12). The viral life cycle can be divided into two stages: in the first, HPV infects keratinocytes in the basal layer of stratified squamous epithelium and maintains its genome as an episome at a low copy number (1, 11–14). In this stage, the early genes (E1, E2, E4, E5, and oncogenes E6 and E7) are expressed from the early viral promoter (13, 15, 16). The second stage occurs once an infected cell rises through the epithelial suprabasal layers and begins to differentiate. This triggers the late stage of the viral life cycle, whose events include vegetative genome amplification, expression of the capsid proteins L1 and L2 from a viral late promoter, and ultimately, complete virion formation (1, 15, 17). As a keratinocyte differentiates and moves up through the layers of stratified squamous epithelium, it expresses a successive series of proteins that commonly serve as differentiation markers. These include Keratins 1 and 10, Involucrin, Loricrin, and Filaggrin (18, 19). Similarly, HPV expresses its genes in a precisely ordered fashion in association with specific epithelial layers. There is thus a tight link between events dictating the expression of cellular genes during differentiation and those dictating expression of HPV genes. Although it is known that terminal differentiation of epithelial cells is required for completion of the viral life cycle, the cellular factors activated during epithelial differentiation, and how these factors signal to induce the appropriate viral promoters, are not fully defined.
Efforts to understand the regulation of differentiation-dependent HPV gene transcription have implicated a wide variety of transcription factors and cellular processes. These include transcription factors specific to differentiating keratinocytes, CDP and EPOC-1, as well as ubiquitously expressed transcription factors whose levels may change during epithelial cell differentiation such as AP-1 family members, Oct1, C/EBP, YY1, and others (20–29). In addition to global changes in levels of transcription factors, mechanisms including shifting ratios of factors (Sp1/Sp3) (30), increased binding to HPV promoters during epithelial differentiation (31), and activation of cellular signaling pathways (protein kinase C) (32) have been associated with HPV transcription. Furthermore, linker scanning mutational studies have mapped cis regulatory elements upstream of the HPV promoters that may be important during different stages of the life cycle (33, 34).
The understanding of how differentiation-dependent HPV gene transcription is regulated is further complicated by a switch from the early viral promoter to the late viral promoter. The late viral promoter does not become active until the host cell rises to the upper granular and cornified layers of the stratified squamous epithelium. Even after infected cells have begun differentiating, there are additional signals required to activate the viral late promoter. Studies have shown a role for SRp20, a cellular splicing factor, and transcription elongation factors (35, 36), but many gaps remain in our knowledge of what controls this transition in HPV transcription. Despite the abundance of transcription factors and complexity of mechanisms shown to be associated with differentiation-dependent HPV transcription, there is still not a complete understanding of the process.
We have previously shown that HR HPV type 16 E6 (16E6) and NFX1-123, a known cellular protein partner of 16E6, collaboratively increase levels of Notch1 (37). Notch1 is a master regulator of cellular growth and differentiation (38, 39). Upon receptor stimulation in these cells, the increased Notch1 expression resulted in an upregulation of its canonical target genes, Hes1 and Hes5, as well as the differentiation markers Keratin 1 and Keratin 10 (40). Blocking Notch1 signaling, however, did not abrogate expression of these differentiation markers, suggesting additional Notch1-independent mechanisms by which 16E6 and NFX1-123 are able to affect keratinocyte differentiation.
In the current study, we further explore the role of NFX1-123 in mediating epithelial differentiation. We utilize targeted differentiation stimuli to further delineate these Notch1 independent mechanisms and demonstrate that NFX1-123, a host cellular protein partner of HPV type 16 E6, is able to regulate differentiation marker expression in HFKs and HPV16-positive cells through JNK signaling. Finally, we show that NFX1-123’s role in mediating epithelial differentiation is ultimately associated with expression of HPV16 L1, pointing to its potential as a cellular factor connecting epithelial differentiation and the differentiation-dependent HPV life cycle.
RESULTS
Expression levels of NFX1-123 increased with epithelial cell differentiation
NFX1-123 is a known cellular protein partner of HR HPV type 16 E6 (16E6) and is endogenously expressed in epithelial cells (41). Although NFX1-123 is increased in cervical cancer cell lines (40), HPV positive cervical precancerous lesions, and HPV-positive cervical cancers (42), the overall expression of NFX1-123 in normal epithelial tissue has not been characterized. To do so, we utilized organotypic raft cultures, which approximate the growth and physiology of stratified squamous epithelium, as well as archived normal cervical epithelial biopsies. Raft cultures were grown using human foreskin keratinocytes (HFKs), HFKs expressing 16E6 and endogenous levels of NFX1-123 (16E6/LXSN HFKs), and HFKs expressing 16E6 and overexpressing FLAG-tagged NFX1-123 (16E6/FN123 HFKs). Expression of 16E6 in HFKs was confirmed by immunoblot documenting a reduction of p53 (Supp Figure 1). These rafts and normal cervical epithelial samples were stained for NFX1-123 via immunohistochemistry (Fig 1A).
Fig 1. NFX1-123 expression increases with keratinocyte differentiation.

(A) Organotypic raft cultures were grown using HFKs, HFKs serially transduced with 16E6 and vector control (16E6/LXSN), and HFKs transduced with 16E6 and FLAG-tagged NFX1-123 overexpression construct (16E6/FN123). These rafts and normal cervical epithelial biopsies were stained for NFX1-123 protein via immunohistochemistry or stained using a rabbit polyclonal IgG as a control. Staining intensity was measured over a linear distance from the basal layer in ImageJ and the average of four plots over two sections is shown. (B and C) 16E6/LXSN and 16E6/FN123 HFKs were differentiated with 1.8mM Ca2+ treatment for 0, 72, or 120 hours (B) or suspension in 1.7% methylcellulose medium (MC) (C) and total mRNA and protein collected. Mean expression of NFX1-123 mRNA was measured by qPCR and compared to 16E6/LXSN 0 hours. Protein levels of NFX1-123 were measured by Western blot using actin or GAPDH as a loading control. Induction of the differentiation marker Keratin 1 indicates successful differentiation. White spaces in Western blots indicate removal of empty or irrelevant lanes from original image for clarity. (D) HFKs transduced with empty vector or with 16E6 were differentiated with 1.8mM Ca2+ treatment for 0, 72, or 120 hours, and total mRNA and protein collected. Mean expression of NFX1-123 mRNA was measured by qPCR and each sample was compared to HFK/vector 0 hours. Protein levels of NFX1-123 were measured by Western blot using actin as a loading control. For B, C, and D, mRNA expression of NFX1-123 for each sample was normalized to the housekeeping gene 36B4, and all error bars represent 95% confidence intervals from replicates. Protein expression of NFX1-123 for each sample was quantified using ImageJ and normalized to the loading control for that sample. One representative experiment is shown from at least three conducted in biologically independent HFK cell lines. Statistical significance was calculated using one-way ANOVA with Bonferroni correction. * p≤0.05, ** p≤0.01, *** p≤0.001, **** p≤0.0001
Matching previous findings that NFX1-123 has primarily cytoplasmic subcellular localization, there was cytoplasmic staining in cells throughout the raft cultures and cervical epithelium, with some perinuclear staining seen in basal layer cells. This perinuclear staining in basal layer cells was sporadic in the raft cultures, but especially evident in the normal cervical epithelium (Fig 1A). In normal cervical epithelium, NFX1-123 staining was predominately perinuclear in basal layers and became predominately cytoplasmic in intermediate and upper layers. Expression of 16E6 did not affect expression of NFX1-123 (HFK vs 16E6/LXSN). Rafts grown with 16E6/FN123 cells that overexpressed NFX1-123 had darker staining of NFX1-123 indicating that NFX1-123 overexpression was maintained.
Interestingly, we noted that expression of NFX1-123 rose overall with increasing distance from the basal layer in the raft cultures and normal cervical epithelium. Indeed, when staining intensity was plotted as a function of distance from the basal layer, there was a direct relationship (Fig 1A, graphs). Because epithelial cell differentiation also progresses with increasing distance from the basal layer, we hypothesized that NFX1-123 expression was associated with cells undergoing differentiation.
To identify a direct link between cellular differentiation and NFX1-123 expression, we induced differentiation in 16E6 HFKs using high-dose calcium or suspension in methylcellulose and quantified NFX1-123 mRNA and protein levels. 16E6/LXSN and 16E6/FN123 HFKs were cultured in media containing 1.8mM Ca2+ and total mRNA and protein were serially collected. There was a significant induction of the differentiation marker Keratin 1 after calcium treatment, indicating effective differentiation (Fig 1B, Western blot). mRNA and protein levels of NFX1-123 were higher after 72 hours of calcium exposure, compared to undifferentiated cells with no calcium exposure, and rose even further after 120 hours (Fig 1B). Induction of NFX1-123 with differentiation was observed even in 16E6/FN123 cells that began with greater expression. To further test the association between epithelial differentiation and NFX1-123 expression, we induced differentiation in 16E6 HFKs using another well-characterized stimulus, suspension in methylcellulose (43–45). Again, induction of Keratin 1 indicated successful differentiation (Fig 1C). With methylcellulose suspension, 16E6/LXSN and 16E6/FN123 cells had rapid and robust increases NFX1-123 levels at 24 hours; these levels were slightly increased or maintained at 48 hours (Fig 1C, Western blot). NFX1-123 expression increased during suspension-induced differentiation in both 16E6/LXSN and 16E6/FN123 cells. Finally, we compared endogenous NFX1-123 expression in HFKs and 16E6 HFKs induced to differentiate with high-dose calcium. NFX1-123 was increased with differentiation in HFKs, and 16E6 did not have any significant impact on its endogenous expression or induction. Altogether, these data indicated that NFX1-123 itself was increased with keratinocyte differentiation, even in cells that began with overexpressed NFX1-123, and that this upregulation was unaffected by 16E6 expression.
NFX1-123 mediated keratinocyte differentiation in both Notch1-dependent and Notch1-independent manners
We have demonstrated previously that greater NFX1-123 affected keratinocyte pathways (37, 40, 46) and increased expression of differentiation markers Keratin 1 and Keratin 10 (40). Because these studies had been conducted in non-differentiating conditions, we were interested in interrogating whether NFX1-123 regulates expression of these markers in the context of full keratinocyte differentiation. 16E6/LXSN and 16E6/FN123 raft cultures were grown and stained for differentiation markers Keratin 1 and Loricrin (Fig 2A). FN123 rafts with greater levels of NFX1-123 had more intense staining of these differentiation markers compared to LXSN rafts. These data confirmed that greater NFX1-123 increased expression of these markers when cells were actively differentiating.
Fig 2. NFX1-123 regulates expression of differentiation markers in 16E6 HFKs.
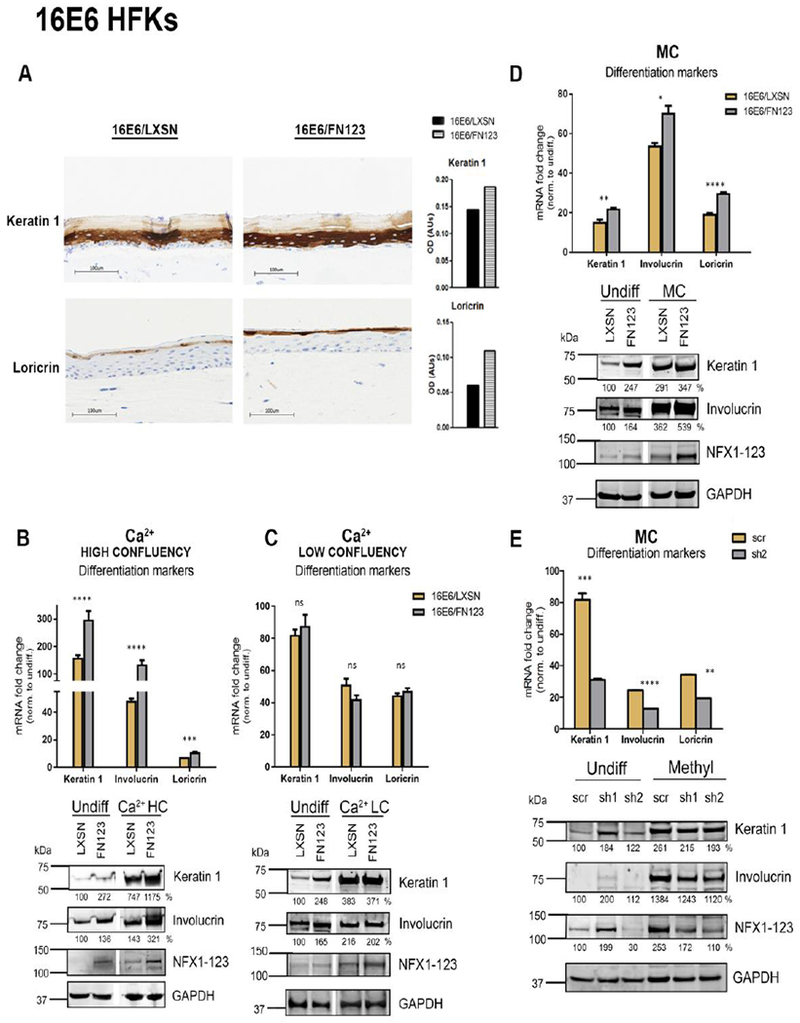
(A) 16E6/LXSN and 16E6/FN123 raft cultures were stained for protein expression of differentiation markers Keratin 1 and Loricrin via immunohistochemistry. Total staining intensity for the section area was calculated in ImageJ. (B and C) 1×106 16E6/LXSN and 16E6/FN123 HFKs (B) or 3×105 16E6/LXSN and 16E6/FN123 HFKs (C) were plated and treated with 1.8mM Ca2+ for 0 or 72 hours, and total mRNA and protein collected. (D) 1×106 16E6/LXSN and 16E6/FN123 HFKs were suspended in methylcellulose (MC) for 0 or 24 hours, and total mRNA and protein collected. (B, C, and D) mRNA levels of Keratin 1, Involucrin, and Loricrin were measured by qPCR and compared to 16E6/LXSN 0 hours. Protein levels of Keratin 1, Involucrin, and NFX1-123 were assessed by Western blot. GAPDH was used as a loading control. Densitometry analysis was done in ImageJ. Protein levels were normalized to the loading control, and all samples compared to undifferentiated 16E6/LXSN. Statistical significance was calculated using unpaired t-tests. White spaces in Western blots indicate removal of empty or irrelevant lanes from original image for clarity. (E) 16E6 HFKs were transduced with short hairpins targeting NFX1-123 (sh1 and sh2) or scramble short hairpin control (scr). 1×106 scr, sh1, and sh2 cells were suspended in methylcellulose (MC) for 0 or 24 hours, and total mRNA and protein collected. mRNA levels of Keratin 1, Involucrin, and Loricrin were measured by qPCR and compared to scr 0 hours. Statistical significance was calculated using one-way ANOVA with Bonferroni correction. Protein levels of Keratin 1, Involucrin, and NFX1-123 were assessed by Western blot. GAPDH was used as a loading control. Densitometry analysis was done in ImageJ. Protein levels were normalized to the loading control, and all samples compared to undifferentiated 16E6/scr. For B, C, D, and E, expression levels of genes of interest were normalized to the housekeeping gene GAPDH, and all error bars represent 95% confidence intervals from replicates. One representative experiment is shown from at least three conducted in biologically independent HFK cell lines. * p≤0.05, ** p≤0.01, *** p≤0.001, **** p≤0.0001
Previous data from our laboratory has shown that NFX1-123 regulated Notch1 expression and consequently affected its canonical pathway targets (37, 40). This effect of NFX1-123 and Notch1 was also linked to keratinocyte differentiation, but regulation of that pathway by NFX1-123 was less direct. Specifically, when Notch1 was activated in 16E6 HFKs overexpressing NFX1-123, canonical and differentiation targets were increased; however, when Notch1 activation was blocked in these same cells, the canonical pathway targets of Notch1 rapidly fell while differentiation pathway targets did not (40). To more deeply interrogate the interplay between Notch1, NFX1-123, and keratinocyte differentiation, we selected methods to stimulate differentiation in epithelial cells that would limit or permit Notch1 signaling. In selecting differentiation stimuli, we leveraged the fact that Notch1 pathway activation requires cell-to-cell contact for signaling to occur (47–49). We additionally wanted to identify how NFX1-123 altered differentiation pathways when 16E6 was co-expressed, to model how NFX1-123 might regulate differentiation in HPV infected cells. NFX1-123 interacts with E6, and our previous work showed synergy of NFX1-123 with 16E6 in increasing differentiation markers (40). Modulating NFX1-123 in the presence of 16E6 allowed us to probe the role of NFX1-123 itself.
First, 16E6/LXSN and 16E6/FN123 HFKs were plated at high confluency and treated with high-dose calcium for 72 hours to induce differentiation. The high confluency allows for ample cell-cell contact and is thus permissive for Notch1 activation and signaling (48, 49). This Notch1 signaling was reflected in the high induction of the canonical Notch1 target gene Hes1 (Supp Fig 2) (47). 16E6/LXSN HFKs had a robust upregulation of differentiation markers Keratin 1, Involucrin, and Loricrin (Fig 2B, gold bars), and there was an even greater induction in the 16E6/FN123 HFKs (Fig 2B, gray bars). This was reflected at the protein level as well; in agreement with our previous studies, we saw a greater expression of differentiation markers with more NFX1-123, even in undifferentiated cells (Fig 2B). Following differentiation, 16E6/FN123 HFKs had greater levels of Keratin 1 and Involucrin compared to 16E6/LXSN (Fig 2B). This confirmed that NFX1-123 played a role in mediating keratinocyte differentiation when Notch1 signaling could occur.
Next, cells were plated at low confluency (~10%), which limited cell-cell contact and therefore limited Notch1 signaling. Accordingly, there was no change in Hes1 expression in these cells (Supp Fig 2). These low confluency 16E6 HFKs were treated with high dose of calcium to induce differentiation. There was again an upregulation of Keratin 1, Involucrin, and Loricrin, indicating epithelial differentiation was triggered; there was, however, no difference in the mRNA or protein levels of differentiation markers in 16E6/LXSN cells compared to 16E6/FN123 (Fig 2C). Without the availability of Notch1 signaling, greater expression of NFX1-123 did not augment differentiation marker induction in 16E6 HFKs stimulated by high-dose calcium alone.
Lastly, 16E6 HFKs were stimulated to differentiate with suspension in semisolid methylcellulose medium. This stimulus virtually eliminates cell-cell contact and Notch1 signaling, similar to low confluency plating, but the intracellular signaling pathways that are activated and lead to differentiation are distinct from those activated by high-dose calcium (44, 50). Again, confirming restricted Notch1 signaling, there was no induction of Hes1 (Supp Fig 2). Examining levels of differentiation markers, 16E6/FN123 HFKs had a greater induction of these markers at both the mRNA and protein level compared to 16E6/LXSN cells, indicating that NFX1-123 was able to mediate keratinocyte differentiation stimulated by suspension and independent of Notch1 (Fig 2D). Overexpression of NFX1-123 alone increased induction of differentiation markers even in the absence of 16E6; however, expression of 16E6 augmented this trend (data not shown). To further confirm the involvement of NFX1-123, 16E6 HFKs expressing short hairpin RNAs targeting NFX1-123 (sh1 and sh2) were differentiated via methylcellulose suspension. Knock down of NFX1-123 reduced induction of differentiation markers at the mRNA level relative to control (scr) (Fig 2E). Although the knock down of NFX1-123 was not evident in undifferentiated sh1 cells, there was still a dose-dependent association between NFX1-123 expression and differentiation marker expression. When NFX1-123 was knocked down, this did affect the low levels of Keratin 1 and Involucrin protein in undifferentiated cells (scr vs. sh2). After methylcellulose suspension, both sh1 and sh2 cells had decreased levels of NFX1-123 and had reduced induction of Keratin 1 and Involucrin compared to scr (Fig 2E). These data collectively identified that NFX1-123 itself was increased during differentiation (Fig 1); that with its increased expression, NFX1-123 augmented differentiation stimulated by methylcellulose suspension (Fig 2D); and that NFX1-123 could achieve this through Notch1-independent signaling pathways.
Intracellular signaling and MAP kinase pathways triggered by differentiation stimuli
Extensive work in the field of epidermal differentiation has characterized the intracellular signaling pathways that are activated by the high-dose calcium and methylcellulose suspension, and how these signaling pathways subsequently lead to induction of differentiation markers (44, 50–55). These complex pathways have been simplified in the schema shown in Fig 3. In addition to their utility for investigating differentiation with or without Notch1 involvement, the stimuli chosen have unique intracellular signaling pathways (Fig 3). Identifying where these signaling pathways overlap and differ may indicate a cellular pathway through which NFX1-123 affects differentiation. NFX1-123 regulated keratinocyte differentiation stimulated by suspension and by calcium in high confluency cells, but not low confluency. Examining the intracellular signaling pathways activated by high confluency calcium and suspension in methylcellulose, we saw that they share components of mitogen-activated protein kinase (MAPK) signaling. In the case of methylcellulose suspension, these are specifically the MAPKs ERK and JNK (50, 56). We hypothesized that these two kinases were therefore potential targets through which NFX1-123 mediated epithelial cell differentiation.
Fig 3. Diagram of intracellular signaling pathways activated by differentiation stimuli.
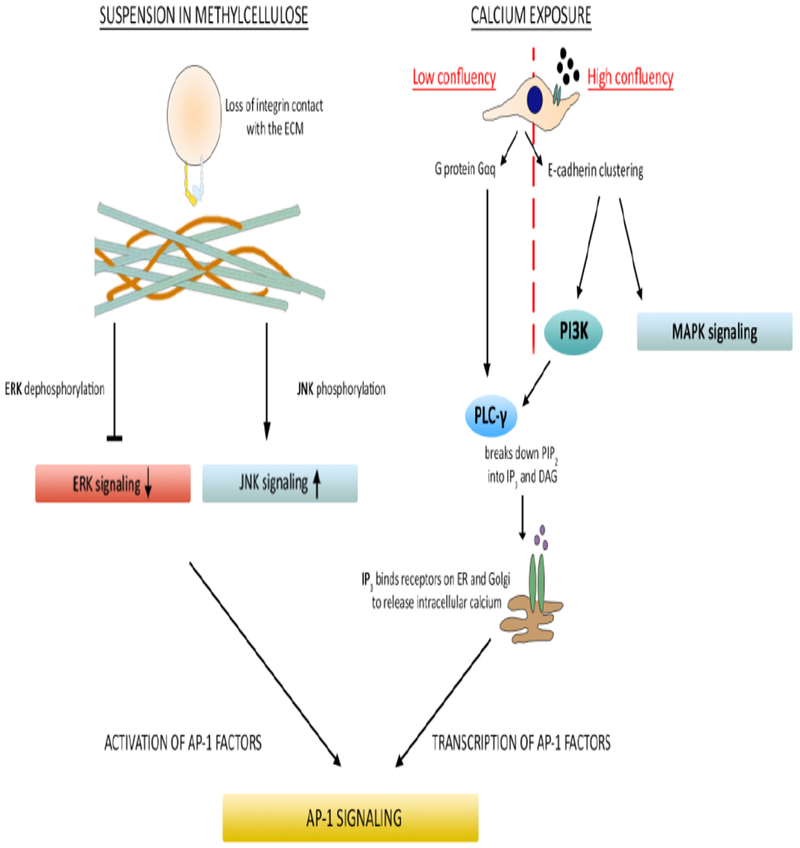
A representation of the intracellular signaling pathways known to be activated by differentiation stimuli used in Figures 1 and 2. Following suspension in methylcellulose, phosphorylation and signaling activity of extracellular signal-regulated kinase (ERK) decreases. Phosphorylation and signaling activity of c-Jun N-terminal kinase (JNK) increases. This leads to activation of AP-1 transcription factors and subsequent transcriptional expression of differentiation markers. For cells plated at low confluency, exposure to extracellular calcium activates the G protein Gαq, which in turn activates phospholipase C gamma (PLC-γ). PLC-γ hydrolyzes the second messenger phosphatidylinositol 4,5-bisphosphate (PIP2) and activates a series of reactions that lead to the transcription of AP-1 subunits, AP-1 signaling and subsequent transcriptional expression of differentiation markers. For cells plated at high confluency, exposure to extracellular calcium leads to E-cadherin clustering, activating mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) activity. PI3K activates PLC-γ and here, the pathway converges with that of low confluency cells treated with calcium terminating in AP-1 signaling and transcription of differentiation markers.
16E6 and NFX1-123 mediate levels of activated JNK through upstream kinases
To investigate whether 16E6 and NFX1-123 alter JNK and ERK signaling, we returned to 16E6 HFKs with endogenous (LXSN) or overexpressed NFX1-123 (FN123) that were suspended in methylcellulose. Upon differentiation, there were no significant differences seen in total protein levels of JNK or ERK, regardless of the amount of NFX1-123 (Fig 4A). Because these kinases are activated by phosphorylation, we also examined levels of phospho-JNK (P-JNK) and phospho-ERK (P-ERK). Suspension-induced differentiation resulted in decreased P-ERK; the small amounts were difficult to quantify and thus differences due to NFX1-123 could not be determined (data not shown). However, upon methylcellulose-induced differentiation, cells with more NFX1-123 had greater levels of P-JNK (Fig 4A). JNK can be phosphorylated by a number of MAP kinase kinases (MKKs), but in particular by MKK4, which is specific for JNK and p38, and MKK7, which is solely JNK-specific (57, 58). Intriguingly, 16E6/FN123 HFKs showed greater amounts of MKK4 and MKK7 compared to 16E6/LXSN HFKs. This greater MKK4 and MKK7 with more NFX1-123 was observed in both undifferentiated and differentiated cells (Fig 4A).
Fig 4. NFX1-123 regulates phosphorylation of JNK through MKK4 and MKK7.
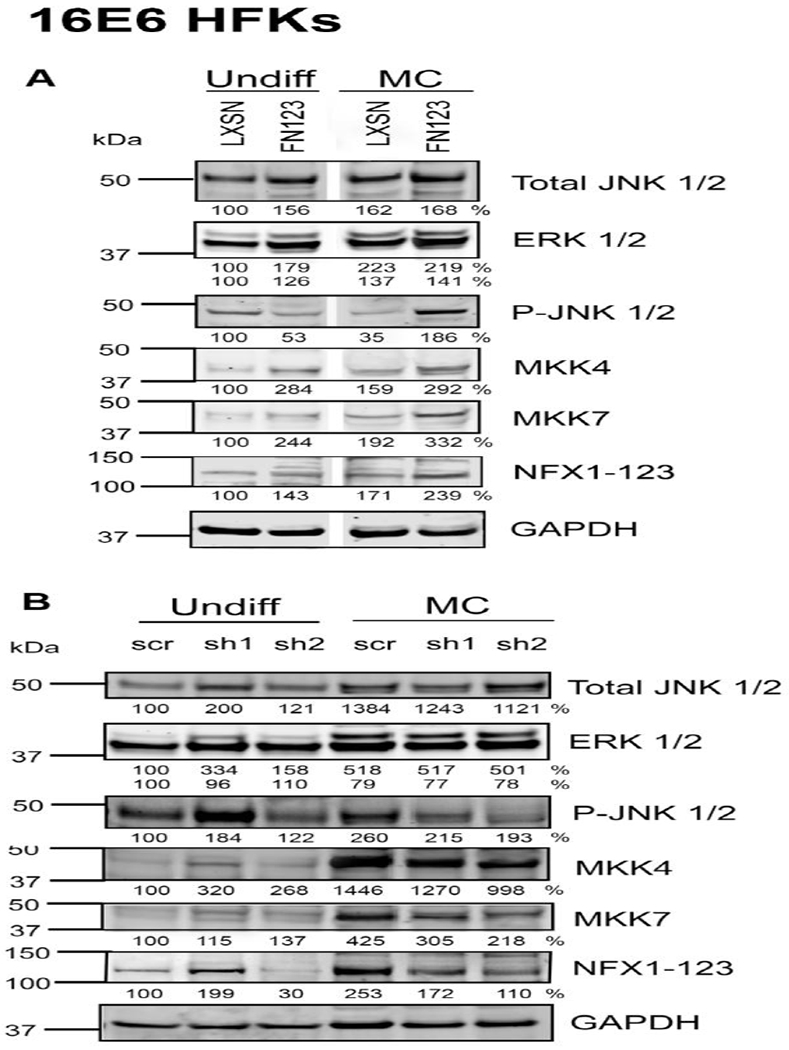
(A) 1×106 16E6/LXSN and 16E6/FN123 HFKs were suspended in methylcellulose (MC) for 0 or 24 hours, and total protein collected. Protein levels of phosphorylated JNK (P-JNK), total JNK, total ERK, total MKK4, and total MKK7 were assessed by Western blot. GAPDH was used as a loading control. Densitometry analysis was done in ImageJ. Protein levels were normalized to the loading control, and all samples compared to undifferentiated 16E6/LXSN. White spaces in Western blots indicate removal of empty or irrelevant lanes from original image for clarity. (B) 16E6 HFKs were transduced with a short hairpin targeting NFX1-123 (sh1 or sh2) or a scramble short hairpin control (scr). 1×106 scr, sh1, and sh2 cells were suspended in methylcellulose for 0 or 24 hours, and total protein collected. Protein levels of P-JNK, total JNK, total ERK, total MKK4, and total MKK7 were assessed by Western blot. Densitometry analysis was done in ImageJ. Protein levels were normalized to the loading control, and all samples compared to undifferentiated scr. For A and B, one representative experiment is shown from at least three conducted in biologically independent HFK cell lines.
To confirm the required role of NFX1-123 in JNK phosphorylation and MKK expression, we examined levels of these signaling proteins in the 16E6 HFKs expressing short hairpins against NFX1-123 that had been differentiated in methylcellulose. When NFX1-123 was knocked down by short hairpin RNA, there were again no differences in total levels of ERK or JNK (Fig 4B). Knockdown of NFX1-123 did, however, result in decreased levels of P-JNK, MKK4, and MKK7 upon differentiation (Fig 4B). These data indicate that NFX1-123 was required for full MKK4 and MKK7 expression and JNK phosphorylation upon suspension-induced differentiation.
NFX1-123 regulated JNK signaling and differentiation in HPV16-positive W12E cells and modulated HPV16 L1 expression
Given that NFX1-123 regulated keratinocyte differentiation marker expression, that this regulation by NFX1-123 was augmented by 16E6 (40), and that HPV late gene transcription is differentiation-dependent, we wished to assess if NFX1-123 impacted and supported these events of the late HPV life cycle. To explore this, we utilized W12E cells, HPV16-positive cells that harbor the full viral genome (59). First, we wanted to confirm that NFX1-123 was endogenously expressed in these cells, and that its expression responded to differentiation in a similar manner as HFKs. Cells were transduced with vector control (LXSN) or NFX1-123 overexpression construct (FN123) and then suspended in methylcellulose for 24 or 48 hours to induce differentiation. Comparable to the results observed in 16E6 HFKs, NFX1-123 mRNA was expressed in W12E cells and increased in both W12E LXSN and W12E FN123 cells with 24 hours of methylcellulose suspension (Fig 5A). These levels were further increased or maintained at 48 hours (Fig 5A). Endogenous protein levels of NFX1-123 in W12E cells appeared to be lower than in HFKs, as NFX1-123 protein could not be detected by immunoblot in either undifferentiated W12E LXSN or W12E FN123 cells. Upon differentiation, however, NFX1-123 protein increased to detectable amounts, and greater protein could be seen in W12E FN123 cells relative to W12E LXSN cells (Fig 5A).
Fig 5. NFX1-123 mediates differentiation and L1 expression in HPV16-positive W12E cells.
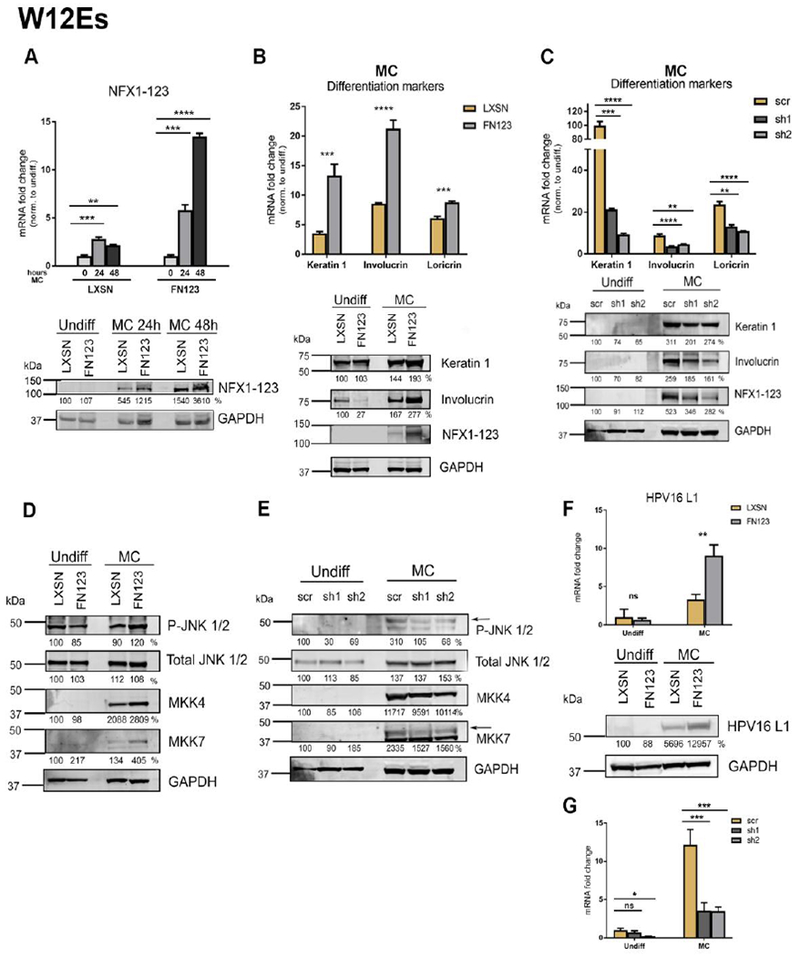
(A) W12E cells were transduced with LXSN control vector (LXSN) or FLAG-tagged NFX1-123 overexpression construct (FN123) and 1×106 cells were suspended in methylcellulose (MC) for 0, 24, or 48 hours. Mean expression of NFX1-123 mRNA measured by qPCR and compared to LXSN 0 hours. Protein levels of NFX1-123 were measured by Western blot using GAPDH as a loading control. (B) 1.5×106 LXSN and FN123 W12E cells were suspended in methylcellulose (MC) for 24 hours and total mRNA and protein collected. mRNA levels of Keratin 1, Involucrin, and Loricrin were measured by qPCR and compared to W12E/LXSN 0 hours. Statistical significance was calculated using unpaired t-tests. Protein levels of Keratin 1, Involucrin, and NFX1-123 were assessed by Western blot. GAPDH was used as a loading control. Protein levels were normalized to the loading control, and all samples compared to undifferentiated LXSN. (C) W12E cells were transduced with a short hairpin targeting NFX1-123 (sh1 or sh2) or a scramble short hairpin control (scr), and 1.5×106 cells differentiated by suspension in methylcellulose for 0 or 24 hours. mRNA expression of Keratin 1, Involucrin, and Loricrin was measured by qPCR and compared to scr 0 hours. Statistical significance was calculated using one-way ANOVA with Bonferroni correction and p values for the difference in means between scr and sh1 or sh2 are shown. Protein levels of Keratin 1, Involucrin, and NFX1-123 were measured by Western blot using GAPDH as a loading control. (D and E) Protein levels of P-JNK, total JNK, total ERK, total MKK4, and total MKK7 were assessed by Western blot in LXSN and FN123 W12E cells (D) or scr, sh1, and sh2 W12E cells (E). (F) mRNA expression levels of HPV16 L1 were measured by qPCR and compared to LXSN or scr W12E cells. Protein levels of L1 were assessed by Western blot in LXSN and FN123 W12E cells. (G) . For all qPCRs, expression levels of the genes of interest were normalized to the housekeeping gene GAPDH, and all error bars represent 95% confidence intervals from replicates. For all Western blots, protein levels were normalized to the loading control GAPDH, and all samples compared to the undifferentiated control (LXSN in 5A, B, D, F; or scr in 5C and E). Densitometry analysis was done in ImageJ. All experiments in Figure 5 were performed three independent times. * p≤0.05, ** p≤0.01, *** p≤0.001, **** p≤0.0001
We next examined whether NFX1-123 also regulated expression of differentiation markers in W12E cells. Overexpression of NFX1-123 in W12E cells recapitulated the results seen in 16E6 HFKs, with greater levels of NFX1-123 resulting in increased induction or differentiation markers at the mRNA and protein level (Fig 5B). As expected, knock down of NFX1-123 resulted in decreased induction of differentiation markers in response to suspension (Fig 5C). Analysis of MAPK proteins revealed that modulating levels of NFX1-123 had no effect on total levels of ERK or JNK, but did alter levels of P-JNK, MKK4, and MKK7 (Fig 5D). P-JNK, MKK4, and MKK7 levels were increased when NFX1-123 expression was increased (Fig 5D, FN123 versus LXSN) and were decreased with knock down of NFX1-123 (Fig 5E, sh1 and sh2 versus scr). In total, these results support what was observed in HFKs: NFX1-123 was required for full induction of epithelial differentiation marker induction, augmented phosphorylation of JNK, and increased expression of its upstream kinases.
To explore whether this regulation of cellular differentiation also impacted the transcription of the HPV late genes that are triggered during differentiation, we quantified the expression of HPV16 L1; L1 encodes the HPV major capsid protein and is a common reading frame of all mRNA products expressed from the viral late promoter. As expected, both W12E LXSN and W12E FN123 cells upregulated L1 mRNA following differentiation (Figs 5F and 5G Undiff versus MC). However, W12E FN123 cells overexpressing NFX1-123 had an even greater induction of L1 mRNA compared to W12E LXSN cells (Fig 5F). This upregulation in mRNA translated to increased protein production as well, with W12E FN123 cells containing more L1 protein than W12E LXSN (Figure 5F). Therefore, increased expression of NFX1-123 affected both cellular differentiation and subsequent differentiation-dependent viral transcription.
To determine if NFX1-123 was required for full induction of L1 during differentiation, W12E were transduced with short hairpin RNAs against NFX1-123 (sh1 or sh2) or a scrambled control (scr) and then differentiated with methylcellulose suspension. W12E scr cells, with endogenous expression levels of NFX1-123, had a robust upregulation of L1 upon differentiation, while W12E cells with NFX1-123 knocked down had a lowered induction of L1 (Fig 5G). This demonstrated that the ability of NFX1-123 to augment epithelial cell differentiation pathways in HPV 16-positive cells, and its requirement for full activation of cellular differentiation, subsequently influenced the differentiation-dependent late gene transcription of HPV16.
DISCUSSION
This study confirms that the cellular protein NFX1-123 is a regulator of epithelial cell differentiation, identifies an intracellular signaling cascade affected by NFX1-123 expression, and subsequently provides insight into the linkage between epithelial differentiation and the HPV life cycle. The data presented here extend our previous findings, which were conducted in keratinocytes grown in non-differentiating media. We now demonstrate that NFX1-123 is able to regulate induction of epithelial differentiation markers in cells actively undergoing differentiation. Previously, we had shown that NFX1-123 was able to increase expression of differentiation markers when Notch1 was stimulated, but also determined that NFX1-123 employed Notch1-independent mechanisms (40). Here, we use targeted differentiation stimuli and modulation of NFX1-123 expression to explore the Notch1-independent mechanisms by which NFX1-123 regulates epithelial differentiation markers. Confirming earlier studies, we show that overexpressed NFX1-123 results in increased expression of differentiation markers when cells are stimulated to differentiate in the context of Notch1 signaling. This is seen both in raft cultures and in high confluency plating with calcium exposure (Fig 2). When Notch1 signaling is limited, however, NFX1-123 has a more selective role. We show that NFX1-123 mediates differentiation downstream of methylcellulose suspension-induced differentiation, but not in low confluency cells treated with calcium (Fig 2). This indicates that NFX1-123 does not have a global effect on differentiation marker expression but rather affects a particular intracellular signaling cascade.
Previous studies in our laboratory have focused on the post-transcriptional regulatory role NFX1-123 has on gene expression, both with and without 16E6. For the case of telomerase, which is activated by 16E6, NFX1-123 post-transcriptionally stabilized the mRNA of hTERT, the catalytic subunit of telomerase (46, 60, 61). For Notch1, NFX1-123 increased its mRNA and protein expression, and like hTERT, it required its RNA binding and regulatory motifs to do so (37). However, in our studies of differentiation marker and pathway regulation, NFX1-123 likely does not regulate target protein expression in the same manner. If post-transcriptional stabilization of differentiation genes were the mechanism by which NFX1-123 increased their expression, we would expect NFX1-123 to affect levels of differentiation markers, irrespective of what had stimulated their expression. We instead identified that NFX1-123 specifically affected the JNK signaling pathway, with greater NFX1-123 expression robustly activating proteins within that cascade.
Further studies are required to elucidate the role of 16E6 in partnering with NFX1-123 in the context of epithelial differentiation. NFX1-123 appears to play a role in mediating keratinocyte differentiation by itself, and data suggest that 16E6 is able to enhance this while protecting against cellular growth arrest (40). This mirrors what we have previously published on the regulation of Notch1 expression; NFX1-123 itself regulated the expression of Notch1, and this role was augmented by 16E6 (37). In contrast, the control of hTERT transcription in keratinocytes required 16E6 expression, and NFX1-123 functioned to augment this activation (60). These studies collectively demonstrate two mechanisms by which 16E6 functions in keratinocytes. 16E6 augments the typical regulation of pathways and genes by NFX1-123, and 16E6 also co-opts NFX1-123 to control genes and pathways it normally would not. This duality in the partnership between 16E6 and NFX1-123 has been seen with HR HPV E6 and other host protein partners, such as E6AP. E6 binds to and co-opts the functional role of E6AP in cells, targeting E6AP to degrade proteins and activate genes (62, 63). These targets are both novel genes and pathways, as well as ones typically controlled by E6AP (64). More work is required to elucidate the exact contributions of E6 and its partnership with NFX1-123 in the context of differentiation. Although we know that 16E6 and NFX1-123 collaborate to modify cellular signaling pathways, and that the HPV late promoter is dependent on and responsive to cellular signaling, we do not yet know the exact molecular mechanism by which E6 is acting.
Ultimately, the data presented in this study adds to previous work demonstrating that NFX1-123, as an endogenous host protein partner, is fundamental to the cellular dysregulation driven by HPV 16E6. Without adequate expression of NFX1-123, the ability of HR HPV to activate, augment, or accelerate key pathways, such as epithelial differentiation, is muted. Our studies utilized L1 expression as one indicator of the late HPV life cycle events that are activated during differentiation. Furthermore, these studies utilized experimental systems of differentiation in order to discern the cellular networks that were activated and altered. Future experiments will further dissect differentiation-dependent events of the late viral life cycle, such as genome amplification and E1^E4 expression, in additional systems that promote the full virus life cycle such as organotypic raft cultures.
While keratinocytes differentiating in squamous cell epithelium would have multiple, overlapping, and concurrently active stimuli and pathways, we applied a single stimulus to cells to isolate unique intracellular signaling pathway cascades. MAPK signaling, specifically involving ERK and JNK, is central to triggering suspension-induced epithelial differentiation (50, 55). Analysis of ERK and JNK demonstrated that after differentiation, NFX1-123 had a significant effect on levels of phosphorylated JNK (Fig 4). Examining the kinases upstream of JNK, we identified that greater NFX1-123 resulted in increased expression of MKK4 and MKK7 (Fig 4). Signal amplification in MAPK cascades can be driven by increased abundance of a kinase within any step of the cascade (58). Cells with more NFX1-123 may have increased baseline levels of MKK4 and MKK7 that, when the cascade is activated, result in increased phosphorylation of JNK. In HPV-positive W12E cells, the regulation of differentiation by NFX1-123 was paralleled by expression of L1, a product controlled by the differentiation-dependent viral late promoter. Although more studies are needed to interrogate whether this regulation of JNK signaling is directly or indirectly controls L1 expression, elevation of P-JNK levels during HPV infection has been documented before (65). In that study, increased P-JNK drove differentiation-dependent genome amplification and transcription of HPV16.
Our study joins others that document HPV’s co-opting and usage of MAPK signaling. MAPK signaling is a central axis around which HPV achieves many of the events required for its life cycle (66): E1 phosphorylation by ERK and JNK promotes its nuclear accumulation, which is critical for viral DNA replication (67); activated p38 and JNK enhances E6 targeting of PDZ domain-containing targets (68, 69); and sustained JNK signaling engendered by E7 prolongs the G2 phase of cells to drive viral DNA replication (70, 71). Alteration of MAPK signaling in the context of HPV infection has been shown previously, but this is the first time that involvement of NFX1-123 has been identified.
Another novel finding was that NFX1-123 expression was itself induced during differentiation, with the highest expression in the upper, more differentiated layers of the epithelium. This is of interest considering that even within differentiating cells, HPV genes expression is restricted to certain layers of stratified squamous epithelium. What controls this tight linkage between HPV gene expression and progressive epithelial differentiation is ill-defined. NFX1-123 may be a candidate for regulating this component of the viral life cycle. Intriguingly, we also observed basal cells with intense perinuclear staining for NFX1-123 in raft cultures and normal cervical samples. In upper layers, NFX1-123 became more predominately cytoplasmic. We see in multiple contexts that greater expression of NFX1-123 can significantly affect cellular processes and engender a milieu beneficial to HR HPV infection. These cellular processes include ones are typically thought of as opposing, such as cellular proliferation and differentiation (37, 40, 42, 46, 60). The subcellular localization of NFX1-123 in different epithelial layers may have a correlation with the cellular processes being affected. We have also observed in this study and others that there is a dose-response relationship between NFX1-123 levels and alterations in these cellular processes, especially in the context of 16E6. HPV may prefer to infect these basal cells that have greater than typical levels of NFX1-123. Additionally, endogenous expression of NFX1-123 may vary from person to person across normal cervical samples. It is therefore possible that upon infection with HR HPV, a person who naturally has higher endogenous levels of NFX1-123 will display more acute changes in infected cells and potentially better harbor HPV infection.
The study presented here extends previous work in which greater NFX1-123 indirectly induced differentiation marker expression through increased Notch1 expression. Now, we show that NFX1-123 itself is increase during differentiation and that it is required for and can augment differentiation marker expression in HFKs and HPV16-positive cells. These data also identify NFX1-123 as a cellular protein partner through which HPV increases MAPK signaling and engenders a cellular milieu favorable to the viral life cycle. This work adds to our understanding of the cellular pathways altered by HPV to promote infection and lends insight into the control of the differentiation-dependent HPV life cycle.
MATERIALS AND METHODS
Cell culture and generation of organotypic raft cultures
Primary human foreskin keratinocytes (HFKs) were cultured as described previously (41). Briefly, HFKs were derived from neonatal foreskins and grown in EpiLife medium supplemented with calcium chloride (60 μM), human keratinocyte growth supplement (Life Technologies, Carlsbad, CA), and penicillin-streptomycin. 293T cells were grown in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific, Grand Island, NY) containing 10% fetal bovine serum and penicillin-streptomycin. W12 clone 20863 cells (W12E), cervical epithelial cells containing full copies of the HPV16 genome, were a gift from the laboratory of Paul Lambert. W12E cells were maintained in E-media supplemented with 5% fetal bovine serum at sub-confluency as published (59). Organotypic HFK raft cultures were grown in E-media containing 5% fetal bovine serum, as published with 6-well transwells (Corning, Corning, NY) in place of wire mesh, also as published (72, 73).
Plasmids
The FLAG-tagged NFX1-123WT (FN123), pBabe-puro 16E6 (16E6), LXSN vector control (LXSN), and c-FUGW plasmids have been described previously (60). Scrambled short hairpin control (scr) and short hairpin 1 RNA (sh1) have also been described previously (60). Short hairpin 2 RNA to NFX1-123 (sh2) was designed specifically to the novel C terminus of NFX1-123. BLAST (Basic Local Alignment Search Tool) was used to confirm no off-target effects of shRNA and scramble sequences. Annealed oligonucleotides were ligated into the C-FUGW vector using BamHI and EcoRI restriction sites.
Retrovirus production and infection
Retrovirus was produced in 293T cells by a transient vesicular stomatitis virus G-pseudotyped virus (VSV-G) production protocol as previously described (74). Lentivirus was also produced as previously described (60). Briefly, short hairpin NFX1-123 (sh1 or sh2) c-FUGW or scramble c-FUGW (scr) constructs were co-transfected with cytomegalovirus-vesicular stomatitis virus G and Δ8.9 plasmids into 293T cells using FuGENE6 (Roche, Alameda, CA), and lentiviral retrovirus was serially collected. Retrovirus was concentrated by ultracentrifuge, mixed with Polybrene (8 μg/mL) (EMD Millipore, Billerica, MA), and incubated with 50 to 60% confluent HFKs or W12E cells. Virus was left on overnight and medium replaced the next day. For VSV-G pseudotyped virus infections (FN123, LXSN vector control, pBabe-puro 16E6) the cells were expanded 24 hours post-transduction, and after 48 hours the cells were placed under neomycin/G418 selection (50 μg/mL) or puromycin selection (0.5 μg/mL). All lentivirus infections (scramble, sh1 and sh2) were confirmed by green fluorescent protein expression.
Immunohistochemistry and histologic analysis
Organotypic HFK raft cultures were formalin fixed and embedded in paraffin (FFPE) following standard procedures. 32 normal cervical epithelium specimen FFPE blocks were obtained from the University of Washington HPV Research Group Specimen Repository. They were deidentified and considered to be not human subjects by the Seattle Children’s Research Institute IRB. Sections 4μm thick were stained for NFX1-123 using a rabbit polyclonal anti-NFX1-123 antibody, a gift from Dr. Ann Roman, a rabbit polyclonal anti-Loricrin antibody (BioLegend, San Diego, CA), a rabbit polyclonal anti-cytokeratin 1 (BioLegend, San Diego, CA), ora rabbit polyclonal IgG control at a 1:1000 dilution. Epitope retrieval was done using citrate for 10 minutes at 100°C. Slides were scanned in brightfield at 20× magnification using the Hamamatsu NanoZoomer Digital Pathology System.
For semiquantitative analysis of NFX1-123, Keratin 1, and Loricrin staining, ImageJ was used. Color deconvolution for hematoxylin and DAB was performed and the deconvolved image displaying only the DAB channel used for subsequent quantitation. For NFX1-123, plot profiles of intensity were obtained over a set linear distance from the basal layer upwards through the apical surface of the raft culture or tissue specimen. For NFX1-123, eight plots over four sections were averaged per sample and are shown as a function of distance from the basal layer. For Keratin 1 and Loricrin, total intensity of a section were obtained.
Differentiation of keratinocytes
Prior to differentiation, HFKs were trypsin harvested from monolayer cultures. For calcium differentiation, HFKs were grown in EpiLife medium without human keratinocyte growth supplements, with 1.8mM calcium chloride and penicillin-streptomycin added. After trypsin harvest, cells were counted using the Bio-Rad TC20 Automated Cell Counter (Bio-Rad, Flercules, CA); for low confluency calcium differentiation, 3×105 HFKs were plated on a 10cm tissue culture dish; for high confluency calcium differentiation, 1×106 HFKs were plated. Cells were tryspin harvested after three days’ of daily calcium treatment. For differentiation in semisolid medium, 1×106 HFKs were plated into 25mL of EpiLife containing 1.7% methylcellulose in a non-treated 10cm dish. Cells were harvested using washing in ice-cold PBS and centrifugation as previously described. Untreated controls for differentiation were cells plated into monolayer in normal EpiLife and collected at the same timepoint as differentiated samples.
Differentiation of W12E cells
W12E cells were differentiated in a similar manner to HFKs. W12E cells were trypsin harvested from subconfluent monolayer cultures. 1.5×106 W12E cells were plated into 25mL of semisolid medium, E-media containing 5% FBS and 1.7% methylcellulose in a non-treated 10cm dish. Cells were harvested using washing in ice-cold PBS and centrifugation as above. Untreated controls for differentiation were cells plated into monolayer in E-media containing 5% FBS.
Quantitative real-time PCR
RNA was isolated with TRIzol reagent (Thermo Fisher Scientific, Grand Island, NY) as previously described (60). Total RNA (1 μg) was DNase treated and cDNA generated using random hexamer primers and Superscript IV reverse transcriptase (Thermo Fisher Scientific, Grand Island, NY). Quantitative real-time PCR (qPCR) was performed using an ABI StepOne Plus system (Applied Biosystems, Foster City, CA). Primer sequences to amplify NFX1-123 and 36B4 were described previously (46, 60). Primers used to amplify HPV16 L1 are as follows: forward primer-5’ GGTGTTGAGGTAGGTCGTGG 3’ (nucleotides 5948-5967) and reverse primer – 5’ CACACCTGCATTTGCTGCAT 3’ (nucleotides 6042-6061) (75). Amplification was carried out in replicative triplicates using Power UP SYBR Green master mix (Thermo Fisher Scientific, Grand Island, NY). Error bars represent 95% confidence intervals. For differentiation markers, amplification was carried out using TaqMan master mix and the following pre-designed Taqman probes: GAPDH (4333764F), KRT1 (Hs00196158_m1), KRT10 (Hs00166289), IVL (Hs00846307_s1), LOR (HS01894962_s1), HES1 (Hs00172878_m1) as published previously (40) according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA). Analyses were done using the ΔΔCt method, with Pfaffl correction for PCR efficiency. GAPDH was used as a reference gene for normalization of cDNA amounts, and then each experimental sample was compared to the undifferentiated control.
Western blot
Whole cell lysates were prepared directly in 2× sample buffer (100 mM Tris pH 6.8, 4% SDS, 20% glycerol). Equal amounts of protein lysates were electrophoresed on a 4-12% gradient SDS-polyacrylamide gel (Thermo Fisher Scientific, Grand Island, NY) and transferred to Immobilon-FL membrane (Millipore Sigma, Burlington, MA). Blots were probed with antibodies: Cytokeratin 1 (PA5-26699) (Thermo Fisher Scientific, Grand Island, NY); p53 (OP03) at 1:1000 (Millipore Sigma, Burlington, MA); Involucrin (SY5) at 1:1000; total JNK(D-2) at 1:500, Actin (I-19) at 1:1000, (Santa Cruz Biotechnology, Santa Cruz, CA); P-JNK/SAPK (81E11) at 1:1000, total SEK1/MKK4 (9153) at 1:1000, total MKK7 (4127S) at 1:1000, (Cell Signaling Technology, Danvers, MA); GAPDH (6C5) at 1:100,000 (Abcam, Cambridge, MA); total ERK (W15133B) at 1:1000, (BioLegend, San Diego, CA). The rabbit polyclonal anti-NFX1-123 antibody was used 1:1000 and generously provided by Dr. Ann Roman.
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported by NIH R01 CA 172742 to R.A.K and NIH T32 AI083203 to J.L. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. 2012. The Biology and Life-Cycle of Human Papillomaviruses. Vaccine 30, Supplement 5:F55–F70. [DOI] [PubMed] [Google Scholar]
- 2.zur Hausen H 2002. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2:342–350. [DOI] [PubMed] [Google Scholar]
- 3.Bosch FX, Lorincz A, Munoz N, Meijer CJLM, Shah KV. 2002. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol 55:244–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muñoz N, Bosch FX, de Sanjosé S, Herrero R, Castellsagué X, Shah KV, Snijders PJF, Meijer CJLM, International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. 2003. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 348:518–527. [DOI] [PubMed] [Google Scholar]
- 5.zur Hausen H 2009. Papillomaviruses in the causation of human cancers — a brief historical account. Virology 384:260–265. [DOI] [PubMed] [Google Scholar]
- 6.Stratton KL, Culkin DJ. 2016. A Contemporary Review of HPV and Penile Cancer. Oncol Williston Park N 30. [PubMed] [Google Scholar]
- 7.Kreimer AR, Clifford GM, Boyle P, Franceschi S. 2005. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol 14:467–475. [DOI] [PubMed] [Google Scholar]
- 8.Chaturvedi AK. 2010. Beyond cervical cancer: burden of other HPV-related cancers among men and women. J Adolesc Health Off Publ Soc Adolesc Med 46:S20–26. [DOI] [PubMed] [Google Scholar]
- 9.Forman D, de Martel C, Lacey CJ, Soerjomataram I, Lortet-Tieulent J, Bruni L, Vignat J, Ferlay J, Bray F, Plummer M, Franceschi S. 2012. Global Burden of Human Papillomavirus and Related Diseases. Vaccine 30, Supplement 5:F12–F23. [DOI] [PubMed] [Google Scholar]
- 10.de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, Plummer M. 2012. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol 13:607–615. [DOI] [PubMed] [Google Scholar]
- 11.Doorbar J, Egawa N, Griffin H, Kranjec C, Murakami I. 2015. Human papillomavirus molecular biology and disease association. Rev Med Virol 25:2–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee C, Laimins LA. 2007. The Differentiation-Dependent Life Cycle of Human Papillomaviruses in Keratinocytes, p. 45–67. In Garcea RL, DiMaio D (eds.), The Papillomaviruses. Springer US, Boston, MA. [Google Scholar]
- 13.Doorbar J 2005. The papillomavirus life cycle. J Clin Virol Off Publ Pan Am Soc Clin Virol 32 Suppl 1 :S7–15. [DOI] [PubMed] [Google Scholar]
- 14.Roden RBS, Stern PL. 2018. Opportunities and challenges for human papillomavirus vaccination in cancer. Nat Rev Cancer 18:240–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham S 2010. Human papillomavirus: gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol 5:1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schiffman M, Doorbar J, Wentzensen N, Sanjose S de, Fakhry C, Monk BJ, Stanley MA, Franceschi S 2016. Carcinogenic human papillomavirus infection. Nat Rev Dis Primer 2:16086. [DOI] [PubMed] [Google Scholar]
- 17.Graham SV. 2017. Keratinocyte Differentiation-Dependent Fluman Papillomavirus Gene Regulation. Viruses 9:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuchs E 1990. Epidermal differentiation: the bare essentials. J Cell Biol 111:2807–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watt FM. 1983. Involucrin and other markers of keratinocyte terminal differentiation. J Invest Dermatol 81:100s–3s. [DOI] [PubMed] [Google Scholar]
- 20.O’Connor MJ, Stünkel W, Koh C-H, Zimmermann H, Bernard H-U. 2000. The Differentiation-Specific Factor CDP/Cut Represses Transcription and Replication of Human Papillomaviruses through a Conserved Silencing Element. J Virol 74:401–410. [PMC free article] [PubMed] [Google Scholar]
- 21.Ai W, Toussaint E, Roman A. 1999. CCAAT Displacement Protein Binds to and Negatively Regulates Human Papillomavirus Type 6 E6, E7, and E1 Promoters. J Virol 73:4220–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yukawa K, Butz K, Yasui T, Kikutani H, Hoppe-Seyler F. 1996. Regulation of human papillomavirus transcription by the differentiation-dependent epithelial factor Epoc-1/skn-1a. J Virol 70:10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan WK, Chong T, Bernard HU, Klock G. 1990. Transcription of the transforming genes of the oncogenic human papillomavirus-16 is stimulated by tumor promotors through AP1 binding sites. Nucleic Acids Res 18:763–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chong T, Apt D, Gloss B, Isa M, Bernard HU. 1991. The enhancer of human papillomavirus type 16: binding sites for the ubiquitous transcription factors oct-1, NFA, TEF-2, NF1, and AP-1 participate in epithelial cell-specific transcription. J Virol 65:5933–5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong X-P, Stubenrauch F, Beyer-Finkler E, Pfister H. 1994. Prevalence of deletions of YY1-binding sites in episomal HPV 16 DNA from cervical cancers. Int J Cancer 58:803–808. [DOI] [PubMed] [Google Scholar]
- 26.Hadaschik D, Hinterkeuser K, Oldak M, Pfister HJ, Smola-Hess S. 2003. The Papillomavirus E2 Protein Binds to and Synergizes with C/EBP Factors Involved in Keratinocyte Differentiation. J Virol 77:5253–5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frattini MG, Lim HB, Doorbar J, Laimins LA. 1997. Induction of human papillomavirus type 18 late gene expression and genomic amplification in organotypic cultures from transfected DNA templates. J Virol 71:7068–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gunasekharan VK, Li Y, Andrade J, Laimins LA. 2016. Post-Transcriptional Regulation of KLF4 by High-Risk Human Papillomaviruses Is Necessary for the Differentiation-Dependent Viral Life Cycle. PLOS Pathog 12:e1005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruesch MN, Stubenrauch F, Laimins LA. 1998. Activation of Papillomavirus Late Gene Transcription and Genome Amplification upon Differentiation in Semisolid Medium Is Coincident with Expression of Involucrin and Transglutaminase but Not Keratin-10. J Virol 72:5016–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Apt D, Watts RM, Suske G, Bernard H-U. 1996. High Sp1/Sp3 Ratios in Epithelial Cells during Epithelial Differentiation and Cellular Transformation Correlate with the Activation of the HPV-16 Promoter. Virology 224:281–291. [DOI] [PubMed] [Google Scholar]
- 31.Carson A, Khan SA. 2006. Characterization of Transcription Factor Binding to Human Papillomavirus Type 16 DNA during Cellular Differentiation. J Virol 80:4356–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bodily JM, Alam S, Meyers C. 2006. Regulation of human papillomavirus type 31 late promoter activation and genome amplification by protein kinase C. Virology 348:328–340. [DOI] [PubMed] [Google Scholar]
- 33.Sen E, Bromberg-White JL, Meyers C. 2002. Genetic Analysis of cis Regulatory Elements within the 5’ Region of the Human Papillomavirus Type 31 Upstream Regulatory Region during Different Stages of the Viral Life Cycle. J Virol 76:4798–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sen E, Alam S, Meyers C. 2004. Genetic and Biochemical Analysis of cis Regulatory Elements within the Keratinocyte Enhancer Region of the Human Papillomavirus Type 31 Upstream Regulatory Region during Different Stages of the Viral Life Cycle. J Virol 78:612–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jia R, Liu X, Tao M, Kruhlak M, Guo M, Meyers C, Baker CC, Zheng Z-M. 2009. Control of the Papillomavirus Early-to-Late Switch by Differentially Expressed SRp20. J Virol 83:167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Songock WK, Scott ML, Bodily JM. 2017. Regulation of the human papillomavirus type 16 late promoter by transcriptional elongation. Virology 507:179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vliet-Gregg PA, Hamilton JR, Katzenellenbogen RA. 2013. NFX1-123 and Human Papillomavirus 16E6 Increase Notch Expression in Keratinocytes. J Virol 87:13741–13750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lefort K, Dotto GP. 2004. Notch signaling in the integrated control of keratinocyte growth/differentiation and tumor suppression. Semin Cancer Biol 14:374–386. [DOI] [PubMed] [Google Scholar]
- 39.Rangarajan A, Talora C, Okuyama R, Nicolas M, Mammucari C, Oh H, Aster JC, Krishna S, Metzger D, Chambon P, Miele L, Aguet M, Radtke F, Dotto GP. 2001. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J 20:3427–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vliet-Gregg PA, Hamilton JR, Katzenellenbogen RA. 2015. Human papillomavirus 16E6 and NFX1-123 potentiate notch signaling and differentiation without activating cellular arrest. Virology 478:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gewin L, Myers H, Kiyono T, Galloway DA. 2004. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev 18:2269–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vliet-Gregg PA, Robinson KL, Levan J, Matsumoto LR, Katzenellenbogen RA. 2019. NFX1-123 is highly expressed in cervical cancer and increases growth and telomerase activity in HPV 16E6 expressing cells. Cancer Lett 449:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Green H 1977. Terminal differentiation of cultured human epidermal cells. Cell 11:405–416. [DOI] [PubMed] [Google Scholar]
- 44.Watt FM, Jordan PW, O’Neill CH. 1988. Cell shape controls terminal differentiation of human epidermal keratinocytes. Proc Natl Acad Sci 85:5576–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rice R, Green H. 1978. Relation of protein synthesis and transglutaminase activity to formation of the cross-linked envelope during terminal differentiation of the cultured human epidermal keratinocyte. J Cell Biol 76:705–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katzenellenbogen RA, Vliet-Gregg P, Xu M, Galloway DA. 2009. NFX1-123 Increases hTERT Expression and Telomerase Activity Posttranscriptionally in Human Papillomavirus Type 16 E6 Keratinocytes. J Virol 83:6446–6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kopan R, llagan MXG. 2009. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 137:216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dotto GP. 2008. Notch tumor suppressor function. Oncogene 27:5115–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai EC. 2004. Notch signaling: control of cell communication and cell fate. Development 131:965–973. [DOI] [PubMed] [Google Scholar]
- 50.Connelly JT, Gautrot JE, Trappmann B, Tan DW-M, Donati G, Huck WTS, Watt FM. 2010. Actin and serum response factor transduce physical cues from the microenvironment to regulate epidermal stem cell fate decisions. Nat Cell Biol 12:711–718. [DOI] [PubMed] [Google Scholar]
- 51.Bikle DD, Xie Z, Tu C-L. 2012. Calcium regulation of keratinocyte differentiation. Expert Rev Endocrinol Metab 7:461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Charest JL, Jennings JM, King WP, Kowalczyk AP, Garcia AJ. 2009. Cadherin-Mediated CellȓCell Contact Regulates Keratinocyte Differentiation. J Invest Dermatol 129:564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pece S, Gutkind JS. 2000. Signaling from E-cadherins to the MAPK Pathway by the Recruitment and Activation of Epidermal Growth Factor Receptors upon Cell-Cell Contact Formation. J Biol Chem 275:41227–41233. [DOI] [PubMed] [Google Scholar]
- 54.MCiller EJ, Williamson L, Kolly C, Suter MM. 2008. Outside-in Signaling through Integrins and Cadherins: A Central Mechanism to Control Epidermal Growth and Differentiation? J Invest Dermatol 128:501–516. [DOI] [PubMed] [Google Scholar]
- 55.Watt FM. 2016. Engineered Microenvironments to Direct Epidermal Stem Cell Behavior at Single-Cell Resolution. Dev Cell 38:601–609. [DOI] [PubMed] [Google Scholar]
- 56.Trappmann B, Gautrot JE, Connelly JT, Strange DGT, Li Y, Oyen ML, Cohen Stuart MA, Boehm H, Li B, Vogel V, Spatz JP, Watt FM, Huck WTS. 2012. Extracellular-matrix tethering regulates stem-cell fate. Nat Mater 11:642–649. [DOI] [PubMed] [Google Scholar]
- 57.Johnson GL, Nakamura K. 2007. The c-Jun Kinase/Stress-activated Pathway: Regulation, Function and Role in Human Disease. Biochim Biophys Acta 1773:1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pearson G, Robinson F, Beers Gibson T, Xu B, Karandikar M, Berman K, Cobb MH. 2001. Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions. Endocr Rev 22:153–183. [DOI] [PubMed] [Google Scholar]
- 59.Doorbar J, Parton A, Hartley K, Banks L, Crook T, Stanley M, Crawford L. 1990. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology 178:254–262. [DOI] [PubMed] [Google Scholar]
- 60.Katzenellenbogen RA, Egelkrout EM, Vliet-Gregg P, Gewin LC, Gafken PR, Galloway DA. 2007. NFX1-123 and Poly(A) Binding Proteins Synergistically Augment Activation of Telomerase in Human Papillomavirus Type 16 E6-Expressing Cells. J Virol 81:3786–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katzenellenbogen RA, Vliet-Gregg P, Xu M, Galloway DA. 2010. Cytoplasmic Poly(A) Binding Proteins Regulate Telomerase Activity and Cell Growth in Human Papillomavirus Type 16 E6-Expressing Keratinocytes. J Virol 84:12934–12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–1136. [DOI] [PubMed] [Google Scholar]
- 63.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75:495–505. [DOI] [PubMed] [Google Scholar]
- 64.Beaudenon S, Huibregtse JM. 2008. HPV E6, E6AP and cervical cancer. BMC Biochem 9 Suppl 1:S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Egawa N, Wang Q, Griffin HM, Murakami I, Jackson D, Mahmood R, Doorbar J. 2017. HPV16 and 18 genome amplification show different E4-dependence, with 16E4 enhancing E1 nuclear accumulation and replicative efficiency via its cell cycle arrest and kinase activation functions. PLOS Pathog 13:e1006282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Satsuka A, Mehta K, Laimins L. 2015. p38MAPK and MK2 Pathways Are Important for the Differentiation-Dependent Human Papillomavirus Life Cycle. J Virol 89:1919–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bergvall M, Melendy T, Archambault J. 2013. The E1 proteins. Virology 445:35–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pim D, Banks L. 2010. Interaction of viral oncoproteins with cellular target molecules: infection with high-risk vs low-risk human papillomaviruses. APMIS 118:471–493. [DOI] [PubMed] [Google Scholar]
- 69.Massimi P, Narayan N, Cuenda A, Banks L. 2006. Phosphorylation of the discs large tumour suppressor protein controls its membrane localisation and enhances its susceptibility to HPV E6-induced degradation. Oncogene 25:4276–4285. [DOI] [PubMed] [Google Scholar]
- 70.Banerjee NS, Wang H-K, Broker TR, Chow LT. 2011. Human Papillomavirus (HPV) E7 Induces Prolonged G2 following S Phase Reentry in Differentiated Human Keratinocytes. J Biol Chem 286:15473–15482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davy CE, Jackson DJ, Raj K, Peh WL, Southern SA, Das P, Sorathia R, Laskey P, Middleton K, Nakahara T, Wang Q, Masterson PJ, Lambert PF, Cuthill S, Millar JBA, Doorbar J. 2005. Human Papillomavirus Type 16 E1AE4-Induced G2 Arrest Is Associated with Cytoplasmic Retention of Active Cdk1/Cyclin B1 Complexes. J Virol 79:3998–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shamir ER, Ewald AJ. 2014. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nat Rev Mol Cell Biol 15:647–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anacker D, Moody C. 2012. Generation of organotypic raft cultures from primary human keratinocytes. J Vis Exp JoVE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bartz SR, Vodicka MA. 1997. Production of High-Titer Human Immunodeficiency Virus Type 1 Pseudotyped with Vesicular Stomatitis Virus Glycoprotein. Methods 12:337–342. [DOI] [PubMed] [Google Scholar]
- 75.Bienkowska-Haba M, Luszczek W, Myers JE, Keiffer TR, DiGiuseppe S, Polk P, Bodily JM, Scott RS, Sapp M. 2018. A new cell culture model to genetically dissect the complete human papillomavirus life cycle. PLOS Pathog 14:e1006846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.